construction and characterization of a recombinant adenovirus vector carrying the human...
TRANSCRIPT

BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS 229, 778–787 (1996)ARTICLE NO. 1880
Construction and Characterization of a Recombinant Adenovirus VectorCarrying the Human Preproinsulin Gene under the Control
of the Metallothionein Gene Promoter
Hiroaki Yajima,* Akinobu Kosukegawa,* M. Mainul Hoque,* Tsukasa Shimojima,*Ken-ichiro Ishizu,* Makoto Takayama,† Yutaka Sasaki,‡ Hideto Sakai,§
Masaki Otsuka,§ Sadaki Inokuchi,Ø and Hiroshi Handa*,1
*Faculty of Bioscience and Biotechnology, Tokyo Institute of Technology, 4259, Nagatsuta-cho, Midori-ku,Yokohama 226, Japan; †Yamanouchi Pharmaceutial Co. Ltd., 2-3-1 Honcho, Nihonbashi, Chuou-ku
Tokyo 103, Japan; ‡The First Department of Medicine, Osaka University School of Medicine,2-2, Yamadaoka, Suita, Osaka 565, Japan; and §Division of Nephrology and Metabolism,Department of Internal Medicine and ØDepartment of Critical and Emergency Medicine,Tokai University School of Medicine, Bohseidai, Isehara City, Kanagawa 259-11, Japan
Received October 30, 1996
A new adenovirus vector carrying human-preproinsulin (h-PPI) genomic DNA, which was placed underthe control of the mouse metallothionein gene promoter, was constructed. In the recombinant virus-infectedcells, h-PPI gene expression increased as a function of ZnSO4 concentration. Reversed-phase high-perfor-mance liquid chromatography analysis revealed that the recombinant adenovirus-infected cells secretedimmature insulin containing proinsulin and incorrectly processed insulin. Tyrosyl phosphorylation of humaninsulin receptor substrate 1 occurred when HepG2 cells were treated with the cultured medium, indicatingthat the h-PPI gene product was functionally active in vitro. We also examined the biological activity ofthe product using diabetic severe combined immunodeficient mice and confirmed that the h-PPI geneproduct reduced the blood glucose concentration in vivo. This study suggests that the adenovirus vectorcan be used to express a foreign gene under the control of an external promoter in various human cells.q 1996 Academic Press
The adenovirus vector is one of the most effective vectors for transferring a heterologousgene into several target cells. Adenovirus is a non-enveloped DNA virus. Nearly 50 serotypesof adenovirus have been reported, but only type 2 and type 5 have been generally utilized ingene therapy studies (1). The adenovirus genome is 36 kilobase (kb) long and is composed ofa linear double-stranded DNA containing four early regions (E1-E4). Recombinant replication-deficient adenovirus vectors in which the E1 region is deleted have been called first generationadenovirus vectors. For the clinical use of the first generation of adenovirus vector, the E1region was replaced with several therapeutic genes (2, 3, 4). The advantages of the adenovirusvector include (i) a large host range (dividing and non-dividing cells), (ii) a low pathogenicityin human, (iii) the possibility to obtain high titer of replication deficient recombinant virus.And because of the low efficiency of usual gene transfer method, such as DNA transfection,it is difficult to analyze transferred gene expression in human primary culture cells. Thus, asfor therapeutic purpose, the adenovirus vector is also appropriate for studying somatic cellgene therapy.
Insulin-dependent diabetes mellitus (IDDM) is caused by the lack of insulin. IDDM resultsin hyperglycemia and death from ketoacidosis (5). In general, the treatment for this diseaseentails monitoring blood glucose concentrations and when necessary, the administration of
1 Corresponding author. Fax: 81-45-923-0380. E-mail: [email protected].
0006-291X/96 $18.00Copyright q 1996 by Academic PressAll rights of reproduction in any form reserved.
778
AID BBRC 5842 / 6914$$$301 12-06-96 06:19:14 bbrca

Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
periodic injections of insulin (6). As an attractive alternative to these methods, somatic cellgene therapy has been considered and some attempts have been made to develop gene therapyfor IDDM. These methods depend on the transformation of cells from a diabetic patient withDNA expressing human or mouse preproinsulin (PPI) and introduction of the insulin or proinsu-lin expressing cells into the desired anatomical location (7). Several methods have been em-ployed to introduce PPI gene DNA into cultured cells and all basically rely on DNA transfectionor the use of a retrovirus vector (8, 9). Therefore, increasing the efficiency of the PPI genetransfer contributes to advance this strategy.
The capacity to regulate transferred therapeutic gene expression is also an important aspectof gene therapy. It has been reported that regulation of PPI gene expression can be achievedin vitro with cyclic AMP or isomethylbutylxanthine when the PPI gene is placed under thecontrol of the metallothionein gene promoter (9, 5). The promoters of metallothionein genesare active for gene expression in a wide variety of animal tissues and cell lines (11, 12) andare controlled by some heavy metals (11, 13), glucocorticoids (14) and certain hormones (15).Sensitivity of this promoter to heavy metal ion concentration has been well studied and heavymetal concentration has been used to regulate metallothionein promoter controlled PPI geneexpression (9, 16).
In this report, we constructed a replication-deficient adenovirus vector designated rAdMTinsfor analysis of prohormone processing and behavior in several cells. The vector contains h-PPI genomic DNA joined with the mouse metallothionein gene promoter in the E1-deletedregion. We show that the major product of the h-PPI gene is proinsulin. The expression ofproinsulin could be induced by addition of ZnSO4 to rAdMTins-infected cell culture mediumand the amount of the product could be regulated by the concentration of ZnSO4. The biologicalactivity of the h-PPI gene product in mice was also examined.
MATERIALS AND METHODSCells and media. For the propagation and titration of adenoviruses, 293 cells (17) were cultured in Dulbecco’s
modified essential medium (DMEM) containing 10% fetal bovine serum (FBS). DMEM was used for the infectionof 293 cell with rAdMTins. HeLa cells (18) were cultured in Eagle’s minimal essential medium (MEM) containing10% FBS. MEM was also used for infection of HeLa cells with rAdMTins. To assay the biological activity of theh-PPI products, HepG2 cells (19) were cultured in MEM containing 10% FBS and non-essential amino acids.
Construction of recombinant adenovirus. The h-PPI gene genomic DNA was amplified by the polymerase chainreaction (PCR). The primers were designed so as to introduce both NcoI and BamHI sites into the 5*-terminus and3*-terminus of the amplified h-PPI gene DNA, respectively. The h-PPI gene DNA was digested with NcoI, and itscohesive termini were filled in with Klenow fragment and then digested with BamHI. The h-PPI gene DNA wasfractionated on an agarose gel, excised and recovered by the electroelution method (20). Mouse metallothionein(mMT) expression vector, mMT-1 (21), was digested with BglII, and its termini were filled in with Klenow fragmentand then digested with BamHI. The 4.7kb fragment of mMT-1 was recovered as described above and ligated withthe BamHI-NcoI fragment of the h-PPI gene DNA to produce a recombinant plasmid, pMTins. This plasmid carrieda chimeric gene termed MTins. pMTins was transfected into HeLa cells by CaPO4 coprecipitation method (22), andexpression of the MTins gene was examined as follows. The plasmid DNA was digested with EcoRI and BamHI.Both termini of the digested DNA were filled in with Klenow fragment, and the MTins gene DNA fragment wasseparated by agarose gel electrophoresis and recovered by the method described above. The MTins gene DNA wasligated with synthesized NheI-HindIII-NheI linker (5*-GCGCTAGCAAGCTTGCTAGCGC-3*) and then digested withHindIII and cloned into the HindIII site of pUC119. E. coli DH5a was the host strain for all the bacterial plasmidsdescribed here. Plasmid DNA was prepared by standard protocols (23).
Replication-deficient adenovirus DE1/X (24), a derivative of adenovirus type 5, was propagated in 293 cells. ADNA-terminal protein complex (DNA-TPC) of DE1/X was prepared from DE1/X-infected 293 cells as previouslydescribed (25). The DE1/X DNA-TPC was digested with XbaI and dephosphorylated with calf intestine alkalinephosphatase (CIAP) to insert MTins gene into the XbaI site. The XbaI-digested DE1/X DNA-TPC (10mg) was ligatedwith 1mg of the MTins gene (NheI) fragment by T4 DNA ligase. To cleave self-ligated DE1/X DNA molecules, theligation mixture was digested again with XbaI. Nick column (Pharmacia) was used for changing reaction buffersaccording to the manufacturer’s recommendation.
The ligated mixture of DNA-TPC was then transfected into 293 cell by the CaPO4 coprecipitation method. After3 days, the transfected cells and the cultured medium were frozen and thawed 4 times to release the recombinant
779
AID BBRC 5842 / 6914$$$302 12-06-96 06:19:14 bbrca

Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
adenovirus. Plaque forming assay (26) was performed to isolate the desired recombinant virus. Each isolated plaquewas propagated in 293 cells. The viral DNA was prepared by the method of Hirt (27), and recombinant virusescontaining the MTins gene were identified by PCR using primers specific for the metallothionein gene and the flankingregion of the adenovirus.
Expression of the MTins gene of rAdMTins. 293 and HeLa cells were used to analyze the expression of the MTinsgene under rAdMTins replicative and non-replicative conditions, respectively. Both cells were cultured as describedabove. The cells were grown until subconfluent (approximately 51106 cells per 100mm tissue culture dish) and thecultured medium was changed with the indicated medium containing 2% FBS. The cells were infected with rAdMTinsat 5 or 25 plaque forming units (PFU) per cells, and incubated for 48 hours. Six hours after infection, ZnSO4 wasadded to the medium to yield the indicated final concentration. After brief centrifugation, production of insulin in thecultured media of infected cells was examined by radioimmunoassay (RIA) using Phadeseph Insulin RIA (Pharmacia)according to the manufacturer’s recommendation. The supernatants were also used for reversed-phase high-performanceliquid chromatography (RP-HPLC) analysis of the MTins gene products.
Analysis of insulin gene products. RP-HPLC analysis was performed by using SMART system (Pharmacia) andthe eluent buffer was 125mM ammonium sulfate (pH 4.0). After samples were loaded onto a Sephacil C18 column(5mm SC 2.1/10, Pharmacia), elution was performed at room temperature with the following two linear acetonitrile(ACN) gradients at 250 ml/min. After elution with the buffer containing 10% ACN for 17min, the first gradient (10%to 20% ACN for 8 min) and the second (20% to 40% ACN for 40 min) were subsequently applied to separateimmature insulin components. After separation, the column was washed by raising the concentration of ACN to 60%for 15 min. Fractions of 0.6 min were collected during elution with the second gradient (150 ml/fraction). Thesecollected fractions were lyophilized and resolved in 75ml of phosphate buffered saline (PBS) containing 1% bovineserum albumin. The samples were then subjected to RIA as described above.
Detection of activity of insulin gene products in vitro. HepG2 cells were grown to 106 cells in a 60mm tissueculture dish and washed twice with MEM containing non-essential amino acids. The cells were incubated overnightin the same medium and then the medium was discarded. To analyze the tyrosyl phosphorylation of human insulinreceptor substrate 1 (IRS-1), 500ml of the following stimulants were added to the HepG2 cells. The stimulants were10-fold-concentrated media from 293 cell and rAdMTins-infected 293 cell cultures. We prepared media from infected293 cell cultures in which MTins gene expression had been induced or not. As positive controls, MEM containing17.5mg of recombinant human insulin (Becton Dickinson) or 7mg of human proinsulin (Sigma) were also prepared.After 1 min stimulation, HepG2 cells were collected and suspended in Triton-lysis buffer containing 50mM Tris-Cl(pH 7.5), 1% Triton X-100, 2mM EGTA, 10mM EDTA, 100mM NaF, 1mM Na4P2O7, 2mM Na3VO4, 100 mg/mlphenylmethylsulfonyl fluoride, 1 mg/ml aprotinin, 1 mg/ml pepstatin A and 1mg/ml leupeptin (28). The suspensionwas then frozen and thawed 3 times. After centrifugation at 4,0001g for 15min at 47C, the protein concentration ofthe supernatant was measured by the Lowry method using the Dc protein assay (Bio-Rad). The lysates (600mg) werethen subjected to immunoprecipitation followed by western blot analysis. The lysates were reacted with 5mg of anti-rat IRS-1 rabbit polyclonal IgG (Upstate Biotechnology Incorporated) at 47C overnight. Then, 20ml of 50% (v/v)suspension of protein A sepharose beads (Pharmacia) was added and incubated at 47C for 3 hours. The beads werewashed 3 times with Triton-lysis buffer, then boiled in 21SDS-sample buffer containing 125mM Tris-Cl (pH6.8),20% glycerol, 0.5% SDS and 290mM b-mercaptoethanol to separate immunoprecipitates from the beads. The immuno-precipitates were subjected to 8% SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane(Immobilon, MILLIPORE). After blocking the membrane with Tris-buffered saline (TBS) containing 50mM Tris-Cl(pH 7.5) and 150mM NaCl supplemented with 0.1% Tween 20 and 5% skim milk, the membranes were incubatedwith anti-phosphotyrosine monoclonal antibody (PY20; Leinco Technologies Inc.). The bound antibody was detectedusing the ECL (enhanced chemiluminescence) detection kit for mouse antibodies (Amersham) according to themanufacturer’s recommendations.
Injection of insulin gene products into diabetic SCID mice. The concentrated medium from rAdMTins-infected 293cell and pMTins-transfected HeLa cell cultures were injected into diabetic severe combined immunodeficient (SCID)mice. To kill endogenous insulin-secreting islet b-cells and to induce a state of insulinopenic diabetes, CB-17 (scid/scid) mice were intraperitoneally injected with streptozotocin (STZ, 100mg/kg). The state of diabetes of STZ-treatedmice was determined by measuring blood glucose concentration. HeLa cells (21107cells) were transfected with 10mgof pMTins, and 293 cells (21107cells) were infected with rAdMTins at MOI 25. Transferred MTins gene expressionwas induced with 100mM ZnSO4 for 42 hours. Then, the media from the transfected HeLa cell cultures and infected293 cell cultures in which MTins gene expression had been induced was lyophilized and resuspended in 10% the initialvolume of distilled water and dialyzed against PBS. This concentrated medium (1ml) was injected intraperitoneally intothe diabetic SCID mice (nÅ6) and the blood glucose concentration of the injected mice was measured.
RESULTSConstruction of rAdMTins
To construct a recombinant adenovirus vector which expresses proinsulin under the controlof the mouse metallothionein gene promoter, the NheI-digested DNA fragment containing the
780
AID BBRC 5842 / 6914$$$302 12-06-96 06:19:14 bbrca

Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
FIG. 1. Construction of the recombinant adenovirus vector rAdMTins. (A) The DNA-TPC of DE1/X was usedfor the construct. The mouse metallothionein gene promoter region is indicated as MT and PPI represents humanpreproinsulin genomic DNA. After a second XbaI digestion, 293 cells were subjected to transfection with the DNA-TPC reaction mixture. (B) The rAdMTins construct. The genome of rAdMTins containing the MTins gene cassetteis shown. The exons and introns of human preproinsulin genomic DNA are indicated separately.
MTins gene was excised from a cloned plasmid and inserted into the XbaI site of DE1/XDNA-TPC. As digestion with NheI produces XbaI-compatible ends, the NheI-digested DNAfragment of the MTins gene can be inserted directly into the XbaI site of DE1/X. Since theligated NheI/XbaI sites cannot be recognized by XbaI, the ligated DNA mixture was digestedagain with XbaI to reduce self-ligated DE1/X DNA-TPC products (Fig. 1A). After a seconddigestion with XbaI, the ligated DNA-TPC was transfected into 293 cells by the CaPO4
coprecipitation method and the cells were allowed to propagate. The recombinant virus wasthen plaque purified using 293 cells. Ten single plaques were isolated. The insertion of theMTins gene and its orientation were analyzed by PCR. The PCR product was detected in threeclones. In one of these clones, the MTins gene was inserted into the XbaI site of DE1/X in adirection opposite to that of E1A (data not shown). This recombinant adenovirus was designatedrAdMTins (Fig. 1B).
Expression of the MTins Gene Introduced by rAdMTins into HeLa and 293 Cells
The expression of the MTins gene was analyzed by RIA following ZnSO4 treatment ofHeLa and 293 cells infected with rAdMTins. The expression in pMTins-transfected HeLa cells
781
AID BBRC 5842 / 6914$$$303 12-06-96 06:19:14 bbrca

Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
TABLE 1Expression of MTins Gene in HeLa and 293 Cells
Concentration of ZnSO4
in cultured medium (mM)
0 25 50 100
rAdMTins infection — a — 3.2 14.5pMTins transfection — — ND 2.4
Note. rAdMTins-mediated MTins gene transfer was moreefficient than the CaPO4 transfection method, and the amountof gene product depended on the concentration of ZnSO4.
a Not detected.
was also analyzed. The MTins gene products were detected in the media from both pMTins-transfected and rAdMTins-infected HeLa cell cultures after the treatment with ZnSO4. Theamount of the product depended on both the concentration of ZnSO4 and the multiplicity ofinfection (MOI). The level of expression of the MTins gene was quite low in pMTins-trans-fected HeLa cells compared to the 6-fold and 77-fold higher levels observed in rAdMTins-infected HeLa and 293 cells, respectively (Tables 1 and 2).
Characterization of MTins Gene ProductsThe anti-human insulin antibody used for RIA crossreacts with proinsulin (information from
the manufacturer). The production of mature insulin requires posttranslational processing. Thissuggests that the MTins gene products from 293 and HeLa cells may be immature insulin.We, therefore, performed RP-HPLC analysis to characterize the MTins gene products. TheRP-HPLC analysis of cultured media of rAdMTins-infected HeLa and 293 cells revealed thatproinsulin was the major MTins gene product along with immature forms of insulin (Fig. 2).In this study, the amount of MTins gene products was calculated according to the standardcurve obtained by RIA for human insulin. Although they were regarded as a immunoreactiveinsulin, they were not mature insulin.
Phosphorylation of IRS-1 by the MTins Gene Product in VitroWe next examined the level of tyrosyl phosphorylation of IRS-1 after incubation in the
presence of the MTins gene product. The lysates of HepG2 cells after treatment with the
TABLE 2
MOI(PFU/cell)
Infected cell 0 5 25
HeLa — a 6 14.5293 — 185 188
Note. The amount of the product synthesizedin 293 cells was 13-fold more than that in HeLacells. Data are indicated in mIU (internationalunit) of immunoreactive insulin per ml of solu-tion.
a The same as indicated in Table 1.
782
AID BBRC 5842 / 6914$$$303 12-06-96 06:19:14 bbrca
Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
FIG. 2. RP-HPLC analysis of the conditioned medium of rAdMTins-infected cell cultures, in which the expressionof the MTins gene was induced. The cultured media of infected 293 cell (l) and HeLa cell (s) were separated onRP-HPLC and each fraction was lyophilized and assayed for immunoreactive insulin. The amount of immunoreactiveinsulin is indicated in international unit (1IU: 42mg of mature insulin). Separation of human insulin (1) and proinsulin(/) is also indicated as a dotted line.
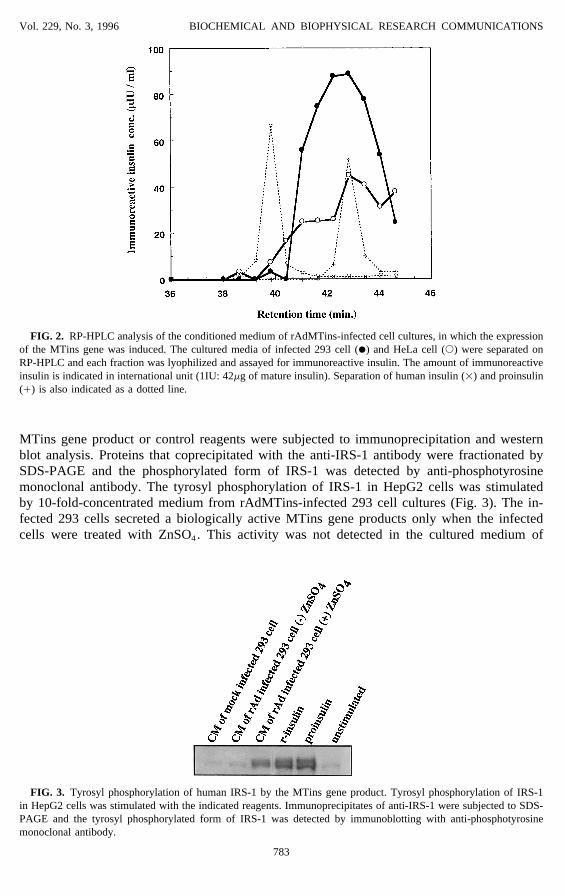
MTins gene product or control reagents were subjected to immunoprecipitation and westernblot analysis. Proteins that coprecipitated with the anti-IRS-1 antibody were fractionated bySDS-PAGE and the phosphorylated form of IRS-1 was detected by anti-phosphotyrosinemonoclonal antibody. The tyrosyl phosphorylation of IRS-1 in HepG2 cells was stimulatedby 10-fold-concentrated medium from rAdMTins-infected 293 cell cultures (Fig. 3). The in-fected 293 cells secreted a biologically active MTins gene products only when the infectedcells were treated with ZnSO4. This activity was not detected in the cultured medium of
FIG. 3. Tyrosyl phosphorylation of human IRS-1 by the MTins gene product. Tyrosyl phosphorylation of IRS-1in HepG2 cells was stimulated with the indicated reagents. Immunoprecipitates of anti-IRS-1 were subjected to SDS-PAGE and the tyrosyl phosphorylated form of IRS-1 was detected by immunoblotting with anti-phosphotyrosinemonoclonal antibody.
783
AID BBRC 5842 / 6914$$$303 12-06-96 06:19:14 bbrca
Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
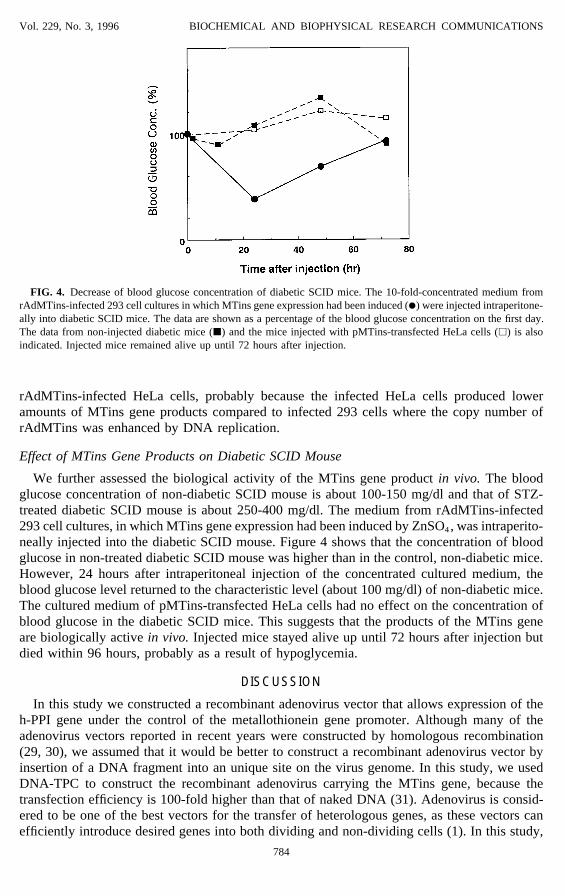
FIG. 4. Decrease of blood glucose concentration of diabetic SCID mice. The 10-fold-concentrated medium fromrAdMTins-infected 293 cell cultures in which MTins gene expression had been induced (l) were injected intraperitone-ally into diabetic SCID mice. The data are shown as a percentage of the blood glucose concentration on the first day.The data from non-injected diabetic mice (j) and the mice injected with pMTins-transfected HeLa cells (h) is alsoindicated. Injected mice remained alive up until 72 hours after injection.
rAdMTins-infected HeLa cells, probably because the infected HeLa cells produced loweramounts of MTins gene products compared to infected 293 cells where the copy number ofrAdMTins was enhanced by DNA replication.
Effect of MTins Gene Products on Diabetic SCID Mouse
We further assessed the biological activity of the MTins gene product in vivo. The bloodglucose concentration of non-diabetic SCID mouse is about 100-150 mg/dl and that of STZ-treated diabetic SCID mouse is about 250-400 mg/dl. The medium from rAdMTins-infected293 cell cultures, in which MTins gene expression had been induced by ZnSO4, was intraperito-neally injected into the diabetic SCID mouse. Figure 4 shows that the concentration of bloodglucose in non-treated diabetic SCID mouse was higher than in the control, non-diabetic mice.However, 24 hours after intraperitoneal injection of the concentrated cultured medium, theblood glucose level returned to the characteristic level (about 100 mg/dl) of non-diabetic mice.The cultured medium of pMTins-transfected HeLa cells had no effect on the concentration ofblood glucose in the diabetic SCID mice. This suggests that the products of the MTins geneare biologically active in vivo. Injected mice stayed alive up until 72 hours after injection butdied within 96 hours, probably as a result of hypoglycemia.
DISCUSSION
In this study we constructed a recombinant adenovirus vector that allows expression of theh-PPI gene under the control of the metallothionein gene promoter. Although many of theadenovirus vectors reported in recent years were constructed by homologous recombination(29, 30), we assumed that it would be better to construct a recombinant adenovirus vector byinsertion of a DNA fragment into an unique site on the virus genome. In this study, we usedDNA-TPC to construct the recombinant adenovirus carrying the MTins gene, because thetransfection efficiency is 100-fold higher than that of naked DNA (31). Adenovirus is consid-ered to be one of the best vectors for the transfer of heterologous genes, as these vectors canefficiently introduce desired genes into both dividing and non-dividing cells (1). In this study,
784
AID BBRC 5842 / 6914$$$303 12-06-96 06:19:14 bbrca

Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
we show that rAdMTins could introduce MTins gene more efficiently than CaPO4 transfectionmethod. Many heterologous genes, such as CFTR, a1AT, and factor IX (4, 32, 33), have beenefficiently transferred into several target cells or tissues by adenovirus vectors. In these cases,expression from the adenovirus-mediated transferred genes is usually under the control of anon-inducible promoter, such as the adenovirus E1a promoter, the adenovirus major latepromoter, or the Rous sarcoma virus long terminal repeat promoter (2, 4, 34). Although, it isimportant for gene therapy to control the expression of transferred genes, relatively little workhas been done in this area (9).
In both 293 and HeLa cells, MTins gene expression was detected only when the infectedcells were treated with ZnSO4. The degree of expression depended on ZnSO4 concentration,suggesting that the expression of h-PPI gene was under the control of the metallothioneinpromoter. We also confirmed that the rAdMTins genome replicated in 293 cells by PCR (datanot shown). Thus, the increased expression of the MTins gene in 293 cells is most likely dueto the increased copy number of the MTins gene.
It has been shown that proinsulin is converted to mature insulin and C-peptide via split-and des-proinsulin intermediates in the b-cell secretory granules by endoproteases, PC2 andPC3 (35). Proinsulin and insulin can be separated by RP-HPLC and the intermediates aregenerally eluted between proinsulin and insulin elution peaks (36). In this study, RP-HPLCanalysis revealed that the major product of the PPI gene was proinsulin. However, in thecultured medium of the infected HeLa cell, products immunoreactive to anti-insulin antibodywere detected in fractions between the mature insulin and proinsulin elution peaks. Theseimmunoreactive products were presumably generated from the incorrect processing of proinsu-lin by cellular proteases because expression of PC2 and PC3 is known to be highly restrictedto neuroendocrine tissues, such as found in the islets of Langerhans, the pituitary and certainregions of the brain (37, 38). The protease activities associated with neuroendocrine tissuesare lacking in both 293 and HeLa cells. Since proinsulin has a weaker biological activity thanmature insulin, cells that can process proinsulin into insulin, such as mouse pituitary AtT20cells (39), are attractive for the further study of the clinical use of rAdMTins. Therefore, weattempted to express the MTins gene in AtT20 cells. When AtT20 cells were infected withrAdMTins at MOI 100, immunoreactive products were detected in the cultured medium. Zincinduction was also required for MTins gene expression in infected AtT20 cells and RP-HPLCanalysis revealed that a small amount of proinsulin had been converted to mature insulin.However, proinsulin was still the major component (data not shown). Since human proinsulinhas been reported to be converted to mature insulin in AtT20 cells that constitutively expressthe human preproinsulin gene (9), the low yield of mature insulin in our system might be dueto the infection by a recombinant adenovirus at high MOI or to the large amount of proinsulinproduced.
In the insulin signal transduction pathway, insulin binds to a cellular receptor (a-submit ofinsulin receptor) and binding induces phosphorylation of the b-subunit. The phosphorylatedb-subunit has tyrosine kinase activity and this results in phosphorylation of tyrosine residuesof the insulin receptor substrate 1 (IRS-1) (40, 41, 42, 43). Although proinsulin is less activethan mature insulin, proinsulin has the same biological activity as mature insulin in this respect(44). In this study, the major MTins gene product found in the cultured medium of infected293 cells was proinsulin and this could stimulate tyrosyl phosphorylation of IRS-1 in HepG2cell, suggesting that the products of the MTins gene are active in vitro.
The anti-human insulin antibody used for RIA in this study crossreacts with proinsulin. Weconfirmed that the crossreactivity of the antibody with proinsulin was about 30% of theimmunoreactivity of mature insulin, and that 1mg of human proinsulin was approximatelyequal to 1mIU of immunoreactive insulin in RIA used here (data not shown). We used 10-fold-concentrated medium from rAdMTins-infected 293 cell cultures to stimulate tyrosyl phos-
785
AID BBRC 5842 / 6914$$$303 12-06-96 06:19:14 bbrca

Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
phorylation of IRS-1. From the result of RIA, the amount of proinsulin in the medium wasestimated to be about 7 mg/ml. Although, the amount of immunoreactive products in thecultured medium used for the assay was one-seventh of the positive control regent (humanproinsulin), the biological activity of the cultured medium estimated from the result (Fig. 3)was about one-third of the control. This suggests that the cultured medium might have containedmore active products than proinsulin.
Here, we show that rAdMTins can introduce MTins gene efficiently into cell culture andthat zinc can induce the expression of the h-PPI gene and regulate its expression in a dosedependent manner. These properties make rAdMTins an attractive tool for studying the stateof both dividing and non-dividing cells expressing insulin gene and for the study of somaticcell gene therapy.
ACKNOWLEDGMENTSThis work was supported by a Grant-in-Aid for Specific Research on Priority Area and a Grant-in-Aid for Cancer
Research from the Ministry of Education, Science, Sports and Culture, Japan.
REFERENCES1. Trapnell, B. C., and Gorziglia, M. (1994) Current Opinion in Biotechnology 5, 617–625.2. Chen, S. H., Shine, H. D., Goodman, J. C., Grossman, R. G., and Woo, S. L. C. (1994) Proc. Natl. Acad. Sci.
USA 91, 3054–3057.3. Ragot, T., Vincent, N., Chafey, P., Vigne, E., Gilgenkrantz, H., Couton, D., Cartaud, J., Briand, P., Kaplan,
J-C., Perricaudet, M., and Kahn, A. (1993) Nature 361, 647–650.4. Rosenfeld, M. A., Yoshimura, K., Trapnell, B. C., Yoneyama, K., Rosenthal, E. R., Dalemans, W., Fukayama,
M., Bargon, J., Stier, L. E., Stratford-Perricaudet, L., Perricaudet, M., Guggino, W., Pavirani, A., Lecocq, J-P.,and Crystal, R. G. (1992) Cell 68, 143–155.
5. Cahill, G. F. (1971) Diabetes 20, 785–798.6. The diabetes control and complications trial research group. (1993) N. Engl. J. Med. 329, 977–986.7. Selden, R. F., Skoskiewicz, M. J., Russell, P. S., and Goodman, H. M. (1987) N. Engl. J. Med. 317, 1067–1076.8. Kolodka, T. M., Finegold, M., Moss, L., and Woo, S. L. C. (1995) Proc. Natl. Acad. Sci. USA 92, 3293–3297.9. Stewart, C., Taylor, N. A., Docherty, K., and Bailey, C. J. (1993) J. Mol. Endocrinol. 11, 335–341.
10. Moore, H. P.-H. Walker, M. D., Lee, F., and Kelly, R. B. (1983) Cell 35, 531–538.11. Durnam, D. M., and Palmiter, R. D. (1981) J. Biol. Chem. 256, 5712–5716.12. Mayo, K. E., and Palmiter, R. D. (1981) J. Biol. Chem. 256, 2621–2624.13. Durnam, D. M., and Palmiter, R. D. (1984) Mol. Cell. Biol. 4, 484–491.14. Durnam, D. M., Hoffman, J. S., Quaife, C. J., Benditt, E. P., Chen, H. Y., Brinster, R. L., and Palmiter, R. D.
(1984) Proc. Natl. Acad. Sci. USA 81, 1053–1056.15. Mayo, K. E., Warren, R., and Palmiter, R. D. (1982) Cell 29, 99–108.16. Hammer, D. H. (1986) Annu. Rev. Biochem. 55, 913–951.17. Harrison, T., Graham, F., and Williams, J. (1977) Virology 77, 319–329.18. Gey, G. O., Coffman, W. D., and Kubicek, G. D. (1952) Cancer Res. 12, 264–265.19. Aden, D. P., Fogel, A., Plotkin, S., Damjanov, I., and Knowles, B. B. (1979) Nature 282, 615–616.20. Moore, D. D. (1994) in Current Protocols in Molecular Biology (Ausubel, F. M., Brent, R., Kingston, R. E.,
Moore, D. D., Seidman, J. G., Smith, J. A., and Struhl, K., Eds.), pp. 2.6.1–2.6.3.21. Stuart, G. W., Searle, P. F., Chen, H. Y., Brinster, R. L., and Palmiter, R. D. (1984) Proc. Natl. Acad. Sci. USA
81, 7318–7322.22. Graham, F. L., and Van Der Eb, A. J. (1973) Virology 52, 456–467.23. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) in Molecular Cloning, 2nd ed., pp. 2.6.1–2.6.3, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, NY.24. Doren, K. V., Hanahan, D., and Gluzman, Y. (1984) J. Virol. 50, 606–614.25. Robinson, A. J., Younghusband, H. B., and D. Bellett, A. J. (1973) Virology 56, 54–59.26. Jones, N., and Shenk, T. (1979) Cell 17, 683–689.27. Hirt, B. (1967) J. Mol. Biol. 26, 365–369.28. Sasaki, Y., Zhang, X. F., Nishiyama, M., Avruch, J., and Wands, J. R. (1993) J. Biol. Chem. 268, 3805–3808.29. Miyake, S., Makimura, M., Kanegae, Y., Harada, S., Sato, Y., Takamori, K., Tokuda, C., and Saito, I. (1996)
Proc. Natl. Acad. Sci. USA 93, 1320–1324.30. Quantin, B., Perricaudet, L. D., Tajbakhsh, S., and Mandel, J.-L. (1992) Proc. Natl. Acad. Sci. USA 89, 2581–
2584.
786
AID BBRC 5842 / 6914$$$303 12-06-96 06:19:14 bbrca

Vol. 229, No. 3, 1996 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
31. Sharp, P. A., Moore, C., and Haverty, J. L. (1976) Virology 75, 442–456.32. Lemarchand, P., Jaffe, H. A., Danel, C., Cid, M. C., Kleinman, H. K., Stratford-Perricaudet, L. D., Perricaudet,
M., Pavirani, A., Lecocq, J-P., and Crystal, R. G. (1992) Proc. Natl. Acad. Sci. USA 89, 6482–6486.33. Smith, T. A., Mehaffey, M. G., Kayda, D. B. Saunders, J. M., Yei, S., Trapnell, B. C., McClelland, A., and Kaleko,
M. (1993) Nature Genet. 5, 397–402.34. Zabner, J., Couture, L. A., Gregory, R. J., Graham, S. M., Smith, A. E., and Welsh, M. J. (1993) Cell 75, 207–
216.35. Steiner, D. F., Smeekens, S. P., Ohagi, S., and Chen, S. J. (1992) J. Biol. Chem. 267, 23435–23438.36. Linde, S., Welinder, B. S., and Nielsen, J. H. (1993) J. Chromatogr. 614, 185–204.37. Seidah, N. G., Marcinkiewicz, M., Benjannet, S., Gaspar, L., Beaubien, G., Mattei, M. G., Lazure, C., Mbikay,
M., and Chretien, M. (1991) Mol. Endocrinol. 5, 111–122.38. Smeekens, S. P., Avruch, A. S., LaMendola, J. S., Chan, S. J., and Steiner, D. F. (1991) Proc. Natl. Acad. Sci.
USA 88, 340–344.39. Buonassisi, V., Sato, G., and Cohen, A. I. (1962) Proc. Natl. Acad. Sci. USA 48, 1184–1190.40. Avruch, J., Nemenoff, R. A., Blackshear, P. J., Pierce, M. W., and Osathanondh, R. (1982) J. Biol. Chem. 257,
15162–15166.41. Ebina, M., Araki, E., Taira, M., Shimada, F., Mori, M., Craik, C. S., Siddle, K., Pierce, S. B., Roth, R. A., and
Rutter, W. J. (1987) Proc. Natl. Acad. Sci. USA 84, 704–708.42. Kwok, Y. C., Nemenoff, R. A., Powers, A. C., and Avruch, J. (1986) Arch. Biochem. Biophys. 244, 102–113.43. Rosen, O. M. (1987) Science 237, 1452–1458.44. Tillili, H., Frank, B. H., Peker, A. H., Broelsch, C., Rubenstein, A. H., and Polonski, K. S. (1990) Endocrinology
127, 2418–2422.
787
AID BBRC 5842 / 6914$$$304 12-06-96 06:19:14 bbrca