controlled epidermal growth factor receptor ligand display
TRANSCRIPT
Controlled Epidermal Growth Factor Receptor Ligand Display onCancer Suicide Enzymes via Unnatural Amino Acid Engineering forEnhanced Intracellular Delivery in Breast Cancer CellsRachel M. Lieser, Wilfred Chen,* and Millicent O. Sullivan*
Department of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, Delaware 19716,United States
*S Supporting Information
ABSTRACT: Proteins are ideal candidates for disease treatmentbecause of their high specificity and potency. Despite this potential,delivery of proteins remains a significant challenge due to the intrinsicsize, charge, and stability of proteins. Attempts to overcome thesechallenges have most commonly relied on direct conjugation of polymersand peptides to proteins via reactive groups on naturally occurringresidues. While such approaches have shown some success, they allowlimited control of the spacing and number of moieties coupled toproteins, which can hinder bioactivity and delivery capabilities of thetherapeutic. Here, we describe a strategy to site-specifically conjugatedelivery moieties to therapeutic proteins through unnatural amino acid(UAA) incorporation, in order to explore the effect of epidermal growthfactor receptor (EGFR)-targeted ligand valency and spacing oninternalization of proteins in EGFR-overexpressing inflammatory breast cancer (IBC) cells. Our results demonstrate theability to enhance targeted protein delivery by tuning a small number of EGFR ligands per protein and clustering these ligandsto promote multivalent ligand−receptor interactions. Furthermore, the tailorability of this simple approach was demonstratedthrough IBC-targeted cell death via the delivery of yeast cytosine deaminase (yCD), a prodrug converting enzyme.
■ INTRODUCTIONLigand display has generated enormous interest in thedevelopment of nanoparticle “carriers” for targeted drugdelivery.1 Ligand valency,2 spacing,3 and orientation4 cansignificantly alter drug biodistribution, targeting efficiency, andtherapeutic efficacy in nanocarriers. Control of ligandclustering is particularly crucial for intracellular cargoes, asligand binding can be used to enhance intracellular deliveryinto specific cells.3,5−7 Recent advances in biological chemistryhave demonstrated the ability to modify proteins in a simpleand site-specific manner, allowing new opportunities forcontrolled display of biomolecules.8 Nonetheless, site-specificligand conjugation has rarely been explored in the context oftherapeutic protein delivery,9−11 and it has never beenexploited to demonstrate the effect of ligand clustering forenhanced targeted uptake of protein therapeutics.Therapeutic proteins are one of the fastest growing sectors
on the pharmaceutical market because of their sophisticatedfunctional properties, capacity for highly specific recognition ofbiological binding partners, and relevance to multiplediseases.12 In fact, biologics represented 35% of all new FDAapprovals between 2000 and 2016, and the market is poised forfurther evolution and growth.13 However, despite enormousinterest and significant investments in developing advancedprotein therapeutics, protein delivery remains a majorlimitation. Stability issues have hindered or halted clinical
advancement of many antibodies.14 Most therapeuticallyrelevant enzymes are incompatible with the extracellularenvironment, leading to aggregation, activity loss, and/orrenal clearance.15 Furthermore, most proteins are membraneimpermeable, and therefore active strategies are necessary todeliver proteins transcellularly or intracellularly, while alsotargeting the correct cells and subcellular compartments. As aresult, the pipeline has a dearth of intracellular protein drugcandidates, even though intracellular proteins comprise over60% of the human proteome and have predicted therapeuticapplications ranging from neurological disorders16 to lysosomalstorage disease17 to cancer.18−22
Engineering efforts to address these issues and improveprotein delivery often rely on modifying proteins throughdirect conjugation of biocompatible polymers or targetingligands that can increase protein stability, alter proteinbiodistribution, and/or improve cellular uptake.23 For example,polyethylene glycol (PEG) has been conjugated to a number ofFDA-approved protein drugs to improve protease resistance
Special Issue: Delivery of Proteins and Nucleic Acids: Achievementsand Challenges
Received: October 29, 2018Revised: December 18, 2018Published: January 7, 2019
Article
pubs.acs.org/bcCite This: Bioconjugate Chem. 2019, 30, 432−442
© 2019 American Chemical Society 432 DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
Dow
nloa
ded
via
UN
IV O
F D
EL
AW
AR
E o
n Fe
brua
ry 2
1, 2
019
at 1
2:50
:17
(UT
C).
Se
e ht
tps:
//pub
s.ac
s.or
g/sh
arin
ggui
delin
es f
or o
ptio
ns o
n ho
w to
legi
timat
ely
shar
e pu
blis
hed
artic
les.

and reduce renal clearance.24 Bioconjugation is typicallyaccomplished using naturally occurring reactive residues (e.g.,lysines) within the protein sequence, or by using genetic fusionto the protein termini.25,26 While both methods have thecapacity to enhance various aspects of delivery as compared toproteins in their native form, the inability to chemically modifyproteins with site-specificity often affects the activity of theprotein and thereby significantly hinders pharmacologicalaction.27 For example, one study conjugated a single PEGmolecule to distinct locations in human growth factor using asite-specific modification technique and observed a 3-folddifference in activity in vivo, depending on the location of thepolymer.9
Non-site-specific modification approaches also do not offercontrol over design variables that can be importantdeterminants of targeting efficacy. In particular, the importanceof ligand spacing and density has been demonstrated formultivalent ligand−receptor interactions via ligand clustering28
and synergistic receptor binding using dual ligand cofunction-alization.29,30 The importance of multivalent effects in deliveryhas been clearly demonstrated using nanocarriers. For example,folate ligands were incorporated in micelles within clusters ofvarying valencies for delivery to folate-overexpressing cancercells. The dissociation constant decreased 2 orders ofmagnitude when micelles had an average of three ligands percluster as compared to micelles with an average of 1.5 ligandsper cluster.6 In other examples involving nanocarriers,clustering cell-binding ligands was shown to increase cell-binding affinity by up to 1000-fold.31,32 Furthermore, optimalligand cluster sizes and densities have been proposed formaximal receptor specificity and affinity.33 These examplespoint out the clear need for new strategies to modify proteinswith well-defined ligand arrays.A potential strategy to accomplish this goal is the use of a
site-specific and multivalent conjugation method for insertionof multiple chemical modification sites into proteins. Previouswork has demonstrated the ability to insert biorthogonalreactive residues into proteins through unnatural amino acid(UAA) incorporation with nonsense codon replacement,enabling direct protein conjugation with simple “click”chemistries.34 To date, over 70 UAAs with an array ofstructures have been successfully incorporated into the geneticcode of multiple organisms.35 UAA incorporation has beenused in a number of applications including protein labeling,biosensing, vaccine development, and antibody-drug conjuga-tion.36−38
Herein, we explored the potential of this approach forimproved intracellular delivery of protein therapeutics. Wespecifically sought to determine whether UAA-modificationcould be used to control the spacing and number of receptor-binding ligands that were inserted into therapeutically relevantenzymes. UAA incorporation was used to attach clusters of thehigh-affinity epidermal growth factor receptor (EGFR)targeting peptide GE1139 into fluorescent proteins and suicideenzymes for delivery to inflammatory breast cancer (IBC)cells. IBC is an aggressive subtype of breast cancer with a lessthan 50% survival rate beyond 5 years,40 and IBC associatedwith EGFR overexpression is particularly aggressive. Currentdrug delivery approaches in IBC are limited due to severeadverse side effects, and hence mechanisms to improvetargeted drug uptake would have significant benefits.We first quantified protein uptake using the fluorescent
model protein mCherry. By varying the spacing and number ofGE11 peptides linked to mCherry via UAA conjugation, wedemonstrated an 18-fold increase in protein uptake when fourGE11 peptides were clustered in the protein as compared toproteins with a single GE11 peptide. In addition, uptake inhealthy breast epithelial cells was found to be 4-fold lower thanuptake in IBC cells, demonstrating the ability to not onlydeliver large amounts of protein, but also selectively target IBCcells. Furthermore, we modularized the GE11-mCherry forattachment to therapeutic enzyme cargoes using SpyCatcher-SpyTag bioconjugation.41 SpyCatcher-SpyTag was used to linkGE11-mCherry to yeast cytosine deaminase (yCD), a suicideenzyme that converts the nontoxic prodrug 5-fluorocytosine(5-FC) into the widely used chemotherapeutic 5-fluorouracil(5-FU). The linkage strategy fully preserved yCD enzymeactivity, and moreover, codelivery of yCD and 5-FC resulted ina 3-fold difference in cell death between the normal andcancerous breast epithelial cells. These results demonstrate thebenefits of UAA incorporation for controlling targeting peptidepresentation and maximizing cargo protein activity, with keybenefits relevant to prodrug therapeutics and a wide range ofother intracellular protein therapies.
■ RESULTS AND DISCUSSION
Unnatural Amino Acid (UAA) Incorporation AllowsSite-Specific Addition of pAzF. Previous studies havedemonstrated successful incorporation of p-Azido-L-phenyl-alanine (pAzF) in E. coli though amber stop codon suppressionwith the pULTRA-CNF suppressor plasmid system.34 Here, 1,
Figure 1. Unnatural amino acid incorporation of pAzF. (A) Expression of full-length mCherry with 1, 2, or 4 pAzF residues on the N-terminus inthe presence (white) or absence (gray) of pAzF. Fluorescence is normalized to lysate concentration determined by Bradford protein assays. Resultsare shown as mean ± standard deviation of three independent experiments. (B) SDS-PAGE analysis of Alexa Fluor 488-alkyne conjugation to azidegroups of mCherry by Coomassie staining to detect total protein (top) and UV transillumination to detect Alexa Fluor 488 fluorescence (bottom).
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
433
2, or 4 pAzF UAAs were incorporated onto the N-terminus ofthe fluorescent protein mCherry, and the resulting proteinswere termed 1Az-, 2Az-, and 4Az-mCherry. Flexible linkersmade up of glycine and serine residues (G4S1) separated theUAAs. The fluorescence intensities of the cell lysates indicatedthat mCherry was preferentially expressed for all threeconstructs only when E. coli cells were grown with pAzFpresent in the culture medium (Figure 1A). Compared to 1Az-mCherry, 1.8-fold and 2.9-fold decreases in expression wereobserved for 2Az-mCherry and 4Az-mCherry, respectively.Decreased expression levels with an increased number of UAAsis a common outcome of UAA-linked protein expression.42
The addition of multiple amber stop codons in the encodinggene increases the possibility of early termination of trans-lation.43 Importantly, expression of mCherry with up to fourUAAs still resulted in significant yields. Growing cells in theabsence of pAzF in the culture medium resulted in low levelsof mCherry expression in any samples, indicating that amajority of the full-length protein in the pAzF-grown sampleslikely contained the UAA (Figure S1A). The three proteinswere purified using His-tag Ni-NTA chromatography (FigureS1B). Incorporation of pAzF was confirmed through copper-catalyzed alkyne−azide cycloaddition (CuAAC) with AlexaFluor 488 alkyne dye. Successful conjugation of the AlexaFluor 488 dye to 1Az-, 2Az-, and 4Az-mCherry was confirmedby sodium dodecyl sulfate polyacrylamide gel electrophoresis(SDS-PAGE), using fluorescence analysis and Coomassiestaining (Figure 1B).CuAAC “Click” Chemistry Conjugates EGFR Targeting
Ligand to Protein. The GE11 targeting peptide was chosento target EGFR, which is overexpressed in a number ofdifferent cancer cells including IBC cells.44,45 The GE11peptide exhibits high affinity toward EGFR (KD = 22 nM) andhas been utilized for delivery of an array of nanoparticles inmultiple types of EGFR-overexpressing cancer cells.39,46−48 Forinstance, liposomes loaded with doxorubicin and function-alized with PEG-GE11 demonstrated a 2.2-fold increase in
A549 tumor cell accumulation in in vivo models as comparedto liposomes functionalized only with PEG.49
Here, GE11 was synthesized with an N-terminal propargyl-glycine through Fmoc solid phase peptide synthesis, purifiedthrough RP-HPLC, and the final product was confirmedthrough MALDI-TOF mass spectrometry (Figure S2).Subsequently, CuAAC was used to conjugate the propargyl-GE11 peptide on to the azido groups of mCherry. The reactionyield of propargyl-GE11 with 1pAzF-, 2pAzF-, and 4pAzF-mCherry was estimated via SDS-PAGE analysis (Figure S3A)and MALDI-TOF mass spectrometry (Figure S3B). Thechange in product size following propargyl-GE11 conjugationto 1Az- and 2Az-mCherry confirmed that a majority of the finalproduct in each protein had all reactive groups modified withthe GE11 peptide. Reaction with 4Az-mCherry indicated amajority of the final product had either three or four GE11peptides. We suspect that the lower modification efficiency of4GE11-mCherry is due in part to limitations in UAAincorporation, i.e., that only three out of the four encodedpAzF groups were actually incorporated in the protein product,as the incorporation of multiple UAAs can increase thelikelihood of amber codon read-through. Additionally, wesuspect that steric hindrance due to the close proximity of theGE11 peptides may be limiting access by additional GE11peptides to the remaining reactive sites in the protein. TheCuAAC reaction itself is unlikely to be the issue as CuAAClinkage typically results in high yields.50 Usage of an E. coli cellline developed specifically for UAA incorporation, rather thanBL21(DE3),51,52 or increasing the spacing of the UAAs, wouldlikely improve the reaction efficiency.
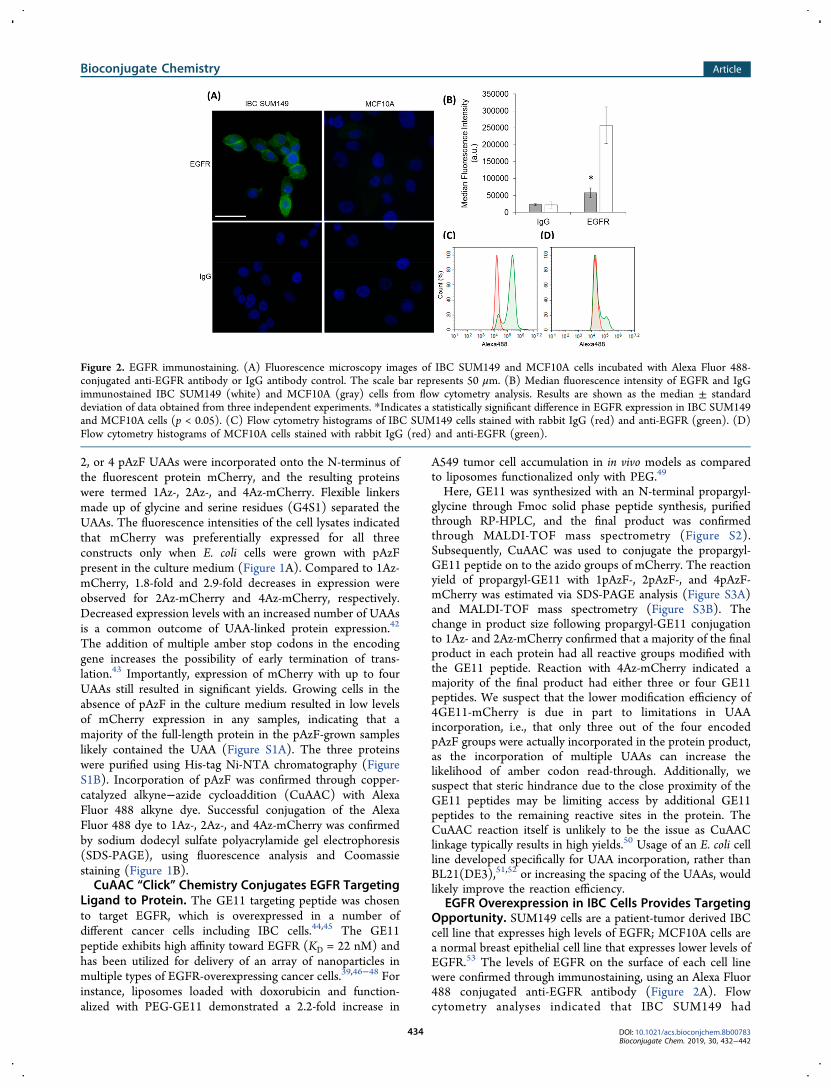
EGFR Overexpression in IBC Cells Provides TargetingOpportunity. SUM149 cells are a patient-tumor derived IBCcell line that expresses high levels of EGFR; MCF10A cells area normal breast epithelial cell line that expresses lower levels ofEGFR.53 The levels of EGFR on the surface of each cell linewere confirmed through immunostaining, using an Alexa Fluor488 conjugated anti-EGFR antibody (Figure 2A). Flowcytometry analyses indicated that IBC SUM149 had
Figure 2. EGFR immunostaining. (A) Fluorescence microscopy images of IBC SUM149 and MCF10A cells incubated with Alexa Fluor 488-conjugated anti-EGFR antibody or IgG antibody control. The scale bar represents 50 μm. (B) Median fluorescence intensity of EGFR and IgGimmunostained IBC SUM149 (white) and MCF10A (gray) cells from flow cytometry analysis. Results are shown as the median ± standarddeviation of data obtained from three independent experiments. *Indicates a statistically significant difference in EGFR expression in IBC SUM149and MCF10A cells (p < 0.05). (C) Flow cytometry histograms of IBC SUM149 cells stained with rabbit IgG (red) and anti-EGFR (green). (D)Flow cytometry histograms of MCF10A cells stained with rabbit IgG (red) and anti-EGFR (green).
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
434

approximately 5-fold higher levels of cell surface EGFR thanMCF10A cells (Figure 2B). Histograms of cellular Alexa Fluor488 fluorescence based on flow cytometry indicated that amajority of the IBC SUM149 cells show high levels of surfaceEGFR (Figure 2C), whereas a majority of MCF10A cells hadonly basal levels of EGFR on their membrane (Figure 2D).The small population of MCF10A cells expressing high levelsof surface EGFR was likely caused either by variations inEGFR expression that occur during the cell cycle54 or byactivation of EGFR, due to the small amount of EGF in theculture medium or antibody-receptor binding, which has beenshown to alter the localization of the receptor to the cellmembrane and enhance EGFR expression in MCF10A cells.53
Control of Ligand Number Provides Tunable Tar-geted Uptake in IBC Cells. To determine if ligand valencyplayed a role in targeted cellular uptake, the GE11-mCherryproteins were delivered to IBC SUM149 cells and MCF10Acells. Fluorescence microscopy was used to visualize cellinternalization (Figure 3A and Figure S5). Corresponding
phase images were used to orient the imaging analyses andconfirm the locations of cell borders (Figure S4). From flowcytometry, 2.9-fold, 13.6-fold, and 40.7-fold increases incellular association were observed in IBC SUM149 cells for1GE11-, 2GE11-, and 4GE11-mCherry as compared tomCherry lacking any GE11, termed 0GE11-mCherry (Figure3B). In addition, GE11-mCherry at all valencies exhibitedhigher association levels in IBC SUM149 cells as compared toMCF10A cells, which express significantly less EGFR. Cellularassociation of 1GE11-mCherry was 1.8-fold higher in IBCSUM149 cells as compared to MCF10A cells, although thisdifference was not statistically significant. The association
levels of 2GE11-mCherry and 4GE11-mCherry were 4.7-foldand 4.1-fold higher in IBC SUM149 cells vs MCF10A cells,respectively. Although association increased in MCF10A cellswith increased GE11 number, the difference in associationbetween IBC SUM149 and MCF10A cells remained significantat higher valencies.We expect that the nonlinearity observed between ligand
number and cellular association/uptake was the result of ligandclustering. Previous studies have demonstrated improveduptake via clustering when targeting EGF receptors. Forexample, affibody molecules against EGFR demonstratedsignificantly higher uptake in EGFR-positive A431 cells whenthe affibodies were clustered through a heptamerizationdomain, as compared to monomeric affibodies.55 An additionalstudy demonstrated a similar phenomenon by clustering HER2ligands on liposomes; uptake was enhanced in both HER2-overexpressing cells and cells with lower HER2 expressionlevels as compared to liposomes with uniformly distributedligands.5
To determine whether UAA-mediated GE11 targetingoffered benefits in cell uptake and cell specificity as comparedwith common cell penetrating peptide (CPP) strategies inproteins, protein internalization levels also were compared touptake levels of a Tat-mCherry fusion protein. Tat is an HIV-derived CPP that is able to elicit high levels of nonspecificcellular uptake due to its positive charge.56 Tat fusions havebeen widely used to stimulate intracellular delivery ofproteins.57 Tat-mCherry showed comparable levels of uptakein IBC SUM149 cells and MCF10A cells demonstrating itsnonspecificity. In addition, Tat-mCherry showed 3.2-fold lowercell association levels as compared to 4GE11-mCherry in IBCSUM149 cells, demonstrating that the UAA/GE11 approachoffered significant improvements in not only targetingspecificity, but also cellular uptake efficiency.
EGF Inhibition Demonstrates EGFR Mediated Uptake.EGF (KD = 2 nM) was used in competitive binding assays toconfirm whether or not GE11-mCherry uptake was EGFRspecific. EGF was incubated with IBC SUM149 cells for 1 hprior to delivery of GE11-mCherry. Fluorescence microscopyand flow cytometry showed significantly reduced uptake levelsof all GE11-mCherry constructs when EGF was preincubatedwith IBC SUM149 cells, suggesting that GE11-mCherryuptake was EGFR mediated (Figure 4A). Specifically, 11.3-fold, 5.4-fold, and 2.6-fold drops in cell association wereobserved when IBC SUM149 cells were preincubated withexcess concentrations of EGF followed by delivery of 1GE11-,2GE11-, or 4GE11-mCherry (Figure 4B), respectively. Theseresults suggest that the number of GE11 peptides has animpact on the ability of EGF to inhibit mCherry internal-ization. One possible explanation for this is ligand clustering,which has been shown to enhance the apparent ligand−receptor binding affinity.58 An alternative possibility is basedon the charge of the GE11 peptide. GE11 has a positive chargeof 1, meaning that the overall charge of GE11-mCherryincreases as the number is increased. Cationic charge has beenshown to elicit cellular uptake, and so the increased chargecould lead to nonspecific mCherry uptake.59 Nonetheless,EGFR appeared to play an important role in the uptakemechanism of the protein.
Ligand Clustering Impacts Cellular Uptake. Todetermine whether ligand clustering influenced EGFR bindingand mCherry uptake, we generated a GE11-mCherry constructin which the GE11 peptide was spaced throughout the protein
Figure 3. GE11-mCherry cellular internalization with varying GE11decoration densities. (A) Fluorescence micrographs of IBC SUM149and MCF10A cells incubated with GE11-mCherry constructs. Thescale bar represents 25 μm. (B) Fluorescence intensity of GE11-mCherry association in IBC SUM149 (white) and MCF10A (gray)cells from flow cytometry analysis. Results are shown as the mean ±standard deviation of data obtained from three independentexperiments. *Indicates a statistically significant difference in uptakebetween IBC SUM149 and MCF10A cells (p < 0.05). **Indicates astatistically significant difference in uptake between mCherryconstructs in IBC SUM149 cells (p < 0.05).
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
435

rather than clustered on the N-terminus. To do this, the GE11peptide was conjugated to mCherry using reactive amines onsurface lysine residues (Figure S7A). First, azidobutyric acidNHS ester was reacted in 10-fold excess with amine groups inmCherry so that azido groups were available on the surface.Second, azido groups were reacted with alkyne-GE11 usingCuAAC to incorporate approximately four GE11 peptides perprotein (Figure S7B). This reaction resulted in mCherryprotein that was highly insoluble, likely due to the loss ofcharge on the mCherry protein resulting from reacting thelysine residues. Applying the randomly modified protein toIBC SUM149 cells resulted in the appearance of protein
aggregates on the surface of the cells, but the protein was notinternalized at high levels (Figure S7C). Extensive optimiza-tion was needed in order to procure protein that was soluble,further highlighting the usefulness of UAA incorporation forligand conjugation.We hypothesized that this decrease in solubility was due to
changing the overall charge of the protein by reacting surfacelysine residues. To improve the solubility of GE11-mCherryconjugated through NHS ester-amine chemistry, azidobutyricacid NHS ester was limited so that only an average of twoazido groups were reacted per protein. Following CuAAC withalkyne-GE11, the protein appeared soluble (Figure S7E). From
Figure 4. GE11-mCherry EGFR Inhibition. (A) Fluorescent microscopy images of GE11-mCherry uptake (-EGF) in IBC SUM149 compared touptake following EGF preincubation (+EGF). Scale bar represents 25 μm. (B) Mean fluorescent intensity of GE11-mCherry association with(gray) and without (white) EGF inhibition from flow cytometry analysis. Results are shown as the mean ± standard deviation of data obtainedfrom three independent experiments. *Indicates a statistically significant difference in uptake between IBC SUM149 cells preincubated or in theabsence of EGF (p < 0.05).
Figure 5. Internalization with nonclustered 2GE11-mCherry. (A) Fluorescence microscopy images of clustered and nonclustered 2GE11-mCherrycellular internalization. Scale bar represents 15 μm. (B) Flow cytometry analyses of nonclustered and clustered 2GE11-mCherry cellularassociation. Results are shown as the mean ± standard deviation of data collected from three independent experiments. *Indicates a statisticallysignificant difference in uptake between clustered and nonclustered 2GE11-mCherry (p < 0.05). (C) Flow cytometry histograms of clustered (red)and nonclustered (green) 2GE11-mCherry uptake in IBC SUM149 cells.
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
436

SDS-PAGE, this protein appeared to have similar numbers ofGE11 per mCherry as compared with 2GE11-mCherry reactedvia the UAA approach (Figure S7D). While there is littlecontrol over GE11 addition using this method, it was expectedthat a majority of the protein would have nonclustered GE11peptides due to the spacing of available lysine residues.Both the clustered and nonclustered 2GE11-mCherry
proteins were administered to IBC SUM149 cells. Cellularinternalization and association was detected with fluorescencemicroscopy (Figure 5A) and flow cytometry (Figure 5B),respectively. The results of these experiments demonstratedthat the clustered 2GE11-mCherry was uptaken at significantlyhigher levels as compared to the nonclustered version, whichdemonstrated comparable uptake levels to 1GE11-mCherry;these results suggest that GE11 ligand clustering plays a clearrole in enhancing EGFR-mediated uptake. These results arecomparable to a study that demonstrated improved DNAdelivery with polyplexes functionalized with a brancheddivalent GE11 ligand as compared with individual GE11peptides.60 It should be noted that while these findings appearto be a result of ligand clustering, because different conjugationtechniques were used to generate the clustered and non-clustered proteins, changes in protein properties, such assolubility, could also be responsible for some of the differencesobserved.Plug-and-Play Incorporation of yCD Suicide Enzyme
with SpyCatcher-SpyTag. The SpyCatcher-SpyTag proteincoupling system was used to facilitate plug-and-play con-jugation of therapeutically relevant enzymes to GE11-mCherry. The lysine on SpyCatcher and aspartic acid onSpyTag are able to form an isopeptide bond within minutesand the reaction demonstrates high yields in diverse solutionconditions.41 A variety of protein cargoes could be delivered bysimply fusing SpyTag, a small 13-amino-acid peptide tag, to achoice therapeutic protein and coupling it to the GE11-mCherry-SpyCatcher constructs. To enable SpyCatcher-SpyTag-mediated enzyme attachment to the GE11-modifiedmCherry delivery constructs, SpyCatcher was fused to the C-terminus of 1Az-, 2Az-, and 4Az-mCherry. Expression levelswere not significantly altered with the addition of theSpyCatcher fusion protein (Figure S8A). Subsequently,GE11 was conjugated to the Az-mCherry-SpyCatcher
constructs using CuAAC chemistry. This reaction showedyields similar to those seen with the Az-mCherry constructs(Figure S8B).To demonstrate the plug-and-play enzyme attachment
strategy, SpyCatcher-SpyTag was used to attach the prodrugconverting enzyme, yCD, to 1GE11-, 2GE11, or 4GE11-mCherry-SpyCatcher. yCD catalyzes the deamination ofcytosine to uracil, enabling rapid conversion of 5-FC, anontoxic prodrug, into 5-FU, a toxic chemotherapeutic.61,62 5-FU is an FDA-approved chemotherapeutic that has been usedwidely in the treatment of colorectal and breast cancers due toits capacity to inhibit DNA replication;63 however, off-targetside effects of 5-FU, such as fatigue, nausea, and cognitiveimpairment,64 have motivated interest in prodrug conversionstrategies. SpyTag was fused to the N-terminus of yCD, andthe SpyTag-yCD protein was coupled to GE11-mCherry-SpyCatcher to form GE11-mCherry-yCD. SDS-PAGE analysisconfirmed that a majority of the full length mCherry proteinwas coupled to yCD (Figure S8B).
Delivery of Suicide Enzyme Elicits IBC-Targeted CellDeath. To assess the internalization of 1GE11-, 2GE11-, and4GE11-mCherry-yCD, uptake experiments were performed inIBC SUM149 cells and MCF10A cells. Flow cytometryexperiments demonstrated that the GE11-mCherry-yCDconstructs showed on average roughly 2-fold lower associationcompared to GE11-mCherry proteins in both cells lines(Figure 6A). This reduction of uptake is likely due to changesin the molecular weight and surface properties from theaddition of the yCD. Despite this difference, the effect of GE11valency remained. Cellular association in IBC SUM149demonstrated 2.4-fold, 7.4-fold, and 19.3-fold increases for1GE11-, 2GE11-, and 4GE11-mCherry-yCD as compared to0GE11-mCherry-yCD. In addition, uptake was higher in IBCSUM149 cells than MCF10A cells for all GE11-mCherryconstructs, again demonstrating the targeting capacity of thisapproach.The activity of yCD was tested in both cell types to
determine whether GE11-mCherry-yCD could selectivelydecrease viability of IBC SUM149 cells when treated withthe prodrug 5-FC. Both types of cells were incubated with theGE11-mCherry-yCD proteins. Extracellular protein wasremoved through multiple wash steps, and 5-FC was
Figure 6. yCD delivery and cell viability. (A) Flow cytometry analyses of mCherry-yCD cellular association in IBC SUM149 (white) and MCF10A(gray) cells. Results are shown as the mean ± standard deviation of data collected from three independent experiments. *Indicates a statisticallysignificant difference in uptake between IBC SUM149 and MCF10A cells (p < 0.05). **Indicates a statistically significant difference in uptakebetween mCherry-yCD constructs in IBC SUM149 cells (p < 0.05). (B) MTT assay to assess the viability of SUM149 (white) and MCF10A (gray)cells following delivery of yCD and treatment with 5-FC. 5-FU viability was set to zero and the results were normalized to 5-FC viability. Resultsare shown as the mean ± standard deviation of data obtained from four independent experiments. *Indicates a statistically significant difference (p< 0.05) in viability between IBC SUM149 and MCF10A cells.
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
437

subsequently administered. Following treatment, cell viabilitywas measured using MTT assays. In the absence of yCD, celldeath did not occur despite the presence of 5-FC, with 91%cell viability in IBC SUM149 cells, and 90% cell viability inMCF10A cells as compared to the same cell types untreated.Direct treatment with 5-FU induced significant decreases inviability for both IBC SUM149 cells and MCF10A cells, withcell viabilities of 45% and 47%, respectively, following 5-FUtreatment. Delivery of yCD proteins without 5-FC did notimpact viability in either cell type (Figure S9).To understand the effect of yCD/5-FC treatment, the cell
viability of 5-FU was set to zero and sample viability wasnormalized to the viability of 5-FC (Figure 6B). In IBCSUM149 cells, a clear reduction in viability was evident as theGE11 valency was increased, with 4GE11-mCherry-yCD/5-FCtreatment inducing toxicity levels that were nearly the same asthe toxicity levels following direct treatment with 5-FU.Treatment of MCF10A cells resulted in a similar effect, in thathigher GE11 valences produced increased levels of off-targetcell death; however, viability at all GE11 densities was higherfor MCF10A cells compared to IBC SUM149 cells.Specifically, 1GE11-, 2GE11-, and 4GE11-mCherry-yCD/5-FC treatment produced 1.4-, 1.7-, and 2.9-fold differences incell viability, respectively, between the two cell types.Treatment with Tat-mCherry-yCD and 5-FC also resulted inelevated levels of cell death in both cell types, but there wasnot a significant difference in viability between IBC SUM149cells and MCF10A cells. These data demonstrate the targetingcapabilities of GE11-mCherry-yCD proteins, and they furtherdemonstrate the ability to control viability selectively in IBCSUM149 cells and MCF10A cells using alterations in GE11valency.
■ CONCLUSIONS
Herein, we demonstrated the ability to control intracellularprotein delivery to IBC cells by controlling the valency ofEGFR targeting peptides through UAA incorporation.Furthermore, the SpyCatcher-SpyTag conjugation systemwas employed for plug-and-play therapeutic cargo proteinattachment. Though this method was applied to prodrugcancer therapy, this approach could be tailored to a multitudeof applications by selecting alternative targeting peptides andtherapeutic proteins. In addition, while the scope of this studyfocused on targeting peptides, site-directed conjugation ofhydrophilic polymers and endosomolytic peptides could befurther explored to address additional challenges associatedwith in vivo cytosolic protein delivery.Using this approach, delivery of the prodrug converting
enzyme, yCD, resulted in significant IBC-targeted cell deathwhen treated with 5-FC, with the levels of cell deathcontrollable through alterations in ligand number. Our resultsdemonstrate the importance of controlling the location ofdelivery molecules conjugated to proteins for improved cellspecificity, delivery efficiency, and pharmacological activity ofprotein drugs, a phenomenon that has not been studiedextensively in proteins due to conjugation limitations. WhileUAA incorporation is currently limited to laboratory scaleproduction, as the ability to site-specifically modify proteinsbecomes more feasible, further understanding of ligand andpolymer display will likely play an important role in improvingdelivery and efficacy of advance protein therapeutics.
■ EXPERIMENTAL PROCEDURES
Materials. All DNA primers used to perform polymerasechain reaction (PCR) were purchased from IDT (Coralville,IA). Restriction enzymes, T4 DNA ligase, and Q5 DNApolymerase for DNA cloning were purchased from NEB(Ipswich, MA). Bacterial culture medium ingredients werepurchased from Fisher Scientific (Pittsburgh, PA). Zyppyplasmid kit was purchased from Zymo Research (Irvine, CA)for DNA purification following digestion or gel electrophoresis.Antibiotics and isopropyl-β-D-1-thiogalactopyranoside (IPTG)were purchased from Sigma-Aldrich (St. Louis, MO). p-Azido-L-phenylalanine (4-azido-L-phenylalanine, ≥98% (HPLC)) waspurchased from Chem-Impex International Inc. (Wood Dale,IL). Reagents for SDS-PAGE were purchased from BIO-RAD(Hercules, CA). Amino acids and resin for peptide synthesiswere purchased from MilliporeSigma (Burlington, MA) andCEM Corporation (Matthews, NC). All solvents for peptidesynthesis were purchased from Fisher Chemical (Fair Lawn,NJ). Dulbecco’s Phosphate Buffered Saline (DPBS, 1×),Ham’s F-12, and Dulbecco’s Modification of Eagle’sMedium/Ham’s F-12 50/50 Mix were purchased fromThermo Fisher Scientific (Grand Island, NY).
Construction of Expression Plasmids. Constructs wereprepared using standard molecular cloning techniques. A genefragment encoding mCherry was amplified with PCR usingprimers amber-mCherry-F and amber-mCherry-R (Table S1),in which an amber codon and NheI cut site were geneticallyfused to the N-terminus of the mCherry gene fragment. ThePCR product was inserted into pET22b-w3-CFP-his665 usingSacII and XhoI sites to yield pET22b-amber-mCherry-his6.Gene fragments encoding amber-G4S1-amber and (amber-G4S1-amber)2, were annealed using primers 2amber-F, 2amber-R, 4amber-F, and 4amber-R (Table S1) followed by T4polynucleotide kinase treatment. The DNA fragments wereinserted into pET22b-amber-mCherry-his6 using SacII andNheI sites to yield pET22b-2amber-mCherry-his6, andpET22b-4amber-mCherry-his6, respectively. All plasmidswere transformed into Escherichia coli NEB5α (NEB, Ipswich,MA) [fhuA2 Δ(argF-lacZ)U169 phoA 23 gln V44 Φ80Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17]. Bacteriawere grown on 25 g/L Luria−Bertani Broth (LB, 10 g/Ltryptone, 5 g/L yeast extract, 5 g/L sodium chloride) and 15g/L agar plates supplemented with 100 μg/mL ampicillin.Positive clones were cotransformed with pULTRA-CNF (a giftfrom Prof. Peter G. Schultz34) into E. coli strain BL21(DE3)(EMD Millipore, Madison, WI) [F- ompT hsdSB(rB- mB-) galdcm (DE3) Δ(srlrecA)306::Tn10 (TetR)]. Plasmid sequencesand primers can be found in the Supporting Information(Figure S10 and Table S1).
Expression and Purification of Proteins. Proteins wereexpressed in Terrific Broth (TB) media (12 g/L tryptone, 24g/L yeast extract, 0.4% (v/v) glycerol, 9.4 g/L monopotassiumphosphate, 2.2 g/L dipotassium phosphate) supplementedwith 100 μg/mL ampicillin and 100 μg/mL spectinomycin.Cultures were inoculated with an overnight culture from asingle colony to an OD600 of 0.05 and allowed to grow at 37°C in a shake flask to an OD600 of 0.6−0.8. Expression wasthen induced with 1 mM IPTG and supplemented with 1 mMpAzF. Cultures were grown overnight at either 20 °C (1Az-mCherry-his6 and Tat-mCherry-his6) or 37 °C (2Az-mCherry-his6 and 4Az-mCherry-his6).
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
438

Cells were pelleted with centrifugation at 4000g for 10 minat 4 °C. Spent media was removed and cells were resuspendedin 1× phosphate buffered saline (PBS, pH 7.4) with 10 mMimidazole to an OD600 of 20. Cells were lysed via sonicationand centrifuged at 10,000g for 15 min at 4 °C to collect solubleprotein. Proteins were purified using His-Bind Ni-NTA resingravity column from Thermo Fisher (Pittsburgh, PA)according to the manufacturer’s protocol. After purification,proteins were dialyzed overnight in 1× PBS.Site-Specific Conjugation of Alexa Fluor 488. Alexa
Fluor 488 Alkyne was purchased from Molecular Probes(Eugene, Oregon). 100 μM Alexa Fluor 488 Alkyne wasreacted to 25 μM azido-mCherry in PBS (pH 6.5) with 250μM CuSO4, 1.25 mM THPTA ligand, and 5 mM sodiumascorbate for 1 h at room temperature.66 Dye conjugation wasconfirmed via SDS-PAGE. Alexa Fluor 488 was detected with aTyphoon laser scanner (Marlborough, Massachusetts) and thegel was subsequently stained with Coomassie Brilliant Blue.Synthesis of GE11 Peptide. GE11 with an N-terminal
linker (HAIYPRHYHWYGYTPQNVI) was synthesized usingsolid phase peptide synthesis.67 A propargyl glycine wasconjugated to the N-terminus for CuAAC. The peptide waspurified by reverse-phase high-performance liquid chromatog-raphy (not shown) and the final product was confirmed withMALDI-TOF mass spectrometry. From MALDI the molecularweight of the final product (2509.47 g/mol) matched theexpected molecular weight (2509.77 g/mol).Site-Specific Conjugation of GE11 Peptide to pAzF-
mCherry Proteins. GE11 was conjugated to mCherryconstructs with CuAAC. Briefly, 150 μM alkyne-GE11 wasreacted to 25 μM azido-mCherry in 1×PBS (pH 6.5) with 250μM CuSO4, 1.25 mM THPTA ligand, and 5 mM sodiumascorbate for 1 h at room temperature.66 The protein-peptideconjugate was then purified with His-Bind Ni-NTA resin anddialyzed overnight in 1× PBS. The products were analyzedwith SDS-PAGE and MALDI mass spectrometry. Sampleswere filtered with a 0.22 μM syringe filter before being used incell studies.Cell Culture. IBC SUM149 cells (a gift from Kenneth van
Golen68) were grown in Ham’s F12 medium supplementedwith 5% FBS, 1% (v/v) penicillin/streptomycin, 1% (v/v)mycoplasma antibiotic supplement, 1% (v/v) glutamine, 5 μg/mL insulin, 2.5 μg/mL transferrin, 200 ng/mL selenium, and 1μg/mL hydrocortisone according to previously establishedmethods.69,70 MCF10A cells, purchased from ATCC (Mana-ssas, Virginia), were grown in 50/50 DMEM/Ham’s F12medium supplemented with 5% FBS, 1% (v/v) penicillin/streptomycin, 50 μg/mL bovine pituitary extract, 10 μg/mLinsulin, 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin,and 20 ng/mL epidermal growth factor.Cellular Internalization of GE11-mCherry Protein.
First, 5 × 104 IBC SUM149 and MCF10A cells were seededin 8-well plates with a collagen film (1.5 mg/mL collagen Ibovine protein in 0.02 M acetic acid in DI water) andincubated for 24 h at 37 °C. Cells were incubated with 1 μM ofprotein for 3 h. Medium was removed and cells were washedthree times in 1× DPBS (pH 7.4). Cells were fixed with 10%formalin for 15 min, treated with DAPI (300 nM) for 10 min,and rinsed three times with 1× DPBS. Internalization wasobserved at 40× magnification on a Leica DM6000fluorescence microscope (Wetzlar, Germany) with 350/50nm excitation and 460/50 nm emission for DAPI and 545/25nm excitation and 605/70 nm emission for mCherry.
Flow cytometry was used as a quantitative analysis of GE11-mCherry association in IBC SUM149 and MCF10A cells. Cellswere seeded in six-well plates at a density of 3 × 105 cells perwell and incubated overnight at 37 °C. Medium was replacedand cells were incubated with 1 μM of protein for 3 h. Cellswere washed three times in 1× DPBS and trypsinized.Following trypsinization, cells were neutralized with theappropriate cell media and centrifuged at 800 rpm for 4 min.Cells were resuspended in cold 1× DPBS and analyzed by flowcytometry (NovoCyte, ACEA Biosciences, Inc., San Diego,CA, USA). Fluorescence intensity of 1 × 104 cells wasmeasured with 488 nm laser and 660 nm emission formCherry. The median fluorescence intensity of three replicateswas reported along with a histogram of one replicate for eachsample (Figure S6).
EGFR Immunostaining. Cells were seeded in 8-well plateswith a collagen film and incubated for 24 h before fixation andDAPI staining as previously described. Following fixation, cellswere blocked with 3% BSA in 1× DPBS for 20 min at roomtemperature then incubated with 2.5 μg/mL Alexa Fluor 488anti-EGFR Antibody, from Thermo Fisher, or Alexa Fluor 488Rabbit IgG Isotype control from Cell Signaling Technology(Danvers, MA) in 1× DPBS for 1 h. Unreacted antibody wasremoved by washing three times with 1× DPBS and labeledcells were imaged at 40× magnification on the fluorescencemicroscope with 488/40 nm excitation and 525/50 nmemission.Flow cytometry was used as a quantitative analysis of EGFR
level in IBC SUM149 and MCF10A cells. Cells were seeded insix-well plates at a density of 3 × 105 cells per well andincubated overnight at 37 °C. Cells were trypsinized and spundown at 800 rpm for 4 min. Medium was removed and cellswere incubated with 10% formalin for 10 min before beingspun down and undergoing one wash step. 2.5 μg/mL of AlexaFluor 488 conjugated anti-EGFR IgG antibody or IgG controlin DPBS were incubated with the cells at room temperature for1 h followed by a wash step and resuspension in cold 1×DPBS. Fluorescence of 1 × 104 cells was measured with a 488nm laser and 532 nm emission for Alexa Fluor 488. Themedian fluorescence intensity of three replicates was reportedalong with a histogram of one replicate for each sample.
EGFR Inhibition. IBC SUM149 cells were seeded at 5 ×104 cells per well in an 8-well collagen coated plate. Followingovernight incubate at 37 °C, medium was replaced and cellswere incubated with 100 μM of EGF for 1 h. Cells werewashed three times with DPBS and incubated with 1 μM ofGE11-mCherry protein for 3 h. Medium was removed andcells were washed three times in 1× DPBS before fixation andDAPI staining. Fluorescence imaging and flow cytometryanalyses were conducted as described above.
SpyTag/SpyCatcher Conjugation of yCD to GE11-mCherry. A gene fragment encoding SpyCatcher wasamplified with PCR using primers SpyCatcher-F andSpyCatcher-R (Table S1). The PCR product was insertedinto pET22b-w3-amber-mCherry-his6 constructs using XhoIand BlpI sites to yield pET22b-amber-mCherry-SpyCatcher-his6. Identical methods of plasmid preparation, proteinexpression, and purification were used as described above forpET22b-amber-mCherry-his6. CuAAC was again used toconjugate GE11 to Az-mCherry-SpyCatcher using the samereactant concentrations as above. Following CuAAC, theprotein was purified and concentrated with his-tag chromatog-raphy and dialyzed in 1× PBS. To couple the SpyTag-yCD, 15
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
439

μM GE11-mCherry-SpyCatcher was reacted with 15 μM ofSpyTag-yCD in 1× PBS (pH 7.4) for 2 h followed again bypurification with his-tag chromatography.Prodrug Treatment and Cell Viability. In a 96-well
plate, cells were seeded at 10,000 cells/well and incubatedovernight. 1 μM of mCherry-yCD protein was added to cellsand incubated for 3 h. Cells were washed three times with 1×DPBS to remove protein that was not internalized, treated with500 μg/mL of 5-FC and incubated for 48 h at 37 °C.Following incubation, MTT cell proliferation assays fromThermo Fisher were performed according to the manufac-turer’s protocol.Statistical Analyses. Results were reported as mean ±
standard deviation except where noted. All experiments werereplicated at least three times with unique protein batches,except for Tat constructs which were repeated three times withtwo unique batches of protein. Statistical significance wasdetermined with an unequal variance t test. Significance wasaccepted at p < 0.05.
■ ASSOCIATED CONTENT
*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.bioconj-chem.8b00783.
SDS-PAGE of Az-mCherry expression and purification;MALDI-TOF mass spectrometry of purified GE11peptide; SDS-PAGE and MALDI-TOF mass spectrom-etry of GE11 conjugation by CuAAC; Phase micrographimages from GE11-mCherry uptake studies; Fluorescentimages of GE11-mCherry uptake in IBC SUM149 cellsat 4× magnification; Flow cytometry histograms ofmCherry and mCherry-yCD internalization; SDS-PAGEand reaction scheme of nonclustered GE11 conjugationvia amine/NHS ester chemistry; Az-mCherry-SpyCatch-er expression and GE11-mCherry-SpyCatcher conjuga-tion to SpyTag-yCD; MTT assay of cell viabilityfollowing yCD/5-FC treatment; DNA oligos andnucleotide sequences used in this study (PDF)
■ AUTHOR INFORMATION
Corresponding Authors*E-mail: [email protected].*E-mail: [email protected].
ORCIDRachel M. Lieser: 0000-0002-7597-0672Wilfred Chen: 0000-0002-6386-6958NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
Thank you to Qirun Li for assisting with gel preparation andprotein expression. RML was supported by grants from theNational Science Foundation (1510817 and 1144726). Anyopinions, findings, and conclusions or recommendationsexpressed in this material are those of the authors and donot necessarily reflect the view of the National ScienceFoundation.
■ ABBREVIATIONS
UAA, unnatural amino acid; pAzF, 4-azido-L-phenylalanine;CuAAC, copper-catalyzed alkyne−azide cycloaddition; IBC,inflammatory breast cancer; EGFR, epidermal growth factorreceptor; yCD, yeast cytosine deaminase; EGF, epidermalgrowth factor; 5-FC, 5-fluorocytosine; 5-FU, 5-fluorouracil;SDS-PAGE, sodium dodecyl sulfate polyacrylamide gelelectrophoresis; MALDI-TOF, Matrix-assisted laser desorp-tion/ionization- time-of-flight; RP-HPLC, Reversed-phasehigh-performance liquid chromatography.
■ REFERENCES(1) Algar, W. R., Prasuhn, D. E., Stewart, M. H., Jennings, T. L.,Blanco-Canosa, J. B., Dawson, P. E., and Medintz, I. L. (2011) TheControlled Display of Biomolecules on Nanoparticles: A ChallengeSuited to Bioorthogonal Chemistry. Bioconjugate Chem. 22, 825−858.(2) Colombo, M., Fiandra, L., Alessio, G., Mazzucchelli, S.,Nebuloni, M., De Palma, C., Kantner, K., Pelaz, B., Rotem, R., andCorsi, F. (2016) Tumour homing and therapeutic effect of colloidalnanoparticles depend on the number of attached antibodies. Nat.Commun. 7, 1 DOI: 10.1038/ncomms13818.(3) Zhang, Q. Y., and Reinhard, B. M. (2018) Ligand Density andNanoparticle Clustering Cooperate in the Multivalent Amplificationof Epidermal Growth Factor Receptor Activation. ACS Nano 12,10473−10485.(4) Mazzucchelli, S., Colombo, M., Verderio, P., Rozek, E., Andreata,F., Galbiati, E., Tortora, P., Corsi, F., and Prosperi, D. (2013)Orientation-Controlled Conjugation of Haloalkane DehalogenaseFused Homing Peptides to Multifunctional Nanoparticles for theSpecific Recognition of Cancer Cells. Angew. Chem., Int. Ed. 52,3121−3125.(5) Sempkowski, M., Zhu, C., Menzenski, M. Z., Kevrekidis, I. G.,Bruchertseifer, F., Morgenstern, A., and Sofou, S. (2016) StickyPatches on Lipid Nanoparticles Enable the Selective Targeting andKilling of Untargetable Cancer Cells. Langmuir 32, 8329−8338.(6) Poon, Z., Chen, S., Engler, A. C., Lee, H. I., Atas, E., vonMaltzahn, G., Bhatia, S. N., and Hammond, P. T. (2010) Ligand-Clustered ″Patchy″ Nanoparticles for Modulated Cellular Uptake andIn Vivo Tumor Targeting. Angew. Chem., Int. Ed. 49, 7266−7270.(7) Moradi, E., Vllasaliu, D., Garnett, M., Falcone, F., and Stolnik, S.(2012) Ligand density and clustering effects on endocytosis of folatemodified nanoparticles. RSC Adv. 2, 3025−3033.(8) Krall, N., da Cruz, F. P., Boutureira, O., and Bernardes, G. J. L.(2016) Site-selective protein-modification chemistry for basic biologyand drug development. Nat. Chem. 8, 102−112.(9) Cho, H., Daniel, T., Buechler, Y. J., Litzinger, D. C., Maio, Z. W.,Putnam, A. M. H., Kraynov, V. S., Sim, B. C., Bussell, S., Javahishvili,T., et al. (2011) Optimized clinical performance of growth hormonewith an expanded genetic code. Proc. Natl. Acad. Sci. U. S. A. 108,9060−9065.(10) Drake, P. M., Albers, A. E., Baker, J., Banas, S., Barfield, R. M.,Bhat, A. S., de Hart, G. W., Garofalo, A. W., Holder, P., Jones, L. C.,et al. (2014) Aldehyde Tag Coupled with HIPS Chemistry Enablesthe Production of ADCs Conjugated Site-Specifically to DifferentAntibody Regions with Distinct in Vivo Efficacy and PK Outcomes.Bioconjugate Chem. 25, 1331−1341.(11) Agarwal, P., van der Weijden, J., Sletten, E. M., Rabuka, D., andBertozzi, C. R. (2013) A Pictet-Spengler ligation for protein chemicalmodification. Proc. Natl. Acad. Sci. U. S. A. 110, 46−51.(12) Lagasse, H., Alexaki, A., Simhadri, V., Katagiri, N., Jankowski,W., Sauna, Z., and Kimichi-Sarfaty, C. (2017) Recent advances in(therapeutic protein) drug development. F1000Research 6, 113.(13) (2017) Biotech products accounted for 35% of all new FDAapprovals in 2000-16. Impact Report: Analysis and Insight intoCritical Drug Development Issues: Tufts Center for the Study of DrugDevelopment.
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
440

(14) Daugherty, A. L., and Mrsny, R. J. (2006) Formulation anddelivery issues for monoclonal antibody therapeutics. Adv. DrugDelivery Rev. 58, 686−706.(15) Estrada, L. H., Chu, S., and Champion, J. A. (2014) ProteinNanoparticles for Intracellular Delivery of Therapeutic Enzymes. J.Pharm. Sci. 103, 1863−1871.(16) Nagahara, A. H., and Tuszynski, M. H. (2011) Potentialtherapeutic uses of BDNF in neurological and psychiatric disorders.Nat. Rev. Drug Discovery 10, 209−219.(17) Grubb, J. H., Vogler, C., and Sly, W. S. (2010) New Strategiesfor Enzyme Replacement Therapy for Lysosomal Storage Diseases.Rejuvenation Res. 13, 229−236.(18) Yang, Y. L., Kitagaki, J., Dai, R. M., Tsai, Y. C., Lorick, K. L.,Ludwig, R. L., Pierre, S. A., Jensen, J. P., Davydov, I. V., Oberoi, P.,et al. (2007) Inhibitors of ubiquitin-activating enzyme (E1), a newclass of potential cancer therapeutics. Cancer Res. 67, 9472−9481.(19) Cramer, S. L., Saha, A., Liu, J. Y., Tadi, S., Tiziani, S., Yan, W.P., Triplett, K., Lamb, C., Alters, S. E., Rowlinson, S., et al. (2017)Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactiveoxygen species and suppresses tumor growth. Nat. Med. 23, 120−127.(20) Li, C., Pazgier, M., Li, C. Q., Yuan, W. R., Liu, M., Wei, G., andLu, W. Y. (2010) Systematic Mutational Analysis of PeptideInhibition of the p53-MDM2/MDMX Interactions. J. Mol. Biol.398, 200−213.(21) Los, M., Panigrahi, S., Rashedi, I., Mandal, S., Stetefeld, J.,Essmann, F., and Schulze-Osthoff, K. (2009) Apoptin, a tumor-selective killer. Biochim. Biophys. Acta, Mol. Cell Res. 1793, 1335−1342.(22) Gaynor, A. S., and Chen, W. (2017) Induced prodrugactivation by conditional protein degradation. J. Biotechnol. 260,62−66.(23) Mitragotri, S., Burke, P. A., and Langer, R. (2014) Overcomingthe challenges in administering biopharmaceuticals: formulation anddelivery strategies. Nat. Rev. Drug Discovery 13, 655−672.(24) Banerjee, S., Aher, N., Patil, R., and Khandare, J. (2012)Poly(ethylene glycol)-Prodrug Conjugates: Concept, Design, andApplications. J. Drug Delivery 2012, 1.(25) Kalkhof, S., and Sinz, A. (2008) Chances and pitfalls ofchemical cross-linking with amine-reactive N-hydroxysuccinimideesters. Anal. Bioanal. Chem. 392, 305−312.(26) Dinca, A., Chien, W. M., and Chin, M. T. (2016) IntracellularDelivery of Proteins with Cell-Penetrating Peptides for TherapeuticUses in Human Disease. Int. J. Mol. Sci. 17, 263.(27) Veronese, F. M., and Mero, A. (2008) The impact ofPEGylation on biological therapies. BioDrugs 22, 315−329.(28) Liu, J. H., Liu, M., Zheng, B., Yao, Z. P., and Xia, J. (2016)Affinity Enhancement by Ligand Clustering Effect Inspired by PeptideDendrimers-Shank PDZ Proteins Interactions. PLoS One 11,e0149580.(29) Mardilovich, A., and Kokkoli, E. (2004) Biomimetic peptide-amphiphiles for functional biomaterials: The role of GRGDSP andPHSRN. Biomacromolecules 5, 950−957.(30) Almeda, D., Wang, B. R., and Auguste, D. T. (2015)Minimizing antibody surface density on liposomes while sustainingcytokine-activated EC targeting. Biomaterials 41, 37−44.(31) Liu, J., Weller, G. E. R., Zern, B., Ayyaswamy, P. S., Eckmann,D. M., Muzykantov, V. R., and Radhakrishnan, R. (2010) Computa-tional model for nanocarrier binding to endothelium validated usingin vivo, in vitro, and atomic force microscopy experiments. Proc. Natl.Acad. Sci. U. S. A. 107, 16530−16535.(32) Thoma, G., Duthaler, R. O., Magnani, J. L., and Patton, J. T.(2001) Nanomolar E-selectin inhibitors: 700-fold potentiation ofaffinity by multivalent ligand presentation. J. Am. Chem. Soc. 123,10113−10114.(33) Elias, D. R., Poloukhtine, A., Popik, V., and Tsourkas, A. (2013)Effect of ligand density, receptor density, and nanoparticle size on celltargeting. Nanomedicine 9, 194−201.(34) Chatterjee, A., Sun, S. B., Furman, J. L., Xiao, H., and Schultz,P. G. (2013) A Versatile Platform for Single- and Multiple-Unnatural
Amino Acid Mutagenesis in Escherichia coli. Biochemistry 52, 1828−1837.(35) Liu, C. C., and Schultz, P. G. (2010) Adding New Chemistriesto the Genetic Code. Annu. Rev. Biochem. 79 (79), 413−444.(36) Niu, W., and Guo, J. T. (2013) Expanding the chemistry offluorescent protein biosensors through genetic incorporation ofunnatural amino acids. Mol. BioSyst. 9, 2961−2970.(37) Wang, N. X., Li, Y., Niu, W., Sun, M., Cerny, R., Li, Q. S., andGuo, J. T. (2014) Construction of a Live-Attenuated HIV-1 Vaccinethrough Genetic Code Expansion. Angew. Chem., Int. Ed. 53, 4867−4871.(38) Axup, J. Y., Bajjuri, K. M., Ritland, M., Hutchins, B. M., Kim, C.H., Kazane, S. A., Halder, R., Forsyth, J. S., Santidrian, A. F., Stafin, K.,et al. (2012) Synthesis of site-specific antibody-drug conjugates usingunnatural amino acids. Proc. Natl. Acad. Sci. U. S. A. 109, 16101−16106.(39) Li, Z. H., Zhao, R. J., Wu, X. H., Sun, Y., Yao, M., Li, J. J., Xu, Y.H., and Gu, J. R. (2005) Identification and characterization of a novelpeptide ligand of epidermal growth factor receptor for targeteddelivery of therapeutics. FASEB J. 19, 1978−1985.(40) Kleer, C. G., van Golen, K. L., and Merajver, S. D. (2000)Molecular biology of breast cancer metastasis - Inflammatory breastcancer: clinical syndrome and molecular determinants. Breast CancerRes. 2, 423−429.(41) Zakeri, B., Fierer, J. O., Celik, E., Chittock, E. C., Schwarz-Linek, U., Moy, V. T., and Howarth, M. (2012) Peptide tag forming arapid covalent bond to a protein, through engineering a bacterialadhesin. Proc. Natl. Acad. Sci. U. S. A. 109, E690−E697.(42) Wals, K., and Ovaa, H. (2014) Unnatural amino acidincorporation in E. coli: current and future applications in the designof therapeutic proteins. Front. Chem. 2, 1 DOI: 10.3389/fchem.2014.00015.(43) Nakamura, Y., and Ito, K. (1998) How protein reads the stopcodon and terminates translation. Genes Cells 3, 265−278.(44) Lev, D. C., Kim, L. S., Melnikova, V., Ruiz, M., Ananthaswamy,H. N., and Price, J. E. (2004) Dual blockade of EGFR and ERK1/2phosphorylation potentiates growth inhibition of breast cancer cells.Br. J. Cancer 91, 795−802.(45) Masuda, H., Zhang, D. W., Bartholomeusz, C., Doihara, H.,Hortobagyi, G. N., and Ueno, N. T. (2012) Role of epidermal growthfactor receptor in breast cancer. Breast Cancer Res. Treat. 136, 331−345.(46) Fan, M. L., Liang, X. F., Yang, D. B., Pan, X. R., Li, Z. H., Wang,H. Y., and Shi, B. Z. (2016) Epidermal growth factor receptor-targeted peptide conjugated phospholipid micelles for doxorubicindelivery. J. Drug Targeting 24, 111−119.(47) Mondal, G., Kumar, V., Shukla, S. K., Singh, P. K., and Mahato,R. I. (2016) EGFR-Targeted Polymeric Mixed Micelles CarryingGemcitabine for Treating Pancreatic Cancer. Biomacromolecules 17,301−313.(48) Chen, J., Ouyang, J., Chen, Q. J., Deng, C., Meng, F. H., Zhang,J., Cheng, R., Lang, Q., and Zhong, Z. Y. (2017) EGFR and CD44Dual-Targeted Multifunctional Hyaluronic Acid Nanogels BoostProtein Delivery to Ovarian and Breast Cancers In Vitro and InVivo. ACS Appl. Mater. Interfaces 9, 24140−24147.(49) Cheng, L., Huang, F. Z., Cheng, L. F., Zhu, Y. Q., Hu, Q., Li, L.,Wei, L., and Chen, D. W. (2014) GE11-modified liposomes for non-small cell lung cancer targeting: preparation, ex vitro and in vivoevaluation. Int. J. Nanomed. 9, 921−935.(50) Liang, L. Y., and Astruc, D. (2011) The copper(I)-catalyzedalkyne-azide cycloaddition (CuAAC) ″click″ reaction and itsapplications. An overview. Coord. Chem. Rev. 255, 2933−2945.(51) Wu, I. L., Patterson, M. A., Desai, H. E. C., Mehl, R. A., Giorgi,G., and Conticello, V. P. (2013) Multiple Site-Selective Insertions ofNoncanonical Amino Acids into Sequence-Repetitive Polypeptides.ChemBioChem 14, 968−978.(52) Johnson, D. B. F., Xu, J. F., Shen, Z. X., Takimoto, J. K.,Schultz, M. D., Schmitz, R. J., Xiang, Z., Ecker, J. R., Briggs, S. P., and
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
441

Wang, L. (2011) RF1 knockout allows ribosomal incorporation ofunnatural amino acids at multiple sites. Nat. Chem. Biol. 7, 779−786.(53) Willmarth, N. E., Baillo, A., Dziubinski, M. L., Wilson, K., Riese,D. J., and Ethier, S. P. (2009) Altered EGFR localization anddegradation in human breast cancer cells with an amphiregulin/EGFRautocrine loop. Cell. Signalling 21, 212−219.(54) Wee, P., and Wang, Z. X. (2017) Epidermal Growth FactorReceptor Cell Proliferation Signaling Pathways. Cancers 9, 52.(55) Kim, D., Yan, Y. T., Valencia, C. A., and Liu, R. H. (2012)Heptameric Targeting Ligands against EGFR and HER2 with HighStability and Avidity. PLoS One 7, e43077.(56) Brooks, H., Lebleu, B., and Vives, E. (2005) Tat peptide-mediated cellular delivery: back to basics. Adv. Drug Delivery Rev. 57,559−577.(57) Becker-Hapak, M., McAllister, S. S., and Dowdy, S. F. (2001)TAT-mediated protein transduction into mammalian cells. Methods24, 247−256.(58) Care, B. R., and Soula, H. A. (2011) Impact of receptorclustering on ligand binding. BMC Syst. Biol. 5, 48.(59) Frohlich, E. (2012) The role of surface charge in cellular uptakeand cytotoxicity of medical nanoparticles. Int. J. Nanomed. 7, 5577−5591.(60) Lee, D., Lee, Y. M., Kim, J., Lee, M. K., and Kim, W. J. (2015)Enhanced tumor-targeted gene delivery by bioreducible polyethyle-nimine tethering EGFR divalent ligands. Biomater. Sci. 3, 1096−1104.(61) Polak, A., Eschenhof, E., Fernex, M., and Scholer, H. J. (2004)Metabolic studies with 5-fluorocytosine-6-C-14 in mouse, rat, rabbit,dog and man. Chemotherapy 22, 137−153.(62) Noordhuis, P., Holwerda, U., Van der Wilt, C. L., VanGroeningen, C. J., Smid, K., Meijer, S., Pinedo, H. M., and Peters, G.J. (2004) 5-fluorouracil incorporation into RNA and DNA in relationto thymidylate synthase inhibition of human colorectal cancers. Ann.Oncol. 15, 1025−1032.(63) Longley, D. B., Harkin, D. P., and Johnston, P. G. (2003) 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev.Cancer 3, 330−338.(64) Wigmore, P. M., Mustafa, S., El-Beltagy, M., Lyons, L., Umka,J., and Bennett, G. (2010) Effects of 5-FU. Adv. Exp. Med. Biol. 678,157−164.(65) Kim, H., and Chen, W. (2016) A non-chromatographic proteinpurification strategy using Src 3 homology domains as generalizedcapture domains. J. Biotechnol. 234, 27−34.(66) Presolski, S., Hong, V. P., and Finn, M. G. (2011) Copper-Catalyzed Azide-Alkyne Click Chemistry for Bioconjugation. Curr.Protoc. Chem. Biol. 3, 153−162.(67) Amblard, M., Fehrentz, J. A., Martinez, J., and Subra, G. (2006)Methods and Protocols of modern solid phase peptide synthesis. Mol.Biotechnol. 33, 239−254.(68) Lehman, H. L., Van Laere, S. J., van Golen, C. M., Vermeulen,P. B., Dirix, L. Y., and van Golen, K. L. (2012) Regulation ofInflammatory Breast Cancer Cell Invasion through Akt1/PKB alphaPhosphorylation of RhoC GTPase. Mol. Cancer Res. 10, 1306−1318.(69) Van den Eynden, G. G., Van Laere, S. J., Van der Auwera, I.,Merajver, S. D., Van Marck, E. A., van Dam, P., Vermeulen, P. B.,Dirix, L. Y., and van Golen, K. L. (2006) Overexpression of caveolin-1and-2 in cell lines and in human samples of inflammatory breastcancer. Breast Cancer Res. Treat. 95, 219−228.(70) Ross, N. L., and Sullivan, M. O. (2016) Overexpression ofcaveolin-1 in inflammatory breast cancer cells enables IBC-specificgene delivery and prodrug conversion using histone-targetedpolyplexes. Biotechnol. Bioeng. 113, 2686−2697.
Bioconjugate Chemistry Article
DOI: 10.1021/acs.bioconjchem.8b00783Bioconjugate Chem. 2019, 30, 432−442
442