cnlac1 is required for extrapulmonary dissemination of ...division of pulmonary and critical care...
TRANSCRIPT

INFECTION AND IMMUNITY, Mar. 2004, p. 1693–1699 Vol. 72, No. 30019-9567/04/$08.00�0 DOI: 10.1128/IAI.72.3.1693–1699.2004Copyright © 2004, American Society for Microbiology. All Rights Reserved.
CNLAC1 Is Required for Extrapulmonary Dissemination ofCryptococcus neoformans but Not Pulmonary Persistence
Mairi C. Noverr,1,2 Peter R. Williamson,3 Ryan S. Fajardo,1 and Gary B. Huffnagle1,2*Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine,1 and Department ofMicrobiology and Immunology,2 University of Michigan Medical School, Ann Arbor, Michigan 48109, and
Division of Infectious Diseases, University of Illinois at Chicago College of Medicine,Chicago, Illinois 606123
Received 22 July 2003/Returned for modification 26 August 2003/Accepted 19 November 2003
The pathogenic yeast Cryptococcus neoformans produces a laccase enzyme (CNLAC1), which catalyzes thesynthesis of melanin in the presence of phenolic compounds. A number of genes have been implicated in theregulation of laccase and melanization, including IPC1, GPA1, MET3, and STE12. Albino mutants derived fromrandom mutagenesis techniques may contain mutations in genes that regulate multiple virulence factors,including CNLAC1. The goal of our study is to investigate the role of CNLAC1 in virulence and evasion ofpulmonary host defenses after infection via the respiratory tract. Using a set of congenic laccase-positive(2E-TUC-4) and laccase-deficient (2E-TU-4) strains, we found that both strains are avirulent at a lower dose(104 CFU/mouse) in mice. After the infectious dose was increased to 106 CFU/mouse, 70% mortality wasobserved in mice infected with 2E-TUC-4 compared to no mortality in mice infected with 2E-TU-4 at day 30postinfection. This observation confirms the requirement for CNLAC1 in virulence. Interestingly, we observedno differences between the two strains in pulmonary growth or in elicitation of cellular immune responses inthe lung. The only measurable defect of 2E-TU-4 was in dissemination to extrapulmonary sites. To examine therole of CNLAC1 in dissemination, mice were infected intravenously. By week 3 postinfection, equal numbers ofstrains 2E-TUC-4 and 2E-TU-4 were recovered from the brain and spleen. This observation indicates thatCNLAC1 facilitates escape from the lung, but not growth in the lungs or brain, and suggests a novel role forCNLAC1 in virulence during an infection aquired via the respiratory tract.
Cryptococcus neoformans is an opportunistic pathogenicyeast acquired via the respiratory tract. Clearance of a pulmo-nary infection requires the development of adaptive immunity.In immunocompromised hosts, C. neoformans can disseminateto extra-pulmonary sites, particularly the central nervous sys-tem (CNS), where infection can lead to fatal meningitis. C.neoformans produces a number of factors that are required forvirulence, including growth at 37°C (13), the presence of poly-saccharide capsule (5), urease (9), phospholipase B (7), andlaccase (31, 33, 39).
C. neoformans produces a phenoloxidase, or laccase (en-coded by the CNLAC1 gene), that catalyzes melanin produc-tion from an exogenous diphenolic or indolic substrate (cate-cholamine, epinephrine, L-dopa, dopamine, and caffeic acid)(26). The resulting heterogeneous pigment is covalently linkedto the cell wall (42). In addition to melanin, a variety of po-tentially toxic by-products are produced by laccase. Both mel-anin and laccase by-products have been detected in vivo (19,23). Therefore, multiple products of the laccase pathway likelyplay a role in virulence.
In vitro studies have identified a number of possible molec-ular mechanisms for the role of laccase during pathogenesis.Melanin provides increased resistance to antifungal drugs (37),antibody-mediated phagocytosis (36), and defensins (10), and
it is an antioxidant both in vitro (12) and in macrophages (18).The goal of our study is to define the role of CNLAC1 inevasion of host defenses in the lung. In vivo, melanin has beenimplicated in inhibition of early recognition events by the im-mune system and interfering with T-cell responses (11). Thesestudies used laccase high- and low-producing strains, limitingdefinitive conclusions on the role of CNLAC1 in vivo. In ad-dition, other studies investigating the role of melanin and/orlaccase in pathogenesis have used albino mutants that maycontain mutations in regulatory genes that control melaniza-tion (2, 15, 31). To specifically investigate the role of CNLAC1,a set of congenic mutants of C. neoformans differing only inCNLAC1 production (2E-TU-4/2E-TUC-4) was used for thepresent study. Strain 2E-TU-4 is a laccase-deficient strain (con-tains mutations and/or deletions in the CNLAC1 gene), and2E-TUC-4 is a laccase-positive reconstituted transformant of2E-TU-4 containing an integrated functional copy of theCNLAC1 gene (33). Using these congenic C. neoformansstrains, we tested the requirement for CNLAC1 in virulenceand in evasion of murine pulmonary immune responses afterinfection via the respiratory tract.
MATERIALS AND METHODS
C. neoformans. Strains 2E-TU-4 and 2E-TUC-4 are laccase-deficient and lac-case-positive congenic strains of C. neoformans, respectively, and were generatedas previously described (33). Briefly, parent strain B-3501 (ATCC 34873) wasmutagenized with ethyl methanesulfonate treatment, and melanin-negative mu-tants were selected on dopamine plates. Albino strain mel2 was then backcrossedonce with strain B-4476. This strain, termed 2E, was transformed with a plasmidcontaining URA5 to generate strain 2E-TU or with a plasmid containing URA5and CNLAC1 to generate strain 2E-TUC. Strains 2E-TU and 2E-TUC were then
* Corresponding author. Mailing address: Division of Pulmonaryand Critical Care Medicine, Department of Internal Medicine, Uni-versity of Michigan Medical School, Ann Arbor, MI 48109-0642.Phone: (734) 936-9368. Fax: (734) 764-4556. E-mail: [email protected].
1693
on June 1, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

backcrossed with strain B-4476 four times to generate strains 2E-TU-4 and2E-TUC-4. Strain 2E-TU-4 was negative for phenoloxidase activity, recessivesterility, and suppression of mutant phenotype by CuSO4. The CNLAC1 genefrom 2E-TU-4 was sequenced and contains six differences at the amino acidlevel, including a His-Tyr substitution in the highly conserved histadine copper-binding site. Southern blot analysis confirmed that transformant 2E-TUC-4 containsan integrated copy of the CNLAC1-containing plasmid. For infection, yeast weregrown to stationary phase (72 h) at 37°C in Sabouraud dextrose broth (1% neopep-tone, 2% dextrose; Difco, Detroit, Mich.) with shaking. The cultures were thenwashed in nonpyrogenic saline (Abbott Laboratories, Chicago, Ill.), counted with ahemocytometer, and diluted to 3.3 � 105 CFU/ml in sterile nonpyrogenic saline.
Mice. Female CBA/J mice (18 � 2 g) were purchased from Jackson Labora-tories (Bar Harbor, Maine) and housed in specific-pathogen-free conditions inenclosed filter-top cages. Food and sterile water were given ad libitum. The micewere maintained by the Unit for Laboratory Animal Medicine at the Universityof Michigan (Ann Arbor, Mich.), and protocols were approved by an animalinstitutional review board.
Intratracheal inoculation. Infection was established via intratracheal inocula-tion with 104 or 106 CFU C. neoformans. Four animals per group per time pointwere infected in two independent experiments for a total of eight mice per groupper time point. Mice were anesthetized with ketamine-xylazine solution (2.5 mgof ketamine/mouse (Fort Dodge Animal Health, Fort Dodge, Iowa)) plus 0.1 gof xylazine/mouse (Lloyd Laboratories, Shenandoah, Iowa) and restrained on asmall board. A small incision was made in the skin over the trachea, and theunderlying tissue was separated. A tuberculin syringe (Monoject, St. Louis, Mo.)was filled with a dilute C. neoformans culture and a 30-gauge needle (BectonDickinson, Rutherford, N.J.) was attached and bent. The needle was insertedinto the trachea and a 30-�l inoculum was delivered. The skin was sutured witha cyanoacrylate adhesive, and the mice recovered with no visible trauma. Ali-quots of the inoculum were analyzed for CFU to monitor the amounts delivered.
Intravenous inoculation. Infection was established via intravenous inoculationwith 106 CFU C. neoformans with 8 to 10 mice per group per time point. Micewere warmed under a heating lamp for 15 min prior to intravenous inoculation.A tuberculin syringe (Monoject) was filled with a dilute C. neoformans culture,and a 30-gauge needle (Becton Dickinson) was attached. The needle was in-serted into the lateral tail vein, and a 250-�l inoculum was delivered. Aliquots ofthe inoculum were analyzed for CFU to monitor the amount delivered.
Harvesting of tissues. Extrapulmonary organs were harvested subsequent toremoval of the lungs. Lung-associated lymph nodes (LALN) were collected byexcising the nodes from the junction of the azygos vein and the superior venacava. Brains were collected by removing the top of the cranium and excising thebrain from the brain stem. Organs were placed in tubes containing 2 ml of sterilewater and homogenized mechanically using a Tissue-Tearor (Biospec Products,Bartlesville, Okla.).
CFU assay. Aliquots of the lungs digests (intratracheal infection), lung ho-mogenates (intravenous infection), and brain, spleen, and LALN homogenateswere plated out on Sabouraud dextrose agar (Difco) in 10-fold dilutions andincubated at room temperature. Colonies were counted 2 to 3 days later, and thenumbers of CFU/organ were calculated.
Lung leukocyte isolation. Mice were euthanized by CO2. Lungs were excised,minced, and enzymatically digested for 30 min at 37°C by using 15 ml/lungdigestion buffer (RPMI, 10% fetal calf serum), antibiotics, 1 mg of collagenase(Boehringer Mannheim Biochemical, Chicago, Ill.)/ml, and 30 �g of DNase(Sigma Chemical Co., St. Louis, Mo.)/ml. Cells were further dispersed by draw-ing the suspension up and down through the bore of a 10-ml syringe. A 100-�laliquot was removed for the CFU assay. The cell suspension was pelleted, anderythrocytes were lysed by incubation in ice-cold NH4Cl buffer (0.829% NH4Cl,0.1% KHCO3, 0.0372% Na2EDTA [pH 7.4]; Sigma). Excess RPMI was added tomake the solution isotonic, and the cells were pelleted and resuspended incomplete medium (RPMI 1640, 10% fetal calf serum; Life Technologies), 5 �10�5 M 2-mercaptoethanol, sodium pyruvate, nonessential amino acids, glu-tamine, and antibiotics (Sigma). Cell concentrations were determined by count-ing cells diluted in trypan blue by using a hemocytometer.
Lung leukocyte culture and cytokine ELISA. Isolated leukocytes (from enzy-matic digests) from individual mice were standardized to 1.5 � 107 cells/3 ml andcultured in complete medium without additional stimulation at 37°C and 5% CO2.Supernatants were harvested at 24 h and assayed for cytokine production by enzyme-linked immunosorbent assay (ELISA; OptEIA; Pharmingen, San Diego, Calif.).
Cell staining. Leukocyte differentials (neutrophils, eosinophils, macrophages,and moncytes or lymphocytes) were visually counted after Wright-Giemsa stain-ing of lung leukocyte samples cytospun onto glass slides (Shandon Cytospin,Pittsburgh, Pa.). The percentage of a leukocyte subset was multiplied by the totalnumber of leukocytes to yield the absolute number of that leukocyte subset.
DTH assay. Mice were tested for the development of delayed-type hypersen-sitivity (DTH) by using a modification of a previously described footpad DTHassay (4). In brief, C. neoformans filtrate antigen (CneF; 20 �l) was injected intothe hind right footpad, and the hind left footpad was injected with 20 �l of 2%bovine serum albumin. After 48 h, the thickness of each footpad was measuredby using a micrometer. The swelling in the right footpad was determined bysubtracting the measurement of the right footpad from the measurement of theleft. Uninfected mice were also challenged as a negative control for the assay.
Histology. After euthanasia and before removal, lungs were perfused with 3 mlof sterile saline to flush out pulmonary blood vessels. The lungs were theninflated with 1 ml of 10% neutral buffered formalin via cannulation of thetrachea. The inflated lungs were tied off, removed, and stored in 10% neutralbuffered formalin. Brains were excised from mice and fixed in 10% neutralbuffered formalin. Organs were then dehydrated and embedded in paraffin.Five-micrometer sections were cut, deparaffinized, and stained with either he-matoxylin and eosin or mucicarmine.
Statistical analysis. The Student t test (two-tailed, unequal variance) was usedto analyze the significance of differences between experimental groups. Data witha P value of �0.05 were considered to be significant.
RESULTS
CNLAC1 and survival in mice. To determine the role ofCNLAC1 in virulence during infection acquired via the respi-ratory tract, we inoculated CBA/J mice intratracheally with 104
CFU of C. neoformans strains 2E-TU-4 or 2E-TUC-4. Inter-estingly, no mortality was observed in mice infected with eitherstrain 2E-TU-4 or 2E-TUC-4 by day 42 postinfection (data notshown). After the infectious dose was increased to 106 CFU/mouse, mice infected with these strains exhibited significantdifferences in mortality (Fig. 1). By day 30 postinfection, 70%of mice infected with the laccase-positive strain 2E-TUC-4died. In contrast, no mortality was observed in mice infectedwith strain 2E-TU-4 by day 63 postinfection. The mean survivaltime of 2E-TU-4-infected mice was �63 days, which was sig-nificantly longer than 2E-TUC-4-infected mice (27.7 � 0.44days) (P � 0.005). This demonstrates that CNLAC1 is requiredfor virulence after infection via the respiratory tract.
CNLAC1 and pulmonary clearance of C. neoformans. Pulmo-nary cryptococcal burden was examined in mice infected at the106-CFU/mouse dose (Fig. 2a). Pulmonary burden remainedbetween 105 and 106 CFU/lung at weeks 1, 2, and 4 postinfec-
FIG. 1. Effect of CNLAC1 on survival of mice after infection.CBA/J mice were infected intratracheally with 104 (a) or 106 (b) CFUof C. neoformans strain 2E-TU-4 (laccase deficient) or 2E-TUC-4(laccase positive). Mice were monitored daily for survival.
1694 NOVERR ET AL. INFECT. IMMUN.
on June 1, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

tion in mice infected with strain 2E-TU-4 or 2E-TUC-4. Datacould not be obtained from mice infected with strain 2E-TUC-4 beyond week 4 postinfection due to mortality. How-ever, in mice infected with strain 2E-TU-4, pulmonary burdendecreased at weeks 5 and 6 postinfection. Overall, no differ-ences were observed in pulmonary clearance in mice infectedwith strain 2E-TU-4 or 2E-TUC-4.
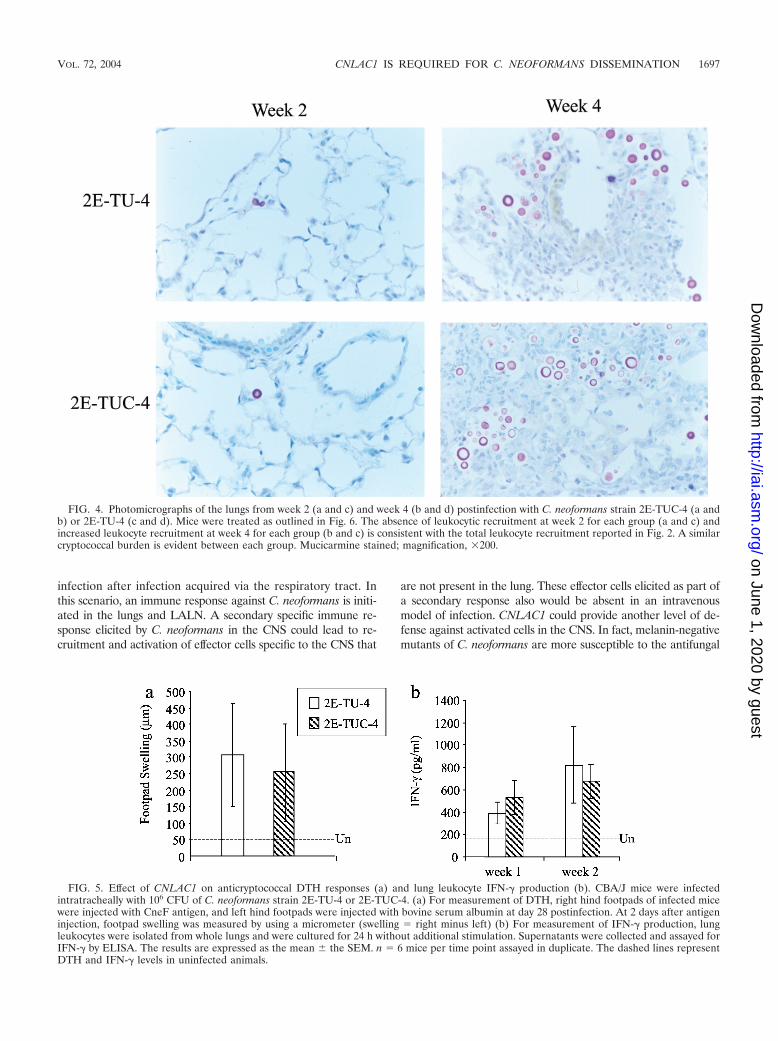
CNLAC1 and cellular immune responses in the lung. Tofurther characterize the infection, pulmonary immune re-sponses were analyzed. CBA/J mice were infected intratrache-ally with 106 CFU of C. neoformans strains 2E-TU-4 or 2E-TUC-4. Lungs were excised at weeks 1, 2, 4, and 5postinfection to examine the kinetics of the pulmonary im-mune responses to these strains. The lungs were digested, andleukocyte recruitment was analyzed quantitatively and qualita-tively. Interestingly, no differences in leukocyte recruitmentwere observed between the two strains at any time point (Fig.2b). Leukocyte subset recruitment was also similar for infec-tion by the laccase-positive and laccase-deficient strains (Fig.3). Histological examination of lungs of mice infected with2E-TU-4 and 2E-TUC-4 taken from weeks 2 and 4 postinfec-tion demonstrated the similarity in inflammatory responses(Fig. 4). At week 2 postinfection, the alveoli of lungs infected
with either strain were devoid of inflammatory cells, and fewcryptococci were detectable. At week 4 postinfection, inflam-matory cells infiltrated the lungs and greater numbers of bothstrains of yeast were present. Overall, there was no differencein pulmonary inflammation after infection with either strain2E-TU-4 or 2E-TUC-4.
We investigated other parameters of the immune responseto these strains. The DTH response was examined by measur-ing footpad swelling after cryptococcal antigen injection (Fig.5a). Mice infected with 2E-TU-4 developed a strong DTHresponse when challenged with cryptococcal antigen (Fig. 5a).However, the levels of footpad swelling in mice infected witheither strain were statistically equivalent (P � 0.05), indicatingthat both strains elicit Th1 responses. Cytokine production wasalso measured in lung leukocyte cultures harvested from in-fected mice (Fig. 5b and data not shown). No difference wasobserved in the production of gamma interferon (IFN-�) atweek 1 or 2 postinfection (Fig. 5b). Interleukin-12 (IL-12),IL-4, and IL-10 levels were low but not significantly differentbetween the two groups (data not shown). Overall, these dataprovide evidence for the development of T-cell responses inmice infected with either strain of C. neoformans but do notidentify differences in host response that correlate with mor-tality in mice infected with laccase-positive strain 2E-TUC-4.
CNLAC1 and dissemination of C. neoformans. Although nomeasurable differences were observed in lungs of mice infectedwith either 2E-TU-4 or 2E-TUC-4, we also examined crypto-coccal burden in extrapulmonary organs in these mice. Signif-icantly greater numbers of 2E-TUC-4 were recovered fromextrapulmonary sites, including the brain and spleen, at week 3postinfection (Fig. 6a). In addition, detectable cryptococciwere recovered from greater numbers of animals infected withlaccase-positive strain 2E-TUC-4 compared to laccase-deficientstrain 2E-TU-4 (11 of 12 mice versus 4 of 13 mice for the brainand 13 of 16 mice versus 6 of 17 mice for the spleen). Significantlygreater numbers of 2E-TUC-4 were also recovered from theLALN [log(6.1 � 0.6) CFU/LALN] compared to 2E-TU-4[log(2.1 � 0.3) CFU/LALN] (data not shown). Thus, CNLAC1plays a role in the ability of C. neoformans to disseminate.
Dissemination is a multistep process, involving escape fromthe lung, emptying into the draining lymph nodes en route tothe lymphatics and bloodstream, and survival and growth inextrapulmonary sites. To determine at which step of dissemi-nation CNLAC1 is involved, mice were infected intravenously.Equal numbers of strain 2E-TUC-4 and 2E-TU-4 were recov-ered from the brain and spleen at week 3 postinfection (Fig.6b). At earlier time points (week 1), similar numbers of lac-case-positive and laccase-negative C. neoformans were recov-ered from the brain (2E-TUC-4, log 5.5 CFU/organ; 2E-TU-4,5.1 CFU/organ) and spleen (2E-TUC-4, 5.1 CFU/organ; 2E-TU-4, log 5.4 CFU/organ) (data not shown). At this time pointboth strains had exited the microcapillaries and were growingin the brain tissue, resulting in tissue damage (Fig. 7). Thisindicates that CNLAC1 does not play a role in (i) survivaland/or growth in extrapulmonary sites, (ii) survival in thebloodstream en route to other destinations, or (iii) survivalwithin the brain. Instead, these results support a role forCNLAC1 in escape from the lungs, which may explain the roleof CNLAC1 in virulence.
FIG. 2. Effect of CNLAC1 on pulmonary cryptococcal burden andleukocyte recruitment. Mice were infected with 106 CFU C. neofor-mans strain 2E-TU-4 or 2E-TUC-4. (a) Pulmonary burden was deter-mined at various time points postinfection. No surviving animals re-mained for mice infected with 2E-TUC-4 after week 4 postinfection.The results are expressed as the mean CFU per organ � the standarderror of the mean (SEM). n 12 to 17 mice per time point pooledfrom two separate experiments. (b) For examination of recruited lungleukocytes, lungs were excised at weeks 1, 2, 4, and 5 postinfection.Leukocytes were isolated from whole lungs by enzymatic digestion andmechanical dispersion. The number of recruited leukocytes in infectedmice was calculated as the total number of leukocytes in infected miceminus the mean number of leukocytes in uninfected mice. The results areexpressed as the mean number of leukocytes per mouse � the SEM. n 7 to 8 mice per time point pooled from two separate experiments.
VOL. 72, 2004 CNLAC1 IS REQUIRED FOR C. NEOFORMANS DISSEMINATION 1695
on June 1, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

DISCUSSION
The goal of our study was to define the role of CNLAC1 inevasion of host defenses in the lung. Increased mortality wasobserved in mice infected intratracheally with laccase-positivestrain 2E-TUC-4, confirming that CNLAC1 is required forvirulence. Surprisingly, we did not observe any differences inthe ability of these strains to grow in the lungs of infected micethrough week 4 postinfection. After week 4, mice infected with2E-TUC-4 began to die, whereas mice infected with 2E-TU-4began to clear the infection in the lungs. No differences wereobserved in leukocyte recruitment or IFN-� production at duringthe course of the infection. However, we observed significantlygreater numbers of laccase-positive organisms in the brain,spleen, and LALN at week 4 postinfection. This indicates thatCNLAC1 plays a role in dissemination of C. neoformans.
Dissemination is a multistep process, involving escape fromthe lung, passage into the draining lymph nodes en route to thelymphatics and bloodstream, and survival and growth in extra-pulmonary sites. Significantly more organisms were recoveredfrom the LALN in mice infected intratracheally. If dissemina-tion from the lungs occurs through the lymphatics, the LALNwould be the first extrapulmonary site infected. These resultssuggest that strain 2E-TUC-4 escapes from the lung with in-creased frequency compared to strain 2E-TU-4. To more spe-cifically address this issue, mice were infected intravenously. Byweek 3 postinfection, equal numbers of each strain were re-covered from the brains and spleens of infected mice, indicat-ing that CNLAC1 does not play a role in growth or survival inextrapulmonary sites or in survival in the bloodstream.
The present study establishes a novel role for CNLAC1 inthe virulence of a C. neoformans infection acquired via therespiratory tract. Our results indicate that CNLAC1 is involvedin promoting escape of C. neoformans from the lung. Escapemay be facilitated by intracellular survival within a cellulardisseminatory vehicle. C. neoformans can grow both intracel-lularly and extracellularly in the lung. However, intracellularsurvival plays a role in escape from the lung (20). CNLAC1 hasbeen shown to play a role in intracellular survival in alveolarmacrophages, which may explain the increased ability of 2E-TUC-4 to disseminate (18). Our results also suggest that in-tracellular survival in alveolar macrophages may not be impor-tant in the growth of C. neoformans in the lung. This idea issupported by the fact that a mutant strain of C. neoformans inApp1, which mediates antiphagocytic activity, exhibits de-creased virulence in mice (20). Therefore, extracellular growthmay be the preferred environment for C. neoformans in thelung for evasion of host defense mechanisms. Overall, we be-lieve the escape of strain 2E-TUC-4 from the lung environ-ment is mediated by a CNLAC1-dependent mechanism andthat subsequent growth within the CNS is the likely cause ofdeath in these studies.
C. neoformans has a unique predilection for the CNS, whereit can cause fatal meningitis. C. neoformans is well known forits propensity to establish a CNS infection (25). Our resultsconflict with the notion that CNLAC1 plays a crucial role in theprotection of cryptococci at the level of the CNS, where sub-strates for the enzyme are more abundant (26). However, it isstill possible that CNLAC1 may facilitate a cryptococcal CNS
FIG. 3. Effect of CNLAC1 on recruitment of leukocyte subsets into the lungs of mice. CBA/J mice were infected intratracheally with 106 CFUof C. neoformans strain 2E-TU-4 or 2E-TUC-4. Lungs were excised at weeks 1, 2, 4, and 5 postinfection. Leukocytes were isolated from whole lungsby mechanical and enzymatic dispersion and then phenotyped by Wright-Giemsa staining of samples cytospun onto slides. Subsets included macrophages(a), grouped lymphocytes and monocytes (Lympho/Mono) (b), neutrophils (c), and eosinophils (d). The percentage of a leukocyte subset was multipliedby the total number of leukocytes to yield the absolute number of that leukocyte subset. The results are expressed as the mean number of leukocytes permouse � the SEM. n 7 to 8 mice per time point pooled from two separate experiments.
1696 NOVERR ET AL. INFECT. IMMUN.
on June 1, 2020 by guesthttp://iai.asm
.org/D
ownloaded from
infection after infection acquired via the respiratory tract. Inthis scenario, an immune response against C. neoformans is initi-ated in the lungs and LALN. A secondary specific immune re-sponse elicited by C. neoformans in the CNS could lead to re-cruitment and activation of effector cells specific to the CNS that
are not present in the lung. These effector cells elicited as part ofa secondary response also would be absent in an intravenousmodel of infection. CNLAC1 could provide another level of de-fense against activated cells in the CNS. In fact, melanin-negativemutants of C. neoformans are more susceptible to the antifungal
FIG. 4. Photomicrographs of the lungs from week 2 (a and c) and week 4 (b and d) postinfection with C. neoformans strain 2E-TUC-4 (a andb) or 2E-TU-4 (c and d). Mice were treated as outlined in Fig. 6. The absence of leukocytic recruitment at week 2 for each group (a and c) andincreased leukocyte recruitment at week 4 for each group (b and c) is consistent with the total leukocyte recruitment reported in Fig. 2. A similarcryptococcal burden is evident between each group. Mucicarmine stained; magnification, �200.
FIG. 5. Effect of CNLAC1 on anticryptococcal DTH responses (a) and lung leukocyte IFN-� production (b). CBA/J mice were infectedintratracheally with 106 CFU of C. neoformans strain 2E-TU-4 or 2E-TUC-4. (a) For measurement of DTH, right hind footpads of infected micewere injected with CneF antigen, and left hind footpads were injected with bovine serum albumin at day 28 postinfection. At 2 days after antigeninjection, footpad swelling was measured by using a micrometer (swelling right minus left) (b) For measurement of IFN-� production, lungleukocytes were isolated from whole lungs and were cultured for 24 h without additional stimulation. Supernatants were collected and assayed forIFN-� by ELISA. The results are expressed as the mean � the SEM. n 6 mice per time point assayed in duplicate. The dashed lines representDTH and IFN-� levels in uninfected animals.
VOL. 72, 2004 CNLAC1 IS REQUIRED FOR C. NEOFORMANS DISSEMINATION 1697
on June 1, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

actions of activated microglial cells, which are effector cells in-volved in controlling an infection in the CNS (3). In addition, thehumoral arm of cell-mediated immune responses (specific anti-body) is also required to mediate antifungal activities of microglialcells (16). Further studies are needed to elucidate the finer detailsof the role of CNLAC1 in defense against innate and acquireddefenses in the CNS.
These studies specifically redefine the role of laccase in
virulence. Previous studies have compared cryptococcal strainswith differences in the ability to melanize (2, 15, 31). Thepresumption that the mutations in these strains were specific tolaccase may need to be reexamined. A number of genes havebeen implicated in the regulation of laccase and melanization,including IPC1 (21), GPA1 (1), MET3 (40), and STE12 (6).Albino mutants derived from random mutagenesis techniquesmay contain mutations in genes that regulate multiple viru-lence factors, including CNLAC1. This may provide an alter-native explanation for the discrepancies between the presentstudy and studies that examined the role of melanization. Thepresent study is the first to use congenic strains differing spe-cifically in laccase to define the role of laccase in cryptococcalpulmonary pathogenesis and dissemination.
We propose that the pathogenesis and tropism of C. neofor-mans is mediated in a stepwise fashion by specific virulencefactors. C. neoformans is an environmental microbe (14, 27, 32)that is initially inhaled (28, 29) and yet exhibits a tropism forthe CNS (17, 30, 34). We believe the dissemination from thelungs to the bloodstream occurs initially through the lymphat-ics draining the lungs. We can detect cryptococci in the LALNprior to the appearance of detectable numbers of organisms inthe spleen or other organ sites (during the first week of infec-tion; data not shown). From the blood, dissemination proceedsto extrapulmonary organs such as the CNS. Polysaccharidecapsule protects C. neoformans from destruction by alveolarmacrophages (22, 35, 38, 41). Phospholipase B and CNLAC1allow the organism to survive inside the alveolar macrophage(18, 24). The mechanism of lung-lymph node dissemination isnot known but is clearly defective in laccase-deficient andphospholipase B-deficient C. neoformans strains (8, 24, 42; thepresent study) and in mice depleted of alveolar macrophages(A. C. Herring et al., unpublished), implicating transport ofintracellular C. neoformans by macrophages into the lymphnodes and subsequently into the bloodstream. Once in thebloodstream, urease appears to facilitate entry into the CNS(24a). The fact that CNLAC1-deficient cells can grow in theCNS but albino mutants cannot (2) suggests that unknownvirulence factors (that are regulated along with CNLAC1) playa role in the neurotropism of C. neoformans. Thus, C. neofor-
FIG. 6. Effect of CNLAC1 on extrapulmonary organ burden. (a)Mice were infected intratracheally with 106 CFU of C. neoformansstrain 2E-TU-4 or 2E-TUC-4, and the brains and spleens were har-vested at week 4 postinfection. n 12 to 17 mice per time point pooledfrom two separate experiments. (b) Mice were infected intravenouslywith 106 CFU of C. neoformans strain 2E-TU-4 or 2E-TUC-4, and thebrains and spleens were harvested at week 3 postinfection. n 9 to 10mice per time point. The results are expressed as the mean CFU perorgan � the SEM. ❋, P � 0.01 (as determined by the Student t test).
FIG. 7. Photomicrographs of brains at week 1 postinfection in intravenously infected mice. Mice were infected with 106 CFU of C. neoformansstrain 2E-TU-4 or 2E-TUC-4, and brains were harvested at week 1 postinfection. Similar cryptococcal burden and tissue destruction is evidentbetween the two groups. Mucicarmine stained; magnification, �100.
1698 NOVERR ET AL. INFECT. IMMUN.
on June 1, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

mans produces a number of factors that function in a stepwisefashion as site-specific virulence factors to promote the dis-semination and virulence of this microbe after inhalation.
ACKNOWLEDGMENTS
We thank Rod McDonald and Rachael Noggle for their work on themurine infections and harvests. We also thank Galen Toews for schol-arly contributions and support.
This study was supported by a New Investigator Award in MolecularPathogenic Mycology from the Burroughs-Wellcome Fund (G.B.H.).M.C.N. was supported by NIAID training grant T32AI07528 andNHLBI training grant T32HL007749. Additional support was providedby the following grants from the National Institutes of Health: RO1-HL65912 (G.B.H.), RO1-HL63670 (G.B.H.), and R01-AI045995(P.R.W.).
REFERENCES
1. Alspaugh, J. A., J. R. Perfect, and J. Heitman. 1997. Cryptococcus neofor-mans mating and virulence are regulated by the G-protein alpha subunitGPA1 and cAMP. Genes Dev. 11:3206–3217.
2. Barluzzi, R., A. Brozzetti, G. Mariucci, M. Tantucci, R. G. Neglia, F. Bistoni,and E. Blasi. 2000. Establishment of protective immunity against cerebralcryptococcosis by means of an avirulent, nonmelanogenic Cryptococcus neo-formans strain. J. Neuroimmunol. 109:75–86.
3. Blasi, E., R. Barluzzi, R. Mazzolla, B. Tancini, S. Saleppico, M. Puliti, L.Pitzurra, and F. Bistoni. 1995. Role of nitric oxide and melanogenesis in theaccomplishment of anticryptococcal activity by the BV-2 microglial cell line.J. Neuroimmunol. 58:111–116.
4. Cauley, L. K., and J. W. Murphy. 1979. Response of congenitally athymic(nude) and phenotypically normal mice to Cryptococcus neoformans infec-tion. Infect. Immun. 23:644–651.
5. Chang, Y. C., and K. J. Kwon-Chung. 1994. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol.Cell. Biol. 14:4912–4919.
6. Chang, Y. C., B. L. Wickes, G. F. Miller, L. A. Penoyer, and K. J. Kwon-Chung. 2000. Cryptococcus neoformans STE12alpha regulates virulence butis not essential for mating. J. Exp. Med. 191:871–882.
7. Chen, S. C., M. Muller, J. Z. Zhou, L. C. Wright, and T. C. Sorrell. 1997.Phospholipase activity in Cryptococcus neoformans: a new virulence factor?J. Infect. Dis. 175:414–420.
8. Cox, G. M., H. C. McDade, S. C. Chen, S. C. Tucker, M. Gottfredsson, L. C.Wright, T. C. Sorrell, S. D. Leidich, A. Casadevall, M. A. Ghannoum, andJ. R. Perfect. 2001. Extracellular phospholipase activity is a virulence factorfor Cryptococcus neoformans. Mol. Microbiol. 39:166–175.
9. Cox, G. M., J. Mukherjee, G. T. Cole, A. Casadevall, and J. Perfect. 2000.Urease as a virulence factor in experimental cryptococcosis. Infect. Immun.68:443–438.
10. Doering, T. L., J. D. Nosanchuk, W. K. Roberts, and A. Casadevall. 1999.Melanin as a potential cryptococcal defense against mcirobicidal proteins.Med. Mycol. 37:175–181.
11. Huffnagle, G. B., G.-H. Chen, J. L. Curtis, R. A. McDonald, R. M. Strieter,and G. B. Toews. 1995. Down-regulation of the afferent phase of T cell-mediated pulmonary inflammation and immunity by a high melanin-produc-ing strain of Cryptococcus neoformans. J. Immunol. 155:3507.
12. Jacobson, E., and S. Tinnell. 1993. Antioxidant function of fungal melanin.J. Bacteriol. 175:7102–7104.
13. Jacobson, E. S., and H. S. Emery. 1991. Temperature regulation of thecryptococcal phenoloxidase. J. Med. Vet. Mycol. 29:121–124.
14. Kwon-Chung, K. J., and J. E. Bennett. 1984. High prevalence of Cryptococ-cus neoformans var. gattii in tropical and subtropical regions. Zentbl. Bakte-riol. Hyg. A 257:213–218.
15. Kwon-Chung, K. J., I. Polacheck, and T. J. Popkin. 1982. Melanin-lackingmutants of Cryptococcus neoformans and their virulence for mice. J. Bacte-riol. 150:1414–1421.
16. Lee, S. C., Y. Kress, D. W. Dickson, and A. Casadevall. 1995. Humanmicroglia mediate anti-Cryptococcus neoformans activity in the presence ofspecific antibody. J. Neuroimmunol. 62:43–52.
17. Levy, R. M., D. E. Bredesen, and M. L. Rosenblum. 1985. Neurologicalmanifestations of the acquired immunodeficiency syndrome (AIDS): expe-rience at UCSF and review of the literature. J. Neurosurg. 62:475–495.
18. Liu, L., R. Tweari, and P. R. Williamson. 1999. Laccase protects Cryptococ-cus neoformans from antifungal activity of alveolar macrophages. Infect.Immun. 67:6034–6039.
19. Liu, L., K. Wakamatsu, S. Ito, and P. R. Williamson. 1999. Catecholamineoxidative products, but not melanin, are produced by Cryptococcus neofor-mans during neuropathogenesis in mice. Infect. Immun. 67:108–112.
20. Luberto, C., B. Martinez-Marino, D. Taraskiewicz, B. Bolanos, P. Chitano,D. L. Toffaletti, G. M. Cox, J. R. Perfect, Y. A. Hannun, E. Balish, and M. D.Poeta. 2003. Identification of App1 as a regulator of phagocytosis and viru-lence of Cryptococcus neoformans. J. Clin. Investig. 112:1080–1094.
21. Luberto, C., D. L. Toffaletti, E. A. Wills, S. C. Tucker, A. Casadevall, J. R.Perfect, Y. A. Hannun, and M. M. Del Poeta. 2001. Roles for inositol-phosphoryl ceramide synthase 1 (IPC1) in pathogenesis of Cryptococcusneoformans. Genes Dev. 15:201–212.
22. Mitchell, T. G., and L. Friedman. 1972. In vitro phagocytosis and intracel-lular fate of variously encapsulated strains of Cryptococcus neoformans. In-fect. Immun. 5:491–498.
23. Nosanchuk, J. D., A. L. Rosas, S. C. Lee, and A. Casadevall. 2000. Melanisationof Cryptococcus neoformans in human brain tissue. Lancet 355:2049–2050.
24. Noverr, M. C., G. M. Cox, J. R. Perfect, and G. B. Huffnagle. 2003. Role ofPLB1 in pulmonary inflammation and cryptococcal eicosanoid production.Infect. Immun. 71:1538–1547.
24a.Olszewski, M. A., M. C. Noverr, G. H. Chen, G. B. Toews, G. M. Cox, J. R.Perfect, and G. B. Huffnagle. Urease promotes Cryptococcus neoformans neu-rotropism by enhancing microcapillary sequestration. Am. J. Pathol., in press.
25. Perfect, J. R., and A. Casadevall. 2002. Cryptococcosis. Infect. Dis. Clin. N.Am. 16:837–874.
26. Polacheck, I., Y. Platt, and J. Aronovitch. 1990. Catecholamines and viru-lence of Cryptococcus neoformans. Infect. Immun. 58:2919–2122.
27. Randhawa, H. S., A. Y. Mussa, and Z. U. Khan. 2001. Decaying wood in treetrunk hollows as a natural substrate for Cryptococcus neoformans and otheryeast-like fungi of clinical interest. Mycopathologia 151:63–69.
28. Randhawa, H. S., and D. K. Paliwal. 1977. Occurrence and significance ofCryptococcus neoformans in the oropharynx and on the skin of a healthyhuman population. J. Clin. Microbiol. 6:325–327.
29. Randhawa, H. S., and D. K. Paliwal. 1979. Survey of Cryptococcus neofor-mans in the respiratory tract of patients with bronchopulmonary disordersand in the air. Sabouraudia 17:399–404.
30. Reblin, T., A. Meyer, H. Albrecht, and H. Greten. 1994. Disseminated cryp-tococcosis in a patient with AIDS. Mycoses 37:275–279.
31. Rhodes, J., I. Polacheck, and K. Kwon-Chung. 1982. Phenoloxidase activityand virulence in isogenic strains of Cryptococcus neoformans. Infect. Immun.36:1175–1184.
32. Ruiz, A., D. Velez, and R. A. Fromtling. 1989. Isolation of saprophyticCryptococcus neoformans from Puerto Rico: distribution and variety. Myco-pathologia 106:167–170.
33. Salas, S. D., J. E. Bennett, K. J. Kwon-Chung, and J. Perfect. 1996. Effect ofthe laccase gene, CNLAC1, on virulence of Cryptococcus neoformans. J. Exp.Med. 184:377–386.
34. Sampaio, R. N., B. Medeiros, M. Milfort, G. F. Alves, C. M. Reis, and I. T.Campbell. 1999. Systemic cryptococcosis with solitary cutaneous lesion in animmunocompetent patient. Int. J. Dermatol. 38:773–775.
35. Vecchiarelli, A., C. Retini, D. Pietrella, C. Monari, and T. R. Kozel. 2000. Tlymphocyte and monocyte interaction by CD40/CD40 ligand facilitates alymphoproliferative response and killing of Cryptococcus neoformans in vitro.Eur. J. Immunol. 30:1385–1393.
36. Wang, Y., P. Aisen, and A. Casadevall. 1995. Cryptococcus neoformans mel-anin and virulence: mechanism of action. Infect. Immun. 63:3131–3136.
37. Wang, Y., and A. Casadevall. 1994. Growth of Cryptococcus neoformans inthe presence of L-dopa decreases its susceptibility to amphoteracin B. An-timicrob. Agents Chemother. 38:2648–2650.
38. Wilder, J. A., G. K. Olson, Y. C. Chang, K. J. Kwon-Chung, and M. F.Lipscomb. 2002. Complementation of a capsule-deficient Cryptococcus neo-formans with CAP64 restores virulence in a murine lung infection. Am. J.Respir. Cell. Mol. Biol. 26:306–314.
39. Williamson, P. R. 1994. Biochemical and molecular characterization of thediphenol oxidase of Cryptococcus neoformans: identification as a laccase. J.Bacteriol. 176:656–664.
40. Yang, Z., R. C. Pascon, A. Alspaugh, G. M. Cox, and J. H. McCusker. 2002.Molecular and genetic analysis of the Cryptococcus neoformans MET3 geneand a met3 mutant. Microbiology 148:2617–2625.
41. Yasuoka, A., S. Kohno, H. Yamada, M. Kaku, and H. Koga. 1994. Influenceof molecular sizes of Cryptococcus neoformans capsular polysaccharide onphagocytosis. Microbiol. Immunol. 38:851–856.
42. Zhu, X., J. Gibbons, J. Garcia-Rivera, A. Casadevall, and P. R. Williamson.2001. Laccase of Cryptococcus neoformans is a cell wall-associated virulencefactor. Infect. Immun. 69:5589–5596.
Editor: T. R. Kozel
VOL. 72, 2004 CNLAC1 IS REQUIRED FOR C. NEOFORMANS DISSEMINATION 1699
on June 1, 2020 by guesthttp://iai.asm
.org/D
ownloaded from