computational studies of polymorphs: principles angelo gavezzotti
DESCRIPTION
Computational studies of polymorphs: principles Angelo Gavezzotti Dipartimento di Chimica Strutturale Università di Milano. molecule intra molecular inter molecular computational box. computational box for succinic anhydride: ideal crystal. - PowerPoint PPT PresentationTRANSCRIPT

Computational studies of polymorphs:principles
Angelo GavezzottiDipartimento di Chimica Strutturale
Università di Milano

molecule intramolecular intermolecular computational box

computational box forsuccinic anhydride: ideal crystal
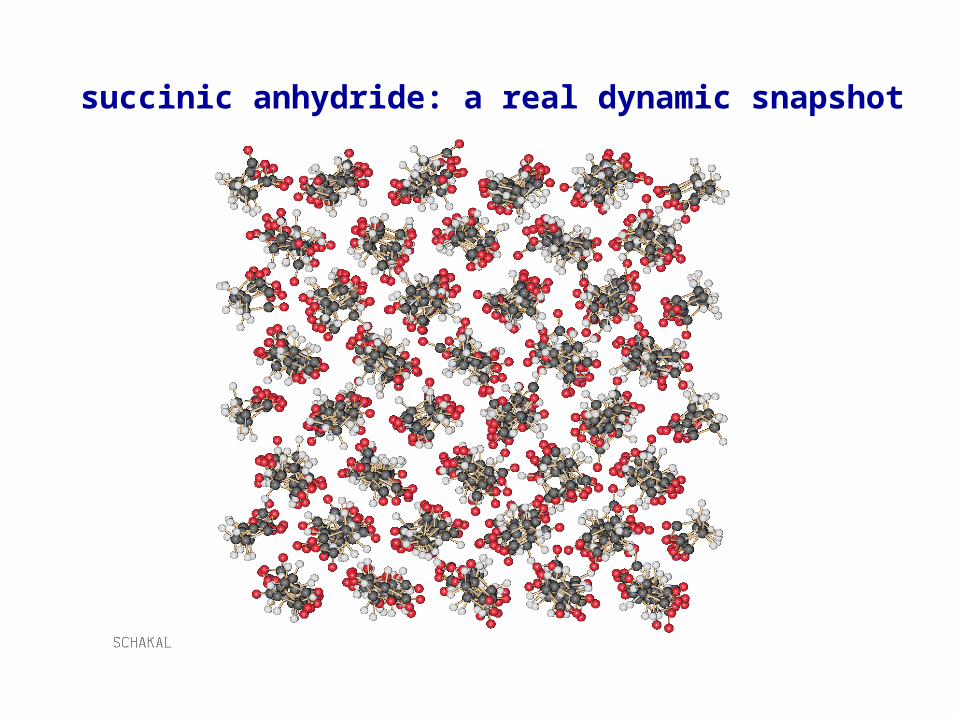
succinic anhydride: a real dynamic snapshot

succinic anhydride: liquid

The key to molecular simulation is the potential
intermolecular model potential:way of representing intermolecular interactions and of calculating quantitatively the energies involved

chemical cohesion:
electrons and nuclei electr(on)ic interactions atoms stick together but solid matter is impenetrable chemical bond(ing)

Richard P.Feynman

The Hellmann-Feynman Electrostatic Theorem:in the Born-Oppenheimer approximation, nuclei seea static, smeared out electron cloud and the forcesacting at nuclei are just coulombic forces exerted by other nuclei and by the electron cloud
if the exact wavefunction is known, any chemical bond or bonding is:electrons between nuclei, more +/- attraction than +/+ and -/- repulsion

Empirical methods do not rely on a wavefunction, but on parameter fitting.
in setting up the potential model, one needs todefine the interaction centers
i.e. , e.g., repulsion between what?

organic crystals

The whole heat of sublimation of a 15-atom moleculeis about 25% of a single C-H bond energy
Average bonding effect in a crystal: 100/15 6 kJ/atom
Strong hydrogen bond: up to 150 kJ/molWeak hydrogen bond: 30-60 kJ/mol (e.g. inamides or carboxylic acids)
How can these weak potentials be described andcalculated?


close-packing...
molecules are held at equilibrium by the balancebetween attraction and repulsion between theirelectron distributions
the arrangement of nuclei of outer atoms is the result, not the cause of this equilibrium
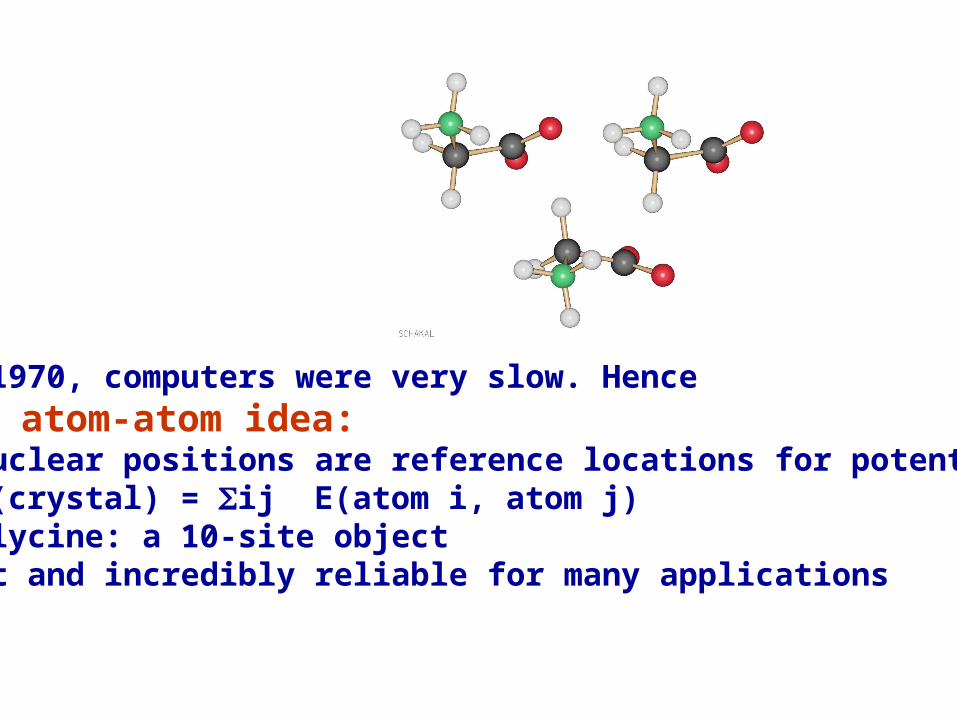
In 1970, computers were very slow. Hence the atom-atom idea:• nuclear positions are reference locations for potentials• E(crystal) = ij E(atom i, atom j)• glycine: a 10-site objectfast and incredibly reliable for many applications

kJ/mol units

Slowly, into chemical thinking creptthe atom-atom 'prejudice':• condensed phases can be understood in termsof localized atom-atom bonds, just like molecules
• that is:whenever the distance between two nuclei is short, they are joined by a chemical bond, and the sum of thesebonds is what determines the crystal structure

an alternative viewmore physical?(more convenient?)

"We shall say that there is a chemical bond between two atoms or two groups of atoms in case that the forces acting between them are such as to lead to the formation of an aggregate with sufficient stability to make it convenient for the chemist to consider it as an independent chemical species.” L.Pauling, The Nature of the Chemical Bond

The PIXEL force field: definition of electron pixels

The PIXEL idea:Evaluate electron density by quantum chemistry for the isolated moleculeEach e-pixel is a siteGlycine: a 7300-site object

Since our basis set is not complete, and our wavefunction is not dynamically adjusted to polarization, and…………….we cannot calculate Hellman-Feynman forces exactly.
We adopt a different point of view:the intermolecular interaction is divided intoa coulombic, a polarization,a dispersion and a repulsion part
Rigid electron densities, no covalent or charge transfer parts

coulombic energy:a sum of coulombic terms over pixels and nuclei(parameter-less)polarization energy:a sum of linear polarization terms over pixels with localpolarizability (one parameter)dispersion energy:a sum over pixel-pixel London-type terms (one parameter)repulsion energy:proportional to the overlap integral between electron densities(one parameter, determined by some meta-parameters)

E(PIXEL,total) = E(coul)+E(pol)+E(disp)+E(rep)
Are these 'the' coulombic, polarization, etc...energies?
No: each method defines its own energy partitioning
But comparisons of the different kinds of energeticcontribution over polymorphs or similar compoundscan be very revealing.

The PIXEL approach shifts the focus of the analysisfrom the nuclei to the electron density
From atom-atom chemical bonds to molecule-molecule chemical bonding
move from qualitative geometrical criteriato quantitative energy criteria

computer modeling of crystal polymorphism:
- analysis of existing crystal structures what are the relevant features?
- generation of new crystal structures a more technical problem
- comparing calculated properties for different polymorphs energy, entropy, density, stress tensors, morphology need accurate potential

ORDERS OF MAGNITUDE distance : 1 angstrom
species against result
Point P q = 1 electron field = 1.44 1011 V m-1
Point P q = 1 electron EPOL = -695 kJ mol-1
1 Å3 polarizability q = 1 electron q = 1 electron ECOUL = +1389 kJ mol-1
Intermolecular interactions are weak only because large energies balance out……!!

kJ/mol units

The OPiX computer program package*
• ZipOpec module: packing analysis (atom-atom energies)
• Prom/Sorter module: polymorph generator (atom-atom energies)
• Pixel module: Pixel calculations for clusters and crystals
*ask me, or write to [email protected]

Explain the crystal structure of naphthalene, naphthoquinone and naphthoic acid
•draw packing diagrams (usually a mess)• look at C...H distances, C-H...…etc• look at dipoles, quadrupoles, simple electrostatic arguments• look at C-H...O distances• look at O-H...O distances• ………

Replace atom-atom analysis by molecule-molecule analysiseach pair of molecules in the crystal is characterized by • a distance between centers of mass• coulombic, dispersion and repulsion energy
Generate many computational crystal structuresfor a given molecule• analyze the crystal energy landscape

naphthalenePIXEL energies
Ecoul+pol Edisp Erep Etot (kJ/mol)
A -8 -24 13 -18B -6 -14 6 -13C -3 -8 5 -6
PS -6 -51 25 -32(Parallel Stack)
A
B
C

Ecoul Epol Edisp Erep Etot (kJ/mol) -23 -11 -96 58 -72 -23 -11 -91 56 -68 -7 -7 -88 42 -61
naphthalene

distance Ecoul+pol Edisp Erep Etot (kJ/mol)
4.07 -5 -46 14 -37 4.38 -3 -37 10 -30
naphthoquinone

E,F
C
O…H distance Ecoul+pol Edisp Erep Etot (kJ/mol)
C 2.52 -18 -9 12 -15 E,F 2.47,2.49 -17 -10 15 -12
naphthoquinone

exptl

2-naphthoic acid
FA
E
Ecoul+pol Edisp Erep Etot (kJ/mol)
A -224 -14 184 -54 E -11 -5 6 -10 F -4 -8 7 -4

B
C
Ecoul+pol Edisp Erep Etot (kJ/mol)
B -9 -38 20 -27 C -6 -17 5 -18
2-naphthoic acid

Naphthoic acid, ab projectionCorrugation to avoid CH…CH electrostatic repulsion

some conventional wisdom:
hydrogen bonds always formtherefore, they are the strongest interactionin a crystal
neighbor molecules are attracted to one anotherthe closer, the more attracted

Ecoul Epol Edisp Erep Etot stack -26 -7 -50 21 -62 H-bond -41 -15 -19 44 -31
H-bond is not most stabilizing interaction
2,6-dinitro-3-acetaminotoluene

A
B
things nearby are not always things stabilizingglycine zwitterion: Ecoul Epol Edisp Erep EtotRef-A -123 -27 -11 83 -78Ref-B +51 -10 -9 12 +44
ref

computational polymorphs of parabanic acid
Ecoul Epol Edisp Erep Etot, Pixel Etot, DMAexptl -107.9 -42.9 -72.0 134.1 -88.7 8 -111.4 1fc5 -106.9 -45.0 -81.4 138.4 -94.8 1 -109.0 2af28 -86.1 -32.1 -87.3 113.7 -91.9 2 -106.8 5
Same total energy for large differences in partial contributions


at the other extreme:large energy differences for small geometry differences
acetic acid hydrogen-bonded dimer

crystal polymorphism:a matter of energy landscapes instead of energy points

caffeine* 1,7-dimethylxanthine
*No ordered anhydrous crystal structure is knownUllrich Griesser knows everything

kJ/mol units

kJ/mol units
1,7-dimethylxanthine 100 crystal structures

THE THERMODYNAMIC RULE free energy: enthalpy and entropy differences
ENTHALPYIntermolecular bonding Electric interactions between molecularelectron densities
ENTROPYif no bonds are broken or formed:mainly from different collective vibration modes

enthalpy-entropy compensation
higher density: better intermolecular contactsenthalpy more stabilizingvibrational modes more energetic, less entropy
lower density:loss of cohesion, enthalpy less stabilizingvibrational modes more accessible, more entropy


Jack Dunitz, Sally Pricefor useful advice on the Pixel method
Ulrich Griesser for data on caffeine
Lucia CarlucciX-ray and DSC work on xanthines
Giuseppe Filippinilattice dynamical results
molecular graphics: SCHAKAL (E. Keller)