
Die neue EU-Verordnung für In-vitro Diagnostika
- Einleitung und State of Play
Birgit Schäfer, VDGH – Verband der Diagnostica-Industrie e.V.
Transferinitiative RLP – Mainz, 28.11.2018
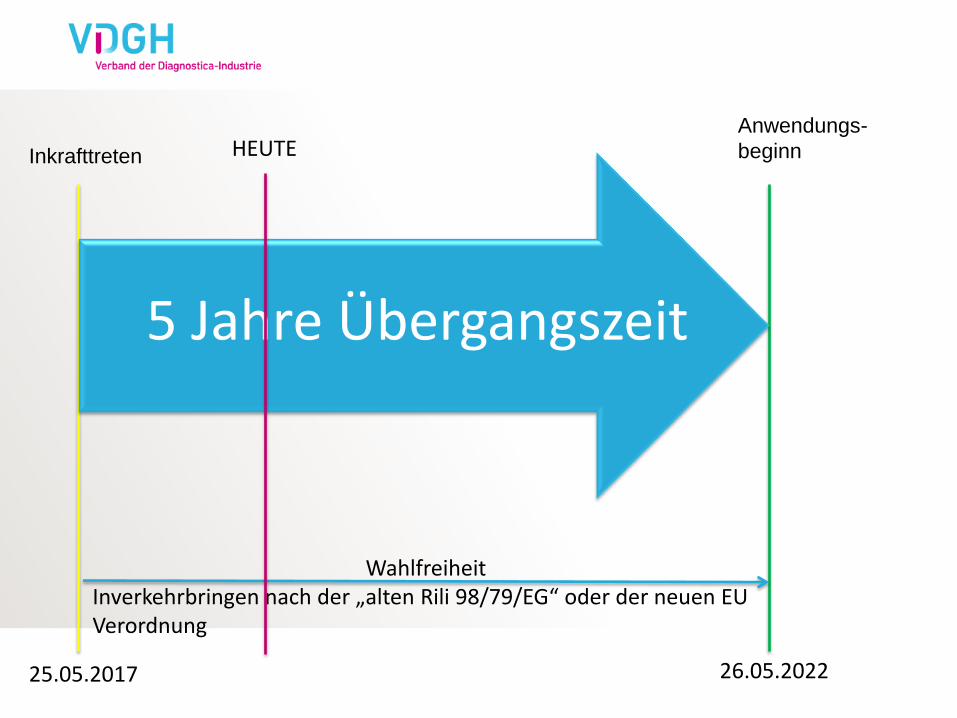
Inkrafttreten
Anwendungs-
beginn
5 Jahre Übergangszeit
Wahlfreiheit Inverkehrbringen nach der „alten Rili 98/79/EG“ oder der neuen EU Verordnung
25.05.2017 26.05.2022
HEUTE
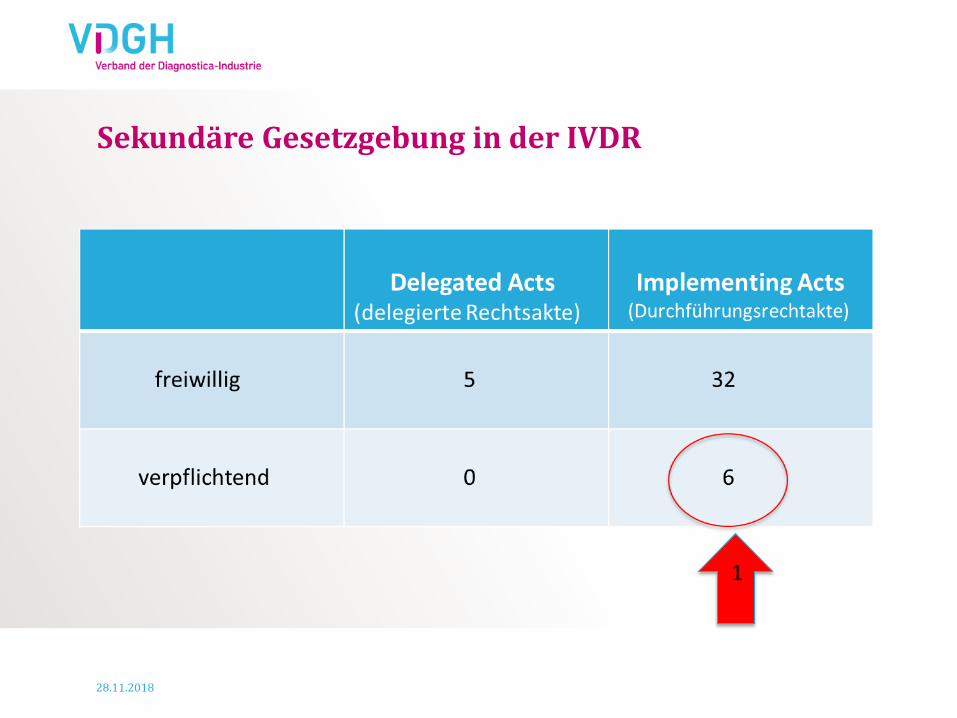
Sekundäre Gesetzgebung in der IVDR
1
28.11.2018

Übergangszeitraum – Art. 110 IVDR
Was ist vor Geltungsbeginn möglich? (25.05.2017 – 25.05.2022)
• Inverkehrbringen IVDD (Rili 98/79) -konformer Produkte √
• Inverkehrbringen IVDR-konformer Produkte
Was gilt zum Stichtag des Geltungsbeginns?
• Notifizierungen nach IVDD der Benannten Stellen sind ungültig
• Aber beachte Art. 110 Abs. 3: Geeignete Überwachung der von Ihnen zertifizierten Produkte muss ermöglicht werden
Was ist nach Geltungsbeginn möglich, sprich ab dem 26.05.2022?
• IVDD-Bescheinigungen behalten bis spätestens 27.05.2024 ihre Gültigkeit
• Deshalb: Inverkehrbringen IVDD (Rili 98/79)-konformer Produkte weiterhin bis 27.05.2024 möglich (siehe Art 110 Abs. 3)
• Inverkehrbringen IVDR-konformer Produkte
28.11.2018

Übergangszeitraum – Art. 110 IVDR
28.11.2018
IVDR-konforme Produkte vor dem 26.05.2022
Art. 110 Abs. 5 - Anforderungen IVDR Einschränkung nach Abs.7 - Produkte, die Art. 48 2+4 unterliegen Abverkaufsregelung: (-), keine Anwendung
Ja, aber…
IVDD-konforme Produkte nach dem 26.05.2022 Art. 110 Abs. 2: - Gültige Bescheinigung BS - KEINE wesentliche Änderung Max. bis 27.05.2024!!
Anforderungen an IVDR (+): - Überwachung - Marktüberwachung - Vigilanz - Registrierung -> Verantwortung BS bleibt bestehen
Betroffenheit gering, da Zertifikat einer BS
erforderlich
Ab
26.05.2022:
„Normalfall“ IVDR-
konforme Produkte
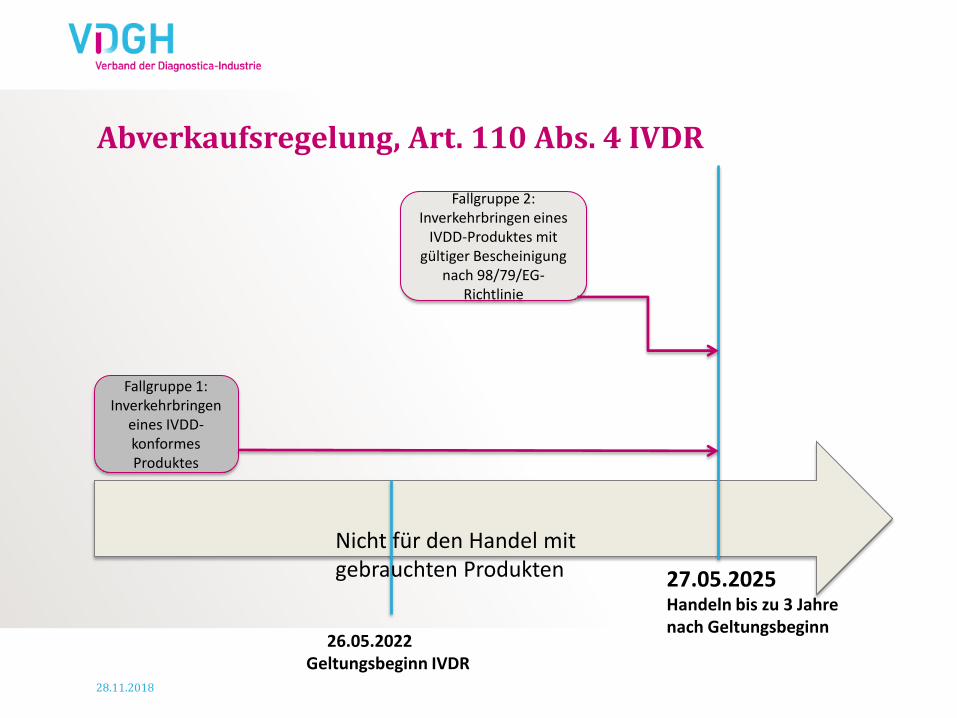
Abverkaufsregelung, Art. 110 Abs. 4 IVDR
28.11.2018
Fallgruppe 1: Inverkehrbringen
eines IVDD-konformes Produktes
26.05.2022 Geltungsbeginn IVDR
27.05.2025 Handeln bis zu 3 Jahre nach Geltungsbeginn
Fallgruppe 2: Inverkehrbringen eines
IVDD-Produktes mit gültiger Bescheinigung
nach 98/79/EG-Richtlinie
Nicht für den Handel mit gebrauchten Produkten

Übergangsfristen zum Nachlesen
• NAKI (Nationaler Implementierungskreis) –
UG 1 Übergangsbestimmungen
https://www.bundesgesundheitsministerium.de/naki.html
• CAMD (Competent Authorities for Medical Devices)
https://www.camd-europe.eu/wp-content/uploads/2018/05/FAQ_IVDR_180117_V1.0-1.pdf
28.11.2018

MPG Reloaded ?
28.11.2018
MedizinproduktebetreiberVO MPSV - Medizinprodukte-Sicherheitsplanverordnung MPKPV – Verordnung über klinische Prüfungen von Medizinprodukte DIMDI-Verordnung MPAV HWG ……
State of Play

Was sind Inhouse-Tests und sind diese auch IVDR konform?
Art. 5 Abs. 5 IVDR vs. § 3 Nr. 22 MPG „Medizinprodukte aus Eigenherstellung“
Ist Inhouse identisch mit RUI (Research only Product) ?
Wer und was ist eine „Gesundheitseinrichtung“ im Sinne der IVDR ?
Neue Anforderungen ?
Nationale Alleingänge möglich? Was macht das BMG?
Was bedeutet „Industrieller Maßstab“?
Wo endet meine Herstellerverantwortung ?
28.11.2018
State of Play
- Für alle Klassen
- nur in einer Einrichtung - QMS ! - ISO 15189 od. vergleichbaren
Standard - Begründung, dass kein CE
markierter Test die benötigten Spezifikationen erfüllt
- Behördliche Überwachung - zugängliche Erklärung über
Inhouse hergestellte Tests
- nationale Einschränkungen
Anbieter laboratoriumsmedizinischer Dienstleistungen aus dem Internet ?

Einmalige Registrierungsnummer gemäß §28 (1+2) in EUDAMED
Art. 10 ff. - Aufgaben der verschiedenen Wirtschaftsakteure
• Hersteller §10
• Bevollmächtigter §11
• Importeure §13
• Händler §14
28.11.2018
Die neue Verordnung sieht verschiedene Wirtschaftsakteure vor:
Haftbar für Schäden gemäß §10 (15),11 (5); Person zur Einhaltung regulatorischer Vor- schriften §15(1),(4)
Erstellen, bzw. Vorhalten und Überprüfen: • der EU-Konformitätserklärung • Register für Vorkommnisse – Weitergabe an Importeur/Bevollmächtigter /Hersteller • IFU, Namen des Herstellers/Importeurs als direkten Abspechpartner • UDI zur Nachverfolgung der Produkte • Kooperation mit den zuständigen Behörden
Beachten Sie Art. 16, Sie könnten „Hersteller“ werden

Herstellerpflichten -Artikel 10 – Was ist neu
OEM Hersteller
Werden Produkte von einem Dritten konzipiert oder hergestellt, sind Angaben zur Identität Bestandteil der zu registrierenden Informationen nach Artikel 27 I
Verpflichtender Versicherungsschutz
Abschluss einer Versicherung abhängig von Produktpalette (Risikoklassen) oder Nachweis, dass ausreichend Deckungssumme vorliegt, für den Fall von Schädigungen, die durch das Produkt entstehen
Person, zuständig für die Einhaltung regulatorischer Anforderungen (Art. 15)
Sicherheitsbeauftragter Medizinproduktebetreiber-VO
• Studium (Recht, Medizin, Pharmazie, Ingenieurswesen oder Ähnliches) + 1 Jahr Erfahrung oder
4 Jahre Erfahrung
• Aufgaben können von mehreren Personen geteilt werden, aber ein Verantwortlicher/Ansprechpartner erforderlich
• Person muss nicht im Unternehmen präsent sein, lediglich erreichbar
28.11.2018
- Was ist OEM/PLM?

Produktregistrierung, Art. 24 IVDR iVm Anhang VI
UDI = UDI-DI + UDI-PI • UDI-DI (Device Identifier) – statische Daten
Das ist die Device-Identifikation eines spezifischen Models und dient in der UDI-Datenbank als „Schlüssel“. Bsp: Alle Kits eines Produktes/ Kitgröße haben die gleiche UDI-DI
• UDI-PI (Production Identifier) – dynamische Daten
Der Produktions-Identifier kennzeichnet jede einzelne Instanz eines Produkts bzw. eines Batches.
Bsp: Jede Abfüllung/Charge hat somit eine eigene UDI-PI
28.11.2018
Eudamed ?
https://ec.europa.eu/growth/sectors/medical-devices/guidance_en
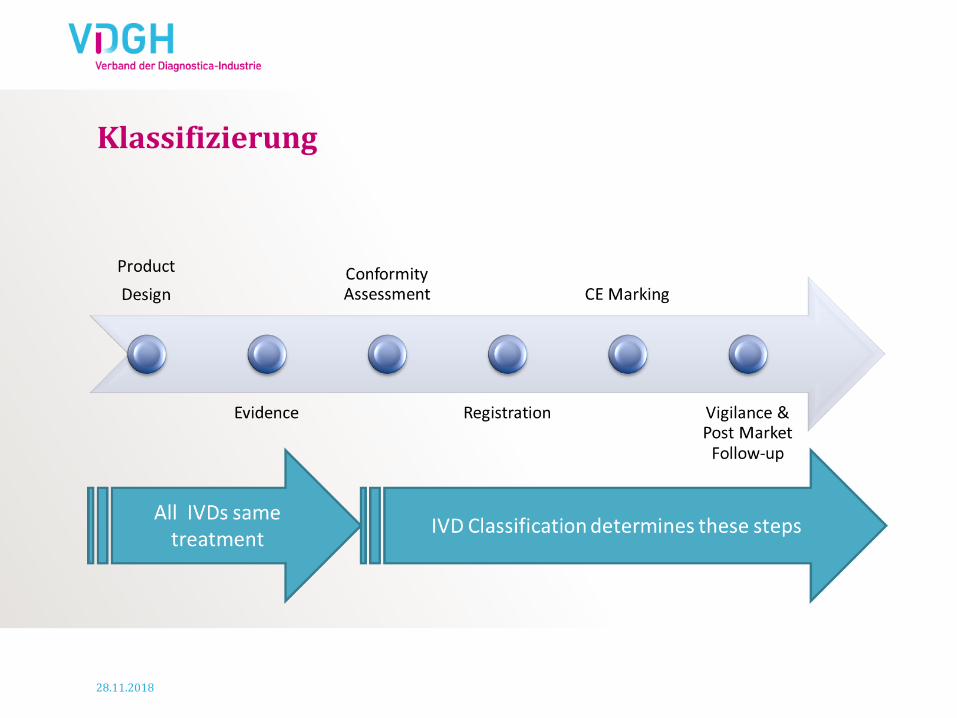
Klassifizierung
28.11.2018
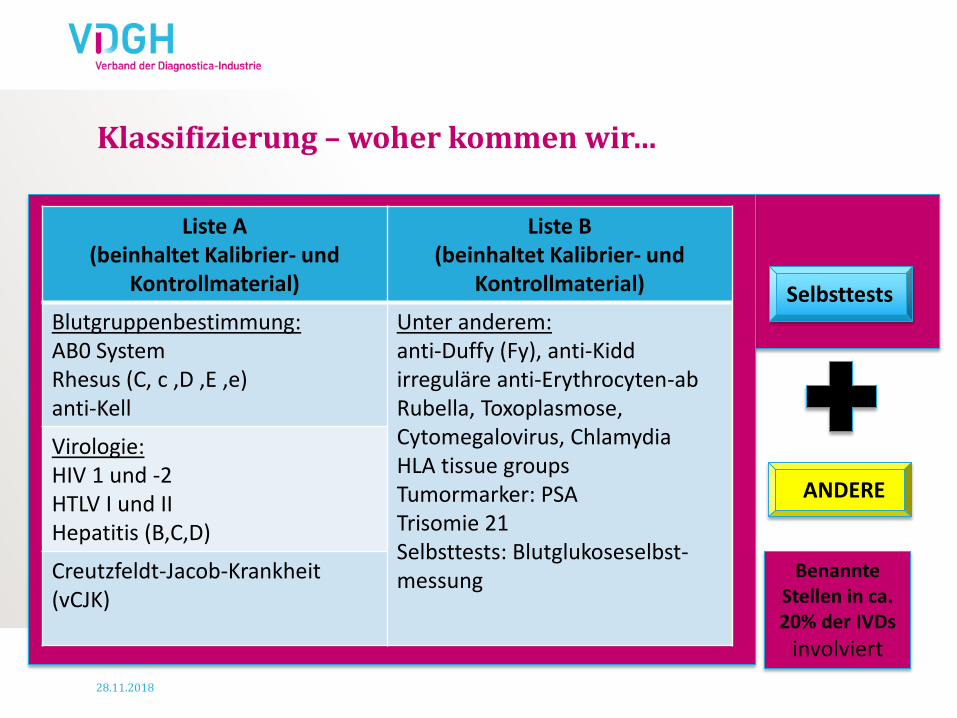
Benannte Stellen in ca. 20% der IVDs
involviert
Klassifizierung – woher kommen wir…
28.11.2018
Liste A (beinhaltet Kalibrier- und
Kontrollmaterial)
Liste B (beinhaltet Kalibrier- und
Kontrollmaterial)
Blutgruppenbestimmung: AB0 System Rhesus (C, c ,D ,E ,e) anti-Kell
Unter anderem: anti-Duffy (Fy), anti-Kidd irreguläre anti-Erythrocyten-ab Rubella, Toxoplasmose, Cytomegalovirus, Chlamydia HLA tissue groups Tumormarker: PSA Trisomie 21 Selbsttests: Blutglukoseselbst-messung
Virologie: HIV 1 und -2 HTLV I und II Hepatitis (B,C,D)
Creutzfeldt-Jacob-Krankheit (vCJK)
Selbsttests
ANDERE
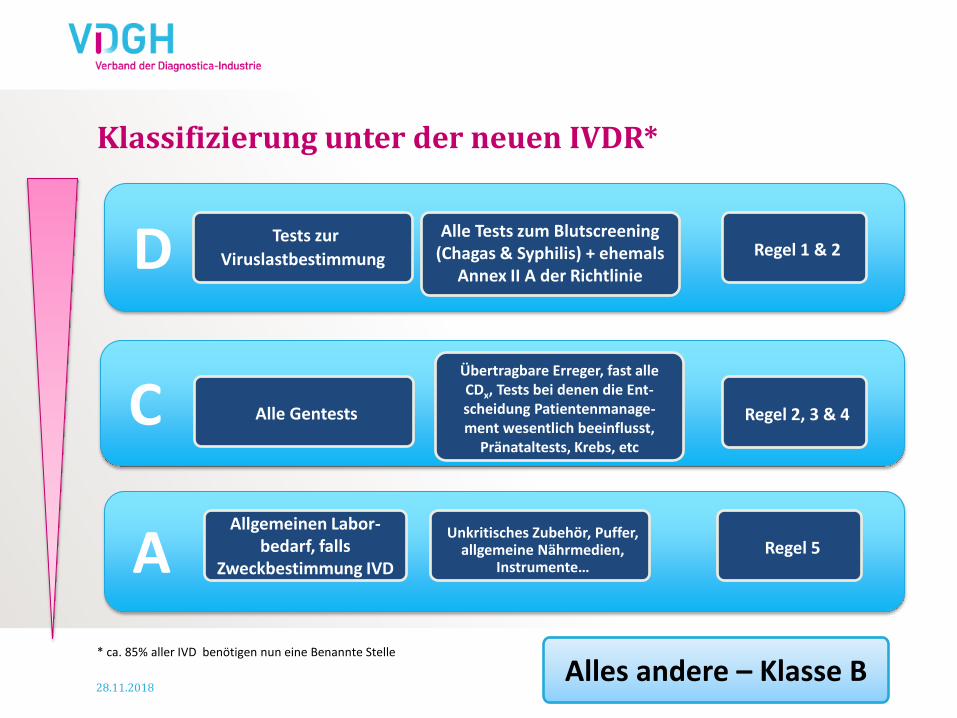
Klassifizierung unter der neuen IVDR*
28.11.2018
D
C
A
Übertragbare Erreger, fast alle CDx, Tests bei denen die Ent-scheidung Patientenmanage-ment wesentlich beeinflusst,
Pränataltests, Krebs, etc
Unkritisches Zubehör, Puffer, allgemeine Nährmedien,
Instrumente…
Alles andere – Klasse B
Tests zur
Viruslastbestimmung
Alle Tests zum Blutscreening (Chagas & Syphilis) + ehemals
Annex II A der Richtlinie
Regel 1 & 2
Alle Gentests Regel 2, 3 & 4
Regel 5
Allgemeinen Labor-bedarf, falls
Zweckbestimmung IVD
* ca. 85% aller IVD benötigen nun eine Benannte Stelle

Klassifizierungsregeln für “spezielle” IVD
Companion Diagnostics:
• Klasse C oder D – Regel 3
Selbsttests:
• Klasse C, außer Tests auf Schwangerschaft, Fruchtbarkeit und Cholesterin und einige Tests bei denen die Ergebnisse aus dem Urin bestimmt werden (Klasse B) – Regel 4
Near Patient Testing:
• Den jeweiligen Regeln entsprechend– Rule 4
Software:
• Standalone („classified in their own right“), sonst mit seinem IVD klassifiziert
Instrumente:
• Klasse A
28.11.2018
Konformitätsbewertung steht hierzu im Widerspruch

Klare Regeln, klare Einstufung ?
28.11.2018
Ergänzungstests
• Alle Regeln sollen auch für „Ergänzungstests“ gelten – Definition? (Regel 1.10)
Unbestimmte Rechtsbegriffe, die einer EU-einheitliche Interpretation benötigen
• Was bedeutet “lebensbedrohlich”?
• Wie ist ein “hohes oder wahrscheinlich hohes Risiko der Verbreitung” definiert?
• Was ist die “state of the art of medicine” in Europa (Irland vs. Deutschland)?
• Welche Produkte fallen (noch) nicht unter die Regeln ? (Regel 6: dann immer B)
EU-Guidance in Arbeit – Ziel: Veröffentlichung Mitte 2019

Konformitätsbewertungsverfahren – Übersicht
28.11.2018
Achtung: Sterile Podukte?

Konformitätsbewertungsverfahren – Übersicht
• Merke: Klassifizierung bestimmt den Weg der Konformitätsbewertung
• Spezielle Bewertungsverfahren für POCT, Selbsttests, Companion Diagnostics (EMA) -> Annex IX Abs. 5 1
• State of Play:
- Durchführungsrechtsakt „Notified bodies scope of designation“ √ (26.11.2017)
- Benennungen: aktuell 0, aber ab Q1/2019 erwartet
- Beratungen auf EU-Ebene laufen, um mögliche Engpässe zu verhindern; Verbände sind in die Diskussion eingebunden
- „Sampling Plan“ ??? (Stichproben für B+C)
- Siehe Art. 48 Abs. 7, 9 IVDR („…Bewertung der technischen Dokumentation gem. den Abschnitten 4.4 - 4.8 des genannten Anhangs, zumindest für ein repräsentatives Produkt pro Produktkategorie bzw. Produktgruppe)
28.11.2018

Konformitätsbewertung– Referenzlaboratorien für IVDs der Klasse D, Art. 100 IVDR
28.11.2018
• „batch release“ - Pflicht
• Einhaltung der „Common Specification“ (CS) wird überprüft (≠ Nicht zwingend heutige CTS)
• Entwicklung von „Common Specifications“ für alle IVDs der Klasse D
(Möglichkeit der Entwicklung von CS auch für IVDs der Klasse C)
• Referenzlaboratorien geben wissenschaftliche Stellungnahmen ab an überwachende Behörden und benannte Stellen
Benennung der Labore erst 18 Monate vor Ende Übergangszeit -> Keine
neuen Klasse D - IVD

…und vergessen Sie nicht die “general safety and performance requirements”
• Elektromagnetische Kompatibilität (e.g. IEC standards)
• Vernetzung von Komponenten (if you have devices interacting)
• Softwaresicherheit (within networks, hospitals, etc.)
• Chemische Sicherheit (any harm for patients, users, staff…)
• Biologische Sicherheit (any harm for patients, users, staff…)
• Physikalische Sicherheit(any harm for patients, users, staff…)
• Gebrauchstauglichkeit (especially for self tests and near patient tests)
• Umweltanforderungen (REACH, disposal route)
• andere zu erfüllende Gesetze/Vorgaben (e.g. machinery directive, REACH, RoHS, etc.)
• ….
28.11.2018

Birgit Schäfer
Rechtsanwältin Stellv. Geschäftsführerin
VDGH e.V. Neustädtische Kirchstr. 8
10117 Berlin [email protected]
www.vdgh.de
28.11.2018







