considerations for protein crystallography (bt chapter 18) 1.growing crystals usually require 0.5mm...
TRANSCRIPT

Considerations for Protein Crystallography (BT Chapter 18)
1. Growing crystals
Usually require 0.5mm in shortest dimension, except if using Synchrotron radiation; Can be “twinned” (two or more crystals growing into each other)
Why X-rays?
The wavelength of radiationis comparable with the lengthof interatomic distances.

Considerations for Protein Crystallography
2. Collecting Diffraction Data
Synchrotron radiation has become routine for structuraldeterminations. The high intensity beam reduces boththe time to collect data and size of crystal needed.

Defining the unit cell of the crystal
Figures 18.6, 18.7 of BT discuss the calculation of the unit cell.
Bragg’s Law 2d(sin) =
is known, and is readily calculated in experimental set-up

Properties of diffracted beam
Each diffracted beam is defined by amplitude, wavelength, and phase.
Amplitude – measured by intensity of recorded spot
Wavelength – set by x-ray source
Phase – major problem in solving crystal structures

Solving the phase
Max Perutz and John Kendrew pioneered molecular isomorphicreplacement (MIR), which utilizes heavy atom derivatives of proteincrystals to introduce new diffraction patterns. Typically use metalssuch as mercury or platinum.
Multiwavelength Anomalous Diffraction (MAD) is extremelypopular but requires synchrotron radiation in addition to a heavymetal derivative. The intensity differences obtained in thediffraction pattern using x-rays of different wavelengths can be usedin a way similar to MIR. The sensitivity permitted by synchrotronradiation allows the use of lighter elements. The most tractable anduseful method has proven to be incorporation of selenomethionineinto expressed proteins.

Structural Genomic Consortia and HTS structure determination
http://www.rcsb.org/pdb/strucgen.html#Worldwide
http://www.stromix.com/

How can I tell if a crystal structure is insightful?
Crystal structures are reported at a variety of resolutions5 Angstroms – can make out secondary structures, but
not individual groups of atoms3 Angstroms – can trace alpha carbon backbone, but
not sidechains1.5 Angstroms – Good resolution
R Factor – Each crystal structure will report this value, which corresponds to error associated with the model; In general,0.2 or lower indicates a well-determined protein structure.
B Factor – Temperature factor, should be 20 or less for good structures. Surface loops or terminal regions often havehigh B values due to flexibility, leading to disorder

Structure from Nuclear Magnetic Resonance
Sample is placed in a strong magnetic field and exposedto radiofrequency radiation.
Energy absorption is characteristic of the nuclei (H1 or C13),and its chemical environment.
Allows structure determination under solution conditions
Some limitation in size, but larger magnets helping
http://www.nmrfam.wisc.edu/

The Future of Cell Biology??
http://www.pnas.org/cgi/reprint/97/26/14245.pdf
Toward detecting and identifying macromolecules
in a cellular context: Template matching applied
to electron tomograms
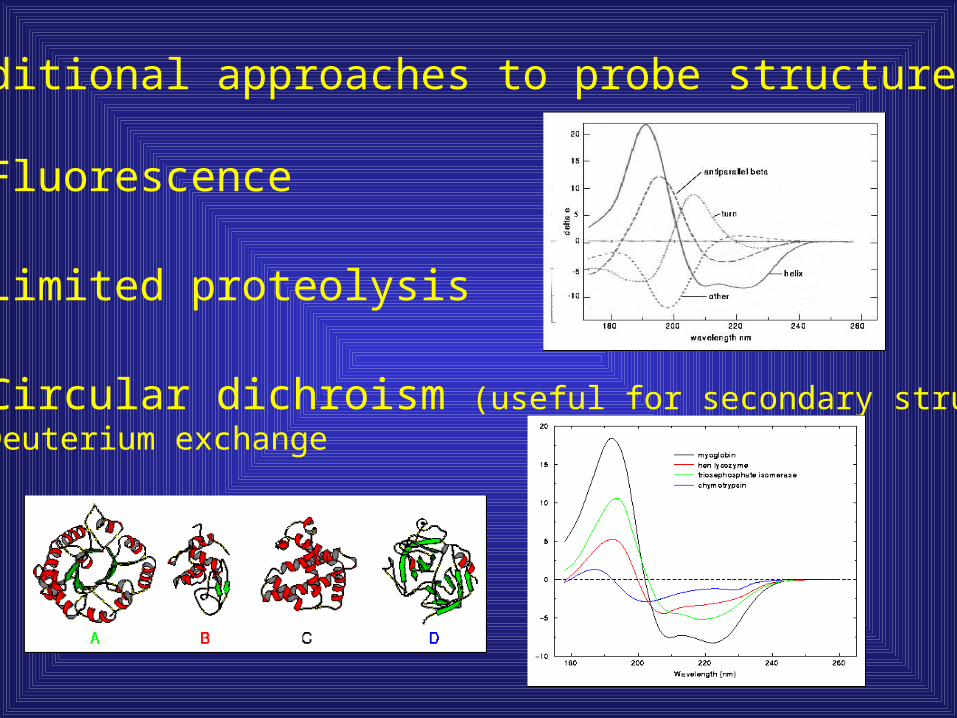
Additional approaches to probe structure:
1) Fluorescence
2) Limited proteolysis
3) Circular dichroism (useful for secondary structure)4) Deuterium exchange

Domains are revealed in protein structures
• Characterized by secondary structure content– All – All / +

Light-harvesting complex was an example of all

Diversity in structures
• Four helix bundle
• Seven membrane spanning regions in proteins involved in signal reception (ie. bacteriorhodopsin)
• Globin fold
• Different folds can affect #residues/turn

Packing helices

Sequence/structure
• All a proteins begin to reveal sequence/structure relationship
• Coiled-coil proteins exhibit periodicity every seventh residue (heptad repeat); also seen in formation of dimers (GCN4)
• Observe hydrophobic moments in membrane proteins
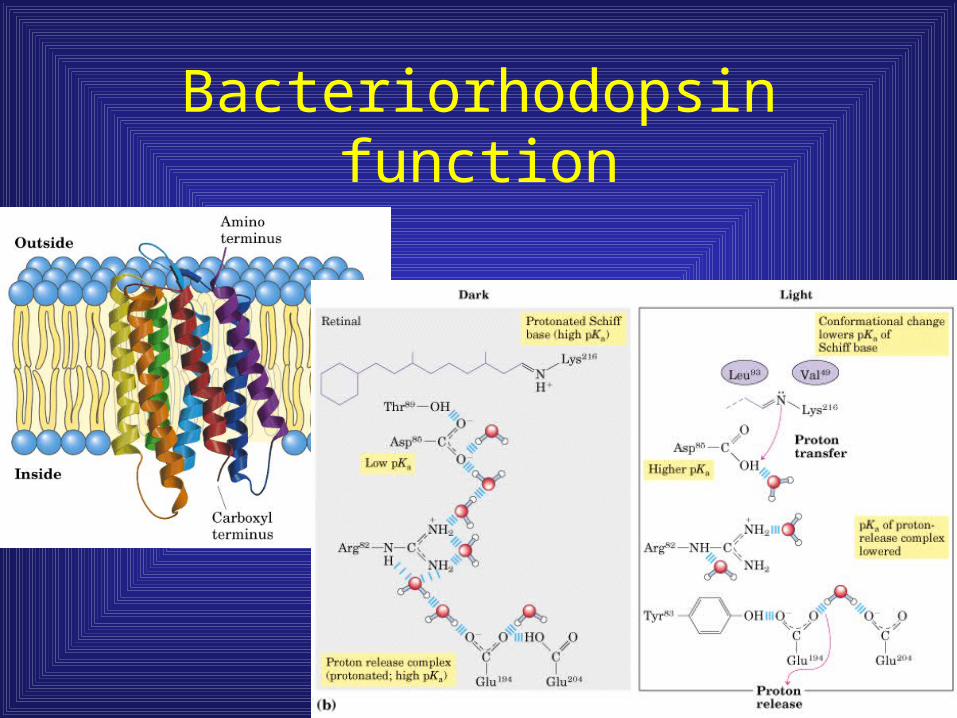
Bacteriorhodopsin function
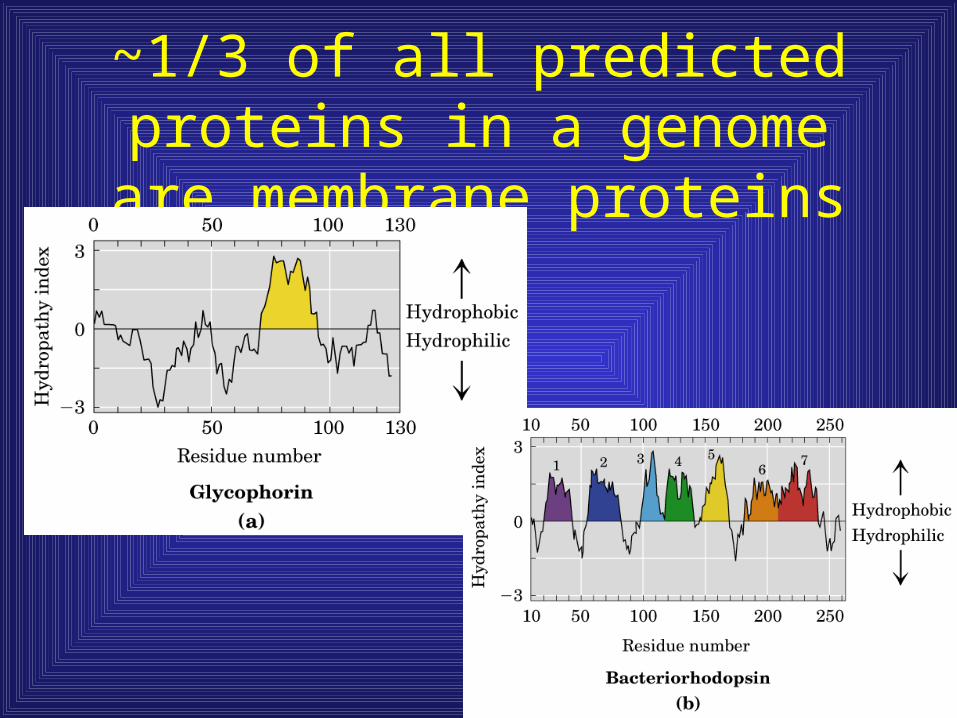
~1/3 of all predicted proteins in a genome are membrane proteins
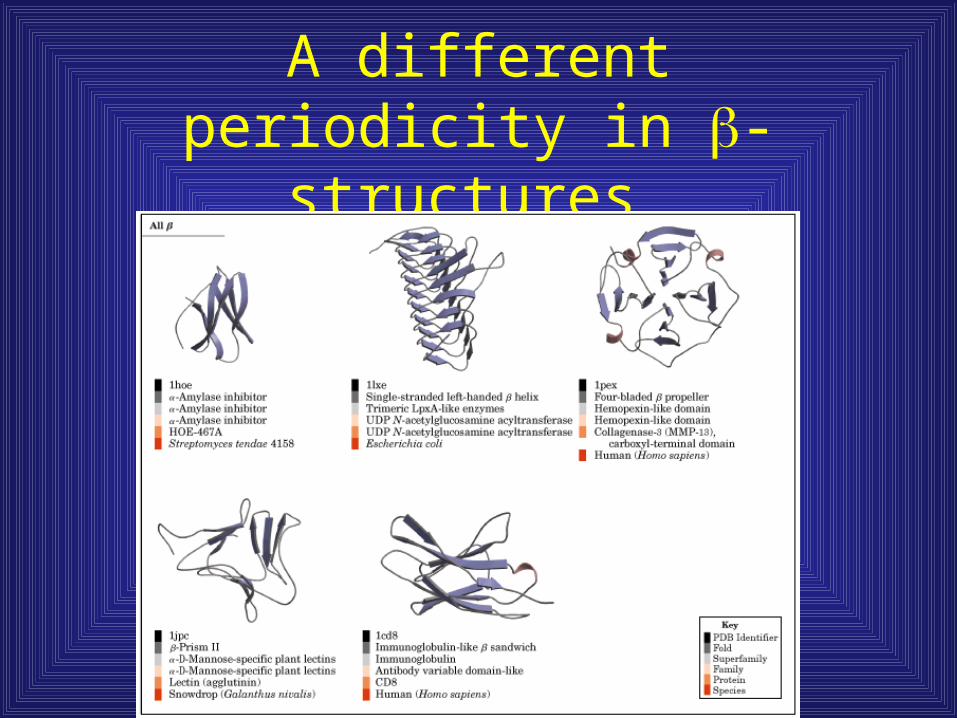
A different periodicity in -structures

Common structures found in structures
• Barrels
• Propellers
• Greek key
• Jelly roll (Contains one Greek key)
• Helix

Barrels – anti-parallel sheets

Anti-parallel structures exhibit every other amino acid periodicity

Propellers
• Variable number of propeller blades
http://info.bio.cmu.edu/courses/03231/ProtStruc/b-props.htm

Propeller blade
• Ninety degree twist
between first and fourth
strand

Quaternary structure of neuraminidase
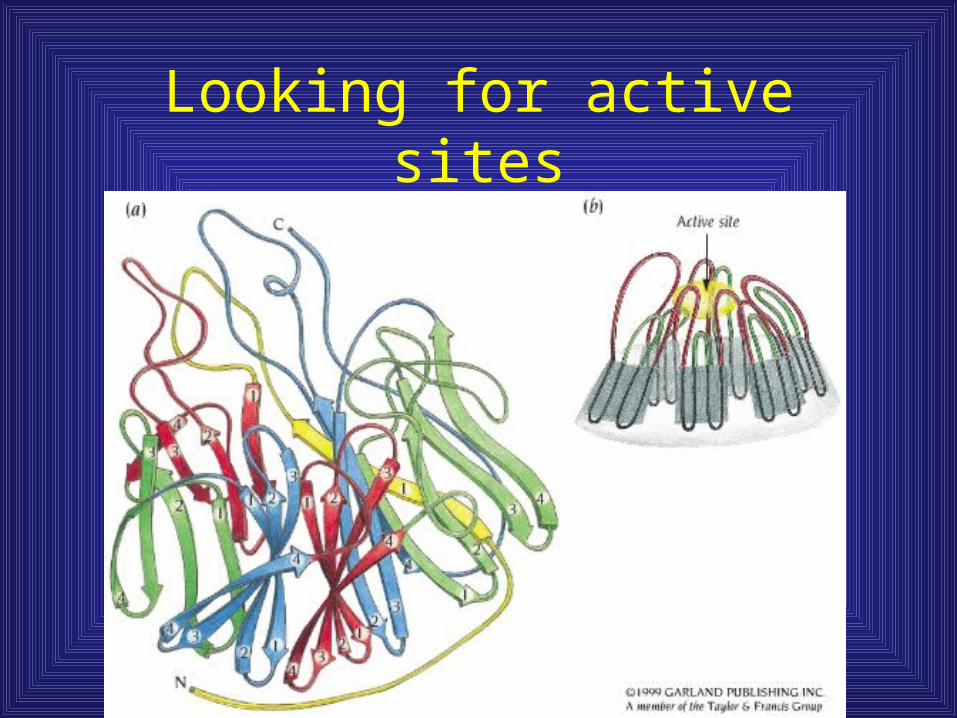
Looking for active sites

Greek key barrels
• Only n + 3 folds are observed
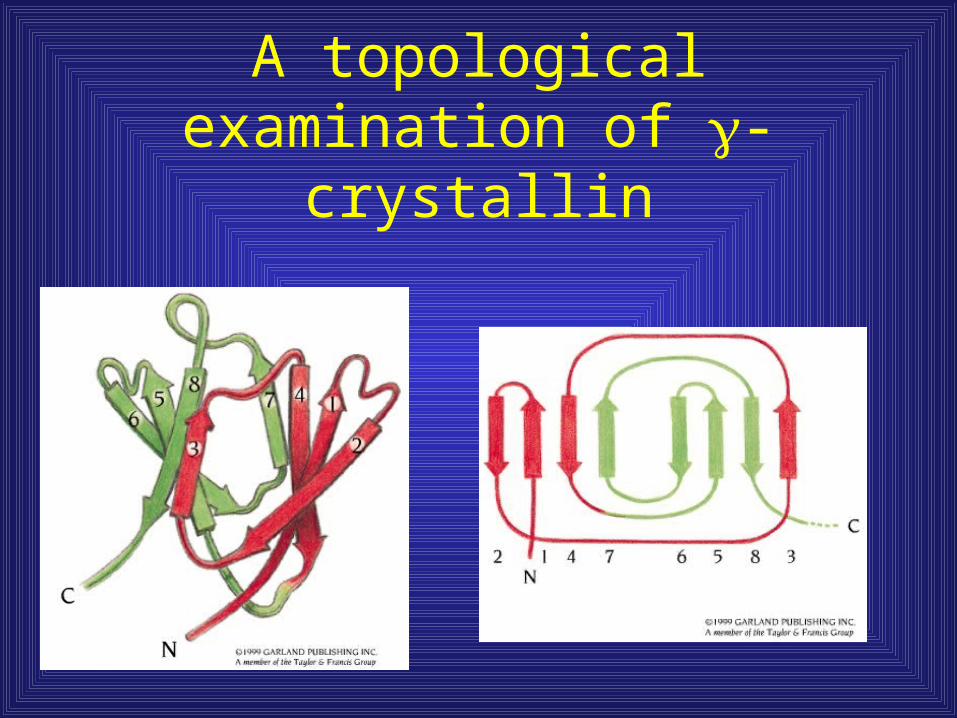
A topological examination of -crystallin

-crystallin has two domains with identical topology
• Protein evolution –
motif duplication and
fusion

Jelly roll motif
• Connections
made over the end
of the barrel
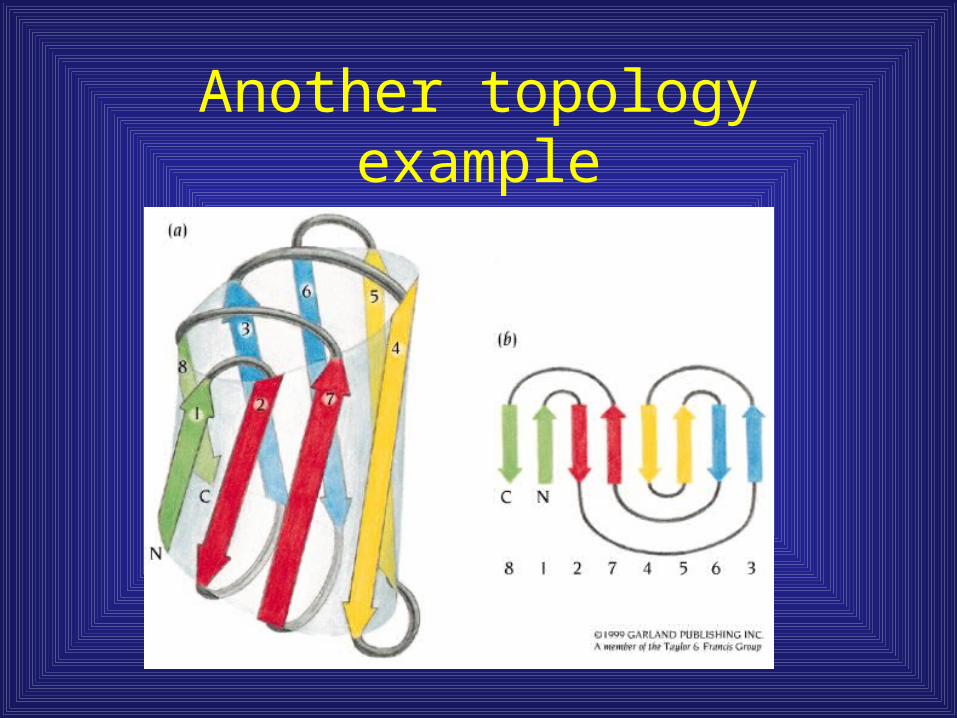
Another topology example
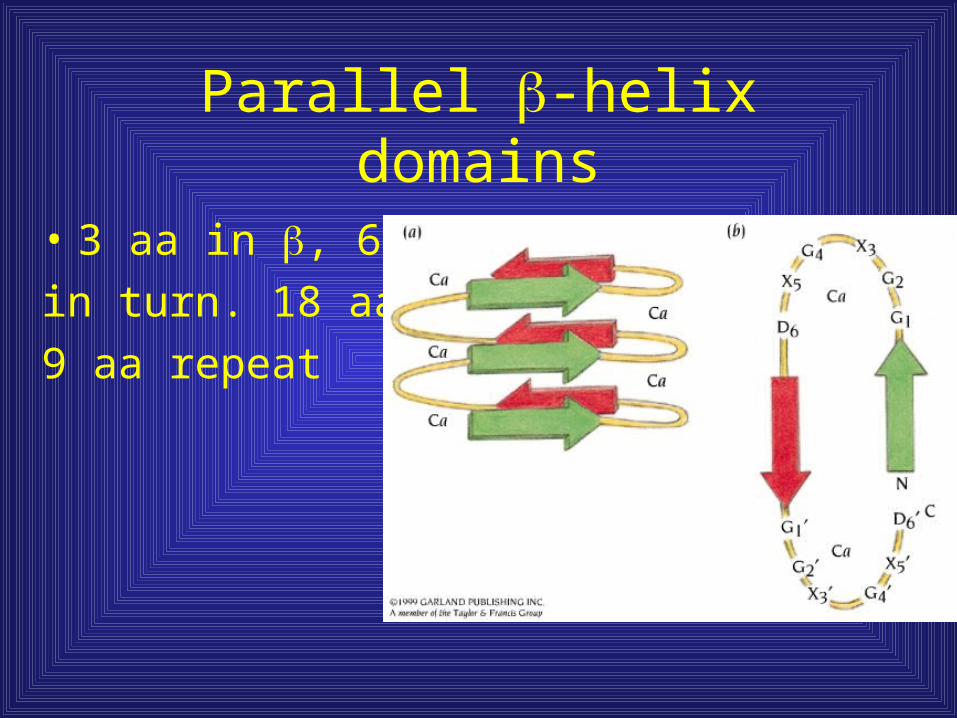
Parallel -helix domains
• 3 aa in , 6 aa
in turn. 18 aa/motif
9 aa repeat
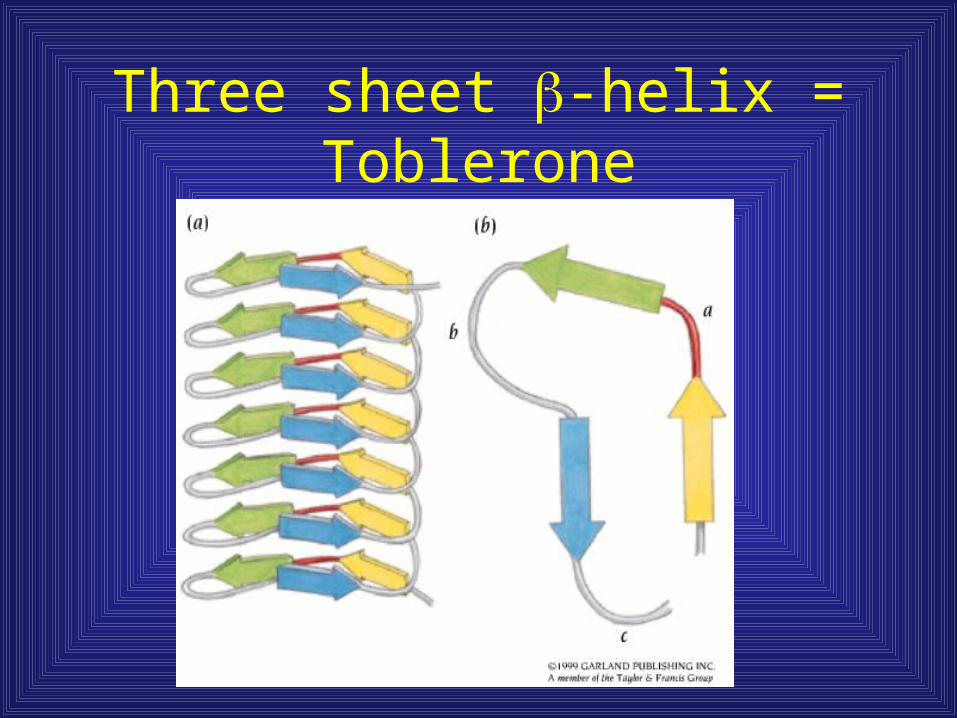
Three sheet -helix = Toblerone

Protein structures containing and
• Distinction between / and + / - Mainly parallel beta sheets (beta-alpha-
beta units) + - Mainly antiparallel beta sheets
(segregated alpha and beta regions)
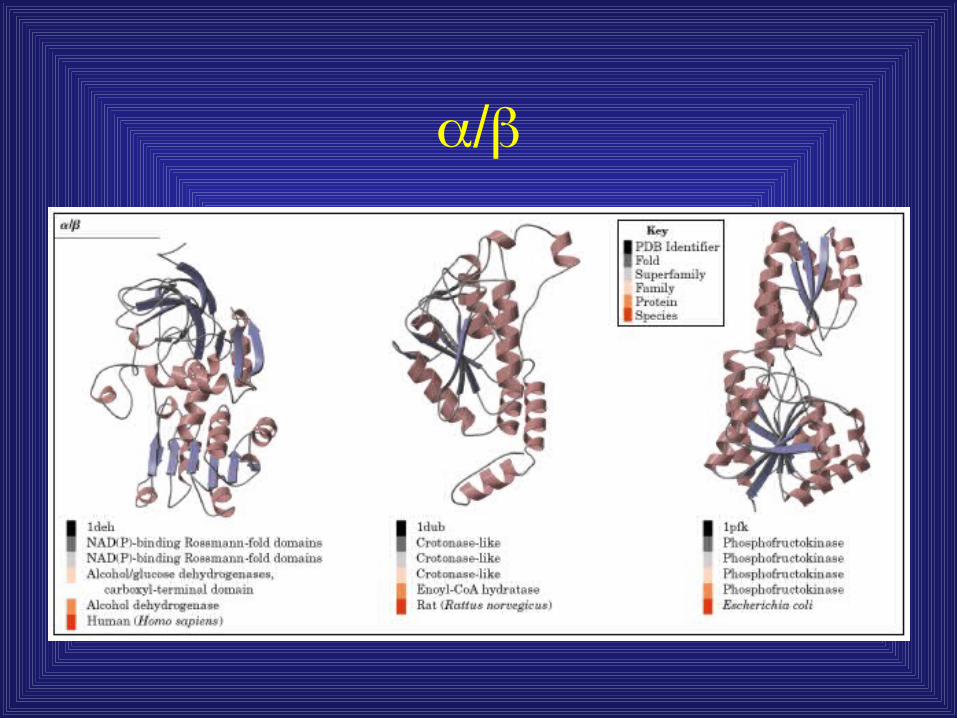

Interspersed and

Generally, a tight hydrophobic core found in barrels

How many folds are there?
To date we know ~8000 protein structures
Within this dataset, 450 folds are recognized
Proteins have a common fold if they have the samemajor secondary structures in the same arrangementand with the same topological connections.
http://scop.mrc-lmb.cam.ac.uk/scop/

How many non-folds are there?
• http://www.scripps.edu/news/press/013102.html
• 30-40% of human genome encodes for “unstructured” native proteins

Think about Domains!