corrections - pnas · corrections biophysics and computational biology correction for “mechanism...
TRANSCRIPT

Mechanism of E-cadherin dimerization probed by NMRrelaxation dispersionYing Lia, Nicole L. Altorellia, Fabiana Bahnaa,b, Barry Honiga,b, Lawrence Shapiroa,c, and Arthur G. Palmer IIIa,1
aDepartment of Biochemistry and Molecular Biophysics, bHoward Hughes Medical Institute, and cEdward S. Harkness Eye Institute, Columbia University,New York, NY 10032
Edited by Adriaan Bax, National Institutes of Health, Bethesda, MD, and approved September 3, 2013 (received for review August 1, 2013)
Epithelial cadherin (E-cadherin), a member of the classical cadherinfamily, mediates calcium-dependent homophilic cell–cell adhesion.Crystal structures of classical cadherins reveal an adhesive dimerinterface featuring reciprocal exchange of N-terminal β-strandsbetween two protomers. Previous work has identified a putativeintermediate (called the “X-dimer”) in the dimerization pathway ofwild-type E-cadherin EC1–EC2 domains, based on crystal structuresof mutants not capable of strand swapping and on deceleration ofbinding kinetics by mutations at the X-dimer interface. In the pres-ent work, NMR relaxation dispersion spectroscopy is used to di-rectly observe and characterize intermediate states without theneed to disrupt the strand-swapped binding interface by mutagen-esis. The results indicate that E-cadherin forms strand-swappeddimers predominantly by a mechanism in which formation ofa weak and short-lived X-dimer–like state precedes the confor-mational changes required for formation of the mature strand-swapped dimeric structure. Disruption of this intermediate statethrough mutation reduces both association and dissociation ratesby factors of ∼104, while minimally perturbing affinity. The X-dimerinterface lowers the energy barrier associatedwith strand swappingand enables E-cadherins to form strand-swapped dimers at a rateconsistent with residence times in adherens junctions.
binding mechanism | domain swapping | protein-protein interaction
Epithelial cadherin (E-cadherin) is a type I cadherin in theclassical cadherin family. E-cadherin mediates calcium-
dependent homophilic cell adhesion and plays important rolesin the formation and maintenance of tissues and organs (1). E-cadherin consists of an ectodomain composed of five similartandemly repeated extracellular cadherin-like (EC) domainsconnected by conserved calcium-binding linker regions, followedby a single transmembrane segment, and an intracellular region.The ectodomain mediates assembly into adherens junctions (2),which are intercellular structures that maintain physical associ-ation between cells (3). The self-assembly of adherens junction isa cooperative process involving both homodimerization in trans(between cadherins from different cell surfaces) and lateraloligomerization in cis (between cadherins from the same cellsurface) (4, 5).The N-terminal outermost extracellular domain (EC1) is di-
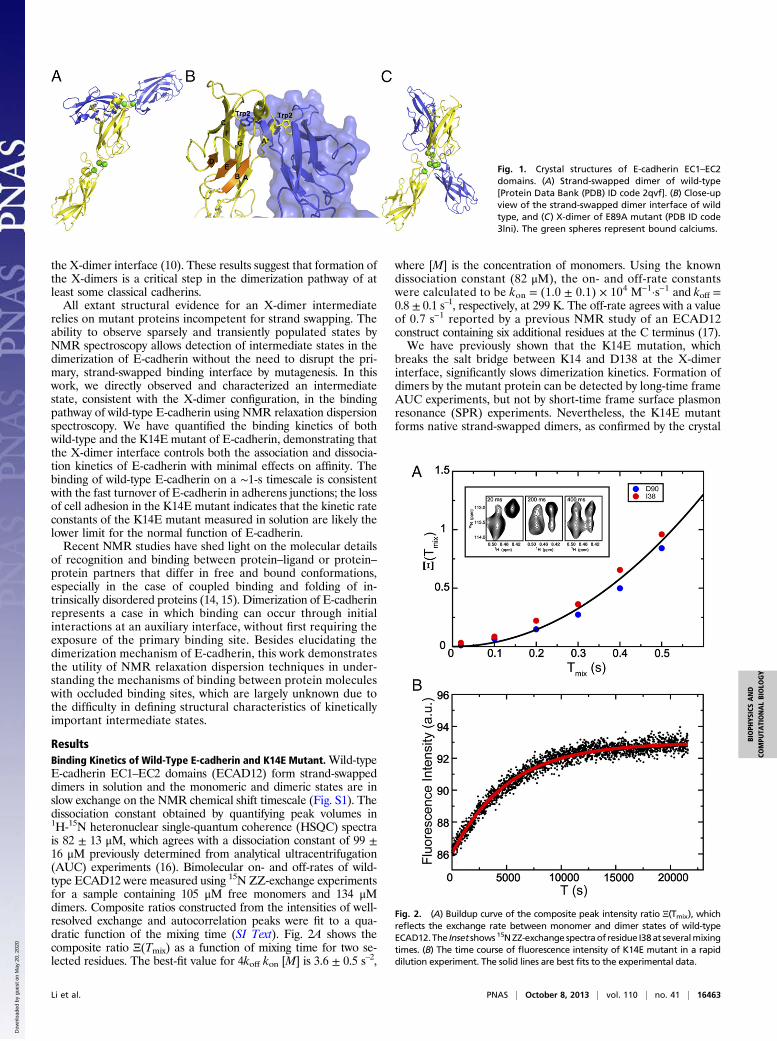
rectly involved in the trans association of E-cadherins from ap-posing cells and dictates the specificity of cell adhesion. Crystalstructures of extracellular domains reveal a dimer interfacecharacterized by reciprocal exchange of the N-terminal half ofthe first β-strand in EC1, termed the A*-strand, from a pair ofprotomers (Fig. 1 A and B) (6). In type I cadherins, a singleconserved and functionally important residue Trp2 acts as ananchor residue for exchange of the A*-strand across the dimerinterface. Monomers assume a closed conformation with Trp2and the A*-strand docked in cis into a hydrophobic pocket onthe same monomer. In dimers, protomers assume a strand flip-ped-out “open” conformation with the hydrophobic pocketavailable for structurally similar binding, but in trans, with thepartner molecule. The strand-swapped interface has been ob-served for all members of classical cadherin family (7) and is
essential for adhesive activity. Strand exchange is an example of“3D domain swapping” (8) and serves as the structural basis forthe low but differential association constants critical for thespecificity of cell adhesion (9).An alternative dimer interface was identified from crystal
structures of site-directed mutants of E-cadherin designed toabolish strand swapping (10). Highly similar X-dimer structures,so named according to the shape of the complexes, were obtainedfor W2A and E89A site-directed mutants and a variant with anAla-Ala extension at the N terminus. The X-dimer interfacecenters around the linker region between the first and second ECdomains (Fig. 1C), does not involve strand swapping, and doesnot overlap substantially with the strand-swapped interface. NMR(11) and single-molecule FRET (12) studies on mutants not ca-pable of strand swapping have detected transiently formed com-plexes, presumably the X-dimers, with association constantssignificantly lower than those of the strand-swapped dimers ofwild-type E-cadherin. At the cellular level, a recent study hassuggested a role of the X-dimer interface in the disassembly ofadherens junctions (13).Our previous work has shown that the K14E mutation, which
destabilizes the X-dimer interface by breaking the salt bridgebetween the K14 side-chain amino group and D138 main-chaincarbonyl group, results in significantly slower binding kineticsthan wild-type E-cadherin and loss of cell adhesion in cell ag-gregation assays (10). However, the K14E mutant forms thesame strand-swapped dimers with similar dissociation constantsas the wild-type protein. The M188D mutant of the type II cad-herin 6 also displays slower binding kinetics, owing to disruption of
Significance
Dimerization of transmembrane protein epithelial cadherin (E-cadherin) molecules extending from apposing cells is a criticalevent in calcium-dependent cell adhesion, a process essentialfor the formation and maintenance of tissues and organs. Thisstudy elucidates the molecular mechanism of E-cadherin di-merization by utilizing nuclear magnetic resonance spectros-copy to detect and characterize sparsely populated molecularspecies. In this mechanism, E-cadherin monomers first interactat an auxiliary interface to form a dimeric intermediate, whichis followed by the conformational changes required to formthe mature dimer. Besides contributing to the understandingof E-cadherin binding and recognition, the mechanism de-scribed in this work may illustrate a more general mechanismby which proteins with occluded binding sites interact witheach other.
Author contributions: Y.L., B.H., L.S., and A.G.P. designed research; Y.L., N.L.A., and F.B.performed research; Y.L. and A.G.P. analyzed data; and Y.L., B.H., L.S., and A.G.P. wrotethe paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314303110/-/DCSupplemental.
16462–16467 | PNAS | October 8, 2013 | vol. 110 | no. 41 www.pnas.org/cgi/doi/10.1073/pnas.1314303110
Dow
nloa
ded
by g
uest
on
May
20,
202
0 D
ownl
oade
d by
gue
st o
n M
ay 2
0, 2
020
Dow
nloa
ded
by g
uest
on
May
20,
202
0 D
ownl
oade
d by
gue
st o
n M
ay 2
0, 2
020
the X-dimer interface (10). These results suggest that formation ofthe X-dimers is a critical step in the dimerization pathway of atleast some classical cadherins.All extant structural evidence for an X-dimer intermediate
relies on mutant proteins incompetent for strand swapping. Theability to observe sparsely and transiently populated states byNMR spectroscopy allows detection of intermediate states in thedimerization of E-cadherin without the need to disrupt the pri-mary, strand-swapped binding interface by mutagenesis. In thiswork, we directly observed and characterized an intermediatestate, consistent with the X-dimer configuration, in the bindingpathway of wild-type E-cadherin using NMR relaxation dispersionspectroscopy. We have quantified the binding kinetics of bothwild-type and the K14E mutant of E-cadherin, demonstrating thatthe X-dimer interface controls both the association and dissocia-tion kinetics of E-cadherin with minimal effects on affinity. Thebinding of wild-type E-cadherin on a ∼1-s timescale is consistentwith the fast turnover of E-cadherin in adherens junctions; the lossof cell adhesion in the K14E mutant indicates that the kinetic rateconstants of the K14E mutant measured in solution are likely thelower limit for the normal function of E-cadherin.Recent NMR studies have shed light on the molecular details
of recognition and binding between protein–ligand or protein–protein partners that differ in free and bound conformations,especially in the case of coupled binding and folding of in-trinsically disordered proteins (14, 15). Dimerization of E-cadherinrepresents a case in which binding can occur through initialinteractions at an auxiliary interface, without first requiring theexposure of the primary binding site. Besides elucidating thedimerization mechanism of E-cadherin, this work demonstratesthe utility of NMR relaxation dispersion techniques in under-standing the mechanisms of binding between protein moleculeswith occluded binding sites, which are largely unknown due tothe difficulty in defining structural characteristics of kineticallyimportant intermediate states.
ResultsBinding Kinetics of Wild-Type E-cadherin and K14E Mutant.Wild-typeE-cadherin EC1–EC2 domains (ECAD12) form strand-swappeddimers in solution and the monomeric and dimeric states are inslow exchange on the NMR chemical shift timescale (Fig. S1). Thedissociation constant obtained by quantifying peak volumes in1H-15N heteronuclear single-quantum coherence (HSQC) spectrais 82 ± 13 μM, which agrees with a dissociation constant of 99 ±16 μM previously determined from analytical ultracentrifugation(AUC) experiments (16). Bimolecular on- and off-rates of wild-type ECAD12 were measured using 15N ZZ-exchange experimentsfor a sample containing 105 μM free monomers and 134 μMdimers. Composite ratios constructed from the intensities of well-resolved exchange and autocorrelation peaks were fit to a qua-dratic function of the mixing time (SI Text). Fig. 2A shows thecomposite ratio Ξ(Tmix) as a function of mixing time for two se-lected residues. The best-fit value for 4koff kon [M] is 3.6 ± 0.5 s–2,
where [M] is the concentration of monomers. Using the knowndissociation constant (82 μM), the on- and off-rate constantswere calculated to be kon = (1.0 ± 0.1) × 104 M–1·s–1 and koff =0.8 ± 0.1 s–1, respectively, at 299 K. The off-rate agrees with a valueof 0.7 s–1 reported by a previous NMR study of an ECAD12construct containing six additional residues at the C terminus (17).We have previously shown that the K14E mutation, which
breaks the salt bridge between K14 and D138 at the X-dimerinterface, significantly slows dimerization kinetics. Formation ofdimers by the mutant protein can be detected by long-time frameAUC experiments, but not by short-time frame surface plasmonresonance (SPR) experiments. Nevertheless, the K14E mutantforms native strand-swapped dimers, as confirmed by the crystal
Fig. 1. Crystal structures of E-cadherin EC1–EC2domains. (A) Strand-swapped dimer of wild-type[Protein Data Bank (PDB) ID code 2qvf]. (B) Close-upview of the strand-swapped dimer interface of wildtype, and (C) X-dimer of E89A mutant (PDB ID code3lni). The green spheres represent bound calciums.
Fig. 2. (A) Buildup curve of the composite peak intensity ratio Ξ(Tmix), whichreflects the exchange rate between monomer and dimer states of wild-typeECAD12.The Inset shows 15NZZ-exchange spectraof residue I38at severalmixingtimes. (B) The time course of fluorescence intensity of K14E mutant in a rapiddilution experiment. The solid lines are best fits to the experimental data.
Li et al. PNAS | October 8, 2013 | vol. 110 | no. 41 | 16463
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
Dow
nloa
ded
by g
uest
on
May
20,
202
0

structure (10). The dissociation constant of the K14E mutantdetermined by NMR spectroscopy is 104 ± 11 μM and agreeswith the value of 117 ± 8 μM previously determined by AUC. Wequantified the binding kinetics of the K14E mutant by moni-toring the time course of dimer dissociation after rapid dilutionby intrinsic tryptophan fluorescence spectroscopy and 1H-15Ntransverse relaxation-optimized spectroscopy (TROSY). Fig. 2Bshows the time dependence of fluorescence intensity. The fluo-rescence-based experiment has much higher sensitivity andallows simpler data analysis than the corresponding NMRexperiment (Fig. S2C). A control fluorescence experiment wasperformed on wild-type ECAD12 to account for effects of photo-bleaching (Fig. S2B and SI Text). Fitting the data to an integratedfirst-order rate equation yielded koff = (2.1 ± 0.2) × 10−4 s–1.Taking into account the 10% underestimation of the rate con-stant due to photobleaching, the corrected koff is 2.3 × 10−4 s–1.Using the known dissociation constant (104 μM), kon was calcu-lated to be 2.2 M–1·s–1. Thus, the binding kinetics of the wild-typeand the K14E mutant differ by approximately four orders ofmagnitude despite their similar binding affinities.
A Dimeric Intermediate State Detected by 1H R2 Relaxation DispersionExperiments. Chemical exchange processes on microsecond-to-millisecond timescales can be effectively detected and charac-terized by transverse relaxation dispersion NMR spectroscopy.In these experiments, kinetic, thermodynamic, and structuralinformation on the exchange process is obtained by measuringtransverse relaxation rate, R2, as a function of the number of 180°pulses, evenly spaced within a constant relaxation time period(18, 19). Amide 15N relaxation dispersion has been extensivelyused to probe chemical exchange processes; however, pre-liminary measurements indicated that the amide 1H is a moresensitive probe of exchange processes in E-cadherin. Accord-ingly, 1H relaxation data were recorded on wild-type ECAD12 attwo protein concentrations, 374 and 97 μM (as representedby the total concentration of monomeric units), using TROSY-selected 1H Carr–Purcell–Meiboom–Gill (CPMG) experiments.The high and low concentration samples contain 105 and 46 μMfree monomers, respectively, at equilibrium. Relaxation disper-
sion curves for all assigned resonances with sufficient sensitivityare shown in Fig. S3.Four residues show substantial (>5 s–1) 1H relaxation disper-
sion, including I7m, E11, K14m, and Q101m (Fig. 3A), where“m” denotes the monomeric state. For E11, monomer andstrand-swapped dimer cross-peaks are degenerate and cannot bedistinguished. The relaxation dispersion profiles are concentra-tion dependent, suggesting that the minor conformational stateis dimeric with a population that depends on protein concen-tration. Data recorded at 600- and 800-MHz 1H frequencies forthe high-concentration sample were globally fit to a two-siteexchange model (Table S1 and Fig. S4). K14m was excludedfrom fitting due to the large uncertainties in the measured re-laxation rates. The best-fit values for the global parameters arekex = 1,890 ± 130 s–1 and pm = 0.025 ± 0.003, where kex is thesum of forward pseudo first-order and reverse kinetic rate con-stants and pm is the population of the minor conformationalstate. Fitting individual residues independently does not statis-tically improve the fit according to an F test. Best-fit values forkex and ΔωH, which is the difference in amide 1H chemical shiftsbetween the major and minor conformational states, determinedfrom the high-concentration sample were used to fit the datarecorded on the low-concentration sample at 600 MHz andyielded pm = 0.017 ± 0.003.All four residues showing significant dispersion are located at
or close to the X-dimer interface defined by X-ray crystallogra-phy (Fig. 3B). K14 and Q101 are directly involved in key contactsat the X-dimer interface: K14 side chain forms a salt bridge withthe main-chain carbonyl group of D138, whereas the Q101 sidechain forms hydrogen bonds with the D100 main chain and theN143 side chain. D138 and N143 are located at or close to theBC loop comprising residues 136–140 in the EC2 domain. Severalresidues in this loop and neighboring regions show broadened15N and 1H resonances (Fig. S5), indicative of microsecond-to-millisecond timescale chemical exchange processes, but signalintensities were too low to permit determination of kineticparameters. The crystal structure reveals an additional saltbridge between R105 and E199; however, these two residuesdo not show relaxation dispersion. This difference could reflect
Fig. 3. (A) 1H relaxation dispersion profiles of wild-type ECAD12 at two different protein concentrations and K14E mutant (blue). The “m” denotes themonomeric state. The total monomer concentrations were 374 μM (red) and 97 μM (purple) for two wild-type samples, respectively; the concentration of theK14E mutant was 375 μM. τcp is the spacing between 180° pulses. For clarity, only data recorded at 600 MHz are shown. (B) X-dimer interface with residuesshowing relaxation dispersion highlighted by stick representation. The green spheres represent bound calciums.
16464 | www.pnas.org/cgi/doi/10.1073/pnas.1314303110 Li et al.
Dow
nloa
ded
by g
uest
on
May
20,
202
0

stabilization of the X-dimer either by crystal packing forces or themutations necessary to abolish strand swapping.Residues E11 and I7 are not involved in direct contacts with
the other protomer. However, E11 is spatially close to the linkerregion and, together with Q101, binds to one of the three cal-cium ions located at the linker region. E11 has been suggested toplay a role in strand swapping by serving as one of the two anchorpoints of the short A/A* strand, introducing conformationalstrain in the monomer structure, and enhancing the relativestability of the strand-swapped dimer conformation (20). I7 islocated right after two prolines, P5 and P6, that serve as thehinge connecting the A*-strand with the remaining portion ofthe A-strand. Formation of X-dimers results in large perturba-tions in the I7 1H chemical shift, ΔωH = 0.45 ppm, as determinedfrom 1H relaxation dispersion data, but not in the 15N chemicalshift, ΔωN ≈ 0, as indicated by lack of significant dispersion in15N experiments. These results suggest that chemical exchangeline broadening reflects changes in the local environment, suchas positioning of aromatic rings, due to X-dimer formation,rather than dihedral angle or other conformational changes,because ΔωH >> ΔωN. In contrast, in the strand-swapped dim-ers, 15N is shifted downfield by 2.28 ppm and 1H is shifteddownfield by 0.30 ppm with respect to the monomer resonances.These results are consistent with conformational changes, be-cause ΔωH ≈ ΔωN, measured in angular frequency units. Overall,the residues showing relaxation dispersion or line broadeningoverlap with those located at the X-dimer interface identifiedfrom crystal structures (Fig. S6), indicating that the minor con-formational state is structurally similar to the X-dimer.
The Intermediate State Is Undetectable in the K14E Mutant. To testwhether the K14E mutation affects the minor conformationalstate observed in the wild-type samples, we performed 1HCPMG experiments on the mutant under the same conditions.The K14E mutant sample contained 116 μM free monomers and129 μM dimers. None of the residues undergoing exchangeprocesses in wild-type ECAD12 shows relaxation dispersion inthe K14E mutant (Fig. 3A), indicating that either the populationof the minor conformational state is below the detection limit of∼0.5% or the exchange process shifts to an NMR chemical shifttimescale too fast to yield detectable line broadening. Changes intimescale can result from either smaller chemical shift differ-ences between exchanging states or increases in the kinetic ex-change rate, which is predominated by the dissociation rate ofthe X-dimer. Both smaller perturbations in chemical shifts andhigher dissociation rates correspond to weaker interactions andlower stability of the minor conformational state. Overall, theresults indicate that the salt bridge between K14 and D138,observed in the crystal structure, is also essential for the stabilityof the minor conformational state observed by NMR, providingadditional support for the conclusion that the minor state ishighly similar to the X-dimer crystal structures.
Kinetic Model for ECAD12 Association and Dissociation. A two-stepmodel has been proposed for E-cadherin dimerization based onthe qualitative difference in binding kinetics of wild-type andK14E mutant ECAD12, detected by AUC and SPR experiments(10). In this model, monomers interact bimolecularly to forma hypothetical X-dimer intermediate followed by a unimolecularrearrangement to form the strand-swapped dimer. The currentstudy elaborates on this model by detecting and characterizingthe X-dimer intermediate state using relaxation dispersion NMRexperiments. Combined with ZZ-exchange NMR experiments,the kinetic parameters were determined at bulk equilibrium.Without loss of generality, ECAD12 dimerization can be
represented by the kinetic scheme shown in Fig. 4. This schemeconsists of the X-dimer–dependent pathway and an alternativepathway collectively representing any other, possibly multistep,
binding mechanisms. The ∼104 reduction in the kinetic rateconstants for the K14E mutant, in which formation of X-dimer isat least partially abrogated, compared with wild-type ECAD12,indicates that X-dimer–dependent pathway is the major if notthe only pathway under our experimental conditions. We notethat, for bimolecular reactions involving multiple pathways andmechanisms, the relative fluxes through each pathway are gen-erally concentration dependent (21).In the major pathway, under steady-state conditions, the
overall association rate constant is as follows:
kon =k1k2
k−1 + k2: [1]
The 1H relaxation dispersion experiments on the wild-typesample at 374 μM yielded 2k1[M] + k−1 = kex = 1,890 s−1, 2k1[M]/kex = pm = 0.025. The ZZ-exchange experiment yielded kon =1.0 × 104 M−1·s–1 and koff = 0.8 ± 0.1 s–1. Combining these resultsgives k1 = 2.3 × 105 M–1·s–1, k–1 = 1.84 × 103 s–1, and k2 = 86 s–1.The value of k1 falls into the 105 to 106 M–1·s–1 range of the theo-retically estimated bimolecular on-rates for protein–protein associ-ation in the absence of long-range forces (22). Because k–1 � k2,
kon ≈�k1k−1
�k2; [2]
namely, the bimolecular on-rate of the overall binding reaction isproportional to the association constant of the intermediatestate, KI = k1/k–1, and the rate of conversion from X-dimers tostrand-swapped dimers, k2. For the dissociation pathway, k–2defines the rate-limiting step; therefore, k–2 ≈ koff = 0.8 s−1.Fig. 5 shows free-energy diagrams for dimerization of wild-
type and K14E mutant ECAD12. No intermediate state wasobserved experimentally by relaxation dispersion for the K14Emutant, confirming that this mutation decreases KI by disruptingthe K14-D138 salt bridge. In addition, the drastic reduction inboth kon and koff without significant change in affinity of theK14E mutant suggests that the K14E mutation perturbs thetransition state associated with the conversion of X-dimers tostrand-swapped dimers with minimal effects on the structuresand relative stability of monomers and strand-swapped dimers.As evident from Fig. 5, the transition state must be perturbed bythe mutation in order for k–2 to decrease, whereas the change ink2 relies on the change in the relative free energy of the X-dimerand the transition state. Destabilization of the intermediate stateand elevation of the energy of the transition state collectivelycontribute to the nearly four orders of magnitude change in kon,whereas the change in koff solely results from decrease in k−2. Theapproximate expression for dissociation constant is Kd = koff /kon ≈(1/KI)(k−2/k2). Destabilization of X-dimer has the exactly oppositeeffects on 1/KI and k–2/k2, and therefore no net effect on Kd, con-sistent with the nearly unchanged Kd of the K14E mutant.For wild-type ECAD12, the binding reaction proceeds in two
steps via major pathway shown in the scheme in Fig. 4. The ki-netically favorable X-dimer state accumulates, because theactivation energy for the second step is larger than that for the
Fig. 4. Pathways of ECAD12 dimerization. M denotes monomers, X-D denotesX-dimer–like intermediates, and SS-D denotes strand-swapped dimers.
Li et al. PNAS | October 8, 2013 | vol. 110 | no. 41 | 16465
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
Dow
nloa
ded
by g
uest
on
May
20,
202
0

essentially diffusion-controlled first step. The molecular mech-anism of diffusion-controlled protein–protein association hasbeen extensively investigated by both experimental and com-putational studies (22, 23). Long-range electrostatic inter-actions contribute to the on-rate (24, 25) and short-rangeinteractions, including hydrophobic and van der Waals inter-actions, hydrogen bonds, and salt bridges, dictate the off-rate.The conformational changes associated with strand swappingoccur during the rate-limiting unimolecular second step. Forthe K14E mutant, neither the binding mechanism nor the rateconstants of each elementary reaction are known. Therefore,the free-energy diagram (Fig. 5B) shows a single overall ac-tivation barrier for simplicity, although intermediate statescould exist at levels undetectable by the relaxation dispersionexperiments.
DiscussionWe have used 1H relaxation dispersion and 15N ZZ-exchangeNMR spectroscopy to investigate the pathway of E-cadherintrans association. The studies were conducted on an E-cadherinconstruct containing the EC1 and EC2 domains and an intactcalcium-binding linker region between the two domains. Weobserved significant relaxation dispersion for residues that lie atthe X-dimer interface, resulting from millisecond timescale chem-ical exchange processes between monomers and the dimeric in-termediate state. In contrast, Trp2 side-chain He resonance doesnot show detectable relaxation dispersion, suggesting that itadopts a conformation similar to the monomer state, i.e., a closedconformation, in the intermediate state. This work has provideddirect evidence for a dimeric intermediate state, with key struc-tural features consistent with crystal structures of X-dimers, in thedimerization pathway for wild-type E-cadherin. It also uncovereda mechanism by which the kinetic barrier associated with strandswapping, or in general, 3D domain swapping, is lowered. In thismechanism, an auxiliary interface is used to form a short-liveddimeric intermediate state, which leads to an energetically fa-vorable transition state that partially resembles the intermediatestate and accelerates overall binding kinetics. Formation of the X-dimer state leads to changes in the conformational energy land-scape, which lower the effective activation barrier associated withstrand swapping by localizing the two β-strands in close proximity.A key factor in this mechanism is that the bimolecular intermediatestate needs to be formed with high enough association constant;otherwise, the low population of the intermediate state becomesthe kinetic bottleneck of the overall reaction (26). This factor isespecially a problem for proteins with occluded binding sites. In thecase of E-cadherin, the X-dimer interface allows formation ofa dimeric intermediate state that is thermodynamically stable
enough for fast overall binding kinetics, yet not so stable as toprevent the formation of the final strand-swapped dimers. TheX-dimer interface does not overlap substantially with the strand-swapped dimer interface, which may allow tuning of bindingkinetics without affecting the association constant of strand-swapped dimers.Although the current data mainly support an X-dimer–
dependent mechanism for wild-type E-cadherin, other mech-anisms cannot be completely excluded, because binding is notcompletely kinetically prohibited for the K14E mutant. Theon- and off-rates of the K14E mutant represent the upper limit ofthe kinetic constants of alternative pathways, although the exactbinding mechanisms are not known for these minor pathways.Previously, a monomeric and A*-strand flipped-out state witha population of 0.032 ± 0.004 was detected and characterized ina construct of mouse type II cadherin 8 containing only the EC1domain, using 15N relaxation dispersion and solvent exchangemeasurements (27). The presence of this sparsely populated mo-nomeric “open” species suggests an alternative binding mecha-nism, at least for cadherin 8, in which two “open” monomerscollisionally interact to form the strand-swapped dimer. However,because the cadherin 8 construct is incapable of forming X-dimerstate owing to the lack of an intact linker region and the EC2domain, the importance of this mechanism in the context of wild-type protein consisting of at least EC1 and EC2 domains, asstudied herein, is not known. No relaxation dispersion was ob-served for side-chain Ne and He resonances of Trp2 in ECAD12monomers, suggesting that the populations of any “flipped-out”states of ECAD12 are below the limit of detection.E-cadherin is a major component of adherens junctions, which
are constantly renewing structures with individual cadherinmolecules entering and exiting the junctions (28). Understandingthe molecular mechanisms underlying the turnover of cadherinsin the adherens junctions requires knowledge about not onlythermodynamics but also kinetics of cadherin association anddissociation. The apparent half-residence times of E-cadherins inadherens junctions have been determined by fluorescence pho-tobleaching experiments to range from 50 s to 4 min dependingon the type of cells used for the measurements (2, 29). The ∼1-stimescale for E-cadherin association and dissociation in solutionallows the trans dimers to form within the lifetime of E-cadherinsin junctions; consequently, strand swapping does not presenta kinetic barrier for the cadherin turnover. The resulting favor-able kinetic properties allow strand-swapped dimers to form anddissociate on a timescale required for physiological functions.The measurements described here are carried out in 3D bulk
solution, whereas intercellular cadherin dimerization occurs inthe quasi-2D environment of a cell surface. We have previouslyderived an equation that relates 3D to 2D binding affinities (30,31). A critical parameter is the thickness of the reaction shellbetween the two cell membranes that defines a region accessibleto the EC1 domains of cadherins located on apposed membranesurfaces (31). The 2D affinity of E-cadherin is ∼100 μm–2 and isin the range of cadherin densities on cell surfaces (9). Thus, theexperimental conditions used in the current work, where con-centrations are in the range defined by the 3D affinity approxi-mate the conditions on cell surfaces.
ConclusionsWe have defined the dimerization pathway of E-cadherin ex-tracellular domains EC1–EC2 by NMR and intrinsic Trp fluo-rescence spectroscopy. An intermediate state with structuralfeatures consistent with previously determined X-dimer struc-tures was detected and characterized by 1H R2 relaxation dis-persion experiments. Mutation of the key residue K14 at theX-dimer interface to E, disrupting the salt bridge between K14and D138, decreases the population of the intermediate to anundetectable level, consistent with the destabilizing effects of the
Fig. 5. The free-energy diagrams of (A) wild type and (B) K14E mutant ofECAD12 at 299 K and 1 M standard concentration. M represents monomericstate, X-D represents X-dimer–like intermediate state, SS-D representsstrand-swapped dimeric state, and TS represents transition state.
16466 | www.pnas.org/cgi/doi/10.1073/pnas.1314303110 Li et al.
Dow
nloa
ded
by g
uest
on
May
20,
202
0

mutation, and dramatically reduces both forward and reversebinding kinetics compared with wild-type protein. These resultssupport an X-dimer–dependent dimerization pathway and definethe rate constants of the elementary reactions. The existence ofX-dimer interface accelerates otherwise extremely slow associa-tion and dissociation kinetics, exhibited by the K14E mutant,allowing E-cadherins to form trans dimers at a rate consistentwith residence times in the adherens junctions. Applying theseexperimental methods to other members of cadherin family willlead to better understanding of the general mechanisms ofcadherin binding and recognition.
Materials and MethodsNMR Samples. The constructs contain 1–213 residues of the mature form ofmouse E-cadherin. Experimental details on protein expression and purifica-tion have been included in SI Text. The NMR buffer comprises 5% (vol/vol)D2O/95% (vol/vol) H2O, 10 mM CaCl2, 120 mM NaCl, 10 mM Tris·HCl, pH 7.9.Protein concentration was determined by absorbance at 280 nm using anextinction coefficient of 21,430 M–1·cm–1.
NMR Spectroscopy. All NMR experiments were performed at 299.2 K unlessstated otherwise and on Bruker 600-, 800-, and 900-MHz spectrometersequipped with triple-resonance z-gradient cryogenic probes. d6-meth-anol was used to calibrate sample temperature (32). Backbone resonanceassignments were obtained using triple-resonance correlation experi-ments with a TROSY detection scheme, including HNCO, HNCA, HN(CO)CA,HNCACB, and HN(CO)CACB, together with 3D 15N-edited NOESY-TROSY(33), on a sample of 2H-, 13C-, 15N-labeled wild-type ECAD12 containing0.8 mM protein. Additional experimental details have been includedin SI Text.
The relative quantity of monomers and dimers for a given sample wasdetermined from the ratio of peak volumes in 1H-15N HSQC spectra acquiredwith 6-s recycle delay. The ratio was corrected for the differential loss of
magnetization for monomers and dimers during magnetization transferperiods in the sequence according to a previously reported method (34).
The pulse sequence for 15N ZZ-exchange experiment is depicted in Fig.S7. The experiments were performed on a sample containing 374 μM 2H-,15N-labeled wild-type ECAD12 at 600 MHz (see SI Text for additionalexperimental details).
The pulse sequence for 1H CPMG experiments is depicted in Fig. S8. Datawere recorded on two 2H-, 15N-labeled wild-type ECAD12 samples at twoprotein concentrations, 374 and 97 μM, respectively, and one 2H-, 15N-labeledK14E sample containing 375 μM protein. For the 374 μM wild-type sample,data were recorded at 600- and 800-MHz 1H frequencies. For other samples,data were recorded only at 600-MHz 1H frequency. The experiments wereperformed with constant relaxation periods of 40 and 20 ms (see SI Text foradditional experimental details).
Fluorescence Experiments. The experimentswere performed at 299K in bufferscontaining 10 mM CaCl2, 120 mM NaCl, 10 mM Tris·HCl, pH 7.9, identical tothose for NMR experiments. The excitation wavelength was 295 nm, and thedetection wavelength was 330 nm. The initial concentration of the K14Esample was 223 μM and the sample was diluted to a final concentration of3 μM rapidly and the fluorescence intensity was recorded every 10 s with 1-ssampling time for 6 h. The experiment was repeated once with a lower finalconcentration, 2 μM. A control experiment was performed on a wild-typesample containing 306 μM protein, which was rapidly diluted to 3 μM.
ACKNOWLEDGMENTS. We thank Oliver Harrison for providing the K14Esample for fluorescence experiments. This work was supported by NationalInstitutes of Health (NIH) Grants GM059273 (to A.G.P.) and GM062270 (toL.S.). A.G.P. and B.H. are members of the New York Structural Biology Center(NYSBC). The data collected at NYSBC were made possible by a grant fromthe New York State Office of Science, Technology and Academic Researchand Office of Research Infrastructure Programs/NIH Facility ImprovementGrant CO6RR015495. The 900-MHz NMR spectrometers were purchased withfunds from NIH Grant P41GM066354, the Keck Foundation, New York StateAssembly, and US Department of Defense. B.H. is an investigator of theHoward Hughes Medical Institute.
1. Takeichi M (1995) Morphogenetic roles of classic cadherins. Curr Opin Cell Biol 7(5):
619–627.2. Hong S, Troyanovsky RB, Troyanovsky SM (2010) Spontaneous assembly and active
disassembly balance adherens junction homeostasis. Proc Natl Acad Sci USA 107(8):
3528–3533.3. Meng W, Takeichi M (2009) Adherens junction: Molecular architecture and regula-
tion. Cold Spring Harb Perspect Biol 1(6):a002899.4. Harrison OJ, et al. (2011) The extracellular architecture of adherens junctions revealed
by crystal structures of type I cadherins. Structure 19(2):244–256.5. Wu Y, et al. (2010) Cooperativity between trans and cis interactions in cadherin-
mediated junction formation. Proc Natl Acad Sci USA 107(41):17592–17597.6. Parisini E, Higgins JM, Liu JH, Brenner MB, Wang JH (2007) The crystal structure of
human E-cadherin domains 1 and 2, and comparison with other cadherins in the
context of adhesion mechanism. J Mol Biol 373(2):401–411.7. Posy S, Shapiro L, Honig B (2008) Sequence and structural determinants of strand
swapping in cadherin domains: Do all cadherins bind through the same adhesive
interface? J Mol Biol 378(4):954–968.8. Bennett MJ, Schlunegger MP, Eisenberg D (1995) 3D domain swapping: A mechanism
for oligomer assembly. Protein Sci 4(12):2455–2468.9. Chen CP, Posy S, Ben-Shaul A, Shapiro L, Honig BH (2005) Specificity of cell-cell ad-
hesion by classical cadherins: Critical role for low-affinity dimerization through beta-
strand swapping. Proc Natl Acad Sci USA 102(24):8531–8536.10. Harrison OJ, et al. (2010) Two-step adhesive binding by classical cadherins. Nat Struct
Mol Biol 17(3):348–357.11. Häussinger D, et al. (2002) Calcium-dependent homoassociation of E-cadherin by
NMR spectroscopy: Changes in mobility, conformation and mapping of contact re-
gions. J Mol Biol 324(4):823–839.12. Sivasankar S, Zhang Y, Nelson WJ, Chu S (2009) Characterizing the initial encounter
complex in cadherin adhesion. Structure 17(8):1075–1081.13. Hong S, Troyanovsky RB, Troyanovsky SM (2011) Cadherin exits the junction by
switching its adhesive bond. J Cell Biol 192(6):1073–1083.14. Wright PE, Dyson HJ (2009) Linking folding and binding. Curr Opin Struct Biol 19(1):
31–38.15. Sugase K, Dyson HJ, Wright PE (2007) Mechanism of coupled folding and binding of
an intrinsically disordered protein. Nature 447(7147):1021–1025.16. Katsamba P, et al. (2009) Linking molecular affinity and cellular specificity in cad-
herin-mediated adhesion. Proc Natl Acad Sci USA 106(28):11594–11599.17. Häussinger D, et al. (2004) Proteolytic E-cadherin activation followed by solution NMR
and X-ray crystallography. EMBO J 23(8):1699–1708.
18. Palmer AG, 3rd, Kroenke CD, Loria JP (2001) Nuclear magnetic resonance methods forquantifying microsecond-to-millisecond motions in biological macromolecules. Meth-ods Enzymol 339:204–238.
19. Mittermaier A, Kay LE (2006) New tools provide new insights in NMR studies ofprotein dynamics. Science 312(5771):224–228.
20. Vendome J, et al. (2011) Molecular design principles underlying β-strand swapping inthe adhesive dimerization of cadherins. Nat Struct Mol Biol 18(6):693–700.
21. Hammes GG, Chang YC, Oas TG (2009) Conformational selection or induced fit: A fluxdescription of reaction mechanism. Proc Natl Acad Sci USA 106(33):13737–13741.
22. Schreiber G, Haran G, Zhou HX (2009) Fundamental aspects of protein-protein asso-ciation kinetics. Chem Rev 109(3):839–860.
23. Schreiber G (2002) Kinetic studies of protein-protein interactions. Curr Opin StructBiol 12(1):41–47.
24. Selzer T, Albeck S, Schreiber G (2000) Rational design of faster associating and tighterbinding protein complexes. Nat Struct Biol 7(7):537–541.
25. Kiel C, Selzer T, Shaul Y, Schreiber G, Herrmann C (2004) Electrostatically optimizedRas-binding Ral guanine dissociation stimulator mutants increase the rate ofassociation by stabilizing the encounter complex. Proc Natl Acad Sci USA 101(25):9223–9228.
26. Bosshard HR (2001) Molecular recognition by induced fit: How fit is the concept?News Physiol Sci 16:171–173.
27. Miloushev VZ, et al. (2008) Dynamic properties of a type II cadherin adhesive domain:Implications for the mechanism of strand-swapping of classical cadherins. Structure16(8):1195–1205.
28. Nishimura T, Takeichi M (2009) Remodeling of the adherens junctions during mor-phogenesis. Curr Top Dev Biol 89:33–54.
29. de Beco S, Gueudry C, Amblard F, Coscoy S (2009) Endocytosis is required for E-cad-herin redistribution at mature adherens junctions. Proc Natl Acad Sci USA 106(17):7010–7015.
30. Wu Y, Vendome J, Shapiro L, Ben-Shaul A, Honig B (2011) Transforming binding af-finities from three dimensions to two with application to cadherin clustering. Nature475(7357):510–513.
31. Wu Y, Honig B, Ben-Shaul A (2013) Theory and simulations of adhesion receptor di-merization on membrane surfaces. Biophys J 104(6):1221–1229.
32. Findeisen M, Brand T, Berger S (2007) A 1H-NMR thermometer suitable for cryop-robes. Magn Reson Chem 45(2):175–178.
33. Zhu G, Xia Y, Lin D, Gao X (2004) TROSY-based correlation and NOE spectroscopy forNMR structural studies of large proteins. Methods Mol Biol 278:57–78.
34. Hu K, Westler WM, Markley JL (2011) Simultaneous quantification and identificationof individual chemicals in metabolite mixtures by two-dimensional extrapolated time-zero 1H-13C HSQC (HSQC0). J Am Chem Soc 133(6):1662–1665.
Li et al. PNAS | October 8, 2013 | vol. 110 | no. 41 | 16467
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
Dow
nloa
ded
by g
uest
on
May
20,
202
0

Corrections
BIOPHYSICS AND COMPUTATIONAL BIOLOGYCorrection for “Mechanism of E-cadherin dimerization probedby NMR relaxation dispersion,” by Ying Li, Nicole L. Altorelli,Fabiana Bahna, Barry Honig, Lawrence Shapiro, and Arthur G.Palmer III, which appeared in issue 41, October 8, 2013, of ProcNatl Acad Sci USA (110:16462–16467; first published September25, 2013; 10.1073/pnas.1314303110).The authors note that the second sentence in the Acknowl-
edgments, “This work was supported by National Institutes ofHealth (NIH) Grants GM059273 (to A.G.P.) and GM062270 (toL.S.)” should instead appear as “This work was supported byNational Institutes of Health (NIH) Grants GM059273 (to A.G.P.)and GM062270 (to L.S.) and by National Science FoundationGrant MCB-0918535 (to B.H.).”
www.pnas.org/cgi/doi/10.1073/pnas.1319465110
DEVELOPMENTAL BIOLOGYCorrection for “A Hox gene controls lateral line cell migrationby regulating chemokine receptor expression downstream ofWnt signaling,” by Marie A. Breau, David G. Wilkinson, andQiling Xu, which appeared in issue 42, October 15, 2013, of ProcNatl Acad Sci USA (110:16892–16897; first published September30, 2013; 10.1073/pnas.1306282110).The authors note that, due to a printer’s error, refs. 33–38 were
numbered incorrectly. The citations to the reference numbers arecorrect in the text. Below is the correct order for refs. 33–38.
33. Gamba L, Cubedo N, Ghysen A, Lutfalla G, Dambly-Chaudière C (2010) Estrogen re-ceptor ESR1 controls cell migration by repressing chemokine receptor CXCR4 in thezebrafish posterior lateral line system. Proc Natl Acad Sci USA 107(14):6358–6363.
34. Westerfield M (1993) The Zebrafish Book (Univ of Oregon Press, Eugene).35. Kwan KM, et al. (2007) The Tol2kit: A multisite gateway-based construction kit for
Tol2 transposon transgenesis constructs. Dev Dyn 236(11):3088–3099.36. Xu Q, Wilkinson DG (1998) In situ hybridization of mRNA with hapten labelled
probes. In Situ Hybridization: A Practical Approach, ed Wilkinson DG (Oxford UnivPress, Oxford), 2nd Ed, pp 87–106.
37. Robu ME, et al. (2007) p53 activation by knockdown technologies. PLoS Genet 3(5):e78.
38. Gerety SS, Wilkinson DG (2011) Morpholino artifacts provide pitfalls and reveala novel role for pro-apoptotic genes in hindbrain boundary development. Dev Biol350(2):279–289.
www.pnas.org/cgi/doi/10.1073/pnas.1319266110
IMMUNOLOGYCorrection for “IFI16 senses DNA forms of the lentiviral replica-tion cycle and controls HIV-1 replication,” by Martin R. Jakobsen,Rasmus O. Bak, Annika Andersen, Randi K. Berg, Søren B.Jensen, Jin Tengchuan, Anders Laustsen, Kathrine Hansen, LarsØstergaard, Katherine A. Fitzgerald, T. Sam Xiao, Jacob G.Mikkelsen, Trine H. Mogensen, and Søren R. Paludan, whichappeared in issue 48, November 26, 2013, of Proc Natl Acad SciUSA (110:E4571–E4580; first published October 23, 2013; 10.1073/pnas.1311669110).The authors note that the author name Jin Tengchuan should
instead appear as Tengchuan Jin. The corrected author line appearsbelow. The online and print versions have been corrected.
Martin R. Jakobsen, Rasmus O. Bak, Annika Andersen,Randi K. Berg, Søren B. Jensen, Tengchuan Jin,Anders Laustsen, Kathrine Hansen, Lars Østergaard,Katherine A. Fitzgerald, T. Sam Xiao, Jacob G. Mikkelsen,Trine H. Mogensen, and Søren R. Paludan
www.pnas.org/cgi/doi/10.1073/pnas.1320190110
MATHEMATICSCorrection for “Violating the Shannon capacity ofmetric graphs withentanglement,” by Jop Briët, Harry Buhrman, and Dion Gijswijt,which appeared in issue 48, November 26, 2013, of Proc Natl Acad SciUSA (110:19227–19232; first published December 24, 2012;10.1073/pnas.1203857110).The authors note that, due to a printer’s error, on page 19227,
left column, first full paragraph, line 6 “Gn” should instead appearas “a∈R”.Also, on page 19227, left column, first full paragraph, line 7
“x∈V ðHnÞ” should instead appear as “x∈ S”. Both the onlinearticle and the print article have been corrected.
www.pnas.org/cgi/doi/10.1073/pnas.1301191110
www.pnas.org PNAS | November 26, 2013 | vol. 110 | no. 48 | 19651–19652
CORR
ECTIONS

ASTRONOMYCorrection for “Prevalence of Earth-size planets orbiting Sun-likestars,” by Erik A. Petigura, Andrew W. Howard, and Geoffrey W.Marcy, which appeared in issue 48, November 26, 2013, ofProc Natl Acad Sci USA (110:19273–19278; first publishedNovember 4, 2013; 10.1073/pnas.1319909110).The authors note that the following statement should be
added as a Note Added in Proof: “Estimates of the occurrence ofEarth analog planets appear in several previous works includingCatanzarite and Shao (25), Traub (26), and Dong and Zhu (27).These estimates, which range from 1% to 34%, were built uponearly catalogs of Kepler planet candidates (based on less than1.3 years of photometry). These estimates did not address surveycompleteness with injection and recovery or uncertain stellarradii with spectroscopy.” The online version has been updated toinclude the following three references:
25. Catanzarite J, Shao M (2011) The occurrence rate of earth analog planets orbitingsun-like stars. Astrophys J 738(2):151–160.
26. Traub W (2012) Terrestrial, habitable-zone exoplanet frequency from Kepler. As-trophys J 745(1):20–29.
27. Dong S, Zhu Z (2013) Fast rise of “Neptune-size” planets (4-8 R_Earth) from P ∼ 10 to∼250 days—Statistics of Kepler planet candidates up to ∼0.75 AU. Astrophys J 778(1):53–63.
www.pnas.org/cgi/doi/10.1073/pnas.1321363110
19652 | www.pnas.org