comparativegenomeanalysisidentifiesthevitamindreceptor...
TRANSCRIPT

Comparative Genome Analysis Identifies the Vitamin D Receptor
Gene as a Direct Target of p53-Mediated Transcriptional Activation
Reo Maruyama,1Fumio Aoki,
3Minoru Toyota,
1,2,5Yasushi Sasaki,
2Hirofumi Akashi,
3Hiroaki Mita,
2
Hiromu Suzuki,1,4Kimishige Akino,
1,2Mutsumi Ohe-Toyota,
2Yumiko Maruyama,
1
Haruyuki Tatsumi,3Kohzoh Imai,
1Yasuhisa Shinomura,
1and Takashi Tokino
2
1First Department of Internal Medicine; 2Department of Molecular Biology, Cancer Research Institute; 3Information Center of ComputerCommunication; and 4Department of Public Health, Sapporo Medical University, Sapporo, Japan; and 5Precursory Researchfor Embryonic Science and Technology, Japan Science and Technology Agency, Kawaguchi, Japan
Abstract
p53 is the most frequently mutated tumor suppressor gene inhuman neoplasia and encodes a transcriptional coactivator.Identification of p53 target genes is therefore key tounderstanding the role of p53 in tumorigenesis. To identifynovel p53 target genes, we first used a comparative genomicsapproach to identify p53 binding sequences conserved in thehuman and mouse genome. We hypothesized that potentialp53 binding sequences that are conserved are more likely tobe functional. Using stringent filtering procedures, 32 geneswere newly identified as putative p53 targets, and theirresponsiveness to p53 in human cancer cells was confirmedby reverse transcription-PCR and real-time PCR. Among them,we focused on the vitamin D receptor (VDR) gene becausevitamin D3 has recently been used for chemoprevention ofhuman tumors. VDR is induced by p53 as well as several otherp53 family members, and analysis of chromatin immunopre-cipitation showed that p53 protein binds to conserved intronicsequences of the VDR gene in vivo . Introduction of VDR intocells resulted in induction of several genes known to bep53 targets and suppression of colorectal cancer cell growth.In addition, p53 induced VDR target genes in a vitamin D3-dependent manner. Our in silico approach is a powerfulmethod for identification of functional p53 binding sitesand p53 target genes that are conserved among humans andother organisms and for further understanding the functionof p53 in tumorigenesis. (Cancer Res 2006; 66(9): 4574-83)
Introduction
The transcriptional coactivator p53 binds to DNA in asequence-specific manner and induces transcription of a varietyof genes involved in cell cycle regulation, apoptosis, inhibition ofangiogenesis, and DNA repair (1, 2). Notably, the gene encodingp53 is the most frequently mutated tumor suppressor gene inneoplasms. Consequently, identification of the transcriptionaltargets of p53 is a potential key to understanding functions ofp53 and its signaling pathways in tumorigenesis, and to exploitingtheir potential use as molecular targets for cancer chemothera-peutic drugs.
Several approaches have been used to successfully identify genesinduced by p53 including differential display, representationaldifference analysis, cDNA microarrays, and serial analysis of geneexpression (3–6). However, identification of p53-inducible targetsby gene expression profiling is highly dependent on the expressionlevel of the target gene, potentially overlooking genes of which theexpression level is relatively low in the tissue analyzed. Alterna-tively, p53 target genes should be identifiable using an in silicoanalysis of p53 response elements (p53RE) as probes (7). However,a complete data set of functional p53REs in the human genome isnot yet available. Comparison of orthologous genomic sequenceshas become a powerful tool for identifying functional elementswithin the genome (8, 9). Using a computational approach, codingregions, regulatory elements, and noncoding RNA conserved indifferent vertebrates have been identified (10). We hypothesizedthat both known and novel functional p53REs important for p53function are likely to exhibit greater sequence conservation thannonfunctional sequences (11). A comparative genomic analysis ofputative p53 binding sequences may thus be a way to identify thedownstream mediators of p53 function.Epidemiologic studies indicate that vitamin D3 and its most
active analogue, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], haveantitumor activity (12, 13). In vitro , vitamin D3 induces cell cyclearrest, differentiation, and apoptosis of cancer cells (14–17). Clinicaltrials are under way to assess the activity of various vitamin Danalogues against tumors. Vitamin D3 exerts its activity throughbinding to the vitamin D receptor (VDR). VDR is down-regulatedduring colorectal tumorigenesis, and absence or low levels of VDRexpression correlate with poor prognosis (18, 19). However, theregulatory mechanism of VDR expression is not fully understood.The aim here was to identify and compare p53 target genes in the
human and mouse genomes using in silico genome scanning forsequence conservation of potential p53 binding sites. Our resultsindicate that this in silico approach is a powerful method foridentification of p53 target genes conserved among humans andother organisms, and help determine new functions of p53 intumorigenesis. Among the genes identified as a putative target ofp53, we examined the induction of VDR by p53 family members. Wealso examined the role of VDR in the regulation of p53 target genes.
Materials and Methods
Construction of p53RE database and comparative analysis of p53target genes. p53REs typically consist of two copies of a 10-bp motif
(RRRCWWGYYY) separated by 0 to 12 bp. We first extracted all putative p53
binding sites in the entire human and mouse genomes. The sequence datawere downloaded from the National Center for Biotechnology Information
(NCBI) Human Assembly 33 (April 10, 2003) and the MGSC Mouse Assembly
3 (February 2003) and then input to the p53RE prediction system. We set the
Note: Supplementary data for this article are available at Cancer Research Online(http://cancerres.aacrjournals.org/).Requests for reprints: Takashi Tokino, Department of Molecular Biology, Cancer
Research Institute, Sapporo Medical University, South 1, West 17, Chuo-ku, Sapporo060-8556, Japan. Phone: 81-11-611-2111, ext. 2386; Fax: 81-11-618-3313; E-mail:[email protected].
I2006 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-05-2562
Cancer Res 2006; 66: (9). May 1, 2006 4574 www.aacrjournals.org
Research Article
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

parameter conditions to include the following restrictions: (i) fewer thanfour mismatches in the 20-nucleotide consensus binding sequence, and (ii)
the spacer between the 10-bp motifs has fewer than 12 bp. The output data,
which consisted of p53 binding site IDs, chromosomal positions, nucleotide
sequences, the number of mismatches, spacer length, and the 200-bpsequences surrounding the binding sites, were stored in the p53RE
database.
We next added the gene annotation information into each of the binding
site entries in the p53RE database. The public database RefFlat provided bythe University of California Santa Cruz was used to determine the distance
between the p53 binding sequences and their closest genes. This data set
contained information from 15,824 human genes and 13,406 mouse genes;
the data for each gene consisted of the chromosome number and thepositions of the transcription start site, the first exon start position and the
first intron start position. We calculated the distance between each p53RE
and the corresponding transcription start site of the closest gene andretrieved only the p53 binding sites located within 10 kb of the transcription
start site.
Cell lines, cell culture, and recombinant adenovirus. The human
cancer cell lines used in this study were purchased from the American TypeCulture Collection (Manassas, VA) or the Japanese Collection of Research
Bioresources (Tokyo, Japan). All cell lines were cultured under conditions
recommended by their individual depositors. The status of the endogenous
p53 expressed in these lines was wild-type for RKO and HCT116, mutant forDLD-1, SW480, and MKN74, and p53-null for Saos-2 and H1299. The
generation and purification of replication-deficient recombinant adenovi-
ruses harboring p53, p73a, p73h, p63g, and the bacterial LacZ gene (Ad-p53,Ad-p73a, Ad-p73h, Ad-p63, and Ad-LacZ, respectively) were previously
described (20). To examine VDR expression induced by endogenous p53,
cancer cells were treated with 0.2 to 0.5 Ag/mL of Adriamycin for 12 to 24
hours and harvested.Knockdown of VDR by short hairpin RNA. Two short hairpin RNA
(shRNA) sequences (sh-VDR20 and sh-VDR32) were designed using
siPRECISE software (B-bridge, Inc., Sunnyvale, CA). Oligonucleotides were
annealed and ligated into pFIV-H1-Puro vector (SBI, Mountain View, CA).
shRNA sequences for VDR are shown in Supplementary Table S1. HCT116
cells were transfected with 2 Ag of sh-VDR20, sh-VDR32, or control pFIV-H1-Puro vector for 4 hours using Lipofectamine 2000 (Invitrogen Inc., Grand
Island, NY) in 2 mL of Opti-MEMmedium (Invitrogen). The culture medium
was replaced with McCoy’s medium containing 1 Ag/mL puromycin and
harvested after 48 hours.Chromatin immunoprecipitation. Chromatin immunoprecipitation
(ChIP) assays were carried out as previously described using a ChIP assay
kit (Upstate Biotechnologies, Lake Placid, NY; ref. 21). Immunoprecipitation
was carried out using mouse antihuman p53 monoclonal antibody (mAb;DO-1, Santa Cruz Biotechnology, Santa Cruz, CA). The primers used were
ChIP-F and ChIP-R; their sequences are shown in Supplementary Table S1.
The amplified products were subjected to agarose gel electrophoresis.
Luciferase assay. To construct luciferase reporter plasmids, oligonucle-otide fragments corresponding to VDR-RE (wt) and VDR-RE (mut;
Supplementary Table S1) were synthesized and inserted upstream of a
basal SV40 promoter in the pGL3-promoter vector (Promega, Madison, WI);the resulting constructs were designated pGL3-VDR-RE (wt) and pGL3-
VDR-RE (mut), respectively. H1299 cells at 50% confluence were transfected
with a reporter and an effector plasmid (total, 100 ng DNA) using Lipofectin
(Life Technologies, Inc., Gaithersburg, MD). After 48 hours, the cells wereharvested for measurement of luciferase activity using a Luciferase Assay
System (Promega). Cell extract was incubated with luciferin and light
emission was measured using a Berthold Luminometer (Berthold Lumat
LB9507, Belthold, Bad Wildbad, Germany).Reverse-transcription PCR. Semiquantitative reverse transcription-
PCR (RT-PCR) was carried out as previously described (6). Briefly, total RNA
was extracted using TRIzol (Invitrogen); after which, a 5-Ag sample was
reverse transcribed using SuperscriptIII (Invitrogen). The primer sequencesused are shown in Supplementary Table S1. The integrity of the cDNA was
confirmed by amplifying glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) as previously described (21). Samples of amplified product were
then subjected to 2.5% Nusieve gel electrophoresis and stained withethidium bromide.
Real-time PCR was carried out using a CYBR Green PCR or TaqMan PCR
Master Mix (Applied Biosystems, Foster City, CA) on an ABI Prism 7000.
Accumulation of PCR product was measured in real time as an increase influorescent signals and analyzed using ABI Prism 7000 SDS Software
(Applied Biosystems). Standard curves relating initial template copy
number to fluorescence and amplification cycle were generated using the
amplified PCR product as a template, and were then used to calculatethe mRNA copy number in each sample. The ratios of the intensities of the
target genes to GAPDH signals were considered to be a relative measure of
the target genes mRNA level in each specimen. The information for TaqMan
primers/probe sets is available in Supplementary Materials and Methods.Northern blot analysis. Ten micrograms of total RNA were separated by
electrophoresis on a 1% agarose gel containing 2.2 mol/L formaldehyde. The
integrity and equal loading of the RNA in each lane were confirmed byethidium bromide staining. The RNA was then transferred onto a
nitrocellulose transfer membrane (Protran, Schleicher and Schuell, Inc.,
Keene, NH) by capillary blotting in 20� SSC; after which, hybridization was
done using radiolabeled probes in 50% formamide, 5� Denhardt solution,3� SSC, 0.1% SDS, and 100 Ag/mL salmon sperm DNA overnight at 42jC.After hybridization, the membranes were washed first in 1� SSC/0.1% SDS
at room temperature and then in 0.25� SSC/0.1% SDS at 60jC. Theintensities of hybridization bands were quantitated using a BAS2000Bioimage Analyzer (Fuji, Tokyo, Japan).
Figure 1. Strategy for identifying putative p53 binding sequences conservedbetween human and mouse using an in silico approach. The algorithm presentedhere was implemented in a computer program written in the C programminglanguage.
VDR Is Up-regulated by p53
www.aacrjournals.org 4575 Cancer Res 2006; 66: (9). May 1, 2006
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

Western blot analysis. Twenty micrograms of cell lysate were subjectedto electrophoresis in a 10% SDS-PAGE; after which, the proteins were
transferred to Immobilon-P membranes (Millipore, Bedford, MA). The
membranes were then blocked with 5% nonfat milk and 0.1% Tween 20 inTBS and probed with anti-VDR (Lab Vision, Fremont, CA) and antiactin
mouse mAbs (Chemicon, Temecula, CA); after which, the hybridization was
visualized using enhanced chemiluminescence (Amersham, Piscataway, NJ).Immunofluorescent staining. Cells grown on coverslips were infected
with Ad-lacZ, Ad-p53, Ad-p63, or Ad-p73. After 48 hours, the cells were fixed
with 4% paraformaldehyde and then incubated for 16 hours at 4jC with
anti-VDR rat mAb (Lab Vision). FITC-conjugated goat anti-rat antibody(Molecular Probes, Eugene, OR) was used as the secondary antibody. Finally,
the nucleus was stained with 4V,6-diamidino-2-phenylindole (DAPI; Vector
Laboratories, Inc., Burlingame, CA) and the cells were examined under a
fluorescence microscope (Olympus, Tokyo, Japan).Geneticin-resistant colony formation assay. Cells were plated in 60-
mm culture dishes to a density of 1 � 105 per plate and incubated for 24
hours; after which, they were transfected with pCMV-VDR or empty pCMVvector (5 Ag each) using Lipofectamine Plus reagent (Invitrogen). Followingtransfection, cells were selected for 14 days in medium containing 0.6 mg/
mL G418 and stained with Giemsa. The colonies were then counted in
triplicate cultures using NIH Image software.Pathway analysis. p53 and VDR target genes were analyzed using
Ingenuity Pathway Analysis software (Ingenuity, Mountain View, CA). To
infer coassociation of common p53 and VDR target genes, this method uses
the gene identities in conjunction with controlled, vocabulary data mining
of literature associations, protein-protein interaction databases, and a
metabolism pathway database. We then combined the two pathways
manually by connecting the genes up-regulated by both p53 and VDR.
Results
In silico identification of p53 target genes. To identify novelp53 target genes within the human genome, we used acomputational approach focusing on p53 binding sites (Fig. 1).The consensus p53 binding sequence consists of two copies ofa 10-bp motif (RRRCWWGYYY) separated by 0 to 12 bases.Because several of the nucleotides comprising the motif areambiguous (R, adenine or guanine; W, adenine or thymine; Y,cytosine or thymine), this sequence has up to 28 � 28 (= 65,536)possible patterns, making generation of comparison patterns aheavy task that requires considerable computer resources andtakes much time to complete the general pattern matching. Wetherefore focused on developing a new algorithm to effectivelydeal with the highly degenerate consensus p53 binding sequenceand the variable spacer length. Instead of performing a genome-wide pattern matching with all possible p53 patterns [patterns =28 � 28 � 13 (spacer) = 851,968], we broke down this consensus
Figure 2. Representative p53 binding sites identified in this study and conserved across multiple species. The consensus p53 binding sequences are indicatedby uppercase letters: R, purines; Y, pyrimidines; W, A or T. Asterisks, sequences conserved across multiple species. Numbers, nucleotide position relative tothe transcription start site. Numbers of mismatched nucleotides in the consensus sequence (p53 ), those conserved between human and mouse sequences(human-mouse), and those conserved across multiple species (human-other species ) are shown below the column.
Cancer Research
Cancer Res 2006; 66: (9). May 1, 2006 4576 www.aacrjournals.org
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

sequence into three units, two 10-bp motifs, and a spacer,making it possible for us to design a strategy to pick up allpossible 10-bp motifs and then to search for integrated p53binding sites with a specific spacer length. We first extracted theentire set of putative p53 binding sequences in the genome. Thetotal numbers of candidate sequences for putative p53 bindingsites in the human and mouse genomes are summarized inSupplementary Tables S2 and S3. If candidate p53REs aredefined as sequences that have four or less mismatches to theconsensus p53 binding sequence along with a 0- to 12-bp spacer,there would be 4,834,075 putative p53REs in the human genome,and a putative p53RE would be present every 620 bp, onaverage. From these, we selected candidate p53REs locatedwithin 10 kb of the transcription start site of a gene; 87,659putative p53REs were estimated to be present in or around theknown genes in the human genome. This suggests that the vastmajority of the p53 binding sequences, defined only by sequenceand proximity to genes, may not be biologically functional.To further refine the set of potentially functional p53REs, we
used a comparative sequence analysis of the human and mousegenome, as the p53REs of several known p53 target genes areconserved in multiple species, including human, mouse, and rat(11), which was confirmed by our computational analysis(Supplementary Fig. S1; Supplementary Table S4). We systemati-cally selected p53REs conserved in orthologous genes betweenhuman and mouse and eliminated repetitive elements. To searchfor p53 binding sites that are conserved in orthologous genesbetween human and mouse, we developed a simple algorithmcomposed of several steps. As a first step, 7,444 genes homologousbetween human and mouse were obtained from the NCBIdatabase. By simple pattern matching, we compared the pair ofp53 binding sites located within each pair of orthologous genes. Wethen extracted 7,530 pairs of putative p53 binding sites withsequence similarity (z70% identity) between the orthologousgenes. As a second step, we eliminated the p53 binding sites thatwere part of repetitive elements. The data on the 200-bp sequencearound the p53 binding sites in the p53RE database were scannedusing RepeatMasker Open 3.0,6 and any conserved segment thatcontained known human repeat motifs within the p53REs wasexcluded from further analysis.Using this approach, we narrowed the number of candidates
for novel p53 target genes to those having conserved p53 bindingsites near their transcription start sites (Fig. 1; SupplementaryTable S5).7 Figure 2 shows six representative p53 bindingsequences that were predicted in this study and that are alsoconserved across multiple mammalian species.Validation of p53 target genes. To assess the validity of the p53
target genes predicted by the in silico analysis described above,their responsiveness to p53 was examined using semiquantitativeRT-PCR and real-time PCR (Supplementary Fig. S2; Table 1). In 17genes, the p53 binding sites contained two mismatches in theconsensus p53 binding sequences and absence of a spacer whereas20 genes contained two mismatches and a 1-bp spacer. In 64 genes,the p53 binding sites contained three mismatches and a 0-bpspacer. When Saos-2 human osteogenic sarcoma cells, which donot express endogenous p53 (22), and DLD-1 colorectal cancercells, which express a mutant form of p53 (23), were infected with
Ad-p53, Ad-p63, Ad-p73a, Ad-p73h, or Ad-LacZ, subsequentsemiquantitative RT-PCR showed that, of 101 genes analyzed, 32were up-regulated and 2 were down-regulated by introduction ofp53, p63, or p73 (Supplementary Table S6). Sixteen genes were notexpressed at all in Saos-2 or DLD-1 cells and the expression of 51was not significantly affected. The p53 target genes with p53REscontaining two mismatches and no spacer tended to show p53responsiveness more frequently than those with p53REs containingthree mismatches and no spacer or two mismatches and a 1-bpspacer (58.8% versus 28.6%, P < 0.05).VDR as a direct transcriptional target of p53. Among the
putative p53 target genes identified by our comparative sequence
6 http://www.repeatmasker.org/.7 http://www.sapmed.ac.jp/freomaru/p53.html.
Table 1. Summary of p53 and p53 family target genesidentified by in silico analysis
Gene name Fold induction
p53 p63 p73a p73h
GAS6 425.0 561.1 112.3 61.6GEM 8.1 25.2 69.3 9.1
VDR 12.1 17.5 10.3 20.2
EFNB1 9.1 3.1 5.6 5.8
SYT13* 8.9 3.3 0.8 0.6LU 5.3 3.1 31.4 14.6
HAS3 5.3 32.5 6.3 21.7
JAG2 7.3 6.7 1.0 5.4IRX5 3.9 17.0 5.9 4.5
LOXL4 2.5 17.0 11.0 15.0
DSIPI* 4.7 14.8 23.9 8.9
CSDA 3.3 12.3 2.0 8.6RABL5 3.1 10.8 3.7 3.7
MDM2 2.6 5.4 1.3 3.8
BDNF* 2.3 4.7 4.4 7.1
TCF12 2.2 3.4 1.8 2.1ACVR2 2.2 2.2 0.7 1.0
AK1 6.4 4.4 2.2 1.7
KCNB1 3.2 1.9 2.3 0.7MITF 3.2 1.3 1.9 0.5
KIAA1536 3.8 0.1 0.1 2.4
EDG4 1.7 128.0 6.9 5.6
LMO4 1.2 10.2 1.2 1.8PC4 0.8 8.7 1.6 5.7
JPH2 0.6 8.1 5.2 1.4
HUS1 0.9 7.2 2.6 4.1
NFE2L1* 0.6 5.0 2.9 0.5CLK3 1.9 4.5 0.9 3.2
KCNJ11 1.2 4.3 4.2 0.7
JMJ* 0.6 3.3 2.5 1.6
ADAMTS4 1.4 3.0 5.5 1.3IL15RA* 1.0 1.2 2.8 5.9
BMP4 0.2 0.0 0.0 0.2
SHOX2 0.1 0.0 0.0 0.1
Control
p21 28.8 36.7 6.8 4.6GADD45A 42.8 98.2 16.8 46.5
*Expression of p53 and p53 family target genes identified by in silico
analysis was examined by real-time PCR in Saos-2 cells or DLD-1 cells.
VDR Is Up-regulated by p53
www.aacrjournals.org 4577 Cancer Res 2006; 66: (9). May 1, 2006
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from
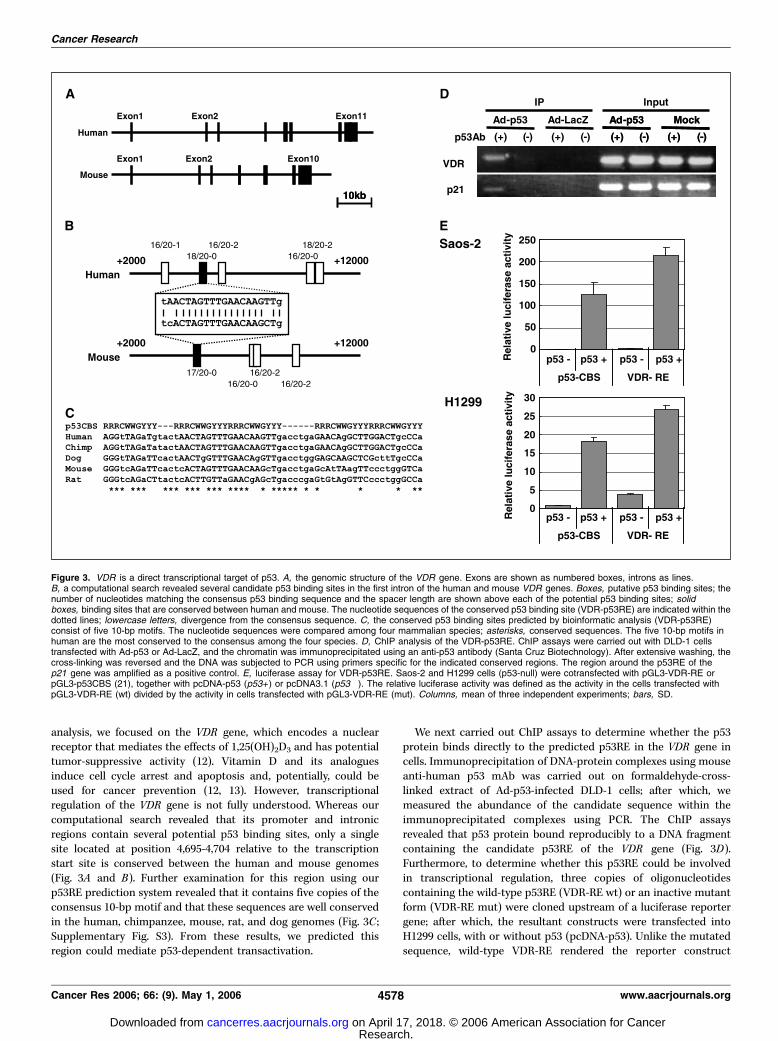
analysis, we focused on the VDR gene, which encodes a nuclearreceptor that mediates the effects of 1,25(OH)2D3 and has potentialtumor-suppressive activity (12). Vitamin D and its analoguesinduce cell cycle arrest and apoptosis and, potentially, could beused for cancer prevention (12, 13). However, transcriptionalregulation of the VDR gene is not fully understood. Whereas ourcomputational search revealed that its promoter and intronicregions contain several potential p53 binding sites, only a singlesite located at position 4,695-4,704 relative to the transcriptionstart site is conserved between the human and mouse genomes(Fig. 3A and B). Further examination for this region using ourp53RE prediction system revealed that it contains five copies of theconsensus 10-bp motif and that these sequences are well conservedin the human, chimpanzee, mouse, rat, and dog genomes (Fig. 3C ;Supplementary Fig. S3). From these results, we predicted thisregion could mediate p53-dependent transactivation.
We next carried out ChIP assays to determine whether the p53protein binds directly to the predicted p53RE in the VDR gene incells. Immunoprecipitation of DNA-protein complexes using mouseanti-human p53 mAb was carried out on formaldehyde-cross-linked extract of Ad-p53-infected DLD-1 cells; after which, wemeasured the abundance of the candidate sequence within theimmunoprecipitated complexes using PCR. The ChIP assaysrevealed that p53 protein bound reproducibly to a DNA fragmentcontaining the candidate p53RE of the VDR gene (Fig. 3D).Furthermore, to determine whether this p53RE could be involvedin transcriptional regulation, three copies of oligonucleotidescontaining the wild-type p53RE (VDR-RE wt) or an inactive mutantform (VDR-RE mut) were cloned upstream of a luciferase reportergene; after which, the resultant constructs were transfected intoH1299 cells, with or without p53 (pcDNA-p53). Unlike the mutatedsequence, wild-type VDR-RE rendered the reporter construct
Figure 3. VDR is a direct transcriptional target of p53. A, the genomic structure of the VDR gene. Exons are shown as numbered boxes, introns as lines.B, a computational search revealed several candidate p53 binding sites in the first intron of the human and mouse VDR genes. Boxes, putative p53 binding sites; thenumber of nucleotides matching the consensus p53 binding sequence and the spacer length are shown above each of the potential p53 binding sites; solidboxes, binding sites that are conserved between human and mouse. The nucleotide sequences of the conserved p53 binding site (VDR-p53RE) are indicated within thedotted lines; lowercase letters, divergence from the consensus sequence. C, the conserved p53 binding sites predicted by bioinformatic analysis (VDR-p53RE)consist of five 10-bp motifs. The nucleotide sequences were compared among four mammalian species; asterisks, conserved sequences. The five 10-bp motifs inhuman are the most conserved to the consensus among the four species. D, ChIP analysis of the VDR-p53RE. ChIP assays were carried out with DLD-1 cellstransfected with Ad-p53 or Ad-LacZ, and the chromatin was immunoprecipitated using an anti-p53 antibody (Santa Cruz Biotechnology). After extensive washing, thecross-linking was reversed and the DNA was subjected to PCR using primers specific for the indicated conserved regions. The region around the p53RE of thep21 gene was amplified as a positive control. E, luciferase assay for VDR-p53RE. Saos-2 and H1299 cells (p53-null) were cotransfected with pGL3-VDR-RE orpGL3-p53CBS (21), together with pcDNA-p53 (p53+ ) or pcDNA3.1 (p53�). The relative luciferase activity was defined as the activity in the cells transfected withpGL3-VDR-RE (wt) divided by the activity in cells transfected with pGL3-VDR-RE (mut). Columns, mean of three independent experiments; bars, SD.
Cancer Research
Cancer Res 2006; 66: (9). May 1, 2006 4578 www.aacrjournals.org
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

responsive to p53, leading to a 20-fold increase in luciferase activity(Fig. 3E ). Taken together, these findings indicate that thenucleotide sequence spanning positions 4,695-4,704 in intron 1 ofthe VDR gene constitutes a bona fide p53RE, and VDR is a directtranscriptional target of p53.Induction of VDR by p53 family genes and genotoxic
stresses. To investigate the effect of p53 on endogenous VDRinduction, the expression levels of VDR mRNA were examined inseveral cell lines. We found that there was significant inductionof VDR mRNA subsequent to infection with Ad-p53 in Saos-2 andHCT116 cells (Fig. 4A, top) and in DLD-1 and SW480 cells(Supplementary Fig. S4A), but no induction in those cellsinfected with Ad-LacZ. Interestingly, induction of VDR is moreprominent in cells transfected with Ad-p63 or Ad-p73, indicatingthat p53 family members are strong inducers of VDR. These
results were confirmed by quantitative real-time PCR, whichshowed that p53 family genes induced a 4- to 10-fold increase inthe expression of VDR mRNA over that seen in cells infectedwith control vector (Fig. 4A, bottom). Furthermore, we confirmedinduction of VDR mRNA in Saos-2, DLD-1, and H1299 cells byNorthern blot analysis, and also induction of VDR protein inSaos-2, HCT116, DLD-1, and RKO cells by Western blot analysis(Fig. 4B and C and Supplementary Fig. S4B and C). In addition,immunocytochemical analysis showed that when Saos-2 cellswere infected with Ad-p53, a significant amount of VDR proteinaccumulated in the nucleus (Fig. 4D). Similar nuclear accumu-lation of VDR protein was seen in Saos-2 cells infected with Ad-p63 and Ad-p73 but not in cells with Ad-lacZ (Fig. 4D) and inDLD-1 cells infected with Ad-p53, Ad-63, and Ad-73 (Supplemen-tary Fig. S4D).
Figure 4. Induction of VDR by p53 family genes. A, RT-PCR (top ) and real-time PCR (bottom ) analysis of VDR expression. RNA was extracted from Saos-2and HCT116 cells infected with Ad-lacZ, Ad-p53, Ad-p63g, Ad-p73a, or Ad-p73h, and tested for expression of VDR mRNA by RT-PCR. For real-time PCR, expressionlevels of VDR mRNA were normalized to those of GAPDH mRNA. Columns, mean of three independent experiments; bars, SD. B, Northern blot analysis ofVDR mRNA in Saos-2 cells infected with Ad-p53, Ad-p73h, or Ad-p63 g. An ethidium-stained gel containing 28S RNA shows the amounts of mRNA loaded in each lane.C, Western blot analysis of VDR protein in Saos-2 and HCT116 cells infected with Ad-LacZ, Ad-p53, Ad-p63g, or Ad-p73h. p21 expression served as a positive controlfor a p53 target; actin expression was used as a loading control. D, immunofluorescence analysis of Ad-p53-induced VDR protein expression. Saos-2 cells wereinfected with Ad-lacZ, Ad-p53, Ad-p63, or Ad-p73h; after which, VDR was detected using an anti-VDR antibody and an FITC-conjugated secondary antibody.The nucleus was stained with DAPI. E, induction of VDR by a chemotherapeutic drug. Expression levels of VDR mRNA in response to Adriamycin (ADR ) in RKO,HCT116, and U2OS cells were analyzed by RT-PCR (top ) and real-time RT-PCR (bottom ). RKO cells were treated with 0.2 Ag/mL Adriamycin for the indicated times(0, 12, and 24 hours). HCT116 and U2OS cells were treated with Adriamycin at the indicated concentration (0-0.5 mg/mL) for 24 hours. F, Western blot analysisof VDR and p53 proteins following treatment with Adriamycin (0-0.5 Ag/mL) for 24 hours.
VDR Is Up-regulated by p53
www.aacrjournals.org 4579 Cancer Res 2006; 66: (9). May 1, 2006
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from
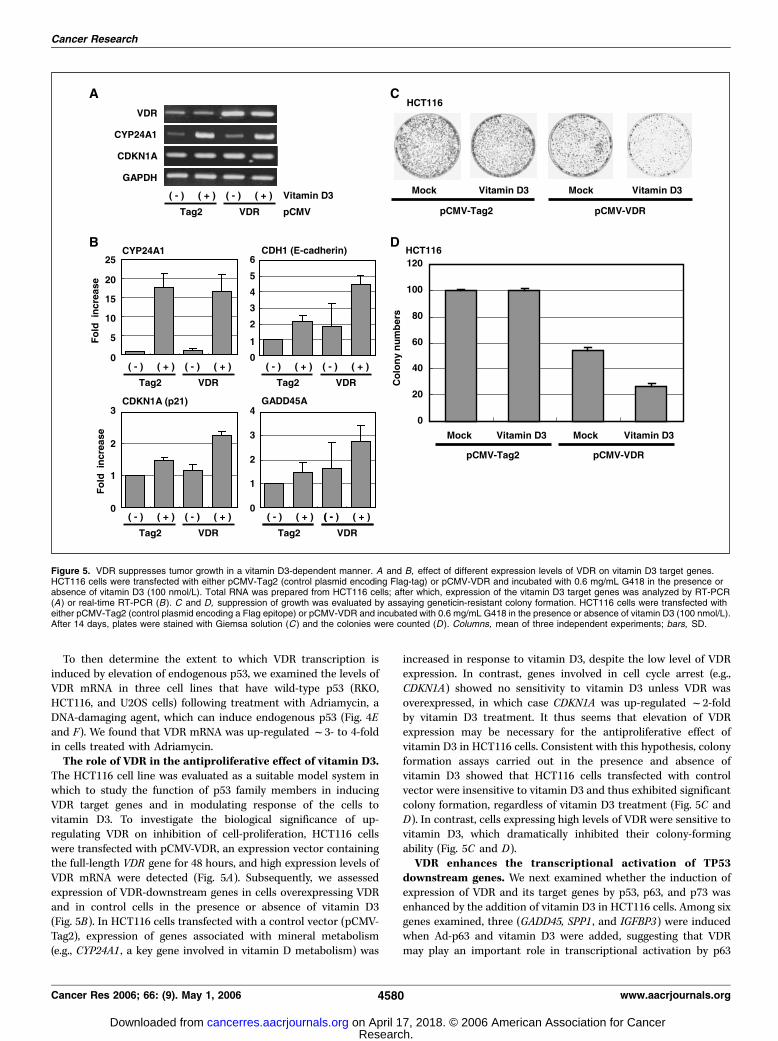
To then determine the extent to which VDR transcription isinduced by elevation of endogenous p53, we examined the levels ofVDR mRNA in three cell lines that have wild-type p53 (RKO,HCT116, and U2OS cells) following treatment with Adriamycin, aDNA-damaging agent, which can induce endogenous p53 (Fig. 4Eand F). We found that VDR mRNA was up-regulated f3- to 4-foldin cells treated with Adriamycin.The role of VDR in the antiproliferative effect of vitamin D3.
The HCT116 cell line was evaluated as a suitable model system inwhich to study the function of p53 family members in inducingVDR target genes and in modulating response of the cells tovitamin D3. To investigate the biological significance of up-regulating VDR on inhibition of cell-proliferation, HCT116 cellswere transfected with pCMV-VDR, an expression vector containingthe full-length VDR gene for 48 hours, and high expression levels ofVDR mRNA were detected (Fig. 5A). Subsequently, we assessedexpression of VDR-downstream genes in cells overexpressing VDRand in control cells in the presence or absence of vitamin D3(Fig. 5B). In HCT116 cells transfected with a control vector (pCMV-Tag2), expression of genes associated with mineral metabolism(e.g., CYP24A1 , a key gene involved in vitamin D metabolism) was
increased in response to vitamin D3, despite the low level of VDRexpression. In contrast, genes involved in cell cycle arrest (e.g.,CDKN1A) showed no sensitivity to vitamin D3 unless VDR wasoverexpressed, in which case CDKN1A was up-regulated f2-foldby vitamin D3 treatment. It thus seems that elevation of VDRexpression may be necessary for the antiproliferative effect ofvitamin D3 in HCT116 cells. Consistent with this hypothesis, colonyformation assays carried out in the presence and absence ofvitamin D3 showed that HCT116 cells transfected with controlvector were insensitive to vitamin D3 and thus exhibited significantcolony formation, regardless of vitamin D3 treatment (Fig. 5C andD). In contrast, cells expressing high levels of VDR were sensitive tovitamin D3, which dramatically inhibited their colony-formingability (Fig. 5C and D).VDR enhances the transcriptional activation of TP53
downstream genes. We next examined whether the induction ofexpression of VDR and its target genes by p53, p63, and p73 wasenhanced by the addition of vitamin D3 in HCT116 cells. Among sixgenes examined, three (GADD45, SPP1 , and IGFBP3) were inducedwhen Ad-p63 and vitamin D3 were added, suggesting that VDRmay play an important role in transcriptional activation by p63
Figure 5. VDR suppresses tumor growth in a vitamin D3-dependent manner. A and B, effect of different expression levels of VDR on vitamin D3 target genes.HCT116 cells were transfected with either pCMV-Tag2 (control plasmid encoding Flag-tag) or pCMV-VDR and incubated with 0.6 mg/mL G418 in the presence orabsence of vitamin D3 (100 nmol/L). Total RNA was prepared from HCT116 cells; after which, expression of the vitamin D3 target genes was analyzed by RT-PCR(A) or real-time RT-PCR (B ). C and D, suppression of growth was evaluated by assaying geneticin-resistant colony formation. HCT116 cells were transfected witheither pCMV-Tag2 (control plasmid encoding a Flag epitope) or pCMV-VDR and incubated with 0.6 mg/mL G418 in the presence or absence of vitamin D3 (100 nmol/L).After 14 days, plates were stained with Giemsa solution (C ) and the colonies were counted (D ). Columns, mean of three independent experiments; bars, SD.
Cancer Research
Cancer Res 2006; 66: (9). May 1, 2006 4580 www.aacrjournals.org
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

(Fig. 6). Expression of CYP24A1 and CDH1 was induced by vitaminD3 but transfection with Ad-p53, Ad-p63, and Ad-p73 did not resultin synergistic enhancement. Expression of CDKN1A was induced byAd-p53, Ad-p63, and Ad-p73 but the effect of vitamin D3 was notsignificant. To further assess the role of VDR in transactivation ofp53 target genes, we examined the effects of disrupting VDRexpression in HCT116 cells using two VDR-specific shRNA (sh-VDR) constructs. Subsequent real-time PCR and Western blotanalysis showed that VDR expression was down-regulated afterpuromycin selection of HCT116 cells that express sh-VDR (Fig. 7A).These results were confirmed using two different shRNA con-structs. We also found that GADD45, SPP1, and IGFBP3, but notCDKN1A, were suppressed by sh-VDR. The results suggest that VDRplays a key role in the transcriptional activation of a subset of p53target genes (Fig. 7B). We next examined whether down-regulationof VDR affects apoptosis induced by Ad-p53 and Ad-p63 (Fig. 7C).Down-regulation of VDR decreased the numbers of apoptoticHCT116 cells induced by Ad-p53 or Ad-p63.The data summarized above suggest that VDR plays a novel role in
the p53 signaling pathways shown in Fig. 8. Under normalphysiologic conditions, VDR induces its downstream genes associ-ated with metabolic pathways, including vitamin D3–dependentCa2+ uptake. Under genotoxic stress, however, VDR induced by p53may serve as an effector to up-regulate expression of genesassociated with cell cycle regulation, differentiation, and apoptosis.
Discussion
In this study, we used a computational approach to predict novelp53 target genes through a genome-wide search for consensus p53binding sequences. Our results indicate that putative p53 bindingsequences are present, on average, every 620 bp in the human
genome, although it is likely that most of them are nonfunctional,at least for transactivation. In this regard, we found thatcomparison of the p53 binding sequences between human andmouse is highly useful for identifying functional p53REs. In severalknown p53 target genes, the sequences surrounding the p53binding sites are highly conserved among different species,suggesting that other unknown elements (e.g., transcription factorbinding sites) are necessary for p53 protein to bind to p53REs andfunction as a transcriptional coactivator. Detailed studies offunctional regulatory elements combined with more sophisticatedcomparative genomics, including comparison across multiplespecies with varying degrees of divergence, should help in resolvingthe flexible regulatory landscape of mammalian genomes. Thereason why the sequences surrounding p53REs are conservedamong species remains unclear. Nevertheless, the informationobtained in the present study should be useful not only foridentifying p53 target genes but also for identifying transcriptionalfactors that function cooperatively with p53. Intriguingly, thesequences surrounding the functional p53RE of the VDR geneare also conserved across species (Supplementary Fig. S3). It is thuspossible that conserved sequences close to p53REs may play a rolein recruiting transcriptional coactivators such as p300, BRCA1,PML, SWI/SNF complex, and IRF-1 to activate gene expression(24–28).We found that expression of the human VDR gene is directly
up-regulated by p53, which confirmed an association between p53and VDR and helps to elucidate one aspect of the biologicalsignificance of elevated VDR expression. Vitamin D analogues,such as 1,25(OH)2D3, can inhibit the growth of various types ofmalignant cells, including breast, prostate, colon, skin, and braincancer cells, as well as myeloid leukemia cells (12, 13, 15, 29).Moreover, 1,25(OH)2D3 shows antitumor activity in animal models
Figure 6. Transcriptional activation of VDR target genes by p53, p63g, or p73. HCT116 cells were infected with Ad-LacZ, Ad-p53, Ad-p63g, and Ad-p73h for48 hours, then either mock treated (white column ) or treated with 100 nmol/L of Vitamin D3 (gray column ) for 48 hours. Total RNA was extracted for expressionanalysis by RT-PCR or real-time PCR. Columns, mean of three independent experiments; bars, SD. An unpaired t test was used to determine statistically significanteffects of vitamin D3 treatment. *, P < 0.05, significance of differences between mock and vitamin D3 treated cells.
VDR Is Up-regulated by p53
www.aacrjournals.org 4581 Cancer Res 2006; 66: (9). May 1, 2006
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

(30) and, accordingly, clinical studies to evaluate the effect ofvitamin D analogues in patients with colorectal cancer and otherneoplasms are under way (12, 13). The molecular mechanisms bywhich 1,25(OH)2D3 suppresses cell growth involve regulation ofthe cell cycle by inducing p21WAF1 and apoptosis mediated byBCL-2 and inhibitor of apoptosis protein (16, 31). In addition,vitamin D3 down-regulates the T-cell factor/h-catenin system (17).VDR has been reported to be down-regulated during tumor
progression although the molecular mechanism is not fullyunderstood. Conversely, VDR is up-regulated by a variety of growthfactors, Sp1 (32, 33), WT1 (34), and vitamin D3, and high levels ofVDR expression are associated with a good prognosis in colorectalcancer (18). In addition, Palmer et al. (19) recently reported thatVDR is down-regulated by the transcriptional repressor SNAIL.They showed that high levels of SNAIL expression are associated
with cell dedifferentiation and a low level of VDR protein. SNAILprotein interacts directly with the VDR promoter, suppressing itsactivity and thereby abolishing induction of E-cadherin and otherVDR target genes by vitamin D. The fact that VDR is a target geneof p53 suggests that alteration of p53 may be one of the causes ofVDR dysregulation in cancer.VDR pathways may partially overlap p53 pathways because
several downstream VDR target genes are also targets of p53 (12,14, 35) and because vitamin D3 can induce cell cycle arrest,differentiation, and apoptosis (Fig. 8). Under normal physiologicconditions, VDR may preferentially induce target genes involvedin Ca2+ uptake and cellular differentiation (e.g., CYP24A1). In thepresence of genotoxic stress however, VDR seems to inducetarget genes associated with cell cycle regulation and apoptosis.This difference in VDR target genes under normal and stressed
Figure 7. Disruption of VDR expression by sh-VDR. A, real-time PCR (top ) and Western blot (bottom ) analysis of VDR. HCT116 cells were transfected with controlshRNA or VDR shRNA (sh-VDR20 or sh-VDR32), cultured with 1 Ag/mL of puromycin for 48 hours, and total RNA and cell lysates were isolated. The amount of VDRmRNA was normalized to GAPDH. For Western blot analysis, membrane was blotted with anti-p53 and antiactin antibodies as control. B, sh-VDR suppressesexpression of p53 target genes. Expression of CDKN1A, GADD45, SPP1, and IGFBP3 was determined by real-time PCR; expression levels were normalized to that ofGAPDH. C, apoptosis induced by Ad-p53 or Ad-p63. HCT116 cells were infected with Ad-p53 or Ad-p63 for 48 hours and apoptotic cells were examined by flowcytometry. *, P < 0.01, apoptosis was observed in cells infected with Ad-p63 more frequently than in Ad-lacZ-infected cells.
Figure 8. Diagram of common p53 andVDR target genes. The pathways weredrawn by Ingenuity Pathway AnalysisSoftware. The p53 target genes (e.g., p21,IGFBP3 , and GADD45a ) can be activatedeither directly by p53 or via activation ofVDR.
Cancer Research
Cancer Res 2006; 66: (9). May 1, 2006 4582 www.aacrjournals.org
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

conditions may be partially explained by the p53-dependent up-regulation of VDR protein expression induced by genotoxicstress.VDR target genes also can be induced by stimulation of vitamin
D3 in the presence of an inactive p53 mutant or in p53-deficientcells (35, 36). Consistent with these findings, we observed that p63and p73 also induce VDR gene expression in cell lines. Althoughboth p63 and p73 can bind to p53REs (37), they specifically bind tothose elements containing three or four copies of the 10-bpconsensus motif separated by spacer sequences (21). This isconsistent with the fact that the p53RE in the VDR gene containsfive copies of the 10-bp motif (Fig. 3C). Recent studies have shownthat both vitamin D and p63 are associated with development anddifferentiation in several organs. Our results indicate that there isan association between p63 and VDR expression, which raises thepossibility that up-regulation of VDR by p63 at appropriate stagesin various cell types might increase their susceptibility to theproapoptotic activity of vitamin D metabolites.
In summary, we have identified novel p53 target genes bycomparative analysis of p53REs. In particular, we also showed thatVDR, which is a transcriptional regulator downstream of p53-dependent cellular signaling, is a direct transcriptional target ofp53 and plays a role in p53-mediated suppression of tumor growth.Our results indicate that this in silico approach is a powerfulmethod for identification of p53 target genes conserved amonghumans and other organisms and could serve to facilitate analysisof the function of p53 in tumorigenesis.
Acknowledgments
Received 7/20/2005; revised 1/31/2006; accepted 2/24/2006.Grant support: Grants-in-Aid for Scientific Research on Priority Areas from the
Ministry of Education, Culture, Sports, Science, and Technology (M. Toyota, K. Imai,and T. Tokino) and a grant from New Energy and Industrial Technology DevelopmentOrganization (M. Toyota).
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Drs. Joseph F. Costello and William F. Goldman for valuable discussion.
References
1. Levine AJ. p53, the cellular gatekeeper for growth anddivision. Cell 1997;88:323–31.2. Vogelstein B, Lane D, Levine AJ. Surfing the p53network. Nature 2000;408:307–10.3. Zhao R, Gish K, Murphy M, et al. Analysis of p53-regulated gene expression patterns using oligonucleo-tide arrays. Genes Dev 2000;14:981–93.4. Yu J, Zhang L, Hwang PM, et al. Identification andclassification of p53-regulated genes. Proc Natl Acad SciU S A 1999;96:14517–22.5. Oda K, Arakawa H, Tanaka T, et al. p53AIP1, apotential mediator of p53-dependent apoptosis, and itsregulation by Ser-46-phosphorylated p53. Cell 2000;102:849–62.6. Adachi K, Toyota M, Sasaki Y, et al. Identification ofSCN3B as a novel p53-inducible proapoptotic gene.Oncogene 2004;23:7791–8.7. Hoh J, Jin S, Parrado T, et al. The p53MH algorithmand its application in detecting p53-responsive genes.Proc Natl Acad Sci U S A 2002;99:8467–72.8. Dermitzakis ET, Reymond A, Lyle R, et al. Numerouspotentially functional but non-genic conserved sequen-ces on human chromosome 21. Nature 2002;420:578–82.9. Dermitzakis ET, Reymond A, Scamuffa N, et al.Evolutionary discrimination of mammalian conservednon-genic sequences (CNGs). Science 2003;302:1033–5.10. Krek A, Grun D, Poy MN, et al. CombinatorialmicroRNA target predictions. Nat Genet 2005;37:495–500.11. el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, apotential mediator of p53 tumor suppression. Cell 1993;75:817–25.12. Lamprecht SA, Lipkin M. Chemoprevention of coloncancer by calcium, vitamin D and folate: molecularmechanisms. Nat Rev Cancer 2003;3:601–14.13. Kumagai T, O’Kelly J, Said JW, Koeffler HP. VitaminD2 analog 19-nor-1,25-dihydroxyvitamin D2: antitumoractivity against leukemia, myeloma, and colon cancercells. J Natl Cancer Inst 2003;95:896–905.14. Liu M, Lee MH, Cohen M, Bommakanti M,Freedman LP. Transcriptional activation of the Cdkinhibitor p21 by vitamin D3 leads to the induced
differentiation of the myelomonocytic cell line U937.Genes Dev 1996;10:142–53.15. Danielsson C, Torma H, Vahlquist A, Carlberg C.Positive and negative interaction of 1,25-dihydroxyvita-min D3 and the retinoid CD437 in the induction ofhuman melanoma cell apoptosis. Int J Cancer 1999;81:467–70.16. Diaz GD, Paraskeva C, Thomas MG, Binderup L,Hague A. Apoptosis is induced by the active metaboliteof vitamin D3 and its analogue EB1089 in colorectaladenoma and carcinoma cells: possible implications forprevention and therapy. Cancer Res 2000;60:2304–12.17. Palmer HG, Gonzalez-Sancho JM, Espada J, et al.Vitamin D(3) promotes the differentiation of coloncarcinoma cells by the induction of E-cadherin and theinhibition of h-catenin signaling. J Cell Biol 2001;154:369–87.18. Evans SR, Nolla J, Hanfelt J, et al. Vitamin D receptorexpression as a predictive marker of biological behaviorin human colorectal cancer. Clin Cancer Res 1998;4:1591–5.19. Palmer HG, Larriba MJ, Garcia JM, et al. Thetranscription factor SNAIL represses vitamin D receptorexpression and responsiveness in human colon cancer.Nat Med 2004;10:917–9.20. Ishida S, Yamashita T, Nakaya U, Tokino T. Adeno-virus-mediated transfer of p53-related genes inducesapoptosis of human cancer cells. Jpn J Cancer Res 2000;91:174–80.21. Sasaki Y, Mita H, Toyota M, et al. Identification of theinterleukin 4 receptor a gene as a direct target for p73.Cancer Res 2003;63:8145–52.22. Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels,functional domains, and DNA damage determine theextent of the apoptotic response of tumor cells. GenesDev 1996;10:2438–51.23. Hirota Y, Horiuchi T, Akahane K. p53 antisenseoligonucleotide inhibits growth of human colon tumorand normal cell lines. Jpn J Cancer Res 1996;87:735–42.24. Zhang H, Somasundaram K, Peng Y, et al. BRCA1physically associates with p53 and stimulates itstranscriptional activity. Oncogene 1998;16:1713–21.25. Guo A, Salomoni P, Luo J, et al. The function of PMLin p53-dependent apoptosis. Nat Cell Biol 2000;2:730–6.
26. Lee D, Kim JW, Seo T, et al. SWI/SNF complexinteracts with tumor suppressor p53 and is necessaryfor the activation of p53-mediated transcription. J BiolChem 2002;277:22330–7.27. Lill NL, Grossman SR, Ginsberg D, DeCaprio J,Livingston DM. Binding and modulation of p53 byp300/CBP coactivators. Nature 1997;387:823–7.28. Tanaka N, Ishihara M, Lamphier MS, et al. Cooper-ation of the tumour suppressors IRF-1 and p53 inresponse to DNA damage. Nature 1996;382:816–8.29. Chen TC, Schwartz GG, Burnstein KL, Lokeshwar BL,Holick MF. The in vitro evaluation of 25-hydroxyvitaminD3 and 19-nor-1a,25-dihydroxyvitamin D2 as therapeu-tic agents for prostate cancer. Clin Cancer Res 2000;6:901–8.30. Harris DM, Go VL. Vitamin D and colon carcinogen-esis. J Nutr 2004;134:3463–71S.31. Chakrabarty S, Wang H, Canaff L, et al. Calciumsensing receptor in human colon carcinoma: interactionwith Ca(2+) and 1,25-dihydroxyvitamin D(3). Cancer Res2005;65:493–8.32. Jehan F, DeLuca HF. The mouse vitamin D receptoris mainly expressed through an Sp1-driven promoterin vivo . Arch Biochem Biophys 2000;377:273–83.33. Wietzke JA, Ward EC, Schneider J, Welsh J. Regula-tion of the human vitamin D3 receptor promoter inbreast cancer cells is mediated through Sp1 sites. MolCell Endocrinol 2005;230:59–68.34. Maurer U, Jehan F, Englert C, et al. The Wilms’ tumorgene product (WT1) modulates the response to 1,25-dihydroxyvitamin D3 by induction of the vitamin Dreceptor. J Biol Chem 2001;276:3727–32.35. Palmer HG, Sanchez-Carbayo M, Ordonez-Moran P,et al. Genetic signatures of differentiation induced by1a,25-dihydroxyvitamin D3 in human colon cancer cells.Cancer Res 2003;63:7799–806.36. Jiang F, Li P, Fornace AJ, Jr., Nicosia SV, Bai W. G2/Marrest by 1,25-dihydroxyvitamin D3 in ovarian cancercells mediated through the induction of GADD45 via anexonic enhancer. J Biol Chem 2003;278:48030–40.37. Levrero M, De Laurenzi V, Costanzo A, et al. Thep53/p63/p73 family of transcription factors: over-lapping and distinct functions. J Cell Sci 2000;113:1661–70.
VDR Is Up-regulated by p53
www.aacrjournals.org 4583 Cancer Res 2006; 66: (9). May 1, 2006
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from

2006;66:4574-4583. Cancer Res Reo Maruyama, Fumio Aoki, Minoru Toyota, et al. Transcriptional ActivationReceptor Gene as a Direct Target of p53-Mediated Comparative Genome Analysis Identifies the Vitamin D
Updated version
http://cancerres.aacrjournals.org/content/66/9/4574
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2006/06/26/66.9.4574.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/66/9/4574.full#ref-list-1
This article cites 37 articles, 17 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/66/9/4574.full#related-urls
This article has been cited by 11 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/66/9/4574To request permission to re-use all or part of this article, use this link
Research. on April 17, 2018. © 2006 American Association for Cancercancerres.aacrjournals.org Downloaded from