

2 main approaches: building a consensus (“overlap–layout–consensus”) generating De Bruijn k-mer graphs
Metagenomics assembly

I _ l i k e _ E B I _ m e t a g e n o m i c s
Genomics assembly: building a consensus
I _ l i k e _ E B I _ m e t a g e n o m i c s
read-depth
high
low
I _ l i k e _ E B I _ m e t
i k e _ E B I _ m e t a g e_ E B I _ m e t a g e n o m
I _ m e t a g e n o m i c s
_ l i k e _ E B I _ m e t a
B I _ m e t a g e n o m i c
Based on ‘word’ overlap
reads
contig

Metagenomics assembly: building a consensus
Issues: read length and repeated sequences
???
. . .
???

Genomics assembly: building a consensus
Practical solution : using coverage / read-depth information
Coverage:ratio between contigs
3 11 1
Allow the elimination of one of the possible assembly:

Genomics assembly: building a consensus
Practical solution : using pair-end reads
Pair-ends:Distance information between sequences
Allow the identification of the correct assembly:

Genomics assembly: De Bruijn k-mer graphs
k-mers
generated by breaking reads into multiple overlapping words of fixed length (k)
I _ l i k e _ E B I _ m e t a g e n o m i c s
k=5
e _ E B Ik e _ E B
i k e _ El i k e _
_ l i k eI _ l i k
_ E B I _E B I _ m
B I _ m eI _ m e t_ m e t am e t a ge t a g et a g e na g e n og e n o m
e n o m in o m i co m i c s

Branches in the graph represent partially overlapping sequences.
T. Brown, 2012
Genomics assembly: using k-mers
Each node represents a 14-mer;Links between each node are 13-mer overlaps
14mer
k=14

Single nucleotide variations cause k-long branches;They don’t rejoin quickly.
Genomics assembly: using k-mers
T. Brown, 2012

Genomics assembly: De Bruijn k-mer graphs
Building the graph is demanding but navigation through is quick and memory efficient.
branches : ambiguity in assembly
short dead-end branches: low coverage
bubbles: sequencing errors or polymorphism ?
converging and diverging paths: repeats
therefore there is a need for biological knowledge and other sequences information to fully reconstruct a genome
J.R. Miller et al. / Genomics (2010)

There is a number of (+/- metagenome-adapted) solutions out there:
MetaVelvet, MetaIDBA and khmer “partition” the assembly de Brujn
graph into sections from different organisms, and then assemble those
individually. This allows them to adjust coverage parameters “locally”.
Genovo uses a 'generative probabilistic model' to identify likely sequence
reconstructions
Euler deals with repeats by identifying an Eulerian path (visiting every
edge only once) in the De Bruijn graph.
and SOAPdenovo (graph), Newbler (for 454, consensus), MetAMOS…
Metagenomics assembly: what to use ?

Butler et al., Genome Res, 2009
Genomics assembly: choosing k-mer
Tools such as Velvet Advisor (http://dna.med.monash.edu.au/~torsten/velvet_advisor/) are available

Judging genomics assembly
parameters 1 parameters 2
measurements:
number of contigs (1)
length of contigs (2)
nucleotides involved (1)
N50 weighted median such that 50% of the entire assembly is contained in contigs equal to or larger than this value
How to judge the better assembly in absence of external information ?

Judging metagenomics assembly
parameters 1 parameters 2
total length: 17contigs: 7
N50 = 3
total length: 15.5contigs: 5
N50 = 2
Therefore the assembly obtained with parameters 2 will be considered the best
Calculating N50: - order the sequences by decreasing length,- add length until 50% of nucleotides reached

Judging metagenomics assembly
parameters 1 parameters 2
For metagenomics, in addition to N50, we can also use the fact that sequences are originating from different species
-% GC will vary between species (20 to 80%) and therefore contigs fromdifferent species could be separated from each others.
-all predicted CDSs from a single contig should be annotated as being from same species (using Blast for example).
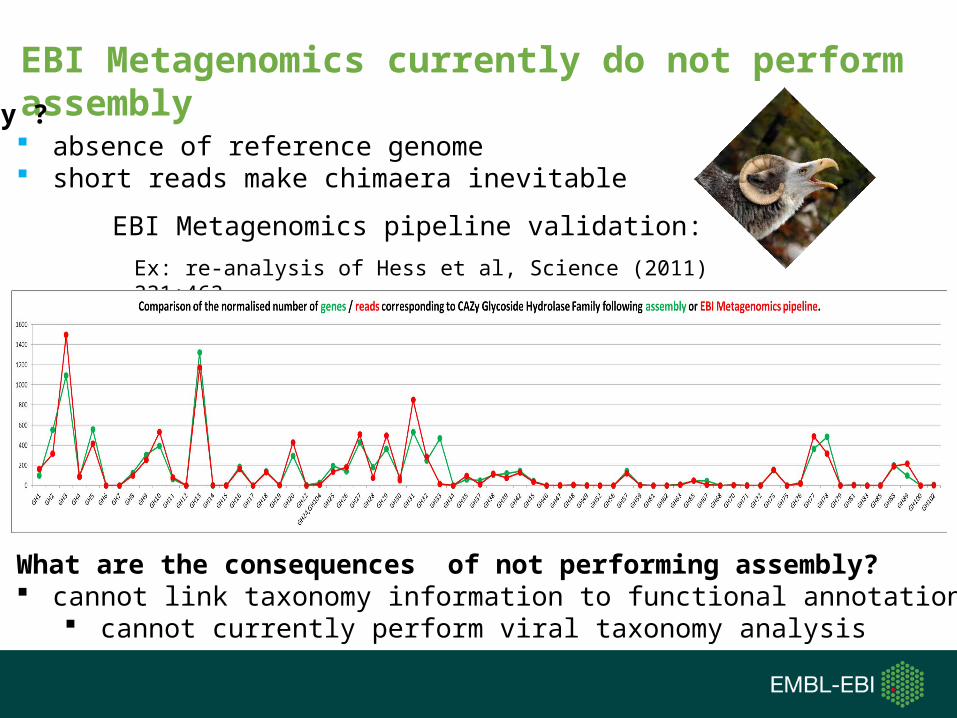
EBI Metagenomics currently do not perform assembly
Why ? absence of reference genome short reads make chimaera inevitable
EBI Metagenomics pipeline validation:
What are the consequences of not performing assembly? cannot link taxonomy information to functional annotations
cannot currently perform viral taxonomy analysis
Ex: re-analysis of Hess et al, Science (2011) 331:463

http://www.ebi.ac.uk/metagenomics/ http://metagenomics.anl.gov/
http://camera.calit2.net/
http://img.jgi.doe.gov/
Public Metagenomics portals
Do not perform assembly but accept assembled data
Perform assembly

Hubert DENISE ([email protected])







