
Bioinformática: Inferência filogenética
WHYDO WE CARE ?
Rita Castilho, [email protected]
What for?
Sistemática Molecular
Evolução
Ecologia
Forense
Medicina (evolução de vírus, vacinas, desenvolvimento de drogas)
Uses of phylogenies: Sistemática• Similar organisms are
grouped together
• Clades share common evolutionary history
• Phylogenetic classification names clades
Source: Inoue, J.G., Miya, M., Tsukamoto, K., Nishida, M. 2003.
Basal actinopterygian relationships: a mitogenomic perspective on the
phylogeny of the “ancient fish”. Molecular Phylogenetics and
Evolution, 26: 110-120.
Pryer et al. 2001
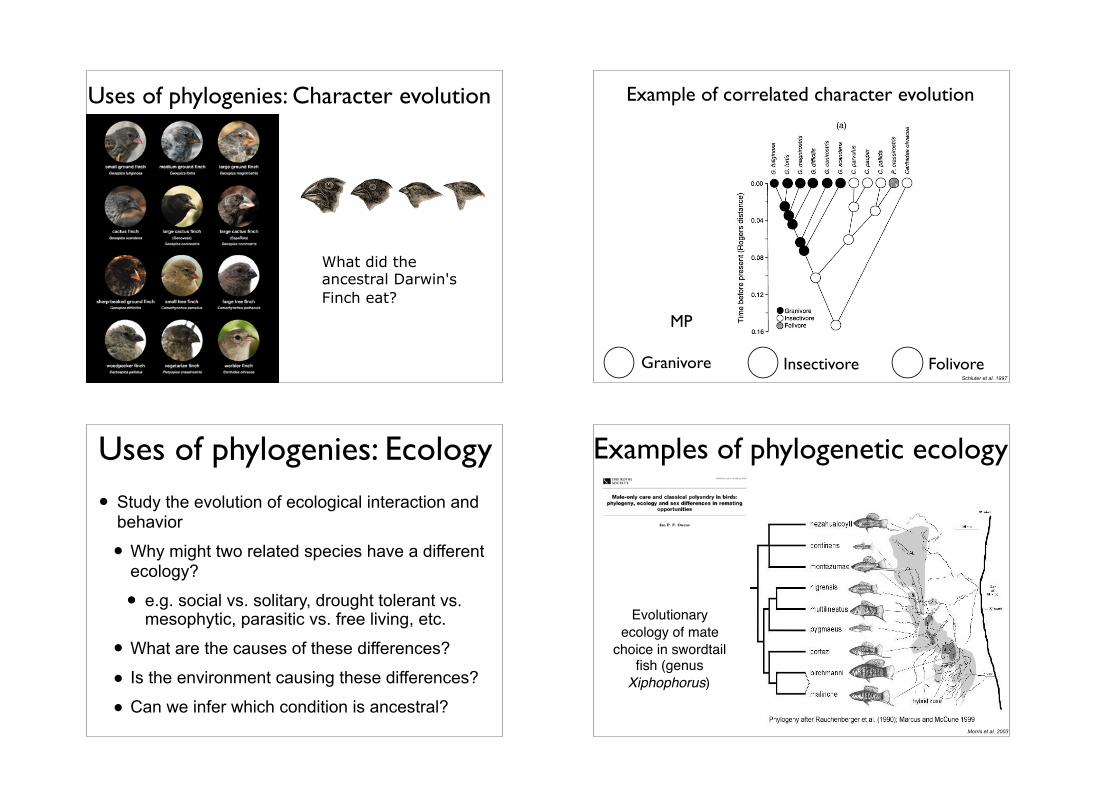
Uses of phylogenies: Character evolution
What did the ancestral Darwin's Finch eat?
Example of correlated character evolution
Granivore Insectivore Folivore
MP
Schluter et al. 1997
Uses of phylogenies: Ecology
• Study the evolution of ecological interaction and behavior
• Why might two related species have a different ecology?
• e.g. social vs. solitary, drought tolerant vs. mesophytic, parasitic vs. free living, etc.
• What are the causes of these differences?
• Is the environment causing these differences?
• Can we infer which condition is ancestral?
Examples of phylogenetic ecology
Evolutionary ecology of mate
choice in swordtail fish (genus Xiphophorus)
Morris et al. 2003
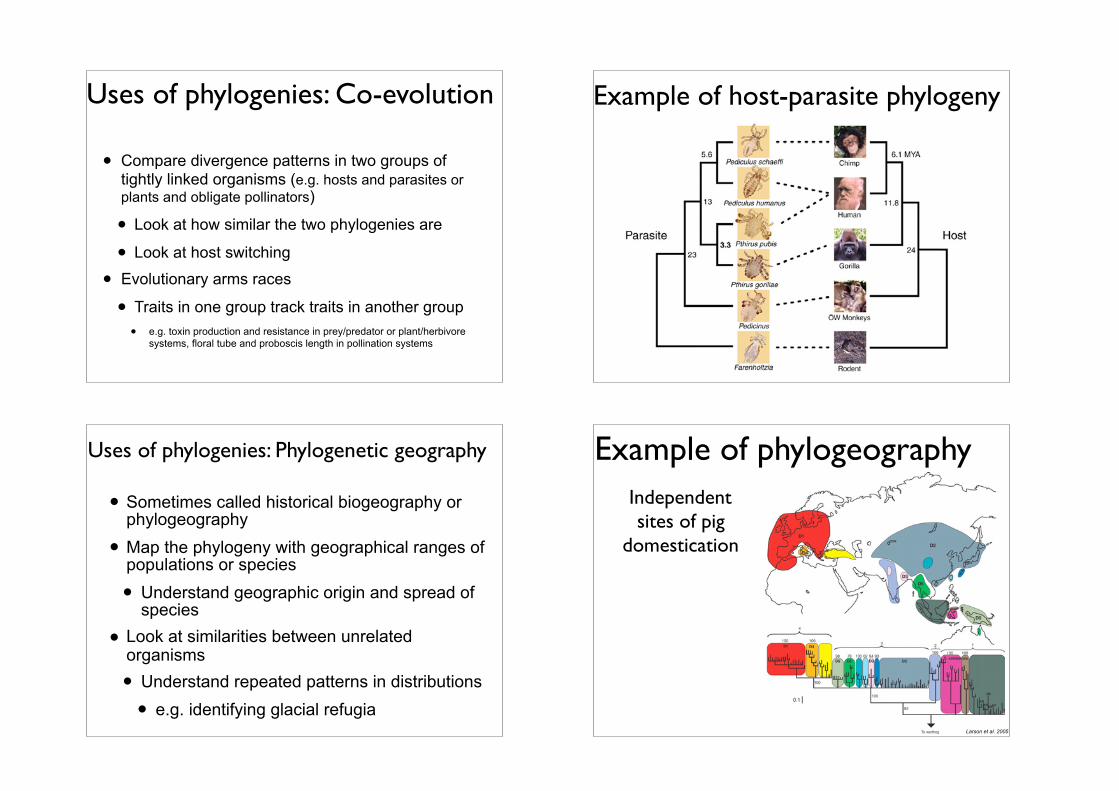
Uses of phylogenies: Co-evolution
• Compare divergence patterns in two groups of tightly linked organisms (e.g. hosts and parasites or plants and obligate pollinators)
• Look at how similar the two phylogenies are
• Look at host switching
• Evolutionary arms races
• Traits in one group track traits in another group • e.g. toxin production and resistance in prey/predator or plant/herbivore
systems, floral tube and proboscis length in pollination systems
Example of host-parasite phylogeny
Uses of phylogenies: Phylogenetic geography
• Sometimes called historical biogeography or phylogeography
• Map the phylogeny with geographical ranges of populations or species
• Understand geographic origin and spread of species
• Look at similarities between unrelated organisms
• Understand repeated patterns in distributions
• e.g. identifying glacial refugia
Example of phylogeographyIndependent sites of pig
domestication
Larson et al. 2005

Example of phylogeography Example of phylogeography
Example of phylogeography
The highly diversified genus Conus includes more then 500 species of venomous marine snails
Example of phylogeographyAll endemic Cape Verde Conus:
are vermivorous (prey on polychaete annelids)
are nonplanktonic lecithotrophic
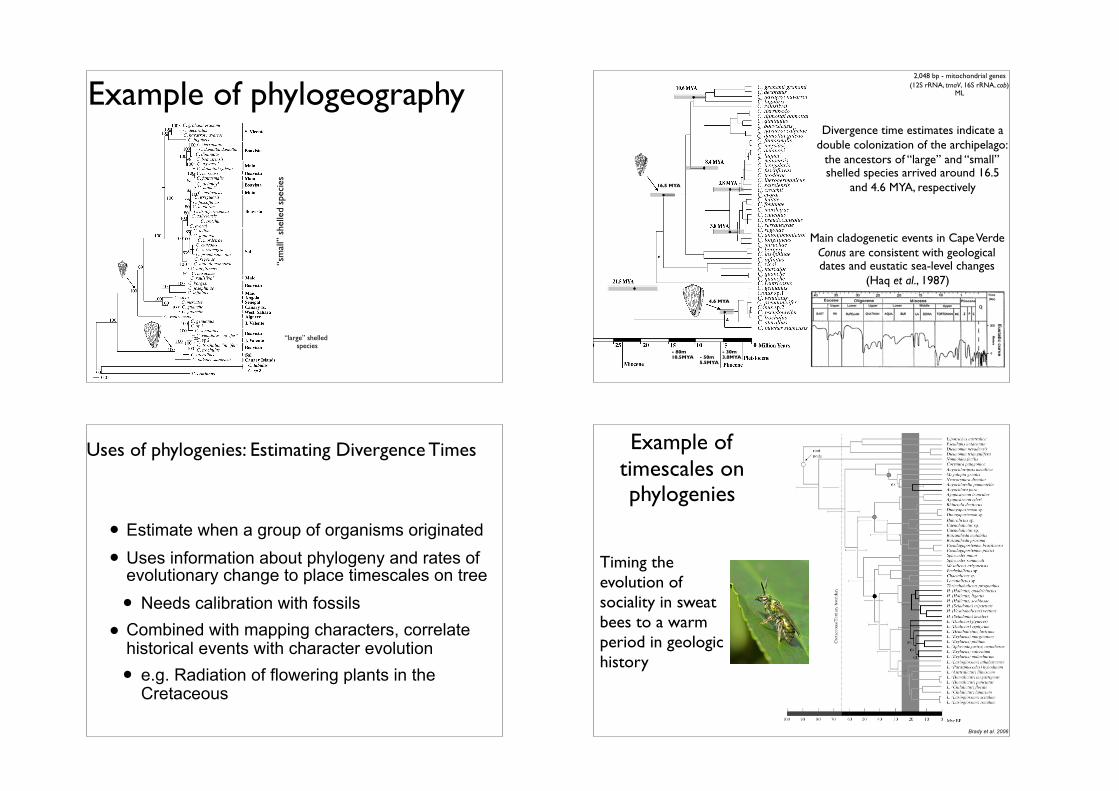
Example of phylogeography
“sm
all”
she
lled
spec
ies
“large” shelled species
Divergence time estimates indicate a double colonization of the archipelago:
the ancestors of “large” and “small” shelled species arrived around 16.5
and 4.6 MYA, respectively
Main cladogenetic events in Cape Verde Conus are consistent with geological dates and eustatic sea-level changes
(Haq et al., 1987)
- 80m10.5MYA - 50m
5.5MYA
- 30m3.8MYA
16.5 MYA
4.6 MYA
2,048 bp - mitochondrial genes (12S rRNA, trnaV, 16S rRNA, cob)
ML
Uses of phylogenies: Estimating Divergence Times
• Estimate when a group of organisms originated
• Uses information about phylogeny and rates of evolutionary change to place timescales on tree
• Needs calibration with fossils
• Combined with mapping characters, correlate historical events with character evolution
• e.g. Radiation of flowering plants in the Cretaceous
Example of timescales on phylogenies
Timing the evolution of sociality in sweat bees to a warm period in geologic history
Brady et al. 2006
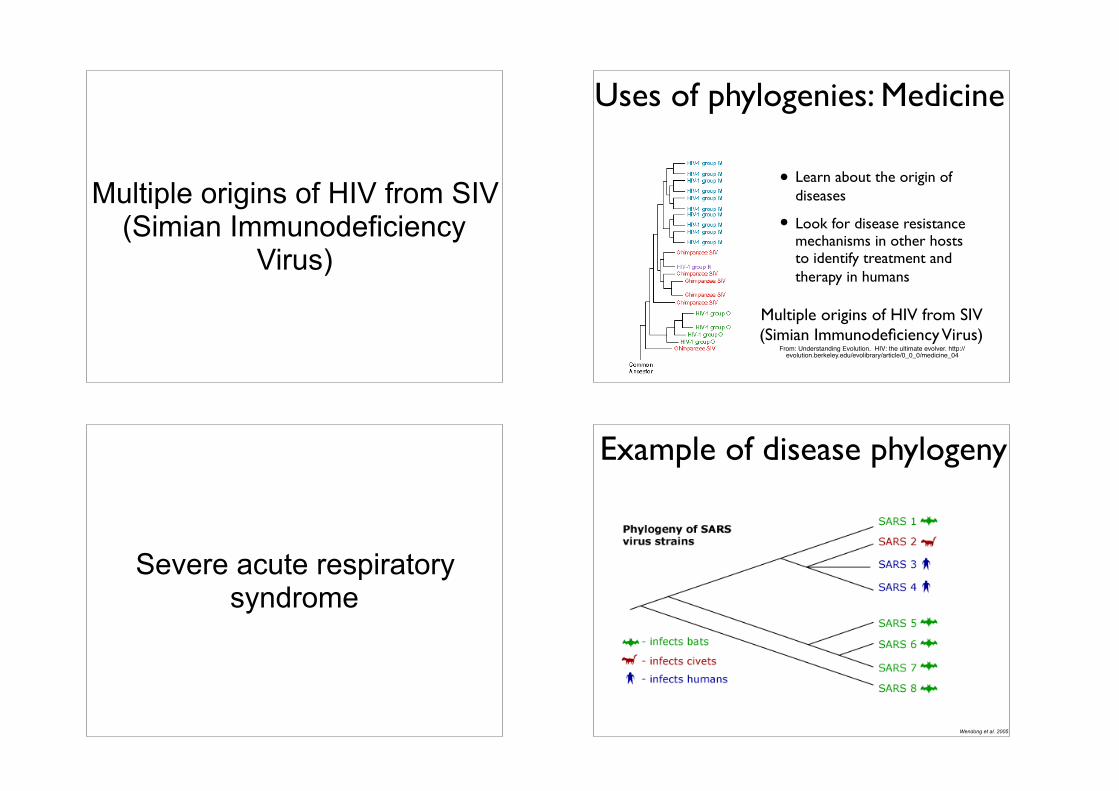
Multiple origins of HIV from SIV (Simian Immunodeficiency
Virus)
Uses of phylogenies: Medicine
• Learn about the origin of diseases
• Look for disease resistance mechanisms in other hosts to identify treatment and therapy in humans
Multiple origins of HIV from SIV (Simian Immunodeficiency Virus)
From: Understanding Evolution. HIV: the ultimate evolver. http://evolution.berkeley.edu/evolibrary/article/0_0_0/medicine_04
Severe acute respiratory syndrome
Example of disease phylogeny
Wendong et al. 2005
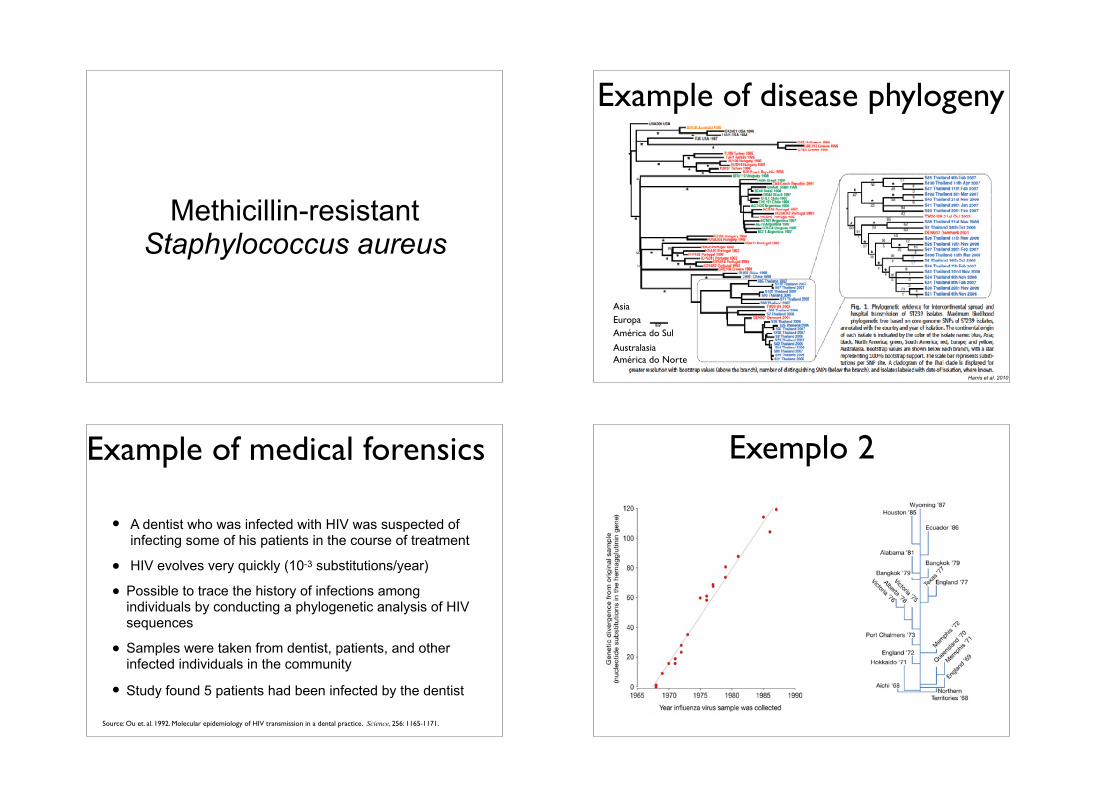
Methicillin-resistant Staphylococcus aureus
AsiaEuropaAmérica do Sul
AustralasiaAmérica do Norte
Example of disease phylogeny
Harris et al. 2010
Example of medical forensics
• A dentist who was infected with HIV was suspected of infecting some of his patients in the course of treatment
• HIV evolves very quickly (10-3 substitutions/year)
• Possible to trace the history of infections among individuals by conducting a phylogenetic analysis of HIV sequences
• Samples were taken from dentist, patients, and other infected individuals in the community
• Study found 5 patients had been infected by the dentist
Source: Ou et. al. 1992. Molecular epidemiology of HIV transmission in a dental practice. Science, 256: 1165-1171.
Exemplo 2

Phylogeny and molecular evolution
Rita Castilho, [email protected]
=
Determining the common origin (ancestral)
What are phylogenies for?

Latimeria
Protopterus
What is the most recent common ancestor of tetrapods?
Latimeria
Protopterus
Coelacanth
What is the most recent common ancestor of tetrapods?
Latimeria
Protopterus
Lungfish
What is the most recent common ancestor of tetrapods?
Latimeria
Protopterus
Teleost
What is the most recent common ancestor of tetrapods?
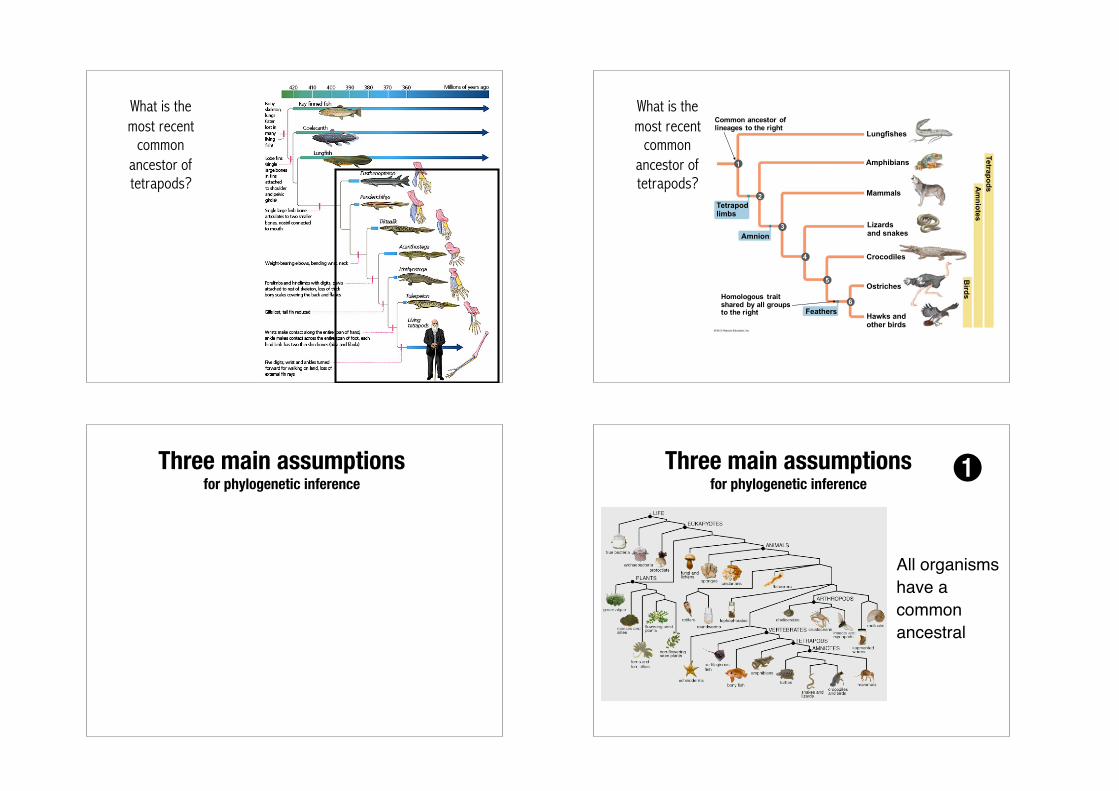
What is the most recent
common ancestor of tetrapods?
What is the most recent
common ancestor of tetrapods?
Tetrapodlimbs
Amnion
Feathers
Lungfishes
Mammals
Amphibians
Lizardsand snakes
Crocodiles
Hawks and other birds
Ostriches
Am
niotesTetrapods
Birds
Common ancestor oflineages to the right
Homologous traitshared by all groupsto the right
2
1
3
4
6
5
Three main assumptions for phylogenetic inference
Three main assumptions for phylogenetic inference
All organisms have a common ancestral
➊
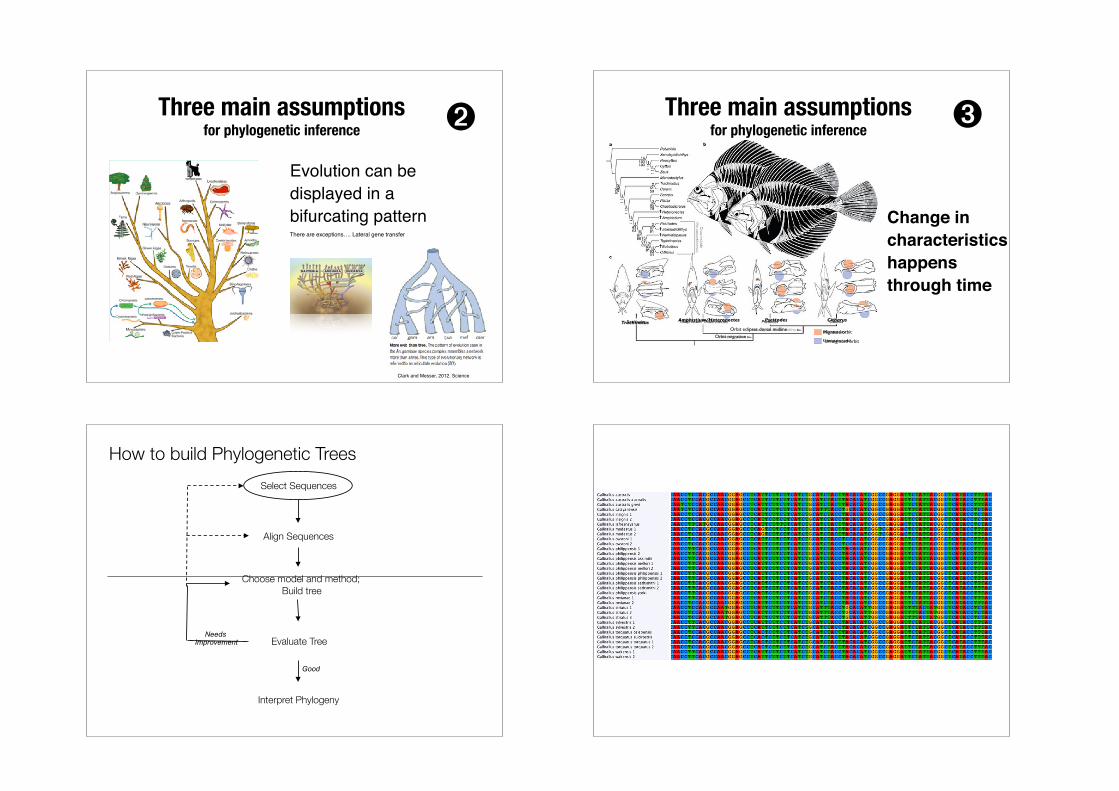
Evolution can be displayed in a bifurcating patternThere are exceptions…. Lateral gene transfer
Three main assumptions for phylogenetic inference ➋
Clark and Messer, 2012. Science
Change in characteristics happens through time
Orbit eclipses dorsal midline
Orbit migration
CitharusPsettodesAmphistium/HeteronectesTrachinatus
Migrated orbit
Unmigrated orbit
Three main assumptions for phylogenetic inference ➌
How to build Phylogenetic TreesSelect Sequences
Align Sequences
Choose model and method; Build tree
Evaluate Tree
Interpret Phylogeny
Good
Needs Improvement
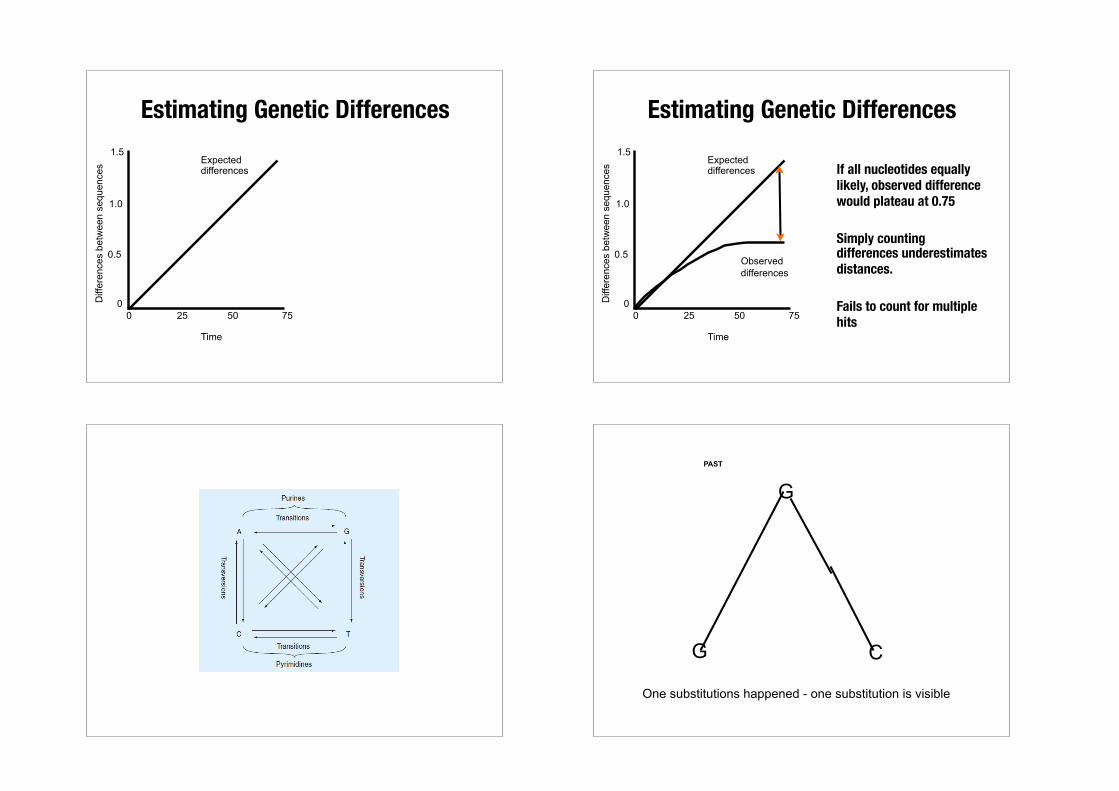
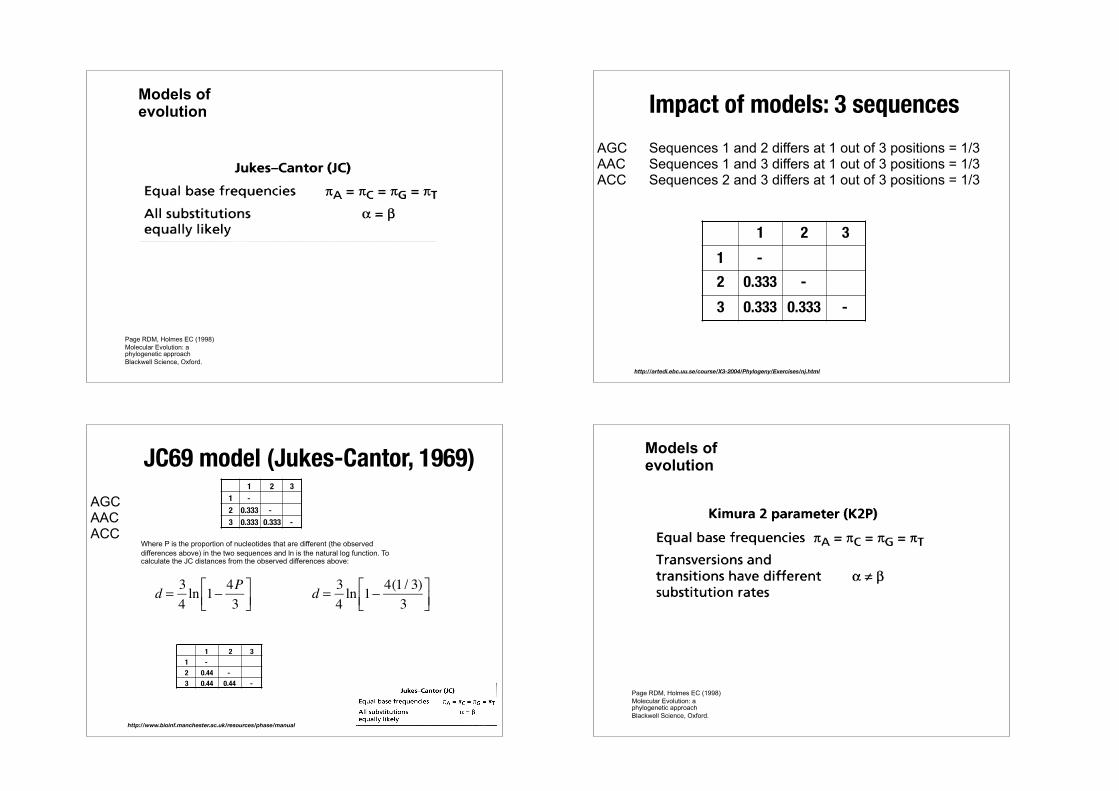
Estimating Genetic Differences
0 25 50 750
0.5
1.0
1.5Expected differences
Time
Diff
eren
ces
betw
een
sequ
ence
sEstimating Genetic Differences
If all nucleotides equally likely, observed difference would plateau at 0.75
Simply counting differences underestimates distances.
Fails to count for multiple hits 0 25 50 75
0
0.5
1.0
1.5Expected differences
Observed differences
Time
Diff
eren
ces
betw
een
sequ
ence
s
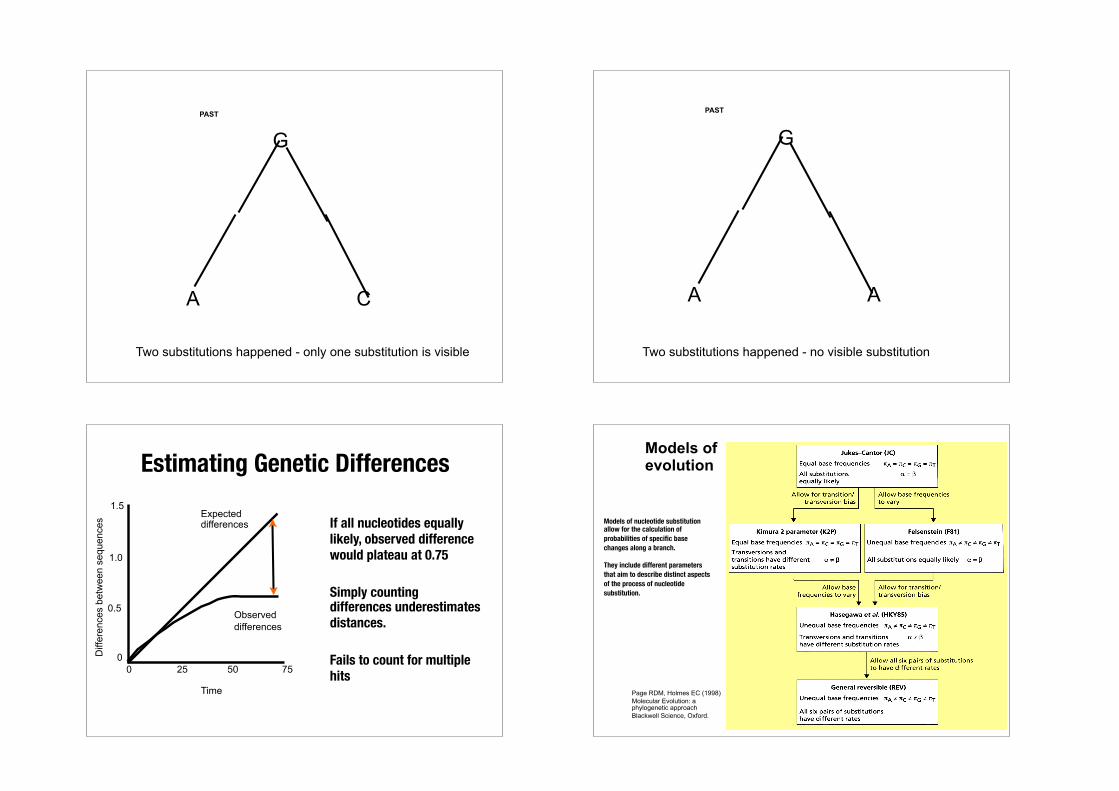
One substitutions happened - one substitution is visible
G
CG
PAST
G
CA
PAST
Two substitutions happened - only one substitution is visible Two substitutions happened - no visible substitution
GPAST
A A
Estimating Genetic Differences
If all nucleotides equally likely, observed difference would plateau at 0.75
Simply counting differences underestimates distances.
Fails to count for multiple hits 0 25 50 75
0
0.5
1.0
1.5Expected differences
Observed differences
Time
Diff
eren
ces
betw
een
sequ
ence
s
Page RDM, Holmes EC (1998) Molecular Evolution: a phylogenetic approach Blackwell Science, Oxford.
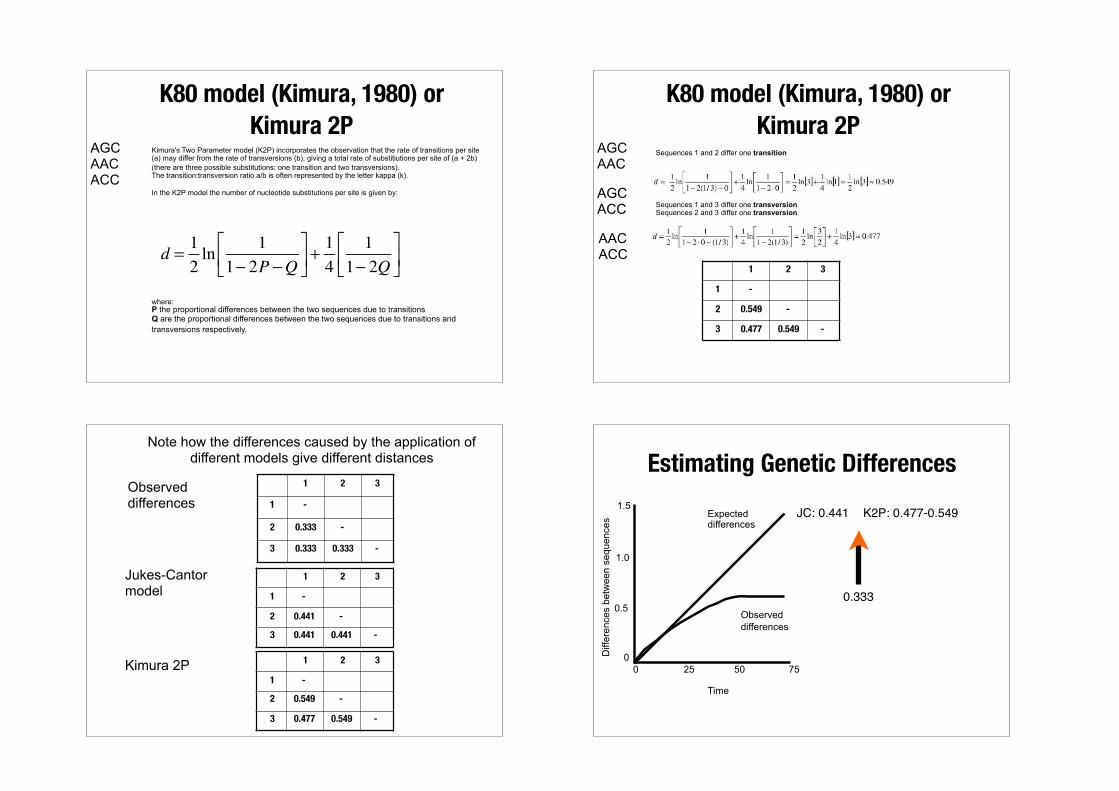
Models of evolution
Models of nucleotide substitution allow for the calculation of probabilities of specific base changes along a branch.
They include different parameters that aim to describe distinct aspects of the process of nucleotide substitution.
Page RDM, Holmes EC (1998) Molecular Evolution: a phylogenetic approach Blackwell Science, Oxford.
Models of evolution Impact of models: 3 sequences
http://artedi.ebc.uu.se/course/X3-2004/Phylogeny/Exercises/nj.html
AGC AAC ACC
Sequences 1 and 2 differs at 1 out of 3 positions = 1/3 Sequences 1 and 3 differs at 1 out of 3 positions = 1/3 Sequences 2 and 3 differs at 1 out of 3 positions = 1/3
1 2 31 -2 0.333 -3 0.333 0.333 -
JC69 model (Jukes-Cantor, 1969)
http://www.bioinf.manchester.ac.uk/resources/phase/manual
Where P is the proportion of nucleotides that are different (the observed differences above) in the two sequences and ln is the natural log function. To calculate the JC distances from the observed differences above:
1 2 31 -2 0.333 -3 0.333 0.333 -
1 2 31 -2 0.44 -3 0.44 0.44 -
AGC AAC ACC
d = 34ln 1− 4P
3⎡⎣⎢
⎤⎦⎥
d = 34ln 1− 4(1 / 3)
3⎡⎣⎢
⎤⎦⎥
Page RDM, Holmes EC (1998) Molecular Evolution: a phylogenetic approach Blackwell Science, Oxford.
Models of evolution
K80 model (Kimura, 1980) orKimura 2P
Kimura's Two Parameter model (K2P) incorporates the observation that the rate of transitions per site (a) may differ from the rate of transversions (b), giving a total rate of substitiutions per site of (a + 2b)(there are three possible substitutions: one transition and two transversions). The transition:transversion ratio a/b is often represented by the letter kappa (k).
In the K2P model the number of nucleotide substitutions per site is given by:
where: P the proportional differences between the two sequences due to transitions Q are the proportional differences between the two sequences due to transitions and transversions respectively.
AGC AAC ACC
d = 12ln 11− 2P −Q⎡⎣⎢
⎤⎦⎥+ 14
11− 2Q⎡⎣⎢
⎤⎦⎥
K80 model (Kimura, 1980) orKimura 2P
Sequences 1 and 3 differ one transversion Sequences 2 and 3 differ one transversion
AGC AAC
Sequences 1 and 2 differ one transition
AGC ACC
AAC ACC
1 2 3
1 -
2 0.549 -
3 0.477 0.549 -
1 2 3
1 -
2 0.549 -
3 0.477 0.549 -
1 2 3
1 -
2 0.441 -
3 0.441 0.441 -
1 2 3
1 -
2 0.333 -
3 0.333 0.333 -
Observed differences
Jukes-Cantor model
Kimura 2P
Note how the differences caused by the application of different models give different distances Estimating Genetic Differences
0 25 50 750
0.5
1.0
1.5Expected differences
Observed differences
Time
Diff
eren
ces
betw
een
sequ
ence
s0.333
JC: 0.441 K2P: 0.477-0.549

Molecular Clock
Proposed that for any given protein, the rate of molecular evolution is approximately constant over time in all lineages.
Molecules reflect evolutionary divergence
0
15
30
45
60
0 275 550 825 1100
Time since divergence (millions of years)
Am
ino
acid
sub
stitu
tions
(p
er 1
00 re
sidu
es) i
n cy
toch
rom
e c
Yeast vs mould
Angiosperms vs animals
Insects vs vertebrates
Fish vs land vertebrates
Amphibians vs birds and mammals
Birds vs mammals
Mammals vs reptiles
Birds vs reptiles
A molecular clock for Cytochrome c

Bioinformática: Inferência filogenética
HOW DO WE DO IT ?
Rita Castilho, [email protected]
ancestor
descendant 1 descendant 2
ASSUMPTION: LIFE IS
MONOPHYLETIC
Time
Reading trees
B C DA
“Root”: common ancestor of organisms in the phylogeny
Reading trees
Nó ancestral ou Raíz da árvore
B C DA
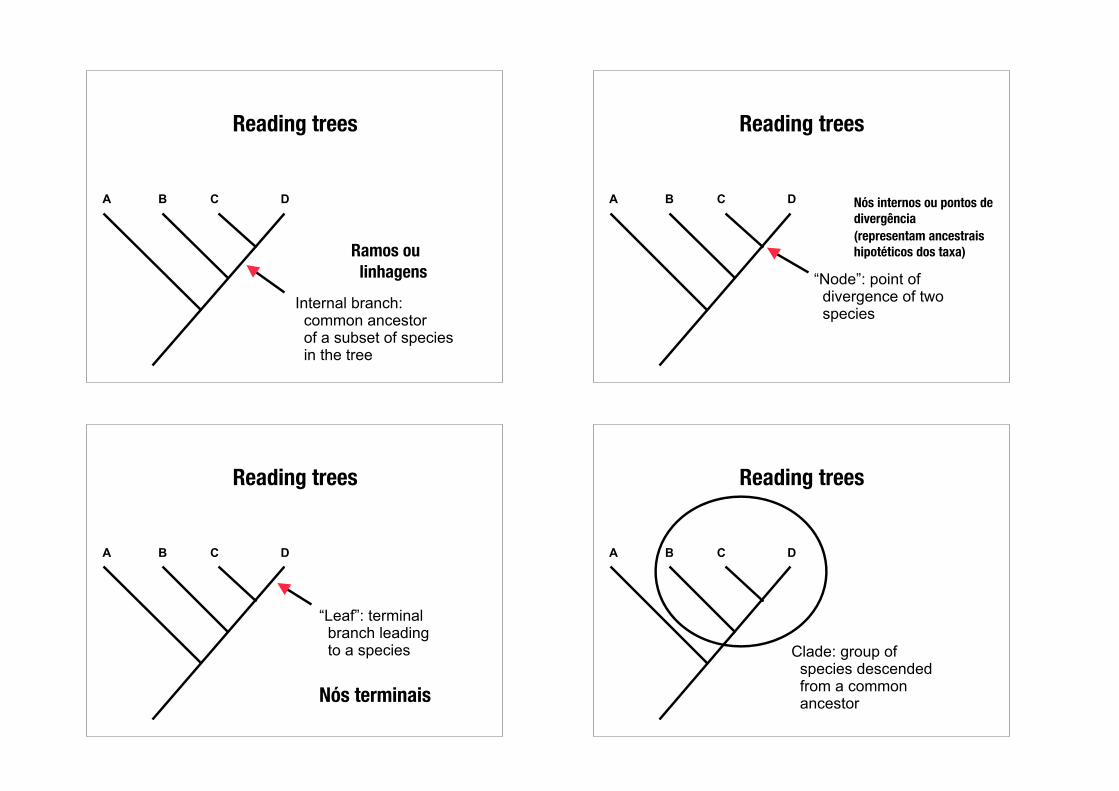
Internal branch: common ancestor of a subset of species in the tree
Reading trees
Ramos ou linhagens
B C DA
“Node”: point of divergence of two species
Reading trees
Nós internos ou pontos de divergência (representam ancestrais hipotéticos dos taxa)
B C DA
“Leaf”: terminal branch leading to a species
Reading trees
Nós terminais
B C DA
Clade: group of species descended from a common ancestor
Reading trees
B C DA
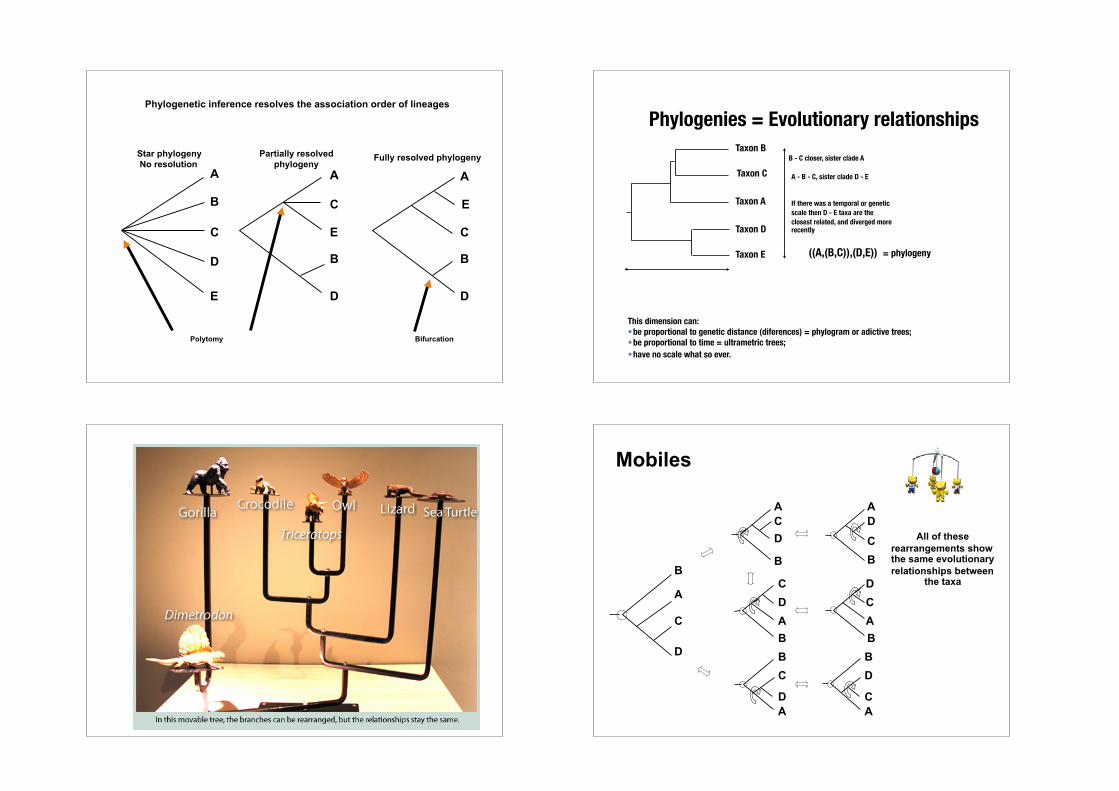
Star phylogeny No resolution
Partially resolved phylogeny
Fully resolved phylogeny
B
B
C
C
C
E
E
E
D
D D
Polytomy Bifurcation
Phylogenetic inference resolves the association order of lineages
A A A
B
Phylogenies = Evolutionary relationships
((A,(B,C)),(D,E)) = phylogeny
B - C closer, sister clade A
Taxon A
Taxon B
Taxon C
Taxon E
Taxon D
This dimension can: •be proportional to genetic distance (diferences) = phylogram or adictive trees; •be proportional to time = ultrametric trees; •have no scale what so ever.
A - B - C, sister clade D - E
If there was a temporal or genetic scale then D - E taxa are the closest related, and diverged more recently
All of these rearrangements show the same evolutionary relationships between
the taxaB
A
C
D
A
B
D
C
B
C
AD
B
D
AC
B
ACD
B
A
C
D
A
B
C
D
Mobiles

A C
B D
Tree 1
A B
C D
Tree 2
A B
D C
Tree 3
Phylogenetic tree building (or inference) methods are aimed at discovering which of the possible unrooted trees is "correct".
We would like this to be the “true” biological tree — that is, one that accurately represents the evolutionary history of the taxa. However, we must settle for discovering the computationally correct or optimal tree for the phylogenetic method of choice.
The number of unrooted trees increases in a greater than exponential manner with number of taxa
# Taxa ( N)
3 4 5 6 7 8 910 . . . .30
# Un rooted trees
1 3 15 105 945 10,935 135,135 2,027,025 . . . . 3.58 x 10 36
(2N - 5)!! = # unrooted trees for N taxa
CA
B D
A B
C
A D
B E
C
A D
B E
C
F
An unrooted, four-taxon tree theoretically can be rooted in five different places to produce five different rooted trees
The unrooted tree 1:
A C
B D
Rooted tree 4
C
D
A
B
4
Rooted tree 3
A
B
C
D
3
Rooted tree 5
D
C
A
B
5
Rooted tree 2
A
B
C
D
2
Rooted tree 1
B
A
C
D
1
These trees show five different evolutionary relationships among the taxa!
1, 2, 3, 4 and 5 possible roots
CA
B D
Each unrooted tree theoretically can be rooted anywhere along any of its branches

A D
B E
C
Each unrooted tree theoretically can be rooted anywhere along any of its branches
CA
B D
A D
B E
C
F
Each unrooted tree theoretically can be rooted anywhere along any of its branches
CA
B D
A D
B E
C
Each unrooted tree theoretically can be rooted anywhere along any of its branches
CA
B D
A D
B E
C
A D
B E
C
F
Taxa Unrooted trees X roots Rooted trees
3 1 3 3
4 3 5 15
5 15 7 105
6 105 9 945
7 945 11 10 395
8 10 935 13 135 125
9 135 135 15 2 027 025
30 3.58 x 1036 57 2.04 x 1038
For 10 sequences there are more than 34 million rooted trees
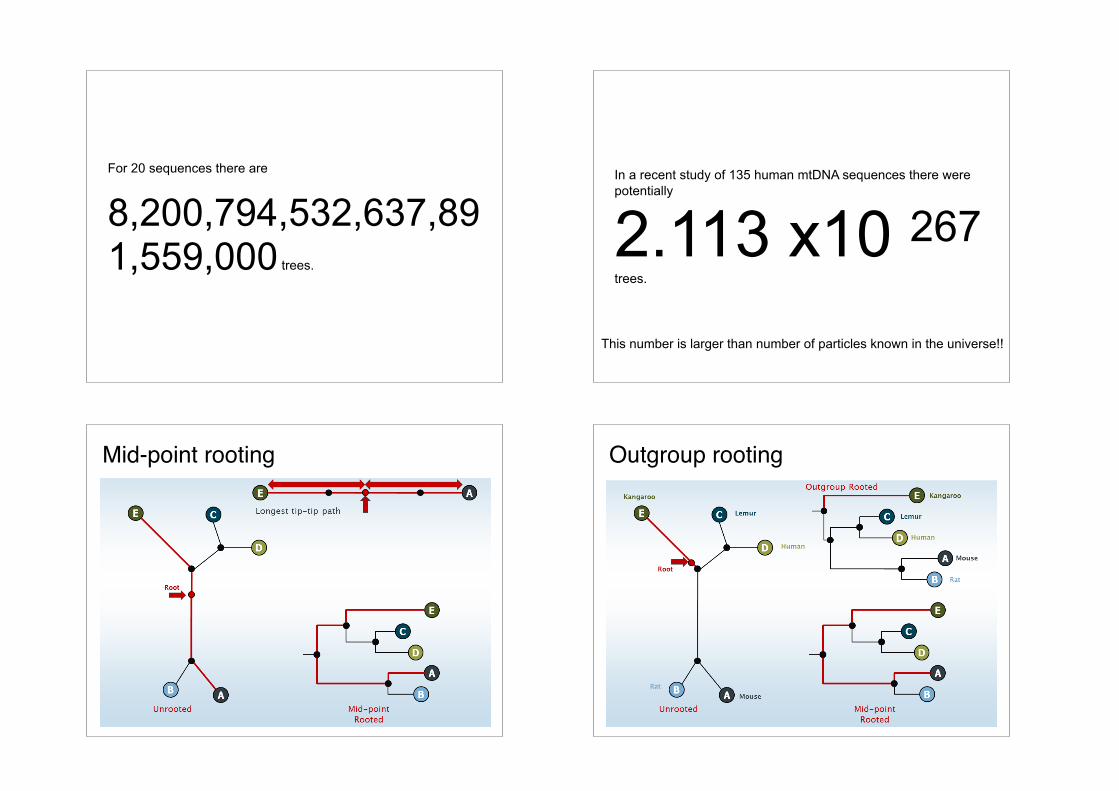
For 20 sequences there are
8,200,794,532,637,891,559,000 trees.
In a recent study of 135 human mtDNA sequences there were potentially
2.113 x10 267 trees.
This number is larger than number of particles known in the universe!!
Mid-point rooting Outgroup rooting

D
C
E
B
G H
F
J
I
K
A
Grouping 2Grouping 1
D
C
E G
F
B
A
J
I
KH D
C
B
E G
F
H
A
J
I
K
Grouping 3
Monophyletic. In this tree, grouping 1, consisting of the seven species B–H, is a monophyletic group, or clade. A mono- phyletic group is made up of an ancestral species (species B in this case) and all of its descendant species. Only monophyletic groups qualify as legitimate taxa derived from cladistics.
Paraphyletic. Grouping 2 does not meet the cladistic criterion: It is paraphyletic, which means that it consists of an ancestor (A in this case) and some, but not all, of that ancestor’s descendants. (Grouping 2 includes the descendants I, J, and K, but excludes B–H, which also descended from A.)
Polyphyletic. Grouping 3 also fails the cladistic test. It is polyphyletic, which means that it lacks the common ancestor of (A) the species in the group. Further-more, a valid taxon that includes the extant species G, H, J, and K would necessarily also contain D and E, which are also descended from A.
Phylogenetic MethodsDistance: • Tree based on pairwise distances between sequences • Evolutionary models applied to pairwise distances to account for multiple substitutions per site and rate heterogeneity among sites
Parsimony: • Minimize the number of substitutions • Assumes sites are independent • Assumes 1 substitution per site
Maximum Likelihood: • Maximize probability of sequences given tree • Evolutionary models applied to each position to account for multiple substitutions per site and rate heterogeneity among sites • Gives single tree with highest likelihood • Assumes sites are independent
Bayesian: • Maximize posterior probability of tree given sequences • Evolutionary models applied to each position to account for multiple substitutions per site and rate heterogeneity among sites • Integrates over all trees • Assumes sites are independent
Molecular phylogenetic tree building methods Are mathematical and/or statistical methods for inferring the divergence order of taxa, as well as the lengths of the branches that connect them. There are many phylogenetic methods available today, each having strengths and weaknesses. Most can be classified as follows:
COMPUTATIONAL METHODClustering algorithmOptimality criterion
DAT
A TY
PE Cha
ract
ers
Dis
tanc
es
PARSIMONY
MAXIMUM LIKELIHOOD
UPGMA
NEIGHBOR-JOINING
MINIMUM EVOLUTION
LEAST SQUARES
Methods of reconstructing phylogenies (evolutionary trees)
Distance matrix methods. Tree that best predicts the entries in a table of pairwise distances among species.
Parsimony methods. Tree that allows evolution of the sequences with the fewest changes is preferred.
Maximum likelihood. Tree that has highest probability that the observed data would evolve.

Distance method
UPGMA(Unweighted Pair Group Method with Arithmetic Mean)
Seq sites1 T T A T T A A2 A A T T T A A3 A A A A A T A
4 A A A A A A T
Distances
Distance-based methods: Transform the sequence data into pairwise distances (dissimilarities), and then use the matrix during tree building.
1 2 3 41 -2 3 -3 5 4 -4 5 4 2 -
1 2 3 41 -2 3 -3 5 4 -4 5 4 2 -
! Métodos principais de filogenia molecular ! UPGMA
Construction of a distance tree using clustering with the Unweighted Pair Group Method with Arithmetic Mean (UPGMA)
From http://www.icp.ucl.ac.be/~opperd/private/upgma.html
A - GCTTGTCCGTTACGATB – ACTTGTCTGTTACGAT
First, construct a distance matrix:
A - GCTTGTCCGTTACGATB – ACTTGTCTGTTACGATC – ACCTGTCCGAAACGATD - ACTTGACCGTTTCCTTE – AGATGACCGTTTCGATF - ACTACACCCTTATGAG
A - GCTTGTCCGTTACGATC – ACCTGTCCGAAACGAT
A B C D E FA -B 2 -C 4 4 -D 6 6 6 -E 6 6 6 4F 8 8 8 8 8 -

! Métodos principais de filogenia molecular ! UPGMA
First round
dist(A,B),C = (distAC + distBC) / 2 = 4 dist(A,B),D = (distAD + distBD) / 2 = 6 dist(A,B),E = (distAE + distBE) / 2 = 6 dist(A,B),F = (distAF + distBF) / 2 = 8
Choose the most similar pair, cluster them together and calculate
the new distance matrix.
A B C D E FA -B 2 -C 4 4 -D 6 6 6 -E 6 6 6 4F 8 8 8 8 8 -
A,B C D E FA,B -C 4 -D 6 6 -E 6 6 4F 8 8 8 8 -
! Métodos principais de filogenia molecular ! UPGMA
Second round
Third round
dist(D,E),(A,B) = (distD(AB) + distE(AB) / 2 = 6 dist(D,E),C = (distDC + distEC) / 2 = 6 dist(D,E),F = (distDF + distEF) / 2 = 8
A,B C D E FA,B -C 4 -D 6 6 -E 6 6 4F 8 8 8 8 -
A,B C D,E F
A,B -
C 4 -
D,E 6 6 -
F 8 8 8 -
! Métodos principais de filogenia molecular ! UPGMA
Forth round
dist(A,B,C),DE = (distABDE + distCDE) / 2 = 6 dist(A,B,C),F = (distABF + distCF) / 2 = 8
A,B C D,E F
A,B -
C 4 -
D,E 6 6 -
F 8 8 8 -
A,B,C D,E F
A,B,C -
D,E 6 -
F 8 8 -
! Métodos principais de filogenia molecular ! UPGMA
Fifth round
Sixth round
Note the this method identifies the root of the tree
A,B,C D,E F
A,B,C -
D,E 6 -
F 8 8 -
(A,B,C)(D,E)(A,B,C)(D,E) -
F 8

! Métodos principais de filogenia molecular ! UPGMA
UPGMA fails when rates of evolution are not constant
A tree in which the evolutionary rates are not equal
From http://www.icp.ucl.ac.be/~opperd/private/upgma.html
A B C D E B 5 C 4 7
D 7 10 7
E 6 9 6 5
F 8 11 8 9 8Método de distância
NJ
! Métodos principais de filogenia molecular ! NJ
The neighbor-joining method of Saitou and Nei (1987). Is especially useful for making a tree having a large number of taxa.
Begin by placing all the taxa in a star-like structure.
Making trees using neighbor-joining
Shortest pairs are chosen to be neighbors and then joined in distance matrix as one OTU.
! Métodos principais de filogenia molecular ! NJ
Tree-building methods: Neighbor-joining
Next, identify neighbors (e.g. 1 and 2) that are most closely related. Connect these neighbors to other OTUs via an internal branch, XY. At each successive stage, minimize the sum of the branch lengths.

! Métodos principais de filogenia molecular ! NJ
Tree-building methods: Neighbor joining
Define the distance from X to Y by
dXY = 1/2(d1Y + d2Y – d12)
! Métodos principais de filogenia molecular ! NJ
The neighbor joining method joins at each step, the two closest sub-trees that are not already joined. It is based on the minimum evolution principle. One of the important concepts in the NJ method is neighbors, which are defined as two taxa that are connected by a single node in an unrooted tree
A B
Node 1
! Métodos principais de filogenia molecular ! NJ
A
B
C
D
E
A
B
5
C
4
7
D
7
10
7
E
6
9
6
5
F 8 11 8 9 8
B
C
D
E
F
A
! Métodos principais de filogenia molecular ! NJ
We have in total 6 OTUs (N=6).
Step 1: We calculate the net divergence r (i) for each OTU from all other OTUs
r(A) = 5+4+7+6+8=30 r(B) = 42 r(C) = 32 r(D) = 38 r(E) = 34 r(F) = 44
A
B
C
D
E
A
B
5
C
4
7
D
7
10
7
E
6
9
6
5
F 8 11 8 9 8

! Métodos principais de filogenia molecular ! NJ
Step 2: Now we calculate a new distance matrix using for each pair of OUTs the formula:
M(ij)=d(ij) - [r(i) + r(j)]/(N-2) or in the case of the pair A,B:
M(AB)=d(AB) -[(r(A) + r(B)]/(N-2) = M(AB)= 5 -[(30 + 42]/(6-2) = -13
A
B
C
D
E
A
B
5
C
4
7
D
7
10
7
E
6
9
6
5
F 8 11 8 9 8
r(A) = 30r(B) = 42r(C) = 32r(D) = 38r(E) = 34r(F) = 44
! Métodos principais de filogenia molecular ! NJ
Step 2: Now we calculate a new distance matrix using for each pair of OUTs the formula:
M(ij)=d(ij) - [r(i) + r(j)]/(N-2) or in the case of the pair A,B:
M(AB)=d(AB) -[(r(A) + r(B)]/(N-2) = M(AB)= 5 -[(30 + 42]/(6-2) = -13
M(AC)= 4 -[(30 + 32]/(6-2) = -11.5 M(AD)= 7 -[(30 + 38]/(6-2) = -10
etc........
A
B
C
D
E
A
B
-13
C
-11.5
-11.5
D
-10
-10
-10.5
E
-10
-10
-10.5
-13
F -10.5 -10.5 11 -11.5 -11.5
r(A) = 30r(B) = 42r(C) = 32r(D) = 38r(E) = 34r(F) = 44
! Métodos principais de filogenia molecular ! NJ
Step 3: Now we choose as neighbors those two OTUs for which Mij is the smallest. These are A and B and D and E. Let's take A and B as neighbors and we form a new node called U ( joining AB).
A
B
C
D
E
A
B
-13
C
-11.5
-11.5
D
-10
-10
-10.5
E
-10
-10
-10.5
-13
F -10.5 -10.5 11 -11.5 -11.5
A
B
C
D
E
A
B
5
C
4
7
D
7
10
7
E
6
9
6
5
F 8 11 8 9 8
r(A) = 30r(B) = 42r(C) = 32r(D) = 38r(E) = 34r(F) = 44
BA
? ?
! Métodos principais de filogenia molecular ! NJ
Now we calculate the branch length from the internal node U to the external OTUs A and B.
S(AU) =d(AB) / 2 + [r(A)-r(B)] / 2(N-2) = S(AU) = 5/2 + [30-42 / 2*(6-2) ] = 1 S(BU) = 5 - S(AU) = 4
A
B
C
D
E
A
B
-13
C
-11.5
-11.5
D
-10
-10
-10.5
E
-10
-10
-10.5
-13
F -10.5 -10.5 11 -11.5 -11.5
A
B
C
D
E
A
B
5
C
4
7
D
7
10
7
E
6
9
6
5
F 8 11 8 9 8
r(A) = 30r(B) = 42r(C) = 32r(D) = 38r(E) = 34r(F) = 44
B
A1
4
U
! Métodos principais de filogenia molecular ! NJ
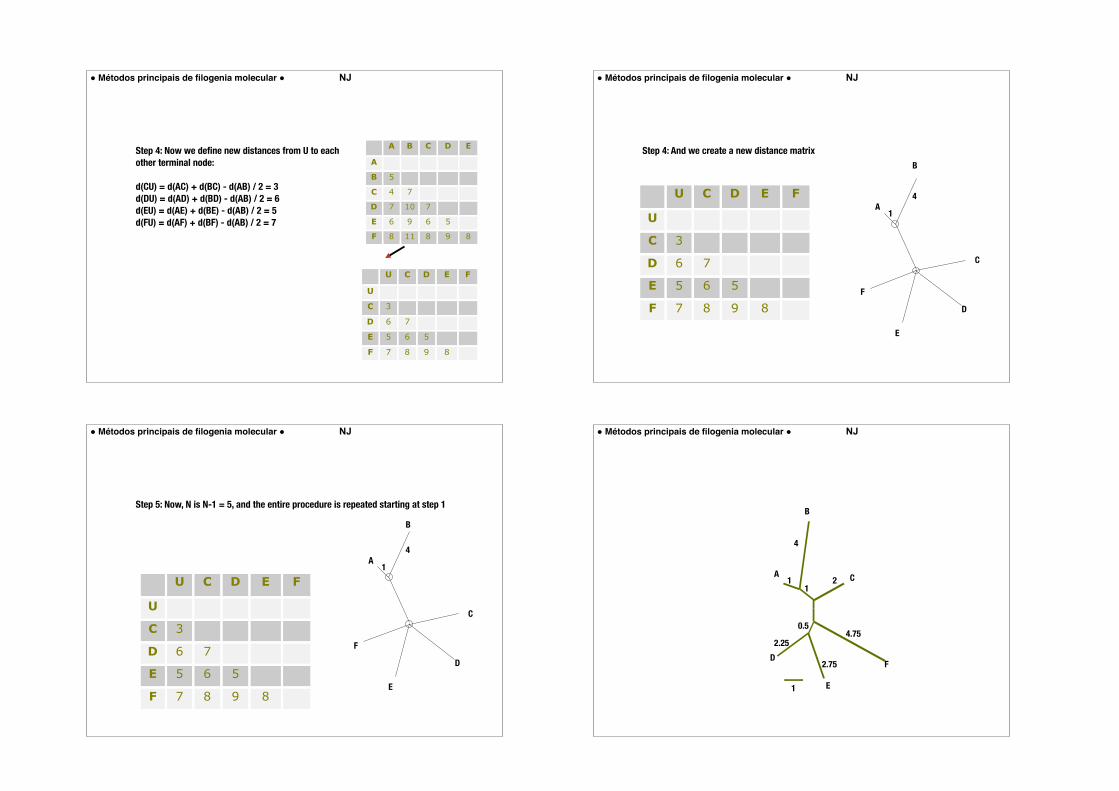
Step 4: Now we define new distances from U to each other terminal node:
d(CU) = d(AC) + d(BC) - d(AB) / 2 = 3 d(DU) = d(AD) + d(BD) - d(AB) / 2 = 6 d(EU) = d(AE) + d(BE) - d(AB) / 2 = 5 d(FU) = d(AF) + d(BF) - d(AB) / 2 = 7
A
B
C
D
E
A
B
5
C
4
7
D
7
10
7
E
6
9
6
5
F 8 11 8 9 8
U
C
D
E
F
U
C
3
D
6
7
E
5
6
5
F
7
8
9
8
r(A) = 30r(B) = 42r(C) = 32r(D) = 38r(E) = 34r(F) = 44
! Métodos principais de filogenia molecular ! NJ
Step 4: And we create a new distance matrix
U
C
D
E
F
U
C
3
D
6
7
E
5
6
5
F
7
8
9
8
B
C
D
E
F
A1
4
! Métodos principais de filogenia molecular ! NJ
Step 5: Now, N is N-1 = 5, and the entire procedure is repeated starting at step 1
U
C
D
E
F
U
C
3
D
6
7
E
5
6
5
F
7
8
9
8
B
C
D
E
F
A1
4
! Métodos principais de filogenia molecular ! NJ
B
C
D
E
F
A
1
1
0.5
4
21
4.752.25
2.75

UPGMA
NJ
ROOT
Comparison of UPGMA and NJ
Neighbor Joining finding shortest (minimum evolution) tree by finding neighbors that minimize the total length of the tree. Shortest pairs are chosen to be neighbors and then joined in distance matrix as one OTU.
the algorithm does not assume that the molecular clock is constant for sequences in the tree. If there are unequal substitution rates, the tree is more accurate than UPGMA.
Distance Methods: evolutionary distances (number of substitutions) are computed for all pairs of taxa.
UPGMA unweighted pairgroup method with arithmetic means.
assumes equal rate of substitutions (therefore is always rooted, as the taxa that has accumulated more sequences is evidently older) (if the substitutions rates are different among taxa, then the tree maybe wrong)
sequential clustering algorithmspairs of taxa are clustered in order of decreasing similarity
Parsimony
Molecular phylogenetic tree building methods Are mathematical and/or statistical methods for inferring the divergence order of taxa, as well as the lengths of the branches that connect them. There are many phylogenetic methods available today, each having strengths and weaknesses. Most can be classified as follows:
COMPUTATIONAL METHODClustering algorithmOptimality criterion
DAT
A TY
PE Cha
ract
ers
Dis
tanc
es
PARSIMONY
MAXIMUM LIKELIHOOD
UPGMA
NEIGHBOR-JOINING
MINIMUM EVOLUTION
LEAST SQUARES

! Métodos principais de filogenia molecular ! Maximum parsimony ! Métodos principais de filogenia molecular ! Maximum parsimony
William of Ockham (or Occam) was a 14th-
century English logician and Franciscan friar
who's name is given to the principle that when
trying to choose between multiple competing theories the simplest one is probably the best.
This principle is known as Ockham's razor.
! Métodos principais de filogenia molecular ! Maximum parsimony
Parsimony: • Minimize the number of substitutions • Assumes sites are independent • Assumes <1 substitution per site
Tree 1
1 (A) 2(G) 3(A) 4 (G)
2 changes
A
Species 1 2 3 4
Data A G A G
Tree 2
1 (A) 2(G) 3(A) 4 (G)
G
2 changes
Fitch (1971) Systematic Zoology 20:406-416
! Métodos principais de filogenia molecular ! Maximum parsimony
Parsimony: • Minimize the number of substitutions • Assumes sites are independent • Assumes <1 substitution per site
Tree 1 and 2 Tree 3
1 (A) 2(G) 3(A) 4 (G)
(A or G) (A or G)
2 changes
(A or G)
1 (A) 2(G)3(A) 4 (G)
(A) (G)
(A or G)
1 change
Species 1 2 3 4
Data A G A G
Fitch (1971) Systematic Zoology 20:406-416

! Métodos principais de filogenia molecular ! Maximum parsimony
Parsimony: • Minimize the number of substitutions • Assumes sites are independent • Assumes <1 substitution per site
Tree 1 Tree 2
1 (A) 2(G) 3(A) 4 (G)
(A or G) (A or G)
2 changes
(A or G)
1 (A) 2(G)3(A) 4 (G)
(A)(G)
(A or G)
1 change
More parsimonious
Fitch (1971) Systematic Zoology 20:406-416
Species 1 2 3 4
Data A G A G
! Métodos principais de filogenia molecular ! Maximum parsimony
Parsimony methods
Optimality criterion: The ‘most-parsimonious’ tree is the one that requires the fewest number of evolutionary events (e.g., nucleotide substitutions, amino acid replacements) to explain the sequences.
Advantages: • Are simple, intuitive, and logical (many possible by ‘pencil-and-paper’). • Can be used on molecular and non-molecular (e.g., morphological) data. • Can tease apart types of similarity (shared-derived, shared-ancestral, homoplasy) • Can be used for character (can infer the exact substitutions) and rate analysis • Can be used to infer the sequences of the extinct (hypothetical) ancestors
Disadvantages: • Not based on statistical properties • Can be fooled by high levels of homoplasy (‘same’ events)
Molecular phylogenetic tree building methods:
Are mathematical and/or statistical methods for inferring the divergence order of taxa, as well as the lengths of the branches that connect them. There are many phylogenetic methods available today, each having strengths and weaknesses. Most can be classified as follows:
COMPUTATIONAL METHODClustering algorithmOptimality criterion
DAT
A TY
PE Cha
ract
ers
Dis
tanc
es
PARSIMONY
MAXIMUM LIKELIHOOD
UPGMA
NEIGHBOR-JOINING
MINIMUM EVOLUTION
LEAST SQUARES
• Parsimony seeks solutions that minimize the amount of change required to explain the data (underestimates superimposed changes)
• ML attempts to estimate the actual amount of change (by specifying the evolutionary model that will account for the data with the highest likelihood)
• Methods that incorporate models of evolutionary change can make more efficient use of the data

! Métodos principais de filogenia molecular ! Maximum likelihood
Maximum likelihood (ML) methodsOptimality criterion: ML methods evaluate phylogenetic hypotheses in terms of the probability that a proposed model of the evolutionary process and the proposed unrooted tree would give rise to the observed data. The tree found to have the highest ML value is considered to be the preferred tree.
Advantages: • Are based on explicit model of evolution. • Usually the most ‘consistent’ of the methods available. • Can be used for character (can infer the exact substitutions) and rate analysis. • Can be used to infer the sequences of the extinct (hypothetical) ancestors. • Can help account for branch-length effects.
Disadvantages: • Are based on explicit model of evolution. • Are not as simple and intuitive as many other methods. • Are computationally very intense (Iimits number of taxa and length of sequence). • Slooooow!!! • Violations of the assumed model can lead to incorrect trees.
! Métodos principais de filogenia molecular ! Maximum likelihood
6 faces 8 faces 12 faces
Ideia from Gavin Naylor
! Métodos principais de filogenia molecular ! Maximum likelihood
Roll the diceHow many points?
Ideia from Gavin Naylor
6 faces 8 faces 12 faces
! Métodos principais de filogenia molecular ! Maximum likelihood
Roll the diceHow many points?
Ideia from Gavin Naylor
6 faces 8 faces 12 faces
14 POINTS

! Métodos principais de filogenia molecular ! Maximum likelihood
Para um resultado de 14, necessitamos de usar dois dados. Qual o par de dados que mais provavelmente originará esse
resultado?
Ideia from Gavin Naylor
6 faces 8 faces 12 faces
For a 14 points results we need 2 dices. Which is the pair of dices that most probably originates that result?
! Métodos principais de filogenia molecular ! Maximum likelihood
Equivalente a: qual a árvore que mais provavelmente terá originado essas sequências?
Ideia from Gavin Naylor
6 faces 8 faces 12 faces
Which tree is most likely to have yielded these sequences?
! Métodos principais de filogenia molecular ! Maximum likelihood
6 + 8
+ + +
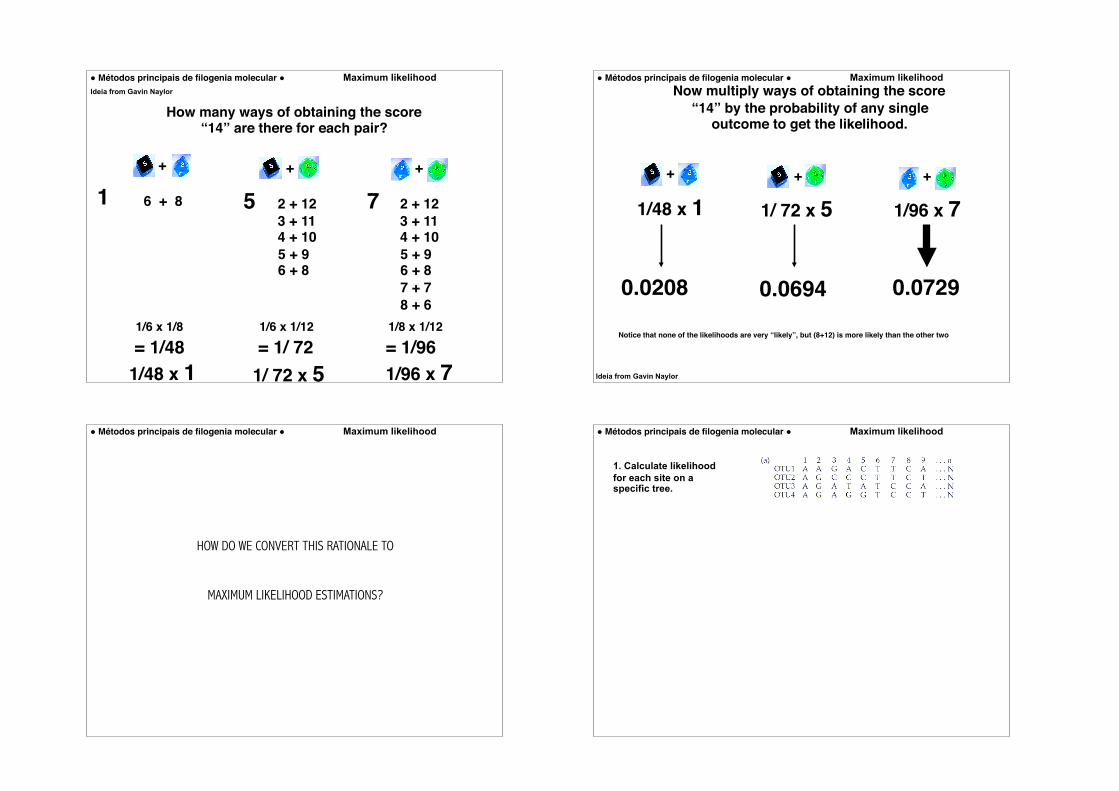
How many ways of obtaining the score “14” are there for each pair?
2 + 12 3 + 11 4 + 10
5 + 96 + 8
1 5 7Ideia from Gavin Naylor
How many possible combinations?
2 + 12 3 + 11 4 + 10
5 + 96 + 87 + 98 + 7
! Métodos principais de filogenia molecular ! Maximum likelihood
6 + 8
+ + +
How many ways of obtaining the score “14” are there for each pair?
2 + 12 3 + 11 4 + 10
5 + 96 + 8
1 5 7
Ideia from Gavin Naylor
2 + 12 3 + 11 4 + 10
5 + 96 + 87 + 98 + 7
1/6 x 1/8
= 1/481/6 x 1/12 1/8 x 1/12
= 1/ 72 = 1/96
5 7
Probability of each combination?
! Métodos principais de filogenia molecular ! Maximum likelihood
6 + 8
+ + +
How many ways of obtaining the score “14” are there for each pair?
2 + 12 3 + 11 4 + 10
5 + 96 + 8
1 5 7
Ideia from Gavin Naylor
2 + 12 3 + 11 4 + 10
5 + 96 + 87 + 78 + 6
1/6 x 1/8
= 1/481/6 x 1/12 1/8 x 1/12
= 1/ 72 = 1/961/48 x 1 1/ 72 x 5 1/96 x 7
! Métodos principais de filogenia molecular ! Maximum likelihoodNow multiply ways of obtaining the score
“14” by the probability of any single outcome to get the likelihood.
+ + +
1/48 x 1 1/ 72 x 5 1/96 x 7
0.07290.06940.0208
Notice that none of the likelihoods are very “likely”, but (8+12) is more likely than the other two
Ideia from Gavin Naylor
! Métodos principais de filogenia molecular ! Maximum likelihood
HOW DO WE CONVERT THIS RATIONALE TO
MAXIMUM LIKELIHOOD ESTIMATIONS?
! Métodos principais de filogenia molecular ! Maximum likelihood
1. Calculate likelihood for each site on a specific tree.

! Métodos principais de filogenia molecular ! Maximum likelihood
1. Calculate likelihood for each site on a specific tree.
A likelihood de uma das posições do alinhamento, neste caso a posição 5, é igual à soma de todas as possíveis reconstruções nos nós 5 e 6.
! Métodos principais de filogenia molecular ! Maximum likelihood
A likelihood da árvore é o produto de todas as likelihoods individuais de todos os sites do alinhamento.
A likelihood é a soma dos logaritmos das likelihoods de cada local
! Métodos principais de filogenia molecular ! Maximum likelihood
1. Calculate likelihood for each site on a specific tree.
2. Sum up the L values for all sites on the tree.
3. Compare the L value for all possible trees.
4. Choose tree with highest L value.








