
MATHIEU DROUIN
EXPRESSION, PURIFICATION ET CARACTÉRISATION
DE L'ENDONUCLÉASE 1-Cmoei
Mémoire
présen te
à la Faculté des Études Supérieures
de l'université Laval
pour l'obtention
du grade de maître ès sciences (M. Sc.)
Département de Biochimie
FACULTE DES SCIENCES ET GGNIE
WIVERSITE LAVAL
NOVEMBRE 1999
OMathieu Drouin, 1999

National Library m * I of Canada Bibliothèque nationale du Canada
Acquisitions and Acquisitions et Bibliogaphic Services seivices bibliographiques
395 Wellington Street 395, rue Wellington Ottawa ON K I A ON4 Ottawa ON K1A ON4 Canada Canada
Your lsb Voire rcihrsnce
Our iYs Notre refdrence
The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sell reproduire, prêter, distribuer ou copies of this thesis in microfom, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfiche/film, de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve La propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts ffom it Ni la thèse ni des extraits substantiels may be printed or othewise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

RÉSUMÉ
Le premier intron du groupe 1 du gène chloroplastique psbA de l'algue verte
Chlamydornonas rnoewusii code une ADN endonucléase à site spécifique contenant le
motif conservé H-N-H. Cette nouvelle endonucléase a été caractérisée au cours de la
présente étude. Elle a été surexprimée chez Escherichia coii sous la forme d'une protéine
flanquée d'une queue de polyhistidines qui a été ajoutée à l'extrémité N-terminale pour
faciliter sa purification. Cette protéine. appelée 1-CmoeI. reconnaît et clive la version
sans intron du gène psbA de la souche d'algue interféconde C. eugametor. 1-Cmorl n'est
active qu'en présence de cations divalents faisant partie de la famille des alcalino-terreux.
1-Cmoel se fixe à son substrat sous forme d'un monomère de 43 kDa. Les paramètres
cinétiques de 1-CmoeI sont de l'ordre de 100 pM pour le Km et de 0.2 min-' pour le km.
Finalement. la mutagenèse dirigée sur le premier résidu histidine du motif H-N-H a
montré que ce dernier est critique pour l'activité de 1-CmocI.

REMERCIEMENTS
Je tiens d'abord à remercier ma directrice Monique Tunnel et mon codirecteur
Claude Lemieux pour m'avoir donné l'opportunité de réaliser ce projet dans leur
laboratoire. J'ai beaucoup apprécié l'intérêt soutenu qu'ils ont démontré tout au long de
mes travaux par le biais de discussions intkressantes.
Je voudrais également remercier les membres de mon comité aviseur Jacques
Lapointe et Lindsay Eltis pour leur support lors des réunions de mon comité aviseur. Je
tiens spécialement à remercier M. Eltis de m'avoir donné l'opportunité d'utiliser l'appareil
AKTA Explorer de son laboratoire de purifier efficacement ma protéine.
Finalement, j'aimerais souligner l'aide appréciée de Christian Otis au niveau
pratique et technique ainsi que les commentaires utiles de mes collègues du laboratoire
Dominique Boudreau, Jean-Philippe Gagné et Ann-Josée Noël. Je voudrais aussi dire un
merci spécial à Nathalie Drouin pour sa précieuse aide au cours de la purification de ma
protéine.

III
TABLE DES MATIÈRES
=SUME .................................. .... ....................................................................................... 1
REMERCIEMENTS ........................................................................................................... II
................................................................................................ TABLE DES MATIGRES III
LISTES DES FIGURES ................................................................................................. VI
LISTE DES TABLEAUX ......................................................................................... VI1
....................................................................................... LISTE DES ABWVIATIONS VI11
CHAPITRE 1
INTRODUCTION .............................................................................................................. 1
Les introns des groupes 1 et II ............................................................................. 1
.................................................................... La mobilité des introns du groupe 1 4
Les endonucléases "horning" .............................................................................. 4
................................. Les endonucléases "homing" de la famille LAGLIDADG 8
Les motifs GIY-YIG et His-Cys Box ............................................................... 12
.............................. Les endonucléases "homing" de la famille à motif H-N-H 13
............................... La protbine à motif H-N-H de Chlamydomonas moewusii 17
Problématique de l'étude .............................................................................. 22
CHAPITRE II
.......... 2.1 Amplification et clonage du gène de I-CmoeI dans le vecteur PET-30a 24
2.2 Transformation des cellules compétentes E . coli STBL2 ................................. 27 2 3 Vérification de la séquence clonée .......................... .. .................................... 27
.......................... . 2.4 Transformation des cellules d'expression E coli BUl(DE3) 27

2.5 Expression de la protéine recombinante ...................................................... 28
2.6 Purification de 1-CmoeI ..............................~...................................................... 28
2.6.1 Préparation et lyse des extraits cellulaires ................................................ 28
2.6.2 Chromatographie d'affinité sur résine de nickel ....................................... 29
2.6.3 Dialyse de l'éluat ....................................................................................... 29
2.6.4 Chromatographie d'échange de cations sur résine Cerarnid Hyper D ....... 29
2.7 SDS-PAGE ................................................................................................. 30
2.8 Clivage du His-Tag par l'entérokinase ......................................................... 30
2.9 Optimisation de I'activité de 1-C'mue1 ............................................................... 31
2.9.1 Effet de la nature et de la concentration du tampon de réaction ............... 33
2.9.2 Effet de la concentration du cofacteur ~ p " ............................................. 33
2.9.3 Effet de la nature du cation divalent agissant comme cofacteur ............... 34
2.9.4 Effet du pH du tampon .............................................................................. 34
2.9.5 Effet de la température de réaction ........................................................... 34
2.9.6 Effet de la force ionique ........................................................................ 3 4
2.9.7 Effet du BSA et du DTT ........................................................................... 35
2.10 Stabilité de 1-CmoeI ....................................~.................................................... 35
2.1 1 Stœchiométne de liaison de 1-Cmoei a son site de reconnaissance .................. 35
2.12 Essai de retardement du substrat sans clivage .................................................. 36
................. 2.13 Détermination des paramètres cinétiques de pseudo premier ordre 36
2.14 Effet du SDS sur la rétention des produits de coupure ..................................... 37
2.15 Turnover de l'enzyme et détermination des paramètres cinétiques selon le
modèle cinétique de Michaelis-Menten ...................................................................... 38
2.16 Amplification et séquençage de I'iniron CrpsbA4 ....................................... 3 9
2.17 Mutant H 154A .................................................................................................. 4û
.... 2.17.1 Mutagenèse dirigée, expression et purification de la protéine mutante 41
2.17.2 Mesure de l'activité du mutant H 154A ................................................ 42
2.17.3 Essai de retardement du substrat par le mutant HIWA ............................ 42
CHAPITRE III
RZSULTATS ................................................................................................................ 43

3.1 Expression et purification de 1-CmoeI et H I S A .......................................... 33
.............................................................. 3.2 Clivage du His-Tag par l'entérokinase 4 5
3.3 Détermination des conditions optimales d'activité ........................................ 49
3.3.1 Nature et concentration du tampon de réaction ........................................ 19 .............................................................. 3.3.2 Concentration du cofacteur Mg" 49
3.3.3 Nature du cation divalent agissant comme cofacteur ............................... 52
3.3.4 Autres paramètres optimisés .............................. ... ............................... 52
Stabilité de I-Cmoel .......................................................................................... 53
.................. Stoechiométrie de liaison de 1-Cm04 à son site de reconnaissance 53
........................... Essai de retardement du substrat sans clivage ... ................ W .......................................... Relâchement des produits de coupure par 1-CmwI 58
Caractérisation des paramètres cinétiques de 1-Cmoel .................................... 61
Séquençage de I'intron CrpsbA4 ..................................................................... 66
Activité et affinité pour le substrat du mutant H 1 H A ...................................... 67
CHAPITRE IV
DISCUSSION ................................................................................................................... 73
CHAPITRE V
S F f RENCES ............................................................................................................ 85

LISTES DES FIGURES
................................................. Figure 1 . Structures secondaires des introns du groupe 1 2
................................. Figure 2 Mécanisme de propagation des endonucléases "homing" 6
Figure 3 . Structure tridimensionnelle de I'endonucléase "homing" à motif LAGLIDADG
.. 1-Crd ...........-..................*....................................................................................... 10
.................................. Figure 4 . Alignement des séquences de protéines à motif H-N-H IJ
Figure 5 . Séquence et structure du motif H-N-H de la colicine E9 ................................ 18
. ............*......... Figure 6 . Organisation du gène psbA chez C nigmetos et C . moawu.sii 20
Figure 7 . Structure et séquence du vecteur pEï-30a ...................................................... 25
Figure 8 . Étapes de purification de I.Cmoel ................................................................. U
..................... Figum 9 . Clivage de 1-Cnioel à différentes concentrations d'entérokinase 47
. Figum 10 Détermination des conditions optimales d'activité pour I.Cmor1 ................. 33
Figum I l . Effet de la préincubation de 1-CrnoeI à 37OC dans le milieu réactionnel
optimisé dépourvu de substrat .................................................................................. 54 ...................................... . Figure 12 Stechiométne de liaison de 1-Cmoel à son substrat 56
. Figure 13 Essais de retardement du substrat en présence de Mn" et dfEDTA .............. 59
. Figure 14 Relâchement des produits de coupure par 1-CmoeI ....................................... 62
. Figure 15 Détermination des paramètres cinétiques de 1-Cmoel ................................... 6J Figure 16 . Structure secondaire de CrpsbA.4 et séquence en acides aminés de la protéine
................................................................................................... codée par cet intron 68
Figure 17 . Analyse de la catalyse et du retardement du substrat par le mutant H141A .. 71

LISTE DES TABLEAUX
Tableau 1. Répertoire de toutes les endonucléases " homing " découvertes jusqu'à
présent.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .9

LISTE DES ABFLÉMATIONS
ADN: Acide désoxyribonucléique
ARN: Acide ribonucléique
A N , : ARN messager
BSA: Bovine Serum Alburnin
CrnpsbA4: Premier intron du gène chloroplastique psbA de l'algue Chfamydornonus rnoe wtrsii
CrpsbAul: Quatrième intron du gène chloroplastique psbA de l'algue Chlam~dornonus reinhardii
[ Y - ~ ~ P ~ ~ A T P : Désoxyadénosine triphosphate. marquée en position y à l'aide de l'isotope radioactif de l'atome de phosphore
[~-"P]~CTP: Désoxycytosine triphosphate. marquée en position a à l'aide de l'isotope radioactif de l'atome de phosphore
dNTPs: Désoxyri bonucléotides
D m : Di thothréi tol
IPTG: Isopropy 1- beta-D-thiogalactopyranoside
M: Molaire
min: minute
ORF: Cadre de lecture ouvert (de l'anglais Open Reading Fracne)
pb: paire( s) de bases
PCR: Polymerase Chain Reaction
psbA: Gène de la protéine de 32 kDa de la membrane thylakoïde
SDS: Sodium Dodecy lsulfate
SDS-PAGE: Polyacrylarnide gel elecirophoresis in the presence of SDS (Blectrophorèse sur gel de polyacrylamide dénaturant en présence de SDS)

INTRODUCTION
Chez tous les organismes vivants, I'information nécessaire pour coder les
protéines se retrouve dans les chromosomes sous forme de gènes. L'organisation de ces
&es varie selon l'origine des organismes. Chez les procaryotes. les gènes sont
habituellement continus. alors que les séquences codant pour les protéines eucaryotes
sont souvent interrompues par des séquences d'ADN supplémentaires nommées introns.
Lon de la transcription des gènes eucaryotes en ARN messager (ARN,), les introns sont
retirés par un ensemble d'événements très précis. I'épissage. Les mécanismes d'épissage
dépendent du type d'intron à retirer du transcrit primaire d'ARN,. Tous les introns
connus jusqutà maintenant peuvent être regroupés en quatre catégories: (1) les introos
insérés dans les gènes des ARN de transfert des archaebactéries (Lykke-Andersen et al.,
199'7). (2) les introns intercalés dans les ARN messagers nucléaires (Guthrie. 1991). (3)
les introns du groupe 1, et (4) les introns du groupe 11 (Cech et Bass. 1986).
1.1 Les introns des gmopes 1 et ï I
Les introns du groupe I présentent plusieurs caractéristiques communes. D'abord.
leur propre épissage se fait par une série de réactions de transestdrifications initiées par
une guanosine (Cech. 1990). Ensuite. ces introns partagent peu d'homologie de
séquence, mais démontrent une conservation au niveau de leun structures secondaires et
tertiaires. Ces stmctures elles-mêmes permettent même à quelques introns du groupe 1 de
s'autoépisser sans nécessiter l'intervention de protéines. Les structures secondaires des
introos du groupe I se caractdrisent par la présence d'une succession d'hélices. nommées
Pl à P10, qui comprennent les élCments de séquences conservées P. Q, R et S (Burke et
al., 1987) (Figure 1A). La région centrale formée des hélices P4, P6 et P7 est
responsable de la structure caractdristique des introns du groupe 1. Certains introns du
groupe 1 peuvent être classifies en sous-groupes par la présence d'une ou deux hélices
supplémentaires entre les éléments R et P3 (figure 1B).

Figure 1. Structures secondaires des introns du groupe 1. Les positions des éléments
de séquences conservées P, Q, R et S sont indiqutes. Les hélices (Pl-HO) de
même que les boucles (Ll-L9) sont désignées selon la nomenclature de Burke
et al. (1987). Les flèches montrent les sites d'dpissage 5' et 3'. Les boîtes
désignent les exons. A) Introns du groupe 1. B) Introns du sous-groupe M.
(Figure tirée de Burke et al., 1987).


Les introns du groupe II se différencient des introns du groupe I par leur structure
secondaire à six domaines et par la formation. lors de I'épissage, d'un lasso intronique
semblable à celui des introns des AM4 messagers nucléaires. Ces deux classes d'introns
partagent toutefois une propriété distinctive: elles contiennent souvent des cadres de
lecture ouverts (ORF) codant pour des protéines basiques (Perlman et Butow. 1989).
Dans le cas des introns du groupe 1, les cadres de lecture ouverts se retrouvent
principalement dans les grandes boucles s'éloignant du noyau de la structure secondaire.
Les protéines introniques sont entre autres responsables de la mobilité de certains introns
du groupe 1 et du groupe II.
1.2 La mobilité des introns do groupe 1
La découverte de protéines codées par des cadres de lecture iatroniques a amené
plusieurs éclaircissements quant a la mobilité des introns du groupe 1 lors de croisements
génétiques (Perlman et Butow, 1989, Mueller, et al.. 1993). Ce phénomène entraîne le
tmsfert d'un intron d'un allèle intron' (possédant un intron) à un allèle intron' (dépourvu
d'un intron). Cette conversion génique unidirectionnelle est très efficace et spécifique.
mais elle occasionne parfois la coconversion des séquences avoisinantes. La mobilité
d'introns est généralement initiée par l'action d'une ADN endonuciéase intronique qui
introduit spécifiquement une coupure double brin dans l'allèle dépourvu d'intron.
L'insertion de I'intron au site de coupure entraîne par le fait même la disparition du site
reconnu par 1 'endonucléase. Le processus d'intégration des introns mobiles initié par les
endonucléases à site spécifique a ét6 désigné par le terme anglais "homing".
1.3 Les endonueMases " h o h g "
La première endonuclbase "homing" rapportde dans la littérature provient de la
levure Saccharomyces cerevisiae. L'intron codant pour cette protéine, nommé W, se situe
dans le gène rnitochondrial de la grande sous-unit6 de I'ARN ribosomique (21s)
(Colleaux et al., 1%). Depuis, les endonucl6ases "homing" ont kt6 identifiées chez les
trois règnes biologiques: archaebactéries, eubactéries et eucaryotes. Chez ces derniers,

elles peuvent être codées autant par le génome nucléaire que par celui des organites. Les
gènes des endonucléases "homing" sont surtout rencontrés dans des introns mais
également dans des séquences codant pour des intéines. c'est-à-dire des séquences codant
pour des précurseurs protéiques à partir desquels sont excisées des endonucléases
"homing" (Belfort et Perlman, 1993, ou encore dans des gènes indépendants. Toutes les
endonucléases "homing" reconnaissent les allèles dépourvus de leur propre élément
génétique et y pratiquent une coupure double brin spécifique qui amorce le mécanisme de
"homing" (Lambowitz et Belfort, 1993). Ce clivage stimule des événements de
recombinaison homologue qui résultent en I'insertion au site de coupure de l'élément
génétique codant pour l'endonucléase (Figure 2) (Belfort et Roberts. 1997). Les
endonucléases "homing" tirent donc leur appellation de leur rôle qui semble directement
relié à leur propre propagation.
Tout comme les enzymes de restriction de type II. par exemple EcoRV et BamHI,
les endonucléases "homing" clivent ['ADN à un site précis et elles requièrent l'ion
magnésium comme cofacteur. Ces deux types d'enzymes diffèrent toutefois quant aux
propriétés de leur site de reconnaissance. de leur rôle, de leur distribution phylogénétique
et de leur classification. D'abord, les endonucléases "homing" recoonaissent de longues
séquences dégénérées d'ADN de 12 à JO paires de bases (pb) comparativement aux
séquences symétriques de 3 à 8 pb reconnues par les enzymes de restriction. Ensuite. le
rôle des endonucléases "homing" concerne leur propre propagation et la conservation des
introns, alors que celui des enzymes de restriction assure la protection de l'hôte en
reconnaissant et clivant l'ADN étranger. La distribution de ces deux types
d'endonucléases est différente puisque les enzymes de restriction sont limitées aux
bactéries et à quelques virus eucaryotiques (Mueller et al., 1993). Finalement, les
enzymes de restriction peuvent difficilement être regroupées en familles selon leur
séquence d'acides aminés (mis à part le motif PDX,,&UD)XK qui est parfois relié à la
fonction endoaucléase de ces enzymes), alors que les endonucldases "homing" peuvent
être classées en quatre grandes familles selon la présence d'un des motifs d'acides aminés
suivants: LAGLiDADG, GIY-YIG. His-Cys box et H-N-H (Belfort et Perlrnan, 1995).

Fignrc 2. Mécanisme de propagation des endonucldases "homing". (A) Lors de
croisements génbtiques entre souches introns* et intron', les endonucléases
"homing", ici exprimees il partir d'un intron ou d'une intéine, recomaissent une
longue séquence d'ADN correspondant à l'allèle d6pourvu d'intron ou d'intéine.
Elles introduisent une coupure double brin qui est suivie d'un évdnement de
recombinaison homologue. (B) Le tout rdsulte en l'intégration spdcifique au
site de coupure de l'intron contenant le cadre de lecture ouvert codant pour
l'endonucléase. De cette manière, les endonucléases "homing" sont
responsables de 1 eur propre propagation. (Figure tirée de Belfort et Roberts,
lm.

INfAON PLUS INtRON OR IMElN INTEIN PLUS M I W
IIL b
b d INTEIN
A L 4
I - - C r &
i I
INTRON Ofi lMElN HOMlNG

1.4 Les endoniicléases " homing" de ia famille LAGLIDADG
La majorité des endonucléases "homing" identifiées jusqu'à maintenant font partie
de la famille à motif LAGLIDADG (Tableau 1) (Belfort et Roberts, 19Y7). Cette famille
compte aussi dans ses rangs des intéines, des maturases rnitochoodriales (Wanng et ai.,
1982) et des protéines codées par des gènes indépendants nucléaires (Kostriken et al.,
198)). Les protéines à motif LAGLIDADG comportent habituellement deux copies du
motif qui s'étendent sur une région d'environ 110 acides aminés. Seules des protéines
introniques chloroplastiques de certaines espèces de Chlamydomonai ne contiennent
qu'une seule copie du motif LAGLIDADG (Belfort et Roberts, 1997).
Des études récentes de cristallographie ont démontré l'implication du motif
LAGLIDADG dans plusieurs aspects de l'activité de deux endonucléases de cette famille.
D'abord, 1-CreI. une endonucléase intronique chloroplastique de Chlanqdomonus
reinhardtii, est composée de deux sous-unités identiques (Heath er al.. 1997; Jurica et al.,
1998). Chaque sous-unité comporte une copie du motif LAGLlDADG qui forme la
majorité de l'interface de dimérisation de la protéine. Lorsque I'enzyme est dimérisée, un
résidu aspartate du motif de chaque monomère se positionne correctement pour former le
site catalytique (Figure 3). Ainsi, le motif LAGLIDADG serait impliqué dans la
dimérisation de la protéine et dans la transformation du substrat.
L'hypothèse de l'implication du motif LAGLIDADG dans la catalyse est soutenue
par des études de mutagenèse et de cristallopenèse de I'endonucléase rnitochondriale
intronique PI-Scel (Gimble et S tephens, 1995: Duan et al., 1997). Cette endonucléase
"homing" à double motif LAGLIDADG, originant de la levure S. ceverisiae. est un
monomère formé de deux domaines: un domaine d'épissage protéique et I'autre
responsable de la fonction endonucldase. Dans ce dernier domaine, les deux copies du
motif LAGLIDADG forment l'axe de symétrie du monomère comme c'est le cas pour le
dimère 1-CreI. Des expériences de mutagenèse dirigée sur le second résidu aspartate de
chaque motif LAGLIDADG ont confirmé l'importance de ces deux résidus dans l'activité
de PI-Scel. Lorsque substitués par un résidu alanine la fonction endonucléase fut totale-

Tableau 1. Répertoire de toutes les endonucléases "homing" découvertes jusqu'8 présent
(tiré de Belfort et Roberts, lm).
Nom Orgtatùsmd Rhgneb LucalïSOfiOn Intronc iUotifd
1-Anii 1-CeuI 1-Chu1 1-CmoeI I-CpaI 1-CpaIl 1-CreI 1-CsmI Mir1 1-Dm01 1-HmuI 1-HmuII 1- LlaI' 1-NuaI 1- P d 1- PpoI 1-ScaI 1-SceI 1-SceII 1-SceIII 1-SceIV 1-SceVe 1-SceVI' 1-SceVII 1-Tevt 1-TevII 1-TevIII PI -PspI PI-SceI PI- Tb1 PI-TliII F-Scei F-SceII F-SUVI F-Tm1
A. nidulans E C. cügwnetos E C. humicoia E C. moewusii E C. pallidostigmutica E C. pallidostigmaiica E C. reinhardtii E C. smithii E Di. iridis E De. mobilis A B. subtilis phage SPOl B B. subtilis phage SP82 B L. lactis B N. andersoni sp. E Pyb. organotrophum A Ph. polycepholum E S. capensis E S. cerevisiae E S. cerevisiae E S. cerevisiae E S. cerevisiae E S. cerevisiae E S. cerevisiae E S. cerevisiae E E. coli phage T4 B E. coli phage T4 B E. coli phage RB3 B Pyc. sp. GB-D A S. cerevisiae A T. Iitoralis A T. iitoralis A S. cerevisiae E S. cerevisiae E S. uvarum E E. coli phage T4 B
Mitochondrie Chloroplaste Chloroplaste Chloroplaste Chloroplaste Chloroplaste Chloroplaste Mitochondrie Noyau Chromosome Phage Phage Chromosome Noyau Chromosome Noyau Mitochondrie Mi tochondne Mi tochondne Mitochondrie Mitochondrie Mitochondrie Mitochondne Mitochondrie Phage Phage Phage Chromosome Chromosome Chromosome Chromosome Mi tocbondne Noyau Mi tochondne Phage
MGWDADG (2) LAGWrDADG ( 1 ) LAGLIDADG (2) H-N-H LAGLIDADG (1) LAGLIDADG (2) MGLIDADG (1) LAGLIDADG (2) His-Cys Box LAGLIDADG (2) H-N-H H-N-H H-N-H His-Cys Box LAGLIDADG (2) His-Cys Box LAGLIDADG (2) LAGLIDADG (2) LAGLIDADG (2) LAGLIDADG (2) LAGLIDADG (2) H-N-H H-N-H LAGLIDADG (2) GIY -Y IG GIY -Y IG H-N-H LAGLIDADG (2) LAGLIDADG (2) LAGLIDADG (2) LAGLIDADG (2) LAGLIDADG (2) LAGLIDADG (2) LAGLIDADG (2) GIY-YIG
F-Tm11 E. coli phage T4 B Phage - GIY -Y IG a A, Aspergillus; B, Bacillus; C, Chlamydomonus; Di, Didymiwn; De, Desulfurococcus; E,
Escherichia; L, Lacrococcus; N, Naegleria; Ph. Physarum; Pyb. Pyrobaculum; o c , Pyrococcus; S. Succharomyces; T, Thermococcus. A, Archaebactéries; B, Eubactéries; E. Eucaryotes. Type d'intron: 1, groupe 1; II, groupe II; A, Archaebacttnes
* ( 1), simple motif LAGLIDADG; (2), double motif LAGLIDADG ' Endonucléase formant un complexe ribonucléoprotéine avec son ARN intronique

F i i r e 3. Structure tridimensionnelle de l'endonucléase "horning" A motif LAGLIDADG
1-CreI. Sur cette vue, l'enzyme est positionnée de manière à reconnaître un
substrat drADN qui serait parallèle au plan de la feuille. Sa forme étendue lui
permet de se lier aux 19 A 24 paires de bases de son site de reconnaissance.
L'encadrd montre l'emplacement des motifs LAGLIDADG qui se situent à
l'interface de dimérisation des deux sous-unités. La figure montre aussi la
position des rtsidus aspartate 20, dsidus importants dans l'activite de cette
enzyme. (Figure tir6e de Belfort et Roberts, 1997).


ment abolie (Gimble et Stephens, 1995). De plus. les données cnstallographique ont
révélé que les résidus aspartate étaient associés à la coordination du Mg', ion essentiel à
l'activité de PI-Scel (Duan et al., 1997). Ainsi, deux copies du motif LAGLIDADG
sont nécessaires afin d'assurer le clivage efficace du substrat. Ceci s'applique sans aucun
doute pour les endonucléases "homing" n'ayant qu'une copie du motif puisque celles-ci
existent toutes sous forme de dimères. Dans cet ordre d'idée. il a été proposé que les
endonucléases "homing" à double motif LAGLIDADG ont évolué à partir de protéines à
simple motif suite à un événement de duplication génétique (Belfort et Roberts, 1997).
1.5 Les motifs GIY-YIG et His-Cys Box
Ces deux familles d'endonucléases "homing" sont moins bien connues, mais elles
possèdent tout de même leurs propres caractéristiques particulières. D'abord. le motif
GIY-YIG est caractérisé par une série de dix ou onze résidus entre les deux triades du
motif consensus ainsi que par une répétition d'acides aminés aux limites du motif
(Cummings ef al., 1989). L'endonucléase 1-TevI. une protéine de 28 kDa codde par
I'intron td du phage T4. fait partie de cette famille. Elle se lie à son substrat sous forme
d'un monomère (Mueller et al., 1995). Son site de reconnaissance est un des plus longs
chez les endonucléases "homing": il s'étend de 35 à 37 pb (Bryk et al.. 1995). Le site de
clivage de cette enzyme n'est pas adjacent au site d'insertion de I'intron qui l'encode. ce
dernier site étant sépare par plus de 23-25 pb du site de clivage (Bell-Peterson er al.,
1991). L'enzyme de 28 kDa fut modélisée en deux domaines unis par une région
charnière flexible: un domaine pour la reco~aissance du site de reconnaissance et l'autre
pour le clivage (Bryk et al., 1995). Le motif GIY-Y IG se retrouverait en N-terminal, soit
dans le domaine catalytique (Derbyshire et al., 1997).
Quant au motif His-Cys Box, il couvre une région d'environ 100 acides aminés
qui est particulièrement riche en résidus histidine et cystéine formant un site de
coordination d'ions mbtailiques (Johansen et al., 1993). 1-401, I'endonucléase "homing"
à motif His-Cys Box la plus étudiée, provient de la moisissure unicellulaire Physmum
polycephahm (Muscarella et al, 1990). La structure tridirnensiomeile de cette enzyme a

récemment été publiée (Fiick et al.. 1998). Cette enzyme est relativement petite (18 à 20
kDa) et se lie à son substrat sous forme de dimère. De plus, 1 - 4 0 1 possède un site de
liaison du zinc formé via des résidus histidine et cystéine qui sont conservés chez toutes
les endonucléases "homing" de la famille His-Cys Box. Chaque monomère possède un
site de coordination du zinc. ce qui veut dire qu'il y a deux cations de zinc lorsque
l'enzyme est liée à son substrat. Ces cations de zinc coordonnés par le motif His-Cys box
seraient impliqués dans la stabilisation de la structure de la protéine plutôt que dans la
fixation à l'ADN, comme c'est le cas pour d'autres protéines contenant deux cations de
zinc qui interagissent avec l'ADN. comme les récepteurs stéroïdiens. le facteur de
transcription GALA et le motif RING finger (Flick et al.. 1998).
1.6 Les endonucl4ases " homing" de la famüle à motif H-N-H
La famille à motif H-N-H regroupe des protéines d'origines très diversifiées. Ce
motif a été identifié grâce à l'alignement de sdquences de protéines codées par des introns
du groupe 1 et du groupe 11. Ceci constitue un des premiers rapprochements entre les
protéines codées par ces deux classes d'introns (Shub et al.. 1994: Gorbalenya, 1994). La
famille à motif H-N-H fut découverte en comparant la séquence d'une endonucléase
codée par un intron du groupe 1 (1-TevllI) avec d'autres séquences protéiques. Une
similitude fut établie entre un segment de 1-TevIII et un domaine semblable à un doigt de
zinc d'une protéine de cyanobactérie codée par un intron du groupe II. En poussant les
comparaisons, plusieurs autres protéines s'ajoutèrent dont des protéines introniques de
bactériophages. des endonucléases bactériennes et des protéines introniques
chloroplastiques (S hub et al., 1994).
La séquence consensus tirée de l'alignement de toutes ces proteines couvre 30 ii
33 acides aminés et comporte une paire de résidus histidine de part et d'autre d'un résidu
asparagine (Figure 4) (Shub et al., 1994). Peu de choses sont connues des endonucléases
introniques de cette famille, mises part quelques propriétbs distinctives de quelques
unes d'entre elles. Par exemple, les deux enzymes du phage de Bacillus sub~ilis, 1-HmuI
et 1-HmuII, clivent seulement un des deux brins d'ADN autant aux cibles intron' qu'in-

Figure 4. Alignement des séquences de protéines à motif H-N-H. L'encadré montre
l'emplacement de la séquence consensus de ce motif. Les rdsidus conformes P
la séquence consensus sont représentés en blanc sur un fond noir. Les résidus
conserv6s à plus de 50% de toutes les skquences sont illustres en lettres
minuscules. alors que ceux conservés 2 plus de 75% sont illustrts en
majuscules. Les chiffres entre accolades indiquent le nombre de résidus
précedant la séquence alignée. La flèche indique la sdquence d'acides aminés
de l'endonucléase 1-CmoeI. Les résidus Cys proposés pour former un motif
similaire a des doigts de zinc sont encadrés. Les numeros d'accès aux bases de
données sont préckdés d'un A ou S s'ils sont de PIR; d'un P s'ils sont de
SWISS-PROT et d'un X s'il sont de EM8UGenbank. Abréviations: Bact.,
Eubactérie; Chir., Chloroplaste; Col., Colicine; endo., endonucieaie; gr..
groupe; Ph., Phage. (Figure tirde de Shub et al., 1994).

A& Na A M 7 7 [q HA L VA I HFCEGYEE* - - - = G X87W [ S ] H R L V A L A F L C R P P G - * - - - K SlOOQ7 ~ Y R K I W I D A N G P I P K D S Q G R T snoto pl K K ~ S E [ ~ H L M F T S E D - - - - i Pn1327 [4tJ H A W V W E O H K G P I P D - - * - * G PO3797 [Id] H I a VWEAANGPIPK- - - - G P03t06 [2t] H A H I Y E E T Y G P V P T - - - - - G PO3785 [a] GK V V Y C HRVMSNAP* . - K G Pa7074 [q YS N I V N SAEHPSKPROTKAG
Pûût53 11291 KN A YN OF I LKRLNOPVT GKU POO79 ( 1 7 ] R E O Y S A D R N S P Q Y N - - - - - L -1 [7i4] VS GL A PYAVPEEHLGSKEK P1WO [24] WV&WIOSREFYYSN- - L R E Pi= [q L I E KGLNAGNOKSL--LTYY Pa#sz [ta7 1 S Q G t A PAARNKOTVGGRRS P W W [140] VS K GY S P F T PKNWVGGRKV PûU19 [SZS] t Q K GU A PFAAKKDQVGGREA Sa453 [526]UKVGKAPKTRTOOVSGKRTS

tron' (Goodrich-Blair et Shub, 19%) et l'endonucléase intronique du phage T4, 1-TevIII,
est la seule endonucléase "homingn qui clive l'ADN double brin en laissant des
extensions 5' au lieu des extensions 3' habituelles (Eddy et Gold, 1991). Aussi, les
endonucléases codées par des introns du groupe 11 rnitochondriaux de levure, 1-SceV et 1-
SceVI, forment des complexes ribonucléoprotéines avec leurs ARN introniques
respectifs. Les deux composantes des complexes sont impliquées dans le clivage de leur
substrat puisqu'en mutant des résidus du motif H-N-H. le clivage d'un des deux brins
d'ADN est bloqué. alors que l'autre brin est tout de même clivé par la composante d'ARP4
(Zimmerly et al., 1995). En ce qui concerne les protéines introniques chloroplastiques de
cette famille. Ia fonction endonucléase de celles-ci reste encore à être étudiée en
profondeur.
La majorité de nos connaissances sur les protéines à motif H-N-H provient des
études de la colicine E9, une endonucléase bactérienne de cette famille. Les travaux
enzyrnologiques effectués sur le domaine DNase de la colicine E9 ont, entre autres.
montré que celui-ci est autant actif en substituant le cofacteur Mg" par des métaux de
traIisitioo (M", Co", ~ n " ) et est même plus actif en prCsence de Ni" (Pommer el al..
1998). Plus récemment, la structure tridimensionnelle du domaine DNase de la colicine
E9 en complexe avec sa protéine d'immunité spécifique (Im9) a été publiée (Kleanthous
et al., 1999). Les données cnstallographiques indiquent que la protéine d'immunité ne se
lie pas au site actif du domaine DNase mais en un site adjacent qui empêche tout de
même la liaison de la protéine à l'ADN et qu'un cation Ni" fait partie du site actif du
domaine endonucléase. En alignant la séquence des résidus du motif H-N-H du domaine
DNase de la colicine E9 avec la séquence consensus de ce motif, les auteurs ont pu
proposer, à l'aide des données cristallographiques. un rôle pour chacun des acides amines
formant le motif H-N-H (Figure 5). Ainsi, le premier résidu histidine du motif serait
impliqué dans la liaison d'un groupement phosphate de l'ADN, l'asparagine stabiliserait
les structures des intermédiaires de réaction lors de la catalyse et finalement, le second
résidu histidine serait le ligand de cations métalliques (Kleanthous et al., 1999).
D'ailleurs, une étude antérieure avait mis en évidence l'implication de ce dernier dans
I'activitd de la colicine E9. Eu mutant cet acide aminé par le résidu neutre alanine,

aucune activité endonucléase n'avait été observée quoique le mutant H575A avait
conservé la capacité de se lier à son site de reconnaissance (Garinot-Schneider et al.,
1 9%).
1.7 La ptothhe à motif B N - H de C h h m y d o m o ~ s moewusii
Certains introns de l'ADN chloroplastique d'algues vertes du genre
Chlumydomonus présentent le phénomène de "homing". C'est le cas d'un intron du
groupe 1 de Chlamydomonar eugametos qui, lors de croisements génétiques entre cette
espèce et l'espèce interféconde C. moewusii, se comporte comme un élément mobile
(Gauthier et al.. 199 1 j. Cet intron. le cinquième du gène de la grande sous-unité dtARN
ribosomique de C. eugametos, est absent dans le même gène chez C. moewusii (Lemieux
et Lee, 1987). 11 contient dans sa séquence un cadre de lecture ouvert codant pour une
protéine à caractère basique de 218 résidus d'acides aminés (Tunnel et al., 1991). Cette
protéine, une endonucléase "homing" nommée I-CeuI, initie le transfert unidirectionnel
de I'intron en clivant I'allèle sans intron du gène de C. moewusii (Gauthier et al.. 1991;
Marshall et Lemieux, 199 1).
Deux autres séquences d'ADN chloroplastique se comportent comme des éléments
mobiles lors de croisements entre ces deux espèces d'algues vertes. II s'agit de segments
d'ADN de 6 et 21 kb présents seulement chez C h l ~ d o r n o n r r s moewusii respectivement
dans les gènes structuraux petD et psbA (Bergeron, 1990; Boulanger. 1988: Lemieux er
al. 1 5 ) . Le gène psbA de C. eugrnetos est continu, dors que chez C. moewusii, il est
composé de trois exons et de deux introns du groupe I de 2.4 et 1.8 kb (Figure 6)
(Boulanger, 1988). 11 a été deteminé que les exons 2 et 3 ainsi que les deux introns font
partie de I'additioddélétion de 21 kb (Tunnel et al., 1989). Chacun de ces introns
contient un cadre de lecture ouvert codant potentiellement pour une protéine de plus de
300 acides aminés (intron 1: 345 a.a.; intron 2: 3 12 a.a.) (Turmel et al., 1989). Ces deux
proidines introniques ont subséquemment été classées dans la famille d'endonucléases
"homing" à motif H-N-H (Shub et al., 1994). D'autre part, le premier intron du

F i i r e 5. Séquence et structure du motif H-N-H de la colicine E9. A) Alignement de
la séquence consensus du motif H-N-H avec la région correspondante du
domaine DNase de la colicine E9. Les rôles respectifs des résidus du motif
conservé sont indiqués. B) Vue en stér6o du motif H-N-H du domaine DNase
de la colicine E9 montrant la position des acides amints importants (Hisl03,
As11118 et Hisl27), de l'ion nickel et de la molCcule de phosphate. (Figure tirée
de Kleanthous et al., 1999).

Phosphate 2" structure Ml' ligand binding stabilisation

Fiiure 6. Organisation du gene psbA chez C. eugametos et C. moewusii. Les régions
noires représentent les exons du gène (E) et les sections grises désignent les
sdquences homologues du gène entre les deux espèces d'algues. En blanc sont
montrés les introns et en hachur6 sont les cadres de lecture introniques. Le
gène est continu chez C. eugametos, alors que chez C. m o e w i i . il est
interrompu par deux introns du groupe 1 qui font partie de I'addition/d6l&ion de
21 kb. (Figure obtenue de Monique Tunnel).

C. eugametos psbA
1 kbp

gène psbA de C. rnoewusii (CmpsbA.1) est inséré à la même position que le quatrième
intron de ce gène chez C. reinhortdii.
Lon d'une étude antérieure réalisée dans notre laboratoire, la protéine codée par
l'intron CmpsbA.1 avait été synthétisée in vitro à partir d'un lysat de réticulocytes de
lapin. Son activité endonucléase avait été démontrée en utilisant comme substrat la
version sans intron du gène psbA de l'ADN chloropiastique de la souche d'algue
interféconde C. eugametos, le clivage double brin de ce substrat d'ADN générant une
extension de quatre nucléotides hydroxylée en 3' (Côté et Tunnel. résultats non- publiés).
La protéine codée par le second intron s'était quant à elle avérée inactive. À la manière
de 1-Ceul, la protéine intronique à activité endonucléase pourrait être l'élément
déclencheur des événements de conversion génique à la base de mobilité de I'intron qui
l'encode et du reste du segment d'ADN constituant I1addition/délétion de 2 1 kb (Bussières
et al.. 19%). La mobilité d'une séquence aussi longue est quelque peu contradictoire au
concept de " boming", mais pourrait être expliquée par l'absence de recombinaison
homologue entre les exons 2 et 3 de C. moewusii et les séquences correspondantes de C.
eugametos. Les exons 2 et 3 seraient trop courts pour permettre la recombinaison
homologue (34 et 272 pb respectivement) (Tunnel et al., 1989) ou encore leurs séquences
seraient trop divergentes de celles correspondantes chez C. eugametos (Bussières et al..
19%).
1.8 Probiématique de l'étude
L'intérêt pour les recherches portant sur les endonucléases "homing" provient du
fait que la plupart de ces enzymes sont responsables d'un phénomène très répandu dans le
monde biologique: la mobilitd d'introns. De plus, ces enzymes sont prisées pour leurs
interactions particulières avec l'ADN. Malgré le nombre croissant d'études sur des
endonucléases "homing" de la famille à motif LAGLIDADG, peu de choses sont connues
du fonctionnement précis de ces endonucléases. Pour ce qui est des endonucléases
"homingn à motif H-N-H, nos connaissances sont encore plus limitées. Ainsi,
l'expression, la purification et la caract6risation d'une telle eodonucléase comportant ce

motif conservé constituerait la première étape de l'étude de cette famille d'endonucléases.
Au moment de débuter ce projet. l'étude de la fonction endonucléase d'une
protéine intronique à motif H-N-H représentait l'une des premières caractérisations de la
fonction endonucléase d'une protéine renfermant ce motif. La protéine codée par I'ORF
du premier intron du gène psbA de C. moewusii fut choisie puisque son activité
endonucléase avait déjà été démontrée et qu'elle comporte le motif H-N-H dans sa
séquence. Aussi, l'étude de cette protéine pourrait suggérer que celle-ci soit à la base de
la mobilité de I'additionMélétion de 21 kb. Afin de purifier et caractériser cette nouvelle
enzyme, nommée 1-Cmoei d'après les règles de nomenclature établies (Belfort et Roberts,
1997), nous avons introduit son gène dans le vecteur p E 3 0 a de Novagen (Figure 7) et
nous avons surexprimé la protéine recombinante dans la souche dlE. coli BL21(DW).
Celle-ci a été purifiée par chromatographie d'affinité sur résine de nickel et par
chromatographie d'éc hauge de cations.
En premier lieu. les conditions optimales d'activité de 1-CmoeI furent déterminées.
Pour ce faire, plusieurs essais enzymatiques furent effectués en utilisant comme substrat
d'ADN l'allèle sans intron provenant de la souche d'algue interféconde C. eugametos.
La stabilité de ['enzyme dans le milieu réactionnel optimisé fut par la suite examinée.
L'endonucléase fut caractérisée en ce qui concerne ses paramètres cinétiques et son
affinité pour I'ADN. La forme sous laquelle i-CmoeI se lie à son site de reconnaissance
fut également étudiée. La mutagenèse dirigée sur le premier résidu histidine du motif H-
N-H fut effectuée afin de démontrer l'importance de ce résidu dans l'activité de I-CmoeI.
Finalement, la séquence d'une endonucldase apparentée présente chez C. r e i n h d i i fut
déterminée dans le but d'obtenir plus d'information sur les résidus essentiels au
fonctionnement des endonucléases H-N-H.

MATÉRIELS ET MÉTHODES
2.1 Amplincation et clonage du gène codant pour 1-CmoeI dans le vatenr PET-
Ma
Le gène codant pour 1-CmoeI, c'est-à-dire I'ORF du premier intron du gène psbA.
fut d'abord amplifié par la réaction de polymérase en chaîne (PCR) à partir de l'ADN
chloroplastique de Chlurnydornonaî rnoewusii. Afin d'insérer les sites de restriction NcoI
et BmHI respectivement en 5' et en 3' de ce gène, les amorces suivantes furent utilisées:
psbAmut.Nco1: 5'-AAmAACAACAAATCCATGGCfCGTAAAAAAAflATAG-3' et
psbAmut.BamH1: 5'-CCATCïGTAGCITGGATCCGGATTGCCATATT'A-3' (les sites
de restriction respectifs sont soulignés). Une quantité de 400 ng de chacune de ces
amorces furent ajoutés à un volume final de 100 pl contenant 10 ng de gabarit d'ADN
chloroplastique de C. moewusii et 0,2 mM de dNTPs (Perkin Elmer). Après un "hot
start" de 5 minutes, l'amplification PCR fut déclenchée par l'ajout de 5 U de T q ADN
polymerase (Stratagene) dans le tampon recommandé par le fabricant. Les 30 cycles de
K R furent réglés ainsi: dénaturation A 94°C. 1 minute; fixation des amorces à 55°C. 1
minute: élongation à 72°C. 2 minutes. La réaction PCR fut terminée avec un cycle
supplémentaire de 2 minutes à 55°C et 25 minutes i 72°C.
Afin de rendre leurs extrémités compatibles. le gène codant pour 1-CmoeI et le
vecteur pET30a (Figure 7) furent successivement digérés par les enzymes de restriction
Ncol et BmHI. Des quantités de 600 ng de vecteur et 500 ng d'insertion furent
séparément incubés pendant une heure à 37°C avec 12,5 U de Ncol (Amersham
Pharmacia Biotech) dans le tampon foumi par le manufacturier. L'ADN de chaque
mélange réactionnel fut purifu5 par extraction au phénol-chloroforme, puis précipité à
l'éthanol 95%. Le lendemain, l'ADN fut centrifugé et resuspendu en vue d'être digéré par
7,5 U BamHl (Gibco B K ) à 37°C pendant 1,s heures. Par la suite, approximativement
50 ng du gène de 1-CmoeI ont ét6 ligaturés avec la même quantité (50 ng) du vecteur
PET-30a linéarisé à l'aide de 12-5 U de la T4 ADN Ligase (Gibco BRL) dans son tampon
Ligase (Gibco BRL) à 16OC pendant une nuit.

Fin! 7. Structure du vecteur pET30a. A) La carte physique du plasmide pET30a
présente les sites de restriction uniques, l'orientation et l'emplacement de
l'origine de réplication (on), de l'origine de replication du phage f 1 (f 1 ongin),
du gène du répresseur lad, du gène de résistance à l'antibiotique kanarnycine
(km) et du site de clonage multiple (flèche noire pleine). B) Sdquence
nuclkotidique et peptidique du site de clonage multiple. Les fltches montrent
l'emplacement du promoteur et de la region de terminaison de I'ARN
polymérase du phage T7, les traits fins indiquent les sites de restriction, et les
traits gras indiquent les séquences necessaires à l'expression. Les séquences
codant pour le His-Tag, le S-Tag ainsi que les sites de reconnaissance de
l'entérokinase et la thrombine sont aussi reprbsentés par des traits gras. La
num6rotation de la séquence est la même que celle convenue pour le plasmide
pBR322. (Figure tirée du site web de Novagen, www.novagen.corn).

BspLUi 1 I(m / Sap I(3t61) Bsîl 1 O7 l (30~) Tai111 l(30221

2.2 Transformation des ceiiules compétentes E. coli STBL2
Le mélange de ligature précédemment obtenu a été utilisé pour transformer des
cellules compétentes E. coli STBU (Life Technologies). La transformation a été
effectuée selon les recommandations du fabricant. Quelques-uns des transformants
obtenus furent utilisés pour inoculer des milieux de culture de type Luria-Bertani
contenant 30 pglrnl de kanamycine (ICN Biomedicals) (LB-km). Chaque culture fut
incubée à 30°C sous agitation constante (225 rpm) pendant une nuit Pour chacune des
cultures, 900 pl furent mélangés à 100 pl de glycérol avant d'être conservés à -70°C.
2.3 Vérif~cation de la séquence clonée
L'ADN plasmidique d'un des clones a ensuite été purifié à l'aide de la trousse
Qiaprep 8 (Qiagen) selon les indications de la compagnie. Ce plasmide, nommé pet30a-
ICmoeI, a été séquencé afin de s'assurer que le gène codant pour 1-Cmoel était depou r~u
de mutation. Un échantillon de 600 ng de ce plasmide fut séquencé par le Service de
Sdquençage du pavillon Charles-Eugène Marchand à l'aide de la trousse AB1 PrismTM
BigDye Terminator Cycle Sequencing Ready Reaction (PE Applied Biosystems).
2.4 Transformation des ceiides d'expression E. coli BLÎI(DE3)
Le plasmide pet30a-ICmoeI fut introduit dans la souche E. coli BL21(DE3)
(Novagen), souche qui contient dans son génome le gène de I'ARN polymérase du phage
T7 dont l'expression est sous le contrôle du promoteur lacUV5 La procedure de
transformation utilisée est celle transmise par la compagnie. Après la transformation. des
clones ont servi à inoculer des milieux de culture LB-km qui ont été incubés à 37OC. 225
rpm pendant une nuit. Des aliquotes de 900 pl des cultures bactériennes d'E. coli
BUl(DE3) transformées ont été mélangées a 100 pl de glycérol ûû% et conservées à -
70°C.

2.5 Expression de la protéine reeombmante
Une préculture de 10 ml dans le milieu LB-kan a été inoculée avec 20 pl de la
culture glycénnée dlE. coli BL21(DE3) et incubée à 30°C, 225 rpm pendant une nuit. Le
lendemain. la préculture de 10 ml fut utilisée comme inoculum pour une culture de LOO0
ml dans le milieu LB-kan. Ce bouillon de culture fut incubé à 37°C. 225 rpm jusqutà ce
que l'absorbante mesurée à 600 nm atteigne 0.6. De I'IPTG fut ajoute pour obtenir une
concentration finale de 1 rnM afin de permettre l'expression de I'ARN polymérase du
phage T7, sa fixation à son promoteur sur le plasmide pET30a et ainsi. la surexpression
d'une protéine recombinante possédant une queue de six résidus histidine (His-Tag) en N-
terminal. L'induction en présence dlIPTG s'est poursuivie durant 2.5 heures sous les
mêmes conditions. Les bactéries ont ensuite ét6 recueillies par centrifugation à 6000 rpm
pendant 7 minutes à 4'C dans le rotor GSA (Sorvall). Les culots des cultures
bactériennes ont été conservés à -70°C.
A moins d'avis contraire, toutes les étapes de la purification se sont déroulées sur
glace ou bien à CC.
2.6.1 Préparetion et lyse des extraits cellulavcs
Les culots de bactéries ont ét6 resuspendus dans 15 ml volume de tampon de lyse.
Le tampon de lyse était ainsi composk NaHzPO, 500 mM, NaHPO, 500 mM. pH 8,5,
NaCl 300 rnM. imidazole 10 mM et I mM de l'inhibiteur de protéase phényl-méthyl-
sulphonyl fluoride (PMSF).
La suspension de bacteries fut lysde par double passage à la Presse de French,
opérée à 20 000 PSI. Une série de deux centrifugations à 10 000 rpm pendant 15 minutes
à 4°C dans le rotor (Sorvdl) nous permit de nous débarrasser des debris cellulaires et des
proteines insolubles pour ainsi ne conserver que la fraction soluble du lysat.

2.6.2 Chmmatogmphie d'affinité sur résine de nickel
La fraction soluble du lysat cellulaire (environ 20 ml) fut ensuite mise en présence
d'une résine de Ni" nous permettant d e purifier la protéine recombinante par
chromatographie d'affinité puisque les groupements imidazole des acides aminés du His-
Tag ont une forte affinité pour le nickel. La fraction soluble fut mélangée à un volume de
4 ml de résine de Ni" Ni-NTA (Qiagen) sédimentée. Le mélange fut incubé à 4OC
pendant 1 heure avec légère agitation par inversion et fut par la suite transféré dans une
colonne jetable (BIO-RAD). Min de se débarrasser des protéines ne se fixant pas à la
résine ~ i " , la résine fut soumise à une série de trois lavages (NaHIPO, 500 rnM,
N+HPO, MO mM. pH 85. NaCl 300 mM, imidazole 20 mM. PMSF 1 mM). chacun des
lavages équivalant à dix volumes de résine sédimentée. Les protéines fixées a la résine
de Ni" furent éluées en effectuant deux lavages correspondant à deux volumes de résine
avec un tampon contenant une forte concentration d'imidazole (NaH,PO, 500 rnM.
N+HPO,MO rnM, pH 8.5, NaCl 300 mM, imidazole 250 mM, PMSF 1 mM).
2.6.3 Dialyse de I'eluat
L'éluat de la chromatographie d'affinité (= 16 ml) fut dialyse contre un tampon
HEPES-NaOH 50 m M pH 7.5 contenant du PMSF I m M dans des membranes à dialyse
Spectra/Por 1 à pores de 6000-8000 Da. L'échantillon fut d'abord dialysé pendant 3
heures à 4°C avec légère agitation dans 1 litre de tampon. puis à nouveau pendant la nuit
dans 1 litre du même tampon dans les mêmes conditions.
2.6.4 Chromatographie d'échange de cations sur résine Ceromid Hyper D
Une chromatographie d'dchange de cations sur une colonne AP-Minicolumn
(Waters) contenant 6,s ml de résine Ceramid Hyper D 50 prn (Amersham Pharmacia
Biotech) fut requise pour purifier efficacement les protéines recombinantes. Installée sur
un appareil AKTA Explorer dans le laboratoire de Dr L. Eltis, la colonne a d'abord étd
équilibrée avec un équivalent de cinq volumes de résine contenant la solution A (HEPES-
NaOH M m M pH 7.9, 10 volumes de fa solution B (solution A ajoutée de NaCl 1 M) et

finalement cinq volumes de la solution A. Ces opérations se sont déroulées à température
pièce à un débit de 4 Mmin par millilitres de résine. La solution de protéines fut alors
appliquée sur la colonne. Un lavage avec l'équivalent de deux volumes de résine
contenant la solution A fut effectué avant d'éluer les protéines fixées à la résine avec un
,-dient salin linéaire forme à partir des solutions A et B. Tout au long du gradient,
l'élution des protéines a été suivie en mesurant I'absorbance à 2&0 nm.
Les fractions de protCines recueillies ont été analysées par électrophorèse sur gel
dénaturant (SDS-PAGE). Celles correspondant à 1-CmoeI ont été regroupées et
concentrées en utilisant une cellule dtultrafiltration Amicon selon Ies recommandations
du fabricant. La solution de protéine ainsi obtenue a été dosée à I'aide de la trousse de
dosage MicroBCA (Pierce), puis elle fut conservée sous deux conditions: à -20°C dans
50% de glycérol et à -70°C avec au préalable un gel-choc à l'azote liquide.
2.7 SDS-PAGE
Des échantillons prélevés en cours de la purification ont été analysés par SDS-
PAGE afin d'évaluer l'efficacité des différentes étapes effectuées. Tous les gels étaient
composés de 12% de polyacrylamide et ont été réalisés selon la procédure établie par
Laemrnli (1970). En plus des échantillons, des marqueurs de poids moléculaire (SDS-
PAGE Molecular Weight Standards Broad Range de BIO-RAD) ont été déposés sur
chacun des gels. Après l'~lectrophorèse, les protéines ont été colorées en faisant d'abord
incuber les gels durant une heure dans une solution colorante (50% méthanol, 10% acide
acétique, 0,1% Coomassie Blue). Les gels ont par la suite été décolorés dans un solution
de méthanol 5% et d'acide acétique 10%. incubés durant une heure dans une solution
d'&han01 10% et glycérol 496, puis séchds entre deux couches de pellicule GelWrap
(BioDesign).
2.8 Clivage da His-Tag par l'entérokinase
Le clivage de la queue de six résidus histidine (His-Tag) située en N-terminal de
1-CmoeI a été tenté à l'aide de l'ent6rokinase. une protéase spécifique qui peut

théoriquement retirer ce His-Tag. Des échantillons de 10 pg de protéine recombinante
ont été mis en présence d'entérokinase (Novagen) pendant 16 heures à 18°C. selon le
même ratio de protéines-cibles : entérokinase recommandé par le fabricant. soit de 10 pg
: 0,2 U. Des clivages ont aussi été effectués avec diffirentes dilutions d'entérokinase
(0,02. 0,002, 0,0002 U) tout en maintenant la quantité de protéine recombinante à 10 pg.
Les réaction de clivage ont été déposées sur gel de polyacrylamide 12% et les protéines
ont été colorées au bleu de Coomassie tel que décrit dans la section précédente.
2.9 Optimisation de l'activité de 1-CrnoeI
Tous les essais enzymatiques d'optimisation ont été effectués avec une
concentration d'enzyme de 2,s nM et une concentration de substrat de 25 PM. Le substrat
d'ADN utilisé était un produit PCR de 274 pb amplifié à partir du gène psbA de C.
eugametos. Ce fragment d'ADN avait été marqué radioactivement par incorporation
uniforme de [ a - " P ] d n ~ lors de I'amplifkation PCR. 200 ng de chacune des amorces
(psbA#2 3-GTTACITCATCTITAATCCGT-3' et psbA#5 5'-
TTGAATCTACAACTGACTGGT-3') ont été ajoutés à 2 ng de préparation dtADNcp de
C. eugameros. Le tout a été placé dans un volume total de 25 ou 50 pi. dépendamment
du marquage radioactif effectué. soit respectivement une concentration de [a-33P]dCï~
représentant 1/25 ou 11 125 de la concentration de d m totale. Dans les deux cas. 25 pM
de dNTPs (Perkin Umer) furent utilisés en compagnie de 10 pCi de [a-j3PJdCTP
(Amenham) et de 2 5 U de l'ADN polymérase AmpliTizq Gold (Perkin Elmer) dans le
tampon fourni par le fabricant. Après avoir chauffé durant 10 minutes à 95°C afin
d'activer la polymérase. les 30 cycles de PCR furent réglés ainsi: d6naturatioa ii 940C. 1
minute: fixation des amorces a 62°C. 1 minute; élongation à 72OC. 2 minutes. La
rbaction PCR fut terminée par un cycle supplémentaire de 2 minutes 62°C et 25 minutes
à 72°C. Après l'amplification. les produits PCR ont étC purifiés par chromatographie sur
des colonnes Microspin S-400 HR (Amersharn Pharmacia Biotech). Le dosage des
produits PCR s'est fait sur gel de 3,5% d'agarose Metaphor (FMC BioRoducts) contenant
le tampon TBE (0,089 M Tris. 0,044 M Borate et 2 m M EDTA), à l'aide des marqueurs

Low DNA Mass Ladder (Gibco BRL).
Les essais enzymatiques se sont déroulés à 37OC sur 10 minutes dans des volumes
de 1300 pl avec des prises d'aliquotes de 200 pl après 1.2.5 et 10 minutes. Les réactions
ont été arrêtées par l'ajout dtEDTA pour obtenir une concentration finale de 1 rnM afin de
chélater les ions magnésium, de SDS à une concentration de 05% pour briser les
complexes ADN-protéine et de 100 pg de Rotéinase K (Boehnnger Mannheim). Chaque
mélange fut incubé pendant 60 minutes à >O°C. Après précipitation de l'ADN à l'éthanol
en présence de 20 kg de glycogène (Boehringer Mannheim) et de 0 3 M d'acétate
d'ammonium. chaque culot a été dissous en ajoutant 3 pl d'eau bidistiilée et 1 pl de
tampon de dépôt pour gels non-dénaturants (Ficoll JOO 8%. EDTA JO mM, bleu de
bromophénol 0.08%). Les échantillons ont été chauffés 5 minutes à 65°C. puis ont été
déposés sur un gel de polyacrylamide 5% (lx TBE) ayant préalablement subi une
préélectrophorèse de 20 minutes à 20 Vlcm à 25°C. Une fois les échantillons déposes sur
le gel. l'électrophorèse a été poursuivie à 20 Vlcm, pendant environ 2 heures et demie à
25°C. Après la migration. le gel a été incubé dans un mélange fixateur (éthanol IO%,
acide acétique 10 %) pendant 30 minutes à température pièce, puis séché dans un four
Pasteur à 18û°F pendant 2 heures. Le gel a ensuite été exposé contre une plaque de
BioImaging, permettant la quantification de la radioactivité émise par le substrat non-
clivé et les produits de coupure. La radioactivité a été révélée par "Phosphor Imagingt' A
I'aide d'un appareil Fujix Bas 1 0 (Fuji). Pour chaque essai cinétique. la proportion du
substrat non-clivé par rapport au substrat total a éte quantifiée sous le mode Profile du
logiciel MacBAS version 2.5 (Fuji) et a été mise en graphique en fonction du temps. On
constate alors une décroissance exponentielle du substrat non-clivé qui obkit 1'6quation
suivante lorsque [SI << [a:
oh [SI est la concentration de substrat au temps t, [SI, est la concentration de substrat
initiale (temps td et k la constante de pseudo premier ordre. Cene constante k a étk
déterminée pour chaque essai par "curve fittingl' de l'équation (1) à l'aide du logiciel

Kaleidagaph. Pour chaque paramètre d'optimisation, le maximum d'activité ( 100%) fut
attribué aux conditions associées avec la plus forte valeur de k. Les niveaux d'activité
obtenus pour les autres conditions fureat exprimées en fonction de cette valeur maximale.
2.9.1 Effet de lo nature et de la concentration do tampon de réaction
Pour vérifier l'effet de la nature et de la concentration du tampon sur l'activité de
I-CmoeI, des concentrations variant de 5 mM à 250 m M furent utilisées pour les tampons
Tris-HCI et Taps-KOH. tous deux à pH 8.5. Le milieu réactionnel contenait aussi 2.5
m M de MgCI,, 0.05 mghl d'aibumine sérique de boeuf (BSA) (Amenharn Phamacia
Biotech) et I mM de dithiothréitol (MT) (Gibco BRL). Les conditions enzymatiques et
les méthodes d'analyses sont celles décrites en 2.9.
2.9-2 Effet de la concentration du cofactenr Mg2*
L'ion magnésium a été ajouté aux réactions enzymatiques sous forme de sel de
chlore (MgCl?). Les concentrations de MgCl? testées s'étendent de O à 25 mM. Les
autres composantes du milieu réactionnel étaient les suivantes: TAPS-KOH 150 mM pH
8.5. BSA 0.05 mglml (Amenham Pharmacia Biotech) et D ï T 1 mM (Gibco BRL). Pour
déterminer si l'ion magnésium est nécessaire pour l'activité de 1-CmoeI. un échantillon de
500 pl de la préparation de protdine a été dialysé dans une microcassette Slide-A-Lyzer
MWCO 3500 Da (capacité 0.1-0.5 ml, Pierce) contre exactement le même tampon
contenu dans la préparation (HEPES-NaOH 50 m M pH 7.5)- mais auquel de I'EDTA fut
ajouté à une concentration finale de 1 mM. La dialyse a été réalisée à 4°C dans 500 ml
de tampon avec légère agitation pendant 3 heures et par la suite pendant la nuit dans 500
ml de tampon frais dans les mêmes conditions. Après avoir récupéré la protéine dialysde
à l'aide d'une seringue, un essai cinetique a été effectué en absence de MgC& mais en
présence de 1 mM EDTA sous les mêmes conditions enzymatiques que celles decrites
précédemment. Les méthodes d'analyses sont aussi les mêmes qu'en 2.9.

2.9*3 Effet de ia nature do cation divalent agissant comme cofacteur
Les neuf cations testés (Mg2+, Ca2+, Mn2+, Cu". NiZ', Zn", Ba", Co2+ et Si') ont
été employés un à un sous forme de sels de chlore à deux concentrations, soit 1 et 10
mM. Les essais ont été réalisés dans un tampon TRIS-HCI 5 rnM pH 83 contenant du
BSA à 0.05 m g / d (Amenham Pharmacia Biotech) et du DIT à 1 mM (Gibco BRL).
Les conditions enzymatiques et les méthodes d'analyses sont les mêmes que celles
décrites en 2.9.
2a9A Effet du pH du tampon
L'intervalle des pH testés s'étend de 7.5 à 11.0. Quatre tampons furent utilisés
pour couvrir cet intervalle afin de respecter les limites du pouvoir tampon de chacun des
tampons. Ceux-ci sont I'HEPPS-KOH (pH 7.5-8.0-8.3, TAPS-KOH (8.5-9.0), CHES-
KOH (9.0-9.1 10.0) et CAPS-KOH ( 10.0- 10.5- 1 1.0). La concentration des tampons était
fixée à 150 rnM avec 2.5 rnM de MgCI,, 0.05 m g / d de BSA (Amenham Pharmacia
Biotech) et 1 mM DTT' (Gibco BRL). Les conditions enzymatiques et les méthodes
d'analyses sont celles décrites en 2.9.
2.9.5 Effet de ia températore de riaetion
Les températures de réaction testées étaient 4. 10, 25. 37, 50 et 60°C. Tous les
essais enzymatiques ont ét6 effectués dans un bain chauffant à rdglage de température
digital TropiCooler. La composition du tampon utilisé était la suivante: Tris-HCI 5 m M à
pH 8.5, MgC1,2.5 rnM. BSA 0,05 mg/ml et DTï 1 mM. Les conditions enzymatiques et
les méthodes d'analyses sont celles décrites en 2.9.
29-6 Effet de la force ionique
L'effet de la force ionique sur l'activité de 1-CmoeI fut étudiC en ajoutant
différentes quantités d'un sel monovalent (NaCl et KCI) dans le milieu r6actiomel. La
concentration de sel variait de O à 120 mM. La composition du tampon utilisé était la
suivante: TAPS-KOH 10 m M pH 8,5, MgCl2 10 mM, BSA 0,01 mg/d et DIT 1 mM.

Les conditions enzymatiques et les méthodes d'analyses sont celles décrites en 2.9.
2.9.7 Effet du BSA et du DTT
Les concentrations suivantes du stabilisateur BSA furent examinées quant à leur
effet sur l'activité de 1-CmoeI: 0,01,0,05,0.1, 0.5 et 1 mg/ml. Pour ces essais, le tampon
TAPS-NaOH 10 rnM pH 8.5 contenant du MgCl, 10 m M et à DTT 1 rnM fut utilisé. Un
essai enzymatique séparé a permis de vérifier i'importance de la présence de l'agent
réducteur DTT dans le milieu réactionnel. Ceci fut accompli en utilisant un tampon
TAPS-NaOH 10 rnM pH 8,5 contenant du MgC12 10 mM et du BSA 0.05 mg/ml. Les
conditions enzymatiques et les méthodes d'analyses sont celles décrites en 2.9.
2.10 Stabilité de 1-CmoeI
Pour évaluer la stabilité de 1-CmoeI dans son milieu réactionnel optimise (TAPS-
KOH 10 mM pH 8.5. MgCl, 10 mM, BSA 0.01 mg/rnl et DTT 1 mM), la préparation
d'enzyme ( 15 nM) fut préincubée à 37OC dans le milieu réactionnel dCpourvu de substrat
en présence ou absence de 10 mM MgCI,. Après différents temps de preincubation (15 à
360 minutes), une aliquote d'enzyme fut prélevée (2.5 nM) et ajoutée au substrat PCR de
274 bp marqué (25 PM) afin de declencher une réaction enzymatique normale dans 1300
pl de mélange réactionnel. Les conditions enzymatiques et les méthodes d'analyses sont
celles décrites en 2.9,
2.11 Stœchiométrie de liaison de 1-Cmoei B son site de reconnaissance
Le substrat d'ADN a été incubé en absence ou en présence de 1-Cmoel dans un
milieu réactionnel de 20 pl contenant un tampon TAPS-KOH 10 mM pH 8-5, MgCl? 10
mM, BSA 1 mg/rnl, DTT' 1 mM, glycérol 10% et polydUdC 0.05 mglm1 (Amersham
Pharmacia Biotech). Les concentrations du substrat et de l'enzyme étaient tous deux de
10 nM. Les mélanges ont été incubés iî temp6rature pièce pendant 15 minutes et ont été
deposés sur des gels de polyacrylamide de differentes concentrations (6,7,8, 9 et 10%)
(TBE 0,îSC) afin de révéler la présence du complexe enzyme-substrat (ES). Sur chaque

p l , 5 pg des protéines standards de poids moléculaires (Sigma) ont été déposées de part
et d'autre des échantillons analysés. Les protéines standards utilisées étaient les
suivantes: a-lactalbumine ( l4,2 kDa); anhydrase carbonique (29,O kDa); albumine d'œuf
de poule (45.0 kDa); le monomère et le dimère de l'albumine sérique de bœuf (66.0 et
132.0 kDa). Les gels de polyacrylamide, ayant subi une préélectrophorèse de 90 minutes
à 12 V/cm à 4°C. ont été soumis à un courant de 12 Vlcm pendant 4 heures et demie à
4°C. Après la migration, les gels ont été placés dans une solution colorante fixatrice
(Bleu de Coomassie 2,5 m%l, méthanol 50%. acide acétique 10%) pendant 60 minutes et
ont été décoloré pendant la nuit dans une solution décolorante (méthanol 10%. acide
acétique 10%). Les gels ont par la suite été séchés sous vide sur une feuille de papier
Whatrnann (3M) à l'aide d'un appareil BIO-RAD. Les gels séchés ont été exposés contre
une plaque de BioImaging, ensuite la radioactivité a été révélée par "Phosphor Imagiag"
a l'aide d'un appareil Fujix Bas 1ûûû (Fuji) et quantiftée à l'aide du logiciel MacBAS
version 2.5 (Fuji). Les distances de migration des complexes ES et des protéines
standards ont été calculées et traitées selon l'analyse de Ferguson ( 1%4).
2.12 Essai de retardement du substrat sans clivage
Les essais de retardement ont été effectués dans le tampon de réaction optimisé
contenant du MnC12 10 rnM au lieu du MgC12. L'enzyme et le substrat étaient présents à
des concentrations équimolaires de 3 à 3000 pM dans un volume total de 20 pl et ils ont
été incubés ensemble pendant 15 minutes à température pièce. Les échantillons ont 6té
déposés sur un gel de polyacrylarnide 5% contenant également du MgC12 10 m M et ont
migré sous les mêmes conditions que celles mentionnées en 2.11. Après la migration. le
gel de polyac~lamide a été placé dans une solution fixatrice (acide acétique 10%,
méthanol 1096, glycéroi 5%) pendant au moins 30 minutes à température pièce. Le gel a
été séch6 et exposé tel que décrit à la section 2.1 1.
213 Détermimation des pammèûw cin4tiques de pseudo premier ordre
Les essais cinétiques de 1-Cm1 ont initialement été réalisés dans des conditions

de réaction de "single turnover partiel", c'est-à-dire dans des conditions où l'enzyme est
en excès par rapport au substrat. Ce modèle a été employé puisque certaines observations
nous laissaient supposer qu'elle demeurait liée à ses produits de coupure.
Considérons la réaction enzymatique suivante:
Dans un mecanisme de "single turnover partiel", pour une enzyme qui demeure
attachée à ses produits de coupure, la réaction enzymatique est incomplète puisqu'elle
s'arrête à l'étape de formation de EP. Dans ces conditions. le Km' peut être défini comme
étant la concentration d'enzyme correspondant à la moitié de la valeur maximale de la
constante de vitesse (k?). Cette valeur maximale de k2 peut être déterminée à partir d'un
araphique illustrant la relation entre k2 et la concentration d'enzyme (Halford el al.. b
1980). La valeur du KmB peut être plus facilement déterminée en appliquant par "curve
fitting" l'équation suivante au graphique decrit précédemment B l'aide du logiciel
Kaleidagrapb:
2.14 EfYct do SDS sur la rétention des produits de coupure
Dans un volume total de 20 pl, 2 5 nM de 1-Cmoei et 25 pM de substrat PCR
marqué ont été incubés pendant 15 minutes à température pièce dans un tampon TAPS-
KOH 10 rnM pH €45 contenant du MgCll 10 mM, BSA 1 mgfml, DTT 1 mM, glycérol
10% et polydYdC 0,05 m g i d (Amershm Phannacia Biotech). La réaction a été arrêtée
sur glace en présence ou en absence de SDS 0,5%. Chaque échantillon a été déposé en
entier sur un gel de polyacrylarnide 5% (1X TBE) de séparer le substrat seul (S) du
complexe 1-CmoeI-substrat (ES) et des produits de coupure (Pl et PZ). L'électrophorèse
s'est déroulée tel que décrit précedement et après le migration, le gel a ét6 fixé dans la

solution fixatrice décrite en 2.12. Le gel a par la suite été séché, exposé et analysé tel que
décrit en 2.1 1.
2.15 Turnover de l'enzyme et détermination des pamrn6tres cinétiques selon le
modèle cinétique de Michaelis-Menton
Un excès de substrat (1000 PM) fut m i s en présence de l'enzyme (100 PM) afin de
réaliser un essai cinétique permettant de vérifier si 1-CmoeI est régénérée au cours de
réaction de manière à couper plus d'une molécule de substrat. L'essai cinétique a été
effectué à 37°C dans un volume total de 520 pl avec des prises d'aliquotes de 100 pl à 10.
20.40,û-û et 160 minutes. Le tampon de réaction optimisé fut utilisé pour ces essais. Les
échantillons ont été soumis au même traitement d'arrêt de réaction que celui décrit en 2.9
et ont été analysés de la même manière qu'en 2.9.
Le substrat utilisé était le même produit PCR de 274 pb déjà décrit à la section 2.9
mais marqué radioactivement à ses extrémités à I'aide de la polynucléotides kinase du
phage T4 (New England BioLabs) et de [y-33PldATP (Amenharn). 2 0 ng de chacune des
amorces (psbAY2 et psbA#5) ont été marqués à l'aide de 50 pCi de [y-"PldATP et de 20
U de polynucléotides kinase dans un volume de 100 pl pendant 30 minutes à 37OC.
Après une incubation de 10 minutes à 65 O C , le mélange fut ajouté à 50 ng d'une
préparation d'ADNcp de C. eugrunetos, de 10 mM de chacun des dNTPs. 5 U de l'ADN
polymérase AmpliTaq Gold (Perkin Elmer) ainsi que le tampon fourni par le fabricant.
Les cycles appliqués furent les mêmes que ceux précisés en 2.9. Après l'amplification. le
produit K R a été purifié à I'aide d'une colonne Microspin S-300 HR (Amersham
Pharmacia Biotecb) selon les directives de la compagnie. La pureté du produit PCR ainsi
que l'efficacité de l'amplification ont ét6 évaluées en faisant migrer une aliquote de l'éluat
de la colonne sur un p l de 3.5% d'agarose MetaPhor (FMC BioRoducts). Le produit
PCR a été dosé à I'aide d'un spectroscope GeneQuant (Amersham Pharmacia Biotech) à
260 nm.

Le modèle cinétique classique utilisé pour caractériser les réactions enzymatiques
est le modèle d'état statiomaire de Michaelis-Menten. Min de répondre à ce modèle, les
réactions enzymatiques doivent respecter certains postulats tel qu'une concentration de
substrat en excès par rapport à la concentration d'enzyme et une régénération de l'activité
de l'enzyme après la catalyse. Le modèle de Michaeiis-Menten repose sur l'équation
suivante:
où V,, est la vitesse de réaction maximale et Km est la constante de Michaelis (c'est-à-dire
la concentration de substrat à laquelle la vitesse de réaction est la moitié de la vitesse
maximale).
de dé t edne r les paramètres cinétiques de 1-Cmoel selon le modèle de
Michaelis-Menten, douze essais enzymatiques ont été effectués en utilisant différentes
concentration de substrat (10 à 200 PM) qui sont toutes en excès par rapport à la
concentration d'enzyme utilisée (1 PM). Les vitesses de réaction (v,) ont été placées en
graphique en fonction de la concentration de substrat. À l'aide du logiciel LEONORA de
Comish-Bowden, des courbes furent tracées par "curve fitting" de l'équation de
Michaelis-Menten (4) afin de déterminer les paramètres Km, V,, et la constante
d'efficacité de réaction k d Km.
2.16 AmpüBcation et séqaenpge de Ifintron CrpsbA.4
L'ORF du quatrième intron du gène psbA de Chlamydomonas reinhord~ii
(CrpsbA.4) fut amplifié par PCR à partir de l'ADN chioroplastique de cette algue. Cette
amplification fut réalisée dans un volume total de 100 pl en utilisant 200 ng de chacune
des amorces (psbAR2 5'-GTïACTT'CATCiTI'AATCCGT-3' et psbAY 10 5'-
GTAmGAAGAnAGACGACC-3'1, 5 ng du gabarit d'ADN, 10 m M de chaque
dNTPs, le tampon fourni par la compagnie (Perkin Elmer) et 2,s U de l'ADN polymdrase
AmpliTuq Gold (Perkin Elmer). Après un chauffage de 10 minutes à 95OC, les 30 cycles

de PCR furent réglés ainsi: dénaturation à 94OC. 1 minute; fixation des amorces à 6û°C, 1
minute: élongation à 72°C. 3 minutes. La réaction PCR fut terminée avec un cycle
supplémentaire de 2 minutes à 60°C et 25 minutes à 72T. Le produit PCR a été purifié
par chromatographie sur une colonne Microspin S a HR (Amersharn Pharmacia
Biotech) et analysé sur un gel de 0,896 d'agarose (Amersham Pharmacia Biotech). Il a
ensuite été dosé à I'aide d'un spectroscope GeneQuant (Amersharn Pharmacia Biotech) à
260 nrn. Après avoir ajusté la concentration du produit PCR à 100 ng/pl, une aliquote a
été envoyée au Service de Séquençage du pavillon Charles-Eugène Marchand afin que la
séquence de I'intron soit déterminée à I'aide des amorces psbAR et psbA#lO (1.5 PM) au
moyen de la technique de séquençage au Big Dye.
Deux autres oligonucléotides ont été synthétisés afin de compléter le séquençage
de l'intron CrpsbA.4. Ces oligos sont les suivants: psbA#ll. 5'-
ATGACCGAAGGTAATAGCAAC-3' et psbA#12, 5'-
GTCCACCTTTAGAAGAAGCGC-3'.
Les séquences ont été assembides à l'aide du logiciel AB1 AUTOASSEMBLER
(version 1.4.0) et ont été analysées en utilisant le groupe d'applications (version 9,l) du
Genetics Cornputer Group (Madison, WI). L'alignement des séquences en acides amines
de la protéine codée par I'intron CrpsbA4 et de 1-CmoeI a éte fait à l'aide du
CLUSTALW (Thompson et al.. 1994).
2.17 Matant HlS4A
Le premier résidu histidine du motif H-WH (position 154 chez 1-CmoeI) fut
choisi pour effectuer les premières études de mutagenèse puisque ce résidu est
entièrement conservé dans l'alignement de séquences des p ro the s de la famille
d'endonucléases "horning" à motif H-N-H. 11 a été substitué par le résidu neutre alanine
dm de visualiser son implication dans l'activité de 1-CmoeI.

2.17.1 Mutagenèse dirigée, expression et purihcation de In protéine mutante
Mh de substituer le résidu histidine par un résidu alanine, la trousse de
mutagenèse dirigée QuickChange (Stratagene) fut utilisée selon les indications fournies
par la compagnie. Après la transformation des cellules compétentes Epicurian coli XL-1
Blue (Stratagene), l'ADN plasmidique fut récupéré à l'aide de la trousse Qiaprep 8
(Qiagen) selon les directives de la compagnie et des échantillons furent envoyés au
Service de séquençage du Pavillon Marchand en vue d'être séquencés tel que décrit en
2.3. Afin de surexprimer la protéine mutante, des cellules compétentes d E culi
BLZl(DE3) (Novagen) furent ensuite transformées selon les directives de la compagnie.
Des clones ont servi à inoculer des milieux de culture LB-km qui ont été incubés à 37"C,
225 rpm pendant une nuit. Des aliquotes de 100 pl des cultures bactériennes d'E. coli
BL2l(DE3) ont été mélangées à 900 pl de plycéroi 80% et conservées à -70°C.
Une préculture de 6 ml dans le milieu LB-kan. inoculée avec environ 10 pi d'une
culture glycérinée d'E. 'oli BL21(DE3), fut incubée 5 37°C. 225 rpm pendant une nuit.
Le lendemain. cette préculture fut utilisée comme inoculum pour une culture de 500 ml
dans le même milieu. Ce bouillon de culture fut incubé sous les mêmes conditions que
celles de la préculture jusqu'à ce que son absorbance à 600 nm atteigne 0.6. De I'IPTG
fut alors ajouté à une concentration de 1 m M pour induire la production de protéines
recombinantes. L'incubation de la culture fut poursuivie durant une période de 2.5 heures
sous les mêmes conditions. Les bacteries ont ensuite été recueillies par centrifugation à
6000 rpm pendant 7 minutes à 4°C dans le rotor GSA (Sorvail). Les culots des cultures
bactériennes ont &té conservées à -70°C.
Le mutant Hl54 fut purifié de la même manière que la protéine sauvage (voir
sections 2.6.1 à 2.6.3) jusqu'a l'étape de chromatographie d'&changes de cations, excepte
que 2 ml de résine sédimentée Ni-NTA (Qiagen) furent utilisés au lieu de 4 ml. Les
fractions de protéines recueillies ont et6 analysées par SDS-PAGE tel que ddcrit en 2.7.
La solution de protéine a été dosée à l'aide de la trousse de dosage MicroBCA (Pierce).
Enfiil. la préparation de protkine purifide fut conservée à -20°C dans 50% de glycérol.

2.17.2 Mes- de l'activité du mutant H154A
Des essais enzymatiques furent effectués en utilisant une concentration du mutant
H154A (23 à 800 nM) et une concentration de substrat de 25 PM. Le substrat utilisé
était le même produit PCR de 274 pb marqué de la même façon que celui employé lors
des essais cinétiques de l'enzyme sauvage. Ces essais ont eu lieu à 37OC dans le même
tampon que celui de l'enzyme sauvage. Des aliquotes prélevées à 5, 15,45 et 90 minutes
ont été soumises au même traitement d'arrêt de réaction et de méthodes d'analyses que
ceux décrits en 2.9.
2.17.3 Essai de retardement du substrat par le mutant H154A
Des concentrations équimolaires d'enzyme et de substrat (03, 1.3. 10 nM) ont été
placées dans le tampon de réaction optimisé contenant 10 rnM de MgCl, pendant 15
minutes à température pièce. Les échantillons ont été déposés sur un gel de
polyacrylarnide 5% contenant également IO rnM de MgCl? et ont migré sous les mêmes
conditions que celles mentionnées en 2.12. Après la migration. le gel a été fixé, séché et
exposé tel que décrit à la section 2.13.

RÉSULTATS
3.1 Expression et purif%cation de 1-Cmoei et H154A
La surexpression de 1-Cmoel à partir du plasmide pet30a-ICrnoeI ne montra
aucun signe de toxicité sur la souche dlE. coli BUl(DE3) . Le niveau relativement élevé
de surexpression de la protéine recombinante témoigne de ce fait après l'induction de
deux heures et demie à I'IPI'G. Ce résultat est présenté à la Figure 8A (puits 1) illustrant
le profil électrophorétique des protéines cellulaires totales d'une culture induite après
analyse par SDS-PAGE. Chaque étape de la purification de 1-CmoeI a d'ailleurs été
suivie sur gel dénaturant. 1-CmoeI est une protéine fortement soluble puisque plus de
95% de la protéine recombinante surexprimée s'est retrouvée dans le surnageant après la
centrifugation du lysat cellulaire (Figure 8A. puits 2). Une période d'incubation de la
préculture à 30°C permet une croissance bactérienne plus lente évitant ainsi une
"vieillissement" prématuré de la préculture et par conséquent une période d'incubation de
culture plus longue avant d'atteindre 0,6 d'absorbance à 600 nm.
Un maximum d'environ 5 mg de protéine recombinante a été retenue par ml de
résine de Ni" lors de la première chromatographie. Malgré la forte spécificité du His-
Tag de 1-CmoeI pour les ions Ni2+. plusieurs contaminants sont restés sur la résine de
nickel en compagnie de 1-CrnoeI après les lavages avec de I'imidazoie 20 m M visant à se
débarrasser des protéines indésirables (Figure SA, puits 3). Après cette étape. 1-CmoeI a
été récupérée à un degré de pureté d'environ 75 à 80% d'homogénéitd.
A f ~ n d'augmenter le degré de pureté de 1-Cmoel, la solution protéique a dQ être
soumise à une dialyse contre un tampon HEPES-NaOH 50 mM à pH 73 avant d'être
l'objet d'une deuxième chromatographie, une chromatographie d'dchange de cations. Au
cours de la dialyse, une faible quantité de protéine s'est déposée contre les parois du sac
de dialyse, amenant ainsi une faible perte de la protbine recombinante. Une résine Mono
S KR 5/5 (Amersharn Pharmacia Biotech) fut d'abord utilisée comme essai avec un
échantillon de 15 ml de solution protbique (0,75 mg de protbine), mais le degré de purifi-

Figure 8. Étapes de purification de 1-CmoeI. A) Des échantillons ont été recueillis au
cours de la purification et analysés par SDS-PAGE. Les gels ont été colorés au
bleu de Coornassie. Les trois premiers puits contiennent chacun un équivalent
de 100 pl de culture bactérienne. Le puits L contient une aliquote de la culture
bactérienne après surexpression de la protéine recombinante; puits 2: la
fraction soluble du lysat cellulaire; puits 3: I'éluat de la résine de Ni'+ NiN-TA
et puits 4: 2.5 pg de la protbioe recombinante purifiée par chromatographie sur
la résine Ceramid Hyper D. Le puits M correspond aux marqueurs de poids
moléculaires Broad Range de BioRad. Les poids moléculaires respectifs sont
indiqués. B) Chromatographie d'échanges de cations. L'axe des x indique le
volume total des fractions recueillies en ml. l'axe des y de gauche montre la
densité optique à 280 om alors que l'axe des y de droite montre le pourcentage
de NaCI. La ligne oblique dans le chrornatogramme représente le profil du
gradient salin qui a été appliqué pour éluer les protéines. Le trait pointillé
délimite les fractions qui ont été regroupées et conservées pour les étapes de
purification suivantes.

volunu das f rac t ions

cation n'a pas été amélioré de beaucoup puisque la majorité des contarninants de l'élution
de la résine de Ni" sont restés. Un autre type de résine échangeuse de cations fut donc
mis à l'essai, soit une résine Ceramid Hyper D (Amenham Pharmacia Biotech). Une
augmentation linéaire de la concentration de NaCl de O à 0,2 M fut nécessaire pour éluer
1-C'mue1 (Figure 8B). Le degré de purification fut nettement amélioré par cette
chromatographie. la protéine recombinante 1-CmoeI représentant 95% des protéines dans
les fractions récupérées (Figure 8A. puits 4). Après la concentration des fractions
protéiques, une quantité toiale d'environ 2,5 mg de protéines (dans une solution de 0,85
rnglrnl) fut récupérée.
La purification du mutant H154A fut effectuée de la même manière que celle de
l'enzyme sauvage. sauf que la chromatographie d'échanges de cations n'a pas été utilisée.
Le degré de purification obtenu pour ce mutant H l H A est d'environ 75% et il correspond
à celui observé au puits 3 de la Figure 8A.
3.2 Clivage du His-Tag par I'ent6rokinase
Quelques problèmes sont survenus lorsque nous avons tenté de retirer le His-Tag
de la protéine recombinante à l'aide de I'entemkinase. 1-Cmoel a subi une forte
dégradation lorsqu'el le fut mise en présence d'entérokinase pendant 16 heures en utilisant
le ratio protéines-cibles : entérokinase (M pg : 1 U) recommandé par le fabricaut (Figure
9, puits 6). 10 pg de protéine recombinante et 0.2 U d'entérokinase furent utilisés pour
réaliser un essai selon ce ratio. D'autres essais de clivage ont été faits en utilisant de plus
faibles concentrations d'entérokinase, soit de 0,02 U à 0,0002 U. À ces concentrations, la
coupure n'a pas été nécessairement plus spécifique puisque des produits de protéolyse ont
quand même étd observés (Figure 9. puits 345). 11 a été vérifié que 1-Cmocl est stable
en absence d'entérokinase lorsqu'elle est incubée dans le même tampon que celui utilisé
pour le clivage avec cette enzyme et ce pendant la même durée de temps (Figure 9, puits
2). Puisqu'un essai enzymatique effectué avec la protéine recombinante possédant le His-
Tag a révé16 une forte activité de coupure, il fut convenu d'61irniner l'étape de clivage à
1 'entérokinase (résultat non-illuseé). Ainsi, dans ce texte, lorsque nous parlons de

Figure 9. Clivage de 1-Cmoel à différentes concentrations d'entérokinase. Des
échantillons de 10 pg de 1-CmoeI ont été mis en présence ou en absence
d'entérokinase. et après 16 heures d'incubation à 18OC. chacun des mélanges
réactionnels a été déposé sur gel de polyacrylamide 12%. Les protéines ont été
colorés au bleu de Coomassie. Dans le puits 1. on voit 1-CmoeI avant le
clivage et l'incubation; dans le puits 2, 1-CmoeI incubée en absence
d'entérokinase; dans le puits 3, 1-CmoeI clivée par 0,0002 U d'entérokinase:
dans le puits 4. 1-CmoeI clivée par 0.002 U d'entérokinase: dans le puits 5. I-
CmoeI clivée par 0.02 U d'entérokinase et dans le puits 6, I-CmoeI clivée par
0.2 U d'entérokinase. Les clivages et les dilutions ont été effectués dans les
tampons fournis par le fabricant (Novagen). Le puits M correspond aux
marqueurs de poids moléculaires Broad Range de BioRad. Les poids
moléculaires de ces marqueurs sont indiqués.


1- Cmoel, il s'agit en fait de la protéine possédant une queue de résidus histidine.
3.3 Détermination des conditions optimaies d'activité
Des études antérieures ont démontré qu'au moins quatre facteurs influencent
l'activité des endonucléases "horning": (1) le pH. (2) les cations divalents. (3) les cations
monovalents et (4) la température (Monteilhet et al.. 1990). Ainsi, au cours d'une série
d'essais enzymatiques, ces facteurs de même que quelques autres ont été modifiés un a un
dans le but de déterminer les conditions optimales d'activité pour 1-Cmorl. Dans tous ces
essais enzymatiques. le substrat utilisé était un fragment PCR de 274 pb contenant le site
de reconnaissance et de coupure de 1-CmoeI. Ce substrat avait été uniformément marqué
au "P lors de l'amplification PCR ce qui nous permet de détecter deux fragments de 176
et 9û pb suite au clivage par 1-CmoeI. La figure 10A montre les résultats typiques d'une
analyse sur gel de polyacrylamide de 5% d'échantillons prélevés à différents temps après
le début d'une réaction de clivage par 1-Cmoel.
33.1 Nature et concentration da tampon de réaction
Deux tampons ont été testés quant à leur habileté à supporter le clivage du
substrat par 1-CmoeI. Il s'agit des tampons Tris-HCI et TAPS-KOH qui ont été utilisés à
concentrations variables, mais tous deux à pH 8,5. L'activité optimale de 1-CmoeI fut
observée dans le tampon Tris-HCI à une concentration de 5 mM (Figure LOB). Cette
concentration est cependant trop faible pour tamponner le milieu réactionnel
convenablement tout au long de la réaction. C'est pourquoi il fut convenu d'utiliser le
tampon Taps à une concentration de 10 mM pour tous les essais subséquents puisque ce
tampon assure un meilleur pouvoir tampon que le Tris et que I'activité y est sensiblement
comparable A celle mesurée dans le Tris 5 rnM (plus de 80% d'activité).
t3.2 Concentmtion du cofacteur
La prbsence du cation divalent MC (ou d'un cation divalent substitut, voir section
3.3.3) s'est avéke essentielle à I'activité de 1-CmoeI puisqu'aucun clivage ne fut observé

Figure 10. Détermination des conditions optimales d'activité pour 1-CmoeI. A) Roduits
d'essais enzymatiques typiques déposés sur gel de polyacrylamide 5%. On y
voit I'évolution de clivage du substrat (SI en produits de coupure (Pl et P2)
dans le tampon de réaction optimisé. Les représentations graphiques illustrent
les effets de la nature et de la concentration du tampon de reaction (B), de la
concentration du cofacteur ~ g " (C). de divers cations divalents a une
concentration de 1 et 10 rnM dans le milieu réactionnel (D) et de la force
ionique (E). Pour chaque graphique. l'activité de 100% représente l'activité
maximale obtenue au cours des essais visant à optimiser le paramètre
correspondant.

1 2 5 10 min 1 00 a % UactMtb dans k TAPS
80
a0
Q %a C
O 0
$0
O 5 10 U) mo ZSQ
Concentration du tampon (mM)

lors d'un essai en absence de M ~ " . II est important de mentionner que cet essai avait été
effectué en présence dEDTA afin de chélater toutes traces d'ions M<' (ou autres cations
divalents) pouvant se trouver dans la solution d'enzyme edou dans le tampon de réaction
puisqu'en absence d'EDTA des traces de coupure avaient été observées. La figure 10C
montre que l'activité endonucléase de 1-Cmoel est à son maximum à une concentration de
10 m . en MgCl, alors qu'une concentration plus forte (25 mM) entraînne une diminution
de I'activité.
3.3.3 Nature du cation divalent agissant comme cofacteur
L'activité enzymatique de 1- CmoeI a été testée en substituant le cofacteur Mg "
par l'un des cations divalents suivants: Ca". Mn", Cu". Ni". Zn", Ba". Co" et Si.'.
Seuls quatre de ces cations ont entraîné le clivage du substrat par 1-Cmoel. Que ce soit à
une concentration de 1 ou 10 mM. I'ion Mg2+ a induit le plus d'activité. suivi des ions
Ca". Si+ et Ba" (Figure IOD). Une concentration de 10 rnM de I'ion Ca" a induit un peu
plus de 80% d'activité comparativement au maximum d'activité mesuré en présence de
Mg". Cinq cations n'ont pas supporté l'activité de 1-Cmocl: ce sont le Mn2+. le Ni2+, le
Zn". le Cu" et le Co".
3.3.4 Antres paramètres optimisés
Les autres paramètres testés sont le pH. la température. la force ionique, l'effet de
la présence d'une protéine inerte comme le BSA et d'un agent réducteur comme le Dm.
Le pH optimal est 8,5, la température optimale est 37°C (résultats non-illustrés).
L'augmentation graduelle de la force ionique par l'ajout de NaCl ou de KCI jusqu'à une
concentration totale de 120 m M en sels monovalents a eu pour effet de diminuer
progressivement le clivage (Figure 10E). Le stabilisant BSA est nécessaire mais en petite
quantite dans le milieu réactionnel; la plus forte activité de 1-CmoeI fut enregistrCe don
que la concentration en BSA n'était que de 0,01 m g h l (résultats non-illustrés). Cagent
réducteur DTï est aussi indispensable afin d'optimiser l'activité enzymatique. En
absence de DTT dans le milieu réactionnef, le clivage du substrat fut diminué d'environ
85% par rapport h la rhction contenant 1 mM de MT (rbsultats non-illustr6s).

Ainsi. en supposant que les divers facteurs étudiés agissent indépendamment les
uns des autres sur la vitesse de réaction, la composition du milieu réactionnel donnant le
clivage optimal pouvait être définie comme suit: TAPS-KOH 10 mM pH 8.5, MgCI, 10
mM, DTT 1 m M et BSA 0.01 mg/ml.
Pour évaluer la stabiiité de 1-CmoeI dans son milieu réactionnel optimisé. la
préparation d'enzyme fut préincubée à 37OC dans le milieu réactionnel dépourvu de
substrat en présence ou absence de Mg" et, à différents temps de préincubation, une
aliquote d'enzyme fut ajoutée à son substrat pour déclencher une réaction enzymatique
standard. La demi-vie de 1-Cmoel, c'est-à-dire le temps de préincubation requis pour que
l'enzyme perde 50% de son activité. est respectivement de 230 et 325 minutes en
prdsence ou en absence de Mg' (Figure 11). Finalement, il est à noter que la protéine
purifiée perd de son activité lorsque conservée à -20°C. Après une période de neuf mois
à -20°C dans une solution contenant du glycérol XI%, 1-CmoeI a perdu environ 75% de
son activité. Une perte d'activité de seulement 20% fut observée à -70°C durant la même
période.
3.5 Stœchiométrie de liaison de 1-CmoeI ib son site de reconnaissance
Une analyse de Ferguson a été effectuée afin de déterminer la forme sous laquelle
1-Cmoel se fixe à son substrat (Ferguson, 1964). Ce type d'analyse consiste à faire migrer
le complexe enzyme-substrat (ES) et le substrat seul (S) sur des gels de différentes
concentrations de polyacrylamide, soit de 6 à 10%. La fixation de l'enzyme à son site de
reconnaissance entraîne un retardement de la migration du complexe par rapport il celle
du substrat seul (Figure 12A). Les conditions utilisées pour cette analyse n'ont pu
empêcher complètement le clivage du substrat: les produits de coupure représentaient
1,596 de l'ADN total marquC (Figure 12A, puits +m. Nous avons fait migrer en parallele
des protéines standards de poids moléculaires sur chacun des gels (voir légende de la

FigUm 11. Effet de la préincubation de 1-CmoeI à 37°C dans le milieu rtactiomel
optimisé dépourvu de substrat. L'enzyme a ét6 préincubée en présence ou en
absence de magnésium dans le mélange réactionnel dépourvu de substrat.
Après 15, 30, 45, 60, 90, 120, 210 et 360 minutes de préincubation, une
aliquote de 1-Cmoel fut prelevée et mise en présence de son substrat afin de
déclencher une réaction enzymatique. Le graphique montre la relation entre le
log du pourcentage d'activitd initiale et le temps du prbincubation.

C I
l
t 1 - 1
y = 86.812 + 4.1 - 'y= 76.16 + 4.080
i 10 ' ' ' ' ' ~ 1 ' ~ ' ~ ' ' ' ' ~ ' ' " " '
O 50 100 150 200 250 300 350 400 Temps de préincubation (min)

Figure 12. Stœchiométrie de liaison de 1-Cmoel à son substrat. A) Gel de
polyacrylamide 8%. Le substrat d'ADN, un produit PCR de 274 pb, est incubé
en absence (-E) et en présence (+E) d'enzyme et déposé sur des geis de
polyacrylamide de 6 à 10% afin de révéler la présence du complexe enzyme-
substrat (ES). Des protéines standards de poids moléculaires sont séparées
dans des puits adjacents sur chacun de ces gels (résultats non-illustrés). B)
Représentation graphique de I'analyse de Ferguson. Une régression linéaire
relie une fonction logarithmique de la mobilité de chacune des protéines
standards (1 à S), du substrat seul (S) et du complexe enzyme-substrat (ES) au
pourcentage de polyacrylamide des gels. Les marqueurs de protéines utilisCs
sont respectivement: 1, a-lactalbumine (143 kDa); 2, anhydrase carbonique
(29,O ma); 3, albumine d'œuf de poule (45.0 kDa); 4 et 5, le monomère et le
dimère de l'albumine serique de boeuf (66,O et 132,O kDa). C) Reprdsentation
araphique de la pente negative (-Kr) obtenue en B) pour chacune des proteines e
standards en fonction du poids moléculaire de celles-ci. Les marqueurs
protéiques 1 à 5 sont ddfinis en B). D) Tableau résumant les résultats de
l'analyse de Ferguson. La valeur n représente le coefficient de stœchiom&ie
de la liaison de 1-CmoeI à son site de reconnaissance.

O h du gel de polyacrylamide
O ' ------- - - . . -A-+---- *... . - - * -.---
O 20 40 80 80 100 120 1 4 0
Poids rnolthulaire (kDa)
-Kr PM de ES PM de S PM théorique PM calculé n de I-Cmoel de E
(-j ( k w w a ) ( k ~ a ) ( k ~ (-j 15,356 192,73 152,16 43,422 40,573 093

Figure 12). Par la suite, les distances de migration des standards et celles de ES et S ont
été mesurées et mises en graphique en fonction du pourcentage de polyacrylamide du
gel (Figure 12B). Il va de soi qu'une pente négative (-Kr) est obtenue puisque plus la
concentration en polyacrylamide du gels augmente, moins les distances parcourues par
les protéines sont grandes. La valeur de cette pente négative est à son tour mise en
graphique en fonction du poids moléculaire de chaque standard de poids moléculaire ce
qui nous donne une droite standard illustrant la distance de migration en fonction du
poids moléculaire (Fig. 12C). À l'aide de cette droite et du Kr obtenu pour le complexe
ES. nous pouvons évaluer le poids moléculaire de ES et ainsi déterminer celui de
I'enzyme en soustrayant le poids moléculaire théorique de S. Le résultat obtenu, 4-57
kDa. nous indique que 1-CmoeI se lie à son substrat sous la forme d'un monomère
puisque le poids moléculaire théorique de cette protéine est de 43.22 kDa (Figure 12D).
Ceci représente un coefficient de stœchiométne de 0.93. valeur qui se rapproche de 1,
c'est-à-dire qu'une "copie" de l'enzyme se fixe au substrat.
3.6 Essai de retardement du substrat sans cîivage
Des essais de retardement ont été effectués afin de mettre au point les conditions
permettant de mesurer l'affinité de l'enzyme envers son substrat par la détermination
d'une constante. la constante de dissociation KD. Pour ce faire, des conditions où
l'enzyme peut lier son site de reconnaissance sans catalyse doivent être employ6es. Un
essai de retardement a été fait en utilisant un des cations divalents n'ayant pas soutenu
l'activité de 1-Cmoei, soit le Mn" (Figure 13A). Un autre essai de retardement a été
effectué en présence d'EDTA din de vérifier si l'absence de clivage du substrat en
présence d'EDTA est due à une inactivation de l'enzyme ou à l'absence d'appariement de
1-Cmoel à son site de reconnaissance (Figure 13B). Dans les deux cas. aucun
retardement du substrat ne fut observé et ce, jusqu'à des concentrations équimolaires
d'enzyme et de substrat de 3 nM.
3.7 Relâchement des produits de coupure par 1-Cmoel
La solution d'arrêt des essais enzymatiques se compose dtEDTA, de SDS et de protdinase

Figum 13. Essais de retardement du substrat en présence A) de Mn" et B) dlEDTA. Les
essais de retardement ont été effectués dans un tampon contenant 10 m M de
MnClz ou 1 rnM dlEDTA. Les échantillons ont été d6posés sur gel de
polyacrylarnide 5% qui contenait également 10 rnM de MnCl, ou I m M
d'EDTA. Les puits 1 à 7 correspondent respectivement aux concentrations
équimolaires de 1-CmoeI et de substrat 3, 10, 30. LOO, 300, 1000 et 3000 PM.
La concentration du substrat S1 est de 100 PM, dors que celle du substrat S2
est de 3000 PM. La position du substrat dans le gel de polyacrylamide après
l'électrophorèse est indiquée.

Substr
Substrat

K. Afin de vérifier si le SDS est nécessaire pour dissocier 1-CmoeI de ses produits de
coupure. un essai de retardement sur gel de polyacrylamide 5% fut effectué. En utilisant
toujours le même produit PCR de 274 pb comme substrat, l'essai fut accompli en
présence de Mg" et l'enzyme fut incubée avec le substrat durant une période de 15
minutes à 25°C. La moitié des réactions de clivage furent arrêtées par l'ajout de SDS
0,5%, tandis que l'autre moitié furent simplement placées à 4OC (Figure 14). Après la
migration sur gel de polyacrylamide 5%. les produits de coupure sont observés autant
pour les échantillons -SDS que +SDS et ce. avec une intensité comparable. 1-CrnoeI
retient donc peu ou pas du tout ses produits de coupure après le clivage du substrat.
3.8 Caractérisation des paramètres cinétiques de ECmoeI
L'activité de 1-Cmoei fut d'abord caractérisée en déterminant les paramètres
cinétiques de pseudo premier ordre suite à des résultats préliminaires suggérant que cette
endonucléase n'était pas disponible pour plus d'une coupure du substrat. L'enzyme restait
fortement liée ses produits de coupure après le clivage du substrat (un plasmide de 10.5
kb contenant le site de coupure de 1-CmoeI) nécessitant l'ajout de protéinase K afin de
libérer les produits de coupure de I-CmoeI. Par la suite. 1-CmoeI fut placée dans des
conditions de Michaelis-Menten afin de déterminer les paramètres cinétiques se
rattachant à ce modèle cinétique.
3.8.1 Paramètres cinétiques de pseudo premier odre
Dans le cas des réactions de clivage de I-Cmoel, le mécanisme de "single turnover
partiel1' implique des réactions enzymatiques effectuées avec une concentration d'enzyme
en excès de celle du substrat (voir section 2.13). Dans ces conditions, le Km' peut être
défini comme étant la concentration d'enzyme correspondant à la moitié de la valeur
maximale de la constante de single turnover (k,) (Halford et al., 1980) (Figure 15A). La
valeur Km' obtenue pour 1-CmoeI fut de 43 pM I 7.
3.8.2 Paramétres cinétiques des coadiaons de Mirimelis-Menten
Puisque nous avons démontré que 1-CmoeI relâche ses produits de coupure après

Figure 14. Relâchement des produits de coupure par 1-CmoeI. Des réactions de clivage
ont été arrêtdes sur glace avec (+) ou sans (-) l'addition de SDS 0,5%. Chaque
échantillon a été dépod sur un gel de polyacrylamide 5% de separer le
substrat seul (S) du complexe 1-CmoeI-substrat (ES) et des produits de coupure
(PI et P2).
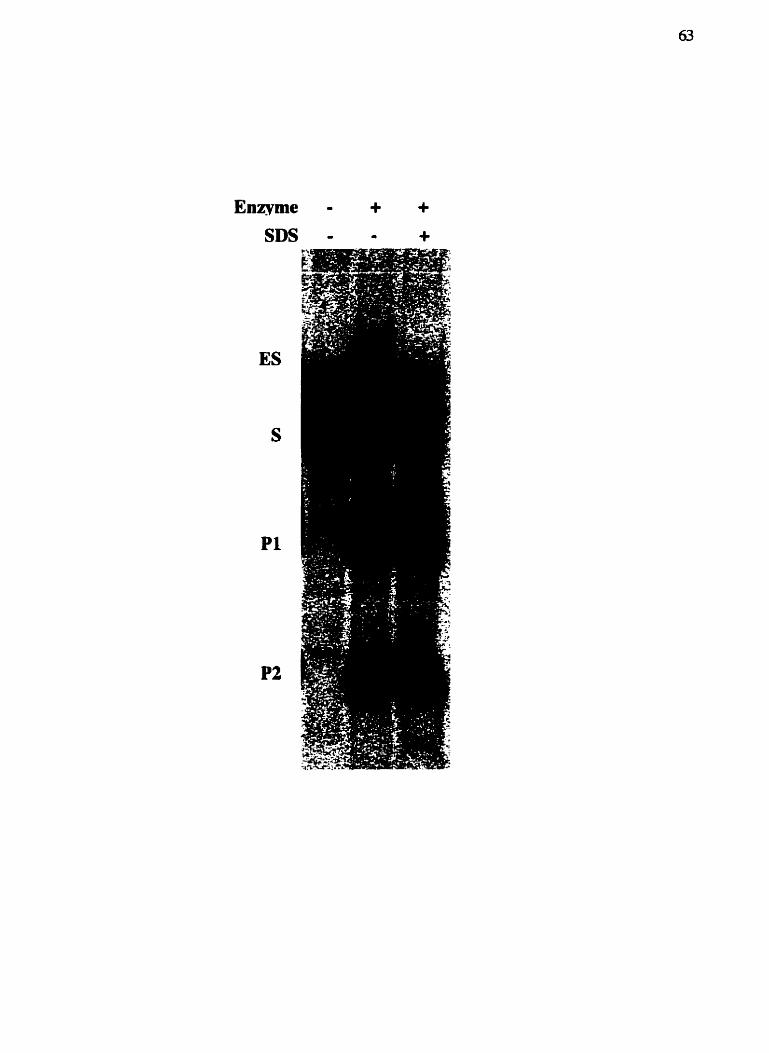
Enzyme - + + I +

Figure 15. A) Détermination des paramètres cinétiques de pseudo premier ordre de I-
CmoeI. B) Détermination des paramètres cinétiques de I-CmeI selon le
modèle cinétique de Michaelis-Menten. C) Résumé des paramètres cinétiques
obtenues pour 1-Cmoei en conditions de pseudo premier ordre et en conditions
de Michaelis-Menten.

O 50 100 150 200 250
Concentration de substrat (PM)
Pararn8tres cinétiques de premier ordre Paramètres cinbtiques de Michaelis-Menten
(PM) (min-') (PM-' min-') (PM) (pmoVmin) (min") (M* 43 0,704 0,00916 101 0,000134 0,256 4,27x 107

la catalyse (voir section 3.7). il était intéressant de mesurer les paramètres cinétiques de
cette endonucléase dans un système correspondant au modèle cinétique dUtat stationnaire
de Michaelis-Menten. II fut d'abord démontré que 1-CmoeI fait preuve de "turnover".
c'est-à-dire qu'une molécule d'enzyme peut participer à plus d'une transformation du
substrat. Après 80 minutes de réaction. à une concentration de substrat 10 fois supérieure
à la concentration d'enzyme. plus de 3% du substrat a été transformé en produits de
coupure. Si 1-Cmod n'avait pas montré de "tumover", seulement 10% de produits de
coupure auraient été observés durant le même période de temps.
Ainsi, des essais cinétiques dans lesquels la concentration de substrat est en excès
par rapport à celle de l'enzyme furent réalisés afin de tracer le graphique de la vitesse de
réaction de chacun de ces essais en fonction de la concentration de substrat de l'essai en
question (Figure 15B). En traçant la courbe obéissant A l'équation de Michaelis-Menten
(équation 3, voir section 2.15) par "curve fitting". nous avons pu déterminer les
paramètres cinétiques Km, V,, de même que la valeur de tumover km. Les valeurs
obtenues pour 1-Cmoel sont présentées dans la figure 15C. L'étendue des concentrations
de substrat mises à l'essai fut restreint car les cinétiques effectuées à des concentrations
de substrat supérieures à 500 pM suggéraient que 1-Cmoel subissait une inhibition par
son substrat. Les vitesses de réaction étaient considérablement réduites à des
concentrations de substrat supérieures à 500 pM lors d'essais enzymatiques faits avec une
concentration de 1 pM d'enzyme.
3.9 Séquenpge de I'iatron CrpsbA.4
Une approche qui pourrait peut-être permettre de mettre à jour les résidus de I-
CmoeI impliqués dans l'activité endonucléase serait d'identifier les acides aminés
conservés entre 1-CmoeI et une endonucléase à motif H-N-H phylogénétiquement
distante, mais partageant le même site de reconnaissance. Puisque le quatrième intron du
gène psbA de C. reinhardtii, (CrpsbA.4, 1,8 kb) (Enckson et al., LW), une espèce très
éloignée de C. moewusii, est iasdré à la même position dans le gène psbA que l'intron
codant pour 1-Cmoei et que sa grande taille suggère qu'il contient probablement un ORF,

la protéine codée par cet intron de C. reinhanhi' pourrait être homologue ii 1-Crnoer. La
séquence de CrpsbA.4 n'étant pas disponible pour confmner ces hypothèses, nous avons
entrepris le séquençage de cet intron.
L'intron CrpsbA4 adopte la même structure secondaire que celle de CmpsbA.1,
soit la structure secondaire classique des introns du groupe 1 (Figure 16A). L'intron de C.
reinhardtii, tout comme CmpsbAel, comporte les éléments de séquences conservées P.
Q, R et S et il peut être classé dans le sous-groupe lA, par la présence d'une hélice entre
R et P3 (Burke et al.. 1987). Après une recherche informatisée. un ORF intronique
codant pour une protéine de 320 acides aminés fut localisé dans la boucle M. C'est aussi
dans cette boucle que se situe I'OW de CmpsbA.1. La séquence de I'ORF de CrpsbA4
est très similaire à celle de I'ORF de CmpsbA.1. La figure 16B présente un alignement
des protéines codées par ces ORFs. Les protéines peuvent être alignées sur toute leur
étendue à l'exception d'un seul trou de 7 acides aminés. Les deux protéines introniques
partagent des niveaux de similarité et d'identité relativement élevés (similaires à plus de
65% et identiques à plus de 57%). 11 est donc impossible d'identifier les résidus
importants qui seraient susceptibles de jouer un rôle dans l'activité de ces protéines.
(Figure 16B). Il serait ainsi délicat de cerner quels résidus pourraient être intéressants
pour enclencher de nouvelles études de mutagenèse puisque les résidus conservés sont
trop nombreux.
3.10 Activité et afenitk pour k substrat du mutant H154A
Le premier résidu histidine du motif H-N-H (position 154 chez 1-Cmoel) fut
choisi pour effectuer les premières études de mutagenèse puisque ce résidu est
entièrement conservé dans l'alignement de séquences des protéines de la famille
d'endonucléases "homing" ii motif' H-N-H (Shub et al., 1994). Nous l'avons substitué par
le résidu neutre alanine afin de déterminer s'il joue un rôle dans le clivage ou la
reconnaissance du substrat. Le degré de purification de H154A est comparable à celui
obtenu pour 1-CmoeI au puits 3 de la Figure 8. On estime que la protéine mutante a étd
purifiée A 75% d1homog6n6it6. Après des essais cinétiques et de retardement, le mutant

Figare 16. A) Structure secondaire de CrpsbA 4. L'emplacement des Béments conservés
P, Q, R et S sont indiqués par des lettres en caractere gras. Les flèches
montrent les sites d'épissage 5' et 3' . B) Alignement des séquences en acides
amines des protéines codées par les inbons CrpsbA4 et CmpsbA.1. Les
residus représentes sur fond noir montrent les acides aminés conservés, alon
que les résidus sur fond gris designent les changements conservateurs d'acides
amines. L'encadré montre l'emplacement de la séquence consensus du motif
H-N-H (Shub et ai., 1994). Les r6sidus du motif H-N-H sont en lettres
majuscules et en minuscules sont les &idus de la séquence consensus
conservds entre 50 et 75% des proteines alignées.

U U u - 4
f?; A - U A - U
U u A - u A - U c - O A - u
:=i n : z * u - A A - U
A - U A r !
c - a A C
i c- - : y :
r - " n
: = a C - A C U U
: -

H154A s'est avéré inactif autant au niveau du clivage du substrat que de la furatioo de
l'enzyme à son site de reco~aissance (Figure 17). En ce qui concerne les essais de
coupure. le mutant est demeuré inactif jusqu'à une concentration de 800 nM dans le
milieu réactionnel (Figure 17A). Quant aux essais de retardement, la plus haute
concentration d'enzyme utilisée fut 10 nM et aucun retardement ne fut observé (Figure
17B).

F i r e 17. Analyse de la catalyse et du retardement du substrat par le mutant H l%A. A)
Essais cinetiques effectués avec le mutant H 154A aux concentrations indiquées
et à une concentration de 0,l nM de substrat. Le substrat utilisé était le même
produit PCR de 274 pb que celui employd Ion des essais cinetiques effectués
avec l'enzyme sauvage. B) Essais de retardement du substrat par le mutant
H154A. Des concentrations dquimolaires d'enzyme et de substrat (S) ont et6
placées dans un tampon contenant 10 mM de magndsium pendant 15 minutes à
température pièce. Les puits 1 à 4 correspondent respectivement aux
concentrations équimolaires de I-CmoeI et de substrat de 03, 1, 3 et 10 nM.
Les kchantillons ont 6tt5 ddposés sur gel de polyacrylamide 5% contenant
également 10 rnM MgCl,. Les fleches montrent la position du substrat seul
après Mec trophorèse des khan ti llons.


Expression et purif~catioa
Les résultats de l'expression de I'endonucléase "homing" 1-CmoeI indiquent que
celle-ci ne cause aucun problème de toxicité envers son hôte bactérien, ce qui lui permet
d'être fortement exprimée dans la souche d'E. coli BL21(DE3). De tels problèmes ont été
observés lors de l'expression chez E. coli de l'endonucléase "homing" à motif
LAGLIDADG 1-CeuI (Gauthier et al., 1991). Le site de reconnaissance de cette
endonucléase se retrouve dans chacun des sept opérons codant pour I'ARN ribosomique
dans le génome dlE. coli, plus précisément dans le gène de codant pour la grande sous-
unité de I'ARN ribosornique ( L i u et al., 1993). La séquence reconnue par 1-Ceul est
parmi les séquences d'ADN les plus conservées dans les génomes d'organismes des trois
règnes biologiques. Puisque le site de coupure de 1-CmoeI se situe dans un gène qui se
retrouve exclusivement dans le génome chloroplastique. aucune coupure létale ne peut
survenir dans le génome de la bactérie E. coli.
Le système d'expression pEï30a a permis de produire 1-CmoeI en ,-de
quantité. Cette protéine recombinante compte pour environ 10% des protéines cellulaires
totales après la surexpression, et se retrouve à plus de 95% dans la fraction soluble des
protéines totales. La première chromatographie sur résine Ni" ne permit pas de purifier
1-CmoeI à homogénéité. Plusieurs contaminants sont restks liés à la résine Ni" en
compagnie de 1-CmoeI. La grande majorité de ces contaminants sont des proidines de
plus faibles poids moléculaire que 1-CmoeI, ce qui aurait pu nous faire croire que celles-
ci étaient le rdsultat de l'action de protéases présentes dans l'ensemble des protéines
bactériennes. Mais cette hypothèse semble peu probable puisque toutes les étapes de la
chromatographie d'affinité sur résine Ni" se sont déroulées en présence du puissant
inhibiteur de protéase PMSF. II n'est cependant pas exclu que des protéases intrinsèques
résistantes à cet agent aient causé la degradation observée. La seconde chromatographie
fut iddale pour une proteine à caractère basique telle que 1-CmoeI (pI=10,02) comme en
témoigne le degré d'homogénéité de plus de 95% de la protéine recombinante puriflée.

Le His-Tag de la protéine recombinante n'a pas été retiré à cause de la grande
sensibilité de 1-CrnoeI face à l'entérokinase. Malgré l'absence apparente de sites
spécifiques à l'entérokinase dans sa séquence. 1-CmoeI a subi une forte dégradation
lorsqu'elle fut mise en présence d'entérokinase en utilisant les concentrations relatives
recommandées par le fabricant (Figure 9, puits 6). D'autres essais de clivage ont été
réalises en utilisant des concentrations d'entérokinase moindres que celle recommandée,
mais la coupure n'a pas été nécessairement plus spécifique, des produits de protéolyse
étaient quand même apparus. L'incubation de 1-CmoeI sans entérokinase durant la même
période de temps et à la même température que les réactions de clivage a permis d'exclure
la possibilité que l'enzyme s'autodégrade lorsqu'elle est en solution durant de longues
périodes de temps ( 16 heures) ou qu'elle subisse une dégradation due à une ou plusieurs
protéases intrinsèques à la préparation de protéine (Figure 9, puits 2). Malgré la présence
de la quarantaine d'acides aminés composant entre autre le His-Tag présent en N-
terminal, la protéine non-clivée démontra une grande activité endonucléase. La position
du His-Tag au sein de la structure tertiaire de la protéine ne doit donc pas se situer près
du site actif de l'enzyme.
La présence d'une telle queue de polyhistidines au sein de 1-CmoeI pourrait peut-
être influencer l'activité enzymatique de cette endonucléase. Par exemple. sachant que le
His-Tag possède une affinité pour des métaux divalents (principalement le Ni2'), celui-ci
pourrait influencer le comportement de 1-CmoeI lors d'essais visant à déterminer l'effet
des cations divalents métailiques sur son activité (voir section Determination des
conditions optimales d'activité). Mais i i a maintes fois été démontré que la présence d'un
His-Tag n'interférait pas avec l'activité enzymatique de protéines recombinante et que,
pour les essais enzymatiques en présence de divers cations divalents. l'influence du His-
Tag fut sans doute minime puisque ces essais ont été effectués à des concentrations de
cations divalents (de l'ordre du rnM) en excès par rapport à la concentration de la protéine
recombinante (de l'ordre du nM).
Détermination des conditions optinuiles d'activité
Lors de la détermination des conditions conférant le maximum d'activité à 1-

Cmoei, il fut observé que l'endonucléase est plutôt sensible à la présence de cations
monovalents. D'abord. le tampon entraînant le plus d'activité, le tampon TAPS, ne
contient qu'une faible concentration de I'ion monovalent K+ comme contre-anion.
Ensuite. 1-CmoeI ne requiert pas de sels monovalents supplémentaires dans le milieu
réactionnel. que ce soit sous forme d'un ion monovalent à grand rayon comme le K+ ou
d'un ion monovalent à petit rayon tel que le Na'. Dès l'ajout de sels monovalents (KCl ou
NaCI). l'activité de 1-CmoeI fut grandement réduite. Des observations similiaires ont été
rapportées pour les endonucléases 1-CreI (Wang et al., 1997), 1-CeuI (Drouin. 1998), I-
Scel (Pemn et al.. 1993) et I-PpoI (Wittmayer et Raines. 19%). Mais contrairement à I-
CmoeI, les endonucléases PI-SceI (Gimble et Thorner, l m ) , I-SceIV (Wemette, 1998)
et 1-Dm01 (Dalgaard ef al.. 1994) tolèrent ou requièrent la présence de sels monovalents.
Les études structurales et biochimiques sur les ADN endonucléases à site
spécifique suggèrent que les cations métalliques jouent un rôle important dans l'activité
de ces enzymes (Lykke-Andersen er al.. 1997). L'endonucléase 1-CmoeI ne ddroge pas à
cette règle puisque comme pour plusieurs autres endonucléases "horning" (Lykke-
Andersen et al.. 1997: Marshall er al., 1994; Monteilhet et ai.. 1990: Wang et al.. 1997;
Wittmayer et Raines, 19%) et enzymes de restriction (Vipond et al., 1995). la présence
du Mg+ ou d'un autre cation divalent dans le milieu réactionnel est essentielle à son
activité. 1-Cmoel montre une affinité relativement forte pour le Mg" puisquien présence
de concentrations extrêmement faibles en Mg2+, elle coupe son substrat. Une dialyse
contre un tampon contenant de I'EDTA fut requise avant d'effectuer l'essai enzymatique
démontrant que le Mg" est requis comme cofacteur. Après la dialyse. 1-CmoeI regagne
son activité endonucléase en présence de Mg2+ dans le milieu réactionnel mais est
inactive en absence de ce dernier. Toutefois, 1-CmoeI nécessite une concentration de
Mg2+ assez élevée dans le milieu réactionnel pour atteindre son maximum d'activité.
D'autres cations divalents peuvent aussi induire le clivage du substrat par I-
Cmoel. Seulement quatre des neuf cations testés, soit le MC? Ca", Si ' et Ba2+ ont
supporté l'activité de 1-CmoeI, don que les ions Mn2+, Cu2+, Zn2+, Ni" et Co" n'ont pas
permis la coupure du substrat. Nous puvons donc affirmer que 1-CmoeI montre une trZs

,-de spécificité relativement aux cations métalliques puisque les quatre cations ayant
induit le clivage font partie de la même famille d'éléments du tableau périodique. En
effet, le magnésium, le calcium, le strontium et la banum font partie de la famille des
alcalino-terreux. C'est donc dire qu'ils possèdent la même configuration électronique et
que seule leur taille diffêre. On peut d'ailleurs remarquer que plus la taille du cation est
importante, plus l'activité enzymatique diminue et ce, autant à une concentration de
cation de t m M que de 10 m M (Fig. IOD). Cette spécificité est pratiquement unique car
peu d'endonucléases " homing " font une telle discrimination quant au choix de leur
cofacteur, ce qui est aussi le cas pour les enzymes de restriction (1-CreI, Wang et al..
1997; 1-DmoI, Aagaard et al., 1997: 1-PorI. Lykke-Andersen et al.. 1997; 1-PpoI,
Wittmayer et Raines, 19%; PI-SceI, Wende et al., 19%: EcoW et EcoRV. Vipond et al..
1995). Même la colicine E9, une endonucléase bactérienne de la famille à motif H-N-H
et une des seules protéines de cette famille à avoir été caractt5risée, ne fait pas preuve
d'une telle spécificité quant au choix des cations divalents pouvant agir comme cofacteur
(Pommer et al., 1998). Outre le Mg+, seuls les cations divalents dérivés des métaux de
transitions tels que le nickel, le cobalt et la manganèse peuvent être utilisés comme
cofacteur par cette endonucléase.
Le fait que les ions Ca" favorisent le clivage du substrat par 1-Cmoel est assez
surprenant car peu d'endonucléases "homing" ni même d'enzymes de restriction montrent
de l'activité en présence de ce cation (Vipood et al., 1995). Une des seules endonucléases
"homing" montrant de I'activitk en presence de Ca2+ est I'endonucldase à motif His-Cys
Box 1-Ppoi (Wittmayer et Raines, 19%). Par ailleurs, la DNase I du pancréas de bovin
nécessite des ions Ca" comme cofacteur structural ainsi qu'un deuxième cation divalent
métallique afin de procéder au clivage de son substrat (Price, 1975). Pour sa part, la
colicine E9 n'est pas activée en présence de Ca" (Pommer et al., 1998). Généralement, la
présence de Ca2+ permet aux ADN endonucldases de se lier à leur substrat sans toutefois
pouvoir le cliver (Vipond et Halford, 1995). Cette propriété est utile lonqu'on effectue
des études d'affinitd pour le substrat car ce dernier ne doit pas être coupé dans ces
expériences.

Il est également surprenant qu.1-CmoeI ne clive pas son substrat en présence de
Mnz+, un cation pouvant efficacement remplacer le M g ' comme cofacteur dans le cas de
plusieurs endonucléases "homing" et enzymes de restriction (Aagaard et al., 1997:
Vipond et al., 1995). L'absence de retardement du substrat en présence de Mn2+ suggère
que ce cation ne supporte pas l'activité de 1-CmoeI parce qu'il n'y a simplement pas
formation du complexe enzyme-substrat. Ce cation n'aurait pas la configuration
électronique nécessaire pour se fixer au site de coordination des ions métalliques reconnu
par les cations divalents ayant engendré le clivage. Ceci doit fort probablement être aussi
le cas pour les quatre autres cations qui n'entraînent pas le clivage du substrat. Comme il
a été mentionné précédemment. 1-CmoeI montre une spécificité unique quant aux cations
divalents pouvant agir comme cofacteur puisque seuls les cations faisant partie de la
famille des alcalino-terreux supportent l'activité de l'enzyme. Les éléments de cette
famille possèderaient donc la conf~guration électronique requise pour lier le site de
coordination de la protkine et modifier celle-ci afin qu'elle se fixe efficacement à son site
de reconnaissance. Ainsi, la taille du cation divalent n'est pas le facteur limitant pour lier
le site de coordination puisque de l'activité fut notée en présence du plus gros cation testé.
soit le Ba" dont le rayon ionique est de - 136 pm (Shannon et Prewitt. 1%9).
1-Cmoei ne se distingue pas des autres endonucléases "homing" en ce qui
concerne le pH et la température. Le clivage du substrat par 1-CmoeI est à son meilleur ii
un pH alcalin de 8.5, ce qui est similaire au pH optimal observé pour 1-CreI (Wang et al.,
1997). 1-Ceul (Drouin. 1998). 1-Dmol (Dalgaard et al., 1994). 1-SceI (Monteilhet et al.,
1990) et PI-SceI (Gimble et Thomer. l m ) . A des pH supérieurs à 9,5, l'activité de I-
CmwI est grandement réduite. La température de clivage optimale de I-CmoeI est de
37°C alors que des températures plus élevées (50 et 60°C) ont eu un effet inhibiteur.
Cette faible activité enzymatique à de hautes températures n'est pas surprenante puisque
C. moewusii n'est pas une algue thennophile. La majorité des endonucléases "homing"
caractérisées j usqu'a présent ont une température d'activité optimale qui tourne autour de
37"C, seule l'endonucléase "homing" d'une archaebactérie thermophile 1-Dm01 possède
une température optimale entre 60 et 70°C (Daigaard, et al., 1994).

Stabiiité de 1-CmoeI
1-CmwI montre une stabilité hors du commun lonqu'on préincube cette enzyme
durant de longues périodes de temps en absence de son substrat avant de l'employer pour
une réaction enzymatique de pseudo premier ordre standard. En présence d'un tampon de
préincubation contenant des ions M c , 1-CmoeI conserve 50% de son activité après plus
de 230 minutes de préincubation. Ceci est fort différent de ce qui a été observé pour les
endonucléases "homing" 1-CreI, 1-CeuI et 1-SceI qui, toujours en présence de Mg", ont
une demi-vie de 18, -10 et 1 minutes à 37OC (Wang et al., lm: Drouin, 1998;
Monteiihet et al., 1990). En absence de M c , 325 minutes de préincubation sont
nécessaires pour que 1-Cmoel perde la moitié de son activité initiale.
Stœcbiométrie de liaison de 1-CmoeI h son site de reconnaissance
Par détermination du poids moléculaire des complexes ADN-protéines à l'aide de
l'analyse de Ferguson. il a été démontré que 1-CmoeI se lie à son substrat sous forme d'un
monomère de 4 3 m a . Quelques autres endonucléases "homing" se lient également à
leur substrat sous fome de monomère. Par exemple. la colicine E9 à motif H-N-H se
comporte aussi comme un monomère en solution, mais la stœchiométrie de la liaison à
son substrat n'a pas été déterminée (Pommer et ai., 1998). De plus. les endonucléases
"homing" à double motif LAGLIDADG 1-SceI (Montheillet et ai.. 1990) et PI-SceI
(Gimble et Thomer. 1993), de même que l'endonucléase "homing" à motif GIY-YIG I-
TevI (Mueller et al., 1995) sont des monomères. Par ailleurs. les endonucléases
"homing" à simple motif LAGLIDADG 1-CreI (Wang et aL, 199'7) et 1-Ceul (Drouin,
1998), et l'endonucléase "homing" A motif His-Cys Box 1-Ppol (Eilison et Vogt, 1993) se
lient à leur substrat sous forme de dimères tout comme les enzymes de restriction EcoRI
et BamHI (McClarin et al., 1986). Par conséquent, dans le cas des endonucléases
"horning" de la famille LAGUDADG, l'état de monomère ou dimère des protéines
dépend du nombre de copies du motif dans la séquence protkique. Puisque les
endonucléases se liant ii leur substrat sous forme de dimères comportent chacune une
seule copie du motif LAGLIDADG, dors que les monomères en comptent deux, ii
semble que deux copies du motif LAGLiDADG sont absolument nCcessaires pour mener
une activité endonucléase (Belfort et Robeas, 1997). On considère d'ailleurs que les

endonucléases "homing" à double motif LAGLIDADG ont évolué par duplication
génétique à partir de protéines à simple motif (Belfort et Roberts, 1997).
Séquençage de I'intron CrpsbA.4
Les résultats du séquençage du quatrième intron du gène psbA de C. reinhardtii
ont démontré que cet intron est très similaire au premier intron du même gène de C.
moewusii. De plus, les protéines codées par chacun des O W introniques sont aussi très
similaires. En utilisant une méthode d'alignement de séquences en acides aminés déjà
utilisée dans notre laboratoire, nous avons voulu cerner les résidus pouvant être
importants pour l'activité de 1-Cmoel (Tunnel et al., 1997). Puisque les deux protéines
introniques sont si similaires, il fut impossible de déterminer quels résidus de 1-CrnoeI
pourraient être importants pour la reconnaissance ou le clivage du substrat outre les
résidus conservés du motif H-N-H. C'est pourquoi nous avons convenu de muter le
premier résidu histidine du motif H-N-H.
Mutant H154A
Le résidu Hl54 de 1-Crnoel, le premier résidu histidine du motif H-N-H, est
critique pour l'activité de cette endonucléase "homing" puisqu'en le substituant par le
résidu neutre alanine. la protéine mutante H154A s'est avérée inactive autant au niveau de
la catalyse de la réaction que de la fixation de l'enzyme à son site de reconnaissance.
D'après ce résultat. il semble que ce résidu histidine soit un élément clé du site actif de
cette enzyme. Ceci était quelque peu attendu puisque ce résidu est entièrement conservé
dans les membres de la famille de proteines à motif H-N-H (Shub et al., 1994). Mais ce
résidu ne doit pas être le seul h jouer un rôle important dans le clivage et la
reconnaissance du substrat. Comme l'indiquent les études de mutagenèse aidatoire
effectuées sur le domaine endonucldase de la coiicine E9, d'autres résidus du motif H-N-
H ont sfirement une implication importante dans l'activité de 1-CmoeI (Garinot-Schneider
et al., 1996). Ces études ont demontré qu'en changeant le deuxième résidu histidine du
motif H-N-H par une alanine, l'activité endonucléase de cette protéine était abolie. Ce
mutant H575A conservait n6anmoins la capacité de se lier à son site de reconnaissance.
Conservé à plus de 75% dans la famille H-N-H, le deuxième résidu histidine est prdsent

dans la séquence en acides aminés de 1-CmoeI et aussi dans celle de la protéine codée par
Ifintron CrpsbAJ. La forte conservation de ce résidu suggère que chez 1-CmoeI, il
jouerait le même rôle que celui proposé pour la colicine E9 (voir ci-dessous).
L'étude divulguant la structure tridimensionnelle de la colicine E9 a permis de
suggérer plus précisément un rôle pour chacun des résidus formant le motif H-N-H
(Kleanthous et al., 1999). Il a été démontré que le deuxième résidu histidine du motif
conservé serait davantage impliqué dans la liaison du cation divalent métallique agissant
comme cofacteur (Kleanthous et al., 1999). Ce modèle est en accord avec l'absence de
clivage observée pour le mutant His575Ala par Garinot-Schneider et al. puisque la
présence d'un cofacteur métallique est essentielle pour l'activité de la colicine E9
(Pommer er al.. 1998). Au lieu du rôle dans la coordination de l'ion métallique, Garinot-
Schneider et aL avaient antérieurement propose que le dernier résidu histidine servait de
base générale pour activer une molécule d'eau liée lors de la catalyse (Garinot-Schneider
et al., 1996). Quant au premier résidu histidine du motif H-N-H. un rôle dans la liaison
du phosphate scissile de l'ADN lui a été proposé, ce qui est en accord avec nos études de
mutagenèse montrant que cet acide aminé est critique pour la liaison de 1-CmoeI à son
substrat (Kleanthous er al., 1999). Finalement, un rôle de stabilisation de structure
transitoire lors du clivage de l'ADN fut proposé pour l'asparagine du motif. Mais les
rôles précis du premier résidu histidine et de l'asparagine restent à être démontrés
expérimentalement.
Par ailleurs. il fut dernièrement démontre que des résidus histidine et asparagine
jouent aussi un rôle déterminant dans I'activite de 1-001, une endonucléase "homing" à
motif His-Cys Box (Friedhoff et al., 1999). Le motif conservé des protéines de cette
famille comporte une série d'environ LOO acides aminés particulièrement riches en résidus
histidine et cystéine (khansen et al., 1993). L'étude de Friedhoff et al. montre qu'un
rCsidu histidine conservé est le résidu clé dans l'activité catalytique de 1-401. Ce résidu
serait impliqué dans la liaison d'une molécuie d'eau menant à l'attaque du lien phosphate
scissile de l'ADN. Un résidu asparagine fait aussi partie du site catalytique de 1-Ppol via
un rôle de coordination du cation Mg2+ au site actif de l'endonucléase (Friedhoff et al..

1999). Ce mécanisme d'action a aussi été proposé pour la nucléase non-spécifique de
Serraria (Miller et al., 1994: Friedhoff et al., 1996). Les résidus histidine et asparagine
les plus importants de 1-401 ont leur réplique dans la nucléase de Serrafia (Fnedhoff et
al., 1999). ce qui indique que ces deux endonucléases ont indépendamment évolué pour
en arriver à posséder un site actif fortement semblable. En comparant le mode d'action de
ces deux enzymes avec celui proposé pour les endonucléases à motif H-N-H, on constate
que les mêmes acides aminés ont des fonctions différentes au sein des protéines de deux
familles distinctes d'endonucléases " homing''.
Paramètres cinétiques de 1-CmoeI
Les paramètres cinétiques de 1-CmoeI ont été déterminés selon deux modèles
cinétiques différents. Le premier modèle. nommé "single turnover partiel". utilisé a été
élaboré par Halford et al. (Halford et al., 1980; Halford et Johnson, 1983) lors des études
des réactions de EcoRi en présence de substrats "non-cognate" (c'est-à-dire des substrats
comportant des sites similaires au site de reconnaissance de EcoRI qui sont reconnus et
clivés par l'enzyme). Ce modèle convient à des réactions enzymatiques pour lesquelles la
concentration d'enzyme est en excès par rapport à celle du substrat. Un tel modèle a aussi
été appliqué pour les essais cinétiques des endonucléases "homing" I-CeuI et 1-CreI
puisqu'il fut démontré que celles-ci restaient fortement liées à leur proauits de coupure
après le clivage du substrat (resultats non-publids de notre laboratoire). Les paramètres
cinétiques qui peuvent être soutirés du graphique dit de Halford (Figure 15A), sont le
k,, et le Km*. Le k, représente la valeur la valeur maximale que prend la constante de
vitesse k? dans un graphique de constante k, en fonction de la concentration de substrat
dans une série d'essais enzymatiques effectués à une concentration d'enzyme fixe. Quant
au rapport ~JK,,,', il représente une constante apparente de deuxième ordre correspondant
à la spécificité de l'enzyme pour son substrat La valeur Km8 de 43 pM obtenue pour I-
CmoeI suggère que cette enzyme a une forte affinité pour son substrat, mais que cette
dernière est beaucoup moins grande comparativement à celle de 1-CreI. une endonucléase
"homing" à motif LAGLIDADG. Le Km' de 1-CreI est de l'ordre de 0.01 pM, soit environ
4000 fois moindre que celui de 1-Cmoeï (résultats non-publiés de notre laboratoire). Très
peu d'endonucl&ises "homing" ni même d'enzymes de restriction ont ét6 caractérisées par

la méthode de "single turnover partiel". ce qui explique la difficulté de comparer les
paramètres cinétiques de 1-CmoeI avec ceux de d'autres enzymes.
Les quelques endonucléases "homing" dont les paramètres cinétiques ont été
publiés dans la litîérature ont toutes été étudiées dans les conditions de Michaelis-
Menten. La majorité d'entre elles font partie de la farniile LAGLIDADG mise à part
L'endonucléase 1-001 de la famille His-Cys box. Puisque l'activité des endonuciéases
"homing" et des enzymes de restriction est grandement influencée par les conditions de
réaction, la comparaison des paramètres cinétiques de différentes endonucltases peut
parfois être difficile à interpréter. Néanmoins, en supposant que les paramètres
cinétiques de toutes les endonucléases étudiées ont été obtenus dans les conditions
optimales de clivage, des comparaisons peuvent être faites entre ces paramètres. La
valeur Km de LOO pM que nous avons observé pour I-Cmoel. indique que cette
endonuclCase "homing" possède une forte affinité pour son substrat. Le Km de 1-CmoeI
est relativement bas comparativement aux valeurs de Km des endonucléases "homing" I-
Scel, I-DmoI. I-Pori et I-SceIV. qui sont respectivement de 34000.6000.4ûûû et 150 pM
(Monteilhet et al, 1990: Dalgaard et al.. 1994; Lykke-Andersen et al.. 1994; Wernette,
1998). Dans le cas des enzymes de restriction EcoRI. EcoRV et BamHI. leurs Km pour le
clivage d'ADN plasmidique sont respectivement de l'ordre de 2500, 500 et 3600 pM
(Halford et Johnson. 1981: Halford et Goodall. 1988: Nardone et Chinkjian. 1987). Seul
le Km de I'endonucléase "homing" à motif His-Cys box avec une valeur de 10 pM est
inférieur à celui de 1-Cmoel (Wittmayer et Raines, 19%). Quant aux taux de turnover.
les km respectifs de 1-Cmoel, I-Ppo1 et 1-SceIV sont de 0.256. 0.046 et 0,003 min-'
comparativement à ceux des enzymes de restriction EcoRi, EcoRV et BamHI qui sont de
7.7,0,9 et 1.5 min". Les faibles taux de turnover des endonucléases "homing" pourraient
suggérer que ces enzymes demeurent liées il leur produits de coupure (ou s'en libèrent
lentement) après le clivage du substrat afin de faciliter les événements de recombinaison
homologue suivant la coupure d'ADN lors du processus de mobilité d'introns. Ceci n'est
toutefois pas le cas pour 1-Cmoei puisque nous avons démontré que cette endonucléase
libère ses produits de coupure après avoir clive son substrat. Ceci explique la valeur
superieure de k, de 1-CmoeI par rapport à celle de 1-SceIV dont la rétention des produits

de coupure après la catalyse a été suggérée (Wernette, 1998).
Les résultats de la détermination des paramètres cinétiques suggèrent que 1-Cmoel
subit une inhibition réversible par son substrat lorsque la concentration de ce dernier dans
le milieu réactionnel est élevée. L'inhibition par le substrat est un phénomène bien
documenté dans la litiérature, ayant même été sujet d'une nouveau modèle cinétique
(Wang ef al.. 1999). Puisqu'une expérience de préincubation de 1-CmoeI en présence et
en absence de substrat a révélé que l'enzyme perdait de l'activité en fonction du temps
(résultats non-illustrés), le calcul des vitesses initiales durant des temps de réaction très
courts devrait être privilégié pour la détermination les paramètres cinétiques en
conditions de Michaeiis-Menten afin de pallier la pene d'activité de 1-CmoeI en fonction
du temps.
Conclusion et perspectives
En somme. cette étude a permis de purifier efficacement une première
endonucléase intronique à motif H-N-H et de déterminer certaines de ses propriétés. Un
protocole efficace d'expression et de purification a étt? mis au point à t'aide du vecteur
PET-30a et des chromatographies d'affinité et d'échanges de cations. Les préparations
homogènes obtenues ont permis de mener à une solide caractérisation enzymologique de
l'endonucléase. 1-Cmod montre un patron unique de spécificité pour les cations divalents
pouvant agir comme cofacteur puisque seuls les cations faisant partie de la famille des
alcalino-terreux ont favorisé le clivage. Contrairement à quelques endonucléases
"homing", 1-CmoeI fait preuve de turnover lors du clivage de son substrat puisque qu'elle
ne demeure pas liée à ses produits de coupure après le clivage de ce dernier. 1-CmoeI
possède une forte affinité envers son substrat comme le montre la valeur de Y, de l'ordre
du picoMolaire. Contrairement 3 quelques endonucléases "homing " et même enzymes de
restriction qui sont des dimères, 1-Cmoeï se lie à son substrat sous forme d'un monomère.
Finalement, le premier résidu histidine du motif' H-N-H est critique pour l'activité de cette
endonucléase comme le prouve l'inactivité totale du mutant H 154A.
En perspective, il serait important de caracteriser le site de reconnaissance de 1-

CmoeI afin de déterminer, par exemple, la longueur minimale du site de reconnaissance
et les bases importantes pour la fixation de l'enzyme. II serait également important de
produire et caractériser d'autres variants du motif H-N-H afin d'obtenir de nouveaux
indices concernant l'implication de ce motif dans I'activité de I'endonucléase. Enfin, il
serait intéressant de tenter des essais de cristallogenèse de 1-CmoeI liée à son substrat a h
de résoudre la structure tridimensionnelle d'une première endonucléase intronique de la
famiHe à motif H-N-H.

Aagaard. C.. Awayez. M.J. et Garrett. R.A. ( 1997) Profiles of the DNA recognition site
of the archeal homing endonuclease 1-Drnol. Nucleic Acids Res. 25. 1523- 1530.
Belfort. M. et Perlman. P.S. ( 1995) Mechanisms of Uitmn mobüity. J. Biol. Chem. 270,
30237-30240.
Belfort. M. et Roberts. R.J. (1997) Homing endonucleases - Keeping the house in
order. Nucleic Acids Res. 35,3379-3388.
Bell-Petenon. D.. Quirk. S.M.. Bryk. M. et Belfort. M. ( 1991) 1-Tevi, the endonuclease
encoded by the mobile id intron, recognizes binding and cieavage domnins on its
DNA target. Proc. Nat. Acad. Sci.. U.S.A. 88.77 19-7723.
Bergeron. A. ( 1990) Analyse srnichirale d'un ADN linéaire de six kilopaires de bases
chez Chhnrydonionas moewusii. Mémoire M . Sc. Université Laval
Boulanger. J. ( 1988) Comparaison des gènes chlomplastiques psbA de
Chlumydo~nonas eugametos et Chlamydomonas ntoewusii. Mémoire M . Sc. Université
Laval
Bryk. M.. Belisle. M.. Mueller. LE. et Belfort. M. ( 1995) Selection of a remote cleavage
site by 1-TevI, the id htmn-encoded endonuclease. J. Mol. Biol. 247. 197-2 10.
Burke. J.M.. Belfort. M.. Cecb. T.R., Davies. R.W.. Schweyen, R.J.. Shub. D.A.. Szostak.
LW. et Tabak. H.F. ( 1981) Structural conventions for group 1 intmns. Nucleic Acids.
Res. l S , Z 17-722 1.
Bussières. J., Lemieux. C., Lee. R.W. et Tunnel. M. ( 19%) Optional elements in the
chloiioplast dans of Chlumydonroiurs eugametos and C. m o e w d : unidireetional gene

conversion and co-conversion of adjacent markers in higb-viability crosses. Curr.
Genet. 30.356-365.
Cech. T.R. ( 1990) Self-splicing of g m i p I introns. Annu. Rev. Biochem. 59,543-568.
Cech. T.R. et Bass. B.L. ( 1986) Biological catalysis by RNA. Annu. Rev. Biochem. 55.
599-629.
Colleaux. L.. d'Auriol. L.. Betennier. M.. Cottarel. G.. Jacquier. A.. Galibert. F. et Dujon.
B. ( 1986) Universal code equivalent of a yeast mitochondrial intmn reading frame
is expressed into E. coli as a specifi double strand endonuclease. CellJ4.32 1-93.
Cumming. D.J.. Michel. F. et McNally. K.L. (1989) DNA sequence analysis of the
apocytochrome b gene of Podospora anserina: a new f a d y of intmnic open reading
frame. Curr. Genet. 16.4û7-418.
Dalgaard. J.Z.. Garret. R.A. et Belfort. M. (19%) Purifkation and chamctezation of
two forms of 1-DmoI, r thermophilic site-specifm ensonuclease encoded by an
archeal intron. J. Biol. Chem. 269,2888528892.
Derbyshire. V.. Kowalski. J.C.. Dansereau. J.T.. Hauer. C.R. et Belfort. M. (1997) Two-
domain structure of the td intron-encoded endonuclease 1-TevI corniates with the
hvo-domain conf~umtion of the homing site. J. Mol. Biol. 265.494-506.
Drouin, N. ( 1998) Rôle des résidus conservés de i'endonucléase 1-CeuI dans la
reconnaissance et le clivage de son substrat. Mémoire M. Sc. Université Laval
Duan. X.. Gimble. F.S. et Quicho. F.A. ( 1997) Crystai structure of PI-SceI, a homing
endonuclease with protein spücing activity. Cell89,555W.
Eddy, S.R. et Gold, L. ( 1991) The phage T4 nrdB Uitron: a deletion matant of a

version found in the wild. Genes Dev. 5. 1032- 1 û41.
Ellison. E.L. et Vogt. V.M. ( 1993) Interaction of the intron-encoded mobiüty
endonuclease 1-PpoI with its target site. Mol. Cell. Biol. 13.791-7539.
Enckson. J.M.. Rahire. M. et Rochaix. J.-D. ( 1984) Chloniydomonas reinhardlii gene
for the 32 000 mol. w t protein of photosystem II contains four large inbons and is
located entirely within the chlomplast inverted repeat. EMBO J. 3.2753-2762.
Ferpuson. K.A. ( 1964) Sbrch gel electrophoresis - application to the classification of
pituitary proteins and polypeptides. Metabolism 13.985- 1 102.
Flick. K.E.. Jurica. M.S. et Monnat. R.J. (1998) DNA binding and cleavage by the
noclear intron-encoded homing endonnclease 1-PpoI. Nature 394. %- 10 1.
Friedhoff. P.. Kolmes. B.. Girnadutdinow. O.. Wende. W.. Knuse. K.L. et Pingoud. A.
(19%) Anabsis of the mechanism of the S e d a nuclease ushg s i te-dkted
mutagenesis. Nucleic Acids Res. 24.2632-2639.
Friedhoff. P.. Franke. 1.. Meiss. G.. Wende. W.. Krause. K.L. et Pingoud. A. ( 1999) A
similar active site for non-spocifc and specüie endonucleases. Nature Struct. Biol. 6.
1 12-1 13.
Garinot-Schneider. C., Pommer. A.J., Moore. G.R., Kleanthous, C. et James. R. (1996)
Identif~cation of putative active-site residues in the DNase domsin of cok in E9 by
random mutagenesis. J. Mol. Biol. 260,73 1-742.
Gauthier. A.. Tunnel. M et Lemieux, C. (1991) A p o p 1 intron in the cbloropiast
sobunit rRNA gene of Chlon?ydomonus eugonietos encodes a double-stmnd
endonuclease that cleaves the homiag site of this intmn. Curr. Genet. 19,4347.

Gimble. F.S. et Stephens. B.W. ( 1995) Substitution in conserved dodecapeptide motifs
that uncouple the DNA binding and DNA cleavage activities of PI-SceI
endonuclease. J. Biol. Chem. 270.5849-5856.
Gimble. F.S. et Thomer. J. ( 1993) Purifkation and charaeterizstion of VDE, a site-
specifie endonuclease €rom the yeast Saccuromyces cerevisioe. J. Biol. Chem. 268.
2184421853.
Goodrich-Blair. H. et Shub. D.A. ( 19%) Beyond homing: cornpetition between intmn-
endonulease confers a selective advantage on flanking genetic markers. Cel184.2 1 1 -
22 1.
Gorbalenya. A.E. (1994) Self-splicing gmup 1 and group II inhons encode
homologous (putative) DNA endonuclease of a new fardy. Protein Sci. 3. 1 1 17- 1 120.
Guthrie. C. ( 1991 ) Messenger RNA splicing in yeast: Clues to why the spliceosome is
a ribonucleoprotein. Science. 253. 157- 1 63.
Halford. S.E.. Johnson, N P . et Grinsted. J. ( 1980) The EcoRI restriction endonuclease
with bacteriophage h DNA Kinetic studies. Biochem. J. 191.B 1-592.
Halford. S.E. et Goodall. AJ. ( 1988) Modes of DNA cleavage by the EcoRV
restriction endonuclease. Biochemistry 27. 177 1 - 1777.
Halford. S.E. et Johnson, N.P. (1983) Single turnovers of the EcoRI restriction
endonuclease. Biochem. J. 211,405-45.
Heath, P.J.. Stephens. KM., Monnat. RJ. et Stoddard. B.L. ( 1997) The stmctiuP of I-
CreI, a p u p I Intron-encded homuig endonuclease. Nature Stmct. Biol. 4.468476.
Johansen, S., Embley, T.M. et Willassen, N.P. (1993) A f a d y of nuclear homing

endonucleases. Nucleic Acids Res. 21, M5-411 .
Jurica. M.S.. Monnat. R.J. et Stoddard. B.L. ( 1998) DNA recognition and cleavage by
the LAGLIDADG homing endonuclease 1-CreI. Mol. Cell. 2.469476.
Kleanthous. C.. Kuhlmann, U.C.. Pommer, A L . Ferguson. N.. Radford. S.E.. Moore.
G.R.. James. R. et Hemmings. A.M. (19991 Structural and mechanistic basis of
immunity toward endonuclease colicins. Nature S tnict. Bi01 .6. 243-252.
Kowalski. J.C.. Belfort. M.. Stapleton. M.A., Holpen. M.. Dansereau. J.T.. Pieirokovski.
S.. Baxter. S.M. et Derbyshire. V. ( 1999) Confqumtion of the catalytic GN-YIG
domain of intmn endonuclease 1-TevI: coincidence of cornputational and molecular
fmdings. Nucleic Acids Res. 27.2 1 15-2 125.
Laemmli. U.K. ( 1970) Cleavage of structural proteins during the assembly of the
head of bacteriophage TJ. Nature 227,680-685.
Lambowitz. A.M. et Belfort. M. (1993) Introns as mobile genetic elements. Annu.
Rev. Biochem. 62. 587-622.
Lemieux. C.. Tumiel. M.. Lee. R.W. et Bellemare. G. (1985) A 21 küobase-pair
deletiodaddition diîference in the inverted repeat sequence of chlomplast DNA
from Chlamydomonas eugametus and C. moewusii. Plant Mol. Biol. 5.77-84.
Lemieux. C. et Lee. R.W. ( 1987) Nomiprocal recombination ktween aileles of the
chlompiast U s rRNA gene in interspeciuc ChIrtmydomoms crosses. Proc. Natl.
Acad. Sci,. U.S.A. 84.41664170.
Liu, S.-L.. Hessel. A. et Sanderson, K.E. (1993) Genomie mapping with 1-CeuI, an
inha-encoded endonuclease s p i & for genes for ribosomal in Sufmonello spp.,
Escherichia d i and other bacteria Roc. Natl. Acad. Sci ., U.S.A. 90,6874-6878.

Lykke-Andersen. J.. Garrett. R.A. et Kjems. i. ( 1997) Mapping metal ions at the
catalytk centres of hvo intron-encoded endonucleases. EMBO J. 16.3272-328 1.
Marshall. P. et Lernieux. C. ( 1991) Cleavage pattern of the horning endonuclease
encoded by the 6fth intmn in the chloroplast large subunit rRNA gene of
Chlamydornonas eugantetos. Gene 104.24 1-245.
Marshall, P.. Davis. T.B. et Lemieux. C. (1994) The 1-CeuI endonuclease: its
purification and its potential role in the evolution of the f ~ t h intron tCeLSU.5) of
the rmL gene of Chlamydornonas eugametos. Eur. J. Biochem. 220.855859.
McClarin. J. A.. Fredenck. C. A.. Wang. B. C.. Greene. P.. Boyer. H. W.. Grable. J. et
Rosenberg, J. M. (1986) Srnichire of the DNA-EcoRI endonuclease recognition
complex at 3 A resoluiion. Science. 234. 1526- 1 4 1 1.
Miller. M.D.. Tanner. J.. Alpaugh. M.. Benedik. M.J. et Krause. K.L. ( 1994) 2.1 A
structure of Serratia endonuclease suggests a mechanism for binding to double-
stranded DNA. Nature Stmct. Biol. 1.46 1 -C68.
Monteiihet. C.. Pemn. A.. Thierry. A.. Colleaux. L. et Dujon. B. ( 1990) Purification and
characterization of the in v i h o activity of 1-SceI, a novel and highly speeitlc
endonuclease encoded by gmup 1 introns. Nucleic Acids Res. 18. lU7- 1413.
Mueller. J.E.. Bryk, M.. Loizos. N. et Belfort, M. ( 1993) Homing endonucleases. In
Linn, S.M., Lloyd, R.S.. et Roberts. R.J. (eds.). Nueleases. Cold Spring Harbor
Laboratory Press. Col spring Harbor W . vol. 2, pp. 1 1 1 - 144.
Mueller. J.E.. Smith. D., Bryk, M. et Belfort, M. ( 1995) Intron-eacoded endonuclease
1-Tevl binds as a monomer to effet seqaencinl ckavage via conformational changes
in the td homing site. EMBO J. 14.57245735.

Muscarella. D.E.. Ellison. E.L.. Ruoff, B.M. et Vogt. V.M. (1990) Characterizstion of
1-Ppol, an intron-encoded endonucîease that mediates homing of a gmup 1 intmn in
the ribosomal DNA of Physarum polycephalum. Mol. Cell. Biol. 10.3386-33%.
Nardone, G. et Chinkjian, J.G. ( 1 9 8 7 ) The enzymes of the BamHI restriction-
modification system. Gene Ampiif. Anal. 5, 147- 1%.
Perlman. P.S. et Butow. R.A. ( 1989) Mobile introns and intron-ecoded proteins.
Science, 246. 1 106- 1109.
Pemn. A.. Buckle. M et Dujon. B. ( 1993) Asymmetrical recognition and activity of the
1-SceI endonuclease on its site and on intmn-exon junctions. EMBO I. 12. 2939-
2947.
Pingoud. A. et Jeltsch. A. ( 1997) Recognition and cleavage of DNA by typo-II
restriction endonucleases. Eur. J. Biochem. 346. t -22.
Pommer, A.J.. Wallis. R.. Moore, GR., James. R et Kleanthous. C. ( 1998)
Enzymological characterization of the nuclease domain €mm the bacterial toxin
c o l i ~ h E9 from Escherichia coli. Biochem. J. 334.387-392.
Price. P.A. ( 1975) The essential role of Ca" in the activity of bovine pancreatic
deoxyribonuclease. J. Biol. Chem. 250. 198 1 - 1986.
Shannon et Prewitt ( 1969) Acta Crystallogr. B25.2939-2947.
Shub, D.A.. Goodrich-Blair. H. et Eddy. S.R. (1994) Amino acid sequence motif of
gmap I intron endonuclease is conserved in open reading frames of groop II
introns. Trends Biochem. 19,4û2-4û4.

Thompson. J.D.. Higgins. D.G. et Gibson. T.J. (1994) CLUSTALW: impmving the
sensitivity of progressive multiple sequence alignment thmugh sequence weighting,
positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22.
4613-4680.
Turmel. M.. Bellemare. G et Lemieux. C. (1987) Pbysical mapping of dinerences
behveen the chloroplast dans of the interfertile algae Chlamydonionas eugametos
and Chlnmydomonus moewusii. Curr. Genet. 11.543-552.
Turmel. M.. Boulanger. J. et Lemieux. C. (1989) Two group 1 introns with long
interna1 open reading frames in the chiomplast psbA gene of Chlamydononas
rnoewusii. Nucleic Acids Res. 17,3875-3887.
Tunnel. M.. Boulanger. J.. Schnare. M N . . Gray. M.W. et Lemieux. C. ( 199 1) Six group
1 introns and three intemal transcribed spacers in the chlompîast large snbunit
ribosomal RNA gene of the green alga Chlumydomonas eugametos. J. Mol. Biol. 218.
293-3 1 1 .
Turmel. M.. Otis. C.. Côté. V. et Lemieux. C. (1997) Evolutionariiy conserved and
functionaUy important residues in the 1-CeuI h o h g endonuclease. Nuclei c Acids
Res. 25.26 10-26 19.
Vipond. I.B. et Halford. S.E. (i995) Specifii DNA recognition by EcoRV restriction
endonuclear induced by calcium ions. Biochemistry 34. 1 1 13- 1 1 19.
Vipond, I.B.. Baldwin. G.S. et Halford. S.E. (1995) Divaient metal ions at the active
sites of the EcoRV and EcoRI restriction endonacleases. Biochemistry, 34,697-704.
Wang, J., Dim. H.-H., Yan, X. et Hemn, DL. (1997) Purifjcation, biochexnicai
characterhation and protein-DNA interactions of the 1-Crel endonaekase p d u c e d
in Eschenchia c d . Nucieic Acids Res. 25,3767-3776.

Wang.. J.S.. Araki. T., Ogawa, T.. Matsuoka, M et Fukuda. H. (1999) A method of
graphicaîiy analyzing su bsbateinhibition kinetics. Biotechnol . Bioeng. 62.1024 1 1.
Wende, W.. Grindl, W.. Christ. F.. Pingoud, A. et Pingoud. V. (19%) Binding, bending
and ckavage of DNA substrates by the homing endonuclease PI-SceI. Nucleic Acids
Res. 34,41234132.
Wernette. C.M. ( 1998) Structure and activity of the mitochondrial intron-encoded
endonuclease, 1-SceIV. Biochem. Biophys. Res. Commun. 148. 127- 133.
Wittmayer. P.K. et Raines. R.T. ( 19%) Susbtrate binding and turnover by the highly
specific 1-PpoI endonuclease. Biochemistry 35, 1076- 1083.







