dissertaÇÃo de mestrado...dissertaÇÃo de mestrado formaÇÃo de partÍculas submicromÉtricas de...
TRANSCRIPT

DISSERTAÇÃO DE MESTRADO
FORMAÇÃO DE PARTÍCULAS SUBMICROMÉTRICAS DE PMMA POR CRISTALIZAÇÃO TÉRMICA DE
SOLUÇÃO POLIMÉRICA
ANDRÉ ANDERSON COSTA PEREIRA
Orientador: Prof.ª Dr.ª Elisa Maria Bittencourt Dutra de Sousa
Coorientador: Prof. Dr. Jackson Araújo de Oliveira
Natal/RN Agosto/2013
Universidade Federal do Rio Grande do Norte Centro de Tecnologia
Departamento de Engenharia Química Programa de Pós-Graduação em Engenharia Química

André Anderson Costa Pereira
Formação de partículas submicrométricas de PMMA por Cristalização térmica de solução polimérica
Agosto/2013 Natal/RN
Dissertaçãode mestrado apresentada ao programa de pós-graduação em Engenharia Química da Universidade Federal do Rio Grande do Norte, comoparte dos requisitos necessários para obtenção do título de mestre em Engenharia Química, sob a orientação da Prof.ª Dr.ª Elisa Maria Bittencourt Dutra de Sousae Prof. Dr. Jackson Araújo de Oliveira.

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 iii
Catalogação da Publicação na Fonte. UFRN / CT / DEQ
Biblioteca Setorial “Professor Horácio Nícolás Sólimo”.
Pereira, André Anderson Costa. Formação de partículas submicrométricas de PMMA por cristalização térmica de solução polimérica / André Anderson Costa Pereira. - Natal, 2013. 76 f.: il. Orientador: Elisa Maria B. D. de Sousa. Co-Orientador: Jackson Araújo de Oliveira. .
Dissertação (Mestrado) - Universidade Federal do Rio Grande do Norte. Centro
de Tecnologia. Departamento de Engenharia Química. Programa de Pós-Graduação em Engenharia Química.
1. Cristalização - Dissertação. 2. Polímero - Dissertação. 3. Solubilização - Dissertação. I. Sousa, Elisa Maria B. D. de. II. Oliveira, Jackson Araújo de. III. Universidade Federal do Rio Grande do Norte. IV. Título.
RN/UF CDU 66.065.5(043.3)

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 v

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 iv
PEREIRA, A. A. C. - Formação de partículas submicrométricas de PMMA por Cristalização térmica de solução polimérica. Dissertação de Mestrado. UFRN, Programa de Pós-Graduação em Engenharia Química, Área de concentração: Engenharia Química, Natal, RN, Brasil, 2013.
Orientador: Prof.aDr.ª Elisa Maria Bittencourt Dutra de Sousa.
Coorientador:Prof. Dr. Jackson Araújo de Oliveira.
RESUMO: Partículas poliméricas formadas em escala nanométrica são de fundamental
interesse atualmente, principalmente quando utilizadas como sistemas carreadores na
liberação controladade fármacos, cosméticos e nutracêuticos, e no recobrimento de materiais
com propriedades magnéticas. O principal objetivo do presente estudo diz respeito à produção
de partículas submicrométricas de poli (metacrilato de metila) (PMMA) através da
cristalização de uma solução poliméricapor resfriamento controlado termicamente. Neste
trabalho, soluções de PMMA em etanol e 1-propanol foram preparadas em diferentes
concentrações (1% a 5% em massa) e cristalizadas em diferentes taxas de resfriamento (0,2 a
0,8 ºC/min) controladas linearmente. Análises de distribuição de partículas (DLS/CILAS) e
microscopia eletrônica de varredura (MEV) foram realizadas com o intuito de avaliar as
características morfológicas das partículas formadas. Os resultados demonstraram que é
possível obter partículas poliméricas submicrométricas com morfologia perfeitamente esférica
utilizando a técnica abordada no presente estudo. Observou-se também que, a depender da
taxa de resfriamento e da concentração da solução polimérica, é possível obter alto
rendimento na formação de partículas submicrométricas. Adicionalmente, foram realizados
testes preliminares com o propósito de verificar a capacidade desta técnica em formar
partículas carreadoras de materiais com propriedades magnéticas. Os resultados permitiram
concluir que a técnica estudada pode ser uma alternativa interessante na obtenção de
partículas poliméricas com propriedades magnéticas.
Palavras-chave: PMMA, cristalização térmica, partículas submicrométricas.

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 vi
PEREIRA, A. A. C. – Formation of submicron particles of PMMA by thermal crystallization of the polymer solution. UFRN, PPGEQ, Natal, RN, Brazil, 2013.
Professor Advisor: Prof.aDr. Elisa Maria B. D. de Sousa.
Co-supervisor: Prof. Dr. Jackson Araújo de Oliveira.
ABSTRACT
Polymer particles in the nanometer range are of fundamental interest today, especially when
used as carrier systems in the controlled release of drugs, cosmetics and nutraceuticals, as
well as in coating materials with magnetic properties. The main objective of the present study
concerns the production of submicron particles of poly (methyl methacrylate) (PMMA) by
crystallization of a polymer solution by thermally controlled cooling. In this work, PMMA
solutions in ethanol and 1-propanol were prepared at different concentrations (1% to 5% by
weight) and crystallized at different cooling rates (0.2 to 0.8 ° C / min) controlled linearly.
Analysis of particle size distribution (DLS / CILAS) and scanning electron microscopy
(SEM) were performed in order to evaluate the morphological characteristics of the produced
particles. The results demonstrated that it is possible to obtain submicron polymer perfectly
spherical particles using the technique discussed in this study. It was also observed that,
depending on the cooling rate and the concentration of the polymer solution, it is possible to
achieve high yield in the formation of submicron particles. In addition, preliminary tests were
performed in order to verify the ability of this technique to form particulated carrier material
with magnetic properties. The results showed that the developed technique can be an
interesting alternative to obtain polymer particles with magnetic properties.
Keywords: PMMA, thermal crystallization, submicron particles.

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 vii
AGRADECIMENTOS
Primeiramente, gostaria de agradecer a Deus por tudo que ocorreu nessa minha trajetória até
aqui. Meus sinceros agradecimentos aos meus orientadores: Prof. Dr. Elisa Maria B. D.
Sousa e Prof. Dr. Jackson Araújo de Oliveira, pela orientação, dedicação, compreensão e
principalmente, pela paciência durante o período da dissertação.
Peço a benção de meus pais e agradeço pelo apoio que recebi ao longo da minha vida
acadêmica e pessoal. Eles são à base da minha vida.
Agradeço a minha querida eterna namorada e amada noiva Suênya Monique de Medeiros
Confessor que me incentivou a ingressar na pós-graduação e tem sempre me motivado a
terminar essa dissertação e também prosseguir na vida acadêmica.
Aos meus primos Alberto Carlos e Ramonn Mateus pela voluntaria ajuda nos experimentos
preliminares. Eles foram muito importantes nesta etapa.
Aos meus amigos do Laboratório de Tecnologia Supercrítica e Biodiesel, pela colaboração e
auxílio em determinadas etapas do projeto da dissertação.
Um agradecimento, especial, ao pessoal dos laboratórios da UFRN, UEM e UNIT, que
viabilizaram as análises apresentadas neste trabalho.
Ao Programa de Pós-Graduação em Engenharia Química pela oportunidade que me foi
concedida para realização deste trabalho e a Coordenação de Aperfeiçoamento de pessoal de
Nível Superior (CAPES) pelo apoio financeiro.
E, por fim, a todas as pessoas que contribuíram para finalização desse projeto.

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 viii
SUMÁRIO
ABSTRACT .............................................................................................................................. vi
AGRADECIMENTOS ............................................................................................................. vii
ÍNDICE DE FIGURAS .............................................................................................................. x
ÍNDICE DE TABELA ............................................................................................................ xiii
NOMENCLATURA/SIGLAS/SÍMBOLOS ........................................................................... xiv
1. Introdução ............................................................................................................................... 2
2. Revisão bibliográfica .............................................................................................................. 6
2.1 – O poli(metacrilato de metila) (PMMA) ......................................................................... 6
2.1.1 – Estrutura e propriedades do PMMA ....................................................................... 6
2.1.2 – Aplicações do PMMA ............................................................................................ 7
2.2 – A solubilidade do poli(metacrilato de metila) (PMMA) ............................................... 8
2.2.1 – Forças moleculares dos polímeros .......................................................................... 9
2.2.2 – Comportamento do polímero em solução ............................................................. 10
2.2.3 – Solubilização de um polímero .............................................................................. 11
2.3 – A Cristalização ............................................................................................................ 13
2.3.1 – Precipitação com anti-solvente ............................................................................. 14
2.3.2 – Cristalização por evaporação do solvente ............................................................ 16
2.3.3 – Cristalização por resfriamento .............................................................................. 17
2.3.4 – Ação da agitação na cristalização ......................................................................... 18
2.3.5 – Obtenção da supersaturação ................................................................................. 18
2.3.6 – Etapa de Nucleação .............................................................................................. 20
2.3.7 – Etapa do Crescimento ........................................................................................... 22
2.3.8 – Zona metaestável .................................................................................................. 23
2.3.9 – Condição θ ............................................................................................................ 27

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 ix
2.4 – Encapsulação de materiais magnéticos........................................................................ 28
3. Materiais e métodos .............................................................................................................. 31
3.1 – Etapa da Solubilização ................................................................................................ 31
3.2 – Etapa do processo de cristalização .............................................................................. 32
3.3 – Etapa de Filtração ........................................................................................................ 34
3.3.1 – Quantificação da fração de partículas submicrométricas ..................................... 35
3.4 – Etapa de Caracterização............................................................................................... 36
3.4.1 – Microscopia Eletrônica de Varredura (MEV) ...................................................... 36
3.4.2 – Distribuição de tamanho de partículas por espalhamento dinâmico de luz (DLS)
.......................................................................................................................................... 36
3.4.3 – Microscopia Óptica (MO) .................................................................................... 37
3.4.4 – Distribuição de tamanho de partículas (DTP) pelo granulômetro ........................ 38
3.5 – Correlação linear das variáveis de entrada e saída ...................................................... 39
4. Resultados e Discussões ....................................................................................................... 41
4.1 – Solubilização do PMMA ............................................................................................. 41
4.2 – Estratégia para obtenção das taxas de resfriamento .................................................... 42
4.3 – Quantificação da fração de partículas submicrométricas ............................................ 45
4.4 – Distribuição de tamanho de partículas da fração retida ............................................... 46
4.5 – Análises por Microscopia Óptica das partículas presentes na fração retida ................ 50
4.6 – Distribuição de tamanho de partículas na fração passante .......................................... 53
4.7 – Análises por Microscopia Eletrônica de Varredura (MEV) ........................................ 57
4.8 – Encapsulação de material magnético ........................................................................... 59
4.9 – Análise de correlação linear ........................................................................................ 61
5.Conclusão .............................................................................................................................. 64
Referências ............................................................................................................................... 67

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 x
ÍNDICE DE FIGURAS Figura 2.1 – Unidade química de repetição do PMMA .............................................................. 7
Figura 2.2 – Representação de pontes de hidrogênio e forças de Var der Waals entre cadeias
de PMMA. .................................................................................................................................. 9
Figura 2.3 – Conformação aleatória ou novelo de uma cadeia polimérica amorfa (Canevarolo
Jr., 2010). .................................................................................................................................. 10
Figura 2.4 – Representação esquemática do volume ocupado por uma cadeia polimérica em
solução quando sujeita a uma variação na temperatura ou no poder do solvente (Canevarolo
Jr., 2010). .................................................................................................................................. 11
Figura 2.5 – Solubilidade do PMMA em misturas de álcool e água. ○, Metanol; ●, Etanol; □,
1-propanol; ■, 2- propanol; ◊, t-butanol (Cowier et al., 1987). ................................................ 12
Figura 2.6 – Representação esquemática da ação das moléculas de água na cadeia polimérica
(Hoogenboom et al, 2010). ....................................................................................................... 12
Figura 2.7 – Solubilização de um polímero mostrando seus dois estágios. ............................. 13
Figura 2.8 – Esquema do processo de precipitação com anti-solvente (Dalvi & Thorat, 2012).
.................................................................................................................................................. 15
Figura 2.9 – Representação da técnica de precipitação por fluido super crítico usando o CO2
como anti-solvente (Garay et al, 2010). ................................................................................... 15
Figura 2.10 – Esquema da cristalização por evaporação utilizada na formação das partículas
(Sarkari et al., 2002). ................................................................................................................ 17
Figura 2.11 – A dependência do tempo com tamanho de partícula e a supersaturação (Kim,
2001). ........................................................................................................................................ 20
Figura 2.12 – Relação da concentração e metaestabilidade (Kim & Petermann, 2001). ......... 24
Figura 2.13 – Diagrama funcional de cristalização: concentração versus temperatura (Merheb,
2009). ........................................................................................................................................ 25
Figura 2.14 – Representação esquemática da exclusão entre volumes ocupados por duas
moléculas poliméricas diferentes (Canevarolo Jr., 2010)......................................................... 28
Figura 3.1 – Grãos de PMMA usado na solubilização. ............................................................ 31
Figura 3.2 – Equipamentos utilizados para a solubilização do polímero. ................................ 32
Figura 3.3 – Equipamentos utilizados para a formação das partículas. .................................... 32
Figura 3.4 – Perfil de perturbação aplicado ao set-point do banho termostático para obtenção
das taxas de resfriamento linearmente controladas. ................................................................. 33

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 xi
Figura 3.5 – Recipientes com o armazenamento das amostras após o processo de cristalização.
.................................................................................................................................................. 34
Figura 3.6 – Sistema de filtração a vácuo. ................................................................................ 35
Figura 3.7 – Analisador de partícula por Espalhamento de Luz Dinâmica (DLS). .................. 37
Figura 3.8 – Microscópio Óptico (Olympus). .......................................................................... 38
Figura 3.9 – Analisador de tamanho de partícula (CILAS). ..................................................... 38
Figura 4.1 – Solubilização do PMMA em solução alcoólica. .................................................. 42
Figura 4.2 – Identificação do processo mediante perturbações realizadas na temperatura do
set-point do banho. Perturbação no set-point e resposta do sistema para amplitudes: (a) de
5°C; (b) de 10°C e (c) de 20°C. ................................................................................................ 43
Figura 4.3 – Perfis de resfriamento típicos praticados durante os experimentos de
cristalização, com taxas de resfriamento de (a) -0,2°C/min.;(b) -0,5°C/min. e (c) -0,8°C/min.
.................................................................................................................................................. 45
Figura 4.4 – Histogramas de DTP das amostras (a) PE1 e (b) P1. ........................................... 47
Figura 4.5 – Histograma de DTP para a amostra PE2 .............................................................. 47
Figura 4.6 – Histogramas de DTP das amostras (a) PE3 e (b) P3. ........................................... 48
Figura 4.7 – Histogramas de DTP das amostras (a) PE4 e (b) P4. ........................................... 48
Figura 4.8 – Histogramas de DTP das amostras (a) PE5 e (b) P5. ........................................... 48
Figura 4.9 – Histogramas de DTP das amostras (a) PE6 e (b) P6. ........................................... 49
Figura 4.10 –Histogramas de DTP das amostras (a) PE7 e (b) P7. .......................................... 49
Figura 4.11 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE1 e (b)P1. ..................... 51
Figura 4.12 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE2 e (b)P2. ..................... 51
Figura 4.13 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE3 e (b)P3. ..................... 52
Figura 4.14 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE4 e (b)P4. ..................... 52
Figura 4.15 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE5 e (b)P5. ..................... 52
Figura 4.16 – Análiseem Microscopia Óptica a 100x; Ensaios (a)PE6 e (b)P6. ...................... 53
Figura 4.17 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE7 e (b)P7. ..................... 53
Figura 4.18 – Curvas de DTP obtidas por análises em DLS para as amostras PE1 e P1. ........ 54
Figura 4.19 – Curvas de DTP obtidas por análises em DLS para as amostras PE2 e P2. ........ 54
Figura 4.20 – Curvas de DTP obtidas por análises em DLS para as amostras PE3 e P3. ........ 55
Figura 4.21 – Curvas de DTP obtidas por análises em DLS para as amostras PE4 e P4. ........ 55
Figura 4.22 – Curvas de DTP obtidas por análises em DLS para as amostras PE5 e P5. ........ 55
Figura 4.23 – Curvas de DTP obtidas por análises em DLS para as amostras PE 6 e P6. ....... 56

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 xii
Figura 4.24 – Curvas de DTP obtidas por análises em DLS para as amostras PE7 e P7. ........ 56
Figura 4.25 – Imagens do Ensaio P3 obtidas no microscópio eletrônico de varredura (MEV).
.................................................................................................................................................. 57
Figura 4.26 – Imagens do Ensaio PE4 obtidas no microscópio eletrônico de varredura (MEV).
.................................................................................................................................................. 58
Figura 4.27 – Imagens do ensaio PE5 obtidas no Microscópio eletrônico de varredura (MEV).
.................................................................................................................................................. 58
Figura 4.28 – Imagens no MEV das partículas do trabalho Kim (2005); Taxas de resfriamento
(a) -0,5 e (b) -1ºC/min para 3%w/w. ........................................................................................ 59
Figura 4.29 – Solução resultante após a filtragem dos ensaios com a Ferrita (a); Suspensão
sobre indução de um campo magnético (b). ............................................................................. 60
Figura 4.30 – Nanoesferas de PMMA + Fe3O4 obtidas via polimerização por emulsão (Cui &
Wang et al., 2010). ................................................................................................................... 60
Figura 4.31 – Microscopia óptica de partículas de PMMA com Ferrita, ensaio FP2. ............. 61
Figura 4.32 – Representação dos valores numéricos dos coeficientes de correlação possíveis
em uma análise de correlação. .................................................................................................. 62

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 xiii
ÍNDICE DE TABELA Tabela 3.1 – Níveis reais e codificados das variáveis do processo de cristalização em estudo.
.................................................................................................................................................. 34
Tabela 4.1 – Parâmetros para a função de perturbação aplicada no set-point. ......................... 44
Tabela 4.2 – Frações de partículas retidas e passantes obtidas da cristalização em solução de
etanol + água. ............................................................................................................................ 45
Tabela 4.3 – Frações de partículas retidas e passantes obtidas da cristalização em solução de
1-propanol + água. .................................................................................................................... 45
Tabela 4.4 – Diâmetros resultantes da fração retida com amostras cristalizadas em etanol +
água. .......................................................................................................................................... 50
Tabela 4.5 – Diâmetros resultantes da fração retida com amostras cristalizadas em 1-propanol
+ água. ...................................................................................................................................... 50
Tabela 4.6 – Diâmetros Médios e polidispersão resultantes das análises em DLS. ................. 57
Tabela 4.7 – Matriz de correlação das variáveis de entrada e saída para um planejamento 22 +
3 pontos centrais. ...................................................................................................................... 61

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 xiv
NOMENCLATURA/SIGLAS/SÍMBOLOS % – Porcentagem
%w/w – Porcentagem em massa
∆Cmax – Supersaturação máxima
∆t – duração
∆Tmax – Largura da zona metaestável
∆ρ – Variação da supersaturação
°C – Graus Celsius
Amp – Amplitude
ATRP – Polimerização Radicalar de Transferência de Átomo
C – Concentração
C* ou Ceq – Concentração de equilíbrio
C*met – Concentração metaestável
CAPES – Coordenação de Aperfeiçoamento de Pessoal de Nível Superior
CoFe2O4 – Ferrita de cobalto
Conc – Concentração polimérica (w/w)
Da – Dalton
DLS – Espalhamento de Luz Dinâmica
Dpe – Diâmetro médio das partículas de etanol
Dpp – Diâmetro médio das partículas de 1-propanol
DTP – Distribuição de tamanho de partícula
exp – Exponencial
FeO – Óxido de Ferro
Fet – Fração submicrométrica de etanol
Fprop – Fração submicrométrica de 1-propanol
g – gramas
h – hora
iPS – Poliestireno isotático
L – Litro
L – Tamanho dos cristais
MEV – Microscopia Eletrônica de Varredura
min. – Minutos

__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 xv
mL – mililitro
mm – milímetros
MO – Microscopia Óptica
mW – miliwalt
Ni – Níquel
nm – nanômetro
P1, P2, P3, P4, P5, P6 e P7 – Ensaios com o solvente 1-propanol
PE – Polietileno
PE1, PE2, PE3, PE4, PE5, PE6 e PE7 – Ensaios com o solvente etanol
PET – Politereftalato de etileno
PMMA – Poli(metacrilato de metila)
PP – Polipropileno
PPGEQ – Programa de Pós-Graduação em Engenharia Química
PVA – Poli-vinil álcool
rxy – Coeficiente de correlação linear
s – Variável de Laplace
Si-ATPR – Polimerização Radicalar de Transferência de Átomo de Superfície Iniciada
Ṫ – Taxa de resfriamento constante
T – Temperatura
T* – Temperatura de equilíbrio
tc – Tempo de detecção dos cristais
Tg – Temperatura de transição vítrea
tind – Período de indução da nucleação
tmax – Período máximo
Tmet – Temperatura metaestável
u(s) – Temperatura de set-point em variáveis desvios no banho termostatizado
UEM – Universidade Estadual de Maringá
UFRN – Universidade Federal do Rio Grande do Norte
UNIT – Universidade Tiradentes
y(s) – Temperatura da solução em variáveis desvios no cristalizador
µm – mícron
Ө – teta

Capítulo 1
Introdução

Introdução
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 2
1. Introdução A cristalização tem sido uma técnica empregada de forma significativa na separação
de espécies químicas contidas em misturas de matérias-primas e produtos de diversos
processos industriais (Giulietti et al., 1996; Jones, 2005; Mendonça, 2008; Merheb, 2009). A
técnica de cristalização, comumente usada para preparação de sólidos cristalinos (Kim, 2000),
também pode ser utilizada como um método alternativo para formar partículas
submicrométricas de polímero. Em geral, as técnicas para a produção de partículas
poliméricas submicrométricas estão baseadas em sistemas de polimerização em mini-emulsão
ou emulsão (Gu, 1998; Noda, 1997), nestas formulações para a polimerização incluem várias
substâncias químicas, tais como: monômero, iniciador, emulsificante e estabilizante, podendo
resultar na presença de impurezas residuais nas partículas formadas (Kim, 2005). Além disso,
condições ótimas de polimerização são necessárias para se obter as partículas no tamanho
desejado. Para tal, são utilizados sistemas de alta agitação ou mesmo ultrassom para quebrar
as gotas em dimensão adequada.
Em comparação com a polimerização, a formação de partículas pelo processo de
cristalização, apresenta algumas vantagens (Kim, 2005). Além da simplicidade de execução, a
principal vantagem desta técnica consiste na não utilização de diversos reagentes, como é o
caso das polimerizações em emulsão e miniemulsão, já que apenas o polímero é solubilizado
em solvente adequado e, após a formação das partículas, as mesmas são separadas e lavadas
durante filtração. Outro ponto positivo pode ser atribuído ao controle do tamanho da partícula
através das condições de cristalização. Este processo se desenvolve por dois mecanismos
simultâneos principais: a nucleação e o crescimento, que juntos com fenômenos secundários
vão gerar a distribuição granulométrica do material, caracterizada por um tamanho e sua
dispersão (Derenzo, 2003).
Nanopartículas poliméricas tem sido utilizadas como sistemas carreadores, sendo de
interesse para uso na liberação controlada de fármacos, cosméticos e nutracêuticos, bem como
na formação de partículas com propriedades magnéticas. No caso particular das
nanopartículas magnéticas, estas têm sido amplamente utilizadas em diversas áreas biológicas
para fins analíticos e clínicos, na imobilização de enzimas, em sequenciamento de ácido
nucléico, na purificação de proteínas, no isolamento de células e no monitoramento celular,
em fotossensibilizantes (Chung, 2008). Para tanto, o polímero deve ser compatível ao corpo
humano, em termos de adaptabilidade (não toxicidade e não-antigenicidade) (Ghosh, 2000). A

Introdução
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 3
aplicação na área biomédica é um novo campo de aplicação destas partículas, desde que estas
apresentem tamanho uniforme, biocompatibilidade e propriedades superparamagnéticas (Fan
et al., 2009). Ainda na área biomédica, uma aplicação bastante relevante corresponde às
nanopartículas carreadoras e de liberação controlada de fármacos anticancerígenos (Puisieux,
1994; Couvreur, 1995; Yoo, 2000) e de antibióticos (Puisieux, 1994; Fresta, 1995; Pinto-
Alphandary, 2000), buscando-se uma administração parenteral (sem utilizar o trato
gastrointestinal) para alcançar uma distribuição mais seletiva dos medicamentos e, assim, um
aumento do índice terapêutico. Cabe ressaltar que o PMMA é um dos polímeros com
excelente característica de biocompatibilidade, sendo usado em formulações de cosméticos e
em próteses para implantes biomédicos ou bioplastia (Hoogenboom et al., 2010).
O presente estudo teve como objetivo principal produzir partículas submicrométricas
de poli(metacrilato de metila) (PMMA) pelo processo de cristalização de solução polimérica
via resfriamento controlado. Entre os objetivos específicos, é possível citar:
(i) a definição do controle do processo de cristalização por meio de um perfil linear de
resfriamento pré-definido;
(ii) o estudo da influência das variáveis do processo de cristalização do polímero (taxa de
resfriamento, concentração de polímero na solução em dois solventes alcoólicos distintos),
avaliando os efeitos sobre a distribuição do tamanho das partículas formadas;
(iii) a inserção de materiais com propriedades magnéticas na estrutura da partícula formada;
(iv) a caracterização das partículas formadas no que diz respeito as suas propriedades
morfológicas (microscopia óptica e eletrônica de varredura, distribuição de tamanho de
partículas).
Constitui também como objetivo deste trabalho a aplicação da técnica de cristalização
por resfriamento linear de solução polimérica para a encapsulação de Ferrita ou Magnetita,
buscando obter partículas submicrométricas com propriedades magnéticas.
É importante reportar que este estudo foi desenvolvido no Laboratório de Tecnologia
Supercrítica e Biodiesel do Departamento de Engenharia Química da UFRN e parte da
caracterização das partículas obtidas foi realizada em laboratórios das universidades UEM e
UNIT, parceiras do Projeto Nanobiotec/CAPES, projeto este na qual este trabalho está
inserido.
O presente documento de dissertação de mestrado está organizado em cinco capítulos,
onde:no Capítulo 2 é apresentada uma revisão bibliográfica sobre os principais assuntos
relativos à cristalização por resfriamento; no Capítulo 3 são descritos o procedimento

Introdução
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 4
experimental para a cristalização da solução polimérica e os procedimentos analíticos
empregados para a caracterização das partículas obtidas e quantificação do rendimento das
frações de partículas submicrométricas; no Capítulo 4 são apresentados os resultados obtidos
com os ensaios e é feita uma discussão dos aspectos gerais observados com o estudo; e por
fim, o Capítulo 5 é destinado à apresentação das conclusões dos estudos realizados e das
considerações gerais sobre o assunto, sendo encerrado com sugestões para a continuidade
desta linha de estudo.

Capítulo 2
Revisão Bibliográfica

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 6
2. Revisão bibliográfica Neste capítulo serão abordados alguns aspectos sobre o polímero PMMA, os
princípios básicos referentes à solubilidade dos polímeros, a cristalização por resfriamento e
os principais fatores que exercem influência na formação das partículas poliméricas.
2.1 – O poli(metacrilato de metila) (PMMA)
A história do poli (metacrilato demetila)(PMMA) se inicia a partir da síntese de seus
monômeros acrílicos em laboratório, em 1843, quando se obteve a primeira síntese do ácido
acrílico (Miles & Bristorn, 1975).
Após este fato, Miles & Bristorn (1975) citam a seguinte sequência de eventos: em
1865 seguiu-se a preparação do metacrilato de etila, por Frankland e Duppa, enquanto que em
1877 Fittig e Paul notaram que este composto químico possuía certa tendência para
polimerizar. Por volta de 1900, a maioria dos acrilatos mais comuns havia sido preparada em
laboratório e ao mesmo tempo já existiam alguns trabalhos sobre a sua polimerização. Em
1901, Rohm, na Alemanha, começou um trabalho sistemático no campo dos acrílicos e mais
tarde tomou parte ativa no desenvolvimento industrial dos polímeros do éster acrílico naquele
país. O PMMA foi o primeiro polímero acrílico produzido industrialmente. Foi vendido como
uma solução de polímero em solvente orgânico e foi usado principalmente em lacas e
formulações para revestimentos superficiais. Mais tarde, o metacrilato de metila e sua
polimerização foi estudada em profundidade, tendo sido desenvolvido um método econômico
para fabricação do monômero.
2.1.1– Estrutura e propriedades do PMMA
O PMMA é completamente amorfo, mas apresenta uma resistência elevada e uma
estabilidade dimensional excelente, devido às cadeias poliméricas rígidas (Tg = 105°C)
(Odian, 2004). Esta característica amorfa se deve aos grupos laterais volumosos que não
possuem uma estereorregularidade, forma com a qual os grupos funcionais R estão dispostos
na molécula (Poli, 1978). Tem excepcional claridade óptica, muito boa resistência às
intempéries, resistência ao impacto e,embora apresente resistência a diversos produtos
químicos, pode ser atacados por solventes orgânicos (Odian, 2004). É um material polar e
possui uma alta resistência à corrente elétrica, ou seja, possui alta constante dielétrica e um
alto fator de potência (Poli, 1978). É o material mais importante do grupo de termoplásticos

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 7
acrílicos (António, 2007), tendo como unidade química de repetição a estrutura apresentada
na Figura 2.1.
Figura 2.1 – Unidade química de repetição do PMMA
O polímero PMMA é aproximadamente 70-75% sindiotático, ou seja,suas cadeias
apresentam grupos assimétricos espaçados regularmente e arranjados em configuração
alternada. Conforme apresentado na Figura 2.1, a ligação dos grupos metila e metacrilato
àcadeia principal, segundo uma estrutura sindiotática (alternante), origina bloqueios espaciais
consideráveis na macromolécula, tornando o polímero relativamente mais rígido e mais
resistente. Este material, dependendo de suas características para utilização, pode ser obtido a
partir dos processos de polimerização em solução, suspensão e emulsão (Odian, 2004).
O poli (metracrilato de metila) é solúvel na maioria das cetonas, dos ésteres, dos
hidrocarbonetos clorados e aromáticos, podendo inchar em contato com o éter. Resiste ao
ataque de ácidos diluídos e de soluções aquosas de sais inorgânicos (Miles & Briston, 1975).
Em relação as suas propriedades térmicas, amolece entre 70-120°C, dependendo das
condições do ensaio, da massa molar e da presença de plastificantes (Poli, 1978).
2.1.2 – Aplicações do PMMA
O PMMA é um termoplástico de grande interesse e amplamente usado nos mais
variados campos devido às suas propriedades, destacando-se excelente transparência e baixa
densidade. É uma matéria-prima para resinas de revestimento de superfície, plásticos, resinas
de troca iônica, prótese dentária. Utiliza-se ainda em substituição ao vidro em aviões e barcos,
iluminação exterior, painéis para anúncios de publicidade, iluminação interior e exterior,
estruturas arquitetônicas, banheiras e sanitários, fibras ópticas para transmissão de luz,
próteses e materiais restauradores odontológicos, lentes de contato (rígida ou flexível), lentes
para automóveis, lentes para óculos de proteção e diversos utensílios domésticos. (António,
2007; Odian, 2004).

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 8
Além disso, é largamente empregado como adesivos quando dissolvido num solvente
apropriado ou no próprio monômero (António, 2007).
Devido a sua resistência à ruptura, é usado na fabricação de lentes de aumento
inquebráveis e algumas partes de instrumentos delicados. Na forma de emulsão e suspensão,
são usados em larga escala no tratamento de tecidos, papel e couro (Miles & Briston, 1975).
A aplicação na área biomédica é um novo campo para aplicação destas partículas. O
material polimérico apresenta uma vasta gama de aplicações que inclui formulações de
cosméticos e biomédicos (Hoogenboom et al., 2010), incluindo objetos de implantes na
bioplastia e na forma de cimento ósseo na artroplastia (Kaufmann et al., 2002).
No âmbito de partículas carreadoras de nanopartículas magnéticas, o PMMA é um
polímero que apresenta biocompatibilidade e pode ser utilizado na encapsulação de
nanopartículas magnéticas, apresentando um ótimo grau de saturação na magnetização
(Feuser, 2010). Estas têm sido amplamente utilizadas em diversas áreas biológicas para fins
analíticos e clínicos, bem como na imobilização de enzimas, sequenciamentos de ácido
nucléico, purificação de proteínas, isolamento de células, monitoramento celular, hipertemia e
fotossensibilizantes (Chung, 2008). Um fator importante na obtenção de tais partículas é que o
polímero deve ser biocompatível, tanto em termos de não toxicidade como não-antigenicidade
(Ghosh, 2000). Uma das áreas promissoras na utilização das nanopartículas é a vetorização de
fármacos anticancerígenos (Puisieux, 1994; Couvreur, 1995; Yoo, 2000) e de antibióticos
(Puisieux, 1994; Fresta, 1995; Pinto-Alphandary, 2000), principalmente através de
administração parenteral (sem utilizar o trato gastrointestinal), almejando uma distribuição
mais seletiva dos mesmos e, assim, um aumento do índice terapêutico. Vale salientar que o
polímero PMMA apresenta uma vasta gama de aplicações em razão das suas propriedades não
tóxicas e antigênicas, o que inclui desde materiais cosméticos até biomédicos (Hoogenboom
et al., 2010), sendo utilizado no campo de implantes (bioplastia), cimentação óssea
(artroplastia), etc.
2.2 – A solubilidade do poli(metacrilato de metila)(PMMA)
A solubilidade, em linhas gerais, consiste na propriedade de certas substâncias serem
dissolvidas quando combinadas com outras, formando uma solução. Quanto maior a
solubilidade, maior é o grau que uma substância se dissolve em outra. A condição de
solubilidade ou saturação pode ser experimentalmente determinada aquecendo-se uma

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 9
suspensão e observando a temperatura em que os sólidos são completamente dissolvidos
(Silva, 2006).
A solubilidade do PMMA em solução alcoólica é extremamente dependente da
temperatura e da proporção de água usada como co-solvente na solução (Hoogenboom et al.,
2010; Shimizu et al., 1998).
2.2.1 – Forças moleculares dos polímeros
Para ocorrer solubilidade de um polímero, tal como o PMMA, as forças moleculares
devem ser vencidas, já que tais forças provocam um grande número de enovelamento entre as
longas cadeias poliméricas. Para tal, a separação das cadeias poliméricas pode ser feita de
uma maneira prática utilizando-se um solvente adequado (Canevarolo Jr, 2010).
As cadeias poliméricas, por serem macromoléculas formadas de unidades de repetição
(meros) contendo geralmente segmentos de cadeia, atraem-se por forças secundárias fracas
chamadas de forças intermoleculares. Tais forças aumentam com a presença de grupos polares
e diminuem com o aumento da distância entre as moléculas, sendo menos intensa que as
forças primárias chamadas de forças intramoleculares (Canevarolo Jr, 2010). As forças
intermoleculares podem ser classificadas em dois tipos principais: forças de Van der Waals
(interação dipolo-dipolo e interação dipolo-dipolo induzido) e pontes de hidrogênio.
O tipo de interação mais comum nos polímeros, em geral, é a interação dipolo-dipolo.
Esta ocorre, segundo Canevarolo Jr. (2010), em polímeros que apresentam moléculas polares
em suas cadeias, como é observada no grupo carbonila do PMMA. Outro tipo de força
secundária envolvendo longas distâncias e baixa energia, também observada neste mesmo
grupo, é a ponte de hidrogênio proveniente das ligações entre o oxigênio da carbonila e o
hidrogênio das ramificações carbônicas. Na Figura 2.2 tem-se uma representação esquemática
das forças intermoleculares que ocorrem entre cadeias de PMMA.
Figura 2.2– Representação de pontes de hidrogênio e forças de Var der Waals entre cadeias de PMMA.

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 10
São estas forças intermoleculares que são decisivamente responsáveis pela maioria das
propriedades físicas dos polímeros, tais como: temperatura de transição vítrea, temperatura de
fusão, solubilidade, cristalinidade, difusão, permeabilidade a gases e vapores, deformação
mecânica e característica de escoamento (Canevarolo Jr., 2010). Quanto mais fortes forem
estas forças, maior a atração entre as cadeias e, portanto, mais difícil todo e qualquer evento
que envolva a separação ou fluxo de uma cadeia sobre a outra. Já as forças intramoleculares
são responsáveis por determinar o arranjo molecular das unidades de repetição das cadeias,
estrutura química e o tipo de cadeia polimérica, bem como sua estabilidade térmica, química,
fotoquímica, etc.
2.2.2 – Comportamento do polímero em solução
Em geral, todas as cadeias poliméricas em solução, apresentam a conformação em
novelo, que é a mais estável (Canevarolo Jr., 2010). Tal conformação é completamente
aleatória, de modo que a cadeia detém mobilidade para enrolar-sena forma de um novelo por
razões termodinâmicas, não tendo qualquer periodicidade definida. Na Figura 2.3, mostra-se
uma ilustração de como a cadeia polimérica tende a apresentar a conformação mencionada.
Figura 2.3 – Conformação aleatória ou novelo de uma cadeia polimérica amorfa (Canevarolo Jr., 2010).
Segundo Canevarolo Jr. (2010), a estabilidade da conformação deve-se à distância
entre as duas extremidades de uma cadeia polimérica. Quanto mais afastadas estão as
extremidades, maior a energia da conformação. Portanto, para a cadeia ficar em um estado de
baixa energia, a mesma deve adquirir uma conformação aleatória ou em novelo, como
apresentado na Figura 2.3. É possível destacar a influência da variação da temperatura e o
poder do solvente no distanciamento destas extremidades provocada pela variação do volume
hidrodinâmico da cadeia polimérica. Na presença de um bom solvente ou em altas
temperaturas, o volume hidrodinâmico da cadeia tende a aumentar. Da mesma forma que na
presença de um solvente pobre ou em baixas temperaturas, o volume ocupado pela molécula
em solução tende a diminuir (Figura 2.4).

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 11
Figura 2.4 – Representação esquemática do volume ocupado por uma cadeia polimérica em solução quando sujeita a uma variação na temperatura ou no poder do solvente (Canevarolo
Jr., 2010).
A conformação polimérica permite evidenciar a capacidade de um solvente ou mistura
de solventes no que se refere à solubilização e, posteriormente, no processo de cristalização,
comprovando a maneira de como uma cadeia polimérica pode se empacotar e formar a
partícula sólida. Este processo é dependente tanto da temperatura quanto da concentração do
polímero no meio solvente.
2.2.3 – Solubilização de um polímero
Alguns trabalhos têm sido publicados sobre a solubilização do PMMA (Shimizu et al.,
1998; Hoogenboom et al., 2010; Jewrajka et al., 2004).Em particular, álcool ou água puros, à
temperatura ambiente,são considerados como não solvente ou solvente pobre (Shimizu et al.,
1998). Por outro lado, uma mistura adequada desses solventes é capaz de solubilizar o
PMMA. Na Figura 2.5, são apresentadas curvas de solubilidade de misturas de alcoóis e água
para diferentes temperaturas. É possível notar que a solubilidade é dependente da fração de
álcool em água e muda com o tipo de álcool. O fenômeno em que uma mistura de solventes
apresente maior capacidade de solubilidade que dos solventes originalmente puros é
conhecido como co-solvência (Cowier et al., 1987). Particularmente, o poder de solubilização
do PMMA aumenta em misturas contendo água e álcool devido à presença de “Shell” de
hidratação em volta da parte hidrofóbica da molécula do álcool (Hoogenboom et al., 2010).
Esta hidratação, conforme relatado por Noskov et al. (2005), forma-se em torno dos grupos
carbonila do polímero e age como uma camada de compatibilização, proporcionando uma

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 12
maior interação entre os segmentos da cadeia polimérica e o solvente,conforme pode ser
observado na Figura 2.6.
Figura 2.5 – Solubilidade do PMMA em misturas de álcool e água. ○, Metanol; ●, Etanol; □, 1-propanol; ■, 2- propanol; ◊, t-butanol (Cowier et al., 1987).
Figura 2.6 – Representação esquemática da ação das moléculas de água na cadeia polimérica (Hoogenboom et al., 2010).
Embora as moléculas de água desempenhem um papel importante na solubilização do
PMMA em álcool (Shimizu et al., 1998), o acréscimo da quantidade de água na mistura é
limitado até certo ponto, já que aumentando ainda mais a quantidade de água ocorre um
aumentona polaridade da mistura de solventes, o que leva à formação de “clusters de água”.
Desta forma, a solubilidade do polímero é diminuída pela necessidade da quebra das ligações

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 13
de hidrogênio água-água para a formação de uma camada de hidratação (Hoogenboom et al.,
2010).
A solubilização de um polímero é um processo físico reversível e que não altera a
estrutura da cadeia polimérica (Canevarolo Jr., 2010). Com isto, ela se diferencia do ataque
químico, que é um processo químico irreversível, levando à degradação da cadeia. A
solubilização é um processo lento que acontece em dois estágios, conforme representados na
Figura 2.7.
Figura 2.7 – Solubilização de um polímero mostrando seus dois estágios.
Como pode ser visto na Figura 2.7, no primeiro estágio, o PMMA em contato com os
solventes tende a inchar devido à difusão das moléculas do solvente para dentro da massa
polimérica, formando um gel inchado. Este estágio só não acontecerá se as estruturas
químicas do polímero e do solvente forem diferentes ou se existirem interações polímero-
polímero e quando estas forem muito mais intensas que as interações polímero-solvente.No
segundo estágio, a entrada de mais solvente leva à desintegração do gel inchado, com a
consequente formação de uma solução. Este estágio poderá ser prejudicado se estivem
presentes na massa polimérica: cristalinidade, pontes de hidrogênio, ligações cruzadas (em
baixas concentrações) e se as interações polímero-polímero forem maiores que as interações
polímero-solvente (Canevarolo Jr., 2010).
2.3 – A Cristalização
A cristalização, também conhecida na área de polímeros como precipitação, consiste
num processo em que, partindo-se de uma mistura líquida (solução), cristais de um dos
componentes da mistura podem ser formados. Para tal processo, criam-se as condições
termodinâmicas que levam as moléculas a aproximarem-se e a agruparem-se em estruturas
altamente organizadas, formando partículas específicas em tamanho e forma (Kim, 2000).
Para tal, é necessária a ocorrência de supersaturação na mistura líquida, ou seja, a existência

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 14
de uma concentração de soluto na solução superior à concentração de saturação (limite de
solubilidade).
A cristalização de solução é uma tecnologia comumente utilizada em indústrias
químicas e afins (Giulietti et al., 1996; Jones, 2005). Os cristais produzidos nesta operação
podem ser desde tamanho nanométrico ou submicrométrico a micrométrico, sejam como
partículas isoladas ou como aglomerados estruturados (Jones, 2005). Este processo é
amplamente utilizado na obtenção de diversos materiais da química fina e seus intermediários,
tais como: sal, carbonato de sódio, catalisador zeólita, absorventes, cerâmica, precursores de
poliéster, detergentes, fertilizantes, alimentos, produtos farmacêuticos e pigmentos (Jones,
2005).
A operação de cristalização pode ser ainda descrita como um processo de
reorganização molecular. Fatores cristalográficos e moleculares são muito importantes, pois
afetam a forma, a pureza e a estrutura dos cristais (Mullin, 2001; Myerson, 1999).
Diversas técnicas de cristalização ou precipitação podem ser utilizadas na obtenção de
cristais ou partículas de um determinado material, tais como: precipitação com anti-solvente
(líquido ou supercrítico), precipitação ou cristalização por evaporação de solvente e
cristalização por resfriamento de solução. Como o presente estudo se baseia na cristalização
por resfriamento, será dada uma maior ênfase a esta ultima técnica de cristalização.
2.3.1 – Precipitação com anti-solvente
No processo de precipitação por adição de um não solvente para o soluto, chamado
anti-solvente, a precipitação do soluto é conseguida através da redução do poder solvente
sobre o soluto dissolvido na solução. Neste caso, é importante o uso de dois solventes
completamente miscíveis, de modo que o soluto a ser precipitado é solúvel no primeiro
solvente, mas insolúvel no segundo (Garay et al., 2010). Esta técnica pode ser utilizada para
uma vasta gama de materiais, tais como: fármacos, compostos inorgânicos, polímeros e
proteínas. Além disso, uma das vantagens deste processo é que pode ser realizado a
temperaturas ambientes e à pressão atmosférica, sem exigência de equipamentos caros (Dalvi
& Thorat, 2012). Na Figura 2.8, apresenta-se um esquema do processo de precipitação de
partículas com anti-solvente, cujas etapas são: mistura de solução e anti-solvente; geração de
supersaturação; nucleação; crescimento por coagulação e condensação, podendo ocorrer ou
não aglomeração. Cabe ressaltar que as etapas de coagulação e condensação estão
relacionadas, respectivamente, com a diminuição da supersaturação e redução do número total

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 15
de partículas formadas (Sun, 2002). Já a etapa de aglomeração está diretamente relacionada
com a densidade do material presente na solução (Jones, 2002).
Figura 2.8 – Esquema do processo de precipitação com anti-solvente (Dalvi &Thorat, 2012).
Dentre as técnicas de precipitação com anti-solvente pode-se citar o processo de
precipitação com adição de líquido não solvente e fluído supercrítico. Nesta primeira técnica,
apenas a adição de um solvente líquido é capaz de precipitar partículas presentes no solvente.
No caso da precipitação supercrítica, a adição do anti-solvente CO2 supercrítico induz
a formação de uma solução contendo os dois solventes, seguido de supersaturação e a
precipitação do soluto (Muhrer et al., 2003), conforme ilustrado na Figura 2.9. Nos últimos
anos, muitas técnicas baseadas nesta tecnologia foram desenvolvidas para a precipitação de
partículas, utilizando diferentes mecanismos de nucleação e de crescimento (Dixon et al.,
1993; Reverchon,1999).
Figura 2.9 – Representação da técnica de precipitação por fluido supercrítico usando o CO2 como anti-solvente (Garay et al, 2010).
Solvente 1 +
Solvente 2 (CO2)

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 16
Tendo como objetivo a formação de partículas, tal técnica apresenta algumas
vantagens (Fages et al., 2004; Miguel et al., 2006; Wang et al., 2004):
a) Permite com flexibilidade a seleção das propriedades e composição das partículas
(polimorfismo do cristal, tamanho e pureza), através da mudança nas condições de
operação;
b) Pode ser utilizada para quase todo tipo de composto, incluindo polímeros;
c) É uma técnica alternativa ideal quando comparada aos processos com anti-solvente
líquido, uma vez que é possível remover completamente o anti-solvente (CO2) através
da redução da pressão;
d) É possível gerar partículas a temperaturas próximas do ambiente, desde que seja feita
uma seleção de anti-solvente adequado.
Vale ressaltar que partículas de polímero obtidas a partir desta técnica se destacam por
serem úteis em diversas aplicações, tais como: adsorventes e suportes de catalisadores, bem
como sistemas carreadores de drogas (Dixon et al., 1993).
2.3.2 – Cristalização por evaporação do solvente
A evaporação do solvente foi o primeiro método desenvolvido para obtenção de
partículas de um polímero (Vanderhoff et al., 1979). Atualmente, esta técnica é empregada na
tecnologia farmacêutica a qual os polímeros biodegradáveis têm sido aplicados na produção
de drogas (Gurny et al., 1981).
Neste método, as soluções de polímero são preparadas em solventes voláteis e as
emulsões são formuladas. Basicamente, a rápida evaporação deste solvente orgânico
proporciona uma alta supersaturação e uma rápida precipitação do material sob a forma de
uma suspensão coloidal (Sarkari et al., 2002).
Um caso particular que usa esta técnica é relatado por Sarkari et al. (2002) em que o
ingrediente farmacêutico ativo é dissolvido num solvente orgânico de baixa ebulição. Esta
solução é bombeada através de um tubo em que é aquecida sob pressão a uma temperatura
superior ao ponto de ebulição do solvente e, em seguida, pulverizado através de um bocal de
atomização para uma solução aquosa aquecida contendo um estabilizante, como ilustrado na
Figura 2.10.
Embora este método possa ser considerado simples, este é um processo demorado e a
possível coalescência das nanogotas durante o processo de evaporação pode afetar o tamanho
e a morfologia das partículas (Rao & Geckelera, 2011).

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 17
Figura 2.8 – Esquema da cristalização por evaporação utilizada na formação das partículas (Sarkari et al., 2002).
2.3.3 – Cristalização por resfriamento
Vários são os artigos e trabalhos que relatam sobre este tipo de cristalização na
separação de espécies químicas contidas em misturas de matérias-primas e produtos de
diversos processos industriais, os quais contemplam a qualidade do cristal, distribuição do
tamanho, forma e a pureza (por exemplo, Mersmann, 1996; Gahn & Mersmann, 1996;
Zacher, 1995; Schubert & Mersmann, 1996; Randolph & Larson, 1988; Nyvlt, 1994;
Mersmann, 1995; Schaaf et al., 1987; Mendonça, 2008; Merheb, 2009). Em muitos trabalhos,
o resfriamento é usado, logo após um determinado tratamento térmico, tanto para estudo
cinético de cristalização de polímeros no estado sólido (amorfo) específico quanto para
estudos na formação de blendas poliméricas, também em estado sólido, (Wellen, 2007; Boyer
& Haudin, 2010; Exner et al., 2010; Piccarolo et al., 2002; Gedde et al., 2013).
Como relatado em vários artigos (Kim, 2000; Kim, 2005; Sang et al., 1997; Linet al.,
1993; Giulietti et al., 1996; Waddon & Karasz, 1992), a cristalização por resfriamento
consiste em uma técnica em que uma solução de determinada concentração (superior á
concentração saturada) é submetida a uma taxa linear de resfriamento,dando origem, por
razões termodinâmicas, a uma distribuição específica de tamanho de partículas (submicro ou
micro) de morfologia esféricas de um determinado material.

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 18
Em geral, este tipo de cristalização envolve o abaixamento de temperatura no decorrer
do processo de formação de partículas, diminuindo a solubilidade do polímero na solução.
Desta maneira, a supersaturação é produzida pela remoção de calor da solução (Wellen,
2007). Neste caso, como relatado em alguns trabalhos, o resfriamento é obtido por troca
térmica com as paredes do cristalizador, em um tanque encamisado, mediante o auxilio de um
banho termostatizado usado para resfriar o fluido da camisa do recipiente.
É importante ressaltar que o tamanho destas partículas está relacionado com taxas de
resfriamento aplicadas na solução. Ou seja, maiores taxas geram uma distribuição de tamanho
de partículas menores que taxas de resfriamentos mais baixas (Lin et al., 1992; Kim 2005).
Em decorrência deste processo de resfriamento, ocorre a formação de núcleos nos
pontos onde pequenas regiões (embaraçadas e aleatórias) das moléculas se tornam ordenadas
e alinhadas (Callister, 2002). Após a nucleação e durante o estágio de crescimento da
cristalização, os núcleos crescem pela continuação da ordenação e do alinhamento de novos
segmentos de cadeias moleculares (Callister, 2002).
2.3.4 – Ação da agitação na cristalização
No cristalizador, a complexidade da cinética faz da agitação uma possível variável de
controle da distribuição granulométrica final. De acordo com Mendonça (2008),a agitação
tem as seguintes funções:
a) Manter os cristais em suspensão homogênea;
b) Promover a transferência térmica;
c) Proporcionar a transferência de matéria para a partícula no decurso do crescimento;
d) Controlar a nucleação;
e) Quebrar as partículas em suspensão;
O problema põe-se pela escolha e grau da agitação do cristalizador que é complexa,
uma vez que ao aumentar a velocidade da agitação aumenta-se o grau de homogeneidade.
Assim, a transferência de calor e matéria é mais uniforme, o que favorece a nucleação e reduz
o tamanho final médio das partículas, contribuindo igualmente para a distribuição
granulométrica (Mendonça, 2008).
2.3.5 – Obtenção da supersaturação
Para ocorrer a cristalização, é necessário criar condições no seio da mistura para as
moléculas se aproximarem e darem origem a partícula (Merheb, 2009). Portanto, a

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 19
concentração de soluto na solução deve ser superior à concentração saturada, ou seja, a
solução deve ser supersaturada.
Esta é uma das variáveis mais importantes do processo de cristalização e é definida
como a diferença entre a concentração real do soluto e a concentração de equilíbrio em
condições idênticas, isto é, sua solubilidade na solução. Considerada por Nývlt et al.(2001)
como a força motriz da cristalização, essa variável merece uma análise mais detalhada quanto
a seus métodos de obtenção.
Em geral, como já comentado no item da cristalização, a supersaturação de uma
solução pode ser obtida de diversas maneiras (Mendonça, 2008):
a) Resfriamento direto;
b) Resfriamento por evaporação a vácuo;
c) Evaporação isotérmica;
d) Mudança do meio, seja pela adição de outra substância sólida, seja na adição de
outro solvente;
e) Reação química, para criar um produto que precipita.
Como o estudo em questão se baseia na cristalização por resfriamento, uma maior
ênfase será dada a este tipo de cristalização. A cristalização por resfriamento é utilizada
quando a variação da solubilidade com a temperatura é importante (Mendonça, 2008).
As dependências do tamanho da partícula e da supersaturação com o tempo
encontram-se esquematicamente representados na Figura 2.11. O período de indução da
nucleação, tind, pode ser definido como o tempo decorrente entre o momento em que o estado
supersaturado é gerado e o tempo (tmáx) em que a supersaturação passa por um valor máximo
(Kim & Mersmann, 2001). A supersaturação (∆ρ) atinge um máximo no ponto de transição
entre a nucleação e o crescimento da partícula. A partir deste tempo, a supersaturação tende a
esmaecer, devido ao crescimento espontâneo dos cristais.

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 20
Figura 2.9 – A dependência do tempo com tamanho de partícula e a supersaturação (Kim, 2001).
2.3.6 – Etapa de Nucleação
Para o entendimento do processo de cristalização é necessário um breve conhecimento
sobre o mecanismo de formação de núcleos cristalinos. Estes podem ser criados através de
diversos mecanismos (Bartosch & Mersmann, 1998),sendo dois deles principais: a nucleação
primária (Homogênia e Heterogênea) e secundária. De acordo com Nývlt et al.(2001), a
nucleação é um processo que define o tamanho dos cristais do produto e, portanto, em última
análise suas propriedades físicas e pureza.
Nývlt et al.(2001) ainda mencionam que provavelmente os diversos mecanismo de
nucleação ocorram simultaneamente, mas a nucleação secundária prevalece fortemente em
cristalizadores.
2.3.6.1 – Nucleação primária
A nucleação primária é caracterizada por mecanismos nos quais o aparecimento dos
núcleos ocorre na ausência de cristais (Nývltet al., 2001), podendo ocorrer através de
diferentes mecanismos, sendo dois deles os principais: nucleação homogênea e nucleação
heterogênea. A nucleação homogênea ocorre em alguns materiais altamente puros, sendo que,

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 21
sob condições específicas, o próprio movimento aleatório dos átomos ou moléculas resulta no
ordenamento dos mesmos. Por outro lado, na nucleação heterogênea o processo de
ordenamento é catalisado pela presença de heterogeneidades, as quais podem ser as mais
variadas possíveis (Manrich et al., 1992). Sendo assim, se a solução é absolutamente pura, a
nucleação ocorre pelo mecanismo de nucleação homogênea, enquanto que, na presença de
substâncias sólidas estranhas ao meio (pó, colóides e paredes do cristalizador), a nucleação
ocorre de forma heterogênea.
É valido dizer que no presente trabalho, o mecanismo de formação de núcleos
cristalinos se dá especificamente através da nucleação primária homogênea. Basicamente, a
solução polimérica é submetida a um tratamento térmico (resfriamento) formar cristais do
polímero em estudo, o PMMA. Sem a presença de substâncias estranhas no meio. Para o
estudo e obtenção de partículas poliméricas carreadoras de materiais magnéticos, a nucleação
preponderante pode ser devido ao mecanismo de nucleação heterogênea.
2.3.6.2 – Nucleação secundária
A nucleação secundária se dá em uma suspensão cristalina. Neste mecanismo a
solução já está supersaturada, ou seja, há formação de cristais em solução, e para
prosseguimento da formação de novos cristais, certa quantia de cristais é depositada (Nývlt et
al.,2001). Vale salientar que este mecanismo é importante tanto na formação de partículas do
polímero em estudo quanto para a agregação de novas substâncias como, por exemplo, um
fármaco ou outro polímero ou até mesmo um material com propriedades magnéticas. Para
esclarecer melhor este fato, neste subitem serão apresentados os mecanismos desta nucleação,
até então conhecidos e que, provavelmente, atuam simultaneamente. Esses mecanismos
podem ser divididos em três classes.
Nucleação secundária aparente
Neste mecanismo, a nucleação ocorre ao submergir um cristal seco na solução
supersaturada. Após a submersão, estes cristais começam a se soltar da superfície aderida,
servindo de núcleos de crescimento (Nývltet al.,2001). A operação consiste da introdução de
cristais de tamanho especifico numa solução supersaturada, o que favorece o crescimento do
núcleo e evita a nucleação primária (Mendonça, 2008).
Nucleação por contato
Para este mecanismo, descrito por Nývlt et al.(2001), a nucleação baseia-se no fato de
que a superfície do cristal não é completamente lisa e contém inúmeras imperfeições de

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 22
diversos tamanhos. O impacto de um corpo sólido nessa superfície quebra essas imperfeições
e estes resíduos podem agir como núcleos cristalinos (Mendonça, 2008).
Nucleação na camada intermediária
Segundo Nývlt et al.(2001), os mecanismos de nucleação na camada intermediária da
superfície do cristal podem ser subdivididas nos seguintes grupos:
- Nucleação vegetativa: próximo à superfície do cristal formam-se clusters ou blocos
que podem ser facilmente removidos da camada intermediária para a solução, escoando do
cristal e gerando novos núcleos cristalinos.
- Força apresentada pela superfície de cristal, que causa mudança na estrutura da
solução aderida, levando a uma diminuição da solubilidade de soluto e a uma supersaturação
local maior na vizinhança do cristal.
-Mecanismo do gradiente de impureza: concentrações relativas de impurezas
próximas à superfície do cristal são menores que aquelas no seio da solução e se o aditivo
retarda a nucleação, as condições próximas à superfície de cristal favorecem o surgimento de
novos núcleos cristalinos.
2.3.7 – Etapa do Crescimento
Após a etapa da nucleação, há o crescimento das partículas, ocorrendo através de dois
estágios:
a. Crescimento ou cristalização primária: a fase de cristalização primária é
caracterizada porum acréscimo das dimensões do núcleo em cada direção, por unidade de
tempo. O crescimento é retardado à medida que os núcleos encontram regiões já cristalizadas
e finalmente cessa quando toda a massa disponível está transformada (fim da cristalização
primária) (Wellen, 2007).
b. Cristalização secundária: a fase de cristalização secundária ocorre no final da
cristalização pela colisão entre os esferulitos nos últimos estágios do crescimento cristalino ou
por posterior perfeição ou reorganização de macromoléculas nas regiões intra e
interesferulíticas, produzindo um aumento na cristalinidade e na espessura das lamelas
cristalinas já formadas, a qual pode em alguns casos ser muito relevante. A ocorrência de
cristalização secundária tem sido observada em diversos polímeros, copolímeros e blendas
poliméricas. Exemplos de sistemas poliméricos que apresentaram cristalização secundária
são: PE (Lorenzo, 2003), PP (Calvert & Ryan, 1984), PET (Bove et al., 1997; Bikiaris &
Karayannidis, 1998; Lu & Hay, 2001), iPS (Liu & Petermann, 2001; Liu, 2003).

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 23
Sendo assim, quando a nucleação é lenta e o crescimento rápido, o resultado é uma
pequena quantidade de esferulitos com grande tamanho, enquanto que uma rápida nucleação
irá conduzir à profusão de esferulitos que terão crescimento limitado. Quando possível, o
controle no tamanho dos esferulitos é conseguido variando-se a taxa de nucleação (Wellen,
2007). Outra forma possível em polímeros, é quando a nucleação ocorre de forma
heterogênea, ou seja, é iniciada através de partículas não-poliméricas, que atuam como
núcleos de cristalização..
2.3.8 – Zona metaestável
A zona metaestável é considerada como uma propriedade característica especifica de
cada sistema de cristalização por resfriamento e de seus mecanismos de nucleação primária
numa solução supersaturada de substâncias solúveis (Kim & Petermann, 2001). Ela depende
principalmente da temperatura, solução, taxa de resfriamento, presença de impurezas, efeitos
mecânico (agitação), etc (Kim & Petermann, 2001). Também é um importante parâmetro para
análise tanto na distribuição de tamanho como na forma dos cristais gerados no processo de
cristalização.
A zona metaestável pode ser demonstrada na Figura 2.12, a qual mostra a solubilidade
de uma substância em relação à temperatura (Kim & Petermann, 2001). Na cristalização por
resfriamento, a concentração é alterada através da linha de operação em função do tempo. O
processo consiste do resfriamento da solução, considerada subsaturada de concentração C e
temperatura T*, em que a solução está em equilíbrio com a fase sólida. Depois de um
resfriamento adicional, a fase sólida ainda não está formada, até certo tempo em que Tmet é
atingido, correspondendo à metaestabilidade da solução. Esta metaestabilidade é expressa
como um subresfriamento máximo (isto é, largura da zona metaestável) ∆Tmax=T*–Tmet,
correspondente à supersaturação máxima ∆Cmax= C–C*met.

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 24
Figura 2.10 – Relação da concentração e metaestabilidade (Kim & Petermann, 2001).
A zona metaestável está localizada entre a zona estável (subsaturada) e a zona lábil
(sobressaturada), conforme ilustra a Figura 2.13. Estas duas últimas zonas podem ser
distinguidas como, zona onde o crescimento do cristal é impossível e zona onde a nucleação é
espontânea, respectivamente, conforme trata Nývlt et al.(2001).

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 25
Figura 2.11 – Diagrama funcional de cristalização: concentração versus temperatura (Merheb, 2009).
De acordo com a Figura 2.13, o resfriamento de uma solução subsaturada parte da
zona estável e passa pelo ponto A pertencente à curva de solubilidade. Para Nývlt et
al.(2001), neste ponto a solução está saturada e deverá estar em equilíbrio com a fase sólida
correspondente, se houver. O resfriamento subsequente leva ao estado em que a concentração
do soluto é maior que a correspondente ao equilíbrio e a solução está supersaturada. Apesar
disso, nenhuma formação espontânea de cristais ocorre de imediato, ou seja, não há formação
até que o ponto D seja atingido. Esse ponto denota o limite da zona metaestável sob dadas
condições e quando ultrapassado, a solução torna-se lábil, formando-se, imediatamente, a fase
sólida. A fronteira da zona metaestável pode ser expressa pelo máximo sub-resfriamento
atingido, ΔTmáx (interseção AD), ou pela correspondente supersaturação máxima atingida,
ΔCmáx(interseção DC). Esta inter-relação é apresentada na Equação 1 (Nývlt et al., 2001):
∆��á� � ∆��á���
(1)
Um método usual de medida da largura da zona metaestável citado por Nývlt et
al.(2001) é o método politérmico. Nesta técnica, a solução saturada de concentração Ceq é
resfriada com uma velocidade ou taxa de resfriamento constante, - Ṫ, até que os cristais
visíveis possam ser detectados no tempo tc.

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 26
2.3.8.1– Efeito da temperatura
Como dito por Nývltet al.(2001), a largura da zona metaestável depende da temperatura
segundo a expressão:
∆�á�
���
���(�����.)
�/� (2)
É possível notar na Equação 2que, com o aumento da temperatura de saturação T da
solução, a largura da zona metaestável diminui.
2.3.8.2 – Efeito da pureza física da solução
Nývlt et al.(2001) expõem que a presença de um certo número de partículas sólidas
ou de poeiras de cristais tem um importante efeito na largura da zona metaestável. A presença
de substâncias isomórficas às que estão cristalizando facilita significativamente a nucleação e
consequentemente estreita a largura da zona. Portanto, o efeito de fases sólidas, representado
por corpos estranhos (nucleação heterogênea) ou cristais (nucleação secundária), é essencial
para a cristalização.
2.3.8.3 – Efeito da história térmica da solução
De acordo com Nývlt et al.(2001), estabeleceu-se que soluções mantidas por diversas
horas em temperaturas suficientemente acima de suas temperaturas de equilíbrio apresentam
zonas metaestáveis mais largas, isto é, possuem velocidades de nucleação menores quando
comparadas a soluções cujas temperaturas não excederam significativamente suas
temperaturas de saturação.
2.3.8.4 – Efeito da ação mecânica na solução
A ação mecânica na solução (agitação, vibração, impacto, ultra-som, etc), como dito
por Nývlt et al.(2001), transfere energia mecânica à solução, diminuindo a largura da zona
metaestável. Assim, soluções estagnadas possuem zonas mais largas que soluções agitadas,
justificando, com isso, a importância da utilização de uma agitação mais limitada.
2.3.8.5 – Efeito de impurezas solúveis
É praticamente impossível predizer o efeito de aditivos solúveis na largura da solução
(Nývlt et al.,2001). Portanto, o solvente deve ser filtrado para se obter o maior nível de pureza
desta substância. Para exemplificar o efeito de impurezas, aditivos que não possuam íons

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 27
comuns à substância que cristaliza, pode afetar a largura da zona metaestável em ambas
direções.
2.3.9 – Condição θ
A condição θ foi definida por Flory (1953) e corresponde à condição na qual, durante
o resfriamento de uma solução polimérica, as cadeias poliméricas estão na iminência da
precipitação. Neste ponto, qualquer variação na temperatura no processo de resfriamento
resulta na precipitação das cadeias, tornando a solução turva. Logo, é possível dizer que acima
da temperatura teta (Tθ) existe uma solução estável, e abaixo de Tθ tem-se a precipitação do
polímero.
Assim, de acordo com Canevarolo Jr. (2010), pode-se também definir a condição θde
um dado polímero como aquela condição instável em que a cadeia polimérica em solução
ocupa o menor volume hidrodinâmico, estando na eminência de precipitação, ao mesmo
tempo em que a interação polímero-polímero desaparece.
Para a determinação da condição teta de dado polímero, Canevarolo Jr.(2010)
menciona o método de turbidimetria. Neste método, o polímero é dissolvido em um solvente
pobre, com aquecimento e agitação. Após completa solubilização, a temperatura da solução
polimérica é reduzida lentamente (por exemplo, a 0,2 ºC/min.) até que ocorra a precipitação
do polímero, gerando uma solução turva, chamada Ponto de Névoa. Essa temperatura é dita
critica (Tc) e depende da massa molar e da fração volumétrica do polímero.
Segundo Canevarolo Jr. (2010), uma molécula em solução tende a excluir todas as
outras do volume que ela ocupa. Deste modo, a redução da temperatura tende a diminuir o
volume excluído, resultante da ocupação de volumes de duas moléculas poliméricas.
Esquematicamente, isso pode ser observado na Figura 2.14. Esta redução de temperatura vai
até uma temperatura mínima, conhecida como ϴ (T = ϴ), a qual o volume excluído é zero,
não havendo mais exclusão, isto é, uma molécula não sente a presença de nenhuma outra, não
havendo mais interação polímero-polímero (volume excluído igual a zero). Assim, para
atingir esta condição, são usados solventes cada vez mais pobres e/ou temperaturas cada vez
mais baixas, pois ambos reduzem o comprimento entre as pontas de uma cadeia polimérica.
Coma redução de temperatura abaixo da Tϴ,ocorre um aumento da atração entre as moléculas
(volume excluído menor que zero) e, consequentemente, um aumento na velocidade de
aproximação entre os segmentos, levando o colapso da estrutura novelar e a precipitação das
cadeias.

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 28
Figura 2.12 – Representação esquemática da exclusão entre volumes ocupados por duas moléculas poliméricas diferentes (Canevarolo Jr., 2010).
2.4 – Encapsulação de materiais magnéticos
Nos últimos anos o desenvolvimento de partículas poliméricas magnéticas tem atraído
o interesse de muitos pesquisadores por representarem uma nova classe de materiais.
Materiais poliméricos orgânicos, com excelentes propriedades ópticas, boa flexibilidade e
resistência são fáceis de processar e podem melhorar a fragilidade dos materiais inorgânicos
(Romio, 2011). Desta forma é possível combinar as vantagens dos polímeros, como
elasticidade, transparência e propriedades dielétricas e também as vantagens das
nanopartículas inorgânicas, por exemplo, absorção específica de luz, efeitos de
magnetorresistência, atividade química e catálise.
Sendo assim, a área de interesse deste trabalho é também movida pela possibilidade de
combinar nanopartículas poliméricas de PMMA a diferentes formas de óxido de ferro que
detém aplicações biológicas, e tem sido o foco de muitos estudos nos últimos anos. Estas
aplicações incluem biosseparação magnética (Bruce et al.,2009), marcação celular e
diagnóstico (Azzazy & Mansour, 2009), agentes de contraste para imagens de ressonância
magnética (Lee et al., 2006), hipertemia magnética para tratamento de tumores (Kim et al.,
2008) e carreadores de fármacos (Qinet al., 2009). Tais aplicações exploram duas vantagens
do uso de nanopartículas de óxido de ferro: sua baixa toxicidade in vivo e a possibilidade de
controlar sua magnetização. Na escala nanométrica, elas não permanecem magnetizadas após
a remoção de um campo magnético externo devido as suas propriedades superparamagnéticas
(He et al., 2005). Sob um campo magnético oscilante, as nanopartículas torna-se poderosas
fontes de aquecimentos, pela transformação da energia do campo magnético em calor
(Chastellain et al., 2004). Para tanto, estas partículas devem apresentar tamanho uniforme, e
biocompatibilidade (Fan et al., 2009).

Revisão bibliográfica
André Anderson Costa Pereira, Agosto/2013 29
Dito isto, conforme Oh & Park (2010) métodos típicos têm sido desenvolvidos para a
preparação de nanomateriais híbridos de polímero contidos de nanopartículas de FeO (óxido
de ferro) superparamagnéticosa qual incluem-se: a modificação direta com polímeros,
superfície iniciada por polimerização controlada, sílica inorgânica/hibridização do polímero,
auto-montagem, auto-associação, e vários métodos de polimerização heterogêneos (mini-
emulsão reversa e polimerização por dispersão). Além disso, juntam-se a estes os métodos
convencionais: polimerização interfacial (Romio, 2011), polimerização em suspensão
(Romio, 2011), polimerização Sol-Gel (Camilo, 2006) e a polimerização em mini-emulsão
(Erdem et al., 2000). É perceptível que a maioria dos métodos apresentados para
encapsulação de partículas magnéticas tem a polimerização como sua principal via de
obtenção destas partículas. Tais técnicas requerem condições ótimas de polimerização para se
obter materiais com propriedades desejadas. A implementação destas condições muitas das
vezes é complexa e apresenta algumas limitações.
Dentre as diversas peculiaridades das técnicas mencionadas acima pode-se mencionar,
um novo método empregado por Chen et al. (2008)a qual prepararam com sucesso partículas
superparamagnéticas de poliestireno/magnetita. Para tanto, foi utilizada a técnica de
polimerização ememulsão inversa com ferrofluido a base de água como fase dispersa
esolvente orgânico e estireno como fase contínua. O preparo das partículas poliméricas
magnéticas foi realizado com a dispersão do ferrofluido na fase óleo, constituída por
querosene, estireno e Span-80. A polimerização em emulsão foi iniciada por radiação de raios
gama de60 Co a temperatura ambiente. As partículas de magnetita foram encapsuladas pelo
poliestireno e apresentaram forma esférica.
Chen et al. (2009) empregaram a técnica de polimerização radicalar de transferência
de átomo de superfície iniciada(si-ATRP), para preparar nanopartículas de Ni-g-polímero.
Nanopartículas de níquel foram modificadas com trietoxisilanocontendo iniciadores ATRP
sem agregação através da combinação e troca de ligantes e reação de condensação. PMMA e
PNiPAM foram enxertados com sucesso a partir das nanopartículas modificadas de Ni pela
técnica si-ATRP, apresentando uma melhora na dispersão e estabilidade em solventes
apropriados.

Capítulo 3
Metodologia

Metodologia
André Anderson Costa Pereira, Agosto/2013 31
3. Materiais e métodos Neste trabalho, as atividades experimentais foram realizadas em quatro etapas
principais: (i) solubilização do polímero PMMA, (ii) processo de cristalização térmica da
solução polimérica; (iii) filtração a vácuo; e (iv) caracterização das partículas poliméricas
obtidas.
3.1 – Etapa da Solubilização
Uma mistura de álcool (etanol, ATOM 99,5% P.A. ou 1-propanol, VETEC 99,5%) e
água destilada, com 15% em massa deste último, foi utilizada para solubilizar o PMMA
(fornecido pela Aldrich, com pureza mínima de 99,5%, massa molar 120000 Da e índice de
polidispersão entre 2,0 e 2,4), ilustrado na Figura 3.1. Para garantir total solubilização do
polímero, a mistura foi aquecida até cerca de 45ºC por meio de um aquecedor com agitação
magnética. Conforme apresentado na Figura 3.2, o sistema de solubilização polimérica foi
formado por: (a) condensador de vidro, para evitar a evaporação do álcool; (b) balão de fundo
chato; (c) banho d’água; (d) termômetro digital e (e) aquecedor com agitador magnético. É
importante destacar que álcool puro não é capaz de solubilizar com facilidade o PMMA. No
entanto, uma mistura de álcool e água em proporção adequada torna o PMMA solúvel
(Shimizu, 1998; Hoogenboom et al., 2010; Jewrajka et al., 2004).
Figura 3.1– Grãos de PMMA usado na solubilização.

Metodologia
André Anderson Costa Pereira, Agosto/2013 32
Figura 3.2 – Equipamentos utilizados para a solubilização do polímero.
3.2 – Etapa do processo de cristalização
Conforme apresentado na Figura 3.3, o sistema de cristalização é formado por: (1)
banho térmico com controle de temperatura; (2) agitador com controle de rotação; (3)
recipiente encamisado em vidro de borosilicato; (4) sensor de temperatura e (5) sistema de
aquisição de dados.
Figura 3.3 – Equipamentos utilizados para a formação das partículas.
A mistura foi resfriada com base no método de resfriamento linear (Kim, 2005), com o
auxilio de um banho termostatizado (modelo Te–184, faixa de temperatura de -10 a 99,9 °C e
precisão de controle de 0,1 °C), usado para resfriar o fluido de refrigeração da camisa do
recipiente. Embora o equipamento apresente um sistema de controle de temperatura, o mesmo
não possui a habilidade de controle automático de taxa de resfriamento. Para isto, foi
necessário desenvolver uma estratégia de atuação sobre o banho, visando o seu controle e a

Metodologia
André Anderson Costa Pereira, Agosto/2013 33
obtenção de taxas lineares durante o resfriamento. A partir de um estudo prévio de resposta
dinâmica do banho, observou-se que o mesmo apresentou um comportamento característico
de um sistema de segunda ordem com atraso. Baseado nesta característica, foi utilizado um
procedimento de perturbações no set-point de temperatura do banho, buscando-se alcançar o
resfriamento em taxas previamente estabelecidas. As perturbações utilizadas seguiram uma
trajetória correspondente a uma sequência de degraus aplicados no set-point de temperatura
do banho, onde a duração (∆t) e a amplitude (Amp) de cada degrau estãorepresentadas
conformea Figura 3.4.
Figura 3.4 – Perfil de perturbação aplicado ao set-point do banho termostático para obtenção das taxas de resfriamento linearmente controladas.
A estratégia de resfriamento foi conduzida iniciando-se em 40°C até temperaturas de
aproximadamente 0°C. A escolha da amplitude (Amp) de perturbação foi correspondente à
taxa de resfriamento pré-definida, sendo: Amp = -0,7°C para a taxa de -0,2°C/min; Amp = -
1,7°C para a taxa de -0,5°C/min e Amp = -3,3°C para taxa de -0,8°C/min. No que se refere ao
intervalo de perturbação, utilizou-se o mesmo intervalo de ∆t = 3 min para todas as taxas de
resfriamento.
É importante ressaltar que o controlador de temperatura do banho térmico foi ajustado
para executar o resfriamento da solução de forma linear de acordo com a taxa estabelecida
pelo planejamento experimental. Durante toda etapa de cristalização, a mistura foi mantida
em agitação branda, constante, de aproximadamente 80 rpm. Cada experimento foi conduzido
de acordo com um planejamento experimental do tipo fatorial com dois níveis e triplicata no
ponto central, como representado na Tabela 3.1. Tendo-se como variáveis: o tipo de álcool
(etanol ou 1-propanol), a concentração do polímero na solução e a taxa de resfriamento linear

Metodologia
André Anderson Costa Pereira, Agosto/2013 34
durante a cristalização. Em todos os experimentos de cristalização, a massa de solução total
utilizada foi de 200 g.
Na Tabela 3.1 são apresentadas as variáveis e as condições de estudo para o processo
de cristalização por resfriamento de solução polimérica, tendo sido aplicado com as duas
misturas de solvente estudadas: água+etanol e água+1-propanol.
Tabela 3.1 – Níveis reais e codificados das variáveis do processo de cristalização em estudo.
Após o processo de cristalização, 300 mL de água destilada contendo 0,6% de PVA
(Poli-vinil-álcool) foram adicionadasà suspensão com o propósito de diluir o álcool da
suspensão, sendo esta levada posteriormente para um ultrassom por 60 min., para evitar a
aglomeração de partículas.As amostras foram armazenadas em garrafas de 1L. Como
apresentado na Figura 3.5, as suspensões contidas nos recipientes apresentam coloração
esbranquiçada (branca),evidenciando a formação de partículas.
Figura 3.5 – Recipientes com o armazenamento das amostras após o processo de cristalização.
3.3 – Etapa de Filtração
Após o tratamento no Ultrasson,a mistura foi levadaa um sistema de filtração a vácuo,
com o objetivo de filtrar e reter as partículas superiores a 3 µm. Cabe ressaltar que tanto a
adição de água quanto o tratamento com o ultrassom foram usados para evitar aglomeração de
partículas. As partículas retidas em filtro foram lavadas em águaresfriada a temperatura de

Metodologia
André Anderson Costa Pereira, Agosto/2013 35
aproximadamente 5oC, obtendo-se uma nova suspensão de partículas maiores, enquanto o
passante pelo filtro formam a suspensão de partículas submicrométricas.
O sistema de filtração a vácuo utilizado para filtrar a solução polimérica e obter as
partículas formadas após o procedimento de cristalização está ilustrado na Figura 3.6, sendo
constituído por: uma bomba a vácuo (modelo TE-058); um conjunto com funil de Buchner e
Kitassato e papel filtro com tamanho de poro de 3µm, diâmetro 9 mm, de marca FUSION.
Figura 3.6 – Sistema de filtração a vácuo.
3.3.1 – Quantificação da fração de partículas submicrométricas
Após a filtração, buscou-se avaliar as frações de partículas retidas e passantes em filtro
de 3µm. O objetivo desta avaliação foi verificar como as condições de cristalização podem
influenciar na formação da fração de partículas submicrométricas ou passantes pelo filtro. A
princípio, a análise consistiu em quantificar a massa polimérica (teor de sólidos) no material
passante e no retido na filtragem a vácuo. Para isto, foi retirada de cada amostra, após a
cristalização, uma quantidade definida de 100 mL (aproximadamente) de suspensão
polimérica formada, sendo, em seguida, submetida à filtração.
Do volume de passante da filtração, uma alíquota de 50 mL foi retirada e levada para
secagem em estufa de recirculação mantida em 30°C. Após esta etapa, o material foi pesado
para quantificação.
Em relação ao material retido em filtro, o mesmo foi lavado para remoção das
partículas, obtendo-se uma suspensão. A partir da suspensão, uma alíquota de 50 mL foi
retirada e levada para secagem em estufa de recirculação mantida a temperatura de 30°C.As
partículas poliméricas secas foram pesadas para quantificação.
Após a quantificação da massa polimérica passante e da retida na filtragem, foi gerada
uma tabela com os resultados das frações de partículas submicrométricas e superiores a 3µm.

Metodologia
André Anderson Costa Pereira, Agosto/2013 36
3.4 – Etapa de Caracterização
A caracterização das partículas poliméricas obtidas, basicamente foram realizadas
análises de distribuição de tamanho de partícula (DTP) e microscopia. No caso das partículas
submicrométricas da suspensão passante na filtração, usou-se Microscópio Eletrônico de
Varredura (MEV) eo DLS (Espalhamento de Luz Dinâmica) para a caracterização. Já no caso
das partículas retidas na filtração, usou-se o Microscópio Óptico (MO) e um Granulômetro do
tipo Nono Sizer (marca CILAS) para análise de DTP.
De acordo com o planejamento experimental e uma avaliação estatística dos
resultados, a análise da distribuição do tamanho de partículas permitiu uma avaliação do
efeito da concentração de polímero na solução e da taxa de resfriamento durante a
cristalização.
3.4.1 – Microscopia Eletrônica de Varredura (MEV)
As análises de MEV foram feitas em um microscópio eletrônico de varredura da
marca Shimadzu (modelo SS550), pertencente ao Laboratório de Análises da Universidade
Estadual de Maringá (UEM).
Para estas análises, foi utilizado um porta amostra de aço inoxidável. Sobre ele foi
colocado uma fita adesiva de carbono e sobre esta última, foi depositada a amostra da
suspensão passante na etapa da filtração. Em seguida, uma fina camada de ouro foi depositada
no material (amostra + porta amostra), utilizando um metalizador IC-50 ION COATER,
marca Shimadzu. Foi empregada uma voltagem de 15 kV com uma intensidade de corrente de
30 µA.
3.4.2 – Distribuição de tamanho de partículas por espalhamento dinâmico de luz (DLS)
A análise de distribuição de tamanho de partícula (DTP), foram realizadas em um
analisador de tamanho de partícula em suspensão coloidal da marca Zeta Plus, modelo 90Plus
(BrookhavenInstruments Corporation), pertencente Laboratório de análises do
NUPEG/UFRN, conforme apresentado na Figura 3.7. Baseado no princípio do Espalhamento
Dinâmico da Luz (DLS), foi utilizado um laser semicondutor de 35 mW com controle de
potência até 3,5 mw (automático ou via software) para otimizar medida, sendo o detetor APD
para maximizar a sensibilidade das partículas com baixo espalhamento.

Metodologia
André Anderson Costa Pereira, Agosto/2013 37
Para estas análises foi usada uma cubeta de vidro óptico quadadra, na qual as amostras
das suspensões passantes na etapa de filtração foram colocadas, sem a necessidade de
diluição. Ao final de cada aferição, as amostras remanescentes foram descartadas e a cubeta
foi lavada com água destilada, antes de ser novamente inserida no equipamento contendo
nova amostra. Em cada análise, o software do equipamento gerou resultados da distribuição
de tamanho de partículas, bem como das propriedades médias, tais como: tamanho médio de
partícula e índice de polidispersão.
Figura 3.7 – Analisador de partícula por Espalhamento de Luz Dinâmica (DLS).
3.4.3 – Microscopia Óptica (MO)
As análises de morfologia da partícula foram realizadas em um microscópio óptico de
marca Olympus (modelo BX 51-P), pertencente ao Laboratório de Análise do PPGEQ/UFRN
(Figura 3.8). Este equipamento é ideal para observações em luz polarizada. Para tal, este
detém 4 (quatro) lentes objetivas (ACHN10xP, ACHN20xP, ACHN50xP e ACHN100xOP)
destinadas para este tipo de luz. Ele também é munido de um condensador especialmente
projetado e filtros de polarização, de alto valor EF (razão de brilhos que geram menor
distorção do sistema óptico).
Para serem levadas ao microscópio, as amostras das partículas retidas na filtração
foram primeiramente diluídas com água destilada em tubos de ensaios. Em seguida foram
dispostas durante 1h no Ultrassom. Este procedimento foi feito visando dispersar as partículas
ao máximo na solução. Após esta etapa, as amostras foram colocadas em lâminas de vidro
com o auxílio de uma pipeta conta gotas e esperou-se a secagem do material em temperatura
ambiente.

Metodologia
André Anderson Costa Pereira, Agosto/2013 38
Figura 3.8 – Microscópio Óptico (Olympus).
3.4.4 – Distribuição de tamanho de partículas (DTP)pelo granulômetro
Em relação à análise granulométrica, para a realização da medida de distribuição de
tamanhos de partículas retidas na filtração, utilizou-se um analisador de tamanho de partícula
CILAS modelo 1180 (Figura 3.9) com “software” CILAS versão 2.56 do Laboratório de
Tecnologia de Materiais – UFRN.
As amostras líquidas resultantes da lavagem do filtro foram adicionadas
cuidadosamente na cuba do granulômetro com auxílio de um Becker, com o intuito de se
obter a concentração ótima de leitura do equipamento. Em geral, o resultado é fornecido pelo
equipamento em termos de massa acumulada (em percentagens) versus diâmetro esférico
equivalente das partículas ou da distribuição modal de tamanhos de partículas, além das
propriedades médias, tais como: tamanho médio e índice de polidispersão.
Figura 3.9 – Analisador de tamanho de partícula (CILAS).

Metodologia
André Anderson Costa Pereira, Agosto/2013 39
3.5 – Correlação linear das variáveis de entrada e saída
No intuito de estudar o grau de relacionamento entre duas ou mais variáveis, isto é,
saber se as alterações sofridas por uma das variáveis são acompanhadas por alterações em
outras, uma análise de correlação linear das variáveis de entrada (Taxa de resfriamento e
Concentração polimérica) com as variáveis de saída (Diâmetros médios e Frações passantes)
foi realizada a partir de uma matriz de correlação calculada com o auxílio do software
computacional Statistica 7.0. Nesta matriz estão representadas as intensidades e a direção do
relacionamento linear entre as duas variáveis (Lira, 2004).
Sendo assim, esta análise de correlação é um indicador que atende à necessidade de se
estabelecer a existência ou não de uma relação entre essas variáveis sem que, para isso, seja
preciso o ajuste de uma função matemática. Não existe a distinção entre a variável explicativa
e a variável resposta, ou seja, o grau de variação conjunta entre X e Y é igual ao grau de
variação entre Y e X (Lira, 2004).
Por fim, esta representação do relacionamento linear, assim como, a análise dos
coeficientes de correlação obtidos entre as variáveis está apresentada no próximo capítulo.

Capítulo4
Resultados e Discussões

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 41
4. Resultados e Discussões Neste capítulo serão descritos os principais resultados da formação das partículas e
caracterização das mesmas. Todas foram formadas através do método de cristalização por
resfriamento linear. Os resultados apresentados serão comparados com os resultados de Kim
(2005), que também realizou a cristalização por resfriamento linear na obtenção de partículas
nano/micrométricas da solução polimérica. Será apresentado uma discussão da estratégia
proposta para implementar o perfil de resfriamento linear, importante para execução dos
experimentos de cristalização.
4.1 – Solubilização do PMMA
É de conhecimento que um bom solvente é aquele que dissolve certa quantidade de
polímero numa faixa de temperatura compreendida entre seu ponto de fusão e seu ponto de
ebulição, e um mau solvente é aquele que não é capaz de intumescer (inchar) um polímero.
No presente trabalho, o comportamento da solubilidade do PMMA foi observado usando-se
os álcoois etanol e 1-propanol em diferentes temperaturas. Para alcançar considerável
solubilidade do polímero, tais solventes foram testados até temperaturas próximas a 70ºC.
Nesta temperatura, constatou-se que o material intumesceu e ficou retido no fundo do
recipiente usado na solubilização. O resultado obtido no presente trabalho contradiz o que foi
relatado por Kim (2005), onde tal autor reporta solubilização do PMMA em 1-propanol a
20ºC. No entanto, o resultado é concordante com o trabalho de Jewrajka et al.(2004), pois
mostra que tais substâncias puras apresentam baixo poder de solubilizar o polímero PMMA
em temperatura ambiente.
Para fazer a solubilização do PMMA, uma mistura de solventes (álcool e água) foi
sugerida, conforme Hoogenboom et al. (2010). Nesta mistura, a água entra com
características de co-solvência (Cowier et al., 1987), facilitando a inserção do álcool na
estrutura polimérica. Com isso, a solubilização polimérica é facilitada a temperaturas
próximas a 45ºC. Porém, o acréscimo de água na mistura é limitado até certo ponto, já que
aumentando ainda mais a quantidade de água, aumenta a polaridade da mistura de solventes e
leva à formação de “clusters de água”. Este fenômeno diminui a solubilidade do polímero
pela necessidade da quebra das ligações de hidrogênio água-água para a formação de um
camada de hidratação (Hoogenboom et al., 2010). Baseado nestes aspectos, estabeleceu-se um
percentual mássico de água para a solubilização do PMMA em torno de 15% de água na

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 42
solução alcoólica, o que permitiu uma adequada solubilização do polímero sem qualquer
degradação. Na Figura 4.1, mostra-se uma típica solubilização do PMMA numa mistura de
álcool + 15% de água em temperatura de aproximadamente 45 °C.
Figura 4.1– Solubilização do PMMA em solução alcoólica.
4.2 –Estratégia para obtenção das taxas de resfriamento
Conforme mencionado no capítulo anterior, para produzir partículas poliméricas
durante a cristalização, foram pré-definidas taxas de resfriamento. O resfriamento da solução
polimérica foi feito a partir da circulação, pela camisa do vaso cristalizador, de um fluido de
refrigeração proveniente de um banho termostatizado. Tal banho apresenta um sistema de
controle de temperatura capaz de estabelecer com eficiência a temperatura num determinado
set-point. O sistema, porém, não é capaz de controlar uma trajetória pré-definida de set-point,
como é o caso em questão, em que se deseja implementar taxas de resfriamento linearmente
controladas. Para isto, foi necessário desenvolver uma estratégia de atuação sobre o banho,
visando a obtenção das taxas lineares durante o resfriamento.
Para avaliar o comportamento dinâmico do cristalizador, foi feita uma análise
dinâmica experimental do sistema. A análise consistiu em realizar perturbações no set-point
de temperatura do banho, verificando a resposta da temperatura na solução dentro do
cristalizador. Com base nos resultados experimentais, um modelo matemático do tipo
entrada/saída, com representação de função de transferência, foi identificado. O modelo mais
descritivo resultou num sistema de segunda ordem com atraso, de acordo com a Equação 3.
�(�) � ,"#
($,%&∙�())�∙ *+),$&∙� ∙ ,(�) (3)
Sendo: y(s) a temperatura da solução em variáveis desvios no cristalizador; u(s) a temperatura
de set-point em variáveis desvios no banho termostatizado; e s a variável de Laplace.

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 43
Na Figura 4.2, são apresentados resultados da identificação do processo mediante
perturbações realizadas na temperatura do set-point do banho.
Figura 4.2 – Identificação do processo mediante perturbações realizadas na temperatura do set-point do banho. Perturbação no set-point e resposta do sistema para amplitudes: (a) de 5°C; (b) de 10°C e (c) de 20°C.
É possível notar na Figura 4.2 que quanto menor a amplitude da perturbação realizada
no set-point do banho uma melhor descrição foi obtida do modelo matemático. A partir desta
(a)
(b)
(c)

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 44
consideração, buscou-se projetar com o modelo matemático uma trajetória de perturbação no
set-point de modo a obter as taxas de resfriamento desejadas. Na Tabela 4.1são apresentadas
as condições encontradas para a implementação da perturbação no set-point,obtendo-se assim
as taxas de resfriamento usadas nos experimentos de cristalização. As perturbações aplicadas
no set-point de temperatura do banho foram na forma de uma sequência de degraus com
duração (∆t) e amplitude (Amp), sendo os valores dados na Tabela 4.1.
Tabela4.1 – Parâmetros para a função de perturbação aplicada no set-point.
∆t Amp Taxa de resfriamento
3 min -0,7°C -0,2°C/min
3 min -1,7°C -0,5°C/min
3 min -3,3°C -0,8°C/min
As respostas experimentais das taxas lineares de resfriamento em relação à
implementação das trajetórias de perturbação definidas, podem ser observadas na Figura 4.3.
É possível notar nos gráficos de taxa de resfriamento (Figura 4.3) que cada linha de tendência
representa significativamente uma reta, com coeficiente de regressão R2 em torno de 0,99. É
importante salientar que foi definida uma temperatura para o início do resfriamento e, logo
após, perturbações de -0,7ºC, -1,7ºC e -3,3ºC foram efetuadas a cada 3 min. no banho
temostatizado, resultando em taxas de aproximadamente -0,2, -0,5 e -0,8ºC/min,
respectivamente.
(a) (b)
R2 = 0,999 R2 = 0,999

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 45
Figura 4.3 – Perfis de resfriamento típicos praticados durante os experimentos de cristalização, com taxas de resfriamento de (a) -0,2°C/min.;(b) -0,5°C/min. e (c) -0,8°C/min.
4.3 – Quantificação da fração de partículas submicrométricas
O objetivo desta análise foi quantificar, por análise de teor de sólidos, a fração de
partículas passantes e retidas num filtro de 3µm das amostras resultantes da cristalização. Nas
Tabelas 4.2 e 4.3 são apresentados os resultados do planejamento experimental referentes às
cristalizações conduzidas a partir da solução em etanol + água e 1-propanol + água,
respectivamente.
Tabela 4.2– Frações de partículas retidas e passantes obtidas da cristalização em solução de etanol + água.
PE1 PE2 PE3 PE4 PE5 PE6 PE7 Taxa de resfriamento (°C/min) -0,2 -0,8 -0,2 -0,8 -0,5 -0,5 -0,5
Concentração de polím. (%w/w) 1 1 5 5 3 3 3 Fração de Retido 0,19 0,08 0,15 0,82 0,46 0,10 0,28
Fração de Passante 0,81 0,92 0,85 0,18 0,54 0,90 0,72 Tabela 4.3 – Frações de partículas retidas e passantes obtidas da cristalização em solução de 1-propanol + água.
P1 P2 P3 P4 P5 P6 P7 Taxa de resfriamento (°C/min) -0,2 -0,8 -0,2 -0,8 -0,5 -0,5 -0,5
Concentração de polím. (%w/w) 1 1 5 5 3 3 3 Fração de Retido 0,06 0,02 0,94 0,35 0,06 0,32 0,01
Fração de Passante 0,94 0,98 0,06 0,65 0,94 0,68 0,99
Do ponto de vista da fração de partículas submicrométricas, ou seja, passantes pelo
filtro de 3µm, é possível notar, comparando os resultados apresentados nas Tabelas 4.2 e 4.3,
(c)
R2 = 0,996

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 46
que na condição de maior taxa de resfriamento (-0,8 °C/min) e menor concentração
polimérica (1 %w/w), uma maior quantidade de partículas submicrométricas é obtida. Este
resultado apresenta característica parecida com os resultados obtidos por Kim (2005), embora
tenha sido obtido em condições experimentais distintas. O uso do 1-propanol proporcionou
maior quantidade de partículas submicrométricas do que o etanol. Tal fato pode ser explicado
devido ao maior poder de solubilidade da mistura 1-propanol + água, já que esta consegue
solubilizar o PMMA em temperaturas mais baixas que a mistura etanol + água.
Consequentemente, o material atinge a supersaturação mais rapidamente para o etanol+água
conferindo um maior crescimento das partículas, enquanto na mistura 1-propanol + água a
supersaturação ocorre em temperaturas mais baixas conferindo um menor período de
crescimento das partículas. No caso particular da cristalização realizada em solução de etanol
+ água, nota-se que o pior resultado de produção de partículas submicrométricas ocorreu na
maior concentração polimérica (5 %w/w) e na maior taxa de resfriamento (-0,8 °C/min),
enquanto que no caso da cristalização em solução de 1-propanol + água o pior resultado
ocorreu também na maior concentração (5 %w/w), mas na menor taxa de resfriamento (-0,2
°C/min). De certa forma, este resultado permite indicar que quanto maior é a concentração
polimérica, maior a dificuldade para a produção de partículas submicrométricas. O efeito da
concentração de cadeias poliméricas em solução pode resultar em dois fenômenos: (i) formar
um maior número de embriões na nucleação e ter um baixo crescimento, obtendo-se
partículas com tamanhos reduzidos; ou (ii) formar um menor número de embriões na
nucleação e ter um elevado crescimento das partículas, obtendo-se partículas de maior
dimensão. Fazendo-se um comparativo mais abrangente, é possível observar que, em geral, os
resultados das frações de partículas submicrométricas foram melhores para os experimentos
de cristalização realizados com a mistura 1-propanol + água. Tal fato parece apontar para a
discussão de que a utilização de diferentes solventes pode influenciar diretamente no tamanho
da partícula formada na cristalização. No que diz respeito aos experimentos realizados no
ponto central do planejamento, observa-se uma significativa dispersão dos resultados,
podendo-se alcançar resultados de medianos a ótimos, quanto à fração de partículas
submicrométricas obtidas.
4.4 – Distribuição de tamanho de partículas da fração retida
Após o processo de cristalização e por seguinte a etapa de filtração, as partículas
retidas em filtro de 3 µm foram lavadas em água destilada e submetidas a um tratamento

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 47
sônico emultrasom para homogenização. Posteriormente, foram realizadas análises de
tamanho de partícula num granulômetro.Ao fim das análises, foram obtidos os diâmetros
percentuais, compreendendo 10%, 50% e 90%,o diâmetro médio de cada amostra e os
histogramas de distribuição de tamanho de partícula (DTP). Avaliando-se os histogramas de
DTP (Figuras 4.4 a 4.10), nota-se que em geral as distribuições apresentam-se como curvas
largas, caracterizando uma ampla faixa de tamanho de partículas formadas, o que é
característico nos processos de cristalização. É importante salientar que, para todos os casos
estudados, as partículas apresentaram tamanho inferior a 100 µm.No caso particular da Figura
4.5, apenas foi apresentado o histograma de DTP da amostra PE2 obtida com a cristalização
em etanol + água, pois a massa da fração retida referente à amostra P2 obtida com a
cristalização em 1-propanol + água foi insuficiente para a análise em questão.
Figura 4.4 – Histogramas de DTP das amostras (a)PE1 e (b)P1.
Figura 4.5 – Histograma de DTP para a amostra PE2
a b
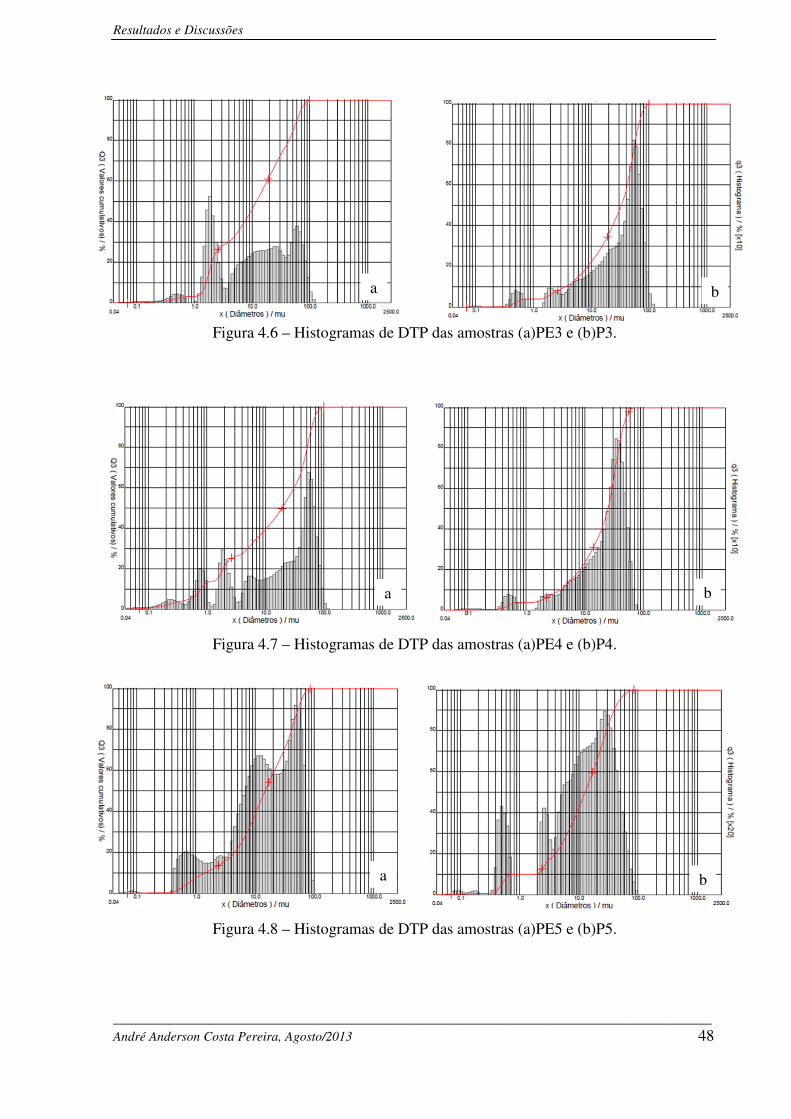
Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 48
Figura 4.6 – Histogramas de DTP das amostras (a)PE3 e (b)P3.
Figura 4.7 – Histogramas de DTP das amostras (a)PE4 e (b)P4.
Figura 4.8 – Histogramas de DTP das amostras (a)PE5 e (b)P5.
a b
a
a
b
b

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 49
Figura 4.9 – Histogramas de DTP das amostras (a)PE6 e (b)P6.
Figura 4.10 – Histogramas de DTP das amostras (a)PE7 e (b)P7.
Conforme as Tabelas 4.4 e 4.5, observa-seque, em geral,os diâmetros das partículas
nas amostras cristalizadas em etanol + água foram inferiores aos diâmetros nas amostras
cristalizadas em 1-propanol + água. Avaliando-se a fração de partículas retidas (Tabelas 4.2 e
4.3), nota-se que as amostras PE4 e P3 foram as que mais produziram partículas retidas. Tal
resultado é corroborado com a análise de tamanho médio de partículas, onde as amostras PE4
e P3 apresentaram os maiores valores, conforme demonstrado nas Tabelas 4.4 e 4.5.
a b
a b

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 50
Tabela 4.4 – Diâmetros resultantes da fração retida com amostras cristalizadas em etanol + água.
Amostra
Diâmetro
10% (µm)
Diâmetro
50% (µm)
Diâmetro
90% (µm)
Diâmetro
médio (µm)
PE1 2,18 12,11 33,52 15,53
PE2 7,91 23,33 43,01 24,64
PE3 1,62 11,79 59,27 21,63
PE4 0,79 19,09 61,3 26,28
PE5 1,41 14,63 52,85 22,06
PE6 0,77 2,86 26,71 8,07
PE7 0,47 2,34 9,08 3,76
Tabela 4.5 – Diâmetros resultantes da fração retida com amostras cristalizadas em1 – propanol + água.
Amostra
Diâmetro
10% (µm)
Diâmetro
50% (µm)
Diâmetro
90% (µm)
Diâmetro
médio (µm)
P1 0,41 22,97 57,82 26,67
P3 3,56 33,06 66,27 34,01
P4 3,82 24,65 44,76 24,37
P5 2 12,71 39,16 17,29
P6 5,41 24,85 43,85 24,71
P7 2,72 16,77 40,31 19,58
4.5 – Análises por Microscopia Óptica das partículas presentes na fração
retida
Amostras da fração retida foram preparadas e dispostas em lâminas de vidro, sendo
analisadas por Microscopia Óptica para avaliar a morfologia das partículas.Nas Figuras 4.12a
4.17 são apresentadas as imagens de microscopia, fazendo-se um comparativo das amostras
cristalizadas em etanol + água com as amostras cristalizadas com 1-propanol + água. Em
geral, é possível perceber que as partículas possuem característica de morfologia esférica com
aspecto aparentemente translúcido, notado pelo brilho intenso provocado pelas partículas em
algumas das imagens obtidas. Nota-se também que as partículas parecem possuir diâmetro
inferior a 10 µm, o que difere dos resultados de tamanho de partícula verificados com a
análise utilizando o granulômetro. Tal diferença pode ser atribuída a possível formação de
aglomerados de partículas, o que pode ter conduzido o granulômetro a apresentar resultados
de tamanho de partículas bem maiores, na faixa de 3 a 100 µm. Nas Figuras 4.12 a 4.16, é
possível notar a ocorrência de aglomerados de partículas, mas os mesmos demonstram serem
formados por partículas com tamanho inferior a 10 µm. Nota-se também que as imagens

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 51
obtidas para as partículas cristalizadas em etanol + água parecem ser mais nítidas e com
tamanhos inferiores às partículas cristalizadas em 1-propanol + água, observação também
registrada na DTP. O tipo do álcool na solução pode causar uma mudança de aspecto na
camada superficial da partícula, tornando-a com menos ou mais capacidade para visualização.
Figura 4.11 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE1 e (b)P1.
Figura 4.12 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE2 e (b)P2.
a
a
b
b

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 52
Figura 4.13 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE3 e (b)P3.
Figura 4.14 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE4 e (b)P4.
Figura 4.15 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE5 e (b)P5.
a b
a b
a b

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 53
Figura 4.16 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE6 e (b)P6.
Figura 4.17 – Análise em Microscopia Óptica a 100x; Ensaios (a)PE7 e (b)P7.
4.6 – Distribuição de tamanho de partículas na fração passante
As amostras passantes em filtro de 3µm foram analisadas em DLS (Espalhamento de
luz dinâmica), permitindo a obtenção das curvas de distribuição de tamanho de partícula
(DTP), dos tamanhos médios de partícula e do índice de polidispersão. Nas Figuras 4.18 a
4.24, apresenta-se um comparativo entre as curvas de DTP para as amostras obtidas na
cristalização com etanol + água e 1-propanol + água. Observa-se que, em geral, as curvas de
DTP estão distribuídas numa faixa compreendida entre 100nm a 3µm, caracterizando estas
partículas como submicrométricas. Este resultado está de acordo com a filtração realizada, já
que as partículas foram passantes em filtro de 3µm. É possível verificar que as curvas de DTP
mais largas ocorreram para as amostras obtidas com os experimentos realizados em menor
taxa de resfriamento (Figuras 4.18 e 4.20). Tal resultado parece indicar que quanto mais lenta
é a taxa de resfriamento, um maior intervalo de tempo é praticado nas etapas de nucleação e
a b
a b

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 54
crescimento, favorecendo uma maior diversificação na organização das partículas e,
consequentemente, permitindo a formação de partículas com maior dispersão em tamanho.
Em geral, as partículas formadas a partir da solução etanol + água tendem a apresentar
tamanhos inferiores àquelas formadas em 1-propanol + água. Este resultado também foi
evidenciado na fração de partículas retidas quando observadas por microscopia óptica,
conforme discutido anteriormente.
Figura 4.18 – Curvas de DTP obtidas por análises em DLS para as amostras PE1 e P1.
Figura 4.19 – Curvas de DTP obtidas por análises em DLS para as amostras PE2 e P2.
0
20
40
60
80
100
120
100 1000 10000
Inte
nsid
ade
Tamanho de partícula (nm)
PE1
P1
0
20
40
60
80
100
120
100 1000 10000
Inte
nsid
ade
Tamanho de partícula (nm)
PE2
P2

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 55
Figura 4.20 – Curvas de DTP obtidas por análises em DLS para as amostrasPE3 e P3.
Figura 4.21 – Curvas de DTP obtidas por análises em DLS para as amostrasPE4 e P4.
Figura 4.22 – Curvas de DTP obtidas por análises em DLS para as amostras PE5 e P5.
0
20
40
60
80
100
120
100 1000 10000
Inte
nsid
ade
Tamanho de partícula (nm)
PE3
P3
0
20
40
60
80
100
120
100 1000 10000
Inte
nsid
ade
Tamanho de partícula (nm)
PE4
P4
0
20
40
60
80
100
120
100 1000 10000
Inte
nsid
ade
Tamanho de partícula (nm)
PE5
P5

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 56
Figura 4.23 – Curvas de DTP obtidas por análises em DLS para as amostras PE 6 e P6.
Figura 4.24 – Curvas de DTP obtidas por análises em DLS para as amostras PE7 e P7.
Em relação ao tamanho médio das partículas, nota-se na Tabela 4.6 que as partículas
obtidas em solução de 1-propanol + água foram mais uniformes, resultando em médias entre
670,3 e 961,8nm. Já as partículas obtidas em solução de etanol + água apresentaram tamanhos
médios mais abrangentes, ficando entre 327,3 a 1952,8 nm. Tais resultados podem estar
relacionados ao fato de que a mistura de solvente etanol + água apresenta mais influência
sobre o processo de cristalização térmica, quando comparado à mistura 1-propanol + água.
Além disso, conforme já discutido, as partículas com um menor tamanho médio foi obtida em
solução de etanol + água. Por outro lado, é importante salientar que as maiores frações de
passantes (partículas submicrométricas) foram encontradas com a mistura 1-propanol + água
(Tabelas 4.2 e 4.3). Em geral, pode-se observar que o índice de polidispersão(Tabela 4.6)
0
20
40
60
80
100
120
100 1000 10000
Inte
nsid
ade
Tamanho de partícula (nm)
PE6
P6
0
20
40
60
80
100
120
100 1000 10000
Inte
nsid
ade
Tamanho de partícula (nm)
PE7
P7

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 57
ficou abaixo de 0,2, indicando que os resultados das análises foram adequados e com boa
confiabilidade. Adicionalmente, tal resultado permite indicar que o método de cristalização
térmica forma partículas submicrométricas com características uniformes quanto ao tamanho.
Tabela 4.6– DiâmetrosMédios e polidispersão resultantes das análises em DLS.
Amostras Diâmetro
Médio(nm) Polidispersão Amostras Diâmetro
Médio(nm) Polidispersão
PE1 1952,8 0,16 P1 741,6 0,007
PE2 603,7 0,005 P2 825 0,017
PE3 327,3 0,304 P3 670,3 0,005
PE4 351,3 0,151 P4 849,3 0,104
PE5 439,5 0,005 P5 961,8 0,043
PE6 393,2 0,063 P6 758,8 0,005
PE7 867 0,027 P7 912,5 0,052
4.7 – Análises por Microscopia Eletrônica de Varredura (MEV)
Na intenção de avaliar morfologicamente as partículas passantes na filtragem, as
amostras obtidas de cada ensaio foram analisadas em microscopia eletrônica de varredura
(MEV). As melhores imagens estão apresentadas da Figuras 4.25a 4.27. Cabe destacar que
não foi possível visualizar com nitidez algumas das amostras através do MEV. Observa-se
que as partículas formadas possuem uma morfologia esférica uniforme com tamanho inferior
a1μm. É importante comentar que estas partículas foram analisadas em suspensão bastante
diluída e, por isso, as mesmas foram observadas de forma bem isolada. Nota-se que as
partículas não se apresentaram em ocorrências de aglomerados, porém parecem coalescidas.
Figura 4.25 – Imagens do Ensaio P3 obtidas no microscópio eletrônico de varredura (MEV).

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 58
Figura 4.26 – Imagens do Ensaio PE4 obtidas no microscópio eletrônico de varredura (MEV).
Figura 4.27 – Imagens do ensaio PE5obtidas no Microscópio eletrônico de varredura (MEV).
Assim como no trabalho de Kim (2005),no presente trabalho foram obtidas partículas
uniformemente esféricas de distribuição específica numa faixa abaixo de 1µm controlando a
taxa de resfriamento do processo de cristalização como relatado por Kim
(2000).Caracterizando um amplo potencial destas partículas poliméricas se apresentarem
como sistemas carreadores (Guterres, 2003).A Figura 4.28 apresenta imagens de MEV do
trabalho de Kim (2005). Nota-se que a imagem de microscopia reportada no seu trabalho
aparece com grande ocorrência de partículas relativamente maiores que as obtidas no presente
estudo, o que permite confirmar que não houve nenhum procedimento de separação das
partículas. Adicionalmente, vale ressaltar que as condições experimentais do trabalho de Kim
(2005) foram diferentes do presente trabalho, coincidindo apenas na condição de taxa de
resfriamento de -0,5°C/min e concentração polimérica da solução de 3% em massa.

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 59
Figura 4.28 – Imagens no MEV das partículas do trabalho Kim(2005); Taxas de resfriamento (a) -0,5 e (b) -1ºC/min para 3%w/w.
4.8 – Encapsulação de material magnético
Na tentativa de adicionar materiais magnéticos à estrutura polimérica foram realizados
testes com a Ferrita (CoFe2O4) na condição considerada neste trabalho, de maior taxa de
resfriamento (-0,8 °C/min.) e menor concentração polimérica (1 %w/w). Foi utilizado 0,25g
deste material em 2 (dois) testes em soluções alcoólicas distintas (etanol e 1-propanol). Sua
adição no sistema de cristalização foi somente realizada depois queo material polímerico, já
solubilizado, foi posto no cristalizador.Após a etapa da formação das partículas, a solução
resultante foi levada ao ultrasom e,em seguida, a solução foi filtrada em um papel filtro de
3µm. Na Figura 4.29(a) está representada a soluçãoobtida após lavagem das partículas retidas
na filtração. Nota-se um aspecto meio escurecido da suspensão, o que pode estar associado à
presença das partículas de ferrita inseridas nas partículas de PMMA. É importante ressaltar
que a solução passante apresentou um aspecto transparente, evidenciando, neste teste, a
ausência de partículas submicrométricas com material magnético encapsulado pelo polímero.
Isto, supostamente, deve-se a moforlogia e tamanhonão-uniformes destas partículas
magnéticas,sendo necessário um melhor tratamento delas para o uso neste processo de
encapsulamento.
Uma indução com campo magnético foi testado no presente trabalho, na suspensão
resultante da cristalização com adição de ferrita, conforme apresentado na Figura 4.29(b).
Nota-se claramente que uma parte significativa do material foi atraída para o lado onde o
campo magnético foi disposto. Tal resposta pode indicar que, de fato, partículas poliméricas
carreadoras de ferrita foram obtidas com os ensaios de cristalização realizados.
a b

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 60
Figura 4.29 – Solução resultante após a filtragem dos ensaios com a Ferrita(a); Suspensão sobre indução de um campo magnético (b).
Aspecto similar foi apresentado no trabalho do Cui& Wang et al. (2010), os quais
obtiveram nanoesferas de PMMA via polimerização por emulsão que continham magnetita
(Fe3O4), conforme apresentado na Figura 4.30, onde é possível notara indução de um campo
magnético sobre o material sintetizado.
Figura 4.30–Nanoesferas de PMMA + Fe3O4 obtidas via polimerização por emulsão (Cui & Wang et al.,2010).
Na tentativa de visualizar imagens das partículas nesta condição de cristalização, as
amostras do material retido foram analisadas em microscopia óptica. Como pode ser
a b

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 61
observado na Figura 4.31, há a presença de partículas esféricas com tonalidade mais escuras
entres as partículas esféricas mais claras. Tal característica pode indicar que as partículas mais
escuras correspondem à inserção de ferrita na partícula polimérica, alterando a sua cor
original.
Figura 4. 31 – Microscopia óptica de partículas de PMMA com Ferrita, ensaio FP2.
4.9 – Análise de correlação linear
Na matriz de correlação representada na Tabela 4.7 é possível observar a variação dos
coeficientes de correlação (rxy). Esta variação, normalmente, pode ser de–1a +1 e sua
interpretação dependerá do valor numérico e do sinal.
Tabela 4.7 – Matriz de correlação das variáveis de entrada e saída para um planejamento 22 + 3 pontos centrais.
*Conc: concentração polimérica (w/w); Fet: fração submicrométrica de etanol; Fprop: fração
submicrométrica de 1-propanol; Dpe: Diâmetro médio das partículas de etanol; Dpp: Diâmetro médio
das partículas de 1-propanol.

Resultados e Discussões
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 62
Basicamente, esta correlação será tanto mais forte quanto mais próxima estiver o
coeficiente de –1 ou +1, e será tanto mais fraca quanto mais próxima o coeficiente estiver de
zero, conforme ilustrado na Figura 4.32.
Figura 4.32 – Representação dos valores numéricos dos coeficientes de correlação possíveis em uma análise de correlação.
Ao Realizar uma análise entre os coeficientes de correlação gerados na matriz de
correlação (Tabela 4.7) pode-se aferir, para uma correlação significativa de grau de confiança
95%, que há uma correlação moderada negativa entre a variável de entrada (taxa de
resfriamento) e as variáveis de saída (Fet e Dpe), indicando que quanto maior a taxa de
resfriamento, menores são a fração de passante e o diâmetro médio das partículas obtidas com
a mistura etanol + água. Já para a mistura de 1-propanol + água, observa-se uma correlação
positiva de fraca a moderada entre a taxa de resfriamento e a fração de passante e a taxa de
resfriamento e o diâmetro médio da partícula (quanto maior a taxa, maiores são o Fpp e o
Dpp).
Em geral, correlações negativas de moderada a forte estão presentes entre a variável de
entrada Conc e as mesmas variáveis de saída comentadas no parágrafo anterior. Neste caso,
quanto maiores às concentrações, menores são as variáveis de saída. Vale ressaltar que esta
aferição condiz com dados obtidos nas análises contidas no tópico anterior (Tabelas 4.6), a
qual foi obtida menores diâmetros de partículas com a mistura etanol + água tanto para maior
taxa de resfriamento quanto para maior concentração. O oposto foi constatado com as
partículas obtidas com 1-propanol + água.

Capítulo 5
Conclusão

Conclusão
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 64
5.Conclusão Neste trabalho, o método de cristalização a partir de solução polimérica via
resfriamento controlado foi estudado com objetivo de obter partículas submicrométricas de
PMMA. Diferentes misturas de solventes (álcool e água) foram testadas e diferentes taxas de
resfriamento e concentração polimérica foram avaliadas.
Foi possível observar que o método de cristalização por resfriamento linear de solução
polimérica é capaz de produzir partículas com estrutura esférica e uniforme do polímero.
Usando exclusivamente as misturas de solventes: etanol+água e 1-propanol+água, as
análises morfológicas mostraram que estas misturas influenciam no tamanho da partícula
formada. A mistura etanol+água, tende a formar partículas menores que a mistura 1-
propanol+água. Além disso, na mistura etanol+água, as propriedades relativas ao tamanho de
partícula do polímero apresentaram maior sensibilidade às mudanças do planejamento
experimental em relação à concentração do polímero e à taxa de resfriamento.
Por outro lado, na mistura 1-propanol+água, maiores frações de partículas
submicrométricas foram obtidas, sendo tais partículas com propriedades mais uniformes.
Concluiu-se também que a taxa de resfriamento e a concentração de polímero na
solução demonstraram influência sobre o tamanho da partícula formada. Após análises, pode-
se afirmar que a condição de maior taxa de resfriamento e menor concentração (P2 e PE2)
apresentou maior formação de partículas de tamanho submicrométrica. Assim como relata
Kim (2005) que maiores taxas e menores concentrações formam partículas ainda menores.
Logo, para as condições contrarias a estas há uma menor produção deste tamanho específico
de partícula, principalmente, com a maior concentração polimérica presente na solução.
Considerando as análises de Microscopia (Ópticas e Eletrônica de Varredura) mostram
que as partículas de todas as amostras apresentaram uma uniformidade na morfologia esférica
semelhantes, como as da literatura. Desta forma, considerado um potencial plausível e
satisfatório para o uso destas partículas como sistemas carreadores de algum material
magnético.
Testes com o material ferrita foram realizados para avaliar o potencial de agregação a
partir da técnica de cristalização estudada. Tais testes foram considerados relativamente
satisfatórios a partir da observação da ferrita no polímero PMMA nas imagens em
microscopia.

Conclusão
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 65
Considerando as formas de análises e resultados obtidos, pode-se concluir, de maneira
geral, que o método avaliado (cristalização por resfriamento controlado) foi satisfatório tanto
para a formação do tamanho das partículas esféricas quanto na formação de partículas
submicrométricas de PMMA, através da mudança nas condições de operação durante a
cristalização. Além da simplicidade de execução (apenas o polímero é solubilizado em
solvente), a sua principal vantagem consiste na utilização de poucos compostos químicos, o
que não ocorre em outros métodos convencionais de obtenção de partículas submicrométricas,
como é o caso das polimerizações em emulsão e miniemulsão.
Na etapa de solubilização do PMMA pode-se aferir, mediante observado na literatura
e comprovado nos ensaios realizados neste trabalho, que a solubilização do polímero é
impedida tanto em água quanto em álcool. Neste último, só alcançado forçando um aumento
considerável da temperatura de solução. Mas com uma mistura adequada de água e álcool,
esta etapa é totalmente facilitada a temperaturas próximas a ambiente. O que diferente do
relato Kim (2005),que afirma ter conseguido solubilização a 20°C usando1-propanol.
Neste sentido, fica como sugestão para futuras pesquisas nesta área, a otimização do
procedimento experimental do método de cristalização térmica via resfriamento, tendo-se
como objetivo a produção de materiais magnéticos (em escala nanométrica) encapsulados
pelo polímero considerado. A idéia é usar estes materiais para nuclear a formação de
partículas poliméricas com propriedades magnéticas. Algumas variáveis poderiam ser
exploradas, tais como: a razão entre a concentração da solução polimérica e concentração do
material magnético na solução e novas taxas de resfriamento.

Capítulo 6
Referências

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 67
Referências
ANTÓNIO, N.N. Estudo dos Mecanismos de Despolimerização Térmica do Poli( Metacrilato
de Metila). 2007. Dissertação ( Mestre em Engenharia Química) -Instituto Superior Técnico,
Universidade Técnica de Lisboa, Portugal, Lisboa.
ASKELANO, O.R. .The Science and Engineering of Materiais.Wadsworth, Inc., Belmont,
USA, p.762, 1984. AZZAZY, H.M.E.; MANSOUR, M.M.H. in vitro diagnostic prospects of
nanoparticules.ClinicaChimicaActa, v.403, p. 1-8, 2009.
BARTOSCH, K.; MERSMANN.A. How to predict the metastable zone width. Journal of
Crystal Growth, v. 183, p. 240-250, 1998.
BIKIARIS, D.N.; KARAYANNIDIS, G.P. Dynamic thermomechanical and tensile properties
of chain-extended poly (ethylene terephthalate).J. Appl. Polym. Sci, v.70, p.797-803, 1998.
BOVE, L.; D'ANIELLO, C.; GORRASI, G.; GUADAGNO, L.; VITTORIA, V.Influence of
ageing on the cold crystallization of glassy poly(ethylene terephthalate). Poly. Bull, v.38,
p.579-585, 1997.
BOYER, S.A.E.; HAUDIN, J.M. Crystallization of polymers at constant and high cooling
rates: A new hot-stage microscopy set-up. Polymer testing, v.29, p.445–452, 2010.
BRUCE, I.J.; TAYLOR, J.; TODD, M; DAVIES, M.J.; BORIONI, E.;SANGREGORIO, C.;
SEM, T. journal of magnetism and magnetic materials, v. 284, p. 145-160, 2009.
CALVERT, P.D.; RYAN, T.G. Reversible secondary crystallization during cooling of
polypropylene. Polymer,v. 25, p.921-926, 1984.
CALLISTER; WILLIAM D. Ciência e engenharia de materiais: uma introdução. Rio de
Janeiro: LTC, 5.ed., 2002.
CAMILO, R. L. Síntese e caracterização de nanopartículas magnéticasde ferrita de cobalto
recobertas por 3-aminopropiltrietoxissiliano para uso como material híbrido em

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 68
nanotecnologia. 2006. Tese (Doutor em ciências na área de tecnologiaNuclear – Materiais) –
Instituto de pesquisas energéticas e nucleares, São Paulo.
COWIER, J.M.G.; MOHSIN, M. A.; MCEWEN, I. J.Alcohol-water cosolvent systems for
poly(methyl methacrylate). Polymer,v. 28. p.1569-1572, 1987.
CANEVAROLO JR., S.V.Ciência dos polímeros: um texto básico para tecnólogos e
engenheiros. São Paulo: Artiber Editora, 3 ed., 2010.
CHASTELAINN, M.; PETRI, A.; GUPTA, A.; RAO, K.V.; HOFMANN, H.
Superparamagnetic sílica-iron oxide composite for application in hyperthermia. Advanced
Engenneering Materials, v. 6, p. 235-241, 2004.
CHEN, R.; MACLAUGHLIN, S.; BOTTON,G.; ZHU, S.; Preparation of Ni-g-polymer core–
shell nanoparticles by surface-initiated atom transfer radical polymerization. Polymer, v. 50,
p. 4293 – 4298, 2009.
CHEN, Y.; QIAN, Z.; ZHANG, Z.; Novel preparation ofmagnetita/polystyrene composite via
inverse emulsion polymerization.Coloids and Surfaces A: Physicochem. Eng. Aspects, v. 312,
p. 209 –213, 2008.
CHUNG, T.H.; LEE, W.C. Preparation of styrene-based, magnetic polymer microspheres by
a swelling and penetration process.Sciense Direct., v. 68, p.1441-1447, 2008.
COUVREUR, P.; DUBERNET, C.; PUISIEUX, F. Controlled drug delivery with
nanoparticles: current possibilities and future trends. Eur. J. Biopharm, v. 41, p. 2-13, 1995.
CUI, X.;WANG, H.;WANGA, C.;YAN, J.; CONG, D. Preparation and characterization of
magnetic hollow PMMA nanospheres via insitu emulsion polymerization. Colloids and
Surfaces A: Physicochem. Eng. Aspects,v.363, p.71–77, 2010.
DALVI, S.V.; THORAT, A. A. Liquid antisolvent precipitation and stabilization of
nanoparticles of poorly water soluble drugs in aqueous suspensions: Recent developments and
future perspective. Chemical Engineering Journal, v.181– 182, p. 1– 34, 2012.

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 69
DERENZO, S. Cristalização de ácido adípio em diferentes solventes. 2003.Tese (doutorado
em Engenharia Química) – Centro de Ciências exatas e tecnologia, Programa de Pós –
Graduação em Engenharia Química,Universidade Federal de São Carlos, São Paulo, São
Carlos.
DIXON,D.J.; JOHNSTON,K.P.; BODMEIER,R.A. Polymeric materials formed
byprecipitation with a compressed fluid antisolvent. AIChE Journal 39 v.1, p. 127–139, 1993.
ERDEM, B.; SUDOL, E. D.; DIMONIE, V. L.; EL-AASSER, M. S.; Encapsulation of
Inorganic Particles via Miniemulsion Polymerization II. Preparation and Characterization of
Styrene Miniemulsion Droplets Containing TiO2 Particles.J. Polym. Sci.: Part A: Polym.
Chem., v. 38, p. 4431, 2000.
EXNER, G.; PEREZ, E.; KRASTEVA, M. N.; KATERSKA, B. Cooling rate effect on the
phase transitions in a polymer liquid crystal: DSC and real-time MAXS and WAXD
experiments. European Polymer Journal,v.46,p.1623–1632, 2010.
FAN et al, L.H. Preparation and characterization of Fe3O4 magnetic composite microspheres
covered by a P(MAH-co-MAA) copolymer. J Nanopart Res., v.40, p. 449-458, 2009.
FAGES, J.; LOCHARD,H.; LETOURNEAU,J-J.; SAUCEAU,M.; RODIER,E. Particle
generation for pharmaceutical applications using supercritical fluid technology, Powder
Technology, v.141 p. 219–226, 2004.
FLORY, P.J. Principles of polymer chemistry, Cornell Univ. Press, 1953.
FRESTA, M. etal., Pefloxacinemesilate-loaded and ofloxacin-loaded polyethylcyanoacrylate
nanoparticles - characterization of the colloidal drug carrier formulation, journal of
pharmaceutical sciences, v. 84, p. 895-902, 1995.
FEUSER, P.E.; SOUZA DE, M.N.Preparação de microesferas contendo metacrilato de metila
(pmma) com nanopartículas magnéticas. . In. 50°Congresso Brasileiro de Química, 2010,
Cuiabá/MT.Trabalhos técnicos... Cuiabá: CBQ, 2010. V.1, p. 1-3.
GAHN, C.; MERSMANN, A. Theoretical prediction and experimental determination of
attrition rates, Trans. Inst. Chern. Eng., v. 75, p.125, 1996.

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 70
GARAY I.; POCHEVILLE, A.; MADARIAGA, L. Polymeric microparticles prepared by
supercritical antisolvent precipitation.Powder Technology,v.197, p. 211–217, 2010.
GEDDE, U.W. ; SANANDAJI, N.; OVASKAINEN, L.; GUNNEWIEK, M. K.; VANCSO,
G.J.; HEDENQVIST, M.S.; YU, S., ERIKSSON, L.; ROTH, S.V. Unusual crystals of poly(ε-
caprolactone) by unusual crystallisation: The effects of rapid cooling and fast solvent loss on
the morphology, crystal structure and melting. Polymer, v.54, p.1497-1503, 2013.
GHOSH, PK. Hydrophilic polymeric nanoparticles as drug carriers.Indian J BiochemBiophys,
v. 37, p. 273-282, 2000.
GIULIETTI, M.; SECKLER, M.M.; DERENZO, S.;VALARELLI,J.V.Changes in copper
sulfate crystal habit during cooling crystallization. Journal of Crystal Growth, v. 166,p. 1089-
1093, 1996.
GU, S.; MOGI, T.; KONNO, M. Preparation of monodispersed, micronsized polystyrene
particles with single-stage polymerization in aqueous media. Journal of Colloid and Interface
Science,v. 207, p. 113– 118, 1998.
GURNY, R.; PEPPAS N.A.; HARRINGTON, D.D.; BANKER, G.S. Development of
biodegradable and injectable lattices for controlled release potent drugs. DrugDevIndPharm,
v.7, p.1–25, 1981.
GUTERRES, S. S.; SCHAFFAZICK, S. R. Caracterização e estabilidade físico-química de
sistemas poliméricos nanoparticulados para administração de fármacos. Quim. Nova, v. 27 p.
726-737,2003.
HE, Y.P.; WANG, S.Q.; LI, C.R.; MIAO, Y.M.; WU, Z.Y.; ZOU, B.S. Synthesis and
characterization of functionalized silica-coated Fe3O4supermagneticnanocristals for biological
application. Jorunal of physics D: Applied physics, v.38, p. 1342-1350, 2005.
HOOGENBOOM, R.; REMZI BECER, C; GUERRERO-SANCHEZ, C; HOEPPENER, S.;
SCHUBERT U. S. Solubility and Thermoresponsiveness of PMMA in Alcohol-Water Solvent
Mixtures. Aust. J. Chem, v.63, p.1173–1178, 2010.

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 71
JASTRZEBSKI, Z.O. .The Nature and Properties of Engineering Materiais. John Wiley &
Sons, Inc., 2nd Ed., New York, 1976.
JEWRAJKA, S. K.; CHATTERJEE, U.; MANDAL, B. M. Homogeneous atom transfer
radical polymerization of methyl methacrylate at ambient temperature in aqueous ethanol.
Macromolecules, v. 37, p. 4325-4328, 2004.
JONES, A.G. Crystallization Process Systems, Butterworth-Heinemann, Oxford, Boston,
2002.
JONES, A; RIGOPOULOS,S.; ZAUNER,R. Crystallization and precipitation
engineering.Computers and Chemical Engineering, v. 29, p. 1159–1166, 2005.
KAUFMANN, T. J.; JENSEN, M.E.; FORD, G.; GILL, L.L.; MARX, W.F; KALLMES, D.
F.. "Efeitos Cardiovasculares de Uso Polimetilmetacrilato em vertebroplastia percutânea."
American JournalofNeuroradiologia v.23, p.601-604, 2002.
KIM, D.H.; NIKLES, D.E.; JOHNSON, D.T.; BRAZEL, C.S. Heat generation aqueously
dispersed CoFe2SO4nanopaticles as heating agents for magnetically activated drug delivery.
Journal of magnetism and magnetic materials, v. 320, p. 2390-2396, 2008.
KIM, K.J. Spherulitic crystallization of 3-nitro-1,2,4-triazol-5-one in water+N-methyl-2-
pyrrolidone, Journal of Crystal Growth , v. 208 p. 569– 578, 2000.
KIM, K.J. Nano/micro spherical poly(Methyl methacrylate) particle formation by cooling
from polymer solution. Powder Technology, p. 156-163. 2005.
KIM K. J., MERSMANN A. Estimation of metastable zone width in different nucleation
processes. Chemical Engineering Science, v. 56, p. 2315-2324, 2001.
LIN, T.; STICKNEY,K. W.; ROGERS,M.; RIFFLE,J. S.; MCGRATH, J. E.; MARANDT, H.
Preparation of submicrometre polyimide particles by precipitation from solution. Polymer, v.
34, 1993.

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 72
LIRA S.A. Análise de correlação: abordagem teórica e de construção dos coeficientes com
aplicações. 2004. Dissertação (Mestre em Ciências) – Universidade Federal do Paraná,
Curitiba, Paraná.
LIU, T. Melting behaviour of isotactic polystyrene revealed by differential scanning
calorimetry and transmission electron microscopy.Eur. Polym. J. v. 39, p. 1311-1317, 2003.
LIU, T.; PETERMANN, J. Multiple melting behavior in isothermally cold-crystallizated
isotactic polystyrene.Polymer v. 42, p. 6453-6461, 2001.
LEE, H; LEE, E; KIM, D.K.; JANG, N.K.; JEONG,Y.Y.; JON, S. Antibiofouling polymer-
coated superparamagnetic iron oxide nanoparticles as pontetial magnetic resonance contrast
agents for in vivo cancer imaging. Journal of American Chemical Society , v.128, p. 7383-
7389, 2006.
LORENZO, M.L. Spherulite growth rates in binary polymer blends. Prog.Polym.Sci., v.28,
p.663-689, 2003.
LU, X.F.; HAY, J.N. Isothermal Crystallization Kinetics and Melting Behaviour of Poly
(ethylene Terephthalate). Polymer, v. 42, p. 9423-9432, 2001.
MANRICH, S.; ZANOTTO, E. D.; HAGE J. E.. Aplicabilidade da Teoria Clássica de
Nucleação Modificada (CO-CNT) à Cristalização de Polímeros. Polímeros: Ciência
Etecnologia,p. 15-20, 1992.
MENDONÇA, R. A. G. Cristalização do clorato de sódio. 2008.103f. Dissertação (mestre em
Engenaria Química e Bioquímica) – Faculdade de Ciências e Tecnologia, Departamento de
Química,Universidade Nova de Lisboa,Portugal,Lisboa.
MERHEB, G. A. Estudo do processo de cristalização da sacarose a partir de soluções
provinientes de cana-de-acúcar por resfriamento controlado. 2009. Dissertação (Mestre em
Engenharia Química), Centro de Ciências exatas e tecnologia, Programa de Pós Graduação
em Engenharia Química, Universidade Federal de São Carlos, São Paulo/SP.
MERSMANN, A. Supersaturation and nucleation, Trans.In st.Chern. Eng. A, v.74 (1) p. 812,
1996.

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 73
MERSMANN, A. General prediction of statistical1y mean growth rates of a crystal collective,
J. Crystal Growth, v.147, p.181, 1995.
MEYERS R. A. Biologia molecular e biotecnologia: uma referência abrangente mesa, Wiley-
VCH, p. 722, 1995.
MILES, D. C.; BRISTORN, J. H. Tecnologia dos polímeros. São Paulo: Polígono S.A, 1975.
MIGUEL,F.; MARTÍN,A.; GAMSE,T.; COCERO,M.J. Supercritical anti solvent
precipitationof lycopene. Effect of the operating parameters, Journal of Supercritical Fluids,
v. 36 p. 225–235, 2006.
MULLIN, J. W. Crystallization (4th ed.). Oxford: Butterworth – Heinemann, 2001.
MUHRER, G.; MAZZOTTI, M.; MÜLLER,M. Gas antisolvent recrystallization of an
organiccompound.Tailoring product PSD and scaling-up.Journal of Supercritical Fluids,
v.27,p.195–203, 2003.
MYERSON, A. S. Molecular modelling applications in crystallization.Cambridge University
Press, 1999.
NODA, M. ISIKAWA, M. YAMAWAKI, Y. SASAKI, A facile preparation of spherical
methylsilsesquioxane particles by emulsion polymerization.InorganicaChimica Acta, v. 263,
p.149– 152, 1997.
NOSKOV, S. Y; LAMOUREUX, G.; ROUX, B..Molecular dynamics study of hydration in
ethanol-water mixtures using a polarizable force field.J. Chem. Phys. B, v.109, p.6705, 2005.
NYVLT, J. Design of agitated crystallizers, Crystal Res. Techno, v. 6, p. 895, 1994.
NÝVLT, J.; HOSTOMSKÝ, J.; GIULIETTI, M. Cristalização.São Carlos, SP:
EdUFScar/IPT, 2001.
ODIAN, G.Principles of polymerization.4° edição.Staten Island, New York: A JOHN WILEY
& SONS, INC., PUBLICATION, 2004

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 74
OH, J. K.; PARK,J. M.Iron oxide-based superparamagnetic polymeric nanomaterials:Design,
preparation, and biomedical application.Progress in Polymer Sciencev.36,p.168–189, 2011.
PAL, T.; PAUL, S.; Biswanath. Polymethylmethacrylate Coated Alginate Matrix
Microcapsules for Controlled Release of Diclofenac Sodium. Pharmacology & Pharmacy,
Kolkata, p. 56-66, 2011.
PICCAROLO, S.; LA CARRUBBA, V.; BRUCATO, V. An experimental methodology to
study polymer crystallization under processing conditions. The influence of high cooling
rates.Chemical Engineering Science, v.57, p.4129 – 4143, 2002.
PINTO-ALPHANDARY, H.; ANDREMONT, A. COUVREUR, P..Targeted delivery of
antibiotics using liposomes and nanoparticles: research and applications.Int. J. Antimicrob.
Agents, v. 13, p.155, 2000.
PUISIEUX, F.; BARRAT, G.; COUARRAZE, G.; COUVREUR, P.; DEVISSAGUET, J-P.;
DUBERNET, C.; FATTAL, E.; FESSI, H.; VAUTHIER,C.; BENITA, S. Polymeric
Biomaterials; Dumitriu, S., ed.; Marcel Dekker: New York, 1994. cap. 16.
POLI, D. C. R. Característica dosimétrica do polimetacrilato de metila (pmma), de nome
comercial clarex, para doses entre lokrad a 10mrad.1978. 56 f. Dissertação (Mestre) –
Instituto de Energia Atomica, São Paulo.
QIN, J.; ASEMPAH, I.; LAURENT, S; FORNARA, A.; MULLER, R.N.; MUHAMMED, M.
Advacedmaterials, v.21, p. 1354-1357, 2009.
RANDOLPH, A.D.; LARSON, M.A. Theory of Particulate Processes, Academic Press, San
Diego, 1988.
RAOA, J. P.; GECKELERA, K. E.Polymer nanoparticles: Preparation techniques and size-
controlParameters.Progress in Polymer Science, v.36, p. 887–913, 2011
ROMIO, A. P. Encapsulação de Nanopartículas de Níquel obtidas a partir da Técnica de
Polimerização em Miniemulsão direta e inversa. 2011. Tese (Doutorado em Engenharia
Química),Centro Tecnológico, Programa de Pós – Graduação em Engenharia Química
Universidade Federal de Santa Catarina, Santa Catarina,Florianópolis.

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 75
REVERCHON,E. Supercritical antisolvent precipitation of micro- and nano-particles, Journal
of Supercritical Fluids,v.15,p. 1–21, 1999.
SA, B.; PAL, T.; PAUL, S. Polymetacrylate coated alginate matrix microcapsules for
controlled release of Diclofenac Sodium. Phamacologyc&Pharmacy,v. 2, p.56-66, 2011.
SARKARI, M.A; BROWN, J.B.; CHEN, X.A.; SWINNEA, S.A.; WILLIAMS III, R.O.B.;
JOHNSTON,K.P.A. Enhanced drug dissolution using evaporative precipitation into aqueous
solution.International Journal of Pharmaceutics, v. 243, p.17–31, 2002.
SANG, Z., LI, Q., & GAO, L. Preparation and properties of nano-TiO2powder. Journal of
Material Science Technology, v. 13, p. 321, 1997.
SCHAAF, P.; LOTZ, B.; WITTMANN, J. C. Liquid-liquid phase separation
andcrystallization in binary polymer systems.Polymer, v.28, 1987.
SCHUBERT, H.; MERSMANN, A. Determination of heterogeneous nucleation rates. Trans.
Inst. Chern. Eng. A, v. 74, p.821, 1996.
SHIMIZU, S. ; AIKI A, Y. ; KURITA, K.; FURUSAKA, M. Small-angle scattering study on
poly(methyl methacrylate) in aqueous alcohol solutions. Physica B,v. 241-243, p.1032-1034,
1998.
SILVA, J.E. Valorização de lamas galvânicas por via hidrometalúrgica. Tese de doutorado
(D.R.); Universidade de Minho, 2006.
SUN,Y.P. Supercritical Fluid Technology in Materials Science and Engineering: Syntheses,
Properties, and Applications, Marcel Dekker, 2002.
VAN ROSMALEN, G.M.; BERMINGMAN, S.;BRUINSMA, D.; KRAMER, H.;
DERENZO, S.; SECKLER, M.; RÉ, M.I.; CEKINSKI, E.; GIULIETTI, M. Tu Delft – IPT
Lectures on Industrial Crystalization e preciptation. 2004.
VANDERHOFF, JW; EL AASSER, MS; UGELSTAD J. Polymer emulsification process. US
Patent 4,177,177 (1979).

Referências
__________________________________________________________________________________________
André Anderson Costa Pereira, Agosto/2013 76
YOO H. S.; LEE K. H.; OH J. E.; PARK T. G. In vitro and in vivo anti-tumor activities of
nanoparticles based on doxorubicin–PLGA conjugates. J. Controlled Release, v. 68, p. 419,
2000.
WADDON, A.J.; KARASZ, F.E. Cristalline and amorphous morphologies of an aromatic
polymide formed on precipitation from solution. Polymer, v. 33, p. 3783-3789, 1992.
WANG,Y.; DAVE,R.N.; PFEFFER,R. Polymer coating/encapsulation of nanoparticles using
a supercritical anti-solvent process, Journal of Supercritical Fluids, v. 28, p.85–99, 2004.
WELLEN, R. M. R.Cristalização a Frio do PET e das Blendas PET/PS e PET/SAN.
2007.Tese(doutorado em Engenharia de Processos), Centro de Ciências e Tecnologia, Pós-
Graduação em Engenharia de Processos, Universidade Federal em Campina Grande,
Paraiba,Campina Grande.
WUNOERLlCH, B. Macromolecular Physics. Crystal Nucleation, Growth, Annealing.
Academic Press, Inc., London, v.2, p.461, 1997.
ZACHER, U. Die Kristall wachstums dispcrsion in einem kontinuierlichen Suspensions
kristallisator, PhD Thesis, Technical University Munich, 1995.
