clinical trials
TRANSCRIPT

Dr. Sirisha1st year PG
CLINICAL TRIALS

DEFINITION• It is a prospective ethically designed investigation in human
subjects to discover/verify/compare the results of two or more
therapeutic measures /drugs.
•WHO definition: Prospectively assigned human participants to
one or more health related interventions to evaluate the effects
on health outcome.

DEFINITIONS
RANDOMIZATION:
Each study subject is randomly assigned to receive
either the study treatment or a control.
PLACEBO:
Pharmacologically inert substance given to please the
subjects

BLIND:
If the study is single blind, the subjects involved in the
study do not know which study treatment they receive.
If the study is double- blind, the researchers also do not
know which treatment is being given to any given subject.
This 'blinding' helps in preventing biases.

TYPES OF CLINICAL TRIALS
Treatment trials
Prevention trials
Diagnostic trials
Screening trials
Quality of life trials

DRUG DISCOVERY SOURCES
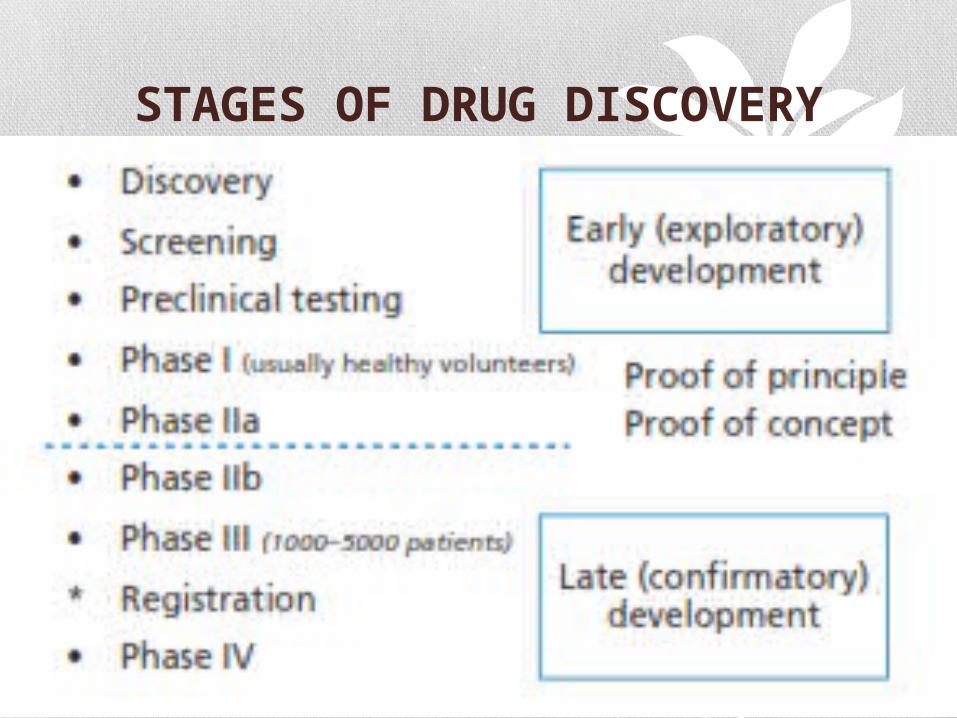
STAGES OF DRUG DISCOVERY

PRE-CLINICAL TRIALS
The testing on animals to expose the pharmacological profile
of a prospective drug is called pre-clinical trials.
The major kinds of information expected from preclinical
toxicity studies are
(1) acute toxicity—effects of large single doses; (LD50)
(2) Sub acute and chronic toxicity—effects of multiple doses,
which are especially important if the drug is intended for
prolonged use in humans

PRE-CLINICAL TRIALS
(3) effects on reproductive functions, including teratogenicity and
postnatal development;
(4) carcinogenicity;
(5) mutagenicity.
(6) Pharmaco dynamics
(7) Pharmaco kinetics
(8) Assessment of safety index

PRE-CLINICAL TRIALS
Tests:
Screening tests
Tests on isolated organs
Tests on bacterial culture
Tests on animal models of human diseases


Importance of pre clinical trials• Triparanol is a drug that lowers the concentration of cholesterol in
plasma.
• It was marketed in the USA in 1959. In 1962, the Food and Drug
Administration (FDA) received a tip-off and undertook an
unannounced inspection. This revealed that toxicology data
demonstrating cataract formation in rats and dogs had been
falsified.
• Triparanol was withdrawn, but some of the patients who had been
taking it for a year or longer also developed cataracts


ETHICSThe U.S. National Institutes of Health notes seven ethical
requirements that must be met before a clinical trial can begin.
These include
• social value,
• scientific validity,
• fair and objective selection of subjects,
• informed consent,

ETHICS
• favourable ratio of risks to benefits,
• approval and oversight by an independent/ institutional
review board (IRB),
• respect for human subjects.

LEGAL ISSUES
• Informed consent
• Institutional review board
•Medical insurance
• confidentiality

INFORMED CONSENT FORM
• Voluntary
• Explained in simple nontechnical, native language
• Comprehensive information regarding the trials like
• Benefit of new therapy over existing ones
• Alternative treatments available
• All possible adverse reactions
• Freedom to withdraw from the trial

Clinical trials must have
a) Good laboratory practice
b) Good clinical practice
c) Good manufacturing practice
d) Good documentation practice
e) Good regulatory practice

IND(Investigational New Drug)
The IND includes
(1) information on the composition and source of the drug
(2) manufacturing information,
(3) all data from animal studies,
(4) clinical plans and protocols,
(5) the names and credentials of physicians who will conduct
the clinical trials.

DRUG REGULATORY AGENCY
A clinical trial can begin only when drug regulatory authority
approves for the conduct
Ex: Food and drug administration(FDA)
Central Standard Drug Control Organization (CDSCO)
Drug Controller General of India(DCGI)


PHASE 0 / EXPLORATORY IND STUDIES• MICRODOSING:
Extremely low, nonpharmacologically active doses of a
drug are used to define the agent’s pharmacokinetic profile in
humans.
•DEFINITION:
Microdosing means use of ‘less than 1/100 of the dose
calculated to yield a pharmacological effect of the test substance
to a maximum dose of <100 micrograms and a maximum micro
dose of <30 nanomoles for protein products.

• FEATURES:
limited number of subjects(10-15)
limited dosing duration(<=7 days)
evaluates pharmacodynamic and pharmacokinetic properties

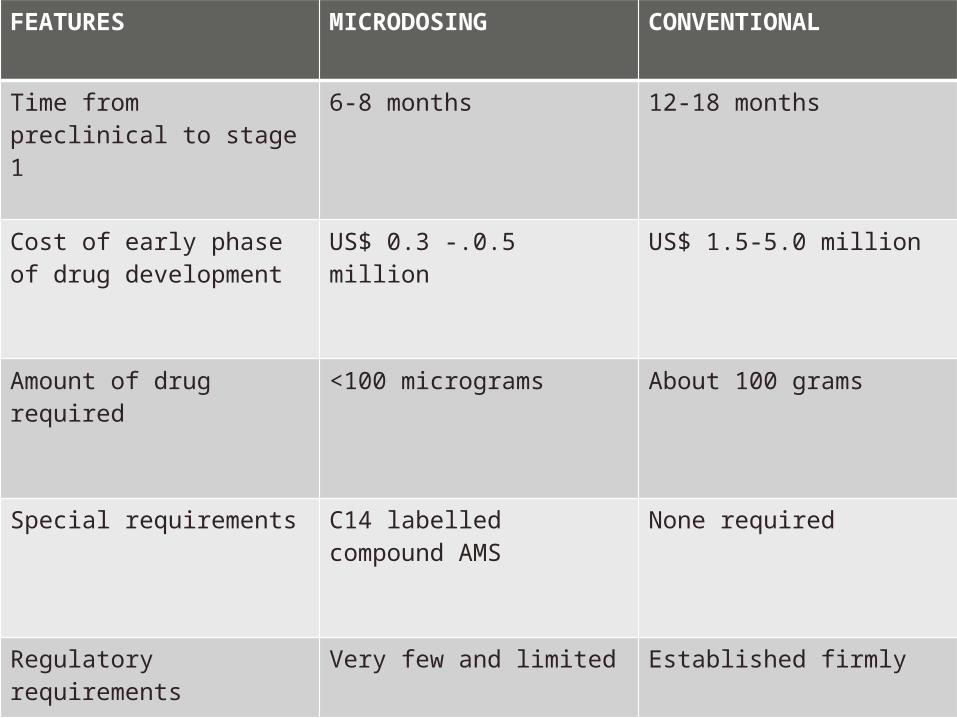
FEATURES MICRODOSING CONVENTIONAL
Time from preclinical to stage 1
6-8 months 12-18 months
Cost of early phase of drug development
US$ 0.3 -.0.5 million US$ 1.5-5.0 million
Amount of drug required <100 micrograms About 100 grams
Special requirements C14 labelled compound AMS
None required
Regulatory requirements Very few and limited Established firmly

Advantages:
• saves investment and resource utilization.
• less time.
• efficiency and success of successive trials can be improved.
•No adverse effects since the dose used has no pharmacological
action
• As per the regulatory requirement, animal studies, at least in one
species, are required to establish micro dose in humans, but at a
much reduced level.

Disadvantages:
• Drugs with non linear kinetics cannot be assessed Ex:warfarin
• False negative results can mislead (compound being rejected)
• No therapeutic or diagnostic conclusions can be made
• false positive results (compound acceptable based on micro
dose data but rejected subsequently when used in
pharmacological doses).
• Compound metabolism and solubility of compound.

Phase 1. Human pharmacology 20-50 subjects, open label, for 1 year
Healthy volunteers or volunteer patients, according to the class
of drug and its safety.
Objectives:
Pharmacokinetics (absorption, distribution, metabolism,
excretion).
Pharmacodynamics (biological effects)
tolerability, safety, efficacy.

Phase 2. Therapeutic exploration 50-300 subjects, open label, 2-3years
Patients.
Trials with inclusion and exclusion criteria
Primary and surrogate end points
Objectives:
Pharmacokinetics and pharmacodynamic dose-ranging
Therapeutic efficacy

• Phase 2a/ Early Phase
• 200 patients
• Single blind
• Potential therapeutic benefits
and side effects
• Phase 2b/ Late Phase
• 400 patients
•Double blind
• Potential therapeutic benefits
and side effects

Phase 3. Therapeutic confirmation
Randomised, double blind, multi-centric controlled trials; 250-
1000+
Cross over design
Patients
Objectives:
Efficacy on a substantial scale; safety;
comparision with existing drugs.
Drug interactions
Guidelines for use of drug

NEW DRUG APPLICATION• If phase 3 results meet expectations, application is made for
permission to market the new agent.
• Marketing approval requires submission of a New Drug
Application (NDA) to the DRA.
• The application contains, often in hundreds of volumes, full
reports of all preclinical and clinical data pertaining to the drug
under review, package inserts.
•median standard approval time was 12.9 months.

Phase 4. Therapeutic use
2000-10000+, post-licensing studies
Periodic Safety Update Report(PSUR)
Objectives:
Surveillance for safety and efficacy
uncommon and long term adverse effects
marketing studies
pharmacoeconomic studies.
additional indications


Phase 5. Therapeutic effectiveness
• The focus of Phase V studies, or "effectiveness" research, is
on determining whether "the therapeutic effect is realized in
day-to-day clinical practice".
• Generally, Phase V research is considered "field research" or
"community-based research" and is designed to test
generalization of the intervention to a larger sample and under
typical and somewhat variable clinical contexts.


PHASE 5
•A central question for Phase V research is whether the effects are
similar to those found in efficacy studies and to determine who
benefits from the treatment.
• It is during this phase of research that questions concerning the
cost-benefit ratio of the intervention can be addressed.

CRITICAL PATH INITIATIVE
• In 2004, a white paper, now known as the Critical Path Initiative
(CPI), is given by FDA that called attention to an alarming
decline in the number of innovative medical products being
submitted for FDA approval.
• This led to the new concept of adaptive phase clinical trials

ADAPTIVE PHASE CLINICAL TRIALS
• Study that includes a prospectively planned opportunity for
modification of one or more specified aspects of the study design
and hypotheses based on analysis of data (usually interim data)
from subjects in the study.
• The purpose is to make clinical trials more flexible, efficient
and fast. Due to the level of flexibility involved, these trial
designs are also termed as “flexible designs.”

NON INFERIORITY CLINICAL TRIALS
•Non-inferiority clinical trials aim to demonstrate that the test
product is no worse than the comparator by more than a
prespecified small amount. This amount is known as the non-
inferiority margin (M), or delta (Δ).

Thank ‘u’