congenital heart disease and pulmonary hypertension n.4... · charles burger, md chair, pulmonary...
TRANSCRIPT

Congenital Heart Disease and Pulmonary Hypertension
ial ournal o t e Pulmonary Hypertension AssociationWinter 2013 Vol 11, No 4
ISSN 1933-088X

Editorial Advisory Board
Editor-in-ChiefErika Berman Rosenzweig, MDAssociate Professor of Clinical Pediatrics
in MedicineDirector, Pulmonary Hypertension CenterColumbia University College of
Physicians and SurgeonsMorgan Stanley Children’s Hospital of
New YorkNew York, New York
Immediate Past Editor-in-ChiefRichard Channick, MDDirector, Pulmonary Hypertension
ProgramMassachusetts General HospitalBoston, Massachusetts
Editor-in-Chief ElectMyung Park, MDAssociate Professor of MedicineDirector, Pulmonary Vascular Diseases
ProgramDivision of CardiologyUniversity of Maryland School of
MedicineBaltimore, MarylandSection Editor
Associate EditorsCharles Burger, MDChair, Pulmonary and Critical Care MedicineAssociate Professor of MedicineMedical Director, PH ClinicMayo Clinic FloridaJacksonville, Florida
Omar A. Minai, MDDepartment of Pulmonary, Allergy and
Critical Care MedicineCleveland ClinicCleveland, Ohio
Fernando Torres, MDDirector, Pulmonary Hypertension ClinicUniversity of Texas Southwestern Medical
CenterDallas, TexasSection Editor
R. James White, MD, PhDAssociate Professor of Medicine,
Pharmacology, and PhysiologyDivision of Pulmonary and Critical Care
MedicineUniversity of RochesterRochester, New York
Editorial BoardKelly Chin, MDAssistant Professor of MedicineUniversity of Texas Southwestern Medical
CenterDallas, TexasSection Editor
Curt Daniels, MDDirector, Adult Congenital Heart Disease
and Pulmonary Hypertension ProgramNationwide Children’s HospitalThe Ohio State UniversityColumbus, Ohio
Harrison Farber, MDProfessor of MedicineDirector, Pulmonary Hypertension CenterBoston University/Boston Medical CenterBoston, Massachusetts
Paul Forfia, MDAssistant Professor of MedicineMedical Director, Pulmonary
Hypertension ProgramHospital of the University of PennsylvaniaPhiladelphia, Pennsylvania
Sean Gaine, MD, PhDDirector, National Pulmonary
Hypertension UnitMater Misericordiae University HospitalUniversity College DublinDublin, Ireland
Dunbar Ivy, MDProfessor of PediatricsUniversity of ColoradoDenver Health Sciences CenterDenver, ColoradoSection Editor
Martha Kingman, NPUniversity of TexasSouthwestern Medical CenterDallas, TexasSection Editor
Richard Krasuski, MDDirector of Adult Congenital Heart
Disease ServicesCleveland ClinicCleveland, Ohio
Deborah Jo Levine, MDAssociate ProfessorPulmonary and Critical Care MedicineLung Transplant PulmonologistDirector of Pulmonary Hypertension CenterDivision of Cardiothoracic SurgeryUniversity of Texas Health Science
Center at San AntonioSan Antonio, TexasSection Editor
Ioana Preston, MDCo-Director, Pulmonary Hypertension
CenterTufts Medical CenterBoston, MassachusettsSection Editor
Sean Studer, MDDirector of Lung Transplantation and
Director of Pulmonary HypertensionNewark Beth Israel Medical CenterNewark, New Jersey
Program DescriptionThe mission of Advances in PulmonaryHypertension is to serve as the premiereforum for state of the art information re-garding diagnosis, pathophysiology, andtreatment of pulmonary hypertension. The2008 Dana Point revision of the WorldHealth Organization Classification servesas a guide to categories of pulmonary hy-pertension addressed in Advances in Pul-monary Hypertension. While focusing onWHO Group 1 PAH, the other categories(Group 2, pulmonary venous hyperten-sion; Group 3, associated with chroniclung disease and/or hypoxemia; Group 4,pulmonary embolic hypertension; Group 5,miscellaneous) are also addressed. Thismission is achieved by a combination ofinvited review articles, roundtable discus-sions with panels consisting of interna-tional experts in PH, and original contri-butions. In addition, a special section inselected issues entitled “Profiles in Pulmo-nary Hypertension” recognizes major con-tributors to the field and serves as an in-spiring reminder of the rich and collegialhistory of dedication to advancing thefield.
Objectives● Provide up-to-date information regard-
ing diagnosis, pathophysiology, andtreatment of pulmonary hypertension.
● Serve as a forum for presentation anddiscussion of important issues in thefield, including new paradigms of dis-ease understanding and investigationaltrial design.
● Recognize and preserve the rich historyof individuals who have made majorcontributions to the field via dedicationto patient care, innovative research, andfurthering the mission of the PH com-munity to cure pulmonary hypertension.
The Scientific Leadership Council of the Pulmonary Hypertension AssociationThe scientific program of the Pulmonary Hypertension Association is guided by the association’s Scientific Leadership Council. The Council includes the following health care professionals.
Richard Channick, MDChair, SLCMassachusetts General HospitalBoston, Massachusetts
Karen A. Fagan, MDChair Elect, SLCUniversity of South AlabamaMobile, Alabama
Vallerie V. McLaughlin, MDImmediate Past Chair, SLCUniversity of MichiganAnn Arbor, Michigan
Charles Burger, MDMayo Clinic College of MedicineJacksonville, Florida
Murali Chakinala, MDWashington University School of
MedicineSt. Louis, Missouri
Serpil Erzurum, MDChair, Research CommitteeCleveland Clinic Lerner College of
Medicine of Case Western ReserveUniversity
Cleveland, Ohio
Marc Humbert, PhD, MDHopital Antoine BeclereClamart, France
Dunbar Ivy, MDUniversity of Colorado Denver Health
Sciences CenterDenver, Colorado
Zhi-Cheng Jing, MDFu Wai Heart HospitalShanghai, China
Dinesh Khanna, MDUniversity of MichiganAnn Arbor, Michigan
James Klinger, MDThe Warren Alpert Medical School of
Brown UniversityProvidence, Rhode Island
Irene M. Lang, MDMedical University of ViennaVienna, Austria
Stephen C. Mathai, MD, MHSJohns Hopkins UniversityBaltimore, Maryland
Michael Mathier, MDChair, PHA Online UniversityUniversity of Pittsburgh Medical CenterPittsburgh, Pennsylvania
John Newman, MDVanderbilt University School of
MedicineNashville, Tennessee
Ronald J. Oudiz, MDChair, Insurance and Advocacy
CommitteeUCLA School of MedicineTorrance, California
Myung Park, MDEditor-in-chief, Advances in Pulmonary
HypertensionUniversity of Maryland Medical CenterBaltimore, Maryland
Ioana Preston, MDTufts Medical CenterBoston, Massachusetts
Tomas Pulido, MDNational Heart InstituteMexico City, Mexico
Erika Berman Rosenzweig, MDColumbia UniversityNew York, New York
Robert Schilz, DO, PhDChair, SLC Education CommitteeCase Western Reserve University School
of MedicineCleveland, Ohio
Virginia Steen, MDGeorgetown University Medical CenterWashington, DC
Duncan Stewart, MDThe Ottawa HospitalOttawa, ON, Canada
Sean Studer, MDNewark Beth Israel Medical CenterNewark, New Jersey
Fernando Torres, MDUT Southwestern Medical CenterDallas, Texas
Terence Trow, MDYale School of MedicineNew Haven, Connecticut
Joel A. Wirth, MDTufts University School of MedicineBoston, Massachusetts
Roham Zamanian, MDStanford School of MedicineStanford, California
LiaisonsTraci Stewart, RN, MSNChair, PH Professional NetworkUniversity of Iowa Hospitals and ClinicsIowa City, Iowa
Melisa Wilson, ARNP, ACNP-BCChair-elect, PH Professional NetworkOrlando Heart Center DowntownOrlando, Florida
Rita OrthPHA Board MemberDanville, California
SLC Distinguished AdvisorsDavid B. Badesch, MDUniversity of Colorado Health Sciences
CenterAurora, Colorado
Robyn Barst, MD†Columbia University, emeritus
Bruce H. Brundage, MDDavid Geffen School of Medicine at
UCLA, emeritusPalm Desert, California
C. Gregory Elliott, MDUniversity of Utah School of MedicineMurray, Utah
Michael D. McGoon, MDMayo ClinicRochester, Minnesota
The mission of the Scientific LeadershipCouncil is to provide medical and scien-tific guidance and support to the PHA for:
● Developing and disseminating knowledgefor diagnosing and treating pulmonaryhypertension.
● Advocating for patients with pulmonaryhypertension.
● Increasing involvement of basic andclinical researchers and practitioners.
More information on PHA’s Scientific Lead-ership Council and associated committeescan be found at www.PHAssociation.org/SLC/
† deceased

154 Editor’s MemoErika Berman Rosenzweig, MD
154 Guest Editor’s MemoRichard Krasuski, MD
157 Article Reviews
162 PHPN: Transitioning the Pediatric Pulmonary HypertensionPatient
165 Advances in Pulmonary Hypertension CME Section
166 Anatomy of Congenital Heart Disease Lesions Associated WithPulmonary Arterial HypertensionTodd L. Kiefer, MD; Thomas Bashore, MD
171 The Essential Role of Imaging in the Evaluation of PatientsWith Pulmonary Arterial Hypertension in Association WithCongenital Heart DiseaseGiancarlo Scognamiglio, MD, PhD; Sonya V. Babu-Narayan, MRCP,PhD; Michael B. Rubens, FRCR; Michael A. Gatzoulis, MD, PhD, FESC,FACC; Wei Li, MD, PhD, FESC, FACC
183 Targeted Pulmonary Arterial Hypertension Therapies and aCombined Medical-Surgical Approach for Congenital HeartDisease PatientsWarren A. Zuckerman, MD; Erika B. Rosenzweig, MD
189 The Role of Catheter-Based and Surgical Treatments inPatients With Congenital Heart Disease and PulmonaryHypertensionJamil A. Aboulhosn, MD, FACC, FSCAI
196 Self-Assessment Examination
198 Pulmonary Hypertension Roundtable
207 Ask the Expert: Congenital Heart Disease With AssociatedPulmonary Arterial Hypertension. Who and When to Operate:A Therapeutic Dilemma
212 News to Use
Cover Image: Fontan procedure.Illustration by Joseph Pangrace. Reprintedwith permission, Cleveland Clinic Center forMedical Art & Photography ©2011-2013.All rights Reserved.
Advances in
Pulmonary HypertensionOfficial Journal of the Pulmonary Hypertension Association Winter 13 Vol 11, No 4
Contents PublisherPulmonary HypertensionAssociationVallerie McLaughlin, MD, Board ChairRino Aldrighetti, President and CEO
PHA OfficePulmonary Hypertension Association801 Roeder Road, Ste 1000Silver Spring, MD 20910301-565-3004; 301-565-3994 (fax)
Publishing OperationsDeborah L. McBride, Managing
EditorMcBride Strategic [email protected]
Copyright ©2013 by Pulmonary Hy-pertension Association. All rightsreserved. None of the contents maybe reproduced in any form whatso-ever without the written permissionof PHA.
Advances in Pulmonary Hyperten-sion is available online atwww.PHAOnlineUniv.org/journal
Advances in Pulmonary Hyperten-sion is circulated to cardiologists,pulmonologists, rheumatologists, andother selected healthcare profession-als by the Pulmonary HypertensionAssociation. The contents of the arti-cles are independently determined bythe Editor-in-Chief and the EditorialAdvisory Board.
Advances in Pulmonary Hypertension: Author GuidelinesGeneral InformationAdvances in Pulmonary Hypertension: Official Journal of the Pulmo-nary Hypertension Association is a quarterly publication directed byan editorial board of renowned experts with the oversight of theAssociation’s Scientific Leadership Council. Its mission is to helpphysicians in their clinical decision making by informing them ofimportant trends affecting their practice and providing an analysis ofthe impact of new findings and current information in the peer-reviewed literature. Each article is reviewed and approved by mem-bers of the Editorial Advisory Board.
While most articles are invited by the editorial board, the followingsubmissions will be considered for publication:
• Reviews that summarize and synthesize peer-reviewed literatureto date on relevant topics
• Letters to the Editor• Clinical case studies
Submitted manuscripts are reviewed by the editorial board and otherexperts in the field. Acceptance of manuscripts is determined byfactors such as quality, relevance, and perceived value to clinicaldecision making.
Manuscript Preparation andSubmission ProcessSubmissions should be sent via e-mail as an attached Word document
to the Editor-in-Chief, Erika Berman Rosenzweig, MD, at [email protected] Manuscripts should be double-spaced and follow AMAstyle. Full-length manuscripts should not exceed 4,000 words includ-ing references. References should be limited to 50 entries. No morethan 5 figures should accompany the manuscript. Acceptable fileformats are .gif, .tif, and .jpg. Each figure should be a separate file andfigure legends should appear at the end of the manuscript. Each figureshould be cited by number in the manuscript. Tables should beself-explanatory and details of the table should not be repeated in themanuscript. Tables should be prepared as part of the Word document.No more than 3 tables should be included with the manuscript.References should conform to AMA style and be numbered consec-utively in the text. Reference numbers should be placed in parenthesesat the end of the relevant sentence.
Accepted manuscripts will be edited for clarity, spelling, punctuation,grammar, and consistency with AMA style.
CopyrightAuthors must confirm they have rights to all material submitted byincluding a copyright release form with the manuscript. The form canbe downloaded from the PHA Web site, www.PHAssociation.org.Authors acknowledge the material has not been previously publishednor is being considered for publication elsewhere simultaneously withconsideration by Advances in Pulmonary Hypertension.
Any previously published figures, tables, etc. must contain a full
credit-line from the copyright owner. Authors are responsible forobtaining permission to reproduce such material and must provide thatmaterial in reproducible form.
Manuscripts are accepted for exclusive publication in Advances inPulmonary Hypertension and will be copyrighted by the PulmonaryHypertension Association.
Conflict of Interest DisclosuresA statement of any and all grant, contract, and industrial support orproprietary interests of the author(s) related to the subject matter mustbe submitted with the manuscript.
ChecklistAuthors should be certain to include the following with themanuscript:
1. Title page listing all authors with their academic degree(s) andaffiliations.
2. Corresponding author contact information including e-mail andphone number.
3. Copyright release form signed by all authors4. Conflict of Interest forms for all authors5. List of approximately 5 key words for indexing purposes6. Summary of the paper not exceeding 250 words
153Advances in Pulmonary Hypertension

Editor’s memo
Pulmonary Hypertension Associated with Congenital HeartDisease: It’s Not All the Same
With improvements inmedical and surgicaltherapeutics over thepast two decades, thenumber of adults livingwith congenital heart
disease now exceeds the number of chil-dren. Whether as a result of excessivepulmonary blood flow in childhood, or re-lated to post-capillary obstruction, manyof these adults have associated pulmonaryhypertension (APAH-CHD) and requireadvanced management strategies. Theevaluation of adults with APAH-CHD,which is often accompanied by complexcardiac lesions including single ventricleanatomy, can be extremely challenging.Presently, with the emergence of noveltargeted PAH agents, medical-surgicalapproaches to APAH-CHD patients are
rapidly evolving. In this edition of Ad-vances, Guest Editor Dr Rich Krasuskicalls upon authors to highlight the latestadvances in the management of PAH inadults with structural heart disease. Fromthe basics on anatomy for the non-congenital heart expert, to imaging, novelmedical and interventional therapeutics,and the importance of transition pro-grams, experts cover it all in this issue.
On a personal note, this edition of Ad-vances represents the final journal pub-lished during my term as Editor-in-Chief.I want to extend a tremendous thanks to theeditorial board and to Deb McBride for theirdedication and assistance during my term. Itis with great pleasure that I am able to handoff the position to a close colleague andfriend, Dr Myung Park. Dr Park’s expertisein the field, and enthusiasm for helping the
PH community, will undoubtedly serve herwell in this new position as Editor-in-Chief.Congratulations, Myung!
Finally, I want to express my deepestgratitude for the years of mentorship byDr Robyn J. Barst who recently lost herown battle with illness, but won so manyfor the PH community. While many of uswill miss her dearly, I am certain thatDr Robyn Barst’s legacy will continueto impact the field for many years tocome.
Signing off,
Erika Berman Rosenzweig, MDDirector, Pulmonary HypertensionCenterColumbia University, College ofPhysicians and Surgeons
Guest Editor’s Memo
This issue of Advancesin Pulmonary Hyper-tension focuses on themanagement of pa-tients with congenitalheart disease and asso-ciated pulmonary hy-
pertension. More than a million adults inthe United States have congenital heartdefects, and adults now outnumber chil-dren with congenital heart defects. Manyof these patients present complex caseswith unique anatomical defects and verycomplicated interplay between pulmonaryblood flow and pulmonary vascular resis-tance. Up to 40% of congenital heart pa-tients are at risk for developing pulmo-nary hypertension and up to 10% actuallydevelop it. Half of these can progress to
Eisenmenger syndrome, a condition re-sulting in profound cyanosis from venousto systemic blood flow, when shunt le-sions go unrecognized and untreated.
The goal of the following articles is toprovide a broad overview of the congen-ital heart lesions most likely to result inpulmonary vascular disease, so the pul-monary hypertension specialist can be-come aware of the presenting features andthe unique management strategies re-quired. A multitude of treatments are nowavailable for these patients, includingmedical therapies targeting the pulmonaryvasculature, percutaneous devices thatcan be used to close abnormal intracardiacand vascular communications, balloonsand stents that can be used to increaseblood flow when the circulation is com-
promised, and even catheter-based valveprostheses that can be implanted withoutrequiring surgery. A variety of surgicalprocedures also exist that can target themany different heart defects and valveabnormalities and dramatically alter thenatural history of these disorders. Oftenwhat is utilized is a hybrid technique withseveral different specialists working to-gether to improve the quantity and qualityof life for this challenging patient popu-lation. It is indeed an exciting time toprovide care for this constantly expandinggroup of patients!
Richard Krasuski, MDDirector of Adult Congenital HeartDisease ServicesThe Cleveland Clinic
154 Advances in Pulmonary Hypertension

INDICATION
Tyvaso is a prostacyclin vasodilator indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to improve exercise ability.
Studies establishing effectiveness included predominately patients with NYHA Functional Class III symptoms and etiologies of idiopathic or heritable
PAH (56%) or PAH associated with connective tissue diseases (33%).
The effects diminish over the minimum recommended dosing interval of 4 hours; treatment timing can be adjusted for planned activities.
While there are long-term data on use of treprostinil by other routes of administration, nearly all controlled clinical experience with inhaled treprostinil
has been on a background of bosentan (an endothelin receptor antagonist) or sildenafil (a phosphodiesterase type 5 inhibitor). The controlled clinical
experience was limited to 12 weeks in duration.
Tyvaso is a registered trademark of United Therapeutics Corporation.
All other trademarks and registered trademarks are the property of their respective owners.
© 2012. United Therapeutics Corporation, Inc. All rights reserved. US/TYV/OCT11/096
ONLY inhaled prostacyclin analogue approved as an
add-on to oral PAH monotherapy1
52% of patients improved 6MWD by greater than 20 m3
Improvement in 6MWD at peak (20 m) and trough (14 m) exposure3
Dosing regimen fits into patients’ schedules
Short treatment sessions: just 2 to 3 minutes, 4x daily2
Set up once daily1,2
— One plastic ampule per day—no need to replace
ampule for each treatment session1
— About 5 minutes a day for device preparation—once in
the morning, and the device is ready to go all day2
Treatment timing can be adjusted for planned activities1
Adverse events
The most common adverse events seen with Tyvaso in ≥4% of
PAH patients and more than 3% greater than placebo in the
placebo-controlled clinical study were cough, headache, throat
irritation/pharyngolaryngeal pain, nausea, flushing, and syncope1
IMPORTANT SAFETY INFORMATION
Tyvaso is intended for oral inhalation only. Tyvaso is approved for use only
with the Tyvaso Inhalation System
The safety and efficacy of Tyvaso have not been established in patients with
significant underlying lung disease (such as asthma or chronic obstructive
pulmonary disease) and in patients under 18 years of age. Patients with
acute pulmonary infections should be carefully monitored to detect any
worsening of lung disease and loss of drug effect
Tyvaso may increase the risk of bleeding, particularly in patients
receiving anticoagulants
In patients with low systemic arterial pressure, Tyvaso may cause
symptomatic hypotension. The concomitant use of Tyvaso with
diuretics, antihypertensives, or other vasodilators may increase the
risk of symptomatic hypotension
Hepatic or renal insufficiency may increase exposure to Tyvaso and
decrease tolerability. Tyvaso dosage adjustments may be necessary if
inhibitors of CYP2C8 such as gemfibrozil or inducers such as rifampin
are added or withdrawn
The most common adverse events seen with Tyvaso in ≥4% of PAH patients
and more than 3% greater than placebo in the placebo-controlled clinical
study were cough (54% vs 29%), headache (41% vs 23%), throat irritation/
pharyngolaryngeal pain (25% vs 14%), nausea (19% vs 11%), flushing (15% vs
<1%), and syncope (6% vs <1%)
Tyvaso should be used in pregnancy only if clearly needed. Caution should
be exercised when Tyvaso is administered to nursing women
Please see brief summary of Full Prescribing Information
on following page. For more information, please see Full
Prescribing Information, Patient Package Insert, and the
Tyvaso Inhalation System Instructions for Use manual.
These items are available at www.tyvaso.com.
6MWD=6-minute walk distance. MLWHF=Minnesota Living With Heart Failure. NYHA=New York Heart Association. WHO=World Health Organization.
References: 1. Tyvaso [package insert]. Research Triangle Park, NC: United Therapeutics Corporation; 2011. 2. Tyvaso [patient package insert]. Research Triangle Park, NC: United Therapeutics Corporation; 2011. 3. McLaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55(18):1915-1922.
www.tyvaso.com www.livingpah.com 1-877-UNITHERRequest a visit from a Tyvaso sales representative by scanning this QR code with your smartphone or by visiting www.tyvasorep.com.To download a QR code reader, visit your smartphone’s app store and search for a QR code reader. A number of code reader apps are available.
STUDY DESIGN: TRIUMPH I was a 12-week, randomized, double-blind, placebo-controlled, multicenter study of patients (N=235) with PAH who were receiving a stable dose of bosentan or sildenafil for 3 months before study initiation. Patients were administered either placebo or Tyvaso in 4 daily treatment sessions with a target dose of 9 breaths (54 mcg) per session over the course of the 12-week study. Primary endpoint was change in 6MWD at 12 weeks. Secondary endpoints included time to clinical worsening, Borg dyspnea score, NYHA functional class, trough 6MWD at week 12 (obtained at least 4 hours after study drug administration), peak 6MWD at 6 weeks, quality of life as measured by the MLWHF questionnaire, and PAH signs and symptoms.3
BRIEF SUMMARY
The following is a brief summary of the full prescribing information
for TYVASO® (treprostinil) Inhalation Solution. Please review the full
prescribing information prior to prescribing TYVASO.
INDICATIONS AND USAGE
TYVASO is a prostacyclin vasodilator indicated for the treatment of
pulmonary arterial hypertension (PAH) (WHO Group 1) to improve
exercise ability. Studies establishing effectiveness included
predominately patients with NYHA Functional Class III symptoms
and etiologies of idiopathic or heritable PAH (56%) or PAH associated
with connective tissue diseases (33%). The effects diminish over the
minimum recommended dosing interval of 4 hours; treatment timing
can be adjusted for planned activities. While there are long-term
data on use of treprostinil by other routes of administration, nearly
all controlled clinical experience with inhaled treprostinil has been
on a background of bosentan (an endothelin receptor antagonist)
or sildenafil (a phosphodiesterase type 5 inhibitor). The controlled
clinical experience was limited to 12 weeks in duration.
CONTRAINDICATIONS
None.
WARNINGS AND PRECAUTIONS
Patients with Pulmonary Disease or Pulmonary Infections–The
safety and efficacy of TYVASO have not been established in patients
with significant underlying lung disease (e.g., asthma or chronic
obstructive pulmonary disease). Patients with acute pulmonary
infections should be carefully monitored to detect any worsening of
lung disease and loss of drug effect.
Risk of Symptomatic Hypotension– Treprostinil is a pulmonary and
systemic vasodilator. In patients with low systemic arterial pressure,
treatment with TYVASO may produce symptomatic hypotension.
Patients with Hepatic or Renal Insufficiency–Titrate slowly in patients
with hepatic or renal insufficiency, because such patients will likely
be exposed to greater systemic concentrations relative to patients
with normal hepatic or renal function.
Risk of Bleeding–Since TYVASO inhibits platelet aggregation, there
may be an increased risk of bleeding, particularly among patients
receiving anticoagulant therapy.
Effect of Other Drugs on Treprostinil–Co-administration of a
cytochrome P450 (CYP) 2C8 enzyme inhibitor (e.g., gemfibrozil)
may increase exposure (both Cmax and AUC) to treprostinil.
Co-administration of a CYP2C8 enzyme inducer (e.g., rifampin) may
decrease exposure to treprostinil. Increased exposure is likely to
increase adverse events associated with treprostinil administration,
whereas decreased exposure is likely to reduce clinical effectiveness.
ADVERSE REACTIONS
The following potential adverse reactions are described in Warnings
and Precautions:
Adverse Reactions Identified in Clinical Trials–Because clinical
trials are conducted under widely varying conditions, adverse
reaction rates observed in the clinical trials of a drug cannot be
directly compared to rates in the clinical trials of another drug
and may not reflect the rates observed in practice. In a 12-week
placebo-controlled study (TRIUMPH I) of 235 patients with PAH (WHO
Group 1 and nearly all NYHA Functional Class III), the most commonly
reported adverse reactions to TYVASO included: cough and throat
irritation; headache, gastrointestinal effects, muscle, jaw or bone
pain, flushing and syncope. Table 1 lists the adverse reactions that
occurred at a rate of at least 4% and were more frequent in patients
treated with TYVASO than with placebo.
The safety of TYVASO was also studied in a long-term, open-label
extension study in which 206 patients were dosed for a mean
duration of one year. The adverse events during this chronic dosing
study were qualitatively similar to those observed in the 12-week
placebo controlled trial. Adverse Events Associated with Route of Administration–Adverse events in the treated group during
the double-blind and open-label phase reflecting irritation to the
respiratory tract included: cough, throat irritation, pharyngeal pain,
epistaxis, hemoptysis and wheezing. Serious adverse events during
the open-label portion of the study included pneumonia in 8 subjects.
There were three serious episodes of hemoptysis (one fatal) noted
during the open-label experience.
DRUG INTERACTIONS
Pharmacokinetic/pharmacodynamic interaction studies have
not been conducted with inhaled treprostinil (TYVASO); however,
some of such studies have been conducted with orally (treprostinil
diethanolamine) and subcutaneously administered treprostinil
(Remodulin®).
Pharmacodynamics–Antihypertensive Agents or Other Vasodilators–Concomitant administration of TYVASO with diuretics,
antihypertensive agents or other vasodilators may increase the
risk of symptomatic hypotension. Anticoagulants–Since treprostinil
inhibits platelet aggregation, there may be an increased risk of
bleeding, particularly among patients receiving anticoagulants.
Pharmacokinetics–Bosentan– In a human pharmacokinetic study
conducted with bosentan (250 mg/day) and an oral formulation
of treprostinil (treprostinil diethanolamine), no pharmacokinetic
interactions between treprostinil and bosentan were observed.
Sildenafil– In a human pharmacokinetic study conducted with
sildenafil (60 mg/day) and an oral formulation of treprostinil
(treprostinil diethanolamine), no pharmacokinetic interactions
between treprostinil and sildenafil were observed. Effect of Cytochrome P450 Inhibitors and Inducers– In vitro studies of
human hepatic microsomes showed that treprostinil does not inhibit
cytochrome P450 (CYP) isoenzymes CYP1A2, CYP2A6, CYP2C8,
CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A. Additionally,
treprostinil does not induce cytochrome P450 isoenzymes CYP1A2,
CYP2B6, CYP2C9, CYP2C19, and CYP3A. Human pharmacokinetic
studies with an oral formulation of treprostinil (treprostinil
diethanolamine) indicated that co-administration of the cytochrome
P450 (CYP) 2C8 enzyme inhibitor gemfibrozil increases exposure
(both Cmax and AUC) to treprostinil. Co-administration of the CYP2C8
enzyme inducer rifampin decreases exposure to treprostinil. It is
unclear if the safety and efficacy of treprostinil by the inhalation
route are altered by inhibitors or inducers of CYP2C8. Effect of Other Drugs on Treprostinil–Drug interaction studies have been carried
out with treprostinil (oral or subcutaneous) co-administered with
acetaminophen (4 g/day), warfarin (25 mg/day), and fluconazole
(200 mg/day), respectively in healthy volunteers. These studies
did not show a clinically significant effect on the pharmacokinetics
of treprostinil. Treprostinil does not affect the pharmacokinetics or
pharmacodynamics of warfarin. The pharmacokinetics of R- and
S-warfarin and the INR in healthy subjects given a single 25 mg dose
of warfarin were unaffected by continuous subcutaneous infusion of
treprostinil at an infusion rate of 10 ng/kg/min.
USE IN SPECIFIC POPULATIONS
Pregnancy—Pregnancy Category B–There are no adequate and
well controlled studies with TYVASO in pregnant women. Animal
reproduction studies have not been conducted with treprostinil
administered by the inhalation route. However, studies in pregnant
rabbits using continuous subcutaneous (sc) infusions of treprostinil
sodium at infusion rates higher than the recommended human sc
infusion rate resulted in an increased incidence of fetal skeletal
variations associated with maternal toxicity. Animal reproduction
studies are not always predictive of human response; TYVASO should
be used during pregnancy only if clearly needed.
Labor and Delivery–No treprostinil treatment-related effects
on labor and delivery were seen in animal studies. The effect of
treprostinil on labor and delivery in humans is unknown.
Nursing Mothers–It is not known whether treprostinil is excreted
in human milk. Because many drugs are excreted in human milk,
caution should be exercised when treprostinil is administered to
nursing women.
Pediatric Use–Safety and effectiveness in pediatric patients have not
been established. Clinical studies of TYVASO did not include patients
younger than 18 years to determine whether they respond differently
from older patients.
Geriatric Use–Clinical studies of TYVASO did not include sufficient
numbers of patients aged 65 years and over to determine whether
they respond differently from younger patients. In general, dose
selection for an elderly patient should be cautious, reflecting the
greater frequency of hepatic, renal, or cardiac dysfunction, and of
concomitant diseases or other drug therapy.
Patients with Hepatic Insufficiency–Plasma clearance of treprostinil,
delivered subcutaneously, was reduced up to 80% in subjects with
mild-to-moderate hepatic insufficiency. Uptitrate slowly when
treating patients with hepatic insufficiency because of the risk of
an increase in systemic exposure which may lead to an increase in
dose-dependent adverse effects. Treprostinil has not been studied in
patients with severe hepatic insufficiency.
Patients with Renal Insufficiency–No studies have been performed
in patients with renal insufficiency. Since treprostinil and its
metabolites are excreted mainly through the urinary route, patients
with renal insufficiency may have decreased clearance of the
drug and its metabolites and consequently, dose-related adverse
outcomes may be more frequent.
OVERDOSAGE
In general, symptoms of overdose with TYVASO include flushing,
headache, hypotension, nausea, vomiting, and diarrhea.
Provide general supportive care until the symptoms of overdose
have resolved.
Manufactured for: United Therapeutics Corporation
Research Triangle Park, NC 27709
Rx only February 2011
www.tyvaso.com
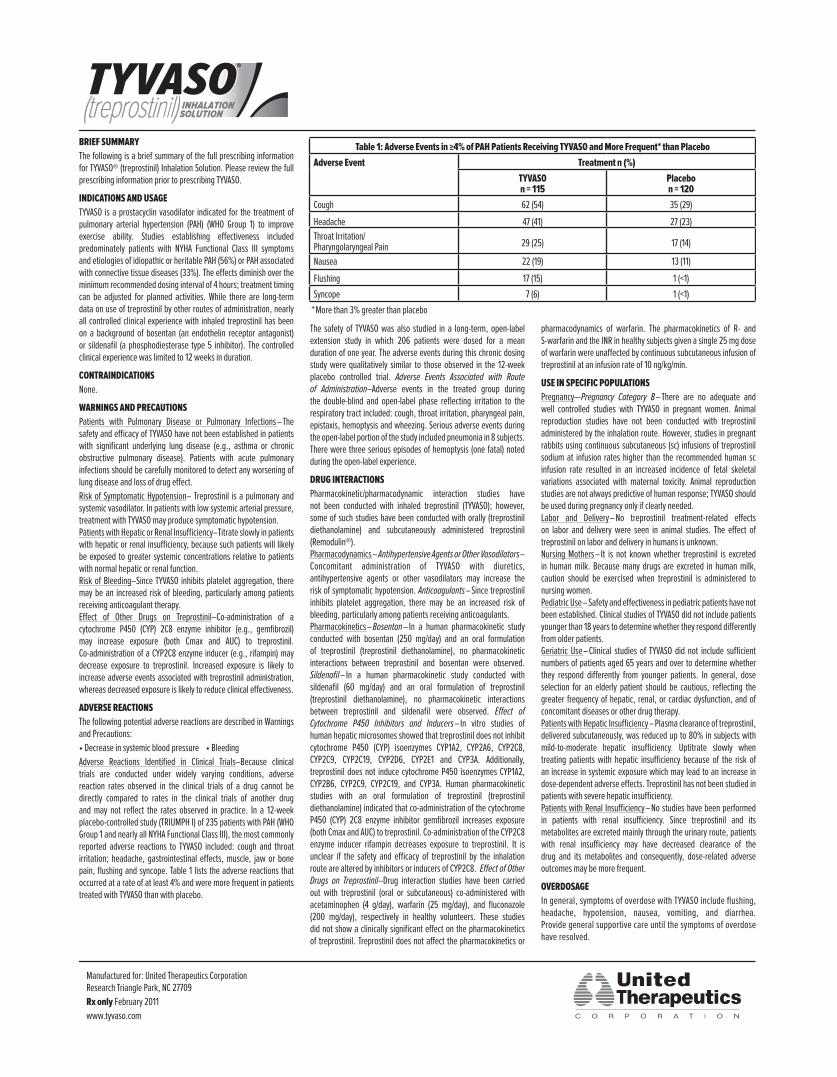
Table 1: Adverse Events in ≥4% of PAH Patients Receiving TYVASO and More Frequent* than Placebo
Adverse Event Treatment n (%)
TYVASO n = 115
Placebo n = 120
Cough 62 (54) 35 (29)
Headache 47 (41) 27 (23)
Throat Irritation/ Pharyngolaryngeal Pain 29 (25) 17 (14)
Nausea 22 (19) 13 (11)
Flushing 17 (15) 1 (<1)
Syncope 7 (6) 1 (<1)
*More than 3% greater than placebo

Article Reviews
Review of the Latest Published Research
Section EditorKelly Chin, MD
Section EditorIoana Preston, MD
Summaries and commentaries from the section editors and invited reviewerspresent a clinical context for practitioners’ application of the latest publishedresearch relevant to the care of patients with pulmonary hypertension. In thisissue, Kelly Chin discusses the role of computed tomography and 6-minutewalk distance in the diagnosis of pulmonary hypertension patients.
Sugiura T, Tanabe N, Matsuura Y, etal. Role of 320-Slice computed tomog-raphy in the diagnostic workup of pa-tients with chronic thromboembolicpulmonary hypertension. Chest. 2012Oct 22. [Epub ahead of print]Sugiura et al evaluated the accuracy of320 slice computed tomography (CT)scanning in patients undergoing evalua-tion for chronic thromboembolic pulmo-nary hypertension (CTEPH). Forty-fourpatients were identified as having proba-ble CTEPH based on VQ scan and echo-cardiography. Patients then underwent320 slice CT scan, right heart catheteriza-tion, and pulmonary angiography. The CTscans were obtained using 0.5 mm slicethickness with ECG gating, and required 2gantry rotations in order to simultane-ously image the lungs and the entire heart.Two independent reviewers read each CTscan, and their results were comparedwith pulmonary arteriogram, set as thegold standard test for the analysis.
Overall, CT performed well. The sen-sitivity of the 320 slice CT scan for de-tecting lobar PEs was 97%, and the spec-ificity was 97.1%, with excellent inter-observer agreement (Kappa�0.91,Table). At the segmental level, the sensi-tivity was 85.8% and the specificity was94.6%, with very good inter-observeragreement (Kappa�0.79). The subseg-mental arteries were not included in theanalysis, mainly due to a lack of an ac-ceptable reference standard. The authorsalso attempted to estimate systolic pulmo-nary arterial pressure (sPAP) by lookingat the curvature of the interventricularseptum, finding a strong correlation be-
tween their measured estimate and theactual sPAP (r�-0.79, P�0.001).
Interestingly, the sensitivity and speci-ficity results were calculated at the levelof the individual vessel. In other words,they calculated the number of correctlyidentified vessels (normal vs clot) in the344 main and lobar arteries and in the 860segmental arteries that were evaluated;the number of correct CTEPH diagnoseswas not reported. Out of 44 patients, only18 subsequently underwent pulmonarythromboendarterectomy; the dispositionof the other patients was not discussed.
The authors concluded that CT pulmo-
nary angiography was a less invasive al-ternative to conventional angiography forthe diagnosis of CTEPH, but acknowl-edged that the study was a retrospective,single-center study of a highly selectedcohort, and that further study was needed.
Sugiura et al provide an interestinglook at how multisclice CT may be usedin the future, though with several caveats.Notably, the patient population includedonly those with “suspicion” for CTEPH,so it is impossible to determine what thesensitivity and specificity of the testwould be in a less select population, nor isit possible to compare its accuracy vs VQscan. Additionally, although the accuracyof the specific vessels involved seems tohave been quite good, they do not discusshow or whether they used this informationin determining “operability,” a critical andchallenging aspect of the CTEPH evalua-
Correspondence: [email protected]
Table: Summary of pathological vascular findings as delineated by CTPA andPDSA, and statistical analysis of findings in CTPA compared to findings in PDSA
CTPA PDSA
Sensi-tivity(%)
Speci-ficity(%)
PPV(%)
NPV(%)
K(95% CI)
Main/lobar arteries(N�344)Number of normal
vessels271 277
Chronic thromboembolicfindings
73 67 97.0 97.1 89.0 99.3 0.91(0.86–0.96)
Segmental arteries(N�860)Number of normal
vessels661 670
Chronic thromboembolicfindings
199 190 85.8 94.6 81.9 95.9 0.79(0.74–0.84)
Definition of abbreviations: CTPA � Pulmonary angiography on 320-slice CT, PDSA �pulmonary digital subtraction angiography, PPV � positive predictive value, NPV �negative predictive value, K � Cohen’s Kappa, 95%CI � 95% confidence interval.Copyright 2012 by the American College of Chest Physicians. Reprinted with permission.
157Advances in Pulmonary Hypertension

tion. Importantly, prior studies have sug-gested that conventional CT angiographyis less sensitive than VQ scan in the di-agnosis of CTEPH.1,2 Further study isneeded prior to more widespread adoptionof 320 slice CT as either a screening orconfirmatory test for CTEPH.
Savarese G, Paolillo S, Costanzo P, etal. Do changes of 6-minute walk dis-tance predict clinical events in patientswith pulmonary arterial hypertension?A meta-analysis of 22 randomized tri-als. J Am Coll Cardiol. 2012;60(13):1192-1201.Savarese et al performed a meta-analysisof 22 clinical trials in pulmonary arterialhypertension (PAH) looking at mortalityand change in 6-minute walk distance(6MWD). They were specifically inter-ested in whether change in 6MWD pre-dicted survival and other outcomes, suchas PAH-related hospital admission. Simi-
lar to prior meta-analyses, they found thatactive treatment led to a reduction in all-cause death (odds ratio [OR] 0.43,P�0.01), and active treatment also re-duced hospitalization for PAH and/orlung transplantation (OR 0.44, P�0.01)and the initiation of PAH rescue therapy(OR 0.56, P�0.01). However, they didnot identify any relationship betweenchange in 6MWD and outcome. They didfind a significant relationship betweenchange in 6MWD and change in pulmonaryvascular resistance (r�-0.63, P�0.01). Inan accompanying editorial, Dr Stuart Richreviewed the history of the 6MWD as aprimary endpoint in PAH phase III clinicaltrials, and suggests that now is the time toconsider novel clinical trial strategies3.
Saverese et al found that change in6MWD does not correlate well with mor-tality and other outcomes. Notably, otherstudies have reported similar findings,suggesting that achieving a particular
walk threshold (�380-440 meters) ismore important than the absolute value ofthe change in 6MWD achieved.4,5
References1. Tunariu, N, Gibbs SJ, Win Z, et al. Ventilation-perfusion scintigraphy is more sensitive than multi-detector CTPA in detecting chronic thromboembolicpulmonary disease as a treatable cause of pulmonaryhypertension. J Nucl Med. 2007;48(5):680-684.2. Soler X, Kerr KM, Marsh JJ, et al. Pilot studycomparing SPECT perfusion scintigraphy with CT pul-monary angiography in chronic thromboembolic pul-monary hypertension. Respirology. 2012;17(1):180-184.3. Rich S. The 6-minute walk test as a primaryendpoint in clinical trials for pulmonary hyperten-sion. J Am Coll Cardiol. 2012;60(13):1202-1203.4. Benza RL, Miller DP, Gomberg-Maitland M, etal. Predicting survival in pulmonary arterial hyper-tension: insights from the Registry to Evaluate Earlyand Long-Term Pulmonary Arterial HypertensionDisease Management (REVEAL). Circulation.2010;122(2):164-172.5. Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primarypulmonary hypertension: prognostic factors and sur-vival. J Am Coll Cardiol. 2002;40(4):780-788.
B u i l d i n g M e d i c a l E d u c a t i o n i n P HA Partnership Initiative to Advance Medical Understanding of Pulmonary Hypertens ion
Building Medical Education in PH (BME) events are designed to foster
partnerships between PHA, PH Centers and medical professionals.
CEU/CME educational events. Participating in PHA’s BME program
medical professionals mailing list, advertising support, educational
materials for distribution to attendees, and more.
To view a full list of education opportunities for medical professionals, visit: www.PHAOnlineUniv.org/Calendar
Upcoming BME events:7th Annual Pulmonary Hypertension SymposiumJune 6, 2013Yale School of Medicine – New Haven, Conn.Register at: www.cme.yale.edu
6th International Conference on Neonatal and Childhood Pulmonary Vascular Disease
Register at: www.KidsWithPHSymposium.com
NYC PH Symposium: Shining A Light on Pulmonary Hypertension
Beth Israel Medical Center – New York, N.Y.
Register at: www.chpnet.org/cme
To partner with PHA in Building Medical Education in PH for your upcoming CME
event, please contact 301-565-3004 x776 or [email protected].
To learn more about this partnership, visit www.PHAssociation.org/BME
158 Advances in Pulmonary Hypertension

to expanding healthcare
options for individuals living with
cardiovascular and pulmonary diseases
through innovative research, access,
and education programs.
Gilead is committed
© 2013 Gilead Sciences, Inc. All rights reserved. UNBP0009 February 2013Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc.

2013 PH Professional Network SymposiumThe Power of Teamwork: 10 Years of Professional Collaboration in PAH
September 26 – 28, 2013Crystal Gateway MarriottArlington, Va.
An educational and networking event for PH-treating healthcare professionals.
This Symposium will feature an extraordinary line-up of speakers and topics highlighting the latest advances and research in pulmonary hypertension. Symposium faculty will include a range of healthcare professionals, such as nurses, pharmacists, respiratory therapists, physician assistants, nurse practitioners and physicians.
PH Professional Network (PHPN) is a recognized network reaching over 1,200 PH-treating healthcare professionals. Members are dedicated to enhancing communication, professional development, research opportunities and education in the medical community.
REGISTRATION OPENS SPRING 2013Don’t miss your chance to register for this valuable educational and networking program! Register online at www.PHAssociation.org/PHPN/Symposium
“Symposium is a great way to
network with peers and gain new
knowledge about the treatment of
PH”
Physicians: Encourage the healthcare professionals in your practice to take advantage of this valuable opportunity. Ensure that the latest advances in the care and treatment of PH patients are incorporated into you practice – consider supporting your staff’s attendance at this educational program!

OPPORTUNITY TO EARN CEUsClose to 30 educational sessions will be presented on topics | including:� Pharmacological management of pediatric PH; � Case studies in PH and methamphetamine use;� Echo interpretation;� Helping patients cope with depression and anxiety;� High �ow oxygen use;� Regenerative medicine;� … and many more!
Sessions will be accredited for nurses, respiratory therapists, pharmacists, physician assistants and social workers.
For additional information, visit: www.PHAssociation.org/PHPN/Symposium or contact 301-565-3004 x761 or [email protected]
CALL FOR ABSTRACTS Want to highlight research conducted at your institution? Consider submitting your research abstract for presentation at the 2013 PH Professional Network Symposium Poster Hall! PH Professional Network welcomes abstract proposals in all areas of practice. While submission of original abstracts is encouraged, submissions do not need to be original work and do not need to be fully executed in practice.
Submission deadline: May 15, 2013
For more information and resources, visit: www.PHAssociation.org/PHPN/Symposium/Abstracts
“This was by far my favorite PH
meeting! The topics were meaningful
and helpful for my practice”
ADVOCACY DAYThursday, September 26, 2013
Make the most of your trip to the D.C. area for Symposium by arriving in time for Advocacy Day! Take this unique opportunity to visit Capitol Hill and educate your Members of Congress about the needs of PH patients and the medical professionals who treat them. Participation is free – just check the “advocacy day” box on your Symposium registration form in spring!

PH Professional Network
Transitioning the Pediatric Pulmonary HypertensionPatient
Section EditorMartha Kingman, NP
Beth A. Coleman, RN, CPNPSenior Instructor, Department of PediatricsDivision of CardiologyPulmonary Hypertension ProgramUniversity of Colorado at Children’s HospitalColoradoAurora, CO
Michelle Calderbank, RN, BSN, CPNClinical Nurse CoordinatorPulmonary Hypertension ProgramChildren’s Hospital ColoradoAurora, CO
Advances in disease awareness, earlier di-agnosis, and additional therapeutic op-tions for treatment of pediatric pulmonaryhypertension (PH) in the last 2 decadeshave dramatically improved survival,leading to the first generation of pediatricpatients surviving to adulthood.1-4 Pediat-ric care is generally a family-centered ap-proach, whereas adult care is morepatient-centered. When patients becomeyoung adults they must move from depen-dency on parental involvement and over-sight to independence and individual ac-countability. It is difficult for young adultpatients to navigate this transition suc-cessfully without coordinated supportfrom their family, pediatric, and adult careproviders.
Recent analyses of pediatric PH withinREVEAL and in the Netherlands Registrydemonstrate the 2 primary subgroups asidiopathic pulmonary arterial hyperten-sion (IPAH) and PH associated with con-genital heart disease. IPAH is seen lessoften in children, while PH associatedwith congenital heart disease is seen moreoften than in adults.4
The proportionately higher incidence ofyoung adults with PH associated withcongenital heart disease that will be re-ceiving care through adult PH programsin the future will require incorporatingadult congenital heart disease (ACHD)–trained physicians into the adult PH careteam. The ACHD community has for-
mally addressed the need to create morestructured programs with a recent publi-cation of a best practice statement formanaging transition to adulthood for ad-olescents with congenital heart disease.5
The adolescent medical community hasrecognized issues around transition formore than 2 decades, including a positionpaper published by the Society of Adoles-cent Medicine in 1993.6 Transition prac-tices can be further modeled from largepediatric chronic disease populations thathave already piloted and implementedthese processes, such as cystic fibrosis andsickle cell disease.7-9
Traditionally, adolescent patients havetransferred to adult programs through a“drift-away” model: an incomplete, vaguetransition from the pediatric team insteadof a clear and comprehensive “handoff” tothe adult care team.10 The “drift-away”model of transfer has been an unsuccess-ful transition, leaving young adults strug-gling to manage their disease and treat-ment well, and frequently resulting inbeing lost to follow-up.
Over the past 5 years, the center atChildren’s Hospital Colorado has experi-enced increasing numbers of adolescentPH patients achieving college acceptance,entering the work force, beginning to liveindependently, and therefore movingaway from their nuclear family and sup-port system. Parallel to this, young adultsare required to transition from a depen-dent role where their parents led interac-tions with health care providers, coordi-nated medication refills and dosing, and,for those on invasive therapies, oftenmixed and changed the infusions daily. Inaddition to the usual challenges of enter-ing college or the work force, these youngadults suddenly have to take ownership oftheir disease process, become indepen-
dent in medication administration, iden-tify changes in clinical symptoms, main-tain medication compliance, and learn toaccess the medical providers and arrangeclinic follow-up appointments. Thesechallenges have led to the development ofa transition program that will assist pro-viders, young adults, and their families inmaking a structured yet individualized,comprehensive, and successful transitionto an adult program.
Barriers to successful implementationof a transition program include insuffi-cient staffing, lack of identified staffmembers responsible for transitions, fi-nancial challenges, institutional accep-tance, and resistance from the adolescentsand their parents in transferring to an adultcenter. Commitment to partnership andopen dialogue between pediatric and adultprograms is vital for smooth and individ-ualized transitions of care.5,10,11
Key aspects of the transition process in-clude: timing; patient, family, and providerreadiness; identification of adult PH careteam; successful completion of transitioncurriculum; and transfer of care (Figure 1).The American Academy of Pediatrics,American Academy of Family Physicians,and the American College of Physicianshave recommended that the transition pro-cess start as early as 12 years of age, withthe physical or absolute transfer of care be-tween 18 and 21 years of age depending ondevelopmental readiness.10 Even before theactual education or curriculum portion ofthe process begins, the concept should bediscussed with the patient and family. Thisdecreases stress of the unknown, as manypediatric patients have built a good rapportwith their pediatrician and pediatric careteam.
After developmental readiness is as-sessed and the patient enters the transitionprocess, the curriculum focuses on basicunderstanding of diagnosis and sequen-tially builds to medication management,
Correspondence: [email protected]
162 Advances in Pulmonary Hypertension
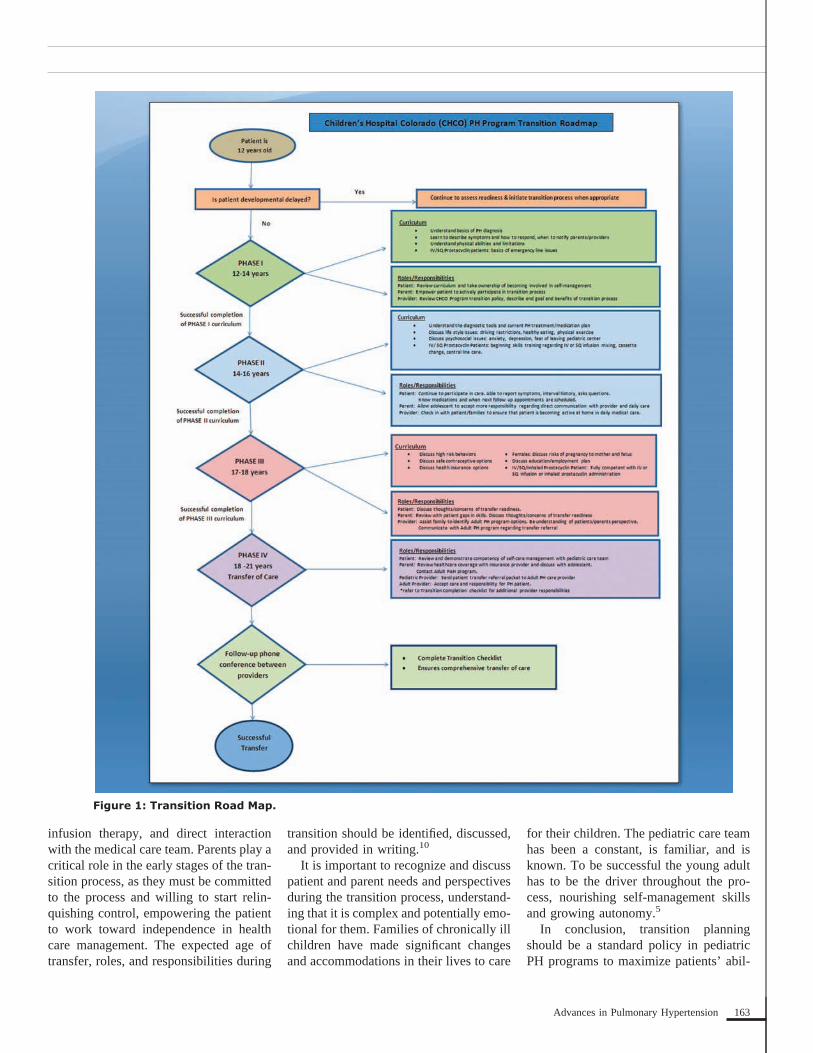
infusion therapy, and direct interactionwith the medical care team. Parents play acritical role in the early stages of the tran-sition process, as they must be committedto the process and willing to start relin-quishing control, empowering the patientto work toward independence in healthcare management. The expected age oftransfer, roles, and responsibilities during
transition should be identified, discussed,and provided in writing.10
It is important to recognize and discusspatient and parent needs and perspectivesduring the transition process, understand-ing that it is complex and potentially emo-tional for them. Families of chronically illchildren have made significant changesand accommodations in their lives to care
for their children. The pediatric care teamhas been a constant, is familiar, and isknown. To be successful the young adulthas to be the driver throughout the pro-cess, nourishing self-management skillsand growing autonomy.5
In conclusion, transition planningshould be a standard policy in pediatricPH programs to maximize patients’ abil-
Figure 1: Transition Road Map.
163Advances in Pulmonary Hypertension

ity to successfully care for their chronichealth needs as they enter adulthood. Rec-ognizing the barriers and forming partner-ships with adult PH programs will result inincreased numbers of young adults success-fully completing the transition processwith transfer of care to the adult center.
References1. Rich S, Dantzker DR, Ayres SM, et al. Primarypulmonary hypertension. A national prospectivestudy. Ann Intern Med. 1987;107(2):216-223.
2. D’Alonzo GE, Barst RJ, Ayers SM, et al. Sur-vival in patients with primary pulmonary hyperten-sion. Results from a national prospective registry.Ann Intern Med. 1991;115(5):343-349.3. Barst RJ, McGoon MD, Elliott CG, ForemanAJ, Miller DP, Ivy DD. Survival in childhood pul-monary arterial hypertension: insights from theregistry to evaluate early and long-term pulmonaryarterial hypertension disease management. Circula-tion. 2012;125(1):113-122.4. van Loon RL, Roofthooft MT, Hillege HL, et al.Pediatric pulmonary hypertension in the Netherlands:epidemiology and characterization during the period
1991 to 2005. Circulation. 2011;124(16):1755-1764.5. Sable C, Foster E, Uzark K, et al; AmericanHeart Association Congenital Heart Defects Com-mittee of the Council on Cardiovascular Disease inthe Young, Council on Cardiovascular Nursing,Council on Clinical Cardiology, and Council onPeripheral Vascular Disease. Best practices in man-aging transition to adulthood for adolescents withcongenital heart disease: the transition process andmedical and psychosocial issues: a scientific state-ment from the American Heart Association. Circu-lation. 2011;123(13):1454-1485.6. Blum RW, Garell D, Hodgman CH, et al. Tran-sition from child-centered to adult health-care sys-tems for adolescents with chronic conditions. A po-sition paper of the Society of Adolescent Medicine.J Adolesc Health. 1993;14(7):570-576.7. Chaudhry SR, Keaton M, Nasr SZ. Evaluationof a cystic fibrosis transition program from pediatricto adult care. Pediatr Pulmonol. 2012 Aug 8. [Epubahead of print]8. McLaughlin MD, Diener-West M, Indurkhya A,Rubin H, Heckmann R, Boyle MP. Improving tran-sition from pediatric to adult cystic fibrosis care:lessons from a national survey of current practices.Pediatrics. 2008;121(5):e1160-e1166.9. DeBaun MR, Telfair J. Transition and sickle celldisease. Pediatrics. 2012;130(5):926-935.10. Cooley WC, Sagerman PJ; American Acad-emy of Pediatrics; American Academy of FamilyPhysicians; American College of Physicians; Tran-sitions Clinical Report Authoring Group. Supportingthe health care transition from adolescence to adult-hood in the medical home. Pediatrics. 2011;128(1):182-200.11. McManus M, Fox H, O’Connor K, Chapman T,MacKinnon J. Pediatric Perspectives and Practiceson Transitioning Adolescents with Special Needs toAdult Health Care. The National Alliance to AdvanceAdolescent Health. Fact Sheet No. 6; October 2008.
Figure 2: Transition Checklist.
164 Advances in Pulmonary Hypertension

Advances in
Pulmonary Hypertension
Program Overview: Pulmonary arterialhypertension (PAH), an incurable disease, ischaracterized by medial hypertrophy, intimal fi-brosis, and in situ thrombi in small muscularpulmonary arteries. PAH was considered a rap-idly fatal illness with a median survival of 2.8years in the 1980s when no evidence-based ther-apies were available. Since then the treatment ofthis disease has made tremendous advances,and in the last 10 years the discovery of newmedications have positively influenced theprognosis and survival of patients with PAH.
This self-study activity is based on 4 articlesthat review the management of patients withcongenital heart disease and pulmonary hyper-tension.
This activity is jointly sponsored by Wash-ington University School of Medicine and thePulmonary Hypertension Association.
Target Audience: This self-study activityis appropriate for cardiologists, pulmonologists,rheumatologists, and other physicians who treatpatients with PH.
Learning Objectives: Upon completionof this activity, participants will be able to:
1. Describe the anatomy of the most commoncongenital heart defects (and their repairs)that are associated with the development ofPAH.
2. Demonstrate the importance of simple andadvanced imaging technique in adults withcongenital heart disease and PH as diagnos-tic and risk stratifying tools.
3. Understand the complex interplay betweenpulmonary blood flow and pulmonary vas-cular resistance and the techniques (medic-inal, catheter-based, and surgical) that areused to modify it.
4. Discuss the clinical studies that have estab-lished the basis for pharmacology therapy,and explore the new therapeutic frontiers inpatients with congenital heart disease and PH.
Self-Assessment Examination: See pages
196 and 197 for self-assessment questions, answer key,
and evaluation form.
FacultyChairRichard A. Krasuski, MD, FACC, FAHADirector of Adult CongenitalHeart Disease
ServicesCardiovascular MedicineCleveland Clinic FoundationCleveland, Ohio
Contributing Authors
Jamil A. Aboulhosn, MD, FACC, FSCAIDirector, Ahmanson/UCLA Adult Congenital
Heart Disease CenterLos Angeles, California
Sonya V. Babu-Narayan, MRCP, PhDRoyal Brompton and Harefield NHS
Foundation TrustNational Heart and Lung InstituteImperial College LondonNIHR Cardiovascular Biomedical Research UnitRoyal Brompton Hospital and Imperial
College LondonLondon, United Kingdom
Thomas Bashore, MDProfessor of MedicineSenior Vice Chief, Division of CardiologyDuke University Medical CenterDurham, North Carolina
Michael A. Gatzoulis, MD, PhD, FESC, FACCRoyal Brompton and Harefield NHS
Foundation TrustNational Heart and Lung InstituteImperial College LondonNIHR Cardiovascular Biomedical Research UnitRoyal Brompton Hospital and Imperial
College LondonLondon, United Kingdom
Todd L. Kiefer, MDAssistant ProfessorDivision of CardiologyDuke University Medical CenterDurham, North Carolina
Wei Li, MD, PhD, FESC, FACCRoyal Brompton and Harefield NHS
Foundation TrustLondon, United Kingdom
Erika B. Rosenzweig, MDColumbia University College of Physicians
and SurgeonsPulmonary Hypertension CenterNew York, New York
Michael B. Rubens, FRCRRoyal Brompton and Harefield NHS
Foundation TrustLondon, United Kingdom
Giancarlo Scognamiglio, MD, PhDRoyal Brompton and Harefield NHS
Foundation TrustLondon, United Kingdom
Warren A. Zuckerman, MD
Columbia University College of Physiciansand Surgeons
Pulmonary Hypertension CenterNew York, New York
Accreditation Statement: This activityhas been planned and implemented in accor-dance with the Essential Areas and Policies ofthe Accreditation Council for Continuing Medi-cal Education (ACCME) through the joint spon-sorship of Washington University School ofMedicine and the Pulmonary Hypertension As-sociation. Washington University School ofMedicine is accredited by the ACCME to pro-vide continuing medical education to physicians.
Credit Designation: Washington Uni-versity School of Medicine designates this en-during material for a maximum of 2.0 AMAPRA Category 1 Credits.™ Physicians shouldclaim only the credit commensurate with theextent of their participation in the activity.
Instructions for Earning Credit: Thisactivity is a self-study program; a self-assessment examination is included on page 196to help physicians review important points. Aform is also included on page 197 for physiciansto evaluate the CME activity. Completion of thisactivity involves reading the journal and com-pleting the self-assessment examination andevaluation form with a passing grade of 70% orhigher, which may take up to 2 hours. Credits forthis self-study program are available from May31, 2013 through April 30, 2014. There is no feefor this program. Please note that this self-studyprogram may also be viewed online at https://cme-online.wustl.edu/pha.
Accreditation Statement: Depart-ment of Continuing Medical Education, Wash-ington University School of Medicine, CampusBox 8063, 660 South Euclid Ave., St. Louis, MO63110
Disclosures: The Accreditation Council forContinuing Medical Education and the Associa-tion of American Colleges have standards andguidelines to ensure that individuals participatingin CME activities are aware of relationships be-tween authors and commercial companies thatcould potentially affect the information pre-
(Continued on page 196)
CM
ESection
165Advances in Pulmonary Hypertension

Anatomy of Congenital Heart Disease LesionsAssociated With Pulmonary ArterialHypertensionTodd L. Kiefer, MDAssistant ProfessorDivision of CardiologyDuke University Medical CenterDurham, NC
Thomas Bashore, MDProfessor of MedicineSenior Vice Chief, Division of CardiologyDuke University Medical CenterDurham, NC
Adult congenital heart disease represents a growing population of patients.Many patients survive to adulthood and lead functional, productive lives. Infact, there are more adults living with congenital heart disease than pediatricpatients. Many adult patients will have had prior surgical repair as children.However, some patients present in adulthood with a new diagnosis of congen-ital heart disease. Furthermore, there are a variety of complications associatedwith each individual congenital lesion and specific surgical repair procedure.
Pulmonary arterial hypertension (PAH) isone of the well-characterized sequelae. Itis particularly common with unrepairedlarge left to right shunt lesions that occurdistal to the tricuspid valve. Despite priorcardiac surgery, some patients have resid-ual defects that contribute to the develop-ment of PAH. The diagnosis of PAH re-quires right heart catheterization and isdefined as a mean pulmonary artery (PA)pressure greater than 25 mm Hg, with apulmonary capillary wedge pressure, leftatrial pressure, or left ventricular end-diastolic pressure less than 15 mm Hg,and a pulmonary vascular resistance(PVR) greater than 3 Wood units.1
It is estimated that overall 5%-10% ofpatients with congenital heart disease andas many as 30% of unrepaired patientshave PAH.2 When PAH does occur inconjunction with congenital heart disease,it is associated with increased morbidityand mortality.2,3 However, the outcomeand response to vasodilator therapies ismuch better for the cohort with congenitalheart disease than for all other etiologiesof PAH.1
The pathophysiology leading to the de-velopment of PAH in congenital heart dis-ease patients is related to increased pres-sure and blood flow in association with aleft to right shunt lesion. This scenario iscommon in a large, uncorrected ventricu-lar septal defect (VSD) or patent ductusarteriosus (PDA), and in surgically con-structed shunts where the pulmonary vas-culature is exposed to aortic systolic pres-sure. Alternatively, pre-tricuspid valve
shunts, which are low-pressure lesions as-sociated with increased volume circulat-ing through the right ventricle (RV) andpulmonary circulation, such as atrial sep-tal defects (ASD) and partial anomalouspulmonary venous return (PAPVR), leadto PAH much less often (Table).
Over time these shunt lesions lead todistinct changes in the pulmonary arteri-oles with the development of plexiformlesions and subsequent increases in PApressures and PVR. This often results inright ventricular dysfunction, and in somecases as the PA pressures increase, rever-sal of shunting from net left to right to netright to left occurs with notable cyanosisand the onset of Eisenmenger syndrome.One rationale for early surgical or percu-taneous repair of congenital cardiac dis-ease is to prevent the onset or avoid theprogression of PAH.
In this review, we will focus on theanatomy of the various congenital cardiaclesions that are associated with PAH.There are several congenital lesions thatproduce pulmonary venous hypertension,such as pulmonary veno-occlusive dis-ease, cor triatriatum sinister, mitral valveabnormalities, and other left-sided ob-structive lesions (coarctation of the aortaand supra-, sub-, and valvular aortic ste-nosis), but these lesions produce a differ-ent pathophysiology and will not be thefocus of this discussion.
A VSD is a common form of congenitalheart disease with an estimated preva-lence of 3 per 1000 in children and 0.3 per1000 adults, as some VSDs close sponta-
neously during childhood into adult-hood.4,5 It is also the most common con-genital cardiac lesion associated withPAH in a Dutch registry.6 There are mul-tiple types of VSDs depending on theirlocation: membranous or perimembra-nous, muscular, inlet and outlet varieties(doubly committed or infundibular) (Fig-ure 1). Often more than 1 defect in theventricular septum is present. Anatomi-cally, a membranous VSD is bordered bythe membranous portion of the ventricularseptum, the aortic valve, and the tricuspidvalve. In some cases, the septal leaflet ofthe tricuspid valve will cover this defectand form a “windsock” deformity withventricular systole. Often the windsock isfenestrated with left to right shunting ofblood from the left ventricle (LV) to theRV. At times, the septal tricuspid leafletcan fuse with the membranous ventricularseptum, leading to closure of the defectand obliteration of shunting.5 MuscularVSD, as the name suggests, is surroundedby myocardium and can be located any-where in the ventricular septum. There areoften multiple VSD sites in a given pa-tient. An inlet VSD is bordered by themitral valve, the tricuspid valve, and themuscular septum. Given this location, it isa part of the spectrum of the atrioventric-ular (AV) canal or AV septal defect, pre-viously referred to as endocardial cushiondefects, and is often associated with tri-somy 21 (Down syndrome). The inletVSD is most commonly associated withPAH, with nearly 40% of such patientsdeveloping PAH.6 Finally, an outlet VSD,also referred to as an infindibular, doublycommitted, or supracristal VSD with itlocation superior to the crista supraven-tricularis, is surrounded by ventricular
Key Words—atrial septal defects, congenital heart disease, Eisenmenger syndrome, patent ductusarteriosus, ventricular septal defectCorrespondence: [email protected]
CM
ESe
ctio
n
166 Advances in Pulmonary Hypertension

septum, aortic valve, and pulmonic valve.It is important to recognize that any typeof VSD may occur either in isolation orwith other congenital abnormalities.
The size of a VSD (small vs large) isclinically often estimated, especially inchildren, by the ratio of the diameter ofthe VSD to the diameter of the aorticannulus.7 Defects that are less than orequal to 25% of the diameter of the aorticannulus (usually less than 1 cm) are des-ignated as small, restrictive defects. Ingeneral, the smaller size limits flow andleft to right shunt magnitude. In this sce-nario, the development of PAH is muchless likely. Conversely, a large defect isdefined as having a diameter greater than75% of the diameter of the aortic annulus(usually greater than 1 cm). Given thelarger defect and lack of restriction toflow, the pulmonary arterial bed is ex-
posed to a greater degree of systemic LVsystolic pressure, and the subsequent de-velopment of PAH is much more com-mon.
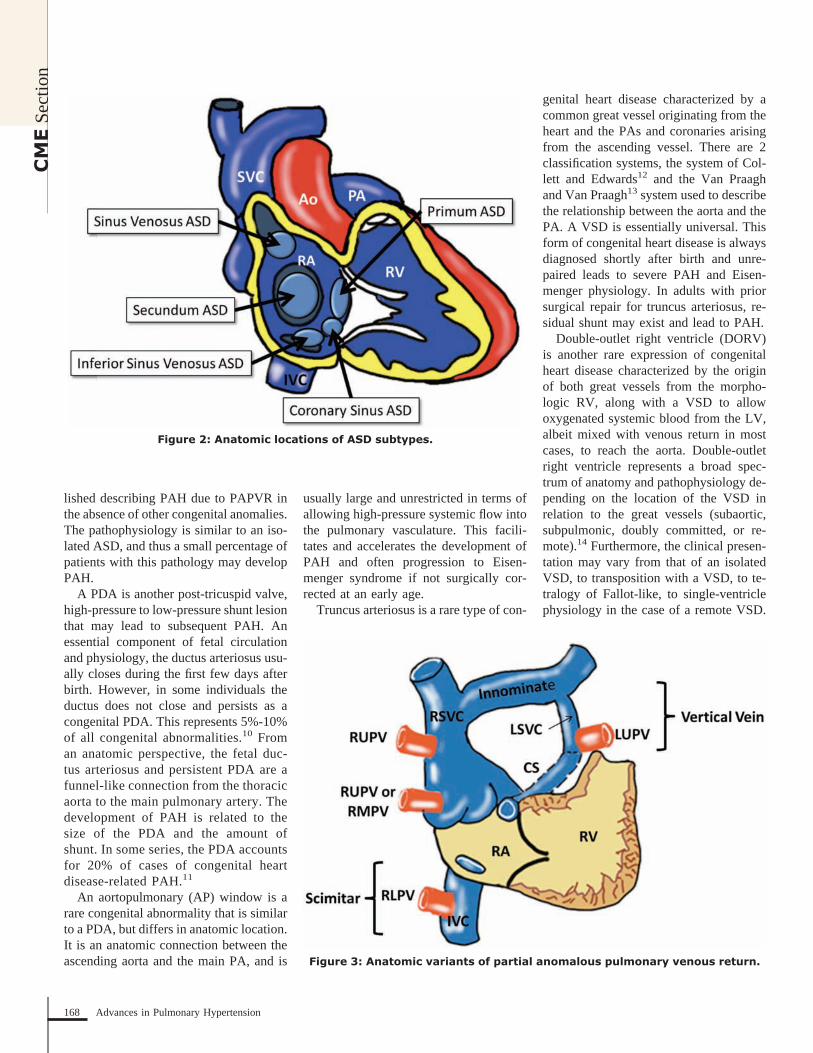
Atrial septal defects are another com-mon form of congenital heart disease.However, PAH develops less commonly(10%) with this lesion than in post-tricuspid valve shunt lesions in which thepulmonary vascular bed is exposed tohigher pressures as well as the increasedshunt volume.8 Five subtypes of ASDhave been described: secundum (75%),primum (15%), and superior sinus veno-sus (10%) are the most common. Lesscommon are the unroofed coronary sinusand the inferior sinus venosus defect (Fig-ure 2).9 A secundum ASD is characterizedby a defect in the fossa ovalis region (gen-erally central region) of the atrial septum.The primum ASD involves the inferior
aspect of the atrial septum near the atrio-ventricular valves. If a concomitant inletVSD is present, then the defect is classi-fied as an AV septal defect (see previoussection on VSD subtypes). In addition, theprimum ASD is often associated with anabnormality in the anterior mitral valveleaflet termed a cleft mitral valve, whichis associated with varying degrees of mi-tral regurgitation. The sinus venosus ASDis divided into 2 anatomic subtypes: asuperior sinus venosus defect and an in-ferior sinus venosus defect. The superiorsinus venosus ASD involves a defect inthe superior aspect of the atrial septum atthe junction of the roof of the atria and theentrance of the superior vena cava into theright atrium (RA). A superior sinus veno-sus ASD is associated with greater than90% of cases with PAPVR of the rightupper pulmonary vein, which aberrantlydrains into the RA instead of the leftatrium. The inferior sinus venosus ASD isdefined by a defect in the interatrial sep-tum inferiorly near the junction with in-ferior vena cava.
Partial anomalous pulmonary venousreturn functions as a low-pressure, pre-tricuspid valve shunt lesion, which servesto volume load the RV and pulmonarycirculation in a similar manner to an ASD.It is also a rare lesion, with autopsy stud-ies demonstrating an incidence of 0.6%-0.8%.17,18 Likewise, in addition to thepreviously discussed association of supe-rior sinus venosus ASD and right upperpulmonary vein anomalous return, there isan association in 5%-10% of cases of se-cundum ASD with PAPVR.19 Anomalouspulmonary venous return also occurs at anincreased frequency in patients withTurner syndrome.
There are multiple variations ofPAPVR in terms of the anatomic locationof the vein and number of veins involvedand location of anomalous venousattachment/drainage. Anomalous veinsmay drain into the right atrium, left in-nominate vein, coronary sinus, superiorvena cava, or the inferior vena cava(Scimitar syndrome) (Figure 3). Giventhat PAPVR is an uncommon lesion, anaccurate population level estimate of as-sociation with PAH is not available. How-ever, multiple case reports have been pub-
Table: Risk for development of PAH with various shunt lesions
Low Risk Intermediate Risk High Risk
• Secundum ASD • Sinus venosus ASD • Large, unrestricted VSDor PDA
• Partial anomalous pulmonaryvenous return
• Cooley-Waterston shunt
• Small, restrictive VSD • Pott’s shunt• Primum ASD• AV septal defect
Figure 1: Anatomic locations of VSD subtypes.
CM
ESection
167Advances in Pulmonary Hypertension
lished describing PAH due to PAPVR inthe absence of other congenital anomalies.The pathophysiology is similar to an iso-lated ASD, and thus a small percentage ofpatients with this pathology may developPAH.
A PDA is another post-tricuspid valve,high-pressure to low-pressure shunt lesionthat may lead to subsequent PAH. Anessential component of fetal circulationand physiology, the ductus arteriosus usu-ally closes during the first few days afterbirth. However, in some individuals theductus does not close and persists as acongenital PDA. This represents 5%-10%of all congenital abnormalities.10 Froman anatomic perspective, the fetal duc-tus arteriosus and persistent PDA are afunnel-like connection from the thoracicaorta to the main pulmonary artery. Thedevelopment of PAH is related to thesize of the PDA and the amount ofshunt. In some series, the PDA accountsfor 20% of cases of congenital heartdisease-related PAH.11
An aortopulmonary (AP) window is arare congenital abnormality that is similarto a PDA, but differs in anatomic location.It is an anatomic connection between theascending aorta and the main PA, and is
usually large and unrestricted in terms ofallowing high-pressure systemic flow intothe pulmonary vasculature. This facili-tates and accelerates the development ofPAH and often progression to Eisen-menger syndrome if not surgically cor-rected at an early age.
Truncus arteriosus is a rare type of con-
genital heart disease characterized by acommon great vessel originating from theheart and the PAs and coronaries arisingfrom the ascending vessel. There are 2classification systems, the system of Col-lett and Edwards12 and the Van Praaghand Van Praagh13 system used to describethe relationship between the aorta and thePA. A VSD is essentially universal. Thisform of congenital heart disease is alwaysdiagnosed shortly after birth and unre-paired leads to severe PAH and Eisen-menger physiology. In adults with priorsurgical repair for truncus arteriosus, re-sidual shunt may exist and lead to PAH.
Double-outlet right ventricle (DORV)is another rare expression of congenitalheart disease characterized by the originof both great vessels from the morpho-logic RV, along with a VSD to allowoxygenated systemic blood from the LV,albeit mixed with venous return in mostcases, to reach the aorta. Double-outletright ventricle represents a broad spec-trum of anatomy and pathophysiology de-pending on the location of the VSD inrelation to the great vessels (subaortic,subpulmonic, doubly committed, or re-mote).14 Furthermore, the clinical presen-tation may vary from that of an isolatedVSD, to transposition with a VSD, to te-tralogy of Fallot-like, to single-ventriclephysiology in the case of a remote VSD.
Figure 2: Anatomic locations of ASD subtypes.
Figure 3: Anatomic variants of partial anomalous pulmonary venous return.
CM
ESe
ctio
n
168 Advances in Pulmonary Hypertension

The subaortic subtype of DORV is mostcommon, accounting for approximately50% of cases.14 The subaortic DORVsubtype also has the strongest associationwith development of PAH given patho-physiology similar to a large VSD. Pul-monary arterial hypertension may also oc-cur in the unrepaired subpulmonic DORVsubtype if there is not RV outflow orpulmonary valve level obstruction to min-imize pulmonary blood flow. In one re-cent series from a database of patientswith adult congenital heart disease, 17%of patients with the diagnosis of DORVwere noted to have PAH.6
Some patients with congenital heartdisease have had a surgical shunt to in-crease flow into the pulmonary circuitwhen the congenital abnormality pre-vented adequate pulmonary perfusion.These are generally palliative shunts as abridge to complete surgical repair. Assuch, surgical shunts would often be li-gated or taken down at the time of subse-quent cardiac surgery. However, it is notuncommon to encounter an adult patientwith a patent surgical shunt. Through the1960s and 1970s, as surgical experiencewith congenital heart defects grew, it wasdiscovered that these palliative high-flow,high-pressure shunts that delivered sys-temic blood flow to the lungs commonlyresulted in PAH.
The first surgical shunt, referred to as aBlalock-Taussig (BT) shunt, was per-formed in 1944.20 The BT shunt was con-structed from connection of the right sub-clavian artery to the right PA (Figure 4).The original BT shunt evolved throughseveral modifications, including use of theleft side and a synthetic conduit (modifiedBlalock shunt) to connect the subclavianartery to the PA, which preserved the sub-clavian artery and circulation to the upperextremity along with control over flow tothe lung via diameter of the conduit.
Subsequently, Dr Willis Potts per-formed a surgical procedure connectingthe descending thoracic aorta to the leftPA, which became known as a Potts shunt(Figure 4).20 In a similar manner, Dr Da-vid Waterston devised a surgery wherebyan anastomosis between the posterior as-pect of the ascending aorta and the rightPA was created (Figure 4).20 This is com-
monly known as a Cooley-Waterston shunt.Less frequently used as a palliative shuntwas the central shunt, or a surgically createdequivalent of the congenital AP window, inwhich an anastomosis was made betweenthe ascending aorta and the main PA. ThePotts and Waterston surgical shunts have amuch stronger propensity to produce PAHthan the Blalock shunts due to less restric-tive flow into the PA from the aorta. For thatreason, they were abandoned in favor of theBlalock approach.
Eisenmenger syndrome is the end-stageresult of long-standing PAH. It has a prev-alence of approximately 8%-10% in pa-tients with congenital heart disease.21 Thepathophysiology involves the progressionof irreversible PVR to the point at whichPA pressures are greater than systemicaortic pressures and a shunt lesion thatwas originally left to right reverses indirection of blood flow right to left. Withthe onset of right to left shunting, cyanosisis apparent. In cases of a PDA, differentialcyanosis may be observed due to the an-atomic location of the shunt with cyanoticlower extremities and normal appearingupper extremities. Moreover, Eisen-menger syndrome is associated with a va-riety of other end-organ system complica-tions. Once PA systolic pressure and/orPVR is greater than two thirds of systemicvalues, and certainly when Eisenmengersyndrome with right to left shunting ispresent, surgical intervention to correctthe underlying cardiac pathology is gen-erally contraindicated.
In conclusion, several anatomic formsof congenital heart disease can lead toPAH. In particular, pressure and volumeloading left to right shunt lesions (post-tricuspid valve) in contradistinction toonly volume loading left to right shuntlesion (pre-tricuspid valve) are muchmore likely to cause PAH. From an epi-demiologic perspective, unrepaired VSDand PDA are 2 of the more common le-sions that will be complicated by pulmo-nary hypertension. The management ofpatients with congenital heart disease iscomplex, and pulmonary hypertension ex-perts should work closely with cardiolo-gists who have specialized training inadult congenital heart disease in order tooptimize outcomes for patients with PAHrelated to congenital heart disease.
References1. McLaughlin VV, Archer SL, Badesch DB, et al.ACCF/AHA 2009 expert consensus document onpulmonary hypertension a report of the AmericanCollege of Cardiology Foundation Task Force onExpert Consensus Documents and the AmericanHeart Association developed in collaboration withthe American College of Chest Physicians; Ameri-can Thoracic Society, Inc.; and the Pulmonary Hy-pertension Association. J Am Coll Cardiol. 2009;53(17):1573-1619.2. Diller GP, Gatzoulis MA. Pulmonary vasculardisease in adults with congenital heart disease. Cir-culation. 2007;115(8):1039-1050.3. Lowe BS, Therrien J, Ionescu-Ittu R, Pilote L,Martucci G, Marelli AJ. Diagnosis of pulmonaryhypertension in the congenital heart disease adultpopulation impact on outcomes. J Am Coll Cardiol.2011;58(5):538-546.4. Warnes CA, Liberthson R, Danielson GK, et al.
Figure 4: Anatomic locations of surgical palliative shunts.
CM
ESection
169Advances in Pulmonary Hypertension

Task force 1: the changing profile of congenital heartdisease in adult life. J Am Coll Cardiol. 2001;37(5):1170-1175.5. Minette MS, Sahn DJ. Ventricular septal de-fects. Circulation. 2006;114(20):2190-2197.6. Duffels MG, Engelfriet PM, Berger RM, et al.Pulmonary arterial hypertension in congenital heartdisease: an epidemiologic perspective from a Dutchregistry. Int J Cardiol. 2007;120(2):198-204.7. Warnes CA, Williams RG, Bashore TM, et al.ACC/AHA 2008 guidelines for the management ofadults with congenital heart disease: a report of theAmerican College of Cardiology/American HeartAssociation Task Force on Practice Guidelines(Writing Committee to Develop Guidelines on theManagement of Adults With Congenital Heart Dis-ease). Developed in Collaboration With the Ameri-can Society of Echocardiography, Heart RhythmSociety, International Society for Adult CongenitalHeart Disease, Society for Cardiovascular Angiog-raphy and Interventions, and Society of ThoracicSurgeons. J Am Coll Cardiol. 2008;52(23):e143-e263.8. Steele PM, Fuster V, Cohen M, Ritter DG, Mc-Goon DC. Isolated atrial septal defect with pulmo-nary vascular obstructive disease–long-term
follow-up and prediction of outcome after surgicalcorrection. Circulation. 1987;76(5):1037-1042.9. Webb G, Gatzoulis MA. Atrial septal defects inthe adult: recent progress and overview. Circulation.2006;114(15):1645-1653.10. Brickner ME, Hillis LD, Lange RA. Congen-ital heart disease in adults. First of two parts. N EnglJ Med. 2000;342(4):256-263.11. Adatia I, Kothari SS, Feinstein JA. Pulmonaryhypertension associated with congenital heart dis-ease: pulmonary vascular disease: the global per-spective. Chest. 2010;137(6 Suppl):52S-61S.12. Collett RW, Edwards JE. Persistent truncusarteriosus; a classification according to anatomictypes. Surg Clin North Am. 1949;29(4):1245-1270.13. Van Praagh R, Van Praagh S. The anatomy ofcommon aorticopulmonary trunk (truncus arteriosuscommunis) and its embryologic implications. Astudy of 57 necropsy cases. Am J Cardiol. 1965;16(3):406-425.14. Lev M, Bharati S, Meng CC, Liberthson RR,Paul MH, Idriss F. A concept of double-outlet rightventricle. J Thorac Cardiovasc Surg. 1972;64(2):271-281.15. Poirier NC, Gatzoulis MA. Double-inlet ven-tricle. In: Gatzoulis MA, Webb GD, Daubeney PEF,
eds. Diagnosis and Management of Adult Congeni-tal Heart Disease. Philadelphia, PA: Saunders;2011.
16. Cook AC, Anderson RH. The anatomy ofhearts with double inlet ventricle. Cardiol Young.2006;16 Suppl 1:22-26.
17. Hoffman JI, Kaplan S. The incidence of con-genital heart disease. J Am Coll Cardiol. 2002;39(12):1890-1900.
18. Healey JE Jr. An anatomic survey of anoma-lous pulmonary veins: their clinical significance.J Thorac Surg. 1952;23(5):433-444.
19. Ellis AR. Partial Anomalous Pulmonary Ve-nous Connections and the Scimitar Syndrome. In:Gatzoulis MA, Webb GD, Daubeney PEF, eds. Di-agnosis and Management of Adult Congenital HeartDisease. 2nd ed. Philadelphia, PA: Saunders; 2011.
20. Waldhausen JA. The early history of congen-ital heart surgery: closed heart operations. Ann Tho-rac Surg. 1997;64(5):1533-1539.
21. Kaemmerer H, Mebus S, Schulze-Neick I, etal. The adult patient with eisenmenger syndrome: amedical update after dana point part I: epidemiology,clinical aspects and diagnostic options. Curr CardiolRev. 2010;6(4):343-355.
CM
ESe
ctio
n
170 Advances in Pulmonary Hypertension

The Essential Role of Imaging in the Evaluationof Patients With Pulmonary Arterial Hypertensionin Association With Congenital Heart Disease
CLASSIFICATION OF PAH-CHDThe heterogeneity of cardiac morphologyand its functional consequences require acustomized classification, including adetailed description of cardiac morphol-ogy, previous surgical and/or catheterintervention/s, and current functional andhemodynamic status of the patient.
LEFT TO RIGHT SHUNTSLeft to right shunts are the most commonsubstrate for PAH-CHD, accounting forapproximately 38% of patients with CHDand pulmonary hypertension (PH) in arecent report from Canada.3 This groupincludes patients with Eisenmenger syn-drome, where the development of PAHoccurs in the presence of a nonrestrictiveleft to right shunt—intracardiac (usuallypost-tricuspid) or extracardiac. In about50%-70% of cases, patients develop irre-versible and progressive pulmonary vas-cular disease in the first few years of life,resulting eventually in reversal of bloodflow through the shunt and cyanosis.4 Ex-amples of these lesions are large ventric-ular septal defects (VSD), patent ductusarteriosus (PDA), complete atrioventricu-lar septal defects (AVSD), truncus arteri-osus, or functionally univentricular heartswithout pulmonary stenosis. In cardiac le-sions with a left to right shunt at thepre-tricuspid level, ie, patients with largeatrial septal defects (ASD) with initialvolume and not pressure overload, the de-velopment of pulmonary vascular diseaseis less frequent and later in life. Only
one-tenth to one-fifth of patients with ahemodynamically important and large in-teratrial communication will eventuallydevelop pulmonary vascular disease,mostly in late adulthood.
FONTAN CIRCULATIONPulmonary arterial hypertension can alsocomplicate other subgroups of CHD, bothin their natural history and after surgicalrepair; such an example of the uniquefeatures of the pulmonary circulation inCHD is represented by the Fontan “circu-lation,” where systemic venous return isdirected to the lungs without an inter-posed ventricular pump (Figure 1). At firstglance, it may appear to be out of line withthe classical definition of PAH (increasedpulmonary vascular resistance [PVR] ofgreater than 3 Wood units per metersquared or MPAP of more than 25 mmHg). However, there is evidence of anabnormal pulmonary vascular bed in thesepatients, as suggested by an abnormal re-sponse to exogenous nitric oxide admin-istration.5 We submit, herewith, that pa-tients with the Fontan circulation shouldbe considered in the PAH-CHD group, aseven a minimal increase in PVR can havea major adverse effect on the pulmonarycirculation and thus cardiac output giventhe absence of a subpulmonary ventricle.Based on these considerations, a more re-fined classification of PAH that coexistswith CHD has been proposed to betteraddress this rather heterogeneous patientgroup that cannot be assumed to be sim-
ilar in terms of pathophysiology and he-modynamics (Table 1).6
ROLE OF IMAGINGMultimodality imaging plays a key role inassessing and managing patients withPAH-CHD. It is important to recognizethe strengths/weaknesses and the comple-mentary nature of different imaging mo-dalities as well as the complex nature ofthe diagnostic questions that need to beaddressed.
In this review, we will focus on theimaging tools commonly employed toevaluate PAH in CHD patients and theirrelative contribution to diagnostic assess-ment, evaluation of the functional and he-modynamic impairment, and longer-termprognostication.
ECHOCARDIOGRAPHYTransthoracic echocardiography (TTE) isthe first-line cardiovascular imaging mo-dality in the assessment of patients withvarious types of PAH because it is easy toapply, relatively inexpensive, and pro-vides accurate information on cardiacanatomy and physiology.
In the setting of PAH-CHD, TTE isparticularly suitable for the real-time in-terrogation of structural abnormalities aswell as hemodynamic disturbances. In themajority of these patients, TTE allows theevaluation of cardiac anatomy (ie, orien-tation and veno-atrial, atrioventricular,and ventriculo-arterial connections), themorphology of cardiac structures, ventric-
Key Words—cardiovascular magnetic resonance, echocardiography, Fontan circulation, myocardial performance index, right ventricular functionCorrespondence: [email protected]: Sonya V. Babu-Narayan is supported by an Intermediate Clinical Research Fellowship from the British Heart Foundation. This project wassupported by the NIHR Cardiovascular Biomedical Research Unit of Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. Thisreport is independent research by the National Institute for Health Research Biomedical Research Unit Funding Scheme. The views expressed in thispublication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
Pulmonary arterial hypertension (PAH) is a hemodynamic and pathophysio-logic condition defined as an increase in mean pulmonary artery pressure(MPAP) of �25 mm Hg at rest measured at right heart catheterization (RHC).1,2
Patients with PAH associated with congenital heart disease (PAH-CHD) are agrowing population consisting of an anatomically and phenotypically hetero-geneous group, where differences among specific cardiac defects, along withtheir varied clinical course and prognosis, influence treatment choices for theindividual patient.
Giancarlo Scognamiglio, MD, PhD,
Sonya V. Babu-Narayan, MRCP, PhD,
Michael B. Rubens, FRCR,
Michael A. Gatzoulis, MD, PhD, FESC, FACC,
Wei Li, MD, PhD, FESC, FACC,Royal Brompton and Harefield NHS FoundationTrust, London, United Kingdom
CM
ESection
171Advances in Pulmonary Hypertension
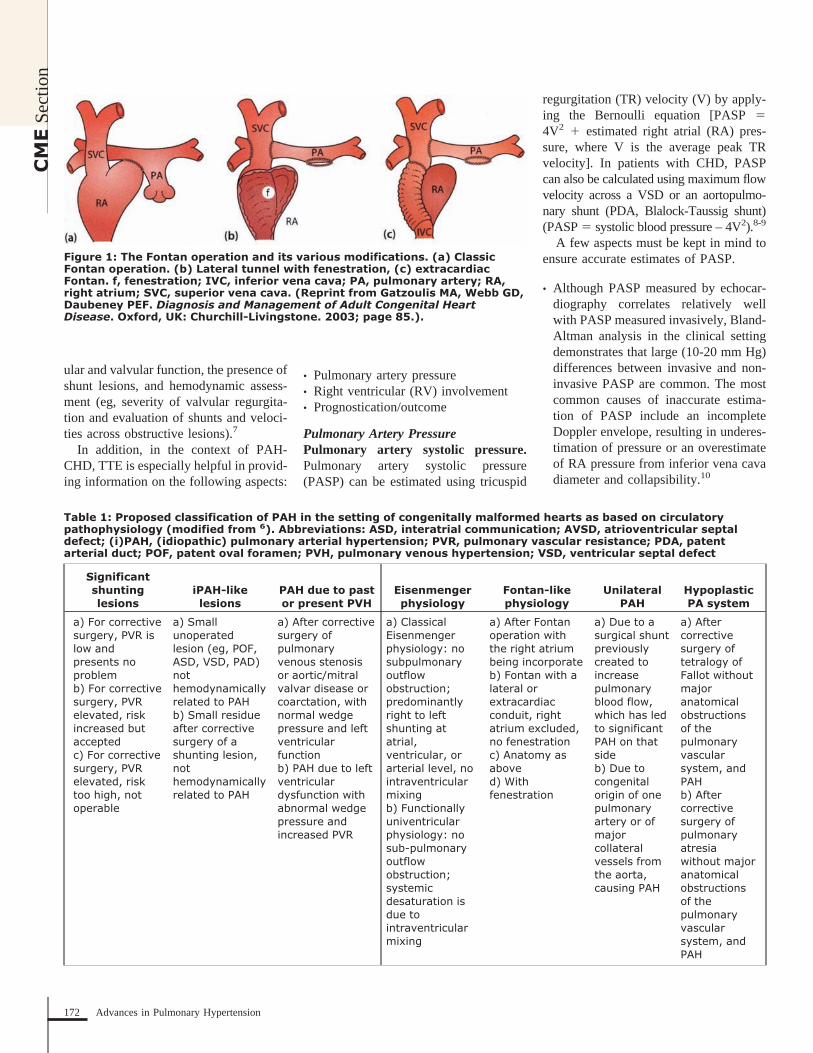
ular and valvular function, the presence ofshunt lesions, and hemodynamic assess-ment (eg, severity of valvular regurgita-tion and evaluation of shunts and veloci-ties across obstructive lesions).7
In addition, in the context of PAH-CHD, TTE is especially helpful in provid-ing information on the following aspects:
• Pulmonary artery pressure• Right ventricular (RV) involvement• Prognostication/outcome
Pulmonary Artery PressurePulmonary artery systolic pressure.Pulmonary artery systolic pressure(PASP) can be estimated using tricuspid
regurgitation (TR) velocity (V) by apply-ing the Bernoulli equation [PASP �4V2 � estimated right atrial (RA) pres-sure, where V is the average peak TRvelocity]. In patients with CHD, PASPcan also be calculated using maximum flowvelocity across a VSD or an aortopulmo-nary shunt (PDA, Blalock-Taussig shunt)(PASP � systolic blood pressure – 4V2).8-9
A few aspects must be kept in mind toensure accurate estimates of PASP.
• Although PASP measured by echocar-diography correlates relatively wellwith PASP measured invasively, Bland-Altman analysis in the clinical settingdemonstrates that large (10-20 mm Hg)differences between invasive and non-invasive PASP are common. The mostcommon causes of inaccurate estima-tion of PASP include an incompleteDoppler envelope, resulting in underes-timation of pressure or an overestimateof RA pressure from inferior vena cavadiameter and collapsibility.10
Figure 1: The Fontan operation and its various modifications. (a) ClassicFontan operation. (b) Lateral tunnel with fenestration, (c) extracardiacFontan. f, fenestration; IVC, inferior vena cava; PA, pulmonary artery; RA,right atrium; SVC, superior vena cava. (Reprint from Gatzoulis MA, Webb GD,Daubeney PEF. Diagnosis and Management of Adult Congenital HeartDisease. Oxford, UK: Churchill-Livingstone. 2003; page 85.).
Table 1: Proposed classification of PAH in the setting of congenitally malformed hearts as based on circulatorypathophysiology (modified from 6). Abbreviations: ASD, interatrial communication; AVSD, atrioventricular septaldefect; (i)PAH, (idiopathic) pulmonary arterial hypertension; PVR, pulmonary vascular resistance; PDA, patentarterial duct; POF, patent oval foramen; PVH, pulmonary venous hypertension; VSD, ventricular septal defect
Significantshuntinglesions
iPAH-likelesions
PAH due to pastor present PVH
Eisenmengerphysiology
Fontan-likephysiology
UnilateralPAH
HypoplasticPA system
a) For correctivesurgery, PVR islow andpresents noproblemb) For correctivesurgery, PVRelevated, riskincreased butacceptedc) For correctivesurgery, PVRelevated, risktoo high, notoperable
a) Smallunoperatedlesion (eg, POF,ASD, VSD, PAD)nothemodynamicallyrelated to PAHb) Small residueafter correctivesurgery of ashunting lesion,nothemodynamicallyrelated to PAH
a) After correctivesurgery ofpulmonaryvenous stenosisor aortic/mitralvalvar disease orcoarctation, withnormal wedgepressure and leftventricularfunctionb) PAH due to leftventriculardysfunction withabnormal wedgepressure andincreased PVR
a) ClassicalEisenmengerphysiology: nosubpulmonaryoutflowobstruction;predominantlyright to leftshunting atatrial,ventricular, orarterial level, nointraventricularmixingb) Functionallyuniventricularphysiology: nosub-pulmonaryoutflowobstruction;systemicdesaturation isdue tointraventricularmixing
a) After Fontanoperation withthe right atriumbeing incorporateb) Fontan with alateral orextracardiacconduit, rightatrium excluded,no fenestrationc) Anatomy asaboved) Withfenestration
a) Due to asurgical shuntpreviouslycreated toincreasepulmonaryblood flow,which has ledto significantPAH on thatsideb) Due tocongenitalorigin of onepulmonaryartery or ofmajorcollateralvessels fromthe aorta,causing PAH
a) Aftercorrectivesurgery oftetralogy ofFallot withoutmajoranatomicalobstructionsof thepulmonaryvascularsystem, andPAHb) Aftercorrectivesurgery ofpulmonaryatresiawithout majoranatomicalobstructionsof thepulmonaryvascularsystem, andPAH
CM
ESe
ctio
n
172 Advances in Pulmonary Hypertension

• The right ventricular systolic pressure(RVSP) calculated from TR velocitymay only be taken as the PASP in theabsence of RV outflow obstruction. InCHD, more care should be taken to ex-clude any obstruction along the pulmo-nary pathway, especially after pulmonicvalve surgery or in patients with previoussystemic to pulmonary shunts. In somecases, peripheral or segmental pulmonarystenosis may also be present; this willrequire complementary imaging such ascardiovascular MRI to delineate.
• Velocity measurements are angle de-pendent. Tricuspid regurgitant jetsshould be taken from multiple acousticwindows (apical 4-chamber views, RVinflow, and off axis if necessary) withaccurate transducer angulation in orderto obtain a parallel intercept angle be-tween the ultrasound beam and jet toavoid underestimation. In some cases oftrivial regurgitant jet and suboptimalcontinuous-wave Doppler spectrum, theinjection of contrast agents (agitated sa-line, sonicated albumin, or air-blood-salinemixture) may be required to achieveclear delineation of the jet envelope.11
• Close relationship between PASP andRV cardiac output exists. In cases of“end-stage” PAH, where both advancedRV dysfunction and increased PVRcause a significant reduction in stroke
volume, PASP may appear “pseudonor-malized” as a consequence of the lowdriving pressure generated by the failingRV.12 Underestimation of RV pressuremay also occur with the development ofdiastolic RV dysfunction, characterizedby high RA pressure and a stiff RV.
• Furthermore, in cases of severe TR thepeak velocity may underestimate thetrans-tricuspid pressure gradient be-cause of early equalization of pressurebetween RA and RV, leading to trunca-tion of the Doppler envelope.12
Pulmonary artery mean and end-diastolic pressure. Mean pulmonary ar-tery pressure and pulmonary end-diastolicpressure (PADP) are especially usefulwhen TR velocity cannot be obtained orwhen further information is required.
A jet of pulmonary regurgitation, pres-ent in the majority of patients with PAH-CHD, permits the measurement of theend-diastolic pulmonary pressure usingthe modified Bernoulli equation:[PADP � 4 � (end-diastolic pulmonaryregurgitant velocity)2 � RA pressure].
Similarly, MPAP can be determinedfrom early peak pulmonic regurgitationvelocity using the modified Bernoulliequation and adding the estimated RApressure.13 Mean pulmonary artery pres-sure can also be estimated by using pul-
monary acceleration time (AT) measuredfrom the onset of RV ejection to peakpulmonary flow velocity (Figure 2). Gen-erally, the shorter the AT, the higher thePVR and hence the pulmonary artery pres-sure. A value �105 ms is suggestive ofPH.14 Mean pulmonary artery pressure canalso be derived by regression formulaswhere: MPAP � 79 � (0.45 � AT). Thesame authors also found that in patientswith ATs �120 ms, the formula [MPAP �90 � (0.62 � AT)] performed better.15
In addition to AT, the shape of the flowwave is of interest, as PH is associatedwith a deceleration of flow in mid systole(notching). In the presence of increasedPVR and low arterial compliance, pulsewave reflection has greater magnitude andpropagates more rapidly, arriving at theright ventricular outflow tract (RVOT)during systole.16
In patients with a Fontan circulation, aspreviously discussed, even a minor in-crease in pulmonary artery pressure mayhave significant hemodynamic conse-quences on the Fontan circulation. Con-ventional diagnostic criteria for PAH can-not be applied in this type of circulation.Information about MPAP in this settingcan be derived from mean flow velocityacross a fenestration between the Fontan ortotal cavopulmonary connection pathwayand the atria; when such a fenestration is
Figure 2: (a) Estimation of mean and diastolic pulmonary artery pressure by continuous-wave Doppler signal ofpulmonary regurgitation (PR). Point 1 indicates maximal early diastolic PR velocity, which can be used to calculateMPAP. Point 2 marks end-diastole PR velocity used to calculate end-diastolic pulmonary artery pressure. (b) Measure-ment of pulmonary AT from the pulsed-wave Doppler signal of pulmonary arterial flow.
CM
ESection
173Advances in Pulmonary Hypertension

present and can be interrogated by echoDoppler [MPAP � 4 V2 � left atrial (LA)mean pressure]. If this value is more than17 mm Hg, it is highly suggestive of PAH.
Comprehensive diagnosis of PAH-CHD should combine Doppler pressuremeasurements with other accompanyingechocardiographic features such as ven-tricular size and systolic function, as it isthe RV that plays the key role in deter-mining clinical presentation and progno-sis in PAH-CHD.
Assessment of RV Morphology andFunctionRight ventricular morphology. Nor-mally the RV is a thin-walled chamber. Inmost forms of PAH, as a result of chronicprogressive pressure loading, progressiveRV remodelling occurs, initially in theform of hypertrophy and later as dilata-tion, along with progressive contractileimpairment and, eventually, RV failure.
Compared to the patients with otherforms of PAH, Eisenmenger syndrome he-modynamics and resulting RV remodellingare distinctly different. In adults with Eisen-menger syndrome with post-tricuspid de-fects and 2 ventricles, the RV often appearsgreatly hypertrophied with no significantdilatation. This unique physiopathologicadaptive model is explained by the preser-vation of a “fetal-like” phenotype withoutloss of RV hypertrophy and the presence ofa ventricular communication, allowing bothventricles to function as a single entity.17
In contrast, adults with PH and a pre-tricuspid shunt (ie, ASD) show greaterLA, RA, and RV dilatation. It can there-fore be postulated that loss of RV hyper-trophy during infancy, lack of a trainingeffect on the RV during childhood, and theabsence of a ventricular communication thatpairs the 2 ventricles functionally likely ex-plain the differing RV response.18
Eccentricity index. In patients withPAH, the high RV pressure may reducethe trans-septal pressure gradient betweenthe 2 ventricles and leads to the frequentlyobserved flattening of the intraventricularseptum (IVS). M-mode analysis, with itshigh temporal resolution, can accuratelyestimate differences in the timing of left-ward IVS shift during the cardiac cycle.Two-dimensional echo permits the quan-
tification of the septal deformation usingthe eccentricity index, measured from aparasternal short axis view at the level ofthe chordae tendineae as the ratio of theleft ventricle (LV) dimension parallel andperpendicular to the IVS respectively. It isusually measured both at end diastole andend systole with a normal value of 1.0,which occurs when the LV cavity main-tains a round and symmetrical configura-tion on short-axis imaging. Mild, moder-ate, and severe septal bowing isrepresented by values of 1.1–1.4, 1.5–1.8,and �1.8.19
LV filling abnormalities. Intraventricu-lar septum deformation also alters LVshape, size, and diastolic filling. Thus, acommon echocardiographic finding inthese patients is blunted early diastolicfilling of the LV, which in this scenario isnot indicative of LA hypertension, butinstead represents a marker of abnormalventriculo-ventricular interaction. In fact,increased RV pressure and prolonged RVsystole cause early diastolic reversal ofthe IVS. As a result, early diastolic trans-mitral filling is reduced and redistributedto late diastole.20
Right ventricular function. Assessmentof RV function is the single most impor-tant aspect of the echocardiographic ex-amination in patients with PAH, becausesymptoms and outcome both depend onthe ability of the RV to adapt to an in-creased pulmonary vascular load.
Right ventricular dysfunction is chal-lenging to quantify on echocardiography.All available acoustic windows and viewsshould be used to provide complementaryinformation and allow for a comprehen-sive assessment. Qualitative assessmentof function based on visual inspection iscommonly used in practice, but is limitedby a significant interobserver variability,which is especially problematic when as-sessing relative changes in RV function inthe same patient.
Tricuspid annular plane systolic excur-sion. Differences in muscle fiber orienta-tion of the RV suggest that longitudinalshortening plays a greater role in RV emp-tying than in the LV. This predominantlylongitudinal contractile pattern of the RVcan be easily obtained.
Tricuspid annular plane systolic excur-sion (TAPSE) is the longitudinal systolicdisplacement of the RV base toward theRV apex and has been shown to correlatestrongly with RV ejection fraction (EF).21
Tricuspid annular plane systolic excursioncan be derived using 2D guided M-mode(Figure 3a), is simple and highly repro-ducible, and has been recommended byAmerican Society of Echocardiography(ASE) guidelines as part of routine echo-cardiographic evaluation.8 Normal valuesvary between 2.3 cm–2.6 cm, with aTAPSE of 2.0 cm likely representing thelowest acceptable normal value. Values inthe range of 1.8 cm–2.0 cm, 1.6 cm–1.8cm, and �1.6 cm are consistent with mild,moderate, and severe RV systolic dys-function.19 A significant limitation ofTAPSE in PAH-CHD is that it is highlyload dependent, such that it may becomepseudonormalized in the presence of sig-nificant ventricular volume loading, suchas with left to right shunting or severeTR.22
Tissue Doppler imaging. Analogous toTAPSE, systolic wave velocity by tissueDoppler imaging (TDI) is a measure oflongitudinal myocardial contraction. Tis-sue Doppler imaging like TAPSE is loaddependent and may become pseudonor-mal under conditions of increased ventric-ular volume loading. Mean value in nor-mal controls is approximately 15 cm/s atthe annulus, with a lower accepted refer-ence limit of normal of 10 cm/s.8
Fractional area change. A more quan-titative approach of assessing RV functionis to measure the RV functional areachange (FAC), [(end-diastolic area – end-systolic area)/end-diastolic area] � 100,which has demonstrated a close correla-tion with RVEF by MRI.23 It is obtainedby tracing areas of the RV at end diastoleand end systole from the apical 4-chamberview beneath the trabeculations. Unfortu-nately, incomplete visualization of the RVcavity, especially when the RV is dilated,as well as difficulties in endocardial def-inition lead to relatively poor reproduc-ibility, thus making it an unreliable toolfor serial assessment.
Myocardial performance index. Themyocardial performance index (MPI),also known as the Tei index, provides a
CM
ESe
ctio
n
174 Advances in Pulmonary Hypertension
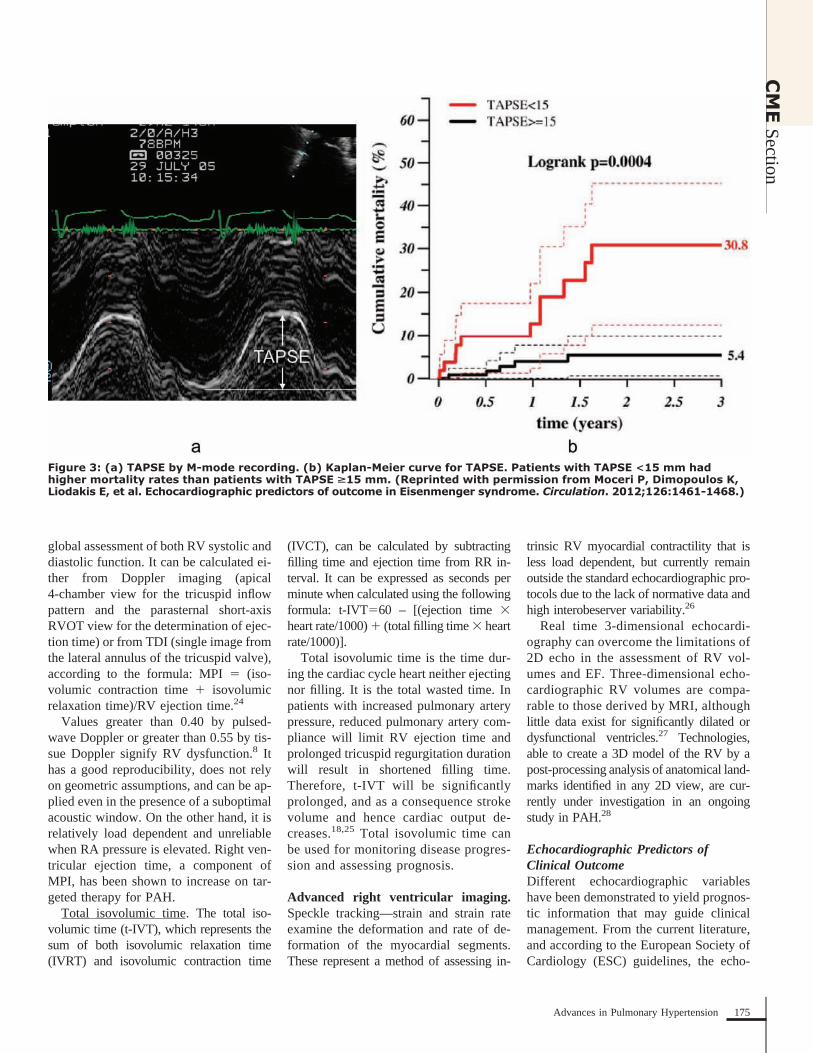
global assessment of both RV systolic anddiastolic function. It can be calculated ei-ther from Doppler imaging (apical4-chamber view for the tricuspid inflowpattern and the parasternal short-axisRVOT view for the determination of ejec-tion time) or from TDI (single image fromthe lateral annulus of the tricuspid valve),according to the formula: MPI � (iso-volumic contraction time � isovolumicrelaxation time)/RV ejection time.24
Values greater than 0.40 by pulsed-wave Doppler or greater than 0.55 by tis-sue Doppler signify RV dysfunction.8 Ithas a good reproducibility, does not relyon geometric assumptions, and can be ap-plied even in the presence of a suboptimalacoustic window. On the other hand, it isrelatively load dependent and unreliablewhen RA pressure is elevated. Right ven-tricular ejection time, a component ofMPI, has been shown to increase on tar-geted therapy for PAH.
Total isovolumic time. The total iso-volumic time (t-IVT), which represents thesum of both isovolumic relaxation time(IVRT) and isovolumic contraction time
(IVCT), can be calculated by subtractingfilling time and ejection time from RR in-terval. It can be expressed as seconds perminute when calculated using the followingformula: t-IVT�60 – [(ejection time �heart rate/1000) � (total filling time � heartrate/1000)].
Total isovolumic time is the time dur-ing the cardiac cycle heart neither ejectingnor filling. It is the total wasted time. Inpatients with increased pulmonary arterypressure, reduced pulmonary artery com-pliance will limit RV ejection time andprolonged tricuspid regurgitation durationwill result in shortened filling time.Therefore, t-IVT will be significantlyprolonged, and as a consequence strokevolume and hence cardiac output de-creases.18,25 Total isovolumic time canbe used for monitoring disease progres-sion and assessing prognosis.
Advanced right ventricular imaging.Speckle tracking—strain and strain rateexamine the deformation and rate of de-formation of the myocardial segments.These represent a method of assessing in-
trinsic RV myocardial contractility that isless load dependent, but currently remainoutside the standard echocardiographic pro-tocols due to the lack of normative data andhigh interobeserver variability.26
Real time 3-dimensional echocardi-ography can overcome the limitations of2D echo in the assessment of RV vol-umes and EF. Three-dimensional echo-cardiographic RV volumes are compa-rable to those derived by MRI, althoughlittle data exist for significantly dilated ordysfunctional ventricles.27 Technologies,able to create a 3D model of the RV by apost-processing analysis of anatomical land-marks identified in any 2D view, are cur-rently under investigation in an ongoingstudy in PAH.28
Echocardiographic Predictors ofClinical OutcomeDifferent echocardiographic variableshave been demonstrated to yield prognos-tic information that may guide clinicalmanagement. From the current literature,and according to the European Society ofCardiology (ESC) guidelines, the echo-
Figure 3: (a) TAPSE by M-mode recording. (b) Kaplan-Meier curve for TAPSE. Patients with TAPSE <15 mm hadhigher mortality rates than patients with TAPSE >15 mm. (Reprinted with permission from Moceri P, Dimopoulos K,Liodakis E, et al. Echocardiographic predictors of outcome in Eisenmenger syndrome. Circulation. 2012;126:1461-1468.)
CM
ESection
175Advances in Pulmonary Hypertension

cardiographic indices most closely asso-ciated with unfavorable outcome, includingRA area index, diastolic eccentricity index,pericardial effusion, MPI, and TAPSE,are all indicators of RV decompensation.
However, prognosis is significantly af-fected by the etiology of PAH. Patientswith Eisenmenger syndrome exhibit a bet-ter prognosis compared with idiopathicPAH and connective tissue disease–associated PAH. Patients may survive de-cades after the initial diagnosis of PAH-CHD, even before the advent of advancedtargeted PAH therapy. As mentioned be-fore, the difference in outcome is thought
to be related to better adaptation of the RVto systemic or high pulmonary arterypressure. In support of this view, we haverecently demonstrated that the longitudi-nal function of the RV is preserved ormildly impaired in the majority of patientswith Eisenmenger syndrome, and thateven though RV dilation was prevalent, itwas less severe than that described in id-iopathic PAH and was not related to ad-verse outcome.29
Right ventricular long axis function(TAPSE). Right ventricular longitudinalcontraction in Eisenmenger patients has
been shown to be an independent prog-nostic factor, both in our cohort, whereeven small reductions in TAPSE were as-sociated with adverse outcome (Figure3b), and in another recent prospectivestudy from Van De Bruaen and colleagueswhere TAPSE �15.9 mm was predictiveof lower event-free survival and higherall-cause mortality.30
Ratio of RV effective systolic to dia-stolic duration. The duration of TR, amarker of impaired adaptation to pressureoverload and of RV failure, is stronglyrelated to outcome. In fact, in these cir-
Figure 4: (a) Measurement of effective RV systolicand diastolic duration on trans-tricuspid Doppler.(b) Measurement of RA area on 2D echocardiogram.(c) Time-dependent receiver operating characteristiccurves at 1.5 years for the echocardiographiccomposite score (TAPSE <15 mm, ratio of RVeffective systolic to diastolic duration >1.5, rightatrial area >25 cm2, RA/LA area ratio >1.5).(Reprinted with permission from Moceri P,Dimopoulos K, Liodakis E, et al. Echocardiographicpredictors of outcome in Eisenmenger syndrome.Circulation. 2012;126:1461-1468.).
CM
ESe
ctio
n
176 Advances in Pulmonary Hypertension
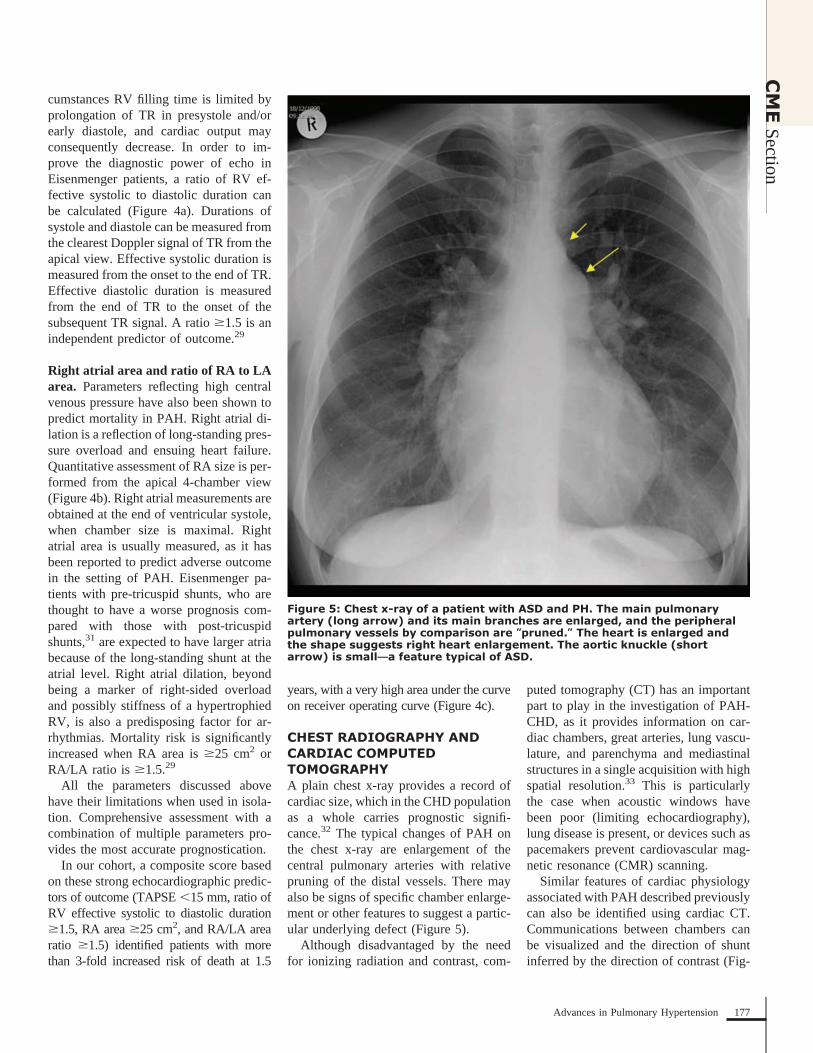
cumstances RV filling time is limited byprolongation of TR in presystole and/orearly diastole, and cardiac output mayconsequently decrease. In order to im-prove the diagnostic power of echo inEisenmenger patients, a ratio of RV ef-fective systolic to diastolic duration canbe calculated (Figure 4a). Durations ofsystole and diastole can be measured fromthe clearest Doppler signal of TR from theapical view. Effective systolic duration ismeasured from the onset to the end of TR.Effective diastolic duration is measuredfrom the end of TR to the onset of thesubsequent TR signal. A ratio �1.5 is anindependent predictor of outcome.29
Right atrial area and ratio of RA to LAarea. Parameters reflecting high centralvenous pressure have also been shown topredict mortality in PAH. Right atrial di-lation is a reflection of long-standing pres-sure overload and ensuing heart failure.Quantitative assessment of RA size is per-formed from the apical 4-chamber view(Figure 4b). Right atrial measurements areobtained at the end of ventricular systole,when chamber size is maximal. Rightatrial area is usually measured, as it hasbeen reported to predict adverse outcomein the setting of PAH. Eisenmenger pa-tients with pre-tricuspid shunts, who arethought to have a worse prognosis com-pared with those with post-tricuspidshunts,31 are expected to have larger atriabecause of the long-standing shunt at theatrial level. Right atrial dilation, beyondbeing a marker of right-sided overloadand possibly stiffness of a hypertrophiedRV, is also a predisposing factor for ar-rhythmias. Mortality risk is significantlyincreased when RA area is �25 cm2 orRA/LA ratio is �1.5.29
All the parameters discussed abovehave their limitations when used in isola-tion. Comprehensive assessment with acombination of multiple parameters pro-vides the most accurate prognostication.
In our cohort, a composite score basedon these strong echocardiographic predic-tors of outcome (TAPSE �15 mm, ratio ofRV effective systolic to diastolic duration�1.5, RA area �25 cm2, and RA/LA arearatio �1.5) identified patients with morethan 3-fold increased risk of death at 1.5
years, with a very high area under the curveon receiver operating curve (Figure 4c).
CHEST RADIOGRAPHY ANDCARDIAC COMPUTEDTOMOGRAPHYA plain chest x-ray provides a record ofcardiac size, which in the CHD populationas a whole carries prognostic signifi-cance.32 The typical changes of PAH onthe chest x-ray are enlargement of thecentral pulmonary arteries with relativepruning of the distal vessels. There mayalso be signs of specific chamber enlarge-ment or other features to suggest a partic-ular underlying defect (Figure 5).
Although disadvantaged by the needfor ionizing radiation and contrast, com-
puted tomography (CT) has an importantpart to play in the investigation of PAH-CHD, as it provides information on car-diac chambers, great arteries, lung vascu-lature, and parenchyma and mediastinalstructures in a single acquisition with highspatial resolution.33 This is particularlythe case when acoustic windows havebeen poor (limiting echocardiography),lung disease is present, or devices such aspacemakers prevent cardiovascular mag-netic resonance (CMR) scanning.
Similar features of cardiac physiologyassociated with PAH described previouslycan also be identified using cardiac CT.Communications between chambers canbe visualized and the direction of shuntinferred by the direction of contrast (Fig-
Figure 5: Chest x-ray of a patient with ASD and PH. The main pulmonaryartery (long arrow) and its main branches are enlarged, and the peripheralpulmonary vessels by comparison are “pruned.” The heart is enlarged andthe shape suggests right heart enlargement. The aortic knuckle (shortarrow) is small—a feature typical of ASD.
CM
ESection
177Advances in Pulmonary Hypertension

ure 6a). Although excellent at providinganatomical detail, it is generally not themodality of choice for providing func-tional data. If necessary, biventricularfunction can be assessed, though this ne-cessitates greater radiation exposure tocapture information throughout the car-diac cycle.
The CT pulmonary angiogram is thetest of choice to assess the proximal anddistal pulmonary arteries noninvasively.In doing so, pulmonary artery dilatationcan be identified as well as the presence ofthrombus, which may form in situ due tosluggish blood flow in an inflamed pul-monary vascular bed rather than beingembolic in origin (Figures 6b and c).
High-resolution CT scanning providesvaluable information on lung paren-chyma, which can be abnormal in patientswith CHD because of bronchiectasis orhypoplasia. It may also detect parenchy-mal changes due to PAH, for example,ground glass changes, nodular opacities,and serpiginous intrapulmonary vessels.In those with chronic thromboembolicPH, it may also identify within the lungtissue hemorrhage, infarction, or a mosaicpattern (due to heterogeneous lung perfu-sion). High-resolution CT has a vital rolein identifying patients with pulmonaryveno-occlusive disease, where advancedtherapies might be harmful. The key fea-tures of this rare entity on CT are inter-
lobular septal thickening, ground glassshadowing, and adenopathy.
When contrast is enhanced, this tech-nique is particularly adept in the assess-ment of extracardiac features, particularlynative or surgically fashioned systemic topulmonary shunts. Collateral vessels arereadily identified. These include dilatedbronchial arteries (a feature commonlyseen in PAH) or bypassing vessels incases of pulmonary venous occlusion.
An analytic technique called fractalanalysis has been studied to determinewhether the degree of branching withinthe pulmonary arteries of children andyoung adults with PAH, half of whom hadCHD, could be used as a noninvasive
Figure 6: Computed tomography pulmonary angiograph of patient with AVSD and PAH. (a) The common AV valve andatrial and ventricular components of the AVSD are seen, the right atrium is dilated, and there is RV hypertrophy. (b)The main pulmonary artery is dilated and larger in cross-section than the ascending aorta. (b and c) Extensivethrombus is present in the branch pulmonary arteries (arrows).
CM
ESe
ctio
n
178 Advances in Pulmonary Hypertension
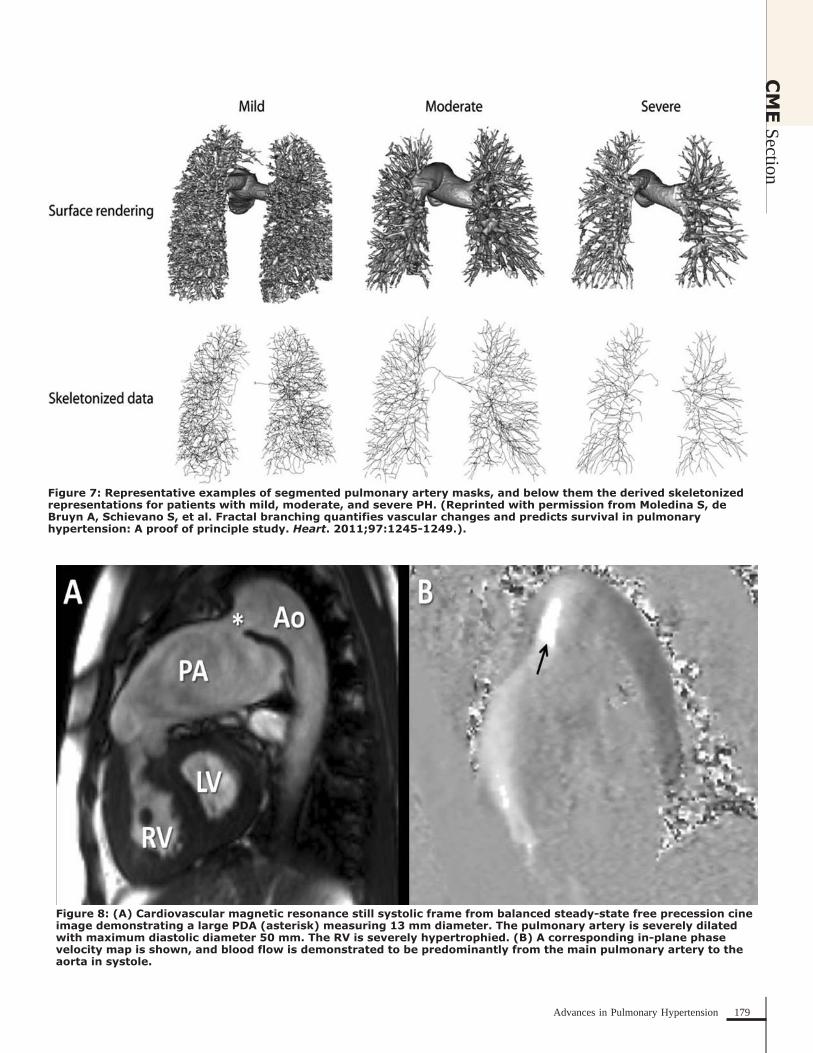
Figure 7: Representative examples of segmented pulmonary artery masks, and below them the derived skeletonizedrepresentations for patients with mild, moderate, and severe PH. (Reprinted with permission from Moledina S, deBruyn A, Schievano S, et al. Fractal branching quantifies vascular changes and predicts survival in pulmonaryhypertension: A proof of principle study. Heart. 2011;97:1245-1249.).
Figure 8: (A) Cardiovascular magnetic resonance still systolic frame from balanced steady-state free precession cineimage demonstrating a large PDA (asterisk) measuring 13 mm diameter. The pulmonary artery is severely dilatedwith maximum diastolic diameter 50 mm. The RV is severely hypertrophied. (B) A corresponding in-plane phasevelocity map is shown, and blood flow is demonstrated to be predominantly from the main pulmonary artery to theaorta in systole.
CM
ESection
179Advances in Pulmonary Hypertension

measure of PH (Figure 7).34 This indexcompared well with conventional markersof disease severity such as functionalclass, 6-minute walk test distance, andindexed PVR, and predicted death amongthe cohort [HR 5.6 (95% CI 1.2 to 25; P �0.027)].
CARDIOVASCULAR MAGNETICRESONANCECardiovascular magnetic resonance hasthe major advantage of being able to im-
age in any plane with high spatial andtemporal resolution without requiring ion-izing radiation. As repeated examinationsbecome common, the latter characteristicis particularly beneficial for patients withPAH-CHD.
In order to optimize image quality,breath holding is required. This may beproblematic for some patients with PAH-CHD. In addition, pacemakers/devicescurrently represent a contraindication toroutine CMR.
CMR acquisition and subsequent anal-ysis of a stack of contiguous cine imagecovering the whole heart from base toapex provides the gold standard assess-ments of right and left ventricular size andfunction,35 especially so for heavily tra-beculated RVs. However, analysis re-mains operator dependent.
Using both cine imaging and in-planeflow mapping, intracardiac shunts can beeasily identified and quantified. By compar-ing the ratio of flow through the pulmonary
Figure 9: Typical flow patterns in the RVOT at different cardiac phases for a patient with manifest PH (A, D, and G), apatient with latent PH (B, E, and H), and a normal subject (C, F, and I). PA indicates main pulmonary artery; PV,pulmonary valve; and RV, right ventricle. At maximum outflow (A through C), flow profiles were distributedhomogenously across the cross sections of the main pulmonary artery in the manifest PH group (A), the latent PHgroup (B), and the normal group (C). In later systole (D through F), a vortex was formed in the manifest PH group(D). No such vortex could be found in the latent PH group (E) or the normal group (F). After pulmonary valve closure(G through I), the vortex in the PH group persisted for some time. In all cases, continuous diastolic blood flowupward along the anterior wall of the main pulmonary artery could be observed. (Reprinted with permission fromReiter G, Reiter U, Kovacs G, et al. Magnetic resonance-derived 3-dimensional blood flow patterns in the mainpulmonary artery as a marker of pulmonary hypertension and a measure of elevated mean pulmonary arterialpressure. Circ Cardiovasc Imaging. 2008;1:23-30.).
CM
ESe
ctio
n
180 Advances in Pulmonary Hypertension

artery to that in the aorta using flow map-ping, Qp:QS can be determined and aorticflow mapping can be used to determine car-diac output. Tricuspid and pulmonary regur-gitation can be assessed with cine imaging,through-plane and in-plane flow.
With the addition of contrast enhancedmagnetic resonance angiography, extra-cardiac shunts (Figures 8a and b) as wellas the pulmonary vasculature can be de-lineated.36 Septal motion can be qualita-tively noted on cine imaging just as inechocardiography.
Late gadolinium enhancement for myo-cardial tissue characterization has been ap-plied to PAH and typically demonstratesareas of enhancement at the RV-LV inser-tion points. Histological data, however, sup-port the concept that the myocardial disar-ray at these sites is a normal feature ofinsertion-region anatomy exaggerated inPH by the hypertrophy of the RV.37
Significant efforts have been given overto noninvasively assessing pulmonarypressures using CMR with indices such asRV mass,38 septal deviation,39 pulmonaryartery stiffness,40 and more recently4-dimensional flow patterns41 (Figure 9).Of the longer established measures, nonehas emerged as a reliable measure in pa-tients with PAH.42 Moreover, none havebeen routinely tested in a CHD populationwhere the presence of pre-existing RVhypertrophy, septal defects, shunts, andarterial abnormalities may confoundmany of these parameters.
In the setting of a hybrid CMR inter-ventional lab, PVR derived from the Fickmethod has been shown to be inaccuratein conditions of high pulmonary bloodflow or increased oxygen concentration.With promising results, the same tech-nique has been used to show that PVR canbe determined noninvasively in a smallcohort with mainly atrial and ventricularseptal defects. A pulmonary flow of 6.05l/min/m2 or a Qp/QS ratio �2.5/1 had aspecificity of 100% for predicting PVR of�3.5 Wood units/m2 on receiver-operatorcharacteristic analysis.43
CMR Assessment of Treatment andPrognosisThe main measure used in clinical prac-tice and as a trial endpoint in PAH is the
6-minute walk test. However, this hasfaced considerable criticism given its lim-itations and failure to demonstrate a rela-tionship with clinical endpoints. This hasspurred investigation into alternative sur-rogates, one of which has been RV massas assessed by CMR.44 This measure,when employed after medical and surgicaltherapies, has been shown to reflect im-provements in indices of remodelling.45
Cardiovascular magnetic resonance hasalso been used to prognosticate in patientswith PAH. Right ventricular dilatation46
and impaired systolic RV function47 as wellas increased degrees of pulmonary arterystiffness are predictors of a poor outcome inPAH. Unfortunately, data specific to PAH-CHD have yet to be published.
CMR in Eisenmenger SyndromeCardiovascular magnetic resonance inEisenmenger syndrome occasionally cor-rectly defines the precise nature and func-tional significance of the underlyingCHD. In surgically palliated patients,CMR can be used to assess the presenceand patency of surgical shunts. It is alsouseful for assessment of other relevantextracardiac features such as PDA or aor-topulmonary collaterals. The central pul-monary arteries should be imaged forpresence of aneurysmal dilatation, poorexpansibility, sluggish flow, and in siturather than thromboembolic pulmonaryarterial thrombus.48
CONCLUSIONIn summary, multimodality imaging playsa major role in assessing patients withPAH-CHD in terms of anatomy, physiol-ogy, presence, extent and progression ofPAH, and RV function. Different imagingmodalities come with strengths and weak-nesses, and physicians and imaging spe-cialists should be aware of their comple-mentary and prognostic role, as to providethe optimal therapy and outcomes for thepatient with PAH-CHD.
References1. McLaughlin VV, Archer SL, Badesch DB, et al.ACCF/AHA 2009 expert consensus document onpulmonary hypertension a report of the AmericanCollege of Cardiology Foundation Task Force onExpert Consensus Documents and the AmericanHeart Association developed in collaboration with
the American College of Chest Physicians; Ameri-can Thoracic Society, Inc.; and the Pulmonary Hy-pertension Association. J Am Coll Cardiol. 2009;53(17):1573-1619.2. Galie N, Hoeper MM, Humbert M, et al. Guide-lines for the diagnosis and treatment of pulmonaryhypertension: the Task Force for the Diagnosis andTreatment of Pulmonary Hypertension of the Euro-pean Society of Cardiology (ESC) and the EuropeanRespiratory Society (ERS), endorsed by the Interna-tional Society of Heart and Lung Transplantation(ISHLT). Eur Heart J. 2009;30(20):2493-2537.3. Lowe BS, Therrien J, Ionescu-Ittu R, Pilote L,Martucci G, Marelli AJ. Diagnosis of pulmonaryhypertension in the congenital heart disease adultpopulation impact on outcomes. J Am Coll Cardiol.2011;58(5):538-546.4. Gatzoulis MA, Alonso-Gonzalez R, Beghetti M.Pulmonary arterial hypertension in paediatric andadult patients with congenital heart disease. EurRespir Rev. 2009;18(113):154-161.5. Khambadkone S, Li J, de Leval MR, Cullen S,Deanfield JE, Redington AN. Basal pulmonary vas-cular resistance and nitric oxide responsiveness lateafter Fontan-type operation. Circulation. 2003;107(25):3204-3208.6. Schulze-Neick I, Beghetti M. Classifying pul-monary hypertension in the setting of the congeni-tally malformed heart–cleaning up a dog’s dinner.Cardiol Young. 2008;18(1):22-25.7. Kilner PJ. Imaging congenital heart disease inadults. Br J Radiol. 2011;84 Spec No 3:S258-S268.8. Rudski LG, Lai WW, Afilalo J, et al. Guidelinesfor the echocardiographic assessment of the rightheart in adults: a report from the American Societyof Echocardiography endorsed by the European As-sociation of Echocardiography, a registered branchof the European Society of Cardiology, and the Ca-nadian Society of Echocardiography. J Am SocEchocardiogr. 2010;23(7):685-713; quiz 786-788.9. Hatle L, Angelsen BA, Tromsdal A. Non-invasive estimation of pulmonary artery systolicpressure with Doppler ultrasound. Br Heart J. 1981;45(2):157-165.10. Fisher MR, Forfia PR, Chamera E, et al. Ac-curacy of Doppler echocardiography in the hemody-namic assessment of pulmonary hypertension. Am JRespir Crit Care Med. 2009;179(7):615-621.11. Bossone E, D’Andrea A, D’Alto M, et al.Echocardiography in pulmonary arterial hyperten-sion: from diagnosis to prognosis. J Am Soc Echo-cardiogr. 2013;26(1):1-14.12. Ling LF, Marwick TH. Echocardiographic as-sessment of right ventricular function: how to ac-count for tricuspid regurgitation and pulmonary hy-pertension. JACC Cardiovasc Imaging. 2012;5(7):747-753.13. Abbas AE, Fortuin FD, Schiller NB, AppletonCP, Moreno CA, Lester SJ. Echocardiographic de-termination of mean pulmonary artery pressure.Am J Cardiol. 2003;92(11):1373-1376.14. Mahan G, Dabestani A, Gardin J, et al.Estima-tion of pulmonary artery pressure by pulsed Dopplerechocardiography. Circulation. 1983;68:367.15. Dabestani A, Mahan G, Gardin JM, et al. Eval-
CM
ESection
181Advances in Pulmonary Hypertension

uation of pulmonary artery pressure and resistanceby pulsed Doppler echocardiography. Am J Cardiol.1987;59(6):662-668.16. Arkles JS, Opotowsky AR, Ojeda J, et al.Shape of the right ventricular Doppler envelope pre-dicts hemodynamics and right heart function in pul-monary hypertension. Am J Respir Crit Care Med.2011;183(2):268-276.17. Hopkins WE. The remarkable right ventricle ofpatients with Eisenmenger syndrome. Coron ArteryDis. 2005;16(1):19-25.18. Tan JL, Prati D, Gatzoulis MA, Gibson D,Henein MY, Li W. The right ventricular response tohigh afterload: comparison between atrial switchprocedure, congenitally corrected transposition ofthe great arteries, and idiopathic pulmonary arterialhypertension. Am Heart J. 2007;153(4):681-688.19. Forfia PR, Vachiery JL. Echocardiography inpulmonary arterial hypertension. Am J Cardiol.2012;110(6 Suppl):16S-24S.20. Galie N, Hinderliter AL, Torbicki A, et al.Effects of the oral endothelin-receptor antagonistbosentan on echocardiographic and Doppler mea-sures in patients with pulmonary arterial hyperten-sion. J Am Coll Cardiol. 2003;41(8):1380-1386.21. Kaul S, Tei C, Hopkins JM, Shah PM. Assess-ment of right ventricular function using two-dimensional echocardiography. Am Heart J. 1984;107(3):526-531.22. Ghio S, Klersy C, Magrini G, et al. Prognosticrelevance of the echocardiographic assessment ofright ventricular function in patients with idiopathicpulmonary arterial hypertension. Int J Cardiol.2010;140(3):272-278.23. Anavekar NS, Gerson D, Skali H, Kwong RY,Yucel EK, Solomon SD. Two-dimensional assess-ment of right ventricular function: an echocardio-graphic-MRI correlative study. Echocardiography.2007;24(5):452-456.24. Tei C, Dujardin KS, Hodge DO, et al. Dopplerechocardiographic index for assessment of globalright ventricular function. J Am Soc Echocardiogr.1996;9(6):838-847.25. Duncan AM, Francis DP, Henein MY, GibsonDG. Limitation of cardiac output by total isovolumictime during pharmacologic stress in patients withdilated cardiomyopathy: activation-mediated effectsof left bundle branch block and coronary arterydisease. J Am Coll Cardiol. 2003;41(1):121-128.26. Puwanant S, Park M, Popovic ZB, et al. Ven-tricular geometry, strain, and rotational mechanics inpulmonary hypertension. Circulation. 2010;121(2):259-266.
27. Grapsa J, O’Regan DP, Pavlopoulos H,Durighel G, Dawson D, Nihoyannopoulos P. Rightventricular remodelling in pulmonary arterial hyper-tension with three-dimensional echocardiography:comparison with cardiac magnetic resonance imag-ing. Eur J Echocardiogr. 2010;11(1):64-73.28. Dragulescu A, Grosse-Wortmann L, FackouryC, et al. Echocardiographic assessment of right ven-tricular volumes after surgical repair of tetralogy ofFallot: clinical validation of a new echocardio-graphic method. J Am Soc Echocardiogr. 2011;24(11):1191-1198.29. Moceri P, Dimopoulos K, Liodakis E, et al.Echocardiographic predictors of outcome in eisen-menger syndrome. Circulation. 2012;126(12):1461-1468.30. Van De Bruaene A, De Meester P, Voigt JU, etal. Right ventricular function in patients with Eisen-menger syndrome. Am J Cardiol. 2012;109(8):1206-1211.31. Engelfriet PM, Duffels MG, Moller T, et al.Pulmonary arterial hypertension in adults born witha heart septal defect: the Euro Heart Survey on adultcongenital heart disease. Heart. 2007;93(6):682-687.32. Dimopoulos K, Giannakoulas G, Bendayan I,et al. Cardiothoracic ratio from postero-anteriorchest radiographs: A simple, reproducible and inde-pendent marker of disease severity and outcome inadults with congenital heart disease. Int J Cardiol.2011 Dec 1. [Epub ahead of print]33. Nicol ED, Kafka H, Stirrup J, et al. A single,comprehensive non-invasive cardiovascular assess-ment in pulmonary arterial hypertension: combinedcomputed tomography pulmonary and coronary an-giography. Int J Cardiol. 2009;136(3):278-288.34. Moledina S, de Bruyn A, Schievano S, et al.Fractal branching quantifies vascular changes andpredicts survival in pulmonary hypertension: a proofof principle study. Heart. 2011;97(15):1245-1249.35. Bradlow WM, Hughes ML, Keenan NG, et al.Measuring the heart in pulmonary arterial hyperten-sion (PAH): implications for trial study size. J MagnReson Imaging. 2010;31(1):117-124.36. Kafka H, Mohiaddin RH. Cardiac MRI andpulmonary MR angiography of sinus venosus defectand partial anomalous pulmonary venous connectionin cause of right undiagnosed ventricular enlarge-ment. AJR Am J Roentgenol. 2009;192(1):259-266.37. Bradlow WM, Assomull R, Kilner PJ, GibbsJS, Sheppard MN, Mohiaddin RH. Understandinglate gadolinium enhancement in pulmonary hyper-tension. Circ Cardiovasc Imaging. 2010;3(4):501-503.
38. Saba TS, Foster J, Cockburn M, Cowan M,Peacock AJ. Ventricular mass index using mag-netic resonance imaging accurately estimates pul-monary artery pressure. Eur Respir J. 2002;20(6):1519-1524.39. Roeleveld RJ, Marcus JT, Faes TJ, et al. In-terventricular septal configuration at mr imaging andpulmonary arterial pressure in pulmonary hyperten-sion. Radiology. 2005;234(3):710-717.40. Bradlow WM, Gatehouse PD, Hughes RL, etal. Assessing normal pulse wave velocity in theproximal pulmonary arteries using transit time: afeasibility, repeatability, and observer reproducibil-ity study by cardiovascular magnetic resonance. JMagn Reson Imaging. 2007;25(5):974-981.41. Reiter G, Reiter U, Kovacs G, et al. Magneticresonance-derived 3-dimensional blood flow pat-terns in the main pulmonary artery as a marker ofpulmonary hypertension and a measure of elevatedmean pulmonary arterial pressure. Circ CardiovascImaging. 2008;1(1):23-30.42. Roeleveld RJ, Marcus JT, Boonstra A, et al. Acomparison of noninvasive MRI-based methods ofestimating pulmonary artery pressure in pulmonaryhypertension. J Magn Reson Imaging. 2005;22(1):67-72.43. Bell A, Beerbaum P, Greil G, et al. Noninva-sive assessment of pulmonary artery flow and resis-tance by cardiac magnetic resonance in congenitalheart diseases with unrestricted left-to-right shunt.JACC Cardiovasc Imaging. 2009;2(11):1285-1291.44. Wilkins MR, Paul GA, Strange JW, et al.Sildenafil versus Endothelin Receptor Antagonist forPulmonary Hypertension (SERAPH) study. Am JRespir Crit Care Med. 2005;171(11):1292-1297.45. Bradlow WM, Gibbs JS, Mohiaddin RH. Car-diovascular magnetic resonance in pulmonary hy-pertension. J Cardiovasc Magn Reson. 2012;14:6.46. van Wolferen SA, Marcus JT, Boonstra A, etal. Prognostic value of right ventricular mass, vol-ume, and function in idiopathic pulmonary arterialhypertension. Eur Heart J. 2007;28(10):1250-1257.47. van de Veerdonk MC, Kind T, Marcus JT, etal. Progressive right ventricular dysfunction in pa-tients with pulmonary arterial hypertension respond-ing to therapy. J Am Coll Cardiol. 2011;58(24):2511-2519.48. Broberg CS, Ujita M, Prasad S, et al. Pulmo-nary arterial thrombosis in eisenmenger syndrome isassociated with biventricular dysfunction and de-creased pulmonary flow velocity. J Am Coll Cardiol.2007;50(7):634-642.
CM
ESe
ctio
n
182 Advances in Pulmonary Hypertension

Targeted Pulmonary Arterial HypertensionTherapies and a Combined Medical-SurgicalApproach for Congenital Heart Disease Patients
Warren A. Zuckerman, MDPulmonary Hypertension CenterColumbia University College of Physiciansand SurgeonsNew York, NY
Erika B. Rosenzweig, MDPulmonary Hypertension CenterColumbia University College of Physiciansand SurgeonsNew York, NY
Pulmonary arterial hypertension (PAH) with increased pulmonary vascularresistance (PVR) is a frequent complication of congenital heart disease (CHD),most commonly occurring with systemic-to-pulmonary shunt lesions. The nat-ural disease progression involves pulmonary endothelial damage due to expo-sure to increased pulmonary blood flow and pressure, and in its most severeform results in Eisenmenger syndrome (ES), in which there is shunt reversaland cyanosis. Due to anatomic and histopathologic similarities of PAH in pa-tients with CHD, as well as those with idiopathic and other forms of Group 1PAH, there is an evolving interest and role for the use of the newer targetedPAH therapies in CHD patients. While early closure of shunt lesions is the bestpreventive measure, the use of targeted medical therapies when a patient istoo high risk for surgery, following surgical repair, and even in those withreversal of shunting due to advanced disease, has emerged over the pastdecade.
Pulmonary arterial hypertension with in-creased PVR is a frequent complication ofCHD, known as associated pulmonary ar-terial hypertension (APAH)-CHD.1,2 Thisresults from pulmonary vascular remod-eling due to nonrestrictive, shunt-relatedincreases in pulmonary blood flow (PBF)and/or exposure to increased pulmonaryartery pressure (PAP) and sheer stress.3
While the currently accepted definition ofPAH no longer includes elevated PVR, itis very important to determine PVR whenevaluating a patient with APAH-CHD, asan isolated elevation in PAP with normalPVR may occur with increased PBF andcan be amenable to surgery as opposed torepresenting true pulmonary vasculopa-thy. Therefore, these authors continue touse the classic definition of PAH whenevaluating APAH-CHD, which includes amean PAP �25 mm Hg with normal left-sided filling pressures (left ventricularend-diastolic pressure or pulmonary cap-illary wedge pressure �15 mm Hg) andan elevated PVR (PVR indexed to bodysurface area [PVRI] �3 Wood units[WUxm2]).4-6 In cases of APAH-CHD, it isalso important to differentiate PAH frompulmonary venous hypertension, in whichthere is increased PAP in the setting ofelevated left-sided filling pressures andnormal PVR, or a mixed picture with el-evation of PAP, left-sided filling pres-sures, and also PVR. It is critical to dis-
tinguish between patients with PAH,pulmonary venous hypertension, andmixed disease (PAH/PVH), as the treat-ments for these various forms of diseasenot only vary, but targeted medical ther-apies may be unsafe in patients with as-sociated postcapillary pulmonary hyper-tension.
As more patients with CHD are surviv-ing into adulthood, APAH-CHD has be-come an important medical managementissue. The development of PAH in thesetting of CHD is partly dependent on thetype and size of the cardiac defect, as wellas other predisposing environmental andgenetic factors. Post-tricuspid valve le-sions such as ventricular septal defect(VSD) and patent ductus arteriosus (PDA)are more prone to the development ofPAH than pre-tricuspid valve lesions suchas atrial septal defect (ASD).7 With pro-gression of disease in these patients, cya-nosis eventually develops as a result ofreversal of left to right shunting, and thisis known as Eisenmenger syndrome.7 It isgenerally believed that in order to avoidthe development of pulmonary vasculardisease (PVD), nonrestrictive post-tricuspid defects such as large VSDs andPDAs should be repaired prior to 1 or 2years of age, while ASDs may be repairedlater in childhood. In addition, there aremore complex cardiac defects that are as-sociated with the early development of
PAH. These include truncus arteriosus,transposition of the great vessels (espe-cially in the presence of a VSD), andcomplete atrioventricular septal defect(especially in the setting of trisomy 21). Ifthese defects are not repaired within thefirst few weeks of life, severe PVD willalmost invariably develop.8
The clinical presentation of APAH-CHD can be divided into 4 physiologicsubtypes (Table). This clinical classifica-tion becomes very important in the man-agement of these patients, as treatmentstrategies are not necessarily the same foreach subtype. The latter 2 categories,PAH in the setting of small defects andPAH after corrective cardiac surgery, arephysiologically analogous to idiopathicPAH (IPAH), with respect to the lack ofan adequate “pop-off” for the failing rightventricle (RV). These patients are likely atincreased risk for more rapidly progres-sive RV failure and worse outcomes thanthose patients with adequate RV “pop-off.” The medical approach in these pa-tients does not differ from that of otherforms of WHO Group 1 PAH, althoughthere are less controlled data on the use oftargeted PAH therapies available for suchpatients.
During the last 2 decades there havebeen significant improvements in thetreatment and outcomes of patients withWHO Group 1 PAH. Nine medicationshave become available in the UnitedStates to target 3 main pathways involvedin the pathophysiology of PAH: the pros-tacyclin, endothelin, and nitric oxide path-
Key Words—associated PAH, Eisenmenger syndrome, endothelin receptor agonist, phosphodiesterase-5inhibitors, prostanoidsCorrespondence: [email protected]
CM
ESection
183Advances in Pulmonary Hypertension

ways, and treatment options now span theoral, inhaled, subcutaneous, and intrave-nous routes. Much of the initial testingoccurred in small observational trials ofAPAH-CHD or in patients with Group 1PAH; however, only a small number ofthe Group 1 patients included in many ofthe early landmark trials were APAH-CHD patients. In 1999, Rosenzweig et alreported on the benefits in functional ca-pacity and hemodynamics of long-termintravenous epoprostenol in patients withAPAH-CHD,9 and Simonneau et al re-ported similar benefits of subcutaneoustreprostinil following a 12-week random-ized, double-blind, placebo-controlledtrial in which 109 of 470 enrolled patientseither had repaired or unrepaired congen-ital shunt lesions.10 The early, random-ized, double-blind, placebo-controlledtrials involving the phophodiesterase-5 in-hibitors sildenafil (SUPER-1 trial) andtadalafil (PHIRST trial) included 7% and12% APAH-CHD patients (either ASD orrepaired shunt lesions).11,12 Although theearlier trials involving the endothelin re-ceptor antagonists bosentan and am-brisentan did not include APAH-CHD pa-tients, the first randomized, double-blind,placebo-controlled drug trial solely on ESpatients was the BREATHE-5 trial in-volving bosentan, published in 2006.13
Due to the similarities in histopathologyand pathophysiology within WHO Group1 PAH patients, including those withIPAH and APAH-CHD, the targeted PAH
therapies are often used in the treatment ofAPAH-CHD patients, as well. However,the absence or small percentage ofAPAH-CHD patients included in thesedrug trials underscores the need for futurestudies designed specifically to study thisheterogeneous patient population. Medi-cal management of APAH-CHD patientswithin the first 2 subtypes of APAH-CHD(Table), those with ES and those withPAH in the setting of unrepaired moderateto large defects, requires additional atten-tion to the impact of residual shunting onmedical management. The clinical picturewithin these groups of patients spans awide range, and at either end of the spec-trum, medical decision making is rela-tively straightforward. At one end, an in-fant with a large nonrestrictive VSD willhave elevated PAP and normal PVRI (�3WUxm2), and can be treated with surgicalclosure of the shunt with little fear thatPAP will remain elevated postoperatively.On the opposite end of the spectrum arepatients with severe pulmonary vascu-lopathy, or ES, demonstrated by shuntreversal, low PBF, and cyanosis in thesetting of elevated PAP and high PVR. Inthese patients with significantly ele-vated PVR and reversal of shunting, sur-gical closure would not be advisable;however, these patients may benefit fromsome of the newer targeted PAH medicaltherapies.13-16 With advances made in tar-geted PAH therapies, the concept of acombined medical-surgical approach has
also become more feasible for subgroup 2patients (PAH with moderate to large de-fects in whom PVR is at least mildlyincreased, systemic-to-pulmonary shunt isstill prevalent, and no cyanosis is presentat rest), with optimization first medicallyprior to consideration of surgery. In addi-tion, the idea of a partial repair hasemerged in which a fenestration, mostcommonly an interatrial communication,is left by the surgeon to serve as a “pop-off valve” for the RV.8 This determinationof operability is one of the more challeng-ing and more important aspects of themanagement of patients with APAH-CHD.
PAH ASSOCIATED WITHCONGENITAL SYSTEMIC TOPULMONARY SHUNTS: PRE-EISENMENGER SYNDROMEFor patients with APAH-CHD in the sec-ond physiologic subgroup (Table) withmildly to moderately elevated PVR andmoderate to large increases in PBF, thedetermination of operability is based onwhether PAH will improve or progressfollowing surgery. While there is no ac-cepted protocol or algorithm to make thisdetermination, a vital tool at the disposalof the cardiologist in such cases is thecardiac catheterization. Although no val-idated criteria exist for predicting postop-erative morbidity and mortality, a com-plete set of hemodynamics should beobtained, along with acute vasodilatortesting and possible balloon occlusion ofthe defect. In IPAH patients, the mostprognostic catheterization data are indicesof right heart function, such as cardiacindex and right atrial pressure,17 and PAPis generally less useful for assessing dis-ease severity and prognosis, as it may infact decrease with worsening right heartfailure, due to the RV’s inability to gen-erate higher pressure. In shunt lesions, it isimportant to realize that by having a non-restrictive VSD or PDA, PAP will be atsystemic levels regardless of the PVR. Inorder to evaluate the degree of PVD, it isimportant to examine PVR, the ratio ofPVR to systemic vascular resistance(SVR), and the ratio of pulmonary to sys-temic blood flow. In pediatrics, by con-vention, PVR is indexed to body surfacearea (PVRI) to allow for a more direct
Table: Clinical classification of congenital systemic-to-pulmonary shuntsassociated with PAH. From Simonneau, et al. 200955
Eisenmenger syndrome Patients with unrepaired systemic-to-pulmonaryshunts resulting from large nonrestrictivedefects leading to a severe, progressiveincrease in PVR, bidirectional shunting, andultimately reversed shunting with centralcyanosis
PAH with moderate to largedefects
PVR is mildly to moderately increased,systemic-to-pulmonary shunt is still present,and no cyanosis is present at rest
PAH with small defects Smaller defects generally include VSD �1 cmand ASD �2 cm, and clinical picture is similarto IPAH
PAH following correctivecardiac surgery
CHD has been corrected, but PAH is presenteither immediately after surgery or recursseveral months or years after surgery in theabsence of significant residual shunts
CM
ESe
ctio
n
184 Advances in Pulmonary Hypertension

comparison of hemodynamics betweendisparately sized patients.18
During evaluation of a systemic-to-pulmonary shunt, if there is no evidenceof pulmonary venous hypertension oncebaseline data are collected and PVRIis �3 WUxm2, acute vasodilator testingshould be performed with inhaled nitricoxide or intravenous epoprostenol, duringwhich another full hemodynamic assess-ment should be performed. Separate as-sessment with 100% oxygen may be per-formed, as well. An extensive review ofhemodynamic parameters in determiningoperability in APAH-CHD was recentlypublished by Giglia and Humpl.19 Theauthors acknowledged limitations tomany of the studies reviewed, and wereclear that any parameters be used as aguide within the context of the completeclinical picture. PVRI values in the rangeof 6 to 8 WUxm2 or lower are generallyconsidered operable.19 The ability tolower PVRI to 6 WUxm2 or lower withvasodilator testing with inhaled nitric ox-ide �/� oxygen has been associated withbetter outcomes, as well.20-22 Addition-ally, a �10% decrease in PVR and PVR/SVR ratio in response to acute vasodilatortesting, with a final PVR/SVR ratio of�0.3 are predictive of better postopera-tive outcomes.23,24
Due largely in part to advances in tar-geted PAH therapies and studies demon-strating improved hemodynamics inAPAH-CHD patients with such therapies,there is an evolving role for a combinedmedical-surgical approach to those pa-tients that are either borderline operable,or in some cases initially inoperable.25-28
It is reasonable to treat with targetedtherapies for a period of months and re-evaluate by catheterization, sometimes re-quiring serial reevaluations. If medicaltreatment of a patient with a shunt andmoderate PVD is effective, PVR will lowerand result in increased PBF. As a result,surgical measures may actually be neces-sary to protect the pulmonary vasculaturefrom the development of further damage.
Cardiac catheterization alone, however,cannot determine operability, since cath-eterization data are often obtained under“ideal” resting conditions. While a patientat rest may seem to be operable, with a
minor respiratory illness and hypoxic va-soconstriction, it may become evident thata shunt should not be closed, or at leastnot completely. A thorough medical his-tory and physical examination, and exer-cise testing when possible, play importantroles in the determination of operability.Important elements of the history includeage, type of CHD, and time of and cir-cumstances surrounding diagnosis. Theimportance of the type of CHD has beenpreviously discussed; and in general, theearlier a shunt lesion is diagnosed, themore likely the patient is operable. In ad-dition, a history of cyanosis and/or dys-pnea with exertion is important. Signs ofcyanosis such as blue lips or nail bedswith exercise, clubbing, and erythrocyto-sis also help provide a complete picture todetermine operability.
EISENMENGER SYNDROMEIn 1897, Viktor Eisenmenger first de-scribed a 32-year-old patient who diedof massive hemoptysis and had a VSDon postmortem examination. The term“Eisenmenger syndrome” was coined byPaul Hamilton Wood in 1958 to define thecondition of increased PAP and PVR inrelation to a VSD with resultant shuntreversal and cyanosis. Subsequently, EShas been used to describe any CHD orshunt between the great arteries with re-sultant increase in PVR and shunt rever-sal.29 Advances in CHD diagnosis andcardiac surgery, especially during infancyand early childhood, have helped to in-crease the number of CHD patients sur-viving into adulthood, and decrease thenumber of patients with ES in the Westernworld. Only around 5% of adults withCHD will develop PAH.30 However, indeveloping countries where patients seekmedical care later in life, ES still remainsa significant problem. The worldwideprevalence of PAH in adults with CHDhas recently been estimated at between1.6 and 12.5 million, with 25% to 50%presenting with ES.31 In Latin America,the prevalence of advanced APAH-CHDrelative to IPAH at cardiovascular centersis between 2:1 and 3:1.32
Although life expectancy is reduced inES, it is significantly better than IPAH,with many patients surviving into their
third and fourth decades,33 and even someinto their seventh decade.33,34 More than40% of subjects are expected to be alive25 years after diagnosis.7,35 There is somebias in these data as many patients werefrom the era prior to targeted PAH thera-pies. As a result, many who died early ofsevere hypoxemia and RV failure due toadvanced ES may not have been included.However, with advances in targeted ther-apies, the hope is for future improvementsto these numbers, supported by a recentstudy predicting 5-year survival of 95.3%in children with ES.36
Conventional Therapies forEisenmenger SyndromeHistorically, treatment options for ES pa-tients had been limited to palliative ther-apies and heart-lung transplantation orlung transplantation with surgical correc-tion of a simple shunt. Currently, conven-tional management is used in combinationwith targeted PAH therapies. Commonlyused conventional therapies may includedigoxin, diuretics, and anticoagulation, aswell as antiarrhythmics when warranted,although none of these agents have beenshown to improve survival in ES. Al-though supporting evidence is not partic-ularly strong, digoxin is generally used forright heart failure.37 Diuretics are oftenemployed in this situation, as well; how-ever, they should be used cautiously, asthey may reduce plasma volume in pa-tients with erythrocytosis, and also lead todehydration. Anticoagulation in ES pa-tients is a debatable subject due to in-creased risks of pulmonary artery throm-bosis as well as hemoptysis, stroke, andhemorrhage.37 Although the benefit of an-ticoagulation in IPAH patients has beendemonstrated,38,39 no such data exist inES patients. Given the potential compli-cations, the decision to anticoagulateshould be made carefully on an individ-ual, case-by-case basis. Long-term use ofoxygen is often employed in ES patients;and while it may be associated with im-provement in subjective status, no sur-vival benefit has been reported.37,40
ES patients that are chronically cya-notic may develop secondary erythrocy-tosis. With hemoglobin above 20 g/dL,hyperviscosity symptoms may develop,
CM
ESection
185Advances in Pulmonary Hypertension

including headache, fatigue, and difficultyconcentrating. Phlebotomy, combinedwith isovolumic fluid replacement, is re-served only for patients with symptomatichyperviscosity and no iron deficiency ordehydration.41,42 Iron deficiency, oftenmissed in this patient population due tothe requirement of a relatively high rest-ing hemoglobin in the setting of chroniccyanosis, is associated with lower event-free survival and higher mortality in ESpatients, and should be treated when rec-ognized.41,43
Targeted Therapies for EisenmengerSyndromeThere are emerging data on the use of all3 main classes of targeted PAH therapies(prostanoids, endothelin receptor antago-nists, and phosphodiesterase-5 inhibitors)for the treatment of ES patients. As op-posed to APAH-CHD with moderatePVD, ES patients are unlikely to experi-ence a significant decrease in PVR thatwould be enough to reverse shunting andallow for surgical correction of CHD. Theaim of targeted therapies in these cases isto improve exercise tolerance, hypox-emia, physical capacity, and ultimatelysurvival.
Intravenous epoprostenol has been usedin pediatric PAH,44 and specifically inAPAH-CHD,9,45 leading to improvementsin functional capacity, hemodynamics,and survival. In 1999, Rosenzweig et alreported the first benefit of PAH-targetedtherapy of any form in this patient popu-lation, with improvements in cardiac in-dex, PVR, and quality of life in 20 pa-tients with APAH-CHD.9 Similarly, in2003, Fernandes et al demonstrated im-provements in functional capacity, oxy-gen saturation, and hemodynamics in 8ES patients using epoprostenol.45 Treat-ment with epoprostenol requires use of apermanent central venous catheter, whichcan be problematic in the setting of rightto left shunt due to the potential for throm-boembolic events. In addition, systemicand local complications may include in-fections, sepsis, and line breakage withdrug interruption. Treprostinil, a similarand longer-acting prostanoid, can be de-livered either intravenously, subcutane-ously, or by inhalation, and offers poten-
tial advantages over epoprostenol in termsof a longer half-life and mode of delivery.Although efficacy and safety has notbeen fully established in this patientpopulation, open-label multicenter trialsinvolving the intravenous and subcuta-neous formulations have included pa-tients with APAH-CHD,10,46 and the in-haled formulation has recently been FDA-approved for use in patients in WHOGroup 1.
The first randomized, double-blind,placebo-controlled study in ES patientswas the BREATHE-5 trial, which inves-tigated the efficacy and safety of the dualendothelin receptor antagonist bosentan inadult ES patients.13 It remains the onlytrial of its kind dedicated solely to the ESpopulation. During the 16-week study,bosentan significantly reduced PVR andimproved PAP and exercise capacitycompared to placebo.13 Longer-term datafrom the follow-up portion of the studydemonstrated continued improvements inexercise capacity and functional classover an additional 24 weeks.15 Safetyfindings were of particular importance,given the potential for worsening of rightto left shunting in the face of decreasedSVR with vasodilator therapies. Therewas no significant difference in oxygensaturation change between the bosentanand placebo groups during the study pe-riod. In addition, there was a worsening ofPVR in the placebo group, underscoringthe progressive nature of untreated ES.Smaller-scale, open-label studies demon-strate sustained effects over longer peri-ods of time using bosentan in the ESpopulation.47-49 The selective endothelinreceptor antagonist ambrisentan offers po-tential advantages over bosentan given itsselectivity for the endothelin-A receptor,which demonstrates vasoconstrictor ef-fects. Although less studied, ambrisentanhas also been noted to be safe and effica-cious in APAH-CHD and ES.28
Oral sildenafil is the most widely usedof the phosphodiesterase-5 inhibitors inthe treatment of PAH, and has been used inpediatrics and APAH-CHD, with benefitson exercise capacity and hemodynamicsdemonstrated.26,50,51 Singh et al performeda randomized, placebo-controlled, double-blind, crossover study in 10 IPAH and 10
ES patients in 2006, and found that silde-nafil significantly improved functionalstatus, exercise capacity, and PAP com-pared to placebo.50 Similarly, a recentprospective, open-label, multicenter studyout of China on 84 ES patients demon-strated safety and improved functionalstatus, exercise capacity, oxygen satura-tion, PAP, and PVR after 12 months ofsildenafil therapy.14 Sildenafil also comesin an intravenous form, and there may bea role for its use perioperatively in theintensive care setting. The longer-acting,once-daily dosed tadalafil is less wellstudied than sildenafil; however, a recentrandomized, placebo-controlled, double-blind, crossover study in ES patients alsodemonstrated safety and short-term im-provements in exercise capacity, func-tional class, oxygen saturation, and hemo-dynamics after 6 weeks of therapy.52
Due to the progressive nature of PAHand the efficacy limitations of each of thedrug classes, one of the mainstays of thetreatment of PAH has become combina-tion therapy. In this manner, drugs withdifferent mechanisms of action may pro-vide an additive effect, or even the sameeffect at lower doses. Although data arelimited in ES patients, in 2010, Iversen etal performed a randomized, placebo-controlled, double-blind, crossover studyevaluating the effect of combination ther-apy with bosentan and sildenafil in 21 ESpatients.53 In this study, patients weretreated with bosentan for 9 months andwere then treated for 3 months with silde-nafil or placebo, followed by a 3-monthcrossover. They found improvements inexercise capacity and hemodynamics aftertreatment with bosentan, but no furtherbenefit after addition of sildenafil, al-though there was an increase in oxygensaturation and the combination was welltolerated. The future of PAH therapy inES patients certainly involves evaluationof such combination therapies, as well asstudies evaluating cutting-edge, targetedPAH therapies such as oral prostacyclin(selexipag), tissue-targeting endothelinreceptor antagonist (macitentan), recep-tor tyrosine kinase antagonist (ima-tinib), and soluble guanylate cyclase in-hibitor (riociguat).54 Over the last decade,the outlook for the ES patient has gone
CM
ESe
ctio
n
186 Advances in Pulmonary Hypertension

from a “hands-off” strategy to a hopefulone, in the form of targeted PAH agentsthat may lead to improvements in func-tional status, exercise capacity, and sur-vival. Further study in clinical trials willbe essential for optimization of medicaltherapies for APAH-CHD over the nextdecade.
CONCLUSIONSWhile PAH associated with CHD is clas-sified with many other subgroups asGroup 1 pulmonary hypertension, thisgroup is very heterogeneous in terms ofanatomic, physiologic, and clinical fea-tures. Improvements in diagnosis and sur-gery for CHD have dramatically im-proved the short- and long-term outlookfor patients with APAH-CHD. Althoughadvancements in noninvasive imagingsuch as echocardiography have helped inthe evaluation of this patient population,the importance of cardiac catheterizationcannot be overstated in helping with man-agement. In addition, the newer targetedPAH therapies appear to have short-termbenefits in these patients, but require fur-ther investigation, and their use in patientswith borderline hemodynamics has pavedthe way for a combined medical-surgicalapproach to management in select pa-tients.
References1. Rose ML, Strange G, King I, et al. Congenitalheart disease-associated pulmonary arterial hyper-tension: preliminary results from a novel registry.Intern Med J. 2012;42(8):874-879.2. Gatzoulis MA, Alonso-Gonzalez R, Beghetti M.Pulmonary arterial hypertension in paediatric andadult patients with congenital heart disease. EurRespir Rev. 2009;18(113):154-161.3. Fratz S, Geiger R, Kresse H, et al. Pulmonaryblood pressure, not flow, is associated with netendothelin-1 production in the lungs of patients withcongenital heart disease and normal pulmonary vas-cular resistance. J Thorac Cardiovasc Surg. 2003;126(6):1724-1729.4. McLaughlin VV, Archer SL, Badesch DB, et al.ACCF/AHA 2009 expert consensus document onpulmonary hypertension a report of the AmericanCollege of Cardiology Foundation Task Force onExpert Consensus Documents and the AmericanHeart Association developed in collaboration withthe American College of Chest Physicians; Ameri-can Thoracic Society, Inc.; and the Pulmonary Hy-pertension Association. J Am Coll Cardiol. 2009;53(17):1573-1619.5. Galie N, Hoeper MM, Humbert M, et al. Guide-
lines for the diagnosis and treatment of pulmonaryhypertension: the Task Force for the Diagnosis andTreatment of Pulmonary Hypertension of the Euro-pean Society of Cardiology (ESC) and the EuropeanRespiratory Society (ERS), endorsed by the Interna-tional Society of Heart and Lung Transplantation(ISHLT). Eur Heart J. 2009;30(20):2493-2537.6. Badesch DB, Raskob GE, Elliott CG, et al.Pulmonary arterial hypertension: baseline character-istics from the REVEAL Registry. Chest. 2010;137(2):376-387.7. Kidd L, Driscoll DJ, Gersony WM, et al. Secondnatural history study of congenital heart defects.Results of treatment of patients with ventricular sep-tal defects. Circulation. 1993;87(2 Suppl):I38-I51.8. Rosenzweig EB, Barst RJ. Congenital heart dis-ease and pulmonary hypertension: pharmacologyand feasibility of late surgery. Prog Cardiovasc Dis.2012;55(2):128-133.9. Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension withassociated congenital heart defects. Circulation.1999;99(14):1858-1865.10. Simonneau G, Barst RJ, Galie N, et al. Con-tinuous subcutaneous infusion of treprostinil, a pros-tacyclin analogue, in patients with pulmonary arte-rial hypertension: a double-blind, randomized,placebo-controlled trial. Am J Respir Crit Care Med.2002;165(6):800-804.11. Galie N, Ghofrani HA, Torbicki A, et al. Silde-nafil citrate therapy for pulmonary arterial hyperten-sion. N Engl J Med. 2005;353(20):2148-2157.12. Galie N, Brundage BH, Ghofrani HA, et al.Tadalafil therapy for pulmonary arterial hyperten-sion. Circulation. 2009;119(22):2894-2903.13. Galie N, Beghetti M, Gatzoulis MA, et al.Bosentan therapy in patients with Eisenmenger syn-drome: a multicenter, double-blind, randomized,placebo-controlled study. Circulation. 2006;114(1):48-54.14. Zhang ZN, Jiang X, Zhang R, et al. Oral silde-nafil treatment for Eisenmenger syndrome: a pro-spective, open-label, multicentre study. Heart. 2011;97(22):1876-1881.15. Gatzoulis MA, Beghetti M, Galie N, et al.Longer-term bosentan therapy improves functionalcapacity in Eisenmenger syndrome: results of theBREATHE-5 open-label extension study. Int J Car-diol. 2008;127(1):27-32.16. Dimopoulos K, Inuzuka R, Goletto S, et al.Improved survival among patients with Eisenmengersyndrome receiving advanced therapy for pulmonaryarterial hypertension. Circulation. 2010;121(1):20-25.17. D’Alonzo GE, Barst RJ, Ayres SM, et al. Sur-vival in patients with primary pulmonary hyperten-sion. Results from a national prospective registry.Ann Intern Med. 1991;115(5):343-349.18. Ofori-Amanfo G, Hsu D, Lamour JM, et al.Heart transplantation in children with markedly el-evated pulmonary vascular resistance: impact ofright ventricular failure on outcome. J Heart LungTransplant. 2011;30(6):659-666.19. Giglia TM, Humpl T. Preoperative pulmonaryhemodynamics and assessment of operability: is
there a pulmonary vascular resistance that precludescardiac operation? Pediatr Crit Care Med. 2010;11(2 Suppl):S57-S69.20. Atz AM, Adatia I, Lock JE, Wessel DL. Com-bined effects of nitric oxide and oxygen during acutepulmonary vasodilator testing. J Am Coll Cardiol.1999;33(3):813-819.21. Bush A, Busst CM, Haworth SG, et al. Corre-lations of lung morphology, pulmonary vascular re-sistance, and outcome in children with congenitalheart disease. Br Heart J. 1988;59(4):480-485.22. Neutze JM, Ishikawa T, Clarkson PM, CalderAL, Barratt-Boyes BG, Kerr AR. Assessment andfollow-up of patients with ventricular septal defectand elevated pulmonary vascular resistance. Am JCardiol. 1989;63(5):327-331.23. Berner M, Beghetti M, Spahr-Schopfer I,Oberhansli I, Friedli B. Inhaled nitric oxide to testthe vasodilator capacity of the pulmonary vascularbed in children with long-standing pulmonary hy-pertension and congenital heart disease. Am J Car-diol. 1996;77(7):532-535.24. Balzer DT, Kort HW, Day RW, et al. InhaledNitric Oxide as a Preoperative Test (INOP Test I):the INOP Test Study Group. Circulation. 2002;106(12 Suppl 1):I76-I81.25. Rosenzweig EB, Ivy DD, Widlitz A, et al.Effects of long-term bosentan in children with pul-monary arterial hypertension. J Am Coll Cardiol.2005;46(4):697-704.26. Humpl T, Reyes JT, Holtby H, Stephens D,Adatia I. Beneficial effect of oral sildenafil therapyon childhood pulmonary arterial hypertension:twelve-month clinical trial of a single-drug, open-label, pilot study. Circulation. 2005;111(24):3274-3280.27. Barst RJ, Ivy D, Dingemanse J, et al. Pharma-cokinetics, safety, and efficacy of bosentan in pedi-atric patients with pulmonary arterial hypertension.Clin Pharmacol Ther. 2003;73(4):372-382.28. Zuckerman WA, Leaderer D, Rowan CA, Mi-tuniewicz JD, Rosenzweig EB. Ambrisentan for pul-monary arterial hypertension due to congenital heartdisease. Am J Cardiol. 2011;107(9):1381-1385.29. Vongpatanasin W, Brickner ME, Hillis LD,Lange RA. The Eisenmenger syndrome in adults.Ann Intern Med. 1998;128(9):745-755.30. Duffels MG, Engelfriet PM, Berger RM, et al.Pulmonary arterial hypertension in congenital heartdisease: an epidemiologic perspective from a Dutchregistry. Int J Cardiol. 2007;120(2):198-204.31. Galie N, Manes A, Palazzini M, et al. Man-agement of pulmonary arterial hypertension associ-ated with congenital systemic-to-pulmonary shuntsand Eisenmenger’s syndrome. Drugs. 2008;68(8):1049-1066.32. Lopes AA, Bandeira AP, Flores PC, SantanaMV. Pulmonary hypertension in Latin America: pul-monary vascular disease: the global perspective.Chest. 2010;137(6 Suppl):78S-84S.33. Diller GP, Dimopoulos K, Broberg CS, et al.Presentation, survival prospects, and predictors ofdeath in Eisenmenger syndrome: a combined retro-spective and case-control study. Eur Heart J. 2006;27(14):1737-1742.
CM
ESection
187Advances in Pulmonary Hypertension

34. Daliento L, Somerville J, Presbitero P, et al.Eisenmenger syndrome. Factors relating to deterio-ration and death. Eur Heart J. 1998;19(12):1845-1855.35. Saha A, Balakrishnan KG, Jaiswal PK, et al.Prognosis for patients with Eisenmenger syndromeof various aetiology. Int J Cardiol. 1994;45(3):199-207.36. Haworth SG, Hislop AA. Treatment and sur-vival in children with pulmonary arterial hyperten-sion: the UK Pulmonary Hypertension Service forChildren 2001-2006. Heart. 2009;95(4):312-317.37. Deanfield J, Thaulow E, Warnes C, et al. Man-agement of grown up congenital heart disease. EurHeart J. 2003;24(11):1035-1084.38. Frank H, Mlczoch J, Huber K, Schuster E,Gurtner HP, Kneussl M. The effect of anticoagulanttherapy in primary and anorectic drug-induced pul-monary hypertension. Chest. 1997;112(3):714-721.39. Fuster V, Steele PM, Edwards WD, Gersh BJ,McGoon MD, Frye RL. Primary pulmonary hyper-tension: natural history and the importance of throm-bosis. Circulation. 1984;70(4):580-587.40. Diller GP, Gatzoulis MA. Pulmonary vasculardisease in adults with congenital heart disease. Cir-culation. 2007;115(8):1039-1050.41. Oechslin E. Hematological management of thecyanotic adult with congenital heart disease. IntJ Cardiol. 2004;97 Suppl 1:109-115.42. Warnes CA, Williams RG, Bashore TM, et al.ACC/AHA 2008 Guidelines for the Management ofAdults with Congenital Heart Disease: a report of
the American College of Cardiology/AmericanHeart Association Task Force on Practice Guidelines(writing committee to develop guidelines on themanagement of adults with congenital heart dis-ease). Circulation. 2008;118(23):e714-e833.43. Van De Bruaene A, Delcroix M, Pasquet A, etal. Iron deficiency is associated with adverse out-come in Eisenmenger patients. Eur Heart J. 2011;32(22):2790-2799.44. Barst RJ, Maislin G, Fishman AP. Vasodilatortherapy for primary pulmonary hypertension in chil-dren. Circulation. 1999;99(9):1197-1208.45. Fernandes SM, Newburger JW, Lang P, et al.Usefulness of epoprostenol therapy in the severelyill adolescent/adult with Eisenmenger physiology.Am J Cardiol. 2003;91(5):632-635.46. Tapson VF, Gomberg-Maitland M, McLaugh-lin VV, et al. Safety and efficacy of IV treprostinilfor pulmonary arterial hypertension: a prospective,multicenter, open-label, 12-week trial. Chest. 2006;129(3):683-688.47. Baptista R, Castro G, da Silva AM, MonteiroP, Providencia LA. Long-term effect of bosentan inpulmonary hypertension associated with complexcongenital heart disease. Rev Port Cardiol. 2013;Epub ahead of print.48. Kaya MG, Lam YY, Erer B, et al. Long-termeffect of bosentan therapy on cardiac function andsymptomatic benefits in adult patients with Eisen-menger syndrome. J Card Fail. 2012;18(5):379-384.49. Williams R, Houser L, Miner P, Aboulhosn J.Efficacy and safety of bosentan in adults with simple
and complex Eisenmenger’s syndrome. CongenitHeart Dis. 2012;7(1):12-15.
50. Singh TP, Rohit M, Grover A, Malhotra S,Vijayvergiya R. A randomized, placebo-controlled,double-blind, crossover study to evaluate the effi-cacy of oral sildenafil therapy in severe pulmonaryartery hypertension. Am Heart J. 2006;151(4):851.e1-5.
51. Raposo-Sonnenfeld I, Otero-Gonzalez I,Blanco-Aparicio M, Ferrer-Barba A, Medrano-Lopez C. [Treatment with sildenafil, bosentan, orboth in children and young people with idiopathicpulmonary arterial hypertension and Eisenmenger’ssyndrome]. Rev Esp Cardiol. 2007;60(4):366-372.
52. Mukhopadhyay S, Nathani S, Yusuf J, ShrimalD, Tyagi S. Clinical efficacy of phosphodiesterase-5inhibitor tadalafil in Eisenmenger syndrome–a ran-domized, placebo-controlled, double-blind cross-over study. Congenit Heart Dis. 2011;6(5):424-431.
53. Iversen K, Jensen AS, Jensen TV, VejlstrupNG, Sondergaard L. Combination therapy withbosentan and sildenafil in Eisenmenger syndrome: arandomized, placebo-controlled, double-blindedtrial. Eur Heart J. 2010;31(9):1124-1131.
54. Yao A. Recent advances and future perspec-tives in therapeutic strategies for pulmonary arterialhypertension. J Cardiol. 2012;60(5):344-349.
55. Simonneau G, Robbins IM, Beghetti M, et al.Updated clinical classification of pulmonary hyper-tension. J Am Coll Cardiol. 2009;54(1 Suppl):S43-S54.
CM
ESe
ctio
n
188 Advances in Pulmonary Hypertension

The Role of Catheter-Based and SurgicalTreatments in Patients With Congenital HeartDisease and Pulmonary Hypertension
Jamil A. Aboulhosn, MD, FACC, FSCAIDirector, Ahmanson/UCLA Adult CongenitalHeart Disease CenterLos Angeles, CA
This manuscript is intended to provide a brief overview of the indications forand outcomes of surgical and transcatheter interventions for congenital heartdisease and pulmonary hypertension (PH). Pulmonary hypertension is fre-quently encountered in children and adults with congenital heart disease and ismost commonly related to large “central” shunts, ie, those occurring at theventricular or great arterial level (Figure 1). If uncorrected early in infancy orchildhood, large central shunts result in increased pulmonary blood flow, leftheart volume overload, PH, and heart failure. If the child survives this initialperiod of volume overload and heart failure, they will very likely developeffacement of the normal pulmonary arterial architecture and severe eleva-tions in pulmonary arterial resistance, eventually resulting in cyanosis andEisenmenger syndrome.1
Pre-tricuspid valve shunts, ie, those at theatrial and/or venous level, are typicallynot associated with severe PH in infancyand childhood, although progressive PHwith age often occurs. Indications for sur-gical or transcatheter closure include ev-idence of right heart volume overload,arrhythmias, mild to moderate PH, anddecreased functional capacity (Table 1).2
Doppler echocardiography is indispens-able as a cost-effective tool for the non-invasive evaluation of hemodynamics andshunt fractions.3,4 Invasive cardiac cathe-terization is reserved for the subset ofpatients in whom inadequate acousticwindows limit the utility of transthoracicechocardiography or those in whom pul-monary vascular resistance or chamberpressures must be measured directly.Cross-sectional imaging techniques usingcomputed tomography or magnetic reso-nance imaging are also widely used in thenoninvasive assessment of anatomy andfunction.5
SURGICAL INTERVENTIONSOperative interventions to palliate or re-pair the congenital lesions were originallydevised to address physiologic issues,specifically to increase or diminish thesupply of blood to the pulmonary circu-lation. The early era of congenital cardiacsurgery is marked by giant leaps forwardin the physiologic treatment of lesions.For example, patients with pulmonaryatresia or single ventricle defects under-
went placement of an arterio-pulmonaryshunt, a wave of surgical innovation ini-tiated by the famed Blalock-Taussigshunt, a subclavian to pulmonary arteryconnection supplying a controlled volumeof arterial blood to the pulmonary arterialcirculation (Figure 2).6 Residual hemody-namic defects are often present in oper-ated patients and are a major cause ofprogressive deterioration that may not be-come evident for decades after surgery.Residual hemodynamic defects may beamenable to further surgery or transcath-eter intervention. Reoperations in adultswith congenital heart disease are commonand provide particular challenges.7 Therisks of reoperation are often greater thanfor the primary procedures, requiringcareful entry into the chest with extensivedissection of scar tissue and longer car-diopulmonary bypass times and greateruse of blood products.8 Careful preopera-
tive planning should include an in-depthunderstanding of the underlying cardio-vascular anatomy and the alterationscaused by previous surgical intervention.Computed tomography or magnetic reso-nance angiography may be utilized to de-termine the anatomic relationships andquantify the proximity of the heart to thesternum; sternal entry is particularly riskywhen a high pressure ventricle, great ar-tery, or conduit lies immediately posteriorto the sternum. In patients with complexcongenital heart disease, specificallythose with cyanotic lesions, definitive“correction” may not be possible until theanatomy and physiology have been opti-
Figure 1: Types and locations ofcongenital cardiac defects.
Key Words—atrial septal defect, congenital heart disease, pulmonary vascular disease, transcatheterinterventions, ventricular septal defectCorrespondence: [email protected]
Figure 2: Tricuspid atresia andpulmonary atresia. Surgical arterio-pulmonary shunts placed to increasepulmonary blood flow. The classic BT(Blalock-Taussig), Waterston, andPott’s shunts are of historicalimportance and may be present inadults, but are no longer in clinicaluse for a variety of reasons,including difficulty in controllingpulmonary blood flow and thedevelopment of PH. The central andmodified BT shunts are made ofsynthetic materials and are used inthe current surgical era.
CM
ESection
189Advances in Pulmonary Hypertension

mized by 1 or more “palliative” proce-dures. In patients with known PH, modu-lation of pulmonary arterial resistancewith inhaled nitric oxide or parenteralprostacyclin therapy is indicated in theperioperative period. The decrease in sys-temic arterial resistance and systemicblood pressure encountered with prosta-cyclin therapy can be counteracted with
selective alpha 1 agonists or vasopressinin hypotensive patients.
Heart and heart-lung (block) transplan-tation are ultimate therapeutic options inpatients who continue to deteriorate withoptimal medical therapy and have noother good reparative surgical or interven-tional options. Compared with adult re-cipients, patients with adult congenital
heart disease experience higher post-hearttransplantation mortality and retransplan-tation.9 Patients with Eisenmenger syn-drome may be offered lung transplanta-tion with repair of the cardiac defect orheart-lung transplantation. The success ofeither approach in these patients has beenlimited.10 Given the advancements in themanagement of PH and the limited suc-cess of these operations, mainly the sick-est patients who fail to stabilize or im-prove on pulmonary arterial vasodilatortherapy are considered candidates. Thepotential roles of ventricular assist de-vices and the total artificial heart in con-genital patients are currently being inves-tigated with promising early results.11,12
TRANSCATHETERINTERVENTIONSMajor advances in percutaneous trans-catheter interventions have been madeover the past 25 years in the field of con-genital heart disease.13 Improvements indevice, imaging, and catheterization tech-nologies and procedural techniques havebrought interventional cardiology to theforefront as a therapeutic intervention thatmay delay or obviate surgery. Adult con-genital cardiac catheterizations today areoften performed solely for reparative orpalliative transcatheter interventions.13,14
Interventional catheterization has largelyreplaced surgery as the treatment ofchoice for a number of congenital cardio-vascular conditions, including secundumatrial septal defect (ASD) (Figures 3 and6), patent ductus arteriosus (Figure 5),and ventricular septal defect (VSD) clo-sure (Figure 4).13,15 Careful patient se-lection and imaging are imperative tothe safety and success of transcatheterprocedures.
SPECIFIC LESIONSAtrial Septal DefectAtrial septal defects are commonly en-countered and occur in one-third of adultswith congenital heart disease. Varioustypes exist: secundum ASD is the mostcommon, accounting for 75% of defects.16
Ostium primum defects, often accompa-nied by endocardial cushion defects andinlet-type VSDs, occur in 20% of cases.Sinus venosus defects (usually superior)
Table 1: Indications for intervention in congenital shunt defects. Rightventricular enlargement (RVE), right atrial enlargement (RAE), pulmonaryartery pressure (PAP), pulmonary vascular resistance (PVR),supraventricular tachycardia (SVT), dyspnea on exertion (DOE), left atrialenlargement (LAE), left ventricular enlargement (LVE), aortic regurgitation(AR), patent ductus arteriosus (PDA), pulmonary blood flow (Qp), systemicblood flow (Qs)
Defect Indications for Intervention
Secundum ASD Qp:Qs �1.5:1RVE, RAEMild-Mod Pulmonary HTN:PAP �2/3 systemicPVR �2/3 systemicParadoxical EmbolismSVT/DOE
Patent Foramen Ovale Paradoxical Embolism (not prevented by antiplatelet/antithrombotic Rx)
Ventricular Septal Defect Qp:Qs �1.5:1LAE, LVEMild-Mod Pulmonary HTN:PAP �2/3 systemicPVR �2/3 systemicDOEAR
Patent Ductus Arteriosus Qp:Qs �1.5:1Mild-Mod Pulmonary HTN:PAP �2/3 systemicPVR �2/3 systemicLAE, LVESVT/DOEAny PDA?
Figure 3: A) Transthoracic echo with color Doppler, modified parasternallong-axis view demonstrating a complex secumdum type atrial septal defect(ASD) (vertical white arrows) that has multiple fenestrations and a left toright shunt. The right atrium (RA) and right ventricle (RV) are dilated. B)Post-transcatheter ASD closure with 2 Amplatzer septal Occluder devices.Left ventricle (LV), tricuspid valve (TV).
CM
ESe
ctio
n
190 Advances in Pulmonary Hypertension

occur in 5% of patients; the rarest type isthe coronary sinus ASD (Figure 1).
Atrial septal defects often go unrecog-nized for the first 2 decades because of theindolent clinical course and benign find-ings on physical examination. Initial di-agnosis in adulthood is common and sur-vival into adulthood is the rule. However,life expectancy is not normal in the unre-paired patient, with mortality increasingby 6% per year after age 40.17,18 Progres-sive symptoms of dyspnea on exertionand palpitations frequently occur in adult-hood and are caused by increasing rightsided chamber enlargement, PH, rightventricular failure, tricuspid regurgitation,and atrial arrhythmias. The degree of leftto right shunt may increase with age asleft ventricular compliance decreases andsystemic arterial resistance increases afterthe fourth decade. Paradoxical embolismmay occur.
Surgical repair has been performed forover 40 years and has been efficacious andsafe provided the pulmonary arterial re-sistance is not severely elevated.16,19 Sev-eral studies have shown improvement infunctional capacity, reduced arrhythmiarisk, and reduced incidence of PH aftersurgical or transcatheter closure, includ-ing in those with small defects (�1 cm),older patients, and asymptomaticindividuals.20-23 Predictors of increasedsurgical mortality include: older age atoperation, advanced heart failure (NYHAIII or IV), Qp:Qs �2.5:1, pulmonary ar-tery systolic pressure �40 mm Hg, and
increased pulmonary arterial resistance.24
The exclusion of patients with severe PHfrom defect closure may eventually beobviated by pulmonary artery vasodilatortherapy with prostaglandins, endothelinblockers, and phosphodiesterase type 5(PDE-5) inhibitors that may reduce pul-monary arterial pressure and resistancepermitting shunt closure in these pa-tients.25,26 Severe PH in patients withASD probably represents the coincidenceof idiopathic PH or PH secondary to an-other process (eg, scleroderma) andASD.1 Unlike patients with large unoper-ated nonrestrictive central shunts (eg,VSD) who experience PH from birth anddevelop pulmonary vascular diseasewithin the first few years, patients with
large ASD of similar shunt magnitude donot necessarily develop severe PH andright to left shunting or the onset of PH isdelayed into late adulthood. That beingsaid, a large ASD may contribute to thedevelopment of PH, but may not be thesole cause of the underlying pulmonaryvascular disease in a cyanotic patient. Pa-tients with trisomy 21 (Down syndrome)may develop accelerated pulmonary vas-cular disease in the presence of ASD (pri-mum or secundum).
Transcatheter device closure of secun-dum type ASD was first performed in1976 by King and Mills.27 Advancementsin biocompatible materials, device design,and catheterization technology have led tothe availability of a variety of occlusion
Figure 4: A) Transesophageal echo with color Doppler demonstrating a membranous ventricular septal defect (VSD)with left to right shunt and partial closure by the septal leaflet of the tricuspid valve (TV). B) Following placement ofAmplatzer muscular VSD (mVSD) transcatheter device occlusion. C) Transthoracic echo, apical 4-chamber viewdemonstrating the mVSD device straddling the membranous septum below the aortic valve (AoV). Right ventricle(RV), right atrium (RA).
Figure 5: A) Transthoracic echo with color Doppler, parasternal short axisview, demonstrating continuous left to right shunting from the descendingaorta (DAO) to the main pulmonary artery (MPA) via a patent ductusarteriosus (PDA). B) Aortic cineangiogram, right anterior oblique viewfollowing transcatheter device closure of the PDA, no residual shunting isseen. Right atrium (RA), right ventricle (RV), right pulmonary artery (RPA).
CM
ESection
191Advances in Pulmonary Hypertension

devices (Figures 3 and 6).28,29 Transcath-eter device closure compares favorablywith surgical closure in terms of long-term outcome and is associated withshorter hospital stays and fewer post-procedural complications.30,31 Appropri-ate patient selection is imperative andmay be accomplished via a variety ofnoninvasive and/or invasive imagingmethods.32-35 Transcatheter device clo-sure techniques have supplanted surgeryat many institutions as the method ofchoice for ASD closure in properly se-lected patients; complications are rare.Short-term complications have includeddevice embolization, aortic root or atrialwall perforation, and cardiac tampon-ade.15 Mid- and long-term complicationsinclude thrombus formation, device ero-sion into the aortic root, atrial dysrhyth-mias, and infective endocarditis. The useof platelet inhibitors for at least 6 monthsfollowing device closure is recommendedto decrease the risk of device thrombo-sis.36 The long-term outcomes of deviceclosure using the Amplatzer septal areequivalent to long-term surgical re-sults.31,37 Older patients with abnormalleft ventricular compliance or restrictivephysiology may have a significant in-crease in left heart filling pressure follow-ing ASD closure. Balloon test occlusionof the ASD with simultaneous measure-ment of pulmonary artery occlusion pres-sure or direct measurement of left ventric-ular diastolic or left atrial pressure may berevealing. Manual fenestration of com-mercially available devices allows for a
small “pop-off” for decompression (Fig-ure 6).38
Ventricular Septal DefectIsolated VSD is the most commonly en-countered form of congenital heart dis-ease in the pediatric population; most aresmall and close spontaneously. The spec-trum of isolated residual VSD encoun-tered in the adult patient usually consistsof:
1) Small restrictive defects or defects thathave closed partially with time. Thepulmonary vascular resistance is notsignificantly elevated and the left toright shunt magnitude is mild (Qp:Qs�1.5:1).
2) Large nonrestrictive defects in cya-notic patients who have developedEisenmenger syndrome, with systemicpulmonary vascular resistance andshunt reversal (right to left).
3) Patients with moderately restrictivedefects (Qp:Qs �1.6:1 and �2:1) whohave not undergone closure for somereason. These patients often have mildto moderate PH.
4) Patients who have had their defectsclosed in childhood. These patientsmay have VSD patch leaks.
Small restrictive defects of the muscularor membranous septum may be watchedconservatively without need for operativeintervention. Six percent of patients withsmall supracristal or perimembranous de-fects may develop aortic valve prolapse
and resultant aortic regurgitation that maybe progressive.39,40 The prolapsing aorticvalve cusp (usually the right coronarycusp) may partially or completely closethe VSD. Aortic valve repair or replace-ment may be necessary in patients withaortic regurgitation who develop exer-tional symptoms or progressive left ven-tricular dilation.41,42 In a long-termfollow-up registry, the overall survivalrate was 87% for all patients with unop-erated VSD at 25 years.43 For patientswith small defects (Qp:Qs �1.5 and lowpulmonary artery pressure), the survivalrate was 96%; patients with moderate andlarge defects fare worse, with 25-year sur-vival of 86% and 61%, respectively.Those with cyanosis (Eisenmenger’scomplex) had a much lower 25-year sur-vival of 41.7%.
In patients with large nonrestrictiveVSD, pulmonary vascular disease beginsat birth or soon afterwards with abnormalvascular remodeling; eventually, if the de-fect is not repaired, the pulmonary arterialresistance exceeds the systemic arterialresistance resulting in right to left shunt-ing and cyanosis, the condition known asEisenmenger syndrome.44 Early attemptsat surgical closure of central shunts inpatients with Eisenmenger syndromewere met with an unacceptably high riskof mortality and the practice was quicklyabandoned. Thereafter, the condition wasdeemed “irreversible”; however, thiscommon wisdom is now being chal-lenged. There is ample evidence thatpulmonary vasodilators result in im-proved pulmonary blood flow, improvedfunctional capacity, and may improve sur-vival in patients with Eisenmengersyndrome.45-48 Although isolated casesand small series of successful defect clo-sure in Eisenmenger syndrome have beenpublished, the majority of cases aredeemed too high risk and closure is con-traindicated.2,49 Larger defects may be re-paired in the absence of severe PH andseverely elevated pulmonary vascular re-sistance, which incurs a high periopera-tive risk (Table 1).2,50,51 Postoperative lifeexpectancy is not normal but has im-proved over the past 50 years with im-proved surgical techniques and experi-ence. Postoperative conduction defects
Figure 6: A) Transesophageal echo with color Doppler demonstrating arestrictive residual shunt through a fenestrated Amplatzer septal occluder.B) 3-D color Doppler viewed from the right atrium (RA) demonstratingresidual shunting through a fenestration. Tricuspid valve (TV), left atrium(LA), aortic value (AoV).
CM
ESe
ctio
n
192 Advances in Pulmonary Hypertension

are common but complete heart block israre in the current era. Transcatheter de-vice occlusion of muscular and perimem-branous VSD is feasible and trials dem-onstrate a good safety and efficacy profile(Figure 4).52-55 Complete heart block hasbeen noted to occur in up to 6% ofchildren and 1% of adults.52,55 Hybridtechniques, those involving surgical andtranscatheter components, are being in-creasingly applied and may obviate theneed for cardiopulmonary bypass. Theyare especially attractive for defects thatmay prove challenging to close via trans-vascular or surgical approaches alone orin infants with concerns over vascular ac-cess.56,57 Patients with small restrictivedefects (Qp:Qs �1.5:1 and low pulmo-nary artery pressure) are generally asymp-tomatic and do not require interventionunless they have aortic regurgitation orinfective endocarditis.2
PATENT DUCTUS ARTERIOSUSThe ductus arteriosus is an essential com-munication during fetal life that (alongwith the foramen ovale) allows oxygen-ated maternal blood to be directed to thesystemic circulation, thus avoiding thehigh resistance, kinked and collapsed, fe-tal pulmonary arterial circulation. Within48 hours of birth, and under the influenceof higher oxygen levels in the newborn ascompared to the fetus in utero, the ductusarteriosus begins to close. In a small sub-set of human beings, occurring eitherspontaneously or more rarely as part of afamily cluster, the ductus arteriosus re-mains open and is appropriately named aPDA. Patent ductus arteriosus is associ-ated with other congenital malformationssuch as VSD or coarctation of the aorta.Those born at high altitude, presumablydue to the lower oxygen tension, have ahigher prevalence of PDA. The conse-quences of a PDA are largely dependenton the size of the duct and the magnitudeof the shunt; very small PDA with negli-gible shunts are rarely problematic and donot result in PH or heart failure, but arerarely associated with endarteritis. Largedefects with a Qp:Qs of �1.5:1 often re-sult in left sided volume overload andprogressive increases in pulmonary arte-rial pressure and resistance. If unrepaired
surgically or via transcatheter techniques,these defects often result in Eisenmengersyndrome with suprasystemic pulmonaryarterial resistance and shunt reversal.Given that the location of the PDA isusually beyond the takeoff of the left sub-clavian artery, the deoxygenated pulmo-nary arterial blood shunts to the lowerbody resulting in differential cyanosis(Figure 7). Indications for surgical ortranscatheter closure are similar to thosefor ASD and VSD (Table 1). Patients withsevere PH without reactivity to pulmo-nary vasodilators or improvement withtransient balloon occlusion are generallynot considered candidates for PDA clo-sure; however, there is a growing body ofevidence that responsiveness to treatmentwith pulmonary vasodilators may facili-tate subsequent defect closure using a va-riety of commercially available Nitinoldevices.58-60
CONCLUSIONPulmonary hypertension is often presentin patients with native or operated con-genital heart disease. The care of thesepatients is often challenging given the de-
gree of heterogeneity of native defects,including variations in defect location,size, shunt magnitude, shunt direction, co-existent conditions, the presence of mul-tiple defects, and a wide spectrum of po-tential anatomic variations. The clinicianseeking to provide care to this populationshould be familiar with the various surgi-cal and transcatheter interventions that arecurrently utilized, their outcomes, poten-tial complications, and expected sequelae.The indications and contraindications tosurgical or transcatheter interventions inpatients with PH and congenital heart dis-ease are outlined. The outcomes of surgi-cal and transcatheter procedures in appro-priately selected patients are usuallyexcellent.
References1. Wood P. The Eisenmenger syndrome or pulmo-nary hypertension with reversed central shunt. BrMed J. 1958;2(5099):755-762.2. Warnes CA, Williams RG, Bashore TM, et al.ACC/AHA 2008 Guidelines for the Management ofAdults with Congenital Heart Disease: a report ofthe American College of Cardiology/AmericanHeart Association Task Force on Practice Guidelines(writing committee to develop guidelines on themanagement of adults with congenital heart dis-ease). Circulation. 2008;118(23):e714-e833.3. Friedberg MK, Rosenthal DN. New develop-ments in echocardiographic methods to assess rightventricular function in congenital heart disease. CurrOpin Cardiol. 2005;20(2):84-88.4. Lytrivi ID, Lai WW, Ko HH, Nielsen JC, Par-ness IA, Srivastava S. Color Doppler tissue imagingfor evaluation of right ventricular systolic functionin patients with congenital heart disease. J Am SocEchocardiogr. 2005;18(10):1099-1104.5. Aboulhosn J, Dinh H, Finn J. Computed tomog-raphy and magnetic resonance imaging in adultswith congenital heart diasease. In: Perloff JK, ChildJ, Aboulhosn J, eds. Congenital Heart Disease inAdults. 3rd ed. Philadelphia, PA: Saunders; 2008.6. Blalock A, Taussig HB. Landmark article May19, 1945: The surgical treatment of malformations ofthe heart in which there is pulmonary stenosis orpulmonary atresia. By Alfred Blalock and Helen B.Taussig. JAMA 1984;251(16):2123-2138.7. Dore A, Glancy DL, Stone S, Menashe VD,Somerville J. Cardiac surgery for grown-up congen-ital heart patients: survey of 307 consecutive oper-ations from 1991 to 1994. Am J Cardiol. 1997;80(7):906-913.8. Stellin G, Vida VL, Padalino MA, Rizzoli G;European Congenital Heart Surgeons Association.Surgical outcome for congenital heart malformationsin the adult age: a multicentric European study.Semin Thorac Cardiovasc Surg Pediatr Card SurgAnnu. 2004;7:95-101.9. Karamlou T, Hirsch J, Welke K, et al. A United
Figure 7: Differential cyanosis in apatient with patent ductusarteriosus, severe PH, and right toleft shunt (Eisenmenger syndrome).Note the severe clubbing andcyanosis of the toes as compared tothe lesser degree of clubbing andcyanosis of the fingers.
CM
ESection
193Advances in Pulmonary Hypertension

Network for Organ Sharing analysis of heart trans-plantation in adults with congenital heart disease:outcomes and factors associated with mortality andretransplantation. J Thorac Cardiovasc Surg. 2010;140(1):161-168.10. Choong CK, Sweet SC, Guthrie TJ, et al. Re-pair of congenital heart lesions combined with lungtransplantation for the treatment of severe pulmo-nary hypertension: a 13-year experience. J ThoracCardiovasc Surg. 2005;129(3):661-669.11. Everitt MD, Donaldson AE, Stehilk J, et al.Would access to device therapies improve transplantoutcomes for adults with congenital heart disease?Analysis of the United Network for Organ Sharing(UNOS). J Heart Lung Transplant. 2011;30(4):395-401.12. Russo P, Wheeler A, Russo J, Tobias JD. Useof a ventricular assist device as a bridge to trans-plantation in a patient with single ventricle physiol-ogy and total cavopulmonary anastomosis. PaediatrAnaesth. 2008;18(4):320-324.13. Aboulhosn J, Levi D, Moore J. Trans-catheterinterventions in adults with congenital heart disease.In: Perloff JK, Child J, Aboulhosn J, eds. CongenitalHeart Disease in Adults. 3rd ed. 2008, Philadelphia,PA: Saunders; 2008.14. Aboulhosn J, Levi DS, Child JS. Commoncongenital heart disorders in adults: percutaneoustherapeutic procedures. Curr Probl Cardiol. 2011;36(7):263-284.15. Schneider DJ, Levi DS, Serwacki MJ, MooreSD, Moore JW. Overview of interventional pediatriccardiology in 2004. Minerva Pediatr. 2004;56(1):1-28.16. Kirklin JK, Barratt-Boyes B. Cardiac Surgery.New York, NY: Wiley; 1986:463-497.17. Perloff JK. Ostium secundum atrial septaldefect–survival for 87 and 94 years. Am J Cardiol.1984;53(2):388-389.18. Campbell M. Natural history of atrial septaldefect. Br Heart J. 1970;32(6):820-826.19. Morriss JH, McNamara DG. Residuae, se-quelae, and complications of surgery for congenitalheart disease. Prog Cardiovasc Dis. 1975;18(1):1-25.20. Engelfriet P, Meijboom F, Boersma E, TijssenJ, Mulder B. Repaired and open atrial septal defectstype II in adulthood: an epidemiological study of alarge European cohort. Int J Cardiol. 2008;126(3):379-385.21. Altindag T, Roos-Hesselink JW, Cuypers JA,et al. Transcatheter device closure of atrial septaldefects in patients aged 40 years and older. NethHeart J. 2010;18(11):537-542.22. Brochu MC, Baril JF, Dore A, Juneau M, DeGuise P, Mercier LA. Improvement in exercise ca-pacity in asymptomatic and mildly symptomaticadults after atrial septal defect percutaneous closure.Circulation. 2002;106(14):1821-1826.23. Attie F, Rosas M, Granados N, Zabal C,Buendıa, Calderon J. Surgical treatment for secun-dum atrial septal defects in patients �40 years old.A randomized clinical trial. J Am Coll Cardiol.2001;38(7):2035-2042.24. Konstantinides S, Geibel A, Olschewski M, et
al. A comparison of surgical and medical therapy foratrial septal defect in adults. N Engl J Med. 1995;333(8):469-473.25. Schwerzmann M, Zafar M, McLaughlin PR,Chamberlain DW, Webb G, Granton J. Atrial septaldefect closure in a patient with “irreversible” pul-monary hypertensive arteriopathy. Int J Cardiol.2006;110(1):104-107.26. Kim YH, Yu JJ, Yun TJ, et al. Repair of atrialseptal defect with Eisenmenger syndrome after long-term sildenafil therapy. Ann Thorac Surg. 2010;89(5):1629-1630.27. Mills NL, King TD. Nonoperative closure ofleft-to-right shunts. J Thorac Cardiovasc Surg.1976;72(3):371-378.28. Banerjee A, Bengur AR, Li JS, et al. Echocar-diographic characteristics of successful deploymentof the Das AngelWings atrial septal defect closuredevice: initial multicenter experience in the UnitedStates. Am J Cardiol. 1999;83(8):1236-1241.29. Walsh KP, Tofeig M, Kitchiner DJ, Peart I,Arnold R. Comparison of the Sideris and Amplatzerseptal occlusion devices. Am J Cardiol. 1999;83(6):933-936.30. Du ZD, Koenig P, Cao QL, Waight D,Heitschmidt M, Hijazi ZM. Comparison of trans-catheter closure of secundum atrial septal defectusing the Amplatzer septal occluder associated withdeficient versus sufficient rims. Am J Cardiol. 2002;90(8):865-869.31. Kutty S, Hazeem AA, Brown K, et al. Long-term (5- to 20-year) outcomes after transcatheter orsurgical treatment of hemodynamically significantisolated secundum atrial septal defect. Am J Cardiol.2012;109(9):1348-1352.32. Abdel-Massih T, Dulac Y, Taktak A, et al.Assessment of atrial septal defect size with 3D-transesophageal echocardiography: comparison withballoon method. Echocardiography. 2005;22(2):121-127.33. Mazic U, Gavora P, Masura J. The role oftransesophageal echocardiography in transcatheterclosure of secundum atrial septal defects by theAmplatzer septal occluder. Am Heart J. 2001;142(3):482-488.34. AboulHosn J, French WJ, Buljubasic N, Mat-thews RV, Budodd MJ, Shavelle DM. Electron beamangiography for the evaluation of percutaneous atrialseptal defect closure. Catheter Cardiovasc Interv.2005;65(4):565-568.35. Budoff MJ. Images in cardiology: Electronbeam angiography of percutaneous atrial septal de-fect closure. Clin Cardiol. 2004;27(12):702.36. Franke A, Kuhl HP. The role of antiplateletagents in the management of patients receiving in-tracardiac closure devices. Curr Pharm Des. 2006;12(10):1287-1291.37. Masura J, Gavora P, Podnar T. Long-term out-come of transcatheter secundum-type atrial septaldefect closure using Amplatzer septal occluders.J Am Coll Cardiol. 2005;45(4):505-507.38. Kretschmar O, Sglimbea A, Corti R, KnirschW. Shunt reduction with a fenestrated Amplatzerdevice. Catheter Cardiovasc Interv. 2010;76(4):564-571.
39. Corone P, Doyon F, Gaudeau S, et al. Naturalhistory of ventricular septal defect. A study involv-ing 790 cases. Circulation. 1977;55(6):908-915.40. Yoshimura N, Hori Y, Horil Y, et al. Compar-ison of magnetic resonance imaging with transtho-racic echocardiography in the diagnosis of ventric-ular septal defect-associated coronary cusp prolapse.J Magn Reson Imaging. 2010;32(5):1099-1103.41. Bonow RO, Carabello BA, Kanu C, et al.ACC/AHA 2006 guidelines for the management ofpatients with valvular heart disease: a report of theAmerican College of Cardiology/American HeartAssociation Task Force on Practice Guidelines(writing committee to revise the 1998 Guidelines forthe Management of Patients With Valvular HeartDisease): developed in collaboration with the Soci-ety of Cardiovascular Anesthesiologists: endorsedby the Society for Cardiovascular Angiography andInterventions and the Society of Thoracic Surgeons.Circulation. 2006;114(5):e84–e231.42. Tatsuno K, Konno S, Sakakibara S. Ventricu-lar septal defect with aortic insufficiency. Angiocar-diographic aspects and a new classification. AmHeart J. 1973;85(1):13-21.43. Kidd L, Driscoll DJ, Gersony WM, et al. Sec-ond natural history study of congenital heart defects.Results of treatment of patients with ventricular sep-tal defects. Circulation. 1993;87(2 Suppl):I38–I51.44. Hall SM, Haworth SG. Onset and evolution ofpulmonary vascular disease in young children: ab-normal postnatal remodelling studied in lung biop-sies. J Pathol. 1992;166(2):183-193.45. Yang-Ting S, Aboulhosn J, Sun XG, Child JS,Sietsema KE. Effects of pulmonary vasodilator ther-apy on ventilatory efficiency during exercise inadults with Eisenmenger syndrome. Congenit HeartDis. 2011;6(2):139-146.46. Post MC, Janssens S, Van de Werf F, Budts W.Responsiveness to inhaled nitric oxide is a predictorfor mid-term survival in adult patients with congen-ital heart defects and pulmonary arterial hyperten-sion. Eur Heart J. 2004;25(18):1651-1656.47. Galie N, Beghetti M, Gatzoulis MA, et al;Bosentan Randomized Trial of Endothelin Antago-nist Therapy-5 (BREATHE-5) Investigators. Bosen-tan therapy in patients with Eisenmenger syndrome:a multicenter, double-blind, randomized, placebo-controlled study. Circulation. 2006;114(1):48-54.48. Dimopoulos K, Inuzuka R, Goletto S, et al. Im-proved survival among patients with Eisenmenger syn-drome receiving advanced therapy for pulmonary ar-terial hypertension. Circulation. 2010;121(1):20-25.49. Batista RJ, Santos JL, Takeshita N, et al. Suc-cessful reversal of pulmonary hypertension in Eisen-menger complex. Arq Bras Cardiol. 1997;68(4):279-280.50. Cartmill TB, DuShane JW, McGoon DC, Kirk-lin JW. Results of repair of ventricular septal defect.J Thorac Cardiovasc Surg. 1966;52(4):486-501.51. Mattila S, Kostiainen S, Kyllonen KE, Tala P.Repair of ventricular septal defect in adults. ScandJ Thorac Cardiovasc Surg. 1985;19(1):29-31.52. Butera G, Carminati M, Chessa M, et al. Trans-catheter closure of perimembranous ventricular sep-tal defects: early and long-term results. J Am Coll
CM
ESe
ctio
n
194 Advances in Pulmonary Hypertension

Cardiol. 2007;50(12):1189-1195.53. El Said HG, Bratincsak A, Gordon BM, MooreJW. Closure of perimembranous ventricular septaldefects with aneurysmal tissue using the AmplazterDuct Occluder I: lessons learned and medium termfollow up. Catheter Cardiovasc Interv. 2012;80(6):895-903.54. Fu YC, Bass J, Amin Z, et al. Transcatheterclosure of perimembranous ventricular septal defectsusing the new Amplatzer membranous VSD oc-cluder: results of the U.S. phase I trial. J Am CollCardiol. 2006;47(2):319-325.55. Masura J, Gao W, Gavora P, et al. Percutane-ous closure of perimembranous ventricular septal
defects with the eccentric Amplatzer device: multi-center follow-up study. Pediatr Cardiol. 2005;26(3):216-219.56. Chen Q, Cao H, Zhang GC, Chen LW, Li QZ,Qiu ZH. Closure of Perimembranous VentricularSeptal Defects with Intraoperative Device Tech-nique: Another Safe Alternative to Surgical Repair.Thorac Cardiovasc Surg. 2012 Jun 8. [Epub aheadof print]57. Aboulhosn J, Levi D, Sopher M, Johnson A,Child JS, Laks H. Perventricular closure of a largeventricular septal defect in congenitally correctedtransposition of the great arteries. Congenit HeartDis. 2010;5(1):60-65.
58. Hokanson JS, Gimelli G, BassJL. Percutane-ous closure of a large PDA in a 35-year-old manwith elevated pulmonary vascular resistance. Con-genit Heart Dis. 2008;3(2):149-154.59. Pac A, Polat TB, Vural K, Pac M. Successfultwo-stage correction of ventricular septal defect andpatent ductus arteriosus in a patient with fixed pul-monary hypertension. Pediatr Cardiol. 2010;31(1):111-113.60. Bhalgat PS, Pinto R, DalviBV. Transcatheterclosure of large patent ductus arteriosus with severepulmonary arterial hypertension: Short and interme-diate term results. Ann Pediatr Cardiol. 2012;5(2):135-140.
CM
ESection
195Advances in Pulmonary Hypertension

Self-Assessment Examination See answer key on next page
1. Aggressive monitoring for the development of PAHshould occur in Fontan patients because:a. They often have a mean pulmonary pressure
�25 mm Hg at right heart catherizationb. An Eisenmenger physiology is associated in most casesc. A conduit obstruction may lead to cardiac
decompensationd. Even slight increase in PVR may have significant
hemodynamic consequences2. Which of the following forms of congenital heart
disease is most likely to lead to the development ofPAH?a. Partial anomalous pulmonary venous return without
repairb. Secundum atrial septal defect without repairc. An unrestricted ventricular septal defect without prior
repaird. Patent foramen ovale without repair
3. A 30-year-old woman is diagnosed with amembranous VSD. She undergoes echocardiographicimaging evaluation and an invasive hemodynamicstudy. The hemodynamic study demonstratespulmonary artery pressures of 110/50 mm Hg. Hercentral aortic pressure is 100/60 mm Hg. Apulmonary venous oximetry sample has a saturationof 95% and her femoral artery oximetry sample hasa saturation of 86%. There is severe pulmonaryregurgitation, as well. Which of the following iscorrect regarding treatment?a. Closure of the VSD is indicatedb. PAH vasodilator therapy only is indicatedc. Heart transplantation should be consideredd. She should have pulmonary valve replacement
4. Which of the following is the most common subtypeof ASD?a. Muscularb. Outletc. Secundumd. Sinus venosuse. Membranous
5. Echocardiographic indicators of poor outcomes inpatients with Eisenmenger syndrome are:a. Pericardial effusion � low TAPSE
b. Low TAPSE � shortened RV filling time � increased RAarea/LA area
c. Pericardial effusion � low TAPSE � shortened RV fillingtime
d. Bi-atrial enlargement6. In Eisenmenger patients, the presence of RV late
gadolinium enhancement at cardiac MRI:a. Is a pathologic finding in any casesb. Is usually located at the apexc. Is a normal feature typically evident at the insertion
pointsd. When present at the insertion points is an indicator of
poor outcome7. Catheter-based interventions are available for all of
the following lesions except:a. Secundum atrial septal defectb. Muscular ventricular septal defectc. Ostium primum atrial septal defectd. Patent ductus arteriosus
8. In a 4-month-old infant with APAH-CHD due to anonrestrictive VSD, what would you most expecthemodynamics to resemble on cardiaccatheterization?a. Elevated pulmonary artery pressure, normal wedge
pressure, elevated pulmonary blood flow, normalpulmonary vascular resistance
b. Elevated pulmonary artery pressure, elevated wedgepressure, normal pulmonary blood flow, normalpulmonary vascular resistance
c. Normal pulmonary artery pressure, normal wedgepressure, normal pulmonary blood flow, normalpulmonary vascular resistance
d. Elevated pulmonary artery pressure, normal wedgepressure, normal pulmonary blood flow, elevatedpulmonary vascular resistance
9. The BREATHE-5 trial involving which endothelinreceptor antagonist was the first randomized,double-blind, placebo-controlled drug trailconducted solely on Eisenmenger patients?a. Sildenafilb. Bosentanc. Ambrisentand. Treprostinil
Disclosures(continued from page 165)sented. To be disclosed to participants are all personal financialrelationships with a com-mercial interest whose products arerelevant to the content of this CME activity. It is the policy ofWashington University School of Medicine, Continuing MedicalEducation, to ensure balance, independence, objectivity, andscientific rigor in all its educational activities. All faculty par-ticipating in this activity are expected to disclose to the audienceany financial interest or other potential conflict. Each author wasasked to complete a disclosure information form for this activity.Disclosures are reported below:
Dr Aboulhosn has no financial relationships to disclose. Dr Babu-Narayan has no financial relationships to disclose. Dr Bashore has nofinancial relationships to disclose. Dr Gatzoulis has no financial rela-tionships to disclose. Dr Kiefer has no financial relationships to dis-close. Dr Li has no financial relationships to disclose. Dr Rubens has nofinancial relationships to disclose. Dr Scognamiglio has no financial
relationships to disclose. Dr Zuckerman has no financial rela-tionships to disclose.
Dr Rosenzweig has served as a consultant/advisory board/steeringcommittee member for United Therapeutics, Actelion, and Gilead; andreceived institutional grant/research support from Actelion, Gilead,United Therapeutics, GSK, Bayer, and Novartis.
Dr Dodson has no financial relationship to disclose. Dr Chakinalareceives research support or serves as a consultant to Actelion, Gilead,United Therapeutics, Lung LLC, Novartis, and Ikaria.
CME Reviewers
W. Edwin Dodson, MD, Associate Vice Chancellor and Associate Deanfor Admissions and Continuing Medical Education, Professor of Pedi-atrics and Neurology, Washington University School of Medicine.
Murali Chakinala, MD, Associate Professor, Division of Pulmonary andCritical Care Medicine, Washington University School of Medicine.
CM
ESe
ctio
n
196 Advances in Pulmonary Hypertension

Congenital Heart Disease and Pulmonary Hypertension
Congenital Heart Disease and PulmonaryHypertensionIndividuals wishing CME credit for this self-study activity shouldread the text, answer the self-assessment examination questions,complete the form below,* and send by US mail to the followingaddress by April 30, 2014. You should receive a score of 70% orhigher for CME credit. Your test will be scored and your partici-pation will be entered into the CME recorded at Washington Uni-versity School of Medicine.
Department Continuing Medical EducationWashington University School of MedicineCampus Box 8063660 South Euclid Ave.St. Louis, MO 63110
Your certificate will be mailed within 3 weeks of request.
Self-Assessment Answer KeyCircle one correct answer.
1. a b c d
2. a b c d
3. a b c d
4. a b c d e
5. a b c d
6. a b c d
7. a b c d
8. a b c d
9. a b c d
Name (Please print)
Degree(s)
Specialty
Street
City
State Zip
E-mail Address
Date Test Completed
Check number of CME credits requestede 1.0 e 1.5 e 2.0
Signature
Evaluation of CME Activity
Poor Satisfactory Excellent
1. Extent to which objectives were met
1 2 3 4 5
2. How would you rate the course overall
1 2 3 4 5
3. How helpful was the information presented
1 2 3 4 5
4. Was the activity free of commercial bias? Yes No
5. After participating in this activity, do you antici-pate changing any of your patient care practices?
Yes No
If yes, what do you plan to change?
6. Do you foresee any barriers to change? Yes No
If yes, what?
Additional comments about this self-study activity:
Suggestions for future topics:
* Self-assessment examination may also be completedonline at: https://cme-online.wustl.edu/pha
CM
ESection
197Advances in Pulmonary Hypertension

Pulmonary Hypertension Roundtable
Management of PAH in Adults withCongenital Heart Disease:Impact and DilemmasGuest editor Richard Krasuski, MD, convened a group of experts by telephone on January 17, 2013, todiscuss current trends in diagnosis and treatment of pulmonary hypertension among patients withcongenital heart disease. Joining the call were Professor Maurice Beghetti, Head of Pediatric Subspe-cialties, Division Head of Pediatric Cardiology Unit, Children’s University Hospital, Geneva, Switzerland;Curt Daniels, MD, Director, Adolescent and Adult Congenital Heart Disease Program Nationwide Chil-dren’s Hospital, The Ohio State University, Columbus, Ohio; Wayne J. Franklin, MD, Texas Children’sHospital, Houston; and Michael J. Landzberg, MD, Associate Director, Adult Pulmonary HypertensionProgram and Director, Boston Adult Congenital Heart Program, Boston Children’s Hospital.
Dr Krasuski: We are here today to discuss the impactand dilemmas in the management of pulmonary arterialhypertension (PAH) in adults with congenital heartdisease (ACHD). There are now up to a million ACHDpatients, and we believe that up to 40% of these patientsare at risk for PAH; 10% of ACHD patients will actu-ally develop PAH. So we’re talking about 100,000 suchpatients existing in the United States alone, and possi-bly up to 40,000 that have developed Eisenmengersyndrome. So it’s a large and growing group and anexciting and challenging field to practice in. It’s veryimpressive in terms of what’s happened in the lastdecade or 2, regarding the development of multifacetedmanagement strategies for these people. I am going tobegin by asking the following question of this presti-gious panel: “how can we best identify those patientswith congenital heart disease who have pulmonary hy-pertension and who might be candidates for some of thetherapeutic interventions that we now have available?”
Dr Daniels: We know a group of patients from thelarge congenital heart disease population, who arehigher-risk patients. Those are patients who have shuntlesions and some of our more complex lesions. So theseare the patients we must be aware of as having thepotential to either have pulmonary hypertension or de-velop pulmonary hypertension. Then we also have to beaware, as an educational point to ourselves and ourcommunity, but also other cardiologists and even pos-sibly pulmonologists who see patients, to rule out pul-monary hypertension or evaluate for pulmonary hyper-tension because patients may be completely repairedand still develop pulmonary hypertension. We know apercentage of patients, even if they have shunts closed,even at what we consider an earlier age, still maydevelop pulmonary hypertension down the road as anadult. There are risk factors that lead to those with shuntlesions and complex congenital heart disease thatmakes them more vulnerable to develop pulmonaryhypertension, such as timing of when a septal defectwas closed, surgical shunts that may have been placed,
length of time with a shunt before they had a completerepair. So we have to be aware that the congenital heartdisease population as a whole is at risk, but there arecertain patients, certain populations within the congen-ital heart disease population, that are at higher risk fordeveloping pulmonary hypertension, and be aware andbe able to evaluate those patients, looking specificallyfor signs, symptoms of pulmonary hypertension.
Dr Landzberg: What Curt identified were findingsthat many of us, as congenital heart disease docs, wouldrecognize, but I wonder if we can extend that a little bit.For the general practitioner or for the internist who isout there, if one is fortunate enough to have the preop-erative history on these folks, we could say that, almosteverybody’s at risk but, in particular, folks who mayhave had preprocedural large-volume shunting. Maybeif you had ventricular dysfunction going into the shuntrepair. If you had a lot of volume coming back to yourleft side of your heart, as well, that’s a sign that some-one may be at increased risk for the development ofpulmonary hypertension after closure. I think many ofus will see arrhythmia as a preoperative arrhythmia orpostoperative arrhythmia as a sign that the patient maybe having increased risk for developing pulmonary hy-pertension. Persistent RV dysfunction, functional de-cline, just in general, if someone’s not doing well withcongenital heart disease afterwards, that puts thethought into my head, could this person have pulmo-nary hypertension, and just age alone. So I think thatanyone that’s not doing well, anyone that’s gettingolder, anyone that has heart muscle dysfunction, I’mthinking has the potential for pulmonary hypertension.
Dr Beghetti: I think you both raise a very importantpoint. So we may ask adult physicians to see thesepatients and refer to “adult congenital heart disease.” Ithink one important point that you both raised is that weneed to know what happened at an early age, especiallyin the ones that had surgery, and then come back to theadult clinic with PH later in life. And I think the
198 Advances in Pulmonary Hypertension

transition and the connection between the pediatriccardiology and the adult cardiology with regard tothese patients is very important, to be sure that wehave the data on what happened and how the decisionwas done to do the surgery, to close the shunt, andexactly what Mike just raised now: if there werespecific problems that can be identified and indeed,the risk factors for this population, and then identifythe risk factors that will allow you to identify thepatients that are at high risk to develop postoperativepulmonary hypertension. Because I think in our data-base now, what we see is that the Eisenmenger pop-ulation is an old population. We should see fewer ofthese patients. But the growing population is patientswith PH after repair. And so we need to identify thesepatients and the risk factors for these patients.
Dr Krasuski: I completely agree. Between a ran-domized trial and several prospective registries, we’veaccumulated quite a bit of data about Eisenmengerpatients. But as time goes on, hopefully we can iden-tify and intervene in these patients early enough toprevent them from developing Eisenmenger physiol-ogy. We have developed trials focused on Eisen-menger syndrome, including the publishedBREATHE-5 study and newly enrolling MAESTROtrial. These are randomized, placebo-controlled trialsexamining the role of pharmacologic therapy. But forpatients with earlier forms of PAH, how can we applythese data to them? Should we be more aggressive atearlier stages of the disease? And how can we applywhat’s been learned in other etiologies of pulmonaryhypertension to our ACHD patients?
Dr Beghetti: Eisenmenger patients and the oneswho present with PH after complete repair may be abit different. For the Eisenmenger patients, as yousaid, the MAESTRO will include patients with, Iwould say, functional class II, which would be con-sidered as mildly symptomatic, and we’ll see whathappens with this population. I think the other groupappears really quite affected with the preliminary re-sult we have. And with the group that presents withPH after repair, it seems, even if we do not have stillall the data, that we need to be a bit more aggressivewith this population, compared to Eisenmenger pa-tients. Even if we think we still need to have more dataon Eisenmenger patients to see also the benefit fromearly aggressive therapy.
Dr Daniels: I agree. I think that finding a patientwith complete repair of a shunt, for instance, who hasdeveloped pulmonary hypertension, has the patho-physiology of advancing pulmonary hypertension.And so far, there is no evidence to the contrary, to
believe that this particular patient population is notgoing to follow a pathway with a closed, repairedshunt, almost similar to an idiopathic PH patient. Ofcourse, we don’t know this and we know the Eisen-menger population has a very different course in termsof their prognosis. But the patient with a closed,repaired shunt, and found later to have pulmonaryvascular disease, we have to believe this is an advanc-ing disease process. We certainly see that as we followpatients now, we’re collecting more information.Therefore, we have to believe that early therapy isquite important and not waiting until they are moresymptomatic, which we all know means that the rightventricle is becoming more dysfunctional from a sys-tolic and diastolic and a compliance standpoint. Soearly treatment certainly seems to be the best courseof action for these patients.
Dr Franklin: I think that the Eisenmenger data forus, specifically in Houston, have been very helpful thepast few years. Because in the past, where I think wewould just start them on maybe one medicine and thatwas all. Often these are Down syndrome patients. Ithink now we’ve been more aggressive to try to getthem on advanced therapies, whether it’s 2 drugs or 3drugs. Usually it’s 2; often they do not tolerate 3drugs. But I also agree with Curt closely that the onesthat we think are repaired, we’re still following everyyear. And I think maybe we should think about start-ing them earlier on therapy. So I think there has beenthe real emphasis on early detection now, as well.
Dr Krasuski: Let me shift gears a little bit and askthe group to briefly discuss what type of workup theydo in the newly diagnosed ACHD-PH patient. So youhave a patient who has a congenital heart lesion anddevelops pulmonary hypertension, though not yetEisenmenger syndrome. What types of studies shouldwe perform to look for other sources of pulmonaryhypertension? Should we be doing a full pulmonaryworkup for these patients, such as VQ scanning andpulmonary function testing? Bloodwork assessing forcollagen vascular disease? Sleep-disordered breathingworkup? What is your standard practice in these pa-tients? Particularly in this era of cost containment, doyou run the whole gamut and follow the same algo-rithm as for any newly diagnosed PH patient? Or doyou focus on what you think the most likely etiologyis?
Dr Landzberg: Before we address this, let me shiftback a little bit. There were a couple of things thatfolks mentioned that gave me a bit of a twitch, onlybecause it underscored that we’re missing some dataor there are some additional data out there that may be
“There are risk
factors that lead
to those with
shunt lesions and
complex
congenital heart
disease that
makes them more
vulnerable to
develop pulmonary
hypertension.”
Dr Daniels
199Advances in Pulmonary Hypertension

helpful in the Down syndrome population. And folkslike Michelle Dalto and others have underscored thatit may be as high as 25-plus percent of the congenitalheart disease population with pulmonary hypertensionthat has Down syndrome. And we have scant data inthis country with regard to the triggers in that popu-lation that make us particularly concerned. Our dataon therapies are limited to cohort studies, so that thereis a whole batch of workup that needs to be done inthis population. I know that we all worry about howlittle data we have in just congenital heart disease aftera repair. The REVEAL dataset makes me worry. It’sclearly in a patient population that came to advancedheart—pulmonary hypertension clinics rather thanadult congenital heart disease clinics, but it was sug-gested a terrible prognosis in that small patient group.But there are 2 ongoing registries in this country thatare going to hopefully define that for us. And as partof those registries, we are mandating exactly what youmentioned, Rich, that we go through the full evalua-tion. Our patients with congenital heart disease havemany other triggers for pulmonary hypertension. SoI’m a big fan of a full and complete evaluation, despitethe fact that somebody had a shunt to begin with anddespite the fact that somebody has congenital heartdisease—there are way too many times that we findother contributors that have their own independenttherapies.
Dr Beghetti: I definitely agree with that, becauseyou can have a congenital heart defect and also haveother triggers or other risk factors. And that’s ex-tremely, extremely important to know. On top of this,it’s extremely important also to re-cath the patients,for the reasons that Mike raised before, to be sure thatthere is not a combination of pre- and postcapillarypulmonary hypertension, and also to see if there areany clots in the lung. Because they had surgery, some-times they have catheters in place for a long time aftersurgery. So I think definitely we need to look ateverything before starting these therapies, becauseotherwise you will blame the therapy for not working,but maybe that’s because the indication was not ex-actly the one you thought. And the complete workupshould help make the diagnosis correctly.
Dr Daniels: I would completely agree. And I thinkto bring it back to a specific patient population that weall see is the atrial septal defect (ASD) patient. Apatient with an ASD that’s been closed or even re-mains open and has pulmonary hypertension, this is apopulation we all see. We’re not sure many times isthe ASD truly causing the pulmonary vascular diseaseor an innocent bystander, or possible a contributor.And so I think this highlights, at least for me in the
evaluation and workup, we do need to perform acomplete workup, even on our congenital heart dis-ease patients, not knowing if this is cause and effect oran innocent bystander. We don’t want to miss a di-agnosis, as Maurice says, go down the wrong pathwayin terms of our therapies when we should have beenlooking in a different direction.
Dr Krasuski: Those are all excellent points. Now,Maurice, you alluded to the importance of heart cath-eterization and potentially repeat heart catheterizationwhile on therapy to assess therapeutic response. Whatabout performing hemodynamic challenges in the cathlab? How often do you do vasodilator challenges, fluidchallenges, and other such studies to assess the phys-iologic response? Do you reserve such procedures tothe first catheterization or is it worth reassessing?
Dr Beghetti: I tend to do complete caths all thetime, including vasoreactivity testing. It’s not becauseI think that I will find the patients becoming reactive,because this is extremely, extremely uncommon. Butbased on some data coming from Belgium, from thegroup of (s/l Vander Butz), and also from (s/lMikhaila Douto) in Italy, this could be a good way toidentify some risk factors for this population. Whenyou still have some reactivity, it seems based on these2 studies that the patient may have a better outcome.They also may have a better response to some of thetherapies, because there is some vasodilatory reserve.So that may be the reason to assess vasodilatoryreserve. I think the fluid challenge, especially if thepatient is older or if there is some history of ventric-ular dysfunction, can unmask diastolic dysfunction,and I think that’s very important to know in ourpopulation. I think maybe we mismanage these kindsof things in some of our patients. In terms offollow-up caths, I think it’s important to do follow-upcaths in the population where there is closed shunt inPH. That will be exactly the same follow-up that youdo in idiopathic PH. In Eisenmenger syndrome, andI’m sure that Mike will have strong ideas on that, theproblem is to reproduce the data properly and reallybe sure that you can compare data from cath to cath.And sometimes it’s not easy because these caths withopen shunts are sometimes a bit difficult, as you allknow. So I think you need also to adapt a little bit tothe population that you follow.
Dr Landzberg: All of us in this group perform cath-eterizations. And have different opinions about howoften to cath, but I think we all probably share theopinion that cath plays a vital role. The number oftimes that we find something unexpected at a cath insomebody who has congenital heart disease and pul-
200 Advances in Pulmonary Hypertension

monary hypertension is far too often. And I would saythat, as we’ve underscored before in terms of themany triggers to pulmonary hypertension develop-ment, our patients are prone to pericardial construc-tion and have reasons to have pulmonary venousstenosis, restrictive myocardial disease, other unex-plained or unexpected pulmonary arterial stenoses.And so I underscore that during the very first cathe-terization, it’s critical to do a full, complete angio-graphic, hemodynamic, multiple maneuver catheter-ization. I have been amazed at the more than rarepatient that is responsive to pulmonary vasodilatoracute therapy, and I know that we all believe slightlydifferently in terms of whether or not somebody canrespond to a calcium antagonist in our population orshould respond. In the same breath, I agree that it’svitally important to know whether or not somebody isresponsive. It tells you something about their progno-sis as well. My toughest point is what about serialcatheterizations? And this applies to the patient withidiopathic disease, as well as to our own patients.There is so much hour-to-hour variation of the hemo-dynamics of our patients, in a normal host, or a patientwith pulmonary hypertension that small differences,even small to moderate differences, don’t necessarilytell us a lot, but there are still key prognosticators thatwe get from hemodynamics. I often repeat caths, butI have no idea how often it should be. Certainly, whenthere is a functional decline, that’s a marker for us togo back and reassess hemodynamics.
Dr Daniels: I completely agree. The first cath iscritical. And this is where the data are so importantthat it’s accurate and done with detail and in anorganized fashion. You know, for the audience thatwill be reviewing this roundtable discussion, manymay not be congenital heart disease experts in per-forming cardiac catheterizations on patients withshunts. And I would emphasize the point of collabo-rating with congenital heart disease experts with car-diac catheterization data, because collecting data in acorrect fashion will make the difference betweenwhich pathway you will go with that particular pa-tient, whether it’s pulmonary hypertension-specifictherapy, whether it’s deciding to close the shunt,whether it’s deciding therapy should be headed to-ward heart failure, diastolic dysfunction. And so it’scritically important that the correct information isobtained, under the right conditions. The oxygen sat-uration data: is the patient on supplemental oxygen?That the vasodilator trial is done correctly. Becausethis is the one shot in the catheterization lab to obtaincorrect information. So I would emphasize, even ifyou are in a center that performs cardiac catheteriza-tion for pulmonary hypertension, but maybe not spe-
cifically for congenital heart disease, collaborationwith congenital heart disease experts is critical toobtain the correct data.
Dr Krasuski: It’s great to hear such a strong con-sensus on the importance of hemodynamically defin-ing the disorder and properly collecting the data. Thisreally sets you on the proper path toward appropriatetherapy. My next question is: “how do you followACHD-PH patients in terms of assessing their diseaseprogression and response to treatment?” The 6-minutewalk has gotten kind of a black eye recently as asurrogate for outcomes in pulmonary hypertension.Do you guys regularly measure the 6-minute walk inyour CHD-PH patients? Do you utilize metabolicstress testing? Do you measure biomarkers? Do youregularly perform echocardiograms? We’ve alreadydiscussed catheterization and the importance of po-tentially repeating it at some point, though we maydiffer perhaps in what we believe the appropriateinterval should be. When you see your patients back inclinic, what are those essential tools that you use toassess how the patient is doing and how successfultherapy has been?
Dr Franklin: It’s interesting, Rich. The 6-minutewalk, as you mentioned, has been controversial lately.But I still use it. I still use it for enrolling patients andstarting therapy and monitoring patient responses. Thetest is easy to do. It’s a good, sustainable test, if youwill. But I also use echo; I use saturations. Some ofour patients that are pretty debilitated are not able todo even a submaximal stress test. So that’s where Ithink the 6-minute walk continues to be very useful. Itwould be interesting to see what the group consensusis about repeat catheterization. I usually will save thatuntil there’s either some unusual response, or thepatient is not responding, or I’m going to start asecond drug, or it’s been a year and the patient may bea surgical candidate, or something like that. But I tendto use more of the noninvasive measures, rather thancathing them more than once or twice.
Dr Beghetti: Yeah, that’s what I said before. I thinkwe should clearly differentiate Eisenmenger and non-Eisenmenger patients. I think repeating caths in Eisen-menger, again it’s very difficult to see a major differ-ence. And there is always some risk to redo the cathin this population; you never know what can happen.So I think I would do exactly what you say. If thepatient is not responding as you expected or you planmaybe to add another drug, that is one of the goodreasons to repeat the cath. But if an Eisenmengerpatient is doing well, I would not do repeated routinecath. This may be different in a closed shunt. Some
“In Eisenmenger
syndrome, the
problem is to
reproduce the
data and really be
sure that you can
compare data
from
catheterization
to
catheterization.”
Dr Beghetti
201Advances in Pulmonary Hypertension

centers are following exactly the same approach if apatient is doing perfectly well with all the noninvasiveassessment, they would keep the cath only for patientsthat have inadequate response to treatment. And Iwould do exactly as you said, a 6-minute walk test,saturation at rest and at end of exercise, BNP, echoand a function class assessment. I still think the func-tional class one can gain from discussing with thepatient is sometimes just as helpful. It’s simple butstill very helpful in these patients. I think the CPETfor the Eisenmenger patient is a bit of a tricky test,even if it becomes a little more used in the otherpopulations. I think in the very blue patient, it’s verydifficult to have reliable data.
Dr Daniels: I would pose to the group that one of theaspects that we are most concerned with is right ven-tricular performance. And certainly, newer data seemto suggest that maybe we should be looking at theright ventricle in a different way. Certainly, we alllook at our patients with echocardiograms on someregular basis. I find the echocardiogram for PH prob-ably to be the least helpful in following patients, justbecause most patients with advanced disease, andcertainly with Eisenmenger patients, we really do notgain much information beyond what everybody’smentioned: how they are feeling clinically, their ox-ygen saturation data, exercise data. But certainly eval-uating the right ventricular systolic function is animportant parameter that we probably should be fol-lowing more closely. And whether or not that allowsus to change our therapy, add therapy, consider othertherapies may be important for the future. In ourEisenmenger patients, we do have a difficult time inthe cath lab, I agree. And a difficult time really withobtaining accurate and consistent data. I’d be inter-ested to see what others think.
Dr Beghetti: MRI should be one of the options.
Dr Krasuski: With regard to MRI and some of theother novel, newer techniques for disease assessment,such as strain imaging on echo, is there anything thatyou all see that will change how we practice in thispatient population? Maybe there are some biomarkersthat are easy to follow? How do you utilize measure-ment of natriuretic peptides?
Dr Landzberg: Let’s focus on MR and anatomy.Our world of congenital heart disease underscores thatthe progressive decline or the ability of the patient tosucceed with pulmonary vascular disease is in partrelated to the pulmonary vascular bed and in partrelated to the supporting structures that mount theflow to the pulmonary vascular bed. And so that thestandard right ventricle in idiopathic pulmonary arte-
rial hypertension or acquired PAH is very different forour patients who often don’t have a normal ventricle,don’t have a normal atrioventricular valve, or don’thave a normal conduit system in terms of passage ofpreload to the subpulmonary ventricle. So I think thatunderstanding how that ventricle is doing, not justfrom a hemodynamic standpoint but from an imagingstandpoint, is particularly valuable in the manage-ment, but also in the primary classification of what’sgoing on. So MR for us, and Curt, I’m glad youunderscored it, is an increasingly valuable aspect ofnot just the management but also in terms of the veryclassification of our patients.
Dr Krasuski: Would someone want to comment onthe routine measurement of natriuretic peptides?
Dr Beghetti: I measure them, but I’m a bit careful.I think the data that has just been published by the (s/lBrompton) group is very interesting. But I think thatsometimes you have to be very careful not to over relyon the BNP values. We still have to learn about howthis works. In patients that have Eisenmenger, somerenal dysfunction, that are using diuretics. Sometimesdoing a BNP during the day, in the morning or in theevening, you may have surprise that the level is a bitdifferent, if it’s before or after Lasix dose, dependingof the renal function in your patient. And so I think weneed to learn a little bit more. But the data coming outfrom some studies are quite interesting. And definitelyin the MAESTRO trial, we would like to measure thatin a standardized way, to see if in a standardized wayin a large cohort of patients, this can be used toaddress if the treatment is or is not working.
Dr Landzberg: Is it reasonable to say, Maurice, thatmost of us will collect natriuretic peptide will use it aspart of the assessment, but none of us will take it inisolation? And I think that the recent data wouldunderscore that it’s an important part of the mix, butnot to be taken in isolation.
Dr Beghetti: Definitely. That’s exactly what Imeant.
Dr Daniels: And I would agree, it’s a part of thefollow-up and evaluation of our patients, a perfectway to say it but not the sole decision maker aboutadding additional therapy or changing therapy, but it’sadded to our process of evaluation.
Dr Krasuski: In our program we collect a lot of dataat each clinical visit. We look at functional status,natriuretic peptides, echoes, and 6-minute walks. Oneof the things that I like to examine is the general trend
202 Advances in Pulmonary Hypertension

in each of these. I’ve found that taking patients to thecatheterization laboratory is most helpful for clarifi-cation of the disease state when there are conflictingdata. If all the data are heading in the wrong direction,I’m fairly confident that our chosen clinical strategy isprobably not right. When there are, for instance, im-provements in the 6-minute walk and functional statebut the natriuretic peptides are increasing, then I’llstart thinking about some of the things that Mauricementioned: maybe it’s the measuring technique or thetime of day when the measurement was made. But ifI have more conflicting data, like evidence that theright heart is failing despite no worsening of thepatient reported function state, then this is the timewhere catheterization may be most helpful—to knowwhich direction the hemodynamics (pressures andpulmonary/systemic blood flows) are going. So whydon’t we now move into how we approach thesepatients therapeutically? Let’s start by reviewing life-style modification. I think one of the areas that hasalways been controversial, and where we’re learningmore that some of the recommendations we made inthe past weren’t correct, is exercise. What do you tellyour patients, particularly the ones that have pulmo-nary hypertension and congenital heart disease, aboutexercise? Do you encourage them to participate inprograms? What kinds of restrictions do you place?
Dr Daniels: Well, we will at our center encouragepatients with new diagnosis of pulmonary hyperten-sion, whether it’s congenital heart disease or not, toinitially be involved in a rehab program. It’s difficultin the United States to have patients approved throughinsurance companies for cardiac rehab, so many of ourpatients will go through pulmonary rehab. With apulmonary hypertension diagnosis, they can proceedwith pulmonary rehabilitation, which I think allowsthem to begin an exercise program, and allows them tohave confidence in what they’ll be able to do. And asthey hopefully improve on therapy, they’ll be able toaccelerate their own exercise performance. So I guessas an opener, I would say that we try to incorporate anexercise program into the pulmonary hypertensionpopulation, and the congenital heart disease patientsfall into this mix.
Dr Beghetti: When you consider again the Eisen-menger and the closed shunt, do you give the samepossibility of exercise to both? Or you would advisethem differently?
Dr Daniels: Well, I would say we’re a little morecautious with Eisenmenger patients, only from thestandpoint of some of the isometrics. There’s alwayssome component of isometrics with a rehab program.
Dr Daniels: I think we are a little less willing tofreely open the door to the isometric program that’spart of rehab with the Eisenmenger population. That isprobably the only difference and caveat. But other-wise, the aerobic performance, the aerobic activity, weprescribe in a similar fashion.
Dr Landzberg: The pulmonary rehab that you men-tion, Curt, is so attuned to what our patients can andshould be doing in terms of their mixed diseases thatare going on, in terms of both pulmonary parenchyma,Bellows, and peripheral musculature. Those programsare often well designed to what our patients need. Itreally is remarkable at how the referring clinicianpopulation frequently is so concerned about our pa-tients going to physical therapy and rehab and yet thepatients so desperately welcome it. And there areaccruing data, not just for the idiopathic pulmonaryarterial hypertension population but also for the con-genital heart disease population, which you under-scored, Curt. It’s now a routine part of all of ourpractices to have patients with congenital heart dis-ease, associated pulmonary hypertension, go for re-hab. It’s one of the first, if not the first things that wedo.
Dr Daniels: Maurice, what do you do?
Dr Beghetti: So, it’s a bit difficult to send them tocardiac rehab. And so the pulmonary hypertensioncenter is run in the adult field by pulmonologists andwe’re working together. So it seems that the possibil-ity from this side is a little bit better. The only concernthat sometimes they have, and as we are not directlyinvolved, is the saturation of the patients. And did youneed to teach a little bit the people taking care of themto not be too scared about the saturation? Because ifyou send them to the pulmonary rehab, where they’reused to stress a little bit if the sats go below 90, justimagine when they have patients satting in the low70s- so that’s more the physician, the nurses, and thetechnicians that you need to teach sometimes aboutthe disease.
Dr Daniels: Yes. Good point. Education for the re-hab program for these particular patients is key.
Dr Krasuski: With regard to oxygen saturations,what are your individual practices in terms of pre-scribing oxygen? Do you recommend it for all patientsgoing through rehabilitation? Focusing on the Eisen-menger patient, I think we all recognize that the datahere are very, very limited and that perhaps we’vebeen overzealous with oxygen. Certainly, we have allexperienced one of our Eisenmenger patients getting
“It’s critical to do
a full, complete
angiographic,
hemodynamic,
multiple
maneuver
catheterization.”
Dr Landzberg
203Advances in Pulmonary Hypertension

admitted to the hospital overnight; and when we walkin room on the following morning, the patient is on100% oxygen by non-rebreather mask. We obviouslyhave work to do in terms of educating our nursingstaff and other physicians that take part in the care ofour patients. What do you recommend in terms ofoxygen prescriptions and the use of supplemental ox-ygen at night and during exercise?
Dr Beghetti: I would like to answer before Michael,because I know that he has strong ideas about that. Iwould say that I don’t know. But what I’m sure is thatthe workup of the lungs of these patients should bedone, because that may help you to decide if thepatient needs or does not need oxygen. Because if heor she has some restrictive disease, restrictive lungdisease, or some gas exchange problem, I think that’svery important to know. Because in this population,there may be a good chance that oxygen may help.And then there’s maybe a population that oxygen maynot help. And that’s why we still have a lot of con-troversy about the use of oxygen in this population.But I’m sure that Michael will comment on that.
Dr Landzberg: All of us respect the work that wasdone by Julio Sandoval in Mexico City, who hastaught us much about pulmonary arterial hyperten-sion. The study that most people refer back to is a verysmall study that relates to nocturnal oxygen use inchronically cyanotic patients with general heart dis-ease. It’s a difficult study to extrapolate from. Andwithout being controversial, I would suggest exactlyas Maurice said. The key here is to assess the under-lying pulmonary parenchymal and Bellows diseasethat so many of our patients have. I don’t restrictoxygen away from our patients, but I put it into themix if they have combined disease.
Dr Daniels: And I think also since our population isdifferent, many of them would have had open-heartsurgery. They would have had surgical procedures, asternotomy. We know now from our congenital heartdisease data that many of our patients have restrictivelung disease. And so they do have limitations of theirlung capacities, which in many cases will lead to areasof lung that do not participate in oxygen exchange ina normal fashion or the capacity at which we considerto be normal. So a workup of lung disease is impor-tant. And I think all of us have found patients, thatsurprisingly, do feel better on oxygen can exerciselonger and do perform better. And at the end of theday, it is all about patients’ feeling better and improv-ing their quality of life. And so it’s important, I think,to assess whether or not patients that are desaturatedneed oxygen, whether they respond to oxygen. Do
they feel better with oxygen? Does it improve theirquality of life? And I guess to be quite frank, we manytimes will allow patients to make that decision. Youknow, if you feel better on oxygen and you’re able toexercise and your quality of life improves, then that’sfine. If you really don’t feel better and the chore ofhaving oxygen, carrying oxygen actually worsensyour quality of life and we really do not see a re-sponse, then obviously we wouldn’t prescribe oxygen.
Dr Beghetti: Yes, we need to remember that some-times they’ve had several surgeries. And so they mayhave chest deformity, not only lungs, chest defor-mity. And it is well known that cyanotic patientssometimes have scoliosis. So if you had the chest de-formity because of the surgery, the scoliosis, and maybesome lung disease, clearly you will find some patientsthat after the complete lung workup, they will requireoxygen.
Dr Franklin: Yes, that’s a good point, too. It’s some-thing that we struggle with, I think, here in Houston.Because I generally don’t necessarily start oxygen.But that said, I have patients who’ve come to me onoxygen and I don’t necessarily stop it, either fromtheir pulmonologist or the prior cardiologist that I’veinherited from. But, to Curt’s point, I’d say yes, somepatients feel better on it. Rich, I think you mentionedthe nocturnal oxygen. Some patients use it at night andthey just feel more rested in the morning. Who knowsif that’s placebo effect or not. But, like Curt said,quality of life is important, too. But I tend to notnecessarily start oxygen if I’m going to start patientson an exercise regimen, per se.
Dr Krasuski: There is a growing body of literature,Wayne, as you’re alluding to, that patients with pul-monary hypertension have sleep-disordered breathing.So it certainly makes sense that the ACHD patientswith pulmonary hypertension behave similarly andmay therefore benefit from nocturnal oxygen. Iwanted to bring up another controversial topic: anti-coagulation. How do we handle that in our ACHD-PHpatients? It’s interesting that this controversy has ex-isted for over 2 decades and I’m not sure we’re anysmarter about this now. I want to know what thisexpert panel thinks in terms of how we should utilizeanticoagulation and what type of impact we makewhen we do so.
Dr Daniels: I personally separate the Eisenmengercyanotic patient from the noncyanotic patient. I guesseven though it is controversial, I still follow somepretty general rules until I see data otherwise. And thegeneral rules that I continue to follow is if they’re a
204 Advances in Pulmonary Hypertension

patient that is cyanotic, this brings a whole host ofother hematologic issues of cyanosis, but if they’re anoncyanotic patient, a congenital heart disease patientwith pulmonary hypertension, then I typically followthe general rules, which is prescribing anticoagula-tion, unless a contraindication. The cyanotic patientbecomes much more difficult and somewhat morecontroversial. I do not prescribe anticoagulation for acyanotic Eisenmenger patient, because of the concern,and certainly we see bleeding diathesis. The cyanoticpatient has polycythemia, and typically bleeding dis-order that aren’t always completely worked out, butcertainly we know that this is the case. And in thosepatients, unless there is a strong reason to prescribeanticoagulation, such as pulmonary emboli or atrialarrhythmias, the bleeding risk outweighs preventiveanticoagulation.
Dr Landzberg: I think for the last 20 years, I’vetaught every fellow that if Curt Daniels says some-thing that you need to totally disregard anything I’veever said and listen to what Curt says.
Dr Daniels: Until now. (laughter)
Dr Landzberg: On the same hand, what’s under-scored in terms of the population of folks who takecare of them, the data that are out there, are quiteinteresting in this very question, because here arecenters that are so aligned with each other. And Iactually thought Curt was going to say the exactopposite of what he said, because in the practice here,I will tend to use anticoagulation as the last therapy toadd onto the patient with a closed shunt who’s notcyanotic, because I think the data are the least there,and I try to get those patients onto every other therapywhere I might have some data or some cohort obser-vational studies, at the least. And it’s the patients withEisenmenger syndrome that I’m the most concernedwith. I know that there are data in terms of prothrom-bosis; those data are strong. Granted, exactly whatCurt said, the data that say that we make a differencewith anticoagulation are unknown, but I worry inparticular about them. So with the same data, we canargue both sides frequently. But I’ll go back to thevery first part of my statement, which was if Curt saidsomething, that’s what I would argue.
Dr Beghetti: I think this underscores the problemwe have. We absolutely do not know. And that’s theproblem. For example, as a pediatrician, for my idio-pathic PH in pediatrics, usually I don’t anticoagulateunless they have a severe RV dysfunction. There is noscience behind that, but the problem is that the risk ofbleeding in a young patient is pretty high because
they’re still very active. And so we rely more onexperience than on real data for this anticoagulationapproach. And I think in both patients, idiopathic PHand Eisenmenger. And I don’t see how we will indeeddesign any study at the current time that would help usto really decide for that, unless some of you have anidea. But I think it will be very difficult now to do thestudy in this population.
Dr Daniels: It certainly will be very important to, asa registry, to try to gather more data on Eisenmengerpatients. Who is on anticoagulation? What is theiroutcome? What is the risk? I mean, it’s an incrediblyheterogeneous population, so it will be very difficultto find information, except for observational registry.But, you know, Mike’s point is, of course, a good one.The risk with Eisenmenger is thromboembolic, butthey also develop hemoptysis. Clinically I have seen agreater incidence of hemoptysis than thromboembolicevents. So it’s an incredibly tough balance. Any sci-entific evaluation of that population says we see clot-ting and we see bleeding. And so it makes it verydifficult to know what is the proper approach.
Dr Franklin: Very good points. Maybe registrieswill be the answer. Because I certainly go both ways,but I probably tend to not anticoagulate. I guess nohard data either way. And I tend to think iatrogenicbleeding, whether it’s hemoptysis or what have you, isprobably worse. And so I tend to not. But hopefully,we’ll get smarter about this as some of these registriescome through.
Dr Krasuski: I would add that one group I regularlyanticoagulate are the Eisenmenger patients with in-dwelling lines for intravenous therapies.
Dr Beghetti: Definitely, yeah.
Dr Krasuski: This also applies to the same group ofpatient with pacemakers or defibrillators. These arepatients in whom I am worried about the risk forparadoxical embolization. All the points on anticoag-ulation are well taken. I particularly like the way thatMike was able to explain how you could use the datato argue each side of whether or not to anticoagulatein Eisenmenger and CHD-PAH patients.
Dr Krasuski: We’ve unfortunately run out of time,though we have covered many of the topics I wantedto discuss. Let me try to partially summarize ourdiscussion for some “take-home” points. We first dis-cussed the at-risk population. That there are certaingroups of patients in whom we’re worried about thedevelopment of pulmonary hypertension. Patients
“I’ve found that
taking patients to
the catheterization
laboratory is most
helpful for
clarification of the
disease state
when there are
conflicting data.”
Dr Krasuski
205Advances in Pulmonary Hypertension

with larger shunts (natural or surgically created), olderpatients, those with ventricular dysfunction early on,and patients who had lesions repaired later in life.These are the patients in whom we don’t want to missthe opportunity to screen for PAH. We should beaware of this complication even in the patients thathave been previously repaired—this is a very impor-tant point for the care providers that don’t routinelyfollow congenital heart patients—having a completeanatomical repair does not always equal a lifetime freeof complications. And pulmonary hypertension is avery important complication. We discussed the ap-proach to workup in these patients. The consensus wasthat a thorough workup is incredibly important, be-cause we have a tendency to lump patients togetherwho may not respond the same way to therapy. Themore we know about certain characteristics such astheir lung status, the better we’ll be able to adapt ourtherapeutic approach and improve outcomes. The im-portance of catheterization was emphasized. We eachcome from the vantage point that all perform heartcatheterization. We all agreed that an initial hemody-namic assessment for any of the patients who is goingto undergo selective pulmonary therapies is absolutelycritical. And that it really sets the patient on the properpath for treatment. We were a little conflicted whendiscussing if and when we should take the patientback to the cath lab. Our agreement was that recath-eterization in the patient with Eisenmenger syndromewho is doing well is unnecessary. For the patient witha corrected shunt, we would be a little bit more likelyto reassess hemodynamics, particularly if there areany conflicting data about their clinical status. Wetalked a little bit about natriuretic peptides. They maybe an exciting marker to follow, as is MRI for theassessment of ventricular function. In terms of ourtherapeutic approach to patients, we all mentionedhow important exercise is and that’s an important firststep in getting patients on the road to feeling better.All of us kind of mentioned the struggle, particularlywith insurance companies, that we’ve each had ingetting patients into cardiac rehab. Pulmonary reha-bilitation may be an alternative pathway for gettingpatients properly regimented to start exercising again.The use of oxygen should depend upon whether un-derlying lung disease is present. There are plenty ofpatients with congenital heart defects who also haverestrictive lung disease and other pulmonary prob-lems, so it’s important we properly assess those pa-tients. We don’t want to necessarily restrict their ox-ygen, particularly if it helps them feel better, butsupplemental oxygen at this point, particularly in the
Eisenmenger patient, remains fairly controversial. Ourdiscussion of anticoagulation illustrated that wehaven’t gotten very far in research in this area, and wehave not come up with any good guidelines for whoshould be anticoagulated. Because the opinion is sostrong among physicians who treat ACHD-PAH, weabsolutely need to collect these data in our registries.Perhaps we won’t be able to do a randomized trial, butthrough the newer registries that all of you are part ofon this board, we may be able to better answer thisquestion in the future. We only talked a little bit aboutselective pulmonary vasodilator therapy for these pa-tients. The trend appears toward more aggressive useof combination therapy at an earlier stage in the dis-ease process. I think you guys did a fabulous job, andmy job as moderator was pretty easy with such aterrific group of panelists. Are there any other finalparting comments or anything that we missed todaythat we should have covered?
Dr Daniels: I would just say, Rich, it was an out-standing discussion and I think important for the au-dience, the topics and the synopsis you just providedreally goes to the importance of this educational ex-perience and this opportunity for those that are goingto be reviewing our roundtable discussion. So thanks,Rich, for putting it together. And I thank my col-leagues for their expert opinion.
Dr Beghetti: I have one additional comment. I thinkthe discussion was very interesting for one more rea-son. You may have noticed, and it’s not to minimizethe role of the new targeted therapies, because I thinkthese therapies have started again to work on thesepatients, but we almost did not discuss these newtherapies. And we discussed that we still need tounderstand what happened to our patients and that weneed to very well work up our patients before usingthese therapies. That’s a very strong message of thisroundtable.
Dr Landzberg: The study of pulmonary hyperten-sion really began with congenital heart disease andits understanding and the pulmonary vasculature. Ipersonally think that our collaboration with ourcolleagues who study solely pulmonary arterial hy-pertension is very rich. Future understanding of thecoupling between the right (pulmonary) ventricleand the other supporting structures in the arterialvasculature is really going to lay the foundation forbetter understanding of this disorder.
206 Advances in Pulmonary Hypertension

Ask the Expert
Congenital Heart Disease With Associated PulmonaryArterial Hypertension. Who and When to Operate: ATherapeutic Dilemma
Section EditorMyung H. Park, MD
Usha Krishnan, MDAssociate Director, Pulmonary HypertensionPediatric CardiologyColumbia University Medical CenterNew York, New York
In countries with easy availability of sur-gical care for congenital heart disease,most patients undergo surgery at an ap-propriate age before development of pul-monary vascular disease. However, thereis an increasing population of childrenand young adults with previously undiag-nosed or unoperated congenital heart dis-ease. The responsibility of deciding whois operable and whether treatment modal-ities can reverse vascular changes enoughto convert a previously “borderline or in-operable” patient to being a surgical can-didate often rests with the pulmonary hy-pertension specialist.1-3 Indeed, studieshave shown that operating on patientswith high pulmonary vascular resistance(PVR) makes the prognosis significantlyworse than the natural history of leavingthem without surgery.
Cardiac catheterization still remains thegold standard for deciding on operabilityof these patients.1,2 One must have a clearmental distinction between elevated pul-monary pressures (as in large left to rightpost-tricuspid shunts) and elevated PVR,which implies distal vascular changes.Children with large left to right shunts andheart failure symptoms with increasedpulmonary blood flow will clearly benefitfrom early surgery. Patients with largedefects and PVR equal to or more thansystemic vascular resistance (SVR) willhave reversed shunts (Eisenmenger syn-drome) and are inoperable. Patients withelevated but subsystemic PVR and there-fore smaller left to right shunts are thesubjects of the therapeutic dilemma. Mustone treat with pulmonary vasodilators firstand then recheck hemodynamics and op-erate if suitable—or just leave them aloneand manage as early Eisenmenger syn-
drome?3 What is the long-term outcomeof survivors of this strategy?
Clinical clues to increasing PVR can besought by careful history taking. An infantwith a large left to right shunt is usuallyundernourished, has feeding problemsand irritability, may become diaphoreticwith crying, and may have recurrent re-spiratory infections. Then, as the childgrows, if pulmonary vascular disease de-velops, these symptoms of increased pul-monary blood flow gradually disappearand the child starts catching up on growthand appetite and becomes less tachy-pnoec. This is the honeymoon period,when the parents are lulled into a falsesense of security that their child is curedof the heart disease, until the shunt re-verses as PVR becomes more than theSVR and the child initially gets blue withexercise, and later is cyanosed all thetime.
Different defects develop pulmonaryvascular disease at different times.1,3
Complex congenital heart defects liketransposition of great arteries, truncus ar-teriosus, and endocardial cushion defects(especially with Down syndrome) almostalways develop increased PVR in in-fancy.3 Many children with large defectsat the ventricular or great artery level de-velop vascular changes in the first 2 yearsof life, unlike atrial defects, where themajority of patients have near normalPVR well into adulthood. Again, there isa subgroup of patients with atrial shuntswho present with elevated PVR at a veryearly age, and are thought to possibly bepatients with idiopathic pulmonary arte-rial hypertension (IPAH) with an associ-ated atrial shunt. The goal in managingpatients with left to right shunts is to di-agnose these lesions early and intervenebefore development of irreversible pul-
monary vascular disease. Patients withsignificant shunts (Qp/Qs �2:1) at theventricular (VSD) or great artery level(PDA or AP Window) should be operatedin infancy to prevent vascular disease. Pa-tients with atrial septal defects (ASD) aremanaged differently. Most patients withmoderate secundum ASDs can be treatedin the interventional cardiac catheteriza-tion laboratory, using devices to close thedefect. Careful preprocedure evaluationinvolves sizing of the defect by priorechocardiography, as well as by balloonsizing using transesophageal echocardio-graphic guidance during cardiac catheter-ization, ensuring adequate rims to seat thedevice and avoiding interference with sur-rounding structures. A major part of thisdecision making can be done by transtho-racic echocardiogram before taking thepatient for the procedure. Patients withsinus venosus, coronary sinus, and pri-mum ASDs are not amenable to transcath-eter closure and need to undergo surgicalrepair. Patients with smaller ASDs withno evidence of right sided volume over-load by echocardiogram are usually not atrisk for developing PAH, endocarditis, orheart failure and do not need to be closed.The only indications for closure of a smallatrial defect or patent foramen ovalewould be in an adult with recurrentstrokes without other etiologies, or peoplewho train to be deep sea divers (to preventreverse shunting and catastrophic events).
Evaluation in the cardiac catheteriza-tion laboratory should involve very care-ful assessment of baseline hemodynamicswith at least 3 complete right and leftheart saturation and pressure runs (Table1). Hemodynamic calculations includepulmonary and systemic blood flow (Qpand Qs), pulmonary and systemic resis-tances (PVR and SVR) at baseline, andwith acute vasodilator testing (AVT) us-ing inhaled nitric oxide (iNO). [Acute va-
Correspondence: [email protected]
207Advances in Pulmonary Hypertension

soresponsiveness is defined as reductionof mean pulmonary artery pressure by atleast 10 mm to a value �40 mm with anincreased or unchanged cardiac index.]4-6
There are several issues that the clini-cian has to be aware of while perform-ing and interpreting cardiac catheteriza-tion data. Snapshot hemodynamic studiesare greatly influenced by sedation, gen-eral anesthesia, airway and lung paren-chymal issues, stress (of intubation),catecholamines, and variations in acid-base balance, especially in young childrenunder general anesthesia. Several studieshave demonstrated benefits of treating
Eisenmenger patients with advanced ther-apies. It is thought that their antiprolifera-tive effects lead to reverse remodeling inthe pulmonary circulation and may even-tually cause reduction in PVR and im-prove right ventricular hypertrophy.1 Per-haps treating borderline patients withpulmonary vasodilators for a finite periodof time and reassessing their hemodynam-ics would reduce their PVR enough tomake them candidates for shunt closure.Since short-term improvements in thesepatients may not translate into long-termsurvival benefit, partial closure of the de-fect in highly selective patients with im-
proved hemodynamics on treatment andcontinuing pulmonary vasodilator therapyseems an attractive prospect and meritsfurther investigation. Larger and longer-term studies using these strategies will berequired before one can be sure of theright approach for these patients, but theavailability of newer drugs holds a prom-ise for the future.
References1. Dimopoulos K, Peset A, Gatzoulis MA. Evalu-ating operability in adults with congenital heart dis-ease and the role of pretreatment with targeted pul-monary arterial hypertension therapy. Int J Cardiol.2008;129(2):163-171.2. Viswanathan S, Kumar RK. Assessment of op-erability of congenital cardiac shunts with increasedpulmonary vascular resistance. Catheter CardiovascInterv. 2008;71(5):665-670.3. Rosenzweig EB, Barst RM. Congenital heartdisease and pulmonary hypertension: pharmacologyand feasibility of late surgery. Prog Cardiovasc Dis.2012;55(2):128-133.4. Douwes JM, van Loon RL, Hoendermis ES, etal. Acute pulmonary vasodilator response in paedi-atric and adult pulmonary arterial hypertension: oc-currence and prognostic value when comparing threeresponse criteria. Eur Heart J. 2011;32(24):3137-3146.5. Hill KD, Lim DS, Everett AD, Ivy DD, MooreJD. Assessment of pulmonary hypertension in thepediatric catheterization laboratory: current insightsfrom the Magic registry. Catheter Cardiovasc In-terv. 2010;76(6):865-873.6. Balzer DT, Kort HW, Day RW, et al. InhaledNitric Oxide as a Preoperative Test (INOP Test I):the INOP Test Study Group. Circulation. 2002;106(12 Suppl 1):I76-I81.
Table 1: Formulae for calculation of shunts and resistances usinghemodynamic data
Qp�VO2/ (PVO2-PAO2)�VO2/(Pulmonary Vein—Pulmonary Artery Saturation)(Oxygen Capacity);Qs�VO2/ (Aortic—Mixed Venous Saturation)(Oxygen Capacity);Qep�VO2/ (Pulmonary Vein—Mixed Venous Saturation)(Oxygen Capacity);L-R Shunt�Qp-Qep; R-L Shunt�Qs-QepQp/Qs�Aortic—Mixed Venous Saturations/Pulmonary Vein—Pulmonary ArterySaturation.While breathing 100% O2, one must include dissolved oxygen in the calculation ofoxygen content, using the equation “dissolved Oxygen”�0.03*PO2.PVR�mPAP-mPVP/QpSVR�MAoP-MRAP/QsOxygen capacity�13.6*Hemoglobin(gm/dl).
Abbreviations: VO2 � oxygen consumption; Qp � pulmonary blood flow; Qs �systemic blood flow; Qep � effective pulmonary flow; PVO2 � pulmonary vein O2content; PAO2 � pulmonary artery O2 content; mPAP � mean pulmonary arterypressure; mAoP � mean aortic pressure; mRAP � mean right atrial pressure; mPVP �mean pulmonary venous (or pulmonary artery wedge) pressure; saturation PVR �pulmonary vascular resistance; SVR � systemic vascular resistance.
208 Advances in Pulmonary Hypertension

Proven PDE-5 inhibition that can help patients with PAH be more active
ADCIRCA once-daily opens up possibilities
The only once-daily PDE-5 inhibitor for PAH1
33-meter placebo-adjusted mean improvement in 6MWD at 16 weeks1
A $20 co-pay for eligible patients on commercial/private insurance plans*
The most common adverse event with ADCIRCA is headache. Other common adverse events include myalgia, nasopharyngitis, flushing, respiratory tract infection, extremity pain, nausea, back pain, dyspepsia, and nasal congestion
* This assistance program is not valid for prescriptions reimbursed under Medicare, Medicaid, TRICARE, state pharmaceutical assistance programs, or other federal or state programs. This assistance program is not valid for patients in the state of Massachusetts with prescription drug coverage.
Important Safety InformationCONTRAINDICATIONS
ADCIRCA should not be used in patients taking medicines that contain nitrates, as the combination could cause a sudden, unsafe drop in blood pressure
Patients with a known serious hypersensitivity to tadalafil should not take ADCIRCA
WARNINGS AND PRECAUTIONS If a patient experiences anginal chest pain after taking ADCIRCA they
should seek immediate medical attention Phosphodiesterase 5 inhibitors (PDE-5i), including tadalafil, have mild
systemic vasodilatory properties that may result in transient decreases in blood pressure. Before prescribing ADCIRCA, physicians should carefully consider whether their patients with underlying cardiovascular disease could be adversely affected by such actions. Pulmonary vasodilators may significantly worsen the cardiovascular status of patients with pulmonary veno-occlusive disease (PVOD) and administration of ADCIRCA to these patients is not recommended
The use of ADCIRCA with alpha blockers, blood pressure medications, and alcohol may lower blood pressure significantly and may lead to symptomatic hypotension (fainting)
Tadalafil is metabolized predominately by CYP3A in the liver. Use of ADCIRCA with potent CYP3A inhibitors, such as ketoconazole and itraconazole, should be avoided. For patients on ADCIRCA therapy that require treatment with ritonavir, ADCIRCA should be discontinued at least 24 hours prior to starting ritonavir. For patients on ritonavir therapy that require treatment with ADCIRCA, start ADCIRCA at 20 mg once a day. Use of ADCIRCA with potent inducers of CYP3A, such as rifampin, should be avoided
The use of ADCIRCA is not recommended for patients with severe renal or hepatic impairment. Please see full prescribing information for dosing recommendations for patients with mild to moderate renal or hepatic impairment
ADCIRCA contains the same ingredient (tadalafil) as Cialis, which is used to treat erectile dysfunction (ED). The safety and efficacy of combinations of ADCIRCA with Cialis or other PDE-5 inhibitors have not been studied. Therefore, the use of such combinations is not recommended
In rare instances, men taking PDE-5 inhibitors (including tadalafil) for ED reported a sudden decrease or loss of vision or hearing, or an erection lasting more than four hours. A patient who experiences any of these symptoms should seek immediate medical attention
ADVERSE REACTIONS The most common adverse event with ADCIRCA is headache (42%
ADCIRCA vs 15% placebo). Other common adverse events (reported by ≥9% of patients on ADCIRCA and more frequent than placebo by 2%) include myalgia (14% vs 4%), nasopharyngitis (13% vs 7%), flushing (13% vs 2%), respiratory tract infection (13% vs 6%), extremity pain (11% vs 2%), nausea (11% vs 6%), back pain (10% vs 6%), dyspepsia (10% vs 2%), and nasal congestion (9% vs 1%)
See how Lung Rx is making strides in pulmonary medicine at www.lungrx.com
Please see brief summary of Full Prescribing Information on following page. Please see Full Prescribing Information and Patient Information available at www.adcirca.com, or call 1-800-545-5979.
© 2011 Lung Rx, LLC All rights reserved. ADC_JA_MAY11v.1
ADCIRCA® (tadalafil) tablets is a phosphodiesterase 5 inhibitor (PDE-5i) indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to improve exercise ability. Studies establishing effectiveness included predominately patients with NYHA Functional Class II–III symptoms and etiologies of idiopathic or heritable PAH (61%) or PAH associated with connective tissue diseases (23%).
Reference: 1. ADCIRCA [package insert]. Indianapolis, IN: Eli Lilly and Company; 2011.
www.adcirca.com 877-UNITHER
ADCIRCA is a registered trademark of Eli Lilly and Company, 2011.

ADCIRCA® (tadalafil) tabletsBRIEF SUMMARYThe following is a brief summary of the full prescribing information on ADCIRCA (tadalafil). Please review the full prescribing information prior to prescribing ADCIRCA.INDICATIONS AND USAGEPulmonary Arterial Hypertension: ADCIRCA is indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to improve exercise ability. Studies establishing effectiveness included predominately patients with NYHA Functional Class II–III symptoms and etiologies of idiopathic or heritable PAH (61%) or PAH associated with connective tissue diseases (23%).CONTRAINDICATIONSConcomitant Organic Nitrates: Do not use ADCIRCA in patients who are using any form of organic nitrate, either regularly or intermittently. ADCIRCA potentiates the hypotensive effect of nitrates. This potentiation is thought to result from the combined effects of nitrates and ADCIRCA on the nitric oxide/cGMP pathway. Hypersensitivity Reactions: ADCIRCA is contraindicated in patients with a known serious hypersensitivity to tadalafil (ADCIRCA or CIALIS). Hypersensitivity reactions have been reported, including Stevens-Johnson syndrome and exfoliative dermatitis.WARNINGS AND PRECAUTIONSCardiovascular Effects: Discuss with patients the appropriate action to take in the event that they experience anginal chest pain requiring nitroglycerin following intake of ADCIRCA. At least 48 hours should elapse after the last dose of ADCIRCA before taking nitrates. If a patient has taken ADCIRCA within 48 hours, administer nitrates under close medical supervision with appropriate hemodynamic monitoring. Patients who experience anginal chest pain after taking ADCIRCA should seek immediate medical attention. PDE5 inhibitors, including tadalafil, have mild systemic vasodilatory properties that may result in transient decreases in blood pressure. Prior to prescribing ADCIRCA, carefully consider whether patients with underlying cardiovascular disease could be affected adversely by such vasodilatory effects. Patients with severely impaired autonomic control of blood pressure or with left ventricular outflow obstruction, (e.g., aortic stenosis and idiopathic hypertrophic subaortic stenosis) may be particularly sensitive to the actions of vasodilators, including PDE5 inhibitors. Pulmonary vasodilators may significantly worsen the cardiovascular status of patients with pulmonary veno-occlusive disease (PVOD). Since there are no clinical data on administration of ADCIRCA to patients with veno-occlusive disease, administration of ADCIRCA to such patients is not recommended. Should signs of pulmonary edema occur when ADCIRCA is administered, the possibility of associated PVOD should be considered. There is a lack of data on safety and efficacy in the following groups who were specifically excluded from the PAH clinical trials:
Use with Alpha Blockers and Antihypertensives — PDE5 inhibitors, including ADCIRCA, and alpha–adrenergic blocking agents are vasodilators with blood pressure-lowering effects. When vasodilators are used in combination, an additive effect on blood pressure may be anticipated. In some patients, concomitant use of these two drug classes can lower blood pressure significantly, which may lead to symptomatic hypotension (e.g., fainting). Safety of combined use of PDE5 inhibitors and alpha blockers may be affected by other variables, including intravascular volume depletion and use of other antihypertensive drugs. Use with Alcohol — Both alcohol and tadalafil are mild vasodilators. When mild vasodilators are taken in combination, blood pressure-lowering effects are increased. Use with Potent CYP3A Inhibitors or Inducers: Co-administration of ADCIRCA in Patients on Ritonavir — In patients receiving ritonavir for at least one week, start ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability. Co-administration of Ritonavir in Patients on ADCIRCA — Avoid use of ADCIRCA during the initiation of ritonavir. Stop ADCIRCA at least 24
hours prior to starting ritonavir. After at least one week following the initiation of ritonavir, resume ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability. Other Potent Inhibitors of CYP3A — Tadalafil is metabolized predominantly by CYP3A in the liver. In patients taking potent inhibitors of CYP3A such as ketoconazole and itraconazole, avoid use of ADCIRCA. Potent Inducers of CYP3A — For patients chronically taking potent inducers of CYP3A, such as rifampin, avoid use of ADCIRCA. Use in Renal Impairment: In patients with mild or moderate renal impairment — Start dosing at 20 mg once daily. Increase the dose to 40 mg once daily based upon individual tolerability. In patients with severe renal impairment — Avoid use of ADCIRCA because of increased tadalafil exposure (AUC), limited clinical experience, and the lack of ability to influence clearance by dialysis. Use in Hepatic Impairment: In patients with mild to moderate hepatic cirrhosis (Child-Pugh Class A and B) — Because of limited clinical experience in patients with mild to moderate hepatic cirrhosis, consider a starting dose of 20 mg once daily ADCIRCA. In patients with severe hepatic cirrhosis (Child-Pugh Class C) — Patients with severe hepatic cirrhosis have not been studied. Avoid use of ADCIRCA. Effects on the Eye: Physicians should advise patients to seek immediate medical attention in the event of a sudden loss of vision in one or both eyes. Such an event may be a sign of non–arteritic anterior ischemic optic neuropathy (NAION), a cause of decreased vision, including permanent loss of vision that has been reported rarely postmarketing in temporal association with the use of all PDE5 inhibitors. It is not possible to determine whether these events are related directly to the use of PDE5 inhibitors or other factors. Physicians should also discuss with patients the increased risk of NAION in individuals who have already experienced NAION in one eye, including whether such individuals could be adversely affected by use of vasodilators such as PDE5 inhibitors. Patients with known hereditary degenerative retinal disorders, including retinitis pigmentosa, were not included in the clinical trials, and use in these patients is not recommended. Hearing Impairment: Physicians should advise patients to seek immediate medical attention in the event of sudden decrease or loss of hearing. These events, which may be accompanied by tinnitus and dizziness, have been reported in temporal association to the intake of PDE5 inhibitors, including ADCIRCA. It is not possible to determine whether these events are related directly to the use of PDE5 inhibitors or to other factors. Combination with Other PDE5 Inhibitors: Tadalafil is also marketed as CIALIS. The safety and efficacy of taking ADCIRCA together with CIALIS or other PDE5 inhibitors have not been studied. Inform patients taking ADCIRCA not to take CIALIS or other PDE5 inhibitors. Prolonged Erection: There have been rare reports of prolonged erections greater than 4 hours and priapism (painful erections greater than 6 hours in duration) for this class of compounds. Priapism, if not treated promptly, can result in irreversible damage to the erectile tissue. Patients who have an erection lasting greater than 4 hours, whether painful or not, should seek emergency medical attention. ADCIRCA should be used with caution in patients who have conditions that might predispose them to priapism (such as sickle cell anemia, multiple myeloma, or leukemia), or in patients with anatomical deformation of the penis (such as angulation, cavernosal fibrosis, or Peyronie’s disease). Effects on Bleeding: PDE5 is found in platelets. When administered in combination with aspirin, tadalafil 20 mg did not prolong bleeding time, relative to aspirin alone. ADCIRCA has not been administered to patients with bleeding disorders or significant active peptic ulceration. Although ADCIRCA has not been shown to increase bleeding times in healthy subjects, use in patients with bleeding disorders or significant active peptic ulceration should be based upon a careful risk-benefitassessment.ADVERSE REACTIONSThe following serious adverse reactions are discussed elsewhere in the labeling:
Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. Tadalafil was administered to 398 patients with PAH during clinical trials worldwide. In trials of ADCIRCA, a total of 311 and 251 subjects have been treated for at least 182 days and 360 days, respectively. The overall rates of discontinuation because of an
adverse event (AE) in the placebo-controlled trial were 9% for ADCIRCA 40 mg and 15% for placebo. The rates of discontinuation because of AEs, other than those related to worsening of PAH, in patients treated with ADCIRCA 40 mg was 4% compared to 5% in placebo-treated patients. In the placebo-controlled study, the most common AEs were generally transient and mild to moderate in intensity. Table 1 presents treatment-emergent adverse events reported by ≥9% of patients in the ADCIRCA 40 mg group and occurring more frequently than with placebo.
TABLE 1: Treatment-Emergent Adverse Events Reported by ≥9% of Patients in ADCIRCA and More
Frequent than Placebo by 2%
Postmarketing Experience: The following adverse reactions have been identified during post-approval use of tadalafil. These events have been chosen for inclusion either because of their seriousness, reporting frequency, lack of clear alternative causation, or a combination of these factors. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate reliably their frequency or establish a causal relationship to drug exposure. The list does not include adverse events that are reported from clinical trials and that are listed elsewhere in this section. Cardiovascular and cerebrovascular — Serious cardiovascular events, including myocardial infarction, sudden cardiac death, stroke, chest pain, palpitations, and tachycardia, have been reported postmarketing in temporal association with the use of tadalafil. Most, but not all, of these patients had preexistingcardiovascular risk factors. Many of these events were reported to occur during or shortly after sexual activity, and a few were reported to occur shortly after the use of tadalafil without sexual activity. Others were reported to have occurred hours to days after the use of tadalafil and sexual activity. It is not possible to determine whether these events are related directly to tadalafil, to sexual activity, to the patient’s underlying cardiovascular disease, to a combination of these factors, or to other factors. Body as a whole — Hypersensitivity reactions includingurticaria, Stevens–Johnson syndrome, and exfoliativedermatitis. Nervous — Migraine, seizure and seizurerecurrence, and transient global amnesia. Ophthalmologic — Visual field defect, retinal vein occlusion, and retinalartery occlusion. Non–arteritic anterior ischemic optic neuropathy (NAION), a cause of decreased vision including permanent loss of vision, has been reported rarelypostmarketing in temporal association with the use of PDE5 inhibitors, including tadalafil. Most, but not all, of these patients had underlying anatomic or vascular risk factors for development of NAION, including but not necessarily limited to: low cup to disc ratio (“crowded disc”), age over 50, diabetes, hypertension, coronary artery disease, hyperlipidemia, and smoking. It is not possible to determine whether these events are related directly to the use of PDE5 inhibitors, to the patient’s underlying vascular risk factors or anatomical defects, to a combination of these factors, or to other factors. Otologic — Cases of sudden decrease or loss of hearing have been reported postmarketing in temporal association with the use of PDE5 inhibitors, including tadalafil. In some of the cases, medical conditions and other factors were reported that may have also played a role in the otologic adverse events. In many cases, medical follow-up information was limited. It is not possible to determine whether these reported events are related directly to the use of tadalafil, to the patient’s underlying risk factors for hearing loss, a combination of these factors, or to other factors. Urogenital — Priapism.DRUG INTERACTIONSPotential for Pharmacodynamic Interactions with ADCIRCA: Nitrates — Do not use ADCIRCA in patients who
significant aortic and mitral valve disease
constriction
congestive cardiomyopathy
left ventricular dysfunction
Patients with life-threatening arrhythmias
coronary artery disease
(<90/50 mm Hg) or uncontrolled hypertension
ADCIRCA ADCIRCA Placebo (%) 20 mg (%) 40 mg (%)EVENT (N=82) (N=82) (N=79)Headache 15 32 42Myalgia 4 9 14Nasopharyngitis 7 2 13Flushing 2 6 13Respiratory Tract Infection (Upper 6 7 13and Lower)Pain in Extremity 2 5 11Nausea 6 10 11Back Pain 6 12 10Dyspepsia 2 13 10Nasal Congestion (Including sinus 1 0 9congestion)

milk may not accurately predict levels of drug in human breast milk. Because many drugs are excreted in human milk, caution should be exercised when ADCIRCA is administered to a nursing woman. Pediatric Use: Safety and effectiveness of ADCIRCA in pediatric patients have not been established. Geriatric Use: Of the total number of subjects in the clinical study of tadalafil for pulmonary arterial hypertension, 28 percent were 65 and over, while 8 percent were 75 and over. No overall differences in safety were observed between subjects over 65 years of age compared to younger subjects or those over 75 years of age. No dose adjustment is warranted based on age alone; however, a greater sensitivity to medications in some older individuals should be considered. Renal Impairment: For patients with mild or moderate renal impairment, start ADCIRCA at 20 mg once daily. Increase the dose to 40 mg once daily based upon individual tolerability. In patients with severe renal impairment, avoid use of ADCIRCA because of increased tadalafil exposure (AUC), limited clinical experience, and the lack of ability to influence clearance by dialysis. Hepatic Impairment: Because of limited clinical experience in patients with mild to moderate hepatic cirrhosis (Child-Pugh Class A or B), consider a starting dose of ADCIRCA 20 mg once daily. Patients with severe hepatic cirrhosis (Child-Pugh Class C) have not been studied, thus avoid use of ADCIRCA in such patients.OVERDOSAGESingle doses up to 500 mg have been given to healthy male subjects, and multiple daily doses up to 100 mg have been given to male patients with erectile dysfunction. Adverse reactions were similar to those seen at lower doses. Doses greater than 40 mg have not been studied in patients with pulmonary arterial hypertension. In cases of overdose, standard supportive measures should be adopted as needed. Hemodialysis contributes negligibly to tadalafil elimination.
Marketed by Lung Rx, LLC, a wholly owned subsidiary of United Therapeutics Corporation.Rx only April 2011 www.adcirca.com
are using any form of organic nitrate. In clinical pharmacology studies ADCIRCA potentiated the hypotensive effect of nitrates. In a patient who has taken ADCIRCA, where nitrate administration is deemed medically necessary in a life–threatening situation, at least 48 hours should elapse after the last dose of ADCIRCA before nitrate administration is considered. In such circumstances, nitrates should still only be administered under close medical supervision with appropriate hemodynamic monitoring. Alpha-Blockers — PDE5 inhibitors, including ADCIRCA, and alpha–adrenergic blocking agents are both vasodilators with blood-pressure-lowering effects. When vasodilators are used in combination, an additive effect on blood pressure may be anticipated. Clinical pharmacology studies have been conducted with coadministration of tadalafil with doxazosin, alfuzosin or tamsulosin. Antihypertensives — PDE5 inhibitors, including ADCIRCA, are mild systemic vasodilators. Clinical pharmacology studies were conducted to assess the effect of tadalafil on the potentiation of the blood–pressure–lowering effects of selected antihypertensive medications (amlodipine, angiotensin II receptor blockers, bendroflumethiazide, enalapril, and metoprolol). Small reductions in blood pressure occurred following coadministration of tadalafil with these agents compared with placebo. Alcohol — Both alcohol and tadalafil, a PDE5 inhibitor, act as mild vasodilators. When mild vasodilators are taken in combination, blood pressure–lowering effects of each individual compound may be increased. Substantial consumption of alcohol (e.g., 5 units or greater) in combination with ADCIRCA can increase the potential for orthostatic signs and symptoms, including increase in heart rate, decrease in standing blood pressure, dizziness, and headache. Tadalafil (10 mg or 20 mg) did not affect alcohol plasma concentrations and alcohol did not affect tadalafil plasma concentrations. Potential for Other Drugs to Affect ADCIRCA: Ritonavir — Ritonavir initially inhibits and later induces CYP3A, the enzyme involved in the metabolism of tadalafil. At steady state of ritonavir (about 1 week), the exposure to tadalafil is similar as in the absence of ritonavir.
Other Potent Inhibitors of CYP3A — Tadalafil is metabolized predominantly by CYP3A in the liver. In patients taking potent inhibitors of CYP3A such as ketoconazole, and itraconazole, avoid use of ADCIRCA. Potent Inducers of CYP3A — For patients chronically taking potent inducers of CYP3A, such as rifampin, avoid use of ADCIRCA. Potential for ADCIRCA to Affect Other Drugs: Cytochrome P450 Substrates — Tadalafil is not expected to cause clinically significant inhibition or induction of the clearance of drugs metabolized by cytochrome P450 (CYP) isoforms (e.g., theophylline, warfarin, midazolam, lovastatin, bosentan). Aspirin — Tadalafil (10 mg and 20 mg once daily) does not potentiate the increase in bleeding time caused by aspirin. P-glycoprotein (e.g., digoxin) — Coadministration of tadalafil (40 mg once daily) for 10 days did not significantly alter digoxin pharmacokinetics in healthy subjects.USE IN SPECIFIC POPULATIONS Pregnancy: Pregnancy Category B — Animal reproduction studies in rats and mice revealed no evidence of fetal harm. There are, however, no adequate and well-controlled studies of tadalafil in pregnant women. Because animal reproduction studies are not always predictive of human response, tadalafil should be used during pregnancy only if clearly needed. Non–teratogenic effects — Animal reproduction studies showed no evidence of teratogenicity, embryotoxicity, or fetotoxicity when tadalafil was given to pregnant rats or mice at unbound tadalafil exposures up to 7 times the maximum recommended human dose (MRHD) of 40 mg/day dur ing organogenesis. In one of two perinatal/postnatal developmental studies in rats, postnatal pup survival decreased following maternal exposure to unbound tadalafil concentrations greater than 5 times the MRHD based on AUC. Signs of maternal toxicity occurred at doses greater than 8 times the MRHD based on AUC. Surviving offspring had normal development and reproductive performance. Nursing Mothers: It is not known whether tadalafil is excreted into human milk. While tadalafil or some metabolite of tadalafil was excreted into rat milk, drug levels in animal breast
Clearing the Fog: PH-Associated Diseases
If your patients are juggling multiple conditions associated with pulmonary hypertension, you know that managing their care can be overwhelming for them and their families.
Now there’s some clarity: In PHA’s new video series, your patients can hear stories from fellow PH patients living with associated diseases and the healthcare professionals who treat them.
Learn more at:www.PHAssociation.org/PHPlus
Scleroderma and PH is not my identity ... If your sickness
becomes your identity, then you don’t have a shot. You have to surround yourself with people
who do not make you feel sick.

News to Use
Help Your Patients andCaregivers Connect to SupportPHA offers monthly telephone supportgroups for patients, caregivers, and par-ents. We make it easy for you to share thisresource with your patients: you can signup for email alerts, download a postcardas a PDF, or request printed postcards foryour clinic at www.PHAssociation.org/TelephonePostcards. Your patients canalso find a support group in their commu-nity by going to www.PHAssociation.org/FindaSupportGroup.
New Resources on PVD andRight Ventricular DysfunctionSessions from the September 2012 Key-stone Symposium, titled “Pulmonary Vas-cular Disease and Right Ventricular Dys-function: Current Concepts and FutureTherapies,” are now on PHA Online Uni-versity. Organized by Drs. Georg Hans-mann of Hannover Medical School, Ste-phen L. Archer of Queen’s University, andMargaret R. MacLean of the University ofGlasgow, this conference gathered basicand clinical researchers in pulmonary vas-cular disease and right ventricular dysfunc-tion. Recordings cover topics such as cellphenotype and function in PAH, right ven-tricle and pulmonary circulation in PAH,right ventricle failure in PAH, and more.The recordings are at www.PHAOnlineUniv.org/KeystoneSymposium.
Global PHCR MembershipsTo increase global membership in PH Cli-nicians and Researchers (PHCR) and fos-ter the sharing of ideas around the world,PHA is offering free first-year member-ship for non-US physicians, researchers,residents, and fellows interested in PH.Benefits of PHCR membership includecase-based learning opportunities by topPH specialists, access to an email group ofa growing number of PHCR members,inclusion in PHA’s Find a Doctor Direc-tory, and more. For medical professionalsin countries that have a gross nationalincome per capita of less than $5000USD, PHCR memberships may be re-newed at no cost each year. Learn more atwww.PHAssociation.org/PHCR.
PHA Launches Major Campaignfor Early DiagnosisLynn Brown, MD, campaign chairImages of zebras and their stripes havebeen cropping up more frequently amongprofessionals who treat pulmonary hyper-tension (PH). They’ve appeared in thisand other issues of Advances, for instance,and on buttons given out at medical meet-ings. Expect the zebra to proliferate indiverse primary and specialty care circles,as well, as PHA carries out its new zebra-themed Sometimes it’s PH campaign forearly diagnosis in the US and globalhealth communities.
Physicians on PHA’s Board of Trusteessuggested the zebra emblem to reverse thelesson young diagnosticians receive intraining: “When you hear hoofbeats, thinkhorses not zebras.” The campaign’s centralmessage is: “Sometimes dyspnea, chestpain, and other widespread symptoms maylead you to conclude it’s asthma, chronicobstructive pulmonary disease (COPD),obesity, or lack of fitness. But sometimesit’s not. Sometimes it’s PH.”
Sometimes it’s PH responds to a call toaction from a November 2011 white pa-per, Pulmonary Arterial Hypertension:Recommendations for Improving PatientOutcomes, written under PHA’s leader-ship by a group of PH experts fromaround the globe. Data cited included afinding from the REVEAL Registry thatthe mean duration between symptom on-set and confirmed diagnosis by right heartcatheterization is 2.8 years and hasscarcely improved in more than 2 de-cades. The paper concluded that delays indiagnosis of PH and referral to specializedcare are now the main barriers to better
patient outcomes. Optimal care, noted inthe paper, is collaboration between pri-mary and specialty care providers.
PHA’s 5-year campaign will teachhealth care professionals to consider a PHdiagnosis when symptoms warrant and toteam up earlier with specialized PH phy-sicians who can confirm diagnosis andoffer a wider range of treatment, clinicaltrials, and patient support networks. Thecampaign will educate medical profes-sionals by publishing articles in profes-sional journals, placing news stories, de-veloping new forums for educating aboutearly diagnosis and referral, exhibitingand speaking at medical and allied healthmeetings, and joining with key profes-sional and advocacy organizations on ed-ucation and communication initiativesthat reach their members.
At this early stage, the campaign’s ac-tivities have included the creation of aWeb site devoted to the campaign, www.SometimesItsPH.org, the release of a 60-second video spot accessible through thatsite, and outreach to health care media.We have formed leadership committeescomposed of PH physicians and alliedhealth professionals. Each committee willguide activities in 1 of the 3 campaignapproaches: educating, communicatingwith health professionals, and formingcollaborative relationships with key pro-fessional associations.
This column will appear in future is-sues of Advances to provide updates onSometimes it’s PH. Keep an eye on thisbold and important new PHA initiative.
212 Advances in Pulmonary Hypertension

asthma
sleep apneaCOPD
obesity
When learning to diagnose, doctors are taught that hoof beats suggest horses, not zebras. But a rare disease like pulmonary hypertension is like a zebra among horses.
Those expecting common diseases often overlook the zebras. As a result, many PH patients have been misdiagnosed repeatedly before getting the treatment they need. Even worse, almost three-quarters of patients have advanced PH by the time they are diagnosed.
The Pulmonary Hypertension Association is taking unprecedented action to change that. Our Sometimes it’s PH campaign promotes early diagnosis among primary and specialty care providers. Join us.
��������������������� �������������������������������������������
www.SometimesItsPH.org

To order additional copies, call or contact PHA at 1-866-474-4742 or www.PHAssociation.org. All issues of Advances in Pulmonary Hypertension are also available online at www.PHAOnlineUniv.org/Journal
Advances in Pulmonary HypertensionPulmonary Hypertension Association801 Roeder Road, Suite Silver Spring, MD 20910-4496
NON-PROFIT ORG.US POSTAGEPAIDLancaster, PAPermit #161
Jointly SponsoredMentored Clinical Scientist Development Award (K08) &
Mentored Patient-Oriented Research Career Development Award (K23)
MECHANISM:Awards in response to the program announcement will use the National Institutes of Health (NIH) K08 or the K23 mechanism.
FUNDING:*The award will be funded by PHA and NHLBI and the K08 and/or the K23 will be awarded in 201 .
PURPOSE: K23• To support career development of investigators who have
made a commitment to focus their research endeavors on patient-oriented research.
• To support a 3 to 5 year period of supervised study and research for clinically trained professionals who have the potential to develop into productive, clinical investigators focusing on patient-oriented research in pulmonary hyper-tension.
• To support patient-oriented research, which is defined as research conducted with human subjects (or on material ofhuman origin, such as tissues, specimens, and cognitive phenomena) for which an investigator directly interacts withhuman subjects.
• To support areas of research that include: 1) mechanisms ofhuman disease; 2) therapeutic interventions; 3) clinical trials;and 4) development of new technologies.
PURPOSE: K08• To support the development of outstanding clinician research
scientists in the area of pulmonary hypertension.• To provide specialized study for clinically trained professionals
who are committed to a career in research in pulmonary hypertension and have the potential to develop into independentinvestigators.
• To support a 3 to 5 year period of supervised research experi-ence that integrates didactic studies with laboratory or clinicallybased research.
• To support research that has both intrinsic research importanceand merit as a vehicle for learning the methodology, theories,and conceptualizations necessary for a well-trained independentresearcher.
Pulmonary HypertensionAssociation (PHA)
National Heart, Lung, andBlood Institute (NHLBI)
Program Announcement:
FOR MORE INFORMATION:Visit: www.PHAssociation.org/MedicalProfessionals/Research
* Restrictions apply. Please see complete announcement at the website listed above.
1000
3
New Application Deadline: June 12, 2013New Application Deadline: October 12, 2013
Resubmission Deadline: July 12, 2013Resubmission Deadline: November 12, 2013

I wou
ld li
ke to
rece
ive
a co
mpl
imen
tary
cop
y of
the
Pulm
onar
y H
yper
tens
ion:
Case
s, C
ontr
over
sies
and
Con
undr
ums
CD-R
OM
.
I w
ould
like
to
rece
ive
a co
mpl
imen
tary
cop
y of
the
2010 P
ulm
onar
y H
yper
tens
ion
Sci
entif
ic S
essi
ons
DVD
.
I w
ould
like
to
rece
ive
a qu
arte
rly, c
ompl
imen
tary
cop
y of
Adv
ance
s in
Pul
mon
ary
Hyp
erte
nsio
n.
Nam
eTi
tle
Aff
iliat
ion
Add
ress
City
Sta
teZI
P
Pho
ne
E-m
ail ad
dres
s
Win
ter 2
013
Ho
st a
Fre
e P
HA
On
-Dem
and
Eve
nt
at Y
ou
r In
stit
uti
on
Brin
g fr
ee P
AH
med
ical
edu
catio
n to
you
r co
mm
unity
. PH
A’s
On-
Dem
and
initi
ativ
e en
able
s m
edic
al p
rofe
ssio
nals
to c
hoos
ea
prog
ram
, top
ic, s
peak
er, f
orm
at, a
nd d
ate,
and
PH
A ta
kes
care
of t
he r
est.
Lear
n m
ore
abou
t the
On-
Dem
and
Pro
gram
: w
ww
.PH
Ass
ocia
tion.
org/
OnD
eman
d.
I w
ould
like
to
rece
ive
notif
icat
ion
whe
n th
e on
line
editi
on o
f Adv
ance
s in
Pul
mon
ary
Hyp
erte
nsio
n is
ava
ilabl
e.
I w
ould
like
to
rece
ive
notif
icat
ion
whe
n ne
w P
H w
ebin
ars
and
onlin
e co
urse
s ar
e of
fere
d.

NO
PO
STA
GE
NE
CE
SS
AR
Y IF
MA
ILE
D
IN T
HE
U
NIT
ED
STA
TES
PO
STA
GE
WIL
L B
E P
AID
BY
AD
DR
ES
SE
E
BU
SIN
ES
S R
EP
LY M
AIL
FIR
ST-C
LASS
MAI
L
PE
RM
IT N
O.
7387
SILV
ER S
PRIN
G,
MD
Hel
p P
atie
nts
Fin
d Y
ou
!
As
PH
A s
triv
es t
o be
tter
ser
ve o
ur c
onst
ituen
ts,
we
are
com
mitt
ed t
o m
akin
g su
re o
ur F
ind
a D
octo
r D
irec
tory
,lo
cate
d at
ww
w.P
HA
ssoc
iatio
n.or
g/F
indA
Doc
tor,
pro
vide
sth
e m
ost c
urre
nt in
form
atio
n fo
r pat
ient
s. T
he F
ind
a D
octo
rD
irect
ory
is P
HA
’s p
rem
ier
reso
urce
for
patie
nts
seek
ing
PH
-tre
atin
g ph
ysic
ians
, and
bei
ng li
sted
in th
e di
rect
ory
is
a be
nefit
ava
ilabl
e on
ly to
mem
bers
of P
H C
linic
ians
and
Res
earc
hers
(P
HC
R).
To
ensu
re y
our
listin
g is
com
plet
ean
d co
rrec
t, m
ake
sure
you
r on
line
prof
ile is
upd
ated
.C
urre
nt m
embe
rs:
To
upda
te y
our
listin
g in
the
Fin
d a
Doc
tor
Dire
ctor
y, p
leas
e vi
sit
ww
w.P
HA
ssoc
iatio
n.or
g/P
HC
R/P
rofil
eUpd
ate.
Laps
ed m
embe
rs:
To
rene
w y
our
mem
bers
hip,
ple
ase
visi
t ww
w.P
HA
ssoc
iatio
n.or
g/P
HC
R/R
enew
.
PU
LM
ON
AR
Y H
YP
ER
TE
NS
ION
AS
SO
CIA
TIO
N80
1 R
OE
DE
R R
OA
DS
UIT
E 1
000
SIL
VE
R S
PR
ING
, MD
209
10