
of June 10, 2018.This information is current as
Costimulation in Primary Murine T CellsMediates Signal Integration of TCR/CD28 p38 Mitogen-Activated Protein Kinase
Cameron, Isabelle Bergerot and Terry L. DelovitchJian Zhang, Konstantin V. Salojin, Jian-Xin Gao, Mark J.
http://www.jimmunol.org/content/162/7/38191999; 162:3819-3829; ;J Immunol
Referenceshttp://www.jimmunol.org/content/162/7/3819.full#ref-list-1
, 34 of which you can access for free at: cites 54 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 1999 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 10, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

p38 Mitogen-Activated Protein Kinase Mediates SignalIntegration of TCR/CD28 Costimulation in Primary MurineT Cells1
Jian Zhang,2* Konstantin V. Salojin, 2* Jian-Xin Gao,* Mark J. Cameron,* † Isabelle Bergerot,*and Terry L. Delovitch 3*†‡
Optimal T cell activation requires two signals, one generated by TCR and another by the CD28 costimulatory receptor. In thisstudy, we investigated the regulation of costimulation-induced mitogen-activated protein kinase (MAPK) activation in primarymouse T cells. In contrast to that reported for human Jurkat T cells, we found that p38 MAPK, but not Jun NH2-terminal kinase(JNK), is weakly activated upon stimulation with either anti-CD3 or anti-CD28 in murine thymocytes and splenic T cells. However,p38 MAPK is activated strongly and synergistically by either CD3/CD28 coligation or PMA/Ca21 ionophore stimulation, whichmimics TCR-CD3/CD28-mediated signaling. Activation of p38 MAPK correlates closely with the stimulation of T cell prolifera-tion. In contrast, PMA-induced JNK activation is inhibited by Ca 21 ionophore. T cell proliferation and production of IL-2, IL-4,and IFN-g induced by both CD3 and CD3/CD28 ligation and the nuclear expression of the c-Jun and ATF-2 proteins are eachblocked by the p38 MAPK inhibitor SB203580. Our findings demonstrate that p38 MAPK 1) plays an important role in signalintegration during costimulation of primary mouse T cells, 2) may be involved in the induction of c-Jun activation and augmen-tation of AP-1 transcriptional activity, and 3) regulates whether T cells enter a state of functional unresponsiveness.The Journalof Immunology,1999, 162: 3819–3829.
T he transmission of extracellular signals to intracellulartargets in various cell types is mediated by several proteinkinases, including the family of mitogen-activated protein
kinases (MAPKs)4 (1–4). MAPKs are serine/threonine kinasesthat include extracellular signal-regulated kinases (ERKs), JunNH2-terminal kinases (JNKs), and p38 MAPK (2, 3). MAPKs areactivated by dual phosphorylation in a Thr-Xaa-Tyr motif, inwhich Xaa corresponds to Glu in ERK, Pro in JNK, and Gly in p38MAPK (2, 4). These dual specificity kinases are relatively specificfor each MAPK subgroup, allowing for their independent regula-tion. Thus, MAPK/ERK kinase-1 (MEK-1) and MEK2 selectivelyphosphorylate and activate ERKs, whereas MAPK kinase-3(MKK-3) and MKK6 selectively phosphorylate and activate p38MAPK. MKK4 does not activate the ERK subgroup, but activatesboth p38 MAPK and JNK (5–9).
ERKs are activated by agonists for tyrosine-encoded receptorsand G protein-coupled receptors that induce mitogenesis or cellu-lar differentiation (2–4). ERKs mediate the effects of these ago-nists by phosphorylating and regulating the activity of several pro-teins, including cytoplasmic enzymes and nuclear factors (1, 2).JNKs phosphorylate the NH2-terminal activation domain of c-Junand activating transcription factor-2 (ATF-2), increasing their tran-scriptional activity (4, 10). JNKs are activated preferentially bycellular stress and inflammatory cytokines, but also by G protein-coupled receptor agonists, growth factors, and cytoplasmic onco-genes (4, 10–16). Similarly, p38 MAPK is activated by cellularstresses, inflammatory cytokines, LPS (14, 17–19), and G protein-coupled receptors (20), and activated p38 MAPK in turn mediatescytokine production, stress responses, and apoptosis (21–26). Thep38 MAPK substrates include MAPK-activating protein kinase-2(MAPKAP kinase-2), ATF-2 (18, 19, 27, 28), cAMP responseelement binding protein (CREB), ATF-1 (29), Elk-1 (25), C/EBP-homologous protein (CHOP) (30), and myocyte-enhancer factor2C (MEF2C) (31).
TCR engagement activates the ERK cascade in T cells (32–34).Analyses of Jurkat human T cells and various activated mouse Tcell clones have suggested that JNK activation stimulated by TCRengagement requires CD28 coligation (33, 34). p38 MAPK may befully activated in mouse T cell clones by signaling via either CD3or CD28, but CD3/CD28 costimulation does not further enhancethe amount of p38 MAPK activation (35). In contrast, stimulationof CD28 fails to activate p38 MAPK, but synergizes with CD3stimulation to fully activate p38 MAPK in preactivated prolifer-ating T cells (36). These results raise the possibility that p38MAPK may mediate CD28 costimulation in primary naive mouseT cells. The latter possibility is supported by reports that, in T cellsfrom MKK-4-deficient mice, CD28-mediated IL-2 production andproliferation are impaired and CD28 costimulation and PMA/Ca21
ionophore-induced signaling can stimulate proliferation and IL-2
*Autoimmunity/Diabetes Group, John P. Robarts Research Institute, London, On-tario, Canada; and Departments of†Microbiology and Immunology and‡Medicine,University of Western Ontario, London, Ontario, Canada
Received for publication August 10, 1998. Accepted for publication December21, 1998.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Vern Bruder Grant from the Canadian DiabetesAssociation and a grant from the Juvenile Diabetes Foundation International (toT.L.D.). J.Z. and K.V.S. were recipients of Juvenile Diabetes Foundation Interna-tional postdoctoral fellowships.2 J.Z. and K.V.S. contributed equally to this work.3 Address correspondence and reprint requests to Dr. Terry L. Delovitch, Autoim-munity/Diabetes Group, John P. Robarts Research Institute, 1400 Western Road,London, Ontario N6G 2V4, Canada. E-mail address: [email protected] Abbreviations used in this paper: MAPK, mitogen-activated protein kinase; ATF-2,activating transcription factor-2; CsA, cyclosporin A; ERK, extracellular signal-reg-ulated kinase; GST, glutathioneS-transferase; hsp, heat-shock protein; JNK, c-JunNH2-terminal kinase; MAPKAP, MAPK-activating protein; MBP, myelin basic pro-tein; MEK, MAP kinase/ERK kinase; MKK, MAP kinase kinase.
Copyright © 1999 by The American Association of Immunologists 0022-1767/99/$02.00
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

production independently of JNK activation (37). In addition, inhuman T cells, the p38 MAPK inhibitor SB203580 blocks CD28-dependent proliferation and IL-2 production (38). Thus, the ques-tion of whether p38 MAPK is activated upon either TCR or CD28stimulation or after TCR/CD28 coligation in primary, unstimu-lated, naive mouse T cells merits further investigation.
In this study, we determined whether differential regulation ofMAPK activation occurs in primary mouse T cells in response toTCR/CD28 or PMA/Ca21 ionophore costimulation. We show thatp38 MAPK activation mediates both TCR- and CD28-induced sig-naling in primary mouse T cells. Ligation of TCR or CD28 resultsin only modest p38 MAPK activation, whereas TCR and CD28synergize upon coligation to elicit enhanced p38 MAPK activa-tion. PMA/Ca21 ionophore costimulation, which mimics TCR/CD28-mediated signaling, fully activates p38 MAPK in primarymouse T cells. Our results demonstrate that p38 MAPK is involvedin both TCR- and CD28-signaling pathways, and that p38 MAPK,but not JNK, is involved in signal integration during costimulationof naive mouse primary T cells.
Materials and MethodsMice
C57BL/6 (B6) and BALB/c mice were purchased from The Jackson Lab-oratory (Bar Harbor, ME), maintained in the Animal Care Facility of theFaculty of Medicine at the University of Western Ontario (London, ON,Canada), and used at 6–10 wk of age.
Abs, proteins, and reagents
The following reagents were purchased from Santa Cruz Biotechnology(Santa Cruz, CA): rabbit polyclonal Abs against mouse ERK-1, JNK-1/2,and p38 MAPK; mouse mAbs against c-Jun, c-Fos, and ATF-2; glutathioneS-transferase (GST)-c-Jun (1–79), GST-ATF-2 (1–505), and glutathione-agarose. MBP, IL-2, cycloheximide, and PMA were each obtained fromSigma (St. Louis, MO). Anti-MAPKAP kinase-2 antiserum and recombi-nant murine heat-shock protein 25 (hsp25) were purchased from UpstateBiotechnology (Lake Placid, NY) and StressGen Biotechnology (Victoria,Canada), respectively. The 145-2C11 anti-CD3 and 37.51 anti-CD28 mAbswere purified by protein G affinity chromatography (Pharmacia Biotech,Uppsala, Sweden) of the supernatants of the B cell hybridomas kindlysupplied by Dr. J. Bluestone (University of Chicago, Chicago, IL) and Dr.J. Allison (University of California, Berkeley, CA), respectively. The PV-1anti-CD28 mAb was kindly supplied by Dr. C. June (Naval Medical Re-search Institute, Bethesda, MD). The p38 MAPK inhibitor 203580 wasgenerously provided by Dr. P. Young (SmithKline Beecham Pharmaceu-ticals, King of Prussia, PA). Cyclosporin A (CsA) was supplied by SandozCanada (Dorval, Quebec, Canada). The mouse anti-human CD3 (OKT3)and CD28 mAbs were obtained from PharMingen (San Diego, CA). PMAand A23187 was purchased from Calbiochem (San Diego, CA), respec-tively. IL-2 and cycloheximide were purchased from Sigma.
Cell isolation and stimulation
B6 and BALB/c thymocytes or splenic T cells were purified (purity$98%as determined by FACS analysis of CD3 cell surface expression) on T cellenrichment columns (R&D Systems, Minneapolis, MN). Murine T cellswere cultured for 5 h at 37°C in complete RPMI 1640 medium supple-mented with 10% heat-inactivated FCS, 10 mM HEPES, 0.1 mg/ml strep-tomycin, 100 U/ml penicillin, 0.05mM 2-ME, and 2 mM glutamine (allpurchased from Life Technologies, Burlington, ON, Canada) before stim-ulation to decrease high basal levels of p38 MAPK activity. Cells werestimulated for various times with either the anti-CD3, anti-CD28, or anti-CD3 plus anti-CD28 mAbs, or with PMA plus the Ca21 ionophoreA23187. Jurkat human T cells were also grown in complete RPMI 1640under the same conditions. After culture, cells (23 107/ml) were resus-pended at 37°C in complete RPMI 1640, washed twice with serum-freeRPMI 1640, and stimulated as above. Where indicated, SB203580 or CsAwas added to the cells 15 min before stimulation. To obtain proliferating Tcells, splenic T cells were cultured (106/ml) in complete RPMI 1640 insix-well plates precoated with anti-CD3 (1mg/ml) and IL-2 (20 U/ml).After 48 h, T cells were removed from the plates and expanded in IL-2.Proliferating T cells were harvested on day 4 of culture, and were thenstimulated with either anti-CD3, anti-CD28, or anti-CD3 plus anti-CD28mAbs.
In vitro kinase assays
After stimulation, cells were lysed in ice-cold lysis buffer containing 1%Triton X-100, 10 mM Tris (pH 7.5), 150 mM NaCl, 2 mM EGTA, 50 mMb-glycerophosphate, 2 mM Na3VO4, 10 mM NaF, 1 mM DTT, 1 mMPMSF, 10mg/ml leupeptin, and 10mg/ml apoptinin. Lysates were clarifiedby centrifugation at 12,000 rpm for 10 min at 4°C, and their protein contentwas determined by the Bradford assay using BSA as a standard. Lysateswere divided into three replicate samples; incubated for 1 h at 4°C witheither anti-ERK-1, anti-JNK-1, or anti-p38 MAPK Abs; and further reactedwith protein G agarose (Santa Cruz) or protein A-Sepharose CL-4B (Phar-macia Biotech, Baie d’Urfe, PQ, Canada) for an additional 1 h at 4°C.Immunoprecipitates were washed three times with lysis buffer and twicewith kinase buffer (20 mM HEPES (pH 7.5), 20 mM MgCl2, 20 mMMnCl2, 2 mM DTT, 25 mMb-glycerophosphate, and 100 nM Na3VO4).Kinase assays were performed using MBP, GST-c-Jun, and GST-ATF-2fusion proteins as substrates for ERK-1, JNK-1, and p38 MAPK, respec-tively. Immunoprecipitates were resuspended in 18ml kinase buffer con-taining 5mg MBP, 1mg GST-c-Jun, and 1mg GST-ATF-2 fusion proteinsin the presence of 20mM cold ATP and 20mCi [g-32P]ATP (AmershamLife Science, Arlington Heights, IL), and incubated for 30 min at 30°C.Solid-phase JNK assays were performed essentially as previously de-scribed (39). Whole cell extracts (23 107 cell equivalents) were preparedand reacted for 4 h at 4°C with a GST-c-Jun (1–79) fusion protein boundto glutathione-agarose beads to immobilize JNK. The washed beads werethen analyzed for their associated kinase activity by incubation for 30 minat 30°C in kinase buffer containing 20mCi [g-32P]ATP. Reactions wereterminated by the addition of SDS sample buffer, samples were boiled, andkinase reaction products were resolved by SDS-PAGE. The MAPKAPkinase-2 assay was performed as described (35), using murine recombinanthsp25 as a substrate. Equal loading of precipitated proteins was confirmedby probing the blots with specific Abs, and phosphorylation of the sub-strates was quantitated using a Molecular Imager System and MolecularAnalyst imaging software (Bio-Rad, Hercules, CA).
T cell proliferation assay
Splenic T cells (106/ml) were resuspended in complete RPMI 1640 me-dium in the absence or presence of various concentrations of SB203580,and then incubated for 15 min at 37°C. Cells were cultured for 48 h at 37°Cin round-bottom 96-well plates (Nunc) precoated with the 145-2C11 anti-CD3e mAb (1 mg/ml) in the presence or absence of the 37.51 anti-CD28mAb (1 mg/ml). [3H]thymidine (1mCi/well; Amersham) was added 24 hbefore the end of culture, and cultures were harvested using a TomtecHarvester 96 cell harvester (Fisher Scientific, Ottawa, ON, Canada). Theextent of T cell proliferation was proportional to the amount of [3H]thy-midine incorporation, which was determined using a Wallac 1450 Micro-beta Plus beta counter (Fisher Scientific).
Cytokine assays
Splenic T cells (106/ml) were pretreated with different concentrations ofSB203580 for 15 min at 37°C, and cultured in round-bottom 96-well platescoated with anti-CD3 (1mg/ml) in the presence or absence of the 37.51anti-CD28 mAb (1mg/ml). Supernatants collected after 48 h were assayedfor their cytokine concentrations by ELISA using a double ligand method.IL-2 concentrations were interpolated from a standard curve using murinerIL-2 captured by the JES6-1A12 mAb and detected by the biotinylatedJES6-5H4 mAb. IL-4 concentrations were measured using recombinantmurine IL-4, the BVD4-1D11 mAb, and biotinylated BVD6-24G2 mAb,while IFN-g concentrations were detected using recombinant murineIFN-g, R4-6A2 mAb, and biotinylated XMG1.2 mAb (all obtained fromPharMingen). Briefly, flat-bottom 96-well microtiter plates were coatedwith 50 ml/well of capture mAb (1mg/ml) in 0.1 M NaHCO3 overnight at4°C. Nonspecific binding sites were blocked with 3% BSA for 2 h at23°C.Standards or samples (50ml) were added, left overnight at 4°C, and thenincubated with 50ml/well of biotinylated detecting mAb (1mg/ml) for45 min at 23°C. Streptavidin-peroxidase conjugates (1mg/ml; Sigma) indiethanolamine buffer were successively added to develop the reaction at23°C. Plates were read at 405 nm in an automated microplate reader(Bio-Rad). Cytokine standard curves were linear in the range of20–20,000 pg/ml.
Cell viability assay
Splenic T cells were stimulated with an anti-CD3 mAb as above in thepresence or absence of SB203580 (100mM). Cells were harvested afterincubation for 48 h, washed in PBS, stained with propidium iodide, andanalyzed by flow cytometry.
3820 COSTIMULATORY ACTIVATION OF p38 MAPK IN PRIMARY T CELLS
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Effect of SB203580 on nuclear expression of c-Fos, c-Jun, andATF-2
Thymocytes (43 107/ml) were pretreated for 15 min with 10mMSB203580, and were then stimulated for 5 or 15 min with anti-CD3(10mg/ml) and anti-CD28 (5mg/ml). Alternatively, thymocytes were stim-ulated for 4 h at37°C with either anti-CD3, anti-CD28, anti-CD3 plusanti-CD28, or PMA (50 ng/ml). The cells were then collected, washed withice-cold PBS, and lysed for 30 min at 4°C in hypotonic lysis buffer (20 mMHEPES (pH 7.5), 5 mM NaCl, 3 mM MgCl2, 1 mM DTT, 5% glycerol,0.4% Nonidet P-40, 2.5 mM PMSF, 40mg/ml aprotinin, 40mg/ml leupep-tin, 2 mM EDTA, 1 mM Na3VO4, and 10 mM NaF). Nuclear extractionwas performed as previously reported (40). Extracts were separated on10% SDS-PAGE, transferred to nitrocellulose membranes, and probedwith anti-c-Fos, anti-c-Jun, and anti-ATF-2 mAbs, and the c-Fos, c-Jun,and ATF-2 proteins were detected by chemoluminescence.
ResultsColigation of TCR and CD28 synergistically activates p38MAPK, but neither JNK nor ERK in murine thymocytes andsplenic T cells
JNK activation requires costimulation by either CD28 or Ca21
ionophore in Jurkat T cells, suggesting that JNK is involved insignal integration during T cell costimulation (33). However, it isnot known whether activation of JNK and p38 MAPK occurs inprimary naive T cells that are not further stimulated in vitro. To
determine whether CD28 regulates MAPK activation followingCD3 ligation, murine thymocytes or purified splenic T cells werestimulated for 15 min with anti-CD3, anti-CD28, or both mAbs.Cells were lysed; ERK-1, JNK-1, and p38 MAPK were immuno-precipitated with specific Abs; and immunoprecipitates were as-sayed for the activities of associated MAPKs by their ability tophosphorylate the MBP, c-Jun, and ATF-2 substrates, respectively.Anti-CD3 stimulation activated ERK-1 in thymocytes and splenicT cells, and this level of activation of ERK-1 was not increasedafter CD3/CD28 costimulation (Fig. 1,A andB, upper panels), asreported (33, 34).
More significantly, stimulation by anti-CD3 or anti-CD28 eitheralone or in combination did not activate JNK (i.e., JNK-1) in mu-rine thymocytes or splenic T cells (Fig. 1,A andB, middle panels).Anti-CD3 or anti-CD28 stimulation induced modest p38 MAPKactivity, which was elevated about fivefold after CD3/CD28 coli-gation (Fig. 1,A and B, lower panels). PMA activated ERK-1,JNK-1, and p38 MAPK in thymocytes and splenic T cells (Fig. 1,A andB). Stimulation of splenic T cells with the Ca21 ionophoreA23187 did not activate ERK-1 or JNK-1, but rather stimulatedp38 MAPK activity and enhanced the level of p38 MAPK activityinduced by PMA alone (Fig. 1B, lower panel). JNK-2 may bindand phosphorylate c-Jun more efficiently than JNK-1 (41, 42). To
FIGURE 1. MAPK activation is differentially regulated in response to stimulation by CD3, CD28, CD3 plus CD28, PMA, or PMA plus Ca21 ionophore.A, Thymocytes (23 107/ml) were either left unstimulated or stimulated for 15 min at 37°C with anti-CD3 (10mg/ml), anti-CD28 (5mg/ml), anti-CD3 plusanti-CD28, or PMA (50 ng/ml). Cell lysates were precleared and immunoprecipitated with anti-ERK-1, anti-JNK-1, or anti-p38 MAPK, respectively. Invitro kinase assays of ERK-1, JNK-1, and p38 MAPK activity associated with the immunoprecipitates were performed using MBP, GST-c-Jun, andGST-ATF-2 as substrates. Lysates were analyzed for protein abundance of ERK-1, JNK-1, and p38 MAPK by immunoblotting (IB).B, Splenic T cells(107/ml) were stimulated with either anti-CD3, anti-CD28, anti-CD3 plus anti-CD28, PMA, or PMA plus A23187 (500 ng/ml). In vitro kinase assays ofERK-1, JNK-1, and p38 MAPK activity were performed as inA. Lysates were analyzed for protein abundance of ERK-1, JNK-1, and p38 MAPK by IB.C, Thymocytes (23 107/ml) were stimulated with anti-CD3, anti-CD28, anti-CD3 plus anti-CD28, or PMA, and JNK-2 activity was assayed as inA.Lysates were analyzed for protein abundance of JNK-2, as above. Alternatively, cell lysates were reacted for 4 h at 4°Cwith 10 mg GST-c-Jun precoupledto glutathione-agarose beads, followed by an in vitro kinase reaction (C, lower panel). Data from one of three reproducible experiments are shown.
3821The Journal of Immunology
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

exclude the possibility that in primary T cells JNK-2 may be solelyresponsible for c-Jun phosphorylation, thymocytes were lysed fol-lowing stimulation with either anti-CD3, anti-CD28, anti-CD3plus CD28, or PMA. JNK-2 activity associated with anti-JNK-2immunoprecipitates was examined by c-Jun phosphorylation in anin vitro kinase assay. As observed for JNK-1, little or no JNK-2activity was detected in response to ligation of either CD3, CD28,or both (Fig. 1C,upper panel). JNK-2 was activated, however, byPMA stimulation. The failure to activate JNK-1 and JNK-2 bycoligation of CD3 and CD28 was also observed in BALB/c splenicT cells (data not shown), suggesting that these observations are notmouse strain dependent. Since several splice variants of JNK-1 andJNK-2 have been found recently (43), it is possible that the anti-JNK Abs used may not precipitate all variant forms of JNK. Toeliminate this possibility, GST-c-Jun precoupled to glutathione-agarose beads was used in a solid-phase JNK assay to precipitateall JNK variants. Similar to the in vitro kinase assay of JNK-1,c-Jun phosphorylation was not increased upon CD3, CD28, orCD3/CD28 ligation, but was induced significantly upon PMAstimulation (Fig. 1C, lower panel).
To further examine the activation of ERK-1, JNK-1, and p38MAPK induced by stimulation of CD3, CD28, or CD3 plus CD28,thymocytes were pretreated with a constant amount (10mg/ml) ofanti-CD3 together with variable amounts of anti-CD28. With in-creasing concentrations of anti-CD28 mAb (0.1–20mg/ml), no fur-ther augmentation of ERK-1 activation was observed compared
with the amount of ERK-1 activity stimulated by anti-CD3 alone(Fig. 2A, upper panel). A similar result was obtained for JNK-1activity. However, p38 MAPK was optimally activated by anti-CD28 at concentrations of 1–5mg/ml (Fig. 2A, lower panel).
To investigate whether the kinetics of ERK-1 and JNK-1 acti-vation differs from that of p38 MAPK, the time courses of acti-vation of ERK-1, JNK-1, and p38 MAPK induced by coligation ofCD3 and CD28 were determined. ERK-1 and p38 MAPK activitypeaked at 15 and 30 min of stimulation, respectively (Fig. 2B,upperand lower panels), but JNK-1 was not activated throughoutthe entire time course (Fig. 2B,middle panel). This effect of CD28costimulation was also analyzed by pretreating thymocytes with anoptimum concentration of anti-CD28 mAb (5mg/ml) plus variableconcentrations of anti-CD3 mAb (0.1–20mg/ml). ERK-1 activa-tion was gradually enhanced by addition of increasing concentra-tions of anti-CD3, with maximal activation observed at 10 and20 mg/ml (Fig. 2C, upper panel). Similarly, p38 MAPK activitywas elevated by increasing concentrations of anti-CD3 and wasevident even at very low concentrations (0.1mg/ml) of added anti-CD3 (Fig. 2C, lower panel). In contrast, JNK-1 was not activatedsignificantly at any concentration of added anti-CD3 (Fig. 2C,mid-dle panel). The inability to detect JNK-1 activation upon CD3/CD28 coligation in primary mouse T cells may be due to a lack ofreactivity of the 37.51 anti-CD28 mAb with the appropriate CD28epitope required for JNK-1 activation. However, the use of anotheranti-CD28 mAb, PV-1, which recognizes a different epitope than
FIGURE 2. p38 MAPK, but not ERK-1or JNK-1, is synergistically activated by CD3/CD28 coligation.A, Thymocytes (23 107/ml) were incubated with control normal ham-ster IgG (NIg, 10mg/ml) or with anti-CD3(10 mg/ml) plus incremental concentrations(0–20 mg/ml) of anti-CD28 for 15 min at37°C. Cell lysates were immunoprecipitatedwith anti-ERK-1, anti-JNK-1, or anti-p38MAPK, and kinase activities were assayed asin Fig. 1A. Lysates were analyzed for proteinabundance of ERK-1, JNK-1, and p38MAPK, as above.B, Thymocytes (23 107/ml) were stimulated with anti-CD3 (10mg/ml) plus anti-CD28 mAb (5mg/ml) for 0, 15,30, 60, and 120 min. ERK-1, JNK-1, and p38MAPK activities associated with immunopre-cipitates were assayed as in Fig. 1A. Lysateswere analyzed for protein abundance ofERK-1, JNK-1, and p38 MAPK, as above.C,Stimulation of thymocytes (23 107/ml) withNIg (10 mg/ml) or with anti-CD28 (5mg/ml)plus incremental concentrations (0–20mg/ml)of anti-CD3, and assay of ERK-1, JNK-1, andp38 MAPK activities were as in Fig. 1A. Ly-sates were analyzed for protein abundance ofERK-1, JNK-1, and p38 MAPK, as above.D,Thymocytes (23 107/ml) were stimulatedwith either NIg (10mg/ml), PV-1 anti-CD28(5 mg/ml), anti-CD3 (10mg/ml) plus incre-mental concentrations (0–20mg/ml) of PV-1anti-CD28, or PMA (50 ng/ml) for 15 min at37°C. Cell lysates were immunoprecipitatedwith anti-JNK-1 polyclonal Abs, and JNK-1 ac-tivities were assayed as in Fig. 1A. The loadingof equal amounts of JNK-1 proteins was con-firmed by anti-JNK-1 immunoblotting. Datafrom one of two reproducible experiments areshown.
3822 COSTIMULATORY ACTIVATION OF p38 MAPK IN PRIMARY T CELLS
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

the 37.51 anti-CD28 mAb as determined by a competitive bindingassay (our unpublished observations), did not activate JNK-1 (Fig.2D). Note that PMA activated JNK-1 in a positive control sample.These data indicate that p38 MAPK, but not JNK, is involved inboth TCR- and CD28-mediated signaling pathways, and may playan important role in signal integration during CD28 costimulationof primary mouse T cells.
Synergistic activation of p38 MAPK requires costimulation byCa21 in primary mouse T cells
In Fig. 1B, we showed that the Ca21 ionophore A23187 (500 ng/ml) activates p38 MAPK, synergizes with PMA to further enhancep38 MAPK activation, and inhibits JNK-1 activity induced byPMA. To analyze the relationship of these findings to the dose ofA23187, splenic T cells were stimulated with PMA (50 ng/ml) plusa range of concentrations (15–1000 ng/ml) of A23187. ERK-1activity was not enhanced at any of the concentrations of A23187used (Fig. 3A,upper panel). JNK-1 activity was inhibited signif-icantly in a dose-dependent manner when A23187 was added toPMA, and concentrations of$125 ng/ml A23187 abolished vir-tually all detectable JNK-1 activity (Fig. 3A,middle panel). Notethat this inhibition of JNK by A23187 was not due to differentkinetics of JNK activation induced by PMA/A23187 versus PMAalone. PMA-induced JNK activation was significantly increased inperipheral T cells after stimulation for 15 min, whereas little or noactivation of JNK was induced by PMA/A23187 (Fig. 3B). In con-trast, p38 MAPK activity was enhanced upon exposure of the Tcells to increasing doses of A23187 and reached a maximum at aconcentration of 60 ng/ml (Fig. 3A, lower panel). Interestingly, thelatter findings on induced p38 MAPK activity correlate closelywith our observations on the stimulation of proliferation of splenicT cells by PMA and variable amounts of A23187 (Fig. 3C). Theobserved inhibition of PMA-induced JNK activation by A23187suggests that Ca21 ionophore may activate some JNK-specificMAPK phosphatases that inactivate JNK.
To further analyze the effect of Ca21 on p38 MAPK activation,splenic T cells were pretreated with CsA (500 ng/ml), which isbelieved to inhibit the calcium-dependent phosphatase calcineurin.Consistent with our previous observation in Fig. 1C, stimulationwith anti-CD3 or anti-CD28 alone resulted in only modest p38MAPK activation, and p38 MAPK was activated synergistically byeither CD3/CD28 coligation or PMA/A23187 treatment (Fig. 3D,upper panel). These induced p38 MAPK activities were inhibitedappreciably by CsA, with the exception that PMA- and CD28-induced p38 MAPK activation was CsA resistant. Interestingly,inhibition of PMA/Ca21 ionophore-induced p38 MAPK activityby CsA pretreatment occurred in a dose-dependent manner (Fig.3D, lower panel). JNK activity was induced only by PMA, and thisPMA-induced JNK activity was inhibited by Ca21 ionophore (Fig.3D, upper panel). However, CsA did not further inhibit JNK ac-tivity induced by PMA/Ca21 ionophore (Fig. 3D,lower panel).Thus, optimal activation of p38 MAPK in primary mouse T cellsrequires two signals, one provided by the TCR/CD3 complex orPMA and another provided by CD28 or Ca21 ionophore.
Different requirements for JNK-1 and p38 MAPK activation inJurkat T cells, proliferating T cells, and primary T cells
JNK activation requires two signals generated by the TCR andCD28 or PMA and Ca21 ionophore in Jurkat T cells and mouse Tcell clones (33, 34). To determine whether p38 MAPK activationhas the same requirements in Jurkat T cells, we compared theactivation of ERK-1, JNK-1, and p38 MAPK in response toligation of CD3, CD28, CD3 plus CD28, PMA, and PMA plusCa21 ionophore in Jurkat T cells. ERK-1 activation was only seen
upon anti-CD3 or PMA stimulation, and no further augmentationwas observed upon CD3/CD28 coligation or exposure to PMAand A23187 (Fig. 4A, upper panel). JNK-1 activation required
FIGURE 3. PMA and Ca21 ionophore synergistically activate p38MAPK, but not ERK-1 or JNK-1.A, Splenic T cells (107/ml) were incu-bated with PMA (50 ng/ml) and variable (0–1000 ng/ml) concentrations ofA23187 in DMSO for 15 min at 37°C. In vitro kinase activities of ERK-1,JNK-1, and p38 MAPK were detected as in Fig. 1A. Lysates were analyzedfor protein abundance of ERK-1, JNK-1, and p38 MAPK.B, Splenic Tcells (107/ml) were either stimulated with PMA (50 ng/ml) or PMA plusA23187 (500 ng/ml) for 1, 5, 10, and 15 min at 37°C, or left unstimulated.Cells were lysed, and a solid-phase kinase assay of JNK was performed asin Fig. 1C. C, Splenic T cells (106/ml) were cultured in the presence ofPMA (50 ng/ml) and incremental concentrations (15–1000 ng/ml) ofA23187 for 48 h at 37°C. Cell proliferation was determined by [3H]thy-midine incorporation. The results of triplicate cultures are expressed as themean values6 SD. D, Splenic T cells (107/ml) were pretreated in theabsence or presence of CsA (500 ng/ml) for 15 min at 37°C, and were thenstimulated with either anti-CD3, anti-CD28, anti-CD3 plus CD28, or PMA.Alternatively, splenic T cells were pretreated with various amounts of CsA(0–500 ng/ml) for 15 min at 37°C, and were then stimulated with PMAplus A23187. p38 MAPK activity was assayed as in Fig. 1A, and a solid-phase JNK assay was performed as in Fig. 1C. Data from one of threereproducible experiments are shown.
3823The Journal of Immunology
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
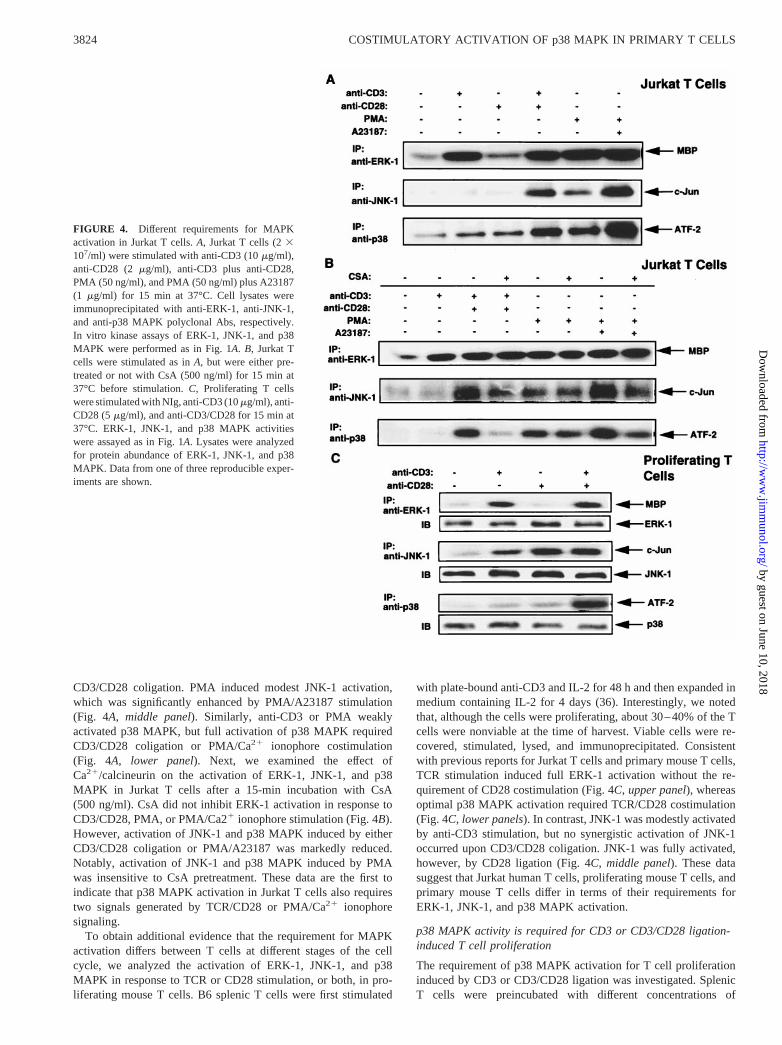
CD3/CD28 coligation. PMA induced modest JNK-1 activation,which was significantly enhanced by PMA/A23187 stimulation(Fig. 4A, middle panel). Similarly, anti-CD3 or PMA weaklyactivated p38 MAPK, but full activation of p38 MAPK requiredCD3/CD28 coligation or PMA/Ca21 ionophore costimulation(Fig. 4A, lower panel). Next, we examined the effect ofCa21/calcineurin on the activation of ERK-1, JNK-1, and p38MAPK in Jurkat T cells after a 15-min incubation with CsA(500 ng/ml). CsA did not inhibit ERK-1 activation in response toCD3/CD28, PMA, or PMA/Ca21 ionophore stimulation (Fig. 4B).However, activation of JNK-1 and p38 MAPK induced by eitherCD3/CD28 coligation or PMA/A23187 was markedly reduced.Notably, activation of JNK-1 and p38 MAPK induced by PMAwas insensitive to CsA pretreatment. These data are the first toindicate that p38 MAPK activation in Jurkat T cells also requirestwo signals generated by TCR/CD28 or PMA/Ca21 ionophoresignaling.
To obtain additional evidence that the requirement for MAPKactivation differs between T cells at different stages of the cellcycle, we analyzed the activation of ERK-1, JNK-1, and p38MAPK in response to TCR or CD28 stimulation, or both, in pro-liferating mouse T cells. B6 splenic T cells were first stimulated
with plate-bound anti-CD3 and IL-2 for 48 h and then expanded inmedium containing IL-2 for 4 days (36). Interestingly, we notedthat, although the cells were proliferating, about 30–40% of the Tcells were nonviable at the time of harvest. Viable cells were re-covered, stimulated, lysed, and immunoprecipitated. Consistentwith previous reports for Jurkat T cells and primary mouse T cells,TCR stimulation induced full ERK-1 activation without the re-quirement of CD28 costimulation (Fig. 4C, upper panel), whereasoptimal p38 MAPK activation required TCR/CD28 costimulation(Fig. 4C,lower panels). In contrast, JNK-1 was modestly activatedby anti-CD3 stimulation, but no synergistic activation of JNK-1occurred upon CD3/CD28 coligation. JNK-1 was fully activated,however, by CD28 ligation (Fig. 4C, middle panel). These datasuggest that Jurkat human T cells, proliferating mouse T cells, andprimary mouse T cells differ in terms of their requirements forERK-1, JNK-1, and p38 MAPK activation.
p38 MAPK activity is required for CD3 or CD3/CD28 ligation-induced T cell proliferation
The requirement of p38 MAPK activation for T cell proliferationinduced by CD3 or CD3/CD28 ligation was investigated. SplenicT cells were preincubated with different concentrations of
FIGURE 4. Different requirements for MAPKactivation in Jurkat T cells.A, Jurkat T cells (23107/ml) were stimulated with anti-CD3 (10mg/ml),anti-CD28 (2 mg/ml), anti-CD3 plus anti-CD28,PMA (50 ng/ml), and PMA (50 ng/ml) plus A23187(1 mg/ml) for 15 min at 37°C. Cell lysates wereimmunoprecipitated with anti-ERK-1, anti-JNK-1,and anti-p38 MAPK polyclonal Abs, respectively.In vitro kinase assays of ERK-1, JNK-1, and p38MAPK were performed as in Fig. 1A.B, Jurkat Tcells were stimulated as inA, but were either pre-treated or not with CsA (500 ng/ml) for 15 min at37°C before stimulation.C, Proliferating T cellswere stimulated with NIg, anti-CD3 (10mg/ml), anti-CD28 (5mg/ml), and anti-CD3/CD28 for 15 min at37°C. ERK-1, JNK-1, and p38 MAPK activitieswere assayed as in Fig. 1A. Lysates were analyzedfor protein abundance of ERK-1, JNK-1, and p38MAPK. Data from one of three reproducible exper-iments are shown.
3824 COSTIMULATORY ACTIVATION OF p38 MAPK IN PRIMARY T CELLS
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

SB203580, a specific inhibitor of p38 MAPK activity, and werethen cultured in complete RPMI 1640 medium with plate-boundanti-CD3 mAb in the presence or absence of anti-CD28 mAb.SB203580 inhibited T cell proliferation significantly and blockedT cell proliferation completely at concentrations of 10 and100 mM, respectively (Fig. 5A). To exclude the possibility thatinhibition of T cell proliferation results from SB203580-inducedcell toxicity, the capacity of viable T cells to survive after exposureto SB203580 was measured. As evaluated by propidium iodidestaining, we found that SB203580 (100mM) completely inhibitedT cell death induced by anti-CD3 stimulation (Fig. 5B). Consistentwith this observation, SB203580 (40–100mM) also significantlyinhibited TCR-induced cell death in the DO11.10 murine T cellhybridoma (data not shown). This ability of SB203580 to promoteT cell survival was not attributable to any nonspecific DMSO sol-vent effects, as pretreatment of T cells with DMSO alone failed toinhibit p38 MAPK activity (data not shown). This ability ofSB203580 to block T cell proliferation correlated closely with itscapacity to inhibit p38 MAPK activity, as activities of p38 MAPKand its downstream target MAPKAP kinase-2 induced by stimu-lation with either anti-CD3 and anti-CD3/CD28 were greatly di-minished by SB203580 in a dose-dependent manner (Fig. 5C).Note that since SB203580 reversibly binds to the ATP binding siteof p38 MAPK, SB203580 was present continuously during thekinase assays.
At concentrations of$10mM, the SB203580 inhibitor may loseits selectivity and also block JNK2b1 and JNK2b2 (44). To con-firm the specificity of this inhibitor, purified splenic T cells werepretreated with various concentrations of SB203580 (0–100mM),then stimulated with either anti-CD3, anti-CD3 plus anti-CD28, orPMA, and lysed. Cell lysates were either reacted with GST-c-Junprecoupled to glutathione-agarose beads or immunoprecipitatedwith an anti-anti-ERK-1 Ab in the presence of SB203580. Theimmobilized kinases were then incubated with substrates andSB203580 (0–100mM) for 30 min at 30°C. ERK-1 and JNK ac-tivities remained unaffected in the presence of increasing concen-trations of SB203580 (Fig. 5D).
SB203580 inhibits cytokine secretions in primary mouse T cells
We next investigated whether SB203580 inhibits cytokine secre-tions induced by CD3 or CD3/CD28 stimulation of primary mouseT cells. Splenic T cells were pretreated with either different con-centrations of SB203580 in DMSO or equivalent amounts ofDMSO as control, and then stimulated as described above. After48 h, supernatants were collected for quantification of IL-2, IL-4,and IFN-g by ELISA. Interestingly, SB203580 inhibited T cellsecretion of of IL-2, IL-4, and IFN-g induced by either CD3 orCD3/CD28 stimulation in a dose-dependent manner (Fig. 6). Sinceinhibition of T cell proliferation and cytokine secretion bySB203580 does not result from cell death, these data suggest that
FIGURE 5. A p38 MAPK inhibitor,SB203580, suppresses T cell proliferation in-duced by CD3 and CD3/CD28 ligation.A,Splenic T cells (106/ml) were pretreated withvariable concentrations (0–100mM) ofSB203580 or DMSO for 15 min at 37°C, andthen cultured with plate-bound anti-CD3 (1mg/ml) in the presence or absence of anti-CD28(1 mg/ml). Cell proliferation was determined by[3H]thymidine incorporation. The results oftriplicate cultures are expressed as the mean val-ues6 SD.B, Splenic T cells (106/ml) were pre-treated with either SB203580 (100mM) inDMSO or an equivalent amount of DMSO, andthen stimulated with anti-CD3 (1mg/ml). Celldeath was detected by propidium iodide stainingand flow-cytometric analysis.C, Splenic T cells(107/ml) were preincubated with SB203580 (1–100 mM) for 15 min at 37°C, and then stimu-lated with anti-CD3 or anti-CD3 plus anti-CD28for an additional 15 min. p38 MAPK activitywas detected as in Fig. 1A. MAPKAP kinase-2activity was performed using recombinant mu-rine hsp25 as a substrate. Note that SB203580was present during the immunoprecipitation andkinase reactions.D, Splenic T cells (107/ml)were preincubated with SB203580 (1–100mM)for 15 min at 37°C, and then stimulated withanti-CD3 or anti-CD3 plus anti-CD28, or alter-natively with PMA for an additional 15 min.ERK-1 activity was assayed as in Fig. 1A, and asolid JNK assay was performed as in Fig. 1C.ERK-1 abundance was detected as in Fig. 1A.Data from one of two reproducible experimentsare shown.
3825The Journal of Immunology
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

treatment with SB203580 induces T cells to become functionallyunresponsive.
SB203580 represses the nuclear expression of c-Jun and ATF-2
Our data suggest that p38 MAPK, but not JNK, is involved insignal integration of TCR- and CD28-mediated signaling pathwaysin primary mouse T cells. However, JNK is a major kinase re-sponsible for c-Jun phosphorylation and p38 MAPK cannot phos-phorylate c-Jun (28, 31). Indeed, using in vitro kinase assays, wefailed to detect c-Jun phosphorylation by p38 MAPK (data notshown). Since phosphorylation of c-Jun and/or ATF-2 is requiredfor c-Jun gene induction, we analyzed whether blockade of p38MAPK activity results in the reduced phosphorylation of c-Jun andATF-2. Thymocytes were preincubated for 15 min at 37°C with10 mM SB203580 or an equal amount of DMSO as control, andwere then either stimulated with anti-CD3 plus anti-CD28 for 5and 15 min, or left unstimulated. Nuclear extracts were blottedwith anti-c-Jun and anti-ATF-2 mAbs, respectively. Phosphoryla-tion of ATF-2, as revealed by its reduced mobility, was evident
after 5 and 15 min of stimulation, and these mobility shifts weresignificantly inhibited by SB203580 (Fig. 7A,upper panel). Incontrast, no c-Jun phosphorylation was observed (Fig. 7A, lowerpanel), consistent with our in vitro data that JNK is not activatedupon CD3 or CD3/CD28 ligation. Note that the failure to detectc-Jun phosphorylation cannot be attributed to the anti-c-Jun mAbused because it clearly recognized the phosphorylated form ofc-Jun induced by PMA stimulation, as reported (45). SB203580can block the induction of the c-Fos and c-Jun genes by diversestimuli (45, 46), indicating that p38 MAPK activity may play arole in the induction of c-Jun gene expression. This may explainour finding of the nuclear expression of the c-Jun and ATF-2 pro-teins at later time points following CD3/CD28 coligation. Inter-estingly, while SB203580 pretreatment did not alter the nuclearexpression of c-Fos after CD3 or CD3/CD28 stimulation (Fig. 7B,upper panel), SB203580 pretreatment significantly reduced thenuclear expression of c-Jun and ATF-2 induced by CD3 orCD3/CD28 ligation (Fig. 7B, middleand lower panels). The nu-clear expression of c-Jun and ATF-2 was enhanced more upon
FIGURE 6. SB203580 inhibits cytokine production induced by CD3 orCD3/CD28 ligation in primary mouse T cells. Splenic T cells (106/ml)were preincubated with different concentrations (0–50mM) of SB203580for 15 min at 37°C, and then stimulated with anti-CD3 (1mg/ml) in thepresence or absence of the 37.51 anti-CD28 mAb (1mg/ml). Supernatantswere collected after 48 h, and were assayed for their concentrations of IL-2,IL-4, and IFN-g by ELISA using a double ligand method. The results oftriplicate cultures are expressed as the mean values6 SD. Data from oneof three reproducible experiments are shown.
FIGURE 7. SB203580 inhibits the nuclear expression of the c-Jun andATF-2, but not c-Fos proteins in primary mouse T cells.A, Thymocytes(4 3 107/ml) were incubated in the presence (1) or absence (2) ofSB203580, and were either left unstimulated or stimulated for 5 or 15 minat 37°C with anti-CD3 plus anti-CD28.B, Thymocytes were preincubatedwith or without SB203580 and then stimulated for 4 h at37°C with anti-CD3, anti-CD3 plus anti-CD28, or PMA.C, Thymocytes were either leftuntreated or pretreated for 15 min with 20mM cycloheximide (CHX), andwere then stimulated for 4 h at 37°C with anti-CD3 or anti-CD3 plusanti-CD28. Nuclear extracts of cell lysates were analyzed by SDS-PAGE.Immunoblotting was conducted using anti-c-Fos, anti-c-Jun, and anti-ATF-2 mAbs, respectively, and the c-Fos, c-Jun, and ATF-2 proteins weredetected by chemoluminescence. Data from one of three reproducible ex-periments are shown.
3826 COSTIMULATORY ACTIVATION OF p38 MAPK IN PRIMARY T CELLS
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

CD3/CD28 coligation than ligation of CD3 alone. The nuclearexpression of c-Fos, c-Jun, and ATF-2 stimulated by PMA was notinhibited by SB203580.
To confirm that the increase of c-Jun expression in the nucleuswas due to enhancement of de novo c-Jun protein synthesis, thesensitivity of c-Jun nuclear expression to cycloheximide, a proteinsynthesis inhibitor, was determined. Although cycloheximide maystimulate JNK activity, pretreatment of thymocytes with 20mMcycloheximide virtually abolished the increased abundance ofc-Jun induced by CD3 and CD3/CD28 ligation (Fig. 7C). Theblockade of c-Jun expression in the nucleus by SB203580 suggeststhat p38 MAPK activity plays a role in the induction of expressionof the c-Jun protein.
DiscussionPrevious studies of MAPK activation in the Jurkat human T cellline and mouse T cell clones in long-term culture suggested thatJNK activation requires costimulatory signals via CD28 or Ca21
ionophore to synergize with TCR- or PMA-driven signals (33, 34).However, the pathways of activation of JNK and p38 MAPK inprimary T cells were not studied extensively and are less wellunderstood. Accordingly, we investigated the regulation of ERK-1,JNK-1, and p38 MAPK activation stimulated by TCR/CD28 orPMA/Ca21 ionophore-mediated signaling in naive mouse thymo-cytes and splenic T cells. Our main findings are: 1) CD3 or CD28stimulation of primary naive thymocytes and T cells activates p38MAPK; 2) CD3/CD28 coligation synergistically activates p38MAPK, but not JNK; 3) activation of p38 MAPK is Ca21 depen-dent; and 4) the p38 MAPK inhibitor, SB203580, blocks CD3 andCD3/CD28 ligation-induced T cell proliferation as well as produc-tion of IL-2, IL-4, and IFN-g. These results suggest that p38MAPK, but not JNK, is involved in signal integration during CD28costimulation of murine primary T cells and may also play animportant role in the induction of T cell anergy.
Recently, it has been shown that CD28 and PMA/Ca21 iono-phore-triggered signaling stimulate thymocytes and lymph node Tcells to proliferate and produce IL-2 independently of JNK acti-vation in MKK-4-deficient mice (37), suggesting that otherMAPK(s) might be involved in primary T cell costimulation. Moreimportantly, impaired CD28-mediated IL-2 production and prolif-eration by T cells were observed in these mice (37). Since MKK-4activates both JNK and p38 MAPK (5–9), this impairment mightbe attributed to defective p38 MAPK activation in MKK-4-defi-cient mice. However, p38 MAPK activity was not analyzed in thelatter report. Nonetheless, deficient expression of MKK4 does notaffect p38 MAPK activation in embryonic stem cells (47), andSB203580 inhibits CD28-dependent proliferation and IL-2 pro-duction in human T cells (38). These two sets of evidence suggesta role for p38 MAPK in the costimulation of primary mouse Tcells. Although T cell proliferation in response to Con A and PMA/ionomycin stimulation is normal in dominant-negative p38 trans-genic mice, TCR/CD28-mediated T cell proliferation and p38MAPK activity in response to different stimuli were not investi-gated in this study (48). Additional studies conducted primarilywith previously activated T cells have shown that p38 MAPK ac-tivation is inducible by CD3, CD28, or CD3/CD28 stimulation (35,36). Interestingly, however, we observed that MAPK activationdiffers between primary naive T cells and proliferating preacti-vated T cells. In proliferating T cells, while ERK-1 and p38MAPK activity are similar to that in primary naive T cells, JNK-1is weakly activated by CD3 stimulation and is fully activated onlyupon CD28 stimulation. It is important to note that a high percent-age of proliferating T cells become apoptotic, perhaps due to the
continuous exposure to IL-2 (49). Thus, p38 MAPK and JNK-1activation in proliferating T cells may result in part from activa-tion-induced apoptosis.
The requirements for p38 MAPK and JNK-1 activation in mu-rine primary T cells were found to differ from those in Jurkat cells,a human T cell line frequently used to identify pathways of T cellsignaling. In Jurkat cells, optimal activation of both p38 MAPKand JNK-1 requires TCR/CD28 coligation or PMA/Ca21 iono-phore costimulation. In primary mouse T cells, however, stimula-tion by either CD28 or Ca21 ionophore activates p38 MAPK, andp38 MAPK can be activated synergistically by CD3/CD28 coli-gation or PMA/Ca21 ionophore stimulation. Whereas high con-centrations of Ca21 ionophore inhibit JNK-1 activity in primary Tcells, these same concentrations of Ca21 ionophore augment p38MAPK activity. This enhancement of p38 MAPK activity corre-lates closely with T cell proliferation induced by optimal concen-trations of PMA and Ca21 ionophore. Furthermore, CsA inhibitedp38 MAPK activation by anti-CD3, anti-CD3 plus CD28, or PMAplus Ca21 ionophore. Although the levels of activation of ERK-1and p38 MAPK in CD3- or CD3/CD28-stimulated proliferatingand naive primary mouse T cells are similar, JNK-1 is fully acti-vated by CD28 ligation alone in proliferating T cells. These datademonstrate that the requirements for full activation of JNK-1 andp38 MAPK differ among primary T cells, Jurkat T cells, and pro-liferating T cells. Such differences may arise from the stage ofactivation of a T cell. Jurkat T cells are transformed activated cellsin continuous growth in culture, and these cells appear to differsomewhat in their signaling pathways from fresh isolated primaryT cells that presumably consist mainly of T cells in a resting state.
As JNK-1 activation was not detected after CD3/CD28 costimu-lation in primary T cells, we further analyzed the reactivity andepitope specificity of the 37.51 anti-CD28 mAb used for T cellstimulation. Costimulation by a wide concentration range of thismAb plus a single dose of anti-CD3 did not induce JNK-1 acti-vation. In contrast, optimum conditions of CD3/CD28 costimula-tion elicited the full activation of p38 MAPK. While recent studiesindicate that JNK-1 may be activated by CD3/CD28 coligation inmouse lymph node T cells, this was achievable only after preac-tivation of the T cells in the presence of plate-bound anti-CD3 andanti-CD28 mAbs for 40 h (50). Replacement of the 37.51 anti-CD28 mAb with the PV-1 anti-CD28 mAb, which differs in itsCD28 epitope specificity from the 37.51 mAb, also did not stim-ulate JNK-1 activation. Furthermore, the 37.51 anti-CD28 mAbelicited full JNK-1 activation in preactivated proliferating mouse Tcells. Importantly, the results obtained from a solid-phase JNKassay were similar to those observed using in vitro kinase assays ofJNK-1 and JNK-2. This suggests that the failure to detect JNKactivation in response to CD3 and CD28 ligation is not due to theinability of the anti-JNK Abs used to immunoprecipitate all iso-forms of JNK. Note that the gel loading of equivalent amounts ofJNK proteins was confirmed by anti-JNK immunoblotting, whichexcludes the possibility that these observations result from unequalamounts of kinase proteins loaded. It is evident therefore that thefailure to detect JNK activation in response to CD3/CD28 coliga-tion in primary mouse T cells is not due to the use of an inappro-priate anti-CD28 mAb. Thus, JNK does not appear to mediateCD28 costimulatory signaling in primary mouse T cells.
Additional supportive evidence for a role for p38 MAPK in Tcell costimulation was derived from the use of SB203580, a highlyspecific pyrinidyl imidazole inhibitor of p38 MAPK (17).SB203580 effectively blocked T cell proliferation, cytokine (IL-2,IL-4, and IFN-g) production, and p38 MAPK activation followingCD3 or CD3/CD28 stimulation. These results lend credence to the
3827The Journal of Immunology
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

idea that p38 MAPK plays an important role in TCR/CD28 co-stimulation of proliferation and cytokine production in primary Tcells. Since inhibition of T cell proliferation and cytokine secretionby SB203580 does not result from T cell death or T cell toxicity,SB203580 treatment appears to induce the functional unrespon-siveness of T cells. The latter observation supports the notion thatp38 MAPK may regulate the entry of T cells into a state of anergy.Consistent with this observation, SB203580 has been shown toinhibit anti-CD3-induced deletion of CD41CD81 thymocytes infetal thymic organ culture (51).
The protein complex that binds to the AP-1 transcriptional ac-tivation site of the IL-2 gene promoter region may comprise eithera c-Jun-c-Jun homodimer or c-Jun-Fos heterodimer, and formationof the AP-1 complex is critical for the regulation of IL-2 geneexpression (33). Decreased binding of the AP-1 protein complex tothe IL-2 promoter has been implicated in the molecular basis of Tcell anergy (52). Phosphorylation of c-Jun at Ser63 and Ser73 ac-tivates c-Jun gene transcription (10, 53), and this phosphorylationis mediated by JNK, but not p38 MAPK (28). We found that CD3/CD28 or PMA/Ca21 ionophore costimulation did not result in ap-preciable JNK-1 or JNK-2 activation in primary mouse T cells;rather, PMA/Ca21 ionophore costimulation inhibited JNK-1 activ-ity in these T cells. Although we did not detect any c-Jun phos-phorylation by p38 MAPK in CD3/CD28-costimulated mouse thy-mocytes, c-Jun protein synthesis induced by CD3 or CD3/CD28ligation was completely inhibited by SB203580. Consistent withour findings, SB203580 is known to block the induction of thec-Fos and c-Jun genes by various stimuli despite the lack of aneffect of SB203580 on c-Jun and ATF-2 phosphorylation (45, 46).This raises the possibility that the phosphorylation of other tran-scription factors by p38 MAPK may be essential for c-Jun geneactivation. We have shown that ATF-2 can be phosphorylated byp38 MAPK in vitro, and phosphorylation of ATF-2 in the nucleusis inhibited by SB203580. ATF-2 may therefore mediate the in-duction of c-Jun gene activation. Furthermore, in monocytes thatmigrate to a site of inflammation, phosphorylation of MEF2C byp38 MAPK increases c-Jun transcription and inhibition of p38MAPK activity diminishes LPS-induced c-Jun transcription (31).Taken together, our data suggest that p38 MAPK may be the majorkinase responsible for the induction of c-Jun activation in primarymouse T cells. Although JNK-3 has been identified (54), JNK-3 isselectively expressed in the nervous system, and it is unlikely thatJNK-3 is involved in the phosphorylation and induction of c-Jungene expression in T cells. The lack of an effect of SB203580 onnuclear c-Fos expression may be explained by the fact that CD3ligation also activates ERK-1, which is not inhibited by SB203580.
In summary, our results suggest that p38 MAPK is involved inboth TCR- and CD28-signaling pathways, and that p38 MAPK,but not JNK, plays an important role in signal integration duringcostimulation of primary mouse T cells. p38 MAPK activity maybe involved in the induction of c-Jun activation, augmentation ofAP-1 transcriptional activity, and stimulation of IL-2, IL-4, andIFN-g production. Inhibition of p38 MAPK activity may lead tothe induction of T cell anergy.
AcknowledgmentsWe thank Drs. S. Kaga and H. Tan for helpful discussions; Drs.J. Bluestone, J. P. Allison, P. Young, and C. June for their kind gifts ofvarious reagents that made this work possible; Ms. A. Leaist for her valu-able assistance in the preparation of this manuscript; and all members ofour laboratory for their advice and encouragement.
References1. Blenis, J. 1993. Signal transduction via the MAP kinases: proceed at your own
RSK. Proc. Natl. Acad. Sci. USA 90:5889.2. Cobb, M. H., and E. J. Goldsmith. 1995. How MAP kinases are regulated.J. Biol.
Chem. 270:14843.3. Davis, R. J. 1993. The mitogen-activated protein kinase signal transduction path-
way. J. Biol. Chem. 286:14553.4. Kyriakis, J. M., and J. Avruch. 1996. Sounding the alarm: protein kinase cascades
activated by stress and inflammation.J. Biol. Chem. 271:24313.5. Deacon, K., and J. L. Blank. 1997. Characterization of the mitogen-activated
protein kinase kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38pathways regulated by MEK kinases 2 and 3.J. Biol. Chem. 272:14489.
6. Derijard, B., J. Raingeaud, T. Barrett, I.-H. Wu, J. Han, R. J. Ulevitch, andR. J. Davis. 1995. Independent human MAP kinase signal transduction pathwaysdefined by MEK and MKK isoforms.Science 267:682.
7. Ellinger-Ziegelbauer, H., K. Brown, and U. Siebenlist. 1997. Direct activation ofthe stress-activated protein kinase (SAPK) and extracellular signal-regulated pro-tein kinase (ERK) pathways by an inducible mitogen-activated protein kinase/ERK kinase kinase 3 (MEKK) derivative.J. Biol. Chem. 272:2668.
8. Raingeaud, J., A. J. Whitmarsh, T. Barrett, B. Derijard, and R. J. Davis. 1996.MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway.Mol. Cell. Biol. 16:1247.
9. Tibbles, L. A., Y. L. Ing, F. Kiefer, J. Chan, N. Iscove, J. R. Woodgett, andN. J. Lassam. 1996. MLK-3 activates the SAPK/JNK and p38/RK pathways viaSEK1 and MKK3/6.EMBO J. 15:7026.
10. Derijard, B., M. Hibi, I. H. Wu, T. Barrett, B. Su, T. Deng, M. Karin, andR. J. Davis. 1994. JNK1: a protein kinase stimulated by UV light and Ha-Ras thatbinds and phosphorylates the c-Jun activation domain.Cell 76:1025.
11. Adler, V., M. R. Pincus, A. Polotskaya, X. Montano, F. K. Friedman, andZ. Ronai. 1996. Activation of c-Jun-NH2-kinase by UV irradiation is dependenton p21ras. J. Biol. Chem. 271:23304.
12. Chen, Y. R., C. F. Meyer, and T. H. Tan. 1996. Persistent activation of c-JunN-terminal kinase 1 (JNK1) ing radiation-induced apoptosis.J. Biol. Chem.271:631.
13. Galcheva-Gargova, Z., B. Derijard, I.-H. Wu, and R. J. Davis. 1994. An osmo-sensing signal transduction pathway in mammalian cells.Science 265:806.
14. Read, M. A., M. Z. Whitley, S. Gupta, J. W. Pierce, J. Best, R. J. Davis, andT. Collins. 1997. Tumor necrosis factora-induced E-selectin expression is acti-vated by the nuclear factor-kB and c-Jun N-terminal kinase/p38 mitogen-acti-vated protein kinase pathways.J. Biol. Chem. 272:2753.
15. Rosette, C., and M. Karin. 1996. Ultraviolet light and osmotic stress: activationof the JNK cascade through multiple growth factor and cytokine receptors.Sci-ence 274:1194.
16. Swantek, J. L., M. H. Cobb, and T. D. Geppert. 1997. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharidestimulation of tumor necrosis factora (TNF-a) translation: glucocorticoids in-hibit TNF-a translation by blocking JNK/SAPK.Mol. Cell. Biol. 17:6274.
17. Cuenda, A., J. Rouse, Y. N. Doza, R. Meier, P. Cohen, T. F. Gallagher,P. R. Young, and J. C. Lee. 1995. SB203580 is a specific inhibitor of a MAPkinase homologue which is stimulated by cellular stresses and interleukin-1.FEBS Lett. 364:229.
18. Raingeaud, J., S. Gupta, J. S. Rogers, M. Dickens, J. Han, R. J. Ulevitch, andR. J. Davis. 1995. Pro-inflammatory cytokines and environmental stress causep38 mitogen-activated protein kinase activation by dual phosphorylation on ty-rosine and threonine.J. Biol. Chem. 270:7420.
19. Rouse, J., P. Cohen, S. Trigon, M. Morange, A. Alonso-Liamazares,D. Zamanillo, T. Hunt, and A. R. Nebreda. 1994. A novel kinase cascade trig-gered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphor-ylation of the small heat shock proteins.Cell 78:1027.
20. Yamauchi, J., M. Nagao, Y. Kaziro, and H. Itoh. 1997. Activation of p38 mito-gen-activated protein kinase by signaling through G protein-coupled receptors:involvement of Gbetagamma and Galphaq/11 subunits.J. Biol. Chem.272:27771.
21. Cuenda, A., P. Cohen, V. Buee-Scherrer, and M. Goedert. 1997. Activation ofstress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses ismediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 andSAPK2 (RK/p38).EMBO J. 16:295.
22. Graves, J. D., K. E. Draves, A. Craxton, J. Saklatvala, E. G. Krebs, andE. A. Clark. 1996. Involvement of stress-activated protein kinase and p38 mito-gen-activated protein kinase in mIgM-induced apoptosis of human B lympho-cytes.Proc. Natl. Acad. Sci. USA 93:13814.
23. Kummer, J. L., P. K. Rao, and K. A. Heidenreich. 1997. Apoptosis induced bywithdrawal of trophic factors is mediated by p38 mitogen-activated protein ki-nase.J. Biol. Chem. 272:20490.
24. Lee, J. C., J. T. Laydon, P. C. McDonnell, T. F. Gallagher, S. Kumar, D. Green,D. McNutty, M. J. Blumenthal, J. R. Heys, S. W. Landvatter, et al. 1994. Aprotein kinase involved in the regulation of inflammatory cytokine biosynthesis.Nature 372:739.
25. Price, M. A., F. H. Cruzalegui, and R. Treisman. 1996. The p38 and ERK MAPkinase pathways cooperate to activate ternary complex factors and c-fos tran-scription in response to UV light.EMBO J. 15:6552.
26. Toyoshima, F., T. Moriguchi, and E. Nishida. 1997. Fas induces cytoplasmicapoptotic responses and activation of the MKK7-JNK/SAPK and MKK6–p38pathways independent of CPP32-like proteases.J. Cell Biol. 139:1005.
3828 COSTIMULATORY ACTIVATION OF p38 MAPK IN PRIMARY T CELLS
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

27. Jiang, Y., C. Chen, Z. Li, W. Guo, J. A. Gegner, S. Lin, and J. Han. 1996.Characterization of the structure and function of a new mitogen-activated proteinkinase (p38b). J. Biol. Chem. 271:17920.
28. Goedert, M., A. Cuenda, M. Graxton, R. Jakes, and P. Cohen. 1997. Activationof the novel stress-activated protein kinase SAPK4 by cytokines and cellularstresses is mediated by SKK3 (MKK6); comparison of its substrate specificitywith that of other SAP kinases.EMBO J. 12:3563.
29. Tan, Y., J. Rouse, Z. Zhang, S. Cariati, P. Cohen, and M. J. Comb. 1996. FGF andstress regulate CREB and ATF-1 via pathway involving RK/p38 MAP kinase andMAPAP kinase-2.EMBO J. 15:4629.
30. Wang, X., and D. Ron. 1996. Stress-induced phosphorylation and activation ofthe transcription factor CHOP (GADD153) by p38 MAP kinase.Science 272:1347.
31. Han, J., Y. Jiang, Z. Li, V. V. Kravchenko, and R. J. Ulevitch. 1997. Activationof the transcription factor MEF2C by the MAP kinase p38 in inflammation.Nature 386:296.
32. Gupta, S., D. Campbell, B. Derijard, and R. J. Davis. 1995. Transcription factorATF2 regulation by the JNK signal transduction pathway.Science 267:389.
33. Su, B., E. Jacinto, M. Hibi, T. Kallunki, M. Karin, and Y. Ben-Neriah. 1994. JNKis involved in signal integration during costimulation of T lymphocytes.Cell77:727.
34. Li, W., C. D. Whaley, A. Mondino, and D. L. Mueller. 1996. Blocked signaltransduction to the ERK and JNK protein kinases in anergic CD41 T cells.Science 271:1272.
35. DeSilva, D. R., E. A. Jones, W. S. Feeser, E. J. Manos, and P. Scherle. 1997. Thep38 mitogen-activated protein kinase pathway in activated and anergic Th1 cells.Cell. Immunol. 180:116.
36. Salmon, R. A., I. N. Foltz, P. R. Young, and J. W. Schrader. 1997. The p38mitogen-activated protein kinase is activated by ligation of the T or B lymphocyteantigen receptors, Fas or CD40, but suppression of kinase activity does not inhibitapoptosis induced by antigen receptors.J. Immunol. 159:5309.
37. Nishina, H., M. Bachmann, A. J. Oliveira-dos-Santos, I. Kozieradzki,K. D. Fischer, B. Odermatt, A. Wakeham, A. Shahinian, H. Takimoto,A. Bernstein, et al. 1997. Impaired CD28-mediated interleukin-2 production andproliferation in stress kinase SAPK/ERK1 kinase (SEK1)/mitogen-activated pro-tein kinase kinase 4 (MKK4)-deficient T lymphocytes.J. Exp. Med. 186:941.
38. Ward, S. G., R. V. Parry, J. Matthews, and L. O’Neill. 1997. A p38 MAP kinaseinhibitor SB203580 inhibits CD28-dependent T cell proliferation and IL-2 pro-duction.Biochem. Soc. Trans. 25:304S.
39. Hibi, M., A. Li, T. Smeal, A. Minden, and M. Karin. 1993. Identification of anoncoprotein- and UV-responsive protein kinase that binds and potentiates thec-Jun activation domain.Genes Dev. 7:2135.
40. Dolmetsch, R. E., R. S. Lewis, C. C. Goodnow, and J. I. Healy. 1997. Differentialactivation of transcription factors induced by Ca21 response amplitude and du-ration.Nature 386:855.
41. Kalluni, T., B. Su, I. Tsigelny, H. K. Sluss, B. Derijard, G. Moore, R. J. Davis,and M. Karin. 1994. JNK2 contains a specificity-determining region responsiblefor efficient c-Jun binding and phosphorylation.Genes Dev. 8:2996.
42. Sluss, H. K., T. Barrett, B. Derijard, and R. J. Davis. 1994. Signal transductionby tumor necrosis factor mediated by JNK protein kinases.Mol. Cell. Biol. 14:8376.
43. Gupta, S., T. Barrett, A. J. Whitarsh, J. Cavanagh, H. K. Sluss, B. Derijard, andR. J. Davis. 1996. Selective interaction of JNK protein kinase isoforms withtranscription factors.EMBO J. 15:2760.
44. Whitmarsh, A. J., S.-H. Yang, M. S.-S. Su, A. D. Sharrocks, and R. J. Davis.1997. Role of p38 and JNK mitogen-activated protein kinases in the activation ofternary complex factors.Mol. Cell. Biol. 17:2360.
45. Hazzalin, C. A., E. Cano, A. Cuenda, M. J. Barratt, P. Cohen, andL. C. Mahadevan. 1996. p38/RK is essential for stress-induced nuclear response:JNK/SAPKs and c-Jun/ATF-2 phosphorylation are insufficient.Curr. Biol.6:1028.
46. Hazzalin, C. A., A. Cuenda, E. Cano, P. Cohen, and L. C. Mahadevan. 1997.Effects of the inhibition of p38/RK MAP kinase on induction of fivefosand jungenes by diverse stimuli.Oncogene 15:2321.
47. Yang, D., C. Tournier, M. Wysk, H.-T. Lu, J. Xu, R. J. Davis, and R. A. Flavell.1997. Targeted disruption of the MKK4 gene causes embryonic death, inhibitionof c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional ac-tivity. Proc. Natl. Acad. Sci. USA 94:3004.
48. Rincon, M., H. Enslen, J. Raingeaud, M. Recht, T. Zapton, M. S.-S. Su,L. A. Penix, R. J. Davis, and R. A. Flavell. 1998. Interferon-g expression by Th1effector T cells mediated by the p38 MAP kinase signaling pathway.EMBO J.17:2817.
49. Lenardo, M. J., S. Boehme, L. Chen, B. Combadiere, G. Fisher, M. Freedman,H. McFarland, C. Pelfrey, and L. Zheng. 1995. Autocrine feedback death and theregulation of mature T lymphocyte antigen responses.Int. Rev. Immunol. 13:115.
50. Calvo, C. R., D. Amsen, and A. M. Kruisbeek. 1997. Cytotoxic T lymphocyteantigen 4 (CTLA-4) interferes with extracellular signal-regulated kinase (ERK)and Jun NH2-terminal kinase (JNK) activation, but does not affect phosphoryla-tion of T cell receptorz and ZAP-70.J. Exp. Med. 186:1645.
51. Sugawara, T., T. Moriguchi, E. Nishida, and Y. Takahama. 1998. Differentialroles of ERK and p38 MAP kinase pathways in positive and negative selectionof T lymphocytes.Immunity 9:565.
52. Kang, S.-M., B. Beverly, A.-C. Tran, K. Brorson, R. H. Schwartz, andM. J. Lenardo. 1992. Transactivation by AP-1 is a molecular target of T cellanergy.Science 257:1134.
53. Minden, A., A. Lin, F. X. Claret, A. Abo, and M. Karin. 1995. Selective acti-vation of the JNK signaling cascade and c-Jun transcriptional activity by thesmall GTPases Rac and Cdc42Hs.Cell 81:1147.
54. Yang, D. D., C. Y. Kuan, A. J. Whitmarsh, M. Rincon, T. S. Zheng, R. J. Davis,P. Rakic, and R. A. Flavell. 1997. Absence of excitotoxicity-induced apoptosis inthe hippocampus of mice lacking the Jnk gene.Nature 389:865.
3829The Journal of Immunology
by guest on June 10, 2018http://w
ww
.jimm
unol.org/D
ownloaded from







