conformational transition pathway in the allosteric …conformational transition pathway in the...
TRANSCRIPT

Conformational transition pathway in the allostericprocess of human glucokinaseJian Zhang†‡, Chenjing Li†‡, Kaixian Chen†, Weiliang Zhu†, Xu Shen†§, and Hualiang Jiang†§¶
†Center for Drug Discovery and Design, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Shanghai Institutes for BiologicalSciences, and Graduate School, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; and ¶School of Pharmacy, East ChinaUniversity of Science and Technology, Shanghai 200237, China
Communicated by William N. Lipscomb, Harvard University, Cambridge, MA, July 10, 2006 (received for review April 18, 2006)
Glucokinase (GK) is an important enzyme for regulating bloodglucose levels and a potentially attractive target for diabetes of theyoung type 2 and persistent hyperinsulinemic hypoglycemia ofinfancy. To characterize the conformational transition of GK fromthe closed state to the superopen state, a series of conventionalmolecular dynamics (MD) and target MD (TMD) simulations wereperformed on both the wild-type enzyme and its mutants. Two10-ns conventional MD simulations showed that, although theallosteric site of GK is �20 Å away from the active site, the activatoris able to enhance the activity of the enzyme through conforma-tional restriction. Fourteen TMD simulations on GK and five of itsmutants revealed a reliably conformational transition pathway.The overall conformational transition includes three stages, andthree likely stable intermediate states were identified by freeenergy scanning for the snapshots throughout the pathway. Theconformational transition feature revealed by our TMD simulationsrationalized several important mutagenesis and kinetic data. Re-markably, the TMD simulations predicted that Y61S, I159A, A201R,V203E, and V452S mutations, which have not been investigated sofar, may facilitate the opening process of GK. These predictionsalso have been verified by mutagenesis and kinetic analyses in thisstudy. These observations are beneficial to understanding themechanism of GK regulation and designing the compounds fortreating metabolic diseases.
molecular dynamics � mutagenesis
G lucokinase (GK) (hexokinase IV or D) is a glycolytic enzymethat plays an important role in blood sugar regulation related
to the glucose utilization and metabolism in the liver and pancreatic�-cells. Serving as a glucose sensor, GK controls plasma glucoselevels (1). GK plays a dual role in reducing plasma glucose levels:Glucose-mediated activation of the enzyme in hepatocyte facili-tates hepatic glucose update and glycogen synthesis, while that inpancreatic �-cells ultimately induces insulin secretion. Both of theseeffects in turn reduce plasma glucose levels (2, 3). Clinical evidencehas shown that GK variants with decreased and increased activitiesare associated with diabetes of the young type 2 (MODY2) andpersistent hyperinsulinemic hypoglycemia of infancy (PHHI), re-spectively (4–7). Therefore, GK might be an important target forthese two metabolic diseases.
GK belongs to the hexokinase family (8). Although GK shares ahigh degree of sequence homology with other family members suchas hexokinase I (�54.4%), it differs from other members in variousaspects, such as binding glucose with lower affinity, positive coop-erative behavior with regard to glucose binding, and lack ofinhibition for glucose-6-phosphate (9). Remarkably, the activityof GK is represented by a sigmoidal glucose kinetic curve insteadof the Michaelis–Menten kinetic curve of nonallosteric hexokinases(10). Structurally, GK is a monomeric enzyme containing a singleactive site. Therefore, it must behave as a distinct allosteric mech-anism differing from that of other well studied allosteric enzymes,which consist of several subunits that are activated and inactivatedin a concerted manner (11). To illustrate the allosteric mechanismof GK, numerous mutagenesis studies have been conducted (9,
12–14). Recently, several activators of GK with the same effect asthe activating mutations were also discovered (15, 16). The x-raycrystallographic determination addressed the crystal structures ofGK at the closed (active) and superopen (inactive) states, implyingthat GK exhibits a global conformational transition between thesetwo states, and such a global alteration in enzyme conformationmay be associated with the special allosteric characteristics of GK(17). Thus, a rigorous mechanistic study of the global conforma-tional transition is critical to understanding the regulation mecha-nism of GK and for development of new therapeutic approaches fortype 2 diabetes and persistent hyperinsulinemic hypoglycemia ofinfancy.
Although the crystal structures of GK have been determined,and facilitated a proposal for a mnemonical mechanism for thecooperativity of GK, several fundamental questions are still unan-swered. The allosteric site is �20 Å away from the active site, so howdoes the activator enhance the GK activity? How does GK makeprogress in the large-scale conformational change from the closedto the superopen state? Why can some mutations activate theallosteric traits? Unfortunately, because it is difficult to catch thestructures of the intermediate states in the pathway for the globalconformational transition of GK, the investigation of the aboveproblems in atomic detail through experimental methods is stillintractable. Computational simulation, with its extremely high timeresolution and atomic level representation, has been increasinglyused in understanding the complex conformational features ofproteins and predicting structural preferences (18–24). To ourknowledge, the global alteration in GK conformation has not beencomputationally simulated.
Here, we report the results of the conformational transition ofGK addressed by a series of conventional molecular dynamics(CMD) and target molecular dynamics (TMD) simulations. Byrunning two 10-ns CMD simulations in aqueous solution, we showthat, although the allosteric site of GK is �20 Å away from theactive site, the activator is able to enhance the activity of the enzymethrough conformational restriction. Afterward, by running 14 TMDsimulations on GK and five of its mutants, we find a reliablyconformational transition pathway from the closed to the supero-pen state. The overall conformational transition includes threestages, and three likely stable intermediate states were identified byfree energy scanning for the snapshots throughout the pathway.The important components relevant to the conformational changeof GK were addressed by analyzing the detailed structures of theTMD trajectories. The simulation results are in accordance with therecent findings of mutagenesis experiments and related kineticstudies. In addition, the TMD simulations predicted that Y61S,I159A, A201R, V203E, and V452S mutations, which have not been
Conflict of interest statement: No conflicts declared.
Abbreviations: GK, glucokinase; MD, molecular dynamics; CMD, conventional MD; TMD,target MD.
‡J.Z and C.L. contributed equally to this work.
§To whom correspondence should be addressed. E-mail: [email protected] [email protected].
© 2006 by The National Academy of Sciences of the USA
13368–13373 � PNAS � September 5, 2006 � vol. 103 � no. 36 www.pnas.org�cgi�doi�10.1073�pnas.0605738103
Dow
nloa
ded
by g
uest
on
Mar
ch 4
, 202
0
investigated so far, may facilitate the opening process of GK.Remarkably, we verified these predictions by mutagenesis andkinetic analyses, which are also reported in this work. Theseobservations are beneficial to understanding the mechanism of GKregulation and designing the compounds for treating metabolicdiseases.
Results and DiscussionThe main goal of this study was to investigate the allosteric processof human GK, which is more associated with the conformationalchange from the closed to the superopen state. To this end, fourmodels for CMD and TMD simulations were designed. In model I,two 10-ns CMD simulations (A1 and A2) were conducted on theclosed state and its complex with an activator of the free protein toprobe how the activator affects the conformational change of GK.In model II, six TMD simulations (B1–B6) were performed by usinga force constant of 0.1 kcal�mol�1�Å�2 with different initial veloc-ities to explore the minimal energy pathway between the closed andsuperopen states. In model III, to verify the reliability of thepathway for the conformational alternation of GK, three additionalTMD simulations (C1–C3) were carried out with force constants of0.2, 0.5, and 1.0 kcal�mol�1�Å�2, respectively. In model IV, K56A,E256A, V62M, E158A, and I159A mutants were constructed forGK, and five TMD simulations were performed on these mutants.In total, 2 CMD and 14 TMD simulations were performed on GKand its mutants. More detailed information for these moleculardynamics (MD) simulations is provided in Table 2, which ispublished as supporting information on the PNAS web site.
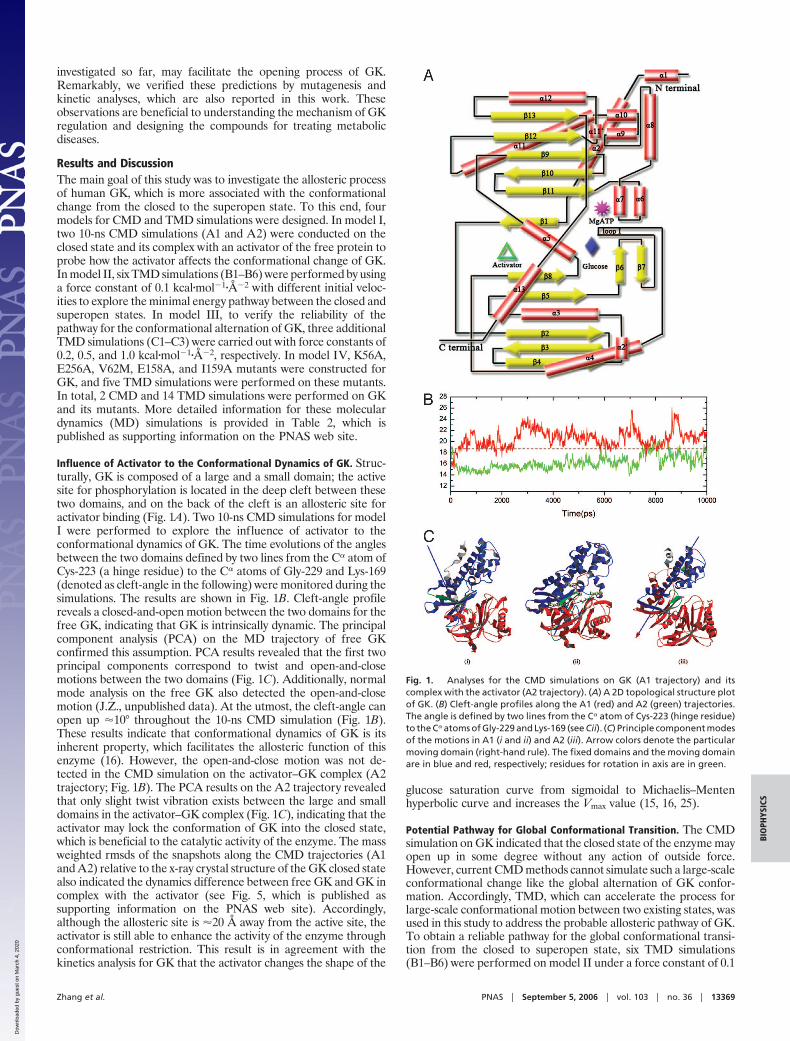
Influence of Activator to the Conformational Dynamics of GK. Struc-turally, GK is composed of a large and a small domain; the activesite for phosphorylation is located in the deep cleft between thesetwo domains, and on the back of the cleft is an allosteric site foractivator binding (Fig. 1A). Two 10-ns CMD simulations for modelI were performed to explore the influence of activator to theconformational dynamics of GK. The time evolutions of the anglesbetween the two domains defined by two lines from the C� atom ofCys-223 (a hinge residue) to the C� atoms of Gly-229 and Lys-169(denoted as cleft-angle in the following) were monitored during thesimulations. The results are shown in Fig. 1B. Cleft-angle profilereveals a closed-and-open motion between the two domains for thefree GK, indicating that GK is intrinsically dynamic. The principalcomponent analysis (PCA) on the MD trajectory of free GKconfirmed this assumption. PCA results revealed that the first twoprincipal components correspond to twist and open-and-closemotions between the two domains (Fig. 1C). Additionally, normalmode analysis on the free GK also detected the open-and-closemotion (J.Z., unpublished data). At the utmost, the cleft-angle canopen up �10° throughout the 10-ns CMD simulation (Fig. 1B).These results indicate that conformational dynamics of GK is itsinherent property, which facilitates the allosteric function of thisenzyme (16). However, the open-and-close motion was not de-tected in the CMD simulation on the activator–GK complex (A2trajectory; Fig. 1B). The PCA results on the A2 trajectory revealedthat only slight twist vibration exists between the large and smalldomains in the activator–GK complex (Fig. 1C), indicating that theactivator may lock the conformation of GK into the closed state,which is beneficial to the catalytic activity of the enzyme. The massweighted rmsds of the snapshots along the CMD trajectories (A1and A2) relative to the x-ray crystal structure of the GK closed statealso indicated the dynamics difference between free GK and GK incomplex with the activator (see Fig. 5, which is published assupporting information on the PNAS web site). Accordingly,although the allosteric site is �20 Å away from the active site, theactivator is still able to enhance the activity of the enzyme throughconformational restriction. This result is in agreement with thekinetics analysis for GK that the activator changes the shape of the
glucose saturation curve from sigmoidal to Michaelis–Mentenhyperbolic curve and increases the Vmax value (15, 16, 25).
Potential Pathway for Global Conformational Transition. The CMDsimulation on GK indicated that the closed state of the enzyme mayopen up in some degree without any action of outside force.However, current CMD methods cannot simulate such a large-scaleconformational change like the global alternation of GK confor-mation. Accordingly, TMD, which can accelerate the process forlarge-scale conformational motion between two existing states, wasused in this study to address the probable allosteric pathway of GK.To obtain a reliable pathway for the global conformational transi-tion from the closed to superopen state, six TMD simulations(B1–B6) were performed on model II under a force constant of 0.1
Fig. 1. Analyses for the CMD simulations on GK (A1 trajectory) and itscomplex with the activator (A2 trajectory). (A) A 2D topological structure plotof GK. (B) Cleft-angle profiles along the A1 (red) and A2 (green) trajectories.The angle is defined by two lines from the C� atom of Cys-223 (hinge residue)to the C� atoms of Gly-229 and Lys-169 (see Cii). (C) Principle component modesof the motions in A1 (i and ii) and A2 (iii). Arrow colors denote the particularmoving domain (right-hand rule). The fixed domains and the moving domainare in blue and red, respectively; residues for rotation in axis are in green.
Zhang et al. PNAS � September 5, 2006 � vol. 103 � no. 36 � 13369
BIO
PHYS
ICS
Dow
nloa
ded
by g
uest
on
Mar
ch 4
, 202
0

kcal�mol�1�Å�2 with different initial velocities (Table 2). The sixTMD simulations almost produced the same conformational tran-sition pathway as demonstrated by the weighted rmsds among thesix trajectories (see Table 3, which is published as supportinginformation on the PNAS web site), indicating that the initialvelocities for TMD simulations did not affect the transition path-way. To probe the influence of the force constant on the confor-mational transition pathway, three additional TMD simulations(C1–C3) were conducted on model III under force constants of 0.2,0.5, and 1.0 kcal�mol�1�Å�2, respectively. In comparison with tra-jectories B1–B6, trajectories C1–C3 did not give qualitatively dif-ferent results (see Table 4 and Fig. 6, which are published assupporting information on the PNAS web site). All these datasuggest that the conformational transition pathway is reliable. In thefollowing, we use trajectory B1 for further discussion.
The TMD-simulated conformational transition pathway of GKfrom the closed to superopen state is shown in Fig. 2. Although thetransition took place continuously, the overall conformationaltransition can be roughly divided into three stages (Fig. 2). Theconformational transition begins with a stage in which the cleft-angle gradually opens up �9° during the time period from 0 to 160ps. Above 10 ns, CMD simulation on free GK also addressed theconformational alternation of this stage (Fig. 1). Because it isdifficult to overcome the energy barrier produced by the stronginteraction of loop I between �4 and �7 in the small domain with�1 and �11 in the large domain (Fig. 1A), the CMD simulation isnot able to go through the whole pathway of the conformationaltransition. As the TMD simulation goes on, the interaction betweenthe small and large domains is ruptured, and the cleft-angle opensup �40°. At the second stage (360–700 ps), the �6 and �7 strandsin the small domain continually relax their positions within therange of local space after GK undergoes a rapid opening process.At �550 ps, the �6 and �7 strands move away from the allostericsite, turn their orientation, and completely drift into the solvent.During this stage, these two �-strands begin to unfold their secondstructures into coils. In the third period (700–1,000 ps), the cleft-
angle reaches the superopen state, and the �6 and �7 strandscompletely convert into coil structures and expose the solvent.Meanwhile, the �13 helix departs from the small domain. Ingeneral, the large domain moves almost as a rigid body during theconformational transition; however, the movement for the smalldomain is more complicated, which is associated with the biologicalfunction of GK as we will discuss below.
Possible Intermediate States in the Transition Pathway. The x-raycrystal structures of GK indicate that the conformational transitionfrom the closed to superopen state is a complicated process.According to the mnemonical mechanism of GK regulation pro-posed by Kamata et al. (17) and the behaviors of other hexokinases(25), stable intermediate states should exist in the conformationaltransition pathway to decrease cost for the large-scale conforma-tional change and increase the opportunity of regulation. Never-theless, such kinds of intermediate states of GK have not beendetected by experimental methods (17). To explore the possibleintermediate states for GK, the free energy landscape along theconformational transition pathway was calculated by using theMM-PBSA-Nmode method encoded in AMBER (Version 7.0)(26). The result is shown in Fig. 3A, which indicates that there arethree energy wells (denoted P1, P2, and P3) in the pathway at timeperiods of �130–160, �370–550, and �695–705 ps. Conformationsin these energy wells are possibly stable intermediate states. Toverify the possibility of these intermediate states further, snapshotswere isolated from the B1–B6 trajectories and superimposed withthe crystal structures of other hexokinases. The result indicated thatconformations in the P1 well are very similar to the x-ray crystalstructure of the open state of hexokinase I (Fig. 3B) (25). This resultshows that the P1 state may be a real stable intermediate state ofGK, i.e., the open state of GK, which is a desired outcome (17).Conformations dropped into the second and third wells (P2 and P3states) might also be the intermediate states in the conformationaltransition pathway, as we will discuss below.
Fig. 2. The conformational transition pathway of GKfrom the closed to the superopen state. The centerimage is the time dependence of the cleft-angle be-tween the two domains. Around the center image, sixsnapshots are extracted from the B1 trajectory at thetimes of 150 (1), 320 (2), 400 (3), 600 (4), and 800 (5) ps,and superopen state (6), respectively. The loop I be-tween �4 and �7 segments is in red; the �6 and �7strands are in cyan; helix �13 is in yellow.
13370 � www.pnas.org�cgi�doi�10.1073�pnas.0605738103 Zhang et al.
Dow
nloa
ded
by g
uest
on
Mar
ch 4
, 202
0

Important Components Along the Transition Pathway and Mutagen-esis Validation. The x-ray crystal structure of the closed stateindicates that there are two direct hydrogen bonds (H-bonds)between the small and large domains, i.e., Lys-56���Asn-166 andGlu-256���Lys-169. TMD simulation revealed that these two H-bonds are stable before 160 ps (the first stage in the pathway ofconformational transition) (Fig. 3Ci and see also Fig. 7, which ispublished as supporting information on the PNAS web site). As theTMD went on, these two H-bonds ruptured, and the cleft of GKopened up more. Interestingly, the point of the H-bonds breaking(�160 ps) exactly corresponds to the predicted intermediate (P1)state, as indicated by the free energy calculation (Fig. 3A). Thisresult highlights that these two H-bonds play important roles in theconformational transition. Most likely, rupture force needed tobreak these two H-bonds caused the energy barrier from P1 to thesuperopen state. To map the functionality of these two H-bonds,two additional TMD simulations (D1 and D2) on K56A and E256Amutants were performed. The results indicated that K56A mutationdid not change the conformational transition pathway, whereasE256A mutation delayed the opening process of the two domains(Fig. 4 A and B). These results are consistent with the mutagenesisstudies, which revealed that K56A has little impact on the kineticproperty of GK, but E256A severely decreases the affinity andcooperativity of GK for glucose (27). Structural analysis indicatedthat, although K56A mutation abolished the H-bond of Lys-56���Asn-166, the H-bond of Glu-256���Lys-169 was still maintainedbefore 160 ps during the TMD simulation. However, E256Amutation changed the local structure of the interaction interfacebetween the two domains. In addition, Asn-166 formed a newH-bond with Ala-256, and the side chain of Lys-169 deeply insertedinto the cleft between the two domains (see Fig. 8, which ispublished as supporting information on the PNAS web site). Thisstructural adjustment enhanced the interaction between the twodomains, slowing down the opening process of the cleft anddiscouraging glucose from binding with GK.
Several experiments indicated that the catalytic cycle of GK is a
slow process, implying that the conformational transition corre-sponding to this cycle goes slowly (28, 29). Remarkably, the freeenergy calculation captured another probable intermediate statefrom the second stage in the conformational transition pathway (P2state in Fig. 3A). Accordingly, the structural details of the P2 statewere carefully analyzed. The result revealed that, at the secondstage, Tyr-61, Val-62, Ala-201, Val-203, and Val-452 form a ‘‘hy-drophobic pocket,’’ and the side chains of Asp-158 and Ile-159 inthe �6 strand insert into the hydrophobic pocket through hydro-phobic interactions, which lock the movement between the twodomains of GK (Fig. 3Cii). Therefore, the energy barrier, caused bythe P2 state, might be the reason underlying the slow process of thecatalytic cycle. To confirm this finding, three additional TMDsimulations (D3–D5) on V62M, D158A, and I159A mutants of GKwere conducted. The first two mutations enhanced the hydrophobicinteractions, and the last mutation reduced the hydrophobic inter-actions between the two domains at the second stage. Indeed,V62M and D158A mutations delayed the opening process from theclosed to the superopen state, as indicated by the profiles of thedistance between Ile-159 and Val-452 vs. simulation time (Fig. 4 Cand D). This result is in agreement with the mutagenesis results,which revealed that V62M and D158A are activating mutationswith decreased S0.5 values (concentration of glucose at which GKshows the half activity of Vmax) (9, 30). TMD simulation predictedthat I159A mutation might facilitate the opening process of GK(Fig. 4E). However, there are no experimental data for thismutation. We constructed the clone of this mutant and obtained thepure protein by expression (detailed procedures for the proteinexpression, purification, and enzymatic assay are described inSupporting Materials and Methods, which is published as supportinginformation on the PNAS web site). Kinetic assay indicated I159Ais an inactivating mutation: Its S0.5 increased �8 fold, and Vmaxdecreased in comparison with the wild-type GK (Table 1). TheTMD simulations also indicate the importance of residues Tyr-61,Ala-201, Val-203, and Val-452 in stabilizing the structure of P2 (Fig.3Cii). Also, no mutation data have been reported for these residues.
Fig. 3. Free energy landscape and intermediate states along the conformational transition pathway. (A) Free energy profiles of the conformations along theB1 trajectory. The shadows indicate three minimal free energy wells. (B) rmsd evolutions of the conformations along the six TMD trajectories (B1–B6) withthe closed (i) and the open (ii) states of hexokinase I. (C) Important interactions for the three possible intermediate states [P1 (i), P2 (ii), and P3 (iii)] on theconformational transition pathway.
Zhang et al. PNAS � September 5, 2006 � vol. 103 � no. 36 � 13371
BIO
PHYS
ICS
Dow
nloa
ded
by g
uest
on
Mar
ch 4
, 202
0

To verify the simulation result, these residues were mutated intopolar amino acids, resulting in Y61S, A201R, V203E, and V452Smutants, for weakening the hydrophobic interactions of Asp-158and Ile-159 with the hydrophobic pocket (Fig. 3Cii). Indeed, kineticdeterminations revealed that all these mutations reduced theenzymatic activity of GK with increased S0.5 and decreased Vmaxvalues (Table 1), suggesting that, similar to I159A, these mutationsfacilitate the opening process of GK. These mutagenesis analysesverified the TMD prediction, demonstrating again the reliability ofthe conformational transition pathway and corresponding interme-diate states derived from the TMD simulations.
The �13 helix is surrounded by �5, �3, �4, and �5, and Tyr-215on �5 serves as a gatekeeper to prevent the �13 helix from escapingthe small domain. This process produced the third intermediatestate in the conformational transition pathway (P3 in Fig. 3C). Asthe cleft opened more after the P2 state, the �6 and �7 strandsgradually departed from the allosteric site, changed orientation,and drifted into the solvent. At this stage, the small domain twisted
mildly relative to the large domain, vacating space for the �13 helixto depart from the small domain. The detailed process of the �13helix being released from the small domain is shown in Fig. 9, whichis published as supporting information on the PNAS web site.During this process, Tyr-215 on �5 prevents the �13 helix fromleaving the small domain, and the residues in the �13 helix thatdirectly contact Tyr-215 are Val-455 and Ala-456. It can be pre-dicted from the TMD results that mutations by residues withbulkier side chains may inhibit the release of �13 helix, thusprolonging the conformational transition process. Two suchmutants of GK (V455M and A456V) were found in the metabolicdisease patients. In line with the TMD results, kinetic analysis ofthese two mutants showed substantially decreased S0.5 values (6, 7).Accordingly, the release of the �13 helix from the small domain,especially the contacts of Val-455 and Ala-456 of �13 with Tyr-215of �5, is also an important component for the conformationaltransition.
Implications in the Allosteric Mechanism. Based on the x-ray struc-tures of GK, Kamata et al. (17) devised a kinetic model for theallosteric mechanism of GK regulation and predicted that inter-mediate states likely exist in the conformational transition pathwaybetween closed and superopen states. However, these intermediatestates of GK have not yet been detected by experimental methods.A very important mechanistic conclusion from the present TMDsimulations of GK is that there are three intermediate states (P1–P3in Fig. 3A) in the conformational transition pathway from the closedto superopen state. The interaction components that caused theenergy barriers corresponding to the intermediate states have beenaddressed by the TMD simulations, and all of them are in agree-ment with the available data of mutagenesis and kinetic analyses.Remarkably, the TMD simulations revealed that Ile-159, which hasnot been investigated before, might play an essential role during theconformational transition of GK, and its mutation is an inactivatingone. By using mutagenesis and kinetic analyses, we verified theTMD prediction.
Based on the present MD simulation analyses, we propose anatomic mechanism for the kinetic model of the GK allostericprocess. As shown in Fig. 3A, GK may exist in three intermediatestates (P1–P3) between the closed and superopen states. The TMDsimulations revealed that GK undergoes conformational transitionfrom closed to superopen state through three stages (Fig. 2). Duringthe first stage, GK opens up from the closed state to P1 state, whichis the open state as predicted by Kamata et al. (17). The 10-ns CMDsimulation indicated that closed–open conformational change be-tween the closed and P1 states is an intrinsic property of GK. Thisprocess corresponds to the ‘‘fast cycle’’ for GK catalytic mechanismproposed by Kamata et al. (17). Two H-bonds formed betweenLys-56 and Asn-166 and between Glu-256 and Lys-169 are themajor components to stabilize the structure of the P1 state. Duringthe second stage, conformation changes from P1 to P2. At thisstage, the important component to produce the energy barrier for
Fig. 4. Impacts of mutations K56A (A), E256A (B), V62M (C), D158A (D), andI159A (E) to the conformation transition pathway of GK. Angles in A and B aredefined in the same way as in Fig. 1B. Distances in C–E are measured between theC� atoms of I159 and V452.
Table 1. Kinetic analyses for GK and its mutants
Mutants
Proteinconc.,mg�ml
Vmax,mOD�min
Km for ATP,mM
S0.5 forglucose, mM nH†
LGK2-WT 2.24 181.51 � 6.78 0.14 � 0.01 11.64 � 0.83 1.40 � 0.12Y61S mutant 2.24 60.33 � 3.83 0.08 � 0.01 116.86 � 7.46 1.23 � 0.10I159A mutant 2.24 23.32 � 1.20 0.05 � 0.01 95.16 � 7.32 0.96 � 0.04A201R mutant 2.24 149.62 � 8.50 0.20 � 0.01 18.14 � 0.26 0.76 � 0.06V203E mutant 2.24 39.26 � 2.85 0.55 � 0.07 201.23 � 18.76 0.58 � 0.04V452S mutant 2.24 86.61 � 1.78 0.23 � 0.01 28.11 � 0.73 0.96 � 0.02
Data are means � SEM for wild-type hLGK2 and hLGK2 mutants. The results are means of kinetic analyses of threeindependent experiments of wild-type hLGK2 and hLGK2 mutants.†nH is the Hill number.
13372 � www.pnas.org�cgi�doi�10.1073�pnas.0605738103 Zhang et al.
Dow
nloa
ded
by g
uest
on
Mar
ch 4
, 202
0

the conformational change is the hydrophobic interactions betweenthe side chains of Asp-158 and Ile-159 and the hydrophobic pocketrounded by Tyr-61, Val-62, Ala-201, Val-203, and Val-452. Thethird stage is the release of the �13 helix from the small domain, andthe energy barrier of this step is mainly from the resistant interac-tion between the �13 helix (Val-455 and Ala-456) and the �5 helix(Tyr-215). The existence of the P2 and P3 states is in agreementwith the conclusion deduced from the glucose phosphorylation thatthe conformational changes between the open and superopen statesare slower than the closed–open conformational change (17).
MethodsSimulation Systems. Initial coordinates for the closed and superopenstates of GK were taken from the x-ray crystal structures (17)(Protein Data Bank ID codes 1V4S and 1V4T). The missingresidues were repaired by using the loop search method in theHomology module of Insight II (Accelrys, San Diego, CA). Forthe simulations of GK (or mutated GK) in aqueous solution, theprotein was first put into a suitably sized box, in which the minimaldistance from the protein to the box wall was 1.5 nm. Then, the boxwas solvated with the SPC water model (31). The protein�watersystem was submitted to energy minimization. Afterward, counte-rions were added to the system to provide a neutral simulationsystem. The whole system was subsequently minimized again. Thecharges of the atoms of activator N-thiazol-2-yl-2-amino-4-fluoro-5-(1-methylimidazol-2-yl)thiobenzamide were calculated by usingthe RESP method (32) encoded in the AMBER program suite (26)at the level of RHF�6-31G*. Covalent and nonbonded parametersfor the activator atoms were assigned by analogy or throughinterpolation from those already present in the AMBER forcefield (33).
CMD and TMD Simulations. CMD simulations were carried out byusing the AMBER package (Version 7.0) with constant tempera-ture, constant pressure (NPT), and periodic boundary conditions.
The Amber Parm99 force field (33) was applied for the proteins.The particle mesh Ewald method (34) was used to calculate thelong-range electrostatics interactions. The nonbonded cutoff wasset to 12.0 Å, and the nonbonded pairs were updated every 25 steps.The SHAKE method (35) was applied to constrain all covalentbonds involving H atoms. Each simulation was coupled to a 300 Kthermal bath at 1.0 atm of pressure (1 atm � 101.3 kPa) by applyingthe algorithm of Berendsen et al. (36). The temperature andpressure coupling parameters were set as 0.2 and 0.05 ps, respec-tively. An integration step of 2 fs was set up for the MD simulations.The TMD simulations were performed by using the Sander moduleencoded in AMBER (Version 7.0) (26). Restraint force was appliedonto each initial structure to bias the trajectories toward eachrespective target structure. The integration step for TMD simula-tions was set to 1 fs. The detailed procedure of TMD simulation canbe found in ref. 37 and in several recent applications (38, 39).
Kinetic Analyses for GK and its Mutants. The DNA sequence ofhuman liver GK isoform2 (hLGK2) was prepared by total genesynthesis (Sangon, Shanghai, China), and then cloned into pQE-30(Qiagen, Valencia, CA) to make the recombinant plasmid pQE-30-hLGK2. The Y61S, I159A, A201R, V203E, and V452S mutantclones were prepared with the QuikChange site-directed mutagen-esis kit (Stratagene, La Jolla, CA) by using pQE-30-hLGK2 as thetemplate. Expression and purification of the recombinant proteinswere preformed according to His�Bind Kits (Novagen, San Diego,CA). The type hLGK2 and mutant proteins were then subjected tokinetic analysis as described by Grimsby et al. (16).
For further details, see Supporting Materials and Methods.
We thank the Shanghai Supercomputing Center and Computer NetworkInformation Center of Chinese Academy of Sciences for allocation ofcomputing time. This work was supported by Special Fund for the MajorState Basic Research Project of China Grant 2002CB512802, NationalNatural Science Foundation of China Grant 30525024, and ShanghaiScience and Technology Commission Grant 03DZ19228.
1. Al-Hasani, H., Tschop, M. H. & Cushman, S. W. (2003) Mol. Interventions 3, 367–370.2. Van Schaftingen, E. (1994) Diabetologia 37, Suppl. 2, S43–S47.3. Matschinsky, F. M., Glaser, B. & Magnuson, M. A. (1998) Diabetes 47, 307–315.4. Vionnet, N., Stoffel, M., Takeda, J., Yasuda, K., Bell, G. I., Zouali, H., Lesage,
S., Velho, G., Iris, F., Passa, P., et al. (1992) Nature 356, 721–722.5. Froguel, P., Zouali, H., Vionnet, N., Velho, G., Vaxillaire, M., Sun, F., Lesage,
S., Stoffel, M., Takeda, J., Passa, P., et al. (1993) N. Engl. J. Med. 328, 697–702.6. Glaser, B., Kesavan, P., Heyman, M., Davis, E., Cuesta, A., Buchs, A., Stanley, C. A.,
Thornton, P. S., Permutt, M. A., Matschinsky, F. M., et al. (1998) N. Engl. J. Med. 338,226–230.
7. Christesen, H. B., Jacobsen, B. B., Odili, S., Buettger, C., Cuesta-Munoz, A.,Hansen, T., Brusgaard, K., Massa, O., Magnuson, M. A., Shiota, C., et al. (2002)Diabetes 51, 1240–1246.
8. Grossbard, L. & Schimke, R. T. (1966) J. Biol. Chem. 241, 3546–3560.9. Gloyn, A. L., Odili, S., Zelent, D., Buettger, C., Castleden, H. A., Steele, A. M.,
Stride, A., Shiota, C., Magnuson, M. A., Lorini, R., et al. (2005) J. Biol. Chem.280, 14105–14113.
10. Storer, A. C. & Cornish-Bowden, A. (1976) Biochem. J. 159, 7–14.11. MacRae, I. J., Segel, I. H. & Fisher, A. J. (2002) Nat. Struct. Biol. 9, 945–949.12. Xu, L. Z., Harrison, R. W., Weber, I. T. & Pilkis, S. J. (1995) J. Biol. Chem. 270,
9939–9946.13. Xu, L. Z., Zhang, W., Weber, I. T., Harrison, R. W. & Pilkis, S. J. (1994) J. Biol. Chem.
269, 27458–27465.14. Moukil, M. A., Veiga-da-Cunha, M. & Van Schaftingen, E. (2000) Diabetes 49, 195–201.15. Brocklehurst, K. J., Payne, V. A., Davies, R. A., Carroll, D., Vertigan, H. L.,
Wightman, H. J., Aiston, S., Waddell, I. D., Leighton, B., Coghlan, M. P., et al.(2004) Diabetes 53, 535–541.
16. Grimsby, J., Sarabu, R., Corbett, W. L., Haynes, N. E., Bizzarro, F. T., Coffey,J. W., Guertin, K. R., Hilliard, D. W., Kester, R. F., Mahaney, P. E., et al. (2003)Science 301, 370–373.
17. Kamata, K., Mitsuya, M., Nishimura, T., Eiki, J. & Nagata, Y. (2004) Structure (London)12, 429–438.
18. Daura, X., Jaun, B., Seebach, D., van Gunsteren, W. F. & Mark, A. E. (1998) J. Mol. Biol.280, 925–932.
19. Duan, Y., Wang, L. & Kollman, P. A. (1998) Proc. Natl. Acad. Sci. USA 95,9897–9902.
20. Simmerling, C., Strockbine, B. & Roitberg, A. E. (2002) J. Am. Chem. Soc. 124,11258–11259.
21. Snow, C. D., Nguyen, H., Pande, V. S. & Gruebele, M. (2002) Nature 420, 102–106.22. Mayor, U., Guydosh, N. R., Johnson, C. M., Grossmann, J. G., Sato, S., Jas, G. S.,
Freund, S. M. V., Alonso, D. O. V., Daggett, V. & Fersht, A. R. (2003) Nature421, 863–867.
23. Zangi, R., de Vocht, M. L., Robillard, G. T. & Mark, A. E. (2002) Biophys. J. 83, 112–124.24. Yechun, X., Jianhua, S., Xiaomin, L., Weiliang, Z., Kaixian, C., Jianpeng, M. &
Hualiang, J. (2005) Proc. Natl. Acad. Sci. USA 102, 5403–5407.25. Aleshin, A. E., Zeng, C., Bartunik, H. D., Fromm, H. J. & Honzatko, R. B. (1998) J. Mol.
Biol. 282, 345–357.26. Case, D. A., Cheatham, T. E., III, Darden, T., Gohlke, H., Luo, R., Merz, K. M., Jr.,
Onufriev, A., Simmerling, C., Wang, B. & Woods, R. (2005) J. Comput. Chem. 26,1668–1688.
27. Pilkis, S. J., Weber, I. T., Harrison, R. W. & Bell, G. I. (1994) J. Biol. Chem. 269,21925–21928.
28. Lin, S. X. & Neet, K. E. (1990) J. Biol. Chem. 265, 9670–9675.29. Neet, K. E., Keenan, R. P. & Tippett, P. S. (1990) Biochemistry 29, 770–777.30. Miller, S. P., Anand, G. R., Karschnia, E. J., Bell, G. I., LaPorte, D. C. & Lange,
A. J. (1999) Diabetes 48, 1645–1651.31. Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F. & Hermans, J. (1981)
in Intermolecular Forces, ed. Pullman, B. (Reidel, Dordrecht, The Netherlands),pp. 331–342.
32. Bayly, C. I., Cieplak, P., Cornell, W. D. & Kollman, P. A. (1993) J. Phys. Chem. 97,10251–10512.
33. Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M.,Spellmeyer, D. C., Fox, T., Caldwell, J. W. & Kollman, P. A. (1995) J. Am. Chem. Soc.117, 5179–5197.
34. Darden, T., York, D. & Pedersen, L. (1993) J. Chem. Phys. 98, 10089–10092.
35. Ryckaert, J. P., Ciccotti, G. & Berendsen, J. C. (1977) J. Comput. Phys. 23, 327–341.36. Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak,
J. R. (1984) J. Chem. Phys. 81, 3684–3690.37. Schlitter, J., Engels, M., Kruger, P., Jacoby, E. & Wollmer, A. (1993) Mol. Simul. 10,
291–308.38. Kong, Y., Shen, Y., Warth, T. E. & Ma, J. (2002) Proc. Natl. Acad. Sci. USA 99,
5999–6004.39. Kong, Y., Ma, J., Karplus, M. & Libscomb W. N. (2006) J. Mol. Biol. 356, 237–247.
Zhang et al. PNAS � September 5, 2006 � vol. 103 � no. 36 � 13373
BIO
PHYS
ICS
Dow
nloa
ded
by g
uest
on
Mar
ch 4
, 202
0