differential inhibitor sensitivity of anaplastic lymphoma ... · differential inhibitor sensitivity...
TRANSCRIPT

DOI: 10.1126/scitranslmed.3002950, 108ra114 (2011);3 Sci Transl Med
, et al.Scott C. BreslerFound in NeuroblastomaDifferential Inhibitor Sensitivity of Anaplastic Lymphoma Kinase Variants
Editor's Summary
baby step toward improving crizotinib therapy in the clinic.aperformed to find the maximum tolerated dose in the pediatric population, this mechanistic study provides more than
mutation. Although careful toxicity studies need to beALKshould improve therapy for patients with this common ALK. These observations suggest that either increasing the dose of crizotinib or engineering higher-affinity inhibitorstherapy, respectively. This reduced sensitivity was caused by a heightened ATP-binding affinity in F1174L-mutated
mutations, F1174L and R1275Q, are unresponsive to and effectively inhibited by crizotinibALKmost common twosubstrate. The authors used human neuroblastoma cell lines and xenografts in mice to show that cancers with the
Crizotinib inhibits kinase activity by competing for binding with the enzyme's adenosine triphosphate (ATP)
mutations.ALKcrizotinib sensitivities of individual . now dissect the molecular mechanisms behind the differentialet alnot appear to respond to crizotinib. Bresler
mutations doALKcarcinomas, and the drug is in early clinical trials for neuroblastoma. However, tumors with certain small cell lung−for the treatment of non−−inhibitor of ALK's tyrosine kinase activity and thus its cell signaling function
a small-molecule−−lymphoma kinase (ALK). The U.S. Food and Drug Administration recently approved crizotinibanaplasticyear of age. Nearly 10% of spontaneous neuroblastoma patients house mutations in the gene that encodes
Neuroblastoma, a malignancy of the autonomic nervous system, is the most common cancer in children under 1
A Boost for Neuroblastoma Therapy
http://stm.sciencemag.org/content/3/108/108ra114.full.htmlcan be found at:
and other services, including high-resolution figures,A complete electronic version of this article
http://stm.sciencemag.org/content/suppl/2011/11/07/3.108.108ra114.DC1.html can be found in the online version of this article at: Supplementary Material
http://www.sciencemag.org/about/permissions.dtl in whole or in part can be found at: article
permission to reproduce this of this article or about obtaining reprintsInformation about obtaining
is a registered trademark of AAAS. Science Translational Medicinerights reserved. The title NW, Washington, DC 20005. Copyright 2011 by the American Association for the Advancement of Science; alllast week in December, by the American Association for the Advancement of Science, 1200 New York Avenue
(print ISSN 1946-6234; online ISSN 1946-6242) is published weekly, except theScience Translational Medicine
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om

R E S EARCH ART I C L E
NEUROBLASTOMA
Differential Inhibitor Sensitivity of AnaplasticLymphoma Kinase Variants Found in NeuroblastomaScott C. Bresler,1,2,3* Andrew C. Wood,4,5* Elizabeth A. Haglund,4,5 Joshua Courtright,4,5
Lili T. Belcastro,4,5 Jefferson S. Plegaria,4,5 Kristina Cole,4,5 Yana Toporovskaya,4,5
Huaqing Zhao,5,6 Erica L. Carpenter,4,5 James G. Christensen,7 John M. Maris,4,5,8
Mark A. Lemmon,1,2† Yaël P. Mossé4,5†
vem
ber
9, 2
011
Activating mutations in the anaplastic lymphoma kinase (ALK ) gene were recently discovered in neuroblastoma, acancer of the developing autonomic nervous system that is the most commonly diagnosed malignancy in the firstyear of life. The most frequent ALK mutations in neuroblastoma cause amino acid substitutions (F1174L andR1275Q) in the intracellular tyrosine kinase domain of the intact ALK receptor. Identification of ALK as an oncogenicdriver in neuroblastoma suggests that crizotinib (PF-02341066), a dual-specific inhibitor of the ALK and Met tyrosinekinases, will be useful in treating this malignancy. Here, we assessed the ability of crizotinib to inhibit proliferationof neuroblastoma cell lines and xenografts expressing mutated or wild-type ALK. Crizotinib inhibited proliferationof cell lines expressing either R1275Q-mutated ALK or amplified wild-type ALK. In contrast, cell lines harboringF1174L-mutated ALK were relatively resistant to crizotinib. Biochemical analyses revealed that this reduced sus-ceptibility of F1174L-mutated ALK to crizotinib inhibition resulted from an increased adenosine triphosphate–binding affinity (as also seen in acquired resistance to epidermal growth factor receptor inhibitors). Thus, thiseffect should be surmountable with higher doses of crizotinib and/or with higher-affinity inhibitors.
n N
o
ostm
.sci
ence
mag
.org
Dow
nloa
ded
from
INTRODUCTION
Neuroblastoma arises in the developing autonomic nervous systemand is the most commonly diagnosed malignancy in the first year oflife. The disease shows a wide range of clinical phenotypes: Althoughtumors regress spontaneously in some patients, most have aggressivemetastatic disease (1). Neuroblastoma remains a leading cause of child-hood cancer mortality despite marked escalations in dose-intensivechemoradiotherapy, and long-term survivors experience significanttreatment-related morbidity (2).
One promising therapeutic target in neuroblastoma is the ana-plastic lymphoma kinase (ALK), an orphan receptor tyrosine kinase(RTK) normally expressed only in the developing nervous system(3). Oncogenic ALK alterations were first described in anaplasticlarge cell lymphoma (4), where a chromosomal translocation leadsto production of a fusion protein with the ALK intracellular regionfused to an N-terminal fragment of nucleophosmin (NPM). OtherALK fusion proteins are potent oncogenic drivers in a subset of non–small cell lung cancers (NSCLCs) (5) and drive inflammatory myo-
1Department of Biochemistry and Biophysics, Perelman School of Medicine at theUniversity of Pennsylvania, Philadelphia, PA 19104–6059, USA. 2Graduate Group inBiochemistry and Molecular Biophysics, Perelman School of Medicine at the Univer-sity of Pennsylvania, Philadelphia, PA 19104, USA. 3Medical Scientist Training Pro-gram, Perelman School of Medicine at the University of Pennsylvania, Philadelphia,PA 19104, USA. 4Division of Oncology and Center for Childhood Cancer Research,Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA. 5Department of Pe-diatrics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia,PA 19104, USA. 6Biostatistics and Data Management Core, Children’s Hospital of Phil-adelphia, Philadelphia, PA 19104, USA. 7Department of Cancer Research, Pfizer GlobalResearch and Development, La Jolla Laboratories, La Jolla, CA 92121, USA. 8AbramsonFamily Cancer Research Institute, Perelman School of Medicine at the University ofPennsylvania, Philadelphia, PA 19104, USA.*These authors contributed equally to this work.†To whom correspondence should be addressed. E-mail: [email protected] (Y.P.M.);[email protected] (M.A.L.)
www.ScienceT
fibroblastic tumors (IMTs) as well as other cancers (6). In neuroblas-toma, germline activating point mutations in the intact ALK genewere revealed by linkage analysis of a set of families with highly pen-etrant autosomal dominant disease (7). In addition, somatic ALKmutations were found in ~10% of sporadic neuroblastoma cases(7–11). The most frequently observed substitutions, together ac-counting for >80% of sporadic ALK mutations in neuroblastomasamples (12), were F1174L and R1275Q, which lie in key regulatoryregions of the ALK receptor kinase domain. Mutations in the intactALK gene have also recently been reported in anaplastic thyroid can-cer (13).
ALK tyrosine kinase activity can be inhibited by crizotinib (PF-02341066), a small-molecule adenosine triphosphate (ATP)–competitiveinhibitor that selectively targets both the ALK and the Met RTKs(14). A recent phase 1 study of crizotinib demonstrated safety and tol-erability in humans, as well as tumor shrinkage or stable disease inmost patients with ALK-dependent NSCLC (15). Crizotinib is alsoin early-phase clinical testing in patients with neuroblastoma. As withother tyrosine kinase inhibitor therapies, acquired resistance to crizotinibis already beginning to emerge (16–18). Understanding how muta-tions affect both kinase activity and inhibitor sensitivity is imperativefor guiding future clinical use of ALK-targeted inhibitors. Here, weexplored the ability of crizotinib to inhibit intact ALK in neuroblas-toma cell line models and analyzed the effects of the two most com-mon activating mutations seen in neuroblastoma on ALK’s tyrosinekinase activity. We found that the F1174L mutation, although activat-ing, reduced ALK sensitivity to crizotinib in xenograft, cell line, andenzymatic assays, consistent with the recent surprising report of thismutation as an acquired resistance mutation in an oncogenic ALKfusion protein (17). Compared with the R1275Q activating mutation,we found that an F1174L substitution increased ATP-binding affinity,leading to crizotinib resistance that should be surmountable with higherdoses of crizotinib or new higher-affinity inhibitors.
ranslationalMedicine.org 9 November 2011 Vol 3 Issue 108 108ra114 1

R E S EARCH ART I C L E
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
RESULTS
The effect of crizotinib on growth of neuroblastoma-derivedcell lines depends on ALK genomic status and thespecific mutationTo assess how the most common ALK mutations in neuroblastoma(F1174L and R1275Q) affect intrinsic ALK activity, we expressed full-length ALK variants in human retinal-pigmented epithelial (RPE)cells immortalized with telomerase reverse transcriptase (hTERT-RPE1). We selected RPE cells because they are derived from humanneural crest–like neuroblastomas but express no endogenous ALK(Fig. 1A). Whereas wild-type ALK expressed in hTERT-RPE1 cellswas not detectably phosphorylated (Fig. 1A), both R1275Q- andF1174L-mutated ALK showed robust autophosphorylation in im-munoblots using an ALK pY1604-specific antibody, regardless ofthe presence of serum in the medium (Fig. 1A, middle panels). Thus,both common neuroblastoma mutations caused constitutive ALK acti-vation to similar extents, as seen in Ba/F3 (10), NIH3T3 (9, 19), andPC12 cells (20), as well as in numerous neuroblastoma-derived celllines (7, 10, 11, 19). Consistent with previous reports (19, 21), two ALKspecies were always observed. Full-length ALK migrates as a 220-kDprotein, and the 140-kD species is a cleavage product lacking partof the extracellular region (21).
We and others have shown that RNA interference (RNAi) knock-down of ALK, or pharmacological inhibition of ALK kinase activity,has an antiproliferative effect in several ALK-mutated neuroblastomacell lines (7, 9–11), associated with G1 arrest and increased apoptosis(10). To exploit this observation clinically, we assessed the cytotoxicityof the ALK/Met inhibitor crizotinib (PF-02341066) in cell models forneuroblastoma. We first analyzed the ability of crizotinib to inhibitsubstrate-adherent growth of a panel of 18 human neuroblastoma–derived cell lines. The 18 cell lines were chosen to represent ALK ge-nomic status in primary tumors, and have all previously been wellcharacterized. Analysis of concentration-response curves across a four-log crizotinib dose range (1 to 10,000 nM) revealed significant dif-ferences in IC50 (median inhibitory concentration) that correlated withALK status. As illustrated in Fig. 1B, cell lines harboring an ALK ab-erration (amplification or mutation) were significantly more sensitiveto growth inhibition by crizotinib than those with wild-type ALK sta-tus (P = 0.001, two-sided exact Wilcoxon-Mann-Whitney test). To en-sure that crizotinib cytotoxicity in this assay reflected ALK inhibition,we confirmed that the drug reduced phospho-ALK (pALK) levels (seebelow). In addition, because crizotinib also inhibits Met kinase activity(14), it was important to exclude Met inhibition as a mechanism. Weconfirmed that none of the crizotinib-inhibited neuroblastoma celllines displayed significant phospho-Met levels (fig. S1). Moreover,RNAi knockdown of Met in a panel of cell lines with altered ALKgenomic status had no growth-inhibitory effect (table S1). The enhancedcrizotinib sensitivity of almost all neuroblastoma cell lines harboringALK mutation or amplification (Fig. 1B) strengthens the argumentfor ALK inhibition as a useful therapeutic strategy in neuroblastoma.
Mutation-specific stratification of crizotinib sensitivity in neuroblas-toma cell lines (Fig. 1B) may have significant implications for clinicaluse of this drug. Cell lines expressing F1174L-mutated ALK were mar-ginally significantly less sensitive to growth inhibition by crizotinibthan those expressing R1275Q-mutated ALK (P = 0.067, two-sidedexact Wilcoxon-Mann-Whitney test), justifying further investigation.This result was initially unexpected, because the F1174L and R1275Q
www.ScienceT
mutations appear to promote similar degrees of constitutive ALKactivation (Fig. 1A), and small interfering RNA (siRNA) knock-down of either variant inhibits growth of the relevant cell lines (7).Assuming a similar oncogenic driver role for both ALK variants, thedata in Fig. 1B suggest that the F1174L mutation may also promotecrizotinib resistance. Indeed, an F1174L mutation was recently foundto cause acquired crizotinib resistance in a patient with an IMT drivenby constitutively active RANBP2-fused ALK (17). The F1174L subs-titution may therefore combine the characteristics of an activating
Fig. 1. Constitutive activity and inhibitor sensitivity of F1174L and R1275ALK mutants. (A) Immunoblots of total ALK and pALK in hTERT-RPE1 cellsinfected with retroviruses directing expression of wild-type (WT) or mu-tated ALK. Lower panel is actin loading control. (B) Proliferation of neuro-blastoma cell lines over 72 hours of incubation with 333 nM crizotinib.Growth inhibition ± SD is reported for at least three independent ex-periments. P values were calculated for marked comparisons with two-sided exact Wilcoxon-Mann-Whitney tests. Cell lines were as follows (leftto right): WT ALK-amplified (NB1); R1275Q (NB1643, LAN5); F1174L (SH-SY5Y,KELLY, NBSD, SMS-SAN); WT ALK, normal copy number (NB1691, NB-EBc1,IMR5, NB16, NLF, IMR32, NBLS, SKNBE2C, NGP, SKNAS, SKNFI).
ranslationalMedicine.org 9 November 2011 Vol 3 Issue 108 108ra114 2

R E S EARCH ART I C L E
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
mutation and a resistance mutation, understanding of which is crucialfor developing ALK-targeted therapy.
Tumor growth driven by F1174L-mutated ALK also showsreduced sensitivity to crizotinib in vivoUsing xenograft models of neuroblastoma, we next asked whetherthe two ALK variants show differential crizotinib sensitivity in vivo.We tested a pharmacologically relevant crizotinib dose (100 mg/kgper day for 4 weeks) against serially passaged human neuroblastomacells xenografted in the flank of CB17 immunodeficient mice (14).As shown in Fig. 2A, crizotinib caused tumor regression within3 weeks in all NB1643 xenografts (R1275Q-ALK), and complete re-gression was sustained over the fourth week of dosing (P < 0.0001).Assessment of pALK levels by immunoblotting NB1643 xenograftlysates harvested after 48 hours of crizotinib administration (4 hoursafter final dose) confirmed inhibition of ALK phosphorylation (Fig. 2A,inset). Consistent with the differential sensitivity suggested in vitro,xenografts harboring F1174L-ALK (SH-SY5Y and NBSD) were sub-stantially less responsive to crizotinib. No tumor regression was ob-served in crizotinib-treated SH-SY5Y xenografts (Fig. 2B), althoughtumor growth was significantly delayed (P < 0.0001) and the timetaken to reach the study endpoint (tumor volume ≥1.5 cm3) wasextended by an average of 7.7 days (P < 0.0001). Crizotinib also failedto reduce tumor volume (P = 0.29) in NBSD (F1174L) xenografts (Fig.2C), although the time to reach the study endpoint was again ex-tended by a mean of 3.7 days (P = 0.04). These results argue that tu-mors driven by F1174L-mutated ALK are significantly less sensitive tocrizotinib in vivo. Crizotinib treatment led to complete tumor regres-sion in NB1 xenografts with amplified, overexpressed wild-type ALK(Fig. 2D), but not in NB-EBc1 or SKNAS xenografts that have noamplification (Fig. 2, E and F). Crizotinib treatment delayed tumorgrowth (P < 0.0001) in NB-EBc1 xenografts (low pALK levels, butno mutation or amplification), but had no detectable effect (P = 0.70)in SKNAS xenografts, which express low levels of wild-type ALK andshow no detectable pALK (Fig. 2F).
The F1174L mutation reduces crizotinib sensitivityof ALK autophosphorylationThe reduced crizotinib sensitivity of cell lines and xenografts harbor-ing F1174L-mutated ALK prompted us to analyze the effects of thisdrug on constitutive ALK activity in representative cell lines: NB1643cells (which express R1275Q ALK) and SH-SY5Y cells (which expressF1174L ALK). Crizotinib treatment abrogated Y1604 phosphorylationof ALK in both cell lines, but at different doses (Fig. 3). In NB1643(R1275Q) cells, ALK phosphorylation was essentially abolished at100 nM crizotinib (IC50, ~10 nM), whereas complete inhibition ofF1174L ALK phosphorylation in SH-SY5Y cells required almost1 mM crizotinib (IC50, ~50 nM). Akt phosphorylation followed simi-lar trends (fig. S2), consistent with a previous report that ALK inhibi-tion promotes apoptosis in neuroblastoma cell lines (10). Crizotinibresistance of cells harboring the ALK F1174L mutation therefore ap-pears to arise from less potent inhibition of constitutive ALK activityand thus of downstream survival signaling.
F1174L and R1275Q mutations promoteautophosphorylation of the ALK kinase domainTo understand why F1174L- and R1275Q-mutated ALK have differ-ent crizotinib sensitivities and how the mutations enhance kinase
www.ScienceT
Fig. 2. Crizotinib activity in vivo for WT and mutated ALK. Subcuta-neously implanted neuroblastoma tumors were monitored in CB17 scidmice treated with crizotinib (solid red lines) or vehicle (dashed bluelines). Tumor volume (left panels) is displayed as mean ± SEM. Studyend points for survival analysis (right panels) were tumor volume ≥1.5cm3
or treatment-related death. (A) NB1643 (R1275Q) xenografts: inset showsimmunoblot of ALK and pALK. (B) SH-SY5Y (F1174L). (C) NBSD (F1174L). (D)NB1 (WT amplified with strong pALK staining). (E) NB-EBc1 (WT, with weakpALK staining). (F) SKNAS (WT, undetectable pALK). Statistical treatmentis described in Materials and Methods.
ranslationalMedicine.org 9 November 2011 Vol 3 Issue 108 108ra114 3

R E S EARCH ART I C L E
activity, we turned to in vitro studies of the ALK tyrosine kinase do-main (ALK-TKD), purified from baculovirus-infected Sf9 cells as ahexahistidine-tagged protein. To generate fully dephosphorylated (in-
www.ScienceT
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
active) ALK-TKD, we used YopH phosphatase treatment to reversespontaneous autophosphorylation arising during production (Supple-mentary Methods). For comparative studies, fully autophosphorylatedALK-TKD was generated by clustering the protein on lipid vesiclesbearing NTA-Ni head groups (DOGS-NTA-Ni), thus imitating ligand-induced ALK dimerization. This method has been used to promoteassembly and activation of other receptor fragments, including epi-dermal growth factor receptor (EGFR)–TKD (22, 23), and markedlyenhances the rate of ALK-TKD autophosphorylation (Fig. 4A).
Our initial investigations with MnCl2-containing assay conditionsused in previous ALK-TKD studies (24–26) suggested only a three-fold increase in kinase activity upon autophosphorylation. This con-trasts starkly with the ~100- to 200-fold activity increase typically seenupon activation loop autophosphorylation of other kinases from theinsulin receptor (IR) family (27–29). The anomaly arises because thepreviously used high (nonphysiological) Mn2+ concentrations both in-crease unphosphorylated ALK-TKD activity (fig. S3, A to C)—as reportedfor other RTKs (30, 31)—and reduce activity of fully autophospho-rylated ALK-TKD (fig. S3D). We therefore used a more physiological10 mM MgCl2 in subsequent studies (and no Mn2+). Under theseconditions, autophosphorylation promoted strong ALK-TKD activa-tion (see below), as did the F1174L and R1275Q mutations (Fig. 4Band fig. S3E).
Native gel electrophoresis (Fig. 4B) showed that autophospho-rylation of dephosphorylated ALK-TKD was substantially accel-erated by the F1174L and R1275Q mutations, confirming that thesemutations activate the isolated kinase domain. Mobility of ALK-TKDin native gels was increased by autophosphorylation, with successiveautophosphorylation events giving rise to four differently phosphoryl-ated species over a period of 0.5 to 20 min at 37°C. Whereas unphos-phorylated wild-type ALK-TKD was still detectable in Fig. 4B after4 min, this species disappeared completely within 1 min for both mu-tated forms. Similarly, the first pALK-TKD species persisted untilat least 8 min for wild-type protein, but only until ~2 min for F1174LALK-TKD and ~3 min for R1275Q ALK-TKD. These results are showngraphically in fig. S4. Using analytical ultracentrifugation, we ruledout the possibility that increased dimerization of mutated TKDs en-hances their autophosphorylation (fig. S5). ALK-TKD remained mono-meric in solution regardless of mutation. The increased ALK-TKDautophosphorylation rates promoted by the F1174L and R1275Q mu-tations therefore reflect elevated basal kinase activity.
Wild-type and mutated ALK-TKDs have similarinhibitor selectivity profilesThe relative crizotinib resistance of F1174L-mutated ALK observedin cell-based and xenograft studies prompted us to compare its inhib-itor selectivity profile with those of wild-type and R1275Q-mutatedALK. We reasoned that the F1174L mutation might alter the drug-binding site so that certain ATP-competitive compounds (such ascrizotinib) are less effective inhibitors of this variant than of R1275Qor wild type. To establish inhibitor selectivity profiles for wild-type,F1174L, and R1275Q ALK-TKD, we assessed the ability of overlap-ping panels of 320 well-characterized kinase inhibitors to inhibit theirin vitro autophosphorylation (see the Supplementary Methods). Asshown in fig. S6, the inhibition profile of F1174L-mutated ALK-TKDwas essentially identical to those for wild-type and R1275Q-mutatedALK-TKD. Thus, the F1174L mutation does not appear to change therelative abilities of different inhibitors to bind ALK-TKD.
Fig. 3. Inhibition of constitutive ALK autophosphorylation by crizotinib.Representative immunoblots of ALK autophosphorylation in neuro-blastoma cell lines after treatment with different crizotinib concentra-tions (0 to 10 mM). (A and B) Whole-cell lysates were immunoblottedfor pALK (using pY1604 antibody), total ALK, and actin (loading con-trol) for (A) NB1643 cells (R1275Q ALK) and (B) SH-SY5Y cells (F1174LALK). Downstream signaling molecules are analyzed in fig. S2. (C) Quan-tification of pALK levels (220-kD species) as a function of crizotinibconcentration.
ranslationalMedicine.org 9 November 2011 Vol 3 Issue 108 108ra114 4

R E S EARCH ART I C L E
Autophosphorylation alone is sufficient formaximal ALK-TKD activationTo better understand the reduced crizotinib sensitivity of F1174L-mutated ALK, we analyzed kinase activity more quantitatively. As abaseline for these studies, it was important first to establish the fullrange of wild-type ALK-TKD activity levels—in fully activated andunactivated states, respectively. We monitored tyrosine phosphoryl-ation of a peptide corresponding to the ALK activation loop, withsequence ARDIYRASYYRKGGCAMLPVK [“YYY” peptide (25)].Table 1 lists kinetic parameters (kcat, Km, peptide, and Km, ATP) forwild-type ALK-TKD in both inactive (unphosphorylated) and ac-tivated (fully autophosphorylated) states.
www.ScienceT
As shown in Fig. 4C and Table 1, autophosphorylation elevatedALK-TKD catalytic efficiency primarily through a 45-fold increase inkcat (P < 0.0001), from 9.32 ± 0.85 min−1 (unphosphorylated) to 424 ±63 min−1 (fully phosphorylated), accompanied by a 1.6-fold decrease inKm, peptide. Values for Km, ATP were higher than reported from Mn2+-containing assays (24–26) and were not significantly altered by ALK-TKD autophosphorylation (Table 1 and Fig. 4D). This contrasts withother RTKs, where activation loop autophosphorylation reduces Km forboth peptide and ATP substrates under similar (MnCl2-free) assay con-ditions (28, 29, 32). Unlike other members of its family, ALK thereforedoes not appear to be autoinhibited by pseudosubstrate-like interactionof its unphosphorylated activation loop with the active site.
ranslationalMedicine.org 9 No
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
Although autophosphorylation in-creases ALK-TKD activity, we wonderedwhether TKD dimerization might playan additional allosteric activating role, asseen with EGFR (23). Whereas clusteringEGFR-TKD on the surface of vesicles con-taining DOGS-NTA-Ni lipids promotedsignificant activation (Fig. 4E), no activat-ing effect was seen when fully phospho-rylated pALK-TKD was similarly treated(Fig. 4F). These data argue against an al-losteric activation mechanism for ALKand suggest that autophosphorylation issufficient for its maximum activation.
F1174L and R1275Q mutationsconstitutively activate ALK-TKD anddisplay different kinetic profilesAs reported in Table 1, the F1174L andR1275Q mutations both cause significantincreases in kcat even without autophos-phorylation. The F1174L mutation in-creased kcat by ~40-fold (P = 0.0013) to365 ± 61 min−1, close to the maximummeasured for fully phosphorylated wild-type protein. The R1275Qmutation had amore modest effect, increasing kcat of un-phosphorylated ALK-TKD by just 12-fold (P = 0.0001). Whereas the R1275Qmutation left Km, peptide unaltered, theF1174L mutation increased Km, peptide byabout threefold. These two neuroblasto-ma mutations therefore promoted similar(10-fold) increases in catalytic efficiency(kcat/Km, peptide) of unphosphorylatedALK-TKD (Fig. 5A and Table 1) in thepresence of saturating ATP. Autophos-phorylation further increased catalytic ef-ficiency (kcat/Km, peptide) for both R1275Qand F1174L ALK-TKD variants (Fig. 5E).For R1275Q, this resulted largely from anincrease (about threefold) inkcat. ForF1174L,phosphorylation reducedKm, peptide by aboutsevenfold (Table 1). In addition to beingconstitutively activated, F1174L ALK-TKDmay be slightly “superactivated” upon full
Fig. 4. Analysis of ALK-TKD activation in vitro. (A) Separation of differently autophosphorylated ALK-TKDspecies by native gel electrophoresis to monitor autophosphorylation at 25°C in the absence (top) andpresence (bottom) of vesicles containing 10% DOGS-NTA-Ni (100 mM total lipid), 10 mM MgCl2, and2 mM ATP. (B) Autophosphorylation of WT and mutated ALK-TKD (10 mM) with saturating ATP (2 mM)and 10 mM MgCl2 at 37°C. Results are quantified in fig. S4. (C) Rate of 32P incorporation at 25°C intosubstrate peptide (see Materials and Methods) for autophosphorylated ALK-TKD (10 nM) and unphos-phorylated ALK-TKD (500 nM) as peptide substrate concentration is increased. ATP concentration was2 mM. (D) Km, ATP determination for autophosphorylated (10 nM) and unphosphorylated (500 nM)ALK-TKD at fixed peptide substrate concentration (1 mM). (E) Enhancement of EGFR-TKD kinase (1 mM)by vesicles containing increasing molar percentages of DOGS-NTA-Ni (10 mM DOGS-NTA-Ni, 100 to200 mM total lipid). (F) Effect on autophosphorylated ALK-TKD (1 mM) activity of adding DOGS-NTA-Nivesicles. Data are shown as means ± SEM from at least three independent experiments. All experimentsexcept those in (B) were performed at 25°C.
vember 2011 Vol 3 Issue 108 108ra114 5

R E S EARCH ART I C L E
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
autophosphorylation for the particular peptide substrate used here(Fig. 5B), with a catalytic efficiency that was ~30% higher than mea-sured for wild-type (P = 0.0342) or R1275Q (P = 0.0327) pALK-TKD.
Reduced Km for ATP explains the relative resistance ofF1174L-mutated ALK to crizotinibThe Km, ATP values listed in Table 1 (and Fig. 5C) suggest one expla-nation for the reduced crizotinib sensitivity of cell lines and xenograftsexpressing F1174L-mutated ALK. Km, ATP for the F1174L mutant was2.3-fold lower than that of R1275Q (P = 0.0007) when autophos-phorylated (Fig. 5, D and F), and 2.6-fold lower (P = 0.0045) in thedephosphorylated form (Fig. 5, C and F). This trend was maintainedwhen the assay was repeated at a higher peptide concentration of 2 mM(fig. S7 and table S2). These data suggest that the F1174L mutationenhances the ATP-binding affinity of ALK, which in turn will reducepotency of any ATP-binding site competitor (such as crizotinib) at cel-lular ATP concentrations, as seen for drug resistance mutations in someother RTKs (33).
To test this hypothesis, we compared the in vitro crizotinib sensi-tivity of the recombinant ALK-TKD variants at two different ATP con-centrations (Fig. 5, G and H). At 0.2 mM ATP (significantly lowerthan cellular levels), IC50 values (measured for 50 nM unphosphoryl-ated protein) were similar for R1275Q and F1174L ALK-TKD (Fig. 5Gand table S3). However, at a more physiological ATP concentrationof 2 mM, F1174L ALK-TKD was significantly less sensitive (P = 0.0128)to crizotinib inhibition (IC50 = 130 nM) than R1275Q ALK-TKD (IC50 =84.6 nM). A similar difference was also seen with autophosphorylatedproteins (table S4), as anticipated from Table 1. Wild-type ALK-TKDresembled the F1174L variant in its relative insensitivity to crizotinibinhibition in vitro (tables S3 and S4), consistent with the finding thatthe drug does not inhibit growth of neuroblastoma cells that expresswild-type ALK at normal levels (Figs. 1B and 2E). However, NB1 cellsthat express the receptor at very high levels because of an ALK am-plification were sensitive to crizotinib (Figs. 1B and 2D), indicating thatinhibitor response may also be affected by issues of trafficking, sub-cellular localization, and expression level (19).
The correlation between the increased ATP-binding affinity ofF1174L-mutated ALK and its reduced sensitivity to crizotinib argues thatthis effect will be general, affecting the response to all ATP-competitiveinhibitors. We tested this hypothesis with two other inhibitors. As shownin fig. S8 and table S5, in vitro inhibition of ALK-TKD by staurosporineis also impaired by the F1174L mutation compared with that seen for
www.ScienceT
R1275Q-mutated ALK-TKD. Moreover, the ALK/insulin-like growthfactor 1 receptor (IGF-1R)/IR inhibitor GSK1838705A (34) was sub-stantially less effective at inducing cytotoxicity in neuroblastoma celllines expressing F1174L ALK than those expressing R1275Q ALK(table S6). Like crizotinib, GSK1838705A only showed significantin vivo activity against R1275Q xenografts (fig. S9). These data, andthe Km, ATP values reported in Table 1, therefore suggest that F1174L-mutated ALK will be less sensitive to all ATP-competitive inhibitors.
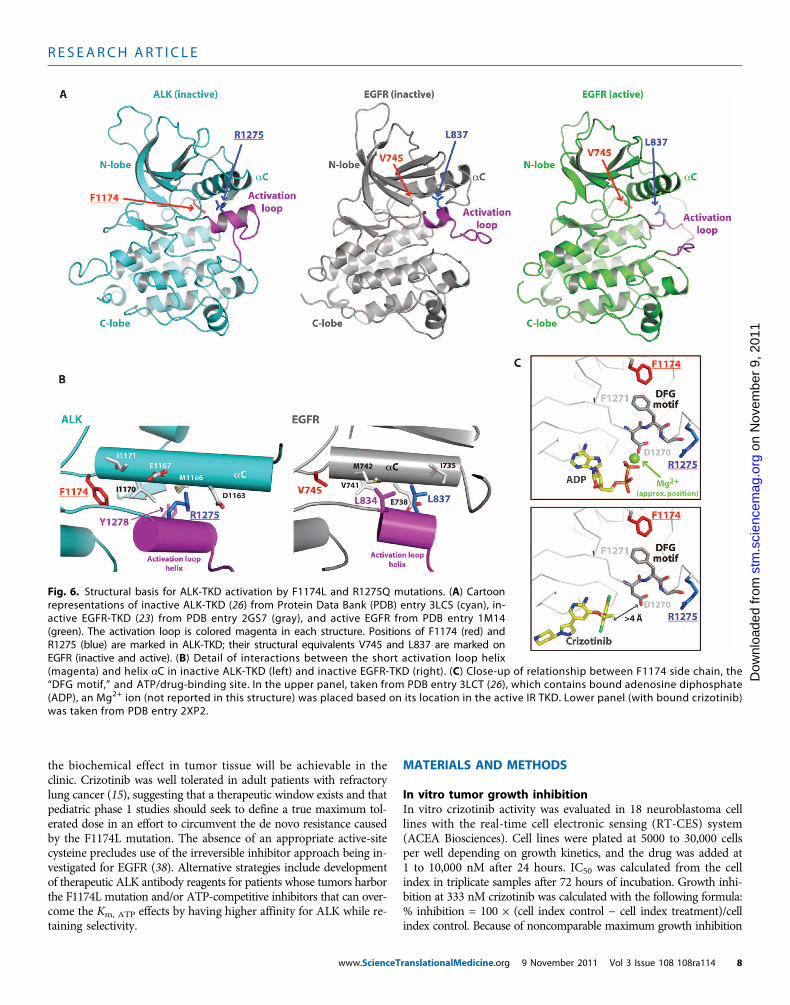
Structural changes can explain the increased ATP-bindingaffinity in F1174L-mutated ALKRecent crystal structures of ALK-TKD (24, 26) revealed unexpectedsimilarities with EGFR-TKD that are useful for understanding andpredicting the consequences of ALK mutations. In the inactive ALKand EGFR TKD structures, key autoinhibitory interactions betweenthe crucial aC helix and a short a helix at the beginning of the ac-tivation loop (magenta in Fig. 6) displace the aC helix from the po-sition it must adopt in the “active” kinase. Intriguingly, the locationof R1275 in ALK coincides closely with that of L837 in EGFR (L861 inpro-EGFR), where mutation to glutamine activates EGFR in NSCLC(35). The R1275 side chain contributes directly to interactions betweenthe magenta activation loop helix and the aC helix (Fig. 6B), stabiliz-ing the autoinhibited ALK-TKD conformation. Replacing R1275 witha glutamine, or phosphorylating nearby Y1278 in the activation loop(Fig. 6B), will disrupt these autoinhibitory interactions and thus acti-vate ALK.
F1174 lies at the C terminus of the aC helix (Fig. 6B), and its sidechain contributes to the small well-packed hydrophobic “core” be-tween aC and the activation loop. A reduction in the size of theF1174 side chain will disrupt this packing, weakening autoinhibitoryinteractions and thus allowing ALK-TKD to adopt its active configu-ration. F1174 also makes another important interaction that may ex-plain its effects on ATP binding. As shown in Fig. 6C, F1174 is in directvan der Waal’s contact with F1271 of the crucial “DFG” motif, the as-partate of which (D1270) coordinates a Mg2+ ion involved in ATP bind-ing. Reducing the size of the F1174 side chain would remove onestructural restraint on the DFGmotif, which may allow D1270 to adopta position with more optimal Mg2+ coordination geometry, slightlyincreasing ATP-binding affinity (that is, reducing Km, ATP). By contrast,crizotinib makes no interactions with the DFG motif (Fig. 6C, lowerpanel) and does not come within 4 Å of the D1270 side chain, soaltering the DFG motif position should not affect crizotinib binding.
Table 1. Kinetic parameters of ALK-TKD. Data are shown as means ± SEM of at least three independent experiments. Details of analysis are de-scribed in the Supplementary Methods. P values quoted in the text were determined with an unpaired t test.
Kinase
kcat(min−1)
Km, peptide(mM)
rans
kcat/Km, peptide
(min−1 mM−1)
lationalMedicine.org 9 Nove
Km, ATP*(mM)
mber 2011 Vol 3 Issue 108
kcat/Km, ATP
(min−1 mM−1)
Wild type
9.32 ± 0.85 2.88 ± 0.42 3.41 ± 0.44 0.134 ± 0.007 69.7R1275Q
119 ± 13 2.56 ± 0.32 46.8 ± 1.9 0.326 ± 0.033 364F1174L
365 ± 61 9.18 ± 1.43 39.7 ± 2.8 0.127 ± 0.011 2870Phospho–wild type
424 ± 63 1.80 ± 0.17 237 ± 35 0.159 ± 0.012 2660Phospho-R1275Q
347 ± 15 1.39 ± 0.10 252 ± 24 0.248 ± 0.015 1400Phospho-F1174L
436 ± 51 1.25 ± 0.20 357 ± 25 0.109 ± 0.001 3980*Determined at 1 mM peptide.
108ra114 6

R E S EARCH ART I C L E
These observations provide a structural hypothesis for how the F1174Lmutation can increase the affinity of ALK-TKD for ATP without af-fecting its affinity for ATP-competitive inhibitors such as crizotinib,
www.ScienceT
staurosporine, and GSK1838705A, leading to relative resistance. Bycontrast, the R1275 side chain is more than 7 Å away from the ATP-binding site, so its mutation should not directly affect ATP binding.
ranslationalMedicine.org 9 No
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
DISCUSSION
Genetic studies have firmly establishedALK as a tractable molecular target inneuroblastoma, as well as several otherhuman malignancies including NSCLC.We established here that neuroblastomacell lines driven by ALK mutation or am-plification were sensitive to crizotinib, anorally bioavailable ATP-competitive ALKinhibitor. Neuroblastoma cell lines andxenografts that express R1275Q-ALK,one of the two most commonly occurringmutations (12), were highly sensitive tocrizotinib. By contrast, those expressingF1174L-mutated ALK (the other of thetwo most common mutations) were re-sistant to crizotinib in vitro and in vivo.We showed that the reduced sensitivityof F1174L-expressing cell lines can be ex-plained, at least inpart, by an increasedATP-binding affinity compared with R1275Q,asmanifested byKm, ATP values, and this re-duces the potency of several ATP-competitiveinhibitors.
The R1275Qmutation in ALK resem-bles oncogenic EGFR mutations found inNSCLC, such as L834R (L858R in pro-EGFR), increasing both kcat and Km, ATP
(36, 37). By contrast, the F1174L mutationin ALK appears to combine the charac-teristics of an activating mutation and aresistance mutation, increasing kcat whilemaintaining a wild-type–like Km, ATP. TheF1174L mutation thus resembles the drug-resistant EGFR L834R/T766M double mu-tation (L858R/T790M in pro-EGFR) thathas a reduced Km, ATP (33, 37). The F1174Lmutation has emerged not only as a built-in resistance mechanism in neuroblasto-ma as described here but also as an escapemechanism in ALK-translocated tumorstreated with crizotinib (17).
Despite the resistance described here,our results indicate that patients harbor-ing the F1174L ALK mutation may ben-efit from treatment with ATP-competitiveALK inhibitors in some circumstances. Forcrizotinib itself, an increase in dosage toovercome the relative difference in Km, ATP
compared with R1275Q-mutated ALK maybe possible, although it remains unclearwhether the exposures necessary to achieve
A B
C D
E F
Unphosphorylated Phosphorylated
0.2 mM ATP 2 mM ATPG H
0.0 0.5 1.0 1.5 2.00
20
40
60
80
min
–1
0.0 0.5 1.0 1.5 2.00.0
0.2
0.4
0.6
0.8
1.0
Frac
tio
n o
f Vm
ax
0.0 0.5 1.0 1.5 2.00.0
0.2
0.4
0.6
0.8
1.0Fr
acti
on
of V
max
Wild-type F1174L R1275Q0.0
0.1
0.2
0.3
0.4
Km
, ATP
(mM
)
–8.0 –7.5 –7.0 –6.5 –6.00.0
0.2
0.4
0.6
0.8
1.0
1.2
Frac
tio
nal
act
ivit
y
–8.0 –7.5 –7.0 –6.5 –6.00.0
0.2
0.4
0.6
0.8
1.0
1.2
Frac
tio
nal
act
ivit
y
0.0 0.5 1.0 1.5 2.00
100
200
300
min
–1
Wild-type F1174L R1275Q0
100
200
300
400
500
k cat/
Km
, pep
tid
e
(min
–1m
M–1
)
F1174LR1275QWild-type
pF1174L
pR1275Q
pwild-type
[Peptide] (mM) [Peptide] (mM)
[ATP] (mM) [ATP] (mM)
F1174LR1275QWild-type
pF1174LpR1275Qpwild-type
UnphosphorylatedPhosphorylated
UnphosphorylatedPhosphorylated
ALK-TKD variant ALK-TKD variant
Log [crizotinib]
F1174L
R1275Q
F1174L
R1275Q
Log [crizotinib]
Fig. 5. Comparison of WT ALK-TKD with F1174L and R1275Q variants in vitro. (A and B) Rates of 32Pincorporation into YYY peptide at saturating ATP (2 mM) for (A) unphosphorylated WT (500 nM) or
mutated (50 nM) ALK-TKD and (B) phosphorylated WT and mutated ALK-TKD (all at 10 nM). (C and D)Km, ATP determination (with YYY peptide fixed at 1 mM) for (C) unphosphorylated WT (500 nM) ormutated (50 nM) ALK-TKD and (D) pALK-TKD variants (all at 10 nM). (E) Comparison of catalytic effi-ciencies (kcat/Km, peptide) for unphosphorylated ALK-TKD and pALK-TKD variants. (F) Comparison ofKm, ATP values. (G and H) Inhibition of unphosphorylated F1174L and R1275Q ALK-TKD (50 nM) bycrizotinib in peptide phosphorylation assays ([peptide] is 0.5 mM) at 0.2 mM ATP (G) and 2 mM ATP(H). All data are shown as means ± SEM from at least three independent experiments. Experiments wereperformed at 25°C.vember 2011 Vol 3 Issue 108 108ra114 7
R E S EARCH ART I C L E
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
the biochemical effect in tumor tissue will be achievable in theclinic. Crizotinib was well tolerated in adult patients with refractorylung cancer (15), suggesting that a therapeutic window exists and thatpediatric phase 1 studies should seek to define a true maximum tol-erated dose in an effort to circumvent the de novo resistance causedby the F1174L mutation. The absence of an appropriate active-sitecysteine precludes use of the irreversible inhibitor approach being in-vestigated for EGFR (38). Alternative strategies include developmentof therapeutic ALK antibody reagents for patients whose tumors harborthe F1174L mutation and/or ATP-competitive inhibitors that can over-come the Km, ATP effects by having higher affinity for ALK while re-taining selectivity.
www.ScienceT
MATERIALS AND METHODS
In vitro tumor growth inhibitionIn vitro crizotinib activity was evaluated in 18 neuroblastoma celllines with the real-time cell electronic sensing (RT-CES) system(ACEA Biosciences). Cell lines were plated at 5000 to 30,000 cellsper well depending on growth kinetics, and the drug was added at1 to 10,000 nM after 24 hours. IC50 was calculated from the cellindex in triplicate samples after 72 hours of incubation. Growth inhi-bition at 333 nM crizotinib was calculated with the following formula:% inhibition = 100 × (cell index control − cell index treatment)/cellindex control. Because of noncomparable maximum growth inhibition
Fig. 6. Structural basis for ALK-TKD activation by F1174L and R1275Q mutations. (A) Cartoonrepresentations of inactive ALK-TKD (26) from Protein Data Bank (PDB) entry 3LCS (cyan), in-active EGFR-TKD (23) from PDB entry 2GS7 (gray), and active EGFR from PDB entry 1M14(green). The activation loop is colored magenta in each structure. Positions of F1174 (red) andR1275 (blue) are marked in ALK-TKD; their structural equivalents V745 and L837 are marked onEGFR (inactive and active). (B) Detail of interactions between the short activation loop helix
(magenta) and helix aC in inactive ALK-TKD (left) and inactive EGFR-TKD (right). (C) Close-up of relationship between F1174 side chain, the“DFG motif,” and ATP/drug-binding site. In the upper panel, taken from PDB entry 3LCT (26), which contains bound adenosine diphosphate(ADP), an Mg2+ ion (not reported in this structure) was placed based on its location in the active IR TKD. Lower panel (with bound crizotinib)was taken from PDB entry 2XP2.ranslationalMedicine.org 9 November 2011 Vol 3 Issue 108 108ra114 8

R E S EARCH ART I C L E
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
depending on ALK status, growth inhibition at a single pharmaco-logically relevant dose was used to compare cell lines. P values were cal-culated with two-sided exact Wilcoxon-Mann-Whitney tests. All lineswere routinelymycoplasma-tested andgenotyped (AmpFISTR Identifilerkit, Applied Biosystems) to verify identity.
In vitro protein and phosphoprotein detectionEach cell line was grown to 70 to 80% confluence, treated with crizotinibat the noted concentration (ranging from 0 to 10,000 nM) for 2 hours,and washed twice with ice-cold phosphate-buffered saline. Whole-celllysates were then analyzed by immunoblotting as described (7) withantibodies to ALK (1:1000; Cell Signaling), pALK Tyr1604 (1:1000; CellSignaling), and actin (1:2000; Santa Cruz). Immunoblots were quan-tified with ImageJ (National Institutes of Health).
In vivo tumor growth inhibitionCB17 scid female mice (Taconic Farms) were used to propagate sub-cutaneously implanted neuroblastoma tumors. Tumor diameters weremeasured twice per week with electronic calipers, and tumor volumeswere calculated with the spheroid formula, (p/6)×d3, where d repre-sents mean diameter. Once tumor volume exceeded 200 mm3, micewere randomized (n = 10 per arm) to receive crizotinib (100 mg/kgper dose) or vehicle (acidified water) daily by oral gavage for 4 weeks.Mice were maintained under protocols and conditions approved byour institutional animal care and use committee. Mice were killed whentumor volume exceeded 1500 mm3. A mixed-effects linear model wasused to assess tumor volume over time between treatment and vehiclegroups, controlling for tumor size at enrollment. Survival analysis wasperformed with the log-rank test with progression defined as tumorvolume exceeding 1500 mm3 or treatment-related death.
In vivo protein and phosphoprotein detectionMice harboring subcutaneously implanted NB1643 neuroblastoma tu-mors were randomized once tumor volume exceeded 300 mm3 (n = 3per arm) to receive crizotinib or vehicle as described above for 2 days.Mice were killed 4 hours after the final dose, and tumors were imme-diately snap-frozen in liquid nitrogen and pulverized for extraction ofwhole-cell lysates with 100 ml of extraction buffer (Invitrogen) con-taining protease inhibitor (Sigma), phosphatase inhibitors (Sigma),and phenylmethylsulfonyl fluoride. Lysates were sonicated, rotated for1 hour at 4°C, and clarified by centrifugation. Protein concentrationwas normalized with the Bradford method, and lysates (200 mg) weresubjected to immunoblotting as outlined above.
Recombinant protein expression and purificationA construct encoding ALK residues 1090 to 1416, together with anN-terminal hexahistidine tag, was used to express hexahistidine-tagged recombinant ALK-TKDs in Sf9 cells as described in the Sup-plementary Methods.
Native gel kinase assaysNative gel electrophoresis was used to monitor autophosphorylationprogress for 10 mM ALK-TKD in 100 mM Hepes (pH 7.4), 150 mMNaCl, 2 mM dithiothreitol, 2 mM ATP, and 10 mMMgCl2, either freein solution or in the presence of lipid vesicles containing 10% DOGS-NTA-Ni (100 mM total lipid). Aliquots (10 ml) were taken at eachtime point, quenched by adding EDTA to 20 mM and placing onice, and then subjected to electrophoresis on 7.5% tris-glycine native
www.ScienceT
gels at 100 V for ~14 hours. Gels were stained with Coomassie bril-liant blue R-250.
Peptide phosphorylation assaysAnalysis of substrate phosphorylation by ALK-TKD used a peptidemimic of the ALK activation loop with the following sequence: biotin-ARDIYRASYYRKGGCAMLPVK (CanPeptide), referred to as YYY pep-tide (25). Assay details (with Mg2+ as divalent cation) and analysis ofinhibition are described in the Supplementary Methods. Spontaneousautophosphorylation is negligible at the ALK-TKD concentrations usedfor assays.
SUPPLEMENTARY MATERIAL
www.sciencetranslationalmedicine.org/cgi/content/full/3/108/108ra114/DC1MethodsFig. S1. Met and pMet protein expression in neuroblastoma cell lines.Fig. S2. Crizotinib inhibits ALK autophosphorylation and downstream signaling in neuroblastomacell lines.Fig. S3. Effects of MnCl2 on ALK-TKD assays.Fig. S4. Progress of ALK-TKD autophosphorylation as assessed in native gels.Fig. S5. Sedimentation equilibrium ultracentrifugation analysis of ALK-TKD variants.Fig. S6. Inhibition of ALK-TKD by inhibitors in the Enzo ScreenWell and EMD InhibitorSelect1 to 3 collections.Fig. S7. Confirmation of Km, ATP values of ALK-TKD variants at different near-saturating peptideconcentrations.Fig. S8. The F1174L mutation also increases IC50 for staurosporine inhibition.Fig. S9. The ALK/IGF-1R inhibitor GSK1838705A promotes regression of xenografts containingR1275Q mutations, but not F1174L or wild-type ALK.Table S1. Effect of siRNA knockdown of ALK and Met on proliferation of neuroblastoma cell lines.Table S2. Dependence of Km, ATP on peptide concentration for unphosphorylated ALK-TKDvariants.Table S3. Crizotinib inhibition of unphosphorylated ALK-TKD in vitro at two different ATPconcentrations.Table S4. IC50 measurements for crizotinib inhibition of phosphorylated ALK-TKD variants (10 nM).Table S5. IC50 measurements for in vitro staurosporine inhibition of ALK-TKD variants.Table S6. IC50 measurements for inhibition of neuroblastoma cell lines by the ALK/InsR/IGF-1Rinhibitor GSK1838705A.References
REFERENCES AND NOTES
1. G. J. D’Angio, A. E. Evans, C. E. Koop, Special pattern of widespread neuroblastoma with afavourable prognosis. Lancet 1, 1046–1049 (1971).
2. W. L. Hobbie, T. Moshang, C. A. Carlson, E. Goldmuntz, N. Sacks, S. B. Goldfarb, S. A. Grupp,J. P. Ginsberg, Late effects in survivors of tandem peripheral blood stem cell transplantfor high-risk neuroblastoma. Pediatr. Blood Cancer 51, 679–683 (2008).
3. T. Iwahara, J. Fujimoto, D. Wen, R. Cupples, N. Bucay, T. Arakawa, S. Mori, B. Ratzkin,T. Yamamoto, Molecular characterization of ALK, a receptor tyrosine kinase expressedspecifically in the nervous system. Oncogene 14, 439–449 (1997).
4. S. W. Morris, M. N. Kirstein, M. B. Valentine, K. G. Dittmer, D. N. Shapiro, D. L. Saltman,A. T. Look, Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’slymphoma. Science 263, 1281–1284 (1994).
5. M. Soda, Y. L. Choi, M. Enomoto, S. Takada, Y. Yamashita, S. Ishikawa, S. Fujiwara, H. Watanabe,K. Kurashina, H. Hatanaka, M. Bando, S. Ohno, Y. Ishikawa, H. Aburatani, T. Niki, Y. Sohara,Y. Sugiyama, H. Mano, Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 (2007).
6. R. H. Palmer, E. Vernersson, C. Grabbe, B. Hallberg, Anaplastic lymphoma kinase: Signallingin development and disease. Biochem. J. 420, 345–361 (2009).
7. Y. P. Mossé, M. Laudenslager, L. Longo, K. A. Cole, A. Wood, E. F. Attiyeh, M. J. Laquaglia,R. Sennett, J. E. Lynch, P. Perri, G. Laureys, F. Speleman, C. Kim, C. Hou, H. Hakonarson,A. Torkamani, N. J. Schork, G. M. Brodeur, G. P. Tonini, E. Rappaport, M. Devoto, J. M. Maris,Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455,930–935 (2008).
ranslationalMedicine.org 9 November 2011 Vol 3 Issue 108 108ra114 9

R E S EARCH ART I C L E
on
Nov
embe
r 9,
201
1st
m.s
cien
cem
ag.o
rgD
ownl
oade
d fr
om
8. H. Carén, F. Abel, P. Kogner, T. Martinsson, High incidence of DNA mutations and geneamplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J.416, 153–159 (2008).
9. Y. Chen, J. Takita, Y. L. Choi, M. Kato, M. Ohira, M. Sanada, L. Wang, M. Soda, A. Kikuchi,T. Igarashi, A. Nakagawara, Y. Hayashi, H. Mano, S. Ogawa, Oncogenic mutations of ALKkinase in neuroblastoma. Nature 455, 971–974 (2008).
10. R. E. George, T. Sanda, M. Hanna, S. Fröhling, W. Luther II, J. Zhang, Y. Ahn, W. Zhou,W. B. London, P. McGrady, L. Xue, S. Zozulya, V. E. Gregor, T. R. Webb, N. S. Gray, D. G. Gilliland,L. Diller, H. Greulich, S. W. Morris, M. Meyerson, A. T. Look, Activating mutations in ALKprovide a therapeutic target in neuroblastoma. Nature 455, 975–978 (2008).
11. I. Janoueix-Lerosey, D. Lequin, L. Brugières, A. Ribeiro, L. de Pontual, V. Combaret, V. Raynal,A. Puisieux, G. Schleiermacher, G. Pierron, D. Valteau-Couanet, T. Frebourg, J. Michon, S. Lyonnet,J. Amiel, O. Delattre, Somatic and germline activating mutations of the ALK kinase receptorin neuroblastoma. Nature 455, 967–970 (2008).
12. S. De Brouwer, K. De Preter, C. Kumps, P. Zabrocki, M. Porcu, E. M. Westerhout, A. Lakeman,J. Vandesompele, J. Hoebeeck, T. Van Maerken, A. De Paepe, G. Laureys, J. H. Schulte,A. Schramm, C. Van Den Broecke, J. Vermeulen, N. Van Roy, K. Beiske, M. Renard, R. Noguera,O. Delattre, I. Janoueix-Lerosey, P. Kogner, T. Martinsson, A. Nakagawara, M. Ohira, H. Caron,A. Eggert, J. Cools, R. Versteeg, F. Speleman, Meta-analysis of neuroblastomas reveals askewed ALK mutation spectrum in tumors with MYCN amplification. Clin. Cancer Res. 16,4353–4362 (2010).
13. A. K. Murugan, M. Xing, Anaplastic thyroid cancers harbor novel oncogenic mutations ofthe ALK gene. Cancer Res. 71, 4403–4411 (2011).
14. J. G. Christensen, H. Y. Zou, M. E. Arango, Q. Li, J. H. Lee, S. R. McDonnell, S. Yamazaki,G. R. Alton, B. Mroczkowski, G. Los, Cytoreductive antitumor activity of PF-2341066, anovel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models ofanaplastic large-cell lymphoma. Mol. Cancer Ther. 6, 3314–3322 (2007).
15. E. L. Kwak, Y. J. Bang, D. R. Camidge, A. T. Shaw, B. Solomon, R. G. Maki, S. H. Ou,B. J. Dezube, P. A. Jänne, D. B. Costa, M. Varella-Garcia, W. H. Kim, T. J. Lynch, P. Fidias,H. Stubbs, J. A. Engelman, L. V. Sequist, W. Tan, L. Gandhi, M. Mino-Kenudson, G. C. Wei,S. M. Shreeve, M. J. Ratain, J. Settleman, J. G. Christensen, D. A. Haber, K. Wilner, R. Salgia,G. I. Shapiro, J. W. Clark, A. J. Iafrate, Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N. Engl. J. Med. 363, 1693–1703 (2010).
16. Y. L. Choi, M. Soda, Y. Yamashita, T. Ueno, J. Takashima, T. Nakajima, Y. Yatabe, K. Takeuchi,T. Hamada, H. Haruta, Y. Ishikawa, H. Kimura, T. Mitsudomi, Y. Tanio, H. Mano; ALK LungCancer Study Group, EML4-ALK mutations in lung cancer that confer resistance to ALKinhibitors. N. Engl. J. Med. 363, 1734–1739 (2010).
17. T. Sasaki, K. Okuda, W. Zheng, J. Butrynski, M. Capelletti, L. Wang, N. S. Gray, K. Wilner,J. G. Christensen, G. Demetri, G. I. Shapiro, S. J. Rodig, M. J. Eck, P. A. Jänne, Theneuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinaseinhibitor in ALK-translocated cancers. Cancer Res. 70, 10038–10043 (2010).
18. R. Katayama, T. M. Khan, C. Benes, E. Lifshits, H. Ebi, V. M. Rivera, W. C. Shakespeare,A. J. Iafrate, J. A. Engelman, A. T. Shaw, Therapeutic strategies to overcome crizotinibresistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK.Proc. Natl. Acad. Sci. U.S.A. 108, 7535–7540 (2011).
19. P. Mazot, A. Cazes, M. C. Boutterin, A. Figueiredo, V. Raynal, V. Combaret, B. Hallberg,R. H. Palmer, O. Delattre, I. Janoueix-Lerosey, M. Vigny, The constitutive activity of theALK mutated at positions F1174 or R1275 impairs receptor trafficking. Oncogene 30,2017–2025 (2011).
20. T. Martinsson, T. Eriksson, J. Abrahamsson, H. Caren, M. Hansson, P. Kogner, S. Kamaraj,C. Schönherr, J. Weinmar, K. Ruuth, R. H. Palmer, B. Hallberg, Appearance of the novelactivating F1174S ALK mutation in neuroblastoma correlates with aggressive tumorprogression and unresponsiveness to therapy. Cancer Res. 71, 98–105 (2011).
21. C. Moog-Lutz, J. Degoutin, J. Y. Gouzi, Y. Frobert, N. Brunet-de Carvalho, J. Bureau,C. Créminon, M. Vigny, Activation and inhibition of anaplastic lymphoma kinase receptortyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin.J. Biol. Chem. 280, 26039–26048 (2005).
22. A. L. Shrout, D. J. Montefusco, R. M. Weis, Template-directed assembly of receptorsignaling complexes. Biochemistry 42, 13379–13385 (2003).
23. X. Zhang, J. Gureasko, K. Shen, P. A. Cole, J. Kuriyan, An allosteric mechanism for acti-vation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149(2006).
24. R. T. Bossi, M. B. Saccardo, E. Ardini, M. Menichincheri, L. Rusconi, P. Magnaghi, P. Orsini,N. Avanzi, A. L. Borgia, M. Nesi, T. Bandiera, G. Fogliatto, J. A. Bertrand, Crystal structuresof anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry49, 6813–6825 (2010).
25. A. Donella-Deana, O. Marin, L. Cesaro, R. H. Gunby, A. Ferrarese, A. M. Coluccia, C. J. Tartari,L. Mologni, L. Scapozza, C. Gambacorti-Passerini, L. A. Pinna, Unique substrate specificity ofanaplastic lymphoma kinase (ALK): Development of phosphoacceptor peptides for theassay of ALK activity. Biochemistry 44, 8533–8542 (2005).
www.ScienceTra
26. C. C. Lee, Y. Jia, N. Li, X. Sun, K. Ng, E. Ambing, M. Y. Gao, S. Hua, C. Chen, S. Kim, P. Y. Michellys,S. A. Lesley, J. L. Harris, G. Spraggon, Crystal structure of the ALK (anaplastic lymphoma kinase)catalytic domain. Biochem. J. 430, 425–437 (2010).
27. M. H. Cobb, B. C. Sang, R. Gonzalez, E. Goldsmith, L. Ellis, Autophosphorylation activatesthe soluble cytoplasmic domain of the insulin receptor in an intermolecular reaction.J. Biol. Chem. 264, 18701–18706 (1989).
28. S. Favelyukis, J. H. Till, S. R. Hubbard, W. T. Miller, Structure and autoregulation of theinsulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 8, 1058–1063 (2001).
29. C. M. Furdui, E. D. Lew, J. Schlessinger, K. S. Anderson, Autophosphorylation of FGFR1kinase is mediated by a sequential and precisely ordered reaction. Mol. Cell 21, 711–717(2006).
30. M. R. Grace, C. T. Walsh, P. A. Cole, Divalent ion effects and insights into the catalyticmechanism of protein tyrosine kinase Csk. Biochemistry 36, 1874–1881 (1997).
31. S. R. Wente, M. Villalba, V. L. Schramm, O. M. Rosen, Mn2+-binding properties of a recom-binant protein-tyrosine kinase derived from the human insulin receptor. Proc. Natl. Acad.Sci. U.S.A. 87, 2805–2809 (1990).
32. J. H. Till, M. Becerra, A. Watty, Y. Lu, Y. Ma, T. A. Neubert, S. J. Burden, S. R. Hubbard, Crystalstructure of the MuSK tyrosine kinase: Insights into receptor autoregulation. Structure 10,1187–1196 (2002).
33. M. J. Eck, C. H. Yun, Structural and mechanistic underpinnings of the differential drugsensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 1804,559–566 (2010).
34. P. Sabbatini, S. Korenchuk, J. L. Rowand, A. Groy, Q. Liu, D. Leperi, C. Atkins, M. Dumble,J. Yang, K. Anderson, R. G. Kruger, R. R. Gontarek, K. R. Maksimchuk, S. Suravajjala, R. R. Lapierre,J. B. Shotwell, J. W. Wilson, S. D. Chamberlain, S. K. Rabindran, R. Kumar, GSK1838705Ainhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase andshows antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 8,2811–2820 (2009).
35. S. V. Sharma, D. W. Bell, J. Settleman, D. A. Haber, Epidermal growth factor receptor mutationsin lung cancer. Nat. Rev. Cancer 7, 169–181 (2007).
36. K. D. Carey, A. J. Garton, M. S. Romero, J. Kahler, S. Thomson, S. Ross, F. Park, J. D. Haley,N. Gibson, M. X. Sliwkowski, Kinetic analysis of epidermal growth factor receptor somaticmutant proteins shows increased sensitivity to the epidermal growth factor receptortyrosine kinase inhibitor, erlotinib. Cancer Res. 66, 8163–8171 (2006).
37. C. H. Yun, K. E. Mengwasser, A. V. Toms, M. S. Woo, H. Greulich, K. K. Wong, M. Meyerson,M. J. Eck, The T790M mutation in EGFR kinase causes drug resistance by increasing theaffinity for ATP. Proc. Natl. Acad. Sci. U.S.A. 105, 2070–2075 (2008).
38. W. Zhou, D. Ercan, L. Chen, C. H. Yun, D. Li, M. Capelletti, A. B. Cortot, L. Chirieac, R. E. Iacob,R. Padera, J. R. Engen, K. K. Wong, M. J. Eck, N. S. Gray, P. A. Jänne, Novel mutant-selectiveEGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 (2009).
39. Acknowledgments: We thank members of the Lemmon, Mossé, and Maris labora-tories for valuable discussions, and Pfizer for their gift of crizotinib. Funding: Thiswork was funded in part by the U.S. Army Peer-Reviewed Medical Research Program(W81XWH-10-1-0212/3 to M.A.L. and Y.P.M.), NIH grant R01-CA140198 (Y.P.M.), theCarly Hillman Fund (Y.P.M.), the Alex’s Lemonade Stand Foundation (J.M.M.), and theAbramson Family Cancer Research Institute (J.M.M.). S.C.B. was supported by an NIHTraining Grant in Structural Biology (T32-GM008275). A.C.W. was supported by a fellow-ship from the St. Baldrick’s Foundation. Author contributions: S.C.B. designed and per-formed in vitro studies of ALK-TKD, analyzed and interpreted the data, and drafted themanuscript. A.C.W. designed and performed cellular and in vivo experiments, analyzedand interpreted the data, and drafted the manuscript. E.A.H., J.C., L.T.B., J.S.P., K.C., Y.T.,and H.Z. made important contributions to cellular and in vivo experiments and/or dataanalysis. E.L.C., J.G.C., J.M.M., Y.P.M., and M.A.L. guided study design, implementation,and interpretation; provided conceptual direction; and wrote the manuscript with S.C.B.and A.C.W. Competing interests: J.G.C. is an employee of Pfizer, the developer of crizotinib.J.M.M. and Y.P.M. are authors on a patent related to the discovery of ALK mutations filedby the Children’s Hospital of Philadelphia (WO/2009/103061 or PCT/US2009/034288:Methods and Compositions for Identifying, Diagnosing, and Treating Neuroblastoma).The other authors declare that they have no competing interests.
Submitted 21 July 2011Accepted 30 September 2011Published 9 November 201110.1126/scitranslmed.3002950
Citation: S. C. Bresler, A. C. Wood, E. A. Haglund, J. Courtright, L. T. Belcastro, J. S. Plegaria,K. Cole, Y. Toporovskaya, H. Zhao, E. L. Carpenter, J. G. Christensen, J. M. Maris, M. A. Lemmon,Y. P. Mossé, Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found inneuroblastoma. Sci. Transl. Med. 3, 108ra114 (2011).
nslationalMedicine.org 9 November 2011 Vol 3 Issue 108 108ra114 10

www.sciencetranslationalmedicine.org/cgi/content/full/3/108/108ra114/DC1
Supplementary Materials for
Differential Inhibitor Sensitivity of Anaplastic Lymphoma Kinase Variants Found in Neuroblastoma
Scott C. Bresler, Andrew C. Wood, Elizabeth A. Haglund, Joshua Courtright, Lili T.
Belcastro, Jefferson S. Plegaria, Kristina Cole, Yana Toporovskaya, Huaqing Zhao, Erica L. Carpenter, James G. Christensen, John M. Maris, Mark A. Lemmon,* Yaël P. Mossé*
*To whom correspondence should be addressed. E-mail: [email protected] (Y.P.M.);
[email protected] (M.A.L.)
Published 9 November 2011, Sci. Transl. Med. 3, 108ra114 (2011) DOI: 10.1126/scitranslmed.3002950
The PDF file includes:
Methods Fig. S1. Met and pMet protein expression in neuroblastoma cell lines. Fig. S2. Crizotinib inhibits ALK autophosphorylation and downstream signaling in neuroblastoma cell lines. Fig. S3. Effects of MnCl2 on ALK-TKD assays. Fig. S4. Progress of ALK-TKD autophosphorylation as assessed in native gels. Fig. S5. Sedimentation equilibrium ultracentrifugation analysis of ALK-TKD variants. Fig. S6. Inhibition of ALK-TKD by inhibitors in the Enzo ScreenWell and EMD InhibitorSelect 1 to 3 collections. Fig. S7. Confirmation of Km, ATP values of ALK-TKD variants at different near-saturating peptide concentrations. Fig. S8. The F1174L mutation also increases IC50 for staurosporine inhibition. Fig. S9. The ALK/IGF-1R inhibitor GSK1838705A promotes regression of xenografts containing R1275Q mutations, but not F1174L or wild-type ALK. Table S1. Effect of siRNA knockdown of ALK and Met on proliferation of neuroblastoma cell lines. Table S2. Dependence of Km, ATP on peptide concentration for unphosphorylated ALK-TKD variants. Table S3. Crizotinib inhibition of unphosphorylated ALK-TKD in vitro at two different ATP concentrations. Table S4. IC50 measurements for crizotinib inhibition of phosphorylated ALK-TKD variants (10 nM). Table S5. IC50 measurements for in vitro staurosporine inhibition of ALK-TKD variants.

Table S6. IC50 measurements for inhibition of neuroblastoma cell lines by the ALK/InsR/IGF-1R inhibitor GSK1838705A. References

2
SUPPLEMENTARY METHODS
DNA constructs and retrovirus production. Mutated and wild-type ALK cDNAs were cloned into the pCMV-XLS vector and subcloned into the retroviral vector MigR1, which also directs EGFP expression. Infection of retinal pigment epithelial cells (RPE1) that express telomerase (hTERT-RPE1) was performed as follows: Phoenix Ampho cells (Oribigen – RVC-10001) were plated ~ 500,000 cells in a 6 well plate in DMEM media with 10% FBS, 1% Pen/Strep, Gentamicin. Twenty-four hours after plating (~50% confluent), Phoenix cells were transfected with the ALK expression constructs using a 6:1 dilution of Fugene:plasmid DNA, and virus-containing medium was harvested 48 hours later. hTERT-RPE1 cells were plated at ~500,000 cells per well in a 6 well plate, and growth medium was replaced with ʻvirus cocktailʼ (2 ml growth medium, 1ml filtered (0.45µm) viral media from Phoenix cells, 4µg/ml Polybrene (Santa Cruz)) and incubated overnight. Viral medium then replaced with fresh growth medium, and hTERT-RPE1 cells were incubated for 48 hours before sorting by flow cytometry for EGFP-positive cells. Production and purification of recombinant ALK-TKD proteins. DNA encoding ALK residues 1090-1416, together with an N-terminal hexahistidine tag, was subcloned into pFastBac-1 (Invitrogen) for expression of histidine-tagged recombinant ALK-TKDs in Sf9 cells. Recombinant baculovirus was generated using the Bac-to-Bac system (Invitrogen). Sf9 cells were infected with recombinant virus for 3 days at 27˚C, harvested by centrifugation, and lysed by sonication. His-tagged protein was recovered from the lysis supernatant using NTA-Ni-agarose beads (Qiagen). After elution from NTA-Ni beads, ALK-TKD was dephosphorylated by incubation with 1µM YopH phosphatase (S1) for 12 hours at 4˚C to reverse spontaneous autophosphorylation that occurs within Sf9 cells during expression. ALK-TKD was then flowed over a cation exchange column (to remove YopH), exchanged into buffer containing 1M (NH4)2SO4, and applied to a butyl sepharose column (GE Healthcare). Protein was eluted with a 20 column-volume linear gradient to 0M (NH4)2SO4. ALK-TKD fractions were pooled and concentrated, and gel filtered using a Superdex 200 column (GE Healthcare) equilibrated in 25mM HEPES pH 7.4, 150mM NaCl, 4mM DTT. Mass spectrometry confirmed that dephosphorylation of the starting material was complete. Protein concentrations were determined by absorbance at 280nm using the calculated extinction coefficient 39440cm-1M-1. Phosphorylated ALK-TKD for peptide-based assays was produced in 100mM HEPES pH 7.4, 150mM NaCl, 2mM DTT,10mM MgCl2, 2mM ATP in the presence of lipid vesicles containing 10% DOGS-NTA-Ni (Avanti Polar Lipids) in a background of dioleoylphosphatidylcholine (Avanti Polar Lipids) prepared as described (S2) with a final concentration of 10µM DOGS-NTA-Ni (100µM total lipid). Full ALK-TKD autophosphosphorylation was achieved within 50mins (Fig. 4A, lower panel) under these conditions at 25˚C. Reactions were quenched where necessary by addition of EDTA to a final concentration of 20mM, followed by desalting under the conditions described above for gel filtration. EGFR-TKD was prepared as described (S2). Mass spectrometry-based phosphopeptide analysis confirmed that ALK-TKD autophosphorylation occurs at the expected sites within the activation loop, as well as additional sites. In vitro protein and phosphoprotein detection. Each neuroblastoma cell line was cultured in ten T75 flasks under standard cell culture conditions. At 70-80% confluence crizotinib was added to cell culture medium to achieve a designated final concentration at one of nine doses ranging from 0nM to 10,000nM. Cells were incubated for 2 hours with drug, then collected, pelleted, and washed twice with ice cold phosphate-buffered saline (PBS). Whole cell lysates were then harvested, separated and

3
immunoblotted as previously described (S3). The following antibodies were used, according to the supplierʼs instructions: anti-ALK (1:1,000; Cell Signaling, 3333), anti-phospho-ALK Tyr 1604 (1:1,000, Cell Signaling, 3341), anti-STAT3 (1:1,000; Cell Signaling, 9132), antiphospho-STAT3 Tyr 705 (1:1,000; Cell Signaling, 9145), anti-AKT (1:1,000; Cell Signaling, 9272), anti-phospho-AKT Ser 473 (1:1,000; Invitrogen, 44-621G), anti-p44/42 MAPK (ERK1/2) (1:1,000; Cell Signaling, 4695), anti-phospho-p44/42 MAPK (ERK 1/2) (1:1,000; Cell Signaling, 9101), anti-actin (1:2,000; Santa Cruz, sc-1616). Quantification of native gels. Quantification of autophosphorylation progress employed ImageJ (NIH). Background was subtracted using a rollerball radius of 50 pixels. Using the gel analysis tool, the intensity of each band in the native gel was determined and normalized to the total intensity in each respective lane. Data were plotted as fraction of total intensity in each lane vs. reaction time. Sedimentation equilibrium analytical ultracentrifugation. ALK-TKD proteins were diluted to 10 and 16.5µM in 25mM HEPES pH 7.4, 150mM NaCl, 2mM DTT, and subjected to sedimentation equilibrium ultracentrifugation at 13,000, 19,000, and 25,000 rpm in a Beckman Optima XL-A ultracentrifuge. Representative data from 13,000 rpm at 10µM are shown in fig. S5. Data are plotted as ln(abs280) vs (r2 -r0
2)/2, where r is the radial distance of the sample and r0 is the radial distance of the meniscus. The slope of the line is proportional to the weight-average molecular weight of the species in the sample. Data were analyzed using Sedphat (http://analyticalultracentrifugation.com). Inhibitor profiling using commercially available kinase inhibitor screens. Kinase inhibitor screens containing 320 (overlapping) commercially available and well-characterized ATP-competitive inhibitors (Enzo Life Sciences ScreenWell and EMD InhibitorSelect Kinase Inhibitor Libraries 1-3) were used to compare the inhibitor selectivities of wild-type, R1275Q, and F1174L ALK-TKD. ALK TKD (2µM) was incubated with ATP (200µM, corresponding to the approximate Km,ATP of ALK-TKD at 10mM MgCl2) in 96-well format with 50µM of each inhibitor (DMSO only in the negative control wells, or TAE-684 in the positive control wells) in a final reaction volume of 10µl. After incubation at room temperature, reactions were quenched with by adding EDTA to a final concentration of 20mM in 25mM HEPES pH 7.4, 150mM NaCl, 4mM DTT, and spotted onto nitrocellulose membranes in triplicate. Membranes were immunoblotted with anti-phosphotyrosine (PY20, 1:1000, Biomol), detecting with HRP-conjugated α-mouse IgG (1:1000, GE Healthcare). In vitro kinase assays. Analysis of substrate phosphorylation by ALK-TKD employed a peptide mimic of the ALK activation loop with sequence: biotin-ARDIYRASYYRKGGCAMLPVK (CanPeptide), referred to as YYY peptide (S4). Autophosphorylated ALK-TKD was used at 10nM in these assays, whereas unphosphorylated kinase was studied at 500nM (wild-type) or 50nM (R1275Q and F1174L), concentrations at which spontaneous autophosphorylation is negligible. Assays monitored incorporation of 32P from γ-32P ATP included in trace amounts (~20µCi per experiment) in the reactions. Comparisons with other peptides confirmed that peptide biotinylation does not affect kinetic parameters, and that reaction rates were linear with respect to both enzyme concentration and time under the assay conditions used (fig. S3E,F). Peptide concentrations were determined by absorbance at 280nm using a calculated extinction coefficient 3960cm-1M-1.

4
Assays were performed in 100mM HEPES pH 7.4, 150mM NaCl, 2mM DTT, 10mM MgCl2, and 0.5mg/mL BSA at 25˚C, and were initiated by mixing equal volumes of solutions containing ALK-TKD and substrates respectively (at twice the desired final concentration). For determination of kcat and Km, peptide, peptide concentration was varied from 0 to 2mM, and ATP was saturating (2mM). For determination of Km, ATP, peptide was present at a fixed concentration of 1 or 2mM, and ATP was varied from 0.015625 to 2mM. Aliquots were taken at each time point, spotted onto pieces of phosphocellulose paper (Upstate Biotechnology), and immediately quenched with 0.5% phosphoric acid followed by extensive washing and drying with acetone. Incorporated radioactivity was measured by scintillation counting with appropriate background correction. Initial rates (determined at <10% product formation) were calculated using measured apparent γ-32P ATP specific activity, normalized to enzyme concentration, and fit to the Michaelis-Menten equation (vo = vmax[S]/(Km+[S])) using GraphPad Prism 4.0. The enzyme was assumed to have one active site per molecule, therefore kcat = vmax at saturating ATP concentrations.
For experiments with added vesicles, which were prepared essentially as described (S2), the appropriate amount of vesicle solution (10µM DOGS-NTA-Ni, 100 or 200µM total lipid depending on the molar percentage of DOGS-NTA-Ni used) was added to the 2x protein mixture, and the reaction was performed and processed as above. Control experiments were performed with vesicles containing only dioleoylphosphatidylcholine (200µM total lipid). Enzyme concentration was held fixed at 1µM, with 2mM peptide and 2mM ATP present. Experiments using EGFR-TKD were performed with poly-Glu4Tyr (Sigma) instead of YYY peptide. Data are reported in Table 1 and tables S2-S5 as the mean of individual experiments (in at least triplicate) ± SEM. p values were determined using an unpaired t-test in GraphPad Prism 4.0.
In vitro analysis of ALK-TKD inhibition. Inhibitor was added to the 2x substrate mixture prior to initiating reactions with ALK-TKD addition, so that final reactions represented a log2 dilution series from 0 to 25,600nM inhibitor. Protein concentration was held at 50nM, and assays were performed as described above. The concentration of ʻYYYʼ peptide was held fixed at 0.5mM, and ATP was present at 0.2mM or 2mM. Background counts from a no-enzyme control were subtracted, the data were normalized to the 0nM inhibitor reaction and were fit to a sigmoidal dose-response (variable slope) equation in GraphPad Prism 4.0. Data are reported as means of individual experiments (in at least triplicate) ± SEM. p values were determined using an unpaired t-test in GraphPad Prism 4.0.

5
Figure S1. Met and pMet protein expression in neuroblastoma cell lines. Immunoblots showing native Met and phospho-Met expression in 7 neuroblastoma cell lines, as well as native HeLa cells and HeLa cells treated with hepatocyte growth factor (HGF), the ligand for Met. F1174L-mutated ALK is expressed in NBSD and KELLY cells. R1275Q ALK is expressed in NB1643 cells. NB1 cells have amplified wild-type ALK, and IMR32 cells express wild-type ALK at normal levels. NLF and SKNAS cells harbor no ALK alteration (and do not appear to express ALK).

6
Figure S2. Crizotinib inhibits ALK autophosphorylation and downstream signaling in neuroblastoma cell lines. Indicated cell lines were treated with the noted crizotinib concentrations and prepared for immunoblotting of whole cell lysates as described in Supplementary Methods. Diminution of pAkt levels approximately parallels that of ALK phosphorylation, with pAkt levels maximally reduced by ~100nM crizotinib in NB1 cells, ~333nM in NB1643 cells, but not until 10,000nM in SH-SY5Y cells. Substantial reduction of pSTAT3 levels requires higher crizotinib concentrations in all three cell lines, and pERK remains detectable even at 10,000nM crizotinib in NB1643 and SH-SY5Y cells. These data are consistent with previous reports that inhibition (or knockdown) of mutated ALK promotes apoptosis of neuroblastoma cell lines (S5). The differences in ʻsharpnessʼ of pAkt and pERK inhibition between NB1 and NB1643/SH-SY5Y cells appear to reflect receptor expression levels (which are very high in NB1)..
7
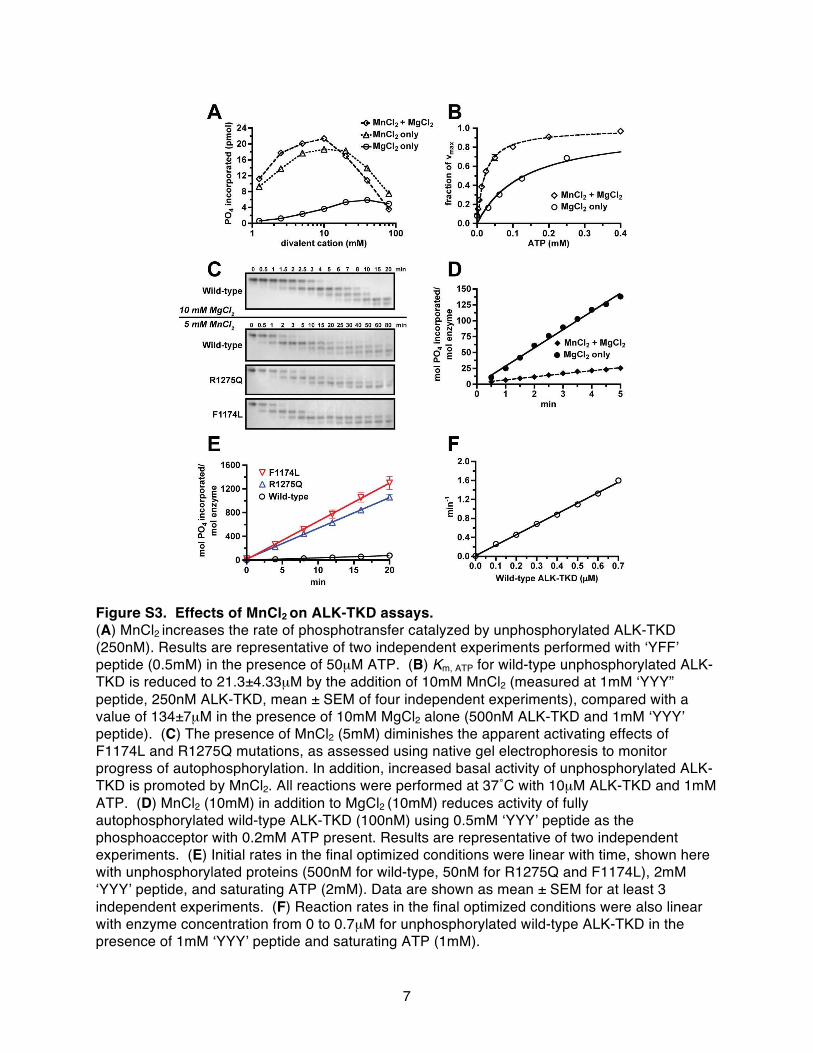
Figure S3. Effects of MnCl2 on ALK-TKD assays. (A) MnCl2 increases the rate of phosphotransfer catalyzed by unphosphorylated ALK-TKD (250nM). Results are representative of two independent experiments performed with ʻYFFʼ peptide (0.5mM) in the presence of 50µM ATP. (B) Km, ATP for wild-type unphosphorylated ALK-TKD is reduced to 21.3±4.33µM by the addition of 10mM MnCl2 (measured at 1mM ʻYYY” peptide, 250nM ALK-TKD, mean ± SEM of four independent experiments), compared with a value of 134±7µM in the presence of 10mM MgCl2 alone (500nM ALK-TKD and 1mM ʻYYYʼ peptide). (C) The presence of MnCl2 (5mM) diminishes the apparent activating effects of F1174L and R1275Q mutations, as assessed using native gel electrophoresis to monitor progress of autophosphorylation. In addition, increased basal activity of unphosphorylated ALK-TKD is promoted by MnCl2. All reactions were performed at 37˚C with 10µM ALK-TKD and 1mM ATP. (D) MnCl2 (10mM) in addition to MgCl2 (10mM) reduces activity of fully autophosphorylated wild-type ALK-TKD (100nM) using 0.5mM ʻYYYʼ peptide as the phosphoacceptor with 0.2mM ATP present. Results are representative of two independent experiments. (E) Initial rates in the final optimized conditions were linear with time, shown here with unphosphorylated proteins (500nM for wild-type, 50nM for R1275Q and F1174L), 2mM ʻYYYʼ peptide, and saturating ATP (2mM). Data are shown as mean ± SEM for at least 3 independent experiments. (F) Reaction rates in the final optimized conditions were also linear with enzyme concentration from 0 to 0.7µM for unphosphorylated wild-type ALK-TKD in the presence of 1mM ʻYYYʼ peptide and saturating ATP (1mM).

8
Figure S4. Progress of ALK-TKD autophosphorylation as assessed in native gels. (A) Species corresponding to unphosphorylated (0P), singly phosphorylated (1P) and those with 2 (2P), 3 (3P) or 4 (4P) phosphates respectively are tracked with time as denoted in the key – for wild-type, R1275Q, and F1174L ALK-TKD (10µM) with 2mM ATP and 10mM MgCl2 as shown in Fig. 4B. In (B), the kinetics of appearance/disappearance of each different phosphospecies are directly compared across the three ALK-TKD variants. All data are shown as mean ± SEM of three independent experiments.

9
Figure S5. Sedimentation equilibrium ultracentrifugation analysis of ALK-TKD variants. Purified wild-type, F1174L, and R1275Q ALK-TKD were subjected to sedimentation equilibrium analytical ultracentrifugation to measure molecular weight and assess propensity to self-associate (which might be affected by activating mutations). Proteins were centrifuged at three different speeds (13,000, 19,000 and 25,000 r.p.m.) at two different concentrations (10µM and 16.5µM). Efforts were made to globally fit data to models assuming either a single species or self-associating species, and best fits suggested a single species with molecular weight of 38.2kDa (wild-type), 42.4kDa (R1275Q) and 39.6kDa (F1174L). Data from representative experiments performed at 10µM ALK-TKD (at 13,000 r.p.m.) are plotted as ln(abs280) against (r2-ro
2)/2. This representation yields a straight line for a single species, with slope proportional to molecular weight. The data (and fits) suggest that – at concentrations in the 10-20µM range – each ALK-TKD variant is monomeric, excluding promotion of dimerization as a mechanism of TKD activation by these neuroblastoma mutations.

10
Figure S6. Inhibition of ALK-TKD by inhibitors in the Enzo ScreenWell and EMD InhibitorSelectTM 1 3 collections. Inhibitor screens using a 96-well plate autophosphorylation assay were performed as described in Supplementary Methods. Several new hits were identified that could be used as scaffolds for future ALK inhibitors. The left-hand arrays correspond to the Enzo ScreenWell panel of 80 kinase inhibitors. Others employed EMD InhibitorSelect Libraries 1-3 as marked. Inhibition was analyzed as described in Supplementary Methods by monitoring autophosphorylation of ALK-TKD variants (2µM) with 0.2mM ATP in anti-pY immunoblots (shown in inverse grey scale). DMSO negative controls are boxed in green. TAE-684 positive inhibition controls are boxed in red. No significant difference in inhibitor selectivity profile is apparent when comparing wild-type, F1174L and R1275Q ALK-TKD, suggesting that the mutations do not substantially alter the nature of the ATP binding site. Components of Enzo Screen-WellTM are listed at http://www.enzolifesciences.com/BML-2832/kinase-inhibitor-library, and those of EMD InhibitorSelectTM are listed at http://www.emdchemicals.com/life-science-research.
to

11
Figure S7. Confirmation of Km, ATP values of ALK-TKD variants at different near-saturating peptide concentrations. (A) Since using a truly saturating level of peptide in Km, ATP assays was not feasible, we determined Km, ATP values for unphosphorylated ALK-TKD variants in the presence of 2mM peptide. Results show a very similar profile to that determined at 1mM peptide (Fig. 5C), with Km, ATP values very slightly increased. The comparative profile for the different ALK-TKD variants (500nM for wild-type, 50nM for mutants) is maintained. (B) Direct comparison of Km, ATP values determined in assays employing 1mM and 2mM peptide. Data are shown as mean ± SEM of three independent experiments, and are also shown in table S2.

12
Figure S8. The F1174L mutation also increases IC50 for staurosporine inhibition. The ability of staurosporine to inhibit ALK-TKD in vitro was analyzed as described for crizotinib in Figure 5 G and H. The IC50 for staurosporine inhibition with 0.2mM ATP (A) is slightly higher for F1174L-mutated ALK-TKD than for the R1275Q variant (see table S5). As with crizotinib, this difference is enhanced when experiments were performed in the presence of 2mM ATP (B), more closely resembling cellular concentrations. All data are shown as the mean ± SEM from at least three independent experiments. The reduced Km,ATP for F1174L ALK-TKD also appears to increase IC50 for staurosporine inhibition.
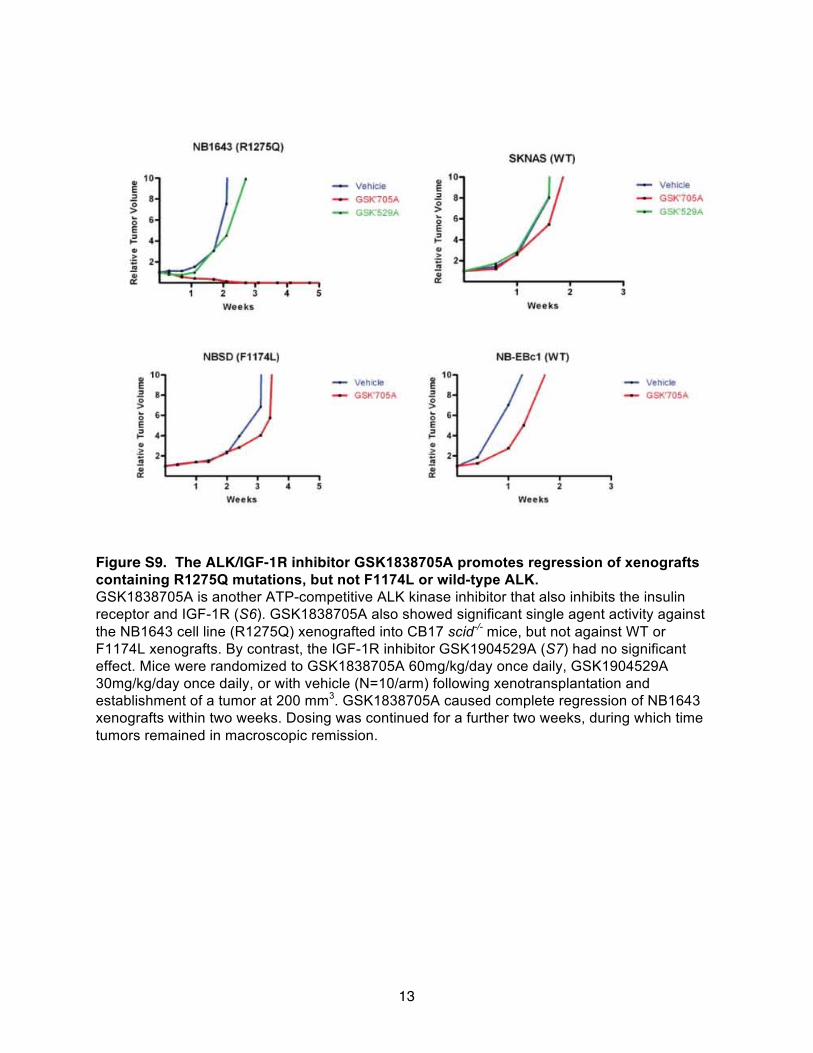
13
Figure S9. The ALK/IGF 1R inhibitor GSK1838705A promotes regression of xenografts containing R1275Q mutations, but not F1174L or wild-type ALK. GSK1838705A is another ATP-competitive ALK kinase inhibitor that also inhibits the insulin receptor and IGF 1R (S6). GSK1838705A also showed significant single agent activity against the NB1643 cell line (R1275Q) xenografted into CB17 scid-/- mice, but not against WT or F1174L xenografts. By contrast, the IGF 1R inhibitor GSK1904529A (S7) had no significant effect. Mice were randomized to GSK1838705A 60mg/kg/day once daily, GSK1904529A 30mg/kg/day once daily, or with vehicle (N=10/arm) following xenotransplantation and establishment of a tumor at 200 mm3. GSK1838705A caused complete regression of NB1643 xenografts within two weeks. Dosing was continued for a further two weeks, during which time tumors remained in macroscopic remission.
-
-
-

14
Table S1. Effect of siRNA knockdown of ALK and Met on proliferation of neuroblastoma cell lines.
Relative proliferation
KELLY (F1174L)
EBC1 (WT)
SKNAS (WT)
NLF (WT)
ALK siRNA 0.190 0.728 0.930 0.729 Met siRNA 0.772 0.421 0.797 0.439
Proliferation of the KELLY cell line harboring a F1174L mutation is inhibited by siRNA knockdown of ALK, but shows less inhibition with Met knockdown. The remaining three cell lines harbor wild-type normal copy number ALK. Cells were plated overnight and then transfected in triplicate with a pool of four siRNA targeting each kinase (Thermo Scientific Kinase siGenome Library), with positive (siPLK1) and negative (GAPD) controls. Substrate adherent growth was monitored for 100 hours using the Roche xCELLigence system. The area under the curve was calculated for each siRNA triplicate and mock transfection. % relative growth = AUC of the siKinase/AUC siControl. Table S2. Dependence of Km, ATP on peptide concentration for unphosphorylated ALK-
TKD variants. (Data are shown as mean ± SEM for at least 3 independent experiments).
kinase
Km, ATP, 1mM peptide (mM)
Km, ATP, 2mM peptide (mM)
WT 0.134 ± 0.007 0.181 ± 0.004 R1275Q 0.326 ± 0.033 0.460 ± 0.024 F1174L 0.127 ± 0.011 0.179 ± 0.022
Table S3. Crizotinib inhibition of unphosphorylated ALK-TKD in vitro at two different
ATP concentrations. (Data are shown as mean ± SEM for at least 3 independent experiments).
kinase
IC50, 2 mM ATP (nM)
IC50, 0.2 mM ATP (nM)
WT 147 ± 6 -- R1275Q 84.6 ± 8.0 80.4 ± 4.8 F1174L 130 ± 10 88.5 ± 6.4
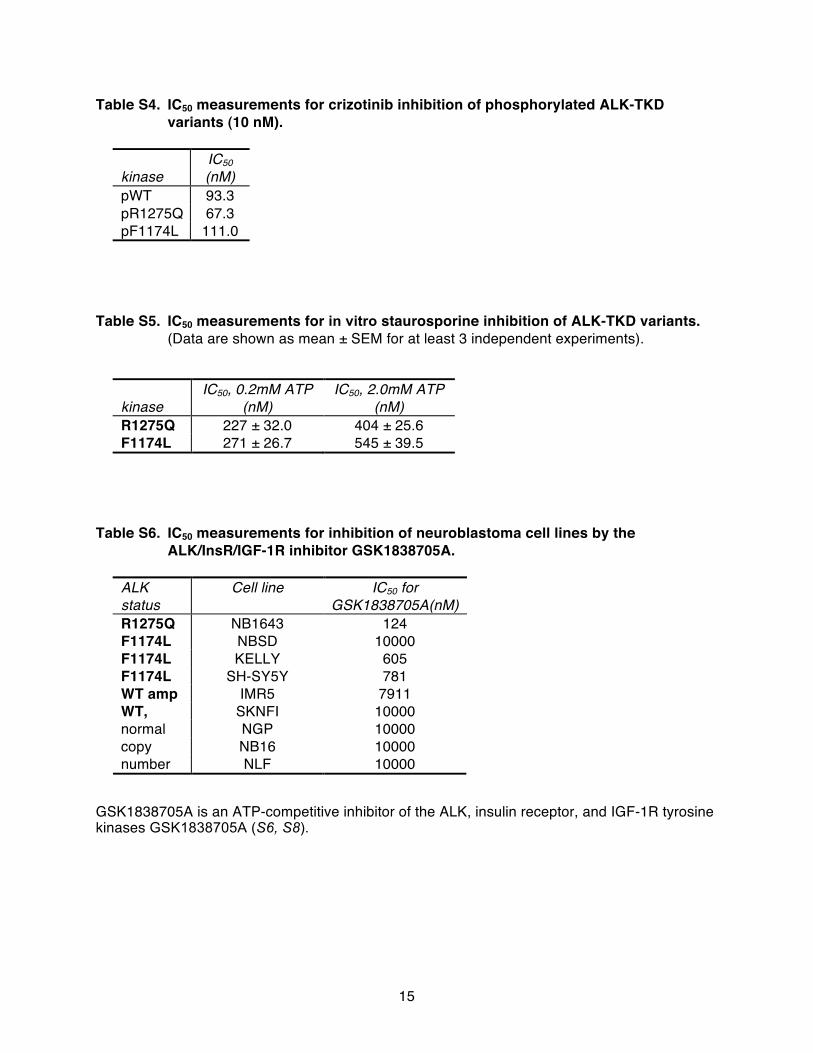
15
Table S4. IC50 measurements for crizotinib inhibition of phosphorylated ALK-TKD variants (10 nM).
kinase
IC50 (nM)
pWT 93.3 pR1275Q 67.3 pF1174L 111.0
Table S5. IC50 measurements for in vitro staurosporine inhibition of ALK-TKD variants. (Data are shown as mean ± SEM for at least 3 independent experiments).
kinase
IC50, 0.2mM ATP (nM)
IC50, 2.0mM ATP (nM)
R1275Q 227 ± 32.0 404 ± 25.6 F1174L 271 ± 26.7 545 ± 39.5
Table S6. IC50 measurements for inhibition of neuroblastoma cell lines by the
ALK/InsR/IGF 1R inhibitor GSK1838705A.
ALK status
Cell line IC50 for GSK1838705A(nM)
R1275Q NB1643 124 F1174L NBSD 10000 F1174L KELLY 605 F1174L SH-SY5Y 781 WT amp IMR5 7911
SKNFI 10000 NGP 10000 NB16 10000
WT, normal copy number NLF 10000
GSK1838705A is an ATP-competitive inhibitor of the ALK, insulin receptor, and IGF 1R tyrosine kinases GSK1838705A (S6, S8).
-
-

16
Supplementary References S1. Z. Y. Zhang, J. C. Clemens, H. L. Schubert, J. A. Stuckey, M. W. Fischer, D. M. Hume,
M. A. Saper, J. E. Dixon, Expression, purification, and physicochemical characterization of a recombinant Yersinia protein tyrosine phosphatase. J. Biol. Chem. 267, 23759-23766 (1992).
S2. X. Zhang, J. Gureasko, K. Shen, P. A. Cole, J. Kuriyan, An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137-1149 (2006).
S3. Y. P. Mossé, M. Laudenslager, L. Longo, K. A. Cole, A. Wood, E. F. Attiyeh, M. J. Laquaglia, R. Sennett, J. E. Lynch, P. Perri, G. Laureys, F. Speleman, C. Kim, C. Hou, H. Hakonarson, A. Torkamani, N. J. Schork, G. M. Brodeur, G. P. Tonini, E. Rappaport, M. Devoto, J. M. Maris, Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930-935 (2008).
S4. A. Donella-Deana, O. Marin, L. Cesaro, R. H. Gunby, A. Ferrarese, A. M. Coluccia, C. J. Tartari, L. Mologni, L. Scapozza, C. Gambacorti-Passerini, L. A. Pinna, Unique substrate specificity of anaplastic lymphoma kinase (ALK): development of phosphoacceptor peptides for the assay of ALK activity. Biochemistry 44, 8533-8542 (2005).
S5. R. E. George, T. Sanda, M. Hanna, S. Frohling, W. Luther, 2nd, J. Zhang, Y. Ahn, W. Zhou, W. B. London, P. McGrady, L. Xue, S. Zozulya, V. E. Gregor, T. R. Webb, N. S. Gray, D. G. Gilliland, L. Diller, H. Greulich, S. W. Morris, M. Meyerson, A. T. Look, Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975-978 (2008).
S6. P. Sabbatini, S. Korenchuk, J. L. Rowand, A. Groy, Q. Liu, D. Leperi, C. Atkins, M. Dumble, J. Yang, K. Anderson, R. G. Kruger, R. R. Gontarek, K. R. Maksimchuk, S. Suravajjala, R. R. Lapierre, J. B. Shotwell, J. W. Wilson, S. D. Chamberlain, S. K. Rabindran, R. Kumar, GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 8, 2811-2820 (2009).
S7. P. Sabbatini, J. L. Rowand, A. Groy, S. Korenchuk, Q. Liu, C. Atkins, M. Dumble, J. Yang, K. Anderson, B. J. Wilson, K. A. Emmitte, S. K. Rabindran, R. Kumar, Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase. Clin. Cancer Res. 15, 3058-3067 (2009).
S8. E. Ardini, P. Magnaghi, P. Orsini, A. Galvani, M. Menichincheri, Anaplastic Lymphoma Kinase: role in specific tumours, and development of small molecule inhibitors for cancer therapy. Cancer Lett. 299, 81-94 (2010).