design, synthesis and analysis of small molecule
TRANSCRIPT

Georgia State University Georgia State University
ScholarWorks @ Georgia State University ScholarWorks @ Georgia State University
Chemistry Dissertations Department of Chemistry
8-8-2017
DESIGN, SYNTHESIS AND ANALYSIS OF SMALL MOLECULE DESIGN, SYNTHESIS AND ANALYSIS OF SMALL MOLECULE
HETEROCYCLIC AROMATIC-BASED CXCR4 MODULATORS HETEROCYCLIC AROMATIC-BASED CXCR4 MODULATORS
Theresa D. Gaines Georgia State University
Follow this and additional works at: https://scholarworks.gsu.edu/chemistry_diss
Recommended Citation Recommended Citation Gaines, Theresa D., "DESIGN, SYNTHESIS AND ANALYSIS OF SMALL MOLECULE HETEROCYCLIC AROMATIC-BASED CXCR4 MODULATORS." Dissertation, Georgia State University, 2017. https://scholarworks.gsu.edu/chemistry_diss/134
This Dissertation is brought to you for free and open access by the Department of Chemistry at ScholarWorks @ Georgia State University. It has been accepted for inclusion in Chemistry Dissertations by an authorized administrator of ScholarWorks @ Georgia State University. For more information, please contact [email protected].

DESIGN, SYNTHESIS AND ANALYSIS OF SMALL MOLECULE HETEROCYCLIC
AROMATIC-BASED CXCR4 MODULATORS
by
THERESA DENISE GAINES
Under the Direction of Suazette Mooring, PhD
ABSTRACT
CXCR4 is a chemokine receptor that has been linked to several disease related pathways
including: HIV-1 proliferation, autoimmune disorders, inflammatory disease and cancer
metastasis. The interaction of the C-X-C chemokine receptor type 4 (CXCR4) with C-X-C
chemokine ligand 12 (CXCL12) plays a key role in triggering these disease related pathways.
Various antagonists for these receptors have been synthesized and tested, but many are not useful
clinically either because of toxicity or poor pharmacokinetics. Some of the most extensive CXCR4
antagonist libraries stem from a class of compounds, p-xylyl-enediamines, which all feature a
benzene ring as the core of the compound. This work focuses on the design and synthesis of a new
class of compounds that show potential as CXCR4 antagonists by using heterocyclic aromatic

rings (2,6-pyridine, 2,5-furan, 2,5-pyrazine and 3,4-thiophene) as the core of the scaffold. After
synthesis, these analogues were probed through a variety of assays and techniques by our
collaborators in the Shim lab at Emory University including: preliminary binding assays, Matrigel
invasion assays, carrageenan mouse paw edema tests, and in silico analysis. In silico analysis also
probed 2,5-thiophene-based analogues previously synthesized. This work has produced the
beginnings of a new library of CXCR4 antagonists and has identified fifteen hit compounds that
are promising leads for further testing and modification.
INDEX WORDS: CXCR4, CXCL12, Chemokine, Chemokine receptor, Inflammation, Irritable
Bowel Disease, Rheumatoid arthritis, Cancer, Cancer metastasis, SAR, Structure activity
relationship, Heterocycle, Small molecule, Synthesis, Drug design, Antagonist,
Modulator, Pyridine, Pyrazine, Furan, Thiophene, Reductive amination, Effective
concentration, Matrigel invasion assay, Carrageenan mouse paw edema, Molecular
modeling, Docking, It1t, 3ODU.

DESIGN, SYNTHESIS AND ANALYSIS OF SMALL MOLECULE HETEROCYCLIC
AROMATIC-BASED CXCR4 MODULATORS
by
THERESA DENISE GAINES
A Dissertation Submitted in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy in Chemistry
in the College of Arts and Sciences
Georgia State University
2017

Copyright by
Theresa Denise Gaines
2017

DESIGN, SYNTHESIS AND ANALYSIS OF SMALL MOLECULE HETEROCYCLIC
AROMATIC-BASED CXCR4 MODULATORS
by
THERESA DENISE GAINES
Committee Chair: Suazette Mooring
Committee: Markus Germann
Maged Henary
Electronic Version Approved:
Office of Graduate Studies
College of Arts and Sciences
Georgia State University
July 2017

v
ACKNOWLEDGEMENTS
I want to give many thanks to everyone who has encouraged me and inspired me to achieve
all I can. Biggest thanks go to my mom, dad and sister, for believing in me and pushing me. I’m
deeply grateful for all the teachers and professors I’ve had through my schooling who invested in
me and guided me, especially Dr. Marvin Miller. I’m thankful to Brett Perkins, the Hendrickson
family, and my ISI, Teen Advisor and NAPC families for providing accountability,
companionship, advice and a loving community.
Thank you to my community of friends from Engi. You’ve encouraged me and helped me
become who I am today since I was thirteen (literally half of my life) and every step of the way
until now. Seriously, thanks for teaching me how to write and I want to give a special thanks to:
Eyad, Merdle, Kana, Kaen, Jami, Buko, Oakapea, WoF, Sushyi, Hitoko, Juushichi and Gransy.
You guys are amazing and I can’t thank you all enough.
I’m thankful to Georgia State University’s Bio-Bus program and to Dr. and Dr. Baumstark
for their mentorship and guidance. I’m grateful to the U.S. Department of Education for funding
this degree through the Graduate Assistance in Areas of National Need Program. I am grateful and
thankful for our collaborators in the Shim laboratory at Emory University for their work and
advice—especially Dr. Renren Bai. Thank you so much for all of the advice you’ve given me.
I’m thankful to my research group—especially Nikita Burrows. Thank you for a wonderful
experience over these five years and for making me a better person and colleague. And finally,
thank you, Dr. Mooring for your guidance, support and confidence over the past five years.

vi
TABLE OF CONTENTS
ACKNOWLEDGEMENTS ............................................................................................. v
LIST OF FIGURES ......................................................................................................... ix
LIST OF SCHEMES ........................................................................................................ 1
LIST OF TABLES ........................................................................................................... 2
1 INTRODUCTION .................................................................................................. 4
1.1 Cancer .................................................................................................................. 4
1.1.1 Hallmarks of Cancer ....................................................................................... 6
1.2 Inflammation ....................................................................................................... 7
2 DESIGN AND SYNTHESIS OF SMALL MOLECULE HETEROCYCLIC-
BASED CXCR4 MODULATORS............................................................................................. 10
2.1 Background ........................................................................................................ 10
2.1.1 Chemokines and CXCR4 .............................................................................. 10
2.1.2 CXCR4 Mechanism of Action ...................................................................... 12
2.1.3 Drug and Disease Related Pathways of CXCR4 .......................................... 13
2.1.4 CXCR4 Antagonists and Modulators ........................................................... 18
2.2 Rationale for Design .......................................................................................... 20
2.3 Results and Discussion ...................................................................................... 26
2.3.1 Chemistry ....................................................................................................... 26
2.3.2 Biology ........................................................................................................... 29

vii
2.4 Summary of Analogues Designed and Synthesized ........................................ 43
3 DOCKING ANALYSIS ........................................................................................... 47
4 CONCLUSIONS AND FUTURE OUTLOOK ...................................................... 59
5 EXPERIMENTAL.................................................................................................... 61
5.1 Biology ................................................................................................................ 61
5.1.1 Primary Binding Affinity Screening ............................................................ 61
5.1.2 Matrigel Invasion Assay ............................................................................... 61
5.1.3 Paw Inflammation Suppression Test ........................................................... 62
5.2 Docking Studies ................................................................................................. 63
5.3 Chemistry ........................................................................................................... 63
5.3.1 General Procedure for the Synthesis of the 2,6-Pyradine Analogues (2). .. 65
5.3.2 General Procedure for the Synthesis of the 2,5-Furan Analogues (4) ....... 72
5.3.3 General Procedure for the Synthesis of the 3,4-Thiophene Analogues (8) 78
5.3.4 General Procedure for the Synthesis of the 2,5-Pyrazine Analogues (14) . 82
REFERENCES ................................................................................................................ 86
APPENDICES ................................................................................................................ 97
NMR Spectra ............................................................................................................... 97
1H NMR Spectra ....................................................................................................... 97
13C NMR Spectra .................................................................................................... 156
Mass Spectra .............................................................................................................. 215

viii

ix
LIST OF FIGURES
Figure 1: This picture shows the six original hallmarks of cancer5. Reprinted from Cell, Volume
144, Issue 5, Hanahan, D., Weinberg, R. A. Hallmarks of cancer: the next generation, page 647,
2011, with permission from Elsevier. ............................................................................................. 7
Figure 2: Structure of CXCR4. Reprinted with permission from Wu, B.; Chien, E. Y. T.; Mol, C.
D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F. C.; Hamel, D.
F.; Kuhn, P.; Handel, T. M., Cherezov, V.; Stevens, R. C. Structures of the CXCR4 chemokine
GPCR with small-molecule and cyclic peptide antagonists. Science. 2010. 330, 1066-1071.
Copyright 2010 American Association for the Advancement of Science. ................................... 11
Figure 3: Structure of CXCL12. Reprinted with permission from Murphy, J. W.; Yuan, H.;
Kong, Y.; Lolis, E. J. Heterologous quaternary structure of CXCL12 and its relationship to the
CC chemokine family. Proteins: Structure, Function, and Bioinformatics. 2009. 78 (5), 1331-
1337. Copyright 2009 Wiley-Liss, Inc. ........................................................................................ 12
Figure 4: Reprinted with permission from Choi, W. T.; Duggineni, S.; Xu, Y.; Huang, Z. W.; An,
J., Drug Discovery Research Targeting the CXC Chemokine Receptor 4 (CXCR4). J Med Chem.
2012, 55 (3), 977-994. Copyright 2017 American Chemical Society. ......................................... 14
Figure 5: General structure for tetrahydroquinoline-based CXCR4 antagonists.92 ...................... 18
Figure 6: Examples of indole-based,115 cyclic pentapeptide-based,116 quinoline-based,117 and
pyrimidine-based CXCR4 antagonists.118 ..................................................................................... 19
Figure 7: General Structure for p-xylyl-enediamine-based CXCR4 antagonists. ........................ 19
Figure 8: Progression of AMD3100 from JM1498...................................................................... 21
Figure 9: Progression from AMD3100 to MSX122 ..................................................................... 22

x
Figure 10: Progression from AMD3100 to the compounds synthesized and analyzed in this
work. The 2,5-thiophene series was synthesized by Francisco Garcia. ........................................ 25
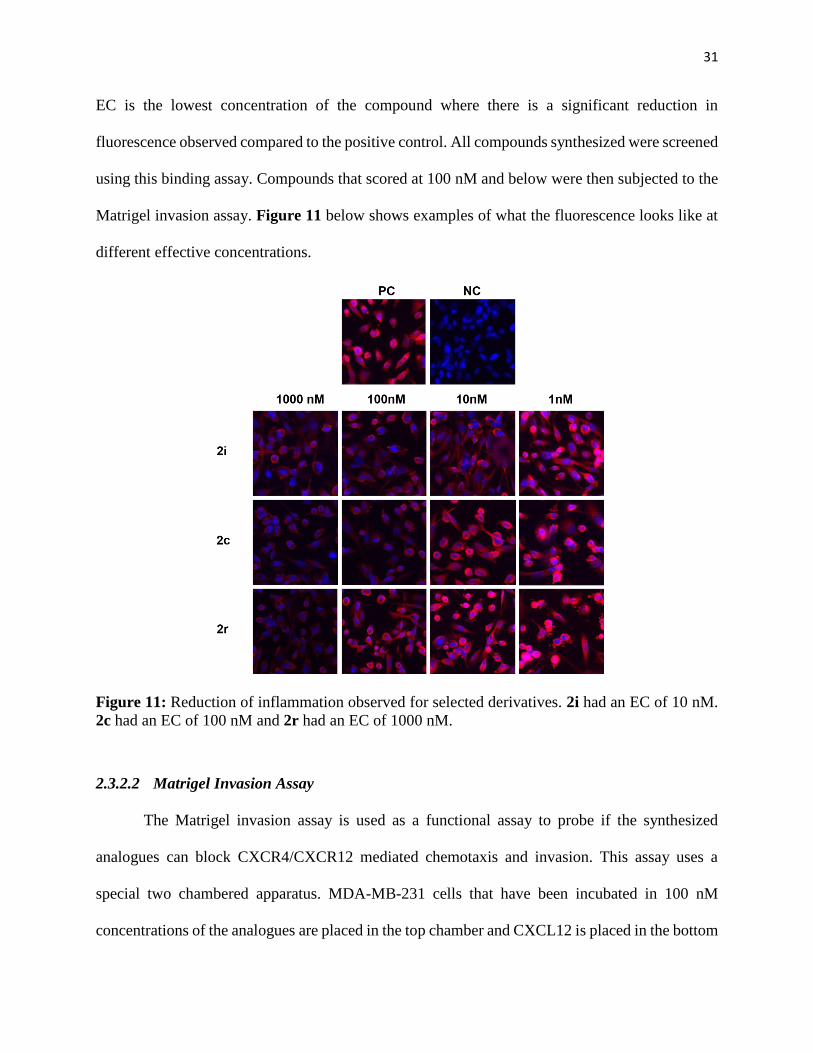
Figure 11: Reduction of inflammation observed for selected derivatives. 2i had an EC of 10 nM.
2c had an EC of 100 nM and 2r had an EC of 1000 nM............................................................... 31
Figure 12: Histological assay of compound 2i. Paw tissue sections were stained with H&E. The
whole tissue slices were scanned/digitized by NanoZoomer 2.0 HT. Software NDP.view 2 was
used to zoom in. ............................................................................................................................ 42
Figure 13: Compound 2i is shown here with the central pyridine protonated. ............................. 49
Figure 14: Protonated 1-methyl piperazine substituent in compound 4u. Pyrrolidine, morpholine
and thiomorpholine substituents also had lower docking scores when protonated. ..................... 50
Figure 15: Compound 2o is shown with pi-pi interactions with TRP94. ..................................... 51
Figure 16: Compound 2h had a low docking score and key residue interactions with TRP 94,
ASP97, and HID113 in silico; however, in vivo failed to show activity. ..................................... 51
Figure 17: Compound 15u received a high docking score of -3.782 kcal/mol, contrary to
promising scores the in vitro results in the binding and Matrigel invasion assays. ...................... 52
Figure 18: The docked pyridine compounds in AutoDock Vina. All compounds are located away
from the key residues. The CXCR4 monomer is shown here in ribbon form; however, the key
residues are highlighted as stick representations. ......................................................................... 54
Figure 19: The docked pyridine compounds shown in AutoDock in the docking gridbox. All of
the docked compounds are centered away from the key residues towards the top of the gridbox.
The CXCR4 monomer is shown here in ribbon form; however, the key residues are highlighted
as stick representations. ................................................................................................................ 55

xi
Figure 20: The docked pyridine compounds in AutoDock Vina using a biased gridbox. All
compounds are located away from the key residues. The CXCR4 monomer is shown here in
ribbon form; however, the key residues are highlighted as stick representations. ....................... 57
Figure 21: The docked pyridine compounds shown in AutoDock in the biased gridbox. All of the
docked analogues are centered away from the key residues towards the top of the gridbox. The
CXCR4 monomer is shown here in ribbon form; however, the key residues are highlighted as
stick representations. ..................................................................................................................... 57

1
LIST OF SCHEMES
Scheme 1: Synthesis of 2,6-pyridine analogues. .......................................................................... 27
Scheme 2: Synthesis of 2,5-furan analogues. ............................................................................... 27
Scheme 3: Synthesis of 3,4-thiophenedicarbaldehyde. ................................................................. 28
Scheme 4: Synthesis of 3,4-thiophene analogues. ........................................................................ 28
Scheme 5: Synthesis of 2,5-pyrazinedicarbaldehyde. ................................................................... 29
Scheme 6: Synthesis of 2,5-pyrazine analogues. .......................................................................... 30

2
LIST OF TABLES
Table 1: Ring structures and their corresponding log P values. aValues from Exploring QSAR.138
bValues from Human Metabolome Database.139-141 ...................................................................... 24
Table 2: Binding and invasion assay results for pyridine analogues synthesized. aThe invasion
assay concentration used for all compounds tested was 100nM. .................................................. 33
Table 3: Binding and invasion assay results for furan analogues synthesized. aThe invasion assay
concentration used for all compounds tested was 100nM. ........................................................... 35
Table 4: Binding and invasion assay results for the 3,4-Thiophene analogues synthesized. aThe
invasion assay concentration used for all compounds tested was 100nM. ................................... 36
Table 5: Binding and invasion assay results for pyrazine analogues synthesized. aThe invasion
assay concentration used for all compounds tested was 100nM. .................................................. 37
Table 6: 2,5-Thiophene analogues synthesized by Francisco Garcia148 which will be analyzed in
the discussion section of this work as well as in silico. ................................................................ 38
Table 7: Assay and test results for hit compounds. aThe concentration used in the invasion assay
was 100 nM. bMice were dosed used 10 mg of compound for every kg the mouse weighed in this
assay. cCompounds with a dash in the carrageenan studies have not yet been submitted. ........... 41
Table 8: Assay and test results for hit compounds. aThe concentration used in the invasion assay
was 100nM. bMice were dosed used 10mg of compound for every kg the mouse weighed.
cCompounds with a dash in the carrageenan studies have not yet been tested. dCompounds
synthesized by Francisco Garcia148 which will be analyzed in the discussion section of this work
as well as in silico. ........................................................................................................................ 45
Table 9: In silico results for hit compounds suing the Schrӧdinger Small Molecule Suite. aThe
compound has two or more interactions with this residue. ........................................................... 48

3
Table 10: In silico results for select compounds using AutoDock Vina....................................... 54
Table 11: In silico results for selected compounds using a biased gridbox in AutoDock Vina. .. 56

4
1 INTRODUCTION
This work is centered around synthesizing and analyzing small molecules to disrupt the
interaction between CXCR4, a protein receptor, and CXCL12, a ligand. This interaction triggers
many pathways in the body—including cancer cell metastasis and inflammation. The precedence
in mitigating these diseases will be explained in two parts below.
1.1 Cancer
A formidable foe and wily enemy, cancer has been a troubling adversary to a many people
from all socio-economic and racial backgrounds. Researchers, doctors, grocers, artists, students,
entrepreneurs, sons and daughters—none are immune to cancer. For the year 2014, cancer was
estimated to have claimed the lives of 585,720 American citizens, which is 1.3 times the population
of Atlanta, Georgia. Furthermore, an estimated 1.6 million people were diagnosed with cancer in
2014 in the United States. According to the CDC, 7.6 million people—just over the population of
Hong Kong—worldwide die from cancer.1 As intimidating as cancer is in the world today—as the
second leading cause of death,2 cancer has been a known disease for centuries, with evidence of
tumors in the bones of mummies from 1600 BC and descriptions of tumor removals in ancient
Egyptian inscriptions.3
Cancer is a broad and multi-faceted disease stemming from numerous mutations from the
host’s DNA genome. Each mutation has the possibility of giving the cancer cell a method of
escaping the body’s immune system or an advantage in order to gain more resources from the
surrounding area to thrive. Because of this, cancer is always different from person to person, and
possibly even from tumor to tumor in a single host’s body. This makes the treatment of cancer

5
difficult—not only are there multiple mutations that can cause a tumor to thrive in a person’s body,
but breast cancer cells from two individuals might not respond to the same medicines or treatments.
Cancer occurs when healthy cells fall out of the healthy cell cycle—which includes cell
death after a certain number of divisions. Cancerous cells continue to grow and divide and do not
undergo programmed cell death.3 Mutated DNA causes these deviations from the healthy cell
cycle. In ordinary cell replication, mutations are not extremely rare; however, organisms contain
enzymes and proteins that check DNA for mutations as well as special enzymes capable of fixing
any mutated strands by replacing the mutated parts before the cell division is completed and normal
daughter cells are still formed. DNA mutation can become the devastating disease we know as
cancer when mutations slip by the body’s processes that aim to prevent the mutations during cell
division. Mutations can be triggered and caused by a number of factors. Mutated DNA can be
given from parents to a child, or environmental factors can cause mutations in a person’s DNA.3
Fortunately, cancer survival rates have increased for the past thirty years. In 1975, newly
diagnosed cancer patients had a 48.9% survival rate over a five-year. In 2010, the five-year survival
rate rose to 68.3%.4 This increased survival rate can be attributed to advances in medicine and
novel strategies to combat cancer.
X-rays were discovered in 1896 and quickly became routine in cancer treatment and
diagnosis in only three years. New technologies such as ultrasound, computed tomography,
magnetic resonance imaging and positron emission tomography have all played an important role
in cancer diagnosis and treatment—providing lower risk and non-invasive methods of analyzing
tumors. Better diagnostic tools as well as new methodologies to destroy or remove them via lasers
have improved surgery procedures. Chemotherapy agents have been synthesized and research over
the years has been effective in reducing side effects. Hormonal and immunotherapy are also paving

6
the way for potential anti-cancer vaccines and drugs that try to reprogram a cancer cells to stop
growing and undergo cell death.3 These new technologies and innovations have changed the fight
against cancer for the better. For most cancer patients, multiple treatment strategies are used to
target the cancer. This is because cancer is able to proliferate through multiple pathways.
1.1.1 Hallmarks of Cancer
Robert Weinberg and Douglas Hanahan proposed the ‘Hallmarks of Cancer,’ which are the
fundamental, distinguishing characteristics that transform normal cells into cancerous cells.5
Cancer is different for every person and classification by treatment would prove to be difficult.
Cancers can grow in different parts of the body and sometimes can metastasize to travel to other
parts of the body. The hallmarks are defined as ‘acquired functional capabilities that allow cancer
cells to survive, proliferate and disseminate.’ Cancerous cells can acquire these hallmarks in
different ways at different times and in different orders. The original six hallmarks of cancer are:
the ability to sustain proliferative signaling, the ability to evade growth suppressors, the ability to
activate invasion and metastasis, the ability to enable replicative immortality, the ability to induce
angiogenesis or new blood vessel generation and the ability to resist cell death. Four new
characteristics have recently been added to the list. There are two abilities—the ability to
deregulate cellular energetics and the ability to avoid immune destruction. The final two have been
called ‘enabling characteristics’ which are tumor-promoting inflammation and genome instability
and mutation.5 The original six hallmarks of cancer are below in Figure 1.

7
Figure 1: This picture shows the six original hallmarks of cancer5. Reprinted from Cell, Volume
144, Issue 5, Hanahan, D., Weinberg, R. A. Hallmarks of cancer: the next generation, page 647,
2011, with permission from Elsevier.
Various cancers can possess various hallmarks, or combination of hallmarks. As hallmarks
are mutated abilities to give cancer extra functionality, a common strategy to treat cancer is to
address the various hallmarks. When undergoing chemotherapy, cancer patients are prescribed
several drugs—each often with a different function—to attack specific aspects or hallmarks of
cancer. Chemotherapy can be combined with other cancer treatment strategies such as radiation or
surgery to achieve a higher success rate of eliminating the cancer.
1.2 Inflammation
Irritable Bowel Disease (IBD) is quickly becoming a global concern; however, the
incidences of IBD vary by location. An estimated 1.2 million Americans6 have been diagnosed

8
with IBD, and between 2.5 and 3 million people in Europe. Another concerning feature is that as
nations become industrialized, incidences of IBD increase.7 Even though 0.5% of the western
world has been diagnosed with IBD,7 incidences are expected to rise exponentially over the next
ten years.8 IBD is a low mortality disease; therefore, even with a steady incidence rate, the amount
of people affected is compounded.
Higher rates of IBD lead to higher costs to treat this disease. Immunosuppressive treatment
plans can cost upwards of $25,000 annually.8 Innovation is needed to prepare for a future where
increasing members of our population need to be treated for this disease and lower the cost of said
treatments.
IBD is comprised of several different diseases—including Crohn’s disease (CD) and
ulcerative colitis (UC)—which are environmentally triggered and appear in genetically susceptible
individuals. The trigger causes an unusual immune response to the individual’s intestinal flora
resulting in gastrointestinal inflammation.9-10 Individuals with IBD have exhibited altered
chemokines and receptors in their epithelial cells, which could disrupt the body’s recognition of
their gut bacteria. Not only are individuals with IBD, unable to recognize enteric microbiota, but
it is also possible that they are unable to regulate the amount of pro-inflammatory chemokines that
are sent as the immune response.11-12
Rheumatoid Arthritis (RA) is an auto-inflammatory disease that affects 1.5 million people
in the United States with about 5-500 new diagnoses per 100,000 people each year.13-14 RA
symptoms usually appear in women between the ages of 40 and 50 and a bit later for men.15 Men
and women from diverse ethnicities and races can all suffer from RA.16 Even though men and
women can both be diagnosed with RA, three times as many women are living with RA than
men.17

9
There is no one test to diagnose RA as the disease seems to both have a genetic component
and environmental triggers.14 Because of this, RA is diagnosed by the symptoms displayed.18 RA
is characterized as inflammation in the joints caused by the immune system attacking the joint
tissue. The inflammatory response causes the synovium to become thick, and leads to swelling of
the joints. As this continues, the inflamed synovium degrades the cartilage and bone and weakens
the surrounding muscles, tendons and ligaments.18 This inflammatory response occurs in the joints
primarily; however, organs can be inflamed as a symptom of RA.19 There is no cure for RA,
however, management and treatment are more successful when it is treated early and
aggressively.20

10
2 DESIGN AND SYNTHESIS OF SMALL MOLECULE HETEROCYCLIC
AROMATIC-BASED CXCR4 MODULATORS
2.1 Background
CXCR4 is a chemokine receptor that has been linked to various disease pathways
including: HIV-1 proliferation, autoimmune disorders, inflammatory disease and cancer
metastasis.21-22 The interaction of the C-X-C chemokine receptor type 4 (CXCR4) with C-X-C
chemokine ligand 12 (CXCL12) plays an important role in cancer metastasis and inflammation.23
Various antagonists for these receptors have been synthesized and tested, but many are not useful
clinically either because of toxicity or poor pharmacokinetics.22, 24 This work focuses on the design
and synthesis of a new class of compounds that show potential as CXCR4 antagonists by using
aromatic heterocyclic rings as the core of the compound.
2.1.1 Chemokines and CXCR4
Cytokines are small proteins (5-20 kDa) involved cell signaling. A large variety of cells
(endothelial cells, macrophages, T cells, B cells) produce these proteins for different functional
purposes. Chemokines are cytokines that chemoattract cells to areas of inflammation by binding
to G protein coupled receptors (GPCRs), chemokine receptors, and creating a chemical gradient.25-
27 Chemokines can belong to four different groups, C, CC, CXC, and CX3C. They are named
based on the location of the first two cystine residues on the N-terminus.28

11
Figure 2: Structure of CXCR4. Reprinted with permission from Wu, B.; Chien, E. Y. T.; Mol, C.
D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F. C.; Hamel, D. F.;
Kuhn, P.; Handel, T. M., Cherezov, V.; Stevens, R. C. Structures of the CXCR4 chemokine GPCR
with small-molecule and cyclic peptide antagonists. Science. 2010. 330, 1066-1071. Copyright
2010 American Association for the Advancement of Science.
Most chemokines can bind to more than one chemokine receptor and receptors to more
than one ligand. These chemokines are referred to as promiscuous; however, six of the eighteen
known chemokine receptors bind to a single chemokine ligand.29 CXCR4 (shown in Figure 2) is
one of these non-promiscuous receptors. CXCR4 is a seven transmembrane GCPR and has been
known by a few other names—CD184, LESTR30 and fusin.31 CXCR4 binds exclusively to the
chemokine ligand, CXCL12 (SDF-1).32-33 CXCL12 has several isoforms in the body and even
though the alpha isoform is the most prevalent,34 CXCR4 seems to bind to each isoform with
similar affinities.35

12
Figure 3: Structure of CXCL12. Reprinted with permission from Murphy, J. W.; Yuan, H.; Kong,
Y.; Lolis, E. J. Heterologous quaternary structure of CXCL12 and its relationship to the CC
chemokine family. Proteins: Structure, Function, and Bioinformatics. 2009. 78 (5), 1331-1337.
Copyright 2009 Wiley-Liss, Inc.
Because CXCR4, is involved in many drug and disease pathways like HIV-131 entry and
cancer metastasis,36 it is one of the most studied chemokine receptors.37 Through knock out mice
studies, it has been shown that CXCR4 is necessary for neural development and hematopoiesis,
organogenesis and vascularization in embryos.38-41 CXCR4 has different functions, in line with
other chemokine receptors, in adults.42 In healthy cells, the interaction between CXCR4 and
CXCL12 triggers several downstream pathways: mobilization of stem cells,43 inflammatory cells
and immune cells,44 cell survival,45 gene transcription,46 intracellular calcium modulation,47 and
cell adhesion.48
2.1.2 CXCR4 Mechanism of Action
The crystal structure of CXCR4 was first reported in 2007 with the protein bound to a small
molecule antagonist, IT1t.49 The crystal structure was incomplete, in that some amino acids in the
N terminus were missing.50 In 2008, an NMR structure of CXCR4’s N terminus in complex with
CXCL12 was reported and it was able fill in some of the missing in pieces and identify some of

13
the interactions between CXCL12 and CXCR4.51 Unfortunately, there is no crystal structure of
CXCR4 and CXCL12 bound together.
CXCL12 binds to CXCR4 in a two-step process. In the first step, the beta sheets, the 50s
loop (the series of amino acids connecting the last beta sheet to the alpha helix), and N loop (the
series of amino acids connecting the N terminus to the first beta sheet) of CXCL12 interact with
the extracellular region on the top of CXCR4.52 The N terminus of CXCR4 then interacts with the
RFFESH loop (residues 12-17) of CXCL12.51, 53 It has been suggested that this first step causes a
conformational change in CXCR4 which exposes the binding pocket in the helices.54-55 This allows
the N terminus of CXCL12 to interact with the residues in the binding pocket near the top of the
helices of CXCR4.53 The crystal structure of CXCR4 reveals that the binding pocket is larger and
closer to the extracellular surface compared to other GCPRs.50
Because of this two-step process, small molecule antagonists may not completely block the
interactions between CXCR4 and CXCL12. Since the CXCR4/CXCL12 interaction is vital for
other necessary physiological processes, and ideal antagonist would block some functionality but
not all function. Particularly, small molecules that block the N terminus of CXCL12 from binding
in the second step would not block interactions in the first step.56 Key residues involved in the
second binding site influence signaling and have been identified as ASP97 (ECL2), ASP187
(TMII), and GLU288 (TMVII).57-58 ASP171, ASP 87 and ASP262 have also been suggested to be
important binding sites for signaling.59
2.1.3 Drug and Disease Related Pathways of CXCR4
CXCR4 is a seven transmembrane GPCR, which is embedded in the cell membrane and
interacts with other proteins from the cell surface.60-62 This receptor was first discovered as a co-

14
receptor for HIV-1 entry and after more studies were conducted, the other functional
responsibilities of CXCR4 were uncovered.61-64 CXCR4 selectively binds to the chemokine ligand,
CXCL1265 and this interaction plays a very important role in the body as knock out mice for both
of these genes do not survive due to a number of defects in the embryo.61-62 The interaction between
CXCR4 and CXCL12 triggers downstream pathways that induce inflammation; therefore, over
expression of CXCR4 has also been linked to inflammatory diseases such as allergic asthma,66
IBD,67 systemic lupus erythematosus (SLE), atherosclerosis68 and rheumatoid arthritis.69 Lastly,
the CXCR4/CXCL12 promotes cancer cell metastasis.70
Figure 4: Reprinted with permission from Choi, W. T.; Duggineni, S.; Xu, Y.; Huang, Z. W.; An,
J., Drug Discovery Research Targeting the CXC Chemokine Receptor 4 (CXCR4). J Med Chem.
2012, 55 (3), 977-994. Copyright 2017 American Chemical Society.

15
Since the CXCR4-CXCL12 axis plays such an important role in these disease related
pathways, blocking this interaction has become a leading strategy in an attempt to reduce the
progression of cancer and other inflammatory conditions.71 Several small molecule antagonists
have had success in blocking this interaction and have been shown to inhibit cancer metastasis,
HIV-1 entry and have anti-inflammatory activity.72-75
2.1.3.1 CXCR4 and Metastasis
Migration of cells is normal in the human body.76 The immune system is comprised of cells
that migrate through the body in order to facilitate healing in infected areas and routine repair and
upkeep inside the body.76-78 This migration inside the body is made possible by two groups of
proteins. The first group—chemokines—is comprised of small proteins, which interact with the
second group of proteins called chemokine receptors.22 In particular, the interaction between the
chemokine ligand, CXCL12, and the chemokine receptor, CXCR4, plays an important role in
triggering pathways to facilitate physiological functions.22, 62, 79 The interaction between CXCR4
and CXCL12 orients hematopoietic stem cells (HSCs), nonhematopoietic tissue-committed stem
cells (TCSCs), pre-B lymphocytes, t lymphocytes and embryonic stem cells (ESCs) to their
appropriate sites in the body.76-77, 80-81
Metastatic cells break from the primary tumor and travel through the body and create
tumors in other parts of the body.76, 82 Metastatic cancer cells take advantage of the
CXCR4/CXCL12 signaling interaction in order to migrate through the body in the same way that
stem cells in the body migrate.70 Many tumors develop hypoxic conditions as it grows due to
changes in cell vasculature.83 HIF-1 is then activated because of the hypoxia and can promote the

16
expression of CXCR4.84-86 It’s also possible that other growth factors contribute to upregulation
of CXCR4 because of the increased surface area of the tumor.37
Over expression of CXCR4 in cancer cells can lead to production of metalloproteases,87-90
which can degrade membranes, enabling the cells to enter into the blood stream.91 Once
circulation, these cells migrate to CXCL12 rich sites in the body—i.e. lymph nodes, lung, liver
and bone marrow.22, 77, 79-80, 92 These cells migrate through chemotaxis, which is a system that pilots
cells through the body.34 CXCL12, that is secreted by cells diffuses away from their source and
this creates a gradient. This gradient extends from the CXCL12 rich sites and becomes more dilute
as distance from the source increases.34 The CXCR4-rich cancer cells will then be guided through
the gradient to the sites that are CXCL12-rich.93 CXCR4 overexpression triggers integrin activity,
enabling the cancerous cells to adhere to the surrounding tissue once they migrate.94-96
Stage IV cancers—metastatic cancers97—usually yield a poor prognosis compared to other
stages98 and CXCR4 expression on a tumor is used to determine the prognosis for many types of
cancers including leukemia, breast cancer, prostate cancer, lung cancer and ovarian cancer.60, 62, 76,
82, 92 As seventy five percent of all cancers over express CXCR4, it is strategic to use CXCR4
expression as marker in cancer diagnosis and prognosis.60
Tissue invasion and metastasis is one of the hallmarks of cancer outlined by Weinberg and
Hanahan.5 Slowing down metastatic cancer could help boost cancer survival rates and keep tumors
localized for other treatments medicines to work more effectively. Targeting the CXCR4/CXCL12
axis to stop metastasis is an alluring tactic because the CXCR4 proteins that are expressed are less
likely to mutate. The CXCR4/CXCL12 interaction is specific; therefore, if the CXCR4 was
changed as response to a drug preventing it from migrating, it would likely no longer bind to

17
CXCL12, and still would not be able to migrate. And since the CXCL12 is produced by healthy
cells, the risk of mutation of the receptor while retaining activity is low.
2.1.3.2 CXCR4 and Inflammation
CXCR4 and CXCL12 are expressed in healthy intestinal epithelial cells (IEC).99-101 Both
CXCR4 and CXCL12 are needed for necessary physiological functions including electrolyte
secretion,102 migration, and upkeep of the epithelial mucosal membrane.103 CXCR4 and CXCL12
expression, however, are both upregulated in the IEC’s of patients with IBD.67, 104 The presence of
extra CXCL12 chemoattracts CD45RO+ T cells, which are rich with CXCR4. This interaction
triggers inflammation in the intestinal membrane and disrupts the intestinal membrane’s
homeostasis.104 It is possible that blocking the CXCR4, CXCL12 interaction can help mitigate
intestinal inflammation for patients with IBD.
In patients with Rheumatoid arthritis, CD+ T cells, neutrophils and macrophages gather
into the synovial tissue in joints and damage surrounding tissue.105-106 Several studies have
suggested that the overexpression of CXCR4 in the synovial tissue, recruits the T-cells to gather
into those areas.72 Furthermore, treatment of RA in mice with CXCR4 antagonists like
AMD3100107 and TN14003,108 a peptide antagonist, yield a reduction in inflammation.
Furthermore, treatment of RA with low-level laser irradiation (LLLI)109 reduces inflammation by
decreasing the expression of CXCR4 in the affected area. Overexpression of CXCR4 has also been
documented in the monocytes, neutrophils and B cells of patients with lupus.110 Additionally,
treatment in mouse models with CXCR4 antagonists have shown reduction of symptoms.111 As
CXCR4 is a key factor in the inflammatory response and as existing small molecule antagonists

18
have shown to be effective in mitigating symptoms of inflammatory diseases, this sets a
precedence for the creation of new antagonists.
2.1.4 CXCR4 Antagonists and Modulators
Since the CXCR4-CXCL12 interaction plays such an important role in these disease related
pathways, blocking this interaction has become a leading strategy in an attempt to mitigate these
pathways or even selectively accelerate normal physiological pathways.22 Several small molecule
antagonists have had success in blocking this interaction and have been shown to inhibit cancer
growth, inhibit cancer metastasis and have anti-RA activity.23, 112-114
Many compounds that have shown potential as CXCR4 antagonists and share the common
central benzene ring. This class of compounds is called p-xylyl-enediamines. There are several
other categories of CXCR4 antagonists, shown in Figures 5, 6 and 7, including: cyclic
pentapeptide-based antagonists, indole based antagonists, quinoline-based antagonists,
pyrimidine-based antagonists and tetrahydroquinoline-based antagonists;92 however, this work
focuses only on the p-xylyl-enediamines.
Figure 5: General structure for tetrahydroquinoline-based CXCR4 antagonists.92

19
Figure 6: Examples of indole-based,115 cyclic pentapeptide-based,116 quinoline-based,117 and
pyrimidine-based CXCR4 antagonists.118
Figure 7: General Structure for p-xylyl-enediamine-based CXCR4 antagonists.
2.1.4.1 AMD3100 and p-xylyl-enediamines
One of the first small molecule inhibitors of CXCR4 to show promise was AMD3100.64
The precursor to this compound was discovered as an impurity that showed anti-HIV activity by
blocking CXCR4, which is one of the receptors HIV-1 uses to enter the cell. This impurity was
composed of two cyclam rings connected by an aliphatic chain. Replacing the aliphatic linker with
a benzene ring increased the potency of the compound—resulting in AMD3100 (Figure 8).119
Other non-aromatic ring derivatives of AMD3100 were synthesized and tested for activity, but
only the compounds featuring benzene rings were active.120

20
AMD3100 was the first FDA approved CXCR4 antagonist; however, even though the
initial purpose for this compound was to block HIV-1 entry into the cells, this drug was not
approved for this purpose.121 In addition to poor oral bioavailability,122-123 AMD3100 is
cardiotoxic.122, 124 The FDA approved AMD3100 for limited use in patients with multiple myeloma
in order to mobilize and harvest stem cells.119, 125-126
It was speculated that the toxicity and poor oral bioavailability of AMD3100 stemmed from
the bicyclam rings. To verify this, new analogues were designed and synthesized in which the
bicyclams rings were replaced. These studies led to the synthesis of a potent p-xylyl-enediamines
derivate, WZ811 (Figure 9).127 There are several other categories of CXCR4 antagonists
including: cyclic pentapeptide-based antagonists, indole based antagonists, and
tetrahydroquinoline-based antagonists.92 Of these classes, several modulators are in clinical
trials,92 where a majority of them are peptide based.128 Many of the p-xylyl-enediamine compounds
have shown CXCR4 activity but very few have gone on to clinical trials primarily due to toxicity
or bioavailability issues.92 Various substituents and side chains have been analyzed in the p-xylyl-
enediamine class of compounds and have been screened for CXCR4 activity. The cyclams and
bicyclams are among the most studied in this group;127 however, there has not been robust
structure-activity relationship (SAR) studies in which the central phenyl ring has been altered to
another aromatic ring.
2.2 Rationale for Design
AMD3100 and other p-xylyl-enediamines are structurally specific drugs, drugs that
interact at a specific site. Changes to the structure of a lead compound can affect the biological
activity of such a type of drug.129 Using this information, it is possible to map out the relation

21
between structure of a compound and activity of a compound by making small changes to a lead
compound—called Structure Activity Relationship (SAR)—and draw conclusions about the active
site where the compound is interacting.
Figure 8: Progression of AMD3100 from JM1498
It was this process that lead to the development of AMD3100 from JM1498 (Figure 8).
JM1657 was discovered as an HIV inhibitor through screening several commercially available
cyclam rings.119 JM1657 was found to be an impurity from synthesizing JM1498 and was then
characterized.119 A series of small modifications were made to the linker between the two cyclam
rings and the lead out of the compound was a benzene ring, connected to the cyclam rings at the 1
and 4 position by a methylene, JM2987 (also known as AMD3100).119
AMD3100 was the first CXCR4 antagonist to become FDA approved;121 however, it was
for one time use to treat multiple myeloma—not HIV. The compound was cardio-toxic.122, 124
AMD3100 was still a good lead to branch off for more SAR studies to mitigate these negative
effects. This gave birth to the class of CXCR4 antagonists called p-xylyl-enediamines. The central
benzene ring was preserved, and at the 1 and 4 positions, different substituents were used to replace
the cyclam rings.92, 127 The active compounds in this class often have benzene-based, pyridine-
based or pyrimidine-based side chains replacing one or more of the cyclam rings.92 Two
forerunners out of this class of compound (Figure 9), were WZ811 (where the cyclam rings have

22
been replaced with a 2-aminopyridinyl group)130 and MSX122 (where the cyclam rings have been
replaced with a 2-aminopyrimidinyl group).131
Figure 9: Progression from AMD3100 to MSX122
The p-xylyl-enediamine compounds which removed the cyclam rings have eliminated the
toxicity issue due to metal chelation;130 however, only MSX-122 has shown promising oral
bioavailability and has made it to clinical trials but it suspended for undisclosed reasons in 2008.92
Many of the other analogues, including WZ811 are not orally bioavailable and have a short half-
life in the body.120
In designing a new class of compounds using WZ811 and MSX122 as leads to make a
more orally bioavailable analogue, the qualities that make a compound orally bioavailable must
be examined. Lipinski’s rule of five132-134 states that compounds with two or more of the following
traits are likely to be poorly orally bioavailable: molecular weights over 500 g/mol, more than five
hydrogen bond donors, more than ten hydrogen bond acceptors and a log P value greater than 5.
Naturally, there are exceptions to this rule and more detailed parameters have been outlined to
account for the exceptions by Ghose et al. Ghose et al. suggests that a log P value between -0.4
and 5.6 and a molecular weight between 180 g/mol and 500 g/mol increase drug-likeness.135 When
examining WZ811 and MSX122 by each of these parameters, they fall well within the parameters
of rules of five. Both leads have molecular weights above 180 g/mol and below 500 g/mol,

23
Hydrogen bond donors are defined as the sum of hydroxyl and amino groups.129 Both leads
have two each and are again, well within Lipinski’s range. Hydrogen bond acceptors are defined
as the sum nitrogen and oxygen atoms.129 WZ811 and MSX122 have four and six, respectively.
The last parameter is a log P value between -0.4 and 5.6 as modified by Ghose et al.135 Log P is
the partition coefficient which is a measure how lipophilic a molecule is and is calculated by
talking the log of the solubility of the molecule in 1-octanol over the solubility of the compound
in water.129 For WZ811 and MSX122, the calculated log P values, using Molinspiration, were 2.65
and 1.65 respectively.136
Since both compounds fall within the range of these rules, it is possible that the lower log
P for MSX122 contributes to its oral bioavailability. In the structure of the p-xylyl-enediamines,
the common motif is the central benzene ring. In previous studies, benzene ring has been altered
to aliphatic rings130 and aliphatic chains137 and both resulted in a loss of activity; however, a 2,6-
pyridine analogue and a 3,5-pyridine analogue based on AMD3100 were synthesized. They were
not as active as AMD3100, but they were still active.137 Pyridine and other heterocyclic aromatic
rings such as pyrazine, furan, and thiophene have lower log P values than benzene as shown in the
Table 1 below.138-141
Ring Structure Log P
Benzene
2.13a
Pyridine
0.65a
Pyrazine
-0.26b

24
Furan
1.34a
Thiophene
1.81a
Table 1: Ring structures and their corresponding log P values. aValues from Exploring QSAR.138 bValues from Human Metabolome Database.139-141
Analogues that change the linker from benzene to other heterocyclic aromatic rings
following the p-xylyl-enediamine motif may retain activity while lowering the log P value. It is
these such analogues that were synthesized in this work (Figure 10). The potential of lowering the
log P value combined with the frequent use of pyridines,142 thiophenes,143 and furans144 and
pyrazines145 in medicine, set a precedence for this work. This work will focus particularly on anti-
metastatic activity and anti-inflammatory activity of these analogues since HIV-1 binds differently
than CXCL12,146 and the analogues synthesized are generally too small to block HIV-1 as a co-
receptor.

25
Figure 10: Progression from AMD3100 to the compounds synthesized and analyzed in this
work. The 2,5-thiophene series was synthesized by Francisco Garcia.

26
2.3 Results and Discussion
2.3.1 Chemistry
2.3.1.1 2,6-Pyridine analogues
The 2,6-pyridine analogues (2a-x) were synthesized via a reductive amination reaction
between 2,6-pyridinedicarbaldehyde (1) and a substituted amine.

27
Scheme 1: Synthesis of 2,6-pyridine analogues.
2.3.1.2 2,5-Furan analogues
The 2,5-furan analogues (4a-u) were synthesized via a reductive amination reaction
between 2,5-furandicarbaldehyde (3) and a substituted amine.
Scheme 2: Synthesis of 2,5-furan analogues.

28
2.3.1.3 3,4-Thiophene analogues
Synthesized by reducing the 3,4-thiophenedicarboxyllic acid (5) to an alcohol using
diisobutylaluminum hydride. The resulting dialcohol (6) was quenched and isolated before it was
oxidized over manganese dioxide to give the dicarbaldehyde product (7).
Scheme 3: Synthesis of 3,4-thiophenedicarbaldehyde.
The 3,4-thiophene analogues (8a-j) were synthesized via a reductive amination reaction
between 3,4-thiophenedicarbaldehyde (7) and a substituted amine (Scheme 4).
Scheme 4: Synthesis of 3,4-thiophene analogues.

29
In synthesizing the 3,4-thiophene analogues, the bis(amino) product is not the major
product. In this reductive amination, when one of the aldehydes reacts with the amine, the same
amine will undergo a second, intramolecular, reductive amination leading to compound 8” shown
above in Scheme 4. This compound is favorable because of the formation of the five-membered
ring. This percent yield of the desired analogue, compound 8, was increased by breaking up the
reaction into a two-step process where the imine is isolated, and then reduced; however, the
cyclized compound is still the major product.
.
2.3.1.4 2,5-Pyrazine analogues
The 2,5-pyrazinedicarbaldehyde starting material was synthesized through a series of
reactions starting from 2,5-dimethylpyrazine. These reactions are shown below in Scheme 5. The
nitrogens on the pyrazine are oxidized under mCPBA. The resulting oxidized compound is
refluxed in acetic anhydride to encourage a substitution of the anhydride followed by a
rearrangement to the methyl group. The resulting compound (11) is then stirred in sodium
hydroxide to yield the dialcohol product (12). This compound was then oxidized using manganese
dioxide to yield the starting aldehyde.
Scheme 5: Synthesis of 2,5-pyrazinedicarbaldehyde.

30
The 2,5-pyrazine analogues (14a-d) were synthesized via a reductive amination reaction
between 2,5-prazinedicarbaldehyde (13) and a substituted amine (Scheme 6).
Scheme 6: Synthesis of 2,5-pyrazine analogues.
2.3.2 Biology
All biological assays were completed by our collaborators at Emory University in Dr.
Hyunsuk Shim’s laboratory at the Winship Cancer Institute.
2.3.2.1 Binding Assay
The derivatives were tested in a semi-quantitative binding affinity assay. It is important to
emphasize that this assay is used as a preliminary screen for potential CXCR4 antagonists that will
warrant further testing. The MDA-MB-231 breast cancer cells are incubated with the compounds
at 1 nM, 10 nM, 100 nM and 1000 nM concentrations. Next, the cells are incubated with a
biotinylated peptide, TN14003 (a known CXCR4 inhibitor), followed by streptavidin-rhodamine.
The fluorescence of the cells is then measured to obtain effective concentration (EC) is obtained.
31
EC is the lowest concentration of the compound where there is a significant reduction in
fluorescence observed compared to the positive control. All compounds synthesized were screened
using this binding assay. Compounds that scored at 100 nM and below were then subjected to the
Matrigel invasion assay. Figure 11 below shows examples of what the fluorescence looks like at
different effective concentrations.
Figure 11: Reduction of inflammation observed for selected derivatives. 2i had an EC of 10 nM.
2c had an EC of 100 nM and 2r had an EC of 1000 nM.
2.3.2.2 Matrigel Invasion Assay
The Matrigel invasion assay is used as a functional assay to probe if the synthesized
analogues can block CXCR4/CXCR12 mediated chemotaxis and invasion. This assay uses a
special two chambered apparatus. MDA-MB-231 cells that have been incubated in 100 nM
concentrations of the analogues are placed in the top chamber and CXCL12 is placed in the bottom

32
chamber as a chemoattractant. The partition between the two chambers is a Matrigel matrix that
the MDA-MB-231 cells can pass through. The measurement gained in this assay is a percentage
of inhibition of chemotaxis. When the assay is complete, the number of cells that migrated through
the matrix is counted. The percent inhibition is the percentage of cells that were prevented from
migrating compared to the negative control. The stronger the inhibitor, the fewer cells that pass
through the membrane.
Only the compounds that showed promise in the binding assay (EC ≤ 100 nm) were tested
in the Matrigel invasion assay. The two compounds used as benchmarks were AMD3100 and
WZ811. An invasion inhibition above 35% was favorable. The results for this assay have been
consolidated with the results from the binding assay for each class of compound in Tables 2
through 6, where AMD3100 and WZ811 were used as benchmarks for comparison.
Compd R Group EC (nM) Invasiona Compd R Group EC
(nM) Invasiona
2a aniline 100 <1% 2m 4-Cl aniline 100 21%
2b 2-Me aniline >1000 -- 2n 2-OMe aniline 1000 <1%
2c 3-Me aniline 100 60% 2o 3-OMe aniline 100 37%
2d 4-Me aniline 1000 -- 2p 4-OMe aniline 100 5%
2e 2-Et aniline 1 59% 2q 2-CF3 aniline >1000 --
2f 3-Et aniline 1000 -- 2r 3-CF3 aniline 1000 15%
2g 4-Et aniline 1 64% 2s 4-CF3 aniline 1 52%

33
2h 2-F aniline 1000 -- 2t 4-SMe aniline >1000 --
2i 3-F aniline 10 50% 2u 3-NO2 aniline >1000 --
2j 4-F aniline
100
5% 2v 4-NO2 aniline >1000 --
2k 2-Cl aniline >1000 -- 2w Morpholine 10 63%
2l 3-Cl aniline 100 34% 2x Pyrrolidine 1 58%
AMD3100131,
147 --- 1000 62%
WZ811127 --- 10 90%
Table 2: Binding and invasion assay results for pyridine analogues synthesized. aThe invasion
assay concentration used for all compounds tested was 100nM.
In the pyridine analogues (Table 2), eight performed well in both assays. Six of these eight
analogues contained aniline-based side chains and the other two were the morpholino analogue
(2w), and the pyrrolidinyl analougue (2x) which had binding assay resutls of 10 nM and 1 nM
respectively, and invasion assay inhibition of 63% and 58% respectively. Out of these compounds,
it seems that analogues with electron withdrawing groups as side chains are not favoured; the nitro
comounds did not have activity and the chloro compounds did not have favourable activity. The
exception is in the trifluoromethyl family, where none of the analogues were active except for the
4-CF3 anilino analogue (2s); where the binding assay concentration was 1 nM and the invasion
inhibition was 52%. Perhaps the CF3 groups in the para position has a favourable interaction in the
pocket that wasn’t favourable for the 4-Me aniline (2d)—as they would take up nearly the same
amount of space. In silico analyses would be required to gain insight into this.
The 3-Fluoro aniline analogue (2i) showed favourable activity with a binding of 10 nM
and an invasion inhhibition of 50%. Halogens are weakly electron withdrawing and have lone pairs
that may be used for donation. This could be another reason why the chloro anilino analogues had

34
some activity but none favourable. This could also be why this fluoro analogue had favourable
activity. Of the methyl analogues, only the 3-Methyl aniline (2c) had faourable activity with an
effective concentration of 100 nM and an invasion inhibition of 60%. The 2-Ethyl (2e) and 4-Ethyl
(2g) aniline analogues showed promising sctivity with effective concentrations of 1 nM for both
and invasion assay inhibitions of 59% and 64% respectively. Lastly, the 3-Methoxy aniline (2o)
showed favourable activity with an effective concentration of 100 nM and an invasion inhibition
of 37%. It is interesting that this analogue had some activity were the 3-Ethyl (2f) did not. It could
be possible that the lone pairs on the oxygen are able to interact with residues or solvent better in
the active site compared to the 3-Ethyl aniline analogue. Where the ortho and para position ethyl
groups have hydrophobic interactions at those positions.
Compd R Group EC
(nM) Invasiona Compd R Group
EC
(nM) Invasiona
4a Aniline >1000 -- 4l 3-Cl aniline >1000 --
4b 2-Me aniline >1000 -- 4m 4-Cl aniline 10 48%
4c 3-Me aniline >1000 -- 4n 2-OMe aniline 100 5%
4d 4-Me aniline 100 75% 4o 3-OMe aniline >1000 --
4e 2-Et aniline 1000 -- 4p 4-OMe aniline >1000 --
4f 3-Et aniline >1000 -- 4q 2-CF3 aniline 100 24%
4g 4-Et aniline >1000 -- 4r 3-CF3 aniline >1000 --
4h 2-F aniline 10 71% 4s 4-CF3 aniline >1000 --
4i 3-F aniline >1000 -- 4t 4-SMe aniline >1000 --

35
4j 4-F aniline 100 53% 4u 1-methylpiperazine 10 82%
4k 2-Cl aniline >1000 --
AMD3100131, 147 --- 1000 62%
WZ811127 --- 10 90%
Table 3: Binding and invasion assay results for furan analogues synthesized. aThe invasion assay
concentration used for all compounds tested was 100nM.
Of the 2,5-furan analogues synthesized (Table 3), five analogues performed well. First was
the 4-Methyl aniline (4d), which had an effective concentration of 100 nM and an invasion
inhibition of 75%. The 2-Fluoro (4h) and 4-Fluro (4j) aniline derivatives had effective
concentrations of 10 and 100 nM respectively and invasion inhibition of 71% and 53%
respectively. The 4-Chloro aniline analogue (4m) had an effective concentration of 10 nM and
invasion inhibition of 48%. The 1-methylpiperazine analogue (4u) had an effective concentration
of 10 nM and inhibited 82% of metastatic cells from invading—the highest percentage of any of
the other compounds synthesized in this work.
It is interesting to note for the 2,5-furanoaniline-based compounds the only compounds
that showed activity were compounds with ortho and para substitutions. Overall, it seems that
weakly electron donating groups like methyl and weakly electron withdrawing groups like the
fluorine and chlorine had the most activity. The 2-CF3 analogue (4q) had some activity with an
effective concentration of 10 nM and an invasion inhibition of 24%; however, it is not enough for
it to pass on to the paw edema test. All other 2,5-furan based compounds had no significant activity.

36
Compd R Group EC (nM) Invasiona Compd R Group EC
(nM) Invasiona
8a Aniline 10 72% 8f 4-F aniline >1000 --
8b 3-Me aniline >1000 -- 8g 3-Cl aniline >1000 --
8c 4-Me aniline >1000 9% 8h 4-Cl aniline >1000 --
8d 4-Et aniline >1000 -- 8i 3-CF3 aniline >1000 --
8e 3-F aniline >1000 -- 8j 4-CF3 aniline >1000 --
AMD3100131,
147 --- 1000 62%
WZ811127 --- 10 90%
Table 4: Binding and invasion assay results for the 3,4-Thiophene analogues synthesized. aThe
invasion assay concentration used for all compounds tested was 100nM.
Of the 3,4-thiophene analogues (Table 4), only the anilino analogue (8a) showed
favourable activity with an effective concentration of 10 nM and an invasion inhibition of 72%.
All other compounds had an effective concentration over 1000 nM and did not show significant
activity.
Compd R Group EC (nM) Invasiona Compd R Group EC
(nM) Invasiona
14a aniline >1000 -- 14c 4-Et aniline >1000 --
14b 3-Me aniline >1000 -- 14d 4-F aniline 10 76%
AMD3100131,
147 --- 1000 62%
WZ811127 --- 10 90%

37
Table 5: Binding and invasion assay results for pyrazine analogues synthesized. aThe invasion
assay concentration used for all compounds tested was 100nM.
Of the 2,5-pyrazine analogues (Table 5), only the 4-Fluoro anilino analogue (14d)
exhibited favorable activity with an effective concentration of 10 nM and an invasion inhibition of
76%. Since only four analogues of this series have been synthesized, there is not enough data to
speculate trends.
Compd R Group EC (nM) Invasiona Compd R Group EC
(nM) Invasiona
15a aniline 1 68% 15n 2-OMe aniline 10 52%
15b 2-Me aniline 10 61% 15o 3-OMe aniline 1000 --
15c 3-Me aniline >1000 -- 15p 4-OMe aniline 10 84%
15d 4-Me aniline 100 49% 15q 2-CF3 aniline >1000 --
15e 2-Et aniline 1000 -- 15r 3-CF3 aniline 100 77%
15f 3-Et aniline >1000 -- 15s 4-CF3 aniline >1000 --
15g 4-Et aniline >1000 -- 15t Morpholine >1000 --
15h 2-F aniline 1000 -- 15u Thiomorpholine 1 88%
15i 3-F aniline >1000 -- 15v Piperidine 1000 --
15j 4-F aniline >1000 -- 15w 4-CF3 Benzylamine 1 97%
15k 2-Cl aniline 10 93% 15x 3-F Benzylamine >1000 --
15l 3-Cl aniline 100 51% 15y 4-Cl Benzylamine >1000 --

38
15m 4-Cl aniline 1 22%
AMD3100131,
147 --- 1000 62%
WZ811127 --- 10 90%
Table 6: 2,5-Thiophene analogues synthesized by Francisco Garcia148 which will be analyzed in
the discussion section of this work as well as in silico.
A detailed summary of the 2,5-thiophene compound’s synthesis and trends can be found
in Francisco Garcia’s thesis;148 however, the most direct and straightforward comparison to be
made is between the 2,5-thiophene analogues and the 2-5-furan analogues. Unlike the furan
aniline-based compounds which only displayed activity for weakly electron donating and weakly
electron withdrawing groups, the thiophene derivatives seem to show more activity across the
board. None of the thiophene fluoroaniline analogues had activity, where the furan derivatives had
activity with the ortho and para fluoroaniline sidechains. The thiophene chloroaniline derivatives
also had activity where only one of the furan-based chlorine substituents had activity. It is possible
that the electronics of the 2,5-thiophene are such that those derivatives are able to interact better
with the residues in the active site compared to the 2,5-furan. It’s also possible that the size
difference between the 2,5-thiophene and the 2,5-furan contribute to the difference between the
two.
Between the 3,4-thiophene and the 2,5-thiophene, it is clear that the 3,4 positioning leads
to loss of activity. It’s possible that the 2,5-thiophene compounds are able to fit better into the
active site because of the geometry of the analogues. This would need to be explored further in
silico.
Overall, there are no strong motifs for substituents across the different core molecules.
Both the pyridine and 2,5-thiophene analogues had a variety of substituents that had activity in the

39
binding and Matrigel invasion assays with no clear pattern. It is worth noting that even though the
2,6-pyridine morpholino analogue (2w) had activity, the 2,5-thiophene morpholino analogue (15t)
did not. Instead, the 2,5-thiophene thiomorpholino derivative (15u) had activity. Compound 4u,
which also had a heterocyclic substituent, 1-methylpiperazine, follows in this trend and had
significant activity. Further probing would need to be done in synthesizing more derivatives as
well as in silico analysis to paint a bigger picture and complete this library.
2.3.2.3 In vivo carrageenan suppression
The mouse paw edema model is used as a proof-of-concept test.127, 131 If these analogues
are able to disrupt the CXCR4-CXCL12 interaction, then it also has an effect on inflammation. If
inflammation were to be induced in the presence of these compounds, a reduction in said
inflammation would be observed for potent CXCR4 antagonists. All compounds that performed
well in both the binding assay and Matrigel invasion assay were submitted for the paw edema test.
This test is also a way to gain insight into the toxicity of our compounds. None of the mice that
were given the synthesized analogues died during this test.
In this test, the hind paws of mice are inflamed using carrageenan, and one paw is treated
with a solution of the analogue (10 mg of analogue per kg), and the other is treated with a saline
solution. Both the saline solution and analogue solution are delivered via injection into the paw.
The paws are then measured at the end of the test using calipers to determine the percent reduction
in swelling. Compounds that score 100 nM or below in the binding assay and above a 35% in the
invasion assay were considered hit compounds and were submitted for further analysis in the paw
edema test. Of the compounds synthesized, fifteen qualified for the mouse paw edema test (shown
in Table 7).

40
Compd R Group EC (nM) Invasiona Carageenanb,c
2c 3-Me aniline 100 60% 20%
2e 2-Et aniline 1 59% 18%
2g 4-Et aniline 1 64% 42%
2i 3-F aniline 10 50% 20%
2o 3-OMe aniline 100 37% 20%
2s 4-CF3 aniline 1 52% 20%
2w Morpholine 10 63% 39%
2x Pyrrolidine 1 58% --
4d 4-Me aniline 100 75% 15%
4h 2-F aniline 10 71% --
4j 4-F aniline 100 53% 31%
4m 4-Cl aniline 10 48% 17%
4u 1-Methylpiperazine 10 82% --
8a Aniline 10 72% --
14d 4-F aniline 10 76% 32%
AMD3100131, 147 1000 62% --
WZ811127 10 90% 40%

41
Table 7: Assay and test results for hit compounds. aThe concentration used in the invasion assay
was 100 nM. bMice were dosed used 10 mg of compound for every kg the mouse weighed in this
assay. cCompounds with a dash in the carrageenan studies have not yet been submitted.
WZ811 is used as the benchmark in this assay, where paw edema test, WZ811 reduced
inflammation by 40% compared to the control. Two compounds, the 4-Ethyl pyridine (2g) and the
morpholino pyridinee (2w) reduced inflammation on par with WZ811 at 42% and 39%
respectively. The para fluoro furan (4j) ands pyrazine derivative (14d) both follow close behind
with inflammation reduction of 31% and 32% respectively.
The tissue slice shown in Figure 12 was taken from a mouse paw that was treated with the
3-fluoro pyridine-based analogue 2i. Even though 2i did not perform the best out of the compounds
selected for the carrageenan study (20%), these tissue slices show that even though the reduction
in swelling was smaller, these compounds are still mitigating the inflammatory response in the
inflamed tissue—where the inflamed dermal papilla is returning to its normal curved and spiked
shape compared to the inflamed shape where it becomes flat and smooth.
In light of this, it is worth noting that several compounds reduced swelling in the range of
2i. The 3-methyl pyridine derivative (2c), reduced swelling by 20%. The 3-methoxy pyridine (2o)
reduced inflammation by 20%. The para trifluoromethyl pyridine analogue (2s) reduced swelling
by 20%. Since tissue slices were not taken for each of these compounds, it could be a stretch to
assume that all of them are showing this activity in the tissue; however, it is promising to know
that some compounds that don’t reduce swelling as dramatically are restoring normal tissue shape.

42
Figure 12: Histological assay of compound 2i. Paw tissue sections were stained with H&E. The
whole tissue slices were scanned/digitized by NanoZoomer 2.0 HT. Software NDP.view 2 was
used to zoom in.
The other remaining compounds, did not have a reduction in swelling above 20% compared
to the control, but some swelling reduction was observed. There is no clear relation between low
effective concentration, high invasion inhibition and inflammation reduction. Several compounds
that scored well in the preliminary assays showed lower edema reduction. 2e and 4d are prime
examples of this, it could be possible that these compounds do not completely block CXCR4 and
CXCL12 is still able to interact with the receptor to trigger inflammation. Bioavailability may be
an issue as these analogues might not stay in circulation long enough to mitigate inflammation
before they are flushed out by the body. Further studies would be needed to determine the

43
pharmacokinetic properties and discern if this is an issue. There are remaining compounds that
need to be submitted for paw edema analysis; however, the data so far lays a good foundation for
analysis and future plans for this work.
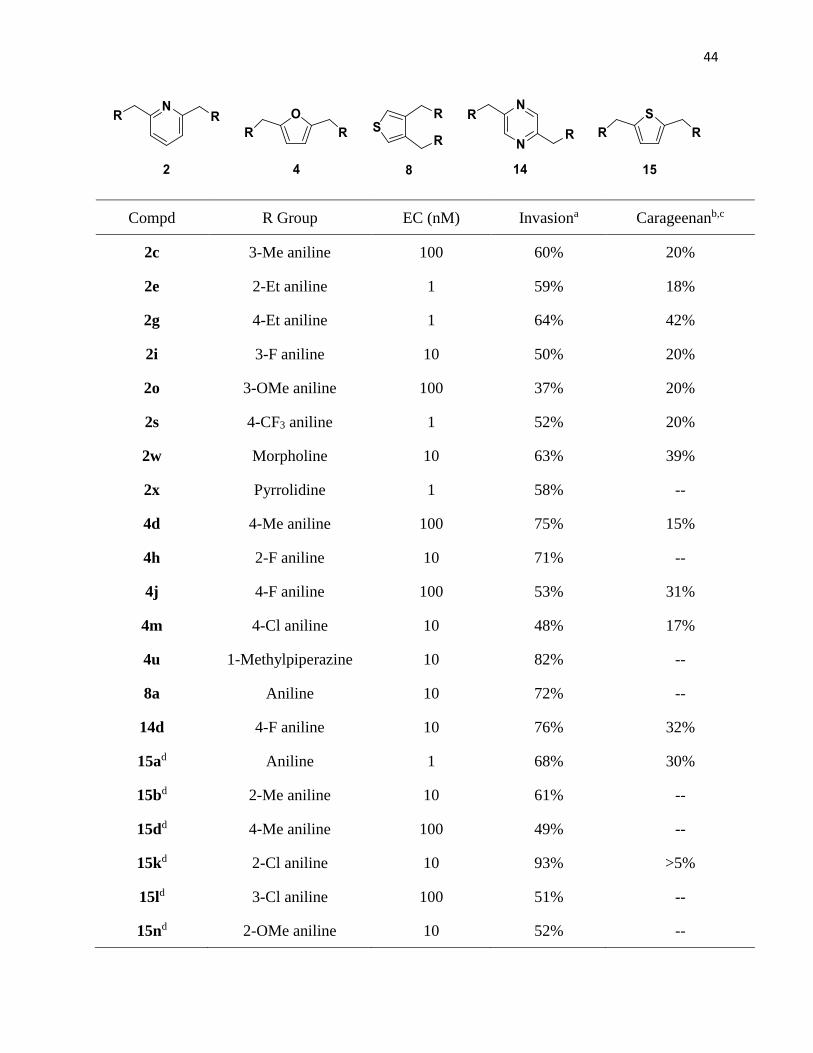
2.4 Summary of Analogues Designed and Synthesized
The compounds shown in Table 8 below show the hit compounds from this work and
Francisco Garcia’s work. Very few of the 2,5-thiophene compounds synthesized have been
submitted for the paw edema test, so it is difficult to make a complete analysis or comparison in
that regard; however out of all the five core molecules that have been synthesized, the 2,5-
thiophenes have had the most active compounds with the best invasion assay inhibitions, where
compounds 15k, 15p, 15u and 15w all inhibited upwards of 80% of had invasion assay results that
high. In the pyridine analogues, the 3-methyl (2c), 4-ethyl (2g), and the morpholino (2w) analogues
all had invasion assay results above 60% inhibition. The 2,5-furan (4d, 4h and 4u) and the 3,4-
thiophene series (8a) both had derivatives with invasion assay inhibition over 70%.
Complete inhibition of invasion—and by extension, the CXCR4/CXCL12 interaction is
undesirable as activity is necessary for normal cell function; however, in probing what about these
2,5-thiophene analogues enables it to block this interaction and prevent metastasis, future
analogues could be used as a lead to further tweak to be a suitable antagonist.
44
Compd R Group EC (nM) Invasiona Carageenanb,c
2c 3-Me aniline 100 60% 20%
2e 2-Et aniline 1 59% 18%
2g 4-Et aniline 1 64% 42%
2i 3-F aniline 10 50% 20%
2o 3-OMe aniline 100 37% 20%
2s 4-CF3 aniline 1 52% 20%
2w Morpholine 10 63% 39%
2x Pyrrolidine 1 58% --
4d 4-Me aniline 100 75% 15%
4h 2-F aniline 10 71% --
4j 4-F aniline 100 53% 31%
4m 4-Cl aniline 10 48% 17%
4u 1-Methylpiperazine 10 82% --
8a Aniline 10 72% --
14d 4-F aniline 10 76% 32%
15ad Aniline 1 68% 30%
15bd 2-Me aniline 10 61% --
15dd 4-Me aniline 100 49% --
15kd 2-Cl aniline 10 93% >5%
15ld 3-Cl aniline 100 51% --
15nd 2-OMe aniline 10 52% --

45
15pd 4-OMe aniline 10 84% --
15rd 3-CF3 aniline 100 77% --
15ud Thiomorpholine 1 88% --
15wd 4-CF3 Benzylamine 1 97% 23%
AMD3100131, 147 1000 62% --
WZ811127 10 90% 40%
Table 8: Assay and test results for hit compounds. aThe concentration used in the invasion assay
was 100nM. bMice were dosed used 10mg of compound for every kg the mouse weighed. cCompounds with a dash in the carrageenan studies have not yet been tested. dCompounds
synthesized by Francisco Garcia148 which will be analyzed in the discussion section of this work
as well as in silico.
Even though the 2,5-thiophene series seem to perform well in the binding and invasion
assay, the preliminary data from the paw edema test shows that they are not reducing inflammation
in vivo as well as they prevented metastasis in vitro compared to the pyridine or furan-based
analogues. Where 15w and 15k, had invasion scores of 93% and 97% respectively, they only
reduced inflammation by 30% and 23% respectively. Compared to 2g which had invasion assay
inhibition of 64% and an inflammation reduction of 42%, it seems that performance in the invasion
assay is not a good predictor for the success of the paw edema test.
As mentioned in the rationale for design, desirable pharmacokinetic properties tend to
increase as the partition coefficient of a potential drug is lowered. Both the pyridine and furan
rings have a lower log P than thiophene. If the 2,5-thiophene analogue’s log P is not low enough,
it may be flushed out of the body too quickly to effectively reduce swelling in this assay. If this
trend continues as more and more 2,5-thiophene analogues are submitted for the paw edema test,
using substituents that lower the overall partition coefficient may yield better results.

46
Similarly, perhaps the pyridine compounds were able to do relatively well in the paw
edema test because of the lower partition coefficient. Even though they did not inhibit invasion of
more than 60% of the metastatic cells compared to the control, perhaps their results in the paw
edema assay are consistently active because of their drug-likeness due to the lowered log P. This
would need to be further evaluated in pharmacokinetic tests.
The 3,4-thiophene analogues overall were not active, with only the aniline derivative (8a)
showing activity. This seems to suggest, that the geometry of the 2,5 positioned five membered
rings may be favorable. This could be analyzed further by synthesizing 3,4-furan-based analogues
for comparison. Since the log P of pyrazine is lower than all of the other heterocyclic aromatic
rings, the pyrazine, further analogue synthesis should be conducted in order to complete this
section of the library; however, the activity of the 4-fluoro pyrazine (14d) is a promising first look.
Overall, analogues 2g, 2w, 4j,14d, and 15a are the five best compounds synthesized and
analyzed so far with favorable effective concentrations, invasion inhibition properties and with
inflammation reduction above 30% compared to the control.

47
3 DOCKING ANALYSIS
3.1 Schrӧdinger Small Molecule Suite
To gain insight as to how these analogues are interacting with the active site, in silico
methods used. In addition to the analogues synthesized in this work, analysis for 2,5-thiophene
(15a-y) derivatives synthesized by Francisco Garcia148 are also provided in this work. Preliminary
docking analysis of several of the 2,6-pyridine compounds were conducted in a previous work by
Dr. Davita Camp. Previous work has identified several key residues that can block the CXCR4-
CXCL12 interaction. Some of these residues include ASP97, GLU288, ASP187, PHE87, ASP171,
and PHE292, where the first three are required for signaling pathways triggered by CXCL12.57-58
Docking scores and key residue interactions for hit compounds are shown in Table 9.
Compd R Group EC (nM) Docking Score
(kcal/mol)
Key Residue
Interactions
2c 3-Me aniline 100 -7.314 GLU288, TRP94,
CYS186
2e 2-Et aniline 1 -5.944 HID113
2g 4-Et aniline 1 -5.716 TRP94, GLU288
2i 3-F aniline 10 -6.822 ASP97a
2o 3-OMe aniline 100 -6.187 TRP94, ASP97a
2s 4-CF3 aniline 1 -6.026 TRP94,a ASP97,
HID113, CYS186
2w Morpholine 10 -7.173 ASP97,a HID113,
TYR116
2x Pyrrolidine 1 -6.400 ASP97, TYR116,
CYS186

48
4d 4-Me aniline 100 -4.809 HID113, ARG188,
GLN200
4h 2-F aniline 10 -4.869 TRP94
4j 4-F aniline 100 -5.524 TRP94, CYS186,
ARG188, GLU288
4m 4-Cl aniline 10 -5.300 HID113, CYS186
4u 1-Methylpiperazine 10 -6.373 TRP94, TYR116,
GLU288
8a Aniline 10 -3.437 ASP97, HID113,
ARG188
14d 4-F aniline 10 -4.600 ASP97, HID113,
ARG188
15ab Aniline 1 -4.773 ASP97
15bb 2-Me aniline 10 -4.354 TRP94, HID113
15db 4-Me aniline 100 -4.506 TRP94, CYS186
15kb 2-Cl aniline 10 -4.840 TRP94, ASP97
15lb 3-Cl aniline 100 -5.975 ASP97
15nb 2-OMe aniline 10 -5.409 ASP97
15pb 4-OMe aniline 10 -4.479 GLU32, ASP97,
ARG183
15rb 3-CF3 aniline 100 -5.944 TRP94, ASP97
15ub Thiomorpholine 1 -3.782 TRP94, GLU288a
Table 9: In silico results for hit compounds suing the Schrӧdinger Small Molecule Suite. aThe
compound has two or more interactions with this residue.
Out of all the compounds docked, the hit pyridine compounds had the lowest docking
scores and out of the hit compounds the pyridine-based compounds had the best scores. The Ligand
Preparation wizard in the Schrödinger program created two different ionized states for each of the
pyridine analogues. The first, where the central ring was deprotonated, and a second where the
nitrogen in the central pyridine was protonated (Figure 13). The protonated pyridine compounds
had lower docking scores compared to the deprotonated version.

49
Figure 13: Compound 2i is shown here with the central pyridine protonated.
Previous studies suggest that small molecules with a positively charged center can interact
with better the acid residues in the active site.59 None of the other heterocyclic cores had an
ionization where the center was protonated; however, certain substituents lead to protonation of
the connecting amine. These substituents—morpholine, thiomorpholine and pyrrolidine have a
generated ionization (Figure 14) where both amines are protonated creating a positive charge in
the active site. These compounds also had a low docking score.

50
Figure 14: Protonated 1-methyl piperazine substituent in compound 4u. Pyrrolidine, morpholine
and thiomorpholine substituents also had lower docking scores when protonated.
Most of the active compounds docked have key residue interactions with ASP97, or
GLU288—two of the three residues mentioned earlier that are necessary for signaling. CYS186,
TRP94, and TYR116, when docked are programmed to freely rotate to allow the ligands to interact
via hydrogen bonding, pi-pi interactions or pi-cation interactions. Compound 2o, shown in Figure
15, displays pi-pi interactions with aromatic residues like TRP94, TYR116 and HID113.
It is worth noting that several ligands which were not active in the binding and Matrigel
assays were docked with low scores. One such example is 2h, shown in Figure 16. This compound
has a docking score of -6.237 kcal/mol and has key residue interactions with TRP94, ASP97, and
HID113. 2h was not active in any of the assays, with an effective concentration of 1000 nM. This
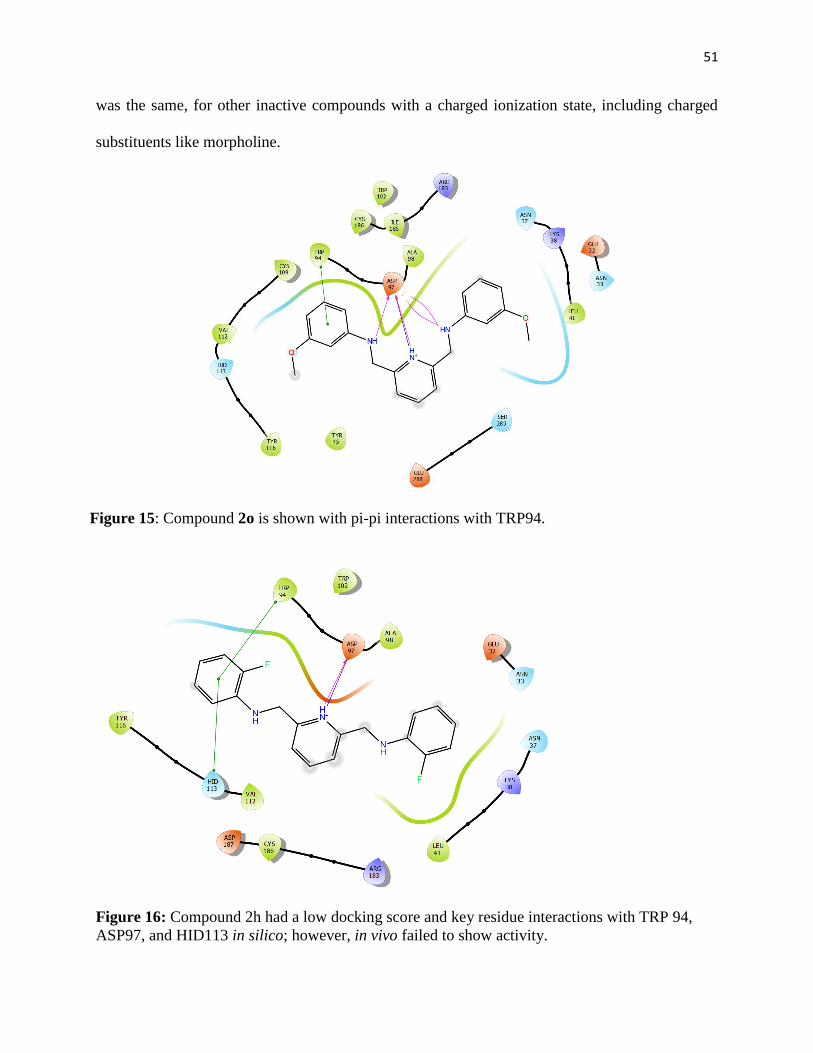
51
was the same, for other inactive compounds with a charged ionization state, including charged
substituents like morpholine.
Figure 15: Compound 2o is shown with pi-pi interactions with TRP94.
Figure 16: Compound 2h had a low docking score and key residue interactions with TRP 94,
ASP97, and HID113 in silico; however, in vivo failed to show activity.

52
Similarly, there were several compounds that had low effective concentrations and high
invasion inhibition in vitro, but obtained a low docking score in silico. Compound 15u (shown in
Figure 17) had an effective concentration of 1 nM and inhibited invasion of metastatic cells by
88%. In silico results yielded a docking score of -3.782 kcal/mol even though it had a key residue
interactions with GLU288. Overall, compared to the activity displayed in the assays, the 2,5-
thiophene analogues received higher docking scores compared to their in vitro activity.
Figure 17: Compound 15u received a high docking score of -3.782 kcal/mol, contrary to
promising scores the in vitro results in the binding and Matrigel invasion assays.
These discrepancies show that the docking model is not perfect; however, there are a few
themes that can be taken away and used to direct future compounds synthesized. Positive charged
ligands are favoured in the active site. All pyridine-based and most furan-based analogues with a
positive charge scored below -5.000 kcal/mol. Synthesis of these analogues with non-aromatic
cyclical amines and heterocyclic as substituents that can be protonated as secondary amines could
yield active compounds.

53
Additionally, as some analogues with aromaticity are able to have pi-pi interactions with
nearby residues, the use of substituted benzylamines, which have an extra sp3 hybridized carbon
linking the substituent to the core molecule, instead of substituted anilines may give more
flexibility to the analogue to fit better inside the pocket to make those pi-pi interactions.
3.2 AutoDock Vina
Due to the ambiguity of the results of the docking analysis using the Schrӧdinger software,
the docking analysis was partially repeated using AutoDock Vina149 on a select subset of the 2,6-
pyridine analogues to determine if the resulting data in these programs yield trends. Ten pyridine
compounds were selected; six had activity in the binding and invasion assays, the other four did
not. The docking scores are displayed below in Table 10. The key residue interactions are not
listed because the AutoDock Vina program does not have an interaction wizard.
Compd R Group EC (nM) Invasion Assay Docking Score
(kcal/mol)
2a Aniline 100 <1% -6.8
2c 3-Me aniline 100 60% -7.3
2e 2-Et aniline 1 59% -6.7
2f 3-Et aniline 1000 -- -6.6
2g 4-Et aniline 1 64% -7.1
2h 2-F aniline 1000 -- -7.2
2i 3-F aniline 100 50% -7.0
2q 2-CF3 aniline >1000 -- -6.3

54
2w Morpholine 1 63% -5.5
2x Pyrrolidine 1 58% -5.3
Table 10: In silico results for select compounds using AutoDock Vina.
Figure 18: The docked pyridine compounds in AutoDock Vina. All compounds are located away
from the key residues. The CXCR4 monomer is shown here in ribbon form; however, the key
residues are highlighted as stick representations.
The docking in this program has no correlation between the assay results compared to in
silico results. The morpholine and pyrrolidine analogues scored the highest; however, all the
other analogues consistently scored between -6.3 and -7.3. The analogues were then visualized in
AutoDock. Figure 18 shows the compounds in relation to the key residues and active site. The
docked analogues are neither interacting with the key residues nor located within the active site.
Figure 19 shows the analogues in relation to the docking gridbox.

55
Figure 19: The docked pyridine compounds shown in AutoDock in the docking gridbox. All of
the docked compounds are centered away from the key residues towards the top of the gridbox.
The CXCR4 monomer is shown here in ribbon form; however, the key residues are highlighted
as stick representations.
In response to the lack of trends in the first set of in silico results, the same selected
pyridine analogues were docked using a biased gridbox. The gridbox was reduced in size and
was re-centered on the active site of the CXCR4 monomer. Docking with a biased gridbox is
investigative in nature as it may help force compounds into a favorable position in the active site
that was not seen in the first gridbox was larger at the same runtime; however, the results from
this set of docking analyses cannot replace the original results from the non-biased docking
analysis. The results of the biased docking analysis can be found in Table 11, below.

56
Compd R Group EC (nM) Invasion Assay Docking Score
(kcal/mol)
2a Aniline 100 <1% -6.8
2c 3-Me aniline 100 60% -3.6
2e 2-Et aniline 1 59% -1.7
2f 3-Et aniline 1000 -- -3.1
2g 4-Et aniline 1 64% -1.8
2h 2-F aniline 1000 -- -5.1
2i 3-F aniline 100 50% -3.8
2q 2-CF3 aniline >1000 -- -1.9
2w Morpholine 1 63% -4.3
2x Pyrrolidine 1 58% -4.4
Table 11: In silico results for selected compounds using a biased gridbox in AutoDock Vina.
The docking scores in the biased gridbox are markedly higher than in the non-biased
gridbox with docking scores ranging from -1.7 to -6.8. Unfortunately the broadness in the range
has not lead to correlation between active compounds and their assay scores. Hit compounds such
as 2e, 2g, and 2w scored -1.7, -1.8 and -4.3 respectively, where inactive compounds such as 2f,
2h, and 2q scored -3.1, -5.1 and -1.9 respectively. The visualization shown in Figure 20 and
Figure 21 show the biased docked compounds and the biased gridbox. Like the unbiased gridbox
first used, the analogues are as far away from the active site as they can be and are clustered in the
top edge of the gridbox.

57
Figure 20: The docked pyridine compounds in AutoDock Vina using a biased gridbox. All
compounds are located away from the key residues. The CXCR4 monomer is shown here in
ribbon form; however, the key residues are highlighted as stick representations.
Figure 21: The docked pyridine compounds shown in AutoDock in the biased gridbox. All of
the docked analogues are centered away from the key residues towards the top of the gridbox.
The CXCR4 monomer is shown here in ribbon form; however, the key residues are highlighted
as stick representations.

58
The docking completed using AutoDock Vina has not yielded any additional information
highlighting how these heterocyclic analogues are interacting with the active site of CXCR4 using
the parameters outlined in this work Further analysis using another program or perhaps more
exhaustiveness may give more information and insight.

59
4 CONCLUSIONS AND FUTURE OUTLOOK
In this work, a small library of heterocyclic-based modulators for CXCR4 mimicking p-
xylyl-enediamines have been synthesized and analyzed in binding assays, Matrigrel invasion
assays and an in vivo paw edema test. Overall, the 2,6-pyridine-based compounds and the 2,5-
thiophene based compounds had the best results in the assays overall; however, more 2,5-thiophen
compounds need to be submitted for the paw edema test. The 3,4-thiophene analogues were largely
inactive, possibly due to the geometry. The 2,5-furan analogues had several with activity; however,
compared to the 2,5-thiophene compounds, they exhibit less activity.
There are several ways to extend this project going into the future. The 2,5-pyraizne library
needs to be completed and analyzed. For the 2,6-pyridine and 2,5-furan compounds, extending the
sidechains by using benzylamines instead of anilines may boost activity as well, potentially
enabling analogues to have pi-pi interactions with nearby residues and granting them more
flexibility in the binding pocket. In addition to benzylamine substituents, more cyclic non-aromatic
analogues need to be synthesized. In silico analysis suggests that these substituents are likely to be
positively charged in physiological conditions and will interact with acidic key residues. The
success of the pyrrolidine, morpholine, thiomorpholine and 1-methylpiperazine compounds set a
precedence to further investigate this trend.
In order to verify if the 3,4-thiophene was largely inactive because of the 3,4 positioning
of the side chains, synthesis of 3,4-furan based derivatives can provide insight into if this is true.
There are a few other core molecules that would contribute to this library as the 2,6-pyridine and
2,5-pyrazine analogues have shown activity. Modeled after the 2,6-pyridine analogues, synthesis

60
of 2,6-pyrazine analogues could possibly increase activity as pyrazine has a lower log P than
pyridine. It would also provide more information as to if the analogues are able to bind better in
the pocket with another nitrogen in the heterocyclic ring can increase activity as in silico analysis
suggests that the pyridine analogues are protonated in the active site and are able to bind differently
be because of the positioning of the cation on the ring instead of on the amine. In silico results did
not suggest protonation for the 2,5-pyrazine cores; however, 2,6-pyrazine should still be explored.
In the same vein, synthesizing 2,5-pyrdine analogues that match the 2,5-pyrazine would be a useful
addition to the library.
For existing core molecules like the 2,5-thiophene and the 2,6-pyridine, using other amines
that have shown to have exception activity in the p-xylyl-enediamine series such as the 2-
aminopyridine and 2-aminopyrimidine and halogen substituted 2-aminopyridine and 2-
aminopyrimidines. Design and synthesis of asymmetrical analogues of these core molecules could
help increase activity by designing a small molecule that can fit better into the active site.

61
5 EXPERIMENTAL
5.1 Biology
All biological assays were completed by our collaborators at Emory University in Dr.
Hyunsuk Shim’s laboratory at the Winship Cancer Institute.
5.1.1 Primary Binding Affinity Screening
The binding affinity assay is a competitive assay where approximately twenty thousand
MDA-MB-231 breast cancer cells are incubated an 8-well slide chamber for two days in 300 μL
of medium. The compounds were also incubated in separate wells at several concentrations (1, 10,
100, 1000 nM) for ten minutes at room temperature. The cells were then fixed in a chilled solution
of 4% paraformaldehyde. After the cells were rehydrated in phosphate-buffered saline (PBS), the
slides were prepared by incubating them with 0.05 μg/mL biotinylated TN14003 for thirty minutes
at room temperature. These slides were washed three times with the PBS solution and were then
incubated for thirty minutes at room temperature in streptavidin-rhodamine (1:150 dilution;
Jackson ImmunoResearch Laboratories, West Grove, PA). The slides were washed again with the
PBS solution and were mounted in an antifade mounting solution (Molecular Probes, Eugene, OR).
A Nikon Eclipse E800 microscope was used to analyze the samples.127, 150
5.1.2 Matrigel Invasion Assay
This assay was performed using a Matrigel invasion chamber (Corning Biocoat; Bedford,
MA). In the bottom chamber, a solution of CXCL12 (200ng/mL; R&D Systems, Minneapolis,
MN) was added to the apparatus. 100nm of the selected compounds (or AMD3100 as a control)

62
were added to the MDA-MB-231. The cells were then placed in the top chamber. The apparatus
was then incubated in a humidified incubator for 22 hours. The remaining cells in the top chamber
were removed using a cotton swab and the invading cells in the bottom chamber were stained
hematoxylin and eosin (H&E) and fixed with methanol. The rate of invasion was calculated by
counting the invading (stained) cells.127, 150
5.1.3 Paw Inflammation Suppression Test
In this test, C57BL/6J does (Jackson Laboratories) are subcutaneously injected with λ-
carrageenan (50 μL in 1% w/v in saline) in the right hind paw to trigger inflammation; the other
hind paw is used as the non-inflammation control. The selected analogues were prepared in 10%
DMSO and 90% of 45% (2-hydroxypropyl)-β-cyclodextrin (CD) in PBS. Doses of the analogues
were set at 10mg/kg and the dose for TN14003 was set at 300 μg/kg. The TN14003 dose was set
lower for this experiment because it was found that 300 μg/kg gave the maximum efficacy at
minimum concentration in breast cancer metastasis in an animal model. The mice were dosed 30
minutes after the carrageenan injection and then once a day following the initial dose. The mice
were sacrificed 74 hours after inflammation was induced and two hours after the last injection of
the selected analogues. The hind paws of the mice were photographed and calipers were used to
measure the thickness of the paw from front to back. To quantify the edema, the measurement
from the untreated paw was subtracted from the volume of the treated paw. The inflammation
suppression percentage was determined by comparing the analogue treated groups to the control
group. Each analogue was tested in quintuplicate using the above procedure.127, 131 Paw tissue slices
were also collected and stained with H&E. Tissue slices were scanned and digitized by
NanoZoomer 2.0 HT. The software NDP.view 2 was used to view the slices in detail.

63
5.2 Docking Studies
5.2.1 Schrodinger Small Molecule Suite
Schrodinger Small Molecule Suite
The structure of the target receptor CXCR4, PDB-ID 3ODU, was retrieved from the RCSB
Protein Data Bank.50 Docking calculations were performed by the Schrodinger suite with default
settings unless otherwise indicated. The Protein Preparation Wizard was used to prepare the
CXCR4 protein for docking. CXCR4 along with its co-crystallized ligand IT1t went through a
preprocess procedure with the additional options to fill in missing side chains and loops using
Prime and removing waters beyond 5Å from heteroatom groups. The heterostate for the co-
crystallized ligand was generated using Epik. The PROPKA feature was utilized to optimize the
hydrogen bond network at a physiological pH. After hydrogen bond optimization, water molecules
were removed with less than 3 H-bonds to non-waters. The protein was then energetically
minimized using a default constraint of 0.3Å RMSD and OPLS 2005 force field. Ligands were
prepared by the LigPrep function of the Schrodinger Suite with default parameters in the gas phase.
Epik generated possible ionization states of the ligands at physiological pH. Receptor grid
generation was performed by inputting the prepared receptor directly from the protein preparation
wizard. Docking was performed using the Extra Precision (XP) feature of the GLIDE program.
Ligands were docked flexibly in the rigid protein devoid of the IT1t ligand and were ranked based
on their GLIDE docking score.

64
5.2.2 Autodock Vina
The structure of the target receptor CXCR4, PDB-ID 3ODU, was retrieved from the RCSB
Protein Data Bank.50 Docking calculations were performed AutoDock Vina and default settings
were used unless otherwise specified. The protein in PDB form was uploaded into the program.
The B dimer of the protein was then selected and removed to create a monomer. The monomer
was then exported as a macromolecule, converting it to a PDBQT file. The gridbox was then
selected; the initial gridbox had the following parameters: center_x = 16.807, center_y = -6.232,
center_z = 69.119, size_x = 28, size_y = 28, size_z = 28, spacing = 1 angstrom. The biased gridbox
had the following parameters: center_x = 19.717, center_y = -6.232, center_z = 69.53, size_x =
22, size_y = 20, size_z = 14, spacing = 1 angstrom. Ligands were uploaded into Autodock as PDB
files. Once inputted, charges were assigned automatically. Ligand was then exported as a PDBQT
file. The PDBQT files were then coded in the configuration files labeled as ligand and receptor
with an exhaustiveness of 16. The compounds were then docked in AutoDock Vina and the
resulting log files were retained.

65
5.3 Chemistry
The 1H NMR (400 MHz) and 13C NMR (100MHz) spectra were recorded on a Bruker Ac
400 FT NMR spectrometer in deuterated chloroform (CDCl3). All chemical shifts were reported
using parts per million (ppm). Mass spectra were recorded on a JEOL spectrometer at Georgia
State University Mass Spectrometry Center. Detailed syntheses for the 2,5-Thiophene analogues
can be found in the thesis of Francisco Garcia.148
5.3.1 General Procedure for the Synthesis of the 2,6-Pyradine Analogues (2).
To a solution of pyridine-2,6-dicarbaldehyde (50 mg, 0.37 mmol) in methanol (2 mL) was
added the aniline derivative of choice (0.81 mmol) and ZnCl2 (100 mg, 0.74 mmol). The solution
was stirred for two hours at room temperature. NaBH3CN (46.5mg, 0.81 mmol) was then added
and the solution as stirred overnight. The crude product was purified by column chromatography.
2,6-Bis(anilinomethyl)pyridine, (2a) Product purified using a 1:1 Hexane/Ethyl Acetate solvent
system and was obtained in 37% yield as an off-white semi-solid. 1H NMR (400 MHz, CDCl3): δ
ppm 4.38 (s, 4H), 6.59 (d, J = 7.8 Hz, 4H), 6.65 (t, J = 7.2 Hz, 2H), 7.06 - 7.15 (m, 6 H), 7.49 (t,
J = 7.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 158.1, 148.0, 137.3, 129.3, 119.9, 117.7,
113.1, 49.3; HRMS: m/z [M + H]+ calcd for C19H19N3: 290.1657, found: 290.1657.
2,6-Bis(2-methylanilinomethyl)pyridine, (2b) The product was obtained in a 7:1 Hexanes/Ethyl
Acetate solvent system and was obtained as an orange oil in 21% yield. 1H NMR (400 MHz,
CDCl3): δ ppm 2.25 (s, 6H), 4.52 (s, 4H), 6.60 (d, J = 7.83 Hz, 2H), 6.68 (t, J = 7.33 Hz, 2H), 7.05
- 7.14 (m, 4H), 7.22 (d, J = 7.83 Hz, 2H), 7.60 (t, J = 7.71 Hz, 1H); 13C NMR (100 MHz, CDCl3):
δ ppm 158.02, 145.84, 137.26, 130.08, 127.13, 122.27, 119.99, 117.22, 110.14, 49.22, 17.59;
HRMS: m/z [M + H]+ calcd for C21H23N3: 318.1965, found: 318.1958.

66
2,6-Bis(3-methylanilinomethyl)pyridine, (2c) The product was purified via flash
chromatography using DCM as a solvent and was obtained as an orange oil in 50% yield. 1H NMR
(400 MHz, CDCl3): δ ppm 2.26 (s, 6 H) 4.42 (s, 4 H) 6.42 - 6.52 (m, 4 H) 6.54 (d, J=7.33 Hz, 2
H) 7.06 (t, J=7.58 Hz, 2 H) 7.17 (d, J=7.58 Hz, 2 H) 7.54 (t, J=7.58 Hz, 1 H); 13C NMR (100 MHz,
CDCl3): δ ppm 158.23, 148.06, 139.06, 137.26, 129.20, 119.86, 118.61, 113.95, 110.23, 49.30,
21.70. HRMS: m/z [M+ Z]+ calcd for C21H23N3 : 318.1965, found: 318.1965.
2,6-Bis(4-methylanilinomethyl)pyridine, (2d) The product was purified via flash
chromatography using DCM as a solvent and was obtained was a yellow-orange solid in 34%
yield. 1H NMR (400 MHz, CDCl3): δ ppm 2.24 (s, 6 H) 4.43 (s, 4 H) 6.59 (d, J=8.34 Hz, 4 H) 6.99
(d, J=8.08 Hz, 4 H) 7.19 (d, J=7.83 Hz, 2 H) 7.56 (t, J=7.71 Hz, 1 H); 13C NMR (100 MHz, CDCl3):
δ ppm 158.36, 145.70, 137.22, 129.78, 126.84, 119.85, 113.27, 49.64, 20.42. HRMS: m/z [M+ Z]+
calcd for C21H23N3 : 318.1965, found: 318.1962.
2,6-Bis(2-ethylanilinomethyl)pyridine, (2e) The product was purified in a 1:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 53% yield as a brown solid. 1H NMR (400 MHz,
CDCl3): δ ppm 1.31 (t, J = 7.45 Hz, 6H), 2.60 (q, J = 7.58 Hz, 4H), 4.52 (s, 4H), 6.61 (d, J = 7.83
Hz, 2H), 6.73 (t, J = 7.33 Hz, 2H), 7.06 - 7.14 (m, 4H), 7.18 - 7.25 (m, 2H), 7.58 (t, J = 7.71 Hz,
1H); 13C NMR (100 MHz, CDCl3): δ ppm 158.17, 145.32, 137.29, 127.89, 127.04, 119.98, 117.43,
110.50, 49.36, 23.98, 12.92. HRMS: m/z [M + H]+ calcd for C23H27N3: 346.2278, found: 346.2276.
2,6-Bis(3-ethylanilinomethyl)pyridine, (2f) The product was purified in a 3:1 Hexanes/Ethyl
Acetate solvent system and was obtained as an orange oil in 23% yield. 1H NMR (400 MHz,
CDCl3): δ ppm 1.18 - 1.24 (m, 6H), 2.56 (d, J = 6.32 Hz, 4H), 4.46 (br. s., 4H), 6.52 (d, J = 14.65
Hz, 4H), 6.59 (br. s., 2H), 7.02 - 7.15 (m, 2H), 7.21 (d, J = 4.04 Hz, 2H), 7.51 - 7.62 (m, 1H); 13C

67
NMR (100 MHz, CDCl3): δ ppm 158.15, 148.10, 145.45, 137.20, 129.19, 119.83, 117.36, 112.80,
110.34, 49.31, 29.02, 15.57; HRMS: m/z [M + H]+ calcd for C23H27N3: 346.2278, found: 346.2285.
2,6-Bis(4-ethylanilinomethyl)pyridine, (2g) The product was purified in a 7:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 11% yield as an orange oil; 1H NMR (400 MHz,
CDCl3): δ ppm 1.18 (t, J = 7.6 Hz, 6H), 2.50 - 2.57 (m, 4H), 4.41 (s, 4H), 6.59 (d, J = 8.3 Hz, 4H),
6.97 (d, J= 8.1 Hz, 2H), 7.01 (d, J = 8.3 Hz, 4H), 7.53 (t, J = 7.7 Hz, 1H); 13C NMR (100 MHz,
CDCl3): δ ppm 158.4, 146.0, 137.3, 133.6, 128.7, 119.9, 115.3, 113.3, 49.7, 28.0, 16.02. HRMS:
m/z [M + H]+ calcd for C23H27N3: 346.2283, found: 346.2291.
2,6-Bis(2-fluoroanilinomethyl)pyridine, (2h) The product was purified in a 3:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 7% yield as an orange oil. 1H NMR (400 MHz,
CDCl3): δ ppm 4.51 (br. s., 4H), 6.59 - 6.74 (m, 4H), 6.88 - 7.07 (m, 4H), 7.24 (d, J = 5.05 Hz,
2H), 7.61 (d, J = 6.06 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 157.81, 152.93, 137.39,
136.33, 124.56, 119.85, 116.96, 114.57, 112.50, 48.90; HRMS: m/z [M + H]+ calcd for
C19H17N3F2: 326.1463, found: 326.1451.
2,6-Bis(3-fluoroanilinomethyl)pyridine, (2i) Product was purified in a 3:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 62% yield as a brown semi-solid. 1H NMR (400 MHz,
CDCl3): δ ppm 4.34 (s, 4H), 6.21 - 6.40 (m, 6H), 6.95 - 7.06 (m, 2H), 7.10 (d, J = 7.8 Hz, 2H),
7.51 (t, J = 7.71 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 165.4, 162.9, 157.5, 149.8, 149.7,
137.4, 130.3,120.1, 109.0, 104.2, 104.0, 99.9, 99.6, 48.9; HRMS: m/z [M + H]+ calcd for
C19H17N3F2: 326.1469, found: 326.1462.
2,6-Bis(4-fluoroanilinomethyl)pyridine, (2j) Product purified using a 3:1 Hexanes/Ethyl Acetate
solvent system and was obtained in 34% yield as a brown semi-solid; 1H NMR (400 MHz, CDCl3):
δ ppm 4.33 (s, 4H), 6.51 (dd, J = 8.72, 4.17 Hz, 4H), 6.81 (t, J = 8.59 Hz, 4H), 7.12 (d, J = 7.58

68
Hz, 2 H), 7.52 (t, J = 7.71 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 158.0, 157.2, 154.8,
137.4, 137.2, 120.0,115.9, 115.7, 115.6, 115.5, 114.3, 113.5, 113.4, 49.9; HRMS: m/z [M + H]+
calcd for C19H17N3F2: 326.1469, found: 326.1462.
2,6-Bis(2-chloroanilinomethyl)pyridine, (2k) The product was purified in a 3:1 Hexane/Ethyl
Acetate solvent system and was obtained in 39% yield as a yellow solid. 1H NMR (400 MHz,
CDCl3): δ ppm 4.54 (d, J = 5.05 Hz, 4H), 6.61 - 6.69 (m, 4H), 7.10 (t, J = 7.45 Hz, 2H), 7.21 (d,
J = 7.58 Hz, 2H), 7.28 (d, J = 7.58 Hz, 2H), 7.61 (t, J = 7.71 Hz, 1H); 13C NMR (100 MHz,
CDCl3): δ ppm 157.64, 143.70, 137.42, 139.18, 137.80, 119.83, 119.49, 117.45, 111.63, 48.94.
HRMS: m/z [M + H]+ calcd for C19H17N3Cl2: 358.0872, found: 358.0877.
2,6-Bis(3-chloroanilinomethyl)pyridine, (2l) Product was purified in a 4:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 14% yield as a yellow semi-solid; 1H NMR (400 MHz,
CDCl3): δ ppm 4.36 (s, 4 H), 6.46 (dd, J = 8.1, 1.0 Hz, 2 H), 6.58 (s, 2 H) 6.61 (d, J = 7.8 Hz, 2
H), 7.01 (t, J = 8.0 Hz, 2 H), 7.12 (d, J = 7.6 Hz, 2 H), 7.54 (t, J = 7.71 Hz, 1 H); 13C NMR (100
MHz, CDCl3): δ ppm 157.4, 149.0, 137.5, 135.1, 130.3, 120.1, 117.5, 112.7, 111.4, 48.9. HRMS:
m/z [M + H]+ calcd for C19H17N3Cl2: 358.0854, found: 358.0864.
2,6-Bis(4-chloroanilinomethyl)pyridine, (2m) Product was purified in a 4:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 15% yield as a yellow solid; mp 116-118°C. 1H NMR
(400 MHz, CDCl3): δ ppm 4.36 (br, s, 4H), 6.51 (d, J = 8.34 Hz, 4H), 7.06 (d, J = 8.34 Hz, 4H),
7.12 (d, J = 7.6 Hz, 2H), 7.54 (t, J = 7.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 157.6,
146.4, 137.4, 129.1, 122.3, 120.0, 114.2, 49.2. HRMS: m/z [M + H]+ calcd for C19H17N3Cl2:
358.0872, found: 358.0864
2,6-Bis(2-methoxyanilinomethyl)pyridine, (2n) The product was purified in a 7:1 Hexane/Ethyl
Acetate solvent system and was obtained in 51% yield as a brown solid; mp 112-114°C. 1H NMR

69
(400 MHz, CDCl3): δ ppm 3.81 (s, 6 H), 4.43 (s, 4 H), 6.49 (d, J = 7.58 Hz, 2H), 6.58 - 6.64 (m,
2H), 6.70 - 6.79 (m, 4H), 7.14 (d, J = 7.8 Hz, 2H), 7.48 (t, J = 7.7 Hz, 1H); 13C NMR (100 MHz,
CDCl3): δ ppm 158.7, 147.0, 137.9, 137.3, 121.3, 119.6, 116.7, 110.3, 109.5, 55.5, 49.4. HRMS:
m/z [M + H]+ calcd for C21H23N3O2: 350.1869, found: 350.1862.
2,6-Bis(3-methoxyanilinomethyl)pyridine, (2o) The product was purified in a 3:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 16% yield as an off white semisolid. 1H NMR (400
MHz, CDCl3): δ ppm 3.76 (br. s., 6H), 4.45 (br. s., 4H), 6.23 (br. s., 2H), 6.30 (m, 4H), 7.09 (d, J
= 3.28 Hz, 2H), 7.20 (m, 2H), 7.59 (m, 1H); 13C NMR (100 MHz, CDCl3): δ ppm 160.79, 157.91,
149.30, 137.28, 130.00, 119.89, 106.18, 102.75, 99.03, 55.08, 49.19. HRMS: m/z [M + H]+ calcd
for C21H23N3O2: 350.1863, found: 350.1854.
2,6-Bis(4-methoxyanilinomethyl)pyridine, (2p) Product was purified in two silica columns. The
fist was purified in a solution of 9:1 DCM/Methanol. The compound was then further purified in
4:5 solution of Hexanes/Ethyl Acetate. Product was obtained in 30% yield as a brown solid; mp
63-65°C. 1H NMR (400 MHz, CDCl3): δ ppm 3.66 (s, 6 H), 4.33 (s, 4 H), 6.55 (d, J = 8.84 Hz, 4
H), 6.70 (d, J = 8.84 Hz, 4 H), 7.12 (d, J = 7.58 Hz, 2 H), 7.49 (t, J = 7.7 Hz, 1 H); 13C NMR (100
MHz, CDCl3): δ ppm 158.39, 152.25, 142.22, 137.20, 119.94, 114.90, 114.36, 55.79, 50.24;
HRMS: m/z [M + H]+ calcd for C21H23N3O2 : 350.1869, found: 350.1865.
2,6-Bis(2-trifluoromethylanilinomethyl)pyridine, (2q) The product was purified in a 7:1
Hexane/Ethyl Acetate solvent system and was obtained in 19% yield as an off white solid. 1H
NMR (400 MHz, CDCl3): δ ppm 4.55 (d, J = 5.05 Hz, 4H), 6.63 - 6.79 (m, 4H), 7.20 (d, J = 7.58
Hz, 2H), 7.32 (t, J = 7.71 Hz, 2H), 7.47 (d, J = 7.58 Hz, 2H), 7.61 (t, J = 7.71 Hz, 1H); 13C NMR
(100 MHz, CDCl3): δ ppm 157.32, 145.14, 137.58, 133.14, 126.72, 126.67, 119.77, 116.27,
113.66, 112.26, 48.69. HRMS: m/z [M + H]+ calcd for C21H17F6N3: 426.1401, found: 426.1399.

70
2,6-Bis(3-trifluoromethylanilinomethyl)pyridine, (2r) The product was purified in a 3:2
Hexanes/Ethyl Acetate solvent system and was obtained in 50% yield as an off white solid. 1H
NMR (400 MHz, CDCl3): δ ppm 4.46 (br. s., 4 H) 6.78 (d, J=8.08 Hz, 2 H) 6.87 (br. s., 2 H) 6.95
(d, J=7.58 Hz, 2 H) 7.19 (d, J=7.83 Hz, 2 H) 7.25 (t, J=1.00 Hz, 2 H) 7.61 (t, J=7.71 Hz, 1 H); 13C
NMR (100 MHz, CDCl3): δ ppm 157.24, 148.06, 137.51, 131.75, 129.71, 125.74, 123.03, 120.20,
116.02, 114.05, 109.23, 48.80. HRMS: m/z [M + H]+ calcd for C21H17F6N3: 426.1399, found:
426.1384.
2,6-Bis(4-trifluoromethylanilinomethyl)pyridine, (2s) The product was purified in a 2:1
Hexanes/Ethyl Acetate solvent system and was obtained in 61% yield as a light orange semi-solid.
1H NMR (400 MHz, CDCl3): δ ppm 4.49 (s, 4 H) 6.65 (d, J=8.34 Hz, 4 H) 7.20 (d, J=7.83 Hz, 2
H) 7.41 (d, J=8.34 Hz, 4 H) 7.63 (t, J=1.00 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ ppm 157.15,
150.28, 137.59, 126.68, 126.32, 123.64, 120.15, 112.21, 48.46. HRMS: m/z [M + H]+ calcd for
C21H17F6N3: 426.1399, found: 426.1381.
2,6-Bis(4-thiomethylanilinomethyl)pyridine (2t) The product was purified in a 3:1
Hexanes/Ethyl Acetate solvent system and was obtained in 14% yield as a white solid; 1H NMR
(400 MHz, CDCl3): δ ppm 2.41 (s, 6 H) 4.44 (br. s., 4 H) 6.61 (d, J=8.34 Hz, 4 H) 7.16 - 7.31 (m,
6 H) 7.59 (t, J=7.71 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ ppm 19.02, 49.12, 113.70, 119.94,
124.61, 131.34, 137.30, 146.70, 157.80. HRMS: m/z [M + H]+ calcd for C21H24N3S2: 382.1206,
found: 382.1404.
2,6-Bis(3-nitroanilinomethyl)pyridine, (2u) The product was purified using a 30:1
DCM/Methanol solvent system and was obtained in 34% yield as a yellow-orange solid; mp 147-
149°C. 1H NMR (400 MHz, CDCl3): δ ppm 4.54 (d, J = 5.05 Hz, 4H), 6.96 (d, J = 8.08 Hz, 2H),
7.24 (br s, 2H), 7.31 (t, J = 8.1 Hz, 2H), 7.49 (br s, 2 H), 7.56 (d, J = 8.08 Hz, 2H), 7.65 - 7.71 (m,

71
1H); 13C NMR (100 MHz, CDCl3): δ ppm 156.8, 148.6, 137.7, 129.8, 120.4, 119.2, 112.4, 106.6,
48.7. HRMS: m/z [M + H]+ calcd for C19H17N5O4: 380.1359, found: 380.1358.
2,6-Bis(4-nitroanilinomethyl)pyridine, (2v) The product was purified using a 10:1
DCM/Methanol soluvent system and was obtained in 10% yield as a yellow semi-solid; 1H NMR
(400 MHz, CDCl3): δ ppm 4.51 (d, J = 5.1 Hz, 4H), 6.56 (d, J = 9.1 Hz, 4H), 7.17 (d, J = 7.8 Hz,
2H), 7.64 (t, J=7.71 Hz, 1H), 8.05 (d, J=9.09 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ ppm 156.1,
152.7, 137.9, 126.4, 120.5, 111.6, 48.9. HRMS: m/z [M + H]+ calcd for C19H18N5O4: 380.1359,
found: 380.1359.
2,6-Bis(morpholinylmethyl)pyridine (2w) The product purified in a 25:1 DCM/Methanol
solvent system and was obtained was a white semi-solid in 1% yield. 1H NMR (400 MHz, CDCl3):
δ ppm 7.73 (1H t), 7.47 (2H d), 5.30 (4H s), 2.60 (8H m), 1.65 (8H m), 1.50 (4H, m); 13C NMR
(100 MHz, CDCl3): δ ppm 152.75, 137.55, 123.65, 120.96, 60.43, 63.80, 25.68, 25.11, 23.78.
HRMS: m/z [M + H]+ calcd for C15H23N3O2: 278.1863, found: 278.1852.
2,6-Bis(pyrrolidinylmethyl)pyridine (2x) The product was purified in a 5:1 DCM:Methanol
solvent system and was obtained was a orangeish oil in 9% yield. 1H NMR (400 MHz, CDCl3): δ
ppm 7.59 (t, J=7.7 Hz, 1H), 7.29 (s, 1H), 7.27 (s, 1H), 3.60 (s, 4H), 2.42-2.44 (m, 8H), 1.55-1.61
(m, 8H), 1.40-1.46 (m, 4H); 13C NMR (100 MHz, CDCl3): δ ppm 158.71, 136.67, 121.20, 65.57,
54.99, 26.20, 24.52. HRMS: m/z [M+H]+ calcd for C15H24N3: 246.1965, found: 246.1956.

72
5.3.2 General Procedure for the Synthesis of the 2,5-Furan Analogues (4)
General Procedure for the Synthesis of the 2,5-furan-based analogues (4). To a solution of
methanol, 50 mg (0.4029 mmol) of furan-2.5-dicarbaldehyde was combined in a dry vial with the
aniline derivative of choice (0.8865 mmol) and 75.963 mg (1.2088 mmol) of sodium
cyanoborohydride (NaBH3CN). The solution was stirred for five minutes at room temperature
before 164.578 mg (1.2088 mmol) of zinc chloride (ZnCl2) was added. The solution was then
stirred for two hours and purified by flash chromatography.
2,5-Bis(anilinomethyl)furan, (4a) Product was purified in a 7:1 Hexanes/Ethyl Acetate solvent
system and was obtained in 38% yield as a yellow oil. 1H NMR (400 MHz, CDCl3): δ ppm ppm
4.27 (s, 4 H) 6.15 (s, 2 H) 6.66 (d, J=7.58 Hz, 4 H) 6.74 (t, J=7.33 Hz, 2 H) 7.18 (t, J=7.96 Hz, 4
H); 13C NMR (100 MHz, CDCl3): δ ppm 152.11, 147.58, 129.22, 118.04, 113.18, 107.80, 41.49.
HRMS: m/z [M + H]+ calcd for C18H19ON2: 279.1492, found: 279.1487.
2,5-Bis(2-methylanilinomethyl)furan, (4b) The product was purified in a 13:1 solvent system
and was obtained as a yellow solid in 35% yield. 1H NMR (400 MHz, CDCl3): δ ppm 2.14 (s, 6
H) 3.84 (br s, 2 H) 4.33 (s, 4 H) 6.17 (s, 2 H) 6.63 - 6.75 (m, 4 H) 7.00 - 7.17 (m, 4 H); 13C NMR
(100 MHz, CDCl3): δ ppm 152.22, 145.56, 130.14, 127.07, 122.34, 117.61, 110.13, 107.83, 41.50,
17.49. HRMS: m/z [M + H]+ calcd for C20H23ON2 : 307.1805, found: 307.1792.
2,5-Bis(3-methylanilinomethyl)furan, (4c) The product was purified in a 3:1 Hexanes/Ethyl
Acetate solvent system and was obtained as a light orange solid in 49% yield. 1H NMR (400 MHz,
CDCl3): δ ppm2.27 (s, 6 H) 4.25 (s, 4 H) 6.13 (s, 2 H) 6.42 - 6.50 (m, 4 H) 6.56 (d, J=7.58 Hz, 2
H) 7.06 (t, J=1.00 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ ppm 152.17, 147.64, 138.98, 129.9,

73
118.97, 114.01, 110.27, 107.73, 41.53, 21.61. HRMS: m/z [M+ Z]+ calcd for C20H23ON2 :
307.1817, found: 307.1810.
2,5-Bis(4-methylanilinomethyl)furan, (4d) The product was purified in a 3:1 Hexanes/Ethyl
Acetate solvent system and was obtained as an orange solid in 49% yield. 1H NMR (400 MHz,
CDCl3): δ ppm 2.22 (s, 6 H) 4.21 (s, 4 H) 6.10 (s, 2 H) 6.56 (d, J=8.08 Hz, 4 H) 6.97 (d, J=7.83
Hz, 4 H); 13C NMR (100 MHz, CDCl3): δ ppm 152.25, 145.33, 129.68, 127.21, 113.38, 107.67,
41.82, 20.40. HRMS: m/z [M+ Z]+ calcd for C20H22ON2Na: 329.1617, found: 329.1624.
2,5-Bis(2-ethylanilinomethyl)furan, (4e) The product was purified in a 9:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 44% yield as a yellow oil. 1H NMR (400 MHz,
CDCl3): δ ppm 1.24 (br d, J=5.56 Hz, 6 H) 2.44 - 2.56 (m, 4 H) 3.94 (br s, 2 H) 4.33 (br s, 4 H)
6.16 (br s, 2 H) 6.66 - 6.78 (m, 4 H) 7.05 - 7.16 (m, 4 H); 13C NMR (100 MHz, CDCl3): δ ppm
152.28, 144.98, 128.02, 127.92, 126.93, 117.81, 110.52, 107.79, 41.59, 23.79, 12.90. HRMS: m/z
[M + H]+ calcd for C22H27ON2: 335.2118, found: 335.2103.
2,5-Bis(3-ethylanilinomethyl)furan, (4f) The product was purified in a 9:1 Hexanes/Ethyl
Acetate solvent system and was obtained as an orange oil in 42% yield. 1H NMR (400 MHz,
CDCl3): δ ppm 1.21 (br d, J=5.31 Hz, 6 H) 2.51 - 2.63 (m, 4 H) 3.93 (br s, 2 H) 4.26 (br s, 4 H)
6.14 (br s, 2 H) 6.45 - 6.54 (m, 4 H) 6.54 - 6.65 (m, 2 H) 7.05 - 7.14 (m, 2 H); 13C NMR (100
MHz, CDCl3): δ ppm 152.20, 147.70, 145.40, 129.15, 117.78, 112.93, 110.47, 107.75, 41.57,
29.00, 15.51. HRMS: m/z [M + H]+ calcd for C22H27ON2: 335.2118, found: 335.2103.
2,5-Bis(4-ethylanilinomethyl)furan, (4g) The product was purified in a 7:1 Hexanes/Ethyl
Acetate solvent system and wass obtained in 27% yield as an orange oil; 1H NMR (400 MHz,
CDCl3): δ ppm 1.18 (t, J=7.58 Hz, 6 H) 2.54 (q, J=7.41 Hz, 4 H) 4.24 (s, 4 H) 6.12 (s, 2 H) 6.60
(d, J=8.34 Hz, 4 H) 7.01 (d, J=8.08 Hz, 4 H); 13C NMR (100 MHz, CDCl3): δ ppm 152.28, 145.58,

74
133.88, 128.52, 113.35, 107.69, 41.83, 27.93, 15.91. HRMS: m/z [M + H]+ calcd for C22H27ON2:
335.2118, found: 335.2111.
2,5-Bis(2-fluoroanilinomethyl)furan, (4h) The product was purified in a 20:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 40% yield as an yellow oil. 1H NMR (400 MHz,
CDCl3): δ ppm 4.28 (br s, 4 H) 6.14 (s, 2 H) 6.59 - 6.78 (m, 4 H) 6.90 - 7.05 (m, 4 H); 13C NMR
(100 MHz, CDCl3): δ ppm 152.84, 151.84, 150.47, 136.04, 135.93, 130.41, 126.93, 124.53,
124.50, 118.58, 117.38, 117.31, 114.90, 114.40, 112.51, 107.94, 41.02. HRMS: m/z [M + H]+
calcd for C18H17ON2F2: 315.13, found: 315.1299.
2,5-Bis(3-fluoroanilinomethyl)furan, (4i) Product was purified in 9:1 Hexanes/Ethyl Acetate
solvent system and was obtained in 35% yield as an orange oil. 1H NMR (400 MHz, CDCl3): δ
ppm 4.23 (s, 4 H) 6.15 (s, 2 H) 6.34 (dd, J=11.49, 1.64 Hz, 2 H) 6.37 - 6.45 (m, 4 H) 7.05 - 7.13
(m, 2 H); 13C NMR (100 MHz, CDCl3): δ ppm 165.22, 162.80, 151.69, 149.35, 149.25, 130.35,
130.25, 109.00, 108.98, 108.07, 104.53, 104.31, 99.95, 99.69, 41.27. HRMS: m/z [M + H]+ calcd
for C18H17ON2F2: 315.1303, found: 315.1292.
2,5-Bis(4-fluoroanilinomethyl)furan, (4j) Product was purified in 3:1 Hexanes/Ethyl Acetate
solvent system and was obtained in 52% yield as an orange oil. 1H NMR (400 MHz, CDCl3): δ
4.22 (s, 4 H) 6.13 (s, 2 H) 6.53 - 6.64 (m, 4 H) 6.88 (m, J=8.70, 8.70 Hz, 4 H); 13C NMR (100
MHz, CDCl3): δ ppm 157.35, 155.00, 152.04, 143.88, 115.77, 115.55, 114.18, 114.11, 107.90,
42.10. HRMS: m/z [M + H]+ calcd for C18H17ON2F2: 315.1294, found: 315.1303.
2,5-Bis(2-chloroanilinomethyl)furan, (4k) The product was purified in a 12:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 6% yield. 1H NMR (400 MHz, CDCl3): δ ppm 4.35
(br s, 4 H) 4.67 (br s, 2 H) 6.17 (br s, 2 H) 6.63 - 6.69 (m, 2 H) 6.73 (br s, 2 H) 7.12 (br s, 2 H)
7.26 (br s, 2 H); 13C NMR (100 MHz, CDCl3): δ ppm 151.72, 143.42, 129.19, 127.76, 119.46,

75
117.89, 111.59, 107.98, 41.16. HRMS: m/z [M + H]+ calcd for C18H17ON2Cl2: 347.0712, found:
347.0509.
2,5-Bis(3-chloroanilinomethyl)furan, (4l) Product was purified in a 7:1 Hexanes/Ethyl Acetate
solvent system and was obtained in 59% yield as an orange oil. 1H NMR (400 MHz, CDCl3): δ
ppm 4.21 (4 H, s) 6.13 (2 H, s) 6.49 (2 H, d, J=8.34 Hz) 6.61 (2 H, br. s.) 6.69 (2 H, d, J=6.82 Hz);
13C NMR (100 MHz, CDCl3): δ ppm 41.18, 108.10, 111.43, 112.79, 114.89, 117.83, 130.21,
148.69, 151.67. HRMS: m/z [M + H]+ calcd for C18H17ON2Cl2: 347.0712, found: 347.0685.
2,5-Bis(4-chloroanilinomethyl)furan, (4m) Product as purified in a 7:1 Hexanes/Ethyl Acetate
solvent system and was obtained in 71% yield as an orange oil. 1H NMR (400 MHz, CDCl3): δ
ppm 4.21 (4 H, s) 6.12 (2 H, s) 6.54 (4 H, d, J=8.59 Hz) 7.09 (4 H, d, J=8.59 Hz). 13C NMR (100
MHz, CDCl3): δ ppm 41.26, 107.80, 114.06, 122.32, 28.85, 146.01, 151.69. HRMS: m/z [M + H]+
calcd for C18H17ON2Cl2: 347.0712, found: 347.0696.
2,5-Bis(2-methoxyanilinomethyl)furan, (4n) The product was purified in a 9:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 29% yield as a white solid; 1H NMR (400 MHz,
CDCl3): δ ppm 7.12 (t, J = 7.71 Hz, 2H), 7.06 (d, J = 7.07 Hz, 2H), 6.64 - 6.72 (m, 4H), 6.17 (s,
2H), 4.33 (s, 4H), 2.14 (s, 6H); 13C NMR (100 MHz, CDCl3): δ ppm 152.22, 145.56, 130.14,
127.07, 122.34, 117.61, 110.13, 107.83, 41.5, 17.49. HRMS: m/z [M + H]+ calcd for
C20H22N2O3Na: 361.1528, found: 361.1537.
2,5-Bis(3-methoxyanilinomethyl)furan, (4o) The product was purified in a 2:1 Hexane/Ethyl
Acetate solvent system and was obtained in 30% yield as an orange oil. 1H NMR (400 MHz,
CDCl3): δ ppm 7.00 - 7.13 (m, 2H), 6.19 - 6.35 (m, 6H), 6.15 (s, 2H), 4.25 (s, 4H), 3.65 - 3.79 (m,
6H); 13C NMR (100 MHz, CDCl3): δ ppm 160.74, 151.99, 148.98, 129.96, 107.87, 106.23, 103.09,
99.20, 55.07, 41.45. HRMS: m/z [M + H]+ calcd for C20H23N2O3: 339.1676, found: 339.1687.

76
2,5-Bis(4-methoxyanilinomethyl)furan, (4p) Product was purified in a 7:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 35% yield as a brown solid. 1H NMR (400 MHz,
CDCl3): δ ppm 3.74 (6 H, s) 4.22 (4 H, s) 6.13 (2 H, s) 6.63 (4 H, d, J=8.84 Hz) 6.78 (4 H, d,
J=8.84 Hz); 13C NMR (100 MHz, CDCl3): δ ppm 152.58, 152.34, 141.77, 116.42, 114.81, 107.72,
55.73, 42.52. HRMS: m/z [M + H]+ calcd for C20H23N2O3 : 339.1701, found: 339.1703.
2,5-Bis(2-trifluoromethylanilinomethyl)furan, (4q) The product was purified in a 13:1
Hexanes/Ethyl Acetate solvent system and was obtained in 9% yield as a yellow oil. 1H NMR (400
MHz, CDCl3): δ ppm 4.37 (br d, J=4.55 Hz, 4 H) 4.72 (br s, 2 H) 6.17 (s, 2 H) 6.70 - 6.88 (m, 4
H) 7.31 - 7.41 (m, 2 H) 7.45 (br d, J=7.58 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ ppm 151.48,
144.97, 133.06, 126.68, 116.66, 114.10, 112.18, 108.02, 41.06. HRMS: m/z [M + H]+ calcd for
C20H17F6N2O: 415.1240, found: 415.1246.
2,5-Bis(3-trifluoromethylanilinomethyl)furan, (4r) The product was purified in a 3:1
Hexanes/Ethyl Acetate solvent system and was obtained in 60% yield as an orange oil. 1H NMR
(400 MHz, CDCl3): δ ppm 7.20 - 7.24 (m, 2H), 6.95 - 6.98 (m, 2H), 6.86 (br. s., 2H), 6.76 - 6.80
(m, 2H), 6.17 (s, 2H), 4.29 (s, 4H); 13C NMR (100 MHz, CDCl3): δ ppm 151.62, 147.67, 129.73,
129.65, 117.97, 116.11, 114.46, 109.34, 108.23, 41.14. MS: m/z [M]+ calcd for C20H16F6N2O:
413.1080.
2,5-Bis(4-trifluoromethylanilinomethyl)furan, (4s) The product was purified in a 3:1
Hexanes/Ethyl Acetate solvent system and was obtained in 33% yield as an orange oil. 1H NMR
(400 MHz, CDCl3): δ ppm 7.41 (d, J=8.08 Hz, 4H), 6.66 (d, J=8.34 Hz, 4H), 6.18 (s, 2H), 4.32
(s, 4H); 13C NM0R (100 MHz, CDCl3): δ ppm 151.54, 149.93, 126.64, 123.53, 119.80, 112.23,
108.23, 40.94. HRMS: m/z [M+ Z]+ calcd for C20H16F6N2ONa: 437.1066, found: 437.1059.

77
2,5-Bis(4-thiomethylanilinomethyl)furan (4t) The product was purified in a 7:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 25% yield as an orange oil. 1H NMR (400 MHz,
CDCl3): δ ppm 2.40 (s, 6 H) 4.25 (s, 4 H) 6.14 (s, 2 H) 6.60 (d, J=8.59 Hz, 4 H) 7.20 (d, J=8.59
Hz, 4 H). 13C NMR (100 MHz, CDCl3): δ ppm 151.94, 146.31, 131.12, 125.17, 113.81, 107.92,
41.48, 18.87. HRMS: m/z [M + H]+ calcd for C20H23N2OS2: 371.1238, found: 371.1246.
2,5-Bis(4-methypiperazin-1-ylmethyl)furan, (4u) The product was purified in a 30:1
DCM/Methanol solvent system and was obtained as a yellow oil in 40% yield. 1H NMR (400 MHz,
CDCl3): δ ppm 6.13 (s, 2H), 3.54 (s, 4H), 2.53 (m, J=16.17 Hz, 16H), 2.30 (s, 6H); 13C NMR (100
MHz, CDCl3): δ ppm 151.09, 109.48, 54.89, 54.76, 52.48, 45.84. HRMS: m/z [M + H]+ calcd for
C16H29N4O: 293.2336, found: 293.2323.

78
5.3.3 General Procedure for the Synthesis of the 3,4-Thiophene Analogues (8)
Procedure for the Synthesis of Thiophene-3,4-dimethanol (6).
The dimethanol compound was prepared according to a literature procedure151 where 500
mg of thiophene-3,4-dicarboxylic acid (2.9mmol) was added to 10mL of dry THF in a dry round
bottom flask and was chilled to 0°C. 17.375mL (17.375mmol) of diisobutylaluminimum hydride
(139mL in 1M hexanes) was added to the flask and the solution was stirred at room temperature
for 16 hours. The reaction was quenched with methanol and water and then 25mL of HCl was
added to break up the solid chunks in the reaction. The solution as then extracted with ethyl acetate,
washed with brine and dried with MgSO4. The solution was then evaporated under reduced
pressure to give an orange oil in 77% yield.
Procedure for the Synthesis of Thiophene-3,4-dialdehyde (7). The thiophene aldehyde
was prepared using an adapted literature procedure152 where 2.0 g of Compound 6 (13.9 mmol) was
added to a 100 mL solution of dry 1,4-dioxane. 6.0 g of dry activated MnO2 (69.00 mmol) was added and
the mixture was refluxed under N2 for forty-five minutes at 96oC. The solution was filtered through a fritted
filter funnel and was concentrated under reduced pressure to yield a light yellow solid in 90% yield. 1H
NMR was used to confirm the structure with the literature characterization
General Procedure for the Synthesis of the 3,4-bis(anilino)thiophene analogues (8).
To a solution of methanol, 50mg (0.3568 mmol) of thiophene-3.4-dicarbaldehyde was combined
in a dry vial with the aniline derivative of choice (0.7849 mmol), 145.68 mg (1.070 mmol) of zinc
chloride (ZnCl2). The solution was stirred at room temperature overnight. The solution was
reduced to dryness and was then dissolved in ethanol, where 67.24 mg (1.070 mmol) of sodium

79
cyanoborohydride (NaBH3CN) was added. The solution was then stirred overnight and purified
by flash chromatography.
3,4-Bis(anilinomethyl)thiophene, (8a) The product was purified in a 7:1 Hexanes/Ethyl Acetate
solvent system and was obtained in this reaction was an brown semisolid in 4% yield. 1H NMR
(400 MHz, CDCl3): δ ppm 7.24 (2H s), 7.18 (4H t), 6.65 (4H d), 6.44 (2H d), 4.31 (4H s); 13C
NMR (100 MHz, CDCl3): δ ppm 147.94, 138.38, 129.30, 124.23, 117.98, 113.21, 42.89. HRMS:
m/z [M + H]+ calcd for C18H19N2S: 291.0956, found: 291.0963.
3,4-Bis(3-methylanilinomethyl)thiophene, (8b) The product was purified in a 20:1
Hexanes/Ethyl Acetate solvent system and was obtained as an orange solid in 26% yield. 1H NMR
(400 MHz, CDCl3): δ ppm 2.34 (6 H, s) 4.77 (4 H, s) 6.81 (2 H, d, J=7.83 Hz) 6.84 (2 H, s) 7.00
(2 H, s) 7.18 - 7.28 (4 H, m); 13C NMR (100 MHz, CDCl3): δ ppm 21.83, 49.59, 114.71, 116.91,
118.75, 120.29, 128.74, 138.62, 140.60, 151.66. HRMS: m/z [M + H]+ calcd for C20H23N2S:
323.1576, found: 323.1561.
3,4-Bis(4-methylanilinomethyl)thiophene, (8c) The product was purified in a 20:1
Hexanes/Ethyl Acetate solvent system and was obtained as an orange solid in 19% yield. 1H NMR
(400 MHz, CDCl3): δ ppm 2.33 (6 H, s) 4.77 (4 H, s) 6.90 (4 H, d, J=8.08 Hz) 7.01 (2 H, s) 7.80
(4 H, d, J=8.34 Hz); 13C NMR (100 MHz, CDCl3): δ ppm 20.82, 49.67, 114.69, 120.05, 121.71,
123.21, 129.47. HRMS: m/z [M + H]+ calcd for C20H23N2S: 323.1576, found: 323.1565.
3,4-Bis(4-ethylanilinomethyl)thiophene, (8d) The product was purified in a 15:1 Hexanes/Ethyl
Acetate solvent system and as obtained as an orange oil in 3% yield. 1H NMR (400 MHz, CDCl3):
δ ppm 1.19 (t, J = 7.45 Hz, 6 H) 2.54 (q, J = 7.58 Hz, 4 H) 4.28 (s, 4 H) 6.60 (d, J = 8.08 Hz, 4 H)
7.02 (d, J = 8.08 Hz, 4 H) 7.23 (s, 2 H); 13C NMR (100 MHz, CDCl3): δ ppm 147.94, 138.38,

80
129.30, 124.23, 117.98, 113.21, 42.89. HRMS: m/z [M + H]+ calcd for C22H27N2S: 351.1889,
found: 351.1888.
3,4-Bis(3-fluoroanilinomethyl)thiophene, (8e) The product was purified in a 7:1 Hexanes/Ethyl
Acetate solvent system and was obtained as an off-white semi-solid in 12% yield. 1H NMR (400
MHz, CDCl3): δ ppm 4.43 (br. s., 4 H) 6.21 - 6.66 (m, 8 H) 7.14 (d, J = 7.07 Hz, 2 H); 13C NMR
(100 MHz, CDCl3): δ ppm 141.68, 137.70, 130.35, 124.53, 114.69, 107.13, 104.13, 99.97, 49.13.
HRMS: m/z [M + H]+ calcd for C18H17N2SF2: 331.1061, found: 331.1075.
3,4-Bis(4-fluoroanilinomethyl)thiophene, (8f) The product was purified in a 6:1 Hexane/Ethyl
Acetate solvent system and was obtained as a yellowish semisolid in 15% yield. 1H NMR (400
MHz, CDCl3): δ ppm 4.25 (s, 4 H) 6.49 - 6.61 (m, 4 H) 6.88 (t, J = 8.72 Hz, 4 H) 7.23 (s, 2 H);
13C NMR (100 MHz, CDCl3): δ ppm 157.32, 144.26, 138.22, 124.40, 115.86, 115.64, 112.16,
43.58. HRMS: m/z [M + H]+ calcd for C18H17N2SF2: 331.1072, found: 331.1075.
3,4-Bis(3-chloroanilinomethyl)thiophene, (8g) Product was purified in a 15:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 31% yield as an off white oil. 1H NMR (400 MHz,
CDCl3): δ ppm 4.26 (4 H, s) 6.48 (2 H, dd, J=8.21, 1.39 Hz) 6.60 (2 H, s) 6.69 (2 H, dd, J=0.80
Hz) 7.06 (2 H, t, J=7.96 Hz); 13C NMR (100 MHz, CDCl3): δ ppm 42.60, 111.47, 112.76, 117.85,
124.53, 130.27, 135.05, 137.64, 148.95. HRMS: m/z [M + H]+ calcd for C18H17N2Cl2S: 363.0484,
found: 363.0466.
3,4-Bis(4-chloroanilinomethyl)thiophene, (8h) Product was purified in a 15:1 Hexanes/Ethyl
Acetate solvent system and was obtained in 23% yield as a brown solid. 1H NMR (400 MHz,
CDCl3): δ ppm 4.26 (s, 4 H) 6.54 (d, J=8.84 Hz, 4 H) 7.11 (d, J=8.59 Hz, 4 H) 7.22 (s, 2 H); 13C
NMR (100 MHz, CDCl3): δ ppm 146.39, 137.82, 129.15, 124.50, 122.67, 114.26, 42.97. HRMS:
m/z [M + H]+ calcd for C18H17N2Cl2S: 363.0408, found: 363.0466.

81
3,4-Bis(3-trifluoromethylanilinomethyl)thiophene, (8i) The product was purified in a 10:1
solvent system of Hexanes/Ethyl Acetate and was obtained in 20% yield as an orange oil. 1H NMR
(400 MHz, CDCl3): δ ppm 4.32 (s, 4 H) 6.74 (d, J=8.08 Hz, 2 H) 6.82 (br. s., 2 H) 6.89 - 7.04 (m,
2 H) 7.23 - 7.33 (m, 4 H); 13C NMR (100 MHz, CDCl3): δ ppm 147.95, 137.47, 131.48, 129.76,
125.63, 125.77, 116.09, 114.50, 109.28, 42.61 HRMS: m/z [M + H]+ calcd for C20H17F6N2S:
431.1011, found: 431.0994.
3,4-Bis(4-trifluoromethylanilinomethyl)thiophene, (8j) The product was purified in a 10:1
Hexanes/Ethyl Acetate solvent system and was obtained in 6% yield as a white semi-solid. 1H
NMR (400 MHz, CDCl3): δ ppm 4.33 (br. s., 4 H) 6.62 (d, J=8.08 Hz, 4 H) 7.25 (s, 2 H) 7.40
(d, J=8.34 Hz, 4 H); 13C NMR (100 MHz, CDCl3): δ ppm 150.17, 137.26, 126.73, 126.69, 126.66,
124.77, 112.21, 42.36. HRMS: m/z [M + H]+ calcd for C20H17F6N2S: 431.1011, found: 431.0995.

82
5.3.4 General Procedure for the Synthesis of the 2,5-Pyrazine Analogues (14)
Procedure for the Synthesis of 2,5-Dimethylpyrazine-1,4-dioxide (10)
The dioxide compound was prepared from a literature procedure152 where 20.20mL
(184.94 mmol) of 2,5-dimethyl pyrazine (9) was added to 50 mL of ethyl acetate. This solution
was combined to a solution of m-CPBA (70% purity, 405.63 mmol) dissolved in 150 mL of ethyl
acetate that had been washed with 150 mL of brine and dried over MgSO4. The mixture was stirred
at room temperature for 24 hours and a white precipitate formed. The precipitate was then filtered
and washed with 100 mL of ethyl acetate in triplicate. This compound was yielded in 90% percent
as a white solid. 1H was used to confirm structure with the literature characterization.
Procedure for the Synthesis of 2,5-Di(acetoxymethyl)pyrazine (11)
The di(acetoxymethyl) compound was prepared from a literature procedure152 where 20 g
of compound 10 (142.72 mmol) was added to 100 mL of acetic anhydride and was refluxed at
158oC for seven hours. The solution was then stirred at room temperature for twelve hours. The
resulting product was a black residue and was acquired by removing the acetic anhydride under
reduced pressure. 500 mL of diethyl ether was added to the black residue and was stirred at room
temperature for two hours. The solution was then filtered and the remaining residue was washed
with 100 mL of diethyl ether. The filtrate was condensed under reduced pressure to yield a dark
yellow residue. The residue was purified via flash column chromatography using a solution of
40% ethyl acetate and 60% hexanes. This yellow product was then recrystallized from ethyl acetate
and hexanes to yield a white solid in 16% yield. 1H NMR was used to confirm the structure with
the literature characterization.

83
Procedure for the Synthesis of 2,5-Di(hydroxymethyl)pyrazine (12)
The di(hydroxymethyl) compound was prepared from a literature procedure152 where 4.0
g of compound 11 (17.84 mmol) was suspended in 100 mL of dry methanol. 0.8 g of NaOMe
(14.80 mmol) was added. The solution was stirred at room temperature for three hours under N2.
105 g of NH4Cl was added to quench the reaction and the methanol was removed under reduced
pressure. The remaining residue was purified by flash chromatography using a solution of 10%
methanol and 90% chloroform to yield a white solid in 91% yield. 1H NMR was used to confirm
the structure with the literature characterization.
Procedure for the Synthesis of 2,5-pyrazinedicarbaldehyde (13)
The di(hydroxymethyl) compound was prepared from a literature procedure152 where 2.0
g of compound 12 (14.27 mmol) was added to a 100 mL solution of dry 1,4-dioxane. 6.0 g of dry
activated MnO2 (69.00 mmol) was added and the mixture was refluxed under N2 for forty-five
minutes at 96oC. The solution was filtered through a fritted filter funnel and was concentrated
under reduced pressure to yield a light yellow solid in 80% yield. 1H NMR was used to confirm
the structure with the literature characterization.
General Procedure for the Synthesis of the 2,5-pyrazinedicarbaldehyde analogues
(14)
To a solution of methanol, 32 mg of compound 13 (0.2351 mmol) was combined in a dry
vial with the aniline derivative of choice (0.5172 mmol) and 96.02 mg (0.7053 mmol) of ZnCl2.
The solution was stirred for two hours at room temperature before 44.32 mg (0.7053 mmol) of

84
NaBH3CN was added. The solution was then stirred overnight and purified by flash
chromatography.
3,4-Bis(anilinomethyl)pyrazine, (14a) The product was purified in a 1:1 Hexane/Ethyl Acetate
solvent system and was obtained as an orange solid in 10% yield. 1H NMR (400 MHz, CDCl3): δ
ppm 4.50 (s, 4 H) 6.68 (d, J=7.83 Hz, 4 H) 6.75 (t, J=7.33 Hz, 2 H) 7.19 (t, J=7.83 Hz, 4 H) 8.60
(s, 2 H); 13C NMR (100 MHz, CDCl3): δ ppm 152.42, 147.42, 142.71, 129.37, 118.17, 113.18,
47.08. HRMS: m/z [M + H]+ calcd for C18H19N4: 291.1604, found: 291.1599.
3,4-Bis(3-methylanilinomethyl)pyrazine, (14b) The product was purified in a 1:1 Hexanes/Ethyl
Acetate solvent system and was obtained as a brown solid in 4% yield. 1H NMR (400 MHz,
CDCl3): δ ppm 8.59 (s, 2H), 7.07 (t, J=7.71 Hz, 2H), 6.23 - 6.67 (m, 6H), 4.49 (s, 4H), 2.27 (s,
6H); 13C NMR (100 MHz, CDCl3): δ ppm 147.49, 146.34, 142.69, 139.18, 129.17, 119.47, 115.92,
112.25, 47.13, 21.44. HRMS: m/z [M + H]+ calcd for C20H23N4: 319.1917, found: 319.1909.
3,4-Bis(4-ethylanilinomethyl)pyrazine, (14b) The product was purified in a 1:1 Hexanes/Ethyl
Acetate solvent system and was obtained as a brown solid in 5% yield. 1H NMR (400 MHz,
CDCl3): δ ppm 1.18 (t, J=7.58 Hz, 6 H), 2.54 (d, J=7.58 Hz, 4 H), 4.48 (s, 4 H), 6.62 (d, J=8.08
Hz, 4 H), 7.03 (d, J=8.08 Hz, 4 H), 8.9 (s, 2 H); 13C NMR (100 MHz, CDCl3): δ ppm 152.60,
145.37, 142.72, 134.05, 128.67, 113.33, 47.45, 27.93, 15.91. HRMS: m/z [M + H]+ calcd for
C22H27N4: 347.2230, found: 347.2225.
3,4-Bis(4-fluoroanilinomethyl)pyrazine, (14d) The product was purified in a 1:1 Hexanes/Ethyl
Acetate solvent system and was obtained as a light orange solid in 5% yield. 1H NMR: δ ppm 4.46
(br. s., 4 H) 6.55 - 6.67 (m, 4 H) 6.90 (m, J=8.20, 8.20 Hz, 4 H) 8.58 (s, 2 H); 13C NMR: δ ppm

85
157.39, 155.05, 152.31, 143.71, 142.73, 115.94, 114.09, 114.02, 47.63. HRMS: m/z [M + H]+
calcd for C18H17N4F2: 327.1416, found: 327.1409.

86
REFERENCES
1. American Cancer Society., Cancer facts & figures 2014. American Cancer Society:
Atlanta, GA.
2. American Cancer Society., Global facts & figures 2nd Edition. American Cancer Society:
Atlanta, GA., 2011.
3. Sudhakar, A., History of Cancer, Ancient and Modern Treatment Methods. J Cancer Sci
Ther 2009, 1 (2), 1-4.
4. Howlader N., N. A. M., Garshell J., Miller D., Altekruse SF., Kosary C.L., Yu M., Ruhl
J., Tatalovich Z., Mariotto A., Lewis D.R., Chen H.S., Feuer E.J., Cronin K.A.,, SEER Cancer
Statistics Review. National Cancer Institute: Bethesda, MD, November 2013.
5. Hanahan, D.; Weinberg, R. A., Hallmarks of cancer: the next generation. Cell 2011, 144
(5), 646-74.
6. Kappelman, M. D.; Moore, K. R.; Allen, J. K.; Cook, S. F., Recent trends in the
prevalence of Crohn's disease and ulcerative colitis in a commercially insured US population.
Dig Dis Sci 2013, 58 (2), 519-25.
7. Molodecky, N. A.; Soon, I. S.; Rabi, D. M.; Ghali, W. A.; Ferris, M.; Chernoff, G.;
Benchimol, E. I.; Panaccione, R.; Ghosh, S.; Barkema, H. W.; Kaplan, G. G., Increasing
incidence and prevalence of the inflammatory bowel diseases with time, based on systematic
review. Gastroenterology 2012, 142 (1), 46-54 e42; quiz e30.
8. Kaplan, G. G., The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol
Hepatol 2015, 12 (12), 720-7.
9. Xavier, R. J.; Podolsky, D. K., Unravelling the pathogenesis of inflammatory bowel
disease. Nature 2007, 448 (7152), 427-34.
10. Scharl, M.; Rogler, G., Inflammatory bowel disease pathogenesis: what is new? Curr
Opin Gastroenterol 2012, 28 (4), 301-9.
11. MacDermott, R. P., Chemokines in the inflammatory bowel diseases. J Clin Immunol
1999, 19 (5), 266-72.
12. Papadakis, K. A.; Targan, S. R., The role of chemokines and chemokine receptors in
mucosal inflammation. Inflamm Bowel Dis 2000, 6 (4), 303-13.
13. Helmick, C. G.; Felson, D. T.; Lawrence, R. C.; Gabriel, S.; Hirsch, R.; Kwoh, C. K.;
Liang, M. H.; Kremers, H. M.; Mayes, M. D.; Merkel, P. A.; Pillemer, S. R.; Reveille, J. D.;
Stone, J. H.; National Arthritis Data, W., Estimates of the prevalence of arthritis and other
rheumatic conditions in the United States. Part I. Arthritis Rheum 2008, 58 (1), 15-25.
14. Scott, D. L.; Wolfe, F.; Huizinga, T. W. J., Rheumatoid arthritis. The Lancet 376 (9746),
1094-1108.
15. Alamanos, Y.; Voulgari, P. V.; Drosos, A. A., Incidence and Prevalence of Rheumatoid
Arthritis, Based on the 1987 American College of Rheumatology Criteria: A Systematic Review.
Seminars in Arthritis and Rheumatism 36 (3), 182-188.
16. Plenge , R. M.; Seielstad , M.; Padyukov , L.; Lee , A. T.; Remmers , E. F.; Ding , B.;
Liew , A.; Khalili , H.; Chandrasekaran , A.; Davies , L. R. L.; Li , W.; Tan , A. K. S.; Bonnard ,
C.; Ong , R. T. H.; Thalamuthu , A.; Pettersson , S.; Liu , C.; Tian , C.; Chen , W. V.; Carulli , J.
P.; Beckman , E. M.; Altshuler , D.; Alfredsson , L.; Criswell , L. A.; Amos , C. I.; Seldin , M.
F.; Kastner , D. L.; Klareskog , L.; Gregersen , P. K., TRAF1–C5 as a Risk Locus for

87
Rheumatoid Arthritis — A Genomewide Study. New England Journal of Medicine 2007, 357
(12), 1199-1209.
17. Gibofsky, A., Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid
arthritis. Am J Manag Care 2012, 18 (13 Suppl), S295-302.
18. Majithia, V.; Geraci, S. A., Rheumatoid Arthritis: Diagnosis and Management. The
American Journal of Medicine 120 (11), 936-939.
19. Turesson, C.; O’Fallon, W. M.; Crowson, C. S.; Gabriel, S. E.; Matteson, E. L., Extra-
articular disease manifestations in rheumatoid arthritis: incidence trends and risk factors over 46
years. Annals of the Rheumatic Diseases 2003, 62 (8), 722-727.
20. Saag, K. G.; Teng, G. G.; Patkar, N. M.; Anuntiyo, J.; Finney, C.; Curtis, J. R.; Paulus, H.
E.; Mudano, A.; Pisu, M.; Elkins-Melton, M.; Outman, R.; Allison, J. J.; Almazor, M. S.;
Bridges, S. L.; Chatham, W. W.; Hochberg, M.; Maclean, C.; Mikuls, T.; Moreland, L. W.;
O'Dell, J.; Turkiewicz, A. M.; Furst, D. E., American College of Rheumatology 2008
recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs
in rheumatoid arthritis. Arthritis Care & Research 2008, 59 (6), 762-784.
21. Horuk, R., Chemokine receptors. Cytokine Growth F R 2001, 12 (4), 313-335.
22. Choi, W. T.; Duggineni, S.; Xu, Y.; Huang, Z. W.; An, J., Drug Discovery Research
Targeting the CXC Chemokine Receptor 4 (CXCR4). J Med Chem 2012, 55 (3), 977-994.
23. Matthys, P.; Hatse, S.; Vermeire, K.; Wuyts, A.; Bridger, G.; Henson, G. W.; De Clercq,
E.; Billiau, A.; Schols, D., AMD3100, a potent and specific antagonist of the stromal cell-
derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-
gamma receptor-deficient mice. J Immunol 2001, 167 (8), 4686-4692.
24. Hendrix, C. W.; Flexner, C.; MacFarland, R. T.; Giandomenico, C.; Fuchs, E. J.;
Redpath, E.; Bridger, G.; Henson, G. W., Pharmacokinetics and safety of AMD-3100, a novel
antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrobial agents and
chemotherapy 2000, 44 (6), 1667-1673.
25. Premack, B. A.; Schall, T. J., Chemokine receptors: gateways to inflammation and
infection. Nat Med 1996, 2 (11), 1174-8.
26. Zlotnik, A.; Yoshie, O., The chemokine superfamily revisited. Immunity 2012, 36 (5),
705-16.
27. Park, W. Y.; Goodman, R. B.; Steinberg, K. P.; Ruzinski, J. T.; Radella, F., 2nd; Park, D.
R.; Pugin, J.; Skerrett, S. J.; Hudson, L. D.; Martin, T. R., Cytokine balance in the lungs of
patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001, 164 (10 Pt
1), 1896-903.
28. Zlotnik, A.; Yoshie, O., Chemokines: a new classification system and their role in
immunity. Immunity 2000, 12 (2), 121-7.
29. Balkwill, F., Cancer and the chemokine network. Nat Rev Cancer 2004, 4 (7), 540-50.
30. Loetscher, M.; Geiser, T.; O'Reilly, T.; Zwahlen, R.; Baggiolini, M.; Moser, B., Cloning
of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in
leukocytes. Journal of Biological Chemistry 1994, 269 (1), 232-237.
31. Feng, Y.; Broder, C. C.; Kennedy, P. E.; Berger, E. A., HIV-1 entry cofactor: functional
cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272 (5263),
872-7.
32. Murphy, P. M.; Baggiolini, M.; Charo, I. F.; Hebert, C. A.; Horuk, R.; Matsushima, K.;
Miller, L. H.; Oppenheim, J. J.; Power, C. A., International union of pharmacology. XXII.
Nomenclature for chemokine receptors. Pharmacol Rev 2000, 52 (1), 145-76.

88
33. Fredriksson, R.; Lagerstrom, M. C.; Lundin, L. G.; Schioth, H. B., The G-protein-coupled
receptors in the human genome form five main families. Phylogenetic analysis, paralogon
groups, and fingerprints. Mol Pharmacol 2003, 63 (6), 1256-72.
34. Chang, S. L.; Cavnar, S. P.; Takayama, S.; Luker, G. D.; Linderman, J. J., Cell, isoform,
and environment factors shape gradients and modulate chemotaxis. PLoS One 2015, 10 (4),
e0123450.
35. Hesselgesser, J.; Liang, M.; Hoxie, J.; Greenberg, M.; Brass, L. F.; Orsini, M. J.; Taub,
D.; Horuk, R., Identification and characterization of the CXCR4 chemokine receptor in human T
cell lines: ligand binding, biological activity, and HIV-1 infectivity. J Immunol 1998, 160 (2),
877-83.
36. Zlotnik, A., Involvement of chemokine receptors in organ-specific metastasis. Contrib
Microbiol 2006, 13, 191-9.
37. Busillo, J. M.; Benovic, J. L., Regulation of CXCR4 signaling. Biochim Biophys Acta
2007, 1768 (4), 952-63.
38. Zhou, N.; Luo, Z.; Luo, J.; Liu, D.; Hall, J. W.; Pomerantz, R. J.; Huang, Z., Structural
and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 co-
receptor by mutagenesis and molecular modeling studies. J Biol Chem 2001, 276 (46), 42826-33.
39. Ma, Q.; Jones, D.; Borghesani, P. R.; Segal, R. A.; Nagasawa, T.; Kishimoto, T.;
Bronson, R. T.; Springer, T. A., Impaired B-lymphopoiesis, myelopoiesis, and derailed
cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci U S A
1998, 95 (16), 9448-53.
40. McGrath, K. E.; Koniski, A. D.; Maltby, K. M.; McGann, J. K.; Palis, J., Embryonic
expression and function of the chemokine SDF-1 and its receptor, CXCR4. Dev Biol 1999, 213
(2), 442-56.
41. Tachibana, K.; Hirota, S.; Iizasa, H.; Yoshida, H.; Kawabata, K.; Kataoka, Y.; Kitamura,
Y.; Matsushima, K.; Yoshida, N.; Nishikawa, S.; Kishimoto, T.; Nagasawa, T., The chemokine
receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 1998, 393
(6685), 591-4.
42. Murphy, P. M., The molecular biology of leukocyte chemoattractant receptors. Annu Rev
Immunol 1994, 12, 593-633.
43. Miller, R. J.; Banisadr, G.; Bhattacharyya, B. J., CXCR4 signaling in the regulation of
stem cell migration and development. Journal of neuroimmunology 2008, 198 (0), 31-38.
44. Agle, K. A.; Vongsa, R. A.; Dwinell, M. B., Calcium Mobilization Triggered by the
Chemokine CXCL12 Regulates Migration in Wounded Intestinal Epithelial Monolayers. The
Journal of Biological Chemistry 2010, 285 (21), 16066-16075.
45. Zhu, B.; Xu, D.; Deng, X.; Chen, Q.; Huang, Y.; Peng, H.; Li, Y.; Jia, B.; Thoreson, W.
B.; Ding, W.; Ding, J.; Zhao, L.; Wang, Y.; Wavrin, K. L.; Duan, S.; Zheng, J., CXCL12
Enhances Human Neural Progenitor Cell Survival through a CXCR7- and CXCR4- mediated
Endocytotic Signaling Pathway. Stem cells (Dayton, Ohio) 2012, 30 (11), 2571-2583.
46. Busillo, J. M.; Benovic, J. L., Regulation of CXCR4 Signaling. Biochimica et biophysica
acta 2007, 1768 (4), 952-963.
47. Princen, K.; Hatse, S.; Vermeire, K.; De Clercq, E.; Schols, D., Evaluation of SDF-
1/CXCR4-induced Ca2+ signaling by fluorometric imaging plate reader (FLIPR) and flow
cytometry. Cytometry A 2003, 51 (1), 35-45.
48. Libura, J.; Drukala, J.; Majka, M.; Tomescu, O.; Navenot, J. M.; Kucia, M.; Marquez, L.;
Peiper, S. C.; Barr, F. G.; Janowska-Wieczorek, A.; Ratajczak, M. Z., CXCR4-SDF-1 signaling

89
is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood
2002, 100 (7), 2597-606.
49. Ryu, E. K.; Kim, T. G.; Kwon, T. H.; Jung, I. D.; Ryu, D.; Park, Y. M.; Kim, J.; Ahn, K.
H.; Ban, C., Crystal structure of recombinant human stromal cell-derived factor-1alpha. Proteins
2007, 67 (4), 1193-7.
50. Wu, B.; Chien, E. Y.; Mol, C. D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.;
Brooun, A.; Wells, P.; Bi, F. C.; Hamel, D. J.; Kuhn, P.; Handel, T. M.; Cherezov, V.; Stevens,
R. C., Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide
antagonists. Science 2010, 330 (6007), 1066-71.
51. Veldkamp, C. T.; Seibert, C.; Peterson, F. C.; De la Cruz, N. B.; Haugner, J. C., 3rd;
Basnet, H.; Sakmar, T. P.; Volkman, B. F., Structural basis of CXCR4 sulfotyrosine recognition
by the chemokine SDF-1/CXCL12. Sci Signal 2008, 1 (37), ra4.
52. Kofuku, Y.; Yoshiura, C.; Ueda, T.; Terasawa, H.; Hirai, T.; Tominaga, S.; Hirose, M.;
Maeda, Y.; Takahashi, H.; Terashima, Y.; Matsushima, K.; Shimada, I., Structural basis of the
interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled
receptor CXCR4. J Biol Chem 2009, 284 (50), 35240-50.
53. Crump, M. P.; Gong, J. H.; Loetscher, P.; Rajarathnam, K.; Amara, A.; Arenzana-
Seisdedos, F.; Virelizier, J. L.; Baggiolini, M.; Sykes, B. D.; Clark-Lewis, I., Solution structure
and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4
activation from binding and inhibition of HIV-1. EMBO J 1997, 16 (23), 6996-7007.
54. Huang, X.; Shen, J.; Cui, M.; Shen, L.; Luo, X.; Ling, K.; Pei, G.; Jiang, H.; Chen, K.,
Molecular dynamics simulations on SDF-1alpha: binding with CXCR4 receptor. Biophys J 2003,
84 (1), 171-84.
55. Xu, L.; Li, Y.; Sun, H.; Li, D.; Hou, T., Structural basis of the interactions between
CXCR4 and CXCL12/SDF-1 revealed by theoretical approaches. Mol Biosyst 2013, 9 (8), 2107-
17.
56. Gupta, S. K.; Pillarisetti, K.; Thomas, R. A.; Aiyar, N., Pharmacological evidence for
complex and multiple site interaction of CXCR4 with SDF-1α: implications for development of
selective CXCR4 antagonists. Immunology Letters 2001, 78 (1), 29-34.
57. Brelot, A.; Heveker, N.; Montes, M.; Alizon, M., Identification of residues of CXCR4
critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J Biol
Chem 2000, 275 (31), 23736-44.
58. Choi, W. T.; Tian, S.; Dong, C. Z.; Kumar, S.; Liu, D.; Madani, N.; An, J.; Sodroski, J.
G.; Huang, Z., Unique ligand binding sites on CXCR4 probed by a chemical biology approach:
implications for the design of selective human immunodeficiency virus type 1 inhibitors. J Virol
2005, 79 (24), 15398-404.
59. Roumen, L.; Scholten, D. J.; de Kruijf, P.; de Esch, I. J.; Leurs, R.; de Graaf, C., C(X)CR
in silico: Computer-aided prediction of chemokine receptor-ligand interactions. Drug Discov
Today Technol 2012, 9 (4), e281-91.
60. Kuhne, M. R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C. L.; Cao, F.;
Niekro, W.; Kempe, T.; Henning, K. A.; Cohen, L. J.; Korman, A. J.; Cardarelli, P. M., BMS-
936564/MDX-1338: A Fully Human Anti-CXCR4 Antibody Induces Apoptosis In Vitro and
Shows Antitumor Activity In Vivo in Hematologic Malignancies. Clin Cancer Res 2013, 19 (2),
357-366.
61. Furusato, B.; Mohamed, A.; Uhlen, M.; Rhim, J. S., CXCR4 and cancer. Pathol Int 2010,
60 (7), 497-505.

90
62. Jacobson, O.; Weiss, I. D., CXCR4 chemokine receptor overview: biology, pathology
and applications in imaging and therapy. Theranostics 2013, 3 (1), 1-2.
63. Choi, W. T.; Kumar, S.; Madani, N.; Han, X.; Tian, S.; Dong, C. Z.; Liu, D.; Duggineni,
S.; Yuan, J.; Sodroski, J. G.; Huang, Z.; An, J., A novel synthetic bivalent ligand to probe
chemokine receptor CXCR4 dimerization and inhibit HIV-1 entry. Biochemistry 2012, 51 (36),
7078-86.
64. De Clercq, E., The bicyclam AMD3100 story. Nat Rev Drug Discov 2003, 2 (7), 581-7.
65. Mukherjee, D.; Zhao, J., The Role of chemokine receptor CXCR4 in breast cancer
metastasis. Am J Cancer Res 2013, 3 (1), 46-57.
66. Lukacs, N. W.; Berlin, A.; Schols, D.; Skerlj, R. T.; Bridger, G. J., AMD3100, a CxCR4
antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol 2002,
160 (4), 1353-60.
67. Mikami, S.; Nakase, H.; Yamamoto, S.; Takeda, Y.; Yoshino, T.; Kasahara, K.; Ueno, S.;
Uza, N.; Oishi, S.; Fujii, N.; Nagasawa, T.; Chiba, T., Blockade of CXCL12/CXCR4 axis
ameliorates murine experimental colitis. J Pharmacol Exp Ther 2008, 327 (2), 383-92.
68. Santos, L. L.; Morand, E. F., Macrophage migration inhibitory factor: a key cytokine in
RA, SLE and atherosclerosis. Clin Chim Acta 2009, 399 (1-2), 1-7.
69. Nanki, T.; Hayashida, K.; El-Gabalawy, H. S.; Suson, S.; Shi, K.; Girschick, H. J.;
Yavuz, S.; Lipsky, P. E., Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions
play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol
2000, 165 (11), 6590-8.
70. Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M. E.; McClanahan, T.;
Murphy, E.; Yuan, W.; Wagner, S. N.; Barrera, J. L.; Mohar, A.; Verastegui, E.; Zlotnik, A.,
Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410 (6824), 50-6.
71. Choi, W. T.; Duggineni, S.; Xu, Y.; Huang, Z.; An, J., Drug discovery research targeting
the CXC chemokine receptor 4 (CXCR4). J Med Chem 2012, 55 (3), 977-94.
72. Matthys, P.; Hatse, S.; Vermeire, K.; Wuyts, A.; Bridger, G.; Henson, G. W.; De Clercq,
E.; Billiau, A.; Schols, D., AMD3100, a potent and specific antagonist of the stromal cell-
derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-
gamma receptor-deficient mice. J Immunol 2001, 167 (8), 4686-92.
73. Huang, E. H.; Singh, B.; Cristofanilli, M.; Gelovani, J.; Wei, C.; Vincent, L.; Cook, K.
R.; Lucci, A., A CXCR4 antagonist CTCE-9908 inhibits primary tumor growth and metastasis of
breast cancer. J Surg Res 2009, 155 (2), 231-6.
74. Richert, M. M.; Vaidya, K. S.; Mills, C. N.; Wong, D.; Korz, W.; Hurst, D. R.; Welch, D.
R., Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone.
Oncol Rep 2009, 21 (3), 761-7.
75. Kwong, J.; Kulbe, H.; Wong, D.; Chakravarty, P.; Balkwill, F., An antagonist of the
chemokine receptor CXCR4 induces mitotic catastrophe in ovarian cancer cells. Mol Cancer
Ther 2009, 8 (7), 1893-905.
76. Ramsey, D. M.; McAlpine, S. R., Halting metastasis through CXCR4 inhibition. Bioorg
Med Chem Lett 2013, 23 (1), 20-25.
77. Tamamis, P.; Floudas, C. A., Elucidating a key component of cancer metastasis:
CXCL12 (SDF-1alpha) binding to CXCR4. J Chem Inf Model 2014, 54 (4), 1174-88.
78. Tanegashima, K.; Suzuki, K.; Nakayama, Y.; Tsuji, K.; Shigenaga, A.; Otaka, A.; Hara,
T., CXCL14 is a natural inhibitor of the CXCL12-CXCR4 signaling axis. FEBS Lett 2013, 587
(12), 1731-5.

91
79. Cavallaro, S., CXCR4/CXCL12 in non-small-cell lung cancer metastasis to the brain. Int
J Mol Sci 2013, 14 (1), 1713-27.
80. Kucia, M.; Jankowski, K.; Reca, R.; Wysoczynski, M.; Bandura, L.; Allendorf, D. J.;
Zhang, J.; Ratajczak, J.; Ratajczak, M. Z., CXCR4-SDF-1 signalling, locomotion, chemotaxis
and adhesion. J Mol Histol 2004, 35 (3), 233-45.
81. Ma, Q.; Jones, D.; Borghesani, P. R.; Segal, R. A.; Nagasawa, T.; Kishimoto, T.;
Bronson, R. T.; Springer, T. A., Impaired B-lymphopoiesis, myelopoiesis, and derailed
cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proceedings of the National
Academy of Sciences of the United States of America 1998, 95 (16), 9448-9453.
82. Chen, H. Y.; Wang, J. M.; Wang, H. Y.; Zhang, Y. X.; Liu, W.; Pan, L.; Wang, W. H.;
Chen, S. F.; Jin, W. G.; Wang, L., Effect of short hairpin RNA-induced CXCR4 silence on
ovarian cancer cell. Biomed Pharmacother 2012, 66 (7), 549-53.
83. Hirota, K.; Semenza, G. L., Regulation of angiogenesis by hypoxia-inducible factor 1.
Crit Rev Oncol Hematol 2006, 59 (1), 15-26.
84. Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S. K.; Doni, A.; Rapisarda, A.;
Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; Mantovani, A.; Melillo, G.; Sica, A.,
Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med 2003, 198 (9), 1391-402.
85. Staller, P.; Sulitkova, J.; Lisztwan, J.; Moch, H.; Oakeley, E. J.; Krek, W., Chemokine
receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 2003,
425 (6955), 307-11.
86. Zagzag, D.; Krishnamachary, B.; Yee, H.; Okuyama, H.; Chiriboga, L.; Ali, M. A.;
Melamed, J.; Semenza, G. L., Stromal cell-derived factor-1alpha and CXCR4 expression in
hemangioblastoma and clear cell-renal cell carcinoma: von Hippel-Lindau loss-of-function
induces expression of a ligand and its receptor. Cancer Res 2005, 65 (14), 6178-88.
87. Fernandis, A. Z.; Prasad, A.; Band, H.; Klosel, R.; Ganju, R. K., Regulation of CXCR4-
mediated chemotaxis and chemoinvasion of breast cancer cells. Oncogene 2004, 23 (1), 157-67.
88. Janowska-Wieczorek, A.; Marquez, L. A.; Dobrowsky, A.; Ratajczak, M. Z.; Cabuhat,
M. L., Differential MMP and TIMP production by human marrow and peripheral blood CD34(+)
cells in response to chemokines. Exp Hematol 2000, 28 (11), 1274-85.
89. Samara, G. J.; Lawrence, D. M.; Chiarelli, C. J.; Valentino, M. D.; Lyubsky, S.; Zucker,
S.; Vaday, G. G., CXCR4-mediated adhesion and MMP-9 secretion in head and neck squamous
cell carcinoma. Cancer Lett 2004, 214 (2), 231-41.
90. Spiegel, A.; Kollet, O.; Peled, A.; Abel, L.; Nagler, A.; Bielorai, B.; Rechavi, G.;
Vormoor, J.; Lapidot, T., Unique SDF-1-induced activation of human precursor-B ALL cells as a
result of altered CXCR4 expression and signaling. Blood 2004, 103 (8), 2900-7.
91. Nabeshima, K.; Inoue, T.; Shimao, Y.; Sameshima, T., Matrix metalloproteinases in
tumor invasion: role for cell migration. Pathol Int 2002, 52 (4), 255-64.
92. Debnath, B.; Xu, S.; Grande, F.; Garofalo, A.; Neamati, N., Small molecule inhibitors of
CXCR4. Theranostics 2013, 3 (1), 47-75.
93. Teicher, B. A.; Fricker, S. P., CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer
Res 2010, 16 (11), 2927-31.
94. Campbell, J. J.; Hedrick, J.; Zlotnik, A.; Siani, M. A.; Thompson, D. A.; Butcher, E. C.,
Chemokines and the arrest of lymphocytes rolling under flow conditions. Science 1998, 279
(5349), 381-4.

92
95. Glodek, A. M.; Honczarenko, M.; Le, Y.; Campbell, J. J.; Silberstein, L. E., Sustained
activation of cell adhesion is a differentially regulated process in B lymphopoiesis. J Exp Med
2003, 197 (4), 461-73.
96. Wright, N.; Hidalgo, A.; Rodriguez-Frade, J. M.; Soriano, S. F.; Mellado, M.; Parmo-
Cabanas, M.; Briskin, M. J.; Teixido, J., The chemokine stromal cell-derived factor-1 alpha
modulates alpha 4 beta 7 integrin-mediated lymphocyte adhesion to mucosal addressin cell
adhesion molecule-1 and fibronectin. J Immunol 2002, 168 (10), 5268-77.
97. Howlader, N. N., A. M.; Krapcho, M.; Garshell, J.; Miller, D.; Altekruse, S. F.; Kosary,
C. L.; Yu, M.; Ruhl, J.; Tatalovich, Z,; Mariotto, A.; Lewis, D. R.; Chen, H. S.; Feuer, E. J.;
Cronin, K. A.; editors. SEER Cancer Statistics Review, 1975-2012; 2014.
98. Kohler, B. A.; Sherman, R. L.; Howlader, N.; Jemal, A.; Ryerson, A. B.; Henry, K. A.;
Boscoe, F. P.; Cronin, K. A.; Lake, A.; Noone, A. M.; Henley, S. J.; Eheman, C. R.; Anderson,
R. N.; Penberthy, L., Annual Report to the Nation on the Status of Cancer, 1975-2011, Featuring
Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J Natl Cancer Inst
2015, 107 (6), djv048.
99. Dotan, I.; Werner, L.; Vigodman, S.; Weiss, S.; Brazowski, E.; Maharshak, N.; Chen, O.;
Tulchinsky, H.; Halpern, Z.; Guzner-Gur, H., CXCL12 is a constitutive and inflammatory
chemokine in the intestinal immune system. Inflamm Bowel Dis 2010, 16 (4), 583-92.
100. Katsuta, T.; Lim, C.; Shimoda, K.; Shibuta, K.; Mitra, P.; Banner, B. F.; Mori, M.;
Barnard, G. F., Interleukin-8 and SDF1-alpha mRNA expression in colonic biopsies from
patients with inflammatory bowel disease. Am J Gastroenterol 2000, 95 (11), 3157-64.
101. Agace, W. W.; Amara, A.; Roberts, A. I.; Pablos, J. L.; Thelen, S.; Uguccioni, M.; Li, X.
Y.; Marsal, J.; Arenzana-Seisdedos, F.; Delaunay, T.; Ebert, E. C.; Moser, B.; Parker, C. M.,
Constitutive expression of stromal derived factor-1 by mucosal epithelia and its role in HIV
transmission and propagation. Curr Biol 2000, 10 (6), 325-8.
102. Dwinell, M. B.; Ogawa, H.; Barrett, K. E.; Kagnoff, M. F., SDF-1/CXCL12 regulates
cAMP production and ion transport in intestinal epithelial cells via CXCR4. Am J Physiol
Gastrointest Liver Physiol 2004, 286 (5), G844-50.
103. Smith, J. M.; Johanesen, P. A.; Wendt, M. K.; Binion, D. G.; Dwinell, M. B., CXCL12
activation of CXCR4 regulates mucosal host defense through stimulation of epithelial cell
migration and promotion of intestinal barrier integrity. Am J Physiol Gastrointest Liver Physiol
2005, 288 (2), G316-26.
104. Werner, L.; Elad, H.; Brazowski, E.; Tulchinsky, H.; Vigodman, S.; Kopylov, U.;
Halpern, Z.; Guzner-Gur, H.; Dotan, I., Reciprocal regulation of CXCR4 and CXCR7 in
intestinal mucosal homeostasis and inflammatory bowel disease. J Leukoc Biol 2011, 90 (3),
583-90.
105. Kohem, C. L.; Brezinschek, R. I.; Wisbey, H.; Tortorella, C.; Lipsky, P. E.;
Oppenheimer-Marks, N., Enrichment of differentiated CD45RBdim,CD27- memory T cells in
the peripheral blood, synovial fluid, and synovial tissue of patients with rheumatoid arthritis.
Arthritis Rheum 1996, 39 (5), 844-54.
106. Morimoto, C.; Romain, P. L.; Fox, D. A.; Anderson, P.; DiMaggio, M.; Levine, H.;
Schlossman, S. F., Abnormalities in CD4+ T-lymphocyte subsets in inflammatory rheumatic
diseases. Am J Med 1988, 84 (5), 817-25.
107. De Klerck, B.; Geboes, L.; Hatse, S.; Kelchtermans, H.; Meyvis, Y.; Vermeire, K.;
Bridger, G.; Billiau, A.; Schols, D.; Matthys, P., Pro-inflammatory properties of stromal cell-

93
derived factor-1 (CXCL12) in collagen-induced arthritis. Arthritis Res Ther 2005, 7 (6), R1208-
20.
108. Tamamura, H.; Fujisawa, M.; Hiramatsu, K.; Mizumoto, M.; Nakashima, H.; Yamamoto,
N.; Otaka, A.; Fujii, N., Identification of a CXCR4 antagonist, a T140 analog, as an anti-
rheumatoid arthritis agent. FEBS Lett 2004, 569 (1-3), 99-104.
109. Zhang, L.; Kajiwara, H.; Kuboyama, N.; Abiko, Y., Reduction of CXCR4 expression in
Rheumatoid Arthritis Rat Joints by low level diode laser irradiation. Laser Therapy 2011, 20 (1),
53-58.
110. Wang, A.; Guilpain, P.; Chong, B. F.; Chouzenoux, S.; Guillevin, L.; Du, Y.; Zhou, X. J.;
Lin, F. M.; Fairhurst, A. M.; Boudreaux, C.; Roux, C.; Wakeland, E. K.; Davis, L. S.; Batteux,
F.; Mohan, C., Dysregulated Expression of CXCR4/CXCL12 in Subsets of Patients With
Systemic Lupus Erythematosus. Arthritis Rheum-Us 2010, 62 (11), 3436-3446.
111. Chong, B. F.; Mohan, C., Targeting the CXCR4/CXCL12 axis in systemic lupus
erythematosus. Expert Opin Ther Targets 2009, 13 (10), 1147-53.
112. Huang, E. H.; Singh, B.; Cristofanilli, M.; Gelovani, J.; Wei, C. M.; Vincent, L.; Cook,
K. R.; Lucci, A., A CXCR4 Antagonist CTCE-9908 Inhibits Primary Tumor Growth and
Metastasis of Breast Cancer. J Surg Res 2009, 155 (2), 231-236.
113. Richert, M. M.; Vaidya, K. S.; Mills, C. N.; Wong, D.; Korz, W.; Hurst, D. R.; Welch, D.
R., Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone.
Oncol Rep 2009, 21 (3), 761-767.
114. Kwong, J.; Kulbe, H.; Wong, D.; Chakravarty, P.; Balkwill, F., An antagonist of the
chemokine receptor CXCR4 induces mitotic catastrophe in ovarian cancer cells. Mol Cancer
Ther 2009, 8 (7), 1893-1905.
115. Ueda, S.; Kato, M.; Inuki, S.; Ohno, H.; Evans, B.; Wang, Z. X.; Peiper, S. C.; Izumi, K.;
Kodama, E.; Matsuoka, M.; Nagasawa, H.; Oishi, S.; Fujii, N., Identification of novel non-
peptide CXCR4 antagonists by ligand-based design approach. Bioorg Med Chem Lett 2008, 18
(14), 4124-9.
116. Inokuchi, E.; Oishi, S.; Kubo, T.; Ohno, H.; Shimura, K.; Matsuoka, M.; Fujii, N., Potent
CXCR4 antagonists containing amidine type Peptide bond isosteres. ACS Med Chem Lett 2011,
2 (6), 477-80.
117. Kim, J.; Yip, M. L.; Shen, X.; Li, H.; Hsin, L. Y.; Labarge, S.; Heinrich, E. L.; Lee, W.;
Lu, J.; Vaidehi, N., Identification of anti-malarial compounds as novel antagonists to chemokine
receptor CXCR4 in pancreatic cancer cells. PLoS One 2012, 7 (2), e31004.
118. Yen, C. F.; Hu, C. K.; Chou, M. C.; Tseng, C. T.; Wu, C. H.; Huang, Y. H.; Chen, S. J.;
King, C. H. R., Pyrimidine compounds. Google Patents: 2009.
119. De Clercq, E., The AMD3100 story: the path to the discovery of a stem cell mobilizer
(Mozobil). Biochem Pharmacol 2009, 77 (11), 1655-64.
120. Zhan, W.; Liang, Z.; Zhu, A.; Kurtkaya, S.; Shim, H.; Snyder, J. P.; Liotta, D. C.,
Discovery of small molecule CXCR4 antagonists. J Med Chem 2007, 50 (23), 5655-64.
121. Lefrancois, M.; Lefebvre, M. R.; Saint-Onge, G.; Boulais, P. E.; Lamothe, S.; Leduc, R.;
Lavigne, P.; Heveker, N.; Escher, E., Agonists for the Chemokine Receptor CXCR4. ACS Med
Chem Lett 2011, 2 (8), 597-602.
122. Scozzafava, A.; Mastrolorenzo, A.; Supuran, C. T., Non-peptidic chemokine receptors
antagonists as emerging anti-HIV agents. J Enzyme Inhib Med Chem 2002, 17 (2), 69-76.
123. Schwarz, M. K.; Wells, T. N., New therapeutics that modulate chemokine networks. Nat
Rev Drug Discov 2002, 1 (5), 347-58.

94
124. Hendrix, C. W.; Flexner, C.; MacFarland, R. T.; Giandomenico, C.; Fuchs, E. J.;
Redpath, E.; Bridger, G.; Henson, G. W., Pharmacokinetics and safety of AMD-3100, a novel
antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents
Chemother 2000, 44 (6), 1667-73.
125. Liles, W. C.; Rodger, E.; Broxmeyer, H. E.; Dehner, C.; Badel, K.; Calandra, G.;
Christensen, J.; Wood, B.; Price, T. H.; Dale, D. C., Augmented mobilization and collection of
CD34+ hematopoietic cells from normal human volunteers stimulated with granulocyte-colony-
stimulating factor by single-dose administration of AMD3100, a CXCR4 antagonist. Transfusion
2005, 45 (3), 295-300.
126. Hatse, S.; Princen, K.; Bridger, G.; De Clercq, E.; Schols, D., Chemokine receptor
inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett 2002, 527 (1-3), 255-62.
127. Zhu, A.; Zhan, W.; Liang, Z.; Yoon, Y.; Yang, H.; Grossniklaus, H. E.; Xu, J.; Rojas, M.;
Lockwood, M.; Snyder, J. P.; Liotta, D. C.; Shim, H., Dipyrimidine amines: a novel class of
chemokine receptor type 4 antagonists with high specificity. J Med Chem 2010, 53 (24), 8556-
68.
128. Portella, L.; Vitale, R.; De Luca, S.; D'Alterio, C.; Ierano, C.; Napolitano, M.; Riccio, A.;
Polimeno, M. N.; Monfregola, L.; Barbieri, A.; Luciano, A.; Ciarmiello, A.; Arra, C.; Castello,
G.; Amodeo, P.; Scala, S., Preclinical development of a novel class of CXCR4 antagonist
impairing solid tumors growth and metastases. PLoS One 2013, 8 (9), e74548.
129. Silverman, R. B., The organic chemistry of drug design and drug action. 2nd ed.;
Elsevier Academic Press: Amsterdam ; Boston, 2004; p xix, 617 p., 12 p. of plates.
130. Zhan, W. Q.; Liang, Z. X.; Zhu, A. Z.; Kurtkaya, S.; Shim, H.; Snyder, J. P.; Liotta, D.
C., Discovery of small molecule CXCR4 antagonists. Journal of Medicinal Chemistry 2007, 50
(23), 5655-5664.
131. Liang, Z.; Zhan, W.; Zhu, A.; Yoon, Y.; Lin, S.; Sasaki, M.; Klapproth, J. M.; Yang, H.;
Grossniklaus, H. E.; Xu, J.; Rojas, M.; Voll, R. J.; Goodman, M. M.; Arrendale, R. F.; Liu, J.;
Yun, C. C.; Snyder, J. P.; Liotta, D. C.; Shim, H., Development of a unique small molecule
modulator of CXCR4. PLoS One 2012, 7 (4), e34038.
132. Lipinski, C. A., Drug-like properties and the causes of poor solubility and poor
permeability. Journal of Pharmacological and Toxicological Methods 2000, 44 (1), 235-249.
133. Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J., Experimental and
computational approaches to estimate solubility and permeability in drug discovery and
development settings. Adv Drug Deliv Rev 2001, 46 (1-3), 3-26.
134. Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J., Experimental and
computational approaches to estimate solubility and permeability in drug discovery and
development settings. Advanced Drug Delivery Reviews 1997, 23 (1), 3-25.
135. Ghose, A. K.; Viswanadhan, V. N.; Wendoloski, J. J., A knowledge-based approach in
designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and
quantitative characterization of known drug databases. J Comb Chem 1999, 1 (1), 55-68.
136. Molinspiration Calculation of Molecular Properties. http://www.molinspiration.com/
(accessed March 15, 2017).
137. De Clercq, E.; Yamamoto, N.; Pauwels, R.; Balzarini, J.; Witvrouw, M.; De Vreese, K.;
Debyser, Z.; Rosenwirth, B.; Peichl, P.; Datema, R.; et al., Highly potent and selective inhibition
of human immunodeficiency virus by the bicyclam derivative JM3100. Antimicrob Agents
Chemother 1994, 38 (4), 668-74.

95
138. Hansch, C.; Leo, A.; Hoekman, D. H., Exploring QSAR. American Chemical Society:
Washington, DC, 1995.
139. Wishart, D. S.; Jewison, T.; Guo, A. C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.;
Mandal, R.; Aziat, F.; Dong, E.; Bouatra, S.; Sinelnikov, I.; Arndt, D.; Xia, J.; Liu, P.; Yallou,
F.; Bjorndahl, T.; Perez-Pineiro, R.; Eisner, R.; Allen, F.; Neveu, V.; Greiner, R.; Scalbert, A.,
HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Research 2013, 41 (D1),
D801-D807.
140. Wishart, D. S.; Knox, C.; Guo, A. C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D. D.;
Psychogios, N.; Dong, E.; Bouatra, S.; Mandal, R.; Sinelnikov, I.; Xia, J.; Jia, L.; Cruz, J. A.;
Lim, E.; Sobsey, C. A.; Shrivastava, S.; Huang, P.; Liu, P.; Fang, L.; Peng, J.; Fradette, R.;
Cheng, D.; Tzur, D.; Clements, M.; Lewis, A.; De Souza, A.; Zuniga, A.; Dawe, M.; Xiong, Y.;
Clive, D.; Greiner, R.; Nazyrova, A.; Shaykhutdinov, R.; Li, L.; Vogel, H. J.; Forsythe, I.,
HMDB: a knowledgebase for the human metabolome. Nucleic Acids Research 2009, 37
(suppl_1), D603-D610.
141. Wishart, D. S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A. C.; Young, N.; Cheng, D.; Jewell,
K.; Arndt, D.; Sawhney, S.; Fung, C.; Nikolai, L.; Lewis, M.; Coutouly, M.-A.; Forsythe, I.;
Tang, P.; Shrivastava, S.; Jeroncic, K.; Stothard, P.; Amegbey, G.; Block, D.; Hau, D. D.;
Wagner, J.; Miniaci, J.; Clements, M.; Gebremedhin, M.; Guo, N.; Zhang, Y.; Duggan, G. E.;
MacInnis, G. D.; Weljie, A. M.; Dowlatabadi, R.; Bamforth, F.; Clive, D.; Greiner, R.; Li, L.;
Marrie, T.; Sykes, B. D.; Vogel, H. J.; Querengesser, L., HMDB: the Human Metabolome
Database. Nucleic Acids Research 2007, 35 (suppl_1), D521-D526.
142. Altaf, A. A. S., A.; Gul, Z.; Rasool, N.; Badshah, A.; Lal, B.; Khan E., A Review on the
Medicinal Importance of Pyridine Derivatives. Journal of Drug Design and Medicinal Chemistry
2015, 1 (1), 1-11.
143. Mishra, R.; Jha, K.; Kumar, S.; Tomer, I., Synthesis, properties and biological activity of
thiophene: A review. Pharma Chemica 2011, 3 (4), 38-54.
144. Banerjee, R., Kumar, K. H. S., Banerjee, M., Medicinal significance of furan derivatives:
A review. Int. J. Rev. Life. Sci. 2012, 2 (1), 7-16.
145. Asif, M., Piperazine and pyrazine containing molecules and their diverse
pharmacological activities. International Journal of Advances in Scientific Research 2015, 1 (1),
5-11.
146. Tian, S.; Choi, W. T.; Liu, D.; Pesavento, J.; Wang, Y.; An, J.; Sodroski, J. G.; Huang,
Z., Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived
factor 1alpha on CXCR4 transmembrane helical domains. J Virol 2005, 79 (20), 12667-73.
147. Bai, R.; Liang, Z.; Yoon, Y.; Liu, S.; Gaines, T.; Oum, Y.; Shi, Q.; Mooring, S. R.; Shim,
H., Symmetrical bis-tertiary amines as novel CXCR4 inhibitors. Eur J Med Chem 2016, 118,
340-350.
148. Garcia, F. Synthesis & evaluation of thiophene derivatives as CXCR4 antagonists.
Georgia State University, 2016.
149. Trott, O.; Olson, A. J., AutoDock Vina: improving the speed and accuracy of docking
with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010,
31 (2), 455-61.
150. Mooring, S. R.; Liu, J.; Liang, Z.; Ahn, J.; Hong, S.; Yoon, Y.; Snyder, J. P.; Shim, H.,
Benzenesulfonamides: a unique class of chemokine receptor type 4 inhibitors. ChemMedChem
2013, 8 (4), 622-32.

96
151. Douglas, J. D.; Griffini, G.; Holcombe, T. W.; Young, E. P.; Lee, O. P.; Chen, M. S.;
Frechet, J. M. J., Functionalized Isothianaphthene Monomers That Promote Quinoidal Character
in Donor-Acceptor Copolymers for Organic Photovoltaics. Macromolecules 2012, 45 (10), 4069-
4074.
152. Das, S. K.; Frey, J., Regioselective double Boekelheide reaction: first synthesis of 3,6-
dialkylpyrazine-2,5-dicarboxaldehydes from DL-alanine. Tetrahedron Lett 2012, 53 (30), 3869-
3872.

97
APPENDICES
NMR Spectra
1H NMR Spectra
TG12-1 FINAL PROTON COPY.ESP
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
4.004.002.086.021.00
M04(d)
M05(s)
M03(t)
M01(t)
M02(m)
7.5
17
.49
7.4
77
.13
7.1
17
.09
6.6
76
.65
6.6
06
.58
4.3
8
1H NMR Spectrum for Compound 2a

98
TG99 proton.esp
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
6.204.192.002.054.001.911.05
M02(d)
M07(m)
M03(t)
M04(d)
M05(s)
M06(s)
M01(t)
7.6
2 7.6
07
.58
7.2
3
7.1
07
.08
6.7
06
.68
6.6
16
.59
4.5
2
2.2
5
1H NMR Spectrum for Compound 2b
99
TG34 last spot proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
6.003.506.052.001.711.06
M03(t)
M02(d)
M07(s)
M04(d)
M06(m)
M05(s)
M01(t)
7.5
6 7.5
47
.52
7.1
8
7.0
67
.04
6.5
5
6.4
96
.47
6.4
5
4.4
2
2.2
6
1H NMR Spectrum for Compound 2c

100
TG32-1 Third spot orange oiL PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70
No
rma
lize
d I
nte
nsity
6.003.153.263.601.640.92
M02(d)
M03(d)
M05(s)
M04(d)
M06(s)
M01(t)
7.5
8 7.5
67
.54
7.2
07
.00
6.9
8
6.6
06
.58
4.4
3
2.2
4
1H NMR Spectrum for Compound 2d

101
TG82PROTON.esp
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
6.073.734.071.821.853.692.041.00
M06(m)
M05(d)
M07(m)
M04(t)
M03(t)
M08(t)
M02(q)
M01(s)7
.66
7.6
4
7.2
87
.27
7.1
87
.16
7.1
4
6.8
16
.79
6.6
86
.66
4.5
8
2.6
92
.67
2.6
62
.64
1.3
91
.37
1.3
5
1H NMR Spectrum for Compound 2e

102
TG100.001.001.1r.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
No
rma
lize
d I
nte
nsity
6.004.073.375.871.460.68
M06(m)
M03(d)
M02(m)
M05(d)M04(m)
M01(br. s.)
7.5
87
.57
7.5
6
7.2
1 7.2
0 6.5
96
.53
6.5
0
4.4
6
2.5
72
.55
1.2
11
.21
1.2
0
1H NMR Spectrum for Compound 2f

103
TG22-4 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
6.004.053.033.833.680.82
M03(d)
M04(s)M06(m)
M05(m)M02(m)
TMS
M01(t)
7.5
57
.53
7.5
1
7.0
06
.98
6.6
06
.58
4.4
1
2.5
42
.52
2.5
12
.51
1.1
81
.16
1H NMR Spectrum for Compound 2g

104
TG98-2 dirty PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
3.733.603.672.221.00
M04(d)
M05(d) M03(m)
M01(br. s.)M02(br. s.)
7.6
2 7.2
47
.23
6.9
96
.97
6.9
5 6.6
6
4.5
1
1H NMR Spectrum for Compound 2h

105
TG13-1 Final proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
4.005.931.971.970.99
M03(d)
M05(s)
M04(m)
M01(t)
M02(m)
7.5
37
.51
7.4
9
7.1
17
.09
7.0
27
.00
6.9
8
6.3
56
.35
6.3
36
.32
6.3
16
.27
6.2
4
4.3
4
1H NMR Spectrum for Compound 2i

106
TG7-1 Final Proton Copy.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
3.923.933.931.881.00
M02(d)
M04(dd)
M05(s)
M01(t)
M03(t)
7.5
2
7.1
3
6.8
1
6.5
2
4.3
3
1H NMR Spectrum for Compound 2j

107
TG83 PROTON.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
4.003.901.991.972.001.00
M03(d)
M02(d)
M05(m)
M06(d)
M01(t)
M04(t)
7.6
37
.61
7.5
97
.29
7.2
77
.22
7.2
07
.10
7.0
8 6.6
66
.65
6.6
2
4.5
54
.54
1H NMR Spectrum for Compound 2k

108
TG16-1 final proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
4.052.001.951.961.991.960.98
M06(s)
M05(d)
M03(d)
M02(t)
M04(t)
M07(dd)
M01(s)
7.5
67
.54
7.5
2
7.1
3 7.0
16
.99
6.6
2
6.5
86
.47
6.4
56
.45
4.3
6
1H NMR Spectrum for Compound 2l

109
TG17-1 final proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
4.003.973.842.021.00
M02(d)
M03(d)
M05(br. s.)
M01(t)
M04(d)7
.56
7.5
47
.52
7.1
37
.07 7.0
5
6.5
26
.50
4.3
6
1H NMR Spectrum for Compound 2m

110
TG24 proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
6.193.662.001.973.992.051.03
M02(s)
M06(m)M04(m)
M01(s)
M05(m)M03(t)
M07(d)
7.4
8
7.1
67
.15
7.1
3
6.7
7
6.7
5 6.7
36
.72
6.6
16
.50
6.4
8
4.4
3
3.8
1
1H NMR Spectrum for Compound 2n

111
TG93 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
5.653.632.003.652.041.680.90
M02(br. s.)
M07(br. s.)
M05(m)
M06(br. s.)
M04(m)
M03(m)
M01(br. s.)
7.5
9
7.2
27
.09
6.3
16
.24
4.4
5
3.7
6
1H NMR Spectrum for Compound 2o

112
TG14-2 Final proton.esp
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
6.023.483.543.931.750.93
M05(s)
M06(s)
M04(d)
M02(d)
M03(d)
M01(t)
7.5
1 7.4
97
.47
7.1
37
.11
6.7
26
.69
6.5
66
.54
4.3
3
3.6
6
1H NMR Spectrum for Compound 2p

113
TG90 PROTON.esp
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
4.094.001.952.022.011.03
M06(d)
M03(t)
M04(d)
M05(t)
M07(m)
M01(d)
7.6
37
.61
7.4
87
.46
7.3
07
.21
7.1
9
6.7
56
.73 6
.70
6.6
8
4.5
54
.54
1H NMR Spectrum for Compound 2q

114
TG29 proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9N
orm
aliz
ed
In
tensity
4.002.041.991.981.961.970.97
M05(d)
M06(t)
M04(br. s.)
M02(d)
M03(d)
M01(t)
M07(br. s.)
7.6
37
.61
7.5
9 7.2
77
.25 7
.20
7.1
86
.94
6.8
76
.79
6.7
7
4.4
6
1H NMR Spectrum for Compound 2r

115
TG28 2nd Spot PROTON.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8N
orm
aliz
ed
In
tensity
4.004.121.934.071.11
M03(d)
M01(t)
M02(d)
M05(s)
M04(d)
7.6
57
.63
7.4
27
.40
7.2
17
.19
6.6
66
.64
4.4
9
1H NMR Spectrum for Compound 2s

116
TG37 dark spot 2.001.001.1r.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
No
rma
lize
d I
nte
nsity
6.034.084.076.001.03
M05(m)
M01(t)
M03(br. s.)M02(d)
M04(s)
7.6
1 7.5
9
7.2
57
.23
7.2
17
.18
6.6
26
.60
4.4
4
2.4
1
1H NMR Spectrum for Compound 2t

117
TG19-2 PROTON Final.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
No
rma
lize
d I
nte
nsity
4.012.081.702.252.022.091.11
M03(br. s.)
M05(br. s.)M07(d)
M02(d)
M06(d)
M01(m)
M04(t)
TMS
7.6
87
.57
7.5
57
.49
7.3
3 7.3
17
.24
6.9
76
.95
4.5
44
.53
1H NMR Spectrum for Compound 2u

118
TG18-1 proton final.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70N
orm
aliz
ed
In
tensity
4.004.711.811.244.60
M04(d)
M05(d)
M01(t)
M03(d)
M02(d)
8.0
68
.04
7.6
57
.64
7.6
2
7.1
87
.16
6.5
76
.54
4.5
14
.50
1H NMR Spectrum for Compound 2v

119
TG33 proton.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70
0.75N
orm
aliz
ed
In
tensity
7.954.408.001.831.05
M05(m)
M04(t)
M02(d)
M01(t)
M03(s)
7.6
5 7.6
37
.61
7.3
37
.31
3.7
53
.73
3.6
7
2.5
32
.52
1H NMR Spectrum for Compound 2w

120
TG39 PROTON.esp
16 14 12 10 8 6 4 2 0 -2 -4
Chemical Shift (ppm)
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.10
0.11N
orm
aliz
ed
In
tensity
8.048.944.272.221.12
M05(t)
M02(m)
M03(s)
M01(t)
M04(t)
7.6
3 7.6
1
7.2
6
3.7
8
2.5
8
1.8
0
1H NMR Spectrum for Compound 2x

121
TG50 PROTON.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
4.171.994.072.014.00
M05(t) M03(d)
M04(t)M02(s)
M01(s)7
.20
7.1
87
.16
6.7
66
.74
6.6
76
.65
6.1
5
4.2
7
1H NMR Spectrum for Compound 4a
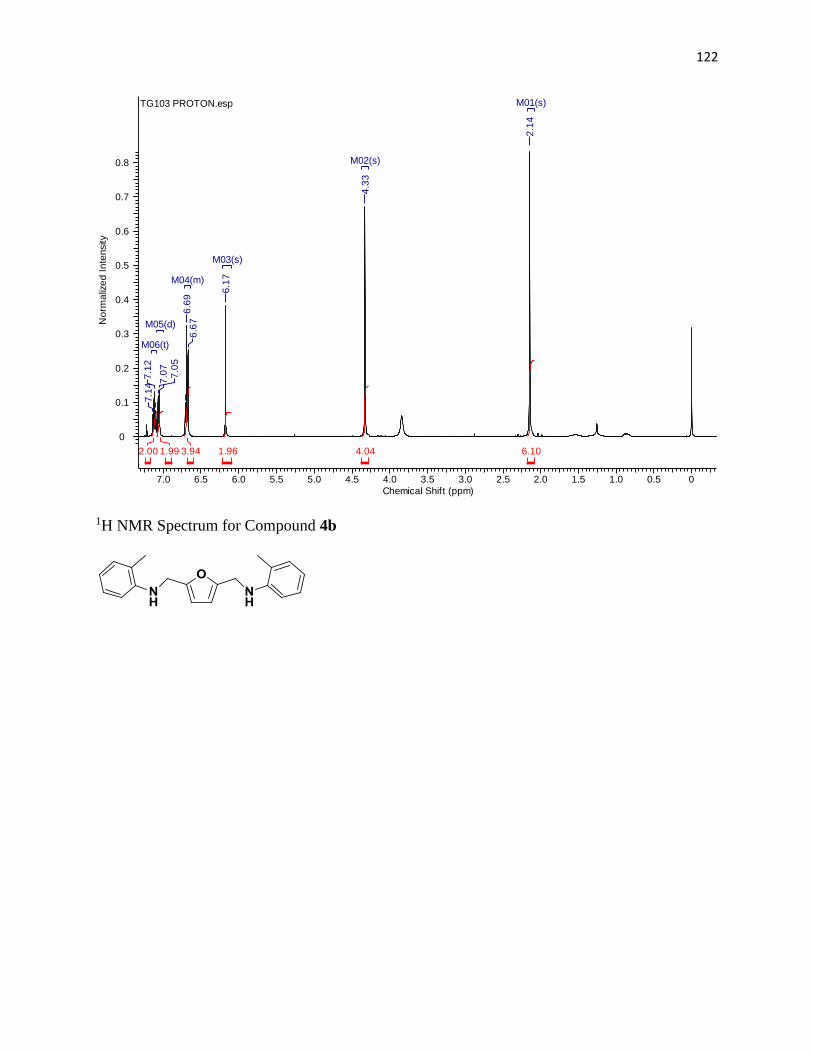
122
TG103 PROTON.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
6.104.041.963.941.992.00
M06(t)
M04(m)
M05(d)
M01(s)
M02(s)
M03(s)
7.1
47
.12
7.0
77
.05
6.6
96
.67
6.1
7
4.3
3
2.1
4
1H NMR Spectrum for Compound 4b

123
TG54 proton.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
6.823.891.824.002.022.22
M06(m)
M05(d)
M03(s)
M04(t)
M01(s)
M02(s)
7.0
87
.06
7.0
4 6.5
7
6.4
86
.46
6.1
3
4.2
5
2.2
7
1H NMR Spectrum for Compound 4c

124
TG55 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
6.594.091.924.204.00
M05(d)M03(s)
M04(d)
M02(s)
M01(s)
6.9
8 6.9
6
6.5
76
.55 6.1
0
4.2
1
2.2
2
1H NMR Spectrum for Compound 4d
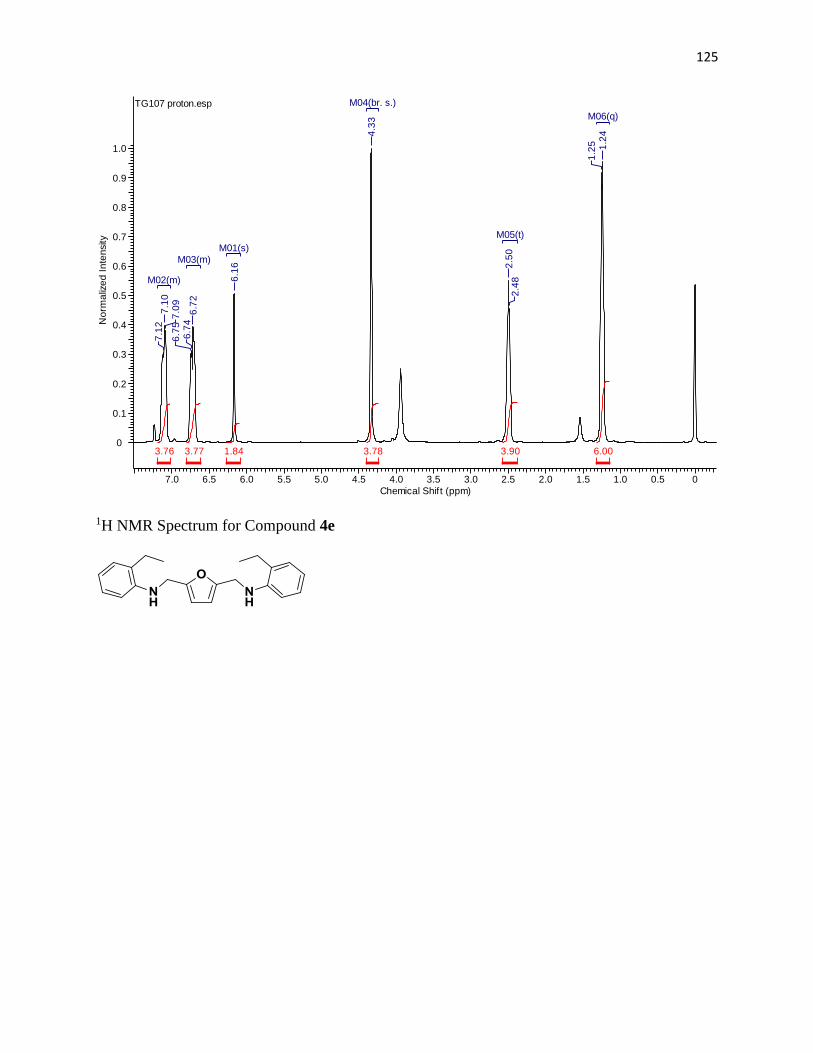
125
TG107 proton.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
6.003.903.781.843.773.76
M04(br. s.)
M02(m)
M01(s)
M06(q)
M03(m)
M05(t)
7.1
27
.10
7.0
96
.75
6.7
46
.72
6.1
6
4.3
3
2.5
02
.48
1.2
5 1.2
4
1H NMR Spectrum for Compound 4e

126
TG108 proton.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
6.114.024.031.925.932.00
M01(br. s.)
M04(t)
M02(br. s.)
M03(q)
M06(m)
M05(m)
7.1
07
.09 6
.60
6.5
0
6.1
4
4.2
6
2.5
72
.55
1.2
2 1.2
0
1H NMR Spectrum for Compound 4f

127
TG51 proton.esp
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
9.504.623.761.803.624.00
M06(d)
M05(d)
M01(t)
M04(s)
M03(s)
M02(q)
7.0
27
.00
6.6
16
.59
6.1
2
4.2
4
2.5
62
.55
2.5
32
.52
2.5
1
1.2
0 1.1
91
.18
1.1
71
.16
1H NMR Spectrum for Compound 4g

128
TG101 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
4.631.522.001.883.17
M03(m)
M02(m)
M04(m)
M05(s) M01(br. s.)
7.0
06
.99
6.9
76
.95
6.9
56
.76
6.7
46
.63
6.6
16
.61
6.1
4
4.2
8
1H NMR Spectrum for Compound 4h

129
TG52 PROTON.esp
8 7 6 5 4 3 2 1 0 -1
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
3.841.806.212.00
M02(s)
M03(dd)
M04(m)
M05(m)
M01(s)
7.1
17
.09
7.0
87
.05 6.4
36
.41
6.4
16
.38
6.3
56
.15
4.2
3
1H NMR Spectrum for Compound 4i

130
TG53 proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
4.061.944.024.00
M03(m)
M01(s)
M02(s)M04(m)
6.9
06
.88
6.8
66
.59
6.5
86
.57
6.1
3
4.2
2
1H NMR Spectrum for Compound 4j

131
TG102 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
3.891.844.001.912.29
M04(m)
M03(m)
M01(br. s.)
M05(m)
M02(br. s.)
7.2
67
.12
6.7
36
.66
6.1
7
4.3
5
1H NMR Spectrum for Compound 4k

132
TG66 proton.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
4.001.882.622.622.66
J(M05)=6.82 Hz
J(M04)=8.34 Hz
M04(d)
M03(br. s.)
M05(d)
M01(s)
M02(s)
7.0
76
.70
6.7
0
6.6
96
.68
6.6
26
.61
6.6
16
.50
6.4
96
.47
6.1
3
4.2
1
1H NMR Spectrum for Compound 4l

133
TG67 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8N
orm
aliz
ed
In
tensity
4.442.034.814.00
M04(d)
M03(d)
M01(s)
M02(s)
7.1
0 7.0
8 6.5
56
.53 6
.12
4.2
1
1H NMR Spectrum for Compound 4m

134
TG60.001.001.1r.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
6.453.891.884.002.221.92
M05(m)
M04(m)
M06(m)
M01(s)
M03(s)
M02(m)
6.8
66
.86
6.7
9 6.7
76
.77
6.7
06
.69
6.6
76
.16
4.3
0
3.8
43
.84
1H NMR Spectrum for Compound 4n

135
TG105 PROTON.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
6.404.022.106.002.05
M03(s)
M01(s)
M06(m)
M02(m)
M04(m)
7.1
07
.08
7.0
6
6.3
16
.29
6.2
26
.15
4.2
5
3.7
73
.75
1H NMR Spectrum for Compound 4o

136
TG74.PROTON.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
6.883.711.764.434.00
M03(s)
M04(s)
M02(d)
M05(s)
M01(d)
6.7
96
.77
6.6
46
.62
6.1
3
4.2
2
3.7
4
1H NMR Spectrum for Compound 4p

137
TG104 PROTON.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
No
rma
lize
d I
nte
nsity
4.632.144.312.002.33
M05(t)
M04(d)
M03(m)
M01(s)
M02(d)7
.46
7.4
47
.35
7.3
3
6.8
0 6.7
86
.75
6.7
4
6.1
7 4.3
84
.37
1H NMR Spectrum for Compound 4q

138
TG58 PROTON.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
3.621.681.931.472.031.99
M03(br. s.)
M04(m)
M01(m)
M06(m)
M05(s)
M02(s)7
.24
7.2
27
.20
6.9
86
.97
6.8
66
.78
6.7
6
6.1
7
4.2
9
1H NMR Spectrum for Compound 4r

139
TG59 proton.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
4.002.134.754.77
M04(s)
M01(s)
M03(d)
M02(d)7
.42 7
.40
6.6
76
.65
6.1
8
4.3
2
1H NMR Spectrum for Compound 4s

140
TG75 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
6.294.271.964.244.00
M05(d) M03(d)
M04(s)
M01(s)
M02(s)
7.2
17
.19
6.6
16
.59 6
.14
4.2
5
2.4
0
1H NMR Spectrum for Compound 4t

141
TG111 april10 PROTON.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5 -1.0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
5.9415.984.251.86
M03(s)
M01(s)
M02(s)
M04(d)
6.1
3
3.5
4
2.5
5 2.5
1
2.3
0
1H NMR Spectrum for Compound 4u

142
TG9-1 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
4.202.101.102.002.08
7.3
57
.33
7.3
1
7.0
26
.80 6
.78
6.6
86
.66
4.4
5
1.6
11
.47
1.2
91
.14
0.9
20
.90
0.8
7
0.7
90
.77
0.7
5
0.1
1
1H NMR Spectrum for Compound 8a

143
TG64-1 proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9N
orm
aliz
ed
In
tensity
M05(s)
M04(d)
M03(s)
M02(s)
M06(m) M01(s)
7.2
7
7.2
17
.19 7
.00
6.8
46
.82
6.8
0
4.7
7
2.3
4
1H NMR Spectrum for Compound 8b

144
TG65-1 proton.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
No
rma
lize
d I
nte
nsity
6.484.104.262.004.03
M02(s)
M05(s)
M04(d)
M03(d)
M01(s)
7.8
17
.79
7.0
1 6.9
16
.89
4.7
7
2.3
3
1H NMR Spectrum for Compound 8c

145
TG36 proton.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
0.045
0.050
0.055
0.060N
orm
aliz
ed
In
tensity
6.003.602.903.283.231.42
M04(s)
M01(s)
M03(d)
M02(d)
M06(t)
M05(q)
7.0
37
.01
6.6
16
.59
4.2
8
2.5
72
.55
2.5
32
.52
1.2
11
.19
1.1
7
1H NMR Spectrum for Compound 8d

146
TG10-1 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
4.003.271.951.04
7.2
87
.24
7.0
3
6.4
86
.46
6.4
4 6.4
26
.39
6.3
56
.32 5
.32
4.4
3
1.5
61
.43
1.2
81
.13
0.9
80
.91
0.8
90
.86
0.7
90
.76
0.7
4
0.1
0
1H NMR Spectrum for Compound 8e

147
tg8 SPOT 2 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50N
orm
aliz
ed
In
tensity
4.194.204.102.00
M04(s)
M01(s)
M02(m)
M03(t)
7.2
3
6.9
06
.88
6.8
66
.58
6.5
76
.56
6.5
56
.54
4.2
5
1H NMR Spectrum for Compound 8f

148
TG68 proton.esp
7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 -0.5
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
No
rma
lize
d I
nte
nsity
4.342.142.032.002.20
M05(dd)
M04(s)
M02(dd)
M03(t)
M01(s)
7.0
87
.06
7.0
4
6.7
0
6.6
06
.49
6.4
96
.47
6.4
7
4.2
6
1H NMR Spectrum for Compound 8g

149
TG69 PROTON.esp
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
No
rma
lize
d I
nte
nsity
4.214.234.082.00
M04(s)
M03(s)
M02(d)
M01(d)
7.2
2
7.1
0
6.5
56
.53
4.2
6
1H NMR Spectrum for Compound 8h
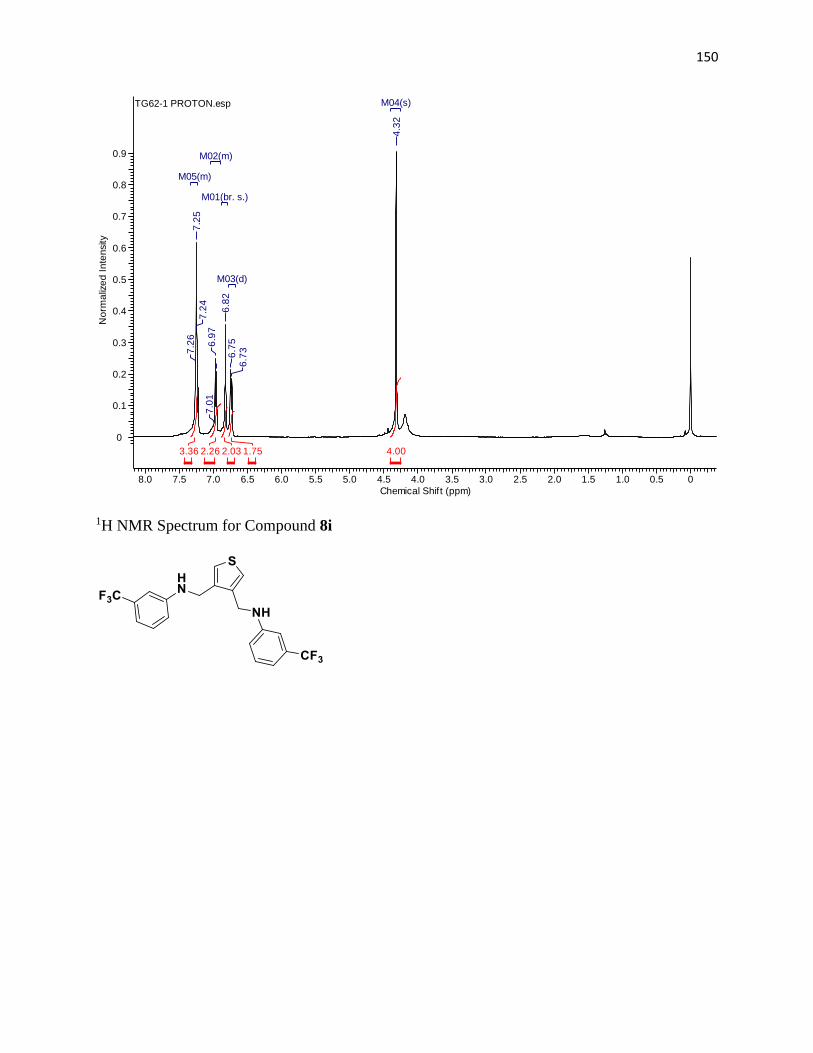
150
TG62-1 PROTON.esp
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
No
rma
lize
d I
nte
nsity
4.001.752.032.263.36
M01(br. s.)
M05(m)
M03(d)
M04(s)
M02(m)
7.2
67
.25
7.2
47
.01
6.9
7
6.8
26
.75
6.7
3
4.3
2
1H NMR Spectrum for Compound 8i

151
TG63-1 PROTON.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70
0.75N
orm
aliz
ed
In
tensity
3.943.632.003.54
M04(s)M03(br. s.)
M01(d)
M02(d)
7.4
17
.39
7.2
5
6.6
36
.61
4.3
3
1H NMR Spectrum for Compound 8j

152
TG42 proton.esp
9 8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70N
orm
aliz
ed
In
tensity
4.364.542.494.592.00
M05(s) M02(d)
M01(s)M04(t)
M03(t)
8.6
0
7.2
1 7.1
97
.17
6.7
5 6.6
96
.67
4.5
0
1H NMR Spectrum for Compound 14a

153
TG46 PROTON.esp
16 14 12 10 8 6 4 2 0 -2 -4
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
6.613.436.472.002.02
M02(s)
M05(t)
M03(s)
M04(m)M01(s)
8.5
9
7.0
97
.07
7.0
56
.50
6.4
8
4.4
9
2.2
7
1H NMR Spectrum for Compound 14b

154
TG43 PROTON.esp
9 8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70
0.75N
orm
aliz
ed
In
tensity
6.003.914.093.803.791.89
M04(s)
M03(d)
M02(d)M06(t)
M01(s) M05(d)
8.5
9
7.0
47
.02
6.6
36
.61 4
.48
2.5
52
.53
1.2
0 1.1
81
.16
1H NMR Spectrum for Compound 14c

155
TG45 Proton.esp
10 9 8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
No
rma
lize
d I
nte
nsity
3.394.003.821.55
M03(m)
M02(br. s.)
M01(s)
M04(m)8
.58
6.9
26
.90
6.8
86
.61
4.4
6
1H NMR Spectrum for Compound 14d

156
13C NMR Spectra
13C NMR Spectrum for Compound 2a
TG12-1 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
8.1
3
14
7.9
6
13
7.2
8
12
9.3
1
11
9.9
1
11
3.1
3
77
.41
77
.09
76
.77
49
.29
29
.75

157
13C NMR Spectrum for Compound 2b
TG99 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
8.0
2
14
5.8
4
13
7.2
6
13
0.0
81
27.1
31
22.2
71
19.9
91
17.2
2
11
0.1
4
77
.33
77
.02
76
.70
49
.22 1
7.5
9

158
TG34 last spot carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
8.2
3
14
8.0
6
13
9.0
61
37.2
6
12
9.2
0
11
9.8
61
18.6
11
13.9
51
10.2
3
49
.30
21
.70
13C NMR Spectrum for Compound 2c
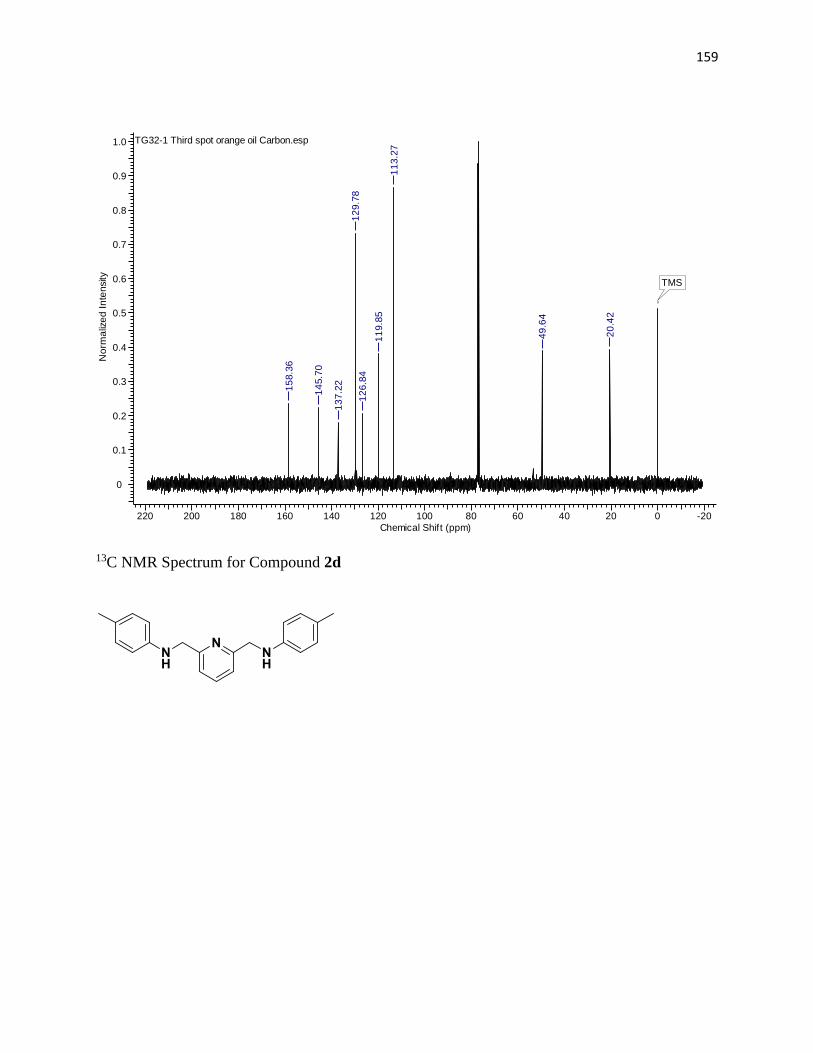
159
TG32-1 Third spot orange oil Carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS1
58.3
6
14
5.7
0
13
7.2
2
12
9.7
81
26.8
4
11
9.8
5
11
3.2
7
49
.64
20
.42
13C NMR Spectrum for Compound 2d

160
TG82CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
8.1
7
14
5.3
2
13
7.2
9
12
7.8
91
27.0
41
19.9
81
17.4
3
11
0.5
0
77
.41 77
.09
76
.77
49
.36 23
.98
12
.92
13C NMR Spectrum for Compound 2e

161
TG100.002.001.1r.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
8.1
5
14
8.0
11
45.4
5
13
7.2
0
12
9.1
9
11
9.8
31
17.3
61
12.8
01
10.3
4
77
.35
77
.04
76
.72
49
.31
29
.71
29
.02
28
.83
15
.57
0.0
0
13C NMR Spectrum for Compound 2f

162
TG22-4 Carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
8.4
1
14
6.0
0
13
7.2
81
33.5
51
28.6
5
11
9.9
1
11
3.3
0
77
.48
77
.17
76
.85
49
.71
28
.05
28
.01
16
.02
13C NMR Spectrum for Compound 2g

163
TG98-2 dirty CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS
15
7.8
1
15
2.9
3
13
7.3
91
36.3
3
12
4.5
6 11
9.8
51
14.5
7 11
2.5
0
77
.34
77
.02
76
.70
48
.90
29
.71
13C NMR Spectrum for Compound 2h

164
TG13-1 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
16
5.3
51
62.9
41
57.5
11
49.7
61
49.6
6
13
7.4
4
13
0.3
2
12
0.0
6
10
9.0
01
04.1
71
03.9
69
9.8
99
9.6
4
77
.41
77
.09
76
.77
48
.99
13C NMR Spectrum for Compound 2i

165
TG7-1 - YELLOW - CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
No
rma
lize
d I
nte
nsity
15
7.9
51
57.1
51
54.8
1
14
4.2
7
13
7.3
81
37.2
0
12
0.0
31
15.8
61
15.6
41
15.5
41
14.3
21
13.4
4
77
.35
77
.03
76
.72
49
.85
49
.26
13C NMR Spectrum for Compound 2j

166
TG83 Carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
7.6
4
14
3.7
0
13
7.4
2
12
9.1
81
27.8
01
19.8
31
17.5
41
11.6
3
77
.34
77
.02
76
.71
48
.94
13C NMR Spectrum for Compound 2k

167
TG16-1 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
7.3
9
14
9.0
2
13
7.4
5
13
0.2
6
12
0.0
91
17.5
31
12.7
41
11.4
2
77
.36
77
.05
76
.73
48
.86
13C NMR Spectrum for Compound 2l

168
TG17-1 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
7.6
4
14
6.4
3
13
7.3
8
12
9.1
21
22.2
9 12
0.0
2
11
4.1
6
77
.34
77
.03
76
.71
49
.21
13C NMR Spectrum for Compound 2m

169
TG24 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
8.6
0
14
6.9
7
13
7.8
5
12
1.2
2
11
6.7
21
10.2
31
09.4
3
77
.31
77
.00
76
.68
55
.44
49
.28
13C NMR Spectrum for Compound 2n

170
TG93 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
No
rma
lize
d I
nte
nsity
16
0.8
51
57.9
7
14
9.3
4
13
7.2
9
13
0.0
2
11
9.9
4
10
6.2
21
02.8
29
9.1
1
77
.35
77
.03
76
.71
55
.10
53
.43
49
.24
0.0
0
13C NMR Spectrum for Compound 2o

171
TG14-2 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
8.3
9
15
2.2
5
14
2.2
2
13
7.2
0
11
9.9
41
14.9
01
14.3
6
77
.39
77
.07
76
.76
55
.79
55
.50
50
.24
29
.73
14
.23
13C NMR Spectrum for Compound 2p

172
TG90 Carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
7.3
2
14
5.1
6
13
7.5
8 13
3.1
4
12
6.7
21
19.7
71
16.2
71
12.2
6
77
.35 7
7.0
37
6.7
1
48
.69
0.0
0
13C NMR Spectrum for Compound 2q

173
TG29 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
7.2
4
14
8.0
6
13
7.5
1
13
1.7
51
29.7
1
12
3.0
31
20.2
01
16.0
21
14.0
51
09.2
3
48
.80
13C NMR Spectrum for Compound 2r

174
TG28 2nd Spot carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
7.1
5
15
0.2
8
13
7.5
9
12
6.6
81
26.3
21
20.1
5
11
2.2
1
48
.46
13C NMR Spectrum for Compound 2s

175
TG37 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
7.8
0
14
6.7
0
13
7.3
0
13
1.3
4
12
4.6
11
19.9
4
11
3.7
0
77
.35
77
.04
76
.72
49
.16
19
.02
0.0
0
13C NMR Spectrum for Compound 2t

176
TG19-2 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.10
0.11
No
rma
lize
d I
nte
nsity
TMS
15
6.7
9
14
8.5
7
13
7.6
9
12
9.8
1
12
0.4
41
19.1
5
11
2.3
5
10
6.6
3
77
.33
77
.02
76
.70
48
.72
13C NMR Spectrum for Compound 2u

177
TG18-1 Second FRaction attempt 2.002.001.1r.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.10
0.11
No
rma
lize
d I
nte
nsity
15
6.1
01
52.6
9
13
7.8
8
12
6.4
1
12
0.4
3
11
1.5
3
77
.32
77
.00
76
.68
48
.07
13C NMR Spectrum for Compound 2v
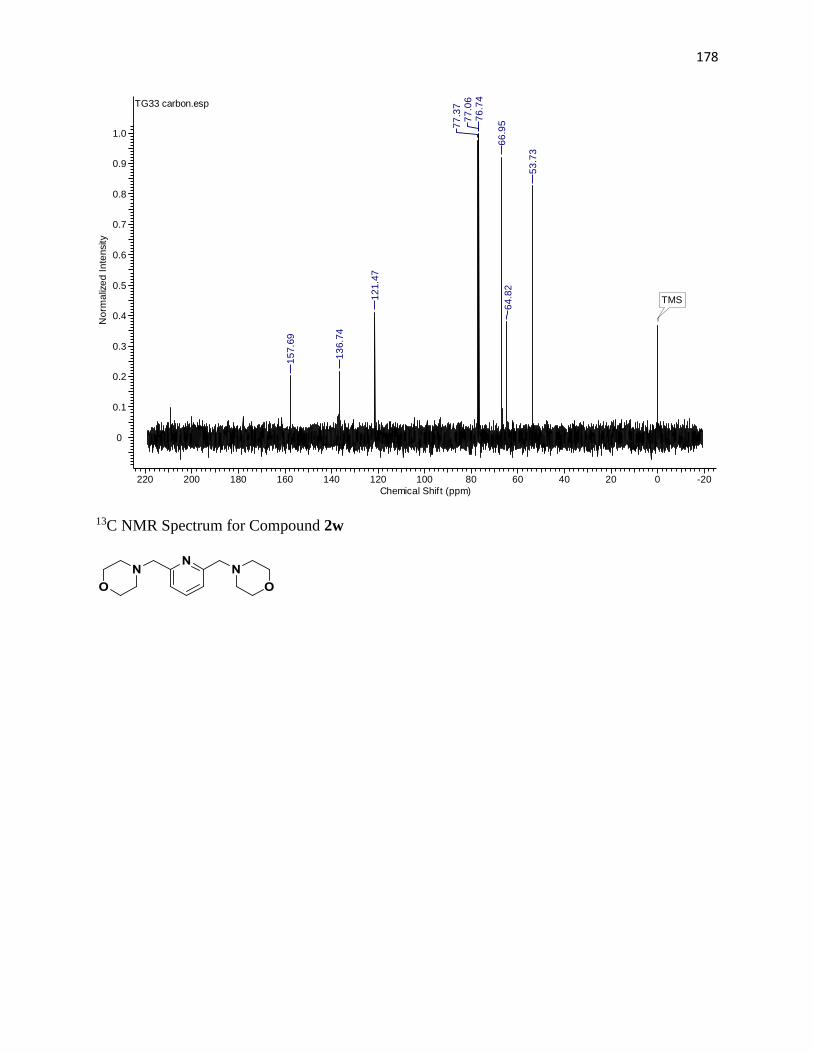
178
TG33 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS1
57.6
9
13
6.7
4
12
1.4
7
77
.37
77
.06
76
.74
66
.95
64
.82
53
.73
13C NMR Spectrum for Compound 2w

179
TG39 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS1
58.8
0
13
6.7
0
12
0.9
3
77
.33 7
7.0
17
6.6
9
62
.35
54
.32
23
.57
13C NMR Spectrum for Compound 2x

180
TG50 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.1
11
47.5
8
12
9.2
2
11
8.0
41
13.1
8
10
7.8
0
77
.34
77
.02
76
.71
41
.49
0.0
0
13C NMR Spectrum for Compound 4a
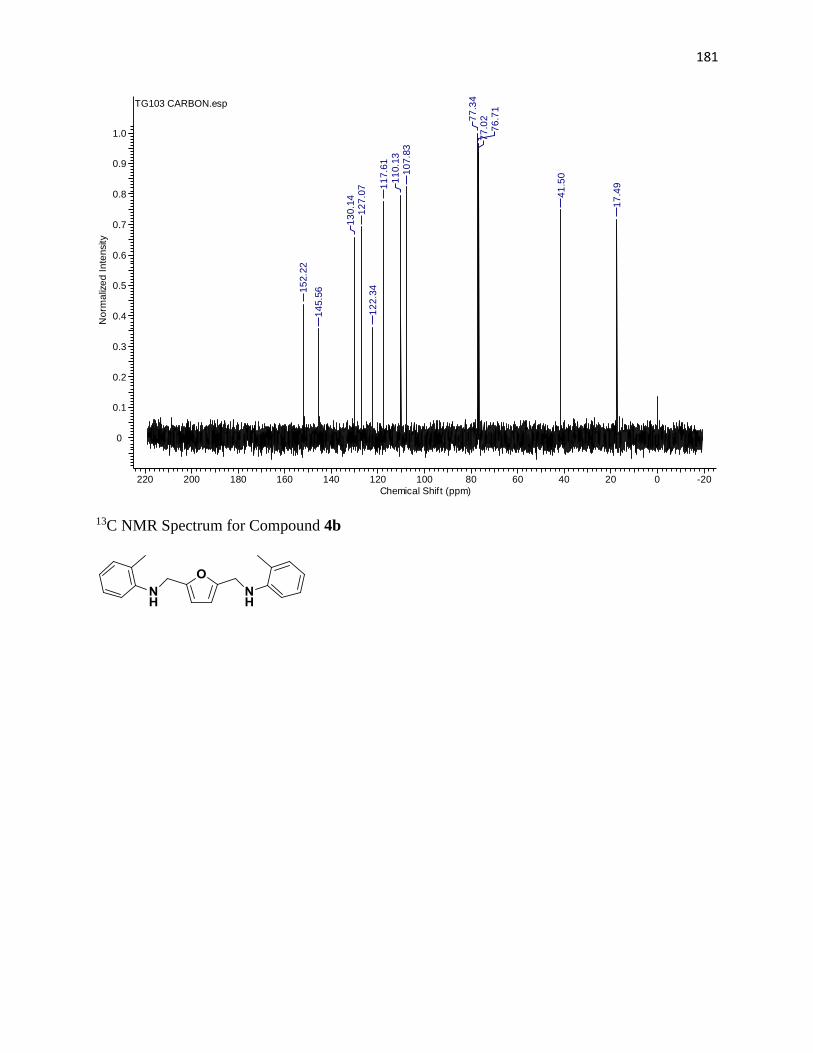
181
TG103 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.2
2
14
5.5
6
13
0.1
41
27.0
71
22.3
41
17.6
1
11
0.1
31
07.8
3
77
.34
77
.02
76
.71
41
.50
17
.49
13C NMR Spectrum for Compound 4b

182
TG54 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.1
71
47.6
4
13
8.9
8
12
9.0
9
11
8.9
7 11
4.0
11
10.2
71
07.7
3
77
.36
77
.04
76
.72
41
.53
21
.61
0.0
0
13C NMR Spectrum for Compound 4c

183
TG55 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.2
5
14
5.3
3
12
9.6
81
27.2
1
11
3.3
8
10
7.6
7
77
.39
77
.07
76
.75
41
.82
20
.40
13C NMR Spectrum for Compound 4d

184
TG107 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.2
8
14
4.9
8
12
8.0
21
27.9
21
26.9
3
11
7.8
1
11
0.5
21
07.7
9
77
.34
77
.03
76
.71
41
.59
23
.79
12
.90
13C NMR Spectrum for Compound 4e

185
TG108 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.2
01
47.7
01
45.4
0
12
9.1
5
11
7.7
81
12.9
31
10.4
71
07.7
5 77
.36
77
.04
76
.72
41
.57
29
.00 15
.51
13C NMR Spectrum for Compound 4f
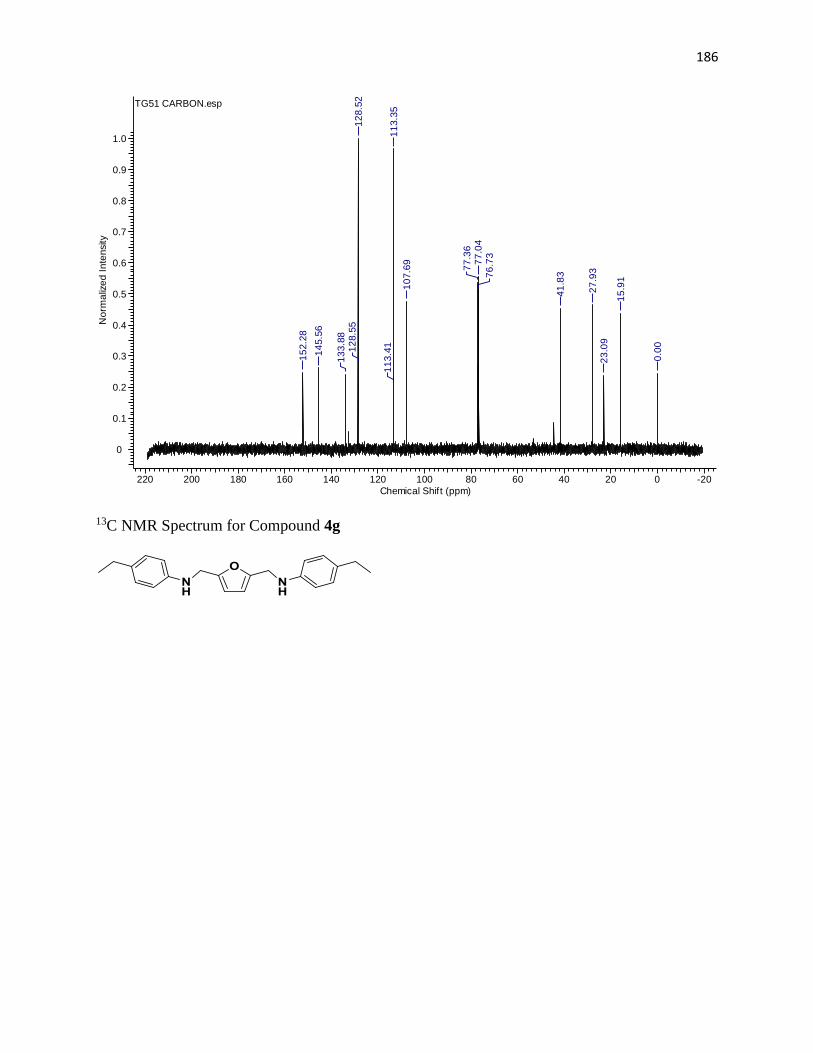
186
TG51 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.2
8
14
5.5
6
13
3.8
81
28.5
51
28.5
2
11
3.4
11
13.3
5
10
7.6
9
77
.36
77
.04
76
.73
41
.83
27
.93
23
.09
15
.91
0.0
0
13C NMR Spectrum for Compound 4g

187
TG101 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.8
41
51.8
41
50.4
71
44.5
3
13
6.0
41
35.9
31
30.4
11
24.5
01
22.2
91
18.5
81
17.3
8
11
2.5
11
07.9
4
77
.37
77
.05
76
.73
41
.02
17
.31
0.0
0
13C NMR Spectrum for Compound 4h

188
TG52 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
16
5.2
21
62.8
0
15
1.6
91
49.3
51
49.2
5
13
0.3
51
30.2
5 10
9.0
01
08.9
81
08.0
71
04.5
3
99
.95
99
.69
77
.36
77
.04
76
.72
41
.27
0.0
0
13C NMR Spectrum for Compound 4i

189
TG53 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
7.3
51
55.0
01
52.0
4
14
3.8
8
11
5.7
71
15.5
51
14.1
8
10
7.9
0
77
.36 77
.04
76
.72
42
.10
0.0
0
13C NMR Spectrum for Compound 4j

190
TG102 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
1.7
2
14
3.4
2
12
9.1
91
27.7
61
19.4
61
17.8
9
11
1.5
91
07.9
8
77
.34
77
.02
76
.70
41
.16
13C NMR Spectrum for Compound 4k

191
TG66 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
1.6
71
48.6
9
13
4.9
5
13
0.2
1
11
8.3
61
17.8
31
12.7
91
11.4
31
08.1
0
77
.43
77
.11
76
.80
60
.44
41
.18
31
.62
22
.69
21
.02
14
.14
13C NMR Spectrum for Compound 4l

192
TG67 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
1.6
9
14
6.0
1
12
8.8
5
12
2.3
2
11
4.0
6
10
7.8
0
41
.26
13C NMR Spectrum for Compound 4m

193
TG60.002.001.1r.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
2.2
1
14
7.0
0
13
7.6
0
12
1.1
71
17.1
11
10.2
81
09.4
71
07.7
0
77
.35
77
.03
76
.72
55
.40
41
.26
0.0
0
13C NMR Spectrum for Compound 4n

194
TG105 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
16
0.7
4 15
1.9
91
48.9
8
12
9.9
6
10
7.8
7
10
3.0
99
9.2
0
77
.36
77
.04
76
.72
55
.07
41
.45
13C NMR Spectrum for Compound 4o

195
TG75 Carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
1.9
4
14
6.3
1
13
1.1
2
12
5.1
7
11
3.8
1
10
7.9
2
77
.36
77
.04
76
.72
41
.48
18
.87
13C NMR Spectrum for Compound 4p

196
TG104 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
1.4
9
14
4.9
8
13
3.0
7
12
6.6
91
26.4
5
11
6.6
71
12.1
81
08.0
3
77
.34
77
.03
76
.71
41
.07
0.0
0
13C NMR Spectrum for Compound 4q

197
TG58 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
1.6
21
47.6
7
12
9.7
31
29.6
5
11
7.9
71
16.1
11
14.4
6
10
8.2
3
77
.35
77
.03
76
.71
41
.15
13C NMR Spectrum for Compound 4r

198
TG59.002.001.1r.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
No
rma
lize
d I
nte
nsity
17
0.1
6
15
8.7
41
56.6
7
15
1.5
21
49.9
1
12
6.6
21
21.8
1
11
2.2
11
08.2
0
81
.08
77
.32
77
.00
76
.68
40
.91
13C NMR Spectrum for Compound 4s
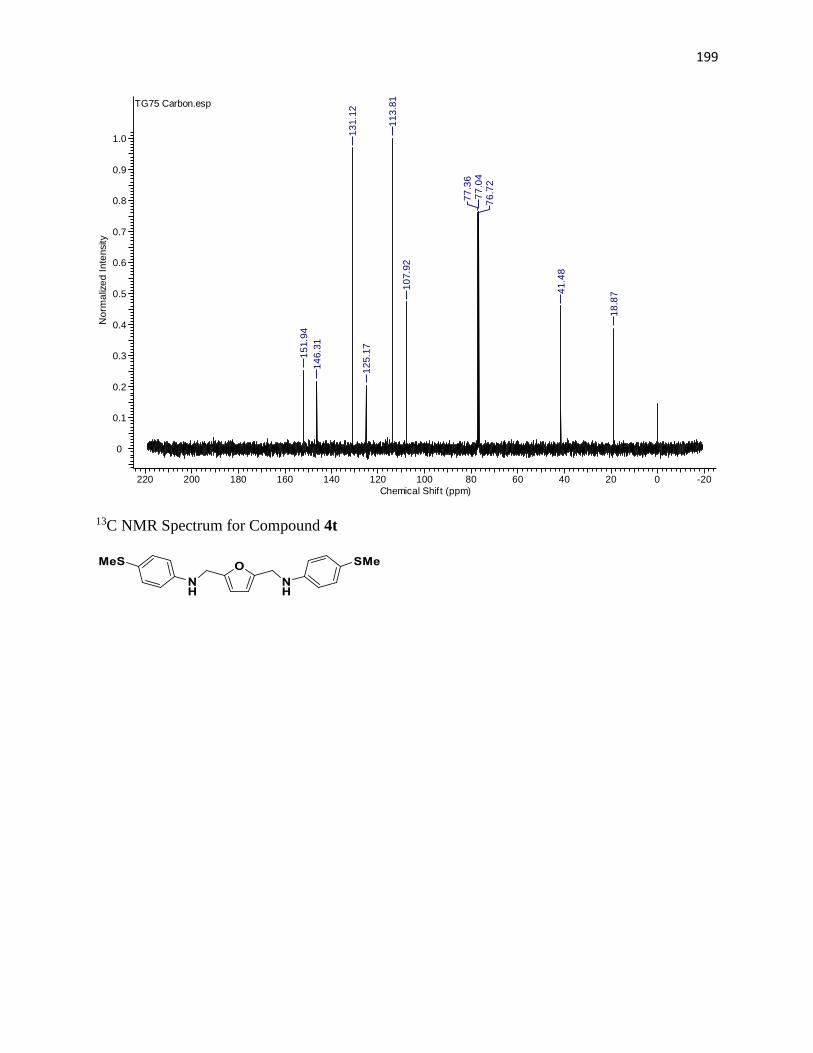
199
TG75 Carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
1.9
4
14
6.3
1
13
1.1
2
12
5.1
7
11
3.8
1
10
7.9
2
77
.36
77
.04
76
.72
41
.48
18
.87
13C NMR Spectrum for Compound 4t

200
TG111 april10 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
1.0
9
10
9.4
8
77
.32
77
.00
76
.69
54
.89
52
.48
45
.84
13C NMR Spectrum for Compound 4u

201
TG9-1 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
14
7.6
6
14
2.2
2
12
9.3
8
11
6.3
41
14.4
31
11.4
1
77
.33
77
.01
76
.69
48
.99
31
.94
29
.71
29
.37
22
.70
14
.12
13C NMR Spectrum for Compound 8a

202
TG64-1 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0N
orm
aliz
ed
In
tensity
TMS
15
1.6
6
14
0.6
01
38.6
2
12
8.7
4
12
0.2
91
18.7
51
16.9
11
14.7
1
49
.59
21
.83
13C NMR Spectrum for Compound 8b

203
TG65-1 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0N
orm
aliz
ed
In
tensity
TMS
12
9.4
7
12
3.2
11
21.7
11
20.0
51
14.6
9
49
.67
20
.82
13C NMR Spectrum for Compound 8c

204
TG63-1 PROTON.esp
8 7 6 5 4 3 2 1 0
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70
0.75N
orm
aliz
ed
In
tensity
3.943.632.003.54
M04(s)M03(br. s.)
M01(d)
M02(d)
7.4
17
.39
7.2
5
6.6
36
.61
4.3
3
13C NMR Spectrum for Compound 8d

205
TG10-1 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.05
0.10
0.15
0.20
0.25
No
rma
lize
d I
nte
nsity
16
5.4
3
14
9.1
7
14
1.6
6
13
0.4
51
30.3
5
11
4.6
8
10
7.1
31
02.9
41
02.7
39
8.5
99
8.3
3
77
.34
77
.02
76
.70
49
.13
31
.95
29
.72
29
.38
22
.71
14
.14
13C NMR Spectrum for Compound 8e

206
tg8 SPOT 2 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0N
orm
aliz
ed
In
tensity
TMS
15
7.3
2
14
4.2
6 13
8.2
2
12
4.4
0
11
5.8
61
15.6
41
14.1
6
43
.58
13C NMR Spectrum for Compound 8f

207
TG68 Carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0N
orm
aliz
ed
In
tensity
14
8.9
5
13
7.6
41
35.0
51
30.2
7
12
4.5
3
11
7.8
5 11
2.7
61
11.4
7
42
.60
13C NMR Spectrum for Compound 8g

208
TG69 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS14
6.3
9
13
7.8
2
12
9.1
51
24.5
01
22.6
7
11
4.2
6
77
.34
77
.03
76
.71
42
.97
31
.60
22
.67
14
.13
13C NMR Spectrum for Compound 8h

209
TG62-1 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
14
7.9
5
13
7.4
7
12
9.7
6
12
4.7
7
11
6.0
91
14.5
01
09.3
2
77
.36
77
.04
76
.72
42
.61
13C NMR Spectrum for Compound 8i

210
TG63-1 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
15
0.1
7
13
7.2
6
12
6.7
3 12
4.7
7
11
2.2
1
77
.34
77
.02
76
.71
42
.36
0.0
0
13C NMR Spectrum for Compound 8j

211
TG42 carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS
15
2.4
21
47.4
21
42.7
1
12
9.3
7
11
8.1
7
11
3.1
8
77
.35
77
.03
76
.71
47
.08
13C NMR Spectrum for Compound 14a

212
TG46 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS
15
2.5
0
14
6.3
4 14
2.6
91
39.1
8 12
9.2
4 12
9.1
7
11
9.4
71
15.9
21
12.2
51
10.3
3
77
.36
77
.04
76
.72
47
.13
21
.63
21
.44
13C NMR Spectrum for Compound 14b
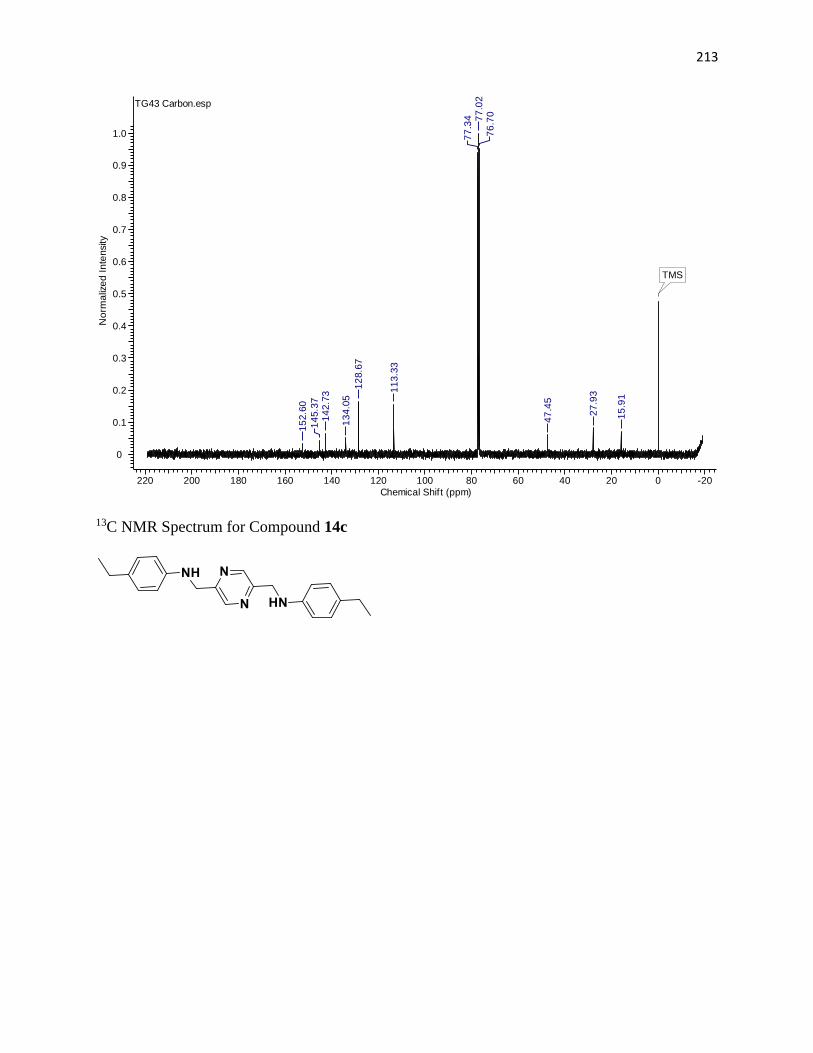
213
TG43 Carbon.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS
15
2.6
0
14
5.3
71
42.7
3
13
4.0
5
12
8.6
7
11
3.3
3
77
.34 77
.02
76
.70
47
.45
27
.93
15
.91
13C NMR Spectrum for Compound 14c

214
TG45 CARBON.esp
220 200 180 160 140 120 100 80 60 40 20 0 -20
Chemical Shift (ppm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
No
rma
lize
d I
nte
nsity
TMS
15
7.3
91
55.0
51
52.3
11
43.7
1 14
2.7
3
11
5.9
41
15.7
11
14.0
91
14.0
2
77
.34
77
.02
76
.71
47
.63
13C NMR Spectrum for Compound 14d

215
Mass Spectra
Mass Spectra for compound 2a
in MeOH 100x dilution 01-Jul-201312:44:58
m/z100 150 200 250 300 350 400 450 500 550 600
%
0
100
THERESA_TG12-1_MOORING-ACCU_07012013_ESI-POS02 64 (1.193) AM (Top,2, Ar,5000.0,0.00,1.00); Sm (SG, 2x3.00); Cm (54:64)1.15e4290.1657
197.112593.1245183.1013
286.1455
291.1728
292.1725379.2231

216
Mass Spectra for compound 2b
TG_99_ESIPOS_MOORING_05202015_150520142143 #213-221 RT: 2.98-3.09 AV: 9 NL: 1.68E9T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
318.1958z=1

217
Mass Spectra for compound 2c
TG34_ESIPOS_MOORING_041317 #217-226 RT: 3.00-3.13 AV: 10 NL: 1.38E8T: FTMS + p ESI Full ms [50.00-1000.00]
305 310 315 320 325 330 335 340 345
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
318.1952z=1
319.1980z=1
314.1650z=1

218
Mass Spectra for compound 2d
Therasa_TG32_ESI_pos_Mooring_04032014_2 #3 RT: 0.01 AV: 1 NL: 3.36E7T: FTMS + c ESI Full ms [100.00-1000.00]
314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
318.1952
319.1984

219
Mass Spectra for compound 2e
Theresa_TG78_ESIPOS_Mooring_04292015_150429152657 #186-198 RT: 2.58-2.75 AV: 13 NL: 2.70E9T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
346.2276z=1

220
Mass Spectra for compound 2f
TG_100_MOORING_ESIPOS_021016_160210142315 #150-160 RT: 2.27-2.40 AV: 11 NL: 2.34E8T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
346.2285z=1
122.0966z=1

221
Mass Spectra for compound 2g
QtofMicro 14-Jan-201416:07:33
m/z100 200 300 400 500 600 700 800 900 1000
%
0
100
THERESA_TG22_MOORING-ACCU_01142014_ESI-POS01 19 (0.354) AM (Cen,2, 80.00, Ar,5000.0,0.00,1.00); Sm (Mn, 2x3.00); Cm (13:19)1.49e5346.2291
225.1465
122.1134
342.2165
226.1571
227.1681
347.2480
368.2348
463.3156 687.4909532.3705
711.4645804.5576

222
Mass Spectra for compound 2h
TG_98_ESIPOS_MOORING_05202015_150520140416 #203-211 RT: 2.83-2.94 AV: 9 NL: 2.02E9T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
326.1457z=1

223
Mass Spectra for compound 2i
in MeOH 100x dilution 01-Jul-201313:00:28
m/z100 150 200 250 300 350 400 450 500 550 600
%
0
100
THERESA_TG13-1_MOORING-ACCU_07012013_ESI-POS01 54 (1.006) AM (Cen,2, 80.00, Ar,5000.0,0.00,1.00); Sm (SG, 2x3.00); Cm (51:60)6.93e3326.1462
215.0945195.0959152.1014 238.0572
327.1527
328.1463 376.1253
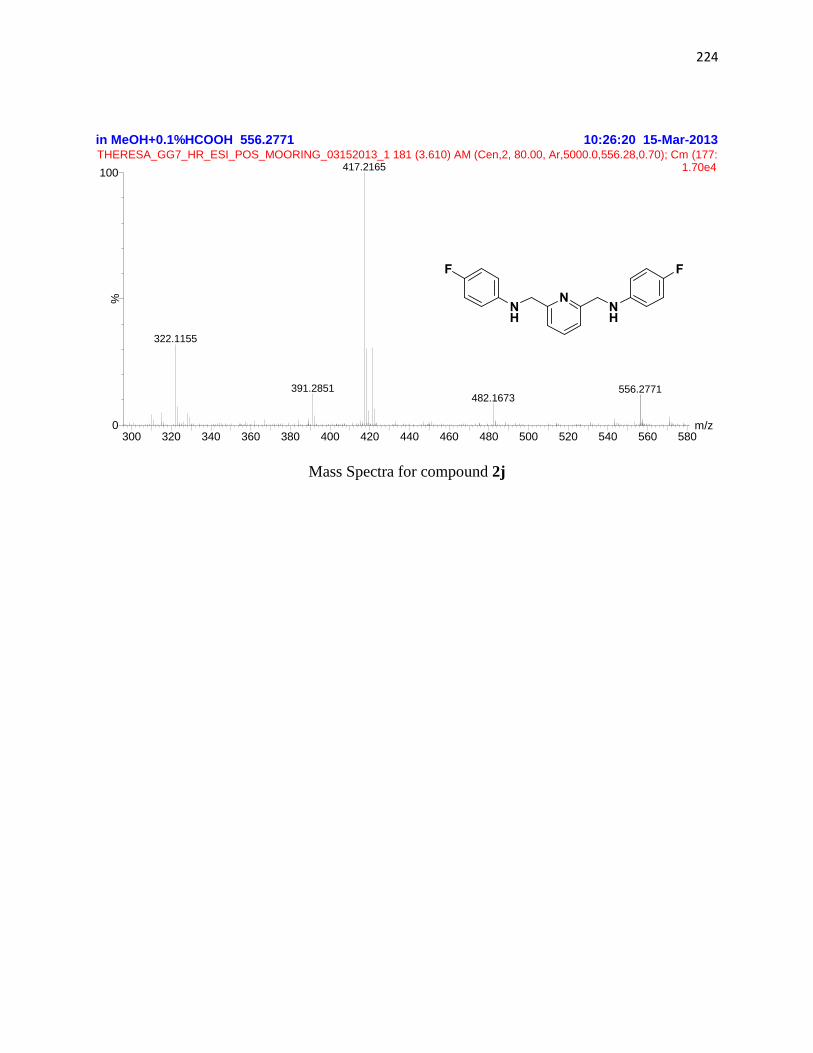
224
Mass Spectra for compound 2j
in MeOH+0.1%HCOOH 556.2771 10:26:20 15-Mar-2013
m/z300 320 340 360 380 400 420 440 460 480 500 520 540 560 580
%
0
100
THERESA_GG7_HR_ESI_POS_MOORING_03152013_1 181 (3.610) AM (Cen,2, 80.00, Ar,5000.0,556.28,0.70); Cm (177:201)1.70e4417.2165
322.1155
391.2851 556.2771482.1673

225
Mass Spectra for compound 2k
Theresa_TG83_ESIPOS_Mooring_04292015 #189-195 RT: 2.57-2.65 AV: 7 NL: 1.18E9T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
358.0877z=1
360.0842z=1
194.0965z=1 238.0865
z=1

226
Mass Spectra for compound 2l
Theresa_TG16-1_Mooring-ACCU_08272013_ESI-POS #165-169 RT: 2.25-2.30 AV: 5 SB: 8T: FTMS + p ESI Full ms [100.00-1000.00]
200 400 600 800 1000
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
358.0864
282.2782
563.5491 815.3322686.0469191.0365 921.4147392.1545

227
Mass Spectra for compound 2m
Theresa_TG17-1_Mooring-ACCU_08272013_ESI-POS_130827185100 #168-173 RT: 2.29-2.36T: FTMS + p ESI Full ms [100.00-1000.00]
200 400 600 800 1000
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
358.0862
282.2781247.0623 686.0467 815.3318571.1619 921.4132
377.0623
455.1868

228
Mass Spectra for compound 2n
QtofMicro 14-Jan-201416:35:55
m/z338 340 342 344 346 348 350 352 354 356 358 360 362 364 366 368
%
0
100
THERESA_TG24_MOORING-ACCU_01142014_ESI-POS01 41 (0.766) AM (Cen,2, 80.00, Ar,5000.0,0.00,1.00); Sm (Mn, 2x3.00); Cm (35:41)1.69e4350.1862
346.1608
342.1349337.1736347.1673
351.1931
364.1717360.1421352.1934
357.1512 365.1776

229
Mass Spectra for compound 2o
TG93_FLUSH_ESIPOS_MOORING_06172015 #183-189 RT: 2.50-2.58 AV: 7 NL: 8.45E7T: FTMS + p ESI Full ms [100.00-1000.00]
200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
350.1854z=1

230
Mass Spectra for compound 2p
in MeOH 100x dilution 01-Jul-201313:05:37
m/z100 150 200 250 300 350 400 450 500 550 600
%
0
100
THERESA_TG14_MOORING-ACCU_07012013_ESI-POS01 86 (1.590) AM (Cen,2, 80.00, Ar,5000.0,0.00,1.00); Cm (75:87)1.69e4350.1865
123.0933
227.1080213.0961
124.0976212.0974
346.1485229.1191
351.1888
469.2160351.2566 470.2405

231
Mass Spectra for compound 2q
Theresa_TG90_ESIPOS_Mooring_04292015 #181-187 RT: 2.49-2.57 AV: 7 NL: 2.48E9T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
426.1401z=1

232
Mass Spectra for compound 2r
Therasa_TG29_ESI_pos_Mooring_04032014_1 #1 RT: 0.01 AV: 1 NL: 1.15E9T: FTMS + c ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
426.1384

233
Mass Spectra for compound 2s
Therasa_TG28_ESI_pos_Mooring_04032014_1 #68 RT: 0.49 AV: 1 NL: 3.11E7T: FTMS + c ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
426.1381

234
Mass Spectra for compound 2t
Theresa_TG37_ESIPOS_Mooring_04232014_3 #120 RT: 1.64 AV: 1 NL: 3.40E8T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
382.1404z=1

235
Mass Spectra for compound 2u
QtofMicro 14-Jan-201416:14:28
m/z376 377 378 379 380 381 382 383 384 385 386 387 388 389 390
%
0
100
THERESA_TG19_MOORING-ACCU_01142014_ESI-POS01 17 (0.317) AM (Cen,2, 80.00, Ar,5000.0,0.00,1.00); Sm (Mn, 2x3.00); Cm (17:78)1.60e4380.1358
378.1206376.1051
379.1213
381.1391
384.1781382.1494 385.1989387.2310 390.1345
388.1492

236
Mass Spectra for compound 2v
QtofMicro 14-Jan-201415:56:23
m/z378 379 380 381 382 383
%
0
100
THERESA_TG18_MOORING-ACCU_01142014_ESI-POS01 5 (0.093) AM (Cen,2, 80.00, Ar,5000.0,0.00,1.00); Sm (Mn, 2x3.00); Cm (3:8)2.06e3380.1359
378.1191 379.0827 379.7990
381.1438
382.1359383.2566

237
Mass Spectra for compound 2w
TG33__ESIPOS_MOORING_072716 #109-184 RT: 1.72-2.82 AV: 76 NL: 5.56E6T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
278.1853z=1
251.1383z=1
209.1277z=1
139.5964z=2

238
Mass Spectra for compound 2x
Theresa_TG39_ESIPOS_Mooring_05142014_2 #71 RT: 0.96 AV: 1 NL: 1.50E7T: FTMS + c ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
246.1956

239
Mass Spectra for compound 4a
Theresa_TG50_ESIPOS_Mooring_061114 #169 RT: 2.39 AV: 1 NL: 2.49E7T: FTMS + p ESI Full ms [200.00-2000.00]
278.2 278.4 278.6 278.8 279.0 279.2 279.4 279.6 279.8 280.0 280.2 280.4 280.6
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
279.1487
280.1519
280.2628
279.2313278.2322279.4247278.8739 280.0693279.0532 280.4678278.7065

240
Mass Spectra for compound 4b
GAINES_103_ESIPOS_MOORING_062415 #158-234 RT: 2.15-3.20 AV: 77 NL: 2.63E5T: FTMS + p ESI Full ms [100.00-1000.00]
285 290 295 300 305 310 315 320 325 330
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
307.1792z=1
305.1638z=1
308.1826z=1
321.1585z=1303.1479
z=1312.3247
z=1

241
Mass Spectra for compound 4c
QtofMicroin ACN+0.1%HCOOH 556.2771 as ITSD 23-Jun-201416:18:29
m/z300 350 400 450 500 550 600 650 700 750
%
0
100
THERESA_TG54_HR_ESIPOS_MOORING_062314 300 (5.578) AM (Cen,4, 80.00, Ar,6000.0,556.28,0.80)2.29e3307.1817
319.1493
321.1647
426.2225
329.1757
*556.2771
578.2488

242
Mass Spectra for compound 4d
TG_55_ESIPOS_MOORING_070516 #136-143 RT: 1.93-2.02 AV: 8 NL: 4.20E5T: FTMS + p ESI Full ms [50.00-1000.00]
310 315 320 325 330 335 340 345 350 355 360
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
329.1617z=1
307.1798z=1 346.2081
z=1
319.1258z=1
330.1654z=1324.1226
z=1 337.1045z=1

243
Mass Spectra for compound 4e
GAINES_107_ESIPOS_MOORING_06242015 #203-210 RT: 2.77-2.87 AV: 8 NL: 1.12E6T: FTMS + p ESI Full ms [100.00-1000.00]
322 324 326 328 330 332 334 336 338 340 342 344 346 348 350 352 354
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
335.2103z=1
336.2136z=1
333.1947z=1

244
Mass Spectra for compound 4f
GAINES_108_ESIPOS_MOORING_06242015 #188-193 RT: 2.56-2.63 AV: 6 NL: 2.01E6T: FTMS + p ESI Full ms [100.00-1000.00]
320 322 324 326 328 330 332 334 336 338 340 342 344 346 348 350 352 354 356
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
335.2103z=1
336.2136z=1
333.1948z=1

245
Mass Spectra for compound 4g
Theresa_BG51_ESIPOS_Mooring_061114 #154 RT: 2.19 AV: 1 NL: 5.38E7T: FTMS + p ESI Full ms [200.00-2000.00]
334.4 334.6 334.8 335.0 335.2 335.4 335.6 335.8 336.0 336.2 336.4 336.6
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
335.2111
336.2143
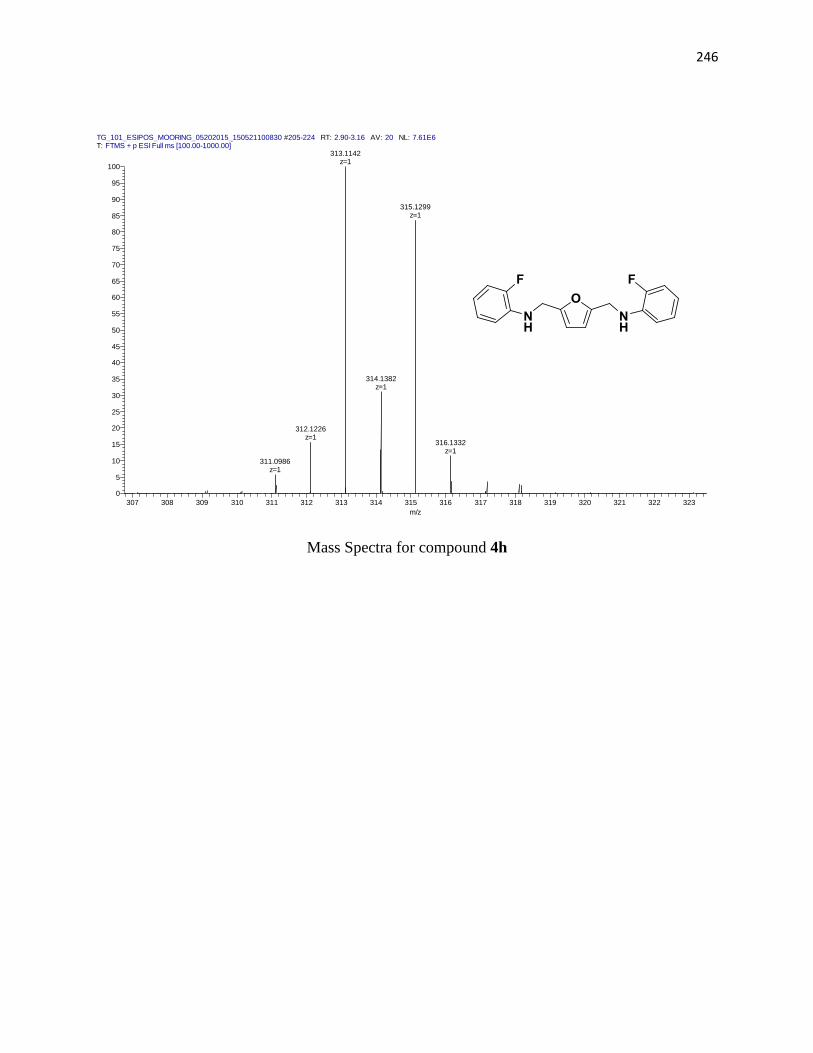
246
Mass Spectra for compound 4h
TG_101_ESIPOS_MOORING_05202015_150521100830 #205-224 RT: 2.90-3.16 AV: 20 NL: 7.61E6T: FTMS + p ESI Full ms [100.00-1000.00]
307 308 309 310 311 312 313 314 315 316 317 318 319 320 321 322 323
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
313.1142z=1
315.1299z=1
314.1382z=1
312.1226z=1
316.1332z=1
311.0986z=1

247
Mass Spectra for compound 4i
Theresa_TG52_ESIPOS_POS_Mooring_061214 #207 RT: 2.91 AV: 1 NL: 9.58E6T: FTMS + p ESI Full ms [100.00-1000.00]
250 300 350 400 450 500 550 600 650 700
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
315.1292
313.1136
629.2512
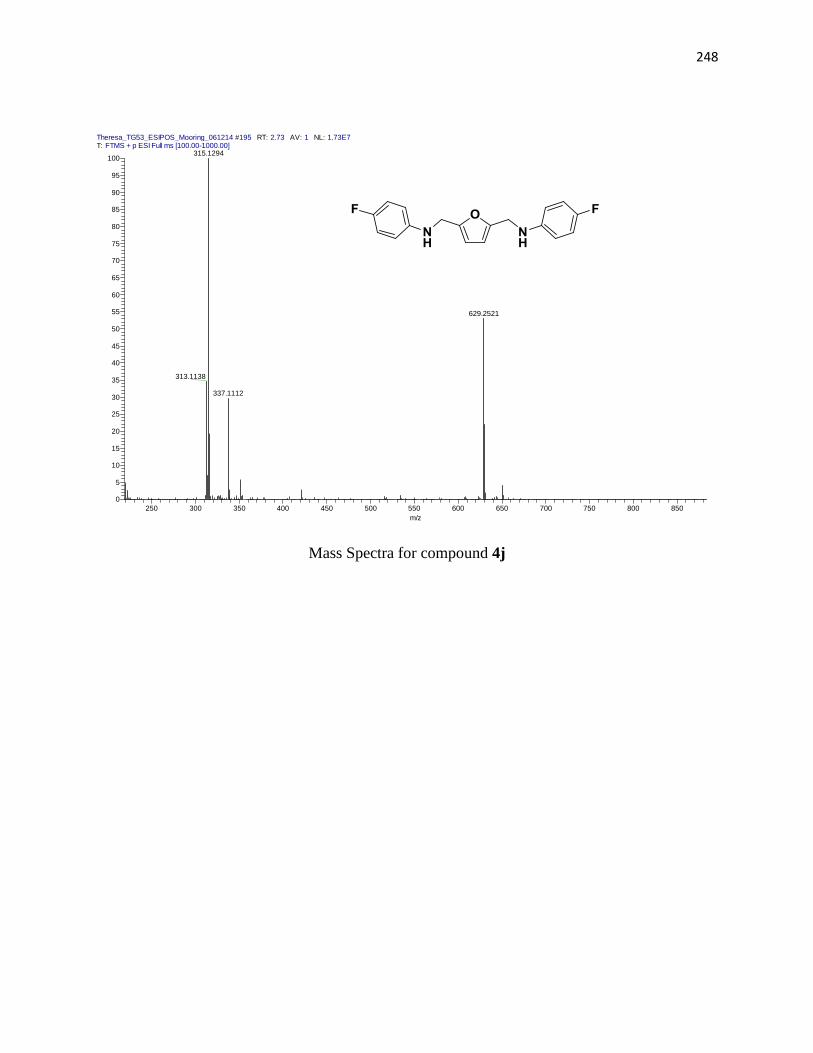
248
Mass Spectra for compound 4j
Theresa_TG53_ESIPOS_Mooring_061214 #195 RT: 2.73 AV: 1 NL: 1.73E7T: FTMS + p ESI Full ms [100.00-1000.00]
250 300 350 400 450 500 550 600 650 700 750 800 850
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
315.1294
629.2521
313.1138
337.1112

249
Mass Spectra for compound 4k
GAINES_102_ESIPOS_MOORING_062415 #168-220 RT: 2.29-3.01 AV: 53 NL: 5.96E5T: FTMS + p ESI Full ms [100.00-1000.00]
330 335 340 345 350 355 360 365 370
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
345.0539z=1
347.0509z=1
354.2832z=1
371.0992z=1
361.0488z=1
355.0680z=1
343.1191z=1
348.0543z=1339.1685
z=1363.0458
z=1369.0511
z=1355.2866
z=1329.0849
z=1359.2386
z=1350.0513
z=1335.1060
z=1364.0490
z=1337.1030
z=1
353.1478z=1

250
Mass Spectra for compound 4l
TGraines_TG66_ESIPOS_Mooring_100182014_1 #171 RT: 2.42 AV: 1 NL: 4.04E6T: FTMS + p ESI Full ms [100.00-1000.00]
300 320 340 360 380 400 420 440 460 480
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
363.0457
347.0685
319.1238
367.0393
377.0788329.0850311.0921 486.0495470.0809445.1154395.3106

251
Mass Spectra for compound 4m
TGraines_TG67_ESIPOS_Mooring_100182014_1 #168 RT: 2.38 AV: 1 NL: 1.65E6T: FTMS + p ESI Full ms [100.00-1000.00]
325 330 335 340 345 350 355 360 365
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
347.0696
349.0667
345.0541
353.2647363.0467
365.0438

252
Mass Spectra for compound 4n
m/z326 328 330 332 334 336 338 340 342 344 346 348 350 352 354 356 358 360 362 364 366 368 370 372 374 376 378 380
%
0
100
TG60_ESIPOS_MOORING_062016_2 420 (7.813) Cm (420:429) TOF MS ES+ 1.78e4362.9202
361.1523
339.1754
340.1850 363.9440

253
Mass Spectra for compound 4o
GAINES_105_ESIPOS_MOORING_062415 #158-199 RT: 2.15-2.72 AV: 42 NL: 1.24E7T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
216.1007z=1
339.1686z=1
124.0749z=1
188.1060z=1

254
Mass Spectra for compound 4p
TG74_ESIPOS_MOORING_041217 #120-137 RT: 1.70-1.93 AV: 18 NL: 1.45E6T: FTMS + p ESI Full ms [50.00-1000.00]
338.6 338.8 339.0 339.2 339.4 339.6 339.8 340.0 340.2 340.4 340.6 340.8 341.0 341.2 341.4 341.6
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
339.1701z=1
340.1737z=1

255
Mass Spectra for compound 4q
TG104_ESIPOS_MOORING_041217 #144-168 RT: 2.03-2.37 AV: 25 NL: 1.91E5T: FTMS + p ESI Full ms [50.00-1000.00]
410 411 412 413 414 415 416 417 418 419 420 421 422 423
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
415.1246z=1
417.1404z=1
416.1279z=1413.1093
z=1419.1565
z=1

256
Mass Spectra for compound 4r
TG58_ESIPOS_MOORING_041217 #163-173 RT: 2.28-2.41 AV: 11 NL: 3.59E6T: FTMS + p ESI Full ms [50.00-1000.00]
390 395 400 405 410 415 420 425 430 435
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
413.1080z=1
411.0925z=1
414.1117z=1
437.1063z=1
393.2976z=1
397.1134z=1

257
Mass Spectra for compound 4s
TG_59_ESIPOS_MOORING_070516 #129-143 RT: 1.83-2.03 AV: 15 NL: 3.10E3T: FTMS + p ESI Full ms [50.00-1000.00]
436.0 436.5 437.0 437.5 438.0 438.5 439.0 439.5
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
437.1066z=?
437.1651z=1
436.3936z=? 438.1096
z=?437.3236z=?
438.4096z=?
436.1713z=1
439.4133z=?

258
Mass Spectra for compound 4t
TG_75_ESIPOS_MOORING_070516 #112-116 RT: 1.59-1.64 AV: 5 NL: 3.36E5T: FTMS + p ESI Full ms [50.00-1000.00]
330 340 350 360 370 380 390 400 410 420 430 440
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
403.1139z=1
371.1238z=1
387.1190z=1
369.1084z=1332.9636
z=1
404.1174z=1
372.1275z=1 425.0963
z=1388.1227
z=1385.1036z=1

259
Mass Spectra for compound 4u
TG111_ESIPOS_MOORING_041217 #114 RT: 1.61 AV: 1 NL: 2.04E7T: FTMS + p ESI Full ms [50.00-1000.00]
280 285 290 295 300 305 310 315 320
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
293.2323z=1
294.2367z=1

260
Mass Spectra for compound 8a

261
Mass spectra for compound 8b
TG_64_ESIPOS_MOORING_071217 #319-325 RT: 4.50-4.59 AV: 7 NL: 4.70E5T: FTMS + p ESI Full ms [50.00-1000.00]
305 310 315 320 325 330 335 340 345 350 355
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
319.1241z=1
323.1561z=1
320.1284z=1

262
Mass spectra for compound 8c
TG_65_ESIPOS_dil_MOORING_071217_1_170713094936 #84-107 RT: 1.19-1.52 AV: 24 NL: 5.18E3T: FTMS + p ESI Full ms [50.00-1000.00]
321.5 322.0 322.5 323.0 323.5 324.0 324.5 325.0 325.5 326.0
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
323.1565z=1
324.1598z=1
325.1722z=1

263
Mass Spectra for compound 8d
Theresa_TG36_ESIPOS_Mooring_04232014_2 #6-7 RT: 0.08-0.09 AV: 2 NL: 9.56E7T: FTMS + p ESI Full ms [100.00-1000.00]
344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
351.1888z=1
352.1919z=1

264
Mass Spectra for compound 8e
TG10_ESIPOS_MOORING_041217 #138-147 RT: 1.95-2.07 AV: 10 NL: 1.62E7T: FTMS + p ESI Full ms [50.00-1000.00]
318 320 322 324 326 328 330 332 334 336 338 340 342 344 346 348
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
331.1061z=1
332.1106z=1

265
Mass Spectra for compound 8f
Theresa_TG8_ESIPOS_Mooring_04232014_2 #75 RT: 1.01 AV: 1 NL: 2.82E8T: FTMS + p ESI Full ms [150.00-2000.00]
150 200 250 300 350 400 450 500 550 600 650 700 750 800 850 900 950
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
331.1077z=1
220.0592z=1

266
Mass Spectra for compound 8g
TGraines_TG68_ESIPOS_Mooring_100182014 #175 RT: 2.48 AV: 1 NL: 1.01E7T: FTMS + p ESI Full ms [100.00-1000.00]
361 362 363 364 365 366 367
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
363.0466
365.0432
364.0496
366.0462 367.0397

267
Mass Spectra for compound 8h
TGraines_TG69_ESIPOS_Mooring_100182014 #162 RT: 2.29 AV: 1 NL: 1.24E7T: FTMS + p ESI Full ms [100.00-1000.00]
356 358 360 362 364 366 368 370 372
m/z
0
10
20
30
40
50
60
70
80
90
100
Re
lative
Ab
un
da
nce
363.0466
365.0433
366.0465

268
Mass Spectra for compound 8i
Theresa_TG62_ESIPos_Mooring_09102014 #183 RT: 2.60 AV: 1 NL: 2.23E7T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
431.0994z=1
270.0548z=1
286.0497z=1 427.0693
z=1133.0522z=1
166.0456z=1

269
Mass Spectra for compound 8j
Theresa_TG63_ESIPos_Mooring_09102014 #195 RT: 2.77 AV: 1 NL: 3.06E7T: FTMS + p ESI Full ms [100.00-1000.00]
100 200 300 400 500 600 700 800 900 1000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
431.0995z=1
270.0548z=1
133.0522z=1 166.0456
z=1 286.0497z=1
270
Mass Spectra for compound 14a
Theresa_TG42_ESIPOS_Nooring_06111401 #104-234 RT: 1.48-3.27 AV: 131 NL: 2.05E8T: FTMS + p ESI Full ms [200.00-2000.00]
200 400 600 800 1000 1200 1400 1600 1800 2000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
291.1599

271
Mass Spectra for compound 14b
Theresa_TG46_ESIPOS_Mooring_061114 #217 RT: 3.02 AV: 1 NL: 7.45E7T: FTMS + p ESI Full ms [200.00-2000.00]
200 400 600 800 1000 1200 1400 1600 1800 2000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
319.1909
422.2328

272
Mass Spectra for compound 14c
Theresa_TG43_ESIPOS_Mooring_06112014 #104-234 RT: 1.48-3.27 AV: 131 NL: 6.65E7T: FTMS + p ESI Full ms [200.00-2000.00]
200 400 600 800 1000 1200 1400 1600 1800 2000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
347.2225
291.1601

273
Mass Spectra for compound 14d
Theresa_TG45_ESIPOS_Mooring_061114 #279 RT: 3.87 AV: 1 NL: 8.33E7T: FTMS + p ESI Full ms [200.00-2000.00]
200 400 600 800 1000 1200 1400 1600 1800 2000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Re
lative
Ab
un
da
nce
327.1409
262.0983