
Investigations of two potential mechanisms which may favour persistence of CDV, the driving force behind the chronic progression of demyelination in canine distemper
Graduate School for Cellular and Biomedical Sciences
University of Bern
PhD Thesis
Submitted by
Dominique Wiener from (Stallikon ZH)
Thesis advisor
Prof. Dr. Andreas Zurbriggen Departement of Clinical Research and Veterinary Public
Health Vetsuisse Faculty of the University of Bern

Accepted by the Faculty of Medicine, the Faculty of Science and the
Vetsuisse Faculty of the University of Bern at the request of the
Graduate School for Cellular and Biomedical Sciences
Bern, Dean of the Faculty of Medicine
Bern, Dean of the Faculty of Science
Bern, Dean of the Vetsuisse Faculty Bern

TABLE OF CONTENTS SUMMARY .......................................................................................................................................... 1
INTRODUCTION ................................................................................................................................ 3
1. Infection and disease caused by canine distemper virus ........................................................ 3
1.1 Natural host range of CDV .................................................................................................... 3
1.2 Route of infection and virus spread ..................................................................................... 3
1.3 Clinical signs and tissues infected ....................................................................................... 4
2. Persistence of CDV in the central nervous system ................................................................... 5
3. Classification and molecular properties of CDV ........................................................................ 7
4. Viral proteins ................................................................................................................................... 9
4.1 Attachment (H) protein .......................................................................................................... 9
4.2 Fusion (F) protein ................................................................................................................. 10
4.3 Matrix (M) protein ................................................................................................................. 10
4.4 Long untranslated region between the M and the F gene (M-F utr) ............................. 11
4.5 Nucleocapsid (N) protein ..................................................................................................... 12
4.6 Large (L) protein ................................................................................................................... 12
4.7 Phosho (P) protein ............................................................................................................... 12
4.8 C protein ................................................................................................................................ 13
4.9 V protein ................................................................................................................................. 13
5. Replication, assembly and release of paramyxoviridae ......................................................... 14
5.1 Replication ............................................................................................................................. 14
5.2 Virion assembly and release ................................................................................................ 14
6. CDV and innate immunity ........................................................................................................... 15
6.1 Interferons ............................................................................................................................. 16
6.2 Antiviral functions of IFN ..................................................................................................... 16
6.3 Interferon induction .............................................................................................................. 17
6.3.1 TLRs: ............................................................................................................................. 17
6.3.2 RNA helicases ............................................................................................................. 18
6.3.3 INF induction ................................................................................................................ 19
6.4 IFN signaling pathways ....................................................................................................... 20
7. Objective of the present study: Investigation of two potential mechanisms which may favor persistence of CDV, the driving force behind the chronic progression of demyelination in canine distemper. ......................................................................................................................... 27
7.1 Investigations about the putative CDV protein M2 .......................................................... 27
7.2 Molecular mechanisms of innate immune control by wild type CDV V protein .......... 27
TABLE OF CONTENTS

8. Discussion and perspectives ...................................................................................................... 30
9. References .................................................................................................................................... 34
CHAPTER ONE ................................................................................................................................ 40
CHAPTER TWO ............................................................................................................................... 75
ACKNOWLEDGEMENTS ............................................................................................................... 91
CURRICULUM VITAE ..................................................................................................................... 92
LIST OF PUBLICATIONS ............................................................................................................... 93
Declaration of Originality ................................................................................................................. 94
TABLE OF CONTENTS

SUMMARY Canine distemper virus (CDV), a morbillivirus of the paramyxovirus family, closely related
to measles virus (MeV), induces a chronic progressive and relapsing demyelinating
disease in dogs, associated with persistence of the virus in the central nervous system.
This naturally occurring demyelinating disease is considered to be a model for multiple
sclerosis in man. Virus persistence in the central nervous system (CNS) appears to play
an essential role in the chronic progression of the disease. The antiviral immune response
leads to virus clearance in the inflammatory lesions. However, CDV can replicate and
persist outside these inflammatory lesions within the brain. Viral persistence is thus the
driving force behind the chronic progression of the disease. The mechanism of
persistence of CDV in the presence of an effective antiviral immune response is not well
understood. In this study two potential mechanisms of CDV persistence were investigated.
We hypothesized that control of persistence could be influenced by the unusually long
“untranslated” region between the M and the F gene of CDV, which is common to all
morbilliviruses. In particular, previous studies revealed a short potential open reading
frame (ORF) situated at the end of the M gene and is referred to as M2. Intriguingly, even
though the sequence of the MF utr differs dramatically between the different CDV strains,
the region of the putative ORF is conserved in current persistent CDV strains. The
conservation of these putative ORF’s within the MF utr indicates, that there must be some
evolutionary pressure in maintaining these ORF’s and suggests a functional role of this
sequence. However, up to now, there is no evidence that the M2 protein is expressed.
In this study, we investigated whether this short polypeptide was expressed from a
putative ORF located within the 3’ utr of the M mRNA of the highly virulent CDV-A75/17
strain. To this purpose, using reverse genetics, we engineered several recombinant
viruses. Even though M2 could efficiently be expressed in transfection experiments,
similar results could not be obtained in the background of full viral infection. Rather,
several biochemical and immunological assays (such as fluorescence microscopy,
1
SUMMARY

immunoblot, flow cytometry and immunoprecipitation) indicated that in viral infection, M2
was not translated. Cell type-specific restriction of M2 expression was unlikely since M2
could not be detected in a range of different cell systems (vero SLAM cells, vero cells,
MDCK SLAM cells, keratinocytes or DBCC’s). All together our results suggest absence of
M2 expression, at least in quantities that could be detected by standard techniques. We
conclude that M2 does not play a role in persistence.
The second mechanism of viral persistence investigated in this study involves immune
evasion of CDV. Morbilliviruses have evolved several strategies to hijack the host cell-
mediated innate immunity. The CDV-V protein has been shown to act as a virulence
factor. Here, we investigated the molecular mechanisms by which the P gene products of
the neurovirulent A75/17-CDV disrupted type I interferons- (IFN-α/β)-mediated antiviral
state. Using recombinant knockout A75/17 viruses, the V protein was identified as the
main antagonist of IFN- α/β -mediated signaling. Importantly, immunofluorescence
analysis illustrated that the latter inhibition correlated with impaired STAT1/STAT2 nuclear
import, though their phosphorylation states were not affected. Co-immunoprecipitation
assays identified the N-terminal region of V (VNT) responsible for STAT1 targeting, which
corroborated with the ability to inhibit the activity of IFN- α/β -mediated antiviral state.
Conversely, while the C-terminal domain of V (VCT) could not function autonomously,
when fused to VNT, it optimally associated with STAT2 and subsequently strongly
suppressed the IFN- α/β -mediated signaling pathway. The latter result was further
supported by a single mutation at position 110 in V resulting in a mutant that lost STAT1
binding, while retaining partial STAT2 association. Taken together, our results identified
VNT and VCT as two essential modules complementing each other to complete IFN- α/β
evasion, which may be involved in CDV-persistence in the CNS. Our experiments also
reveal a novel mechanism of IFN-α/β evasion among the morbilliviruses.
2
SUMMARY

INTRODUCTION
1. Infection and disease caused by canine distemper virus
CDV causes in dogs a chronic, demyelinating, progressive or relapsing neurological
disease, because it persists in the CNS (Vandevelde and Zurbriggen, 2005). CDV is a
model for multiple sclerosis in man (Appel M.J. and Gillespie J.H., 1972). A
spontaneous MS-like disease with multifocal demyelinating lesions is rare in domestic
animals. Primary demyelination in domestic animals has only been observed for Visna,
a lentivirus infection in sheep and in CDV infection. While demyelination is a rare
complication in Visna, it occurs with high frequency in distemper (Vandevelde and
Zurbriggen, 2005).
1.1 Natural host range of CDV
The host spectrum of CDV is widespread and includes numerous families in the order
of Carnivorae like Canidae, Procyonidae, Mustelidae, Mephtidae, Hyaenidae,
Ailuridae, and Viverridae (Beineke et al., 2009). In addition, in the last years, several
outbreaks occurred in large felids (Appel et al., 1994; Roelke-Parker et al., 1996) and
in collard peccaries (Appel et al., 1991). Furthermore, besides infection with the
closely related phocine distemper virus, seals can become infected by CDV (Kennedy
et al., 2000; Kuiken et al., 2006). Recent outbreaks in St.Gallen, Graubünden,
Appenzell, Liechtenstein and Zürich in foxes and badgers show the presence of CDV
in Switzerland (Basler Zeitung, 17.7.2009; Schweiz Magazin, 12.3.2010).
1.2 Route of infection and virus spread
The incubation period may vary from 1-4 weeks and depends on viral strain, age and
immune status of the host. Disease manifestation ranges from virtually no clinical
signs to severe disease with approximately 50% mortality (Appel, 1970). The virus is
3
INTRODUCTION

shed primarily by oro-nasal secretion (but any discharge and secretion can carry the
virus). CDV infects susceptible dogs primarily by inhalation of airborne virus or via
infective aerosol droplets, followed by virus replication in lymphoid-tissue of the
respiratory tract (Beineke et al., 2009). Tissue macrophages and monocytes located in
or along the respiratory epithelium and in tonsils represent the first cell type to pick up
and propagate the virus (Appel, 1970). Then, the virus is disseminated by lymphatics
and blood to distant hematopoietic tissues during the first viraemic phase. After initial
infection of the immune system (lymph nodes, spleen, thymus, bone marrow, mucosa-
associated lymphatic tissues), the second viraemia follows several days later,
frequently associated with high fever, and results in infection of parenchymal and
tissue cells throughout the body (Beineke et al., 2009), including the skin and CNS
where it establishes a persistent infection (Vandevelde and Zurbriggen, 2005; Gröne
et al., 2004).
1.3 Clinical signs and tissues infected
First clinical signs are characterized by lethargy, dehydration, anorexia, weight loss,
development of a biphasic fever, diarrhea, vomiting, mucopurulent and oculo-nasal
discharge, coughing, respiratory distress and possible loss of vision (Beineke et al.
2009; McGavin and Zachary, 2007). Weeks later, nervous signs including ataxia,
paralysis, convulsions, or monoclonus (muscle twitches, tremors and “tics”) occur.
Hard pad disease represents an uncommon cutaneous manifestation of distemper and
is characterized by hyperkeratosis of the footpads and nasal planum. Though the
pathogenesis of this unusual manifestation remains undetermined, it seems that CDV
causes a disturbance of keratinocyte differentiation (Gröne et al., 2003 and 2004;
Beineke et al., 2009).
Canine distemper virus has also the tendency to affect developing tooth buds and
ameloblasts, causing enamel hypoplasia in dogs that recover from infection (McGavin
et al., 2007). A persistence of primary spongiosa in the metaphysic of long bones also
4
INTRODUCTION

termed metaphyseal osteosclerosis or growth retardation lattice has been described in
young dogs suffering from systemic canine distemper (Baumgärtner et al., 1995a and
1995b). Of all distemper lesions, demyelinating encephalomyelitis, which develops
late, is the most devastating (McGavin et al., 2007). The virus causes in the CNS
multifocal lesions in the gray and white matter. Generally, demyelinating lesions
prevail. The predilection sites are the white matter of the cerebellum, the
periventricular white matter (especially around the 4th ventricle), the optic pathways
and the spinal cord. CDV enters the brain by infected mononuclear cells penetrating
the blood barrier and more importantly by circulating in the central spinal fluid and
fusing with the ependymal lining of ventricles, hence the supial and periventricular
location of the lesions (Vandevelde and Zurbriggen, 2005). Respiratory manifestation
results in serous to mucopurulent rhinitis, interstitial pneumonia and necrotizing
bronchiolitis, which is often complicated by a suppurative bronchopneumonia due to
secondary bacterial infection. Enteral infection leads to catarrhal enteritis with
depletion of Peyer’s patches. In naturally infected dogs, a pustular dermatitis, also
termed distemper exanthema, of thighs, ventral abdomen and the inner surface of ear
pinnae can be found. Additionally, a generalized depletion of lymphoid organs and an
associated immunosuppression represents an important and common manifestation of
canine distemper (Beineke et al., 2009).
2. Persistence of CDV in the central nervous system The persistence of CDV in the CNS is related to selective spread of the virus from cell
to cell with limited budding and very limited cytolysis, thus delaying immune detection
of the virus. The initial myelin lesion develop during a period of severe
immunosuppression and are not inflammatory, since perivascular cuffs are entirely
lacking and it was shown that demyelination coincides with replication of CDV in glial
cells. Therefore the demyelination in the acute stages of the disease is virus-induced.
In chronic stages of the disease, demyelination of the CNS is compounded by
5
INTRODUCTION

immunopathological reactions related to persistence of the virus (Vandevelde and
Zurbriggen, 2005). Infection with the virulent, persistent canine distemper wild-type
strain, called A75/17, is associated with a very limited cytopathic effect, limited
budding, selective cellular spread and with very limited cell-cell fusion in canine
footpad keratinocytes and primary canine brain cells (Zurbriggen et al., 1995). This
phenotype of infection is related, at least in part, to viral genetic factors. Comparison
between an attenuated, cytolytic CDV strain (Onderstepoort) and a virulent, persistent
CDV strain (A75/17) showed profound differences in the way the two viruses spread in
culture. The attenuated CDV spreads randomly to immediately adjacent cells, whereas
persistent CDV spreads selectively to more-distant cells by way of cell processes,
enabling the virus to invade the central nervous system without the need of releasing
much virus into the extracellular space (Zurbriggen et al., 1995). The attenuated
Onderstepoort (OP) CDV strain induces the formation of large multi-nucleated cells
(syncytia), followed by subsequent cytolysis. In addition, OP-CDV has been shown to
bud very efficiently from many different kind of cells, including primary canine brain
cells. These marked differences between the wild and attenuated CDV strains, have
been used to investigate viral molecular determinants related to persistent infection
(Vandevelde and Zurbriggen, 2005). Another study, comparing the two strains,
suggests that both cell-cell spread and limited production of infectious virus are related
to reduced expression of fusogenic complexes on the cell membrane, such as the
fusion (F) and attachement (H) proteins on the cell surface. F and H proteins
colocalized strongly in the cytolytic infection of the attenuated strain, but not in the
persistent strain (Meertens et al., 2003). In lymphoid tissue, however, the wild type
strain causes a severe cytolytic infection and the virus cannot persist. Therefore, the
persistence is, in addition to viral factors, also dependent on the infected tissue. The
lymphotropism of CDV is presumably based on the binding of the H protein of CDV to
the SLAM (signaling lymphocyte activation molecule) receptor, also called CD150,
followed by entry of CDV into the cell. SLAM is consitutively expressed in a variety of
6
INTRODUCTION

organs (such as lung, gastrointestinal tract, transitional epithelium) and cells (such as
lymohocytes and macrophages). Upregulation of SLAM expression was observed in
infected dogs, indicating a possible strategy to increase virus amplification in the host
(Wenzlow et al. 2007). Interestingly, SLAM could not be identified either in the footpad
keratinocytes (Wenzlow et al., 2007) or in the brain (Wyss-Fluehmann et al., 2010).
3. Classification and molecular properties of CDV CDV is a non-segmented, negative-stranded, enveloped RNA-virus and belongs to the
morbillivirus genus in the paramyxovirus family. The family of Paramyxoviridae has
been classified into 2 subfamilies, the Paramyxovirinae and the Pneumovirinae (Table
1.). At the present, the subfamily of Paramyxovirinae contain 5 genera (Respirovirus,
Rubulavirus, Henipavirus, Avulavirus and Morbillivirus), while the subfamily
Pneumovirinae contains two (Pneumovirus and Metapneumovirus) (Fontana et al.,
2008). The biologic criteria for this classification are (a) antigenic cross-reactivity
between members of a genus and (b) the presence (Respirovirus, Rubulavirus,
Avulavirus) and absence (Morbillivirus and Henipavirus) of neuraminidase activity
(Lamb et al., 2001). Morbilliviruses in general have been grouped together by their
sequence relatedness and lack of neuraminidase activity. It was found that measles
virus (MeV), CDV, and rinderpest virus (RPV) strains could use SLAMs of their
nonhost species as receptors, albeit at reduced efficiencies. Despite sequence
differences, the structure required for the interaction with morbillivirus H proteins may
be well conserved among SLAMs of many different species. Therefore, the use of
SLAM as a cellular receptor may be included in the characteristic properties of
morbilliviruses (Tatsuo et al., 2001).
7
INTRODUCTION

Table 1. Genera and representative species of the family Paramyxoviridae (Fontana et
al., 2008)
8
INTRODUCTION

4. Viral proteins The CDV genome consists of six genes, coding for 6 structural proteins, i.e. N
(nucleocapsid), M (matrix), F (fusion), H (attachment), P (phospho) and L (large)
protein, which together form the polymerase complex as well as 2 accessory non-
structural proteins, C and V (Lamb et al., 2001). In addition, in between the M and the
F gene, there is an unusual long untranslated region (MF-utr) with a short putative
open reading frame (ORF) of 52 amino acids (Stettler et al., 1997).
4.1 Attachment (H) protein Attachment of virus particles to receptors on host cell membranes is mediated by the
H protein, an integral membrane glycoprotein on the envelope of the virus. In addition
to the receptor binding function, the attachment protein of the Genera Respirovirus,
Morbillivirus and Avulavirus also agglutinate red blood cells, and the attachment
protein of the Genera Respirovirus, Rubulavirus and Avulavirus possess
neuraminidase activity, whereas Morbilliviruses and Henipavirus lack neuraminidase
activity (Fontana et al., 2008; Lamb et al., 2001). Respiroviruses and rubulaviruses
bind to sialic acid-containing proteins or lipids, while pneumoviruses attach to
glycosaminogylcans containing heparin sulphate and chondroitinsulfate. In contrast,
morbilliviruses and henipaviruses bind specific receptor proteins on the cell surface.
For MeV, two receptors have been identified: CD46 and SLAM. CDV and RPV bind to
canine and bovine homologs of SLAM, respectively. Henipaviruses use Ephrin B2 and
Ephrin B3 as cellular receptors (Fontana et al., 2008). After binding to the SLAM
receptor, the H protein interacts with the F protein. This interaction leads to a
rearrangement of the structure of the F protein, which allows subsequently the viral
genome to enter the host cell (von Messling et al., 2004).
9
INTRODUCTION

4.2 Fusion (F) protein Following attachement, the F glycoprotein mediates fusion of the viral envelope with
the plasma membrane of the host cell and release of the nucleocapsid into the
cytoplasm. The F proteins of paramyxoviruses are synthesized as inactive precursors
(F0) that are proteolytically cleaved to biologically active F1 and F2 proteins (Fontana
et al., 2008). The F proteins, are type I integral membrane proteins, which span the
membrane once and contain at their N-terminus a cleavable signal sequence, that
targets the nascent polypeptide chain synthesis to the membrane of the endoplasmatic
reticulum (ER). At their C-termini, a hydrophobic stop-transfer domain anchors the F
protein (as well as the H protein) in the membrane, leaving a short cytoplasmic tail.
Later in infection, the F proteins expressed at the plasma membrane of infected cells
can mediate fusion with neighbouring cells to form syncytia formation (cytopathic
effect, that can lead to tissue necrosis in vivo) (Lamb et al., 2001). Co-expression of
both, the F and the H protein, are necessary and sufficient to induce cellular fusion
(Stern et al., 1995) and both the wild-type F and H proteins as well as the canine
SLAM receptor act in concert to determine the phenotype of infection (Plattet et al.,
2005).
4.3 Matrix (M) protein The M protein is localized beneath the viral lipid bilayer of the envelope and is thought
to be peripherally associated with the membrane and therefore is not an intrinsic
membrane protein. It is considered to be a central organizer of viral morphogenesis,
interacting with the cytoplasmic tails of the integral membrane proteins, the lipid
bilayer and the nucleocapsid (Lamb et al., 2001). In Sendai and Measles virus, the
interaction of the M protein with the two surface glycoproteins and the nucleocapsid
has an influence on cell–cell fusion (Cathomen et al., 1998a and 1998b). Interestingly,
it has been published that, in the absence of the measles M protein, nucleocapsids
were not transported to the cell surface, suggesting that M can drag nucleocapsids
10
INTRODUCTION

from inclusion bodies to the plasma membrane (Runkler et al., 2007). Our previously
published study suggests that very limited fusogenicity in virulent CDV infection
(favouring persistence by limiting cell destruction) involves complex interactions
between all viral structural proteins. Fusion efficiency may be determined by the
structure of the viral fusion protein per se but also by its interaction with other
structural proteins of CDV. This was studied by combining genes derived from
persistent and non-persistent CDV strains in transient transfection experiments. It was
found that fusion efficiency was markedly attenuated by the structure of the fusion
protein of the neurovirulent A75/17-CDV and that the interaction of the surface
glycoproteins with the M protein of the persistent strain greatly influenced fusion
activity. Site directed mutagenesis showed that the C-terminus of the M protein is of
particular importance in this respect. Interestingly, although the nucleocapsid protein
alone did not affect F/H-induced cell–cell fusion, maximal inhibition occurred when the
latter was added to combined glycoproteins with matrix protein (Wiener et al., 2007).
4.4 Long untranslated region between the M and the F gene (M-F utr) In between all the genes of CDV untranslated regions of about 100 – 200 nucleotides
can be found, however in between the M and the F gene an unusual long untranslated
region of about 1000 nucleotides, exists in MeV and all other morbilliviruses (Heider et
al., 1997). Its precise function, if any, is unknown but deletion of this region in a ferret
CDV strain led to loss of neurovirulence (Anderson and von Messling, 2008). Previous
studies in CDV revealed a short potential open reading frame (ORF) situated at the 3’
end of the M1 within this untranslated region (referred as M2) just downstream of the
M1 ORF (Stettler et al., 1997). This small putative ORF was also found in other CDV
strains such as the tissue culture adapted strains Rockborn and vero-adapted strain
A75/17v, as well as in Snyder-Hill (derived from a natural case of distemper, passaged
in dog brains). The presence of this additional open reading frame appeared to
correlate with the ability to cause a spontaneous persistent infection in vitro (Stettler et
11
INTRODUCTION

al., 1997). It is not yet known whether this ORF is produced by the CDV strains and so
far there is no evidence that the M2 protein is expressed.
4.5 Nucleocapsid (N) protein The N protein has several functions in viral replication, including encapsidation of the
genome RNA into an RNAse-resistant nucleocapsid, association with the P-L
polymerase during transcription and replication and interaction with the M protein
during virus assembly (Lamb et al., 2001). Cherpillod et al. (2000) showed, that it was
sufficient to inoculate the nucleocapsid and the glycoproteins of the virulent strain
A75/17 to protect dogs against distemper by inducing an efficient humoral and cellular
immune response
4.6 Large (L) protein The L protein is the least abundant of the structural proteins. The L gene is the most
promoter-distal in the transcriptional map and thus the last to be transcribed. There
are five short regions in the L gene of high homology near the center that are also
conserved in the RNA-dependent RNA polymerases (RNAP) of other virus families.
Mutational analysis of these highly conserved regions indicates that these regions are
essential for RNAP activity. The P and the L proteins form a complex, and both are
required for polymerase activity (Lamb et al., 2001).
4.7 Phosho (P) protein The P gene represents an extraordinary example of a virus compacting as much
genetic information as possible into a small gene. Apart from the P protein, there are
also the 2 non-structural proteins V and C that can be expressed from the P gene.
Expression of CDV-V depends on the insertion of a non-templated guanine nucleotide
at a precise location, named “editing site”, which generates a messenger RNA that
differs from the one of P by one or two nucleotides. This generates an mRNA with an
12
INTRODUCTION

altered ORF downstream of the editing site, and thus, due to this specific mechanism,
the N-terminal domain of P and V are identical, while their C-terminal domains are
unique. The P protein is the only P gene product that is essential for viral RNA
synthesis. It is an essential component of the viral RNAP and the nascent chain
assembly complex. Although the L protein is thought to contain all viral RNAP catalytic
activities, L binds to the N:RNA template via the P protein (Lamb et al., 2001)
4.8 C protein The function of the C protein is not well known, but there is evidence that in measles
virus it plays a role as an infectivity factor (Devaux et al., 2004) and it antagonizes the
proapoptotic and antiviral activities of protein kinase (PKR) (Toth et al., 2009,).
4.9 V protein The V protein is encoded within the P gene. The N-terminus of the V protein is
identical to the P protein, the C-terminal domain of V (VCT) is unique and is known to
contain a conserved cysteine-rich region (Paterson et al., 1995; Thomas et al., 1988)
and recent X-ray studies confirmed that VCT folds into a zinc finger conformation (Li et
al., 2006).The V protein has been identified as the main inhibitor of the IFN-induced
antiviral state, though various molecular mechanisms were unraveled (Fontana et al.,
2008).
13
INTRODUCTION

5. Replication, assembly and release of paramyxoviridae
5.1 Replication Intracellular replication of paramyxoviruses begins immediately after release of the
nucleocapsid into the cytoplasm and is catalyzed by the viral RNA-dependent RNA
polymerase (vRNAP). RNA synthesis begins at the 3’ end of the genome, transcribing
the genes into mRNAs in a sequential manner by terminating and reinitiating at each
of the gene junctions. The RNAP occasionally fails to reinitiate the downstream mRNA
at each junction, leading to a loss of further-downstream genes. Hence there is a
gradient of mRNA synthesis that is inversely proportional to the distance of the gene
from the 3’ end of the genome. After primary transcription and translation, when
sufficient amounts of unassembled N protein are present, the viral RNA synthesis
becomes coupled to the concomitant encapsidation of the (+) nascent RNA chain.
Under these conditions, vRNAP ignores all the junctions, to produce an exact
complementary antigenome chain, in a fully assembled nucleocapsid. (Lamb et al.,
2001).
5.2 Virion assembly and release The assembly of viral particles requires cessation of genome replication, preparation
of completed nucleocapsids for packaging and accumulation of genomes and
nucleocapsids at the plasma membrane for budding. Polymerase complexes remain
associated with the packaged nucleocapsid and serve to initate the next circle of
infection. While the nucleocapsid is assembled in the cytoplasm, the glycoproteins are
synthesized in the endoplasmatic reticulum (eR) and undergo maturation during their
transport through the Golgi network to the cell membrane (Fontana et al., 2008). The
folding of the glycoproteins is not a spontaneous event, but it is assisted by numerous
folding enzymes and chaperons. Only correctly folded and assembled proteins are
generally transported out of the eR. As mentioned before, the M protein plays a major
14
INTRODUCTION

role in bringing the assembled ribonucleoprotein core to the plasma membrane to form
a budding virion. The glycoprotein cytoplasmic tails make important contacts with the
M protein, which, in turn, associates with the nucleocapsid (Lamb et al., 2001). In
sendai virus (SV) it was shown that expression of SV M protein induced the budding
and release of virus-like particles that contained the M protein only. Expression of the
F protein caused release of virus-like particles as well, but the release was less
efficient. Cells that expressed only the haemagglutinin-neuraminidase (HN) protein
released no HN-containing particles. Coexpression of F and M proteins enhanced the
release of F protein (Takimoto et al., 2001). In measles it was shown, that M stability
and accumulation at the intracellular membranes is a prerequisite for M and
nucleocapsid co-transport to the plasma membrane and for subsequent virus
assembly and budding process. This was found by creating recombinant viruses that
had a mutated M protein and therefore had an increased intracellular turnover but no
defective binding to other proteins. This recombinant virus was barely released from
infected cells, showing, that the defect in assembly was not due to a defective M
binding to other proteins but rather due to a reduced ability to associate with the
cellular membranes, accompanied by a deficient transport of nucleocapsids to the cell
surface (Runkler et al., 2007).
6. CDV and innate immunity The innate immunity refers to defence mechanisms of the host that are present even
before infection and have evolved to specifically recognize microbes and protect the
host against infections. Innate immunity is the first line of defence, because it is
always ready to prevent and eradicate infections. The major components of innate
immunity are epithelial barriers that block entry of environmental microbes, phagocytic
cells (mainly macrophages and neutrophils), natural killer cells and several plasma
proteins (Abbas, 1999).
15
INTRODUCTION

6.1 Interferons The interferons (IFN) are a group of secreted cytokines that elicit antiviral effects.
They are grouped in 3 classes called type I, II and III IFNs. Type I interferon comprise
a large group of molecules. Mammals have multiple distinct IFN-α and one to three
IFN-β genes (and other genes, such as IFN-ω, -ε, -δ and –κ). The IFN-α and –β genes
are induced directly in response to viral infection, whereas the other IFNs play less
well-defined roles. Thus IFN type I is rather called IFN-α/β. Type III IFNs have been
described more recently and comprise IFNλ1,-λ2 and -λ3, also referred to as IL-29, IL-
28A and IL-28B, respectively. These cytokines are also induced in direct response to
viral infection and appear to use the same pathway as the IFN-α/β genes to sense
viral infection. Type II IFN has a single member, also called IFN-γ or ‘immune IFN’,
and is secreted by mitogenically activated T cells and natural killer (NK) cells, rather
than in direct response to viral infection (Randall et al., 2008).
6.2 Antiviral functions of IFN Although IFN-α/β, IFN-γ, and IFN-λ share no obvious structural homology, they all
exhibit the ability to generate an ‘antiviral state’ in target cells. The establishment of an
anti-viral state by IFNs involves the induction of a large number of IFN-stimulated
genes (ISGs) encoding cytokines and enzymes that interfere with viral and cellular
processes to block viral replication. For example, double stranded RNA (dsRNA)-
dependent protein kinase (PKR) recognizes dsRNA that is produced during viral
replication and activates itself via autophosphorylation. Activated PKR inhibits protein
synthesis by phosphorylating the subunit of eukaryotic initiation factor 2 (eIF2a) and
also acts on numerous other substrates within the cell to establish an anti-viral state.
Another important way the IFNs limit infection is by inhibiting cell growth and
promoting programmed cell death, or apoptosis, in target cells. In addition to their
direct anti-viral and anti-proliferative properties, the IFNs also play an important role in
regulating host immunity, which can profoundly impact the ability of the host to control
16
INTRODUCTION

infection. Both IFN-α/β and IFN-γ promote the upregulation of major histocompatibility
complex (MHC) class I molecules. Because many viruses downregulate the
expression of MHC class I molecules, this is an important function of IFN that
enhances the ability of CD8+ T cells to recognize and kill virally infected cells. IFNs
also directly regulate the activities of cells participating in the innate and adaptive
immune responses. For example, IFN- α/β is critical for the enhancement of NK-cell
cytotoxicity by upregulating levels of perforins and indirectly influencing NK-cell
proliferation, and it has been shown to promote the maturation of dendritic cells. IFN-
α/β also promotes the proliferation of antigen-specific CD8+ T cells, while
simultaneously inhibiting the proliferation of naïve CD8+ T cells (Fontana et al., 2008).
6.3 Interferon induction Most pathways required for the induction of IFN- α/β are linked to interactions between
viral pathogen-associated molecular patterns (PAMPs) and host pattern-recognition
receptors (PRRs). Two major types of proteins are currently recognized as the cellular
PRRs involved in the induction of IFN- α/β: Toll like receptors (TLRs) and RNA
helicases (Fontana et al., 2008).
6.3.1 TLRs: TLRs are membrane molecules that function in cellular activation by a wide range of
microbial pathogens. In general, TLRs 1,2,4 and 6 recognize bacterial products that
are found on the cell surface, and TLRs 3,7,8, and 9 are involved in viral detection and
nucleic acid recognition within endosomes (Snyder, 2007). The primary ligand for
TLR3 is dsRNA, which is a replication intermediate for many viruses. TLR7, TLR8, and
TLR9 are classified in the TLR9 subfamily due to similarities in their amino acid
sequences, but they recognize different ligands and display different expression
patterns. While TLR7 and TLR8 both recognize viral ssRNA, TLR9 recognizes
unmethylated CpG DNA of bacteria and viruses (Table 2.). The TLRs are most
17
INTRODUCTION

commonly expressed in cellular endosomal compartments. This particular subcellular
localization is important, because it means that the cell does not need to be infected to
produce IFN- α/β, but rather it can recognize viral RNA from inactivated virus particles
or from dead cells as they are taken into the endosomal compartment (Fontana et al.,
2008). All TLRs contain an extracellular domain characterized by a leucin-rich repeat
motif flanked by a cystein-rich motif (Fig. 1). They also contain a conserved
intracellular signaling domain, Toll/interleukin (IL)-1 receptor (TIR). TLRs and their
pathogen-associated ligands are important recognition molecules for the innate
immune system and trigger a number of antimicrobial and inflammatory responses.
Although the individual TLRs exhibit ligand specificity, they differ in their cellular
expression patterns and the signal pathways they activate. There are constitutively
and inducibly expressed TLRs in different tissues. TLRs regulate cell-recruitment to
sites of infection through up-regulation of the expression of adhesion molecules,
chemokines and chemokine receptors during inflammatory response. TLRs activate
leukocytes and epithelial, endothelial and hematopoietic cells. TLRs are also
hypothesized to be essential for linking the innate immune response to the adaptive
immune response (Snyder, 2007).
6.3.2 RNA helicases In contrast to TLRs, RNA helicases are cytosolic and provide a TLR-independent
mechanism for detecting viral nucleic acids generated in the cytoplasm of an infected
cell by viral replication. The two best studied RNA helicases are retinoic acid inducible
gene I (RIG-I) and melanoma differentiation-associated gene-5 (mda-5). Each of these
proteins contains a domain, which recognizes dsRNA, and two amino-terminal
caspase-recruiting domain (CARD)-like regions, which are responsible for recruiting
downstream signaling molecules. Although both RIG-I and mda-5 recognize dsRNA,
these proteins differ in their recognition of various RNA viruses and of specific RNA
structures. For example, RIG-I but not mda-5 recognizes uncapped, unmodified
18
INTRODUCTION

5’triphosphate RNA, which allows detection of MeV and other members of the order
Mononegavirales. In contrast, mda-5 is required to mediate IFN- α/β responses to
polyriboinosinic:polyribocytidylic acid (polyI:C), the synthetic analog of viral dsRNA,
and to encephalomyocarditis (EMC) picornavirus infection in vivo (Fontana et al.,
2008).
6.3.3 INF induction In response to their respective ligands, TLR3, TLR7, TLR8, and TLR9, RIG-I, and
mda-5 each initiate a unique signaling cascade that results in the activation of
transcription factors that promote the induction of IFN- α/β. The ‘classical pathway’ is
most commonly induced by signaling through TLR3, RIG-I, or mda-5, which results in
activation of the main IFN regulatory transcription factors, IFN regulatory factor-3 (IRF-
3) and nuclear factor kB (NF-kB). TLR3 recruits an adapter protein called Toll-IL-1-
receptor domain containing adapter inducing IFN-β (TRIF), which acts as a scaffolding
protein to recruit additional components of two downstream signaling pathways that
result in the activation and translocation to the nucleus of IRF-3 and NF-kB,
respectively.
Upon translocation to the nucleus, these transcription factors bind to the IFN-β
promoter cooperatively with the c-jun/ATF-2 transcription factor to form the
‘enhanceosome,’ which is required for optimal transcription of the IFN-β gene.
IFN-β production that is induced in this way positively feeds back on the cell to
upregulate the IRF-7 transcription factor. IRF-7 is also able to bind to the IFN-β
promoter and enhance the production of IFN-β, or it can activate a ‘second wave’ of
IFN production by promoting the transcription of IFN-α genes (Figure 2A.)
While most cells are able to induce a modest IFN response to viral infection that can
be enhanced through the IRF-7- mediated positive feedback mechanism described
above, plasmacytoid dendritic cells (pDCs) can mount a rapid and extremely robust
IFN response without the need for positive feedback, due to their constitutive
19
INTRODUCTION

expression of IRF-7 (Figure 2B). IFN is induced in these cells through TLR7-, TLR8-,
and TLR9-mediated signaling pathways involving the recruitment of the myeloid
differentiation factor 88 (MyD88) adapter protein. Upon activation, MyD88 recruits IL-1
receptor-associated kinase 1 (IRAK-1) and IRAK-4 into a complex that acts as a
scaffold to recruit additional signaling components responsible for activating IRF-7 and
NF-kB. The activation of these transcription factors enables them to translocate to the
nucleus, where they promote transcription of the IFN-β gene and of multiple IFN-α
genes (Fontana et al., 2008). Similarly, RIG-I and mda-5 recruit an adapter protein
called CARD adapter inducing IFN-β (Cardif), which leads to independent activation of
both IRF-3 and NF-kB (Figure 3.) (Randall et al., 2008).
6.4 IFN signaling pathways The IFN-α/β receptor complex consists of two subunits, IFN receptor (IFNAR) 1 and 2,
which are associated with the ‘Janus’ tyrosine kinases, Tyk2 and Jak1, respectively.
Upon binding of IFN- α/β to the receptor, the subunits dimerize and bring these
kinases within sufficiently close proximity to activate each other by
transphosphorylation. Activated Tyk2 phosphorylates tyrosine 466 on IFNAR1, which
serves as a docking site for signal transducer and activator of transcription (STAT)
molecule, STAT2. Tyk2 then phosphorylates STAT2 on tyrosine 690 to recruit STAT1,
which is subsequently phosphorylated at tyrosine 701. The phosphorylated STATs
form a stable heterodimer, which results both in the creation of a novel nuclear
localization signal (NLS) and in the masking of an intrinsic nuclear export signal (NES)
in the carboxyl-terminus of STAT2. It is currently thought that the STAT1/STAT2
heterodimer next translocates to the nucleus, where it associates with the DNA-
binding protein IRF- 9, to form the IFN-stimulated gene factor 3 (ISGF3) complex. The
ISGF3 complex binds to IFN-stimulated response elements (ISREs) within the
promoters of IFN- α/β -inducible ISGs and activates transcription (Figure 4.) (Fontana
et al., 2008).
20
INTRODUCTION

TLR LIGAND MICROBIAL SOURCE
TLR2 Lipoproteins
Peptidoglycan
Zymosan
LPS
GPI Anchor
Lipoarabinomannan
Phosphatidylinositol-
Dimannoside
Bacteria
Gram-positive bacteria
Fungi
Leptospira
Trypanosomes
Mycobacteria
Mycobacteria
TLR3 Double-stranded RNA Viruses
TLR4 LPS
HSP60
Gram-negative bacteria
Chlamydia
TLR5 Flagellin Various bacteria
TLR6 CpG DNA Bacteria, protozoans
TLR7 Single-stranded RNA Viruses
TLR8 Single-stranded RNA Viruses
TLR9 CpG DNA Bacteria, viruses
Table 2. Toll-like Receptors (TLRs) and TLR Ligands and their microbial source.
Modified from Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of
disease, ed 7, Philadelphia, 2005, Saunders. CpG, Cytosine and guanine-linked
oligonucleotide; GPI, glycosyl phosphatidyl inositol; HSP60, heat shock protein 60;
LPS, lipopolysaccharide
21
INTRODUCTION

Figure 1. Signaling by a prototypic TLR, TLR4, in response to bacterial LPS.
An adapter protein links the TLR to a kinase, which activates transcription factors such
as NF-κB and AP-1. TIR, Toll/IL-1 receptor domain. From Robbins & Cotran pathologic
basis of disease, ed 7, Philadelphia, 2005, Saunders.
22
INTRODUCTION

Figure 2. Pathways involved in the induction of IFNα/β.
TLR3 recognizes extracellular or endosomal dsRNA and recruits the TRIF adapter
protein. Likewise, RIG-I and mda-5 intracellular RNA helicases recognize cytoplasmic
dsRNA and recruit the Cardif/VISA/MAVS/IPS-1 adapter protein. TRIF and
Cardif/VISA/MAVS/IPS-1 each acts as a scaffold to recruit other signaling molecules
23
INTRODUCTION

that are responsible for activating the NF-kB and IRF-3 transcription factors. NF-kB
and IRF-3 bind cooperatively with c-jun/ATF-2 to the IFN-β promoter in order to induce
the ‘first wave’ of IFN-β expression. IFN-β signaling induces the upregulation of the
IRF-7 transcription factor, which both positively feeds back on IFN-β expression and
activates the transcription of a ‘second wave’ of IFN-α expression. (B) The pDC
pathway. TLR7, TLR8, and TLR9 located within the endosomal compartment of pDCs
recognize viral ssRNA or unmethylated CpG DNA upon endocytosis of viral particles.
The MyD88 adapter protein is subsequently recruited and forms a complex with IRAK-
1 and IRAK-4, which acts as a scaffold to recruit other signaling molecules that are
responsible for activating the NF-kB and IRF-7 transcription factors. NF-kB and IRF-7
bind cooperatively with c-jun/ATF-2 to the IFN-β promoter and induce or enhance its
expression. IRF-7 can also directly induce the expression of IFN-α in pDCs without the
need for positive feedback (Fontana et al., 2008).
24
INTRODUCTION

Figure 3. Mda-5 and RIG-I-dependent signaling.
Viral RNA, generated in the cytoplasm by uncoating, transcription or replication,
activates the RNA helicases mda-5 and RIG-I. Mda-5 and RIG-I are both activated by
dsRNA, whilst RIG-I can also be activated by RNA molecules with 5’ triphosphates.
Both helicases have N-terminal CARD domains that recruit the adaptor
Cardif/VISA/MAVS/IPS-1. This adaptor, in turn, acts as a scaffold to recruit signaling
components that feed into either the IRF-3 or the NF-kB pathways. Although the
details of these downstream signalling pathways remain incomplete, for Cardif/VISA/
MAVS/IPS-1 activation, they seem very similar to those events described in Fig. 2
downstream of TRIF (Randall et al., 2008).
25
INTRODUCTION

Figure 4. Signalling pathway activated by IFN-α/β.
The biological activities of IFN-α/β are initiated by binding to the type I IFN receptor.
This leads to the activation of the receptor associated tyrosine kinases JAK1 and
Tyk2, which phosphorylate STAT1 on tyrosine 701 and STAT2 on tyrosine 690.
Phosphorylated STAT1 and STAT2 interact strongly with each other by recognizing
SH2 domains, and the stable STAT1–STAT2 heterodimer is translocated into the
nucleus, where it interacts with the DNA-binding protein IRF-9. The IRF-9– STAT1–
STAT2 heterotrimer is called ISGF3 and it binds to a sequence motif (the IFN
stimulated response element or ISRE) in target promoters and brings about
transcriptional activation. In addition to the phosphorylation of tyrosine, STAT1 also
requires phosphorylation on serine 727 for function (Randall et al., 2008).
26
INTRODUCTION

7. Objective of the present study: Investigation of two potential mechanisms which
may favor persistence of CDV, the driving force behind the chronic progression of
demyelination in canine distemper.
7.1 Investigations about the putative CDV protein M2 As mentioned before, the CDV genome contains an unusual long “untranslated” region
between the M and the F gene, which is common to all morbilliviruses. Studies by
others (Anderson and von Messling, 2008) in a ferret CDV strain have shown that
deletion of this region leads to marked attenuation of the infection in vivo. Previous
studies in our lab revealed a short potential open reading frame (ORF) situated at the
end of the M gene and is referred to as M2. However, its expression has never been
shown. In the present study we performed transient transfection experiments with
several plasmids encoding the M2 ORF, or the M2 ORF fused to an HA-tag or fused to
a red fluorescent protein (RFP) which could show the expression of M2. To validate
this result in the background of full viral infection, several recombinant viruses were
generated with the purpose of detecting expression of M2 by various methods as well
as the production of M2 knock out mutants. Several immunological and biochemical
assays could not reiterate the result of the transfection experiments, therefore
indicating that M2 was not translated. Knocking out M2 did not alter viral growth
kinetics. Altogether, our data provide evidence that M2 is not expressed by CDV, at
least in sufficient amounts to be detected by standard techniques.
7.2 Molecular mechanisms of innate immune control by wild type CDV V protein A pathogen has to evade the innate immune system in order to establish an infection
in the host. In this study we focused on Interferon (IFN) type I. This cytokine is
produced by almost all cells upon viral infection and induces an antiviral state.
27
INTRODUCTION

Recently, it has been documented that a virulent CDV strain (5804P) genetically
modified to inactivate V, was attenuated in ferrets, whereas a C-defective CDV was
fully immunosuppressive (von Messling et al., 2006). These findings demonstrate that
wild type CDV suppressed IFN induction but whether additional modulation of IFN-
mediated signaling sustains viral attenuation remains to be determined. In addition,
recent work done with C- and V-deficient MeV and RPV recombinant viruses, indicated
that V and to some extent P contribute to the final control of IFN-α/β-mediated
signaling pathway. However, to analyze the functions of the P gene products, these
recombinant viruses were based on the genetic background of vaccine strains
(Devaux et al., 2007; Nanda and Baron, 2006). Nevertheless, the paramyxovirus V
protein has been identified as the main inhibitor of the IFN-induced antiviral state,
though various molecular mechanisms were unraveled (Horvath, 2004a and 2004b).
In this study, we investigated the role of the P gene products of the highly virulent
A75/17-CDV strain in counteracting the IFN-α/β-mediated signaling pathway.
Importantly, this strain was isolated from a naturally infected dog and subsequently
kept amplified only in dogs, where it has been reported to maintain its virulence
(Cherpillod et al., 2000). Therefore, this virus has never been adapted to any cell lines.
However, the generation of recombinant virus stocks (rA75/17) with sufficient titers to
work with requires two to three passages in Vero-SLAM cells after virus rescue from
primary full-length cDNA-transfected cells (Rivals et al., 2007). Because these limited
amplification steps might already select viral variants, the entire genome of rA75/17
has been compared by direct sequencing to the one of the parental A75/17 strain and
exhibited no nucleotide difference (Rivals et al., 2007), thereby validating the unique
opportunity to investigate the molecular mechanisms of virus-host cell interactions
based on a demyelinating morbillivirus strain. Hence, recombinant A75/17 viruses and
expression plasmids were generated to investigate the role of the P gene products in
mediating IFN evasion. Infection and transfection experiments were performed in Vero
cells stably expressing the SLAM receptor for CDV. Indeed, Vero-SLAM cells are
28
INTRODUCTION

defective in IFN production and thus not only provide an optimal tool to exclusively
study IFN signaling independently of IFN induction, but they also support very efficient
A75/17-CDV replication.
Our results demonstrate that the V protein was the main viral factor responsible for
disrupting the IFN-α/β-mediated signaling pathway. The latter inhibition was neither
due to STAT1 or STAT2 degradation nor to an impairment of their phosphorylation
states upon IFN-α/β treatment. Rather, the CDV-V protein efficiently associated with
both STAT1 and STAT2, which correlated with complete inhibition of both transcription
factors’ nuclear import. Furthermore, transient expression experiments of engineered
V proteins identified both the N-terminal and the C-terminal domains as two
interdependent modules necessary to exhibit optimal IFN evasion.
29
INTRODUCTION

8. Discussion and perspectives CDV induces a multifocal demyelinating disease in dogs, similar to multiple sclerosis
in man (Vandevelde and Zurbriggen, 2005). Initial demyelination is associated with
viral replication in the white matter of the CNS. The antiviral immune response, with
invasion of immune cells in the CNS, leads to viral clearance within the inflammatory
lesion. However, despite an apparently effective immune response, CDV can persist
outside of the lesions and continues to replicate and spread to other areas. As a result
of viral persistence, a chronic progressive and relapsing disease develops (Zurbriggen
et al., 1995). Understanding of the mechanisms by which CDV is able to evade the
immune response of the host hence establishing persistence, is essential to design
therapeutic measures and improve prevention of this disease. The present study
focused on two potential mechanisms of persistence of CDV.
We first focused on the untranslated region between the M and F CDV gene. Previous
studies showed that the whole M-F untranslated region of CDV modulates virulence by
controlling F protein expression (Anderson and von Messling, 2008). The latter study,
was done with a ferret-adapted CDV strain (5804PeH) constructed by von Messling et
al. (2003). In MeV, which is closely related to CDV Takeda et al. (2005) found, that the
long utr per se was not essential for MeV replication, but that it regulated MeV
replication and cytopathogenicity by modulating the production of the M and F.
Previous studies in our lab on virulent A75/17CDV revealed a short potential open
reading frame (ORF) situated at the 3’ end of the M1 within this untranslated region
(utr) just downstream of the M1 ORF. Intriguingly, even though the sequence of the
MF utr differs dramatically between the different CDV strains, the region of the putative
ORF is conserved in the current persistent CDV strains. The conservation of these
putative ORF’s within the MF utr indicates, that there must be some evolutionary
pressure in maintaining these ORF’s and suggests a functional role of this sequence
(Stettler et al., 1997). Moreover, it is intriguing that the attenuated cytolytic OP CDV
strain lacks the M2 ORF in contrast to the CDV Rockborn, Snyder Hill and Vero
30
INTRODUCTION

adapted A75/17 strains, which in vitro all induce a persistent phenotype. In the present
study we investigated if the M2 protein is expressed and if there is a potential function
of this small protein. Even though M2 could efficiently be expressed in transfection
experiments, the same results could not be obtained in the background of full viral
infection. Several biochemical and immunological assays (such as fluorescence
microscopy, immunoblot, flow cytometry and immunoprecipitation) could not
demonstrate that in the context of a viral infection, M2 is translated. Cell type-specific
restriction of M2 expression was unlikely since M2 could not be detected in a variety of
cellular environments (vero SLAM cells, vero cells, MDCK SLAM cells, keratinocytes
or DBCC’s). In addition, the in vitro behavior of M2 knock out A75/17 Virus did not
differ from that of the parent virus. Thus these negative findings do not support a role
of the M2 ORF in persistence. On the other hand they do not exclude that the MF-utr
region may still play a role maybe not in terms of protein translation but in terms of
stability or controlling up- and/or downstream proteins. It remains intriguing why CDV
conserved this region and even more why it maintained a small putative ORF of 52
amino acids.
Persistence of CDV strongly depends on the ability of the virus to suppress the
immune response of the host. It has recently been described that V knockout CDV
(based on the 5804P virulent strain) was attenuated in infected ferrets, which was
associated, at least in part, with inhibition of IFN-α/β induction in PBMCs (von
Messling et al., 2006). In the present study we showed that the V protein of the highly
virulent A75/17-CDV strain plays a critical role in counteracting innate immunity by
additionally disrupting the IFN-α/β-dependent signaling. Detailed molecular analysis
enabled us to demonstrate that V specifically ablated STATs nuclear import without
affecting their activated phosphorylation states. Furthermore, inhibition of IFN-α/β-
dependent signaling correlated with the capacity of the V protein to efficiently interact
with STAT1 and STAT2. Finally, we identified both the N-terminal and the C-terminal
regions of V as playing a synergistic role in IFN evasion.
31
INTRODUCTION

Initial attempts to map the domains of the V protein that interact with STAT1 and
STAT2 revealed that the N-terminal domain of V (VNT) could function as an
autonomous domain interfering with IFN-α/β-induced signaling. Importantly, co-IP
experiments indicated that VNT retained association with STAT1 but failed to co-purify
STAT2. Since the full length V protein (Vwt) could efficiently co-precipitate both STATs
molecules, this suggests that the VNT domain is very likely responsible for STAT1
interaction, whereas VCT is necessary to target STAT2. Anyway, we cannot exclude
that VCT, when fused to VNT, determines a specific conformational state of VNT that
confers the capacity of the N-terminal region to target both STATs molecules.
Nevertheless, we propose that CDV-VNT and -VCT are two interdependent modules
that function synergistically to allow proper folding of the full-length V protein. In turn,
Vwt gains the ability to efficiently interact with STAT1 via its N-terminal region and
STAT2 through its C-terminal domain, which consequently offers optimal conditions to
prevent nuclear import of both STAT molecules and, consequently, to control IFN-α/β-
mediated signaling. Combined, while further work is required to support or reject this
model, this hypothesis is in excellent agreement with our results demonstrating that
CDV-V is not affecting STAT1 and STAT2 phosphorylation.
In addition to V, the CDV-P protein (sharing the identical N-terminal region with V) also
exhibited slight interaction with STAT1, which correlated with partial suppression of
IFN-α/β-mediated signaling. This is consistent with many negative strand RNA virus P
proteins, where a role in regulating innate immunity has been suggested (e.g. MeV or
RPV) (Devaux et al., 2007; Nanda and Baron, 2006;). Nevertheless, our results
obtained in a co-IP assay indicated that P bound STAT1 very inefficiently, suggesting
that a high amount of P is presumably required to act as an effective antagonist of
IFN-mediated signaling.
The peculiar molecular mechanism by which the A75/17-CDV-V protein inhibits the
IFN-α/β-mediated response differs from studies performed with other morbilliviruses.
To our knowledge, all studies performed with MV-V, except the one of Palosaari et al.
32
INTRODUCTION

(2003), have reported an inhibition of the phosphorylation of STAT1 (Caignard et al.,
2007; Takeuchi et al., 2003; Yokota et al., 2003) and STAT2 (Caignard et al., 2009;
Devaux et al., 2007; Takeuchi et al., 2003). The reasons for the differences between
CDV and MeV remain unclear. In addition to genuine biological differences between
these two morbilliviruses, the origin and passaging histories of the strains used to
study evasion from IFN action may be a factor. Indeed, we studied a highly virulent
viral strain not adapted to cultured cells, whereas the strain of MeV was attenuated
(Devaux et al., 2007), or persistently infected cells were investigated (Yokota et al.,
2003). Taken together, our results shed light into a unique molecular mechanism
sustained by the neurovirulent A75/17-CDV among the Paramyxovirus family in
controlling IFN-α/β-mediated signaling.
While we do not exclude additional molecular mechanisms, the exclusive strategy
applied by the demyelinating morbillivirus A75/17 strain in controlling the IFN-α/β-
induced response may contribute to the development of viral persistence in the CNS
and ensuing progressive tissue destruction. We are convinced that understanding the
various molecular mechanisms employed by different viruses, even among the same
genus, could inevitably raise the opportunity to develop new virus-specific therapeutic
strategies. Hence, while the precise V peptide domains responsible for STAT1 and
STAT2 association need to be defined, this might represent ideal targets for the
development of antiviral compounds that may be employed for treatment and/or to
improve current vaccines.
33
INTRODUCTION

9. References
Abbas A.K. (1999) Diseases of immunity. In: Robbins and Cotran, Pathologic basis of
disase. 7th edition, pp. 87-118
Anderson D.E. and von Messling V. (2008) Region between the canine distemper virus M
and F gene modulates virulence by controlling fusion protein expression. J.Virol. 82,
10510-10518
Appel M.J. (1970) Pathogenesis of canine distemper. J. Am. Vet. Med. Assoc. 156,
1681-1182
Appel M.J. and Gillespie J.H. (1972) Canine distemper virus. In: Virology Monographs.
Springer Verlag, Vienna, New York, pp. 1-96
Appel M.J., Reggiardo C., Summers B.A., Pearce-Kelling S., Maré C.J., Noon T.H.,
Reed R.E., Shively J.N., Oervell C. (1991) Canine distemper virus infection and
encephalitis in javelinas (collard peccaries). Arch. Virol. 119, 147-152
Appel M.J., Yates R.A., Foley G.L., Bernstein J.J., Santinelli S., Spelman L.H., Miller
L.D., Arp L.H., Anderson M., Barr M. (1994) Canine distemper epizootic in lions, tigers,
and leopards in North America. J. Vet. Diagn. Invest. 6, 277-288
Baumgärtner W., Boyce R.W., Alldinger S., Axthelm M.K., Weisbrode S.E., Krakowka S.,
Gaedke K. (1995a) Metaphyseal bone lesions in young dogs with systemic canine
distemper virus infection. Vet Microbiol. 44(2-4),201-9
Baumgärtner W., Boyce R.W., Weisbrode S.E., Aldinger S., Axthelm M.K., Krakowka S.
(1995b) Histologic and immunocytochemical characterization of canine distemper-
associated metaphyseal bone lesions in young dogs following experimental infection.
Vet.Pathol. 32(6), 702-9
Beineke A., Puff C., Seehusen F., Baumgärtner W. (2009) Pathogenesis and
immunopathology of systemic and nervous canine distemper. Veterinary immunology
and immunopathology. 127, 1-18
34
INTRODUCTION

Caignard G., Guerbois M., Labernardiere J.L., Jacob Y., Jones L.M., Wild F., Tangy
F., Vidalain P.O. (2007) Measles virus V protein blocks Jak1-mediated
phosphorylation of STAT1 to escape IFN-alpha/beta signaling. Virology 368, 351-362
Caignard G., Bourai M., Jacob Y., Tangy F., Vidalain P.O. (2009) Inhibition of IFN-
alpha/beta signaling by two discrete peptides within measles virus V protein that
specifically bind STAT1 and STAT2. Virology 383, 112-120
Cathomen T., Mrkic B., Spehner D., Drillien R., Naef R., Pavlovic J., Aguzzi A., Billeter
M.A., Cattaneo R. (1998a) A matrix-less virus is infectious and elicits extensive cell
fusion: consequences for propagation in the brain. EMBO J. 17, 3899-3908
Cathomen T., Naim H.Y., Cattaneo R. (1998b) Measles virus with altered envelope
protein cytoplasmic tails gain cell fusion competence. J.Virol. 72, 1224-1234
Cherpillod P., Tipold A., Griot-Wenk M., Cardozo C., Schmid I., Fatzer R., Schobesberger
M., Zurbiggen R., Bruckner L., Roch F., Vandevelde M., Zurbriggen A. (2000) DNA
vaccine encoding nucleocapsid and surface protein of wild type canine distemper virus
protects its natural host against distemper. Vaccine 18, 2927-2936
Devaux P., Cattaneo R. (2004) Measles virus phosphoprotein gene products:
Conformational flexibility of the P/V protein amino terminal domain and C protein
infectivity factor function. J. Virol. 78(21), 11632-11640
Devaux P., von Messling V., Sonsungthong W., Springfeld C., Cattaneo R. (2007)
Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1
phosphorylation. Virology 360, 72-83
Fontana J.M., Bankamp B., Rota P.A. (2008) Inhibition of interferon induction and
signaling by paramyxoviruses. Immunological Reviews. 225, 46-67
Gröne A., Engelhardt P., Zurbriggen A. (2003). Canine distemper virus infection:
proliferation of canine footpad keratinocytes. Vet.Pathol. 40(5), 574-8
Gröne A., Doherr M.G., Zurbriggen A. (2004) Canine distemper virus infection of canine
footpad epidermis. Vet. Dermatol. 15(3), 159-67
35
INTRODUCTION

Heider A., Santibanez S., Tischer A., Gerike E., Tikhonova N., Ignatyev G., Mrazova M.,
Enders G., Chreier E. (1997) Comparative investigation of the long non-coding M-F
genome region of wild type and vaccine measles viruses. Arch. Virol. 142, 2521-2528
Horvath C.M. (2004a) Silencing STATs: lessons from paramyxovirus interferon evasion.
Cytokine Growth Factor Rev. 15, 117-127
Horvath C.M. (2004b) Weapons of STAT destruction. Interferon evasion by paramyxovirus
V protein. Eur.J. Biochem. 271, 4621-4628
Kennedy S., Kuiken T., Jepson P.D., Deaville R., Forsyth M., Barrett T., van de Bildt
M.W., Osterhaus A.D., Eybatov T., Duck C., Kydyrmanov A., Mitrofanov I., Wilson S.
(2000) Mass die-Off of Caspian seals caused by canine distemper virus. Emerg Infect Dis.
6(6), 637-9.
Kuiken T., Kennedy S., Barrett T., Van de Bildt M.W., Borgsteede F.H., Brew S.D., Codd
G.A., Duck C., Deaville R., Eybatov T., Forsyth M.A., Foster G., Jepson P.D., Kydyrmanov
A., Mitrofanov I., Ward C.J., Wilson S., Osterhaus A.D. (2006) The 2000 canine distemper
epidemic in Caspian seals (Phoca caspica): pathology and analysis of contributory
factors. Vet Pathol. 43(3), 321-38
Lamb R.A. and Kolakofsky D. Paramyxoviridae: The viruses and their replication. In:
Fields Virology, Vol 1, 4th edition, pp. 1305-1340
Li T., Chen X., Garbutt K.C., Zhou P., Zheng N. (2006) Structure of DDB1 in complex with
a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitinin ligase. Cell. 124,
105-117
McGavin M.D. and Zachary J.F. Canine distemper virus. In: Pathologic basis of veterinary
disease, 4th Edition, pp. 541-542
Meertens N., Stoffel M.H., Cherpillod P., Wittek R., Vandevelde M., Zurbriggen A. (2003)
Mechanism of reduction of virus release and cell-cell fusion in persistent canine distemper
virus infection. Acta Neuropathol. 106, 303-310
Nanda S.K. and Baron M.D. (2006) Rinderpest virus blocks type I and II interferon action:
role of structural and nonstructural proteins. J.Virol. 80, 7555-7568
36
INTRODUCTION

Palosaari H., Parisien J.P., Rodriguez J.J, Ulane C.M., Horvath C.M. (2003) STAT protein
interference and suppression of cytokine signal transduction by measles virus V protein.
J.Virol. 77, 7635-7644
Paterson R.G., Leser G.P., Shaughnessy M.A., Lamb R.A. (1995) The paramyxovirus
SV5 V protein binds two atoms of zinc and is a structural component of virions. Virology.
208, 121-131
Plattet P., Rivals J.P., Zuber B., Brunner J.M., Zurbriggen A., Wittek R. (2005) The fusion
protein of wild-type canine distemper virus is a major determinant of persistent infection.
Virology. 337, 312-326
Randall R.E. and Goodbourn S. (2008) Interferons and viruses: an interplay between
induction, signalling, antiviral responses and virus countermeasures. Journal of General
Virology. 89, 1-47
Rivals J.P., Plattet P., Currat-Zweifel C., Zurbriggen A., Wittek R. (2007) Adaptation of
canine distemper virus to canine footpads keratinocytes modifies polymerase activity and
fusigenicity through amino acids substitutions in the P/V/C and H proteins. Virology 359,
6-18
Roelke-Parker M.E., Munson L., Packer C., Kock R., Cleaveland S., Carpenter M.,
O'Brien S.J., Pospischil A., Hofmann-Lehmann R., Lutz H., Mwamengele G.L., Mgasa
M.N., Machange G.A., Summers B.A., Appel M.J. (1996) A canine distemper epidemic in
Serengeti lions (Panthera Leo). Nature, 379(6564). 441-5. Erratum in: Nature. 2010,
464(7290), 942. Nature 1996, 381(6578), 172.
Runkler N., Pohl C., Schneider-Schaulies S., Klenk H.D., Maisner A. (2007) Measles virus
nucleocapsid transport to the plasma membrane requires stable expression and surface
accumulation of the viral matrix protein. Cellular Microbiology 9(5), 1203-1214
Snyder P.W. (2007) Diseases of immunity. In: McGavin and Zachary. Pathologic basis of
veterinary disease. 4th edition, pp. 193-251
37
INTRODUCTION

Stern L.B., Greenberg M., Gershoni J.M., Rozenblatt S. (1995) The hemagglutinin
envelope protein of canine distemper virus (CDV) confers cell tropism as illustrated by
CDV and measles virus complementation analysis. J.Virol. 69(3), 1661-8
Stettler M., Beck K., Wagner A., Vandevelde M., Zurbiggen A. (1997) Determinants of
persistence in canine distemper viruses. Veterinary Microbiology 57, 83-93
Takeda M., Ohno S., Seki F., Nakatsu Y., Tahara M., Yanagi Y. (2005) Long untranslated
region of the measles virus M and F genes control virus replication and cytopathogenicity.
J.Virol. 79, 14346-14354
Takeuchi K., Kadota S.I., Takeda M., Miyajima N., Nagata K. (2003) Measles virus V
protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting
STAT1 and STAT2 phosphorylation. FEBS Lett. 545, 177-182
Takimoto T., Murti K.G., Bousse T., Scroggs R.A., Portner A. (2001) Role of the matrix
and fusion proteins in budding of sendai virus. J.Virol. 75(23), 11384-11391
Tatsuo H., Ono N., Yanagi Y. (2001) Morbilliviruses use signaling lymphocyte activation
molecules (CD150) as cellular receptors. J.Virol. 75(13), 5842-5850
Thomas S.M., Lamb R.A., Paterson R.G. (1988) Two mRNAs that differ by two
nontemplated nucleotides encode the amino coterminal proteins P and V of the
paramyxovirus SV5. Cell. 54, 891-902
Toth A.M., Devaux P., Cattaneo R., Samuel C.E. (2009) Protein kinase PKR mediates
the apoptosis induction and growth restriction phenotypes of C protein-deficient
measles virus. J.Virol. 83(2), 961-968
Vandelde M. and Zurbriggen A. (2005) Demyelination in canine distemper virus
infection: a review. Acta Neuropathol. 109, 56-68
Von Messling V., Milosevic D., Devaux P., Cattaneo R. (2004) Canine distemper virus and
measles virus fusion glycoprotein trimers: Partial membrane-proximal ectodomain
cleavage enhances function. J. Virol. 78, 7894-7903
38
INTRODUCTION

Von Messling V., Svitek N., Cattaneo R. (2006) Receptor (SLAM(CD150)) recognition and
the V protein sustain swift lymphocyte-based invasion of mucosal tissue and lymphatic
organs by a morbillivirus. J. Virol. 80, 6084-6092
Wenzlow N., Plattet P., Wittek R., Zurbriggen A., Gröne A. (2007)
Immunohistochemical demonstration of the putative canine distemper virus receptor
CD150 in dogs with and without distemper. Vet Pathol. 44, 943-948
Wiener D., Plattet P., Cherpillod P., Zipperle L., Doherr M.G., Vandevelde M.,
Zurbriggen A. (2007) Synergistic inhibition in cell-cell fusion mediated by matrix and
nucleocapsid protein of canine distemper virus. Virus Research. 129, 145-154
Wyss-Fluehmann G., Vandevelde M., Zurbriggen A., Plattet P. (2010) Canine
distemper virus persistence in demyelinating encephalitis by swift intracellular cell-cell
spread in astrocytes is controlled by the viral attachment protein. Acta Neuropathol.
119(5), 617-30
Yokota S., Saito H., Kubota T., Yokosawa N., Amano K., Fujii N. (2003) Measles virus
suppresses interferon-alpha signaling pathway: suppression of Jak 1 phosphorylation
and association of viral accessory proteins, C and V, with interferon-alpha receptor
complex. Virology 306, 135-146
Zurbriggen A., Graber H.U., Wagner A., Vandevelde M. (1995) Canine distemper virus
persistence in the nervous system is associated with noncytolytic selective virus
spread. J. Virol. 69(3), 1678-1686
39
INTRODUCTION

Investigation of a unique short open reading frame within the 3’
untranslated region of the canine distemper virus matrix gene
Dominique Wiener1, Marc Vandevelde2, Andreas Zurbriggen1 and
Philippe Plattet1*
1Department of Clinical Research and Veterinary Public Health and 2Division of
Neurology, Vetsuisse faculty, University of Bern, Switzerland.
*To whom correspondence should be addressed, Bremgartenstrasse 109a, 3001 Bern,
Switzerland. Phone: +4131 631 26 48. Fax: ++4131 631 25 38. E-mail:
Abstract: 250
Text: 5347
Running Title: Investigation of the expression of a novel putative CDV open reading
frame
Keywords: wild-type morbillivirus, matrix gene, 3’ untranslated region, open reading frame, translation
Submitted in Virus Research
40
CHAPTER ONE

Abstract
Increasing evidence suggest that the long “untranslated” region (UTR) between
the matrix (M) and the fusion (F) genes of Morbilliviruses has a functional role. In
canine distemper virus (CDV), the F 5’ UTR was recently shown to code for a long F
signal peptide (Fsp). Subsequently, it was reported that the M/F UTRs combined with
the long Fsp were synergistically regulating the F gene and protein expression, thereby
modulating virulence. Unique to CDV, a short putative open reading frame (ORF) has
been identified within the wild-type CDV-M 3’ UTR (termed M2). Here, we
investigated whether M2 was expressed from the genome of the virulent and
demyelinating A75/17-CDV strain. An expression plasmid encoding the M2 ORF
tagged both at its N-terminal (HA) and C-terminal domains (RFP), was first
constructed. Then, a recombinant virus with its putative M2 ORF replaced by HA-M2-
RFP was successfully recovered from cDNA (termed recA75/17green-HA-M2-RFP). M2
expression in cells infected with these mutants was studied by immunoprecipitation,
immunofluorescence, immunoblot and flow cytometry analyses. Although fluorescence
was readily detected in HA-M2-RFP-transfected cells, absence of red fluorescence
emission in several recA75/17green-HA-M2-RFP-infected cell types suggested lack of
M2 biosynthesis, which was confirmed by the other techniques. Consistent with these
data, no functional role of the short polypeptide was revealed by infecting various cell
types with HA-M2-RFP over-expressing or M2 knock-out recombinant viruses. Thus,
in sharp contrast to the CDV-F 5’ UTR reported to translate a long Fsp, our data
provided evidence that the CDV-M 3’ UTR does not express any polypeptides.
41
CHAPTER ONE

1. Introduction
Canine distemper virus (CDV) causes a chronic, demyelinating, progressive or
relapsing neurological disease in dogs, because it persists in the CNS (Vandevelde and
Zurbriggen, 2005). CDV is a non-segmented, negative-stranded, enveloped RNA virus
which belongs to the morbillivirus genus in the paramyxovirus family (Lamb and
Kolakofsky, 2001). Six independent messenger RNAs (mRNAs) are generated by a so-
called “stop-start” sequential transcriptional mechanism driven by the viral RNA-
dependent RNA polymerase (vRdRp) (Lamb and Kolakofsky, 2001). While five out of
the six mRNAs contain 5’ and 3’ untranslated regions (UTR) of short sizes, the
mRNAs encoding the matrix protein (M) and the fusion protein (F) contain unusually
long 3’ UTR and 5’ UTR, respectively (referred to as M 3’ UTR and F 5’ UTR). Since
it is well known that viruses tend to evolve to contain high capacity-encoding genomes
of minimal size to the advantage of efficient viral replication (Domingo and Holland,
1997) the long M/F UTR within the CDV genome can be assumed to offer some
functional advantages to the virus. Indeed, it has been recently documented that the F
5’ UTR of CDV, formerly considered to be truly untranslated, is in fact essential to
translate an unusually long F signal peptide (von Messling and Cattaneo, 2002).
In line with these results, it has recently been shown that ferrets infected with a
recombinant virulent CDV strain (5804P) in which the entire M/F UTR was replaced
by the shorter N/P UTR remained only partially virulent. When the M/F UTR region
was removed including the region coding for the signal peptide of the F protein (Fsp),
the corresponding recombinant virus was fully attenuated (Anderson and von Messling
2008). However, the role of the CDV-M 3’ UTR was not investigated individually and
the results obtained in the latter study could therefore be attributed either to a combined
deletion effect of the Fsp sequence with the M 3’ UTR, the Fsp sequence with the F 5’
42
CHAPTER ONE

UTR or the Fsp sequence with both UTRs. Moreover, it remains also possible that the
length and specific sequences of both M/F UTRs may not be of primary importance.
Indeed, the N/P cis-acting sequences controlling the vRdRp-dependent upstream gene
transcriptional stop and downstream gene re-initiation signals are different from the
one found in the M/F UTR and may have result in the observed unbalanced F gene
transcriptional control. In the related Morbillivirus, measles virus (MeV), in vitro
studies demonstrated that the long M 3’ UTR and long F 5’ UTR were nonessential for
virus replication, but that they regulate MeV replication and cytopathogenicity by
modulating the production of the M and F proteins. The latter finding suggests a role of
this region in MeV-induced pathogenesis, although in vivo experiments were not
performed (Takeda et al., 2005).
The CDV-M mRNA contains about 1500 nucleotides, from which the M-protein
is translated by a classical ribosomal scanning mechanism. Interestingly, previous
sequence analyses revealed the existence of short putative open reading frame (ORF)
located within the 3’ UTR of the M mRNA of the A75/17-CDV strain and will be
referred in this study as M2 (Stettler et al., 1997). While it seems unlikely that the
ribosomes would efficiently scan 1250 nucleotides before initiating translation at the
first potential M2 initiation codon, unusual translation mechanisms are often utilized by
many viruses to encode proteins, including internal ribosome entry sites, ribosome
shunting, and coupled translation termination/initiation process, all of which are
dependent on sequences and structures present within the mRNA molecule that direct
ribosomal initiation at the desired location (Ahmadian et al., 1999; Hemmings-
Mieszczak and Hohn, 1999; Jang et al., 1988; Latorre et al., 1998; Pelletier and
Sonenberg, 1988; Yueh and Schneider, 1996). Moreover, the putative M2 ORF was
found in the wild-type A75/17-CDV, which exhibits a persistent phenotype in vitro.
Conversely, the M2 ORF was revealed to be absent from the M 3’ UTR of the highly
43
CHAPTER ONE

attenuated cytolytic Onderstepoort CDV strain (Stettler et al., 1997). We therefore
speculated that M2 could be a hitherto unidentified viral determinant that may
synergize with the Fsp to control CDV-persistence and/ or virulence.
In order to explore this hypothesis, five specific recombinant viruses were
engineered and successfully recovered from cDNA. Subsequently, their corresponding
phenotypes were investigated in different cell types, which were expressing, or not, the
universal Morbillivirus receptor CD150/SLAM. Combined, our results provided strong
evidence that M2 was not expressed from the unusually long M 3’ UTR of the virulent
A75/17-CDV strain, at least not in sufficient amounts to be detected by standard
fluorescence and biochemical techniques. Thus, we here formally demonstrated that the
CDV-M 3’ UTR, as opposed the CDV-F 5’ UTR, does not express any polypeptides,
thereby suggesting different putative mechanism(s) of action.
44
CHAPTER ONE

2. Materials and Methods
2.1 Cells and viruses
Vero-SLAM cells (kindly provided by V. von Messling, INRS-Institute
Armand-Frappier, University of Quebec, Laval, Quebec, Canada), MDCK-SLAM cells
(Rothlisberger et al., 2010), dog brain cell cultures (Zurbriggen and Vandevelde, 1983),
Vero cells and Bsr-T7 cells (Buchholz et al., 1999) were grown in Dulbecco’s Modified
Eagle Medium (Invitrogen) supplemented with 10% fetal calf serum (FCS), penicillin
and streptomycin. Canine footpad keratinocytes were cultured as described (Engelhardt
et al., 2005). They are maintained in William's Emedium (Bioconcept, Allschwil, CH)
including antibiotic/antimytotic solution (Gibco BRL, Basel, CH), 10% fetal bovine
serum, L-glutamine 2 mM (Bioconcept,Allschwil, CH), 10−10 M cholera toxin (Sigma,
Buchs, CH), and 10 ng/ml of epidermal growth factor EGF (Sigma). The selection of
Vero cells expressing the SLAM receptor was maintained by adding ZeocinTM
(Invitrogen). All cells were kept at 37°C in the presence of 5% CO2. The infection
experiments were performed with recombinant viruses based on the wild-type A75/17-
CDV strain (obtained from M. Appel, Cornell University, Ithaca, NY).
2.2 Antibodies
The anti-M2 antibody was produced against a synthetic peptide (nh2-
PRTCPISSYH-cooh) corresponding to amino acids 18-27 of the putative open reading
frame (M2) located at the untranslated region between the M and the F gene (M/F
UTR) of the A75/17 CDV strain (Primm srl, Custom Antibodies, Milano, Italy). The
anti-HA-tag affinity matrix rat monoclonal antibody (Clone 3F10, Roche) and the anti-
HA-tag mouse monoclonal antibody (Clone 16B12, Covance) were used for
immunoprecipitation (IP) (3F10), western blot and immunofluorescence (16B12). The
monoclonal anti-N antibody (D110) was used to perform western blots.
2.3 Generation of plasmids and recombinant viruses
45
CHAPTER ONE

In order to generate a suitable control for the different experiments, several
plasmids were generated. First, the pCI-M2 plasmid was generated by amplifying by
PCR (Expand High FidelityPLUS PCR System, Roche) the M2 ORF from the A75/17
full-length cDNA plasmid and cloned into the RsrII-cleaved pCI expression vector. The
HA-tag peptide (YPYDVPDYA) was next added, by PCR technology, in frame to the
N-terminal domain of the pCI-M2 ORF, thus generating pCI-HA-M2. Finally, the
plasmid pCI-HA-M2-RFP was created by adding a linker and a red fluorescence
marker gene in frame with HA-M2 (tD tomato, (Wyss-Fluehmann et al., 2010)) by
PCR technology.
The recombinant viruses are based on a neurovirulent wild-type isolate CDV-
A75/17 (Summers et al., 1984). The recombinant virus recA75/17red was previously
described (Rivals et al., 2007). The latter contains an additional red fluorescence
marker located between the M and F genes. To generate the full-length modified
genomic vector, a shuttle vector was first produced by amplifying a segment containing
the M 3’ UTR and subsequently cloned into the pCI-digested plasmid. Then, PCR
amplicons from vector pCI-HA-M2 and pCI-HA-M2-RFP were cloned into the shuttle
vector. Finally, full-length genomic plasmids were re-formed by cloning the fragments
from the shuttle vector into pA75/17red, thus producing pA75/17green-HA-M2 and
pA75/17green-HA-M2-RFP, respectively. To produce an M2 knock-out plasmid, we
mutated the three potential start codons of M2 into stop codons. The mutations were
obtained applying the QuikChange® Site-Directed Mutagenesis Kit (Stratagene) to the
aforementioned shuttle vector. The pA75/17red-M2ko plasmid was re-generated by
cloning the modified fragment from the shuttle vector into cleaved version of
pA75/17red. All full length cDNA clones respected the rule of six (Calain and Roux,
1993). To rescue the recombinant viruses we proceeded as previously described (Plattet
et al., 2004). Briefly, Bsr-T7 cells were co-transfected with the plasmids containing the
46
CHAPTER ONE

relevant full-length cDNA and with helper plasmids coding for the N, P and L proteins
of CDV. After two days the transfected Bsr-T7 cells were co-cultured with Vero-SLAM
cells. When a cytopathic effect had developed the cells were lysed by two freeze and
thaw cycles and the viruses (recA75/17red, recA75/17green-HA-M2, recA75/17green-HA-
M2-RFP, recA75/17green-M2Ko and recA75/17HA-M2-RFP) were stored at -80°C. Finally,
the recombinant viruses were amplified in two passages and titrated by limiting
dilution assay. The viral titers were determined by counting red fluorescent single cells
or syncytia as infectious particles per ml.
2.4 Immunofluorescence
Vero cells and keratinocytes were grown on cover slips in 6-well plates and the
following day infected with the recombinant viruses (recA75/17red, recA75/17green-HA-
M2, recA75/17green-HA-M2-RFP, recA75/17green-M2Ko and recA75/17 HA-M2-RFP) at an
MOI of 0.04. At 4 days post infection, cells were fixed with ETOH:acetic acid (95:5)
for 5 minutes at -20°C and washed in PBS. Blocking of unspecific binding sites was
performed in PBS supplemented with 2% FCS for 1 h at RT. Then the first antibody
(anti-F PAb and anti-HA MAb) diluted in PBS (1:500 and 1:1000, respectively) was
added on the cover slips and incubated 1 h at 37°C. Then the slides were washed in
PBS and incubated for 30 min at room temperature with the second antibody (alexa
fluor anti rabbit 555 and alexa fluor anti mouse 488, Invitrogen) diluted in PBS
(1:500). The cells were washed again and the nuclei were stained with TOTO-3
(Invitrogen) following the manufacturer’s advice. The slides were evaluated with a
confocal fluorescence laser microscope (Olympus).
2.5 Western blotting
Immunoblotting was performed as previously described (Plattet et al., 2007).
Vero cells were cultured in 6-well plates and infected (with the various recCDVs) at an
MOI of 0.04 one day later, or left uninfected. At 4-6 days post infection, the cells were
47
CHAPTER ONE

washed with cold phosphate buffered saline (PBS) and lysed at 4°C in RIPA buffer
(150mM NaCl (500mM NaCl to extract the C protein), 1% NP-40, 0.5% Na-
deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50mM Tris-Cl, pH 7.4) containing
HaltTM Protease Inhibitor Cocktail (SOCOCHIM) and Phosphatase Inhibitor Cocktail
Set II (MERCK) to prevent degradation of the proteins. After centrifugation, Laemmli
Sample Buffer (BIO-RAD) supplemented with DL-dithiothreitol (Fluka) was added to
the samples, they were boiled for 5 min and separated by SDS-polyacrylamide gel
electrophoresis. Afterwards, the proteins were transferred to nitrocellulose membranes
(HybondTM-ECLTM, Amersham Biosciences) which subsequently were blocked in Tris-
buffered saline containing 0.1% Tween 20 (TBS-T) and 5% nonfat dry milk for 1 h.
The membranes were incubated in blocking buffer containing the first antibody at 4°C
over night or 1 h at RT, respectively. Then the membranes were washed three times in
TBS-T and incubated with the horseradishperoxidase-conjugated second antibody for 1
h at RT. After three times washing, ECL (Amersham Biosciences) was used as
substrate and the chemiluminescence signals were detected with a LAS-3000 camera
(FUJIFILM).
2.6 Immunoprecipitation
Vero cells were seeded in 6-well plates and the next day infected at an MOI of
0.04 with recA75/17red, recA75/17green-HA-M2, recA75/17green-HA-M2-RFP,
recA75/17green-M2Ko and recA75/17HA-M2-RFP or left uninfected. In parallel experiments
different expression plasmids were transfected (PCI-HA-M2 and PCI-HA-M2-RFP). At
6 days post infection or 1 day post transfection, the cells were washed with cold PBS
and lysed at 4°C in RIPA buffer (150mM NaCl (500mM NaCl to extract the C protein),
1% NP-40, 0.5% Na-deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50mM Tris-Cl,
pH 7.4) containing HaltTM Protease Inhibitor Cocktail (SOCOCHIM) and Phosphatase
Inhibitor Cocktail Set II (MERCK) to prevent degradation of the proteins. After 10 min
48
CHAPTER ONE

centrifugation at 13000 rpm at 4°C, aliquots for total protein analyses were separated
and the remaining supernatants were incubated with the anti-HA-tag affinity matrix rat
monoclonal antibody (Clone 3F10, Roche) at 4°C over night. After 1 min
centrifugation at 13000 rpm at 4°C the pellets were washed 4 times with the same
buffer used before and finally dissolved directly in Laemmli sample buffer for western
blotting performed as described above.
2.7 Flow cytometry
Vero cells were infected with the various fluorescent protein-expressing
recCDVs with an MOI of 0.04. After 6 days post infection, cells were washed twice
with PBS and subsequently detached from the wells with PBS-EDTA (50mM) for 45
min at 37°C. The mean fluorescence intensity of 100’000 cells was then measured by
using a FACSCalibur flow cytometer.
2.8 Growth kinetics
To determine growth kinetics, cell-associated progeny viruses from different
recCDVs were taken at the indicated time post-infection. Cell-associated viruses were
recovered by scraping the cells into 1 ml of Opti-MEM (Gibco) followed by two cycles
of freeze-thaw. Cell debris were removed by centrifugation at 2000 rpm for 8 min at 4
°C. For titration, limiting dilutions of the stocks of cell-associated viruses were
performed and subsequently inoculated into Vero-SLAM cells. Two hours later, cells
were washed and DMEM-agar (1%) was applied on each wells. The numbers of
fluorescent syncytia induced by the various recCDVs were counted using a confocal
fluorescence laser microscope three days after initial infection.
49
CHAPTER ONE

3. Results
3.1 Sequence alignment of the unusually long Morbillivirus M 3’ UTR revealed a
unique ORF in several virulent CDV strains
A sequence alignment of the M 3’ UTR among several CDV strains and other
morbilliviruses was conducted. Figure 1A illustrates the alignment of the M 3’ UTR of
five virulent (5804P, 98-2646, A75/17, Snyder-Hill and CDV3) as well as one highly
attenuated (Onderstepoort) CDV strains. Clearly, the entire M 3’ UTR is very well
conserved among all tested CDV strains. However, while the putative short M2 ORF
(52aa) is present in the five virulent strains, it is absent in the attenuated one (Fig. 1A).
When the A75/17-CDV M 3’ UTR was aligned with three other morbillivirus
sequences (measles virus -MeV-, rinderpest -RPV- and peste-des-petits-ruminants virus
-PPRV-), it turned out that the high degree of conservation observed among the
different CDV strains was totally absent in the three other morbilliviruses (Fig. 1B).
Moreover, we did not find any other significant putative ORFs within the entire M 3’
UTR of MeV, RPV and PPRV. As could be anticipated, however, the only well
conserved sequence among all Morbillivirus M 3’ UTR was the transcriptional stop
motif specifically recognized by their vRdRp (Fig. 1B). Taken together, our sequence
analysis provided evidence that the presence of a putative short ORF within the 3’ UTR
of the M mRNA is restricted to potentially virulent CDV strains.
3.2 Construction of a double tagged M2 protein
In order to facilitate the detection of the short putative M2 ORF, we first
engineered a new M2-expressing plasmid by fusing at its N-terminal domain the well
known HA-tag sequence. In addition, the red fluorescent protein (RFP) was fused to
the C-terminal region of M2. A short linker peptide was also added in between the M2
and RFP proteins in order to limit the possibility that physical constrains impede both
protein functions. Importantly, red fluorescence was readily detected throughout the
50
CHAPTER ONE

cytosol and the nucleus of HA-M2-RFP-expressing Vero cells (not shown).
Importantly, no difference in cellular localization compared to RFP-transfected cells
was observed (not shown). While we could not directly assess whether the double
tagged M2 construct behaves properly, the above-mentioned results nevertheless
demonstrated that the engineered fusion protein did not dramatically impair the
fluorescence properties of the RFP.
3.3 Generation of recombinant A75/17 viruses
We next engineered five recombinant viruses in order to accurately investigate
whether A75/17-CDV indeed expresses M2 from its M 3’ UTR (Fig. 2). The first virus
consists of the A75/17-CDV genome, which additionally expresses the green
fluorescent protein from a supplementary transcription cassette located between the M
and F genes (recA75/17green). Importantly, in this virus, the M 3’ UTR remained
unaltered. The second virus bears an HA-tag fused to the N-terminal domain of the
putative M2 polypeptide (recA75/17green-HA-M2). In the third virus, in addition to the
HA-tag, the red fluorescent protein (RFP) was fused to the C-terminal part of HA-M2,
thus generating recA75/17green-HA-M2-RFP. The fourth virus was engineered to
potentially over-express the HA-M2-RFP fusion protein. To this purpose, the HA-M2-
RFP gene was removed from its natural position and subsequently cloned in place of
the GFP gene, which is expressed from the additional transcription cassette
(recA75/17HA-M2-RFP). Finally, we modified the A75/17-CDV genome in order to close
the expression of the putative M2 open reading frame (recA75/17green-M2ko). This was
achieved by substituting the three potential initiation codons into stop codons (Fig. 1A)
in order to limit the possibility that modification of the M mRNA 3’ UTR per se causes
indirect influence on viral replication. Then, using reverse genetics technology (Plattet
et al., 2004), all viruses were successfully recovered from their corresponding cDNA
51
CHAPTER ONE

plasmid. The genetic integrity of the five recombinant viruses was subsequently
confirmed by automated sequencing analysis (not shown).
3.4 Evidence for the absence of M2 expression in several different cellular
environments
To investigate whether M2 was expressed from the A75/17-CDV genome, Vero-
SLAM and MDCK-SLAM cells were initially infected with recA75/17HA-M2-RFP and
recA75/17green-HA-M2-RFP with an MOI of 0.01 and 0.03, respectively (titrated in
Vero-SLAM cells). While the first virus should consistently emit red fluorescence by
over-expressing HA-M2-RFP, the second should indicate whether M2 is expressed or
not. Both viruses exhibited a typical Morbillivirus-mediated cytopathic effect (CPE) in
both cell types within 24 hours. The CPE consisted of massive syncytia formation,
which invariably resulted in cytolysis (Fig. 3). Indeed, syncytium formation is
systematically induced by CDV in cells expressing the universal Morbillivirus receptor
CD150/SLAM. The identical phenotype was observed in the parental recA75/17green-
infeced Vero-SLAM and MDCK-SLAM cells (not shown). Importantly, and in an
anticipated manner, the control virus (recA75/17HA-M2-RFP) readily expressed HA-M2-
RFP since red fluorescence emission was repeatedly captured by confocal laser
fluorescence microscopy (Fig. 3). These results additionally confirmed that the RFP
when fused to the C-terminal domain of HA-M2 also remained fully functional in the
context of a viral infection. However, in striking contrast, no red fluorescence at all
could be monitored in recA75/17green-HA-M2-RFP-infected Vero-SLAM and MDCK-
SLAM cells, whereas bright green fluorescence emission was detected in all syncytia
(Fig. 3). Nonetheless, even though wild-type A75/17-CDV very efficiently replicates in
Vero-SLAM and, to a lesser extent, in MDCK-SLAM cells, they do not represent
natural targets of the virus (the parental Vero and MDCK cells are poorly susceptible to
52
CHAPTER ONE

wild-type A75/17-CDV replication). Thus, absence of M2 expression in SLAM stably-
transfected cells might be due to an unsuitable cellular environment.
Therefore, two primary SLAM-negative but CDV-susceptible cell types were
infected with both recombinant viruses (canine epithelial keratinocytes and canine glial
cells). Indeed, these cells not only support efficient A75/17-CDV replication but also
represent natural targets of wild-type CDV throughout a course of infection (skin and
brain) (Appel, 1969; Grone et al., 2003; von Messling et al., 2004). Consistent with our
previous results, recA75/17HA-M2-RFP exhibited strong red fluorescence emission in
infected cells, whereas recA75/17green-HA-M2-RFP did not (not shown). In the case of
recA75/17green-HA-M2-RFP-infected cells, however, GFP-expressing cells in both cell
types were easily observed. Immunofluorescence staining (using the anti-F PAb,
(Cherpillod et al., 1999)) confirmed that both viruses efficiently replicated in canine
keratinocytes and glial cells by mediating a typical non-cytolytic cell-to-cell lateral
spread (not shown) (Rivals et al., 2007; Wyss-Fluehmann et al., 2010; Zurbriggen et
al., 1995). Since both viruses bear an HA-tagged M2 protein, indirect
immunofluorescence staining using an anti-HA MAb was additionally performed on
fixed and permeabilized cells. Consistent with our previous results obtained in direct
fluorescence monitoring, only recA75/17HA-M2-RFP-infected cells exhibited positive
staining. Similarly, no stained cells could be detected in recA75/17green-HA-M2-
infected cells (not shown). Taken together, these results strongly suggested that M2
was not translated in these two additional relevant cellular environments.
3.5 Prolonged infection does not induce M2 expression
Since Vero cells do not express high avidity-binding receptors for wild-type
CDV strains, they are poorly susceptible to A75/17-CDV. Therefore, inoculation in
Vero cells with wild-type CDV leads to infection of only few cells, in which the virus
replicates very well without obvious syncytium formation and very limited viral spread.
53
CHAPTER ONE

Nevertheless, this experiment allowed us to observe infected cells over a long period
(up to 6 days) and to investigate whether M2 expression would perhaps occur over
time, albeit only transiently.
Since the two above mentioned recCDVs either express GFP or RFP from their
additional transcription unit, confocal laser fluorescence microscopy analysis was first
used to monitor viral replication in Vero cells. In an anticipated manner, the percentage
of GFP-expressing Vero cells upon infection with recA75/17green-HA-M2-RFP
remained very low but all infected cells exhibited strong green fluorescence emission,
indicating full replication but very slow viral spread (Fig. 4). Similar results were
obtained with recA75/17HA-M2-RFP but infected cells were emitting red fluorescence.
With both viruses few clusters of a maximum of 5-20 infected cells at 4 dpi could be
observed (Fig. 4). Here again, in almost all green fluorescence-emitting cells,
recA75/17green-HA-M2-RFP did not produce any detectable red fluorescence,
throughout the entire period of incubation (up to 6 days) (Fig. 4). Intriguingly,
however, in extremely rare cells, and only after 4-6 days post infection, very faint red
fluorescence emission could be detected (not shown). To further investigate the
possibility that M2 might be expressed in few selected cells, flow cytometry analysis
was undertaken from infected cells. The results did not support the fluorescence
microscopical findings, since absolutely no red fluorescence signal was reproducibly
recorded (not shown). We thus speculate that the very faint red fluorescence observed
in extremely few infected cells may be due to unspecific emission of recA75/17green-
HA-M2-RFP in Vero cells at late time points post-infection.
3.6 Failure to detect M2 using sensitive biochemical techniques
We next sought to verify the expression of M2 using biochemical methods, such
as protein immunoprecipitation and western blotting. However, all our attempts to
generate functional anti-M2 polyclonal antibodies failed (as controlled with the
54
CHAPTER ONE

different M2 expression plasmids, see material and methods section). Moreover, the
SLAM molecule used to generate the stable Vero-SLAM and MDCK-SLAM cell lines
bears an HA-tag, thus precluding the use of anti-HA MAbs to accurately assess M2
biosynthesis in these cells. Immunoprecipitation (IP) experiments were thus carried out
from infected Vero cells. In this set of experiments, the parental recombinant viruses
(recA75/17green), the HA-tagged M2-expressing virus (recA75/17green-HA-M2) and the
M2 knockout virus (recA75/17green-M2ko) were additionally included in the analysis.
Hence, cells were infected with the various recCDVs with an MOI of 0.04 (as titrated
in Vero-SLAM cells) and 4 days post-infection cells were lysed using the stringent
RIPA buffer. A rat anti-HA MAb directly coupled to beads was next added to total
protein extracts, thereby allowing for potential HA-tagged proteins
immunoprecipitation. Subsequently, proteins were boiled and loaded on a SDS
polyacrylamide gel, ran and transferred onto a nitrocellulose membrane. Finally, HA-
tagged proteins were revealed using a mouse anti-HA MAb. Results shown in figure 5
(upper panel) indicate that no protein could be immunoprecipitated from Vero cells
infected with recA75/17green and recA75/17green-M2ko, the two untagged viruses.
Conversely, a band which migrated corresponding to proteins with a molecular weight
of about 70 kDa was readily detected in Vero cells infected with the control HA-M2-
RFP over-expressing recombinant virus (recA75/17HA-M2-RFP) (Fig. 5, upper panel).
Importantly, the identical band was not detected using protein extracts derived from
recA75/17green-HA-M2-RFP-infected Vero cells (Fig. 5, upper panel). Immunoblots
were additionally performed from infected Vero cells using the anti-N MAb (D110),
and a band of about 60 kDa was detected in all rCDVs-infected cells, thus confirming
that all recombinant viruses could enter and replicate sufficiently in Vero cells to
express detectable amounts of N protein by western blot (Fig. 5, middle panel). Note
that the intensity of the N-protein revealed by western blotting remained extremely
55
CHAPTER ONE

faint. This is very likely due to the very limited number of infected cells observed upon
wild-type recCDVs inoculation in Vero cells. Furthermore, direct immunoblotting
using the anti-HA MAb never revealed any bands (also in case of the M2 over-
expressing virus) presumably due to the limit of detection of the western blot assay as
compared to the IP assay (not shown).
3.7 Growth kinetics comparison between recA75/17green and recA75/17green-M2ko in
different cell types
Although the above data suggested that M2 is not expressed in A75/17-CDV-
infected cells, we wished to exclude the possibility that M2 could nevertheless be
translated in a very limited amount (as potentially suspected only in Vero cells), below
the limit of detection of conventional IP and immunoblot assays. It cannot be excluded
that even small amounts of M2 could have an effect on the infection. However,
although we could not formally demonstrate that M2 kept its natural putative function
within the HA-M2-RFP fusion construct, over-expressing M2 did not modify
substantially the phenotype of infection mediated by recA75/17green, and this in all
tested cell types. Figure 6A illustrates a typical example of similar phenotypes of
infection in Vero cells induced by the M2 over-expressing, the M2 knockout and the
wild-type recombinant viruses.
To further validate these results, titration experiments in several cell lines were
performed using the parental recA75/17green virus and the M2-knockout virus
(recA75/17green-M2ko). Strikingly, viral titers of both cell-free (not shown) and cell-
associated viruses remained largely unaltered for both recombinant viruses in the four
cell types tested in this study (Vero-SLAM, MDCK-SLAM, Vero and canine epithelial
keratinocytes) (Fig. 6). In addition, both viruses produced very similar CPE in Vero-
SLAM and MDCK-SLAM cells and induced an indistinguishable non-cytolytic cell-to-
cell spread type of infection in primary canine keratinocytes and glial cells (not
56
CHAPTER ONE

shown). Taken together, our results provided strong evidence that neither over-
expressing nor deleting M2 from the CDV genome, resulted in detectable phenotypic
differences. This further validated the notion that M2, should it be expressed in very
small amounts, has no obvious effect on viral replication and spread.
57
CHAPTER ONE

4. Discussion
Viruses have evolved to minimize their genetic information, establishing unique
strategies to translate different viral proteins from the same nucleic acid sequence to
the advantage of rapid and efficient replication (Domingo and Holland, 1997). Previous
studies in CDV revealed a short potential open reading frame (M2) located within an
unusually long UTR at the 3’end of the M mRNA (Stettler et al., 1997). This finding
offered a potential explanation of why CDV has retained the long untranslated region
throughout evolution. Although the M2 ORF location within the M 3’ UTR very likely
excludes classical ribosome-scanning as the main mechanism for its translation, unique
strategies to express proteins have often been observed in different viruses (Ahmadian
et al., 1999; Hemmings-Mieszczak and Hohn, 1999; Jang et al., 1988; Latorre et al.,
1998; Pelletier and Sonenberg, 1988; Yueh and Schneider, 1996). Typically,
paramyxoviruses express at least three proteins from their P gene (P, C and V). While
the P-protein is encoded via a common ribosomal-scanning mechanism, production of
C protein depends on a ribosomal-leaky scanning process that initiates translation at an
alternative, out-of-frame (+1), AUG. Furthermore and unique to paramyxoviruses, an
“editing” mechanism that occurs at a precise location within their P gene, generates the
V protein. In Sendai virus (SeV) infection, it has been reported that the unique Y
proteins (also encoded within the SeV-P gene) were produced through an additional so-
called “ribosomal-shunt” translational mechanism (Latorre et al., 1998). These notions
combined with the fact that the CDV-F 5’ UTR formerly believed to be truly
untranslated has recently been demonstrated to be mostly translated into a long F signal
peptide, prompted us to investigate whether the putative M2 ORF within the CDV-M 3’
UTR is indeed translated. This objective was further supported by sequence analyses,
which revealed that, despite a high degree of “GC” content, the M 3’ UTR were highly
variable among several related morbilliviruses, all of which but CDV lack the M2 ORF.
58
CHAPTER ONE

However, in sharp contrast, among various CDV strains, the entire M 3’ UTR was
found to be well conserved. Intriguingly, the M2 ORF was present in all sequences
derived from the virulent CDV strains but not in the attenuated strain suggesting that
M2 might be related to viral-induced persistence and/or virulence.
It is now well accepted that CDV, and morbilliviruses in general, mediate a
cytolytic infection in a cellular environments where the universal Morbillivirus
receptor CD150/SLAM is expressed. In contrast, in infection with the wild-type
A75/17-CDV, no obvious cell-cell fusion and subsequent cytolysis is observed in
different cell types lacking SLAM expression. In the latter cellular environments,
A75/17-CDV spreads from cell-to-cell in a non-cytolytic manner with extremely poor
extracellular progeny virus production (Grone et al., 2003; Rivals et al., 2007; Wyss-
Fluehmann et al., 2010; Zurbriggen et al., 1995). In this study, we therefore
intentionally selected several cellular environments representing these two
fundamentally different phenotypes of infection. In order to demonstrate M2
expression in the present study, we based our strategy on the generation of several
recombinant viruses. However, all our attempts to demonstrate M2 expression from the
viral genome of the highly virulent and demyelinating A75/17-CDV were suggestive
for the absence of M2 translation in all cellular environments tested. Several lines of
evidence support the latter conclusion. First, we tagged both the C-terminal (RFP) and
N-terminal (HA) domains of M2 within the viral genome to allow for efficient
immunoprecipitation and assessment of M2 expression in living cells by fluorescence
microscopy. Even though HA-M2-RFP biosynthesis by recA75/17-CDV was
investigated in several cell types, significant red fluorescence emission was never
detected neither by direct fluorescence emission, indirect immunofluorescence staining
nor flow cytometry analysis. In contrast, all these techniques readily detected the
presence of the fluorescent fusion protein in M2 over-expressing recA75/17HA-M2-RFP-
59
CHAPTER ONE

infected cells. Secondly, due to the presence of the HA-tag, we confirmed by standard
immunoprecipitation and western blot analysis that M2 was indeed not produced in
recA75/17green-HA-M2-RFP-infected cells. Thirdly, because it could not be ruled out
that small amounts of M2 were expressed below the limit of detection of the methods
used, we performed infection experiments with M2 over-expressing, M2 knockout and
wild-type viruses in different cell systems in order to detect any potential differences in
growth kinetics, type of spread and CPE. However, since no differences could be
observed, we concluded that the putative M2 polypeptide, should it be expressed in low
quantities, has no noticeable effect on CDV infection.
It has been clearly documented that the whole CDV-M/F untranslated region
combined with the Fsp modulated virulence in ferrets by controlling F transcription and
protein expression (Anderson and von Messling, 2008). However, Anderson and
colleagues did not determine whether the M 3’ UTR and/or the F 5’ UTR acted
synergistically with the Fsp to attenuate CDV. While the latter question merits further
investigation, the results obtained in our study appear to exclude that the putative ORF
within the M 3’ UTR plays a role in CDV infection. Interestingly, although in the
closely related measles virus (MeV) no long signal peptide is translated from the F 5’
UTR, the extremely long M/F UTR per se was not essential for MeV replication but
could indirectly regulate MeV replication and cytopathogenicity by modulating the
levels of M and F protein synthesis (Sidhu et al., 1995; Takeda et al., 2005). In
particular, the M 3’ UTR seemed to be responsible for stabilizing the mRNA in turn
enhancing the production of M, which was associated with enhanced replication.
Further studies are therefore required to demonstrate whether i) yet unidentified
specific regulating sequences, ii) transcriptional cis-acting sequences, or iii) the length
of the CDV-M 3’ UTR per se are of primary importance for synergizing with the CDV-
Fsp to modulate the infection. In any case, these mechanisms may well be important in
60
CHAPTER ONE

modifying the CDV-M protein expression level, which in addition to F, would in turn
regulate CDV-mediated replication and disease induction.
In conclusion, while we cannot exclude that M2 is selectively translated in cells
that were not tested in this study, our data provided evidence that M2, potentially
translated from a unique and short open reading frame located within the M 3’ UTR of
the virulent A75/17-CDV strain, is not a viral determinant synergising with the Fsp to
control virulence. The intriguing question remains why CDV evolved to retain more
than 400 untranslated nucleotides within the M mRNA, in its otherwise very compact
and tightly tuned genome .
Acknowledgments
We are grateful to Veronika von Messling for having provided the Vero-SLAM
cells.
61
CHAPTER ONE

Figure legends
Figure 1. A) Sequence alignment of the M 3’ UTR among several CDV strains.
GenBank numbers for each virus sequence are: AY386316.1 (CDV, 5804P strain),
AY542312.2 (CDV, 98-2646 strain), AF164967.1 (CDV, A75/17 strain), GU138403.1
(CDV, Snyder Hill strain), EU726268.1, (CDV, CDV3 strain), AF305419.1 (CDV,
Onderstepoort strain). B) Sequence alignment of the M 3’ UTR among several
morbilliviruses. GenBank numbers for each virus sequence are: AF164967.1 (CDV,
A75/17 strain), AB016162.1 (measles virus, ICB strain), NC_006296.2 (Rinderpest
virus, Kabete O strain), EU267273.1 (Peste-des-petits-ruminants virus, ICV89 strain).
Red boxes highlight the putative M2 ORF. The different potential initiation codons are
also represented in red. Furthermore, the black box in figure 1B illustrates the
transcriptional stop cis-acting sequence. Stars below the sequences show identical
residues in all sequences.
Figure 2. Schematic representation of the five generated recombinant A75/17-CDVs.
All viruses bear an additional expressing unit coding for the indicated fluorescence
protein located between the M and F genes. The different constructs (shown above each
genome) that were cloned in place of the natural putative M2 ORF are also represented.
Below every genome is shown the putatively entire generated M mRNA. The length of
each genome is also indicated below its corresponding viral genome’s drawing. RFP:
red fluorescent protein, GFP: green fluorescent protein, M2: putative M2 ORF located
within the M 3’ UTR, HA: influenza hemagglutinin-specific tag sequence. Drawings
are not in scale.
Figure 3. Monitoring of the fluorescence induced by the indicated recombinant viruses.
Vero-SLAM and MDCK-SLAM cells were infected with an MOI of 0.01 and 0.03,
62
CHAPTER ONE

respectively. Two days post-infection infected cells were screened for green and/or red
fluorescence emission by confocal laser fluorescence microscopy. Moreover, phase
contrast images of representative fields of view illustrating the typical cytopathic effect
induced in both cell types by the recCDVs, are shown.
Figure 4. Monitoring of the fluorescence induced by the indicated recombinant viruses.
Vero cells were infected with an MOI of 0.04. Two days post-infection infected cells
were screened for green and/or red fluorescence emission by confocal laser
fluorescence microscopy. Moreover, phase contrast images of representative fields of
view illustrating no obvious cytopathic effect induced by the recCDVs, are shown.
Figure 5. Investigation of M2 translation by immunoprecipitation assay. Vero cells
were infected at an MOI of 0.04 with the various recCDVs or left uninfected, and
subsequently lysed 4 dpi for immunoprecipitation assay. To assess M2 expression, HA-
M2-RFP was immunoprecipitated (IP) with an anti-HA monoclonal antibodies (rat)
directly coupled to G sepharose beads. Then, western blot analysis, using an anti-HA
monoclonal antibody (mouse) to detect the HA-M2-RFP fusion protein, was performed
(upper panel). Total cell lysates (TL) were subjected to immunoblot analysis to
investigate the endogenous the viral N-protein expression (middle panel). As a loading
control, we revealed the actin protein using an anti-actin MAb (bottom panel).
Figure 6. No phenotypic differences are observed in the M2 over-expressing and the
knockout virus-infected cells as compared to the parental wild-type rA75/17green virus.
A) recCDVs lateral spread in Vero cells. Vero cells were infected with recA75/17green-
M2ko, recA75/17HA-M2-RFP and rA75/17green with an MOI of 0.04. Fluorescence emission
from the marker protein expressed from the additional transcription unit was captured
63
CHAPTER ONE

by confocal laser fluorescence microscopy 1, 3 and 6 days post-infection. B) Growth
kinetics of the recCDVs in various cell types. Vero-SLAM, MDCK-SLAM,
keratinocytes and Vero cells were infected with recA75/17green and recA75/17green-
M2ko. Viral titers of cell associated viruses, taken at the indicated time post-infection,
were determined by limiting dilution assay.
64
CHAPTER ONE

Reference List Ahmadian, G., Chambers, P., Easton, A.J., 1999. Detection and characterization of
proteins encoded by the second ORF of the M2 gene of pneumoviruses. J. Gen. Virol. 80, 2011-2016.
Anderson, D.E., von Messling, V., 2008. Region between the canine distemper virus M and F genes modulates virulence by controlling fusion protein expression. J. Virol. 82, 10510-10518.
Appel, M.J., 1969. Pathogenesis of canine distemper. Am. J. Vet. Res. 30, 1167-1182.
Buchholz, U.J., Finke, S., Conzelmann, K.K., 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73, 251-259.
Calain, P., Roux, L., 1993. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J. Virol. 67, 4822-4830.
Cherpillod, P., Beck, K., Zurbriggen, A., Wittek, R., 1999. Sequence analysis and expression of the attachment and fusion proteins of canine distemper virus wild-type strain A75/17. J. Virol. 73, 2263-2269.
Domingo, E., Holland, J.J., 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151-178.
Engelhardt, P., Wyder, M., Zurbriggen, A., Grone, A., 2005. Canine distemper virus associated proliferation of canine footpad keratinocytes in vitro. Vet. Microbiol. 107, 1-12.
Grone, A., Engelhardt, P., Zurbriggen, A., 2003. Canine distemper virus infection: proliferation of canine footpad keratinocytes. Vet. Pathol. 40, 574-578.
Hemmings-Mieszczak, M., Hohn, T., 1999. A stable hairpin preceded by a short open reading frame promotes nonlinear ribosome migration on a synthetic mRNA leader. RNA 5, 1149-1157.
Jang, S.K., Krausslich, H.G., Nicklin, M.J., Duke, G.M., Palmenberg, A.C., Wimmer, E., 1988. A segment of the 5' nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 62, 2636-2643.
Lamb, R.A., Kolakofsky, D., 2001. Paramyxoviridae: the viruses and their replication, p. 1305-1340. In B. N. Fileds, D. M. Knipe, P. M. Howley, and D. E. Griffin (ed), Fields virology, 4th ed. Lippincott-Raven Publishers, Philadelphia, PA.
Latorre, P., Kolakofsky, D., Curran, J., 1998. Sendai virus Y proteins are initiated by a ribosomal shunt. Mol. Cell Biol. 18, 5021-5031.
Pelletier, J., Sonenberg, N., 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334, 320-325.
65
CHAPTER ONE

Plattet, P., Cherpillod, P., Wiener, D., Zipperle, L., Vandevelde, M., Wittek, R., Zurbriggen, A., 2007. Signal peptide and helical bundle domains of virulent canine distemper virus fusion protein restrict fusogenicity. J. Virol. 81, 11413-11425.
Plattet, P., Zweifel, C., Wiederkehr, C., Belloy, L., Cherpillod, P., Zurbriggen, A., Wittek, R., 2004. Recovery of a persistent Canine distemper virus expressing the enhanced green fluorescent protein from cloned cDNA. Virus Res. 101, 147-153.
Rivals, J.P., Plattet, P., Currat-Zweifel, C., Zurbriggen, A., Wittek, R., 2007. Adaptation of canine distemper virus to canine footpad keratinocytes modifies polymerase activity and fusogenicity through amino acid substitutions in the P/V/C and H proteins. Virology 359, 6-18.
Rothlisberger, A., Wiener, D., Schweizer, M., Peterhans, E., Zurbriggen, A., Plattet, P., 2010. Two domains of the V protein of virulent canine distemper virus selectively inhibit STAT1 and STAT2 nuclear import. J. Virol. In press.
Sidhu, M.S., Chan, J., Kaelin, K., Spielhofer, P., Radecke, F., Schneider, H., Masurekar, M., Dowling, P.C., Billeter, M.A., Udem, S.A., 1995. Rescue of synthetic measles virus minireplicons: measles genomic termini direct efficient expression and propagation of a reporter gene. Virology 208, 800-807.
Stettler, M., Beck, K., Wagner, A., Vandevelde, M., Zurbriggen, A., 1997. Determinants of persistence in canine distemper viruses. Vet. Microbiol. 57, 83-93.
Summers, B.A., Greisen, H.A., Appel, M.J., 1984. Canine distemper encephalomyelitis: variation with virus strain. J. Comp Pathol. 94, 65-75.
Takeda, M., Ohno, S., Seki, F., Nakatsu, Y., Tahara, M., Yanagi, Y., 2005. Long untranslated regions of the measles virus M and F genes control virus replication and cytopathogenicity. J. Virol. 79, 14346-14354.
Vandevelde, M., Zurbriggen, A., 2005. Demyelination in canine distemper virus infection: a review. Acta Neuropathol. 109, 56-68.
von Messling, V., Cattaneo, R., 2002. Amino-terminal precursor sequence modulates canine distemper virus fusion protein function. J. Virol. 76, 4172-4180.
von Messling, V., Milosevic, D., Cattaneo, R., 2004. Tropism illuminated: lymphocyte-based pathways blazed by lethal morbillivirus through the host immune system. Proc. Natl. Acad. Sci. U. S. A 101, 14216-14221.
Wyss-Fluehmann, G., Zurbriggen, A., Vandevelde, M., Plattet, P., 2010. Canine distemper virus persistence in demyelinating encephalitis by swift intracellular cell-to-cell spread in astrocytes is controlled by the viral attachment protein. Acta Neuropathol. 119, 617-630.
Yueh, A., Schneider, R.J., 1996. Selective translation initiation by ribosome jumping in adenovirus-infected and heat-shocked cells. Genes Dev. 10, 1557-1567.
66
CHAPTER ONE

Zurbriggen, A., Graber, H.U., Wagner, A., Vandevelde, M., 1995. Canine distemper virus persistence in the nervous system is associated with noncytolytic selective virus spread. J. Virol. 69, 1678-1686.
Zurbriggen, A., Vandevelde, M., 1983. Canine distemper virus-induced glial cell changes in vitro. Acta Neuropathol. 62, 51-58.
67
CHAPTER ONE

CDV-5804P ATCATTGGTTCATGAACTAAAAATCAAATGCCTTGGTGGCATTGTCCAGGATCTCTTAAT 60
CDV-98 2646 ATCATTGGTTCATGAACTAAAACTCAAATGACTTGGTGGCATTGTCCAGGATCCCTTAAT 60
CDV-A75/17 ATCATTAGTTCATGAACTAAAACTCAAATGACTTGGTGGCATTGTCCAGGATCCCTTAAT 60
CDV-SH ATCATTAGTTCATGAACTAAAACTCAAACGCCTTAGTAGCATTGCCCAAGATCCCTTGAT 60
CDV-CDV3 ATCATTAGTTCATGAACTAAAACTCAAACGCCTTAGTAGCATTGCCCAAGATCCCTTGAT 60
CDV-OP ATCATCAGTTCATGAACTTAAAAGCAAACGCCTTAGTAGCACTGCCCAAGATCCCTTGAT 60
***** *********** *** **** * *** ** *** ** *** **** *** **
CDV-5804P CCCCTCAGACAAGAATTGAGGCTACAAGTATCAACTGTCTCGATGTCGCTCCTGCACTTT 120
CDV-98 2646 CCCCTCAGACAAGAATTGAGGCTACAAGTATCAACTGTCTCGATGTTGCTCCTGCATTTT 120
CDV-A75/17 CCCCTCGAACAAGGATTGAGGCTACAAGTATCAACTGTCTCGATGTTGCTCCTGCATTTT 120
CDV-SH CCCCGCAAGCGAGGATTGAGGGTATAAATATCGACTGTCTAGATGTTGCTCCTGCATTTT 120
CDV-CDV3 CCCCGCAAGCGAGGATTGAGGGTATAAATATCGACTGTCTAGATGTTGCTCCTGCATTTT 120
CDV-OP CCCCGCAAGCGAGGATTGAGGGTATAAGCACCGACCATCCAGACGTTGCTCCTGCATTTT 120
**** * * ** ******* ** ** * * ** ** ** ** ********* ***
CDV-5804P AAGCGTGTTCTATAGGTTTCTAAACTGCTCTTTCGTGCCTACTACTCTGGTGGCTCTGCA 180
CDV-98 2646 AAGCGTGTTCTATAGGTTTCTAAACTGCTCTTTCGTGCCTACTACTCTGGTGGCTCTGCA 180
CDV-A75/17 AAGCGTGTTCTATAGTTTTCTAAGCTGCTCGTTCGTGCCTGCTATTCTGGTGACTCTGCA 180
CDV-SH GAGCGTGGCCTATAGGTTTCTAAACTGCTCATCCGTGCCCACAATTCCAGTGACGCCTCA 180
CDV-CDV3 GAGCGTGGCCTATAGGTTTCTAGACTGCCCATCCGTGCCCACAATTCCAGTGACGCCTCA 180
CDV-OP GAGTGTGTCCCATAAGCCTCCAAACCGCTCACTCGTGCCCACAACTCCAGTGACGCCTCG 180
** *** * *** ** * * ** * ****** * * ** *** * * *
CDV-5804P ATATGAAGACAGCTGAATCAAACCAATTCATGCCTAAGAGTAGGTTGATCATTATCGGAC 240
CDV-98 2646 ATATGAAGACAGCTGAATCAAACCAATTTATGCCTAAGAGTAGGTTGATCATTATCGGAC 240
CDV-A75/17 ATATGAAGACAGCTGAATCAAACCAATTCATGCCTAAGAGTAGGTTGATCATTATCGGAC 240
CDV-SH ATATGTGAAAATAGCTGAATCAAAACAGTTCTTGCTTAAGATTAGGTTGATCATTATCGGAC 240
CDV-CDV3 ATATGATGAAAATAGCTGAATCAAAACAGTTCTTGCTTAAGATTGGGTTGATCATTATCGGAC 240
CDV-OP ATACGAAAGCATCCGAACCAAAACAGCTCTTGCCCAAGATTAGGTTGATCATTATCGGAC 240
*** *** * * *** **** ** * *** **** * ******************
CDV-5804P CAAGAAATTTATGGATGCTTGGGGTTTTGAACTTCGCCTCTAGGAATCTCACTTTAACAA 300
CDV-98 2646 CAAGAAATTTATGATGGATGCTTGGGGTTTTGAACTTCGCCTCTAGGAATCTCACTTTAACAA 300
CDV-A75/17 CAAGTAATGTATGGATGCTTGGGGTTTTGAACTTCGCCTCTAGGAATCTCACTTTAACAA 300
CDV-SH CAAGAAATGAATGGATGCCTGGGGTTTTGAGCTTCGCTTCTAGGATTCTCACTTTAACAA 300
CDV-CDV3 CAAGAAATGAATGGATGCCTGGGGTTTTGAGCTTCGCTTCTAGGAATCTCACTTTAACAG 300
CDV-OP CAAGAAATGAATGGATGCCTGGGGTTTTTAGCTTCGCTTCTAGGTATCTCACTTTAACAA 300
**** *** ******** ********* * ****** ****** *************
CDV-5804P TTATACTTCCACGCACTTGCCCGAGCTCAAACTATCACTAGTAGTCCTGTTTCACGAAAT 360
CDV-98 2646 TTATACCTCCACGCACTTGCCCGATCTCAAGCTATCACTAGTAGTCTTGTTTCACGAAAT 360
CDV-A75/17 TTATACCTCCACGCACTTGCCCGATCTCAAGCTATCACTAGTAGTCTTGTTTCACGAAAT 360
CDV-SH TTATACTCCCACGCACTTGCCTGATCTCAAGCCATCACTAGTAGTCTTGTTTCACGGAGT 360
CDV-CDV3 TTATACTCCCACGCACTTGCCTGATCTCAAGCCATCACTAGTAGTCTTGTTTCACGGAGT 360
CDV-OP TTATACTCCCACGCACTTGCCTGATCTCAAGCTATCACTAGTAGTCCTGTTTCACGGAAC 360
****** ************* ** ***** * ************* ********* *
CDV-5804P TGTGACTGTCTATCTTTCTATCACCAATCGTTAATAATTAATCAAAA 407
CDV-98 2646 TATGACTGTCTATCTTTCTATCACCAATCGTTAATAATTAATCAAAA 407
CDV-A75/17 TATGACTGTCTATCTTTCTATTACCAATCGTTGATAACTAATCAAAA 407
CDV-SH TATGACTGTCCATCTTTCTATCACAGCTCATTAATAATTAATCAAAA 407
CDV-CDV3 GATGACTGTCCATCTTTCTATCACAGCTCATTAATAATTAATCAAAA 407
CDV-OP TATGACTGTCCATCTTTCTATCACAGCTCATTAATAATTAATCAAAA 407
******** ********** ** ** ** **** *********
Figure 1
A
68
CHAPTER ONE

CDV-A75/17 -----------------------ATCATTAGTTCATGA--ACTAAAACTCAAATGACTTG 35
MeV-ICB -----------ACCGCAGTGCCCAGCAATACCCGAAAACGACCCCCCTCATAATGACAGC 49
RPV-kabeteO ---------------ACTCACCCAGCATCACTCCA------CCCCCGCCGCACCGTCCCG 39
PPRV-ICV89 ACCGGTCGCCGGACAATAGACCCACCTTCAAGAAGTGGCTCCCCCTTCCCCCGTCCAAAA 60
* * * *
CDV-A75/17 GTGGCATTGTCCAGGATCCCTTAATCCCCTCGAACAAGG-------ATTGAGGCTACAAG 88
MeV-ICB CAGAAGGC-CCGGACAAA--AAAGCCCCCTCCAGAAGACTCCACGGA-CCAAGCGAGAGG 105
RPV-kabeteO CAGAGACC-CACAGCAGGGCAGAACGCCCGACACCAGCCTCAACCCAGCCGAACAACAAG 98
PPRV-ICV89 GACACACC-CCAAGACCCCCCAAAACAGCTCCAACATGGCCATCCTCCCAACGGGACGGG 119
* * * * * *
CDV-A75/17 TA--TCAACTGTCTCGATGTTGCTCCTGCATTTTAAGCGTGTTCTATAGTTTT--CTAAG 144
MeV-ICB CCAGCCAGCAGCCGACAGCAAGTGTGGACACCAGGCGGCCCAAGCACAGAACAGCCCCGA 165
RPV-kabeteO GC--CTAGACCCCCACAGCGCAGCCCCCCACCCCGAACTCAACACACGGAGCA--CCCAG 154
PPRV-ICV89 GCCGACCTCCCCCACGAACCCGGTCCGGCAGGAGGGGCCCCCCCCGCAACCCA--CCGGG 177
* * ** *
CDV-A75/17 C--TGCTCGTTCGTGCCTGCTATTCTGGTGACTCTGCAATATGATGAAGACAGCTGAAT---- 198
MeV-ICB CACAAGGC-----CACCACC-AGCCATCCCAATCTGCGTCCTCCTCGTG---GGAC---C 213
RPV-kabeteO CTCTGGGCA---GCACCCGC-ACCCCACCTGCCCTGCACCCCCACCTGGTCCGGAC---C 207
PPRV-ICV89 CGCCGACCGCGTGGGCCAGC-AGGACGCCCCCAAGAGGGCCCGACCGGGACCGGGCACCC 236
* * ** * * *
CDV-A75/17 -CAAACCAATTCATGATGCCTAAGAGTAGGTTGATCATTATCGGACCAA---GTAATGTATGATGG 254
MeV-ICB CCCGAGGACCAACCCCGAAGGTCGCT-CCGAACACAGACCACCAACCGCATCCCCACAGC 272
RPV-kabeteO TCCCCACGGCTCCGCCCACCGCCGCAGCCGACCAAGGCCCGACCAACACGCGGCCGCTGC 267
PPRV-ICV89 TCCCCCCAAAAAAACCCCCCGACACATCCGAGGCCAGGC-GCCAG--GCACTCCCAATCC 293
* * * ** * *
CDV-A75/17 ATGCTTGGGGTTTTGAACTTCGCCTCTAGGAATCTCACTTTAACAATTATACCTCCACG- 313
MeV-ICB TCCCGGGAAAGGA-ACCCCCAGCAACTGGAAGGCCCCTCCCCCCCTCC--CCCAACGCAA 329
RPV-kabeteO ACATCCAGAGCGC-ACACCCGACAAC-----AACCCCGACAGTCCCCT--ACCGAAAGGA 319
PPRV-ICV89 CGCCACCGGGGGG-ACAAGCAGCCAAGACAGGGCCCCCCCACCCAAACGGACCGCCAGGG 352
* * * * **
CDV-A75/17 CACTTGCCCGATCTCAAGCTA------TCACTAGTAGTCTTGTTTCACGAAATTATGACT 367
MeV-ICB GAA-CCCCACA-ACCGAACCGCACAAGCGACCGAGGTGACCCAACCGCAGGCATCCGACT 387
RPV-kabeteO CAA-CCCCAGACACCCAACCA---GGGCCAACAGAGGAAGGAAACCACAGGAACCAGACA 375
PPRV-ICV89 GAGGCCCCACCGACCCAGCACAGACCCGGCCCAAACAAAGGAGACTCCAAAGACGAAACC 412
* ** * * * * **
CDV-A75/17 GTCT---ATCTT--TCTATTACCAATCGTTGATAACTAATCAAAA 407
MeV-ICB CCTT--AGACAG---ATCCTCTCCCCCCGGCAT-ACTAAACAAAA 426
RPV-kabeteO CCCCCGAGACGAGGCAACCTACCCACCATGAAT-ACCAAACAAAA 419
PPRV-ICV89 GCCC---AGCGC---ACCCTACTCATC-------ATCAAACAAAA 444
* * * * ** *****
Figure 1
B
69
CHAPTER ONE
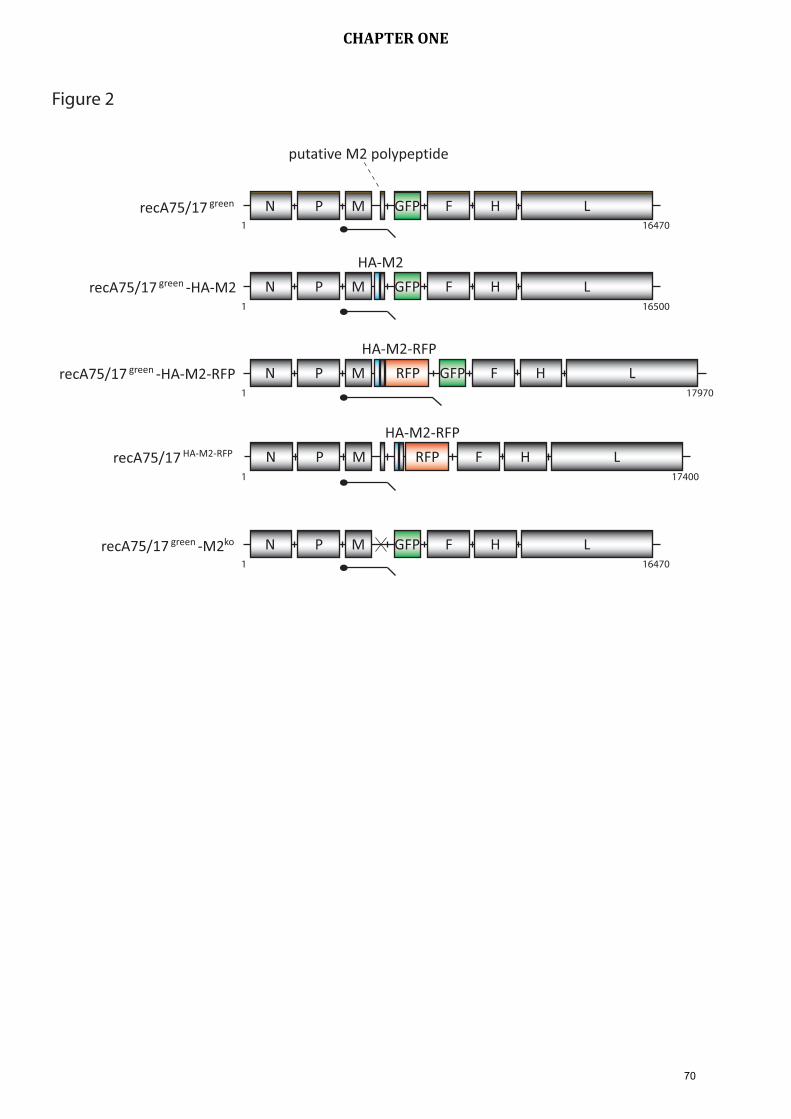
putative M2 polypeptide
N P M F H LGFP
N P M F H LGFP
HA-M2
recA75/17 green
recA75/17 -HA-M2green
N P M F H LGFP
HA-M2-RFP
RFPrecA75/17 -HA-M2-RFPgreen
N P M F H L
HA-M2-RFP
RFPrecA75/17 HA-M2-RFP
N P M F H LGFPrecA75/17 -M2green ko
Figure 2
1 16470
1 16500
1 17970
1 16470
1 17400
70
CHAPTER ONE

Vero-SLAM cells
recA75/17 -HA-M2-RFPgreenrecA75/17 HA-M2-RFP
Figure 3G
FP e
mis
sio
np
has
e co
ntr
ast
MDCK-SLAM cells
recA75/17 HA-M2-RFP recA75/17 -HA-M2-RFPgreen
RFP
emis
sio
n
71
CHAPTER ONE

Vero cells
recA75/17 -HA-M2-RFPgreenrecA75/17 HA-M2-RFP
Figure 4
GFP
em
issi
on
ph
ase
con
tras
tRF
P em
issi
on
72
CHAPTER ONE

recA
75/17
-H
A-M2-R
FP
green
recA
75/17
HA-M2-R
FP
Figure 5
uninfe
cted
IP
recA
75/17
green
recA
75/17
-M
2
green
ko
HA-M2-RFP
recA
75/17
-H
A-M2
green
TL
TLN
actin
73
CHAPTER ONE

Vero-SLAM MDCK-SLAM
Vero Keratinocytes
1
2
3
4
5
6
log
i
nfe
ctio
us
un
its/
ml
10
1
2
3
4
5
6
log
i
nfe
ctio
us
un
its/
ml
10
24h 36h 48h 24h 36h 48h
24h 48h 72h 96h 120h 72h 120h 168h
Figure 6
B
recA
75/17
green
recA
75/17
-M
2
green
recA
75/17
HA-M2-R
FP
1 dpi
3 dpi
6 dpi
A ko
74
CHAPTER ONE

JOURNAL OF VIROLOGY, July 2010, p. 000 Vol. 84, No. 130022-538X/10/$12.00 doi:10.1128/JVI.01878-09Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Two Domains of the V Protein of Virulent Canine Distemper VirusSelectively Inhibit STAT1 and STAT2 Nuclear Import�
Anne Rothlisberger,1† Dominique Wiener,1† Matthias Schweizer,2 Ernst Peterhans,2Andreas Zurbriggen,1 and Philippe Plattet1*
Department of Clinical Research and Veterinary Public Health, Vetsuisse Faculty, University of Bern, Switzerland,1
and Institute of Veterinary Virology, Vetsuisse Faculty, University of Bern, Switzerland2
Received 4 September 2009/Accepted 18 April 2010
Canine distemper virus (CDV) causes in dogs a severe systemic infection, with a high frequency of demy-elinating encephalitis. Among the six genes transcribed by CDV, the P gene encodes the polymerase cofactorprotein (P) as well as two additional nonstructural proteins, C and V; of these V was shown to act as a virulencefactor. We investigated the molecular mechanisms by which the P gene products of the neurovirulent CDVA75/17 strain disrupt type I interferon (IFN-�/�)-induced signaling that results in the establishment of theantiviral state. Using recombinant knockout A75/17 viruses, the V protein was identified as the main antagonistof IFN-�/�-mediated signaling. Importantly, immunofluorescence analysis illustrated that the inhibition ofIFN-�/�-mediated signaling correlated with impaired STAT1/STAT2 nuclear import, whereas the phosphory-lation state of these proteins was not affected. Coimmunoprecipitation assays identified the N-terminal regionof V (VNT) responsible for STAT1 targeting, which correlated with its ability to inhibit the activity of theIFN-�/�-mediated antiviral state. Conversely, while the C-terminal domain of V (VCT) could not functionautonomously, when fused to VNT it optimally interacted with STAT2 and subsequently efficiently suppressed theIFN-�/�-mediated signaling pathway. The latter result was further supported by a single mutation at position 110within the VNT domain of CDV V protein, resulting in a mutant that lost STAT1 binding while retaining a partialSTAT2 association. Taken together, our results identified the CDV VNT and VCT as two essential modules thatcomplement each other to interfere with the antiviral state induced by IFN-�/�-mediated signaling. Hence, ourexperiments reveal a novel mechanism of IFN-�/� evasion among the morbilliviruses.
Virulent canine distemper virus (CDV) causes a severe sys-temic infection in dogs that is characterized by high fever,diarrhea, and pneumonia. Large-scale immunosuppression is ahallmark of infection, and some virus strains additionally in-vade the central nervous system to cause chronic demyelinat-ing encephalitis. The molecular mechanisms differentiating vir-ulent from attenuated strains are poorly understood. However,the fact that dogs can be protected from infection with virulentCDV by vaccination with attenuated strains suggests that reli-able induction of adaptive immunity is possible, provided thatthe critical early stage of infection is successfully mastered bythe host. During the early stage of infection, host defensedepends on the innate immune system, which is also respon-sible for generating signals that activate the adaptive immuneresponse (27). The interferons of type I (IFN-I, e.g., IFN-�/�)are a critical element of the innate immune defense againstviruses (13, 36, 41). Virtually all nucleated cells are capable ofsensing viral infection by receptors such as Rig-I, MDA-5, orToll-like receptor-3 (16). Activation of these receptors initiatesa signal cascade that results in transcription, translation, andrelease from the cells of IFN-�/�. This part of the IFN defenseis referred to as the induction stage. IFN action is initiated bythe binding of IFN to type I IFN receptors that activates the
receptor-associated tyrosine kinases JAK1 and Tyk2, which, inturn, phosphorylate the signal transducers and activators oftranscription (STATs) (21, 41). Subsequently, the activatedSTAT1 and STAT2 together with IFN regulatory factor 9(IRF9) form a complex, the IFN-stimulated gene factor 3(ISGF3), which, once translocated to the nucleus, binds theIFN-stimulated response element (ISRE) sequence (39, 45).This initiates the expression of well over 100 proteins which areresponsible for the antiviral effect of IFN (36). In recent years,gene products targeting specific steps of IFN induction oraction have been found in virtually all viruses studied, indicat-ing the crucial role of IFN evasion in any successful interactionof viruses with their hosts.
CDV, a Morbillivirus of the Paramyxoviridae, contains a non-segmented, single-stranded, negative-sense RNA genome. Thegenome consists of six genes expressing the structural nucleo-capsid (N), matrix (M), fusion (F), hemagglutinin (H), andlarge (L) proteins and the phospho-protein (P) (20). The P andthe L proteins together form the RNA polymerase. In additionto the P protein, the nonstructural C and V proteins are alsoexpressed from the P gene (20). Recently, it has been docu-mented that a virulent CDV strain (5804P) genetically modifiedto inactivate V was attenuated in ferrets, whereas a C-defectiveCDV was fully immunosuppressive (47). These findings demon-strate that wild-type (wt) CDV suppressed IFN induction, butthe issue of whether additional modulation of IFN-mediatedsignaling sustains viral attenuation remains to be determined.In addition, recent work done with C- and V-deficient recom-binant measles virus (MV) and rinderpest virus (RPV) indi-cated that V and, to some extent, P contribute to the final
* Corresponding author. Mailing address: Department Clinical Re-search and Veterinary Public Health, Bremgartenstrasse 109a, 3001Bern, Switzerland. Phone: 4131 631 26 48. Fax: 4131 631 25 09. E-mail:[email protected].
† A.R. and D.W. contributed equally to this study.� Published ahead of print on ●●●●●●●●.
175
CHAPTER TWO

control of the IFN-�/�-mediated signaling pathway. However,to analyze the functions of the P gene products, these recom-binant viruses were based on the genetic background of vac-cine strains (8, 25). Nevertheless, the paramyxovirus V proteinhas been identified as the main inhibitor of the IFN-inducedantiviral state though various molecular mechanisms wereunraveled (12, 14, 15). Expression of the CDV V protein(CDV-V) depends on the insertion of a nontemplated guaninenucleotide at a precise location, called an “editing site,” whichgenerates an mRNA which differs from that of P by one or twonucleotides. This produces an mRNA with an altered open read-ing frame (ORF) downstream of the editing site, and, thus, due tothis specific mechanism, the N-terminal domain of P and V areidentical, whereas their C-terminal domains are unique. The C-terminal domain of V (VCT) is known to contain a conservedcysteine-rich region (31, 43), and recent X-ray studies confirmedthat VCT folds into a zinc finger conformation (22).
In this study we investigated the role of the P gene productsof the highly virulent CDV A75/17 strain in counteracting theIFN-�/�-mediated signaling pathway. Importantly, this strainwas isolated from a naturally infected dog and subsequentlykept amplified only in dogs, where it has been reported tomaintain its virulence (6). Therefore, this virus has never beenadapted to any cell lines. However, the generation of recom-binant virus stocks (rA75/17) with sufficient titers to work withrequires two to three passages in Vero cells expressing signal-ing lymphocyte activation molecule (Vero-SLAM) after virusrescue from primary full-length cDNA-transfected cells (37).Because these limited amplification steps might already selectviral variants, the entire genome of rA75/17 was compared bydirect sequencing to that of the parental A75/17 strain andshowed no nucleotide differences (37), thereby validating theunique opportunity to investigate the molecular mechanisms ofvirus-host cell interactions based on a demyelinating morbilli-virus strain. Hence, recombinant A75/17 viruses and expres-sion plasmids were generated to investigate the role of the Pgene products in mediating IFN evasion. Infection and trans-fection experiments were performed in Vero cells stably ex-pressing the SLAM receptor for CDV. IFN production fromVero-SLAM cells is defective, and thus they not only providean optimal tool to exclusively study IFN signaling indepen-dently of IFN induction but also support very efficient CDVA75/17 replication.
Our results demonstrate that the V protein was the mainviral factor responsible for disrupting the IFN-�/�-mediatedsignaling pathway. The latter inhibition was neither due toSTAT1 or STAT2 degradation nor to an impairment of theirphosphorylation states upon IFN-�/� treatment. Rather, theCDV-V protein efficiently associated with both STAT1 andSTAT2, which correlated with a complete inhibition of thenuclear import of both transcription factors. Furthermore,transient expression experiments of engineered V proteinsidentified both the N-terminal and the C-terminal domains astwo interdependent modules necessary to exhibit optimal IFNevasion.
MATERIALS AND METHODS
Cells and viruses. Vero-SLAM cells (kindly provided by V. von Messling,INRS-Institute Armand-Frappier, University of Quebec, Laval, Quebec, Can-ada), MDCK-SLAM cells, and Bsr-T7 cells(stably expressing T7 RNA polymer-
ase) (3) were grown in Dulbecco’s modified Eagle medium (Invitrogen) supple-mented with 10% fetal calf serum (FCS), penicillin, and streptomycin. Theselection of Vero cells expressing the SLAM receptor was maintained by addingzeocin (Invitrogen). All cells were kept at 37°C in the presence of 5% CO2. Thetransfection and all the infection experiments were performed in Vero-SLAMcells, which are easily infected by CDV. The infection experiments were per-formed with recombinant viruses based on the wild-type CDV A75/17 strain.
Antibodies. In Western blotting the viral proteins were detected by a rabbitanti-P, anti-V, and anti-C antibody and by monoclonal mouse anti-N antibodyD110 (2). The first two antibodies were formerly produced in our laboratory.Briefly, the P-specific and the V-specific domains (both C-terminal) were pro-duced in bacteria, purified, and injected in rabbits to produce P and V antisera.The anti-C antibody was produced against a mix of two synthetic peptides(nh2-RSAASETKPATQARRMEPQACRK-cooh and nh2-RQSSPLKMTSNQDLE-cooh) corresponding to amino acids (aa) 24 to 46 and 85 to 99 of the Cprotein (Primm srl; Custom Antibodies, Milano, Italy). The cellular STAT pro-teins were detected by anti-STAT1 (Cell Signaling), anti-STAT2 (A-7; SantaCruz Biotechnology), anti-phospho-STAT1 (Tyr 701; 58D6; Cell Signaling), andanti-phospho-STAT2 (Tyr 690; Cell Signaling) antibodies. For immunofluores-cence staining the anti-N antibody D110, STAT1� p91 (C-24; Santa Cruz Bio-technology), STAT2 (C-20; Santa Cruz Biotechnology), and anti-phospho-STAT1 (Tyr 701; 58D6; Cell Signaling) were used in combination with AlexaFluor 555 and 488 (Invitrogen) anti-mouse and anti-rabbit antibodies, respec-tively. The coimmunoprecipitations (co-IPs) were performed with either anti-STAT1 p84/p91 (C-136; Santa Cruz Biotechnology) antibody or anti-STAT2(A-7; Santa Cruz Biotechnology). In selected co-IP experiments, the anti-hem-agglutinin (HA) tag monoclonal antibody (MAb) coupled with Sepharose beads(3F10; Roche) was used. Anti-�-actin (Sigma) served as a loading control. Theanti-HA tag monoclonal antibody 16B12 (Covance) was used for immunoblot-ting and immunofluorescence analyses.
Generation of recombinant viruses and plasmids. The recombinant virusesare based on a neurovirulent wild-type isolate, CDV A75/17. The mutations toknock out either the C (Cko) or the V (Vko) protein or both proteins (CVko) wereperformed on a shuttle vector containing the sequences of the N, P, and L genesand produced from a full-length cDNA clone (pFL-A75/17, [18]). The mutationswere obtained by applying a QuikChange Site-Directed Mutagenesis Kit (Strat-agene). After digestion with NotI and PmeI, the M, F, and H genes of CDVA75/17 and a red fluorescence marker gene (tD-tomato; kindly offered by D.Garcin, University of Geneva, Switzerland) (23) placed between the first twogenes were cloned into the shuttle vectors, which contained the mutated P genes,using T4 DNA ligase (New England BioLabs). To rescue the recombinant viruseswe proceeded as previously described (33). Briefly, Bsr-T7 cells were cotrans-fected with plasmids containing the relevant full-length cDNA and helper plas-mids coding for the N, P, and L proteins of CDV. After 3 days the transfectedBsr-T7 cells were cocultured with Vero-SLAM cells. When a cytopathic effecthad developed, the cells were lysed by two freeze-thaw cycles, and the viruses(referred to as rA75/17red, rA75/17red Cko, rA75/17red Vko, and rA75/17red CVko)were stored at �80°C. Finally, the recombinant viruses were amplified in twopassages and titrated by a plaque assay in Vero-SLAM cells, starting at a mul-tiplicity of infection (MOI) of 0.02. The virus titers were determined by countingred fluorescent single cells or syncytia as infectious particles per ml.
To generate the expression plasmids encoding the P, V, and C proteins (calledpCI-P, pCI-V, and pCI-C, respectively), the three relevant sequences of therA75/17red cDNA P gene were amplified by using an Expand High Fidelity PlusPCR System (Roche). The PCR products were digested with RsrII and cloned ina pCI mammalian expression vector (Promega) by applying the T4 DNA ligase(New England BioLabs). To silence the C gene, the same mutations as for theknockout viruses were performed on the plasmids pCI-P and pCI-V, and theadditionally required G was introduced at the editing site in plasmid pCI-V bysite-directed mutagenesis (Stratagene). To generate pCI-RFP (where RFP is redfluorescent protein), the marker gene was amplified from the cDNA clone byPCR and cloned into the pCI expression vector previously digested with theidentical restriction sites as described above. Then, a small linker (SGGSGGTG)and the HA-tagged peptide (YPYDVPDYA) were added by PCR technology inframe to the C-terminal domain of the RFP ORF (pCI-RFP-Linker-HA). Next,pCI-RFP-HA-Vwt (where Vwt is the wt, or full-length, V protein), pCI-RFP-HA-VNT, and pCI-RFP-HA-VCT plasmids were generated by PCR amplifica-tion of the different V domains from the pCI-V vector and subsequently clonedinto the pCI-RFP-Linker-HA-cleaved plasmid. Finally, the single substitutionY110D in V was generated by site-directed mutagenesis (Stratagene), thus pro-viding the pCI-Vwt Y110D or pCI-RFP-HA-Vwt Y110D expression plasmids.HA-tagged versions of the following plasmids were produced by PCR technol-ogy: pCI-HA-Vwt, pCI-HA-P, pCI-HA-C, pCI-HA-VNT, pCI-HA-VCT, and
2 ROTHLISBERGER ET AL. J. VIROL.
76
CHAPTER TWO

pCI-HA-Vwt Y110D. The green fluorescent protein (GFP) gene was amplifiedby PCR technology from a plasmid pgA75/17-V cDNA clone (33) and sub-sequently digested and ligated into the pCI-cleaved plasmid, thus generatingpCI-GFP.
Luciferase assay and transient transfections. Vero-SLAM cells were grown in24-well plates and transfected the following day with pISRE-Luc (encoding anIFN-inducible firefly luciferase), pTK-RL (coding for a constitutively expressedRenilla luciferase as a transfection control; both plasmids were kindly providedby D. Garcin, University of Geneva, Switzerland), and pCI-P, -V, -C (or thederivative RFP constructs), the empty pCI, or the control plasmid pCI-GFPusing Lipofectamine 2000 (Invitrogen) and Opti-MEM (Invitrogen). The nextday the cells were treated (or left untreated) with 1,000 IU/ml universal IFN-�/�(IFN type I; PBL) for 6 h. Then the cells were lysed, and the luciferase activitywas measured by applying a dual-luciferase reporter assay system (Promega)according to the manufacturer’s recommendation. The luminescence signals ofthe firefly and the Renilla luciferase were measured with a TD-20/20 Luminom-eter (Promega), and their ratio was called relative luciferase activity, with theratio of the empty vector pCI set to 1. For MDA5 signaling assays, cells weretransfected with a FLAG-tagged MDA5 construct, p�-IFN-fl-lucter (both vec-tors kindly provided by D. Garcin, University of Geneva), and pTK-RL as wellas with an RFP-expressing plasmid or one of the different V protein-expressing
plasmids. After 24 h of transfection, the cells were stimulated with 1.5 �g ofpoly(I:C)/ml (Sigma) by transfection with Fugene HD (Roche). The cells wereharvested after 15 h and assayed for firefly and Renilla luciferase activity asdescribed above, with the ratio of the pCI-RFP-expressing vector set to 1.
Immunofluorescence. Vero-SLAM or Vero cells were grown on cover slides insix-well plates, and the following day cells were either infected with the recom-binant viruses (rA75/17red, rA75/17red Cko, rA75/17red Vko, and rA75/17red
CVko) at an MOI of 0.02 or transfected with the desired expression plasmids. At24 h postinfection or transfection, cells were treated (or left untreated) with1,000 IU/ml IFN-�/� for 30 min at 37°C and then fixed with phosphate-bufferedsaline (PBS) containing 4% paraformaldehyde (PFA). After permeabilizationwith PFA and 0.1% Triton X-100, the cells were washed in PBS. Blocking ofnonspecific binding sites and incubation with the first antibody were performedin PBS supplemented with 2% FCS for 1 h at room temperature and for 1.5 h at37°C, respectively. Then, the slides were washed in PBS and incubated for 1 h atroom temperature with the second antibody diluted in PBS. The cells werewashed again, and the nuclei were stained with TOTO3 (Invitrogen) according tothe manufacturer’s protocol. Pictures were taken with a confocal microscope(Olympus).
Western blotting. Immunoblotting was performed as previously described(32). Briefly, Vero-SLAM cells were cultured in six-well plates and infected (with
FIG. 1. Generation of expression plasmids and recombinant viruses to assess the A75/17 CDV-P gene products responsible for inhibitingIFN-�/�-mediated signaling. (A) Schematic representation of the full-length A75/17 CDV cDNA clone bearing an additional transcription unitencoding RFP. (B) Schematic presentation of the mutations performed on the P gene of rA75/17red to enable V and P expression without C. Thestart codon for the C was mutated, and an additional stop codon was introduced (performed to obtain rA75/17red Cko, rA75/17red CVko, pCI-P,and pCI-V). To inactivate V expression, three nucleotides were changed at the V editing site (applied for rA75/17red Vko and rA75/17red CVko).All mutations were performed without influencing the P open reading frame. (C) The V protein substantially inhibits the IFN-induced activationof the ISRE promoter in a dual luciferase assay. Vero-SLAM cells were transfected with the expression plasmids for P, V, C, an irrelevant GFPcontrol plasmid, and the empty pCI as well as the two plasmids coding for the IFN-inducible and the constitutively expressed luciferases. At 24 hpost transfection they were treated (or left untreated) with IFN-�/� for 6 h and subsequently lysed for luciferase analysis. (D) P, V, and Cexpression control by SDS-PAGE immunoblotting of Vero-SLAM cells at 2 days posttransfection with the expression plasmids pCI-P, pCI-V,pCI-C, and an empty plasmid (pCI). Each plasmid expresses only one protein (detected by polyclonal antibodies). (E) The recombinant virusesexpress the expected P gene products. Vero-SLAM cells were either infected with rA75/17red, rA75/17red Cko, rA75/17red Vko, and rA75/17red CVko
at an MOI of 0.02 or left uninfected and lysed 2 days postinfection for subsequent Western blot analysis. The viral proteins were detected byimmunoblotting with three polyclonal antibodies. (F) The four recombinant viruses show similar growth curves in Vero-SLAM cells. The diagramindicates the titers of cell-associated viruses at 8, 24, 32, and 48 h postinfection in infectious particles per ml.
VOL. 84, 2010 MECHANISMS OF CDV-V AS IFN ANTAGONIST 3
77
CHAPTER TWO

the various recombinant CDVs [rCDVs]) at an MOI of 0.02 1 day later ortransfected with the different expression vectors. At 2 days postinfection orposttransfection, the cells were washed with cold PBS and lysed at 4°C inradioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl [500 mM NaCl toextract the C protein], 1% NP-40, 0.5% Na-deoxycholate, 0.1% sodium dodecylsulfate]SDS], 50 mM Tris-Cl, pH 7.4) containing Halt Protease Inhibitor Cock-tail (Socochim) and Phosphatase Inhibitor Cocktail Set II (Merck) to protectphosphorylated STAT proteins. After centrifugation, Laemmli sample buffer(Bio-Rad) supplemented with DL-dithiothreitol (Fluka) was added to the sam-ples, and they were boiled for 5 min and separated by SDS-polyacrylamide gelelectrophoresis. Afterwards, the proteins were transferred to nitrocellulosemembranes (Hybond-ECL; Amersham Biosciences), which subsequently wereblocked in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) and 5%nonfat dry milk (or bovine serum albumin for detection of the C protein) for 1 h.The membranes were incubated in blocking buffer containing the first antibodyat 4°C overnight. Then the membranes were washed three times in TBS-T andincubated with the horseradish peroxidase-conjugated second antibody for 1 h atroom temperature. After the membranes were washed three times, ECL sub-strate (Amersham Biosciences) was used, and chemiluminescence signals weredetected with an LAS-3000 camera (Fujifilm). Relative protein amounts werecalculated with AIDA Image Analyzer software (Raytest Schweiz AG, Wetzikon,Switzerland).
Coimmunoprecipitation. Vero-SLAM cells were seeded in six-well plates, andthe next day cells were infected at an MOI of 0.02 with rA75/17red or rA75/17red
Vko or left uninfected. In parallel experiments different expression plasmids weretransfected. At 2 days postinfection or 1 day posttransfection, the cells weretreated (or left untreated) with 1,000 IU/ml IFN-�/� for 30 min at 37°C and thenwashed with cold PBS and incubated on ice with 50 mM Tris, 150 mM NaCl, 2mM EDTA, and 1% NP-40 (pH � 7.5) complemented with Halt Protease
Inhibitor Cocktail (Socochim) for 45 min until complete lysis. After a 10-mincentrifugation at 5,000 � g at 4°C, aliquots for total protein analyses wereseparated, and the remaining supernatants were incubated for 3 h with the firstantibody (anti-STAT1, anti-STAT2, anti-V, or anti-HA) or without antibody,followed by the addition of protein G-Sepharose beads at 4°C overnight. After a1-min centrifugation at 5,000 � g at 4°C, the pellets were washed four times withthe same buffer used earlier and finally dissolved directly in Laemmli samplebuffer for Western blotting performed as described above.
Generation of MDCK cells constitutively expressing the CDV receptor CD150/SLAM. MDCK cells were transduced with pRRL lentivirus vectors at an MOI of5. Subsequently, a highly SLAM-expressing clone was selected by limiting dilu-tion and was used for further experiments.
The lentivirus vector pRRL as been described elsewhere (7) (kindly providedby Patrick Salomon, University of Geneva). Stock of lentivirus vectors wasgenerated in 293T/17 cells as previously described (7).
Production of cIFN-�. Standard reverse transcription-PCR (RT-PCR) tech-niques were employed to amplify and clone the canine IFN-� (cIFN-�) cDNAinto the pCI vector (primers are available upon request). To express cIFN-�,293T cells were transfected with pCI-cIFN-� for 3 days. Then, the supernatantwas harvested, filtered through a 0.45-�m-pore-size filter, and concentratedusing 10-kDa size exclusion Centricon columns (Millipore).
RESULTS
The A75/17 CDV-V protein inhibits the IFN-�/�-mediatedsignaling pathway. In order to investigate any potential role ofthe CDV-P gene products in modulating the IFN-�/�-medi-
FIG. 2. Neither degradation nor inhibition of STAT1 and STAT2 phosphorylation occurs after infection with the various rCDVs. (A) Cyto-pathic effect induced by the different rCDVs in Vero-SLAM cells. Vero-SLAM cells were infected at an MOI of 0.02, and representative fieldsof view were then photographed at 2 days postinfection. (B) To assess whether the various rCDVs affected the endogenous expression of STAT1and STAT2, Vero-SLAM cells were infected with rA75/17red, rA75/17red Cko, rA75/17red Vko, and rA75/17red CVko at an MOI of 0.02. Two dayspostinfection, cells were treated (or left untreated) with IFN-�/� for 30 min and subsequently lysed. Cell lysates were then subjected to immunoblotanalysis using specific anti-STAT1 or anti-STAT2 antibodies. To assess the phosphorylated state of both STAT molecules in the presence orabsence of IFN, Vero-SLAM cells were infected with rA75/17red, rA75/17red Cko, rA75/17red Vko, and rA75/17red CVko at an MOI of 0.02. At 2days postinfection, cells were treated (or left untreated) with IFN-�/� for 30 min and subsequently lysed. Cell lysates were then subjected toWestern blot analysis using specific anti-phospho-STAT1 or anti-phospho-STAT2 monoclonal antibodies.
4 ROTHLISBERGER ET AL. J. VIROL.
78
CHAPTER TWO

FIG. 3. rCDVs expressing the V protein inhibit STAT1 and STAT2 nuclear translocation. (A) Immunofluorescence images of Vero-SLAMcells stained for STAT1. Vero-SLAM cells were infected with the various rCDVs or left uninfected. At 1 day postinfection, cells were treated for
VOL. 84, 2010 MECHANISMS OF CDV-V AS IFN ANTAGONIST 5
79
CHAPTER TWO

ated signaling pathway, we first generated expression vectorsencoding the P, V, and C proteins. In addition, specific nucle-otides in the P and V genes were mutated to close the openreading frame (ORF) of the C protein without affecting theORFs of P and V (Fig. 1B, top). Western blot analysis con-firmed that all proteins were efficiently expressed and migratedaccording to their expected molecular weights in SDS-PAGE(Fig. 1D). Next, a dual luciferase assay was performed to assessthe role of each single protein in controlling IFN-�/�-inducedsignaling. As expected, after treatment with IFN-�/�, the rel-ative luciferase activity clearly increased in cells transientlytransfected with both an empty control plasmid (pCI) and witha plasmid expressing the irrelevant control green fluorescentprotein (GFP) (Fig. 1C). This observation confirmed the bio-activity of the recombinant human IFN in Vero-SLAM cells. Incontrast, the activation of the ISRE promoter was strikingly
inhibited in cells transfected with the V expression plasmid.Interestingly, while we did not observe any effect at all in cellsexpressing the C protein, we noticed a partial inhibition in cellstransiently expressing the P protein (Fig. 1C). Taken together,these results demonstrated that the CDV-V protein potentlyinhibited the activation of the ISRE promoter, whereas the Pprotein could only slightly control the IFN-�/�-mediated sig-naling pathway.
CDV infection neither induces STAT degradation nor inhib-its STAT phosphorylation. We then investigated at which stepthe CDV-V protein was able to affect the IFN-�/�-mediatedsignaling pathway. However, conclusions drawn from single-protein overexpression experiments may differ from resultsobtained with the same protein in the context of a viral infec-tion (25). In order to overcome this problem, we generatedrecombinant C and/or V knockout viruses based on the highly
30 min with IFN-�/� and subsequently fixed, permeabilized, and stained for STAT1 localization using an anti-STAT1 antibody. Then, an AlexaFluor 488-conjugated secondary antibody was employed (green), and images were captured with a scanning confocal laser microscope (Olympus).The nuclei were counterstained with TOTO3 (blue). Infected cells were localized by the expression of RFP. (B) Immunofluorescence images ofVero-SLAM cells stained for STAT2. Experimental settings were identical to those of panel A. Filled arrowheads indicate nuclei without STAT1(or STAT2) accumulation, and open arrowheads indicate nuclei with STAT1 (or STAT2) accumulation. (C) Nuclear translocation of STAT1 andSTAT2 in Vko and CVko recombinant A75/17red viruses is dependent on IFN treatment. Immunofluorescence images show Vero-SLAM cellsstained for STAT1 and STAT2. Experimental settings were identical to those of panel A with the exception that IFN was not added to the cells.In all panels, specific fields of view of the cell monolayer were selected to illustrate infected and noninfected cells in the identical areas.
FIG. 4. The A75/17 CDV-V protein inhibits STAT1 nuclear import. (A) Subcellular localization of the NES-GFP-SV5-V fusion construct in thepresence and absence of leptomycin B (LMB). Vero-SLAM cells were transfected with the fusion protein NES-GFP-SV5-V and treated (or leftuntreated) with LMB for 3 h. Subsequently, cells were fixed and permeabilized, and nuclei were counterstained with TOTO3. Green and bluefluorescence emissions were captured with a scanning confocal laser microscope. (B) Vero-SLAM cells were infected with rA75/17red or rA75/17red Vko
for 1 day. Then, cells were treated (or left untreated) with LMB for 3 h and with IFN-�/� for the last 30 min. Subsequently, cells were fixed andpermeabilized, and nuclei were counterstained with TOTO3. Moreover, STAT1 localization was investigated using an anti-STAT1 antibody followed byan Alexa Fluor 488-conjugated secondary antibody. Green, blue, and red fluorescence emissions were captured with a scanning confocal laser microscope.Filled arrowheads indicate nuclei without STAT1 accumulation, and open arrowheads indicate nuclei with STAT1 accumulation.
6 ROTHLISBERGER ET AL. J. VIROL.
80
CHAPTER TWO

virulent CDV strain A75/17 (Fig. 1A). Mutations ablating theC ORF were identical to those applied in the P and V expres-sion vectors. Furthermore, specific nucleotide substitutionswere performed at the P gene editing site, which preserved theP reading frame while silencing the production of the VmRNA (Fig. 1B, bottom). All viruses were successfully rescuedfrom cDNA, and subsequently the expression of the P geneproducts in cells infected with rA75/17red, rA75/17red Cko,rA75/17red Vko, and rA75/17red CVko was confirmed by immu-noblot analysis. As shown in Fig. 1E all recombinant virusesexpressed the expected pattern of P, V, and C proteins.Growth kinetics indicated that the absence of the C and Vproteins scarcely affected the efficiency of viral replication inVero-SLAM cells since all the recombinant viruses showedvery similar growth curves (Fig. 1F).
Different molecular mechanisms were documented for sev-eral viruses among the family Paramyxoviridae in order toevade the IFN-�/�-induced antiviral state. For instance, the Vproteins of rubulaviruses are responsible for a direct degrada-tion of the pool of STAT molecules in the cytoplasm, whereasthe V proteins of MV (depending on the viral strain) and RPVinhibit the phosphorylation of STAT1 and STAT2 upon IFNtreatment (5, 8, 25, 30, 42). In line with these results, we nextexamined by immunoblotting whether the CDV-V-mediatedinhibition of the IFN-�/�-mediated signaling pathway was dueto either STAT degradation or inhibition of STAT phosphor-ylation. Figure 2A documents that after 2 days of infection inVero-SLAM cells, all rCDVs mediated a typical cytopathiceffect, with the formation of syncytia involving about 80% ofthe cells. Even though only about 20% of the cells remainednoninfected, the expression and stability of both STAT1 andSTAT2 were not modulated in infected cells compared tomock-treated cells (Fig. 2B). Furthermore, none of the recom-binant viruses was able to block the phosphorylation of STAT1and STAT2. Indeed, immunoblotting revealed no major dif-ferences in the amount of phospho-STAT molecules detectedin cells infected with any of the recombinant viruses comparedto mock-infected, IFN-�/�-treated, cells (Fig. 2B). Taken to-gether, these results indicate that the aforementioned role of Vin inhibiting an IFN-�/�-dependent response is due neither tothe degradation of STAT1 and STAT2 nor to the inhibition oftheir phosphorylation.
CDVs expressing the V protein inhibit the nuclear accumu-lation of STATs. Upon IFN-�/� treatment, accumulation ofSTAT1 and STAT2 in the cytoplasm rather than in the nucleuswas reported to occur in cells infected with measles and rinder-pest morbilliviruses (MV and RPV, respectively) (8, 25, 28).Thus, taking advantage of our newly generated recombinantviruses, we next assessed by immunofluorescence analysiswhether the nuclear accumulation of STAT1 and STAT2 wasmodulated by these viruses. In mock-infected Vero-SLAMcells, STAT1 staining exhibited the anticipated shift betweencytoplasmic to mainly nuclear localization upon treatment withIFN-�/� (Fig. 3A, mock). The identical phenotype was ob-served in cells infected with rA75/17red Vko and rA75/17red
CVko. Strikingly, however, cells infected with the rA75/17red
and rA75/17red Cko viruses (the two viruses expressing the Vprotein) exhibited a clear accumulation of STAT1 in the cyto-plasm (Fig. 3A). Identical results were observed for STAT2under corresponding conditions (Fig. 3B). Next, to verify
whether STAT1 nuclear accumulation was dependent on IFNaddition, STAT1 cellular localization was assessed in infectedbut non-IFN-treated cells. In the absence of IFN treatment,rA75/17red Vko and rA75/17red CVko (data not shown) couldnot mediate STAT1 nuclear translocation (Fig. 3C). Rather,STAT1 was readily detected in the cytoplasm of rA75/17red-infected but non-IFN-treated cells, thereby validating the no-tion that STAT1 is not degraded in rA75/17red-infected cells.The identical phenotypes were observed when the cellular lo-calization of STAT2 was assessed (Fig. 3C). These observa-tions are in agreement with the results found in our transienttransfection assay and confirmed a key regulating role of theCDV-V protein in inhibiting IFN-�/�-dependent activation ofinnate immunity.
STAT1 cytoplasmic accumulation is caused by a nuclear im-port inhibition mechanism. Upon IFN-�/� treatment, STATmolecules are first phosphorylated, then translocate into thenucleus, and finally relocate to the cytoplasm through theirnuclear export signal (NES) (1, 24). Hence, CDV-V-depen-dent STAT cytoplasmic accumulation may result either fromnuclear import inhibition or from an accelerated nuclear ex-port mechanism. To discriminate between these two possibil-ities, the Crm1-dependent nuclear export inhibitor leptomycinB (LMB) was used. Immunofluorescence analysis was thenperformed to assess the cellular localization of STAT1 in cellsinfected with rA75/17red or rA75/17red Vko. As a control, weused the simian virus 5 (SV5) V protein ([SV5-V] known to betargeted to the nucleus [34]) fused at its N-terminal region withan NES-tagged green fluorescent protein (NES-GFP-SV5-V).As expected, upon LMB treatment, the fluorescent fusion pro-
FIG. 5. The rA75/17 CDV-V protein associates with endogenousSTAT1 and STAT2. Vero-SLAM cells were infected at an MOI of 0.02with rA75/17red or rA75/17red Vko or left uninfected and subsequentlylysed 2 days postinfection for coimmunoprecipitation assay. STAT1 orSTAT2 was first immunoprecipitated (IP) with an anti-STAT1 or anti-STAT2 monoclonal antibody, which was followed by addition of proteinG-Sepharose beads. Then, Western blot analysis using an anti-V poly-clonal antibody to detect any potential association or anti-STAT1 and-STAT2 antibodies to control for direct IP, was performed. Total celllysates taken prior to IP (TL) were subjected to immunoblot analysis(WB) to investigate the endogenous STAT1 and STAT2 expressions orthe viral V protein expression. In addition, immunoprecipitations wereperformed either with an anti-HA monoclonal antibody or in the absenceof any monoclonal antibody (no antibody) to control the co-IP assay.
VOL. 84, 2010 MECHANISMS OF CDV-V AS IFN ANTAGONIST 7
81
CHAPTER TWO

tein remained accumulated mainly in the nucleus while it wasclearly relocated in the cytoplasm in the absence of the drug(Fig. 4A). Importantly, in IFN- and LMB-treated cells, STAT1was found to be strongly accumulated in the nucleus in recom-binant V knockout virus-infected cells. Conversely, in cellsinfected with the wild-type rA75/17red virus, STAT1 could notbe detected in nuclei (Fig. 4B). Together, these results indicatethat cytoplasmic accumulation of STAT1 in cells infected withCDVs expressing the V protein is caused by nuclear importinhibition rather than by an accelerated export mechanism.
The A75/17 CDV-V protein efficiently binds to endogenousSTAT1 and STAT2. Coimmunoprecipitation (co-IP) from in-fected cells was next performed to assess whether the A75/17CDV-V protein may influence STAT nuclear accumulation bybinding to endogenous STAT1 and/or STAT2. Thus, STAT1from lysates of infected and noninfected cells was immunopre-cipitated with an anti-STAT1 monoclonal antibody (MAb),followed by immunoblotting using an anti-CDV-V poly-
clonal antibody. The identical strategy was employed usingan anti-STAT2 MAb to determine whether V can associatewith endogenous STAT2. An anti-HA MAb immunoprecipi-tation or immunoprecipitation in the absence of antibodywas performed in parallel to validate the co-IP assay. In-deed, in rA75/17red-infected cells, V could be efficientlycopurified after STAT1 and STAT2 immunoprecipitation(Fig. 5). Moreover, these interactions were formed indepen-dently of the activation of the signaling pathway since V wascoprecipitated in both the presence and the absence of IFN-�/� treatment (data not shown). As expected, V was not co-precipitated in rA75/17red Vko-infected cells or when the anti-HAMAb or no MAb was used for immunoprecipitation (Fig. 5).Western blotting performed with cell lysates taken prior toimmunoprecipitation revealed the expected pattern of V ex-pression. Indeed, V was produced by rA75/17red but not byrA75/17red Vko (Fig. 5). Finally, Western blotting using anti-STAT1 and anti-STAT2 antibodies demonstrated that under
FIG. 6. Identification of V domains responsible for inhibiting the IFN-�/�-mediated signaling pathway. (A) Schematic diagram of the A75/17CDV-V protein and engineered variants. (B) Sequence alignment of several canine distemper virus P genes. (C) Effect of the V protein andvariants in inhibiting the IFN-induced activation of the ISRE promoter in a dual luciferase assay. Vero-SLAM cells were transfected with thevarious expression plasmids at two different concentrations (500 ng and 100 ng) or with an empty plasmid (pCI) as well as with the two plasmidscoding for the IFN-inducible and the constitutively expressed luciferases. At 24 h posttransfection, cells were treated (or left untreated) withIFN-�/� for 6 h and subsequently lysed for luciferase analysis. (D) Schematic diagram of the A75/17 CDV-V protein and variants fused to RFP.(E) Experimental settings were identical to those in panel C but with the RFP-fused proteins.
8 ROTHLISBERGER ET AL. J. VIROL.
82
CHAPTER TWO

all conditions both endogenous transcription factors wereexpressed in very similar amounts (Fig. 5). These resultsstrongly suggest that the A75/17 CDV-V protein controlsIFN-�/�-induced signaling by efficiently forming a complexwith STAT1 and STAT2.
Both the N-terminal and C-terminal domains of V are nec-essary to completely inhibit IFN-�/�-induced activity. To ini-tiate the mapping of the CDV-V-dependent STAT-interactingdomains, various HA-tagged expression vectors were engi-neered (Fig. 6A). A first construct, composed of the sharedN-terminal domain between P and V, was produced (VNT; 240aa) (Fig. 6A). The second encompassed the cysteine-rich C-terminal domain of V (VCT; 78 aa) (Fig. 6A). Next, a singlesubstitution (Y110D) was introduced into the N-terminal re-gion of the full-length HA-tagged V protein (308 aa) (Fig. 6A).This tyrosine is, indeed, highly conserved among morbillivi-ruses and has been shown for MV-V to be responsible forspecific STAT1 binding (4, 5, 8, 28, 35). Interestingly, sequencecomparison between several wild-type CDV strains and thelarge plaque-forming variant of the Onderstepoort (OP) vac-cine strain of CDV revealed a substitution at that preciselocation (Y110D) (Fig. 6B). Finally, we also fused the HA tagsequence to the N-terminal part of the P gene construct toallow immunoprecipitation under similar conditions (Fig. 6A).
In order to verify the activity of the HA-tagged full-lengthconstructs and to determine the role of each individual V subdo-main in inhibiting IFN-�/�-mediated signaling, ISRE luciferasereporter gene assays were performed from IFN-treated Vero-SLAM cells transfected with the various expression plasmids.Two different plasmid concentrations were used for transfectionin order to control for the amount of protein expressed. As ex-pected, HA-Vwt strongly suppressed IFN-�/�-mediated signalingin a concentration-dependent manner, whereas HA-P caused apartial inhibition at its higher concentration only (Fig. 6C).Similarly, the HA-VNT and the HA-Vwt Y110D mutantsshowed partial inhibition as well when expressed at highconcentrations. Conversely, the HA-VCT construct alone wasnot sufficient to control IFN-�/�-mediated signaling.
To confirm the expression of the various mutants, immuno-blot analysis from total cell extract of transfected Vero-SLAMcells was undertaken using an anti-HA MAb to detect thevarious HA-tagged proteins. Figure 7A (bottom panel) docu-ments that all proteins, except HA-VCT, were correctly ex-pressed and migrated according to their expected molecularweights in the SDS polyacrylamide gel. It is possible that theextremely small size of HA-VCT affected proper expressionand/or stability. Thus, to overcome these putative defects, Vwt,Vwt Y110D, VNT, and VCT were fused to RFP. In addition,these fusion proteins were designed to contain both a smalllinker peptide and the HA tag sequence to retain both proteinfunctionalities and to facilitate detection and immunoprecipi-tation, respectively (Fig. 6D). Indeed, using this strategy, allengineered proteins were properly expressed, as demonstratedby immunoblot analysis (Fig. 7B, bottom panel). ISRE lucif-erase reporter gene assays were performed in order to assessthe ability of these fusion proteins to control IFN-�/�-medi-ated signaling. Figure 6E illustrates that all proteins modulatedthe IFN-induced activity to the same extent as the identicalnonfused V mutant proteins (Fig. 6C). These results indicatethat the CDV-VCT module alone lacks the capacity to con-
trol IFN-�/�-induced activity, whereas VNT was able tofunction as an autonomous module, albeit to a limited ex-tent compared to Vwt.
To verify the proper folding of the C-terminal region of V inHA-VCT- and RFP-HA-VCT-expressing cells, their ability todisrupt signaling by the RNA helicase protein MDA5 wasinvestigated. Indeed, it has been previously reported that themeasles virus VCT domain was sufficient to suppress theMDA5-mediated signaling pathway (35). The results shown inFig. 7C indicate that HA-VCT presumably does not fold into abiologically active conformation since this domain was not ableto control the MDA5-dependent signaling pathway to theIFN-� promoter reporter gene. In contrast, RFP-HA-VCT wasfully able to ablate MDA5-mediated signaling, as were Vwt and
FIG. 7. Only Vwt efficiently interacts with both STAT molecules.Results of coimmunoprecipitation of endogenous STATs with V pro-tein fragments (A) or with the corresponding RFP-fused proteins(B) are shown. Vero-SLAM cells were transfected to express the var-ious HA-tagged V proteins, and cell extracts were prepared for immu-noprecipitation (IP) with an anti-HA MAb, followed by overnightincubation with protein G-Sepharose beads. The eluates were evalu-ated by STAT immunoblotting. The middle and bottom panels illus-trate total lysate results of samples taken prior to immunoprecipitation(total; representing 1/10 of total cell extracts), whereas the upperpanels indicate the results obtained after HA immunoprecipitation(coIP; representing 9/10 of total cell extracts). (C) Effect of the differ-ent V constructs on the suppression of the MDA5-mediated signalingpathway. Vero-SLAM cells were transfected with p�-IFN-fl-lucter andpTK-RL reporter genes along with the different V-expressing plasmidsand the FLAG-tagged MDA5 construct. At 24 h posttransfection, cellswere left untreated or were additionally transfected with 1.5 �g ofpoly(I:C)/ml (15 h) prior to luciferase assays.
VOL. 84, 2010 MECHANISMS OF CDV-V AS IFN ANTAGONIST 9
83
CHAPTER TWO

Vwt Y110D, but not VNT, with or without fusion to RFP (Fig.7C). We thus concluded that VCT alone was very likely misfoldedand thus not functional but that it could fold into an active con-formation if fused to an irrelevant, stabilizing protein. Impor-tantly, this result furthermore confirms the selective incapacity ofCDV-VCT to control IFN-�/�-induced activity on its own.
Only Vwt efficiently associates with STAT1 and STAT2. Acoimmunoprecipitation assay was used to investigate the abilityof the different V domain mutants to bind the endogenousSTAT1 and STAT2 proteins (in the absence of IFN treat-ment). The anti-HA 3F10 monoclonal antibody was used toimmunoprecipitate the different V constructs, followed by im-munoblot analysis using anti-STAT1 or anti-STAT2 antibodiesfor detection. Figure 7A and B document that, independentlyof whether Vwt was fused to the RFP, both STAT1 and STAT2could efficiently be copurified (Fig. 7A and B, upper panels).Interestingly, although VNT retained some interaction withSTAT1, it displayed significantly reduced binding to STAT2,whereas VCT, even when fused to RFP, showed no bindingactivity at all to either STAT molecule. The latter results are inagreement with the data obtained in the ISRE luciferase re-porter gene assay. Moreover, while the single point mutantVwt Y110D almost completely lost the capacity to associatewith STAT1, we detected a slight interaction with STAT2 (Fig.7A and B, upper panels). Finally, the ability of the P and Cproteins in binding STAT1 and STAT2 was also investigated(HA-tagged versions were constructed). The C protein was
unable to bind both STAT1 and STAT2, whereas the P proteinweakly interacted with STAT1 only (Fig. 7A). Immunoblotanalysis of STAT1 and STAT2 prior to immunoprecipitationrevealed that both transcription factors were expressed to verysimilar amounts under all conditions. These results suggestthat VCT, when fused to VNT, confers the capacity of Vwt toenhance STAT1 binding and to associate with STAT2.
Selective inhibition of nuclear import of STAT molecules byVNT and Vwt Y110D. We next assessed whether the differen-tial binding capacities of the V mutants correlated with thenuclear import inhibition mechanism described above. To thispurpose, Vero-SLAM cells were transfected with the variousexpression plasmids and treated with IFN-�/�, and STAT1/STAT2 cellular localization was subsequently investigated byimmunofluorescence analysis (Fig. 8 and 9, for STAT1 andSTAT2, respectively). Clearly, while HA-Vwt efficiently inhib-ited STAT1 and STAT2 nuclear import, HA-P partially re-tained STAT1 but not STAT2 in the cytoplasm (Fig. 8A and9A). Interestingly, the extent of STAT1 nuclear import inhibi-tion seemed to correlate with the level of P expression intransfected cells (Fig. 8A). Conversely, HA-C did not at allsuppress IFN-�/�-induced STAT1 and STAT2 nuclear accu-mulation (Fig. 8A and 9A), confirming the data obtained in theco-IP assay (Fig. 7).
In order to assess the ability of VNT, VCT, and Vwt Y110Dto modulate STAT1 and STAT2 nuclear translocation, theidentical experiments were performed using the RFP fusion
FIG. 8. Vwt and VNT inhibit STAT1 nuclear translocation. Vero-SLAM or Vero cells were transfected with the various expression vectors.One day posttransfection, cells were treated with IFN-�/� for 30 min. Subsequently, cells were fixed and permeabilized, and STAT1 localizationwas investigated using an anti-STAT1, which was followed by addition of an Alexa Fluor 488-conjugated secondary antibody (green) (A). The viralHA-tagged proteins were stained using an anti-HA monoclonal antibody, followed by addition of an Alexa Fluor 555-conjugated (red) secondaryantibody. STAT1 localization was investigated with an anti-STAT1 antibody, followed by addition of an Alexa Fluor 488-conjugated (green)secondary antibody, and RFP fusion proteins were directly visualized for red fluorescence emission (B). Nuclei were counterstained with TOTO3(blue). Green, blue, and red fluorescence emissions were captured with a scanning confocal laser microscope. Filled arrowheads indicate nucleiwithout STAT1 accumulation, and open arrowheads indicate nuclei with STAT1 accumulation.
10 ROTHLISBERGER ET AL. J. VIROL.
84
CHAPTER TWO

proteins. Here again, only RFP-HA-Vwt efficiently inhibitedboth STAT1 and STAT2 nuclear import (Fig. 8B and 9B).Intriguingly, whereas RFP-HA-VNT was able to efficiently re-tain STAT1 in the cytoplasm, STAT2 mainly localized in thenucleus in the presence of the same construct though theseexperiments were performed in the presence of IFN. Remark-ably, the exact reverse correlation was observed in RFP-HA-Vwt Y110D-transfected Vero-SLAM cells. In this case, STAT1efficiently translocated to the nucleus in the presence of the Vmutant, whereas STAT2 exhibited a clear cytoplasmic accumu-lation. In contrast, and in agreement with the co-IP assay,RFP-HA-VCT did not function autonomously since bothSTAT molecules were located in the nucleus in IFN-�/�-treated cells (Fig. 8B and 9B). These results demonstrate thatVNT and VCT need to be fused to efficiently control nuclearimport of both STAT molecules.
Evidence for cytoplasmic accumulation of phospho-STAT1in CDV-V-expressing cells. We next confirmed the notion thatthe phosphorylation state of STAT1 is not affected by theCDV-V proteins. To this aim, immunofluorescence analysesusing an anti-phospho-STAT1 MAb were performed in cellstransfected with RFP-HA-Vwt, -VNT, -VCT, and -Vwt Y110Dand treated with type I IFN. As expected, nuclear translocationof phospho-STAT1 was observed in IFN-treated and RFP-transfected cells. Importantly, in RFP-HA-Vwt- and RFP-HA-VNT-transfected cells, phospho-STAT1 accumulated in thecytoplasm to a greater extent than in nontransfected cells of
the same area (Fig. 10A). In contrast, phospho-STAT1 wasclearly detected in the nucleus of RFP-HA-VCT- and RFP-HA-Vwt Y110D-transfected cells (Fig. 10A).
The identical experiments were repeated in rCDV-infectedcells. Hence, in rA75/17red- and rA75/17red Cko-infected cells,phospho-STAT1 could be detected in the cytoplasm, whereasnuclear staining was observed in rA75/17red Vko- and rA75/17red CVko-infected cells (Fig. 10B). We noticed less stainingof phospho-STAT1 in the cytoplasm of infected cells than intransfected cells, probably as a result of the formation of largesyncytia by the different recombinant viruses. Taken together,the above data clearly validate the notion that the CDV-Vprotein inhibits STAT1 nuclear import without affecting itsphosphorylation state both in transfected and infected cells.
Viruses lacking V expression are more sensitive to IFN-�/�treatment. Taken together, the above results suggest that V isessential in counteracting IFN-�/�-dependent signaling al-though the P protein was able to exert partial control. To verifywhether the different phenotypes described above correlatewith differences in growth kinetics, Vero-SLAM cells wereinfected with the various recombinant CDVs (Fig. 11). Next,cells were treated (or left untreated) with IFN-�/� at 3, 12, 24,and 36 h postinfection. Finally, at 48 h postinfection, virustiters of cell-associated viruses were determined by limitingdilution assay. Interestingly, all viruses had reduced viral titerscompared to those obtained in IFN-untreated cells (Fig. 9).This is probably because we used an MOI of 0.02 and treated
FIG. 9. Vwt and Vwt Y110D inhibit STAT2 nuclear translocation. Vero-SLAM or Vero cells were transfected with the various expressionvectors. One day posttransfection, cells were treated with IFN-�/� for 30 min. Subsequently, cells were fixed and permeabilized, and STAT2localization was investigated using an anti-STAT2, which was followed by addition of an Alexa Fluor 488-conjugated secondary antibody (green)(A). The viral HA-tagged proteins were stained using an anti-HA monoclonal antibody, followed by addition of an Alexa Fluor 555-conjugated(red) secondary antibody. (B) STAT2 localization was investigated with an anti-STAT2 antibody, followed by addition of an Alexa Fluor488-conjugated (green) secondary antibody, and RFP fusion proteins were directly visualized for red fluorescence emission. Nuclei werecounterstained with TOTO3 (blue). Green, blue, and red fluorescence emissions were captured with a scanning confocal laser microscope. Filledarrowheads indicate nuclei without STAT2 accumulation, and open arrowheads indicate nuclei with STAT2 accumulation.
VOL. 84, 2010 MECHANISMS OF CDV-V AS IFN ANTAGONIST 11
85
CHAPTER TWO

the cells with IFN as early as 3 h postinfection. Thus, most ofthe uninfected cells had probably established an antiviral statebefore being infected, in turn affecting proper viral growtheven in the case of V-expressing viruses. Nevertheless, viruseslacking V expression had approximately 10 times less progenyvirus production, confirming that V is crucial for counteractingthe IFN-�/�-mediated antiviral state. It is important to note
that the concentration of IFN used in these experiments (1,000units/ml) was 10 to 50 times higher than the minimal concen-tration required to completely inhibit a VSV-induced cyto-pathic effect in Vero-SLAM cells (data not shown). Neverthe-less, albeit not to the same extent, all viruses were able to grow,which suggested that a viral component(s) in addition to V mayprovide partial control of innate immunity.
FIG. 10. Phospho-STAT1 is accumulated in the cytoplasm of both CDV-V-transfected cells and rA75/17red-infected cells. (A) Vero-SLAM cellswere transfected with the various expression vectors. One day posttransfection, cells were treated with IFN-�/� for 30 min. Subsequently, cells werefixed and permeabilized, and phospho-STAT1 localization was investigated using an anti-phospho-STAT1 MAb, which was followed by additionof an Alexa Fluor 488-conjugated secondary antibody (green). RFP fusion proteins were directly visualized for red fluorescence emission.(B) Vero-SLAM cells were infected with the corresponding recombinant viruses at an MOI of 0.02. Phospho-STAT1 localization was investigatedwith an anti-phospho-STAT1 MAb, followed by addition of an Alexa Fluor 488-conjugated (green) secondary antibody, and the RFP expressedby the various viruses was directly visualized for red fluorescence emission. In both panels, nuclei were counterstained with TOTO3 (blue). Green,blue, and red fluorescence emissions were captured with a scanning confocal laser microscope. Filled arrowheads indicate nuclei withoutphospho-STAT1 accumulation, and open arrowheads indicate nuclei with phospho-STAT1 accumulation. Specific fields of view of the cellmonolayer were selected to illustrate infected and noninfected cells in the identical areas.
12 ROTHLISBERGER ET AL. J. VIROL.
86
CHAPTER TWO

The CDV-V protein inhibits STAT1 nuclear import in ca-nine cells. To confirm the findings that CDV-V controls IFN-induced STAT1 nuclear import not only in Vero but also incanine cells, we sought to determine the effect of CDV-V inMDCK cells. Indeed, these cells are functional in both IFN in-duction and IFN action (data not shown). First, a cell line stablyexpressing the universal morbillivirus receptor CD150/SLAM wasproduced in order to allow the virulent virus to replicate. Second,since none of the tested recombinant IFN (rIFN) molecules wasfunctional in these cells (human, bovine, feline, and universalrIFN), we cloned the canine IFN-� obtained from primary caninekeratinocytes. Finally, the recombinant canine IFN-� was pro-duced in 293T cells. Figure 12 illustrates that upon addition ofcIFN-�, STAT1 was, indeed, readily translocated into the nucleusof the engineered MDCK-SLAM cells.
As expected, in cells infected with rA75/17red (Fig. 12A) andrA75/17red Cko viruses (data not shown), STAT1 nuclear im-port was inhibited, whereas in rA75/17red Vko-infected cells(Fig. 12A and B) and rA75/17red CVko-infected cells (data notshown), STAT1 was located in the nucleus. The identical phe-notypes were observed for STAT2 (data not shown). Thus, themain conclusion obtained from Vero-SLAM cells correlatedwith results in canine cells, which are characterized by an intactIFN response system. In addition, we performed titration exper-iments at 2 days postinfection in the presence and absence ofcanine IFN-� in rA75/17red- and rA75/17red Vko-infected cells.The Vko virus grew less efficiently than rA75/17red (Fig. 12C) evenin the absence of IFN, presumably because the Vko virus is defi-cient in suppressing both the IFN action and induction signalingpathways. Nevertheless, IFN treatment reduced the replication ofthe Vko virus about 2 orders of magnitude, whereas the V-ex-pressing virus was reduced only about 10-fold.
DISCUSSION
It has recently been reported that V knockout CDV (basedon the 5804P virulent strain) was attenuated in infected ferrets,which was associated, at least in part, with inhibition of IFN-�/� induction in peripheral blood mononuclear cells (PBMCs)(47). We now show that the V protein of the highly virulent
CDV A75/17 strain also counteracts IFN action by additionallydisrupting the IFN-�/�-dependent signaling. Importantly, thisdoes not seem to be valid in only Vero-SLAM cells as prelim-inary experiments in canine MDCK-SLAM cells providedstrong evidence that the effects observed in Vero cells are alsoactive in canine cells. Detailed molecular analysis enabled us todemonstrate that CDV-V specifically ablated the nuclear im-port of STAT1 and STAT2 without affecting their activatedphosphorylation states. Furthermore, inhibition of IFN-�/�-dependent signaling correlated with the capacity of the V pro-tein to efficiently interact with both STAT molecules. Finally,we identified both the N-terminal and the C-terminal regionsof V as playing a synergistic role in IFN evasion.
Initial attempts to map the domains of the V protein thatinteract with STAT1 and STAT2 revealed that the N-terminalregion of V was able to function as an autonomous domaininterfering with IFN-�/�-induced signaling. Importantly, co-IPexperiments indicated that VNT retained association withSTAT1 but failed to copurify STAT2. Since the full-length Vprotein (Vwt) efficiently coprecipitated both STAT molecules,this suggests that VNT is very likely responsible for STAT1interaction, whereas VCT is necessary to target STAT2. Inagreement with this notion, the single-amino acid mutant VwtY110D retained slight STAT2 binding but almost completelylost STAT1 interaction. Nevertheless, we cannot exclude thepossibility that VCT, when fused to VNT, determines a specificconformational state of VNT that confers the capacity of theN-terminal region to target both STAT molecules. Recentwork done with MV is consistent with the former hypothesissince the N-terminal domain of MV-V was assigned to STAT1binding, whereas STAT2 has been discovered to be the maintarget of the MV-VCT module (4, 5, 35). The main differencethat we observed in this study between both morbillivirus Vprotein functionalities is that the CDV-VCT domain expressedalone could not disrupt both the IFN-�/�-mediated and theMDA5-mediated signaling pathways, thus suggesting improperfolding and/or protein degradation. Remarkably, when stabi-lized by an irrelevant protein, the VCT domain selectivelysuppressed MDA5-mediated signaling but not signaling in-duced by IFN-�/�. A sequence alignment of the MV- andCDV-VCT domains shows only about 50% amino acid iden-tity, which may explain the different functions (Fig. 13). It maybe possible that VCT, when fused to RFP but not in the wtprotein, adopts a conformational state that remains functionalin inhibiting the MDA5-mediated signaling pathway but losesits intrinsic ability to bind STAT1 and/or STAT2 to suppressthe IFN-�/�-mediated signaling. Alternatively, VCT may foldsimilarly when fused to the RFP than in the wt protein. In thiscase, the CDV-VCT domain may be responsible for (i) dis-rupting the MDA5-mediated signaling pathway and (ii) con-ferring proper folding to the VNT domain, which consequentlywill efficiently engage STAT1 and STAT2 to control IFN-�/�-mediated signaling. Since Vwt elicited enhanced binding avid-ity to STAT1 compared to VNT alone, this indeed suggeststhat VNT’s conformational state is modulated by the presenceof VCT. Taking these observations together, we propose thatCDV-VNT and -VCT are two interdependent modules thatfunction synergistically to allow proper folding of the full-length V protein. In turn, Vwt gains the ability to efficientlyinteract with STAT1 and STAT2, which offers optimal con-
FIG. 11. Viruses lacking V expression showed enhanced sensitivityto IFN-�/� treatment. Vero-SLAM cells were infected with the differ-ent rCDVs at an MOI of 0.02. Then, IFN-�/� was added (or not) at 3,12, 24, and 36 h postinfection. Virus titers of cell-associated viruseswere determined by limiting dilution assay at 48 h postinfection.
VOL. 84, 2010 MECHANISMS OF CDV-V AS IFN ANTAGONIST 13
87
CHAPTER TWO

ditions to prevent nuclear import of the two STAT mole-cules and, consequently, to control IFN-�/�-mediated sig-naling. These results contrast with those obtained with thehenipavirus family, where the C-terminal region of V was to-tally dispensable for STAT binding and IFN evasion (38, 40).The requirement of both the N- and C-terminal domains of V
to evade innate immunity is consistent with the studies of therubulavirus family, though in that case both domains of V werenecessary to mediate STAT1 (9, 19, 22, 30, 49), STAT2 (17, 18,26, 29, 30, 50), or STAT3 (44) proteasomal degradation ratherthan STAT nuclear import inhibition without affecting theirphosphorylation states.
FIG. 12. STAT1 nuclear import is suppressed in MDCK-SLAM cells after treatment with canine interferon-� (cIFN-�). (A) Immunofluores-cence images of MDCK-SLAM cells stained for STAT1. MDCK-SLAM cells were infected with the rA75/17red and rA75/17red Vko viruses or leftuninfected. One day postinfection, cells were treated (or left untreated) for 30 min with IFN-�/� and subsequently fixed, permeabilized, and stainedfor STAT1 localization using an anti-STAT1 antibody. Then, an Alexa Fluor 488-conjugated secondary antibody was employed (green), and imageswere captured with a scanning confocal laser microscope (Olympus). The nuclei were counterstained with TOTO3 (blue). Infected cells werelocalized by the expression of RFP. (B) Close-up view of the nuclear STAT1 translocation in rA75/17red Vko-infected cells. Staining is identical tothat described in panel A. (C) Titration experiments in the presence or absence of canine IFN-� in rA75/17red- and rA75/17red Vko-infected cells.MDCK-SLAM cells were infected with both rCDVs at an MOI of 0.02. IFN-�/� was added (or not) at 9 h postinfection, and virus titers ofcell-associated viruses were determined by limiting dilution assay 48 h postinfection in Vero-SLAM cells.
14 ROTHLISBERGER ET AL. J. VIROL.
88
CHAPTER TWO

Results obtained in ISRE luciferase reporter gene assaysclearly demonstrated the capacity of both VNT and VwtY110D to interfere with IFN activity although to a limitedextent compared to Vwt. Remarkably, immunofluorescenceanalysis of STAT1 and STAT2 intracellular localizations in thepresence of IFN and in the presence of these two V mutantsrevealed an unanticipated reverse localization of both tran-scription factors. Indeed, while VNT specifically disruptedSTAT1 (but not STAT2) nuclear import, Vwt Y110D impairedSTAT2 (but not STAT1) nuclear localization. These resultsseem to be in contradiction with the current model of theIFN-�/�-induced signaling pathway, which states that phos-phorylated STAT1 and STAT2 dimerize upon IFN treatmentprior to being translocated to the nucleus. We therefore hy-pothesize that the association of VNT with STAT1 and theassociation of Vwt Y110D with STAT2 compete for the dimer-ization of phosphorylated STAT1 and STAT2. Subsequently,in the presence of VNT and IFN, phosphorylated STAT1 ac-cumulates in the cytoplasm, whereas phosphorylated STAT2would translocate and accumulate in the nucleus. Conversely,in the presence of Vwt Y110D, which exhibits STAT1 bindingimpairment but efficient STAT2 association, the reverse pheno-type for STAT cellular localization is observed. This hypothesis isin excellent agreement with our results demonstrating thatCDV-V does not affect STAT1 and STAT2 phosphorylation, butfurther experiments are required to consolidate this model.
In addition to V, the CDV-P protein (sharing the identicalN-terminal region with V) was found to retain weak interac-tion with STAT1, which correlated with partial suppression ofIFN-�/�-mediated signaling. The biological relevance of thislimited interaction with STAT1 was supported by the fact thatalthough the growth of all rCDVs was reduced, they were notcompletely inhibited in the presence of IFN. This effect of theCDV-P is consistent with observations made with P proteins ofother negative-strand RNA viruses, e.g., MV, RPV, Nipahvirus, and rabies virus (8, 10, 25, 46). Nevertheless, our resultsobtained in the co-IP assay indicated that P bound STAT1rather inefficiently, suggesting that a high concentration of Pmay be required for effective inhibition of IFN signaling. Sup-porting this notion, we noticed that at late time points afterinitial infection (48 h postinfection) all knockout viruses wereultimately able to disrupt nuclear import of STATs (data notshown). It is noteworthy that a differential effect of MV-P inIFN evasion was recently documented. Indeed, results suggestedthat the origin of the virus strain determined the extent of the Pfunctionality (11). Thus, further investigations are required todemonstrate whether the extent by which the CDV-P proteincounteracts the IFN-�/� response is also regulated by the originof the strain and/or by the host cell environment.
The mechanism by which the A75/17 CDV-V protein inhib-
its the IFN-�/�-mediated response differs from that of othermorbilliviruses. To our knowledge, all studies performed withMV-V, with the exception of that by Palosaari et al. (28), havereported an inhibition of the phosphorylation of STAT1 (5, 42,48) and STAT2 (4, 8, 42). The reasons for the differencesbetween CDV and MV remain unclear. In addition to genuinebiological differences between these two morbilliviruses, theorigin and passaging histories of the strains used to studyevasion from IFN action may be a factor. Indeed, we studied ahighly virulent viral strain not adapted to cultured cells,whereas the strain of MV was attenuated (8) or persistentlyinfected cells were investigated (48). There are also similari-ties, however, between CDV and other morbilliviruses. Con-sistent with the findings in CDV, MV-V was shown to be morepotent in inhibiting IFN-�/� signaling than the C protein, andthis observation was equally true for all strains independent oftheir virulence (11). Similarly, using recombinant knockoutviruses based on a vaccine strain, RPV-V was shown to blockIFN-mediated phosphorylation of STAT1 and STAT2 withoutcausing the degradation of these proteins (25).
Taken together, our results shed light on a unique molecularmechanism by which a highly virulent CDV strain interfereswith IFN action by disrupting signaling for the synthesis ofantiviral proteins. While the mechanism of CDV virulence islikely to be complex and may involve different host cells andinteractions between cells, disruption of the IFN defense maydeprive the host of an early mechanism known to limit viralreplication and spread at a critical stage of infection. Under-standing the precise mechanisms of IFN evasion may also leadto the rational design of vaccines that combine optimally bal-anced stimulation of the innate immune system, which isknown to be essential for activation of an effective adaptiveimmune response (27).
ACKNOWLEDGMENTS
We thank D. Garcin for offering the red fluorescent marker protein,the luciferase plasmids pISRE-Luc, p�-IFN-fl-lucter, and pTK-RL,and the NES-GFP-SV5-V and FLAG-tagged MDA5-expressing plas-mids, and we thank V. von Messling for providing the Vero-SLAMcells. We are grateful to Ruth Parham for linguistic improvement ofthe manuscript.
REFERENCES
1. Begitt, A., T. Meyer, R. M. van, and U. Vinkemeier. 2000. Nucleocytoplasmictranslocation of Stat1 is regulated by a leucine-rich export signal in thecoiled-coil domain. Proc. Natl. Acad. Sci. U. S. A. 97:10418–10423.
2. Bollo, E., A. Zurbriggen, M. Vandevelde, and R. Fankhauser. 1986. Caninedistemper virus clearance in chronic inflammatory demyelination. Acta Neu-ropathol. 72:69–73.
3. Buchholz, U. J., S. Finke, and K. K. Conzelmann. 1999. Generation ofbovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is notessential for virus replication in tissue culture, and the human RSV leaderregion acts as a functional BRSV genome promoter. J. Virol. 73:251–259.
4. Caignard, G., M. Bourai, Y. Jacob, F. Tangy, and P. O. Vidalain. 2009.
FIG. 13. Sequence alignment of amino acids 233 to 300 (VCT domain) in the carboxyl-terminal segment of V proteins of two wild-type CDVstrains and two MV strains. GenBank numbers for each virus sequence are as follows: AB016162.1 (measles, ICB strain), AB254456.1 (measles,subacute sclerosing panencephalitis strain Kobe-1), AY386316.1 (canine distemper, A75/17 strain), BAA01203.1 (canine distemper, 5804P strain).Gray boxes represent identical residues in all four sequences.
VOL. 84, 2010 MECHANISMS OF CDV-V AS IFN ANTAGONIST 15
89
CHAPTER TWO

Inhibition of IFN-alpha/beta signaling by two discrete peptides within mea-sles virus V protein that specifically bind STAT1 and STAT2. Virology383:112–120.
5. Caignard, G., M. Guerbois, J. L. Labernardiere, Y. Jacob, L. M. Jones, F.Wild, F. Tangy, and P. O. Vidalain. 2007. Measles virus V protein blocksJak1-mediated phosphorylation of STAT1 to escape IFN-alpha/beta signal-ing. Virology 368:351–362.
6. Cherpillod, P., A. Tipold, M. Griot-Wenk, C. Cardozo, I. Schmid, R. Fatzer,M. Schobesberger, R. Zurbriggen, L. Bruckner, F. Roch, M. Vandevelde, R.Wittek, and A. Zurbriggen. 2000. DNA vaccine encoding nucleocapsid andsurface proteins of wild type canine distemper virus protects its natural hostagainst distemper. Vaccine 18:2927–2936.
7. Dayer, A. G., B. Jenny, M. O. Sauvain, G. Potter, P. Salmon, E. Zgraggen, M.Kanemitsu, E. Gascon, S. Sizonenko, D. Trono, and J. Z. Kiss. 2007. Ex-pression of FGF-2 in neural progenitor cells enhances their potential forcellular brain repair in the rodent cortex. Brain 130:2962–2976.
8. Devaux, P., M. von, V. W. Songsungthong, C. Springfeld, and R. Cattaneo.2007. Tyrosine 110 in the measles virus phosphoprotein is required to blockSTAT1 phosphorylation. Virology 360:72–83.
9. Didcock, L., D. F. Young, S. Goodbourn, and R. E. Randall. 1999. The Vprotein of simian virus 5 inhibits interferon signaling by targeting STAT1 forproteasome-mediated degradation. J. Virol. 73:9928–9933.
10. Eaton, B. T., C. C. Broder, and L. F. Wang. 2005. Hendra and Nipah viruses:pathogenesis and therapeutics. Curr. Mol. Med. 5:805–816.
11. Fontana, J. M., B. Bankamp, W. J. Bellini, and P. A. Rota. 2008. Regulationof interferon signaling by the C and V proteins from attenuated and wild-type strains of measles virus. Virology 374:71–81.
12. Fontana, J. M., B. Bankamp, and P. A. Rota. 2008. Inhibition of interferoninduction and signaling by paramyxoviruses. Immunol. Rev. 225:46–67.
13. Goodbourn, S., L. Didcock, and R. E. Randall. 2000. Interferons: cell sig-nalling, immune modulation, antiviral response and virus countermeasures.J. Gen. Virol. 81:2341–2364.
14. Horvath, C. M. 2004. Silencing STATs: lessons from paramyxovirus inter-feron evasion. Cytokine Growth Factor Rev. 15:117–127.
15. Horvath, C. M. 2004. Weapons of STAT destruction. Interferon evasion byparamyxovirus V protein. Eur. J. Biochem. 271:4621–4628.
16. Kawai, T., and S. Akira. 2006. Innate immune recognition of viral infection.Nat. Immunol. 7:131–137.
17. Kawano, M., M. Kaito, Y. Kozuka, H. Komada, N. Noda, K. Nanba, M.Tsurudome, M. Ito, M. Nishio, and Y. Ito. 2001. Recovery of infectioushuman parainfluenza type 2 virus from cDNA clones and properties of thedefective virus without V-specific cysteine-rich domain. Virology 284:99–112.
18. Kozuka, Y., Y. Yamashita, M. Kawano, M. Tsurudome, M. Ito, M. Nishio, H.Komada, and Y. Ito. 2003. Identification of amino acids essential for thehuman parainfluenza type 2 virus V protein to lower the intracellular levelsof the STAT2. Virology 317:208–219.
19. Kubota, T., N. Yokosawa, S. Yokota, and N. Fujii. 2001. C terminal CYS-RICH region of mumps virus structural V protein correlates with block ofinterferon alpha and gamma signal transduction pathway through decreaseof STAT 1-alpha. Biochem. Biophys. Res. Commun. 283:255–259.
20. Lamb, R. A., and D. Kolakofsky. 2001. Paramyxoviridae: the viruses and theirreplication, p. 1305–1340. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A.Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4thed. Lippincott Williams & Wilkins, Philadelphia, PA.
21. Levy, D. E., and J. E. Darnell, Jr. 2002. Stats: transcriptional control andbiological impact. Nat. Rev. Mol. Cell Biol. 3:651–662.
22. Li, T., X. Chen, K. C. Garbutt, P. Zhou, and N. Zheng. 2006. Structure ofDDB1 in complex with a paramyxovirus V protein: viral hijack of a propellercluster in ubiquitin ligase. Cell 124:105–117.
23. Marq, J. B., A. Brini, D. Kolakofsky, and D. Garcin. 2007. Targeting of theSendai virus C protein to the plasma membrane via a peptide-only mem-brane anchor. J. Virol. 81:3187–3197.
24. McBride, K. M., C. McDonald, and N. C. Reich. 2000. Nuclear export signallocated within the DNA-binding domain of the STAT1 transcription factor.EMBO J. 19:6196–6206.
25. Nanda, S. K., and M. D. Baron. 2006. Rinderpest virus blocks type I and typeII interferon action: role of structural and nonstructural proteins. J. Virol.80:7555–7568.
26. Nishio, M., M. Tsurudome, M. Ito, M. Kawano, H. Komada, and Y. Ito. 2001.High resistance of human parainfluenza type 2 virus protein-expressing cellsto the antiviral and anti-cell proliferative activities of alpha/beta interferons:cysteine-rich V-specific domain is required for high resistance to the inter-ferons. J. Virol. 75:9165–9176.
27. Palm, N. W., and R. Medzhitov. 2009. Pattern recognition receptors andcontrol of adaptive immunity. Immunol. Rev. 227:221–233.
28. Palosaari, H., J. P. Parisien, J. J. Rodriguez, C. M. Ulane, and C. M.Horvath. 2003. STAT protein interference and suppression of cytokine sig-nal transduction by measles virus V protein. J. Virol. 77:7635–7644.
29. Parisien, J. P., J. F. Lau, J. J. Rodriguez, B. M. Sullivan, A. Moscona, G. D.
Parks, R. A. Lamb, and C. M. Horvath. 2001. The V protein of humanparainfluenza virus 2 antagonizes type I interferon responses by destabilizingsignal transducer and activator of transcription 2. Virology 283:230–239.
30. Parisien, J. P., J. F. Lau, J. J. Rodriguez, C. M. Ulane, and C. M. Horvath.2002. Selective STAT protein degradation induced by paramyxoviruses re-quires both STAT1 and STAT2 but is independent of alpha/beta interferonsignal transduction. J. Virol. 76:4190–4198.
31. Paterson, R. G., G. P. Leser, M. A. Shaughnessy, and R. A. Lamb. 1995. Theparamyxovirus SV5 V protein binds two atoms of zinc and is a structuralcomponent of virions. Virology 208:121–131.
32. Plattet, P., P. Cherpillod, D. Wiener, L. Zipperle, M. Vandevelde, R. Wittek,and A. Zurbriggen. 2007. Signal peptide and helical bundle domains ofvirulent canine distemper virus fusion protein restrict fusogenicity. J. Virol.81:11413–11425.
33. Plattet, P., C. Zweifel, C. Wiederkehr, L. Belloy, P. Cherpillod, A. Zurbrig-gen, and R. Wittek. 2004. Recovery of a persistent Canine distemper virusexpressing the enhanced green fluorescent protein from cloned cDNA. VirusRes. 101:147–153.
34. Precious, B., D. F. Young, A. Bermingham, R. Fearns, M. Ryan, and R. E.Randall. 1995. Inducible expression of the P, V, and NP genes of theparamyxovirus simian virus 5 in cell lines and an examination of NP-P andNP-V interactions. J. Virol. 69:8001–8010.
35. Ramachandran, A., J. P. Parisien, and C. M. Horvath. 2008. STAT2 is aprimary target for measles virus V protein-mediated alpha/beta interferonsignaling inhibition. J. Virol. 82:8330–8338.
36. Randall, R. E., and S. Goodbourn. 2008. Interferons and viruses: an interplaybetween induction, signalling, antiviral responses and virus countermeasures.J. Gen. Virol. 89:1–47.
37. Rivals, J. P., P. Plattet, C. Currat-Zweifel, A. Zurbriggen, and R. Wittek.2007. Adaptation of canine distemper virus to canine footpad keratinocytesmodifies polymerase activity and fusogenicity through amino acid substitu-tions in the P/V/C and H proteins. Virology 359:6–18.
38. Rodriguez, J. J., C. D. Cruz, and C. M. Horvath. 2004. Identification of thenuclear export signal and STAT-binding domains of the Nipah virus V proteinreveals mechanisms underlying interferon evasion. J. Virol. 78:5358–5367.
39. Schindler, C., X. Y. Fu, T. Improta, R. Aebersold, and J. E. Darnell, Jr. 1992.Proteins of transcription factor ISGF-3: one gene encodes the 91- and 84-kDa ISGF-3 proteins that are activated by interferon alpha. Proc. Natl.Acad. Sci. U. S. A. 89:7836–7839.
40. Shaw, M. L., A. Garcia-Sastre, P. Palese, and C. F. Basler. 2004. Nipah virusV and W proteins have a common STAT1-binding domain yet inhibit STAT1activation from the cytoplasmic and nuclear compartments, respectively.J. Virol. 78:5633–5641.
41. Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D.Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem.67:227–264.
42. Takeuchi, K., S. I. Kadota, M. Takeda, N. Miyajima, and K. Nagata. 2003.Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBSLett. 545:177–182.
43. Thomas, S. M., R. A. Lamb, and R. G. Paterson. 1988. Two mRNAs thatdiffer by two nontemplated nucleotides encode the amino coterminal pro-teins P and V of the paramyxovirus SV5. Cell 54:891–902.
44. Ulane, C. M., J. J. Rodriguez, J. P. Parisien, and C. M. Horvath. 2003.STAT3 ubiquitylation and degradation by mumps virus suppress cytokineand oncogene signaling. J. Virol. 77:6385–6393.
45. Veals, S. A., C. Schindler, D. Leonard, X. Y. Fu, R. Aebersold, J. E. Darnell,Jr., and D. E. Levy. 1992. Subunit of an alpha-interferon-responsive tran-scription factor is related to interferon regulatory factor and Myb families ofDNA-binding proteins. Mol. Cell. Biol. 12:3315–3324.
46. Vidy, A., M. Chelbi-Alix, and D. Blondel. 2005. Rabies virus P proteininteracts with STAT1 and inhibits interferon signal transduction pathways.J. Virol. 79:14411–14420.
47. von Messling, V., N. Svitek, and R. Cattaneo. 2006. Receptor (SLAM[CD150]) recognition and the V protein sustain swift lymphocyte-basedinvasion of mucosal tissue and lymphatic organs by a morbillivirus. J. Virol.80:6084–6092.
48. Yokota, S., H. Saito, T. Kubota, N. Yokosawa, K. Amano, and N. Fujii. 2003.Measles virus suppresses interferon-alpha signaling pathway: suppression ofJak1 phosphorylation and association of viral accessory proteins, C and V,with interferon-alpha receptor complex. Virology 306:135–146.
49. Young, D. F., N. Chatziandreou, B. He, S. Goodbourn, R. A. Lamb, and R. E.Randall. 2001. Single amino acid substitution in the V protein of simian virus5 differentiates its ability to block interferon signaling in human and murinecells. J. Virol. 75:3363–3370.
50. Young, D. F., L. Didcock, S. Goodbourn, and R. E. Randall. 2000. Paramyxo-viridae use distinct virus-specific mechanisms to circumvent the interferonresponse. Virology 269:383–390.
16 ROTHLISBERGER ET AL. J. VIROL.
90
CHAPTER TWO

ACKNOWLEDGEMENTS I would like to express a genuine gratitude to my supervisor, Dr. Philippe Plattet, for
his support, patience, flexibility and friendliness during the entire PhD work.
I am sincerely grateful to my supervisor, Prof. Dr. Andreas Zurbriggen, for the
opportunity to accomplish my PhD thesis in his research group, for his kind support
and his excellent scientific advices
I would like to thank my co-referee, Prof. Dr. Ernst Peterhans for his great scientific
propositions and for evaluating my PhD work.
I would like to thank my mentor, Prof. Dr. Peter Bütikofer, for the involvement in my
scientific development.
I would like to thank Prof. Dr. Marc Vandevelde for his good scientific suggestions and
for his instructional, congenial neurological teaching.
Many thanks to Ljerka Zipperle for giving me technical help, for a day-to-day support
and an enjoyable, friendly atmosphere.
Also my thanks go to all the people of my department, for being always friendly and
making fun.
Special thank to Anne Röthlisberger for the nice and enjoyable co-work and for being
my friend.
My deepest thank to my mother and father, for always having time for me and their
understanding and caring comportment. Without you, I would never have
succeeded.
Thanks to all my friends and family for your support and an enjoyable and amusing
distraction every once a while.
ACKNOWLEDGEMENTS

CURRICULUM VITAE First Name Dominique
Surname Wiener
Date of birth 02.08.1977
Native place Stallikon (ZH)
Education
3.1.2007 – currently PhD student
Department for Clinical Research and Veterinary
Public Health
Vetsuisse Faculty
University of Bern, Switzerland
Thesis title: “Investigation of two potential
mechanisms which may favor persistence of CDV,
the driving force behind the chronic progression of
demyelination in canine distemper”
1.7.2005 – currently Residency/PhD program in animal pathology
Vetsuisse Faculty
University of Bern, Switzerland
1.2.2004 – 31.6.2005 Doctoral thesis
Department for Clinical Research and Veterinary
Public Health
Vetsuisse Faculty
University of Bern, Switzerland
Thesis title: “Effect of structural proteins derived
from cytolytic and persistent canine distemper virus
strains on cell-cell fusion in co-transfection studies”
15.2.1998 – 5.12.2003 Academic study of veterinary medicine
University of Bern
92
CURRICULUM VITAE

LIST OF PUBLICATIONS
Röthlisberger A., Wiener D., Schweizer M., Peterhans E., Zurbriggen A., Plattet P.
(2010) Two domains of the V protein of virulent canine distemper virus selectively
inhibits STAT1 and STAT2 nuclear import, J. Virol. Apr 28. [Epub ahead of print]
Brachelente C., Wiener D., Malik Y., Huessy D. (2007) A case of necrotizing fasciitis
with septic shock in a cat caused by Acinetobacter baumannii. Vet Dermatol.
18(6):432-8.
Pfister P., Geissbuehler U., Wiener D., Hirsbrunner G., Kaufmann C. (2007)
Pollakisuria in a dwarf goat due to pathologic enlargement of the uterus. Vet Q.
29(3):112-6.
Wiener D., Plattet P., Cherpillod P., Zipperle L., Doherr M.G., Vandevelde M.,
Zurbriggen A. (2007) Synergistic inhibition in cell-cell fusion mediated by the matrix
and nucleocapsid protein of canine distemper virus. Virus Res. 129(2):145-154.
Plattet P., Cherpillod P., Wiener D., Zipperle L., Vandevelde M., Wittek R., Zurbriggen
A. (2007) Signal peptide and helical bundle domains of virulent canine distemper virus
fusion protein restrict fusogenicity. J Virol. 81(20):11413-11425.
93
LIST OF PUBLICATIONS

Declaration of Originality Last name, first name: Wiener Dominique Matriculation number: 97-119-499 I hereby declare that this thesis represents my original work and that I have used no
other sources except as noted by citations.
All data, tables, figures and text citations which have been reproduced from any other
source, including the internet, have been explicitly acknowledged as such.
I am aware that in case of non-compliance, the Senate is entitled to divest me of the
doctorate degree awarded to me on the basis of the present thesis, in accordance with
the “Statut der Universität Bern (Universitätsstatut; UniSt)”, Art. 20, of
17 December 1997.
Place, date Signature
……………………………………… …………………………………………………
94







