
Neuropathology
Dr Olaf Ansorge
Oxford
Oxford Pathology Course 2010For FRCPath
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð 40 cases
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 1
Clinical data:M, 47, single seizure, no neurological deficit, non-
enhancing frontal lesion
Slides: Biopsy HE, GFAP, MIB-1, p53
Diagnosis:Diffuse astrocytoma , WHO grade II
Comment:Mild hypercellularity and nuclear atypia only. No
mitoses or vascular proliferation. GFAP expression,low MIB-1, strong p53 overexpression
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 1
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

GFAP
Neuropathology Ð Neoplasia: Case 1
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

MIB-1
Neuropathology Ð Neoplasia: Case 1
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

p53
Neuropathology Ð Neoplasia: Case 1
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 2
Clinical data:M, 56, one week of headaches, one seizure, ring-
enhancing left-parietal lesion
Slides: Biopsy HE, GFAP, MIB-1
Diagnosis:Glioblastoma multiforme, WHO grade IV
Comment: Very hypercellular and pleomorphic. Many mitoses and
vascular proliferation. Necrosis with pseudopalisading,high MIB-1
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 2
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 2
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 2
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

GFAP
Neuropathology Ð Neoplasia: Case 2
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

MIB-1
Neuropathology Ð Neoplasia: Case 2
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 3
Clinical data:M, 9, increasing lethargy over the past year, cystic
posterior fossa tumour
Slides:CSF MGG, smear methylene blue, biopsy HE
Diagnosis: Pilocytic astrocytoma , WHO grade I
Comment:Compact and reticular areas, Rosenthal fibres,
vascular proliferation (not indicative of malignancy inthis entity!), no mitoses
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Arrow: Rosenthal fibre on smear
Neuropathology Ð Neoplasia: Case 3
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 3
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 3
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Vascular proliferation
Neuropathology Ð Neoplasia: Case 3
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Many Rosenthal fibres
Neuropathology Ð Neoplasia: Case 3
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 4
Clinical data: M, 31, seizures x1 per week, frequency recently increasing. MRI Ð
large diffuse cortical and white matter lesion, non-enhancing.
Slides: Biopsy HE, GFAP
Diagnosis: Oligodendroglioma, WHO grade II
Comment: This is a very classical example. Note relative monomorphism and
roundness of cells and nuclei, ÔhalosÕ, and delicate reticular capillarynetwork. Oligos usually show some form of GFAP expression but thepattern is generally distinct from that of astrocytomas
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 4
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 4
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

GFAP
Neuropathology Ð Neoplasia: Case 4
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 5
Clinical data: M, 35, progressive leg numbness and incontinence.
Slides: Biopsy HE x1, SYN, CD34
Diagnosis: Myxopapillary ependymoma, WHO grade I
Comment: This type of ependymoma occurs virtually always at the conus / filum
terminale of the spinal cord. Note the myxoid matrix and elongatedspindle cells with some perivascular rosetting. Vessels are oftenhyalinised. It is the only grade I ependymoma and usually well-encapsulated rather than infiltrating.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 5
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 5
PAS-Alcian blue
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 6
Clinical data: F, 15, short history of headache, left frontal tumour close to lateral
horn.
Slides: Biopsy HE x2, GFAP, EMA
Diagnosis: Anaplastic ependymoma, WHO grade III
Comment: This example contains all features of neoplastic aberrant ependymal
differentiation: perivascular pseudorosettes, true ependymal rosetteswith a lumen, and ependymal canals/free surfaces. GFAP isaccentuated around the perivascular rosettes, ependymal lumina andsurfaces are EMA positive. Widespread necrosis, mitoses andvascular proliferation make it grade III
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 6
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust
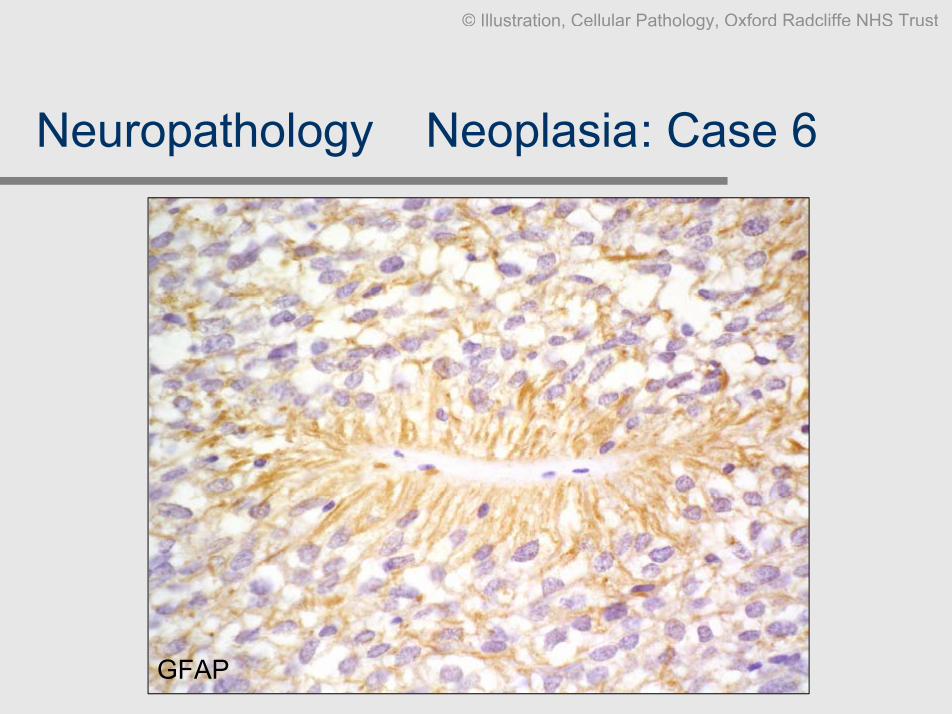
GFAP
Neuropathology Ð Neoplasia: Case 6
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 6
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

EMA
Neuropathology Ð Neoplasia: Case 6
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 7
Clinical data: M, 1, rapidly progressive lethargy, vomiting. Large frontal tumour on
imaging.
Slides: HE, SYN, CD34
Diagnosis: Ependymoblastoma, WHO grade IV
Comment: The ependymoblastoma is a rare tumour and an example of the
supratentorial PNET family. Characteristic are the ÔtrueÕependymoblastic rosettes without a central vessel and multilayeredrosetting cells with mitoses. There is often synaptophysinexpression, characteristic of PNETs, rather than GFAP, which isseen in classical grade I (myxopapillary) and grade II and IIIependymomas.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 7
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 7
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 8
Clinical data: M, 1, lethargy, bouts of projectile vomiting. Large posterior fossa
tumour on imaging.
Slides: Smear methylene blue, biopsy HE, PM cerebellum HE
Diagnosis: Medulloblastoma, WHO grade IV
Comment: The medulloblastoma shares appearances of Ôsmall blue round cellÕ
tumours. It can be viewed as a PNET of the cerebellum. Significantnodularity (= increased differentiation) may confer a betterprognosis. Synaptophysin or GFAP may be expressed
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 8
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 8
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 8
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 8
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 9
Clinical data: F, 34, 6/12 headache/vomiting, worse over last week. Papilloedema.
Lateral ventricle tumour.
Slides: HE, GFAP, synaptophysin, NeuN (neuronal nuclei)
Diagnosis: Central neurocytoma, WHO grade II
Comment: Central neurocytomas virtually always arise within the lateral
ventricles close to the foramen of Monro. They are usuallymonomorphic and may be mistaken for oligodendrogliomas.Immunocytochemistry to confirm neuronal lineage is key.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 9
GFAP
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 9
Synaptophysin NeuN
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 10
Clinical data: F, 13, 4 years of temporal lobe epilepsy, partially cystic tumour left
mesial temporal lobe.
Slides: HE, GFAP, chromogranin
Diagnosis: Ganglioglioma, WHO grade I
Comment: The temporal lobe is a typical site for ganglioglioma. Note the
atypical, occasionally multinucleate ganglion cells mixed withneoplastic astrocytes. The lymphocytic inflammation and calcificationis typical. Mitoses are usually absent. Some ganglioid cells co-express glial and neuronal markers. The prognosis is very good andprogression to high-grade lesions rare.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 10
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 10
Chromogranin GFAP
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 11
Clinical data: 2 different cases. F, 58, occipital tumour (extrinsic). M, 27, convexity
enhancing tumour, well-defined.
Slides: Smear methylene blue, biopsies HE x2, EMA
Diagnosis: a. Meningioma (classical), b. meningioma (microcystic), both WHO
grade I
Comment: Plump spindle cells with whorls are classic. There are many variants
without clinical significance, but they can be diagnosticallychallenging. Most however contain a focus of the classical pattern,EMA can help
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Case (a)
Neuropathology Ð Neoplasia: Case 11
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust
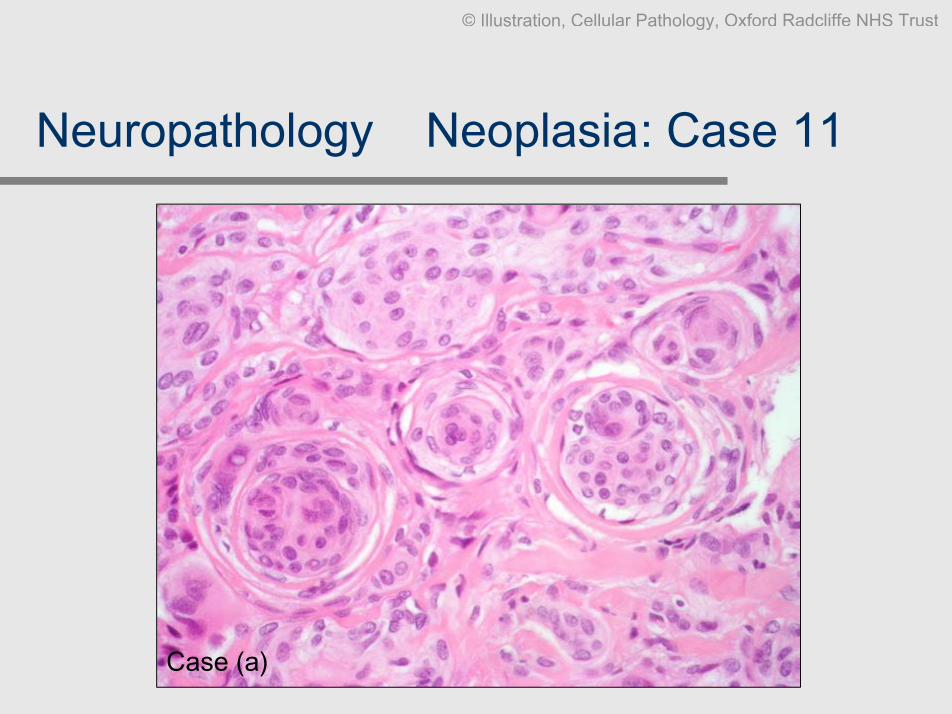
Case (a)
Neuropathology Ð Neoplasia: Case 11
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Case (b)
Neuropathology Ð Neoplasia: Case 11
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust
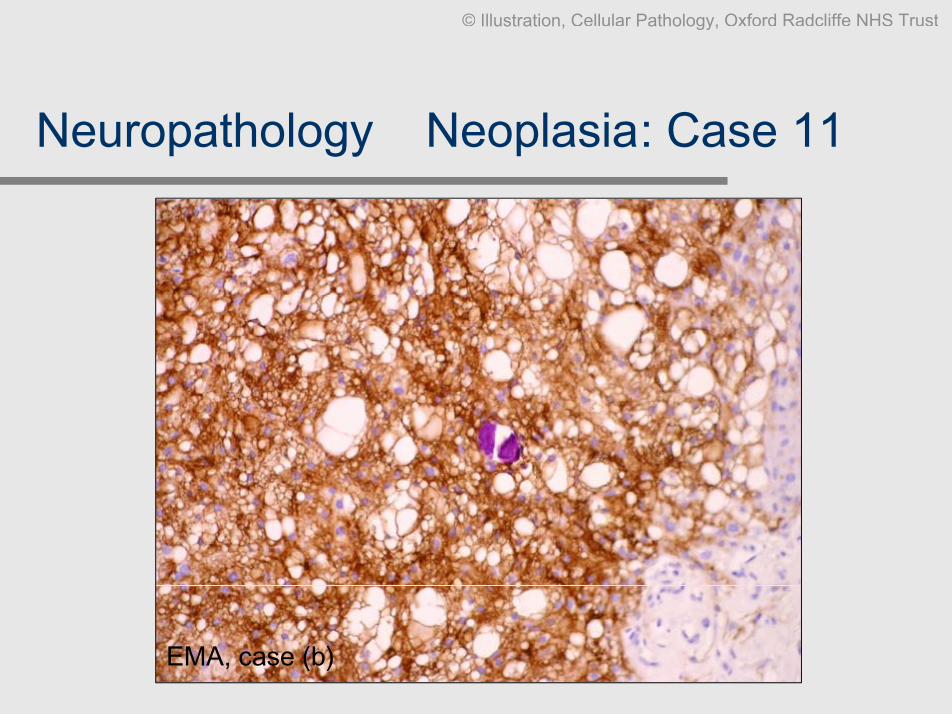
EMA, case (b)
Neuropathology Ð Neoplasia: Case 11
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 12
Clinical data: M, 62, 9/12 slowly progressive weakness. Enhancing parietal lesion
with dural base.
Slides: HE, GFAP
Diagnosis: Atypical meningioma, WHO grade II
Comment: Criteria for atypia in meningiomas include: loss of architecture
(ÔsheetingÕ), macronucleoli, multifocal necrosis, brain invasion, and aÔhighÕ mitotic rate (>4/10HPF according to the WHO). Usually acombination of these factors is taken into account when making thisdiagnosis. Some rare meningioma variants are primarily grade II orhigher (e.g. clear cell, chordoid variants).
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 12
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 12
GFAP
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 13
Clinical data: F, 52, previous dx meningioma (8/12 ago); now very large recurrence
left parietal.
Slides: HE, CD34
Diagnosis: Solitary fibrous tumour of the meninges
Comment: SFTs sometimes have been mistaken for fibroblastic meningiomas.
However, the focally ÔgapingÕ vessels, alternating strands of collagen-rich and myxoid areas, plumper cytology, and, most significantly,diffuse CD34-positivity combined with EMA-negativity are obviouswhen this entity is considered. The main differential diagnosis ishaemangiopericytoma (see case 12).
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 13
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 13
CD34
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 14
Clinical data: M, 43, 4/12 headache latterly confusion. Papilloedema, large frontal
dural-based tumour.
Slides: HE, reticulin, CD34
Diagnosis: Haemangiopericytoma of the meninges
Comment: Haemangiopericytomas can be distinguished from SFTs by their
cytology and more ÔcrowdedÕ sheet-like architecture lacking thefibrous strands and myxoid/hypocellular areas of SFTs. ClassicalÔstag-hornÕ vessels are present and there is a dense pericellularreticulin network. CD34 expression is usually weak and patchy.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 14
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 14
Reticulin CD34
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 15
Clinical data: M, 16, 4/12 headache/vomiting, worse over last week. Papilloedema,
cerebellar signs. Cystic post fossa lesion.
Slides: Smear methylene blue, biopsy HE
Diagnosis: Cerebellar haemangioblastoma, WHO grade I
Comment: Haemangioblastomas smear poorly; there is a mixture of dense
capillaries and plump vacuolated cells. Mast cells may be present.The HE shows the neoplastic ÔfoamyÕ cells much better. Thecapillaries are induced, not neoplastic. Haemangioblastoma mayindicate VHL syndrome.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 15
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 15
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 16
Clinical data: M, 52, slowly progressive deafness left ear, recently unsteadiness,
cerebello-pontine angle tumour.
Slides: HE
Diagnosis: Schwannoma (WHO grade I)
Comment: Cranial nerve schwannomas do not show classical Verocay bodies
as commonly as peripheral nerve tumours. There may be significantdegenerative change manifesting as cellular pleomorphism, cysts,hyalinised vessels and haemosiderin deposition.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 16
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 17
Clinical data: M, 18, occipital scalp lump, patient has NF1.
Slides: Biopsy HE, S100
Diagnosis: Malignant peripheral nerve sheath tumour (MPNST).
Comment: Note origin within a peripheral nerve and involvement of multiple
fascicles. A ÔschwannianÕ growth pattern is preserved in some areas;however appearances are mostly that of a highly malignant spindlecell tumour. S100 expression is surprisingly well preserved. If thereare no morphological features pointing to a specific lineagedifferentiation, an immunopanel for malignant soft tissue tumours isindicated.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 17
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 17
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust
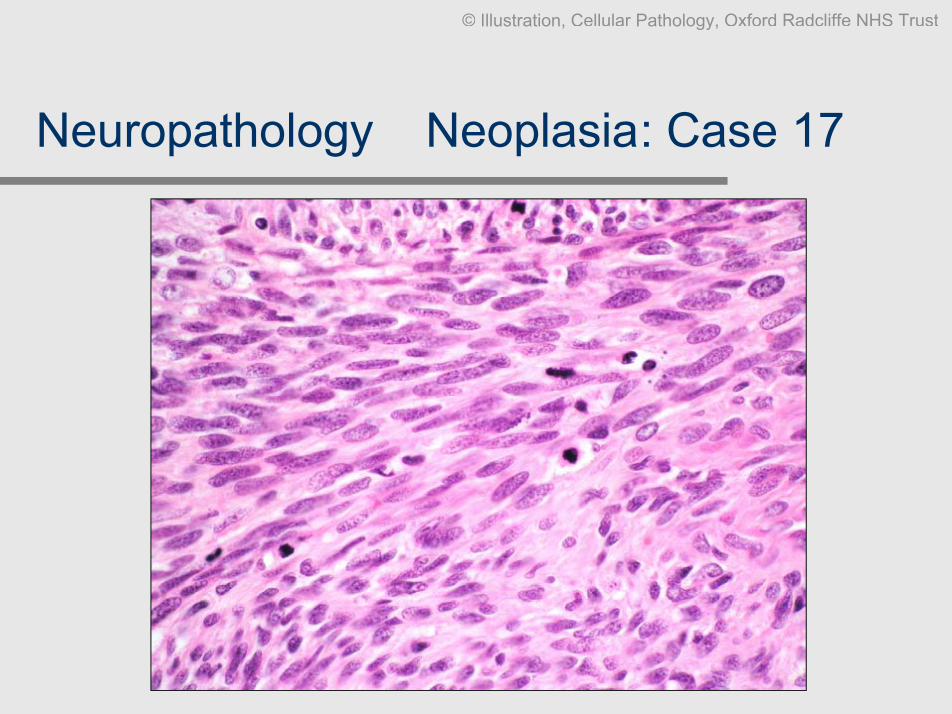
Neuropathology Ð Neoplasia: Case 17
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

S100
Neuropathology Ð Neoplasia: Case 17
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 18
Clinical data: M, 52, nosebleed, headache; tumour in sphenoid/clivus. ENT biopsy
transnasally.
Slides: Biopsy HE, CK, synaptophysin, prolactin
Diagnosis: Prolactinoma, no aggressive features (WHO grade I)
Comment: Pituitary adenomas may sometimes present to ENT departments
where this diagnosis is not suspected. Pituitary hormoneimmunohistochemistry and synaptophysin are diagnostic. Some (notthis one) may be strongly CK positive. Review of the imaging in thiscase demonstrated a link to the sella, however ectopic tumours canoccur in the nasopharynx
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 18
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Synaptophysin
Neuropathology Ð Neoplasia: Case 18
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Prolactin
Neuropathology Ð Neoplasia: Case 18
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 19
Clinical data: M, 24, hemianopia, panhypopituitarism, cystic suprasellar tumour.
Slides: Cyst fluid cytospin MGG, Biopsy HE
Diagnosis: Adamantinomatous craniopharyngioma, WHO grade I
Comment: Foamy macrophages and negative imprints of Ôstate of UtahÕ
cholesterol crystals are typical in cytospins. The reticular pattern withpalisading basal epithelium and dead keratin is diagnostic. If youhave seen one, you have seen them all. The papillary variant isexceedingly rare.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 19
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 19
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 20
Clinical data: M, 69, visual disturbances, headaches. Cystic tumour in the pituitary
fossa invading the skull base.
Slides: HE, PAS/AB, CK, S100.
Diagnosis: Chordoma.
Comment: Chordomas are related to remnants of the notochord and occur
mostly at the proximal and distal ends of the neuraxis. They are softtumours that are relentlessly invasive. PAS/AB highlights the intra-and extracellular mucins. Note the physalipherous cells (with giantvacuoles) and the combination of CK and S100 positivity(chondrosarcoma, the main DD, is CK negative).
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 20
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 20
PAS-OG CK
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 21
Clinical data: F, 10, diabetes insipidus since 12 months; slightly thickened
pituitary stalk
Slides: Biopsy HE, PLAP, c-kit.
Diagnosis: Suprasellar germinoma, WHO grade IV
Comment: A strong immune response to these tumours may lead to waxing
and waning symptoms and puzzling MRI findings. This is a classicexample with strong lymphocytic response with aberrant follicleformation. C-kit may be helpful because PLAP is often very weak
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 21
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 21
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

PLAP
Neuropathology Ð Neoplasia: Case 21
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 22
Clinical data: M, 6, parents noted skull swelling, osteolytic lesion on CT.
Slides: HE, CD1a, S100.
Diagnosis: Eosinophilic granuloma (Langerhans cell histiocytocis).
Comment: The diagnosis is often not suspected clinically. This is a classical
example with well-formed CD1a-positive tumour cell aggregatesintermingled with eosinophils and a rim of lymphocytes at theperiphery. Extensive sampling may be necessary in cases withwidespread fibrosis.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 22
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 22
CD1a
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 23
Clinical data: M, 51, visual disturbances, headaches, vomiting. Occipital contrast-
enhancing lesion. Otherwise well. ?glioblastoma.
Slides: Biopsy HE, CK7, CK20, TTF-1.
Diagnosis: Metastatic adenocarcinoma, c/w lung primary.
Comment: So-called Ômetastatic carcinomas of unknown originÕ are quite
common in surgical neuropathology. A limited immunopanel guidedby morphology can often pinpoint the likely primary site. This patientproved to have a large pulmonary mass (which was obvious on plainchest x-ray but missed due to the dramatic neurologicalpresentation).
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 23
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

CK7
Neuropathology Ð Neoplasia: Case 23
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

CK20
Neuropathology Ð Neoplasia: Case 23
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

TTF-1
Neuropathology Ð Neoplasia: Case 23
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 24
Clinical data: F, 75, Ovarian Ca resected and chemo 8 years ago. Now drowsiness,
confusion. CT brain and abdomen: normal.
Slides: CSF MGG, CK; PM cerebellum, spinal cord.
Diagnosis: Meningeal carcinomatosis (c/w primary adeno Ca).
Comment: Meningeal carcinomatosis presents with non-specific diffuse CNS
dysfunction and is often not suspected. CSF is diagnostic but cavesampling bias. Often there is also a significant inflammatory reactionwhich may be misleading if there are only rare malignant cells.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

CSF - MGG
Neuropathology Ð Neoplasia: Case 24
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

CK - CSF
Neuropathology Ð Neoplasia: Case 24
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

CK, cerebellum
Neuropathology Ð Neoplasia: Case 24
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 25
Clinical data: M, 23, 6 weeks of somnolence, confusion, coma. MRI
hyperintensities hypothalamus and mesial temporal lobe.
Slides: (a) Amygdala HE, GFAP; (b) testes x2, PLAP.
Diagnosis: Paraneoplastic encephalitis (anti-MA2-positive) due to malignant
testicular intratubular germ cell tumour.
Comment: This is unlikely to be a real-life exam case. However, it emphasizes
the sometimes severe remote effects on the CNS of systemic, non-metastatic cancer. Strongly immunogenic Ôonconeuronal antigensÕare responsible for this, destroying the brain and keeping the cancerin check (it is often occult)
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

(a) Temporal lobe
Neuropathology Ð Neoplasia: Case 25
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

(a) Temporal lobe, inflammation, gliosis (GFAP)
Neuropathology Ð Neoplasia: Case 25
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

(b) Testis (neoplastic)
Neuropathology Ð Neoplasia: Case 25
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

(b) Testis (neoplastic), PLAP
Neuropathology Ð Neoplasia: Case 25
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 26
Clinical data: M, 62, weakness and sensory deficits at several spinal cord levels
developing over a few weeks. No diagnosis during life.
Slides: HE, LFB-CV, CD20.
Diagnosis: Intravascular B-cell lymphoma leading to multifocal ischemic lesions
of the spinal cord.
Comment: The CNS is often clinically affected by intravascular lymphoma due
to vasoocclusive disease. The diagnosis can be missed if patientsundergo biopsy during life. Virtually all intravascular lymphomas areof B-cell type. The myelin pallor of the dorsal columns in this case isprobably the result of the severe involvement of the dorsal roots.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 26
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuropathology Ð Neoplasia: Case 26
CD20
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuroinfection Ð Case 27
Clinical data: M, 39, HIV +ve for 9 years. 8 weeks confusion and occasional
seizures. Multiple lesions on MRI.
Slides: PM cerebellum, neocortex, basal ganglia (HE and toxoplasma
immunostain).
Diagnosis: HIV-associated CMV encephalitis, cryptococcosis, toxoplasmosis.
Comment: Note the bizarre CMV-transformed cells with nuclear virus (usually
close to the ventricular surface). The cryptococcus lesion is mainlynecrotic. There are both free as well as cystic forms of toxoplasma.As always, viable organisms are best appreciated at the edge of alesion.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

CMV
Neuroinfection Ð Case 27
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

TOXO (low-power, edge of lesion, cysts – arrows)
Neuroinfection Ð Case 27
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

TOXO(immunostain labels both forms, cyst – arrow)
Neuroinfection Ð Case 27
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuroinfection Ð Case 28
Clinical data: F, 23, drowsiness, mild fever, headaches. CT cortical nodule. Gap
year in Brazil.
Slides: CSF MGG, biopsy: cyst, brain parenchyma, HE.
Diagnosis: CSF: inflammatory with eosinophilia.
Biopsy: cysticercosis.
Comment: CSF eosinophilia is rare and may indicate a parasitic infection.
Biopsy: typical appearance of the cysticercus. The brain showsreactive changes Ð these are often the cause of the symptoms,particularly if the parasite dies or the cyst ruptures. Many cysticercimay remain ÔdormantÕ (clinically silent) throughout a patients life!
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

CSF
Neuroinfection Ð Case 28
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuroinfection Ð Case 28
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuroinfection Ð Case 28
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Reactive brain
Neuroinfection Ð Case 28
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurodegeneration Ð Case 29
Clinical data: F, 76, 7 months history of rapid-onset dementia and lethargy. Nil
else.
Slides: PM neocortex, cerebellum, HE and KG9 (anti PrP).
Diagnosis: Sporadic Creutzfeldt-Jakob disease, Codon 129 M/M.
Comment: This is the most common form of CJD. Note the patchy distribution
of spongiosis and PrP deposition. The pattern is diffuse / synaptic /perivacuolar and typical ÔKuruÕ type plaques are not present. In thisform of CJD the cerebrum is more affected than the cerebellum, andthere is no lymphoreticular infectivity (please compare with the nextcase (19)).
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurodegeneration Ð Case 29
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurodegeneration Ð Case 29
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurodegeneration Ð Case 30
Clinical data: M, 31, 16 months of depression and irritability, followed by ataxia and
finally severe dementia.
Slides: PM neocortex, cerebellum, HE and KG9 (PrP) antibody.
Diagnosis: Variant Creutzfeldt-Jakob disease, Codon 129 M/M.
Comment: The ÔfloridÕ amyloid plaque with a corona of vacuoles is characteristic of
vCJD. The PrP load is usually high and shows plaque, diffuse andperivacuolar patterns. The cerebellum is severely affected and PrP ispresent systemically. So far all symptomatic vCJD patients are Codon129 MM, however 2004 saw the first MV case with systemic only PrPdeposition after a blood transfusion.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurodegeneration Ð Case 30
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Cluster-plaques
Neurodegeneration Ð Case 30
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurodegeneration Ð Case 31
Clinical data: M, 71, 10 years progressive slowness, stiffness, left-sided tremor.
Recently dementia.
Slides: PM midbrain, amygdala HE, alpha-synuclein.
Diagnosis: Lewy-body ParkinsonÕs disease with limbic/neocortical
involvement.
Comment: Lewy-bodies are intraneuronal eosinophilic inclusions composed
mainly of alpha-synuclein. They occur mainly in the nigra but alsoin limbic neocortical systems where they may correlate with PD-associated dementia.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Nigra with Lewy bodies - arrow
Neurodegeneration Ð Case 31
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Nigra, alpha-synuclein positive Lewy body - arrow
Neurodegeneration Ð Case 31
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Amygdala with Lewy bodies - arrows
Neurodegeneration Ð Case 31
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurodegeneration Ð Case 32
Clinical data: M, 54, Trisomy 21.
Slides: Mesial temporal lobe, HE, beta-A4, phospho-tau (AT8).
Diagnosis: AlzheimerÕs disease pathology in Trisomy 21.
Comment The triplicated region of chromosome 21 in DownÕs syndrome
includes the gene for βAPP. ÔOverexpressionÕ of βAPP in thiscondition leads to βA4 amyloid plaque deposition and amyloidangiopathy. This is associated with formation of tau-containingneurofibrillary tangles. The neuropathology is essentially identical tothat of sporadic AlzheimerÕs disease.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Plaques
Neurodegeneration Ð Case 32
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Plaques, bA4
Neurodegeneration Ð Case 32
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Tangle
Neurodegeneration Ð Case 32
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Tangle and neurites, AT8-tau
Neurodegeneration Ð Case 32
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuroinflammation Ð Case 33
Clinical data: F, 53, long history of relapsing-remitting neurological illness
affecting brain, brainstem and spinal cord.Slides:
HE, LFB-CV, CD68, Neurofilament, Bielschowsky silver.Diagnosis:
Multiple sclerosis.Comment:
As illustrated in this section, MS plaques are often concentratedaround CSF spaces (ventricles). Note the sharp demarcation ofthe demyelinated area and the presence of myelin debris (bluegranules) within macrophages at the periphery of the lesion (LFB-CV stain). Note the relative preservation of neurons and axonswithin the demyelinated area (in an infarct both neuronal as wellas oligodendroglial/myelin components would be lost).
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuroinflammation Ð Case 33
LFB-CV CD68
Note: pale demyelinatedarea corresponding tomacrophage-rich area.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neuroinflammation Ð Case 33
LFB-CV CD68
myelin-ladenmacrophages
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurotrauma Ð Case 34
Clinical data: M, 72, acute fracture of the spinal cord, survived 3 days.
Slides: HE, beta-APP, Bielschowsky silver.
Diagnosis: Acute traumatic axonal injury of the spinal cord.
Comment: Diffuse traumatic axonal injury may be the only histological manifestation
of a severe head / spinal injury. The earliest changes are dystrophicaxons that accumulate proteins that are axonally transported (antibodiesto beta-amyloid precurser protein (beta-APP) are the most commonlyused surrogate markers since these changes are rarely visible on H&E).The classical axonal retraction bulbs or axonal spheroids only becomeapparent after 12-36 hours of post-traumatic survival (note that this is nota fool proofed method to determine the time of the injury). The axonalspheroids are apparent on H&E and can be highlighted by silver stainsand immunocytochemical techniques.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Neurotrauma Ð Case 34
Axonal retraction bulbs - arrows
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust
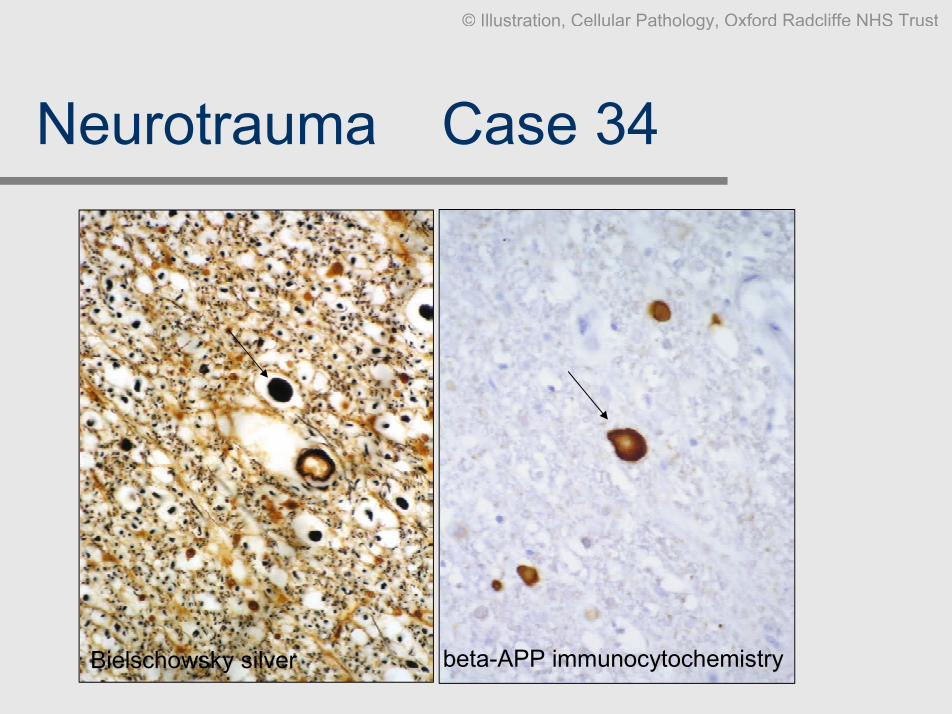
Neurotrauma Ð Case 34
Bielschowsky silver beta-APP immunocytochemistry
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Muscle disease Ð Case 35
Clinical data: M, 66, known peripheral motor-sensory neuropathy.
Slides: HE, NADH.
Diagnosis: Muscle with longstanding denervation and reinnervation.
Comment: Note the increased fat and the relatively well-defined groups of
grossly atrophic fibres alternating with groups of hypertrophicfibres. The latter contain central ring-like structures and are calledÔtarget-fibresÕ; thought to be a sign of re-innervation. The groupatrophy in this example is severe, resulting in nuclear aggregatesand rounded fibres. Early atrophic fibres are often angulated.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Muscle disease Ð Case 35
Group atrophy / hypertrophy / increased fat
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Muscle disease Ð Case 35
Target fibres
NADH
PAS
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Muscle disease Ð Case 36
Clinical data: M, 52, progressive proximal lower and upper limb weakness.
Slides: HE, Gomori trichrome, succinate dehydrogenase (SDH),
cytochrome c oxidase (COX).Diagnosis:
Mitochondrial myopathy.Comment:
The mitochondrial cytopathies can manifest with a wide range ofneurological and non-neurological symptoms. Muscle biopsy is astandard diagnostic approach to these disorders. Not allmitochondrial cytopathies show the classical Ôragged-red fibresÕ(RRF) on Gomori stain; mitochondrial genetics may be necessary.The classical RRFÕs contain aggregated mitochondria, increasedSDH staining and absent COX staining, as in this example.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Muscle disease Ð Case 36
HE can often be unremarkable Gomori trichrome Ð ragged-red fibres
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Muscle disease Ð Case 36
SDH COX
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Miscellaneous CSFs Ð Case 37
Clinical data: F, 30, MS and psychosis.
Slides:CSF MGG, UCHL1, CD20.
Diagnosis: Lymphoplasmacytoid pleocytosis (reactive).
Comment:Most reactive CSF lymphocytic pleocytoses are T-cell
driven, most lymphoid neoplasms are of B-cell lineage.Acute MS may show a dramatic pleocytosis (generally,CSF cytology is NOT indicated in MS).
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

UCHL-1
Miscellaneous CSFs Ð Case 37
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

CD20
Miscellaneous CSFs Ð Case 37
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Miscellaneous CSFs Ð Case 38
Clinical data: M, 1, known AML M4, drowsy.
Slides: CSF MGG.
Diagnosis: Intrathecal dissemination of AML.
Comment: AML and other haematological neoplasms may disseminate via
CSF spaces, particularly in children. Limited phenotyping ispossible but often not necessary because the underlyinghaematooncological entity is already known.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Miscellaneous CSFs Ð Case 38
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Miscellaneous CSFs Ð Case 39
Clinical data:M, 37, headaches, papilloedema.
Slides:CSF MGG, PAS, mucicarmine.
Diagnosis:Cryptococcus neoformans meningitis.
Comment:Not uncommon in severely immunosuppressed patients,
particularly AIDS. May be missed if the CSFinflammatory pleocytosis is significant. PAS/mucicarmineis best and shows the capsule of the yeast forms.
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Miscellaneous CSFs Ð Case 39
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Miscellaneous CSFs Ð Case 39
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Miscellaneous CSFs Ð Case 40
Clinical data: F, 35, breast CA.
Slides: CSF MGG.
Diagnosis: Meningeal carcinomatosis, c/w adenocarcinoma (breast)
primary.
Comment: This is a barn-door case. Note the bizarre vacuoles of the
carcinoma cells.
I hope all exam cases will be like this !
Good luck!
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Miscellaneous CSFs Ð Case 40
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust

Note:The electronic imagescontained within this
document are for personaluse only!
© Illustration, Cellular Pathology, Oxford Radcliffe NHS Trust







