design and synthesis of amine building blocks and - diva portal
TRANSCRIPT

Design and Synthesis ofAmine Building Blocks and
Protease Inhibitors
Susana Ayesa Alvarez
Stockholm University

© Susana Ayesa Alvarez, Stockholm 2008ISBN 978-91-7155-690-5Printed in Sweden by Universitetsservice US-AB, Stockholm 2008Department of Organic ChemistryStockholm University

To my family


Abstract
The first part of this thesis addresses the design and synthesis of aminebuilding blocks accomplished by applying two different syntheticprocedures, both of which were developed using solid-phase chemistry.
Chapter 1 presents the first of these methods, entailing a practical solid-phase parallel synthesis route to N-monoalkylated aminopiperidines andaminopyrrolidines achieved by selective reductive alkylation of primaryand/or secondary amines. Solid-phase NMR spectroscopy was used tomonitor the reactions for which a new pulse sequence was developed.
The second method, reported in Chapter 2, involves a novel approach tothe synthesis of secondary amines starting from reactive alkyl halides andazides. The convenient solid-phase protocol that was devised made use ofthe Staudinger reaction in order to accomplish highly efficient alkylations ofN-alkyl phosphimines with reactive alkyl halides.
The second part of the thesis describes the design and synthesis of threeclasses of protease inhibitors targeting the cysteine proteases cathepsins Sand K, and the serine protease hepatitis C virus (HCV) NS3 protease.
Chapter 4 covers the design, solid-phase synthesis, and structure-activityrelationships of 4-amidofurane-3-one P1-containing inhibitors of cathepsin Sand the effects of P3 sulfonamide groups on the potency and selectivitytowards related cathepsin proteases. This work resulted in the discovery ofhighly potent and selective inhibitors of cathepsin S.
Two parallel solid-phase approaches to the synthesis of a series ofaminoethylamide inhibitors of cathepsin K are presented in Chapter 5.
Finally, Chapter 6 reports peptide-based HCV NS3 protease inhibitorscontaining a non-electrophilic allylic alcohol moiety as P1 group and alsooutlines efforts to incorporate this new template into low-molecular-weightdrug-like molecules.


List of Papers
This thesis is based on the following papers, which are referred to in the textby their Roman numerals:
I. An Expeditious Library Synthesis of N-MonoalkylatedAminopiperidines and –pyrrolidinesSusana Ayesa, Dimitris Argyopoulus, Tatiana Maltseva, ChristianSund, and Bertil SamuelssonEur. J. Org. Chem., 2004, 2723-2737.
II. A One-Pot, Solid-Phase Synthesis of Secondary Amines fromReactive Alkyl Halides and an Alkyl AzideSusana Ayesa, Bertil Samuelsson and Björn ClassonSynlett, 2008, 1, 97-99.
III. Solid-Phase Synthesis and SAR of 4-Amidofurane-3-one Inhibitorsof Cathepsin S: the Effect of Sulfonamides at P3 on Potency andSelectivitySusana Ayesa, Charlotta Lindquist, Tatiana Agback, Kurt Benkestock,Björn Classon, Ian Henderson, Ellen Hewitt, Katarina Jansson, AndersKallin, Dave Sheppard and Bertil SamuelssonSubmitted
IV. Preparation and Characterization of Aminoethylamide Inhibitors ofthe Cysteine Proteinase Cathepsin KSusana Ayesa, Jinq-May Chen, Björn Classon, Jose Gallego, UrszulaGrabowska, Ian Henderson, Narinder Heyer, Tony Johnson, JussiKangasmetsä, Mark Liley, Magnus Nilsson, Kevin Parkes, LaszloRakos, Matthew J. Tozer, and Michelle WilsonSubmitted
V. Investigation of Allylic Alcohols in the P1 Position of Inhibitors ofHepatitis C Virus NS3 ProteaseSusana Ayesa, Tatiana Maltseva, Laszlo Rakos, Elizabeth Hamelink,Björn Classon, and Bertil SamuelssonManuscript

I have also contributed to the following article, which is not included in thisthesis. This publication reports further development of the researchdescribed in paper V.
VI. Novel potent macrocyclic inhibitors of the hepatitis C virus NS3protease: Use of cyclopentane and cyclopentene P2-motifsBaeck, M.; Johansson, P.-O.; Waangsell, F.; Thorstensson, F.;Kvarnstroem, I.; Ayesa, S.; Waehling, H.t; Pelcman, M.l; Jansson, K.;Lindstroem, S.; Wallberg, H.; Classon, B.; Rydergaard, C.; Vrang, L.;Hamelink, E.; Hallberg, A.; Rosenquist, A.; Samuelsson, B.Bioorganic & Medicinal Chemistry, 2007, 15(22), 7184-7202
Papers I and II were reprinted with the kind permission from the publishers.

Abbreviations
Ac acetylAPC antigen-presenting cellsBAP borane pyridine complexBMD bone mass densityBOC, Boc t-butoxycarbonylCha β-cyclohexylalanineCPMG Carr-Purcell-Meiboom-Gill T2-dependent spin-echo sequenceCOSY correlation spectroscopyDCM dichloromethaneDIC diisopropylcarbodiimideDIEA N,N-diisopropylethylamineDMAP 4-(dimethylamino)pyridineDMF dimethylformamideDQF Double-quantum filterEDC 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
hydrochlorideELSD evaporative light scattering detectorFmoc 9-fluorenylmethyloxycarbonylFTIR fourier transformation infrared spectroscopyh hour(s)HATU N-[(dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-yl-
methylene]-n-methylmethanaminium hexafluorophosphonateN-oxide
HBTU N-[(1H-benzotriazole-1-yl)-(dimethylamino)methylene]-N-methylmethanaminium hexafluorophosphate N-oxide
HCV hepatitis C virusHIV human immunodeficiency virusHLA human leukocyte antigenHMBC heteronuclear multiple bond correlationHOBt 1-hydroxybenzotriazoleHRMAS high-resolution magic-angle spinningHSQC heteronuclear single-quantum correlationHTS high throughput screeningHPLC high pressure liquid chromatographyIi invariant chainIL interleukin

K i inhibitory constant/dissociation constant for inhibitor bindingLCMS liquid chromatography mass spectroscopyMeOH methanolMHC major histocompatibility complexNMM 4-methylmorpholineNMP N-methylpyrrolidinoneNMR nuclear magnetic resonanceNS non-structuralNS3 non-structural protein 3NTPase nucleoside triphosphataseNva norvalinePyBOP benzotriazol-1-yloxytris(pyrrolidino)phosphonium
hexafluorophosphateRT room temperatureSAR structure-activity relationshipSLP spin lock pulseSPE solid-phase extractionRAPiD rational approach to protease inhibitor designTES triethylsilaneTMOF trimethylorthoformateTMS trimethylsilylTNFa tumor necrosis factor alphaTFA trifluoroacetic acidTOCSY total correlation spectroscopy

Table of Contents
Part I. Synthetic Methods for the Preparation of AmineBuilding Blocks
1 Solid-Phase Parallel Synthesis of N-MonoalkylatedAminopiperidines and Aminopyrrolidines (Paper I)
1.1 Introduction ............................................................................................... 1
1.1.1 Solid Phase Parallel Synthesis (SPPS) ............................................1
1.1.2 NMR Spectroscopy .......................................................................... 2
1.1.3 Aim of the Study................................................................................4
1.2 Solid-Phase Synthesis: Resin Selection .................................................. 4
1.3 Reductive Alkylation of Primary Amines ...................................................5
1.3.1 Solid-Phase Chemistry .....................................................................5
1.3.1.1 Coupling to the Solid Support .................................................. 6
1.3.1.2 CPMG-T2/diffusion-Filtered gHSQC Experiments ................10
1.4 Reductive Alkylation of Secondary Amines ............................................11
1.4.1 Solid-Supported Chemistry and HRMAS NMR ..............................11
1.5 Conclusions.............................................................................................13
2 A One-Pot, Solid-Phase Synthesis of Secondary Amines fromReactive Alkyl Halides and an Alkyl Azide (Paper II)
2.1 Introduction .............................................................................................15
2.1.1 Preparation of Secondary Amines .................................................15
2.1.2 Utility of the Azide Moiety...............................................................15
2.1.3 Alkylation of Iminophosphoranes: Background .............................16
2.1.4 Aim of the Study.............................................................................17
2.2 Solid-Phase Alkylation of Iminophosphoranes .......................................17
2.2.1 Scope and Limitations....................................................................18
2.3 Conclusions.............................................................................................20

Part II. Design and Synthesis of Inhibitors Targeting theCysteine Proteases Cathepsins S and K and the SerineHCV NS3 Protease
3 General introduction
3.1 Proteases: Hydrolysis of Peptide Bonds.................................................21
3.2 Cysteine and Serine Proteases: Mechanism of Action
and Specificity.........................................................................................22
3.2.1 Catalytic Mechanism of Serine Proteases.....................................22
3.2.2 Catalytic Mechanism of Cathepsin Cysteine Proteases................23
3.2.3 Specificity of Proteases .................................................................24
3.3 Protease Inhibitors ..................................................................................25
3.3.1 Serine Protease Inhibitors .............................................................26
3.3.2 Cathepsin Cysteine Protease Inhibitors ........................................27
4 Solid-Phase Parallel Synthesis and SAR of 4-Amidofuran-3-one P1Containing Inhibitors of Cathepsin S: Effect of Sulfonamides P3Substituents on Potency and Selectivity (Paper III)
4.1 Introduction .............................................................................................29
4.1.1 Antigen Presentation and the Immune Response.........................29
4.1.2 Cathepsin S as Potential Therapeutic Target ...............................32
4.1.3 Background Data and Aim of the Study ........................................33
4.2 Chemistry................................................................................................35
4.2.1 Solid-Phase Chemistry of Dihydro-2(3H)-5-ethyl Furanones........35
4.2.2 Synthesis of P3 acid Capping Groups...........................................37
4.3 Tables: Inhibitory Activity and Selectivity ................................................38
4.4 Structure-Activity Relationships and Modeling .......................................43
4.4.1 Enzyme Inhibitory Activity..............................................................44
4.4.2 Selectivity Profile ...........................................................................47
4.4.3 Cellular Potency ............................................................................48
4.5 Conclusions.............................................................................................49

5 Preparation and Characterization of Aminoethylamide Inhibitors ofthe Cysteine Protease Cathepsin K (Paper IV)
5.1 Introduction .............................................................................................51
5.1.1 Cathepsin K and Osteoporosis......................................................51
5.1.2 Aminoethylamide Cathepsin K Inhibitors: Aim of the Study ..........55
5.2 Solid-Phase Synthesis of Aminoethylamides .........................................56
5.2.1 Aminoethylamide Template ...........................................................56
5.2.2 Aldehyde Resin-Based Synthesis .................................................56
5.2.3 Sulfonamide Resin-Based Synthesis ............................................57
5.3 Inhibitory Activity and Kinetic Studies .....................................................59
5.4 Photodegradation ....................................................................................63
5.5 Conclusions.............................................................................................66
6 Investigation of Allylic Alcohols in the P1 Position of Inhibitors ofHepatitis C Virus NS3 Protease (Paper V)
6.1 Introduction .............................................................................................67
6.1.1 Hepatitis C Virus ............................................................................67
6.1.2 HCV NS3 Protease Inhibitors........................................................68
6.1.3 Aim of the Study ............................................................................69
6.2 Synthesis of P1 Nva Allylic Alcohols .......................................................70
6.3 Isomerically Pure Allylic Alcohols............................................................71
6.4 Synthesis of Hexapeptides .....................................................................72
6.5 Synthesis of Tetrapeptides .....................................................................74
6.6 Synthesis of Tripeptides..........................................................................75
6.7 Tables: Inhibitory Activity ........................................................................75
6.8 Structure-Activity Relationships ..............................................................77
6.9 Conclusions.............................................................................................77
ReferencesAcknowledgmentsAppendices


1
Part I:
Synthetic Methods for the Preparation of AmineBuilding Blocks
1. Solid-Phase Parallel Synthesis of N-Monoalkylated Aminopiperidines andAminopyrrolidines (Paper I)
1.1 Introduction
1.1.1 Solid-Phase Parallel Synthesis
In recent years, numerous reports have been published that describe thedevelopments in solid-phase synthesis1,2 and combinatorial chemistryadapted to polymeric supports,3–5 technologies that are now widely appliedto generate drug-like libraries for high-throughput screening (HTS) against amultitude of drug targets. The idea of such approach is to couple a scaffoldmolecule to a polymer-based resin, after which the scaffold can bechemically manipulated in several ways before it is cleaved off from theresin. The reagents, which are in solution, can be removed after eachreaction step by simple filtration and washing of the resin. Accordingly,workup and purification procedures are not required between steps, and thereagents can be used in excess, which helps to drive the reactions tocompletion. Unfortunately, in general solution-phase reaction conditionsmust be modified and optimized to be used in the solid-phase approach, thusmethod development in order to transfer solution-phase conditions to solid-phase conditions is often required.
There are still several challenges that need to be addressed in the designand synthesis of compound libraries. One such consideration is the choice oflinker, which has to be stable under the reaction conditions employed andmust be selectively cleaved under mild conditions after the synthesissequence is completed in order to yield pure product. The appropriateness ofa linker is often dictated by the functionality present in the specific class ofmolecules of interest.
A major general drawback of solid-support reactions has been thedifficulties related to analysis of the reaction products on the solid phase. To

2
circumvent this problem, analytical control is frequently carried out insolution after cleavage from the polymer, which is a time-consumingstrategy and results in loss of material. Consequently, recent developmentshave focused on techniques for non-destructive on-bead analysis that areadapted to solid-phase chemistry6 and utilize both single-bead FTIR andhigh-resolution magic-angle spinning (HRMAS) NMR spectroscopy.7–9
Thus it is apparent that use of solid-phase chemistry can be tedious andtime consuming, but, once such a method is developed, this approach is veryefficient.
1.1.2 NMR Spectroscopy
More than fifty years have passed since the first observations of nuclearmagnetic resonance (NMR) were made. The seminal step occurred duringthe early 1950s, when it was realized that the resonant frequency of anucleus is affected by its chemical environment, and that one nucleus caninfluence the resonance of another through intervening chemical bonds.NMR spectroscopy is now an important tool employed to determine thestructure of organic compounds. The molecule to be analyzed is dissolved ina suitable solvent and placed in a tube, which is then subjected to a strongmagnetic field, and a target nucleus in the sample experiences this externalfield. A range of one-dimensional (1D) experiments can be carried out toassign the structure of the molecule of interest. However, when consideringlarge structures, the chemical shifts have a higher degree of overlap in the1D spectrum, and hence in such cases it is necessary to perform advanced1D and 2D hetero- and homonuclear experiments.
High-Resolution Magic-Angle Spinning (HRMAS) NMRSpectroscopy
HRMAS NMR spectroscopy, is one of the most powerful nondestructiveanalytical techniques used to monitor reactions on solid support.7–9 Aproblem that can be encountered with 1D and 2D homo- and heteronuclearHRMAS NMR experiments concerns the multitude of non-relevant signalsthat arise from the resin, residual solvents, and from organics encapsulated inthe beads, which make interpretation of the spectra non trivial. There aresome reports describing the use of 1D techniques to remove such unwantedsignals.10–14
An application of high-resolution MAS NMR spectroscopy tocompounds attached to Merrifield resin faces a challenge due to the shortlength of the resin linker.15 The resulting slow mobility of the attachedcompound and the very fast T2-relaxation of the protons give rise to abroadening of the signals in the spectrum.

3
CPMG-T2 /Diffusion-Filter gHSQC Experiments
Two approaches can be applied to remove the residual resin signals in high-resolution NMR: (a) the use of presaturation to saturate the resin signals andallow the signals of interest to appear; (b) the use of CPMG-T2 type filters tosimultaneously suppress all the resin signals. A disadvantage of the firstmethod is that only a small given number of resin peaks can be presaturated,and the presaturation will also remove any useful signals that are hidden bythe resin signals. The use of multiple frequency presaturation with SLP typepulses to remove all the resin signals does not solve this problem, because it“erases” a greater region of the spectrum.
The basis of the second method is that the resin molecules, in essencebecause they are in the solid phase, have much shorter T2 relaxation times.Therefore, if the magnetization is retained for a while in the XY plane afterthe initial excitation pulse, the intensity of the resin signals will decreasemuch faster than the signals from the bound molecules, which are more orless in the liquid state. In this way, all the resin signals will be attenuated,allowing the signals of the bound species to appear. A CPMG-T2 type filter(delay–180o–delay)n can be used to achieve simultaneous removal of theresidual resin signals in high-resolution NMR spectra. This approachemanates from the differences in mobility exhibited by the resin part of themolecule and the resin-bound moieties, which still retain reasonably freerotational diffusion.13 This means that a nucleus belonging to the resin partshould have a considerably shorter T2 relaxation time compared to the nucleiof the resin-bound moieties. Filtering times of 20–80 ms are usuallyemployed. A few reports have described the use of such filters but mainly incombination with 1D techniques.10–14 The T2 parameters must be carefullyselected, since the signals of interest are also attenuated, albeit to a lesserdegree. Another problem that can arise, particularly when using long T2
filtering times and many 180o pulses, is related to B1 inhomogeneity. Thepulses are not precisely 180o over the entire spectral window, and thereforethe repeated use of them causes signals to lose intensity, especially in theedges of the spectrum.
When moving from simple 1D techniques to modern 2D experiments,difficulties can also occur due to J-evolution during the time spent in the XYplane. That effect results in severely phase-distorted multiplets in the 1Dspectra and rather unpredictable behavior of the system with all 2Dtechniques that use 1H decoupling in the F2 dimension. The 2D experimentsmost often applied are DQF-COSY, TOCSY, HSQC and HMBC. However,in all such experiments, with the exception of when using HSQC, what isknown as the mixing or magnetization transfer time (i.e., the time it takes forthe system to obtain the desired properties to detect) is usually in the sameorder of magnitude as the T2 relaxation times of the resin signals.Accordingly, in these techniques, the solution is provided by the experiment

4
NY
NH2
XRN
YNH
XH R
NY
NH2
XH
NY
NH2
XN Y
NH
XH
X, Y= bond, CH2
A B
itself, as a side effect, and there is no need for a dedicated T2 filter. However,when applying HSQC, the transfer times are very small, usually not morethan 10–15 ms, which is not sufficient for the resin signals to die out, andthus in this case it would be appropriate to conduct a CPMG-T2 diffusion-filtered experiment.
1.1.3 Aim of the Study
N-Monosubstituted aminopiperidines and aminopyrrolidines are importantbuilding blocks for the synthesis of small-molecule directed compoundlibraries. The aim of the study reported in Paper I was to find a practicalsolid-supported parallel synthesis route that would supply these keycomponents. It was also desired to achieve a facile monitoring of thesynthesis steps by use of non-destructive methodologies such as FTIR orHRMAS NMR. Two synthetic routes (A and B, depicted schematicallybelow) were proposed and investigated that were intended to accomplishderivatization of the primary or the secondary amino moiety by reductivealkylation. The suggested routes can be readily applied to an extensive poolof commercially available aromatic and aliphatic aldehydes for conversioninto the corresponding N-monoalkylated diamino templates. This procedurenicely complements methodologies entailing amide reductions and reversedreductive aminoalkylations.16–18
1.2 Solid-Phase Synthesis: Resin Selection
The p-nitrophenyl carbonate linker was chosen for development of the twosolid-supported synthetic strategies described above. Its functionality is idealfor the attachment of amines onto the solid support. Specifically,nucleophilic amines are able to form a carbamate bond with the solidsupport, which displaces the stable p-nitrophenol from the resin and therebygives a characteristic yellow color to the reaction. The newly formedcarbamate bond is quite stable in relation to most reaction conditions, exceptfor acidic treatments. The acid liability of the resin is dependent on thenature of the linker.

5
OO
O
NO2
ON
O
NHR
ON
O
NH2
HNNH
R
ON
O
NR
1 2
4 5
3
ba
c d
1 2
3
45
6
78
9
1 2
3
45
6
7 8
9
Merrifield- and Wang-type p-nitrophenylcarbonate resins were investigatedin the current study. No differences between the two were found in terms ofyields and purities when considering the first route (A), in which theaminopiperidines are attached to the resin through the secondary aminemoiety. However, there were substantial differences in the yields obtainedby the second route (B), which involves attachment of the aminopiperidinesthrough the primary amine moiety. In this route, the amines used as startingmaterials are Boc-protected at the secondary amine moiety. Once attached tothe solid support an acidic treatment is performed to remove the protectinggroup, which, in the case of the Wang-type resin, causes cleavage of a largeproportion of the amines at this step, thus lowering the yields of the targetcompounds. By comparison, the Merrifield-type resin is less acid labile, andno cleavage of the amines is observed at the low acidic deprotectiontreatment used; instead, the final cleavage of the amine products from theresin requires a stronger acidic treatment.
1.3 Reductive Alkylation of Primary Amines
1.3.1 Solid-Phase Synthesis
The synthetic route investigated is depicted in Scheme 1. The p-nitrophenylcarbonate Merrifield- (0.8 mmol/g loading) and Wang-type (1mmol/g loading) resins were used as polymeric supports. Attachment of thestarting amine to resin 1 (step a) was performed at 20 C in DMF for 24 h,and the products were analyzed by single-bead FTIR, the ninhydrin test,19,20
and HRMAS NMR.
Scheme 1. Reagents and conditions: (a) 4-(aminomethyl) piperidine (5 equiv. in dry DMF),RT, overnight; (b) RCHO (21) (5 equiv. in TMOF; see scheme 3), RT, overnight; (c) BH3-pycomplex (4 equiv.), DCM:MeOH:AcOH 2: 2: 1, RT, overnight; (d) 75% TFA in DCM, RT, 1h.

6
Typically, during step a, the absorption bands at 1,760 and 1,350 cm-1 in theFTIR spectra, corresponding to the carbonate group and the nitro group,respectively, were transformed into a new band at 1,720 cm–1, assigned tothe carbonyl group of the carbamate function. From the FTIR data, it was notpossible to determine whether the attachment to the resin occurred throughthe secondary or the primary amino moiety, and neither could the absorptionband observed at 3,400 cm–1 (attributed to NH vibration10) beunambiguously assigned to one or the other.
The primary amine 2 on solid phase (Scheme 1) was subjected toalkylations with aldehydes (21) in solution by performing a two-stepprocedure to minimize dialkylations.21–22 The imine-forming step proceededreadily with the amine on the solid phase and the aldehyde in solution.23
Trimethylorthoformate (TMOF) was employed as both solvent anddehydrating agent,24–25 and the reaction was monitored by FTIR, whichshowed a characteristic –N=C– absorption band at 1,645 cm–1. Reduction ofthe imine functionality of resins 3 to the corresponding amine moiety inresins 4 was achieved using borane-pyridine complex26 (BAP) at roomtemperature overnight. Final compounds 5 were obtained as bistrifluoroacetate salts in good yields and purities after TFA treatment of resins4.
1.3.1.1 Coupling to the Solid Support
Indirect Method
To investigate the attachment of aminopiperidines to the solid support and toverify whether the coupling occurred through the primary or secondaryamino moiety, an indirect approach was developed at the beginning of thisstudy. It was envisaged that acylation of the remaining amino group(Scheme 2, step b) would provide the required evidence of the selectivitythat was obtained. Experiments were performed using solution-phase NMRanalysis of the hydrolysis products 9 and 10 (Scheme 2).
To ensure that the reaction of unprotected 4-aminomethylpiperidine withresin 1 had taken place through the secondary amine to give 2 and not 6, anamide coupling experiment (Scheme 2) was performed. In short, 4-azidobenzoic acid was coupled to the amine through a traditional PyBOPpeptide-coupling method, which could readily be followed by FTIR on theresin through a characteristic azide absorption band at 2,120 cm–1.Nevertheless, FTIR could not provide information about the mode ofattachment to the resin (7 or 8).

7
OO
O
NO2
OO
N
N
ON3
OO
N
N
ON3
OO
N
N
OO
N
NH2
N
H2N
O
N3
HNHN
O
N3
or b
c
1
a 6
2
or9
10
12
3
4
56
9
7
8
or
7 89
87
1 234
56
1 0
11
12
1 3
1 415
Scheme 2. Reagents and conditions: (a) 4-(aminomethyl) piperidine (5 equiv. in dry DMF),RT, overnight; (b) 4-azidobenzoic acid (4 equiv.), PyBOP (4 equiv.), DIEA (8 equiv.), dryDMF, RT, overnight; (c) 75% TFA in DCM, RT, 1.5 h.
One of the options to explicitly establish that the reaction proceeded throughthe secondary amine was to fully assign the NMR spectra of the cleavedproduct. In the gCOSY spectrum of the cleaved product, the J-couplingconnectivity starting from the 8H proton and continuing to 7H2 4H 3H22H2 protons was identified that fits with both possible structures 9 and 10(for atom numbering, see Scheme 2). The assignment of the 7H2, 3H2, and2H2 protons of the methylene groups was confirmed by a gHSQCexperiment. The cross peaks between carbonyl carbon, 9C methyleneprotons 7H2 and 9C 8H, were detected in the HMBC spectrum. Thisobservation supports that the cleaved product has structure 10 (Scheme 2),which shows that the amide-forming reaction proceeded through the primaryamine 2 to give 8.
Direct Method: Use of HRMAS NMR Spectroscopy to Monitor theReactions
The ideal situation for the monitoring of the synthesis steps would be to usea more direct method that does not require previous cleavage from the resin,or alternatively to apply an indirect approach involving formation ofreference compounds, as described in the previous section (Indirect Method).During the development time of this study, it was possible to find a solutionby conducting several HRMAS NMR experiments, and we explored thepossibility of applying CPMG-T2 type filters to remove all the polymersignals and enhance the signals of interest. HRMAS NMR was used tomonitor the reductive alkylation steps on the resin (Scheme 1), and this is

8
ppm23456789
2a6a
16
4
2b6b
3a5a
3b5b
9
7
(a)
(b)
(c)
(d)
(e)
2a6a
16
4
2b6b
3a5a
3b5b
97
2a6a
42b6b
3a5a
3b5b
7
2a6a
16
4
2b6b
3a5a 3b
5b
97
2a6a
164
2b6b
3a5a
3b5b
97
17
17
1513 14
F1011
12
13
14
15
16
O
1011
12
1314
15
16
17
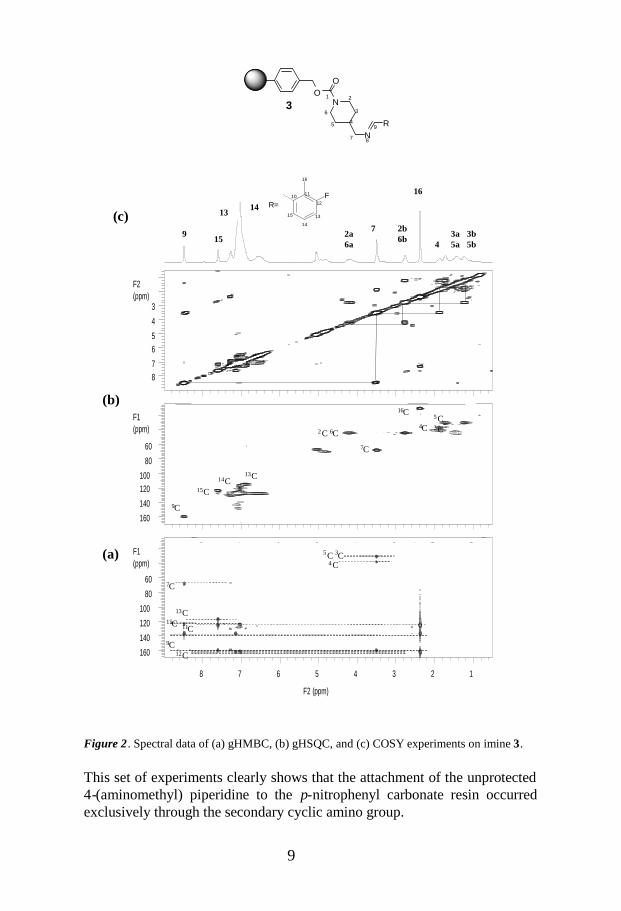
exemplified by two aldehydes in Figure 1. It is noteworthy that the protonsignals of the piperidine fragment of compounds 3 were much broader thanthose of the imine fragment, similar to amines 2 (Fig. 1, a, b, and d).In the gHMBC spectrum of compound 3 (Fig. 2a), a cross peak was clearlyobserved between the protons of the methylene group 7H2 and the carbon C9,which further supports formation of the primary amine 2 in Scheme 1. Thefull assignment of compound 3 was achieved through gHMBC, gHSQC, andCOSY experiments (Fig. 2, a–c). In the gHMBC spectrum (Fig. 2a) ofcompound 3, the protons belonging to the piperidine and resin fragments didnot have any cross peaks with carbons two or three bonds away due to shortT2 relaxation. However, in the HSQC experiments the transfer times are veryshort, usually not more than 10–15 ms, which is not enough time for theresin signals to die out (Fig. 2b).
Figure 1. 1H spectra of the products presented in Scheme 1, obtained using T2 and diffusionfilters: (a) 2; (b) 3, and (c) 4,; (d) 3, and (e) 4.
ON
O
NH2
ON
O
N
2 (Fig .1a )
3 (Fig . 1(b))
1 2
3
45
6
7 8
9
F ON
O
NH
1 2
3
45
6
78
9
F
10
1112
13
1415
16
10
1112
13
1415
16
4 (Fig. 1(c))
ON
O
N3 (Fig. 1(d))
1 2
3
45
6
7 8
9
o10
1112
13
1415
16 17
ON
O
NH4 (Fig. 1(e))
1 2
3
45
6
78
9
o10
11 12
13
1415
16 17
9
F1 (ppm)
12345678
F2(ppm)
3
4
56
78
F2 (ppm)
12345678
F1(ppm)
60
80
100
120
140
160
F2 (ppm)
12345678
F1(ppm)
60
80
100120
140
160
5C 3C4C
7C
13C15C 11C9C
12C
5C3C4C2C 6C
7C
14C13C
15C9C
2a6a
16
4
2b6b 3a
5a3b5b
9 715
13 14(c)
(b)
(a)
16C
FR=
10 11
12
13
14
15
16
ON
O
NR
31 2
3
45
6
7 8
9
Figure 2. Spectral data of (a) gHMBC, (b) gHSQC, and (c) COSY experiments on imine 3.
This set of experiments clearly shows that the attachment of the unprotected4-(aminomethyl) piperidine to the p-nitrophenyl carbonate resin occurredexclusively through the secondary cyclic amino group.

10
F2 (ppm)
1234567
F1
(ppm)
5060708090
100110120130140
F2 (ppm)
1234567
F1
(ppm)
5060708090
100110120130140
5C3C
4C2C6C
14C 13C15C
9C
16C
7C
4C
14C 13C15C
9C
16C
7C
(b)
2a6a
164
2b6b 3a
5a
3b5b
9 715
13 14
(a)
ON
O
NHR
4
1 2
3
45
6
78
9
1.3.1.2 CPMG-T2/Diffusion-Filtered gHSQC Experiments
CPMG-T2 type filters (delay–180o–delay)n were used to remove the residualresin signals from the high-resolution NMR spectra. The T2 filter parametershave to be carefully selected, since the signals of interest are also attenuated,although to a lesser degree. Additionally, a diffusion filter was used toseparate out the signals from the unbound molecules in the sample.10,13
Figure 3 shows two HSQC spectra of compound 4 (Scheme 1) obtainedby a conventional gHSQC experiment (Fig. 3a) and a modified CPMG-T2/diffusion-filtered gHSQC experiment (Fig. 3b). With careful selection ofthe T2 filter parameters and diffusion parameters, most of the polymersignals, as well as those belonging to the methylene fragments 2H2,
3H2,5H2
and 6H2 of piperidine, were removed and gave rise to the signals of interest,group R. The monitoring of the imine reduction was possible by detection ofnew carbon and proton signals of the –N8H-9CH2 fragment of compound 4(Fig. 3b) and disappearance of the –N8 = 9CH fragment of compound 3 (Fig.2b). The proton chemical shifts of the methylene group 7H2 in compound 4had moved upfield compared to the corresponding signal from compound 3(i.e., from δ= 3.5 to 2.3ppm), and this was due to the reduction of the iminemoiety. Full data with proton and carbon assignment of compound 4 werebased on the gCOSY, gHMBC, gHSQC, and modified gHSQC experiments.
Figure 3. 2D 1H-13C correlation experiments on compound 4 performed using (a)conventional gHSQC and (b) modified CPMG-T2/diffusion-filtered gHSQC.
F10 11
12
1314
15
16
R=

11
OO
O
NO2
NH 2N
O
O
N O
O
H 2N
OH
O
O
HNO
H 13
14
O
H O
O
H
O
OHN
O
Y
NX
R
O
HCl
Cl
NO
HNH
O
H
O
H
S
OH
N O
OH2 N
OH
YH2N
15
13
OHN
O
Y
14
N RX
H
O
O
H
O
O
H
N O
O
H2N
NX
OO
1315
10
O
O
HF
16
O
1 1
N O
OH2N
12
13
OHN
O
Y
15
10
O
H
O11
O
H
O
NX
H
O
H
N
N
H2N
O
O
16
12
17
20
a b c d e f
21
b c f
j k
q r
12
ts
g
9
10
1112
i
9
10
1112
13
14
1 516
p
13
a
9
10
1 112
13
1 4
15
16
17
18
19
n
9
1011
12 13
1415
16
a
11
b
12
14-1913
c d
x, y = bond, CH2
m
9
1 0
1112
13
14
11
12
13
9
10
9
1011 12
13
1415
16
9
1 011
910
11
12
d
910
1112
e
h
9 1 011
12 9
10
11
12
13
l
9
10
1112
13
14 1 5
o
910
11
12 13 14
15
16
1718
9 1011
12
1314
15
9
10
11 12 13
1
14
14
[a ]
1.4 Reductive Alkylation of Secondary Amines
1.4.1 Solid- Supported Synthesis and HRMAS NMR
The synthetic route investigated is depicted in Scheme 3. The primaryamines were attached to the solid support by using the corresponding Boc-protected secondary amines as starting material.
Scheme 3. Reagents and conditions: (a) Boc-protected amines (20) (5 equiv. in anh. DMF),RT, overnight; (b) selective Boc deprotection with 10% TFA in DCM, RT, 2 h; (c) RCHO(21) (10 equiv. in TMOF:DMF/EtOH (3/1) 1:1, Borane-pyridine complex (BAP) 10 equiv.,RT, 4 days ; (d) 75% TFA in DCM, RT, 4 h.
[a] Merrifield-type resin gave better yields than Wang-type resin did.

12
ppm2345678
Boc
(a)
(b)
(c)
(d)
816
8 16
17
F1011
12
13
14
15
16
R=
O
R=10
11
12
1314
15
16
17
ON
O
NR
1312
34 5
67
8
ON
O
NBoc
1112
34 5
67
Selective BOC group deprotection of compounds 11 was examined byapplying various acidic conditions (Fig. 4a). The best results were obtainedusing 10% TFA in dichloromethane (DCM),26 by which essentiallyquantitative yields of compounds 12 (Fig. 4b) were achieved, as estimatedfrom the CPMG-T2/diffusion-filtered 1D-1H NMR-spectra (Fig. 4). After thedeprotection step, the resulting secondary amines were subjected toreductive alkylation with aldehydes 21 (Scheme 3). This was achieved insitu using BAP,27 which led to clean reactions and high yields of products.Other reducing reagents examined, including NaBH3CN and NaBH(OAc)3
were found to be less effective.28–30 As an illustration, alkylation products 13(Scheme 3) were analyzed by HRMAS NMR shown in Figure 4 (c and d).The assignment of resonances 8 and 16 of compound 13 with R 21e andresonances 8, 16, and 17 of compound 13 with R 21r is based on CPMG-T2
and diffusion filters gHSQC experiments.
Figure 4. 1H spectra with T2 and diffusion filtering of the products of the reaction presentedin Scheme 3: (a) 11, (b) 12, (c) 13 [with R = 21r and amine 20e, Scheme 3], (d) 13 [with R =21e and amine 20e, Scheme 3].

13
Compounds 14–19 were obtained as bis trifluoroacetate salts in good overallyields and with high LCMS purities after cleavage from the solid support byuse of a solution of 75% TFA/DCM.
1.5 Conclusions
A practical solid-phase synthesis of N-monosubstituted aminopiperidinesand aminopyrrolidines was developed. Selective alkylation of the primary orsecondary amine moiety was accomplished by use of the two syntheticroutes outlined in this study. The products were isolated in good yields andpurities after simple filtration steps, which enabled easy parallel processing.
The non-destructive analytical methodologies solid-phase NMR and FTIRwere used to monitor the reactions. Beside the use of 1D techniques toremove unwanted signals, an extension of these techniques for 2D, gHSQCspectra was reported. Thus it seems that the use of 2D NMR CPMG-T2
filtered gHSQC experiments together with the present solid-phasemethodology aimed at procuring novel diamine building blocks will furtherbroaden the scope of solid-phase approaches and aid the preparation of noveldrug-like compounds.

14

15
2. A One-Pot, Solid-Phase Synthesis of SecondaryAmines from Reactive Alkyl Halides and anAlkyl Azide (Paper II)
2.1 Introduction
2.1.1 Preparation of Secondary Amines
There are numerous methods reported in the literature describing thepreparation of secondary amines, and the majority of them entail eitherreductive aminations or alkylations on primary amines. Although the yieldsare usually good, it is generally difficult to avoid the over-alkylation that isassociated with the use of reactive alkyl halides, which leads to formation ofthe corresponding tertiary amines and quaternary ammonium salts. Otherapproaches to prepare secondary amines include reduction of thecorresponding amides and aza-Wittig procedures.31–33
Both solution-phase and solid-phase methods have been developed formost of the indicated alternatives, but the solid-phase strategies offer theimportant advantage of ease of purification by use of simple filtration steps.However, the emergence of commercially available solid-supported reagentsand scavengers has increased the use of the solution-phase approaches byfacilitating the work-up procedures so that in many cases the use ofchromatography can be avoided.
2.1.2 Utility of the Azide Moiety
It is easy to synthesize alkyl azides starting materials by applying a numberof reported procedures, such as nucleophilic substitution of bromide withsodium azide34 or conversion from amines through diazo transferreactions.35,36 The use of Amberlite® azide exchange resins offers furtherpossibilities for the synthesis of azides.
Furthermore, organic azides have recently attracted considerableinterest, partly due to their popularity in the Cu I-catalyzed reactions betweenazides and terminal alkynes (Scheme 4A),37a and use of the azide functionalgroup for synthesis of 1,2,3-triazoles is well known (Huisgen reaction).
As protecting groups, azides can help avoid many of the concernsassociated with amides and carbamates, and they provide the option of amild reduction (Staudinger reaction). The conversion of azides into amines isa standard synthetic procedure for chemists. The Staudinger reaction(Scheme 4B),37b–d involves the treatment of an alkyl azide withtriphenylphosphine to produce a phosphoazide that rapidly eliminates

16
R-N3
PPh3
NN
NR
P+
PhPh
Ph
NP N
NR
PhPhPh RNH2
-Ph3PO
R-N3 N
N N
R R'
R'
R NHCO2R' ' R NHCOR' '
-N2Ph3P=NR
H2O
CuI
(A) Huisgen reaction
(B) Staudinger reaction
Ph3P=N-Li
NR
HR
NR
RPh3P I
NH3H2N-R1
HO-/H2O HO- /H2O
R1 I, THF, r.t. R2 I, THF, 650CPh3P=NR1 +
3 steps
-
1
1
2
2
nitrogen to afford an iminophosphorane. This intermediate can be readilyhydrolyzed in a protic solvent to give the corresponding primary amine, aprocess that has been transferred to polymer support.38 Iminophosphoranesare useful intermediates in synthesis of a variety of nitrogen-containingcompounds (Scheme 4B), including amines and carbamates, and in the aza-Wittig reactions.39,40
Scheme 4. Transformations of organic azides.
2.1.3 Alkylation of Iminophosphoranes: Background
The use of a phosphonium diaza-diylide or a phosphonium aza-yldiide as areagent for the synthesis of primary and secondary amines (Scheme 5) in astepwise solution-phase dialkylation of an aza-yldiide has been reported byother investigators.41 That approach led to good yields of N-alkylphosphimines or disubstituted aminophosphonium salts, which by hydrolysisprovided the corresponding primary or secondary amines. However, thementioned methodology requires the use of a strong base.
Scheme 5. Synthesis of amines via alkylation of iminophosphoranes.

17
PPh2
R1N3P
Ph
Ph
N R1R2X
P
Ph
Ph
N
R2
R1
N
R1
R2
P
Ph
Ph
HN
R1 R2
2223
27
24 26
25
26
(a) (b)
X X
2.1.4 Aim of the Study
The aim of the study presented in Paper II was to develop a new method thatinvolves preparation of secondary amines through alkylation with reactivehalides in order to avoid the problem of over-alkylation. Such a methodshould be easy to apply and, if possible, should not require chromatographicpurification.
There are reports describing procedures for conversion of azides intosecondary amines that do not involve iminophosphorane or imineintermediates.42 However, whilst methods via an intermediateiminophosphorane have been developed, they entail aza-Wittig reactionsstarting from azides and aldehydes.31,43 Thus it seemed that an attractive newmethodology for the synthesis of secondary amines would be to use theStaudinger reaction to accomplish alkylations of N-alkyl phosphimines or N-aryl phosphimines with reactive alkyl halides, and that was the objective ofthe present study.
2.2 Solid-Phase Alkylation of Iminophosphoranes
The investigation focused on combining the Staudinger reaction with thealkylation of iminophoshoranes to achieve exclusive delivery of secondaryamines starting from azides and alkylating agents. A solid-phase approachwas undertaken to provide ease of purification. Scheme 6 depicts the solid-phase protocol that was developed.
Scheme 6. Reagents and conditions: (a) filtration and washing of the resin; (b) KOH-MeOH,65 C̊; filtration.
The method thus proceeded via a Staudinger reaction employing polymer-supported triphenylphosphine (22)44 and an azide (23) to give thecorresponding iminophosphorane (24). The resulting suspension wasagitated at room temperature for 3–4 h. During development of the protocol,the reactions were conveniently monitored by using FTIR to follow thedisappearance of the signal corresponding to the azide in the solution phase(at 2,045 cm–1) as a new P=N absorption band in the solid phase (at 1,112cm–1). Iminophosphorane 24 was subsequently alkylated in situ with alkyl,

18
N3
Cl
Cl
Br Cl
Cl
N
+
allyl, or benzyl halides (25) to afford the corresponding disubstitutedaminophophonium salts (26). The best results were obtained when thealkylation was allowed to proceed for 16 h at ambient temperature.Prolonged reaction times or heating did not improve the overall yields. Afterwashing the resin, hydrolytic cleavage of the secondary amines from thesolid support was performed using one equivalent of methanolic KOH. Thecleavage time was optimized to 3–4 h at 65 C̊ to furnish the correspondingsecondary amine (27) in solution and resin-bound triphenylphosphine oxide,which was removed by filtration. Microwave heating was attempted for 10–40 minutes at temperatures ranging from 100 to 140 0C without anysignificant improvement of the outcome. After concentration, the resultingsecondary amines were isolated by using dichloromethane and ethyl acetatefor extraction of the aqueous phase containing the potassium salts. Thepolymer-supported triphenylphosphine can be regenerated by reducing thepolymer-supported phosphine oxide with trichlorosilane.45
Table 1 shows the azides and alkylating agents used, as well as the isolatedyields of products and the purities. All products were characterized byLCMS, NMR, and HRMS. No primary amine byproducts from incompletealkylation or any other byproducts were observed. The advantage ofavoiding dialkylation with this new method is most clearly observed in thecases where MeI was used (entries 1 and 6), which, in contrast to othermethylation techniques, did not result in any dimethylated products.
2.2.1 Scope and Limitations
The yields obtained (Table 1) were generally good (78–87%), except for thephenylazide (entry 5), and offered excellent purities (> 95%). Anexplanation for the low yield of entry 5 might be due to slower reactionkinetics between the phenylazide and the polymer-supportedtriphenylphosphine, as has previously been observed in a study concerningthe reduction of phenylazide to aniline.46
A similar finding was made when using 3,4-dichloro phenylazide asstarting azide and 4-tert-butyl benzyl bromide as alkylating agent (Fig. 5). Inthat case, the expected product could not be found.
Figure 5
Although not part of the objective of this study, attempts were made to usealkylating agents that are less reactive than the ones shown in Table 1 in

19
Entry Azide 23 R2X 25 Product 27 Yield(%)a Purity (%)b
1 N3MeI NH 81 95/95
2 N3
NC
Br
NH
CN
82 95/95
3N3
Br
NH
87 94/95
4 N3
Br
NH 78 97/95
5N
3
O
O
Br
NH
O
O
21 97/95
6NH
O O
N3
MeINH
NH
O O
85 97/94
7NH
O O
N3
Br
NH
NH
O O
82 96/95
I
Fm ocNHO
SO O
N3
Azide 23: R2X 25:
order to evaluate the scope of the method developed. For example, branchedhalides and tosylates (Fig. 6) were examined in combination with benzylazide, but without success.
Figure 6
Table 1. Preparation of secondary amines 27 from azides and reactive halides
aYield of pure compound fully characterized by LCMS, NMR, and HRMS with respect to theinitial loading of the resin.bPurity was determined by LCMS/NMR, except for entry 6, which was determined byELSD/NMR.

20
2.3 Conclusions
In summary, a convenient and efficient polymer-supported protocol for thesynthesis of secondary amines was developed that affords the targetmolecules in good overall yields and purities from the corresponding azidesand reactive alkyl halides without the necessity of isolation or purification ofany intermediates. This procedure complements existing methods of aminealkylations, reductive alkylations, and aza-Wittig procedures. Moreover, thepresent results indicate that the new methodology developed works very wellwhen using alkyl azides and benzyl azides as starting materials incombination with reactive halides, as shown in Table 1. Regarding thelimitations of the procedure, it can be deduced that it is generally not suitablefor use with phenyl azides and that the alkylating agent should be reactive.
To broaden the scope of this approach, further work can be envisionedwhich would allow the use of phenyl azides as starting materials by adaptingthe procedure to solution-phase parallel synthesis employing a fluorous-tethered PPh3 in combination with FluoroFlashTM SPE.

21
NH
CH
R
CHN
O
CH
R'
C NH
O
NH
CH
R
C O
O
H3N CH
R'
C NH
O
+
H2O
acid amine
O
NH
O
NH
NH
O
O
NH
NH
O
N terminusC terminus
Scissilepeptide bond
S3
S2 S1'S3'
S1S2'
EnzymeNonprime subsite Prime subsite
P3
P2
P1
P1'
P2'
P3'
Part II:Design and Synthesis of Inhibitors Targeting theCysteine Proteases Cathepsins S and K and theHCV NS3 Protease
3. General Introduction
3.1 Proteases: Hydrolysis of Peptide Bonds
The enzymes known as proteases (protein hydrolases) catalyze amide(peptide) bond hydrolysis in protein or peptide substrates inside and outsideliving cells, and they are involved in the control of dynamics of proteinturnover. Protease-catalyzed rates of hydrolysis are in the range of up to 10-billion-fold faster than non-enzymatic rates. Hydrolysis is an energeticallyfavorable reaction, and therefore the induced proteolysis is an irreversibleprocess that must be stringently controlled.
Proteases can be classified in two ways: (1) as exopeptidases(aminopeptidases and carboxypeptidases) or endopeptidases based on theirsite of action; (2) as serine/threonine, cysteine, aspartyl, or zinc (metallo)proteases by considering their reaction mechanisms and the nature of theactive-site residues involved in the catalytic cleavage. The side chains of thesubstrate/inhibitor recognized by the protease are designated P3, P2, P1, P1',P2' and P3' and the corresponding subsites of the protease S3, S2, S1, S1', S2'and S3' as depicted in Fig. 7.47
Figure 7. Prime and nonprime substrate peptidyl residues (P and P') and enzyme subsites (Sand S') oriented according to the peptide bond cleaved within the substrate.

22
R NHR '
O R NHR '
X-Enz
O
R'NH2
RCOOH + NH 2R'
R X-Enz
O
Acyl-Enz
Tetrahedraladduct
H2O
Enz-X
RCOOH
R N HR'
O
OH
R OH
X-Enz
O
Proteases play important roles in many physiological processes, such asgrowth, signal transduction, cell proliferation, cell death, immune defense,coagulation, fertilization, and wound healing.48 These catalytic proteins arealso involved in many pathological conditions and disorders, includingcancers,49–51cardiovascular diseases,52 autoimmune diseases,53 osteoporosis,Alzheimer’s disease and infectious diseases, e.g. malaria, HIV and HCV.54–56
Obviously, this means that manipulation of the activity (in most casesinhibition) of proteases can provide an opportunity for therapeuticintervention.
3.2. Cysteine and Serine Proteases: Mechanism ofAction and Specificity
Serine and cysteine proteases have nucleophilic moieties that act directly onthe substrate to generate a covalent acyl-enzyme intermediate. Aspartyl andmetalloproteases operate indirectly by activating water molecules as thenucleophilic species attacking the peptide bond (Fig. 8).
Figure 8. Activation mechanisms of proteases.
3.2.1 Catalytic Mechanism of Serine Proteases
Serine proteases constitute the largest class of proteases, and their catalytictriad is characteristically composed of His57, Ser195, and Asp102. Ser195 isthe active nucleophile that attacks the peptide bond as a serine alkoxideequivalent; His57 acts in sequential steps, first as a base and then an acidcatalyst for proton transfer; Asp102 orients the His57 side chain to form anH-bond with Ser195. The backbone amide NH groups of Ser195 and Gly193form the oxyanion hole. In this way, the protease accelerates peptide bondhydrolysis by 109-fold. All steps occur under the promotion of generalacid/base catalysis of His57 and Asp102, and through stabilization of thenegatively charged tetrahedral intermediates by the oxyanion hole.

23
The mechanism is divided in two half reactions: enzyme acylation andenzyme deacylation (Fig. 9). In enzyme acylation, Ser195 attacks first thepeptide bond in the bound substrate to form a tetrahedral adduct, andthereafter an acyl-enzyme bond resulting in the release of the amine product(Fig. 9a). In enzyme deacylation, attack of an OH– on the acyl enzyme leadsto formation of a tetrahedral adduct which collapses delivering the acidproduct (Fig. 9b).
(a)
(b)
Figure 9. The first (a) and second (b) steps of the catalytic mechanism of serine proteases.
3.2.2 Catalytic Mechanism of Cathepsin Cysteine Proteases
The name cathepsin (from the Greek kathepsein, to digest) was initially usedto describe any group of intracellular peptide hydrolases, although it waslater discovered that several proteins also have extracellular functions.Cathepsins are further defined as being cysteine, aspartic, or serineproteases, according to their catalytic mechanism. Cathepsins have beenidentified in a wide array of organisms, ranging from viruses, bacteria, andplants to mammals. To date, 11 human cathepsins are known. Lysosomalcysteine proteases belong to a subgroup of the cathepsin family that isessential for normal cellular functions, such as antigen processing, boneremodeling, and general protein turnover.

24
NH
N
His
H S
Cys
NH
NH
His
S
Cys
NH
O
NHR''
RR'
NH
NH
His
NH
N
His
OH
HNH
NH
His
NH
N
HisS
Cys
O
NHR''
ROH
NH
NHR''
RR'S O
Cys
S
NHR''
R
Cys
OOH
NHR''
RS O
Cys
+ -
+
R'NH2
+-
The same fundamental mechanism of serine proteases is valid for cysteineproteases, in which the cysteine thiolate replaces the serine alkoxide as theactive nucleophile, and the acyl-enzyme intermediate is peptidyl-S-enzyme.The active triad comprises Cys25, His159, and Asn175 (papain numberingsystem), and it shows optimal activity at a pH of ~5.5, which is the pH inlysosomes. Loss of activity at pH down to 3.8 or a neutral pH can be due tostructural instability of the active enzymes, with the exception of cathepsinS, which maintains its activity in a neutral pH range.57-58
The enzymatic mechanism of the cysteine cathepsins involves anucleophilic thiol and general acid-base catalysis (Fig. 10).
Figure 10. Catalytic mechanism of lysosomal cysteine proteases.
The negatively charged Cys25 nucleophile is stabilized by the positiveHis159 and by the positive dipole of the central helix in which it resides.59
The thiazole-imidazolium ion pair is characterized by an unusually low pKavalue of the reactive thiolate group (~2.5–3.5).59 The cysteine thiol groupattacks the carbonyl of the target peptide bond to form a tetrahedralintermediate that collapses to the thiol ester, resulting in the release of thefirst peptide product. A second tetrahedral intermediate is formed uponwater-catalyzed hydrolysis of the thiol ester, which then collapses to releasethe second peptide product and thereby regenerate the active site.
3.2.3 Specificity of Proteases
Given that the catalytic triad inventory of all serine and cysteine proteasesexhibit conserved composition and a common mechanism of action, thequestion arises as to how these enzymes achieve their specificity. This isachieved through the differences manifest in the subpockets, specificitypockets, of the protease. Many proteases have extensive specificity subsitesfor recognizing n+1, n+2, and/or n-1, n-2 residue side chains on proteinsubstrates. For example, the Asp-His-Ser catalytic triad in chymotrypsin,

25
trypsin, and elastase is essentially identical but the specificity pockets theycontain differ in that they accommodate a hydrophobic aryl side chain, acationic side chain, or a small aliphatic side chain respectively (Fig. 11).
Figure 11. Specificity pockets of chymotrypsin, trypsin, and elastase.
In general, the cathepsins show broad specificity for their peptide substrates,although some do prefer certain amino acids over others in the targetsequence, and S2, S1, and S1' substrate binding sites confer the greatestdegree of specificity.47,60 For example, cathepsin K prefers a hydrophobicamino acid/Pro at P2, whereas cathepsin S favors Leu/Val at P2 andcathepsin L a hydrophobic residue at P1 and P3.
3.3. Protease Inhibitors
There are several types of protease inhibitors, and three main methods havebeen used to create new inhibitors: (1) rational design of peptidomimeticinhibitors using substrates as starting points; (2) screening of naturalproducts and corporate compound libraries as starting points and subsequentrefinement of the substances that are identified; (3) if accessible, use of dataon the three-dimensional structure of the active site of the enzyme (obtainedby NMR studies or X-ray crystallography) to apply rational design for thedevelopment of new protease inhibitors.
Inhibitors and substrates generally adopt an extended β-strandconformation in which the linear extended β-strand backbone givesmaximum exposure of its side chains to the binding pockets of the protease.This conformation precludes intramolecular hydrogen bonding in the peptideand exposes the peptide bonds to catalytic hydrolysis.

26
NH
O
NH
X
O
NH
O
NH
X
O
O
NH
NH
Gly193Ser195
Serine protease
X= H, CF3, C2F5, CONHR, COOR, COR, Heterocycle
Serine-trap inhibitor Inhibited serine protease
P2
P1
P2
P1Ser195
3.3.1 Serine Protease Inhibitors
The serine protease inhibitors can be divided into two categories: those thatare covalent (reversible and irreversible) and those that are non-covalent.One of the categories comprises inhibitors derived from the nonprime side ofthe peptide substrate, wherein the scissile amide bond is replaced by anelectrophile that interacts with the catalytic serine residue.48,61–63 However,there are also several potent prime-side inhibitors, which indicates thatinteraction with the prime sites of the enzyme can enhance the bindingaffinity of an inhibitor.61,64,65 The main reason for using an electrophilicgroup to achieve inhibition is to increase the affinity through formation of acovalent tetrahedral intermediate with the catalytic residue, thus resemblingthe transition state of a normal substrate hydrolysis (Fig. 12).66,67
Figure 12. Examples of electrophilic groups that can serve as serine trap inhibitors byforming a tetrahedral intermediate with the enzyme and thus inhibiting the enzyme fromfurther activity.
Typical electrophiles are nitriles, trifluoromethyl ketones, pentafluoroethylketones, aldehydes, α-ketoamides, α-ketoester, α-diketones, α-heterocycles,organoboranic acids, and organophosphonate esters.
Examples of inhibitors that interact with the enzyme via an irreversiblecovalent bond are alkylating agents such as monohalomethylketones orepoxides, or acylating agents such as β-lactams.61,62,68,69a There is a potentialdisadvantage in using covalent inhibitors, since reactivity of the electrophilicmoiety with endogenous proteins or off-targets may result in immuneactivation and other adverse events. Another potential issue is low chemicalstability of the inhibitors. For these reasons, non-covalent inhibitors are oftenpreferred over covalent inhibitors.63 Hydrogen bonds, hydrophobic,electrostatic, and/or van der Waals interactions deliver the binding affinity ofnon-covalent inhibitors.

27
RNH
R'
O
NH
O
F
R''NH2 N
H
NH2+
NH
O
NH
OO
CO2H
O
N NH
O
O
NH
Ph
SPh
O O
Halomethyl ketone-based inhibitors E-64
Vinyl sulphone: LHVS
NH
NH
R
O
R'
R'
R
CN
NHO
R'R
N
ONR
OR'
RNH
R'
O
NH
O
R''
X X
O
R
R' R''
Cyclic ketonesn =1-3
Aminoethyl amidesNon-covalent inhibitors
Nitriles B-Lactams Azepanones
X= H, CO2Me, CONHR
n
3.3.2 Cathepsin Cysteine Protease Inhibitors
The mode of action of enzyme inhibitors can be subdivided into distinctcategories.68 The earliest reported cathepsin inhibitors were irreversibleinhibitors, and they included compounds such as those based on the epoxideE-64, halomethylketones, and azapeptides, as well as those emanating fromMichael acceptors (e.g., the vinyl sulfone LHVS), which were particularlyuseful in elucidation of the pharmacology of cathepsin S inhibition (Fig.13).55 These compounds react with the free thiol of the cysteine in thecathepsin active site and bind in an irreversible manner to inhibit theproteolytic activity of the enzyme.
Figure 13. Classical irreversible cysteine protease inhibitors.
Later, chemists developed inhibitors that form a reversible covalent bondwith the enzyme, most of which have an electron-deficient carbonyl groupand low steric hindrance. Various ‘warheads’ that form such a bond with theactive site cysteine have been described, including peptide aldehydes,nitriles, α-ketones, and α-ketoamides, as well as cyclic ketones, β-lactams,azepanones, and non-covalent aminoethylamides (Fig. 14).69
Figure 14. Examples of reversible cysteine protease inhibitors.

28

29
Antigen
MHCClass II
InvariantChain
Peptide/MHCComplex
Denaturation
Proteolysis
PeptideLoading
Cathepsin S
CD4 T-cell
4. Solid-Phase Parallel Synthesis and SAR of 4-Amidofuran-3-one P1-Containing Inhibitors ofCathepsin S: Effect of Sulfonamides P3Substituents on Potency and Selectivity(Paper III)
4.1 Introduction
4.1.1 Antigen Presentation and the Immune Response
Antigen-presenting cells (APCs) degrade foreign proteins to small peptidesand display these peptides bound to a major histocompatibility complex(MHC) protein on the cell surface, and this process is known as antigenprocessing and presentation (Fig. 15).70 APCs, in particular primarilydendritic cells, B-lymphocytes, macrophages, and microglial cells take upantigens from the extracellular environment. The internalized proteinantigens are processed by use of endosomal or lysosomal proteases togenerate peptides that become associated with MHC class II protein.Peptide-loaded MHC-II complexes are subsequently transported to the cellsurface to be displayed to CD4+ T-cells. The biosynthesis of mature MHC-IIis controlled by interaction with the protein called invariant chain (Ii). Inshort, Ii prevents loading of endogenous peptides onto newly synthesizedMHC-II within the endoplasmic reticulum and facilitates the sorting of theMHC-II-Ii complex into the endosomal system. Once in the lysosomes, aconsortium of proteases degrades Ii to enable the peptide loading.
Figure. 15. Antigen processing and presentation.

30
Ii is processed in several steps (Fig.16) by several proteases, the exactidentity and number of which are unknown. Nevertheless, it is clear that, indendritic cells and B-cells, cathepsin S is the enzyme responsible for thefinal proteolytic step where it degrades the remaining Ii p10 oligomerfragment into a small peptide called CLIP. Finally, the CLIP peptide isremoved from MHC-II through an interaction with endosomal/lysosomalresident protein HLA to permit binding of antigenic peptides.
Figure 16. Processing of the invariant chain.
Recognition of the MHC-II-peptide complexes triggers the activation ofantigen-specific CD4+ T lymphocytes,71a which in turn stimulate othercomponents of the immune system, such as B-cells, macrophages, and CD8+T-cells (Fig. 17). These cellular reactions are a crucial part of the body’sresponse to pathogens, but they are also responsible for the development andsymptoms of allergies and autoimmune diseases.
The purpose of an immune response is to attack a foreign target, and inthat context it is extremely important to be able to distinguish ‘self’ from‘non-self,’ because failure of the immune system to recognize ‘self’ willresult in an autoimmune disease. Examples of current therapies for suchdisorders include TNFa blockers (e.g., Enbrel, Remicade and Humira) forthe treatment of rheumatoid arthritis (RA); interferon β, glatiramer acetate,mitoxantrone hydrochloride, and corticosteroids, which are oftenaccompanied by undesirable effects.71b Other immuno-based treatments likecyclosporine and cytokine inhibitors target the effector molecules ininflammatory reactions, and hence it may be possible to provide a novel
Leu-Ile Met-Leu 80 103
Ii N CCytoplasm Transmembrane CLIP Lumen
Ii p23 N
Ii p10 N
Cathepsin S
CLIP
MHC Class IIIi associated
CLIP
MHC Class IIPeptide complex

31
CytokinesDisease
Pathology
B cellActivated
CD4 T cellCD4 T cell
APC
CD8 T cell
Macrophage
MHC Class II
form of immunotherapy by focusing on the antigen presentation pathwayresponsible for triggering autoimmune diseases.71c
Figure 17. Role of T-cells in the immune response.
Cathepsin S is a 24-kD monomeric cysteine protease that belongs to thepapain super family, and it is expressed in many professional APCs as wellas ‘non-professional’ APCs (e.g., epithelial cells).72 Cathepsin S wasoriginally identified in bovine lymph nodes in 1975,73 and the human form ofthe protein was cloned in 1992.74-75
The most well-characterized function of cathepsin S is related to its rolein antigen presentation to CD4+ T-cells, which involves participation in theproteolysis of the Ii chaperon molecules that is associated with the MHC-IIcomplex,71a, 76a-b a crucial step in the immune response mediated by CD4+ T-cells. Therefore, a selective inhibition of cathepsin S will block thedegradation of the p10 invariant chain (Ii-p10) and disrupt the presentationof antigens, and thereby act in an immunosuppressive manner. Suchselective inhibition of cathepsin S represents a potential for modulatingautoantigen-driven immune responses in MHC-II-restricted autoimmunediseases.
While the role of cathepsin S in MHC-II-mediated antigen presentationhas received the most interest, the potent elastinolytic and collagenolyticactivity of secreted cathepsin S (by macrophages and microglia) additionallymakes the inhibitors of this protein as attractive alternatives for modulatingconnective tissue matrix degradation.
The crystal structure of cathepsin S is highly similar to the structures ofother cysteine proteases in the papain super family, especially cathepsins Kand L.76c The substantial sequence homology between these three cathepsinsoffers considerable challenges in designing selective cathepsin S inhibitors.The tertiary structure of cathepsin S comprises an N-terminal domain thatconsists primarily of α-helices and a C-terminal domain that contains β-sheets. Between these two domains lies the catalytic triad formed by Cys25,

32
His164, and Asn184 (corresponding to Cys25, His159, and Asn175 inpapain), from which a substrate-binding site extends out on both sides.77
Unlike most other cathepsins, which are inactive at neutral pH, humancathepsin S achieves its optimal activity at pH 6.5 and remains active frompH 4.5 to 7.8, consequently increasing its range of activity from acidiclysosomes to neutral or slightly alkaline extracellular environments.78
4.1.2 Cathepsin S as a Potential Therapeutic Target
Cathepsin S is a potential target for the treatment of autoimmune diseases.Substances that achieve selective inhibition of CD4+ T-cells should providea therapeutic advantage over non-selective immunosuppressive compounds,since the function of CD8+ T-cells would remain largely intact. Deletion orinhibition of cathepsin S in some disease-relevant animal models has beenshown to be efficacious. An example of this is a study of collagen-inducedarthritis in cathepsin S knockout mice,79a in which it was found that theseverity of the disease was diminished relative to that seen in wild-typemice. The available animal model data and knowledge regarding theinvolvement of CD4+ T-cells in the disease pathogenesis, as well as themedical need for effective treatment options, have made rheumatoid arthritisa lead indication for therapy using cathepsin S inhibitors. In the case ofrheumatoid arthritis, inhibition of cathepsin-S-mediated degradation ofextracellular matrix might also be beneficial. Cathepsin S might alsorepresent a new pain-relief strategy as reported in a study concerning theinhibition of spinal microglial cathespin S for the reversal of neuropathicpain. 79b-c
In addition to rheumatoid arthritis, it seems that cathepsin S may be asuitable target for the treatment of atherosclerosis and Sjögren’s syndrome,based on evidence provided by animal models using inhibitors and knockoutmice. Moreover, it is possible that cathepsin S inhibitors can be beneficial ina number of autoimmune and allergic conditions in which CD4+ T-cells areimportant factors in the pathogenesis, ranging from type I diabetes tomultiple sclerosis. Cathepsin S has been found to be a key enzyme in theprocessing of human myelin basic protein (MBP) in vitro80 where MBPappear to function as an autoantigen in the development of multiple sclerosis(MS). Cathepsin S inhibition might thus be a viable therapeutic approach forMS disease.
Involvement of cathepsin S in the pathogenesis of psoriasis has alsobeen explored.80b-81 Since the majority of psoriasis patients have elevatedinterferon-gamma-producing T-cells in their epidermis, it was speculatedthat upregulation of cathepsin S could be a factor in induction of the disease.Furthermore, a study has shown that cathepsin-S-positive neurons arepresent in the brains of patients with Alzheimer’s disease (AD) and Down’s

33
O
NH
NH
O O
O
S
28
syndrome,82 and the presence of Aβplaques and vascular deposits in thebrain are considered to be characteristics of AD. Cathepsin S has also beenfound to facilitate the formation of Aβfrom its precursor protein in a kidney239 cell line, which suggests that cathepsin S can be a target for AD therapy.Finally, cathepsin S has even been implicated in some cancers, for instancesubstantial expression of this protein has been observed in prostatecarcinoma.83
4.1.3 Background Information and Aim of the Study
We have previously described novel 4-amidofuran-3-ones as potentcathepsin S inhibitors,84 represented by compound 28, which has a Ki valueof 31 nM. The analogs of this series are equipotent against mouse and ratcathepsin S, enabling proof-of-principle studies to be carried out in a rodentmodel of human disease, such as multiple sclerosis (MS),76c,85 andrheumatoid arthritis (RA). The distal region of the S3 subsite of cathespin Sis defined by backbone Gly62 and the side chains of Lys64 and Phe70 (usingthe numbering from the PDB entry 1MS6 for cathepsin S). Analysis ofstructural data from cathepsin S in complex with 28 implied that there werepotential for hydrogen bond interactions in the S3 subpocket, and it seemedthat the carbonyls of Gly62 and Asn67 were especially likely candidates ashydrogen bond acceptors. Lys64 was also a possible hydrogen bond donor,but with less certainty due to the flexibility of the side chain (Fig. 18).
Figure 18. Compound 28 modeled in the active site of cathepsin S. The subpockets aredenoted S1, S2, and S3.
Some recent studies have indicated that structural differences observed in theS3 pockets of cathepsins S, K, and L give different preferences for the P3substituents.77,86,87 The reduced size of the S3 pocket in cathepsin S haspreviously been recognized from the crystal structures of cathepsin Sinhibitor complexes,88 and the preference for a larger P3 substituent in S3 ofcathepsin K has also been established.89 However, in other experimental

34
O
ON
O
N
O
NSOO
R3
R2
R1
P3
P2
P1
O
NNH
O O
O
NS
OO
H
H
29
S3
S2
S1
G62
K64G69
endeavors,86 receptor optimization calculations revealed that a unique Lys64residue residing in the cathepsin S S3 pocket can re-orient its side chain toaccommodate an extended P3 moiety of the inhibitor. This feature providesnew opportunities for targeting selectivity when considered in combinationwith the selectivity requirements of the S2 pocket, which, in cathepsin S,accepts significantly larger groups compared to the S2 pocket in cathepsin K.The use of structure-based drug design led us to the design of compound 29(Fig. 19) with methyl cyclopentylalanine as preferred P2 side chain and asulfonamide hydrogen donating P3 moiety and as an extended P3 motif.
Figure 19. Schematic representation of the predicted binding mode of inhibitor 29 within theactive site of human cathepsin S. The sulfonamide moiety is assumed to bind in the S3 pocket,the cyclopentyl group within the hydrophobic S2 binding pocket and the C5-ethyl fuarone inthe S1 site.
Compound 29 showed very good inhibitory activity against cathepsin S (K i =2.6 nM), with a 10-fold increase in potency compared to compound 28,although with both P2 and P3 groups altered (Table 2), as well as acceptableselectivity over cathepsin K (K i = 290 nM) and good selectivity towardscathepsin L (Ki = 12,000 nM). These encouraging properties of compound 29supported a more detailed exploration of this class of inhibitors, focusing inparticular on the S3 pocket of cathepsin S.The aim of the study presented in Paper III was to explore the S3 pocket ofcathepsin S and to investigate the scope of the sulfonamide moiety at the P3residue of the C5-ethyl furanones. The intention was to consider theindicated moiety as an extended P3 substituent, while keeping the P2 and P1residues constant (Fig. 20), in order to see how it affects the potency and theselectivity profile.
Figure 20. Schematic representation of the intended P3 variations (R1, R2 and R3) in the C5-ethyl furanone inhibitors of cathepsin S.

35
X
HN
OH
O
SR1
O O
R2
O
O
FmocHN
HN
O
HN NH NH
O
CNH
OO
HN
O HN
O
NH NH NH
O
CNH
OO
Fm ocHN
HN
O
FmocHN NH NH
O
CNH
O
H2N NH NH
O
COOH
O
HN
O
HN
S
NH
O
O
2
3
a, b
c , d
c, e
h, i
xTFA
c, f-h
43, 68-70
O
R1
O
X= C, N
X
HNS
O O
R1R2
X= C, N
O
O
HN
OHN
O
X
HNS
O O
R1R2
X= C, N
29, 37-42, 44-67, 71-81
30 31 32
36 35: 34
4.2 Chemistry
4.2.1 Solid-Phase Chemistry of Dihydro-2(3H)-5-ethylFuranones
Scheme 7 shows the solid-phase parallel synthesis of dihydro-2(3H)-5-ethylfuranones 29 and 37–81 containing sulfonamide moieties as P3 motifs (seestructures in Tables 2–5).
Scheme 7. Solid-phase synthesis of dihydro-2(3H)-5-ethyl furanone inhibitors.Reagents and conditions: (a) MeOH, 70 o C 2 h, then RT 2 h, 85%; (b) amino resin,91 HBTU,HOBt, NMM, DMF, 16 h; (c) 20% piperidine in DMF, 1 h; (d) Fmoc-(methylcyclopentyl)-alanine-OH (33),92 HBTU, HOBt, NMM, DMF, 16 h; (e) P3 acid (35), HBTU, HOBt, NMM,DMF; (f) 4-amino benzoic acid, HBTU, HOBt, NMM, DMF, 16 h; (g) R1SO2Cl, DMAP, Py,DCM, 16 h; (h) 95%TFA/H2O, 1 h; (i) additional step for comp 65: H2, Pd/C, MeOH, 2 h,91%.
Dihydro-(4S-amino-[N-Fmoc])-5S-ethyl-3(2H)-furanone (30) synthesized aspreviously reported84 was treated with acid 3190 in MeOH. The resultingsemicarbazone acid linker was obtained in 85% yield. This linkage not onlytemporarily blocks the reactive carbonyl functionality, but also serves as ananchor for construction of the inhibitor via parallel synthesis. Subsequently,the P1 resin 32 was obtained by treating the linker with the aminomethylfunctionalized polymer support91 (0.24 mmol/g) in the presence of N-[(1H-benzotriazole-1-yl)-(dimethylamino) methylene]-N-methylmethanaminiumhexafluorophosphate N-oxide (HBTU), 1-hydroxybenzotriazole (HOBt), andN-methyl morpholine (NMM) in DMF. Deprotection of resin 32 with 20%

36
O
NH
SO
O
OH
O
NH
SO
O
OH
O
NH
SO
OS
OH
O
NH
SO
O
OHO
S
OO
NH
OH
O
O H
NHS
O O
OS
NH
O O
OH
O
NH
SN
O
O
OHO
NHS
O
O O
OH
O
NS
O
O
OH
O
NHS
O
O
OH
O
NHS
O
O
OH
O
NHS
O
O
OHO
NH
SO
O
OH
O
NH
SO
O Cl
OH
O
NH
SO
OO
OH
O
NH
SO
O
OH
O
NH
SO
O F
O HO
NHS
O
O
OHO
O
NH
SO
O F
OH
O
NH
SO
O
F
OH
O
NHS
O
O N
N
OH
O
NH
SO
O
OH
O
NH
SO
O
F
OH
O
NHS
O
O
OH
O
NHS
O
O
ClOH
O
NH
SO
O O
OH
O
NHS
O
O
O
OH
O
NNHS
O
O
OH
O
NNHS
O
O
OH
O
NHS
O
O
NO 2
OH
O
NHS
O
O
NCl
OHO
NHS
O
O
OH
O
NHS
O
O
O H
O
NHS
O
O
N
O H
O
NHS
O
O
N
O H
O
NH
SO
OS
N
OH
O
NHS
O
O S
N
OH
O
NHS
O
O
N
O H
O
NHS
O
ON
OH
O
NHS
O
O S
N
OH
O
NH
SO
O S
N
OH
35a 35b 35c 35d 35e 35f 35g
35h 35i 35j 35k 35l 35m 35n
35o 35p 35q 35r 35s 35t
35v
35u
35w 35x 35y 35z 35aa 35ab
35ac 35ad 35ae 35af 35ag 35ah 35ai
35aj 35ak 35al 35am 35an 35ao 35ap
piperidine in DMF followed by standard amino acid coupling of the Fmoc-protected methyl cyclopentane alanine amino acid92 33 by use of HBTU,HOBt, and NMM in DMF provided P2-P1 resin 34. After standard removalof the Fmoc group, the P3 acids 35 (Fig. 21), which had been preparedpreviously were coupled using the same coupling conditions as describedabove to furnish P3-P2-P1 resins 36.
A slightly different procedure was employed for target furanones 43(Table 3) and 68–70 (Table 5), for which coupling of the P3 acid residue wasperformed in two steps: attachment of 4-amino benzoic acid onto de-protected resin 34 under standard amino acid coupling conditions (HBTU,HOBt, NMM), and subsequent acylation with the corresponding sulfonylchloride using DMAP and pyridine in DCM.
Final acidic hydrolysis with 95% aqueous TFA cleaved the targetketones 29 and 37–81 (Tables 2-5) from the Webb-linker-bound resin in 75–80% yields. Catalytic hydrogenation using H2 in the presence of Pd/C wasrequired to provide target 65 (Table 5).
Figure 21. Structures of the commercially and non-commercially available P3 acids (35) thatwere utilized in the synthesis.

37
X
H2N
o
o
R2
X
HN
oo
SR1
o o
R2
X
HN
OH
o
SR1
o o
R2
oSo
ClR1
a b2
3 2 23 3
35X=C,N
NS
OO
35ai-35alN
Ar3P=Oa
b
H2N
O
O NH
O
O
S
O O
NNH
OH
O
S
O O
N
c
R= H, Me
R
R R
O
4.2.2 Synthesis of P3 acid Capping Groups
Scheme 8 illustrates the general procedure for the synthesis of the non-commercially available P3 acids 35. Briefly, 4-amino benzoic acid methylester derivatives were reacted with diverse sulfonyl chlorides using pyridineand a catalytic amount of DMAP in DCM to provide the corresponding P3capping group methyl esters. Basic hydrolysis of the methyl ester functionwith 2.5M LiOH in THF:MeOH for 30 min at 110 oC in a microwave cavityfurnished the P3 acids in 3–98% overall yields.
Scheme 8 . General procedure for the synthesis of P3 acids 35 (Fig. 21). Reagents andconditions: (a) Py, DMAP, DCM; (b) LiOH (2.5M), THF:MeOH 2:1, microwave 110 oC, 30min.
Scheme 9 shows an alternative approach93a to the synthesis of the pyridineP3 acids 35 (35ai, 35aj, 35ak, and 35al) (Fig. 21) that were utilized togenerate target compounds 71–74 (Table 5). Trifluoromethane sulfonic acidanhydride was reacted with polymer-supported triphenylphosphine oxide93b
in DCM followed by addition of the cold pyridinium salt of thecorresponding pyridine sulfonic acid and subsequent addition of thecorresponding 4-amino benzoic acid derivative to afford the correspondingpyridine P3 capping group methyl esters. Basic hydrolysis with LiOH asdescribed above furnished the pyridine P3 acids 35ai–35al in 13–25%overall yields.
Scheme 9. Synthesis of P3 acids 35ai–35al (Fig. 21). Reagents and conditions: (a)(CF3SO2)2O, DCM, 1 h (b) 16 h; (c) LiOH (2.5M), THF:MeOH 2:1, microwave 110 oC, 30min.

38
Cmpd Structure Ki (nM)Cath S
28
O
ONH
O
NH
O
S31
29O
ONH
O
NH
O
NH
S
O
O
2.6
37O
NH
O
NH
O
O
NH
S
OO
12
38O
NH
ONH
O O
NS
OO
110
39O
NH
ONH
O O
SNH
OO
140
40O
NH
O
NH
O O
SNH
OO190
4.3 Tables: Inhibitory Activity and Selectivity
Table 2. Inhibition constant (Ki) values against the cathepsin S protease

39
O
ON
O
N
O
NH
SOO
R1
Cmpd P3 acid R1 Ki (nM)Cath S
41 35ag Et 1.9
42 35ah n-But 3.4
43F
FF 8.3
44 35m 2.9
45 35q 2.4
46 35a 180
47 35i N 5.3
48 35b 0.82
49 35c 31
Table 3. Effect of R1 variations

40
O
ON
O
N
O
X
NH
SOO
3
2
R1
R 2
Compd P3 acid R1 R2 X Ki (nM)Cath S
50 35j Me 2-OMe C 26
51 35p Me 3-OMe C 330
52 35l Me 2-Me C 4.9
53 35n Me 3-Me C 30
54 35o Me 2-Cl C 5.6
55 35r Me 2-F C 16
56 35s Me 2-COMe C 57
57 35y Ph 2-Me C 4
58 35z Ph 2-Cl C 12
59 35ac Ph H N 56
60 35ad Me H N 63
Table 4. Effect of variations at the central ring

41
O
ON
O
N
O
NH
SOO
R 1
Cmpd P3 acid R1 Ki (nM)Cath S
Cmpd P3 acid R1 Ki (nM)Cath S
61 35uF
2 72 35aiN
2.1
62 35tF
3.4 73 35ajN
2.3
63 35xF
3.8 74 35alN 0.93
64 35w 3.9 75 35af N
Cl
5.9
65 35ae NH 21.3 76 35d
S2.3
66 35ab O 3.3 77 35v N
N
4.5
67 35aaO
0.99 78 35anS
N 13
68NC
1.1 79 35apS
N 5.7
69NC
0.64 80 35amS
N 0.69
70CN
0.79 81 35aoS
N8.6
71 35akN
2.7
Table 5. Effect of R1: aromatic substituents and heteroaromatic rings

42
Cath S Cath K Cath LCmpdKi(nM) Ki(nM) Ki(nM)
Cath K/Cath S
28 31 318 10000 1029 2.6 290 12000 11037 12 1500 2500 12038 110 98 11000 139 140 420 27000 340 190 23000 15000 12041 1.9 470 17000 25042 3.4 1500 21000 44043 8.3 1200 15000 14044 2.9 500 15000 17045 2.4 440 12000 18046 180 950 64000 547 5.3 300 16000 6048 0.83 430 11000 52049 31 1700 15000 5050 26 560 7200 2051 330 5000 29000 1052 4.9 240 2000 5053 30 1900 23000 6054 5.6 840 970 15055 16 700 7800 4056 57 750 9500 1057 4 730 2300 18058 12 1600 1100 13059 56 2200 31000 4060 63 1200 14000 2061 2 370 5900 18062 3.4 520 7800 15063 3.8 980 11000 26064 3.9 550 8400 14065 1.3 950 9100 73066 3.3 500 11000 15067 0.99 270 14000 27068 1.1 950 7900 86069 0.64 730 6800 114070 0.79 820 8500 104071 2.7 1100 13000 40072 2.1 570 14000 27073 2.3 450 13000 19074 0.93 260 8700 28075 5.9 800 23000 13076 2.3 1000 13000 43077 4.5 640 24000 14078 13 3000 27000 23079 5.7 1700 9100 30080 0.69 1800 16000 261081 8.6 2300 17000 270
Table 6. Selectivity assay: comparison of inhibitory activity of the compounds in humancathepsin S, K and L
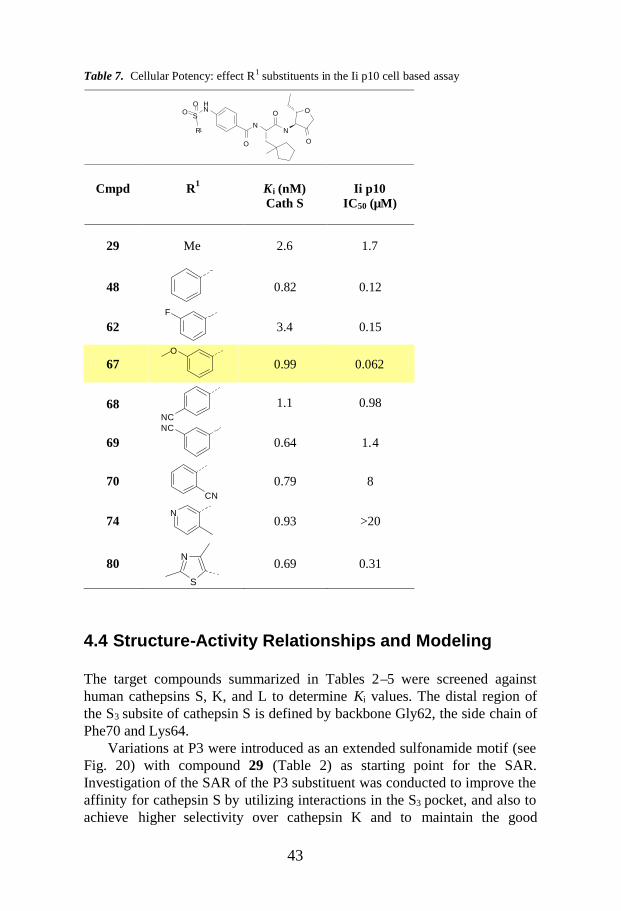
43
O
O
N
O
N
O
NH
SOO
R1
Cmpd R1 K i (nM)Cath S
Ii p10IC50 (µM)
29 Me 2.6 1.7
48 0.82 0.12
62F
3.4 0.15
67O
0.99 0.062
68NC
1.1 0.98
69NC
0.64 1.4
70CN
0.79 8
74N
0.93 >20
80S
N0.69 0.31
Table 7. Cellular Potency: effect R1 substituents in the Ii p10 cell based assay
4.4 Structure-Activity Relationships and Modeling
The target compounds summarized in Tables 2–5 were screened againsthuman cathepsins S, K, and L to determine Ki values. The distal region ofthe S3 subsite of cathepsin S is defined by backbone Gly62, the side chain ofPhe70 and Lys64.
Variations at P3 were introduced as an extended sulfonamide motif (seeFig. 20) with compound 29 (Table 2) as starting point for the SAR.Investigation of the SAR of the P3 substituent was conducted to improve theaffinity for cathepsin S by utilizing interactions in the S3 pocket, and also toachieve higher selectivity over cathepsin K and to maintain the good

44
selectivity towards cathepsin L. Modeling was done to rationalize the SARobserved for the various P3 extensions and to explain the selectivity overcathepsin K.
4.4.1 Enzyme Inhibitory Activity
Table 2 shows the SAR around the initial lead compound 29. In compound37, the sulfonamide moiety was placed at the meta position in the P3 phenylring, which resulted in a slight drop in activity (K i = 12 nM), indicatingpreference for the substitution at the para position. Methylation of thenitrogen of the sulfonamide 38 was detrimental to enzyme activity (50-folddrop), as was also the case for compounds 39 and 40 with reversedsulfonamides. These results indicate the importance of the NH group, thephenyl substitution pattern and the orientation of the sulfonyl group for theinteractions in the S3 pocket.
Compound 29 was modeled in the active site of cathepsin S (Fig. 22)using an in-house crystal structure as the starting point. This inhibitor bindsthrough a covalent reversible interaction with the side chain of Cys25 and ahydrogen bond with His164. The backbone hydrogen bonds of the inhibitorare between the P1 NH and the main chain carbonyl of Asn165, and betweenthe carbonyl and the NH of the P2 and the Gly69 main chain NH andcarbonyl, respectively. The P3 carbonyl interacts with solvent only, whereasthe P2 side chain is in close interaction with Phe70, Met71, Gly137, andVal162. The P3 phenyl ring has an edge on aromatic interaction with Phe70and is in close contact with Gly68 and Gly69 in the S3 subpocket. Thesulfonamide adds stability and activity to the binding of this series through ahydrogen bond from the NH to the carbonyl of Gly62 and via hydrogenbonds from the sulfone group to the side chain of Lys64.
Figure 22. Compound 29 modeled in the active site of cathepsin S. The binding mode of thesulfonamide in particular gives this series increased activities against cathepsin S. Thesubpockets are denoted S1, S2, and S3.

45
The slight drop in activity exhibited by compound 37, which has thesulfonamide in meta position, depends on a different hydrogen bond pattern.The NH of the sulfonamide participates in a hydrogen bond with thebackbone of Asn67 instead of Gly62, whereas the sulfone still interacts withLys64. The methylation of the nitrogen as in compound 38 makes thehydrogen bond with Gly62 impossible, and hence there is a large drop inactivity. The reversed sulfonamides in compounds 39 and 40 lead to lessoptimized interactions with Gly62 and Lys64 and therefore to lower potency.
We also investigated the effect of the R1 substituent on the sulfonamidemoiety, as shown in Table 3. Analysis of the data indicate a preference forthe phenyl group (48), with a considerable increase in enzyme activity (Ki =0.82 nM). Small unbranched alkyl chains (41, 42) and small branched alkylchains and rings (44, 45) were well tolerated, whereas larger rings (46) had adetrimental effect. A slight drop in activity was observed for other groups(43, 47). Elongation of the phenyl substituent (49) led to substantialreduction in activity compared to compound 48. From these observations, itcan be concluded that the phenyl substituent at R1 was the preferred one(48), with a threefold increase in potency over the parent compound 29, dueto aromatic stacking between Phe70 and both P3 phenyls, while maintaininggood hydrogen bonds with Gly62 and Lys64 (Fig. 23).
Figure 23. Compound 48 modeled in the active site of cathepsin S. The additional aromaticstacking with Phe70, and maintained optimal interactions with the sulfonamide, increased theactivity of compound 48 compared to compound 29.
Table 4 shows the SAR around the variations at the central phenyl ring.Diverse R2 substituents at the 2- and 3- positions of the ring and theexchange of the central phenyl for a pyridine ring were explored, keeping R1
as methyl or phenyl. A loss of inhibitory activity was observed for all thesecompounds. Substitutions at the 2 position of the phenyl ring were better

46
tolerated than at the 3 position (50 vs. 51, 52 vs. 53). Small substituents likea methyl group in targets 52 and 57 (K i values 4.9 and 4 nM, respectively)were better tolerated than larger groups (50, Ki = 26 nM; 56, Ki = 57 nM).Despite the fact that the 2 position was the best tolerated in terms ofsubstitution pattern, in general, decorations of the central phenyl ring byintroduction of R2 substituents did not have a favorable effect in terms ofpotency, the corresponding unsubstituted compounds 29 (Table 2) and 48(Table 3) were more potent (K i values 2.7 and 0.82 nM, respectively).Pyridine compounds 59 and 60 were considerably less potent (Ki values 56and 63 nM, respectively) than their phenyl analogs 29 and 48. Mostsubstitutions alter the position of the central phenyl ring, which is importantfor the aromatic stacking with Phe70 and for allowing the optimal hydrogenbond pattern of the sulfonamide with Gly62 and Lys64 residues on the S3
pocket of cathepsin S.
The encouraging results obtained for compound 48 (Table 3) prompted us tofurther explore the development of the SAR around this new lead. Table 5shows the changes that were made at the distal phenyl ring of thesulfonamide. Substitutions were carried out at the different positions aroundthe phenyl ring using electron-withdrawing groups (61–63, 68–70), electron-donating groups (64, 66, 67), and an amino group (65). Replacement of thedistal phenyl ring with heterocycles such as pyridines (71–75), thiophene(76), imidazole (77), and thiazoles (78–81) was also approached in order tofurther extend the SAR.
The majority of these inhibitors showed good affinity for cathepsin S,with Ki values in the same range as the parent lead compound 48. From thesefindings, it can be deduced that a cyano substituent in the meta position atthe distal phenyl ring afforded the most potent compound (69), resulting in aK i value of 0.64 nM, which represented a further increase in potency (> 4 x)compared to initial compound 29 and an increase of 48 times respect tocompound 28 (Table 2). Pyridine rings were well tolerated (71–74, with K i
values of 0.93 to 5.9 nM). However, chloro-substituted pyridine 75 led to adrop in activity (Ki = 5.9 nM), and methyl-substituted pyridine 74 was thepreferred compound of this type (Ki = 0.93 nM). Replacement of the distalphenyl ring with a thiophene (76) also provided a compound with goodinhibitory activity against cathepsin S (K i = 2.3 nM), whereas otherheteroaromatic rings such as imidazole (77) and 2,5-thiazoles (78, 79, 81)led to a 5–10-fold decrease in activity compared to compound 48.Surprisingly, the 2,4-thiazole 80 proved to be a highly potent compound,with a Ki of 0.69 nM. The increased activity of the compounds with anaromatic R1 group in P3 (Table 5) can be explained by the additionalaromatic stacking with Phe70 together with preservation of the favorablebinding mode of the sulfonamide, as described above for compound 48.

47
4.4.2 Selectivity Profile
A selectivity assay was performed to measure the Ki values for all of thesynthesized compounds in relation to human cathepsins K and L. As shownin Table 6, all compounds were highly selective for cathepsin L (> 1000 x),and they had affinity varying from 98 to > 20,000 nM for cathepsin K. Thestarting compound 28 had poor selectivity over cathepsin K (10-fold), andcompound 29 showed a selectivity of 100-fold over cathepsin K, which iswithin the limit of acceptance for our target minimum selectivity ratio. Twoof the target compounds (38, 39) displayed no selectivity between cathepsinsS and K, and several other targets (46, 47, 49–53, 55, 56, 59, and 60) hadlost selectivity (by more than 100-fold).
However, a very interesting finding was made when examining the data: theenhanced activity attained in compound 48 (> threefold) was accompaniedby a fivefold increase in selectivity over cathepsin K (> 500-fold) comparedto initial compound 29. Moreover, the same trend was found for the cyano-phenyl compounds 69 and 70 and the 2,4-thiazole 80, which showed afurther increase in affinity for cathepsin S (Ki values of 0.64, 0.79, and 0.69nM, respectively) compared to the phenyl compound 48 (> fourfold increasewith respect to 29) and also exhibited a greater increase in selectivity overcathepsin K (> 1,000-fold for 69 and 70, and > 2,600-fold for 80).Compound 80 proved to be the most selective of all the compounds of thisserie, showing a 24-fold and 240-fold increase in selectivity over cathepsinK compared to compounds 29 and 28, respectively, and fivefoldimprovement in selectivity ratio over cathepsin L compared to compounds28 and 29.
Besides the use of a cathepsin S favored P2 group, this selectivity wasmainly due to the lack of specific interactions with the sulfonamide in the S3
subpocket of cathepsin K. The hydrogen bond that is formed with thebackbone carbonyl of Gly62 in cathepsin S does not exist with thecorresponding Glu59 in cathepsin K due to a different loop conformation.The hydrogen bonds with the side chain of Lys64 in cathepsin S could not bereplaced by the corresponding Asp61 in cathepsin K.
Even more interesting was the increased selectivity of the inhibitorsdisplaying an aromatic substituent on the sulfonamide, a characteristic thatcan be explained by both greater activity towards cathepsin S and decreasedactivity towards cathepsin K. The aromatic group cannot form a similarstable binding mode in cathepsin K due to the lack of anchoring points of thesulfonamide, and therefore the additional aromatic group is flexible withoutspecific interactions.

48
A comparison of the active sites of cathepsins K and S is presented in Figure24, which shows a model of compound 80 in the active site of cathepsin S.
Figure 24. Comparison of the active sites of cathepsins K (left) and S (right) using thenumbering from PDB entry 1MS6 for cathepsin S and entry 1BG0 for cathepsin K. A modelof compound 80 is shown in the active site of cathepsin S. The different shapes of primarilyS2 (purple) and S3 (orange) are visible. The most important interactions exhibited by thisseries that are lost in cathepsin K are the hydrogen bonds with the sulfonamide.
4.4.3 Cellular Potency
Selected compounds were tested for cellular potency using the Ii-p10cellular assay, which measures inhibition of the cleavage of Ii in (human)9001 B-lymphocytes (Table 7). That method is comparable to the assaydescribed by Thurmond et al.,94 which determines the accumulation of thep10-Ii fragment (a natural cathepsin S substrate).
Some of the compounds (48, 62, 67, 68, and 80) showed markedlyimproved cellular potencies compared to compound 29 (IC50 = 1.7 µM), asindicated by their IC50 values in the range 0.32–0.062 µM. Despite the goodenzymatic inhibitory activity obtained for the cyano-phenyl compounds 69and 70, we did not observe a corresponding increase in their cellularpotency, and indeed the cellular activity of the pyridine analog 74 wasabolished. The reasons for the decreased cellular potency of 70 as comparedto the isomers 68 and 69 and for the low cellular potency of 74 are notreadily apparent. Factors that might influence cellular activity includeoverall cell permeability, access to subcellular compartments containingcathepsin S, and protein binding.
Notably, compound 67, with a K i value of 0.99 nM for cathepsin S,conferred the best cellular potency, showing an IC50 value of 62 nM, whichrepresents a 27-fold increase relative to compound 29.

49
We also tested the compounds in a pseudo-functional cellular assay thatmeasures T-cell activation by assessing the release of the cytokineinterleukin-2 (IL-2) into the tissue culture medium (data not shown). Theresults correlated well with the findings of the Ii-p10 assay, which furthersupports the findings and demonstrates highly efficient inhibition ofcathepsin S.
4.5 Conclusions
The solid-phase parallel synthesis and SAR of a series of inhibitors with asulfonamide moiety in the P3 position that are reported here resulted in thediscovery of highly potent and selective 4-amidofuran-3-one inhibitors of thecysteine protease cathepsin S. The findings of this study show that it ispossible to improve the inhibitory enzyme activity and the cellular potencyof this class of compounds towards cathepsin S by inducing interactions inthe S3 subsite of the enzyme. Furthermore, despite previous reportsindicating that the S3 pocket in cathepsin S is smaller than that in cathepsinK, our results involving extended P3 sulfonamides show that inhibitors withlarge P3 groups can be accommodated in the S3 pocket of cathepsin S. Thepotency of the synthesized inhibitors towards cathepsin S is due to thehydrogen bond network between the sulfonamide and two residues in thedistal region of the S3 pocket.
Although the main selectivity determinant for cathepsins S and K resides inthe S2 pocket, in the present study improved selectivity was achievedthrough favorable interactions between the inhibitor and the S3 pocket ofcathepsin S (Table 6). The incorporation of aromatic substituents on thesulfonamide moiety proved to be useful for increasing the enzyme andcellular inhibitory potency against cathepsin S and the selectivity overcathepsin K. Several members of this series, notably one carrying the P3thiazole sulfonamide moiety (80), display very low (single-digit) nanomolarK i values against cathepsin S and also exhibit an excellent selectivity profileover cathepsins K and L. In addition, compound 67 has an IC50 value of 62nM, which represents a 27-fold increase in cellular potency compared toinitial compound 29.

50

51
5. Preparation and Characterization ofAminoethylamide Inhibitors of the CysteineProtease Cathepsin K (Paper IV)
5.1 Introduction
5.1.1 Cathepsin K and Osteoporosis
Osteoporosis has a significant impact on the quality of life, morbidity, andmortality of ageing populations, and is most commonly found inpostmenopausal women.95a -bThis condition involves skeletal deteriorationthat leads to bone fractures, and it constitutes the second largest healthproblem in the world, being a major cause of death, disability, and medicalexpenses. Statistics show that one in two women and one in eight men willsuffer osteoporosis-related fractures. Indeed, 1.7 million hip fractures werereported in 1990, and it has been estimated that that number will increase to6.3 million by 2050.
The human skeleton consists of 80% cortical bone and 20% trabecularbone. Cortical bone is the dense, compact outer part, and trabecular bonemakes up the inner meshwork. Bone is a form of connective tissue thatcontains type I collagen fibrils and hydroxyapatite. Calcium phosphatecrystals in the form of hydroxyapatite [Ca10(PO4)6(OH)2] are deposited in theosteoid (uncalcified bone matrix) and converted into hard bone. The bonemass undergoes continual remodeling that is performed by osteoblasts andosteoclasts. The osteoclasts are large multinucleated cells responsible forbone resorption, whereas the role of osteoblasts is in rebuilding or formationof bone (Fig. 25).
Figure 25. The process of bone remodeling.

52
A cycle of bone remodeling starts with recruitment of osteoclasts bycytokines (e.g., IL-6). The osteoclasts adhere to and move along an area oftrabecular bone, digging a pit by secreting hydrogen ions and proteolyticenzymes, and this gradually liberates factors such as IGF-1 that areembedded in the bone. These released factors recruit and activate successiveteams of osteoblasts, which have developed from precursor cells uponactivation by parathyroid hormone and calcitriol. The osteoblasts invade thesite and synthesize and secrete the organic bone matrix (osteoid). Thestimulated osteoblasts secrete IGF-1 and eventually other cytokines such asIL6, which in turn recruit osteoclasts and thereby in a very simplisticdescription bring the cycle back to its beginning.95c-d
When bone resorption by osteoclasts outpaces bone formation byosteoblasts, an overall fragility occurs in the microarchitecture of the bone.This imbalance is clinically manifested as osteoporosis, a diseasecharacterized by an increased risk of bone fractures resulting from areduction in bone mass. Patients suffering from osteoporosis have very lowbone density and increased curvature of the spine (Fig. 26).
(A) (B)
(C)
Figure 26. (A) Micrographs comparing normal (left) and osteoporotic (right) bone tissue. (B)Graph showing bone mass in relation to age for women and men. (C) Radiographs showingthe development of osteoporosis in the spine.
Cathepsin K is a papain-like cysteine protease96–97 that shows 73%homology to cathepsin S and 76% to cathepsin L, and it is highly expressedduring bone resorption. Also, it is preferentially expressed in osteoclasts,which, together with its ability to degrade type I collagen, suggests that

53
cathepsin K plays a specific and pivotal role in bone resorption during thecourse of remodeling. Osteoclastic degradation of bone involves twoprocesses: demineralization of the inorganic components of bone anddegradation of the organic matrix. These two functions occur sequentially bytwo separate mechanisms (Fig. 27).96–101
Figure 27. The two phases of osteoclastic bone resorption: demineralization (a) anddegradation (b), reprinted with the kind permission from Elsevier Science Ltd.
In the first phase of resorption (Fig. 27a), the bone matrix is demineralizedby the production of an acidic environment in the sealed compartment in theosteoclast-bone interface. The acidic environment is generated by the activetransport of hydrogen atoms into the lacunae by vacuolar H+ATPase. Thehydrogen atoms are produced through the spontaneous dissociation ofH2CO3 which, in turn, is generated by action of the enzyme carbonicanhydrase II (CAII). The pH within the osteoclast is maintained by a passiveCl–/HCO3– exchanger at the resorptive surface of the cell. The acidicdegradation leaves behind a demineralized region of bone beneath theosteoclast, and then in the second phase the organic component (comprisingmainly collagen) is removed by cathepsin K protease-mediated digestion(Fig. 27b). Cathepsin K is stored as a pro-enzyme in the lysosomes of theosteoclast. Release of cathepsin K into the resorption lacunae stimulatesconversion of the enzyme to its mature form, a process that has been shownto be autocatalytic under acidic conditions.
The most striking evidence of the involvement of cathepsin K in the boneremodeling process and in bone disorders is seen in patients withpycnodysostosis, a disorder that is associated with an inactivating mutationin the coding sequence of the cathepsin K gene and is characterized by facialhypoplasia, premature closure of long bone growth, and bones that arebrittle, dense, and long and show severe osteosclerosis.102 A role for

54
cathepsin K in bone resorption is further exemplified by the observation thatmice deficient in this enzyme display an osteopetrotic phenotype.103,104
Evaluation of osteoclast resorption sites in bones of -/- mice revealedosteoclasts apposed to regions of demineralized bone, demonstrating that theextracellular matrix had undergone normal demineralization, but thedegradation of the demineralized matrix was retarded.103 In contrast, it hasbeen found that transgenic mice that overexpress cathepsin K have lowertrabecular bone volume as a result of higher bone resorption.105
Thus there is now overwhelming evidence of a critical role for cathepsin Kin bone resorption. Consequently, inhibition of this enzyme can no doubtprovide a therapeutic option in disorders such as osteoporosis that are causedby an increase in bone turnover, normally at menopause. Cathepsin Kinhibitors developed elsewhere have shown efficacy in vitro and in vivo.105-
107 For example, it has been reported that peptide aldehyde inhibitors ofcathepsin K reduced bone resorption in a human osteoclast resorption assayand in the ovariectomized (OVX) rat model.106 Furthermore, azepanone-based inhibitors have been found to lower levels of C-telopeptide (CTx), abiomarker of bone resorption in the serum and urine, by 77% in cynomolgusmonkeys.106 Cathepsin K inhibitors have also shown efficacy against boneresorption measured by BMD in Phase II clinical trials.107
The treatments for osteoporosis that are currently available are associatedwith adverse events and there is a need for additional anti-resorptives whichare safe and with a mode of action that is independent of calcitonin,estrogen, selective estrogen receptor modulators (SERMs) andbisphosphonates. Importantly, bisphosphonates are also known to reducebone formation, which does not seem to be the case for cathepsin Kinhibitors.108 Therefore, it is expected that cathepsin K inhibitors willattenuate bone resorption, and, by also allowing bone formation to continue,that they will cause re-equilibration of the bone remodeling process andthereby lead to healthier bone.
Who is at greatest risk of osteoporosis? People who are slender or thinboned, those with a fair complexion, persons with a family history ofosteoporosis, individuals with a diet low in calcium, those who do notexercise regularly, smokers, adults who require long-term use of drugs likecortisone, anti-seizure medications, or thyroid hormone, and finally adultswith excessive alcohol consumption.

55
O NH
O
NH
O
NH
OMe
S3
S2S1'
5.1.2 Aminoethylamide Cathepsin K Inhibitors: Aim of theStudy
Most of the work on cathepsin K inhibitors have focused on reversiblecovalent binding compounds containing reactive groups such as aldehydes,nitriles, ketones, or azepanones.69b-c However, non-covalent inhibitors areconsidered to represent the therapeutic golden standard, because they entailthe lowest risk of adverse effects related to aspects of selectivity or to animmune response induced by covalent modification of a host protein.Recently, several companies have independently describedaminoethylamides as non-covalent inhibitors of cathepsin K.109–114 In one ofthose studies, Altmann et al.110 proposed the concept of binding of theleucine side chain into the hydrophobic S2 pocket and the terminal aminecomponent extending into the S1' groove (Figure 28). In such an inhibitor,van der Waals interactions make up most of the binding interactions. Thebinding affinity of this type of non-covalent inhibitors is partly achievedthrough lipophilic P1' interactions of the aminoethylaniline moiety, whereelectron-donating substituents on the aniline are required for potency.114
Figure 28. Schematic representation of the assumed binding mode of a modelaminoethylamide inhibitor within the active site of human cathepsin K. The Cbz group isbelieved to bind in the S3 subsite, the isobutyl group of the leucine is within the hydrophobicS2 pocket, and the 4-methoxy-phenyl ring extends into the S 1'site.
Smyht115 and Marquis116 have suggested that aminoethylamides might act asslow turnover substrates for cathespin K.
Accordingly, the aim of the study reported in Paper IV was to furtherinvestigate the utility of aminoethylamides as potential non-electrophiliccathepsin K inhibitors.
At the outset of Medivir’s cathepsin K inhibitor program, libraries weredesigned to exploit information generated by screens performed using theRational Approach to Protease Inhibitor Design (RAPiD), which is aMedivir platform technology for obtaining data on enzyme pocketrequirements.117 These initial studies led us to believe that, unlike otherrelated cathepsins, cathepsin K selectivity and potency can be harnessed byinhibitors spanning across the prime site of S1' and S2'. To explore this

56
NH
O
NH
O
NR2
R1
R4
R3
NH
O
NH
O
N OH
O
O
NH2OH NH2 N+ +R2
R1
R4
R3
R2
R1
R4
R3
hypothesis, synthesis of a small focused library of aminoethylamides wasundertaken on solid phase as part of a larger program aimed at developingnovel cathepsin K inhibitors. My task in this study was to design animproved solid-support synthesis route to the aminoethylamides that wouldbe compatible with molecules containing groups sensitive to reduction andacidic conditions. It was also important that the route could be applied to abroad range of amides outside the aminoethylamide category in order toachieve a more general synthetic route to this type of protease peptideinhibitors.
5.2 Solid-Phase Synthesis of Aminoethylamides
5.2.1 Aminoethylamide Template
Representation of the aminoethylamide core structure:
From a retrosynthetic standpoint, one way to obtain the core structure mightbe to divide the skeleton into three parts by breaking the amide bonds at theedges of the central amino acid part of the skeleton, as follows:
This provides the three building blocks that are needed: an amino acid, anacid, and a primary aminoethylamine. Different solid-phase syntheticapproaches can be designed, depending on which diversity group it is ofinterest to vary.
5.2.2 Aldehyde Resin-Based Synthesis
The solid-supported synthetic route depicted in Scheme 10 was establishedat the onset of the project. The route was extended to various amines otherthan aminoethyl amines. Diversity at the P2 was achieved by using differentamino acids, variation at the P1-P1' was introduced by using diverse amines(R3NH2), and variation at the P3 was accomplished by introducing differentacids (R2COOH).

57
OMe
CHO
OMe
NNHR3
O
R1
Fmoc
O Me
NO
O
R1
H
R2
NH
O
R1
NHR3
O
OMe
NO
O
R1
Fmoc
OMe
NOH
O
R1
Fmoc
a b
c d e,f,g
82 83 84
85 86 87
Scheme 10. Reagents and conditions: (a) TosHNCHR1COOAllyl, NaBH3CN, DMF, 18 h,RT; (b) Fmoc-Cl, NMM, DMF, 18 h, RT; (c) Tetrakis (triphenylphosphine)Pd(0), PhSiH3,DCM, 30 min, RT; (d) R3NH2, PyBOP, HOBt, DIEA, DMF, 18 h, RT; (e) 20%piperidine/DMF, 2 h, RT; (f) R2COOH, DIC, DCM, 1 h, 0 °C, then filtrate and addition ofresin from previous step, 24 h, RT; (g) TFA/TES 95/5, 2 h, RT.
Allylester-protected amino acids were coupled to the aldehyde-based resin82 to yield 83. Fmoc protection of the amine of 83 yielded 84, anddeprotection of the acid of 84 yielded 85. Subsequently, coupling on theappropriate amine gave 86. Deprotection of the amine of 86 , coupling withthe appropriate acid, and acidic cleavage from the resin afforded thecorresponding products 87. Overall yields of 60–80% were obtained.
Although this route worked for most of the targeted compounds, it had somedrawbacks:
- It required two additional reactions (besides the loading andcleavage standard reactions): protection and de-protection of theamino functionality of the amino acid.
- It was not advisable for reducion sensitive functionalities and acidlabile functionalities in the molecule due to the acidic cleavageprocedure.
Thus, an improved solid-phase methodology was required to cope with therequirements. A shorter synthetic pathway would be an additional asset.
5.2.3 Sulfonamide Resin-Based Synthesis
A sulfonamide resin would be a very suitable solid-phase support for thesynthesis of the targeted aminoethylamides. Acylation of the sulfonamidesupport by the amino acid provides a support-bound N-acylsulfonamide thatis highly stable to strong bases as well as to strongly acidic conditions priorto activation. After the activation step, the product is released from the solidsupport by the attack of a nucleophile, in our case a primary amine, which isparticularly useful because additional diversity is introduced in the cleavagestep. Furthermore, no reducing agents or acidic conditions are employed in

58
+ FmocHNOH
O
R1
Fm ocHN
HN
O
R1
S
NH
O
O O
NH
HN
O
R 1
S
NH
O
O O
O
R2 NH
HN
O
R 1
R3
O
R 2
a
b, c d, e
88 89 90
91 87
So o
NH2
HN
(CH2)3
O
SNH2
OO
HN
O
SNH2
O O
92 93
this synthetic pathway. The synthetic route illustrated in Scheme 11 wasdesigned and investigated.
Scheme 11. Reagents and conditions: (a) PyBOP, HOBt, DIEA, resin 88, DCM, RT, 16 h; (b)20% piperidine in DMF, 2 h, RT; (c) R2COOH, PyBOP, HOBt, DIEA, DCM, RT, 24 h; (d)ICH2CN, DIEA, NMP, 24 h, RT; (e) R3NH2, THF, 12 h, RT.
Fmoc-protected amino acids 89 were coupled to resin 88 to yield 90. Initialexperiments were performed using phenylsulfonamide linker 92 and loadingof the amino acid by a preformation of the symmetrical anhydride of theamino acid by DIC in DCM at 0 °C.118 However, production of the librarywas done using 4-sulfamylbutyryl linker 93119 and a loading procedureinvolving standard peptide coupling PyBOP conditions.
Deprotection of the terminal amine of 90 and coupling of the appropriateacid yielded 91. Activation of the sulfonamide provided a highly activatedlinker, which could be readily cleaved by nucleophilic displacement withcommercially available amines (R3NH2, aminoethyl amines among them) toafford the corresponding products 87.
Activation of the acylsulfonamide linker was more efficient whenachieved by iodoacetonitrile treatment than by treatment with TMS-diazomethane (TMS-CH2N2) or bromoacetonitrile. The reaction steps weremonitored by FTIR and the ninhydrin test.19 Overall yields of 40–80% wereobtained. This route did not make use of any acidic treatment, and thus thenew sulfonamide solid-support route that was developed was more generaland shorter than the previous approach in order to synthesize a broad rangeof aminoethylamides.

59
Cathepsin K Cathepsin SStructureKi (nM)a Ki (nM)b
94a
O
NH
O
O
NH
NH
2500 4592
94bNH
HN
NO
O
O
650 3200
94cNH
O
O
NH
NH
O
12 2452
94di
NH
O
O
NH
NH
O9 2597
94e
O
NH
O
O
NH
N
17 39110
94fO N
H
O
O
NH
N300 15392
94gii
O NH
O
O
NH
NH
N 180 662
5.3 Inhibitory Activity and Kinetic Studies
From the initial screen of the library of synthesized compounds, it wasconsidered that 94a and 94b (produced according to Scheme 11), with K i
values of 2.5 and 0.65 M, respectively, against human cathepsin K (Table8), were an encouraging start. This led to a larger program aimed atdeveloping the series and addressing issues such as the modest selectivityover cathepsin S (Table 8).
Table 8. Examples of activity values for measurements of inhibition of human cathepsins Kand S by aminoethylamides.
aInhibition of recombinant human cathepsin K activity in a fluorescence assay using 70 M (=KM) D-Ala-Leu-Lys-AMC as substrate in 100 L of 100 mM NaH2PO4, 1 mM EDTA, 1 mMDTT, and 0.1% PEG400 at pH 6.5 in 96-well microtiter plates read in a Fluoroskan Ascentinstrument.bInhibition of the activity of recombinant human cathepsin S in a fluorescence assay using100 M Boc-Val-Leu-Lys-AMC as substrate in 100 mM NaH2PO4, 1 mM DTT, and 100 mMNaCl at pH 6.5.iThis compound is 7h in Altmann et al. (Ref. 109).iiThis compound is 3 in Kim et al. (Ref. 111) and compound 4 in Kim et al. (Ref. 114).

60
It was felt that the poor selectivity against cathepsin S could be addressed bymodification of the P3-P2 left hand side of the molecule. Based on the initialSAR that was generated, amino acid 89 was fixed as L-leucine. A variety ofsolution-phase routes were developed, which gave us greater flexibility andscaling-up possibilities, and also helped us avoid the risk of partialracemization of R1 that has been observed in the final step of similarmethodologies.120 Scheme 2 in Paper IV shows representative results ofsome of these approaches, which yielded compounds 94c–f. An example ofthe solution-phase methods used is illustrated in Scheme 12. Products 94dand 94g are literature reference compounds. The aminoethylamide 94g wasprepared using the route described by Kim et al. 111
NH
O
NH
O
O
NH
NH
O
NH
NH
O
ONH
NH2
94a
a b, c
Scheme 12. Reagents and conditions: (a) Boc-L-Leu, EDC, HOBt, TEA, DCM:DMF 2:1, 12h, 80%; (b) 50% TFA/DCM, 1 h, then (c) 3-furoic acid, EDC, HOBt, TEA, DCM:DMF 2:1,12 h, 85%.
However, as we developed the series, we discovered that the measured Ki
values were not reproducible. More specifically, at times there weresubstantial differences between separate batches of the same sample and,more importantly, between the same sample on different occasions. Forexample we recorded activities ranging from 0.49 to 11 µM for compound94b and from 26 nM to 25 µM for compound 94f. This was clearly a verydisturbing observation and may account for the difficulties we encounteredin trying to develop the SAR of the series.
To better understand what was happening, we undertook a kinetic study ofthe inhibition by measuring the on and off rates, using aminoethylamide 94cas an exemplar of this series (Fig. 29). Close inspection of the assay kineticsindicated that this compound displayed time-dependent inhibition, as shownin the progress curve from cleavage of the substrate D-Ala-Leu-Lys-AMC,which revealed that at 1 µM 94c the initial native rate of cleavage declinedas the portion of uninhibited enzyme dropped (Fig. 29, trace b). However, inthe dilution experiment in which the inhibitor and the enzyme were pre-incubated at high concentration and then diluted into the enzyme, it wasexpected that there would initially be low activity that would steadilyincrease as the inhibitor dissociated, and end up at the same cleavageequilibrium rate as is obtained when the enzyme and the inhibitor weremixed at the start of the experiment. This was clearly not the case for thisinhibitor (Fig. 29, traces c and d). Rough calculations based on the observedenzyme velocities and the second order rate constant at 1 µM inhibitor

61
N
N
O
O
NN
O
O
95a
(~5,300 M–1s–1) gave rise to an off rate of approximately 1 x 10–4 s–1,regardless of the mechanism assumed. This allowed simulation of theexpected results of the dilution experiment (trace e) and highlighted theunexpected nature of the observations in trace c in Figure 29.
Figure 29. The kinetics of inhibition of cathepsin K by 94c. The curves illustrate thefollowing: (a) no inhibitor control; (b) 1 µM 94c and 0.5 nM cathepsin K; (c) 10 nM 94c and0.5 nM cathepsin K; (d) 1 µM 94c and 50 nM cathepsin K incubated for 1 h and then dilutedinto the assay; (e) simulation of the expected results of the dilution experiment in (c). Theexperiments were carried out in a volume of 0.5 mL in a quartz cuvette in a Hitachi F4500fluorometer. The substrate was used at a concentration of 100 µM.
A plausible explanation for these results is the presence of a very minorcontaminant that irreversibly inhibited cathepsin K. We hypothesized that,during the pre-incubation, the minor component inactivated some of theenzyme, resulting in a reduced cleavage rate upon dilution compared to thecontrol. Additional support for this theory was gained from measurements ofthe inhibitory activity of aminoethylamide 94c at a concentration of 100 nMagainst varying concentrations of cathepsin K (Fig. 30).
Figure 30. Titration of cathepsin K against buffer control (●), 10 nM furanone 95a (■), 100nM 94c (▲) and 10 nM E64 (▼). The concentration of furanone 95a was selected to givesimilar levels of inhibition as caused by 100 nM 94c. The substrate was used at aconcentration of 100 µM.

62
The data from this experiment clearly show an x-intercept at 2.5 nMcathepsin K, as would be expected if a minor contaminant had inactivatedsome of the enzyme during the pre-incubation. In the same experiment, wefound that E-64, an irreversible inhibitor of cathepsin K, and at an enzymeconcentration of 10 nM gave an x-intercept at 10 nM, whereas no suchintercept was seen when using the fully reversible inhibitor furanone 95a.Therefore, knowing the concentration of inhibitor present and the level ofirreversible inhibition from the x-intercept, we can calculate that theconcentration of the contaminant in the aminoethylamide 94c was 2.5 mol%.This experiment was performed on several compounds (94a, 94b, and 94c),and the results revealed that those with a low K i value tended to have a highlevel of the impurity.
These results seem to disagree with the findings of dialysis experimentsconducted by Altmann110 and Kim114 and coworkers, which suggested thatinhibitors of this class act as reversible competitive inhibitors. However,there is not necessarily a conflict between the observations. When a highconcentration of enzyme is incubated with a concentration of inhibitor thatshould totally block the activity of the enzyme, there could still be aninsufficient amount of the minor component to completely inactivate theenzyme. If that is the case, then dialysis and dilution will lead to recovery ofmost of the activity. Since there is always some degree of self-degradationassociated with this class of enzymes, complete recovery of activity wouldnot be expected, and recovery of the majority of the activity would suggestthat we are dealing with a reversible inhibitor. It is perhaps pertinent to notethat, in our hands, fresh samples of 94g (the compound used in the kineticstudies performed by Kim’s group) had a Ki of 1.5 μM and only approachedthe reported 10 nM activity after irradiation (vide infra).
Smyth115 and Marquis116 have also discussed the possibility that thesecompounds act as slow-turnover substrates for cathepsin K. For that to be thecase, the leucine side chain would have to bind in the S1 subsite, which weconsider unlikely since we found that cathepsin K does not cleave thesubstrates H-Leu-AMC and Z-Gly-Gly-Leu-AMC. Lack of binding of Leuin S1 is further corroborated by our RAPiD™ data.

63
5.4 Photodegradation
Thus it appears that the activity of these compounds can be attributed to animpurity, but what is this contaminating substance, and where does it comefrom? The fact that the problem was encountered in samples ofaminoethylamides that were prepared using distinct synthetic routes andwere purified according to different protocols (importantly 94g, wasprepared using the route described by Kim et al.), along with the fact thatother laboratories have reported activity for some of these compounds,115,116
suggests that it is not a question of a synthetic impurity or by-product.Since the active component was present at levels that were too low to
enable structure determination of the degradation products from either NMRor MS studies of the parent inhibitors, we attempted to isolate the componentby reverse-phase gradient HPLC. Fractions collected from a semi-preparative purification of aminoethylamide 94e were subjected to bothHPLC reanalysis and measurement of their rate of inactivation of cathepsinK. Figure 31 shows that the rate of inactivation paralleled the amount ofparent compound present in each sample, suggesting that either the impurityco-eluted or it was a degradation product generated directly from the parentin each fraction.
Figure 31. Analysis of HPLC fractions of 94e. Each fraction was re-chromatographed, andthe amount of parent compound was measured (solid bars). Each fraction was also assayedagainst cathepsin K, and the data were fitted to obtain kobs (solid circles). Substrateconcentration was 100 µM. The rates for the most active fractions, 7 and 8, were probablyunderestimated, since they required 16-fold and 4-fold dilutions, respectively, to enabledetermination.
We had noticed that there was a tendency for samples of aminoethylamidesto gain potency over time and also that assay stock solutions preparedfreshly from solid stock initially had rather low potency. This led us toconsider whether the active component was being formed by degradation orphotodegradation of the parent material. To test this possibility, we prepared
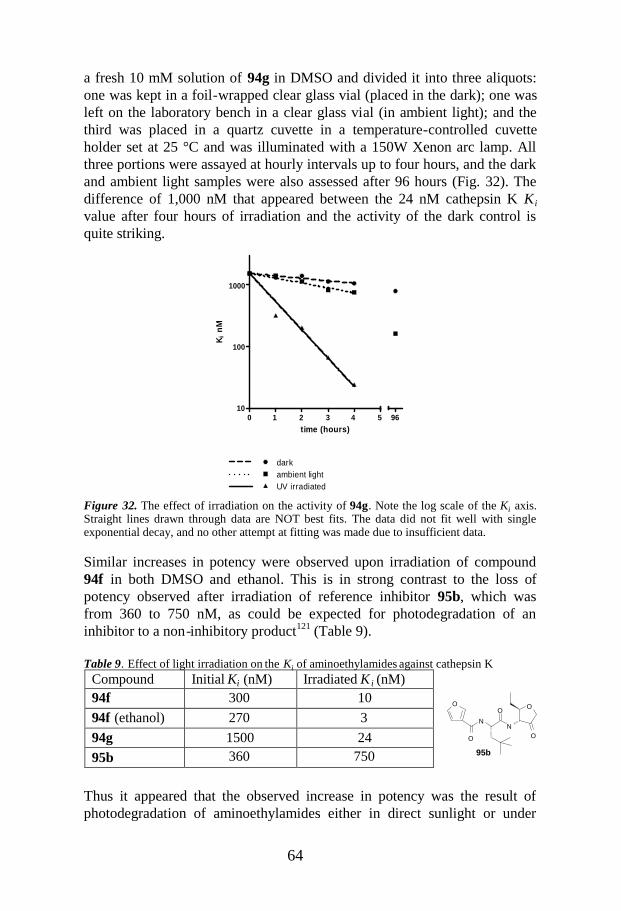
64
O
O
NN
O
O
O
95b
a fresh 10 mM solution of 94g in DMSO and divided it into three aliquots:one was kept in a foil-wrapped clear glass vial (placed in the dark); one wasleft on the laboratory bench in a clear glass vial (in ambient light); and thethird was placed in a quartz cuvette in a temperature-controlled cuvetteholder set at 25 °C and was illuminated with a 150W Xenon arc lamp. Allthree portions were assayed at hourly intervals up to four hours, and the darkand ambient light samples were also assessed after 96 hours (Fig. 32). Thedifference of 1,000 nM that appeared between the 24 nM cathepsin K K i
value after four hours of irradiation and the activity of the dark control isquite striking.
0 1 2 3 4 510
100
1000
96
darkambient lightUV irradiated
time (hours)
Ki
nM
Figure 32. The effect of irradiation on the activity of 94g. Note the log scale of the Ki axis.Straight lines drawn through data are NOT best fits. The data did not fit well with singleexponential decay, and no other attempt at fitting was made due to insufficient data.
Similar increases in potency were observed upon irradiation of compound94f in both DMSO and ethanol. This is in strong contrast to the loss ofpotency observed after irradiation of reference inhibitor 95b, which wasfrom 360 to 750 nM, as could be expected for photodegradation of aninhibitor to a non-inhibitory product121 (Table 9).
Table 9. Effect of light irradiation on the Ki of aminoethylamides against cathepsin K
Thus it appeared that the observed increase in potency was the result ofphotodegradation of aminoethylamides either in direct sunlight or under
Compound Initial Ki (nM) Irradiated K i (nM)94f 300 1094f (ethanol) 270 394g 1500 2495b 360 750

65
HNN N
OCOOH
C2H5
F
Norfloxacin
NNH
R3
O
R2R1
HNN
R3
R2O
R1
N N
R2 OH
R3R1
N N R3R1
R2
+
X-
97 9998
+ OH-
96
fluorescent light. When protected from light in amber vials, the degradationoccurred at a slower rate but nonetheless continued. Our initial hypothesisfor this increase of potency with the aminoethylamides was based on an N toN’ acyl migration in aminoethylamides, as documented by Perillo et al.,122
but this was not linked to photodegradation. The proposed mechanism forthe migration of the acyl from 97 to 99 is shown in Scheme 13.
Scheme 13. N to N’acyl migration.
The cyclic hemiorthoamide 98 or some cationic species such as 96 couldpotentially react irreversibly with the cysteinyl protease, which wouldaccount for the irreversible inhibitor component in the aminoethyl amides.However, neutral or alkaline medium is normally required for thisrearrangement to proceed, and so far we have seen no evidence of therearranged product, although such information might be difficult to obtainwith only a small amount of irreversible component present. Still, eventhough it is possible for aminoethylamide 94a to undergo suchrearrangement, a trisubstituted nitrogen like that present in the otheraminoethylamides synthesized (94b and 94e) clearly cannot, which suggeststhat some other mechanism is responsible for formation of the irreversibleimpurity.
The only other information found in the literature concerns norfloxacin,a broad spectrum antibacterial quinolone that contains an aminoethylaminefunctionality. Norfloxacin has been shown to be a thermostable butphotosensitive drug, especially in solution, which leads to the formation ofaminoethylamine degradates.123,124 Nonetheless, the cited studies focusedmore on the method of analysis than on the structural implications of thephotolysis.

66
5.5 Conclusions
Two solid-supported synthetic routes to afford aminoethylamide compoundswere developed. The initial aldehyde-resin-based route provided good resultsbut was not compatible with reducing sensitive groups and acid labilefunctionalities inherent in some of the target molecules. In order to solvethese problems, an optimized sulfonamide-resin-based method wasdeveloped to obtain a more general and shorter route to parallel synthesis ofthe aminoethylamide compounds.
The results of the kinetic studies performed using several members of theaminoethylamide inhibitor class suggest that the bulk of the observedinhibition of human cathepsin K may be due to the presence of a very minorcomponent that irreversibly inhibits the enzyme. Furthermore, the inhibitoryactivity was found to increase over time and was associated with a numberof structurally different aminoethylamides prepared by different routes andpurified by different techniques, which implies that the inhibitory componentis a degradation product and not a synthetic impurity. The hypothesis wecurrently favor is that the active constituent is a photodegradation product ofthe aminoethylamides formed either in direct sunlight or under fluorescentlight, because the rate of degradation was significantly lower in samplesprotected from light in amber vials.
Without an isolated sample of the degradation product, it is difficult topropose a mechanism of formation or to suggest how this weakness in theseries might be avoided. However, we believe that the results are still ofsubstantial interest to other researchers working in this area. As aconsequence of the mentioned findings, we decided that theaminoethylamide template was unsuitable for further drug development.

67
6. Investigation of Allylic Alcohols in the P1Position of Inhibitors of Hepatitis C Virus NS3Protease (Paper V)
6.1 Introduction
6.1.1 Hepatitis C Virus
The hepatitis C virus (HCV) was first identified in 1989, and since then ithas been estimated that more than 170 million people are infected with HCVworldwide.125 In the Western world, HCV infection is the main cause ofchronic hepatitis, a disease that is a major health problem that leads tocirrhosis of the liver and hepatocellular carcinoma126 and therefore alsoconstitutes the primary reason for liver transplantation.127 Approvedtherapies involve administration of interferon-α(IFN-α) or pegylated IFN-α,either alone or in combination with ribavarin.128 Unfortunately, theseregimens are far from optimal,129–133 because they require prolongedtreatment that is associated with a generally modest sustained virologicalresponse rate (~50%) and significant adverse side effects that result in poortolerance in some patient populations.134,135 Consequently, new and moreeffective therapies are urgently needed.128,136–140 In addition, there iscurrently no vaccine available for HCV, since development of such anapproach has been hampered by the existence of different HCV genotypes,the high replication rate and the low fidelity of the proofreading of the viralRNA.136
HCV is a small, enveloped virus of the Flaviviridae family, and it contains asingle positive-strand RNA genome of approximately 9.6 kb, which carriesone long open-reading frame that encodes a polyprotein of about 3,000amino acids. The HCV polyprotein is composed of structural proteins in itsN-terminal portion and of non-structural (NS) proteins in the remainder ofthe molecule, and it is cleaved co- and post-translationally by cellular andviral proteases into at least 10 different products.141 Practically every step inthe life cycle of HCV—from entry into a host cell to the replication andformation of the new virion particles—is a potential target for antiviraldrugs. The various prospective targets have been comprehensivelyreviewed.128,142,143

68
6.1.2 HCV NS3 Protease Inhibitors
One of the most promising molecular targets for HCV therapy is the non-structural serine protease NS3,143,144 which is one of the best-characterizedHCV enzymes. The structure of HCV NS3 consists of a serine proteasedomain in the N-terminal third of the polyprotein and an RNAhelicase/NTPase domain in the C-terminal portion.145 NS3 protease mediatesproteolytic cleavage of the non-structural proteins NS3, NS4A, NS4B,NS5A, and NS5B. The NS4A protein is an integral structural component ofNS3 and is required to enhance its serine protease activity,146–150 which isessential for the viral life cycle and represents one of the primary targets forthe development of new treatments against HCV.151–157
X-ray crystal structures of HCV NS3 protease have shown that the bindingsite of the enzyme is remarkably shallow and solvent exposed, thus makingthe design of small-molecule inhibitors a challenge.158,159 Despite thisobstacle, several NS3 protease inhibitors have been reported to date.151–
153,160,161 One category of such compounds comprises the classical serine-trapinhibitors, which are characterized by an electrophilic P1 carbonyl group thatforms a reversible covalent bond with the hydroxyl group of the serine in theactive site of the enzyme. Examples of these inhibitors include aldehydes, α-keto amides, α-keto esters, α-diketones, boronic acids, and α-keto acids.162–
176 A second category of non-covalent NS3 protease inhibitors incorporatestructures such as P1 amides,177 P1-P1’ acylsulfonamides,178 or P1carboxylic acids. The P1 carboxylic acids are characterized as product-basedinhibitors,173,179–185 and they display higher specificity for the HCV NS3protease than for other serine proteases.161,186 As deduced from 3D structuremodeling, the carboxylate in the active site binds to the oxyanion cavity (NHof Gly137 and Ser139) and to the catalytic histidine.187 The helicase domainalso appears to participate in the binding of the inhibitors in the native full-length NS3 protein (protease-helicase/NTPase).188
Early but impressive clinical proof-of-concept data were recently disclosedfor BILN-2061 (Ciluprevir; Boehringer Ingelheim),152,153,161,189 which is amacrocyclic non-covalent mimic of the peptide product inhibitor. BILN-2061 was the first HCV protease inhibitor to enter clinical trials, and theinitial results were promising. However, subsequent clinical trials werehalted, apparently due to cardiac toxicity observed in rhesus monkeys givenhigh doses. At present, the two covalent product-based linearpeptidomimetics VX-950 (Telapravir) and SCH 503034 (Boceprevir) arereported to be in Phase III and Phase IIb/III clinical trials, respectively190–196
and the non-covalent macrocyclic peptidomimetic TMC435350 is in Phase IIclinical trials. Medivir licensed its HCV PI program to JNJ/Tibotec

69
NH
OH
100
Ac-Asp-D-Glu-Leu-Ile-Cha
NHN
OH
O
OO
NHO
O
O
N
MeO
S
N HN
BILN 2061
NNH
O
O
ONH2
ONHHN
O
SCH 503034
HN
NH
O
NO
O
O
N
M eO
S
N
TMC435350
SO O
N
HH
O
NH
O
O
NH
ONH
O
NH
O
N
N
VX-950
Pharmaceuticals Ltd in 2004 and TMC435350 was discovered through thisresearch collaboration.
Preliminary investigational work recently published by our group (PaperVI) led to the discovery of a lead compound and subsequently selection ofTMC435350 as the clinical candidate drug. In a Phase 1 trial, it was foundthat TMC435350 was well tolerated during five days of dosing and exhibitedstrong and rapid antiviral activity in patients infected with HCV genotype 1.The results of that trial, which included both healthy volunteers and HCVpatients, formed the basis for the Phase IIa trial that is now underwaythroughout Europe.197
6.1.3 Aim of the Study
Our group has previously described a P1 allylic alcohol representing a newclass of non-electrophilic inhibitors with a Ki value of 0.98 µM (100).198
We were intrigued by the discovery of this class of molecules, because it canenable exploration of the S1’ pocket of the HCV NS3 protease, which,according to recent studies,178,179,199,200 appears to provide additionalfavorable binding interactions with the enzyme. Moreover, it seems that NS3protease is highly flexible in the mentioned region, which should also makeit possible to gain additional binding interactions.134,178,199–202
The study presented in Paper V was conducted to explore P1 allylicalcohols and their interactions with the S1’ pocket in NS3 protease in orderto achieve affinity that would allow truncation at the amino terminus and

70
NH
P1
OH
RAc-Asp-D-Glu-Leu-Ile-Cha
N OH
O
O
N
O
N OH
O
O
N
O
101 102
Ac-Cha-Val Boc-Val
BocHN
O
OMe BocHN
O
P
O
OMe
OMeBocHN
O
BocHN
OH
dimethylmethyl-phosphonate,BuLi, THF
PhCHOLiOH, Ether
106
NaBH4
107
104 10594% 85%
98%*
ds 2:1
also to be able to move from the hexapeptides towards more drug-likemolecules.
The study had two main objectives:
1. To determine how the P1 allylic alcohol hexapeptides bind to theactive site of the HCV NS3 protease and to examine the importancein that context of the alcohol and the double bond moieties.
2. To assess this new P1 series in shorter, more drug-like peptides. Forthat purpose, two known sequences were selected: Ac-Cha-Val-Pro(quinoline)-OH (101)203 and Boc-Val-Pro(quinoline)-OH(102).204
6.2 Synthesis of P1 Nva Allylic Alcohol
A model P1 allylic alcohol was synthesized from -norvaline (Nva) so that itcould be incorporated into a pentapeptide Ac-Asp(OtBu)-D-Glu(OtBu)-Leu-Ile-Cha-OH (103), a tripeptide Ac-Cha-Val-Pro(Quinoline)-OH (101), and adipeptide Boc-Val-Pro (quinoline)-OH (102).
Scheme 14. Synthesis of P1 Nva allylic alcohol.

71
NO
O
OH
ONH
OON
OO
O
ON
OO
O
NO
O
OH
NO
O
OH
NH2
OH
88%
93%
107
108
93%
66%2:1 ds
2:1 cis : trans ds
**
KtOBu/THF 1.NaH, Boc2O/THF
Cs2CO3
MeOH/aq
2.Column chromatography
109
110
(S,S)-107
(S,R)-107
TFA/DCM/H2O
*
+
100%109
110
Cs2CO3
MeOH/aqTFA/DCM/H2O
100%
2:1 ds
111
TFA x
The synthesis was performed by reacting Boc-protected amino acid 104(Boc-Nva-OMe) with 6 equivalents of the lithium salt ofdimethylmethylphosphonate at –78 C̊ in THF205 to give the phosphonate105 in 94% yield (Scheme 14). The trans-enone 106 was obtained in 85%yield using Horner-Wadsworth-Emmons conditions with powdered LiOH indry ether and benzylaldehyde. The allylic alcohol 107 was obtained in 98%yield as a diastereomeric mixture (2:1) by using sodium borohydride orpolymer-bound borohydride to reduce the ketone.
To better understand the role of the alcohol in the binding to HCV NS3protease, attempts were made to obtain the isomerically pure allylic alcohols.One of the methods attempted was separation of the diasteromeric mixture107 by HPLC. However, a clean separation could not be achieved, andselective reduction of the ketone 106 by use of 9-BBN derivatives, R-AlpineBorane, or DIP-Chloride also failed to deliver pure diastereomers.
6.3 Isomerically Pure Allylic Alcohols
To be able to isolate both isomers of alcohol 107, these were exposed tobasic conditions (KtOBu in dry THF)206 to convert them into theoxazolidinone mixture 108 (Scheme 15), which gave a 93% yield of a 2:1mixture. The mixture 108 was subsequently protected using Boc-anhydridein the presence of NaH in dry THF to provide the Boc-protectedoxazolidinones in 66% yield, which were then separated by columnchromatography to furnish a 1:2 ratio of trans 109 and cis 110. Basiccleavage of oxazolidinones 109 and 110 using cesium carbonate inmethanol:water 5:1 afforded (S,S)-107 and (S,R)-107 in 88% and 93% yield,respectively.
Scheme 15. Synthesis to afford isomerically pure allylic alcohols

72
R´HN
OH
Ph
R´´HNP h
R´´HN
OH
Ph
R´HN
O
PhR´´HN
O
Ph
R´´HN
O
Ph
R´´HN
OH
Ph
R´´HNPh
1. TFA/DCM2. 103,HATU,DIEA,DMFDMF
112
113 114
Dess-Martinreagent,DCM25%
1.H2, Pd/CMeOH
TFA/DCM 2. TFA/DCM118 119
TFA/TES/DCM
TFA/DCM/H2O
115
R´= Ac-Asp(OtBu)-D-Glu(OtBu)-Leu-Ile-Cha-
R´´= Ac-Asp-D-Glu-Leu-Ile-Cha-
89%
100% 100%
100%
100%
116
117
107100%
H2 , Pd/CMeOH
100%
H2 , Pd/CMeOH
This methodology constituted a convenient way to obtain isomerically purealcohols (S,S)-107 and (S,R)-107. However, epimerization occurred uponcleavage of the Boc group under standard deprotection conditions(TFA/DCM/H2O or TFA/DCM), which resulted in the 2:1 mixture 111 whenstarting from either alcohol (S,S)-107 or (S,R)-107. This indicates that theallylic alcohol is acid labile and prone to isomerization under theseconditions (i.e., conditions used to achieve cleavage of the Boc-protectinggroup).
Due to the unsuccessful results of the attempt to procure isomerically pureP1 allylic alcohols, the diasteromeric mixture was used for the subsequentcoupling with the hexa-, tri-, and tetrapeptide models. Columnchromatography was carried out to separate the diasteromeric mixture ofhexapeptides but was not successful.
6.4 Synthesis of Hexapeptides
Synthesis of the pentapeptide Ac-Asp(OtBu)-DGlu(OtBu)-Leu-Ile-Cha-OH(103)198 was performed using Fmoc solid-phase methodology.207–209 Bocdeprotection of 107 with TFA/DCM/H2O (70:25:5) and subsequent couplingof the amine with the pentapeptide 103 using N-[(dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-yl-methylene]-n-methylmethanaminiumhexafluorophosphonate N-oxide (HATU) and N,N-diisopropylethylamine(DIEA) in DMF furnished hexapeptide 112 (89% yield, Scheme 16).
Scheme 16. Synthesis of hexapeptides.
Also, to be able to study the role of the alcohol and the double bond in theinteraction with the enzyme, the corresponding unsaturated ketone 118,

73
NH +
saturated ketone 119, and saturated alcohol 114 were synthesized from thehexapeptide 112.
Oxidation of compound 112 to ketone 117 (25% yield, Scheme 16) wasperformed using Dess-Martin periodinane in DCM.210 Cleavage of the t-butyl esters with TFA/DCM afforded compound 118 in a quantitative yield.Hydrogenation of 117 over palladium on carbon in MeOH, followed bycleavage of the ester functions, delivered ketone 119 in a quantitative yield.As a reference compound, Ac-Asp-D-Glu-Leu-Ile-Cha-Nva-OH (120, Table10) was synthesized by coupling of t-butyl L-α-aminopentyrate (H-Nva-OtBu. HCl) with pentapeptide 103 using HATU and DIEA in DMF, whichgave the protected hexapeptide Ac-Asp(OtBu)-D-Glu(OtBu)-Leu-Ile-Cha-Nva-OtBu in 52% yield. The t-butyl esters were cleaved usingTFA/DCM/Et3SiH, furnishing compound 120 in a quantitative yield.
Deprotection of the t-butyl esters in compound 112 usingTFA/DCM/Et3SiH211 afforded compound 115, not compound 113 asanticipated. The target compound 113 was instead obtained by a non-reductive cleavage procedure using aqueous TFA/DCM. This reduction wasobserved only when the double bond was in conjugation with an aromaticsystem.
A proposed intermediate for the obtained result is a carbocation stabilizedthrough conjugation with the phenyl ring:
The same kind of intermediate could give rise to the epimerized product 111,the reduced product 115, and product 113. Hydrogenation of the doublebonds in compounds 113 and 115 was achieved by use of palladium oncarbon in MeOH, which afforded compounds 114 and 116 (Scheme 16) inquantitative yields.

74
RH N
OH
Ph
RHNPh RHN
O
Ph
RHN
OH
Ph
N
O
O
N
O
N
O
O
N
OH
O
103
R=
107
1. 30%TFA/DCMor HCl/EtOAc
2. 101, HATU,DIEA, DMF 121 46% 122
Dess-Martinreagent, DCM
124
TFA/TES/DCM
123
1.H2, Pd/CMeOH
100%95%
100%
Ac-Cha-ValAc-Cha-Val
6.5 Synthesis of Tetrapeptides
Synthesis of P2-P4 tripeptide Ac-Cha-Val-Pro(Quinoline)-OH (101) wasperformed according to procedures described in the literature.203 Bocdeprotection of 107 with TFA/DCM/H2O (70:25:5) and subsequent couplingof the amine with the tripeptide 107 using HATU and DIEA in DMFfurnished tetrapeptide 121 (46% yield, Scheme 17). The unsaturated alcohol121 was hydrogenated over palladium on carbon in MeOH to deliver aquantitative yield of the saturated alcohol 122, which was oxidized to ketone124 (95% yield) using Dess-Martin periodinane in DCM. Treatment ofcompound 121 with TFA/DCM/Et3SiH afforded compound 123 (Scheme 17)in quantitative yield.
Scheme 17 . Synthesis of tetrapeptides.
The reference compound Ac-Cha-Val-Pro(Quinoline)-OH-Nva-OH (125,Table 11) was synthesized by coupling methylnorvalinate (H-Nva-OMe.HCl) with tripeptide 101 using HATU and DIEA in DCM, which gave themethyl ester tetrapeptide in 87% yield. Subsequent hydrolysis of the ester bytreatment with lithium hydroxide in methanol-water afforded the targetcompound 125 (41%).

75
NH
R
P 1
A c-A sp-D -G lu-Leu-Ile -C ha
Cmpd P1 R % inh at10 μM
% inh at1 μM
K i (μM)
120 n-PrO H
O
95 71 0.137
118 n-PrO
98 92 0.107
119 n-PrO
69 13 3.4
113 n-Pr
OH
91 34 2.28
114 n-PrOH
27 9 >10
115 n-Pr 44 52 5.64
116 n-Pr 25 18 >10
100 EtOH
32.5 6.7 13.3 (0.98)*
*Published data 198
N
O
O
N
OH
O
N
O
O
N
NH
OH
O107
1. 30%TFA/DCMor HCl/EtOAc
2. 102, HATU,DIEA, DMF
126 36%
Boc-Val
102
Boc-Val
6.6 Synthesis of Tripeptides
Synthesis of the P2-P3 dipeptide Boc-Val-Pro (quinoline)-OH (102) wasperformed according to procedures in the literature.204 Boc deprotection of107 with TFA/DCM/H2O (70:25:5) and subsequent coupling of the aminewith the dipeptide 102 using HATU and DIEA in DMF furnished the modeltripeptide 126 (36% yield, Scheme 18).
Scheme 18. Synthesis of the tripeptides.
6.7 Tables: Inhibitory Activity
Table 10. Percent inhibition at 10μM and 1μM and Ki inhibition constant for hexapeptidesscreened against full-length NS3 (protease-helicase/NTPase) protease

76
N N
O
O
N
O
R
P1
Ac-Cha-Val
Cmpd P1 R % inh at10 µM
% inh at 1µM
Ki (µM)
125 n-PrOH
O70 24 0.48
121 n-Pr
OH
21 0 nd
122 n-Pr
OH
17 0 nd
123 n-Pr 13 0 nd
124 n-Pr
O
20 0 nd
nd = not determined
N N
O
O
N
O
R
P1
Boc-Val
Cmpd P1 R % inh at10 μM
% inh at1 μM
Ki (μM)
127* n-PrOH
O98 83 0.167
126 n-Pr
OH
28 21 8.3
*Reference compound provided by the University of Uppsala and assessed in our system 212
Table 11. Percent inhibition at 10µM and 1µM and Ki inhibition constant for tetrapeptidesscreened against full-length NS3 (protease-helicase/NTPase) protease.
Table 12 . Percent inhibition at 10μM and 1μM and Ki inhibition constant for tripeptidesscreened against full-length NS3 (protease-helicase/NTPase) protease

77
6.8 Structure-Activity Relationships
Examination of the three classes of inhibitors presented in Table 10 indicatedthat the most potent compound was the α,β-unsaturated ketone 118, whichhad a K i value of 0.107 μM, approximately equipotent with the referenceproduct inhibitor 120 (Ki value of 0.137 μM). The inhibitory activity of theα,β-unsaturated alcohol 113 (Ki 2.28 μM) found in this study was lower thanthat of the reference product inhibitor 120 but represented a 1.5-fold and a5.8-fold increase compared to the activities of 119 and the reference allylicalcohol compound 100, respectively. The inhibitory activity of compound113 was dramatically decreased when the double bond was reduced, as seenin compound 114 (Ki > 10 μM), or when the alcohol function was notpresent, as in compounds 115 (Ki = 5.64 μM) and 116 (K i >10 μM). It can beconcluded that both the alcohol function and the double bond in compound113 are integral functional moieties that contribute to the binding affinity forthe enzyme.
In Table 11, the reference tetrapeptide compound 125 exhibited a 3.5-folddecrease in activity compared to reference hexapeptide product inhibitor120. However, relevant inhibitory activities against NS3 protease were notfound for the tetrapeptide derivatives 121, 122, and 123 (corresponding tothe hexapeptides 113, 114 and 115), or the saturated ketone 124. Themoderate activity found for the α,β-unsaturated alcohol hexapeptide 113could not be transferred to the tetrapeptide 121.
In Table 12, the corresponding α,β-unsaturated alcohol tripeptide 126showed some inhibitory activity (Ki 8.3 μM), although the level representeda 3.6-fold decrease compared to the activity of the α,β-unsaturated alcoholhexapeptide 113.
6.9 Conclusions
Considering the SARs of the hexapeptides (Table 10), the unsaturated ketone118 can be seen as the most potent compound, and it is apparent that boththe alcohol moiety and the double bond were necessary for the inhibitoryactivity exhibited by the α,β-unsaturated alcohol 113. Furthermore,shortening of the α,β-unsaturated alcohol hexapeptide to a tetrapeptide andtripeptide (121 and 126 respectively) resulted in loss of inhibitory activity.Notwithstanding, even though this limited set of P1’ substituents did notgenerate promising low molecular weight NS3 protease inhibitors, it cannotbe excluded that a broader collection of P1’ groups would result insubstantially more potent and promising inhibitors.

References
(1) Thompson, L. A.; Ellman, J. A., J. Am. Chem. Rev., 1996, 96, 555-600.(2) Hermkens, P. H. H.; Ottenheijm, H. C. J.; Rees, D. C., Tetrahedron,
1997, 53, 5643-5678.(3) Hermkens, P. H. H.; Ottenheijm, H. C. J.; Rees, D. C., Tetrahedron,
1996, 52, 4227-4554.(4) Wilson, S. R.; Czarnik, A. W., Combinatorial Chemistry-Synthesis and
Application; Eds. Wiley: New York, 1997.(5) (a) Acc. Chem. Res., 1996, 29, 111-170 (special issue on combinatorial
chemistry). (b) Chem. Rev. 1997, 97, 347-510 (special issue oncombinatorial chemistry).
(6) Egner, B. J.; Bradley, M., Anal. Biochem. Rev., 1997, 2(3), 102-109.(7) Chin, J.; Fell, B.; Shapiro, M. J.; Tomesch, J.; Wareing, J.R.; Bray, A.M.,
J. Org. Chem., 1997, 62, 538-539.(8) Fitch, W. L.; Detre, G.; Holmes, C. P., J. Org. Chem., 1994, 39, 7955-
7956.(9) Luo,Y; Ouyang X.; Armstrong R. W.; Murphy M. M., J. Org. Chem.,
1998, 63, 8719-8722.(10) Chin, J. A.; Chen, A.; Shapiro, M. J., J. Comb. Chem., 2000, 2, 293-
296.(11) Ganapathy, S.; Badiger, M. W.; Rajamohanan, P. R.; Maskelkar, R. A.;
Macromolecules, 1989, 22, 2023-5.(12) Garigipati, R. S.; Adams, B.; Adams, J. L.; Sarkar, S. K.; J. Org. Chem.,
1996, 61, 2911-4.(13) Warrass, R.; Wieruszeski, J-M.; Lippens, G.; J. Am. Chem. Soc., 1999,
121, 3787-88.(14) (a) Ruhland, T.; Andersen, K.; Pedersen, H.; J. Org. Chem., 1998, 63,
9204-9211. (b) Gotfredsen, C. H.; Grotli, M.; Willert, M.; Meldal, M.;Duus, J. O.; J.Chem. Soc., Perkin Trans.1, 2000, 1167-1171. (c) Riedl,R.; Tappe, R.; Berkessel, A.; J. Am. Chem. Soc., 1998, 120, 8994-9000.
(15) Wehler, T.; Westman, J.; Tetrahedron Lett., 1996, 27, 4771-74.(16) Salvino, J.M.; Gerard, B., J. Comb. Chem., 2003, 5(3), 260-266.(17) Fernandez-Forner, D.; Casals G.; Navarro, E.; Ryder,H.; Albericio, F.
Tetrahedron Letters, 2001, 42 4471-4474.(18) Pelletier, J.C.; Khan,A.; Tang, Z. Organic Letters, 2002, 4(26), 4611-
461.3(19) Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I., Anal. Biochem.,
1970, 34, 595-598.(20) Bunnin, B. A.; The Combinatorial Index, 1998; p 214.(21) Aoki, Y.; Kobayashi, S., J. Comb. Chem., 1999, 1, 371-372.

(22) Abdel-Magid, A. F.; Carson, K. G.; Harris, B. D.; Maryanoff, C. A.;Shah, R. D., J. Org. Chem., 1996 , 61, 3849-3862.
(23) Bunnin, B. A.; The Combinatorial Index, 1998 , 84.(24) Sarantakis, D.; Bicksler, J., Tetrahedron Lett., 1997, 38, 7325-7328.(25) Swayze, E. E., Tetrahedron Lett., 1997 , 38, 8465-8468.(26) Pande, C. S.; Gupta, N.; Bhardwa, J., J. Appl. Polym. Sci., 1995, 56,
1127-1131.(27) Khan, N. M.; Vijayalakshmi, A. ; Balasubramanian, S., Tetrahedron
Lett., 1996, 37(27), 4819-4822.(28) Pelter, A.; Rooser, R. M. J. Chem. Soc. Perkin Trans. 1, 1984, 717-720.(29) Bomann, M. D.; Guch, I. C.; Dimare, M. J. Org. Chem., 1995 , 60,
5995-5996.(30) Moormann, A. E.; Synth. Commun., 1993, 23, 789-795.(31) Hemming, K.; Bevan, M. J.; Loukou, C.; Patel, S. D.; Renaudeau, D.;
Synlett, 2000, 11, 1565-1568.(32) (a) Salvatore, R. N. Yoon, C. H.; jung, K. W. Tetrahedron, 2001, 57,
7785-7811. (b) Olsen, C.A.; Witt, M.; Jarosewsli, J. W.; Franzyk, H.Org. Lett., 2004, 6, 1935.
(33) (a) Borch, R. F.; Berstein, M. D.; Durst, H. D.; J. Am. Chem. Soc.,1971, 93, 2891. (b) Cho, B. T.; Kang, S. K. Tetrahedron, 2005, 61,5725-5734. (c) Lu, Z-H.; Bhongle, N.; Su, X.; Ribe, S.; Senenayake,C. H. Tetrahedron Lett ., 2002, 43, 8620.
(34) (a) Foucaud, A.; Guemmout, F.E., Bull. Soc. Chim. Fr., 1989, 403-408;(b) Nayak, S. K.; Thijs, L.; Zwaneburg, B., Tetrahedron Lett., 1999,40, 981-984; (c) Screedlar, B.; Reddy, P. S.; Kumar, N. S.,Tetrahedron Lett., 2006, 47, 3055-3058; (d) Chandrasekhar, S.; Basu,D.: Rambau, Ch., Tetrahedron Lett., 2006, 47, 3059-3063; (e) Patra,A.; Roy, A. K.; Batra, S.; Bhaduri, A. P., Synlett, 2002, 1819-1822; (e)Sá, M. M.; Ramos, M. D.; Fernades, L., Tetrahedron 2006, 62, 11652-11656; (f) Andersen, J.; Bolvig, S.; Liang, X., Synlett, 2005, 19, 2941-2947. (f) Andersen, J.; Madsen, U.; Bjorkling, F.; Liang, X., Synlett,2005, 14, 2209-2213. (g) Varma, R. S.; Naicker, K. P.; Aschberger, J.,Synth. Commun., 1999, 29(16), 2823-2830; (h) Vanek, P.; Klan, P.,Synth. Commun., 2000, 30(8), 1503-1507.
(35) Yan, R.-B.; Yang, F.; Wu, Y.; Zhang, L.-H.; Ye, X.-S., TetrahedronLett., 2005, 46(52), 8993-8995.
(36) Goddard-Borger, E. D.; Stick, R. V., Organic Letters, 2007, 9(19),3797-3800.
(37) (a) Bock, V. D.; Hiemstra, H.; Van Maarseveen, J. H., Eur. J. Org.Chem., 2006, 51-68; (b) Staudinger, H; Meyer, J., Helv.Chim. Acta,1919, 2, 635-646; (c) Gololobov, Y. G.; Zhumurova, I. N.; Kashukin,L. F., Tetrahedron, 1981, 37, 437-472; (d) Vaultier, M; Knouzi, N.;Carrie, R., Tetrahedron Lett., 1983, 24, 763-764.

(38) (a) Holletz, T.; Cech, D., Synthesis, 1994, 789; (b) Shoetzau, T.;Holletz, T.; Cech, D., Chem. Commun., 1996, 387.
(39) (a) Horner, L. and Gross, A. Liebigs, Ann. Chem., 1955, 591, 117; (b)Messmer A.; Pinter I. and Szego, F., Angew. Chem., 1964, 3, 228.
(40) (a) Tsuge, O.; Kanemasa, S.; Matsuda, K., J. Org. Chem., 1984, 49,2688; (b) Anderson, W. K. and Milowski, A. S., J. Org. Chem., 1986,29, 2241; (c) Molina, P.; Arques, A.; Cartagena, I.; Obón, R.,Tetrahedron Lett., 1991, 32, 2521; (d) Zhu, Z.; Espensaon, J. H., J.Am. Chem. Soc., 1996, 118, 9901. (e) Bosch, I.; González, A.; Urpí,F.; Vilarrasa, J., J. Org. Chem., 1996, 61, 5638-5643; (f) Ariza, X.;Urpí; Vilarrasa, J., Tetrahedron Lett., 1999, 40, 7515-7517.
(41) Cristau, H. J.; Garcia, C.; Kadoura, J.; Torreilles, E., Phosphorous,Sulfur, and Silicon, 1990, 49/50, 151-154.
(42) Sampath Kumar, H. M.; Anjaneyulu, S.; Subba Reddy, B.V.; Yadav, J.S., Synlett, 1999, 551.
(43) (a) Bravo, P.; Crucianelli, M.; Vergani, B.; Zanda, M., TetrahedronLett., 1998, 39, 7771; (b) Knapp, S.; Hale, J. J.; Margarita, G.; Gibson,F. S., Tetrahedron Lett., 1990, 31 (5), 2109; (c) Golding, B. T.;O’Sullivan, M. C.; Smith, L.L., Tetrahedron Lett., 1998, 29(50), 6651.
(44) We purchased this reagent fron the Argonaut Technologies, P/N800379.
(45) Bernard, M.; Ford, W. T., J. Org., Chem., 1983, 48, 326.(46) (a) Lindsley, C. W.; Zhao, Z.; Newton, R. C.; Leister, W. H.; Strauss,
K. A., Tetrahedron Lett., 2002, 43, 4467-4470; (b) Irgolic, K. J. InHouben-Weyl, 4th ed., Vol. E12b; Klamann, D., Ed.; Thieme:Stuttgart, 1990, 150.
(47) Schechter, I.; Berger, A., Biochem. Biophys. Res. Commun., 1967 , 27,157.
(48) Leung, D.; Abbenante, G.; failie, D. P., J. Med. Chem., 2000, 43, 305.(49) Beckett, R. P.; Davidson, A. H.; Drummond, A. H.; Whittaker, M.,
Drug Discovery Today, 1996, 1, 16.(50) Johnson, L. L.; Dyer, R.R.; Hupe, D. J., Curr. Opin. Chem. Biol., 1998,
2, 466.(51) Yan, S.; Sameni, M.; Sloane, B. F., Biol. Chem. 1998, 379, 113.(52) Stubbs, M. T.; Bode, W., Thromb. Res. 1993,, 62, 543.(53) (a) Rebecca Roberts, Drug News Perspects, 2005, 18(10), 605-614; (b)
Vasiljeva, O.; Reinheckel, T.; Peters, T., Turk, D.; Turk, V.; Turk, B.,Current Pharm. Design, 2007, 13, 387-403.
(54) Rosenthal, P. J., Emerg. Infect. Dis., 1998, 4, 49.(55) Wlodawer, A.; Erickson, J. W., Annu. Rev. Biochem ., 1993, 62, 543.(56) Gibson, W., Hall, M. R., Drug Des. Discov., 1997, 15, 39.(57) Kirschke, H.; Wiederanders, B.; Bromme, D., Rinne, A., Biochem J.,
1989, 264, 467-473.

(58) Pinitgland, S.; Watts, A. B.; Patel, M. et al. , Biochemistry, 1997, 36,9968-9982.
(59) Driessen, C.; Bryan, R. A.; Lennon-Dumenil, A. M., J. Cell Biol., 1999,147, 775-790.
(60) Berger, A., Schchter, I., Philos. Trans. R. Soc. Lond. Biol. Sci., 1970,257, 249-164.
(61) Walker, B., Lynas, J. F., Cell. Mol. Life Sci. , 2001, 58, 596-624.(62) Babine, R.; E.; Bender, S. L., Chem. Rev., 1997, 97, 1359-1472.(63) Sanderson, P. E. J., Med. Res. Rev., 1999, 19, 179-197.(64) Deadman, J., J. peptide Sci., 2000, 6, 421-431.(65) Ingallinella, P.; Bianchi, E.; Ingenito, R.; Koch, U.; Steinkühler, C.;
Altamura, S.; Pessi, A., Biochemistry, 2000, 39, 12898-12906.(66) Cannon, W. R.; Benkovic, S. J., J. Biol. Chem., 1998, 273, 26257-
26260.(67) Menger, F. M., Biochemistry, 1992, 31, 5368-5373.(68) Powers, J. C.; Asgian, J. L.; Ekici, O. D.; James, K. E., Chem. Rev.,
2002, 102, 4639-4750.(69) (a) Demuth, H. U., Enzyme Inhib., 1990 , 3, 249-278. For recent reviews
of cathepsin K inhibitors, see: (b) Black, W. C.; Percival, M. D.,Annual Reports in Medicinal Chemistry, 2007, 42, 111-127 and (c)Grawoska, U. B.; Chambers, T. J.; Shiroo, M.; Curr. Opin. DrugDiscovery & Development, 2005, 8(5), 619-630.
(70) Germain, R. N., Cell, 1994, 76, 287-299.(71) (a) Unamune, E. R., Annu. Rev. Immunol., 1984, 2, 395-428. (b)
Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G., N. Engl. J. Med., 2000, 343, 938-952; (c) Gupta, S.; Singh, R. K.;Dastimar, S.; Ray, A., Expert Opin. Ther.Targets, 2008, 12(3), 291-299.
(72) (a) Shi, G.P.; Munger, J. S.; Meara, J. P.; Rich, D. H.; Chapman, H. A.,J. Biol. Chem, 1992, 267 (11), 7258-7262.; (b) Shi, G. P.; Webb, A.C.; Foster, K. E.; Knoll, J. H. M.; Lemere, C. A.; Munger, J. S.;Chapman, H. A., J. Biol. Chem., 1994, 269 (15), 11530-11536.; (c)Morton, P. A., Zacheis, M. L.; Giacoletto, K. S.; Manning, J. A.;Schwartz, B. D., J. Immunol., 1995, 154 (1), 137-150.; (d) Beers, C.;Burich, A.; Klejmeer, M. J.; Griffith, J. M.; Wong, P.; Rudensky, A.Y., J. Immunol., 2005, 174 (3), 1205-1212.
(73) Turnsek, T.; Kregar, I.; Lebez, D., Biochim. Biophys. Acta, 1975, 403,514-520.
(74) Wiederanders, B.; Brömme, D.; Kirschke, H., et al., J. Biol. Chem.,1992, 267, 13708-13713.
(75) Shi, G. P.; Munger, J. S.; Meara, J. P., et al., J. Biol. Chem., 1992, 267,7258-7262.
(76) (a) Nakagawa, T. Y.; Rudensky, A. Y. Immunol. Rev. 1999, 172, 121;(b) Villandangos, J. A.; Bryant, R. A.-M.; Riese, R. J.; Roth, W.;

Saftig, P.; Shi, G-P.; Chapman, H.A.; Peters, C.; Ploegh, H.L.;Immunol. Rev. 1999, 172, 109; (c) Thurmond, R. L.; Sun, S.; Karlsson,L.; Edwards, J.P. Current Opinion in Investigational Drugs, 2005,6(5), 473-482.
(77) Pauly, T. A.; Sulea, T.; Ammirati, M., et al., Biochemistry, 2003, 42,3203-3213.
(78) Brömme, D.; Bonneau, P. R.; Lachance, P. et al., J. Biol. Chem., 1993,286, 4832-4838.
(79) (a)Liu, W.; Spero, D. M., Drug News Perspect, 2004, 17 (6), 357-363;(b) Clark, A. K.; Yip, P. J.; Grist, J.; Gentry, C.; Staniland, A. A.;Marchand, F.; Dehvari, M.; Wihterspoon, G.; Winter, J.; Ullah, J.;Bevan, S.; Malcagio, M. PNAS, 2007, 104(25), 10655-10660; (c)Hoyos Flight, M., Nature Reviews Drug Discovery, 2007, vol.6.
(80) (a) Beck, H.; Schwarz, G.; Schroter, C. J. et al., Eur. J. Immunol., 2001,31, 3726-3736: (b) For a recent review of cathepsin S asimmunomodulatory target see: Gupta, S.; Singh, R. K.; Dastidar, S.;Ray, A., Expert Opin. Ther. Targets, 2008, 12(3), 291-299.
(81) Schwarz, G.; Boehncke, W., H.; Braun, M. et al., J. Invest. Dermatol.,2002, 119, 44-49.
(82) Lemere, C. A.; Munger, J. S.; Shi, G. P. et al., Am. J. Pathol., 1995,146, 848-860.
(83) Fernandez, P. L.; Farre, X.; Nadal, A. et al., Int. J. Cancer, 2001, 95,51-55.
(84) Quibell, M.; Taylor, S.¸Grabowska, U.; Nilsson, M.; Morrison, V. PCTInt. Appl. US-20030203900 (2003).
(85) Proof of principle in research on treatment of multiple sclerosis (MS):Medivir AB, Huddinge, Sweden (2002).http://www.medivir.se/v3/se/ir_media/press_releases.aspx?id=57
(86) (a) Liu, H.; Tully, D.C.; et al., Med. Chem. Lett. 2005, 15, 4979; (b)Patterson, A.W.; Wood, J.L.W.; Horsby, M.; Lesley, S.; Spraggon, G.;Ellman, J. A. J. Med. Chem. 2006 , 49, 6298-6307. (c) Li, C S.;Deschenes, D.; et. al. Bioorg. Med. Chem. Lett., 2006, 16, 1985.
(87) Tully, D.C.; Liu, H.; et. al., Bioorg. Med. Chem. Lett. 2006, 16, 1975.(88) Chapman, H., A.; Riese, R. J.; Shi, G. P., Annu. Rev. Phys ., 1997, 59,
63.(89) Gauthier, J. Y.; Black, W. C.; Courchesne, I. et al., Biorg. Med. Chem.
Lett., 2007, 17, 4929-4933.(90) Murphy, A.M.; Dagnino, R., Jr.; Vallar, P. L.; Trippe, A. J.; Sherman,
S. L. Lumpkin, R.H.; Tamura, S.Y.; Webb, T. R., J. Am. Chem. Soc.,1992, 114, 3156.
(91) Material purchased from Novabiochem Art.no. 01-64-0043.(92) (a) Preparation of amidino derivatives as cysteine protease inhibitors:
Graupe, M.; Lau, A.; Jiayao, L.; John, O.; Mossman, C. J., WO2005063742 (2005); (b) Liu, H.; Alper, P. B.; Mutnick, D.;

Karanewsky, D. S., WO 2005039496 (2005); (c) Carrillo-Marquez, T.;Caggiano, L.; Jackson, R. F.; Grabowska, U.; Rae, A.; Tozer, M. J.,Organic & Biomoecular Chemistry, 2005, 22(3), 4117-4123.
(93) (a) Caddick, S.; Wilden, J. D.; Judd, D. B., J. Am. Chem. Soc., 2004,126, 1204-1205; (b) Material purchased from Polymer Laboratories.Part No. : 3424-3689.
(94) Thurmond et al., J. Pharmacol. and Experimental Therapeutics, 2004,308, 268-276.
(95) (a) Hernandez, A. A. and Roush, W. R., Curr Opin Chem Biol, 2002,6(4): 459-465; (b) Veber, D. F., Thompson, S.K., Current Opinion inDrug Discovery & Development, 2000, 3(4): 362-369; (c) Alper, J.,Science, 1994, 263, 323-324; (d) Whitfield, J. M.; Morley, P., TrendPharmacol. Sci., 1995, 16, 382-385.
(96) Bossard, M. J., et al., J Biol Chem, 1996. 271(21): 12517.(97) Bromme, D., et al., J Biol Chem, 1996. 271(4): 2126.(98) Yamashita, D. S. and Dodds, R. A., Curr Pharm Des, 2000. 6(1): 1.(99) Zaidi, M., et al., J Bone Miner Res, 2001. 16(10): 1747.(100) (a) Yasuda, Y., Kaleta, J., and Bromme, D., Adv Drug Deliv Rev,
2005. 57(7): 973; (b) Lazner, F.; Gowen, M.; Kola, I., MolecularMedicine Today, 1999, 5, 413-414.
(101) Gelb, B. D., et al., Science, 1996. 273(5279): 1236.(102) Saftig, P., et al., Proc. Natl. Acad. Sci. U S A, 1998. 95(23): 13453.(103) Gowen, M., et al., J. Bone Miner. Res., 1999. 14(10): 1654.(104) Kiviranta, R., et al., J. Bone Miner. Res., 2001. 16(8): 1444.(105) Lark, M. W., et al., Bone, 2002. 30(5): 746.(106) Stroup, G. B., et al., J Bone Miner. Res., 2001. 16(10): 1739.(107) Stoch S, Wagner J. Clinical Pharmacology & Therapeutics, 2008, 83
(1) 172-176.(108) Inaoka, T., et al., Biochem. Biophys. Res. Commun., 1995, 206(1): 89.(109) Altmann, E., J. Renaud, J. Green, D. Farley, B. Cutting, and W.
Jahnke, J. Med. Chem., 2002. 45(12): p. 2352-4.(110) Altmann, E.; Green; J.; Tintelnot-Blomley, M., Bioorg. Med. Chem.
Lett., 2003. 13(12): p. 1997-2001.(111) Kim, T.S., Hague ; A. B., Lee, Lian ; T.I., B., Tegley, C. M. ; Wang,
X. ; Burgess, T.L. ; Qian,Y. X. ; Ross, S. ; Tagari, P. ; Lin, C. H. ;Mayeda, C. ; Dao, J. ; Jordan, S. ; Mohr, C. ; Cheetham, J. ;Viswanadhan, V. and Tasker, A.S., Bioorg. Med. Chem. Lett., 2004.14(1): p. 87-90.
(112) Oballa, R.M., PCT Int. Appl. WO 0270517, 2002.(113) Naito, S.; Yamane, T.; Shinotsuku, T., JP 2004256525 A2, 2004.(114) Kim T.S.; Tasker A.S.: Non-covalent cathepsin K inhibitors for the
treatment of osteoporosis, Curr. Top. Med. Chem., 2006, 6(4), 355-360.
(115) Smyth, T. P., Bioorg. Med. Chem., 2004, 12(15): 4081.

(116) Marquis, R. W., Annual Reports in Medicinal Chemistry, 2004, 39: 79.(117) (a) Quibell, M.; Johnson, T.; Hart, T., WO 9742216, 1997; (b) Hart, T.:
Rapid responses, Chem. Br., Oct 1998, 46-48.(118) Bunnin, B. A., The Combinatorial Index, 1998; p.49.(119) Backes, B. J.; Ellman, J. A., J. Org .Chem., 1999, 64, 2322-2330.(120) Backes, B.J.; J.L. Harris, F.; Leonetti, C.S. Craik, and J.A. Ellman,
Nat. Biotechnol., 2000, 18(2), 187-193.(121) Quibell, M., Taylor, S., WO 2000069855. 2000.(122) Reverdito, A.M., Garcia, M., Salerno, A., Blanco, M., Perillo, I. A.,
Spectroscopy Letters, 2001. 34(3): p. 335-343.(123) Florey, K., Analytical Profiles of Drug Substances, 1992, 20: p. 588-
590.(124) Cordoba-Diaz, M., C.-B., M., Cordoba-Diaz, D., Journal of
Pharmaceutical and Biomedical Analysis, 1998, 18, 865-870.(125) White, P. W.; Llinas-Brunet, M.; Bos, M. Prog. Med. Chem. 2006,
44, 65–107.(126) Rehermann, B.; Nascimbeni, M. Nat. Rev. Immunol. 2005, 5, 215–
229.(127) Tan, S. L.; Pause, A.; Shi, Y. U.; Sonenberg, N. Nat. Rev. Drug
Discov. 2002 , 1, 867–881.(128) Gordon, C. P.; Keller, P. A. J. Med. Chem. 2005, 48, 1–20.(129) McHutchison, J.G. N. Engl J Med. 1998, 339, 1485-1492.(130) Poynard, T.; Marcellin, P.; Lee, S.S. et al. Lancet 1998, 352, 1426-
1432.(131) Manns, M. P.; McHutchison, J. G.; Gordon S.C., et al. Lancet 2001,
358, 958-965.(132) Fried, M. W.; Schiffman, M. L.; Reddy, K. R., et al. N. Enhl. J. Med.
2002, 347, 975-982.(133) Ronald C. Griffith, Lillian Lou, Christopher D., Roberts, and Uli
Schm. HCV Annual reports in Med. Chem. 2005, 223-237.(134) Strader, D. B.; Wright, T.; Thomas, D. L.; Seeff, L. B., Hepatology
2004, 39, 1147–1171;(135) Chander, G.: Sulkowski, M. S. Jenkes, M.W.; Torbenson, M. S.;
Herlong, H. F.; Bass, E. B.; Gebo, K. A. Hepatology 2002, 36, 135-144.
(136) Tan, S. L.; Pause, A.; Shi, Y.: Soneberg, N. Nat. Rev. Drug. Discov.2002, 1, 867-881.
(137) Dymock, B. W., Emerg. Drugs, 2001, 6, 13-42.(138) Report of a WHO Consultation organized in collaboration with the
Viral Hepatitis Prevention Board, A., Belgium J. Viral Hepat. 1999, 6,35-47.
(139) Papatheodoridis G. V.; Cholongitas, E., Journal of Viral Hepatitis2004, 11:4, 287-296

(140) Moradpour, D.; Brass, V.; Gosert, R.; Rainer, Wolk, B.; Blum, H.E.Trends Mol. Med. 2002, 8, 476.
(141) Bartenschlager, R.; Lohmann, V., J. Gen. Virol., 2000, 81, 1631-1648.
(142) Zhang, X., IDrugs 2002, 5, 154-158.(143) Dymock B.W.; Jones, P. S. and Wilson, F. X., Anitviral Chem.
Chemoth., 2000, 11, 79-96.(144) Steinkühler, C.; Koch, U.; Narjes, F., and Matassa, V. G., Curr. Med.
Chem. 2001, 8, 919-932.(145) Wardell, A. D., Errington, W., Ciaramell, G., Merson, J. and
McGarvey, M. J., Journal of General Virology 1999, 80, 701-709.(146) Bartenschlager, R. J. Viral Hepat. 1999, 6 165-181.(147) Schechter, I.; Berger, A. Biochem. Biophys. Res. Commun. 1967, 27,
157-162.(148) Bianchi, E., Pessi, A. Biopolymers (Peptide Science), 2002, 66, 101-
104.(149) Failla, C.; Tomei, L.; De Francesco, R. J., Virol. 1994, 68, 3753-
3760.(150) De Francesco, R.; Steinkühler, C. Curr. Top. Microbiol. Immunol.
2000, 242, 149-169.(151) Poupart, M. A.; Cameron, D. R.; Chabot, C.; Ghiro, E.; Goudreau, N.;
Goulet, S.; Poirier, M.; Tsantrizos, Y. S. J. Org. Chem. 2001, 66,4743–4751.
(152) Goudreau, N.; Brochu, C.; Cameron, D. R.; Duceppe, J. S.; Faucher,A. M.; Ferland, J. M.; Grand-Maitre, C.; Poirier, M.; Simoneau, B.;Tsantrizos, Y. S. J. Org. Chem. 2004, 69, 6185–6201.
(153) Rancourt, J.; Cameron, D. R.; Gorys, V.; Lamarre, D.; Poirier, M.;Thibeault, D.; Llina`s-Brunet, M. J. Med. Chem. 2004, 47, 2511–2522.
(154) Bartenschlager, R., Intervirology 1997, 40, 378.(155) Myles, D. C., Curr. Opin. Drug Discov. Devel., 2001, 4, 411.(156) Beaulieu, P. L.; Llinàs-Brunet, M. Curr. Med. Chem.: Anti-Infective
Agents 2002, 1, 163.(157) Pause, A.; Kukolj, G.; Bailey, M.; Dô, F.; Halmos, T.; Lagacé, L.;
Maurice, R.; Marquis, M.; McKercher, G.; Pellerin, C.; Pilote, L.;Thibeault, D.; Lamarrel, D. Journal of Biological Chemistry 2003,278:22, 20374-20380.
(158) Love, R.; Parge, H. E.; Wickersham, J. A.; Hostomsky, Z.; Habuka,N.; Moomaw, E. W,; Adachi, T.; Hostomska, Z. Cell 1996, 87, 331.
(159) Kim, j.L.; Morgenstern, K. A.; Griffith, J. P.; Dwyer, M. D.;Thomson, J. A.; Murcko, M.A.; Lin, C.; Caron, P.R. Structure 1998,6, 89.

(160) Llinàs-Brunet, M.; Bailey, M. D.; Ghiro, E.; Gorys, V.; Halmos, T.;Poirier, M.; Rancourt, J.; Goudreau, N., J. Med. Chem. 2004, 47,6584–6594.
(161) Llinàs-Brunet, M.; Bailey, M.D.; Bolger, G.; Brochu, C.; Faucher, A.-M.; Ferland, J.M.; Garneau, M.; Ghiro, E.; Gorys, V. J. Med.Chem.2004, 47, 1605-1608.
(162) Beevers, R.; Carr,M.G.; Jones, P.S.; Jordan, S.; Kay, P.B.; Lazell,R.C.; Raynham, T. M. Bioorg. Med. Chem. Lett. 2002 , 12, 641.
(163) Priestley, E. S.; Decicco, C.P. Org. Lett. 2002, 2, 3095.(164) Priestley, E.; De Lucca, I.; Ghavimi, B.; Erickson-Viitanen, S.;
Decicco, C. Bioorg. Med. Chem. Lett. 2002, 12, 3199.(165) Nizi, E.; Koch, U.; Ponzi, S.; Matassa, V.G.; Gardelli, C. Bioorg.
Med.Chem. Lett. 2002, 12, 3325.(166) Colarusso, S.; Gerlach, B.; Koch, U.; Muraglia, E.; Conte, I.;
Stansfiled, I.; Matassa, V.G.; Narjes, F. Bioorg. Med. Chem. Lett.2002, 12, 705-708.
(167) Narjesa, F.; Brunetti, M.; Colarusso, S.; Gerlach, B.; Dock, U.;Biasiol, G.; Fattori, D.; De Francesco, R.; Matassa, V.G.; Steinkühler,C. Biochemistry 2000, 39, 1849.
(168) Narres, F.; Koehler, K. F.; Koch, U.; Gerlach, B.; Colrusso, S.;Steinkühler, C.; Brunetti, M.; Altamura, S.; De Farncesco, R.;Matassa, V.G. Bioorg. Med. Chem. Lett. 2002, 12, 701-704.
(169) Bennett, J. M.; Campbell, A. J.; Carr, M. G.; Dunsdon, R. M.;Greening, J. R.; Hurst, D. N.; Jennings, N. S.; Jones, P. S.; Jordan , S.;Kay, P. B.; O´Brien, M. A.; King-Underwood, J.; Raynham, T. M.;Wilkinson, C. S.; Wilkinson, T. C. I.; Wilson, F. X. Bioorg. Med.Chem. Lett. 2001, 11, 355.
(170) Dunsdon, R. M.; Greening, J. R.; Jones, P. S.; Jordan, S.; Wilson, F.X. Bioorg. Med.Chem. Lett. 2000, 10, 1577.
(171) Han, W.; Hu, Z.; Jinag, X.; Decicco, C. P.; Bioorg. Med. Chem. Lett.2000, 10, 711.
(172) Attwood, M. R.; Bennett, J. M.; Campbell, A. J.; Canning, G. G. M.;Carr, M. G.; Conway, E.; Dunsdon, R. M.; Greening, J. R.; Hurst, D.N.; Jennings, N. S.; Jones, P. S.; Jordan , S.; Kay, P. B.; Handa, P. B.;O´Brien, M. A.; Overton, H. A.; King-Underwood, J.; Raynham, T.M.; Stenson, K.; Wilkinson, C. S.; Wilkinson, T. C. I.; Wilson, F. X.Antiviral Chem. Chemother. 1999, 10, 259.
(173) Llinàs-Brunet, M.; Bailey, M.D.; Deziel, R.; Fazal, G.; Gorys, V.;Goulet, S.; Hlamos, T.; Maurice, R.; Poirier, M.; Poupart, M. A.;Rancourt, J.; Thibeault, D.; Wernic, D.; Lamarre, D. Bioorg. Med.Chem. Lett. 1998, 8, 2719-2724.
(174) Ede, N. J.; Eagle, S. N.; Wickham, G.; Bray, A. M.; Warne, B.;Schoemaker, k.; Rosenberg, S., J. Peptide Sci. 2000, 6, 11.

(175) Zhang, R.; Durkin, J. P.; Windsor, W.T., Bioorg. Med. Chem. Lett.2002, 12, 1005.
(176) Zhang, A. J.; Russell, D. H.; Zhu, J. and Burgess, K. Tetrahedron Lett.1998, 39, 7439-7422.
(177) Colarusso, S.; Koch, U.; Gerlach, B.; Steinkühler, C.; De Francesco,R.; Altamura, S.; Matassa, V.G.; Narjes, F. J. Med. Chem. 2003, 46:3,345-348.
(178) Johansson, A.; Poliakov, A.; Åkerblom, E.; Wiklund, G.; Lindeberg,G., Winiwarter, S.; Samuelsson, B.; Danielsson, U. H. and Hallberg,A. Bioorg. Med. Chem. 2003, 11, 2551-2568.
(179) Llinàs-Brunet, M.; Bailey, M.D.; Fazal, G.; Goulet, S.; Hlamos, T.;Laplante, S.; Maurice, R.; Poirier, M.; Poupart, M. A.; Thibeault, D.;Wernic, D.; Lamarre, D. Bioorg. Med. Chem. Lett. 1998, 8, 1713-18.
(180) Steinkühler, C.; Biasiol, G.; Brunetti, M.; Urbani, A.; Koch, U.;Cortese, R.; Pessi, A. Biochemistry 1998, 37, 1353.
(181) Poupart, M.-A.; CameronD.; Chbot, C.; Ghiro, E.; Goudreau, N.;Goulet, S.; Poirier, M. and Tsantrizos, Y. S. J. Org. Chem. 2001, 66,4743-4751.
(182) Steinkühler, C.; Biasiol, G.; Brunetti, M.; Urbani, A.; Koch, U.;Cortese, R.; Pessi, A. and De Francesco, R., Biochem. 1998, 37, 8899-8905.
(183) Ingallinella, P. ; Altamura, S.; Bianchi, E.; Taliani, M.; Ingenito, R.Cortese, R.; De francesco, R.; Steinkühler, C.; and Pessi, A. Biochem.1998, 37, 8906-8914.
(184) Llinàs-Brunet, M.; Bailey, M.D.; Fazal, G.; Ghiro, E.; Goulet, S.;Hlamos, T.; Laplante, S.; Maurice, R.; Poirier, M.; Poupart, M.-A.;Rancourt, J.; Thibeault, D.; Wernic, D. and Lamarre, D. Bioorg. Med.Chem. Lett. 2000, 10, 2267-2270.
(185) Rancourt, J.; Cameron, D. R.; Gorys, V.; Lamarre, D.; Poirier, M.;Thibeault, D. and Llinàs-Brunet, M. J. Med. Chem. 2004, 47, 2511-2522.
(186) Lamarre, D.; Bailey, M.; Bolger, G.; Cameron, D.; Cartier, M.;Faucher, A.-M.; Goudreau, N.; Kulkolj, G.; Lagacé, L.; Pause, A.;Rancourt, J.; Thibeault, D.; Tsantrizos, T.; Llinàs-Brunet, M.Hepatology 2002 , 36, 4 part 2, 279A (Abstract 464), See also Abstract800 (page 363A) and Abstract 866 (379A).
(187) Yao, N.; Reichert, P.; Taremi, S. S.; Prosise, W. W.; Weber, P. C.Structure 1999, 7, 1353.
(188) Johansson, A.; Hubatsch, I.; Åkerblom, E.; Lindeberg, G., Winiwarter,S.; Danielsson, U. H. and Hallberg, A. Bioorg. Med. Chem. Lett. 2001,11:2, 203-206.
(189) Lamarre, D.; Anderson, P. C.; Bailey, M.; Beaulieu, P.; Bolger, G.;Bonneau, P.; Bos, M.; Cameron, D. R.; Cartier, M.; Cordingley, M.G.; Faucher, A. M.; Goudreau, N.; Kawai, S. H.; Kukolj, G.; Lagace,

L.; LaPlante, S. R.; Narjes, H.; Poupart, M. A.; Rancourt, J.; Sentjens,R. E.; St George, R.; Simoneau, B.; Steinmann, G.;Thibeault, D.;Tsantrizos, Y. S.; Weldon, S. M.; Yong, C. L.; Llina`s-Brunet, M.Nature 2003, 426, 186–189; (b) Tsantrizos, Y. S.; Bolger, G.;Bonneau, P.; Cameron, D. R.; Goudreau, N.; Kukolj, G.; LaPlante, S.R.; Llina`s-Brunet, M.; Nar, H.; Lamarre, D. Angew. Chem., Int. Ed.2003, 42, 1355–1360. (c) Reiser, M.; Hinrichsen, H.; Benhamou, Y.;Reesink, H. W.; Wedemeyer, H.; Avendano, C.; Riba, N.; Yong, C. L.;Nehmiz, G.; Steinmann, G. G. Hepatology 2005, 41, 832–835.
(190) Lin, C.; Gates, C. A.; Rao, B. G.; Brennan, D. L.; Fulghum, J. R.;Luong, Y. P.; Frantz, J. D.; Lin, K.; Ma, S.; Wei, Y. Y.; Perni, R. B.;Kwong, A. D. J. Biol. Chem. 2005, 280, 36784–36791.
(191) Lin, K.; Perni, R. B.; Kwong, A. D.; Lin, C. Antimicrob. AgentsChemother. 2006, 50, 1813–1822.
(192) Reesink, H. W.; Zeuzem, S.; Weegink, C. J.; Forestier, N.; van Vliet,A.; de Rooij, J. V.; McNair, L. A.; Purdy, S.; Chu, H. M.; Jansen, P. L.Hepatology 2005 , 42, 234A–235A.
(193) Malcolm, B. A.; Liu, R.; Lahser, F.; Agrawal, S.;Belanger, B.;Butkiewicz, N.; Chase, R.; Gheyas, F.; Hart,A.; Hesk, D.; Ingravallo,P.; Jiang, C.; Kong, R.; Lu, J.; Pichardo, J.; Prongay, A.; Skelton, A.;Tong, X.; Venkatraman,S.; Xia, E.; Girijavallabhan, V.; Njoroge, F. G.Antimicrob. Agents Chemother. 2006, 50, 1013–1020.
(194) Lin, C.; Lin, K.; Luong, Y. P.; Rao, B. G.; Wei, Y. Y.; Brennan, D. L.;Fulghum, J. R.; Hsiao, H. M.; Ma, S.; Maxwell, J. P.; Cottrell, K. M.;Perni, R. B.; Gates, C. A.; Kwong, A. D. J. Biol. Chem. 2004, 279,17508–17514.
(195) Tong, X.; Chase, R.; Skelton, A.; Chen, T.; Wright-Minogue, J.;Malcolm, B. A. Antiviral Res. 2006, 70, 28–38.
(196) Venkatraman, S., et. al., J. Med. Chem. 2006, 49, 6074–6086.(197) (a) Posters presented at the 58th Annual Meeting of the American
Association for the Study of Liver Diseases, Boston, MA, 2-6 Nov,2007: Verloes, R. et al., “Results of a Phse I Placebo-Controlled Trialin healthy Volunteers to Examine the Safety, Tolerability and PK ofthe HCV Protease Inhibitor TMC435350 after Single and RepeatedDosing” and Simmen, K. et al.,“In Vivo Activity and PreclinicalPharmakokinetics of the HCV Protease Inhibitor TMC435350”. (b)Poster presented at the 43 rd Annual meeting for the EASL, Milan, 23-27 April, 2008: Gerben, A.E. Van’t Klooster et al., “Once-DailyRegimens of the HCV NS3/4A-Protease Inhibitor TMC435350 ArePredicited to provide Therapeutic Exposure in Plasma and Liver”; (c)Oral presentation at the Cambridge Healthtech Institute’s ThrirdAnnual Drug Discovery Chemistry, 2008, April 28-29, San Diego,USA: Raboisson, P., “Discovery of the Macrocyclic HCV ProteaseInhibitor TMC435350”.

(198) Oscarsson, K.; Poliakov, S.; Oscarson, S.; Danielsson, U. H.; Hallberg,A.; Samuelsson, B. Bioorg. Med. Chem. 2003, 11, 2955-2963.
(199) Casbarra, A.; Dal Piaz, F.; Ingallinella, P.; Orrù, S.; Pucci, P.; Pessi,A.; Bianchi, E. Protein Sci. 2002 , 11:9, 2101-2112.
(200) Liu, Y.; Stoll, V. S.; Richardson, P. L.; Saldivar, A.; Klaus, J.L.;Molla, A.; Kohlbrenner, W.; Kati, W. M. Arch. Biochem. Biophys.2004, 421: 2, 207-216.
(201) Perni, R. B., Pitlik, J.; Britt, S. D.; Court, J. J.; Courtney, L. F.;Deininger, D. D.; Farmer, L. J.; Gates, C. A.; Harbeson S. L.; Levin,R. B.; Lin, C.; Lin, K.; Moon, Y.-P.; O’Malley, E. T.; Rao, B. G.;Thompson, J. A.; Tung, R. D., Van Drie, J. H.; Wei, Y. Bioorg. Med.Chem. Lett. 2004, 14:6, 1441-1446.
(202) Orvieto, F.; Koch, U.; Matassa, V. G.; Muraglia, E., Bioorg. Med.Chem. Lett., 2003, 13:16, 2745-2748.
(203) Ref. patent comp 101: Llinas-Brunet, M.; Bailley, M.; Cameron, D.;Ghiro, E.; Goudreau, N.; Poupart, M.-A.; Rancourt, J.; Tsantrizos, Y.PCT, WO 00/09558 , 2000.
(204) Ref. patent comp 102: Campell, J. A.; Good, A. et al. WO2002060926. PCT Int. Appl. 2002, 240pp.
(205) Chakravarty, P. K.; Greenlee, W. J.; Parsons, W. H.; Patchett, A. A.;Combs, P.; Roth, A.; Busch, R. D.; Melin T. N. J. Med. Chem. 1989,32, 1886-1890.
(206) Benedetti, F.; Maman, P.; Norbedo, S., Tetrahedron Lett. 2000, 41,10071-10074.
(207) Katrinsky, A. R.; Toader, D.; Watson, K.; Kiely, J. S., TetrahedronLett., 1997, 38, 7849-7850.
(208) Sieber, P. Tetrahedron Lett., 1987, 28, 6147-6150.(209) Mergler, M.; Nyfeler, R.; Tanner, R.; Gosteli, J.; Grogg, P.
Tetrahedron Lett., 1988, 29, 4009-4012.(210) Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155-4156.(211) Mehta, A.; Jaohari, R.; Benson, T. J. Douglas, K. T. Tetrahedron Lett.
1992, 33, 5441-5444.(212) The reference compound was synthesized using the procedures and the
P1 and P3 building blocks described in Boehringer Ingelheim'sinternational patent application no.WO99/07733. The P2 buildingblock was as described in Boehringer Ingelheim's international patentapplication no. WO00/09558.

Acknowledgements
The present investigations were carried out at Medivir AB in Huddinge,Sweden.
I would like to express my sincere gratitude to all those who inspired,motivated, and supported me in different ways over the years and made thisthesis possible.
Special thanks to the following people:
Professor Bertil Samuelsson for giving me the opportunity to embark onPh.D. studies, for enabling completion of this research and for providingexcellent working facilities.
Docent Björn Classon for his support, for sharing his knowledge with meand for creating a positive working atmosphere.
Professor Stefan Oscarson for accepting me in his research group atStockholm University during my initial years of graduate studies, and myuniversity colleagues Jenny U., Jenny A., and Catta.
Dr. Ellen Hewitt and Dr. Urszula Grabowska for all their help, support, andvaluable discussions regarding the work on cathepsins S and K.
Dr. Tatiana Maltseva for all assistance with NMR spectroscopy, support andfor fruitful collaboration.
Katarina Jansson, for all the nice molecular models, help andencouragement, and for being such a good colleague and friend.
My many co-authors for productive teamwork.
All the people at Medivir for spreading such friendliness and making thedaily work interesting and pleasant. Special thanks to the past and presentmembers of my group—Daniel, Karolina, Laszlo, Lotta, and Stina—, aswell as Oscar for their support, friendship, and interest in my research.
My former colleagues at Pharmacia, especially Ronny Lundin and myprevious manager Marianne Nilsson, for introducing me to the world ofpharmaceutical research, and for sharing their knowledge with me.
Patricia Ödman, for excellent revision of the English in this thesis.

I would also like to thank all of those who contributed indirectly to this workby helping me on a personal level by giving me courage and support.
Above all, my family, to whom this book is dedicated: my husband Johan,for his love and enthusiasm and our two daughters Mikaela and Annika, porser tan increiblemente maravillosas, mina änglar, ni är livets glädje! Theyare my constant source of love, inspiration, encouragement, energy, insight,pride and fun.
My parents José y Carmen and my brothers Rubén y Nacho: Por vuestrocariño, apoyo, ilusión, esfuerzo y por hacerme sentir orgullosa. Os quiero,siempre en mi corazón.
My in-laws Svante och Bibbi, and Kristina och Gösta, för allt stöd ochuppmuntran.
All my friends outside the world of chemistry, my “faraway” friends, Lauraand Geles, por una duradera y entrañable amistad allí en la distancia, mySpanish girlfriends in Stockholm, in particular my “really close” friendsEncarni y María, por vuestro apoyo incondicional y vuestra amistad, andmina trevliga grannar for their companionship and for all the good times wehave shared.

Appendix A
This appendix contains experimental details, and/or spectroscopic data ofcompounds 8 and 10 described in chapter 1.
General. p-Nitrophenylcarbonate Merrifield resin (0.8mmol/g) waspurchased from Novabiochem. Reactions were performed in 4 and 8 mlWheaton vials with screw caps. IR spectra on solid support were recorded ona Perkin-Elmer FTIR spectrophotometer. LCMS analysis was taken on aWater-Micromass instrument, using Waters 2790 Alliance, Waters 996photodiode array detector and Micromass ZMD (API ES pos and neg) massdetector. Synergi MAX-RP 50x4.6 mm 80A, 4u from Phenomex was used asreverse phase column for the LCMS analysis, and acetonitrile / water0.1%TFA were used as mobile phases. Ninhydrin test was performedfollowing the procedure described by Kaiser9,27 using aminomethylatedpolystyrene resin as positive control. NMR spectra were acquired using aVarian Unity Inova spectrometer at a magnetic field strength of 11.7 Toperating at 499.84 MHz for 1H, and 125,67 MHz for 13C.
Resin 8. Amide coupling (Scheme 2, step b). Dry resin bound 4-(aminomethyl) piperidine (250 mg, 0.2 mmol) obtained as described abovewas suspended in anhydrous DMF (2 mL). A solution of 4-azidobenzoicacid (130 mg, 0.8 mmol), PyBOP (416 mg, 0.8 mmol) and DIEA (280 uL,1.6 mmol) dissolved in anhydrous DMF (3 mL) were added to the resin. Thereaction was performed in a screw capped 8mL vial placed in the shaker atroom temperature over 24 hours. The resin was filtered through a frittedcolumn at the VacMaster and washed with DMF (2 x 10 mL), DCM (2 x 10mL), MeOH (2 x 10 mL), DCM (2 x 10mL) and MeOH (2 x 10 mL) anddried at the vacuum oven. FTIR of the resin: 3347cm-1 broad (NH-), 2121cm-
1 sharp (N3), 1689cm-1 broad (O-CO-N- and –NH-CO-). In order to elucidateif the resin obtained was 7 or 8, high resolution MAS NMR experiment wereperformed and the structure confirmed with standard tube NMR results fromthe cleaved product 10.
Compound 10. 4-azido-N-(4-piperidinylmethyl) benzamide. Resin 8 fromthe previous step was cleaved using a solution of 75% TFA in DCM (3 mL).The reaction was performed in a screw capped 8mL vial placed at the shakerduring 1.5hours. The resin was filtered and the cleaved solution was kepttogether with the washes in DCM (2 x 2 mL). The solvent was evaporated atthe Chiron equipment. The residue was re-dissolved in acetonitrile / waterand then lyophilized to afford 10 (48.4 mg, 96% pure by LCMS, 90% yield).1H NMR (DMSO-d6), (for atom numbering see Scheme 2), δ8.56 (t, 1H)(N)H-8, δ7.87 (2, J=8.2 Hz, 2H) H-11, H-15, δ7.16 (2, J=8.2 Hz, 2H) H-12,

H-14,δ3.24 (m, 2H) H-2’, H-6’, δ3.15 (t, 2H) H-7’, H-7”, δ2.82 (q, 2H) H-2”, H-6”, δ1.80 (m, 3H) H-4, H-3’, H-5’, δ1.33 (m, 2H) H-3”, H-5”. 13CNMR (DMSO-d6) δ166.15 C-9, 142.91 C-10, 131.70 C-13, 129.75 C-11, C-15, 119.48 C-12, C-14, 44.58 C-7, 43.63 C-2, C-6, 34.30 C-4, 26.99 C-3, C-5. MS (ESI): m/z 259.9M + H+.
Appendix B
This appendix contains experimental details, and/or spectroscopic data of thestarting material azide 23 (entry 7, Table 1) described in chapter 2.
(S)-(-)-1,1-Dimethylethyl[1-(Azidomethyl)-3-methylbutyl]carbamate (23,entry 7, Table 1). N-Boc-L-Leucinol (5 g, 23 mmol) was dissolved in DCM(100 mL) and cooled to 0 C̊. DIEA (9.6 mL, 55 mmol) was added to thesolution followed by the addition of methanesulfonyl chloride (2.1 mL, 27.5mmol). The resulting solution was stirred for 16h and the temperature wasslowly allowed to reach RT, and then treated with brine. The organic phasewas separated, dried, filtered and concentrated in vacuo to a brown stickysolid (6.5 g, 22 mmol, 96% yield). The mesylate obtained was dissolved inDMF (35 mL) and treated with sodium azide (4.28 g, 66 mmol). Thereaction mixture was stirred at 80 C̊ for 24h, cooled down to roomtemperature, and diluted with water. The product was extracted with severalportions of ethyl ether, and the extracts were combined, dried, and filteredand concentrated in vacuo. The crude product was purified using silica gel(elution with hexane: DCM 10:1) to afford the titled compound as a whitesolid (3.9 g, 16.1 mmol, 73% yield). 1H NMR (400 MHz, CDCl3) δ4.55-4.44 (m, 1H), 3.85-3.75 (m, 1H), 3.54-3.42 (m, 1H), 3.37-3.25 (m, 1H),1.71-1.59 (m, 1H), 1.45 (s, 9H), 1.42-1.25 (m, 2H), 0.93 (d, J=6.6 Hz, 6H).13C NMR (100 MHz, CDCl3) δ155.4, 79.3, 54.9, 48.5, 41.0, 28.2, 24.6, 22.9.FTIR: azide absorption band at 2100 cm-1. LCMS (ESI): m/z calcd forC11H22N4O2: 242.32; found: 243.19 (MH+).
Appendix C
This appendix contains experimental details, and/or spectroscopic data ofcompounds 83, 84, 86, 90-91, and illustrative examples of the finalcompounds 87 obtained by Scheme 10 and 11 (94a) described in chapter 5.
General. Aldehyde resin (0.9 mmol/g) was purchased from Novabiochem(01-64-0384), 4-sulfamylbenzoyl AM resin (0.89 mmol/g) was purchasedfrom Novabiochem (01-64-0121) and 4-sulfamylbutyryl AM resin(1.1mmol/g) was purchased from Novabiochem (01-64-0152). Reactions

were performed in 4 and 8 ml Wheaton vials with screw caps. IR spectra onsolid support were recorded on a Perkin-Elmer FTIR spectrophotometer.LCMS analysis was taken on a Water-Micromass instrument, using Waters2790 Alliance, Waters 996 photodiode array detector and Micromass ZMD(API ES pos and neg) mass detector. Synergi MAX-RP 50x4.6 mm 80A, 4ufrom Phenomex was used as reverse phase column for the LCMS analysis,and acetonitrile / water 0.1%TFA were used as mobile phases. Ninhydrintest was performed following the procedure described by Kaiser9,27usingaminomethylated polystyrene resin as positive control. NMR spectra wereacquired using a Varian Unity Inova spectrometer at a magnetic fieldstrength of 11.7 T operating at 499.84 MHz for 1H, and at 125.67 MHz for13C.
Preparation of Resins 83. General procedure for the reductiveamination (Scheme 10, step a). Aldehyde resin (3g, 2.7 mmol) wassuspended in DMF (50 mL). Tosylate salt of aminoacid allyl ester (13.5mmol, 4.64g of TosLeu-Oallyl as example), and sodium cyano borohydride(NaBH3CN) (848 mg, 13.5 mmol) were added to the resin. The reaction wasgentle stirred for 18h. and after that the resin was filtered , washed withDMF (2 x 40mL), DCM (2x 40mL), and MeOH (2x 40 mL) and dried toafford the yellow resin 83 (3.79g).
Preparation of Resins 84. General procedure (scheme 10, step b). Resin83 (3.47g, 3.41 mmol) was suspended in DMF (15 mL). FmocChloroformate(4.41 g, 17.35 mmol) dissolved in DMF (10 mL) was added. The reactionwas allowed to go for 15 min. under gentle stirring. NMM (1.72 mL, 17.35mmol) was added dropwise. After gentle stirring for 18h., the resin wasfiltered , washed with DMF (2 x 40mL), DCM (2x 40mL), and MeOH (2x40 mL) and dried to afford yellow resin 84 (3.2 g, 57% loading calculated byFmoc spectrophotometrical method).
Preparation of Resins 86. General procedure (scheme 10, steps c-d).Resin 84 (3.31g) was subjected to allyl removal by treatment with a solutionof Tetrakis (triphenylphosphine) Pd (0) ( 1.63g) and phenylsilane (5.6 g) inDCM (25 mL) for 5 min without any stirring. The procedure was performedtwice. The resin was washed with DCM (2x 50 mL), then 10 times with asolution of sodium diethyl-dithiocarbamate trihydrate 0.5% w/v and DIEA0.5 % v/v in DMF and dried to afford resin 85. Resin 85 (150 mg, 0.135mmol) was suspended in DMF (3 mL). A solution of PyBOP (281 mg, 0.54mmol), HOBt (73 mg, 0.54 mmol) and amine R3 NH2 (0.54 mmol) in DMF(3 mL) followed by DIEA (188 uL, 1.08 mmol) was added. The suspensionwas stirred for 18h at RT. The resin was filtered and washed with DMF (2 x40mL), DCM (2x 40mL), and MeOH (2x 40 mL) and dried to afford resin86.

O
NH
ONH
O
N
Peptide coupling and cleavage from the resin to afford final compounds87. General procedure (scheme 10, steps e-g). Resin 86 (150 mg, 0.135mmol) was treated with a solution of piperidine/DMF (20%v/v) for 2 hr,filtrated, washed several times with DMF, DCM and MeOH and dried. Thecorresponding acid building block R2COOH (2.7 mmol) was dissolved in asolution of 20%DMF/DCM, and colled to 0°C. Diisopropylcarbidiimide(DIC) was added (18.9 mmol). The reaction was quickly shaked and thenleft for 45 min at O°C. The urea prcipitate was filtrered off and the soluionadded to the resins (150 mg). The suspentions were stirred for 48h. Theresins were filtered, washed several times with DMF (2 x 4 mL), DCM (2x 4mL), and MeOH (2x 4 mL) and dried. The final compounds 87 wereobtained after treatment of the resins with a solution of 5%TES in TFA for2h. After filtration of the resins, the solutions were concentrated and thecrudes purified by prep LCMS.
Example of compounds 87 produced by Scheme 10 as described above:
Furan-3-carboxylic acid [3-methyl-1-(2-pyrrolidin-1-yl-ethylcarbamoyl)-butyl]-amide. Synthetic procedure as described above(sheme 9) to afford the title compound (45mg, 54% yield). >95% LCMSpurity. MS (ESI): m/z mass calcd for C17H27N3O3, 321.42, found: 322.19(MH+). 1H NMR (500 MHz, CDCl3) δ8.02 (s, 1H), 7.45 (s, 1H), 6.75 (s,1H), 4.62 (m, 1H), 3.5 (m, 2H), 3.38 (m, 2H), 2.7-2.8 (m, 4H), 1.75-1.8 (m,4H), 1.6-1.7 (m, 3H), 0.98 (m, 6H).

O
NH
ONH
O
OH
Furan-3-carboxylic acid [1-(2-hydroxy-1-phenyl-ethylcarbamoyl)-3-methyl-butyl]amide. Synthetic procedure as described above (sheme 9) toafford the title compound (31mg, 75% yield). >95% LCMS purity. MS(ESI): m/z mass calcd for C17H27N3O3, 344.41, found: 345.09 (MH+), 367.05(MNa+) . 1H NMR (500 MHz, DMSO) δ8.5 (d, NH), 8.3 (s, 1H), 8.2 (d,NH), 7.76 (s, 1H), 7.35-7.25 (m, 4H), 7.24-7.2(m, 1H), 6.97 (m, 1H), 4.9 (t,OH), 4.82 (m, 1H), 4.62 (m, 1H), 3.58 (m, 2H), 1.6-1.5 (m, 3H), 0.96 (m,6H). 13C NMR (500 MHz, DMSO), δ172.48, 162.15, 146.15, 144.59,141.98, 128.81, 128.72, 127.66, 127.42, 123.21, 109.93, 79.88, 65.38, 55.73,51.70, 41.45, 25.02, 23.73, 22.21.
Preparation of Resins 90. General procedure to amino acid attachementonto resin (Scheme 11, steps a-b). 4-sulfamylbutyrryl AM resin 93 (8.5g,9.35 mmol) was suspended in DMF (50 mL). A solution of thecorresponding Fmoc-aminoacid (46.7 mmol, 13.2g of L-LeuNHFmoc as inthe illustrative example 94a, PyBOP (19.5g, 46.7 mmol) and HOBt (5g, 46.7mmol) in DMF (40 mL) was added followed by addition of DIEA (13 mL,93.5 mmol). After the stirring of the suspention for 18h at RT the resin wasfiltered, washed with DMF (2 x 40mL), DCM (2x 40mL), and MeOH (2x 40mL) and dried to afford resin 90. Solid support FT-IR: intense absorptionband at 1713 cm-1corresponding to the carbamide from the Fmoc group.Negative ninhydrin test (the starting resin 93 gave pale blue color beads,while the produced resin 90 gave colorless beads). The resin was then treatedwith a solution of 20% pip/ DMF for 2h at RT, washed, and dried to affordFmoc-deprotected resin 90 (9.84 g). Solid support FT-IR: absence of theabsorption band at 1705 cm -1. Positive ninhydrin test (the produced resingave dark blue beads).
Preparation of Resins 91. General procedure to peptide coupling(Scheme 11, step c). Resin form previous step (9.84 g, 10.8 mmol) wassuspended in DMF (30mL). The corresponding acid R2COOH (43.2 mmol,3-furoic acid, 4.85g, as in the illustrative example 94a was added to the resinfollowed by PyBOP (22.5 mg, 43.2 mmol), HOBt (5.8g, 43.2 mmol) nadDIEA (15 ml, 86.4 mmol). The suspention was stirred for 18 h at RT. Theresin was filtrered, washed with DMF (2 x 40mL), DCM (2x 40mL), and

MeOH (2x 40 mL) and dried to afford resin 91 (8.49 g, 95% yield). Negativeninhydrin test.
Activation of resins 91. General procedure (Scheme 11, step d). Resin 91(8.49 g, 9.3 mmol) was suspended in NMP (70 mL). Iodoacteonitrile (31.2 g,186 mmol) was added followed by DIEA (8 mL, 74.4 mmol). Thesuspension was stirred for 18 h at RT. After filtration, the resin was washedseveral times with NMP, followed by washes of THF and then dried toafford highly activated resin 91 (8.87g).
Nucleophilic cleavage to afford compounds 87. General procedure(Scheme 11, step e). Activated resin 91 from previous step (300 mg, 0.33mmol) was suspended in anhydrous THF (4 mL). The corresponding amineR3NH2 (0.26 mmol, 36 mg of N-phenylethylenediamine as in the illustrativeexample 94a in THF (2 mL) was added. The suspention was allowed to reactfor 18h at RT. The resin was filtered and the solution concentrated. Thecrude was dissolved in MeOH and then purified by prep LCMS to affordcompound 87.
Examples of compounds 87 produced by Scheme 11 as described above:
O
NH
ONH
O
NH
Furan-3-carboxylic acid [3-methyl-1-(2-phenylamino-ethylcarbamoyl)-butyl]-amide (94a). Synthetic procedure described above as an illustrativeexample to afford compound 64 (53mg, 60% yield). >95% LCMS purity.MS (ESI): m/z mass calcd for C19H25N3O3, 343.43, found: 344.16 (MH+). 1HNMR (500 MHz, CDCl3) δ7.98 (s, 1H), 7.42 (s, 1H), 7,18 (m, 2H), 6.70 (m,1H), 6.60-6.55(m, 3H), 6.27 (d, NH), 4.58 (m, 1H), 4.05 (broad s, NH), 3.5(m, 2H), 3.28 (m, 2H), 1.78-1.6 (m, 3H), 0.98 (m, 6H).

O
O
NH
ONH
NH
Benzofuran-2-carboxylic acid [3-methyl-1-(2-phenylamino-ethylcarbamoyl)-butyl] amide. Synthetic procedure as described above toafford the tittle compound (48mg, 65% yield). >95% LCMS purity. MS(ESI): m/z mass calcd for C19H25N3O3, 393.49, found: 394.16 (MH+). 1HNMR (500 MHz, CDCl3) δ7.68, 7.66 (d, 1H), 7.52-7.51(d, 1H), 7.48-7.44(m, 2H), 7.33-7.39 (m, 1H), 7.15-7.12 (m, 2H), 6.95, 6.93 (d, NH), 6.68-6.66(m, 1H), 6.60-6.55 (m, 2H), 6.44 (m, NH), 4.63-4.58 (m, 1H), 3.54-3.49 (m,2H), 3.32-3.29 (m, 2H), 1.83-1.75 (m, 3H), 0.98-0.96 (m, 6H).