dna damage response suppresses epstein-barr virus-driven
TRANSCRIPT

i
v
i
v
DNA Damage Response Suppresses Epstein-Barr Virus-Driven Proliferation
of Primary Human B Cells
by
Pavel Nikitin
Department of Molecular Genetics and Microbiology
Duke University
Date:
Approved:
___________________________
Micah Luftig, Supervisor
___________________________
Sandeep Dave
___________________________
Thomas Petes
___________________________
David Pickup
___________________________
Nancy Raab-Traub
Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor
of Philosophy in the Department of Molecular Genetics and Microbiology in the
Graduate School of Duke University
2012

ABSTRACT
DNA Damage Response Suppresses Epstein-Barr Virus-Driven Proliferation
of Primary Human B Cells
by
Pavel Nikitin
Department of Molecular Genetics and Microbiology
Duke University
Date:
Approved:
___________________________
Micah Luftig, Supervisor
___________________________
Sandeep Dave
___________________________
Thomas Petes
___________________________
David Pickup
___________________________
Nancy Raab-Traub
An abstract of a dissertation submitted in partial fulfillment of the requirements for the
degree of Doctor of Philosophy in the Department of Molecular Genetics and
Microbiology in the Graduate School of Duke University
2012

Copyright by
Pavel A. Nikitin
Павел А. Никитин
2012

iv
Abstract
The interaction of human tumor viruses with host growth suppressive pathways
is a fine balance between controlled latent infection and virus-induced oncogenesis. This
dissertation elucidates how Epstein-Barr virus interacts with the host growth
suppressive DNA damage response signaling pathways (DDR) in order to transform
infected human B lymphocytes.
Here I report that the activation of the ATM/Chk2 branch of the DDR in hyper-
proliferating infected B cells results in G1/S cell cycle arrest and limits viral-mediated
transformation. Similar growth arrest was found in mitogen-driven proliferating of B
cells that sets the DDR as a default growth suppressive mechanism in human B cells.
Hence, the viral protein EBNA3C functions to attenuate the host DDR and to promote
immortalization of a small portion of infected B cells. Additionally, the pharmacological
inhibition of the DDR in vitro increases viral immortalization of memory B cells that
facilitates the isolation of broadly neutralizing antibodies to various infectious agents.
Overall, this work defines early EBV-infected hyper-proliferating B cells as a new stage
in viral infection that determines subsequent viral-mediated tumorigenesis.

v
Dedication
Моей маме To my mom

vi
Contents
Abstract ......................................................................................................................................... iv
List of Tables .................................................................................................................................. x
List of Figures ............................................................................................................................... xi
Acknowledgements .................................................................................................................. xiii
1. Introduction ............................................................................................................................... 1
1.1. Human tumor viruses associate with multiple malignancies ................................... 1
1.2. Epstein-Barr virus infection cycle determines viral latency programs .................... 1
1.2.1. EBV infection in vitro results in poor transformation efficiency .......................... 7
1.3. The tumor suppressive DNA damage responsive signaling pathway .................... 8
1.3.1. Activated oncogenes induce replicative stress ....................................................... 8
1.3.2. Ataxia telangiectasia mutated (ATM) regulates a response to DNA double-
stranded breaks ..................................................................................................................... 9
1.3.3. Innate tumor suppression limits oncogene-induced replicative stress ............ 10
1.3.4. Chronic DNA damage foci form DNA segments with chromatin alterations
reinforcing senescence (DNA-SCARS) ............................................................................ 11
1.4. The DNA damage response in viral-induced cellular transformation .................. 12
1.4.1. Viral gene expression provokes the tumor suppressive DDR ........................... 13
1.4.1.1. Viral oncoproteins drive cellular hyper-proliferation and thus, induce a
tumor suppressive DDR ............................................................................................... 14
1.4.1.2. Viral proteins directly induce a beneficial DDR, including the DNA repair
and activation of checkpoints ....................................................................................... 18
1.4.1.3. DDR activation through viral oncoprotein-mediated mitotic effects ........ 19

vii
1.4.1.4. Tumor viruses activate the DDR through induction of reactive oxygen
species .............................................................................................................................. 20
1.4.2. Tumor viruses modulate the activated DDR to promote tumorigenesis ......... 21
1.4.2.1. Tumor virus suppression of downstream DDR signaling components ... 21
1.4.2.2. Viral oncoproteins directly target DDR checkpoint kinases ....................... 23
1.4.2.3. Viral oncoproteins perturb mitotic checkpoint signaling ........................... 24
2. Results ....................................................................................................................................... 27
2.1. An ATM/Chk2-Mediated DNA Damage-Responsive Signaling Pathway
Suppresses Epstein - Barr virus Driven Proliferation of Primary B cells ..................... 27
2.1.1. Contributions ............................................................................................................ 27
2.1.2. Introduction ............................................................................................................... 28
2.1.3. Results ........................................................................................................................ 29
2.1.3.1. Epstein-Barr virus infection of primary B cells activates a cellular DNA
damage response ............................................................................................................ 29
2.1.3.2. The EBV-induced DNA damage response in primary B cell infection is
not associated with viral episomes or lytic replication ............................................ 33
2.1.3.3. The EBV-induced DNA damage response is associated with a transient
period of hyper-proliferation ....................................................................................... 34
2.1.3.4. Proliferation and DNA damage responsive genes are highly induced
early after EBV infection, then attenuated during LCL outgrowth ........................ 39
2.1.3.5. The EBV-induced hyper-proliferation associated DNA damage response
is growth suppressive.................................................................................................... 43
2.1.3.6. ATM and Chk2 kinases suppress EBV-mediated transformation and
initial B cell proliferation .............................................................................................. 45
2.1.3.7. ATM and Chk2 suppress B cell growth 4-8 days after EBV infection ....... 47

viii
2.1.3.8. EBV latent gene expression changes and consequences in early infected
cell divisions ................................................................................................................... 48
2.1.3.9. EBNA3C is required to attenuate the EBV-induced DNA damage
response ........................................................................................................................... 51
2.1.4. Discussion .................................................................................................................. 57
2.2. Mitogen-Induced B Cell Proliferation Activates Chk2-dependent G1/S Cell Cycle
Arrest ...................................................................................................................................... 58
2.2.1. Contributions ............................................................................................................ 58
2.2.2. Introduction ............................................................................................................... 58
2.2.3. Results ........................................................................................................................ 60
2.2.3.1. Mitogen stimulation of primary B cells results in a period of robust
proliferation .................................................................................................................... 60
2.2.3.2. Mitogen stimulation activates the ATM signaling pathway in hyper-
proliferating cells ........................................................................................................... 61
2.2.3.3. EBV and mitogen-induced B-cell proliferation is suppressed by Chk2 .... 63
2.2.3.4. B-cell mitogens induce caspase 3/7-dependent apoptosis independent of
Chk2 ................................................................................................................................. 65
2.2.3.6. Mitogen-induced hyper-proliferation of human B cells induced a Chk2-
dependent G1/S cell cycle arrest .................................................................................. 66
2.2.3.7. Activated Chk2 induces expression of the CDK inhibitor p21 ................... 69
2.2.4. Discussion .................................................................................................................. 69
2.3. Enhanced Method of Epstein-Barr Virus Mediated Transformation of B Cells
Used for Generation of Human Monoclonal Antibodies ............................................... 73
2.3.1. Contributions ............................................................................................................ 73
2.3.2. Introduction ............................................................................................................... 73

ix
2.3.3. Results ........................................................................................................................ 75
2.3.3.1. Pharmacological inhibition of ATM or Chk2 increases EBV-mediated
transformation of low numbers of B cells ................................................................... 75
2.3.3.2. Inhibition of Chk2 coupled with TLR9 stimulation additively enhances
EBV-mediated transformation of memory B cells from normal donors ................ 77
2.3.3.3. Combined treatment with Chk2i and TLR9 ligand increases EBV-
mediated transformation of memory B cells from a chronic HIV-infected patient
.......................................................................................................................................... 78
2.3.4. Discussion .................................................................................................................. 80
3. Conclusions and future directions ........................................................................................ 81
3.1. The source of DNA damage ......................................................................................... 82
3.2. DDR signaling in transformed cells ............................................................................ 85
3.3. Implication in vivo ........................................................................................................ 86
3.4. Distinct expression of viral latency proteins in EBV infected primary B cells ...... 88
3.4.1. How is EBNA3C activity regulated in infected cells? ......................................... 88
3.4.2. What is the role of LMP1 in restricting the DDR and why is its expression is
delayed? ............................................................................................................................... 89
3.4.3. How EBNA-LP length and expression control EBV-driven oncogenesis? ...... 89
3.5. A proposed model of EBV infection of primary B cells ........................................... 90
References .................................................................................................................................... 92
Biography ................................................................................................................................... 118

x
List of Tables
Table 1: Human oncogenic viruses and their interactions with the host DNA damage
response (adapted from (Nikitin and Luftig, 2012)). ............................................................. 13

xi
List of Figures
Figure 1: The model of EBV initial and persistent infection (from (Thorley-Lawson and
Allday, 2008)). ................................................................................................................................ 4
Figure 2: Interplay between viral oncoproteins and the host DNA damage response
(from (Nikitin and Luftig, 2012)). ............................................................................................. 16
Figure 3: Nuclear genome deposition and the analysis of viral and host gene expression
following primary B cell infection with EBV B95-8, UV-inactivated B95-8, P3-HR1 (A-C).
EBV genomes in infected primary B cells are independent of -H2AX foci (D-G). ........... 30
Figure 4: EBV induced a DNA damage response in primary B cells. .................................. 32
Figure 5: EBV induced a period of hyper-proliferative early after infection that was
associated with activation of the DNA damage response..................................................... 35
Figure 6: CFSE-based kinetic analysis of EBV-induced proliferation. ................................. 37
Figure 7: Transcriptional changes correlate with an EBV-induced early period of hyper-
proliferation and DNA damage response followed by attenuation upon LCL outgrowth.
....................................................................................................................................................... 40
Figure 8: Schematic diagram of GSEA analysis performed on B cells, Proliferating cells,
and LCL microarray data with ATM/p53 target genes. ........................................................ 42
Figure 9: Growth suppression and DNA damage enrichment in early cell divisions. ..... 44
Figure 10: Inhibition of ATM and Chk2 kinases increased EBV transformation efficiency
and proliferation of B cells during a critical period 4-8 days post infection. ...................... 46
Figure 11: EBV latency and consequential host gene expression changes from initial B
cell proliferation through LCL outgrowth. ............................................................................. 50
Figure 12: Expression of viral latency genes during early cell divisions through LCL
outgrowth. .................................................................................................................................... 52
Figure 13: EBNA3C attenuates the EBV-induced DNA damage response. ....................... 54
Figure 14: Characterization of EBNA-3A KO and EBNA-3C KO viruses. ......................... 56

xii
Figure 15: Multiple B cell mitogens induce hyper-proliferation. ......................................... 60
Figure 16: B cell mitogens induce the ATM signaling pathways in hyper-proliferating
cells. ............................................................................................................................................... 62
Figure 17: Chk2 inhibition increase proliferation of human B cells. .................................... 64
Figure 18: B-cell mitogens, but not EBV induces caspases 3/7-dependent apoptosis. ...... 66
Figure 19: Mitogen stimulation or EBV infection of human B cells activates Chk2-
dependent cell cycle arrest. ........................................................................................................ 68
Figure 20: Pharmacological inhibition of DDR kinases increases EBV transformation of
low numbers of total B cells derived from a normal donor. ................................................. 76
Figure 21: Chk2 inhibitor and TLR ligand CpG additively increase EBV-mediated
transformation of human memory B cells. .............................................................................. 77
Figure 22: Enhanced EBV transformation of memory B cells derived from a chronic HIV
patient. .......................................................................................................................................... 79

xiii
Acknowledgements
First of all, I would like to thank my PhD advisor, Dr. Micah Luftig, for his
incredible support during the time spent in graduate school. His invaluable guidance
shaped me as a scientist. This thesis work would not have succeeded without the help of
my committee members, Drs. Raab-Traub, Pickup, Petes and Dave whose critical
reviews facilitated my professional growth. Further, I would like to specially thank all
the present and past members of Luftig lab.
I would like to acknowledge our collaborators from Imperial College London,
Rob White and Martin Allday for their expertise and critical revision of our work and for
EBV viral mutants they kindly provided. I would like to thank Duke’s Dave lab,
Crawford lab, Hauser lab and Human Vaccine Institute members for our productive
collaborations. In addition, I would like to thank Cullen lab members for their advices
and for reagents they shared. I would like to acknowledge Lynn Martinek, Nancy
Martin and Mike Cook from DCI Flow Cytometry core facility for their enormous
support. Finally, I would like to thank my family, including my wife Mayya Shveygert
and my little daughter Masha, for their constant support.

1
1. Introduction
1.1. Human tumor viruses associate with multiple malignancies
Approximately 20% of all cases of human cancer have an infectious etiology,
with ~80% of those being viral (Bouvard et al., 2009) (Table 1). To date there are five
bone-fide human DNA tumor viruses and at least one RNA tumor virus that cause
various malignancies (discussed in details below). A unique aspect of tumor virus
infection is the interaction with the host immune system and cell-intrinsic tumor
suppressive mechanisms. Recent studies identified the tumor suppressive DNA damage
responsive signaling pathway (DDR) as one of the major barriers to viral-driven
tumorigenesis, reviewed below. In this dissertation I focus on Epstein-Barr virus (EBV)-
driven tumorigenesis and report ATM/Chk2-mediated DNA damage response restricts
viral immortalization of primary human B lymphocytes in-vitro.
1.2. Epstein-Barr virus infection cycle determines viral latency programs
EBV is a large double stranded DNA γ-herpesvirus that establishes a latent
infection in more than 95% of the adult population worldwide (Rickinson and Kieff,
2006). EBV infection is associated with various tumors of lymphoid and epithelial origin
including Burkitt’s lymphomas and gastric carcinoma, Hodgkin’s disease and
nasopharyngeal carcinoma, and AIDS-associated lymphomas and post-transplant
lymphomas (Kieff and Rickinson, 2006; Raab-Traub, 2002).

2
Histologically and molecularly diverse EBV-associated malignancies arise from
different stages of viral infectious cycle and reflect dynamic viral latent gene expression.
The initial asymptomatic EBV infection occurs in the oral-pharyngeal epithelium during
the childhood period (Figure 1 and (Thorley-Lawson and Allday, 2008)). In epithelial
cells, EBV undergoes lytic replication and amplification resulting in secretion of new
virions that infect B cells and establish a latent infection.
Viral expression in primary B cells is well studied in a convenient in vitro system
(Alfieri et al., 1991). After infection of B cells, the EBV genome enters the nucleus and
circularizes into an episome (Hurley and Thorley-Lawson, 1988) which allows viral
expression from the W-promoter ((Arrand and Rymo, 1982)) (Woisetschlaeger et al.,
1990). The initial Wp-dependent transcript includes two viral proteins, EBV nuclear
antigen – leader protein (EBNA-LP) and EBNA2 (Alfieri et al., 1991). EBNA2 binds to
host transcription factors, such as RBP-Jk, and upregulates cellular expression of growth
promoting genes (Henkel et al., 1994; Johannsen et al., 1996; Robertson et al., 1996; Wang
et al., 1991). Next, EBNA2 initiates viral transcription of the remaining EBV nuclear
antigens, including EBNA1, that maintains viral episome, EBN3A, 3B and 3C (Zimber-
Strobl et al., 1993) from the C-promoter (Cp), and latent membrane proteins 1 (LMP1),
LMP2A and LMP2B (Kieff and Rickinson, 2006; Wang et al., 1990). In addition to viral
proteins, latently infected cells display a distinct set of viral non-coding RNAs, including
abundant EBV-encoded small RNAs (EBERS) (Arrand and Rymo, 1982; Pathmanathan

3
et al., 1995; Swaminathan et al., 1991), and viral microRNAs encoded within BART and
BHRF regions (Cai et al., 2006; Forte and Luftig, 2011; Grundhoff et al., 2006; Pfeffer et
al., 2004; Skalsky et al., 2012). Intriguingly, expression of LMP-1, a viral protein required
for immortalization of B cells (Kaye et al., 1993; Kulwichit et al., 1998), is regulated by a
separate promoter (Chang et al., 1997; Fennewald et al., 1984; Sadler and Raab-Traub,
1995) and temporally separated in vitro (Price et al., 2012). Expression of all nine latent
proteins and non-coding RNAs leads to immortalization of infected B cells into
lymphoblasts and is termed latency III program.

4
Figure 1: The model of EBV initial and persistent infection (from (Thorley-
Lawson and Allday, 2008)).

5
In vivo, there are at least three possible destinies for the infected proliferating B
cell to progress. The first and the most common pathway includes the elimination of the
infected cell by cytotoxic T and NK-cells. Particularly, EBNA-3 viral proteins and, to a
lesser extent, EBNA-2 induce the potent cytotoxic cell-mediated immune response
(Khanna et al., 1992; Murray et al., 1992). Interestingly, EBV-infected cells expressing the
complete set of nine viral latent proteins are predominantly found in
immunosuppressed individuals, such as transplant or AIDS-infected patients (Raab-
Traub, 2007).
However, EBNA3s binding to RBP-Jk modulates the expression of EBNA2 and
Cp-driven transcripts (Henkel et al., 1994; Robertson et al., 1996) and restricts viral
expression, hence escaping from the immune control and providing the alternative way
to progress. Consistently, in immunocompetent individuals EBV-associated tumors of
epithelial and lymphoid origins express only EBNA-1 and viral non-coding RNAs or
additional latent membrane proteins LMP1 and LMP2. The former expression program
is termed latency I and found in Burkitt’s lymphoma and gastric carcinoma (Rowe et al.,
1987) while the latter is termed latency II and found in germinal center-derived
Hodgkin’s lymphoma and epithelial nasopharyngeal carcinoma (Brooks et al., 1992;
Rowe et al., 1992). While latency III tumors arise in tumor suppression, latency II and I
tumors are most likely promoted by environmental and genetic factors (Raab-Traub,
2012). Further, as latency II tumors were revealed in germinal-center derived B cells, the

6
initial Cp expression in activated lymphoblasts is likely silenced in infected cells
migrated to germinal center. In germinal center infected B cells mimic the normal B-cell
maturation process and differentiate into memory-like B cells (Babcock et al., 1998).
EBNA1 expressing memory-like B cells that persist in peripheral blood are found in
normal individuals and therefore thought to be a viral reservoir (Thorley-Lawson and
Allday, 2008).
Finally, the third way for an infected naïve B-cell to turn into a memory-like B
cell was recently proposed by the Rickinson and Bell group. According to this report,
EBV-driven expression of activation-induced cytidine deaminase (AID) results in
mutagenesis within Ig locus and thus drives the infected cell towards non-switched
memory-like phenotype. Moreover, infected cells with edited Ig loci have an advantage
during in vitro outgrowth (Heath et al., 2012). Therefore, if the proposed mechanism
takes place in vivo, EBV-infected naïve lymphocytes may bypass the germinal center on
their path to a memory-like phenotype. The resulted infected memory B cell circulates in
the peripheral blood and upon stress undergoes differentiation into a plasma cell
driving EBV lytic reactivation (Thorley-Lawson and Allday, 2008).
Overall, a distinct pathway of the host B cell to the final differentiated state
determines dynamic changes in viral gene expression that, in turn, regulates the cellular
growth.

7
1.2.1. EBV infection in vitro results in poor transformation efficiency
As mentioned above, our understanding of EBV latent gene expression changes
predominantly comes from viral infection of primary human B cells in vitro (reviewed in
(Kieff and Rickinson, 2006)). While having limitations (Thorley-Lawson and Allday,
2008), in vitro system allows investigating the EBV latency III program in primary cells at
physiological concentrations of viral proteins, non-coding viral RNAs and miRNAs. In
this system peripheral blood mononuclear cells (PBMC) are latently infected with EBV
in the presence of T-cell suppressants, such as cyclosporine A (Borel et al., 1977).
Infected B cells are induced to proliferate and undergo a growth transformation into
lymphoblastoid cells (LCL). However, only a small percentage of infected cells become
indefinitely proliferating lymphoblasts (Henderson et al., 1977; Sugden and Mark, 1977).
Therefore, there may be intrinsic tumor suppression at play that prevents cellular
proliferation and/or suppresses the immortalization of proliferating B cells.
The study of EBV-induced innate tumor suppressor pathways has been limited.
In immortalized lymphoblasts EBNA-3A and 3C epigenetically silence p16 and prevent
activation of Rb that promotes G1/S transition (Maruo et al., 2011; Skalska et al., 2010).
However, it remains unclear what the role of p16 is in primary infection of lymphocytes.
Aside from p16, EBV infection of primary B cells induces the p53 protein concomitant
with EBNA-LP expression early after infection (Szekely et al., 1995). However, it remains
unclear whether this innate response to EBV-induced proliferation has any long-term

8
functional consequence or what pathways activate p53. Therefore, investigation of cell-
intrinsic mechanisms to suppress tumorigenesis will inform about barriers the virus has
to overcome and allow us to better understand interactions between the virus and the
host.
1.3. The tumor suppressive DNA damage responsive signaling pathway
In the last decade the DNA damage responsive signaling pathway (DDR) was
recognized as a barrier to tumorigenesis in oncogene-expressing cells and in
precancerous lesions. The Chapter 1.3 briefly overviews activation signals, components
and consequences of tumor suppressive DDR.
1.3.1. Activated oncogenes induce replicative stress
Replicative stress is a condition describing the presence of stalled replicative
forks, whether due to a replication block or an impetuous increase in a number of new
origins, resulting in inhibition of DNA synthesis (reviewed by (Halazonetis et al., 2008;
Osborn et al., 2002)). Such collapse of replicative forks often occurs at common places in
the genome, termed fragile sites, and requires activation of recombination and formation
of DNA double-stranded breaks (DSB) to complete the replication.
Activated oncogenes in mammalian cells increase the number of origins of
replication and presumably collapse of replication forks, resulting in the formation of
aberrant DNA structures, such as stretches of single stranded DNA and formation of the
DNA DSB (Bartkova et al., 2006; Di Micco et al., 2006). Upon formation of DSBs, a

9
cascade of events is initiated to activate checkpoints and initiate repair or apoptosis
(Khanna and Jackson, 2001). Therefore, oncogene-induced DSBs initiate a tumor
suppressive cellular response (Bartkova et al., 2005; Gorgoulis et al., 2005).
1.3.2. Ataxia telangiectasia mutated (ATM) regulates a response to DNA double-stranded breaks
ATM serves as the key regulator of the cellular response to DNA double-
stranded breaks and upregulates cellular checkpoints to promote DNA repair or initiate
programmed cell death. The function of ATM and its yeast homolog Tel1 in maintaining
the chromosome stability was established in two initial works (Greenwell et al., 1995;
Savitsky et al., 1995). While mutations in Tel1 led to telomere shortening (Greenwell et
al., 1995) and thus to chromosomal instability, human cells derived from patients with
ataxia telangiectasia (AT), a recessive autosomal disorder characterized by
predisposition to cancer and checkpoints abnormalities, carried a mutant gene named
AT-mutated or ATM (Savitsky et al., 1995). ATM was found to be a serine/threonine
protein kinase with hydrophobic-X-hydrophobic-[S/T]-Q consensus motif (Kim et al.,
1999), and is a member of the phosphoinositide 3-kinase-related protein kinase (PIKK)
family. Later works identified ATM and its downstream kinase Chk2 as regulating
replication checkpoints in response to ionizing irradiation-induced DNA DSBs (Falck et
al., 2001; Matsuoka et al., 1998). Currently, ATM is believed to induce G1/S, intra-S-
phase and G2/M cell cycle checkpoints through the following mechanisms: ATM
phosphorylation of p53 upregulates p21 expression resulting in inhibition of Cyclin-

10
E/CDK2 complex and blocking G1/ S transition. Additionally, an intra-S-phase
checkpoint is regulated by ATM through activation of NBS1, BRCA1, SMC1, FANCD2
and Chk2 kinase. Activated Chk2 kinase induces phosphorylation and a subsequent
degradation of CDC25A. As CDC25A normally activates Cyclin-E/CDK2 complex, its
degradation blocks G1/S transition. Furthermore, CDC25A degradation results in
elevated level of CDK2 phosphorylation and its destabilization slows down the
progression through S-phase. Finally, ATM and Chk2 play a role in the G2/M checkpoint
to promote repair through Rad17 and Artemis (reviewed in (Derheimer and Kastan,
2010; Harper and Elledge, 2007; Shiloh, 2003)).
1.3.3. Innate tumor suppression limits oncogene-induced replicative stress
Innate tumor suppression in response to oncogenic stress includes the well-
characterized alternative reading frame to 16 (ARF) tumor suppressor, also known as
p14, CDKN2A)-mediated activation and stabilization of p53 (Christophorou et al., 2006;
Efeyan et al., 2006; Zindy et al., 2003) and the cellular DNA damage response (DDR) that
is activated following oncogene-induced replicative stress (Halazonetis et al., 2008). As
first recognized by Halazonetis, Bartek and colleagues, tumor cells often display an
activated DDR as evidenced by foci of DDR signaling proteins such as 53BP1 and
activated ATM (DiTullio et al., 2002). Subsequent works demonstrated that acute over-
expression of oncogenes caused replicative stress sensed by the ATM and Rad3-related
kinase (ATR) signaling pathway as well as double-stranded breaks recognized by the

11
ATM pathway (Bartkova et al., 2005; Gorgoulis et al., 2005). Not long after the initial
characterization of these pathways, the functional significance of the DDR activation
was revealed by genetic studies indicating that ATM and Chk2 were critical tumor
suppressors downstream of oncogenes including H-RasV12, Mos, Cdc6, and cyclin E
(Bartkova et al., 2006; Di Micco et al., 2006; Hong et al., 2006; Stracker et al., 2008).
Mechanistically, these data linked the well-studied DDR response to DNA double
stranded breaks and known tumor suppressor functions of its components, including
activation of checkpoints and p53-mediated apoptosis and senescence, to an oncogene-
induced replicative stress.
1.3.4. Chronic DNA damage foci form DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS)
As discussed above, activated oncogenes in cell lines or in precancerous lesions
were found to activate ATM/Chk2-dependent signaling. However, the question remains
whether the activation signal comes from continuous DNA damage or may persist in
proliferating cells after the damage is repaired. Recent works elucidate the difference
between acute DDR signaling and chronic chromatin changes induced by initial
aberrations during DNA synthesis (Di Micco et al., 2011; Rodier et al., 2009). The
Campisi group found chronic DDR signaling foci that lacked the DNA repair proteins
replication protein A (RPA) and RAD51 as well as active DNA replication, but instead
contained activated ATM-downstream components, such as localized Chk2 kinase and
phospho p53-Ser15. Such foci were termed “DNA segments with chromatin alterations

12
reinforcing senescence” or DNA-SCARS. Notably, normally senescent DNA-SCARS
positive cells continued to proliferate after experimental inactivation of p16 (Rodier et
al., 2011) or knockdown of ATM (Rodier et al., 2009). Consistently, d’Adda di Fagagna
group found senescence-associated heterochromatin foci (SAHF) associated with
senescence that required activated p16 (Di Micco et al., 2011).
DNA-SCARS and SAHF presumably mark separate oncogene-induced changes
of the chromatin, as the former co-localizes with activated ATM-signaling molecules and
γH2AX, while the latter excludes γH2AX and depends on ATR. However, both
chromatin alterations occur upon oncogene-induced senescence and persist in
proliferating cells with abrogated checkpoints.
1.4. The DNA damage response in viral-induced cellular transformation
In the last decade, multiple studies have found the tumor suppressive role of the
DDR in response to viral oncoproteins (Table 1). A unique aspect of these interactions is
the interplay between the virus and the host with respect to virus replication versus
aberrant induction of growth control genes and inhibition of apoptosis. Chapter 1.4 will
focus on complex interactions between tumor viruses and the host DNA damage
response and outcomes that promote or prevent virus-induced tumorigenesis.

13
Table 1: Human oncogenic viruses and their interactions with the host DNA
damage response (adapted from (Nikitin and Luftig, 2012)).
Oncogenic
Virus
Tumors associated with
virus infection
Oncoproteins involved
in DDR
Reference(s)
EBV Burkitt’s lymphoma,
Post-transplant lymphoma,
Non-hodgkin’s/Diffuse
large B cell lymphomas,
Nasopharyngeal carcinoma,
Gastric carcinoma
EBNA1 ROS DDR
EBNA3C Chk2, p53
EBNA3C G2/M
checkpoint
LMP1 ATM
(Gruhne et al., 2009a)
(Choudhuri et al.,
2007; Yi et al., 2009)
(Gruhne et al., 2009b;
Parker et al., 2000)
(Gruhne et al., 2009b)
KSHV Kaposi’s sarcoma, primary
effusion lymphoma
v-cyclin ATM
v-cyclinOIS
LANA myc DDR
LANA:p53
v-FLIP OIS
(Koopal et al., 2007)
(Liu et al., 2007)
(Chen et al., 2010;
Friborg et al., 1999;
Leidal et al., 2012)
HPV Cervical cancer, ovarian
cancer
E6,E7 repl stress
DDR E7:pATM
E6 p53
(Bester et al., 2011)
(Moody and Laimins,
2009)
(Scheffner et al.,
1990)
HBV Hepatocellular carcinoma HBV ATR
HBx Ras DDR
HBx p53
(Wang et al., 2008;
Zhao et al., 2008a)
(Klein and Schneider,
1997)
(Wang et al., 1994)
HTLV I Adult T cell leukemia (ATL) Tax DNA-PK
Tax Chk1/Chk2
Tax p53
(Durkin et al., 2008)
(Park et al., 2004;
Park et al., 2006)
(Ariumi et al., 2000;
Pise-Masison et al.,
2000)
1.4.1. Viral gene expression provokes the tumor suppressive DDR
The replication of tumor viruses is intrinsically linked to their ability to drive cell
proliferation. Most of these viruses infect quiescent cells driving re-entry into the cell

14
cycle to promote an environment conducive for viral nucleic acid replication. The
consequences of such aberrant induction of cell proliferation include increased
replicative stress, similar to that of cellular oncogene activation, leading to induction of
the DNA damage response. However, direct viral oncoprotein activation of the DDR
also occurs through multiple mechanisms discussed below.
1.4.1.1. Viral oncoproteins drive cellular hyper-proliferation and thus, induce a tumor
suppressive DDR
Small DNA tumor viruses antagonize the transcriptionally repressive Rb family
of proteins to promote E2F-driven cellular proliferation. Uncontrolled E2F activity has
been shown to activate an ATM-dependent growth-suppressive DDR (Powers et al.,
2004; Rogoff et al., 2004). HPV E7 and SV40 large T antigen are classic examples of viral
oncoproteins targeting Rb by direct disruption of the interaction with E2F thereby
increasing S-phase- promoting E2F family members to drive cellular DNA replication
(Cheng et al., 1995; DeCaprio et al., 1988; Dyson et al., 1989; Munger et al., 1989; Zalvide
and DeCaprio, 1995). In recent work, E6 and E7 over-expression has been shown to
induce replicative stress in primary keratinocytes suggesting that potent loss of growth
control through E7- mediated Rb antagonism drives uncontrolled origin firing leading to
damaged DNA that may contribute to cervical cancer pathogenesis (Bester et al., 2011).
Similarly, SV40 large T antigen is sufficient to activate an ATM-induced DDR (Figure 1)
(Boichuk et al., 2010). However, as described later, SV40 large T antigen activates the
DDR through multiple mechanisms including those independent of Rb interaction

15
(Boichuk et al., 2010). While the polyomavirus SV40 does not cause human cancer,
Merkel cell carcinoma polyomavirus (MCV) expresses a truncated large T antigen in
these tumors lacking the capacity to replicate viral DNA (Shuda et al., 2008). These
mutant large T antigens still retain the ability to perturb cell growth through Rb
antagonism and likely activate the DDR providing selective pressure for mutations in
DDR genes and downstream signaling leading to MCC.
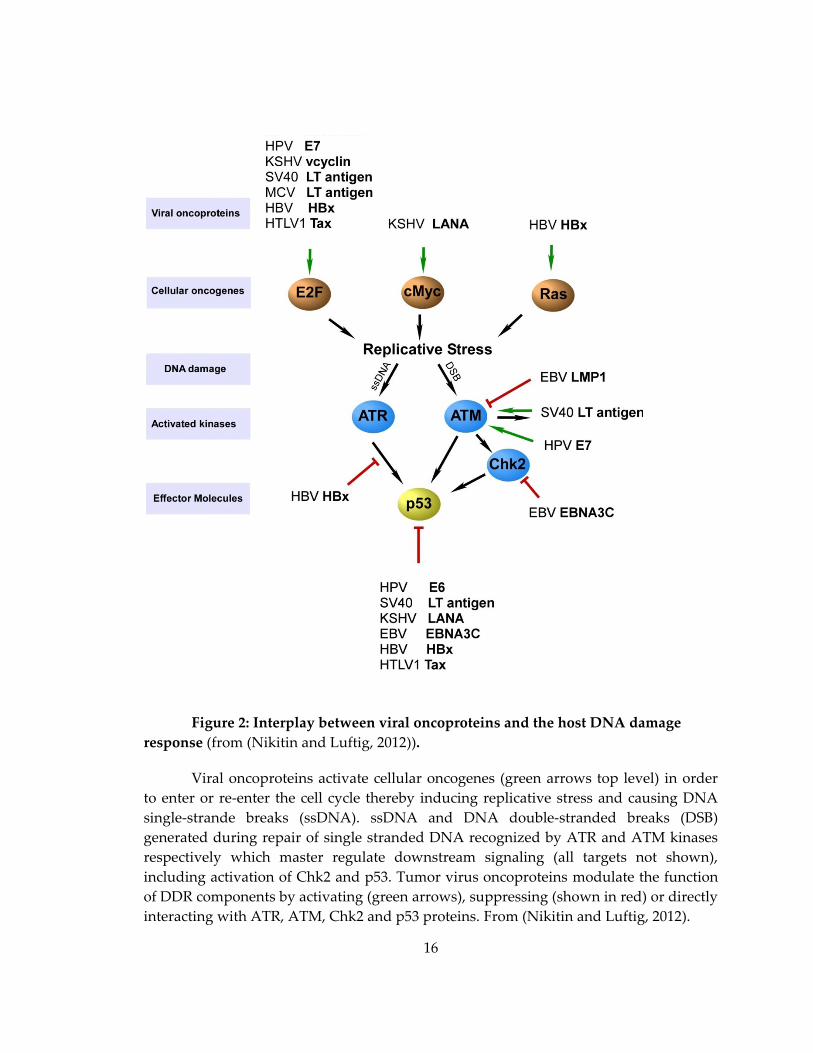
16
Figure 2: Interplay between viral oncoproteins and the host DNA damage
response (from (Nikitin and Luftig, 2012)).
Viral oncoproteins activate cellular oncogenes (green arrows top level) in order
to enter or re-enter the cell cycle thereby inducing replicative stress and causing DNA
single-strande breaks (ssDNA). ssDNA and DNA double-stranded breaks (DSB)
generated during repair of single stranded DNA recognized by ATR and ATM kinases
respectively which master regulate downstream signaling (all targets not shown),
including activation of Chk2 and p53. Tumor virus oncoproteins modulate the function
of DDR components by activating (green arrows), suppressing (shown in red) or directly
interacting with ATR, ATM, Chk2 and p53 proteins. From (Nikitin and Luftig, 2012).

17
Latent infection of large DNA tumor viruses drives robust proliferation of
infected cells and thus, activates a tumor suppressive DDR. -herpesvirus KSHV
encodes a host cyclin-D homolog, viral cyclin (v-cyclin), that binds to CDK6 and drives
cellular proliferation (Verschuren et al., 2004). Furthermore, v-cyclin ectopic expression
alone is sufficient to induce the DNA damage response (Koopal et al., 2007; Leidal et al.,
2012). Moreover, KSHV infection of immortalized endothelial cells in vitro induces the
ATM signaling pathway (Koopal et al., 2007). Consistently, latently infected primary
human foreskin fibroblasts display elevated levels of γ-H2AX and 53BP1 foci (Leidal et
al., 2012). Furthermore, investigation of KS tumors revealed activation of the DDR in
early (patch), but not late (nodular), KS lesions (Koopal et al., 2007). Finally, the
consequences of the activated DDR include the induction of oncogene-induced
senescence and autophagy (Leidal et al., 2012).
Hepatitis B virus (HBV) promotes cellular proliferation and the DDR through the
pleiotropic oncoprotein HBx. Heterologous expression of HBx increases cytosolic Ca2+
levels leading to activation of Pyk2 and c-Src kinases (Klein and Schneider, 1997) and,
ultimately, activation of Ras/Raf/MEK/ERK pathways. HBx expression can also promote
p38MAPK pathway activation which up-regulates E2F-dependent gene expression
(Wang et al., 2008). Constitutive activation of these signaling pathways leads to
activation of the ATR arm of the DDR pathway (Wang et al., 2008).The consequences of

18
this activation are actually beneficial for virus replication despite being tumor
suppressive (Zheng et al., 2011).
1.4.1.2. Viral proteins directly induce a beneficial DDR, including the DNA repair and
activation of checkpoints
Beyond the growth suppressive functions of the DDR, DNA repair and
activation of checkpoints may be beneficial for the replication of tumor viral genomes.
Oncogenic viruses have therefore developed mechanisms to activate specific
components of the DDR pathway, while strictly preventing downstream induction of
apoptosis. Recent work indicates that SV40 large T antigen can serve as both a substrate
for the ATM kinase as well as its direct upstream activator through binding the Nbs1
component of the ATM-activating Mre11/Rad50/Nbs1 complex (Boichuk et al., 2010; Wu
et al., 2004). ATM activation is actually necessary for viral DNA replication (Zhao et al.,
2008b). However, as discussed below, the growth-suppressive consequences of ATM
activation are attenuated by large T antigen downstream enabling SV40-infected cell
survival.
HPV-infected cells display increased, but non-canonical ATM pathway
activation. In particular, HPV oncoprotein-expressing undifferentiated keratinocytes
display an activated DDR characterized by ATM, Chk1, Chk2, and H2AX
phosphorylation (Moody and Laimins, 2009). However, upon differentiation of these
cells, which increases viral genome replication, an additional set of ATM targets is

19
phosphorylated including Nbs1 (Moody and Laimins, 2009). Interestingly, E7 was
demonstrated to associate with the activated Ser1981-phoshorylated form of ATM
independent of differentiation or other viral proteins (Moody and Laimins, 2009).
Therefore, direct association between E7 and phospho-ATM, HPV episome
amplification, and viral-induced replicative stress are all capable of activating the DDR
and it remains unclear which of these activities is critical in regulating HPV
pathogenesis (Moody and Laimins, 2009).
1.4.1.3. DDR activation through viral oncoprotein-mediated mitotic effects
Tumor viruses perturb normal cell cycle control in order to establish a
constitutive S phase-like environment in which cellular factors present are required for
viral replication. One consequence of this constitutive S-phase induction is inappropriate
entry into mitosis which activates DDR checkpoints including those triggered by Chk2
(Sato et al., 2010; Stolz et al., 2010). It was previously shown that KSHV v-cyclin
expression promotes polyploidy and cytokinesis defects (Verschuren et al., 2002) and it
was confirmed later by Ojala and colleagues that v-cyclin expression promotes
amplification of centrosomes and intra-S-phase growth arrest (Koopal et al., 2007).
Moreover, chemical inhibition of ATM/Chk2 led to aberrant mitoses and mitotic
catastrophe in v-cyclin-expressing cells (Koopal et al., 2007).

20
In order to successfully transform cells, SV40 large T antigen targets the spindle
assembly checkpoint component Bub1 leading to ATM/ATR activation (Cotsiki et al.,
2004; Hein et al., 2009). Similarly, the high-risk HPV16 E6 and E7 proteins have been
well documented to increase genomic instability by deregulating mitosis through the
induction of multipolar spindles and centrosome duplication (Duensing et al., 2000).
Specifically, E7 binding to nuclear mitotic apparatus protein 1 appears to deregulate
normal chromosome alignment during prometaphase (Nguyen and Munger, 2009).
More recently, E7 was observed to up-regulate Polo-like kinase 4 (PLK4) expression
leading to centriole multiplication (Korzeniewski et al., 2011). Therefore, multiple viral
oncoproteins perturb mitosis through diverse mechanisms leading to an activated DNA
damage response.
1.4.1.4. Tumor viruses activate the DDR through induction of reactive oxygen species
Elevated levels of reactive oxygen species (ROS) can activate DDR pathways and
may result in mutagenesis during oncogenic virus infection promoting tumorigenesis.
Several tumor virus oncoproteins have been shown to increase ROS levels. For example,
HTLV-1 Tax expression in fibroblasts or T cells induced a ROS-dependent DDR,
although the mechanism by which ROS was induced remains unknown (Kinjo et al.,
2010). Recently, Masucci and colleagues identified the EBV protein EBNA1, which is
required for viral episome maintenance and therefore expressed in every EBV-positive
tumor, as an inducer of ROS and consequent ATM-dependent DDR activation and

21
ultimately chromosomal aberrations (Gruhne et al., 2009a). Interestingly, EBNA1
induced ROS through up-regulation of the mRNA encoding the catalytic subunit of the
leukocyte NADPH oxidase NOX2, which directly promotes ROS accumulation (Gruhne
et al., 2009a). A more recent study suggests that this EBNA1-driven ROS accumulation
may promote telomere dysfunction, another known molecular signal for DDR activation
(Kamranvar and Masucci, 2011).
1.4.2. Tumor viruses modulate the activated DDR to promote tumorigenesis
With the explicit purpose of providing an environment for virus replication,
several tumor virus oncoproteins mitigate the growth suppressive function of the DNA
damage response through altering downstream signaling events. However, the
consequences of suppressing the DDR include aneuploidy and increased mutagenesis
which are major drivers of tumorigenesis. Tumor viruses have been well characterized
to antagonize the function of the p53 tumor suppressor and more recently several
viruses have been shown to target upstream checkpoint kinases as well.
1.4.2.1. Tumor virus suppression of downstream DDR signaling components
The small DNA tumor viruses SV40 and HPV have been well characterized for
their ability to transform cells through perturbing activation of the DDR downstream
target p53 (Kress et al., 1979; Lane and Crawford, 1979; Scheffner et al., 1993; Scheffner et
al., 1990). This activity is thought to be a requirement for cell survival following aberrant

22
S phase induction due to Rb antagonism by T Ag and E7 as described above. While large
DNA tumor viruses generally do not directly promote p53 degradation or abolish its
function, the KSHV latent protein LANA and EBV latent protein EBNA3C have been
shown to modulate p53 activity through direct association (Chen et al., 2010; Friborg et
al., 1999; Yi et al., 2009). Other tumor viruses also directly antagonize p53 function
including the HBV oncoprotein HBx, which both inhibits p53 DNA binding activity and
sequesters p53 in the cytoplasm thereby suppressing apoptosis (Elmore et al., 1997;
Takada et al., 1997; Wang et al., 1994; Wang et al., 1995). HTLV-1 Tax suppresses p53 by
directly antagonizing its trans-activating function through both NF B-dependent and
NF B-independent pathways (Ariumi et al., 2000; Miyazato et al., 2005; Pise-Masison et
al., 2000). While many tumor virus oncoproteins have been shown to associate with p53,
the extents to which these activities contribute to pathogenesis remain unclear.
In addition to targeting p53-downstream signaling, γ-herpseviruses evolved
different strategies to escape from the consequence of the activated DDR. EBV EBNA3C
latent protein downregulates the speed of proliferation and thus attenuates the extent of
the activated DDR, as demonstrated by our group and is discussed below. Unlike EBV,
closely-related KSHV rather actively targets the DDR downstream signaling.
Specifically, KSHV encoded v-FLIP suppresses v-cyclin-driven oncogene-induced
senescence, presumably via blocking autophagy (Leidal et al., 2012). Therefore, the

23
remaining DNA damage promotes mutagenesis and selection for mutations in the DDR
pathway in advanced KS lesions allowing tumor cell survival.
1.4.2.2. Viral oncoproteins directly target DDR checkpoint kinases
Upstream of p53 and cell cycle checkpoints are a series of DNA damage sensing
and signal relaying kinases (Figure 1). Several viral oncoproteins directly target these
upstream kinases through a number of mechanisms ultimately attenuating their
function. For example, the HTLV-1 Tax oncoprotein directly binds to and inhibits
signaling downstream of both Chk1 and Chk2 checkpoint kinases (Gupta et al., 2007;
Park et al., 2004; Park et al., 2006) as well as the upstream DNA damage sensing DNA-
PK (Durkin et al., 2008). Interestingly, Tax was also demonstrated to sequester the DDR
components MDC1, DNA-PK and BRCA1 at artificial Tax-induced foci of pseudo-DNA
damage as a unique mechanism to perturb endogenous DDR signaling pathways
(Belgnaoui et al., 2010). Not unexpectedly, Tax expression attenuated ATM-downstream
signaling leading to faster release of the G1/S checkpoint in response to ionizing
radiation (Chandhasin et al., 2008).
Under circumstances where EBV oncoproteins are aberrantly expressed, as
evidenced in heterologous expression studies in EBV-negative B cells, the DDR
pathways can be directly attenuated. Specifically, Robertson and colleagues have
observed a direct interaction between EBNA3C and Chk2 leading to decreased Chk2

24
activity, which may also contribute to DDR attenuation during primary B cell outgrowth
(Choudhuri et al., 2007). Another study identified the latent membrane protein LMP1 as
an inhibitor of ATM signaling due to transcriptional down-regulation of ATM upon
LMP1 over-expression (Gruhne et al., 2009b). Under certain circumstances, such as in
Hodgkin’s lymphoma or nasopharyngeal carcinoma where LMP1 is expressed at high
levels and may be important for cell survival, this activity may contribute to
tumorigenesis due to the inability of ATM to trigger checkpoints and mediate efficient
DNA repair.
1.4.2.3. Viral oncoproteins perturb mitotic checkpoint signaling
Mitotic checkpoints are often provoked by viral oncoprotein promotion of cell
cycle progression as discussed above. Therefore, in order for these viruses to replicate in
the infected cell, signaling downstream of the G2/M checkpoint must be attenuated.
Several oncogenic viruses encode proteins that precisely target this checkpoint with
potentially catastrophic consequences on the karyotype of surviving cells. HTLV-1 Tax
expression abolishes cellular mitotic checkpoints through directly targeting and
prematurely activating the anaphase promoting complex (Liu et al., 2005) as well as
suppressing the spindle assembly checkpoint protein Mad 1 (Jin et al., 1998) resulting in
highly aneuploid ATL cells. Similarly, the EBV EBNA3 proteins are capable of inhibiting
the canonical G2/M checkpoint through suppression of p27 levels or activity depending
on the cell type (Knight and Robertson, 2004; Parker et al., 2000; Wade and Allday, 2000).

25
Specifically, EBNA3C is capable of suppressing the effects of mitotic poisons in part
through decreasing the levels of the spindle assembly checkpoint protein BubR1
(Gruhne et al., 2009b; Leao et al., 2007). The consequence of bypassing the mitotic
checkpoint and DDR signaling downstream is the accumulation of aneuploid cells that
can promote tumorigenesis through copy number amplification of oncogenes or loss of
tumor suppressors.
In summary, the DDR can be activated directly by aberrant expression of
oncoproteins, cellular or viral, or as a consequence of cellular proliferation-induced
replicative stress. DNA tumor virus-driven cellular transformation occurs as a by-
product of the virus promoting the cell cycle in order to establish an appropriate
environment with the requisite DNA replication machinery and repair factors necessary
for viral DNA replication. Similarly, viruses such as HTLV-1 must activate the infected T
cell in order to promote a favorable environment for proviral DNA integration.
However, in the inadvertent setting such as following aberrant integration of viral
genomes where loss of normal viral replication function occurs or other changes lead to
increased viral oncoprotein expression, a constitutively activated DDR is triggered. DDR
signaling typically limits viral oncogenesis, but also provides selective pressure for
mutations in DDR signaling components that promote tumorigenesis. The delicate
balance between virus replication, latency, and the extent of activation of the DDR

26
ultimately dictates whether an infected cell will give rise to a productive cycle
generating progeny virions or a tumor.

27
2. Results
2.1. An ATM/Chk2-Mediated DNA Damage-Responsive Signaling Pathway Suppresses Epstein - Barr virus Driven Proliferation of Primary B cells
2.1.1. Contributions
The following Chapter 2.1, including text and figures, is adapted from (Nikitin et
al., 2010) with permission from Cell Press. This paper was published in co-first
authorship between Pavel Nikitin and Christopher Yan. Micah Luftig, Pavel Nikitin and
Christopher Yan designed and performed experiments with significant help from Jason
Tourigny, Eleonora Forte and Sandeep Dave. P.N. designed the method to sort
populations of EBV infected B cells and discovered the early proliferating cells
phenotype. C.Y. demonstrated the activation of the DDR by immunofluorescent
microscopy and revealed the role of viral DNA replication by fluorescent in situ
hybridization. E.F. revealed the initial activation of the DDR by Western and prepared
samples for microarray. M.L., J.T. and S.D. analyzed and confirmed the expression
changes. Martin Allday and Rob White kindly shared viral mutants and critically
discussed the manuscript. P.N. and C.Y. partially wrote the manuscript, and M.L. wrote
the final version of the paper. Finally, I would like to thank our co-authors Alessio
Bocedi, Amee Patel, William Kim, Katherine Hu, Jing Guo, David Tainter and Elena
Rusyn for their invaluable help in performing experiments, analyzing expression data
and running the lab in 24/7 schedule.

28
2.1.2. Introduction
As mentioned above, a convenient in vitro system allows identifying early event
upon EBV infection of primary human B cells. Precluding observation that only few
proliferating cells are transformed into indefinitely proliferating lymphoblasts
(Henderson et al., 1977; Sugden and Mark, 1977) led us to hypothesize that an active
host tumor suppressive mechanism prevents the EBV tumorigenesis.
Innate tumor suppressor responses have been better characterized in other
systems. The DNA damage response, overviewed in Chapter 1.3., is triggered by
aberrant replication structures generated by activated oncogenes attempting to
constitutively fire new origins and inappropriately enter S phase (Halazonetis et al.,
2008). The DDR limits aberrant proliferation by mediating oncogene-induced senescence
and apoptosis (Bartkova et al., 2006; Di Micco et al., 2006). Signaling downstream of
oncogenic stress involves activation of the single-stranded DNA-dependent ATR
pathway and the double-stranded break-induced ATM pathway. These DDR kinases
relay downstream signals to critical repair factors and other checkpoint kinases
including Chk1 and Chk2 with extensive cross-talk ultimately resulting in suppression
of oncogene-induced proliferation (Halazonetis et al., 2008; Stiff et al., 2006). Genetic
experiments have identified critical roles for ATM and Chk2 in mediating oncogene-
induced senescence and tumor suppression (Bartkova et al., 2006; Pusapati et al., 2006;
Stracker et al., 2008). Given these observations and the low efficiency of EBV

29
transformation, the intriguing question remains as to whether the host DNA damage
response senses EBV-induced oncogenic stress and, importantly, if this is responsible for
the block to long-term outgrowth of the majority of infected cells.
2.1.3. Results
2.1.3.1. Epstein-Barr virus infection of primary B cells activates a cellular DNA
damage response
We first sought to determine whether EBV infection of primary B cells might
drive an oncogenic stress leading to the activation of the DNA damage response.
Purified CD19+ B cells were infected with the prototypical transforming EBV strain B95-
8 at a multiplicity of infection (MOI) of ~5. Nearly all cells were EBV genome positive as
determined by fluorescence in situ hybridization (FISH) (Fig. 3A).

30
Figure 3: Nuclear genome deposition and the analysis of viral and host gene
expression following primary B cell infection with EBV B95-8, UV-inactivated B95-8,
P3-HR1 (A-C). EBV genomes in infected primary B cells are independent of -H2AX
foci (D-G).
(A) Fluorescence in situ hybridization (FISH) of viral genomes 2 days post B cell
infection with EBV B95-8, UV-inactivated B95-8 (UV B95-8), and P3HR1 (P3) at an MOI
of ~5. (B) Quantitative RT-PCR of the interferon responsive mRNAs ISG15 and IFIT4 24h
following PBMC infection with B95-8 and UV-inactivated B95-8. mRNA levels were
normalized to GAPDH and primers were as described previously (Martin et al., 2007)
(C) Quantitative RT-PCR for Wp-initiated mRNAs 48h after PBMC infection with B95-8,
UV-B95-8, or P3 virus strains. Analysis of the relative levels of W0 – W1/W2 containing
mRNAs indicate that infection was equivalent between B95-8 and P3, while UV-
inactivated virus was essentially unable to produce Wp-initiated transcripts. mRNAs
were normalized to GAPDH and primers for these experiments were previously
described (Bell et al., 2006). (D) Fluorescence in situ hybridization (FISH) of EBV
genomes (green) in B95-8 Z-HT cells (Johannsen et al., 2004). Uninduced cells (-HT)

31
mostly contained latent episomes (left). A representative cell undergoing lytic
replication following exposure to 4-hydroxytamoxifen (+HT) is shown on the right. (E)
EBV FISH in B cells 5 days and 14 days after infection. Intense lytic staining was rarely
observed 5 days after infection (left). Approximately 1-5% of cells were undergoing lytic
DNA replication by FISH at 14 days (right). (F) Left, Representative images of EBV
genomic FISH in primary CD19+ B cells at 1 and 5 days post infection with B95-8 (MOI
~5), steady state 8-10 genomes in a LCL, and 50 genomes per Raji cell. Right, Episome
number as determined by FISH at different times after infection. The y-axis represents
the average number of episomes per cell, the x-axis represents days after infection when
cells were collected and fixed for hybridization (5, 7, 10, 14, 21, and 35 days). Vertical
dashed lines show the beginning and end of the period in which the DNA damage
response (DDR) was observed. Error bars represent SEM. These data were collected
from greater than 50 nuclei at each time point for three independent normal donors. (G)
Left, IF/FISH of H2AX (red) and EBV genomes (green) in B cells 7 days post infection.
Left cell shows IF/FISH, right cell shows only IF for H2AX with white arrows
representing EBV genome location. The right panels are additional representative
IF/FISH and IF images.
Infected cells were initially assayed for the expression of the earliest viral latency
gene product, EBNA-LP, and the DNA damage marker, -H2AX, at different times post
infection. -H2AX activation was not evident prior to 4 days post infection, was robust
from 4 to 7 days post infection, and declined after 7 days to the low levels observed in
LCLs (Fig. 4A and data not shown). Approximately 60% of the infected cells were -
H2AX positive at 7 days post infection. Corroborating our findings of -H2AX
activation, EBV infection induced additional hallmarks of the DDR including auto-
phosphorylation of the H2AX kinase ATM (pATM Ser1981), and punctate localization of
the damage adaptor 53BP1 (Fig. 4B and 4C).
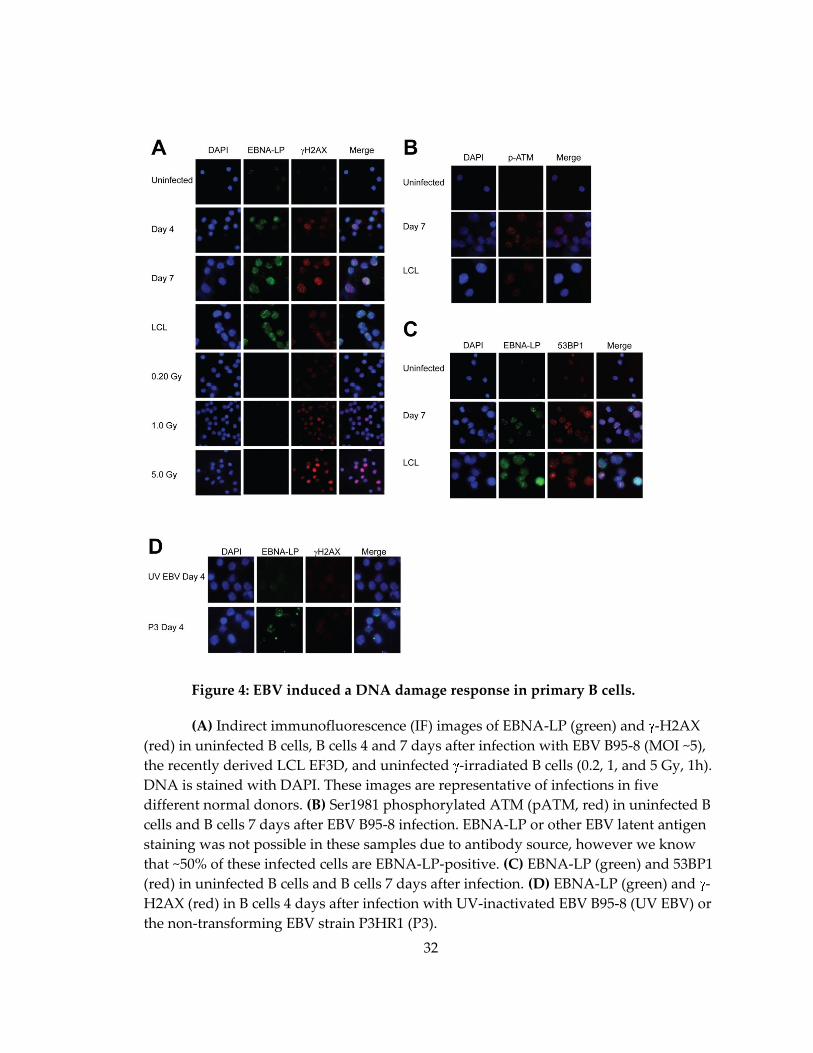
32
Figure 4: EBV induced a DNA damage response in primary B cells.
(A) Indirect immunofluorescence (IF) images of EBNA-LP (green) and -H2AX
(red) in uninfected B cells, B cells 4 and 7 days after infection with EBV B95-8 (MOI ~5),
the recently derived LCL EF3D, and uninfected -irradiated B cells (0.2, 1, and 5 Gy, 1h).
DNA is stained with DAPI. These images are representative of infections in five
different normal donors. (B) Ser1981 phosphorylated ATM (pATM, red) in uninfected B
cells and B cells 7 days after EBV B95-8 infection. EBNA-LP or other EBV latent antigen
staining was not possible in these samples due to antibody source, however we know
that ~50% of these infected cells are EBNA-LP-positive. (C) EBNA-LP (green) and 53BP1
(red) in uninfected B cells and B cells 7 days after infection. (D) EBNA-LP (green) and -
H2AX (red) in B cells 4 days after infection with UV-inactivated EBV B95-8 (UV EBV) or
the non-transforming EBV strain P3HR1 (P3).

33
EBV gene expression was important for virus-induced DDR activation. Cells
infected with UV-inactivated B95-8 virus did not show -H2AX staining at any point
within the first week after infection (Fig. 4D and data not shown). Importantly, UV-
inactivated EBV B95-8 genomes reached the nucleus and these infections induced
interferon-responsive genes (Fig. 3A and B). EBNA2 and latency III gene expression was
specifically necessary to induce the DDR as B lymphocytes infected with the EBNA2
deleted, transformation-incompetent P3HR1 strain of EBV did not contain -H2AX foci
(Fig. 4D) despite similar levels of infection compared to B95-8 (Fig. 3A-C). These data
collectively demonstrate that EBV latent gene expression rather than simply virion
binding or nucleic acid deposition into the nucleus was required to induce -H2AX
activation.
2.1.3.2. The EBV-induced DNA damage response in primary B cell infection is not
associated with viral episomes or lytic replication
We reasoned that either viral or cellular DNA may activate the DNA damage
response. Since evidence in the literature suggested that either viral lytic DNA
replication (Kudoh et al., 2005) or latent viral episome replication (Dheekollu et al., 2007)
may be capable of inducing a DDR, we first assayed viral DNA as a possible source of
the damage. Incoming linear viral DNA was not the source of the damage since UV-
irradiated and EBNA2-deleted P3HR1 virus infections did not induce the DDR (Fig. 4).
We next used a FISH based assay to assess the possible role of lytic DNA replication.

34
The B95-8 Z-HT cell line was used as a positive control where lytic EBV DNA was
recognized as a brightly staining FISH signal rather than the punctate foci of episomal
genomes (Fig. 3D). Less than 1% of EBV-infected cells contained evidence of lytic viral
DNA 5 days post infection, while approximately 1-5% of infected cells were
spontaneously undergoing lytic replication by 14 days similar to that found in LCLs
(Fig. 3E and (Kieff and Rickinson, 2006)). Since far greater than 1% of EBV-infected cells
were -H2AX positive early after infection, we conclude that viral lytic DNA replication
is not responsible for DDR activation.
Next we assessed the possibility that latent viral episomes activate the
DNA damage response. The mean episome number per cell as assessed by FISH did not
increase during the period when -H2AX activity was high early after infection (Fig. 3F).
Furthermore, we failed to observe significant co-localization of EBV episomes with -
H2AX foci in these cells (Fig. 3G). In fact, the number of -H2AX foci per cell was
consistently much greater than the number of EBV genomes (Fig. 3G). Therefore, our
data collectively suggest that the observed EBV-induced DDR is not activated by viral
DNA.
2.1.3.3. The EBV-induced DNA damage response is associated with a transient period
of hyper-proliferation
We next focused our studies on changes in cellular DNA that may induce a DDR.
The period of time post infection when the DDR was active correlates with the initiation
of B cell proliferation (Kieff and Rickinson, 2006). Analysis of CD19+ B cells using the
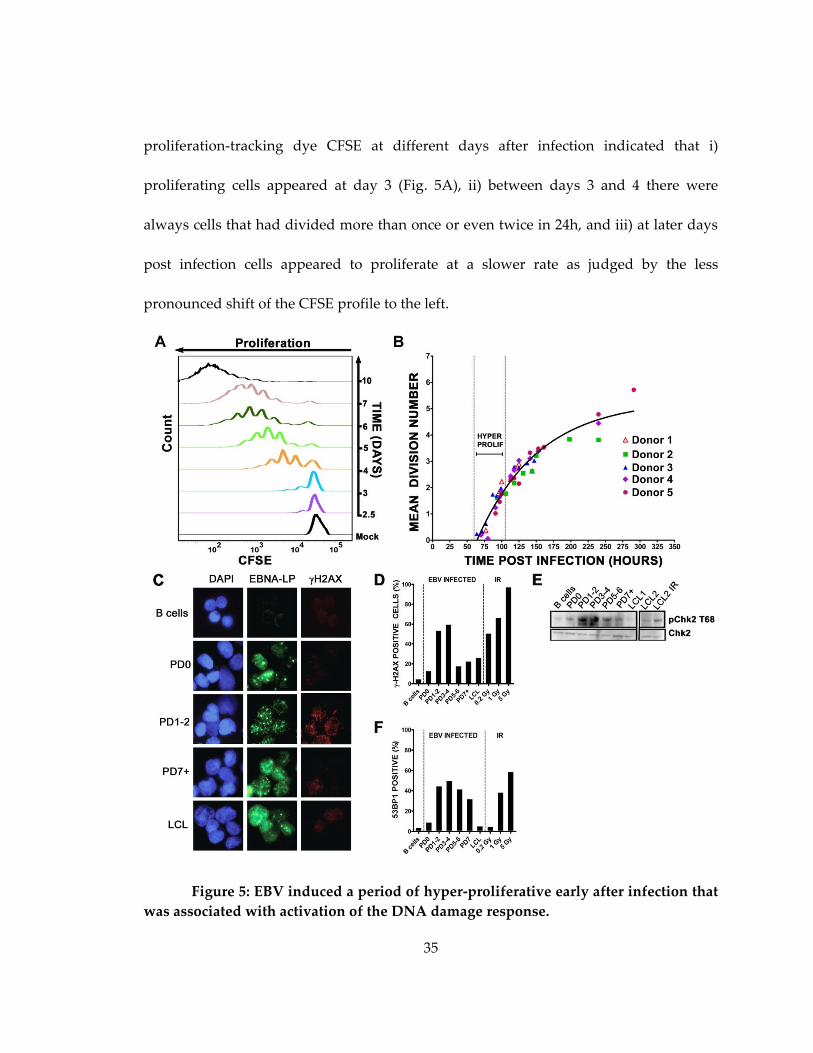
35
proliferation-tracking dye CFSE at different days after infection indicated that i)
proliferating cells appeared at day 3 (Fig. 5A), ii) between days 3 and 4 there were
always cells that had divided more than once or even twice in 24h, and iii) at later days
post infection cells appeared to proliferate at a slower rate as judged by the less
pronounced shift of the CFSE profile to the left.
Figure 5: EBV induced a period of hyper-proliferative early after infection that
was associated with activation of the DNA damage response.

36
(A) Histograms show CD19+ B cell division as measured by CFSE staining at
different days after EBV infection. Mock, mock infected cells. (B) The mean division
number based on precursor cohort analysis for EBV-infected B cells is plotted at
different times post infection. Vertical dashed lines estimate the hyper-proliferation
period. Data are presented from 5 normal donors. (C) IF of -H2AX (red) and EBNA-LP
(green) of uninfected cells, infected cells that have yet to divide (PD0), infected cells after
1 or 2 divisions (PD1-2), or 7 or more divisions (PD7+) and LCLs. DNA is stained with
DAPI. (D) The percentage of EBNA-LP positive cells with -H2AX signal >5X over
background is graphed from uninfected B cells, sorted PDs, and LCLs. Uninfected B
cells following 0.2, 1, and 5 Gy (1hr) -irradiation are also shown as a positive control.
These data are representative of similar experiments from three independent normal
donors. (E) Immunoblot of p-Chk2 Thr68 and Chk2 in sorted cells as in (D) including an
LCL following 5 Gy -irradiation (1hr). (F) The percentage of EBNA-LP positive cells
containing 4 or more 53BP1 foci per cell in sorted populations as in (D) are shown along
with uninfected irradiated B cell controls. PD3-4 contained significantly more 53BP1 foci
per cell than uninfected B cells (p<0.0001), PD0 (p<0.0001), PD7 (p<0.01), and LCL
(p<0.0001).
A more rigorous kinetic analysis of EBV-induced B cell expansion highlighted
the biphasic nature of the proliferation rate (Fig. 5B). Infected CD19+ B cell CFSE profiles
from five normal donors were analyzed at time points prior to and during the first seven
cell divisions. The mean division number (MDN) at each time point was determined by
fitting the precursor-normalized number of cells in each division to a Gaussian
distribution (Fig. 6A and (Hawkins et al., 2007a)).

37
Figure 6: CFSE-based kinetic analysis of EBV-induced proliferation.
(A) Analysis of EBV-infected PBMC stained by CFSE was performed by FACS at
different time of infection (141 hour post infection point is shown, left). CFSE-based
population doublings of CD19+ B cells were determined using the “Proliferation” tool in
the FlowJo program. Number of cells in each PD (N) was normalized to averaged
division number i (i=i+0.5) and fit to a Gaussian distribution for each time point. Mean
division number was determined as shown (MDN=3.09 for 141 hour p.i. for a given
donor, middle). Then, MDN over time post infection plot was generated for each time
point (right). (B) CD19+ cells from population doublings 0, 1, 2 and 3 were double-
sorted by flow-cytometry and incubated separately for 48 hours. Then, proliferative
potency of cells from each population doubling was measured by FACS as a percent of
cells divided more than once, twice or thrice. The data shown is the average for 2
donors. Error bars represent standard error.
The slope of the function relating MDN to time post infection inversely correlates
with the proliferation rate. Consistent with the data in Fig. 5A, we observed that EBV
induced an early phase of hyper-proliferation that was attenuated over time (Fig. 5B).
The proliferation rate of initially proliferating cells was approximately once per 8-12h

38
while later cycles were ~24-30h similar to the ~24-28h rate of LCLs. These findings were
corroborated by cell sorting experiments where cells from earlier divisions proliferated
more quickly than those in later divisions (Fig. 6B). Thus, EBV-mediated B cell
expansion proceeds through an initial period of hyper-proliferation followed by slower
cell divisions typical of emergent LCLs.
We next asked whether the DNA damage response was activated specifically
during the hyper-proliferative divisions independent of time post infection. EBV-
infected B cells sorted based on population doubling (PD) were subjected to
immunofluorescence for EBNA-LP and -H2AX (Fig. 5C). Sorted cells were >85% EBNA-
LP positive in cells not yet dividing (PD0) and >95% EBNA-LP positive in all later PDs.
We observed a robust increase in LP+/ -H2AX+ cells during the early PDs (1-2 and 3-4)
relative to uninfected cells or infected cells not yet proliferating (PD0) (Fig. 5C and 5D).
Importantly, this response was attenuated through later PDs and in LCLs. Moreover, -
H2AX intensity per cell was significantly higher in PD3-4 than PD0 (p<0.0001) and LCL
(p<0.0001). We also observed a transient activation and attenuation of the ATM-specific
phosphorylation of Chk2 on Thr68 (Fig. 5E) as well as accumulation of 53BP1 into DDR
foci (Fig. 5F). These data strongly support the notion that the EBV-induced DDR is
caused by an early period of hyper-proliferation and is attenuated during LCL
outgrowth.

39
2.1.3.4. Proliferation and DNA damage responsive genes are highly induced early
after EBV infection, then attenuated during LCL outgrowth
Our cell-based findings were corroborated by mRNA microarray studies of i)
uninfected B cells, ii) EBV-infected early proliferating cells (Prolif), and iii) monoclonal
LCLs from four normal donors (Fig. 7). We first asked in an unbiased manner which
genes were significantly changed upon proliferation and then, subsequently, during
LCL outgrowth (2-way ANOVA, p<0.01). As expected, the most enriched gene ontology
(GO) category for genes induced from resting B cells to EBV-infected, proliferating B
cells was ‘Cell Proliferation’ (Fig. 7A; GO:0008283, Bayes factor: 51, p<0.0001 (Chang and
Nevins, 2006)).

40
Figure 7: Transcriptional changes correlate with an EBV-induced early period
of hyper-proliferation and DNA damage response followed by attenuation upon LCL
outgrowth.
(A) Heatmap of average expression data across four normal donors for the gene
ontology (GO) category “Cell Proliferation” in uninfected resting B cells (B cell), EBV-
infected early proliferating B cells (Prolif), and monoclonal LCLs (LCL). The genes
presented were derived from GATHER analysis of all genes with significant expression
changes (2-way ANOVA, p<0.01) where the expression level increased from B cell to
Prolif at least 1.5-fold and decreased from Prolif to LCL at least 1.2-fold (left). Heatmap
of individual samples of top 20 “Cell Proliferation” genes (right). (B) Heatmap of
“Response to DNA Damage Stimulus” GO genes across individual samples. (C) Gene
Set Enrichment Analysis (GSEA) of known DNA damage induced ATM and p53-
depending genes in the context of B-Prolif-LCL expression data. The reference list of

41
ATM/p53 target genes was derived from Clusters 2 and 3 of (Elkon et al., 2005) and
compared with a pre-ranked list (by fold) of global average gene expression changes
from B cell to Prolif (left) and Prolif to LCL (right). Statistical scores are inset into the top
right of analysis images (NES: Normalized enrichment score and FWER: Familywise
error rate).
Genes associated with the ‘Response to DNA Damage Stimulus’ were also highly
induced (Fig. 7B; GO:0006974, Bayes factor: 17, p<0.0001). Notably, we observed that the
majority of genes involved in cell proliferation and the DNA damage response were
consistently repressed as cells transitioned from early proliferating to established LCLs
(Fig. 7A, Cell Proliferation, Bayes factor: 63, p<0.0001 and Fig. 7B, Response to DNA
Damage Stimulus, Bayes factor: 22, p<0.0001). Consistently, the expression of genes in an
independently derived set of DNA damage responsive and ATM-dependent p53 targets
(Elkon et al., 2005) was also increased in early proliferating cells and subsequently
attenuated during LCL outgrowth (Fig. 7C and 8).

42
Figure 8: Schematic diagram of GSEA analysis performed on B cells, Proliferating
cells, and LCL microarray data with ATM/p53 target genes.
We imported our average mRNA expression changes from either B to
Proliferating (Prolif) or Prolif to LCL as molecular profiles. Then we assigned the ATM
and p53 target genes as identified from (Elkon et al., 2005) as a gene set into GSEA. The
enrichment of this gene set relative to 1000 random permutations within the B to Prolif
samples was plotted in the data in Fig. 4C, which indicated that EBV-induced
proliferation was associated with an activation of the ATM/p53 gene expression
signature, while from proliferation through LCL outgrowth this gene set was depleted
indicating attenuation of ATM/p53 target gene expression.
Collectively, these global gene expression analyses corroborate our findings of a
period of hyper-proliferation and activation of an ATM-dependent DNA damage
response early after infection that is attenuated during LCL outgrowth.

43
2.1.3.5. The EBV-induced hyper-proliferation associated DNA damage response is
growth suppressive
To further analyze the consequences of the activated DDR in early rapidly
proliferating cells, we designed a sorting strategy to assess the relative growth potential
and DDR activation in cells derived from early or late divisions after infection (Fig. 9A).
We initially stained cells with the proliferation tracking dye PKH26 and sorted cells after
infection for PD1-4 and PD6+ populations (Fig. 9B).
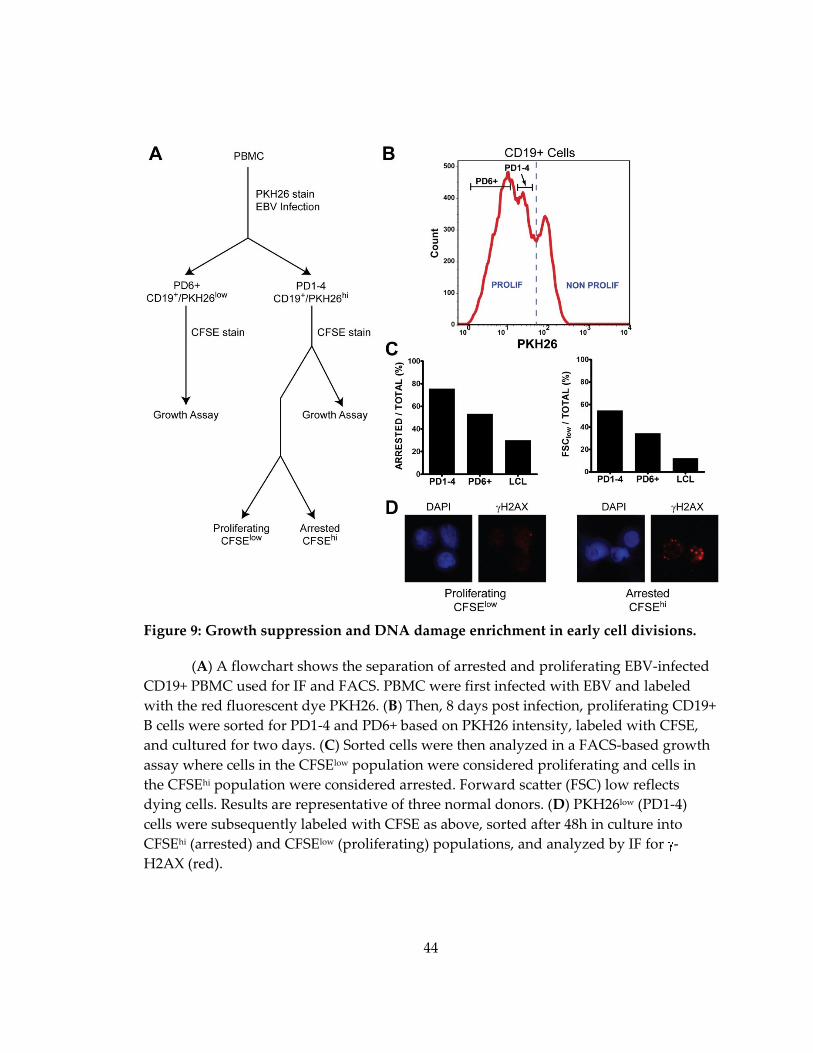
44
Figure 9: Growth suppression and DNA damage enrichment in early cell divisions.
(A) A flowchart shows the separation of arrested and proliferating EBV-infected
CD19+ PBMC used for IF and FACS. PBMC were first infected with EBV and labeled
with the red fluorescent dye PKH26. (B) Then, 8 days post infection, proliferating CD19+
B cells were sorted for PD1-4 and PD6+ based on PKH26 intensity, labeled with CFSE,
and cultured for two days. (C) Sorted cells were then analyzed in a FACS-based growth
assay where cells in the CFSElow population were considered proliferating and cells in
the CFSEhi population were considered arrested. Forward scatter (FSC) low reflects
dying cells. Results are representative of three normal donors. (D) PKH26low (PD1-4)
cells were subsequently labeled with CFSE as above, sorted after 48h in culture into
CFSEhi (arrested) and CFSElow (proliferating) populations, and analyzed by IF for -
H2AX (red).

45
Subsequent staining with CFSE enabled the analysis of proliferation from these
populations. Supporting our hypothesis, the cells in early hyper-proliferating divisions
(PD1-4) were, in fact, more prone to growth arrest and cell death than those in later
divisions (PD6+) and LCLs (Fig. 9C). Consistently, arrested PD1-4 cells displayed
significantly more intense -H2AX staining than their proliferating counterparts (Fig.
9D).
2.1.3.6. ATM and Chk2 kinases suppress EBV-mediated transformation and initial B
cell proliferation
To determine if the activation of the DDR restricts EBV-mediated long-term
outgrowth, we simultaneously infected peripheral blood mononuclear cells (PBMC)
with EBV and treated them with an inhibitor of either ATM (ATMi (Hickson et al.,
2004)) or its downstream effector kinase Chk2 (Chk2i (Arienti et al., 2005)); both are
critical kinases in the DDR checkpoint responding to DNA double-stranded breaks and
oncogenic stress. EBV-mediated B cell transformation efficiency increased in a dose-
dependent manner in response to the inhibitors where 2 M ATMi increased efficiency
by approximately 2-fold and 5 M ATMi by 6-fold over DMSO control treated cells (Fig.
10A). Similarly, Chk2 inhibition increased EBV transformation efficiency approximately
3-fold for 2 M Chk2i and 9-fold for 5 M Chk2i (Fig. 10B). Therefore, an ATM and Chk2
dependent DDR restricts EBV transformation.
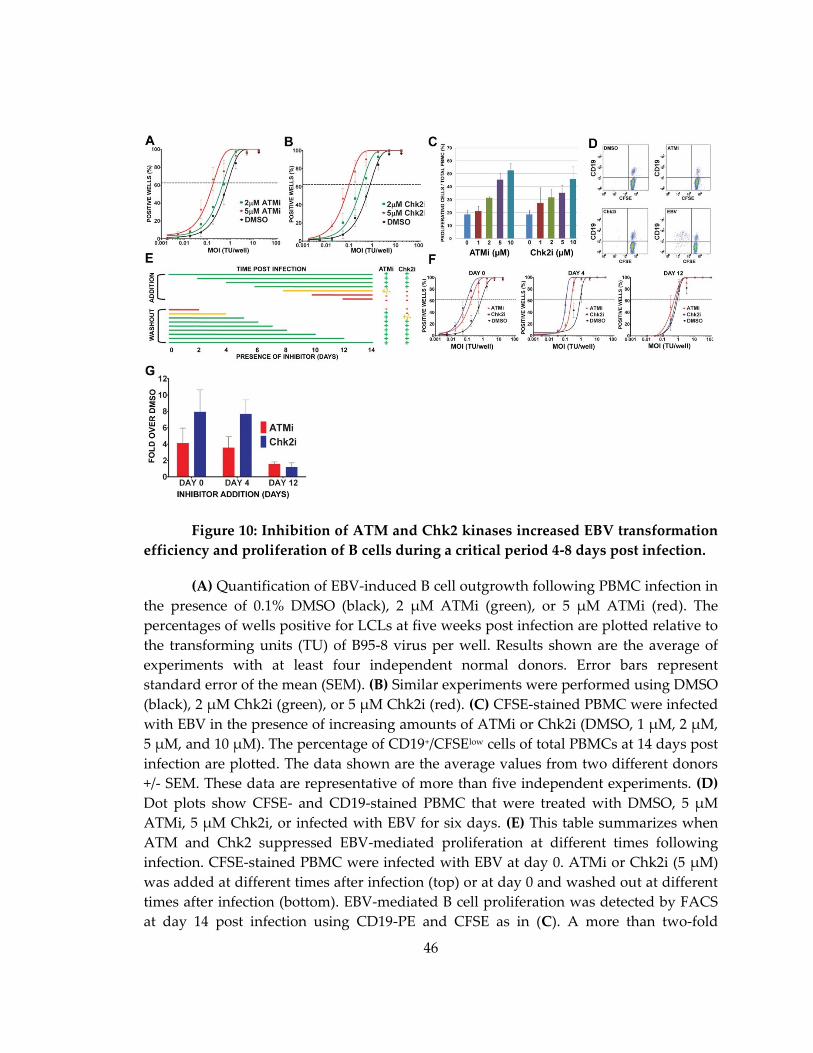
46
Figure 10: Inhibition of ATM and Chk2 kinases increased EBV transformation
efficiency and proliferation of B cells during a critical period 4-8 days post infection.
(A) Quantification of EBV-induced B cell outgrowth following PBMC infection in
the presence of 0.1% DMSO (black), 2 μM ATMi (green), or 5 μM ATMi (red). The
percentages of wells positive for LCLs at five weeks post infection are plotted relative to
the transforming units (TU) of B95-8 virus per well. Results shown are the average of
experiments with at least four independent normal donors. Error bars represent
standard error of the mean (SEM). (B) Similar experiments were performed using DMSO
(black), 2 µM Chk2i (green), or 5 µM Chk2i (red). (C) CFSE-stained PBMC were infected
with EBV in the presence of increasing amounts of ATMi or Chk2i (DMSO, 1 µM, 2 µM,
5 µM, and 10 µM). The percentage of CD19+/CFSElow cells of total PBMCs at 14 days post
infection are plotted. The data shown are the average values from two different donors
+/- SEM. These data are representative of more than five independent experiments. (D)
Dot plots show CFSE- and CD19-stained PBMC that were treated with DMSO, 5 µM
ATMi, 5 µM Chk2i, or infected with EBV for six days. (E) This table summarizes when
ATM and Chk2 suppressed EBV-mediated proliferation at different times following
infection. CFSE-stained PBMC were infected with EBV at day 0. ATMi or Chk2i (5 μM)
was added at different times after infection (top) or at day 0 and washed out at different
times after infection (bottom). EBV-mediated B cell proliferation was detected by FACS
at day 14 post infection using CD19-PE and CFSE as in (C). A more than two-fold

47
increase in treated cells versus DMSO is represented by a green plus; a less than two-
fold increase is represented by a yellow plus; and no increase is represented by a red
dash. The lines indicate the period of incubation and are colored with the proliferation
phenotype after ATMi and Chk2i treatment. Average values from two independent
donors are shown. (F) EBV-induced outgrowth following PBMC infection was measured
as in (A) in the presence of 5 μM Chk2i (blue), 5 μM ATMi (red), or DMSO (black) added
at day 0, day 4 or day 12 after EBV infection. Results shown are the average of four
independent normal donors +/- SEM. (G) Efficiency of EBV outgrowth from (F) was
calculated and the average ratio of inhibitor-treated to DMSO-treated infections +/- SEM
for four normal donors is plotted.
We next assessed whether ATM and Chk2-mediated suppression of EBV
transformation was due to limiting initial B cell proliferation. The continuous presence
of either ATM or Chk2 inhibitor led to a dose dependent increase in B cell number at
two weeks post infection (Fig. 10C). Importantly, ATM or Chk2 inhibitor did not induce
B cell proliferation in the absence of EBV suggesting that these compounds act to
alleviate a block to proliferation rather than stimulating B cells per se (Fig. 10D).
2.1.3.7. ATM and Chk2 suppress B cell growth 4-8 days after EBV infection
Since the DDR peaked during the first week after infection, we assessed when
ATM and Chk2 inhibition enhanced proliferation and transformation. To that end,
PBMC infected with EBV were transiently exposed to ATMi and Chk2i from the start of
infection or the compounds were added at different days post infection. EBV-induced B
cell proliferation was most sensitive to the inhibitors between 4 and 8 days after
infection when cells were present in the hyper-proliferative period (Fig. 10E). For
example, when either inhibitor was added within the first 4 days of infection we

48
observed as pronounced an effect on proliferation as if the inhibitor was added at day 0.
However, if we added inhibitors after day 8, there was no effect on proliferation.
Conversely, if the inhibitors were removed prior to 4 days after infection, then increased
proliferation was not observed.
Similar results were obtained in long-term transformation assays.
Addition of either compound within 4 days of infection increased transformation
efficiency, while adding the compounds at 12 days post infection had little effect (Fig.
10F and 10G). The inhibitors also did not increase LCL growth rates at normal or
limiting density (data not shown). Therefore, during a critical period approximately 4-8
days following infection, EBV induced an ATM and Chk2-dependent growth
suppressive signaling pathway that limited initial B cell proliferation and, consequently,
long-term outgrowth into lymphoblastoid cell lines.
2.1.3.8. EBV latent gene expression changes and consequences in early infected cell
divisions
The dynamic changes in proliferation and DDR associated gene expression
support our cell-based assays indicating an early period of ATM/Chk2-mediated growth
suppression that is attenuated in later divisions enabling long-term LCL outgrowth.
However, to determine whether these changes correlated with viral gene expression, we
queried viral transcripts and proteins associated with the latency III growth program in
sorted population doublings after infection (Fig. 11 and 12). Wp-associated transcripts
were expressed at a markedly higher level than Cp transcripts prior to the first infected

49
cell division (PD0) (Fig. 11A). However, this ratio shifted such that Cp levels were
greater after 3-4 cell divisions and through LCL outgrowth consistent with previous
observations (Fig. 11B and (Schlager et al., 1996; Woisetschlaeger et al., 1989;
Woisetschlaeger et al., 1990)). The consequence of the high Wp/Cp ratio was heightened
levels of EBNA-LP protein as well as a heightened EBNA-LP to EBNA3A and 3C protein
ratio in early divisions that waned through LCL outgrowth (Fig. 11C-D). Thus, the initial
cell divisions characterized by hyper-proliferation display a distinct EBNA gene
expression equilibrium that may affect target gene expression.
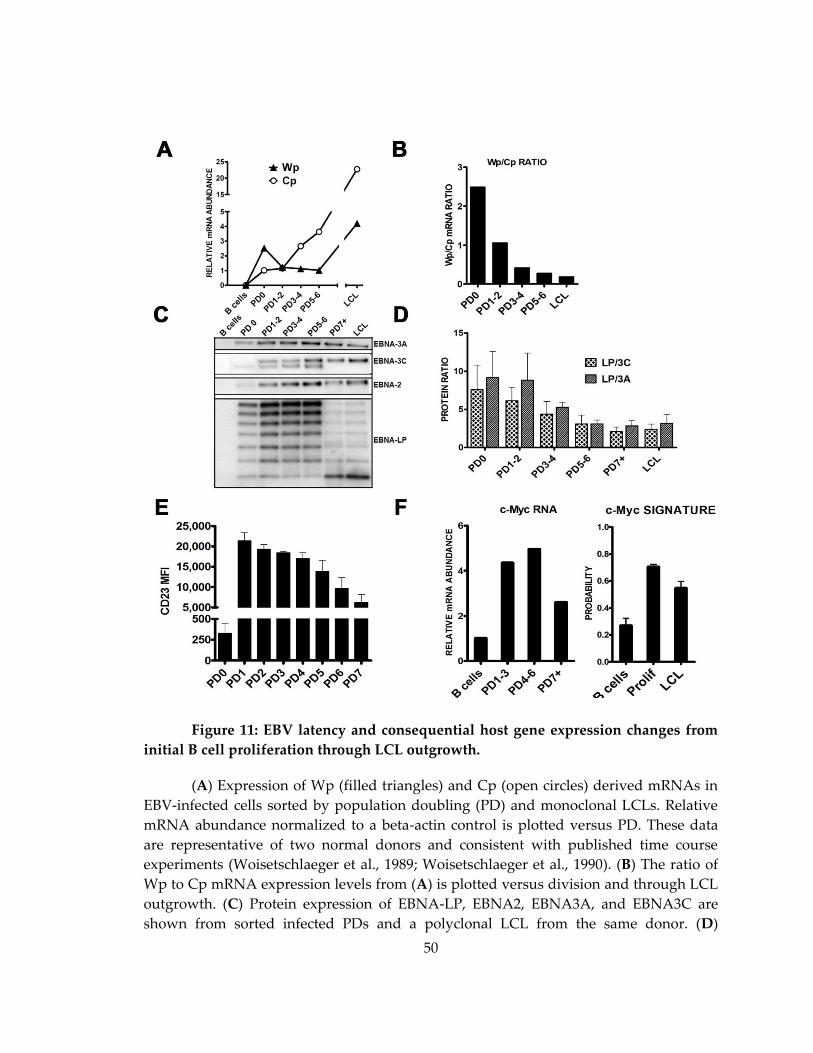
50
Figure 11: EBV latency and consequential host gene expression changes from
initial B cell proliferation through LCL outgrowth.
(A) Expression of Wp (filled triangles) and Cp (open circles) derived mRNAs in
EBV-infected cells sorted by population doubling (PD) and monoclonal LCLs. Relative
mRNA abundance normalized to a beta-actin control is plotted versus PD. These data
are representative of two normal donors and consistent with published time course
experiments (Woisetschlaeger et al., 1989; Woisetschlaeger et al., 1990). (B) The ratio of
Wp to Cp mRNA expression levels from (A) is plotted versus division and through LCL
outgrowth. (C) Protein expression of EBNA-LP, EBNA2, EBNA3A, and EBNA3C are
shown from sorted infected PDs and a polyclonal LCL from the same donor. (D)

51
Proteins detect by Western blotting from three independent normal donors similar to
those in panel (C) were quantified. The average ratio of total EBNA-LP protein (i.e., all
isoforms) relative to total EBNA3A or EBNA3C +/- SEM is plotted versus PD through
LCL. (E) Average CD23 surface expression as mean fluorescence intensity (MFI) is
plotted versus PD +/- SEM for two donors. (F, left) The expression level of c-Myc mRNA
is plotted versus sorted PD. (F, right) The activity of the c-Myc target gene expression
signature (Bild et al., 2006) is plotted from the average expression of targets in
microarray samples from four independent donors of resting B cells (B), early
proliferating B cells (Prolif), and monoclonal LCLs. Error bars represent SEM.
To more rigorously assess this, we analyzed EBNA2 targets including
CD23 (Wang et al., 1991) and c-Myc (Kaiser et al., 1999). In both cases, these EBNA2
targets were highly induced in early cell divisions and then attenuated through LCL
outgrowth, still remaining significantly higher than resting B cell levels (Fig. 11E-F). The
consequences of the transient increase in c-Myc mRNA was manifested in an increase in
the c-Myc target gene expression signature (Bild et al., 2006) during early proliferating
cells that was attenuated in LCLs, though still greater than resting B cell levels (Fig. 11F).
Given the importance in titrating this potentially genotoxic oncoprotein and the known
role of ATM in suppressing c-Myc oncogenesis (Hong et al., 2006; Murphy et al., 2008),
these findings strongly support a model of acute oncogenic stress early after EBV
infection that is modulated through the well described Wp to Cp switch enabling
modest EBNA2 activity critical for indefinite EBV-infected cell outgrowth.
2.1.3.9. EBNA3C is required to attenuate the EBV-induced DNA damage response
While the induction of the DNA damage response after EBV infection requires
latent gene expression and proliferation, a definitive role for viral latent genes in
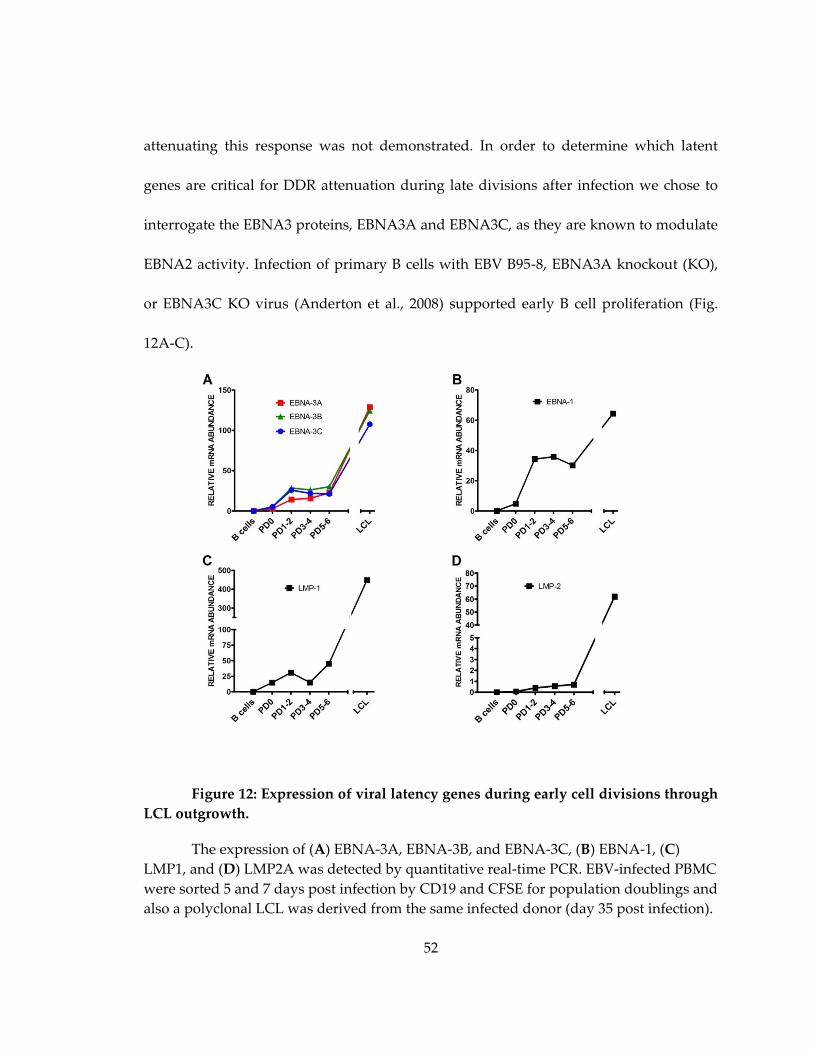
52
attenuating this response was not demonstrated. In order to determine which latent
genes are critical for DDR attenuation during late divisions after infection we chose to
interrogate the EBNA3 proteins, EBNA3A and EBNA3C, as they are known to modulate
EBNA2 activity. Infection of primary B cells with EBV B95-8, EBNA3A knockout (KO),
or EBNA3C KO virus (Anderton et al., 2008) supported early B cell proliferation (Fig.
12A-C).
Figure 12: Expression of viral latency genes during early cell divisions through
LCL outgrowth.
The expression of (A) EBNA-3A, EBNA-3B, and EBNA-3C, (B) EBNA-1, (C)
LMP1, and (D) LMP2A was detected by quantitative real-time PCR. EBV-infected PBMC
were sorted 5 and 7 days post infection by CD19 and CFSE for population doublings and
also a polyclonal LCL was derived from the same infected donor (day 35 post infection).

53
RNA was extracted and SYBR green-based qRT-PCR was performed using primers
spanning exons as previously described (EBNA1 Y3/U/K, LMP1 exon 2/3, and LMP2A
exon 1/2 from (Bell et al., 2006) and EBNA-3A (exon1/2), EBNA-3B (exon 1/2), and
EBNA-3C (exon 1/2) from (Sengupta et al., 2006).Data from one normal donor are shown
in the graphs which are representative of three independent infections. The relative
mRNA abundance is plotted normalized to an HPRT mRNA control (primers from
(Ranuncolo et al., 2007)). We note that for each mRNA, an increase is observed from
PD5-6 through LCL outgrowth, which is likely due to an increase in genome copy
number.
However, upon sorting these early proliferating cells we observed that EBNA3C
KO virus infected cells displayed increased activation of the DNA damage response,
while EBNA3A KO infected cells were similar to WT B95-8 infection in DDR activation
(Fig. 13A-B). Indeed, greater than 80% of EBNA3C KO-infected cells were -H2AX
positive relative to ~50% of WT or EBNA3A KO-infected cells (Fig. 13C).
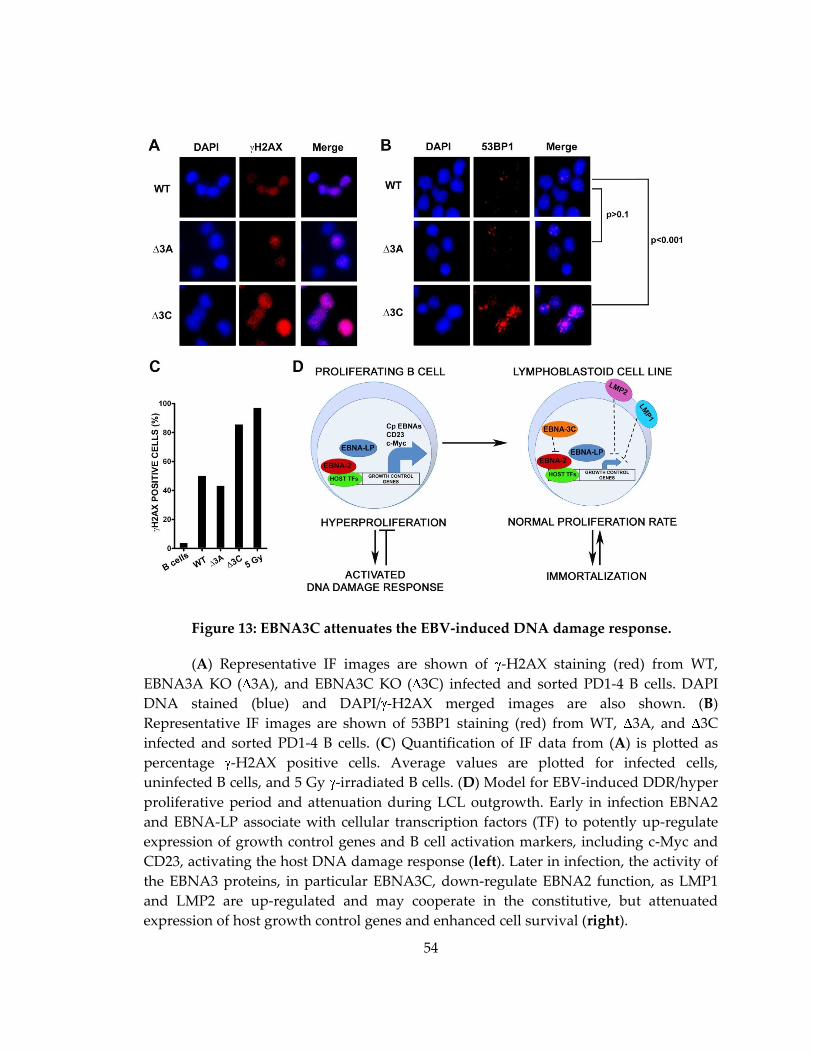
54
Figure 13: EBNA3C attenuates the EBV-induced DNA damage response.
(A) Representative IF images are shown of -H2AX staining (red) from WT,
EBNA3A KO ( 3A), and EBNA3C KO ( 3C) infected and sorted PD1-4 B cells. DAPI
DNA stained (blue) and DAPI/ -H2AX merged images are also shown. (B)
Representative IF images are shown of 53BP1 staining (red) from WT, 3A, and 3C
infected and sorted PD1-4 B cells. (C) Quantification of IF data from (A) is plotted as
percentage -H2AX positive cells. Average values are plotted for infected cells,
uninfected B cells, and 5 Gy -irradiated B cells. (D) Model for EBV-induced DDR/hyper
proliferative period and attenuation during LCL outgrowth. Early in infection EBNA2
and EBNA-LP associate with cellular transcription factors (TF) to potently up-regulate
expression of growth control genes and B cell activation markers, including c-Myc and
CD23, activating the host DNA damage response (left). Later in infection, the activity of
the EBNA3 proteins, in particular EBNA3C, down-regulate EBNA2 function, as LMP1
and LMP2 are up-regulated and may cooperate in the constitutive, but attenuated
expression of host growth control genes and enhanced cell survival (right).

55
Similarly, EBNA3C KO-infected cells accumulated 53BP1 DDR foci to a greater
extent than WT or EBNA3A KO-infected cells (p<0.001, 3C KO v. WT; p>0.1, 3A KO v.
WT). Thus, while B cells infected with either EBNA3A KO or EBNA3C KO virus were
crippled for long-term outgrowth (Fig. 14B-C), these experiments define a critical role
for EBNA3C in attenuating the host DNA damage response to EBV infection early after
infection. These data strongly support our model of a latent gene expression triggered
hyper-proliferation induced DDR, followed by proper expression of the EBNA3
proteins, in particular EBNA-3C, in order to attenuate a potentially genotoxic and
growth suppressive signaling pathway (Fig. 13D).
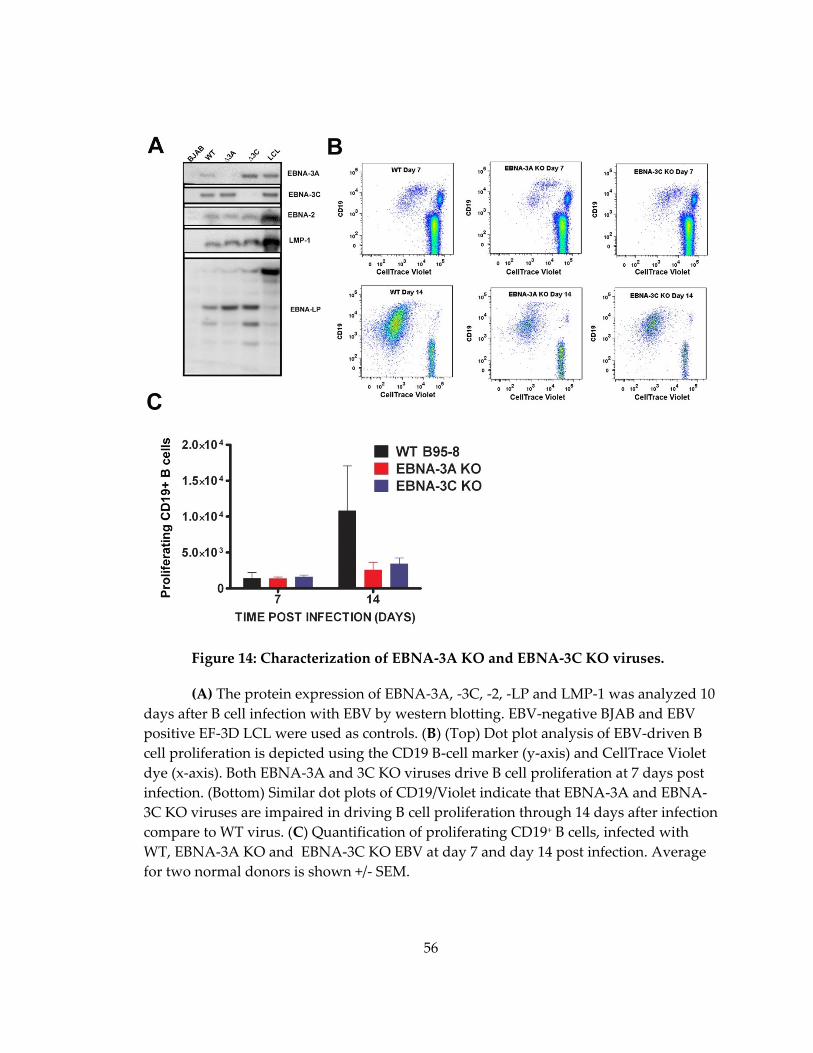
56
Figure 14: Characterization of EBNA-3A KO and EBNA-3C KO viruses.
(A) The protein expression of EBNA-3A, -3C, -2, -LP and LMP-1 was analyzed 10
days after B cell infection with EBV by western blotting. EBV-negative BJAB and EBV
positive EF-3D LCL were used as controls. (B) (Top) Dot plot analysis of EBV-driven B
cell proliferation is depicted using the CD19 B-cell marker (y-axis) and CellTrace Violet
dye (x-axis). Both EBNA-3A and 3C KO viruses drive B cell proliferation at 7 days post
infection. (Bottom) Similar dot plots of CD19/Violet indicate that EBNA-3A and EBNA-
3C KO viruses are impaired in driving B cell proliferation through 14 days after infection
compare to WT virus. (C) Quantification of proliferating CD19+ B cells, infected with
WT, EBNA-3A KO and EBNA-3C KO EBV at day 7 and day 14 post infection. Average
for two normal donors is shown +/- SEM.

57
2.1.4. Discussion
It has long been recognized that Epstein-Barr virus transformation efficiency is
on the order of 1-10% of infected primary human B cells (Henderson et al., 1977; Sugden
and Mark, 1977). However, little is known about the molecular mechanism responsible
for this low efficiency. We hypothesize that a robust innate tumor suppressor response
is activated by latent viral oncoproteins and blocks outgrowth of the majority of infected
cells. Recent evidence suggests that activated oncogene expression is sufficient to trigger
a growth-suppressive DNA damage responsive signaling pathway (Halazonetis et al.,
2008) and other oncogenic viruses have been shown to induce the DDR through
multiple means (reviewed in Chapter 1.4 and (Nikitin and Luftig, 2012). Therefore, in
this study we asked whether EBV was capable of inducing a DNA damage response in
primary B cells and, importantly, whether this response resulted in the low
transformation efficiency. We observed that as EBV-infected cells initiated proliferation,
a transient DNA damage response (DDR) was activated as evidenced by
phosphorylation of ATM Ser1981, H2AX Ser139 ( -H2AX), Chk2 Thr68, and
accumulation of 53BP1 in nuclear foci. Modulation of this signaling pathway by
chemical antagonism of ATM and its downstream target Chk2 markedly increased EBV-
mediated B cell polyclonal expansion and transformation efficiency thereby
demonstrating that the DDR contributes to an EBV-induced innate tumor suppressor

58
pathway. This is the first molecular pathway to be identified that restricts EBV
transformation.
2.2. Mitogen-Induced B Cell Proliferation Activates Chk2-dependent G1/S Cell Cycle Arrest
2.2.1. Contributions
M.L. and P.N. designed experiments and P.N. performed them. C.Y. made an
initial observation of elevated marks of the DDR in CpG-induced B cells. P.N. wrote the
chapter.
2.2.2. Introduction
In the first part of Results, I reported the fast division of Epstein-Barr virus-
infected human B cells, termed hyperproliferation, activating ATM and its downstream
kinase Chk2-dependent signaling. However, whether non-viral activators of B cell
hyper-proliferation activate the DDR remains unknown. Further, the downstream
effectors that mediated the DDR signaling in B lymphocytes are yet to be investigated.
Therefore, in the second part of Results I will discuss the role of the DDR in regulating
mitogen-induced B cell proliferation and the mechanism of Chk2-dependent
suppression of proliferation.
B cell mitogens. Two established ways to drive the proliferation of primary
human B cells in vitro include stimulation of the TLR9 receptor or activation with CD40
ligand coupled with cytokines (Hawkins et al., 2007a; Rousset et al., 1991; Tangye et al.,
2003). The human B cell TLR9 receptor may be potently and selectively activated with a

59
single-stranded oligonucleotide enriched for GC content that mimics the bacterial DNA
(termed “CpG”) (Cunningham-Rundles et al., 2006; Hartmann and Krieg, 2000). CpG
induces T cell-independent activation and accelerates proliferation of B cells that
recognize common pathogen patterns, such as bacterial DNA (Cunningham-Rundles et
al., 2006; Hartmann and Krieg, 2000) the (Fagarasan and Honjo, 2000)to . CpG DNA
binding activates the TLR9 dimer, which results in the recruitment of MyD88 (Hacker et
al., 2000; Latz et al., 2007). MyD88 interacts with IRAK4 and TRAF6 to induce mitogen-
activated protein kinase (MAPK) p38 and NF B transcription factors p50/p65, activator
protein (AP)-1, and activating transcription factor (ATF)-2 (Hartmann and Krieg, 2000;
Peng, 2005; Yi et al., 1998). In addition, MyD88 recruitment initiates a Pyk2-Src-Syk-
STAT3 downstream cascade that is required for cellular proliferation in a DOCK8-
dependent manner (Jabara et al., 2012). TLR9 stimulation of B cells in vitro potently
initiates proliferation of human and murine B cells. The kinetics of CpG-induced
proliferation of B cells has beenbeen well-studied by Phillip Hodgkin’s group (Hawkins
et al., 2007a; Turner et al., 2008). Intriguingly, computational analysis predicted two
distinct factors to control B-cell proliferation in early and later division, with the latter
being programmed cell death while the former remains unknown (Markham et al.,
2010).
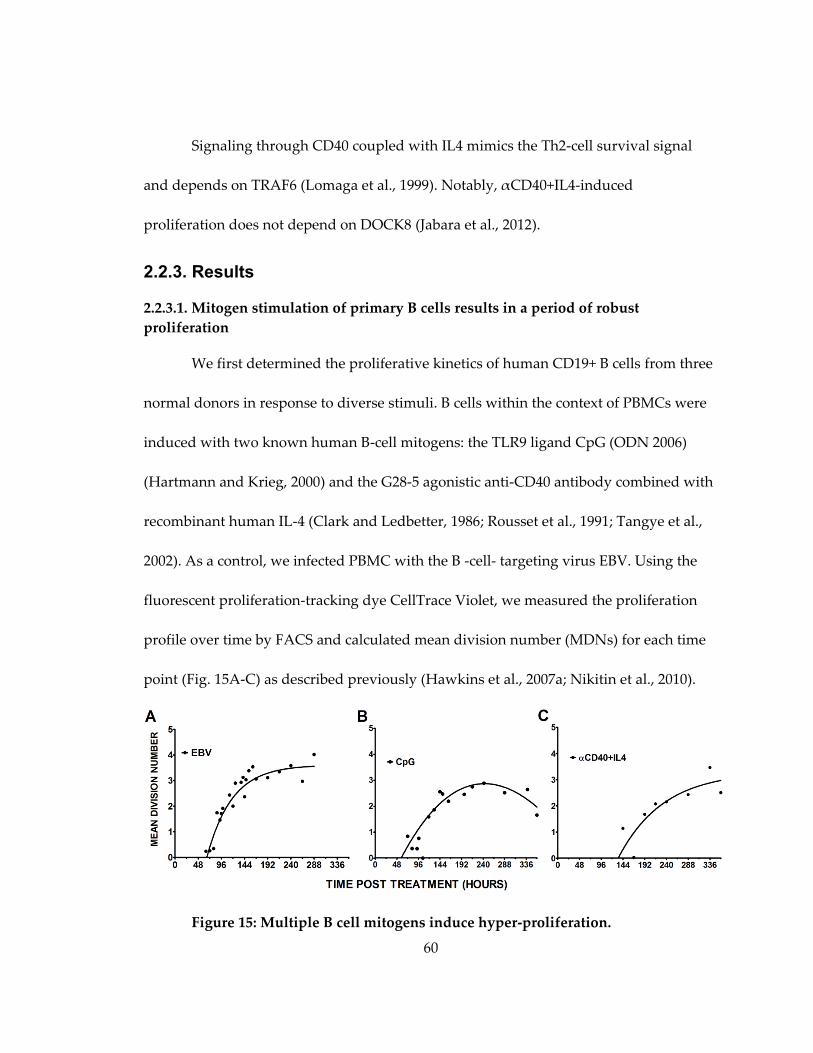
60
Signaling through CD40 coupled with IL4 mimics the Th2-cell survival signal
and depends on TRAF6 (Lomaga et al., 1999). Notably, αCD40+IL4-induced
proliferation does not depend on DOCK8 (Jabara et al., 2012).
2.2.3. Results
2.2.3.1. Mitogen stimulation of primary B cells results in a period of robust
proliferation
We first determined the proliferative kinetics of human CD19+ B cells from three
normal donors in response to diverse stimuli. B cells within the context of PBMCs were
induced with two known human B-cell mitogens: the TLR9 ligand CpG (ODN 2006)
(Hartmann and Krieg, 2000) and the G28-5 agonistic anti-CD40 antibody combined with
recombinant human IL-4 (Clark and Ledbetter, 1986; Rousset et al., 1991; Tangye et al.,
2002). As a control, we infected PBMC with the B -cell- targeting virus EBV. Using the
fluorescent proliferation-tracking dye CellTrace Violet, we measured the proliferation
profile over time by FACS and calculated mean division number (MDNs) for each time
point (Fig. 15A-C) as described previously (Hawkins et al., 2007a; Nikitin et al., 2010).
Figure 15: Multiple B cell mitogens induce hyper-proliferation.

61
Kinetics of B cell proliferation in response to Epstein-Barr virus infection (A), constant
stimulation with TLR9 ligand CpG (2.5µg/mL) (B), and anti-CD40 antibody G28-5
(1µg/mL) coupled with interleukin-4 (20 pg/mL)treatment (C). Mean division number is
calculated as in Fig. 5 for PBMC from 3-6 normal human donors.
In EBV-infected cells we observed a bi-phasic proliferation of EBV-infected
PBMC, including a faster hyper-proliferative period followed by a slower stage of
proliferation (Nikitin et al., 2010) (Fig.15A). In CpG and αCD40/IL4-induced cells we
observed a similar initial period of robust proliferation rate as cells began to divide,
although the time to first division differed among these mitogens (Fig. 15B,C). Notably,
αCD40/IL4 stimulation induced a slower proliferation rate compared to EBV and CpG
(Fig. 15C).
2.2.3.2. Mitogen stimulation activates the ATM signaling pathway in hyper-
proliferating cells
Aberrant oncogene and growth factor induced cellular proliferation has been
shown to activate DNA replicative stress and a subsequent growth-suppressive DNA
damage responsive signaling pathway (Bartkova et al., 2006; Di Micco et al., 2006;
Halazonetis et al., 2008). We previously revealed activated hallmarks of the DDR,
including formation of γH2AX foci and ATM-downstream signaling in EBV-driven
proliferation (Fig. 4). Moreover, the DDR activation did not depend on viral DNA
replication, but rather correlated with the speed of cellular proliferation and was present
in early proliferating cells (Nikitin et al., 2010). In order to separate mitogen-induced
cycling (“proliferating”) and quiescent (“non-proliferating”) cells, we employed FACS

62
sorting. We observed elevated levels of γH2AX in proliferating B cells in all three
conditions (Fig. 16A-B).
Figure 16: B cell mitogens induce the ATM signaling pathways in hyper-
proliferating cells.
(A) Immunofluorescent microscopy of sorted early proliferating B cells. Non Prolif, non-
proliferating sorted B cells; Prolif, proliferating sorted B cells. DAPI, nuclei stain; H2AX,
phosphorylated histone H2AX; B cells, untreated primary B cells; IR, γ-irradiation. (B)
Number of cells with γH2AX intensity >5X over background plotted as a ratio from total
from three normal donors. Erorr bars are SEM. (C) Western blot analysis of Chk2 and its
ATM-phosphorylation site at Thr68. Protein lysates normalized to a total protein
concentration. P, proliferating; NP, non-proliferating cells; Cntr, untreated cells of
Burkitt’s lymphoma cell line 2 (BL2); IR, 5Gy irradiated BL2. Shown blot is a
representative analysis from three normal donors.
Consistent with previous work, ATM-dependent phosphorylation of Chk2 at
threonine-68 increased, suggesting the role of ATM/Chk2 kinases in the mediation of the

63
signal (Fig. 16C). Finally, we observed higher phosphorylation of pChk2-Thr68 in sorted
proliferating EBV-infected or CpG-induced cells, compared to αCD40/IL4-driven
proliferating cells.
2.2.3.3. EBV and mitogen-induced B-cell proliferation is suppressed by Chk2
Previously reported work has identified a role for ATM and its downstream effector
Chk2 in suppressing lymphomagenesis (Hirao et al., 2002; Maclean et al., 2008;
McPherson et al., 2004; Nikitin et al., 2010; Tort et al., 2002). Given our previous findings
indicating that EBV-induced B -cell proliferation was suppressed by Chk2 (Fig. 17A,
left), we assessed the role of Chk2 in suppressing mitogen-stimulated B-cell growth.
Consistently, treatment with a potent and specific small molecule inhibitor of Chk2
(Arienti et al., 2005) increased the number of dividing human B lymphocytes stimulated
with either CpG or CD40/IL4 (Fig. 17).
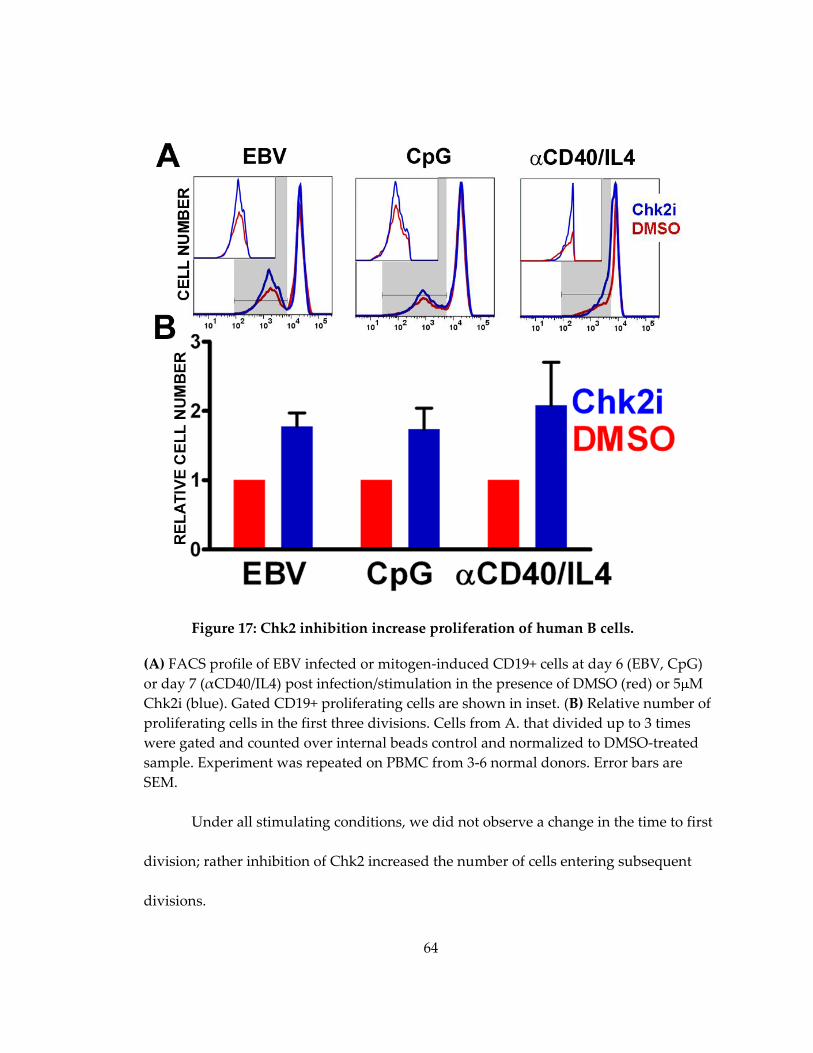
64
Figure 17: Chk2 inhibition increase proliferation of human B cells.
(A) FACS profile of EBV infected or mitogen-induced CD19+ cells at day 6 (EBV, CpG)
or day 7 (αCD40/IL4) post infection/stimulation in the presence of DMSO (red) or 5 M
Chk2i (blue). Gated CD19+ proliferating cells are shown in inset. (B) Relative number of
proliferating cells in the first three divisions. Cells from A. that divided up to 3 times
were gated and counted over internal beads control and normalized to DMSO-treated
sample. Experiment was repeated on PBMC from 3-6 normal donors. Error bars are
SEM.
Under all stimulating conditions, we did not observe a change in the time to first
division; rather inhibition of Chk2 increased the number of cells entering subsequent
divisions.

65
2.2.3.4. B-cell mitogens induce caspase 3/7-dependent apoptosis independent of Chk2
Chk2 downstream signaling induces apoptosis and cell cycle arrest (Sato et al.,
2010). Therefore, we assayed the role of Chk2 kinase in both of these processes during B-
cell mitogen driven hyper-proliferation. Both CpG and αCD40/IL4 stimulation of B cells
induced apoptosis, revealed by increased signal from a fluorescently labeled caspase 3/7
substrate DEVD-FAM (Fig. 18A, B) and by Western analysis of cleaved PARP and
caspase 3 (Fig. 18C). However, inhibition of Chk2 did not prevent the activated
apoptosis in proliferating cells upon CpG and αCD40/IL4 stimulations (compare Fig.18A
top to bottom panels). Surprisingly, we did not observe apoptotic cells in EBV-infected
early proliferating B cells (Fig.18. A-C). Unexpectedly, a modest induction of apoptosis
was observed in EBV-infected non-proliferating cells (Violethi) but not in proliferating
cells (Violetlo).

66
Figure 18: B-cell mitogens, but not EBV induces caspases 3/7-dependent apoptosis.
(A) Activated apoptosis in proliferating B cells treated with DMSO (top panels) or Chk2i
(bottom panels) and measured with DEVD-FAM, a fluorescently labeled soluble caspase
3/7 inhibitor. FACS profiles of CD19+ single cells. (B) Percent of apoptotic cells in non-
proliferating (NP) or proliferating (Prol) CD19+ cells treated with DMSO (blue) or Chk2i
(red). Activation of caspases was compared to LCL and BL2 cells treated with 10µM
Nutlin-3 or 15µM camptothecin. (C) Cleavage of caspase targets (PARP and self-
processing of caspase-3) revealed by western blot analysis of sorted proliferating (Prol)
or non-proliferating (NP) EBV-infected or mitogen-stimulated B cells. Casp3, cleaved
form of Caspase 3.
Therefore, Chk2 inhibition does not relieve activation of caspases 3/7 in early
proliferating B cells.
2.2.3.6. Mitogen-induced hyper-proliferation of human B cells induced a Chk2-
dependent G1/S cell cycle arrest
As discussed in the introduction, Chk2 phosphorylation stabilizes p53, thus
promoting G1/S phase arrest, and activates CDC25s to induce an intra-S phase and
G2/M checkpoints (Falck et al., 2001; Hirao et al., 2000; Matsuoka et al., 1998). We have

67
assayed the cell cycle profile of early proliferating EBV-infected or mitogen-stimulated
cells following a 2 hours pulse of the thymidine analog BrdU. We revealed that Chk2-
inhibition increased the number of cells entering S-phase in CpG-induced or EBV-
infected proliferating cells. Further, αCD40/IL4-stimulation led to a high number of cells
entering the first S-phase that was mildly increased upon Chk2i treatment (Fig. 19A,
left). Finally, in all three stimuli, we did not observe a significant accumulation of cells in
G2/M stage or a release of BrdU-negative cells (intra-S phase checkpoint) upon
inhibition of Chk2. Strikingly, EBV-transformed lymphoblastoid cell lines with
attenuated expression of proliferation-driven genes (Nikitin et al., 2010) were not
responsive to inhibition of Chk2 and had a normal cell cycle profile. Therefore, we
conclude that Chk2 induced a G1/S checkpoint in human B-cells.
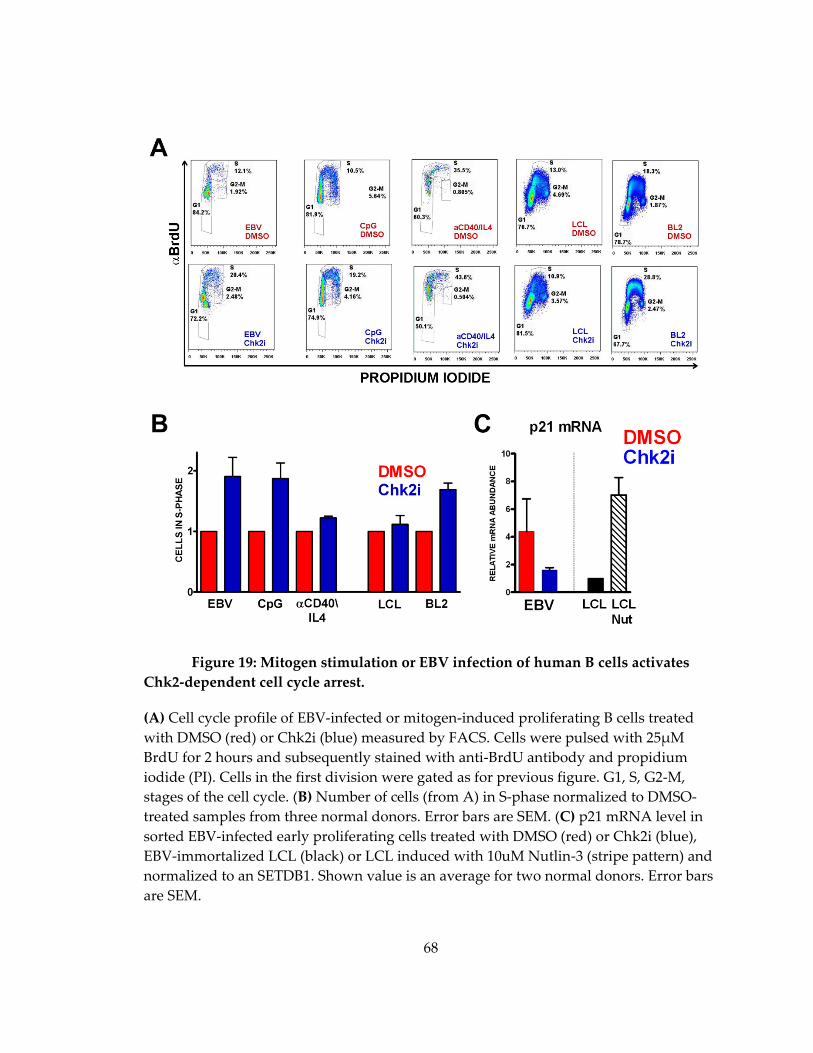
68
Figure 19: Mitogen stimulation or EBV infection of human B cells activates
Chk2-dependent cell cycle arrest.
(A) Cell cycle profile of EBV-infected or mitogen-induced proliferating B cells treated
with DMSO (red) or Chk2i (blue) measured by FACS. Cells were pulsed with 25µM
BrdU for 2 hours and subsequently stained with anti-BrdU antibody and propidium
iodide (PI). Cells in the first division were gated as for previous figure. G1, S, G2-M,
stages of the cell cycle. (B) Number of cells (from A) in S-phase normalized to DMSO-
treated samples from three normal donors. Error bars are SEM. (C) p21 mRNA level in
sorted EBV-infected early proliferating cells treated with DMSO (red) or Chk2i (blue),
EBV-immortalized LCL (black) or LCL induced with 10uM Nutlin-3 (stripe pattern) and
normalized to an SETDB1. Shown value is an average for two normal donors. Error bars
are SEM.

69
2.2.3.7. Activated Chk2 induces expression of the CDK inhibitor p21
As we have observed a Chk2-dependent G1/S phase arrest in EBV-infected or
mitogen-induced normal human B-cells, we next assayed the expression of p53-
dependent effector p21 likely to play a role in limiting the proliferation of human B cells.
We assayed p21 mRNA levels in early proliferating cells treated with DMSO or Chk2i by
qPCR. We found that p21 expression was decreased in Chk2i treated EBV-infected early
proliferating cells by 2-5 folds in two donors (Fig. 19C) to the level of LCL. As a positive
control we used previously reported Nutlin-3 treatment of LCL, that upregulates the p21
level (Forte and Luftig, 2009).
2.2.4. Discussion
We identified a Chk2-dependent G1/S phase cell cycle checkpoint as a limit to
mitogen-induced proliferation of human B-cells. The activated checkpoint was
uncoupled from apoptosis in mitogen-induced or EBV-infected B-cells. Notably, the
pharmacological inhibition of Chk2 increased the number of dividing cells, but did not
change the time to first division nor did it affect the rate of cellular division. Instead,
Chk2 inhibition of EBV-infected early proliferating cells resulted in decreased expression
of G1/S checkpoint effector p21.
EBV infection activates the DDR-dependent G1/S cell cycle arrest and
immortalizes B cells with inactivated p16. Consistent with previously reported findings
(Nikitin et al., 2010), we observed DDR–dependent growth suppression of EBV-driven

70
proliferation of B cells. Surprisingly, we did not detect apoptosis in dividing cells; rather
we observed the DDR-dependent expression of p21 and induction of G1/S phase cell
cycle arrest. In addition, we detected the activation of the senescence-associated beta-
galactosidase in arrested, but not in proliferating cells (data not shown).Notably, we did
not detect an active apoptosis in infected cells (Fig. 18). Expression of viral BCL2
homologs likely prevents activation of apoptosis in EBV-infected cells ((Altmann and
Hammerschmidt, 2005) and Micah Luftig and Jay Tourigny, unpublished).
Further, the DDR-dependent G1/S arrest was readily detected in early
proliferating B cells but not in indefinitely transformed LCL, despite the latter retained
thea low level of γH2AX positivity (Nikitin et al., 2010). Recent reports have begun to
address how the initial DNA damage results in the formation of persistent
heterochromatin foci with active DDR signaling (Di Micco et al., 2011; Rodier et al.,
2011). Specifically, (Rodier et al., 2011) identified DNA-SCARS as foci with persistent
ATM-downstream signaling in dividing cells with inactivated p16. EBNA-3C attenuates
the DDR-mediated growth arrest (Nikitin et al., 2010) and induces epigenetic
inactivation of p16 in transformed LCL (Maruo et al., 2011; Skalska et al., 2010).
Therefore, we hypothesize that EBNA2-dependent transcription of host growth
promoting genes, such as c-Myc, initiates the oncogene-induced replicative stress and
activation of ATM/Chk2-dependent G1/S growth arrest of the majority of infected
proliferating cells. Further, EBNA2 driven EBNA3C attenuates the expression of

71
oncogenes and inactivates p16 allowing subsequent cellular proliferation despite the
presence of active ATM signaling.
DNA damage response in mitogen-induced B cells. Chk2-dependent G1/S cell
cycle arrest was detected in the first three cell divisions of CpG-induced or EBV-infected
cells; however αCD40/IL4-induced cells demonstrated the most apparent response to
Chk2 inhibition during the first division only. Considering the observed decline in the
proliferation speed of αCD40/IL4-induced cells (Fig. 15 and BrdU profiles not shown),
we conclude that Chk2 activates G1/S in hyper-proliferating cells in the first division
upon stimulation with this mitogen. Notably, we previously reported similar transient
activation of Chk2 in EBV-infected hyper-proliferating cells that decreased with the slow
proliferation rate (Nikitin et al., 2010).
TLR9 stimulation of B cells activates two independent suppressive
mechanisms. We have demonstrated a bi-phasic profile of human B cell proliferation in
response to TLR9 stimulation with CpG in vitro. Particularly, during the first three
divisions proliferating B cells were free of apoptotic markers and displayed Chk2-
dependent and reversible G1/S cell cycle arrest. However, divisions four and higher
displayed apoptotic markers, including activated Caspases 3 and 7 and cleaved PARP.
Consistent with our findings, CpG-induced dividing murine BCL2 transgenic B cells
remained alive while arrested in higher divisions (Hawkins et al., 2007b). Therefore,
conclude that the two factors regulating TLR9-induced proliferation of human B-cells in

72
vitro (Markham et al., 2010) are, first, a DDR-dependent G1/S cell cycle arrest and,
second, a DDR-independent proliferation associated apoptosis.
Implications in vivo. TLR9 ligand stimulation, but not αCD40/IL4 treatment,
requires DOCK8 (Jabara et al., 2012), a guanine nucleotide exchange factor that activates
Cdc42 and promotes cellular proliferation (Cote and Vuori, 2002; Harada et al., 2012). In
a recently proposed model Goodnow et al. suggest that DOCK8 upregulates MDM2
through the activation of Akt and therefore silence p53 activity in the germinal center B
cells (reviewed in (Goodnow et al., 2010)). However, DDR signaling activates ATM and
Chk2 which in turn stabilize p53 by direct phosphorylations on Ser 15 and 20 (Hirao et
al., 2000). Strikingly, the ATR/Chk1 shoulder of the DDR is potently down-regulated,
while the expression of pro-survival BCL6 is upregulated in germinal center B cells
(Ranuncolo et al., 2007). Therefore, degraded p53 and down-regulated ATR likely
protect hyper-proliferating B-cells from cell cycle arrest in germinal center, while G1/S
checkpoint may restrict an extra-follicular proliferation of B cells.
Overall, we have identified a Chk2-dependent growth arrest upon EBV-driven or
mitogen-induced proliferation of B-cells in the absence of T cells. Chk2 kinase inhibition
resulted in increased DNA synthesis, but did not affect the induction of apoptosis. We
therefore conclude that Chk2-dependent DDR may limit extrafollicular proliferation of
human B cells.

73
2.3. Enhanced Method of Epstein-Barr Virus Mediated Transformation of B Cells Used for Generation of Human Monoclonal Antibodies
2.3.1. Contributions
The following section of Results has been presented at CFAR retreat by P.N. and
included in patent application (Kwan-Ki Hwang, 2009). Micah Luftig, Barton Haynes
initiated and designed the project. P.N., M.L. and Kwan-Ki Hwang designed the
experimental approach, and P.N. and K.H. performed experiments.
2.3.2. Introduction
Broadly neutralizing monoclonal antibody (BnAB) therapy is a novel promising
method to control chronic infection and to prevent de novo infection with highly
pathogenic agents. The vastly growing pool of isolated human monoclonal antibodies
includes BnAbs to anthrax (Smith et al., 2009), chikungunya virus (Warter et al., 2011),
cytomegalovirus (Macagno et al., 2010), dengue virus, HIV (reviewed in (Haynes et al.,
2012)), influenza virus (Corti et al., 2010; Krause et al., 2012; Yu et al., 2008) (including
“avian” strain H5N1 (Hu et al., 2012; Simmons et al., 2007)), respiratory syncytial virus
(Collarini et al., 2009), rotavirus (Di Niro et al., 2010; Tian et al., 2008), SARS (Traggiai et
al., 2004) and tetanus (Poulsen et al., 2011) (summarized in (Wilson and Andrews,
2012)).
In the case of infectious agents with low immunogenicity of conserved epitopes,
such as HIV, influenza and HCV, inefficient vaccination fails to provide broad and long-

74
lasting protection (Haynes et al., 2012). For instance, the first limited neutralizing Ab
response to HIV gp120 appears by 16 weeks after transmission (Richman et al., 2003;
Wei et al., 2003). Moreover, only ~5% of chronically HIV-infected patients develop
broadly neutralizing antibodies (BnAB) through a presumably atypical maturation
process that results in BnAB with high affinity to host self-antigens and extensive
somatic hyper-mutation (Haynes et al., 2005; Haynes et al., 2012). Therefore, isolation
and investigation of patient-derived BnAbs not only has the potential to generate
therapeutics, but also to provide information for vaccine design.
Currently, the four most common methods to isolate human nAB, include: (1)
phage display, (2) in vitro proliferation and immortalization of memory B cells, (3)
single B cell expression cloning with antigen baiting and (4) plasmablasts expression
cloning after the exposure to an antigen. However, each method suits different needs
and has limitations. Particularly, selection through a phage display leads to bacterial
processing of resulted heavy or light chain; single B cell expression cloning requires a
known and immunogenic baiting antigen; while a single-cell expression cloning of
plasmablasts transiently and briefly detected after the antigen exposure (Wilson and
Andrews, 2012). Finally, the immortalization of memory B cells, usually with EBV, has
long been used as an option to immortalize antibody-secreting cells (Kozbor and Roder,
1981; Steinitz et al., 1977). However, as discussed in previous Chapters, the efficiency of
viral transformation remains below 1-10%. Introduction of B-cell mitogens, such as the

75
TLR9 ligand CpG, increased viral transformation (Iskra et al., 2010; Traggiai et al., 2004).
Particularly, this approach was used to isolate BnAB against SARS coronavirus, H5N1
“avian” influenza virus, and dengue virus from convalescent patients as well as recently
various BnAB to HIV from chronic HIV-infected individuals (Friberg et al., 2012; Hicar
et al., 2010; Hu et al., 2012; Simmons et al., 2007; Traggiai et al., 2004).
In the two previous chapters we identified the DDR as one of the major growth-
suppressive mechanisms to control EBV-driven or CpG-induced proliferation of human
B cells. Therefore, we reasoned that the inhibition of key kinases ATM and Chk2 would
facilitate the isolation of patient-derived human monoclonal antibodies following EBV
infection of memory B cells.
2.3.3. Results
2.3.3.1. Pharmacological inhibition of ATM or Chk2 increases EBV-mediated
transformation of low numbers of B cells
We first identified the minimal number of total primary B cells sufficient to
achieve long-term outgrowth in a single well of a 96-well plate following EBV infection.
To provide supportive growth signals we incubated EBV-infected B cells with irradiated
feeder cells, including autologous PBMC from the same donor or mouse macrophages
J774.A1. We plated EBV-infected total B cells at 1-1000 cells per well density. Five weeks
post infection we detected EBV-transformed clones in 10% of wells plated with 30
cells/well and in all wells plated with 1000 cells/well (Fig. 20, black circles).
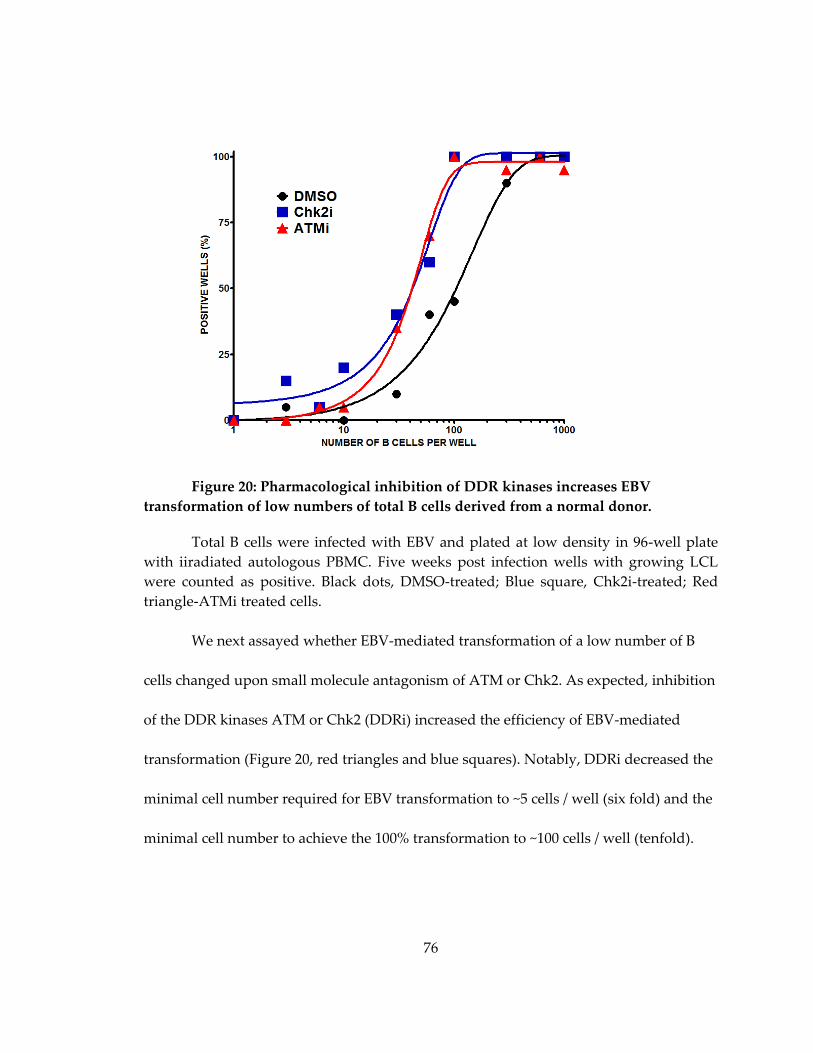
76
Figure 20: Pharmacological inhibition of DDR kinases increases EBV
transformation of low numbers of total B cells derived from a normal donor.
Total B cells were infected with EBV and plated at low density in 96-well plate
with iiradiated autologous PBMC. Five weeks post infection wells with growing LCL
were counted as positive. Black dots, DMSO-treated; Blue square, Chk2i-treated; Red
triangle-ATMi treated cells.
We next assayed whether EBV-mediated transformation of a low number of B
cells changed upon small molecule antagonism of ATM or Chk2. As expected, inhibition
of the DDR kinases ATM or Chk2 (DDRi) increased the efficiency of EBV-mediated
transformation (Figure 20, red triangles and blue squares). Notably, DDRi decreased the
minimal cell number required for EBV transformation to ~5 cells / well (six fold) and the
minimal cell number to achieve the 100% transformation to ~100 cells / well (tenfold).

77
2.3.3.2. Inhibition of Chk2 coupled with TLR9 stimulation additively enhances EBV-
mediated transformation of memory B cells from normal donors
We aimed to improve the EBV-mediated immortalization of IgG-producing cells.
We next focused on memory B cells derived from a normal donor. Previously, TLR9
stimulation with CpG DNA was shown to significantly improve EBV-transformation
efficiency (Traggiai et al., 2004). Therefore, we assayed EBV-transformation efficiency in
the presence of CpG and Chk2 inhibitor. We sorted CD19hi CD27hi IgDlo memory B cells
from a normal donor (Fig. 21, left) and infected them with EBV.
Figure 21: Chk2 inhibitor and TLR ligand CpG additively increase EBV-
mediated transformation of human memory B cells.
Left. CD19 B cells were sorted from a marked gate by FACS. Right. Sorted
CD19+ CD27+IgD- memory B cells were EBV infected and treated with DMSO (black),
CpG (blue), Chk2i (brown) or CpG combined with Chk2 (red) and plated at 300
cells/well density in 96-well plates over irradiate mouse macrophages. Five weeks later
the percent of positive wells was determined as in previous figure.

78
Both CpG and Chk2i treatment increased the EBV transformation of memory B
cells plated at 300 cells per well. EBV-infection or TLR9 stimulation of B cells activates
cell cycle arrest (Chapter. 2.2); however, whether the combined stimulation may
overcome cell cycle arrest remained unknown. We found that the combined treatment
additively elevated EBV transformation efficiency from 70% (EBV+CpG) to 90%
(EBV+CpG+Chk2i) (Fig. 21, right panel). We thus referred to CpG and Chk2i dual
treatment as an enhanced EBV transformation method.
2.3.3.3. Combined treatment with Chk2i and TLR9 ligand increases EBV-mediated
transformation of memory B cells from a chronic HIV-infected patient
As mentioned above, the efficient selection and immortalization of BnAB-
producing memory B cells will facilitate the production of Ig-based therapeutics and
inform vaccine design. In order to identify such BnAB it is important to identify patients
that have recovered or chronically infected with a pathogen. Therefore, we tested our
method of enhanced EBV transformation on B cells derived from a HIV chronically -
infected patient in collaboration with Bart Haynes group at the Duke Human Vaccine
Institute
Thawed PBMC from the chronic HIV patient were FACS sorted as shown above
and infected with EBV in the presence of TLR9 ligand CpG. Five weeks after EBV
infection we observed transformation in ~20% of wells plated with 3000 PBMC and in
~25% of wells plated with 100 memory B cells (Fig. 22, blue bars).

79
Figure 22: Enhanced EBV transformation of memory B cells derived from a
chronic HIV patient.
Frozen PBMC sample from CHAVI’s chronic HIV patient was thawed, FACS
sorted as for previous figure and EBV infected. Unsorted PBMC and sorted memory B
cells were treated with CpG (blue bars) or CpG combined with Chk2i (red bars) and
plated at 3000 cells per well and 100 cells per well density over irradiated macrophages
in 96-well plate. Transformation was determined as in previous figures. c/w, cells per
well.
Strikingly, upon inhibition of Chk2 EBV transformation efficiency increased to
~60% for PBMC (three fold over CpG alone) and to ~55% (two fold over CpG alone) for
Memory B cells.

80
2.3.4. Discussion
We have identified the DNA damage response as a growth suppressive
mechanism to restrict EBV-mediated transformation of low numbers of total B and
memory B cells. Further, we found that EBV transformation during pharmacological
inhibition of the DDR can be performed with as low as 3 B cells per well. Finally, in a
proof-of-concept experiment we demonstrated that TLR9 stimulation coupled with EBV
infection and inhibition of Chk2 kinase increased transformation of memory B cells
derived from a chronic HIV patient.
The results of our work were included in a patent application filed in 2009
(Kwan-Ki Hwang, 2009). Since that time, the inhibition of the DDR kinase Chk2 (Nikitin
et al., 2010), along with TLR9 stimulation (Traggiai et al., 2004), became a common way
to increase EBV mediated transformation of patient-derived memory B cells.
Particularly, this combined treatment was used to isolate HIV-1 envelope-specific BnAB
and study the V2/V3 conformational epitope presentation mechanism (Bonsignori et al.,
2011), to select BnAB specific to a conserved epitope on influenza H1N1 hemagglutinin
(Krause et al., 2011), to isolate weak nAB and study the antibody-mediated enhancement
of secondary dengue virus infection (Smith et al., 2012) and identify early BnAB to HIV-
1 that induce viral immune escape (Bar et al., 2012).

81
3. Conclusions and future directions
We identified the first tumor suppressive pathway to limit EBV immortalization
of primary human B cells. Particularly, we revealed that a transient, viral-driven
elevated expression of growth control genes induces the cellular hyper-proliferation and
activates the growth suppressive ATM/Chk2-dependent DDR resulting in G1/S cell cycle
arrest in most of early proliferating B cells. However, viral protein EBNA3C down-
regulates the host DDR and promotes immortalization of infected cells into
lymphoblasts.
Our work also provides a new paradigm for EBV infection of primary B cells de
novo, elucidating the intermediate period between resting and transformed cells
happened early in infection. Early proliferating cells display an atypically high level of
EBNA-LP expression, robust proliferation rate and activated hallmarks of the DDR
resulting in p21-mediated G1/S cell cycle arrest.
However, there are several questions remained. We found the activated state of
DDR signaling pathway, but we did not detect DNA damage in sorted proliferating cells
by comet assay (not shown). Therefore, whether initial hyper-proliferation leads to a
replication stress and double-stranded breaks, exposed telomeres or induces a formation
of heterochromatin foci remains unknown. Next question is whether transformed
lymphoblastic cells still contain such heterochromatin foci or any traces of DNA
damage. The rationale for this question is the presence of low, but detectable γH2AX

82
signal in LCL. Further, what is the significance of early proliferating phase in vivo?
Replicative stress and high Myc level may facilitate the mutagenesis and selection for
clones with constantly high Myc expression. This process coupled with negative
selection for EBNA3s due to a high immunogenicity would favor the Myc translocation
under Ig promoter and drives early proliferating cells towards latency I Burkitt’s
lymphoma. Finally, we demonstrated an asynchronous proliferation of infected cells.
Does it reflect heterogeneity of latency proteins expression in different cells? If so, which
cells have higher chance to be transformed into latency III in vitro, and which cells
would give raise to latency I lymphomas in vivo?
3.1. The source of DNA damage
The first question is what activates the DNA damage response. Towards
characterizing the EBV-induced DNA damage signal, we reasoned that either viral or
cellular DNA was important for ATM activation. In addition to oncogenic stress,
replication intermediates of DNA viruses and retroviruses contain double-stranded
DNA ends that activate ATM (Lilley et al., 2007). In fact, EBV lytic replication induces a
DDR which is suppressed by inhibition of downstream transcriptional activation of p53
(Kudoh et al., 2005; Mauser et al., 2002). In our studies of primary B cell infection,
however, we found no evidence of viral lytic DNA replication or viral DNA associated
with DDR activation. First, we did not observe DDR activation within the first 3 days
after EBV infection nor did we observe activation using UV-inactivated virus or the non-

83
transforming EBV variant P3HR1 suggesting that the incoming linear DNA genome and
tegument proteins within the virion were not responsible for this signal. Second, lytic
viral DNA was not responsible for DDR activation as less than 1% of infected cells were
undergoing lytic DNA replication when greater than 50% of infected cells were -H2AX
positive. Third, we asked whether the DNA damage signal was derived from viral
episomes since DNA repair factors are recruited to the episome to ensure proper
resolution of Holliday junctions following episome replication (Deng et al., 2002;
Dheekollu et al., 2007). We observed little increase in episome number per cell and
found that viral episomes and -H2AX did not co-localize during the period of DDR
activation. These data collectively demonstrate that viral DNA is not the source of DNA
damage. Our experiments cannot rule out the possibility, however, that viral lytic gene
expression downstream of BZLF1 in the absence of lytic DNA replication (Kalla et al.,
2010) plays a role in the transient DDR early after infection. Despite this possibility, we
inferred from our data that viral latent gene expression causes an oncogenic stress
response leading to cellular DNA damage.
The initiation of cell proliferation defines the period after EBV infection when
ATM and Chk2 were active in suppressing transformation. Rigorous analysis of infected
cell division rates uncovered a period of hyper-proliferation where early population
doublings (PDs) were every 8-12h leading to DDR activation, while later divisions
displayed an attenuated rate of ~24-30h per division similar to LCLs and had little

84
evidence of DDR activation. Microarray analysis of gene expression during the
transition from resting B cell to early EBV-induced hyper-proliferation and through LCL
outgrowth strongly supported our cell-based observations. Specifically, genes involved
in proliferation and the DDR, including ATM/p53-dependent targets (Elkon et al., 2005),
were highly induced early after infection and then attenuated during the transition to
LCL. We propose that aberrant induction of cellular DNA replication early after EBV
infection activates a DNA damage response that is dependent on EBNA2 and EBNA-LP
mediated up-regulation of S phase promoting oncoproteins including c-Myc, cyclin D2,
and E2F1 ((Kaiser et al., 1999; Sinclair et al., 1994) and Fig. 11). Indeed, we observed
increased expression of c-Myc and its gene activation signature in hyper-proliferating
cells relative to LCLs. Furthermore, EBNA-LP protein levels and Wp-derived transcripts
were heightened during this early period relative to EBNA3 proteins and Cp transcripts
consistent with previous analysis of the initial cascade of viral latent gene expression at
different days post infection (Schlager et al., 1996; Woisetschlaeger et al., 1989;
Woisetschlaeger et al., 1990). Finally, EBNA3C, but not EBNA3A, deleted virus-infected
cells displayed a significantly stronger DDR during early proliferation. Thus, while both
EBNA3A and EBNA3C likely mitigate growth arrest in LCLs through p16 suppression
(Hertle et al., 2009; Skalska et al., 2010), during early outgrowth EBNA3C is also
required to modulate the DNA damage response. Collectively, our data support a model
that initial EBV-driven hyper-proliferation leads to an oncogenic stress that is ultimately

85
attenuated as EBNA3 proteins moderate EBNA2 driven c-Myc and its genotoxic and
growth suppressive consequences. This ultimate balance in viral and host gene
expression enables constitutive S phase induction without driving selection of cells with
genomic instability.
Furthermore, in our system, we did not observe increased transformation in the
presence of anti-oxidants including N-acetyl cysteine and citric acid (P.N. and M.L. data
not shown) suggesting that ROS was not responsible for the EBV-induced DDR. We also
did not observe changes in BubR1 or ATM expression through LCL outgrowth (data not
shown). However, we anticipate that genomic instability may ensue in the setting of
aberrant latent oncoprotein expression that may exist in BL and other EBV-associated
tumors. Consistent with this notion and our findings, a recent report suggests that while
LCLs maintain a stable karyotype, early DNA damaging events may lead to non-clonal
chromosomal aberrations including telomere fusions (Lacoste et al., 2009). This report
supports our findings of an early hyper-proliferation associated oncogenic stress that
may induce such structures leading to ATM activation (Karlseder et al., 1999) and
suppression of long-term outgrowth. Thus, only cells with the ability to maintain a
stable karyotype emerge as LCLs.
3.2. DDR signaling in transformed cells
Second question is why EBV-transformed LCL display a residual level of γH2AX
positivity. EBV infection of immortalized lymphoma cells or modulation of expression

86
of viral latent genes in LCL revealed different aspects of viral interaction with the DDR
(summarized in Chapter 1.4). However, we did not observe overt genomic aberrations in
LCL that constitutively express nine viral latent proteins at the physiological level.
Furthermore, while LCL displayed low levels of DDR hallmarks such as γH2AX, we did
not detect the downstream consequences of the DDR activation, such as cell cycle arrest
or apoptosis (Fig. 18 and 19). In contrast, Burkitt’s lymphoma-derived cell line BL2 that
has a wild type p53 readily displayed a Chk2-dependent G1/S cell cycle arrest (Fig. 19).
Observed residual level of γH2AX activation in LCL may reflect the persistent
chromatin changes formed upon DNA damage (Di Micco et al., 2011; Rodier et al., 2011).
Further, DDR-dependent heterochromatin foci display ATM-downstream signaling and
growth arrest in p16-intact cells, but continue to proliferate in the absence of p16 (Rodier
et al., 2011). Intriguingly, in EBV-transformed LCL viral protein EBNA3C epigenetically
represses p16 (Maruo et al., 2011; Skalska et al., 2010). Therefore, we suggest that EBV-
driven cellular hyper-proliferation causes DNA damage and induces formation of
persistent heterochromatin foci with localized ATM signaling. However, EBNA3C
affects the cause of DDR and mediates cell cycle checkpoint through attenuation of the
cellular proliferation and repression of p16.
3.3. Implication in vivo
The third question is how early proliferating cells progress in vivo? Our findings
of an intermediate state early in infection has implications to the germinal center model

87
for EBV infection (Roughan and Thorley-Lawson, 2009) in the context of B cell
lymphomagenesis. In particular, our observed hyper-proliferative phase early after
infection in vitro may be similarly induced by EBV in vivo and is reminiscent of B cell
proliferation rates in the germinal center (MacLennan, 1994). However, there are
important differences between B cell proliferation in germinal center and due to EBV
infection. First, Bcl-6 down-regulation of the DDR mitigates the consequences of
centroblast hyper-proliferation in the germinal center (Ranuncolo et al., 2007), while
EBV potently suppresses Bcl-6 early after infection leaving DDR checkpoints intact
(Siemer et al., 2008). Second, high MDM2 expression in GC cells silences p53 (Goodnow
et al., 2010), while viral protein EBNA3C attenuates DDR later in infection that prevents
p53 activation (Nikitin et al., 2010). Therefore, EBV-infected B cells may not only migrate
into GCs as suggested previously (Thorley-Lawson and Allday, 2008), but may
additionally promote extra-follicular B cell maturation by-passing the germinal center
(Heath et al., 2012). However, a critical balance must be struck between the aberrant
latent oncoprotein-driven proliferation early after infection and the stable proliferative
signals found in LCLs to maintain an activated, immortalized state. Perturbations in this
balance in vivo may select for mutations driving lymphomagenesis. For example, the
IgH/c-myc translocation common in Burkitt’s lymphoma (BL) may be the consequence of
high EBNA-LP expression in the absence of EBNA3C. Given our findings, it is plausible
that imbalances in EBV latent gene expression may provide a milieu of cells with an

88
increased potential for genomic instability. Recent work in BL cell lines suggests that this
is likely the case.
3.4. Expression of viral latency proteins in early proliferating B cell
Finally, we investigated a bulk population of proliferating cells, whether arrested
or continued to proliferate. However, whether EBV latency proteins are expressed at
different levels in different cells is poorly understood. While we demonstrated dynamic
changes in EBNA-LP to EBNA3s expression ratio, we have not investigated the
abundance of these viral proteins in cells that either entered a cell cycle and arrested or
continued to proliferate. Therefore, the key question to be addressed is whether EBV
infection generates distinct subsets of cells with different viral gene expression and thus, potency
to induce oncogenesis.
3.4.1. How is EBNA3C activity regulated in infected cells?
We and others demonstrated the role of EBNA3C in deactivating host tumor
suppression. Particularly, EBNA3C downregulates the DDR (Nikitin et al., 2010) and
epigenetically inactivates p16 (Maruo et al., 2011; Skalska et al., 2010). However, the
expression pattern of EBNA3C per single cell remains unknown. Western blot analysis
detected two isoforms of EBNA3C in early proliferating cells, but not in LCL (Nikitin et
al., 2010). Further, our group revealed host genome-wide alterations in alternative exon
isoform usage in infected cells ((Robinson et al., 2012) and Nicholas Homa and M.L. in
preparation). The genome-wide alternations in initiation and splicing found in EBV

89
transformed cells suggest fundamental changes in mRNA processing machinery
affecting both host and viral gene expression early and late in B cell infection. Therefore,
the functional activity of EBNA3C is likely attributed to a particular isoform expressed
in a given cell, which controls the survival of this cell.
3.4.2. What is the role of LMP1 in restricting the DDR and why is its expression is delayed?
LMP1 expression is delayed in infected B cells in vitro (Price et al., 2012). Notably,
LMP1 expression during latency III is controlled by its own promoter and depends on
EBNA2 and EBNA3C activity (discussed in introduction). Therefore, expression of
LMP1 likely depends on progression through cell divisions and occurs in cells that have
already escaped the DDR-imposed growth arrest, i.e. in cells expressing an active
EBNA3C with attenuated c-Myc level and inactivated p16. LMP1 exclusive expression in
cells with active EBNA3C may particularly explain the stable karyotype in LCL, as only
cells experienced low oncogenic stress progress far enough to receive LMP1-mediated
survival stimuli. Finally, such division-linked regulation may prevent the competition
between DDR-induced p53 and LMP1-driven NF-kB downstream signaling (reviewed
by (Perkins, 2012)) and provide the required survival stimulus to cycling cells.
3.4.3. How EBNA-LP length and expression control EBV-driven oncogenesis?
EBNA-LP co-operates in transcriptional activation of EBNA2 target genes
(Rickinson and Kieff, 2006). We detected a transient up-regulation of EBNA-LP

90
expression, EBNA2 activity and c-Myc mRNA levels in early proliferating infected cells.
Furthermore, and consistent with previous work, we detected up to nine EBNA-LP
isoforms due to the initiation from or splicing through a variable number of W repeats
used early in infection, followed by selection of 1-2 isoforms in transformed cells.
Intriguingly, the number of W repeats found in vivo determines EBV transformation
efficiency (Tierney et al., 2011). Therefore, it is highly possible that newly infected B cells
in vivo express EBNA-LP that contain different combinations of repeats, and therefore
display variable levels of EBNA2 activity. Further, the heterogeneity EBNA-LP/EBNA2
activity results in subsets of cells expressing different levels of growth control genes,
such as c-Myc, and therefore experiencing different extend of oncogenic stress. Finally,
subsets of cells with different levels of expressed oncogenes are likely a basis for
mutagenesis, such as c-Myc translocation under Ig promoter, found in Burkitt’s
lymphomas.
3.5. A proposed model of EBV infection of primary B cells
In summary, I propose that EBV infection results in subsets of B cells expressing
atypical combinations of latent proteins and their isoforms. Therefore, instead of the
linear sequence of events upon infection, schematically written as:
EBNA-LPEBNA2 activityEBNA3CLMP1 transformation
viral infection results in early proliferating cells expressing combinations of proteins :
(EBNA-LPW1-9 and EBNA2 and/or EBNA3Con/off)LMP1early/late

91
Further, considering the tumor suppressive role of EBNA3B (White et al., 2012), the
subset of early proliferating EBV infected cells is likely described by the following
schematic:
[(EBNA-LPW1-9 and EBNA2 and/or (EBNA3Con/off + EBNA3Bon/off)]LMP1early/late.
In addition, each infected cell contains EBNA1 and may contain EBNA3A and LMP2s.
Finally, EBV infected early proliferated cells may express different subsets of viral non-
coding RNAs (not discussed in this dissertation, but emerge as important regulators of
viral latency (Skalsky et al., 2012)).
Hence, viral infection results in subsets of cells with various levels of EBNA-LP
that determines the oncogenic potential of this cell, EBNA3C that promotes
transformation but induces the strong cytotoxic immune response and LMP-1 that likely
is expressed later in infection and provides the required survival stimuli. Each latency
variant expressed in early proliferating B cells may be beneficial in different conditions,
such as immunosuppression, environmental stress or additional pathogens infection.
Overall, EBV-infected early proliferating B cells likely consist of multiple
populations expressing different levels of latency components and provide a broad
range of variants for in vivo selection by the host immune response and cell-intrinsic
growth suppression.

92
References
Alfieri, C., Birkenbach, M., and Kieff, E. (1991). Early events in Epstein-Barr virus
infection of human B lymphocytes. Virology 181, 595-608.
Altmann, M., and Hammerschmidt, W. (2005). Epstein-Barr virus provides a new
paradigm: a requirement for the immediate inhibition of apoptosis. PLoS biology 3,
e404.
Anderton, E., Yee, J., Smith, P., Crook, T., White, R.E., and Allday, M.J. (2008). Two
Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the
proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt's lymphoma.
Oncogene 27, 421-433.
Arienti, K.L., Brunmark, A., Axe, F.U., McClure, K., Lee, A., Blevitt, J., Neff, D.K.,
Huang, L., Crawford, S., Pandit, C.R., et al. (2005). Checkpoint kinase inhibitors: SAR
and radioprotective properties of a series of 2-arylbenzimidazoles. J Med Chem 48, 1873-
1885.
Ariumi, Y., Kaida, A., Lin, J.Y., Hirota, M., Masui, O., Yamaoka, S., Taya, Y., and
Shimotohno, K. (2000). HTLV-1 tax oncoprotein represses the p53-mediated trans-
activation function through coactivator CBP sequestration. Oncogene 19, 1491-1499.
Arrand, J.R., and Rymo, L. (1982). Characterization of the major Epstein-Barr virus-
specific RNA in Burkitt lymphoma-derived cells. Journal of virology 41, 376-389.
Babcock, G.J., Decker, L.L., Volk, M., and Thorley-Lawson, D.A. (1998). EBV persistence
in memory B cells in vivo. Immunity 9, 395-404.
Bar, K.J., Tsao, C.Y., Iyer, S.S., Decker, J.M., Yang, Y., Bonsignori, M., Chen, X., Hwang,
K.K., Montefiori, D.C., Liao, H.X., et al. (2012). Early low-titer neutralizing antibodies
impede HIV-1 replication and select for virus escape. PLoS Pathog 8, e1002721.

93
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., Guldberg, P., Sehested,
M., Nesland, J.M., Lukas, C., et al. (2005). DNA damage response as a candidate anti-
cancer barrier in early human tumorigenesis. Nature 434, 864-870.
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., Vassiliou,
L.V., Kolettas, E., Niforou, K., Zoumpourlis, V.C., et al. (2006). Oncogene-induced
senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints.
Nature 444, 633-637.
Belgnaoui, S.M., Fryrear, K.A., Nyalwidhe, J.O., Guo, X., and Semmes, O.J. (2010). The
viral oncoprotein tax sequesters DNA damage response factors by tethering MDC1 to
chromatin. The Journal of biological chemistry 285, 32897-32905.
Bell, A.I., Groves, K., Kelly, G.L., Croom-Carter, D., Hui, E., Chan, A.T., and Rickinson,
A.B. (2006). Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt's
lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time
PCR assays. The Journal of general virology 87, 2885-2890.
Bester, A.C., Roniger, M., Oren, Y.S., Im, M.M., Sarni, D., Chaoat, M., Bensimon, A.,
Zamir, G., Shewach, D.S., and Kerem, B. (2011). Nucleotide deficiency promotes
genomic instability in early stages of cancer development. Cell 145, 435-446.
Bild, A.H., Yao, G., Chang, J.T., Wang, Q., Potti, A., Chasse, D., Joshi, M.B., Harpole, D.,
Lancaster, J.M., Berchuck, A., et al. (2006). Oncogenic pathway signatures in human
cancers as a guide to targeted therapies. Nature 439, 353-357.
Boichuk, S., Hu, L., Hein, J., and Gjoerup, O.V. (2010). Multiple DNA damage signaling
and repair pathways deregulated by simian virus 40 large T antigen. Journal of virology
84, 8007-8020.
Bonsignori, M., Hwang, K.K., Chen, X., Tsao, C.Y., Morris, L., Gray, E., Marshall, D.J.,
Crump, J.A., Kapiga, S.H., Sam, N.E., et al. (2011). Analysis of a clonal lineage of HIV-1
envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and
their inferred unmutated common ancestors. Journal of virology 85, 9998-10009.

94
Borel, J.F., Feurer, C., Magnee, C., and Stahelin, H. (1977). Effects of the new anti-
lymphocytic peptide cyclosporin A in animals. Immunology 32, 1017-1025.
Bouvard, V., Baan, R., Straif, K., Grosse, Y., Secretan, B., El Ghissassi, F., Benbrahim-
Tallaa, L., Guha, N., Freeman, C., Galichet, L., et al. (2009). A review of human
carcinogens--Part B: biological agents. Lancet Oncol 10, 321-322.
Brooks, L., Yao, Q.Y., Rickinson, A.B., and Young, L.S. (1992). Epstein-Barr virus latent
gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1,
and LMP2 transcripts. Journal of virology 66, 2689-2697.
Cai, X., Schafer, A., Lu, S., Bilello, J.P., Desrosiers, R.C., Edwards, R., Raab-Traub, N.,
and Cullen, B.R. (2006). Epstein-Barr virus microRNAs are evolutionarily conserved and
differentially expressed. PLoS pathogens 2, e23.
Chandhasin, C., Ducu, R.I., Berkovich, E., Kastan, M.B., and Marriott, S.J. (2008). Human
T-cell leukemia virus type 1 tax attenuates the ATM-mediated cellular DNA damage
response. Journal of virology 82, 6952-6961.
Chang, J.T., and Nevins, J.R. (2006). GATHER: a systems approach to interpreting
genomic signatures. Bioinformatics 22, 2926-2933.
Chang, M.H., Ng, C.K., Lin, Y.J., Liang, C.L., Chung, P.J., ChenMl, Tyan, Y.S., Hsu, C.Y.,
Shu, C.H., and Chang, Y.S. (1997). Identification of a promoter for the latent membrane
protein 1 gene of Epstein-Barr virus that is specifically activated in human epithelial
cells. DNA and cell biology 16, 829-837.
Chen, W., Hilton, I.B., Staudt, M.R., Burd, C.E., and Dittmer, D.P. (2010). Distinct p53,
p53:LANA, and LANA complexes in Kaposi's Sarcoma--associated Herpesvirus
Lymphomas. Journal of virology 84, 3898-3908.
Cheng, S., Schmidt-Grimminger, D.C., Murant, T., Broker, T.R., and Chow, L.T. (1995).
Differentiation-dependent up-regulation of the human papillomavirus E7 gene
reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes
Dev 9, 2335-2349.

95
Choudhuri, T., Verma, S.C., Lan, K., Murakami, M., and Robertson, E.S. (2007). The
ATM/ATR signaling effector Chk2 is targeted by Epstein-Barr virus nuclear antigen 3C
to release the G2/M cell cycle block. Journal of virology 81, 6718-6730.
Christophorou, M.A., Ringshausen, I., Finch, A.J., Swigart, L.B., and Evan, G.I. (2006).
The pathological response to DNA damage does not contribute to p53-mediated tumour
suppression. Nature 443, 214-217.
Clark, E.A., and Ledbetter, J.A. (1986). Activation of human B cells mediated through
two distinct cell surface differentiation antigens, Bp35 and Bp50. Proceedings of the
National Academy of Sciences of the United States of America 83, 4494-4498.
Collarini, E.J., Lee, F.E., Foord, O., Park, M., Sperinde, G., Wu, H., Harriman, W.D.,
Carroll, S.F., Ellsworth, S.L., Anderson, L.J., et al. (2009). Potent high-affinity antibodies
for treatment and prophylaxis of respiratory syncytial virus derived from B cells of
infected patients. J Immunol 183, 6338-6345.
Corti, D., Suguitan, A.L., Jr., Pinna, D., Silacci, C., Fernandez-Rodriguez, B.M., Vanzetta,
F., Santos, C., Luke, C.J., Torres-Velez, F.J., Temperton, N.J., et al. (2010). Heterosubtypic
neutralizing antibodies are produced by individuals immunized with a seasonal
influenza vaccine. The Journal of clinical investigation 120, 1663-1673.
Cote, J.F., and Vuori, K. (2002). Identification of an evolutionarily conserved superfamily
of DOCK180-related proteins with guanine nucleotide exchange activity. Journal of cell
science 115, 4901-4913.
Cotsiki, M., Lock, R.L., Cheng, Y., Williams, G.L., Zhao, J., Perera, D., Freire, R.,
Entwistle, A., Golemis, E.A., Roberts, T.M., et al. (2004). Simian virus 40 large T antigen
targets the spindle assembly checkpoint protein Bub1. Proceedings of the National
Academy of Sciences of the United States of America 101, 947-952.
Cunningham-Rundles, C., Radigan, L., Knight, A.K., Zhang, L., Bauer, L., and
Nakazawa, A. (2006). TLR9 activation is defective in common variable immune
deficiency. J Immunol 176, 1978-1987.

96
DeCaprio, J.A., Ludlow, J.W., Figge, J., Shew, J.Y., Huang, C.M., Lee, W.H., Marsilio, E.,
Paucha, E., and Livingston, D.M. (1988). SV40 large tumor antigen forms a specific
complex with the product of the retinoblastoma susceptibility gene. Cell 54, 275-283.
Deng, Z., Lezina, L., Chen, C.J., Shtivelband, S., So, W., and Lieberman, P.M. (2002).
Telomeric proteins regulate episomal maintenance of Epstein-Barr virus origin of
plasmid replication. Molecular cell 9, 493-503.
Derheimer, F.A., and Kastan, M.B. (2010). Multiple roles of ATM in monitoring and
maintaining DNA integrity. FEBS letters 584, 3675-3681.
Dheekollu, J., Deng, Z., Wiedmer, A., Weitzman, M.D., and Lieberman, P.M. (2007). A
role for MRE11, NBS1, and recombination junctions in replication and stable
maintenance of EBV episomes. PLoS One 2, e1257.
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., Schurra, C.,
Garre, M., Nuciforo, P.G., Bensimon, A., et al. (2006). Oncogene-induced senescence is a
DNA damage response triggered by DNA hyper-replication. Nature 444, 638-642.
Di Micco, R., Sulli, G., Dobreva, M., Liontos, M., Botrugno, O.A., Gargiulo, G., dal Zuffo,
R., Matti, V., d'Ario, G., Montani, E., et al. (2011). Interplay between oncogene-induced
DNA damage response and heterochromatin in senescence and cancer. Nature cell
biology 13, 292-302.
Di Niro, R., Mesin, L., Raki, M., Zheng, N.Y., Lund-Johansen, F., Lundin, K.E.,
Charpilienne, A., Poncet, D., Wilson, P.C., and Sollid, L.M. (2010). Rapid generation of
rotavirus-specific human monoclonal antibodies from small-intestinal mucosa. J
Immunol 185, 5377-5383.
DiTullio, R.A., Jr., Mochan, T.A., Venere, M., Bartkova, J., Sehested, M., Bartek, J., and
Halazonetis, T.D. (2002). 53BP1 functions in an ATM-dependent checkpoint pathway
that is constitutively activated in human cancer. Nature cell biology 4, 998-1002.
Duensing, S., Lee, L.Y., Duensing, A., Basile, J., Piboonniyom, S., Gonzalez, S., Crum,
C.P., and Munger, K. (2000). The human papillomavirus type 16 E6 and E7 oncoproteins

97
cooperate to induce mitotic defects and genomic instability by uncoupling centrosome
duplication from the cell division cycle. Proceedings of the National Academy of
Sciences of the United States of America 97, 10002-10007.
Durkin, S.S., Guo, X., Fryrear, K.A., Mihaylova, V.T., Gupta, S.K., Belgnaoui, S.M.,
Haoudi, A., Kupfer, G.M., and Semmes, O.J. (2008). HTLV-1 Tax oncoprotein subverts
the cellular DNA damage response via binding to DNA-dependent protein kinase. The
Journal of biological chemistry 283, 36311-36320.
Dyson, N., Howley, P.M., Munger, K., and Harlow, E. (1989). The human papilloma
virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science (New
York, NY 243, 934-937.
Efeyan, A., Garcia-Cao, I., Herranz, D., Velasco-Miguel, S., and Serrano, M. (2006).
Tumour biology: Policing of oncogene activity by p53. Nature 443, 159.
Elkon, R., Rashi-Elkeles, S., Lerenthal, Y., Linhart, C., Tenne, T., Amariglio, N., Rechavi,
G., Shamir, R., and Shiloh, Y. (2005). Dissection of a DNA-damage-induced
transcriptional network using a combination of microarrays, RNA interference and
computational promoter analysis. Genome Biol 6, R43.
Elmore, L.W., Hancock, A.R., Chang, S.F., Wang, X.W., Chang, S., Callahan, C.P., Geller,
D.A., Will, H., and Harris, C.C. (1997). Hepatitis B virus X protein and p53 tumor
suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci U S A 94,
14707-14712.
Fagarasan, S., and Honjo, T. (2000). T-Independent immune response: new aspects of B
cell biology. Science (New York, NY 290, 89-92.
Falck, J., Mailand, N., Syljuasen, R.G., Bartek, J., and Lukas, J. (2001). The ATM-Chk2-
Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410,
842-847.

98
Fennewald, S., van Santen, V., and Kieff, E. (1984). Nucleotide sequence of an mRNA
transcribed in latent growth-transforming virus infection indicates that it may encode a
membrane protein. Journal of virology 51, 411-419.
Forte, E., and Luftig, M.A. (2009). MDM2-dependent inhibition of p53 is required for
Epstein-Barr virus B-cell growth transformation and infected-cell survival. Journal of
virology 83, 2491-2499.
Forte, E., and Luftig, M.A. (2011). The role of microRNAs in Epstein-Barr virus latency
and lytic reactivation. Microbes and infection / Institut Pasteur 13, 1156-1167.
Friberg, H., Jaiswal, S., West, K., O'Ketch, M., Rothman, A.L., and Mathew, A. (2012).
Analysis of human monoclonal antibodies generated by dengue virus-specific memory
B cells. Viral immunology 25, 348-359.
Friborg, J., Jr., Kong, W., Hottiger, M.O., and Nabel, G.J. (1999). p53 inhibition by the
LANA protein of KSHV protects against cell death. Nature 402, 889-894.
Goodnow, C.C., Vinuesa, C.G., Randall, K.L., Mackay, F., and Brink, R. (2010). Control
systems and decision making for antibody production. Nature immunology 11, 681-688.
Gorgoulis, V.G., Vassiliou, L.V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T.,
Venere, M., Ditullio, R.A., Jr., Kastrinakis, N.G., Levy, B., et al. (2005). Activation of the
DNA damage checkpoint and genomic instability in human precancerous lesions.
Nature 434, 907-913.
Greenwell, P.W., Kronmal, S.L., Porter, S.E., Gassenhuber, J., Obermaier, B., and Petes,
T.D. (1995). TEL1, a gene involved in controlling telomere length in S. cerevisiae, is
homologous to the human ataxia telangiectasia gene. Cell 82, 823-829.
Gruhne, B., Sompallae, R., Marescotti, D., Kamranvar, S.A., Gastaldello, S., and Masucci,
M.G. (2009a). The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via
induction of reactive oxygen species. Proceedings of the National Academy of Sciences
of the United States of America 106, 2313-2318.

99
Gruhne, B., Sompallae, R., and Masucci, M.G. (2009b). Three Epstein-Barr virus latency
proteins independently promote genomic instability by inducing DNA damage,
inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 28, 3997-4008.
Grundhoff, A., Sullivan, C.S., and Ganem, D. (2006). A combined computational and
microarray-based approach identifies novel microRNAs encoded by human gamma-
herpesviruses. RNA 12, 733-750.
Gupta, S.K., Guo, X., Durkin, S.S., Fryrear, K.F., Ward, M.D., and Semmes, O.J. (2007).
Human T-cell leukemia virus type 1 Tax oncoprotein prevents DNA damage-induced
chromatin egress of hyperphosphorylated Chk2. The Journal of biological chemistry 282,
29431-29440.
Hacker, H., Vabulas, R.M., Takeuchi, O., Hoshino, K., Akira, S., and Wagner, H. (2000).
Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker
88 and tumor necrosis factor receptor-associated factor (TRAF)6. The Journal of
experimental medicine 192, 595-600.
Halazonetis, T.D., Gorgoulis, V.G., and Bartek, J. (2008). An oncogene-induced DNA
damage model for cancer development. Science (New York, NY 319, 1352-1355.
Harada, Y., Tanaka, Y., Terasawa, M., Pieczyk, M., Habiro, K., Katakai, T., Hanawa-
Suetsugu, K., Kukimoto-Niino, M., Nishizaki, T., Shirouzu, M., et al. (2012). DOCK8 is a
Cdc42 activator critical for interstitial dendritic cell migration during immune responses.
Blood 119, 4451-4461.
Harper, J.W., and Elledge, S.J. (2007). The DNA damage response: ten years after.
Molecular cell 28, 739-745.
Hartmann, G., and Krieg, A.M. (2000). Mechanism and function of a newly identified
CpG DNA motif in human primary B cells. J Immunol 164, 944-953.
Hawkins, E.D., Hommel, M., Turner, M.L., Battye, F.L., Markham, J.F., and Hodgkin,
P.D. (2007a). Measuring lymphocyte proliferation, survival and differentiation using
CFSE time-series data. Nature protocols 2, 2057-2067.

100
Hawkins, E.D., Turner, M.L., Dowling, M.R., van Gend, C., and Hodgkin, P.D. (2007b).
A model of immune regulation as a consequence of randomized lymphocyte division
and death times. Proceedings of the National Academy of Sciences of the United States
of America 104, 5032-5037.
Haynes, B.F., Fleming, J., St Clair, E.W., Katinger, H., Stiegler, G., Kunert, R., Robinson,
J., Scearce, R.M., Plonk, K., Staats, H.F., et al. (2005). Cardiolipin polyspecific
autoreactivity in two broadly neutralizing HIV-1 antibodies. Science (New York, NY
308, 1906-1908.
Haynes, B.F., Kelsoe, G., Harrison, S.C., and Kepler, T.B. (2012). B-cell-lineage
immunogen design in vaccine development with HIV-1 as a case study. Nature
biotechnology 30, 423-433.
Heath, E., Begue-Pastor, N., Chaganti, S., Croom-Carter, D., Shannon-Lowe, C., Kube,
D., Feederle, R., Delecluse, H.J., Rickinson, A.B., and Bell, A.I. (2012). Epstein-Barr virus
infection of naive B cells in vitro frequently selects clones with mutated immunoglobulin
genotypes: implications for virus biology. PLoS Pathog 8, e1002697.
Hein, J., Boichuk, S., Wu, J., Cheng, Y., Freire, R., Jat, P.S., Roberts, T.M., and Gjoerup,
O.V. (2009). Simian virus 40 large T antigen disrupts genome integrity and activates a
DNA damage response via Bub1 binding. Journal of virology 83, 117-127.
Henderson, E., Miller, G., Robinson, J., and Heston, L. (1977). Efficiency of
transformation of lymphocytes by Epstein-Barr virus. Virology 76, 152-163.
Henkel, T., Ling, P.D., Hayward, S.D., and Peterson, M.G. (1994). Mediation of Epstein-
Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa.
Science (New York, NY 265, 92-95.
Hertle, M.L., Popp, C., Petermann, S., Maier, S., Kremmer, E., Lang, R., Mages, J., and
Kempkes, B. (2009). Differential gene expression patterns of EBV infected EBNA-3A
positive and negative human B lymphocytes. PLoS pathogens 5, e1000506.

101
Hicar, M.D., Kalams, S.A., Spearman, P.W., and Crowe, J.E., Jr. (2010). Emerging studies
of human HIV-specific antibody repertoires. Vaccine 28 Suppl 2, B18-23.
Hickson, I., Zhao, Y., Richardson, C.J., Green, S.J., Martin, N.M., Orr, A.I., Reaper, P.M.,
Jackson, S.P., Curtin, N.J., and Smith, G.C. (2004). Identification and characterization of a
novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res
64, 9152-9159.
Hirao, A., Cheung, A., Duncan, G., Girard, P.M., Elia, A.J., Wakeham, A., Okada, H.,
Sarkissian, T., Wong, J.A., Sakai, T., et al. (2002). Chk2 is a tumor suppressor that
regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an
ATM-independent manner. Molecular and cellular biology 22, 6521-6532.
Hirao, A., Kong, Y.Y., Matsuoka, S., Wakeham, A., Ruland, J., Yoshida, H., Liu, D.,
Elledge, S.J., and Mak, T.W. (2000). DNA damage-induced activation of p53 by the
checkpoint kinase Chk2. Science (New York, NY 287, 1824-1827.
Hong, S., Pusapati, R.V., Powers, J.T., and Johnson, D.G. (2006). Oncogenes and the
DNA Damage Response: Myc and E2F1 Engage the ATM Signaling Pathway to Activate
p53 and Induce Apoptosis. Cell Cycle 5.
Hu, H., Voss, J., Zhang, G., Buchy, P., Zuo, T., Wang, L., Wang, F., Zhou, F., Wang, G.,
Tsai, C., et al. (2012). A human antibody recognizing a conserved epitope of H5
hemagglutinin broadly neutralizes highly pathogenic avian influenza H5N1 viruses.
Journal of virology 86, 2978-2989.
Hurley, E.A., and Thorley-Lawson, D.A. (1988). B cell activation and the establishment
of Epstein-Barr virus latency. The Journal of experimental medicine 168, 2059-2075.
Iskra, S., Kalla, M., Delecluse, H.J., Hammerschmidt, W., and Moosmann, A. (2010). Toll-
like receptor agonists synergistically increase proliferation and activation of B cells by
epstein-barr virus. Journal of virology 84, 3612-3623.
Jabara, H.H., McDonald, D.R., Janssen, E., Massaad, M.J., Ramesh, N., Borzutzky, A.,
Rauter, I., Benson, H., Schneider, L., Baxi, S., et al. (2012). DOCK8 functions as an

102
adaptor that links TLR-MyD88 signaling to B cell activation. Nature immunology 13,
612-620.
Jin, D.Y., Spencer, F., and Jeang, K.T. (1998). Human T cell leukemia virus type 1
oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell 93, 81-91.
Johannsen, E., Luftig, M., Chase, M.R., Weicksel, S., Cahir-McFarland, E., Illanes, D.,
Sarracino, D., and Kieff, E. (2004). Proteins of purified Epstein-Barr virus. Proc Natl
Acad Sci U S A 101, 16286-16291.
Johannsen, E., Miller, C.L., Grossman, S.R., and Kieff, E. (1996). EBNA-2 and EBNA-3C
extensively and mutually exclusively associate with RBPJkappa in Epstein-Barr virus-
transformed B lymphocytes. Journal of virology 70, 4179-4183.
Kaiser, C., Laux, G., Eick, D., Jochner, N., Bornkamm, G.W., and Kempkes, B. (1999). The
proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2.
Journal of virology 73, 4481-4484.
Kalla, M., Schmeinck, A., Bergbauer, M., Pich, D., and Hammerschmidt, W. (2010). AP-1
homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the
epigenetic state of the viral genome. Proc Natl Acad Sci U S A 107, 850-855.
Kamranvar, S.A., and Masucci, M.G. (2011). The Epstein-Barr virus nuclear antigen-1
promotes telomere dysfunction via induction of oxidative stress. Leukemia 25, 1017-
1025.
Karlseder, J., Broccoli, D., Dai, Y., Hardy, S., and de Lange, T. (1999). p53- and ATM-
dependent apoptosis induced by telomeres lacking TRF2. Science (New York, NY 283,
1321-1325.
Kaye, K.M., Izumi, K.M., and Kieff, E. (1993). Epstein-Barr virus latent membrane
protein 1 is essential for B-lymphocyte growth transformation. Proceedings of the
National Academy of Sciences of the United States of America 90, 9150-9154.

103
Khanna, K.K., and Jackson, S.P. (2001). DNA double-strand breaks: signaling, repair and
the cancer connection. Nature genetics 27, 247-254.
Khanna, R., Burrows, S.R., Kurilla, M.G., Jacob, C.A., Misko, I.S., Sculley, T.B., Kieff, E.,
and Moss, D.J. (1992). Localization of Epstein-Barr virus cytotoxic T cell epitopes using
recombinant vaccinia: implications for vaccine development. The Journal of
experimental medicine 176, 169-176.
Kieff, E., and Rickinson, A. (2006). Epstein-Barr virus and its replication. In Fields
Virology, D.M. Knipe, and P.M. Howley, eds. (Philadelphia: Lippincott, Williams, and
Wilkins), pp. 2603-2654.
Kim, S.T., Lim, D.S., Canman, C.E., and Kastan, M.B. (1999). Substrate specificities and
identification of putative substrates of ATM kinase family members. The Journal of
biological chemistry 274, 37538-37543.
Kinjo, T., Ham-Terhune, J., Peloponese, J.M., Jr., and Jeang, K.T. (2010). Induction of
reactive oxygen species by human T-cell leukemia virus type 1 tax correlates with DNA
damage and expression of cellular senescence marker. Journal of virology 84, 5431-5437.
Klein, N.P., and Schneider, R.J. (1997). Activation of Src family kinases by hepatitis B
virus HBx protein and coupled signaling to Ras. Molecular and cellular biology 17, 6427-
6436.
Knight, J.S., and Robertson, E.S. (2004). Epstein-Barr virus nuclear antigen 3C regulates
cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. Journal of
virology 78, 1981-1991.
Koopal, S., Furuhjelm, J.H., Jarviluoma, A., Jaamaa, S., Pyakurel, P., Pussinen, C.,
Wirzenius, M., Biberfeld, P., Alitalo, K., Laiho, M., et al. (2007). Viral oncogene-induced
DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog 3,
1348-1360.

104
Korzeniewski, N., Treat, B., and Duensing, S. (2011). The HPV-16 E7 oncoprotein
induces centriole multiplication through deregulation of Polo-like kinase 4 expression.
Mol Cancer 10, 61.
Kozbor, D., and Roder, J.C. (1981). Requirements for the establishment of high-titered
human monoclonal antibodies against tetanus toxoid using the Epstein-Barr virus
technique. J Immunol 127, 1275-1280.
Krause, J.C., Tsibane, T., Tumpey, T.M., Huffman, C.J., Albrecht, R., Blum, D.L., Ramos,
I., Fernandez-Sesma, A., Edwards, K.M., Garcia-Sastre, A., et al. (2012). Human
monoclonal antibodies to pandemic 1957 H2N2 and pandemic 1968 H3N2 influenza
viruses. Journal of virology 86, 6334-6340.
Krause, J.C., Tsibane, T., Tumpey, T.M., Huffman, C.J., Basler, C.F., and Crowe, J.E., Jr.
(2011). A broadly neutralizing human monoclonal antibody that recognizes a conserved,
novel epitope on the globular head of the influenza H1N1 virus hemagglutinin. Journal
of virology 85, 10905-10908.
Kress, M., May, E., Cassingena, R., and May, P. (1979). Simian virus 40-transformed cells
express new species of proteins precipitable by anti-simian virus 40 tumor serum.
Journal of virology 31, 472-483.
Kudoh, A., Fujita, M., Zhang, L., Shirata, N., Daikoku, T., Sugaya, Y., Isomura, H.,
Nishiyama, Y., and Tsurumi, T. (2005). Epstein-Barr virus lytic replication elicits ATM
checkpoint signal transduction while providing an S-phase-like cellular environment.
The Journal of biological chemistry 280, 8156-8163.
Kulwichit, W., Edwards, R.H., Davenport, E.M., Baskar, J.F., Godfrey, V., and Raab-
Traub, N. (1998). Expression of the Epstein-Barr virus latent membrane protein 1
induces B cell lymphoma in transgenic mice. Proceedings of the National Academy of
Sciences of the United States of America 95, 11963-11968.
Kwan-Ki Hwang, M.L. (2009). MONOCLONAL ANTIBODY PRODUCTION IN B
CELLS AND USES THEROF, USPTO, ed. (USA: Duke University).

105
Lacoste, S., Wiechec, E., Dos Santos Silva, A.G., Guffei, A., Williams, G., Lowbeer, M.,
Benedek, K., Henriksson, M., Klein, G., and Mai, S. (2009). Chromosomal
rearrangements after ex vivo Epstein-Barr virus (EBV) infection of human B cells.
Oncogene.
Lane, D.P., and Crawford, L.V. (1979). T antigen is bound to a host protein in SV40-
transformed cells. Nature 278, 261-263.
Latz, E., Verma, A., Visintin, A., Gong, M., Sirois, C.M., Klein, D.C., Monks, B.G.,
McKnight, C.J., Lamphier, M.S., Duprex, W.P., et al. (2007). Ligand-induced
conformational changes allosterically activate Toll-like receptor 9. Nature immunology
8, 772-779.
Leao, M., Anderton, E., Wade, M., Meekings, K., and Allday, M.J. (2007). Epstein-barr
virus-induced resistance to drugs that activate the mitotic spindle assembly checkpoint
in Burkitt's lymphoma cells. Journal of virology 81, 248-260.
Leidal, A.M., Cyr, D.P., Hill, R.J., Lee, P.W., and McCormick, C. (2012). Subversion of
autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced
senescence. Cell host & microbe 11, 167-180.
Lilley, C.E., Schwartz, R.A., and Weitzman, M.D. (2007). Using or abusing: viruses and
the cellular DNA damage response. Trends Microbiol 15, 119-126.
Liu, B., Hong, S., Tang, Z., Yu, H., and Giam, C.Z. (2005). HTLV-I Tax directly binds the
Cdc20-associated anaphase-promoting complex and activates it ahead of schedule.
Proceedings of the National Academy of Sciences of the United States of America 102,
63-68.
Liu, J., Martin, H.J., Liao, G., and Hayward, S.D. (2007). The Kaposi's sarcoma-associated
herpesvirus LANA protein stabilizes and activates c-Myc. Journal of virology 81, 10451-
10459.
Lomaga, M.A., Yeh, W.C., Sarosi, I., Duncan, G.S., Furlonger, C., Ho, A., Morony, S.,
Capparelli, C., Van, G., Kaufman, S., et al. (1999). TRAF6 deficiency results in

106
osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 13, 1015-
1024.
Macagno, A., Bernasconi, N.L., Vanzetta, F., Dander, E., Sarasini, A., Revello, M.G.,
Gerna, G., Sallusto, F., and Lanzavecchia, A. (2010). Isolation of human monoclonal
antibodies that potently neutralize human cytomegalovirus infection by targeting
different epitopes on the gH/gL/UL128-131A complex. Journal of virology 84, 1005-1013.
Maclean, K.H., Dorsey, F.C., Cleveland, J.L., and Kastan, M.B. (2008). Targeting
lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse
models of lymphomagenesis. The Journal of clinical investigation 118, 79-88.
MacLennan, I.C. (1994). Germinal centers. Annu Rev Immunol 12, 117-139.
Markham, J.F., Wellard, C.J., Hawkins, E.D., Duffy, K.R., and Hodgkin, P.D. (2010). A
minimum of two distinct heritable factors are required to explain correlation structures
in proliferating lymphocytes. Journal of the Royal Society, Interface / the Royal Society 7,
1049-1059.
Martin, H.J., Lee, J.M., Walls, D., and Hayward, S.D. (2007). Manipulation of the toll-like
receptor 7 signaling pathway by Epstein-Barr virus. Journal of virology 81, 9748-9758.
Maruo, S., Zhao, B., Johannsen, E., Kieff, E., Zou, J., and Takada, K. (2011). Epstein-Barr
virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing
p16INK4A and p14ARF expression. Proceedings of the National Academy of Sciences of
the United States of America 108, 1919-1924.
Matsuoka, S., Huang, M., and Elledge, S.J. (1998). Linkage of ATM to cell cycle
regulation by the Chk2 protein kinase. Science (New York, NY 282, 1893-1897.
Mauser, A., Saito, S., Appella, E., Anderson, C.W., Seaman, W.T., and Kenney, S. (2002).
The Epstein-Barr virus immediate-early protein BZLF1 regulates p53 function through
multiple mechanisms. Journal of virology 76, 12503-12512.

107
McPherson, J.P., Lemmers, B., Hirao, A., Hakem, A., Abraham, J., Migon, E., Matysiak-
Zablocki, E., Tamblyn, L., Sanchez-Sweatman, O., Khokha, R., et al. (2004). Collaboration
of Brca1 and Chk2 in tumorigenesis. Genes Dev 18, 1144-1153.
Miyazato, A., Sheleg, S., Iha, H., Li, Y., and Jeang, K.T. (2005). Evidence for NF-kappaB-
and CBP-independent repression of p53's transcriptional activity by human T-cell
leukemia virus type 1 Tax in mouse embryo and primary human fibroblasts. Journal of
virology 79, 9346-9350.
Moody, C.A., and Laimins, L.A. (2009). Human papillomaviruses activate the ATM
DNA damage pathway for viral genome amplification upon differentiation. PLoS
Pathog 5, e1000605.
Munger, K., Werness, B.A., Dyson, N., Phelps, W.C., Harlow, E., and Howley, P.M.
(1989). Complex formation of human papillomavirus E7 proteins with the
retinoblastoma tumor suppressor gene product. The EMBO journal 8, 4099-4105.
Murphy, D.J., Junttila, M.R., Pouyet, L., Karnezis, A., Shchors, K., Bui, D.A., Brown-
Swigart, L., Johnson, L., and Evan, G.I. (2008). Distinct thresholds govern Myc's
biological output in vivo. Cancer Cell 14, 447-457.
Murray, R.J., Kurilla, M.G., Brooks, J.M., Thomas, W.A., Rowe, M., Kieff, E., and
Rickinson, A.B. (1992). Identification of target antigens for the human cytotoxic T cell
response to Epstein-Barr virus (EBV): implications for the immune control of EBV-
positive malignancies. The Journal of experimental medicine 176, 157-168.
Nguyen, C.L., and Munger, K. (2009). Human papillomavirus E7 protein deregulates
mitosis via an association with nuclear mitotic apparatus protein 1. Journal of virology
83, 1700-1707.
Nikitin, P.A., and Luftig, M.A. (2012). The DNA damage response in viral-induced
cellular transformation. British journal of cancer 106, 429-435.
Nikitin, P.A., Yan, C.M., Forte, E., Bocedi, A., Tourigny, J.P., White, R.E., Allday, M.J.,
Patel, A., Dave, S.S., Kim, W., et al. (2010). An ATM/Chk2-mediated DNA damage-

108
responsive signaling pathway suppresses Epstein-Barr virus transformation of primary
human B cells. Cell Host Microbe 8, 510-522.
Osborn, A.J., Elledge, S.J., and Zou, L. (2002). Checking on the fork: the DNA-replication
stress-response pathway. Trends in cell biology 12, 509-516.
Park, H.U., Jeong, J.H., Chung, J.H., and Brady, J.N. (2004). Human T-cell leukemia virus
type 1 Tax interacts with Chk1 and attenuates DNA-damage induced G2 arrest
mediated by Chk1. Oncogene 23, 4966-4974.
Park, H.U., Jeong, S.J., Jeong, J.H., Chung, J.H., and Brady, J.N. (2006). Human T-cell
leukemia virus type 1 Tax attenuates gamma-irradiation-induced apoptosis through
physical interaction with Chk2. Oncogene 25, 438-447.
Parker, G.A., Touitou, R., and Allday, M.J. (2000). Epstein-Barr virus EBNA3C can
disrupt multiple cell cycle checkpoints and induce nuclear division divorced from
cytokinesis. Oncogene 19, 700-709.
Pathmanathan, R., Prasad, U., Chandrika, G., Sadler, R., Flynn, K., and Raab-Traub, N.
(1995). Undifferentiated, nonkeratinizing, and squamous cell carcinoma of the
nasopharynx. Variants of Epstein-Barr virus-infected neoplasia. The American journal of
pathology 146, 1355-1367.
Peng, S.L. (2005). Signaling in B cells via Toll-like receptors. Current opinion in
immunology 17, 230-236.
Perkins, N.D. (2012). The diverse and complex roles of NF-kappaB subunits in cancer.
Nature reviews Cancer 12, 121-132.
Pfeffer, S., Zavolan, M., Grasser, F.A., Chien, M., Russo, J.J., Ju, J., John, B., Enright, A.J.,
Marks, D., Sander, C., et al. (2004). Identification of virus-encoded microRNAs. Science
(New York, NY 304, 734-736.

109
Pise-Masison, C.A., Mahieux, R., Radonovich, M., Jiang, H., Duvall, J., Guillerm, C., and
Brady, J.N. (2000). Insights into the molecular mechanism of p53 inhibition by HTLV
type 1 Tax. AIDS Res Hum Retroviruses 16, 1669-1675.
Poulsen, T.R., Jensen, A., Haurum, J.S., and Andersen, P.S. (2011). Limits for antibody
affinity maturation and repertoire diversification in hypervaccinated humans. J
Immunol 187, 4229-4235.
Powers, J.T., Hong, S., Mayhew, C.N., Rogers, P.M., Knudsen, E.S., and Johnson, D.G.
(2004). E2F1 uses the ATM signaling pathway to induce p53 and Chk2 phosphorylation
and apoptosis. Molecular cancer research : MCR 2, 203-214.
Price, A.M., Tourigny, J.P., Forte, E., Salinas, R.E., Dave, S.S., and Luftig, M.A. (2012).
Analysis of Epstein-Barr Virus-Regulated Host Gene Expression Changes through
Primary B-Cell Outgrowth Reveals Delayed Kinetics of Latent Membrane Protein 1-
Mediated NF-kappaB Activation. Journal of virology 86, 11096-11106.
Pusapati, R.V., Rounbehler, R.J., Hong, S., Powers, J.T., Yan, M., Kiguchi, K., McArthur,
M.J., Wong, P.K., and Johnson, D.G. (2006). ATM promotes apoptosis and suppresses
tumorigenesis in response to Myc. Proceedings of the National Academy of Sciences of
the United States of America 103, 1446-1451.
Raab-Traub, N. (2002). Epstein-Barr virus in the pathogenesis of NPC. Seminars in
cancer biology 12, 431-441.
Raab-Traub, N. (2007). EBV-induced oncogenesis. In Human Herpesviruses: Biology,
Therapy, and Immunoprophylaxis, A. Arvin, G. Campadelli-Fiume, E. Mocarski, P.S.
Moore, B. Roizman, R. Whitley, and K. Yamanishi, eds. (Cambridge).
Raab-Traub, N. (2012). Novel mechanisms of EBV-induced oncogenesis. Current opinion
in virology 2, 453-458.
Ranuncolo, S.M., Polo, J.M., Dierov, J., Singer, M., Kuo, T., Greally, J., Green, R., Carroll,
M., and Melnick, A. (2007). Bcl-6 mediates the germinal center B cell phenotype and

110
lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR.
Nature immunology 8, 705-714.
Richman, D.D., Wrin, T., Little, S.J., and Petropoulos, C.J. (2003). Rapid evolution of the
neutralizing antibody response to HIV type 1 infection. Proceedings of the National
Academy of Sciences of the United States of America 100, 4144-4149.
Rickinson, A., and Kieff, E. (2006). Epstein-Barr virus In Fields Virology, D.M. Knipe,
and P.M. Howley, eds. (Philadelphia: Lippincott, Williams, and Wilkins), pp. 2603-2654.
Robertson, E.S., Lin, J., and Kieff, E. (1996). The amino-terminal domains of Epstein-Barr
virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa). Journal of virology 70,
3068-3074.
Robinson, T.J., Forte, E., Salinas, R.E., Puri, S., Marengo, M., Garcia-Blanco, M.A., and
Luftig, M.A. (2012). SplicerEX: a tool for the automated detection and classification of
mRNA changes from conventional and splice-sensitive microarray expression data.
RNA 18, 1435-1445.
Rodier, F., Coppe, J.P., Patil, C.K., Hoeijmakers, W.A., Munoz, D.P., Raza, S.R., Freund,
A., Campeau, E., Davalos, A.R., and Campisi, J. (2009). Persistent DNA damage
signalling triggers senescence-associated inflammatory cytokine secretion. Nature cell
biology 11, 973-979.
Rodier, F., Munoz, D.P., Teachenor, R., Chu, V., Le, O., Bhaumik, D., Coppe, J.P.,
Campeau, E., Beausejour, C.M., Kim, S.H., et al. (2011). DNA-SCARS: distinct nuclear
structures that sustain damage-induced senescence growth arrest and inflammatory
cytokine secretion. Journal of cell science 124, 68-81.
Rogoff, H.A., Pickering, M.T., Frame, F.M., Debatis, M.E., Sanchez, Y., Jones, S., and
Kowalik, T.F. (2004). Apoptosis associated with deregulated E2F activity is dependent
on E2F1 and Atm/Nbs1/Chk2. Molecular and cellular biology 24, 2968-2977.
Roughan, J.E., and Thorley-Lawson, D.A. (2009). The intersection of Epstein-Barr virus
with the germinal center. Journal of virology 83, 3968-3976.

111
Rousset, F., Garcia, E., and Banchereau, J. (1991). Cytokine-induced proliferation and
immunoglobulin production of human B lymphocytes triggered through their CD40
antigen. The Journal of experimental medicine 173, 705-710.
Rowe, M., Lear, A.L., Croom-Carter, D., Davies, A.H., and Rickinson, A.B. (1992). Three
pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B
lymphocytes. Journal of virology 66, 122-131.
Rowe, M., Rowe, D.T., Gregory, C.D., Young, L.S., Farrell, P.J., Rupani, H., and
Rickinson, A.B. (1987). Differences in B cell growth phenotype reflect novel patterns of
Epstein-Barr virus latent gene expression in Burkitt's lymphoma cells. The EMBO
journal 6, 2743-2751.
Sadler, R.H., and Raab-Traub, N. (1995). The Epstein-Barr virus 3.5-kilobase latent
membrane protein 1 mRNA initiates from a TATA-Less promoter within the first
terminal repeat. Journal of virology 69, 4577-4581.
Sato, K., Ohta, T., and Venkitaraman, A.R. (2010). A mitotic role for the DNA damage-
responsive CHK2 kinase. Nature cell biology 12, 424-425.
Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D.A.,
Smith, S., Uziel, T., Sfez, S., et al. (1995). A single ataxia telangiectasia gene with a
product similar to PI-3 kinase. Science (New York, NY 268, 1749-1753.
Scheffner, M., Huibregtse, J.M., Vierstra, R.D., and Howley, P.M. (1993). The HPV-16 E6
and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53.
Cell 75, 495-505.
Scheffner, M., Werness, B.A., Huibregtse, J.M., Levine, A.J., and Howley, P.M. (1990).
The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the
degradation of p53. Cell 63, 1129-1136.
Schlager, S., Speck, S.H., and Woisetschlager, M. (1996). Transcription of the Epstein-
Barr virus nuclear antigen 1 (EBNA1) gene occurs before induction of the BCR2 (Cp)

112
EBNA gene promoter during the initial stages of infection in B cells. Journal of virology
70, 3561-3570.
Sengupta, S., den Boon, J.A., Chen, I.H., Newton, M.A., Dahl, D.B., Chen, M., Cheng,
Y.J., Westra, W.H., Chen, C.J., Hildesheim, A., et al. (2006). Genome-wide expression
profiling reveals EBV-associated inhibition of MHC class I expression in nasopharyngeal
carcinoma. Cancer Res 66, 7999-8006.
Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity.
Nature reviews Cancer 3, 155-168.
Shuda, M., Feng, H., Kwun, H.J., Rosen, S.T., Gjoerup, O., Moore, P.S., and Chang, Y.
(2008). T antigen mutations are a human tumor-specific signature for Merkel cell
polyomavirus. Proceedings of the National Academy of Sciences of the United States of
America 105, 16272-16277.
Siemer, D., Kurth, J., Lang, S., Lehnerdt, G., Stanelle, J., and Kuppers, R. (2008). EBV
transformation overrides gene expression patterns of B cell differentiation stages. Mol
Immunol 45, 3133-3141.
Simmons, C.P., Bernasconi, N.L., Suguitan, A.L., Mills, K., Ward, J.M., Chau, N.V., Hien,
T.T., Sallusto, F., Ha do, Q., Farrar, J., et al. (2007). Prophylactic and therapeutic efficacy
of human monoclonal antibodies against H5N1 influenza. PLoS medicine 4, e178.
Sinclair, A.J., Palmero, I., Peters, G., and Farrell, P.J. (1994). EBNA-2 and EBNA-LP
cooperate to cause G0 to G1 transition during immortalization of resting human B
lymphocytes by Epstein-Barr virus. The EMBO journal 13, 3321-3328.
Skalska, L., White, R.E., Franz, M., Ruhmann, M., and Allday, M.J. (2010). Epigenetic
repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of
EBNA3A and EBNA3C with CtBP. PLoS Pathog 6, e1000951.
Skalsky, R.L., Corcoran, D.L., Gottwein, E., Frank, C.L., Kang, D., Hafner, M., Nusbaum,
J.D., Feederle, R., Delecluse, H.J., Luftig, M.A., et al. (2012). The viral and cellular
microRNA targetome in lymphoblastoid cell lines. PLoS pathogens 8, e1002484.

113
Smith, K., Garman, L., Wrammert, J., Zheng, N.Y., Capra, J.D., Ahmed, R., and Wilson,
P.C. (2009). Rapid generation of fully human monoclonal antibodies specific to a
vaccinating antigen. Nature protocols 4, 372-384.
Smith, S.A., Zhou, Y., Olivarez, N.P., Broadwater, A.H., de Silva, A.M., and Crowe, J.E.,
Jr. (2012). Persistence of circulating memory B cell clones with potential for dengue virus
disease enhancement for decades following infection. Journal of virology 86, 2665-2675.
Steinitz, M., Klein, G., Koskimies, S., and Makel, O. (1977). EB virus-induced B
lymphocyte cell lines producing specific antibody. Nature 269, 420-422.
Stiff, T., Walker, S.A., Cerosaletti, K., Goodarzi, A.A., Petermann, E., Concannon, P.,
O'Driscoll, M., and Jeggo, P.A. (2006). ATR-dependent phosphorylation and activation
of ATM in response to UV treatment or replication fork stalling. The EMBO journal 25,
5775-5782.
Stolz, A., Ertych, N., Kienitz, A., Vogel, C., Schneider, V., Fritz, B., Jacob, R., Dittmar, G.,
Weichert, W., Petersen, I., et al. (2010). The CHK2-BRCA1 tumour suppressor pathway
ensures chromosomal stability in human somatic cells. Nature cell biology 12, 492-499.
Stracker, T.H., Couto, S.S., Cordon-Cardo, C., Matos, T., and Petrini, J.H. (2008). Chk2
suppresses the oncogenic potential of DNA replication-associated DNA damage. Mol
Cell 31, 21-32.
Sugden, B., and Mark, W. (1977). Clonal transformation of adult human leukocytes by
Epstein-Barr virus. Journal of virology 23, 503-508.
Swaminathan, S., Tomkinson, B., and Kieff, E. (1991). Recombinant Epstein-Barr virus
with small RNA (EBER) genes deleted transforms lymphocytes and replicates in vitro.
Proc Natl Acad Sci U S A 88, 1546-1550.
Szekely, L., Pokrovskaja, K., Jiang, W.Q., Selivanova, G., Lowbeer, M., Ringertz, N.,
Wiman, K.G., and Klein, G. (1995). Resting B-cells, EBV-infected B-blasts and established
lymphoblastoid cell lines differ in their Rb, p53 and EBNA-5 expression patterns.
Oncogene 10, 1869-1874.

114
Takada, S., Kaneniwa, N., Tsuchida, N., and Koike, K. (1997). Cytoplasmic retention of
the p53 tumor suppressor gene product is observed in the hepatitis B virus X gene-
transfected cells. Oncogene 15, 1895-1901.
Tangye, S.G., Avery, D.T., and Hodgkin, P.D. (2003). A division-linked mechanism for
the rapid generation of Ig-secreting cells from human memory B cells. J Immunol 170,
261-269.
Tangye, S.G., Ferguson, A., Avery, D.T., Ma, C.S., and Hodgkin, P.D. (2002). Isotype
switching by human B cells is division-associated and regulated by cytokines. J
Immunol 169, 4298-4306.
Thorley-Lawson, D.A., and Allday, M.J. (2008). The curious case of the tumour virus: 50
years of Burkitt's lymphoma. Nature reviews Microbiology 6, 913-924.
Tian, C., Luskin, G.K., Dischert, K.M., Higginbotham, J.N., Shepherd, B.E., and Crowe,
J.E., Jr. (2008). Immunodominance of the VH1-46 antibody gene segment in the primary
repertoire of human rotavirus-specific B cells is reduced in the memory compartment
through somatic mutation of nondominant clones. J Immunol 180, 3279-3288.
Tierney, R.J., Kao, K.Y., Nagra, J.K., and Rickinson, A.B. (2011). Epstein-Barr virus
BamHI W repeat number limits EBNA2/EBNA-LP coexpression in newly infected B cells
and the efficiency of B-cell transformation: a rationale for the multiple W repeats in
wild-type virus strains. Journal of virology 85, 12362-12375.
Tort, F., Hernandez, S., Bea, S., Martinez, A., Esteller, M., Herman, J.G., Puig, X.,
Camacho, E., Sanchez, M., Nayach, I., et al. (2002). CHK2-decreased protein expression
and infrequent genetic alterations mainly occur in aggressive types of non-Hodgkin
lymphomas. Blood 100, 4602-4608.
Traggiai, E., Becker, S., Subbarao, K., Kolesnikova, L., Uematsu, Y., Gismondo, M.R.,
Murphy, B.R., Rappuoli, R., and Lanzavecchia, A. (2004). An efficient method to make
human monoclonal antibodies from memory B cells: potent neutralization of SARS
coronavirus. Nature medicine 10, 871-875.

115
Turner, M.L., Hawkins, E.D., and Hodgkin, P.D. (2008). Quantitative regulation of B cell
division destiny by signal strength. J Immunol 181, 374-382.
Verschuren, E.W., Jones, N., and Evan, G.I. (2004). The cell cycle and how it is steered by
Kaposi's sarcoma-associated herpesvirus cyclin. The Journal of general virology 85,
1347-1361.
Verschuren, E.W., Klefstrom, J., Evan, G.I., and Jones, N. (2002). The oncogenic potential
of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in
vivo. Cancer Cell 2, 229-241.
Wade, M., and Allday, M.J. (2000). Epstein-Barr virus suppresses a G(2)/M checkpoint
activated by genotoxins. Molecular and cellular biology 20, 1344-1360.
Wang, F., Kikutani, H., Tsang, S.F., Kishimoto, T., and Kieff, E. (1991). Epstein-Barr virus
nuclear protein 2 transactivates a cis-acting CD23 DNA element. Journal of virology 65,
4101-4106.
Wang, F., Tsang, S.F., Kurilla, M.G., Cohen, J.I., and Kieff, E. (1990). Epstein-Barr virus
nuclear antigen 2 transactivates latent membrane protein LMP1. Journal of virology 64,
3407-3416.
Wang, W.H., Hullinger, R.L., and Andrisani, O.M. (2008). Hepatitis B virus X protein via
the p38MAPK pathway induces E2F1 release and ATR kinase activation mediating p53
apoptosis. The Journal of biological chemistry 283, 25455-25467.
Wang, X.W., Forrester, K., Yeh, H., Feitelson, M.A., Gu, J.R., and Harris, C.C. (1994).
Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional
activity, and association with transcription factor ERCC3. Proceedings of the National
Academy of Sciences of the United States of America 91, 2230-2234.
Wang, X.W., Gibson, M.K., Vermeulen, W., Yeh, H., Forrester, K., Sturzbecher, H.W.,
Hoeijmakers, J.H., and Harris, C.C. (1995). Abrogation of p53-induced apoptosis by the
hepatitis B virus X gene. Cancer research 55, 6012-6016.

116
Warter, L., Lee, C.Y., Thiagarajan, R., Grandadam, M., Lebecque, S., Lin, R.T., Bertin-
Maghit, S., Ng, L.F., Abastado, J.P., Despres, P., et al. (2011). Chikungunya virus
envelope-specific human monoclonal antibodies with broad neutralization potency. J
Immunol 186, 3258-3264.
Wei, X., Decker, J.M., Wang, S., Hui, H., Kappes, J.C., Wu, X., Salazar-Gonzalez, J.F.,
Salazar, M.G., Kilby, J.M., Saag, M.S., et al. (2003). Antibody neutralization and escape by
HIV-1. Nature 422, 307-312.
White, R.E., Ramer, P.C., Naresh, K.N., Meixlsperger, S., Pinaud, L., Rooney, C.,
Savoldo, B., Coutinho, R., Bodor, C., Gribben, J., et al. (2012). EBNA3B-deficient EBV
promotes B cell lymphomagenesis in humanized mice and is found in human tumors.
The Journal of clinical investigation 122, 1487-1502.
Wilson, P.C., and Andrews, S.F. (2012). Tools to therapeutically harness the human
antibody response. Nature reviews Immunology 12, 709-719.
Woisetschlaeger, M., Strominger, J.L., and Speck, S.H. (1989). Mutually exclusive use of
viral promoters in Epstein-Barr virus latently infected lymphocytes. Proceedings of the
National Academy of Sciences of the United States of America 86, 6498-6502.
Woisetschlaeger, M., Yandava, C.N., Furmanski, L.A., Strominger, J.L., and Speck, S.H.
(1990). Promoter switching in Epstein-Barr virus during the initial stages of infection of
B lymphocytes. Proc Natl Acad Sci U S A 87, 1725-1729.
Wu, X., Avni, D., Chiba, T., Yan, F., Zhao, Q., Lin, Y., Heng, H., and Livingston, D.
(2004). SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes
Dev 18, 1305-1316.
Yi, A.K., Chang, M., Peckham, D.W., Krieg, A.M., and Ashman, R.F. (1998). CpG
oligodeoxyribonucleotides rescue mature spleen B cells from spontaneous apoptosis and
promote cell cycle entry. J Immunol 160, 5898-5906.

117
Yi, F., Saha, A., Murakami, M., Kumar, P., Knight, J.S., Cai, Q., Choudhuri, T., and
Robertson, E.S. (2009). Epstein-Barr virus nuclear antigen 3C targets p53 and modulates
its transcriptional and apoptotic activities. Virology 388, 236-247.
Yu, X., Tsibane, T., McGraw, P.A., House, F.S., Keefer, C.J., Hicar, M.D., Tumpey, T.M.,
Pappas, C., Perrone, L.A., Martinez, O., et al. (2008). Neutralizing antibodies derived
from the B cells of 1918 influenza pandemic survivors. Nature 455, 532-536.
Zalvide, J., and DeCaprio, J.A. (1995). Role of pRb-related proteins in simian virus 40
large-T-antigen-mediated transformation. Molecular and cellular biology 15, 5800-5810.
Zhao, F., Hou, N.B., Yang, X.L., He, X., Liu, Y., Zhang, Y.H., Wei, C.W., Song, T., Li, L.,
Ma, Q.J., et al. (2008a). Ataxia telangiectasia-mutated-Rad3-related DNA damage
checkpoint signaling pathway triggered by hepatitis B virus infection. World J
Gastroenterol 14, 6163-6170.
Zhao, X., Madden-Fuentes, R.J., Lou, B.X., Pipas, J.M., Gerhardt, J., Rigell, C.J., and
Fanning, E. (2008b). Ataxia telangiectasia-mutated damage-signaling kinase- and
proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-
infected primate cells. Journal of virology 82, 5316-5328.
Zheng, Z., Li, J., Sun, J., Song, T., Wei, C., Zhang, Y., Rao, G., Chen, G., Li, D., Yang, G., et
al. (2011). Inhibition of HBV replication by theophylline. Antiviral Res 89, 149-155.
Zimber-Strobl, U., Kremmer, E., Grasser, F., Marschall, G., Laux, G., and Bornkamm,
G.W. (1993). The Epstein-Barr virus nuclear antigen 2 interacts with an EBNA2
responsive cis-element of the terminal protein 1 gene promoter. The EMBO journal 12,
167-175.
Zindy, F., Williams, R.T., Baudino, T.A., Rehg, J.E., Skapek, S.X., Cleveland, J.L., Roussel,
M.F., and Sherr, C.J. (2003). Arf tumor suppressor promoter monitors latent oncogenic
signals in vivo. Proceedings of the National Academy of Sciences of the United States of
America 100, 15930-15935.

118
Biography
Pavel Nikitin was born in 1983 in Pavlodar, a city in the USSR that is now in
Kazakhstan. In 1999 Pavel was invited to attend a physics and math magnet high school
at Novosibirsk State University, Russia. In 2000 he went to Novosibirsk State University,
where he studied molecular biology and defended a “specialist” Diploma with honors in
2005. During his undergraduate school Pavel received a Potanin federal scholarship and
was awarded with the U.S. Civilian Research & Development Foundation grant to study
methods of inactivation of influenza virus.
In 2006 Pavel moved to the United States for an internship at Reproductive
Genetics Institute, a medical company in Chicago which specialized in pre-implantation
genetic diagnosis. Further, pursuing his interest in virology and genetics, Pavel went to
Duke’s Department of Molecular Genetics and Microbiology (MGM) for graduate
school. At Duke Pavel joined the recently formed laboratory of Dr. Micah Luftig to study
the host growth suppressive mechanisms that restrict Epstein-Barr virus-mediated
tumorigenesis. In Dr. Luftig’s group Pavel published one paper, two reviews and
contributed to one patent. During his time at Duke Pavel received a best oral
presentation award at the EBV Society meeting in Birmingham, UK in 2010 and was
awarded with several travel grants from the MGM Department and Duke’s Graduate
School.