
Understanding Heterogeneous Nucleation in Binary, Solution-Processed, Organic Semiconductor Thin FilmsStephanie S. Lee,† Srevatsan Muralidharan,‡ Arthur R. Woll,§ Marsha A. Loth,∥ Zhong Li,∥
John E. Anthony,∥ Mikko Haataja,‡ and Yueh-Lin Loo*,†
†Department of Chemical and Biological Engineering and ‡Department of Mechanical and Aerospace Engineering, PrincetonUniversity, Princeton, New Jersey 08544, United States§Cornell High Energy Synchrotron Source, Cornell University, Ithaca, New York 14853, United States∥Department of Chemistry, University of Kentucky, Lexington, Kentucky 40506, United States
*S Supporting Information
ABSTRACT: Heterogeneous nucleation is often the precursor to crystallizationin solution-processed organic semiconductor thin films. Here, we study theefficacy of a series of nine small-molecule organic semiconductor additives inseeding the crystallization of solution-processable triethylsilylethynyl anthradi-thiophene (TES ADT). By systematically varying the concentrations of theadditives in TES ADT thin films, we found the tendency of the additives tocrystallize, their solubility in the casting solvent, and their similarity in chemicalstructure to TES ADT, to determine the nucleation and resulting density ofnuclei. Tracking the crystallization process further yields information about themechanism of nucleation. While pure TES ADT nucleates instantaneously at theonset of crystallization, nucleation transitions to a distributed process occurringthroughout crystallization with the incorporation of increasing amounts ofadditives.
KEYWORDS: heterogeneous nucleation, organic semiconductor, Avrami kinetics, dopant, solution processing
■ INTRODUCTION
Blending two organic semiconductors to form thin films is apromising strategy to bring about unique or enhancedelectronic properties in the active layers of organic electronicdevices.1 Bulk-heterojunction organic solar cells (OSCs), forexample, utilize blends of two organic semiconductors withdifferent energy levels to efficiently dissociate excitons forharvesting light.2 Doping an organic semiconductor “host” withfractional amounts of another organic semiconductor, or“guest”, has also been successfully employed to improve themobility of organic thin-film transistors (OTFTs),3,4 theconductivity of charge transport layers in organic light-emittingdiodes (OLEDs),5,6 and the luminescence of light-emittinglayers in OLEDs.7−9 In choosing organic semiconductor pairsfor such blends or guest-host systems, it is important toconsider the respective electronic properties of the constituentcomponents. Equally imperative is a comprehensive under-standing of the morphological development of such systems.10
The presence of an additive, for example, can inducecrystallization and improve the crystallinity of the host organicsemiconductor in the active layers of OTFTs, improving devicemobility.4,11 Aggregation of emissive dopants at high loadinglevels in guest-host OLEDs, on the other hand, can decreasethe overall luminescence of the light-emitting layer.8,9 In lightof the importance of the active layer morphology indetermining overall device performance, it is critical that we
understand how the physical parameters of one compound inbinary, organic semiconductor thin films influences the finalfilm morphology.In single-component, organic semiconductor systems,
structural development occurs through nucleation and growth.Heterogeneous nucleation on foreign objects, like defects anddust particles on the substrate surface,12 determines thenucleation density and thus the final size of crystalline domains,with boundaries between these domains acting as barriers tocharge transport.13−15 In binary organic semiconductorsystems, structural development is more complicated, withboth phase separation and constituent crystallization occurringsimultaneously. Depending on the chemical compatibility of thetwo organic semiconductors and their respective interactionswith the underlying substrates, phase separation can occur bothlaterally and vertically in these thin films.10,16−19 Furthercomplicating the structural development of two-componentsystems is the fact that the organic semiconductor constituentscan individually crystallize and one can aid in nucleating4 orsuppressing20 crystallization of the other. Cho and co-workers,for example, found the incorporation of a small-moleculedopant to poly(3-hexyl thiophene) (P3HT) thin filmsin
Received: April 6, 2012Revised: July 5, 2012Published: July 6, 2012
Article
pubs.acs.org/cm
© 2012 American Chemical Society 2920 dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−2928

addition to increasing the carrier concentrationto seed P3HTcrystallization.4 Both the presence of the dopant and thisimproved crystallinity are reported to increase device mobilityby 30-fold compared to OTFTs comprising pristine P3HT asactive layers. At high blend ratios of P3HT and [6,6]-phenyl-C61-butyric acid methyl ester (PCBM), on the other hand, thecrystallization of both P3HT and PCBM is hindered by thepresence of the other component.20 Postprocessing steps, suchas thermal annealing21 and solvent-vapor annealing,16 or theincorporation of solvent additives to the cosolution prior todeposition,22 have thus been employed to enhance crystal-lization and to improve the efficiencies of OSCs comprisingblends of P3HT and PCBM in the active layers. For maximumdevice performance, it is commonly accepted that suchcrystallization should result in domains on the order of tensof nanometers, a length scale that is commensurate with thecharacteristic exciton diffusion length.23
In this manuscript, we examine how the presence of small-molecule additives affects the structural development of thehost organic semiconductor by studying their effectiveness inheterogeneously nucleating triethylsilylethynyl anthradithio-phene (TES ADT).24 We chose TES ADT as the host organicsemiconductor for several reasons. When spun cast ontosubstrates, TES ADT forms largely amorphous films, allowingus to controllably induce crystallization with subsequentsolvent-vapor annealing.25 During exposure to 1,2-dichloro-ethane (DCE) solvent vapors, TES ADT crystallizes throughnucleation and growth of spherulites. Because each spheruliteresults from a single nucleus, tracking spherulite formationallows us to infer information about nucleation events.Furthermore, the diameters of TES ADT spherulites rangefrom 30 μm to 3 mm, and as such are easily identifiable viaoptical microscopy. By measuring the average spherulitediameter, L, in TES ADT thin films after crystallization iscomplete, we can calculate the average number density ofspherulites, ⟨S⟩, and thus extract the average number of nucleiin TES ADT thin films using the following equation:
π⟨ ⟩ =S
1L4
2
(1)
by approximating spherulites as circles.We have chosen nine different small-molecule organic
semiconductors as additives in TES ADT thin films. We haveselected the additives to be electrically active and usedconcentrations ranging from 0 to 10 mol % relative to TESADT so the findings of our study would be relevant to guest-host OTFT and OLED systems.3−6 Establishing structure-function relationships between the morphology in the activelayer and overall device performance in organic electronics firstrequires a fundamental understanding of structural develop-ment during processing. Central to this manuscript is thus howthe presence of additives affects the overall structure of TESADT thin films. The electrical characterization of these two-component, organic semiconductor thin films is currentlyunderway and will be reported subsequently. By comparing theextent with which these additives can heterogeneously nucleateTES ADT during solvent-vapor annealing, we can determinethe key attributes of the additives that influence their ability toact as nuclei. By tracking the kinetics of crystallization duringDCE solvent−vapor annealing, we further elucidate how thepresence of additives alters the mechanism with which TESADT thin films are nucleated.
■ EXPERIMENTAL METHODSFilm Formation. The 300 nm thermally grown SiO2 on doped-Si
wafers purchased from Process Specialties were used as substrates forTES ADT thin films. The wafers were rinsed sequentially with acetone,isopropyl alcohol, and deionized water and then dried with housenitrogen prior to spin coating TES ADT solutions. TES ADT,24
fluorinated 5,11-bis(triethylsilylethynyl) anthradithiophene (F-TESADT),26 chlorinated 5,11-bis(triethylsilylethynyl) anthradithiophene(Cl-TES ADT),27 brominated 5,11-bis(triethylsilylethynyl) anthradi-th iophene (Br-TES ADT), 27 and iod inated 5 ,11-b i s -(triethylsilylethynyl) anthradithiophene (I-TES ADT),27 diethyl-5,11-bis(triethylsilylethynyl) anthradithiophene (ethyl-TES ADT),28 triiso-propylsilylethynyl pentacene (TIPS pen),29 and triisobutylsilylethynylpentacene (TIBS pen)30 were synthesized according to previouslypublished procedures. The procedure to synthesize a fullerenederivative that is functionalized with triethylsilylethynyl tetracene(TES Tet Fu) is included in the Supporting Information. PCBM waspurchased from Nano-C. To form TES ADT thin films, TES ADT wasfirst dissolved in toluene at a concentration of 2 wt %. In a separatevial, a small-molecule additive was dissolved in toluene atconcentrations between 1 − 2 wt %. The appropriate volume of theadditive solution was then added to TES ADT solutions in order toachieve the desired additive molar concentration relative to TES ADT.Given that TES ADT photobleaches easily, the solution was onlyallowed to sit for <4 min prior to spin coating. Fresh solutions weremade for each subsequent spin coating. Approximately 100 μL of thesolutions were placed onto precleaned-SiO2/doped-Si substrates. Thesubstrates were then spun at 1000 rpm for 60 s to form 100-nm thickfilms, then thermally annealed at 90 °C for 2 min to drive off residualtoluene.
Solvent-Vapor Annealing. Exposure to DCE solvent vapor wascarried out using a solvent-vapor annealing setup described else-where.31 During solvent-vapor annealing, structural development ofthe films was tracked under an optical microscope. For films in whichthe total time for crystallization exceeded 20 s, a Nikon DXM 1200camera was used to take sequential frames of optical micrographs ofthe films in 5 s intervals. For all other samples, a high-speed SonyXCD-710CR camera was used to collect sequential frames of opticalmicrographs every 0.5 s. Image processing to calculate the fractionalarea crystallized in each image was performed in ImageJ software(version 1.45). Because film thicknesses can vary near the edges of thesubstrates as a consequence spin coating, we avoided these regionswhen collecting images for analyses.
Atomic Force Microscopy (AFM) Experiments. The 50/50molar ratio blends comprising TES ADT and TES ADT derivativeswere formed by codissolving the two compounds in toluene for a totalcombined concentration of 2 wt %. The solutions were spun cast at1000 rpm for 60 s onto precleaned SiO2/doped Si substrates. Thefilms were then annealed at 90 °C for 2 min to drive off residualtoluene. These films were not solvent-vapor annealed. Subsequentsolvent-vapor annealing does not incur any substantial structuraldifferences. AFM height micrographs were collected in tapping modeusing a Veeco Dimension 3100 NanoMan AFM.
Grazing Incidence X-ray Diffraction (GIXD) Experiments.GIXD was performed at station G2 at the Cornell High EnergySynchrotron Source (CHESS) with X-rays at 8.64 ± 0.01 keV (λ =0.1435 nm), selected using a Be single-crystal monochromator.Motorized slits were used to define a 0.2 × 3 (V × H) mm2 beam,with a typical flux of 2 × 1010 photons/s. The data were collected usinga 640-element 1D diode-array, of which each element incorporates itsown pulse counting electronics capable of count rates of ∼105photons/s. A set of 0.1° Soller slits were used on the detector armto provide an in-plane resolution of 0.16°. The scattering geometry isdescribed in detail elsewhere.32 GIXD patterns were collected on thesame films that were previously characterized via AFM.
Solubility Measurements. To measure the solubilities of theadditives in toluene, we first weighed out known quantities, between 3and 4 mg, of the compound and then slowly added toluene until thepowders were completely dissolved. The solubility was measured for
Chemistry of Materials Article
dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−29282921
three samples for each additive to estimate the error associated withthe measurements.
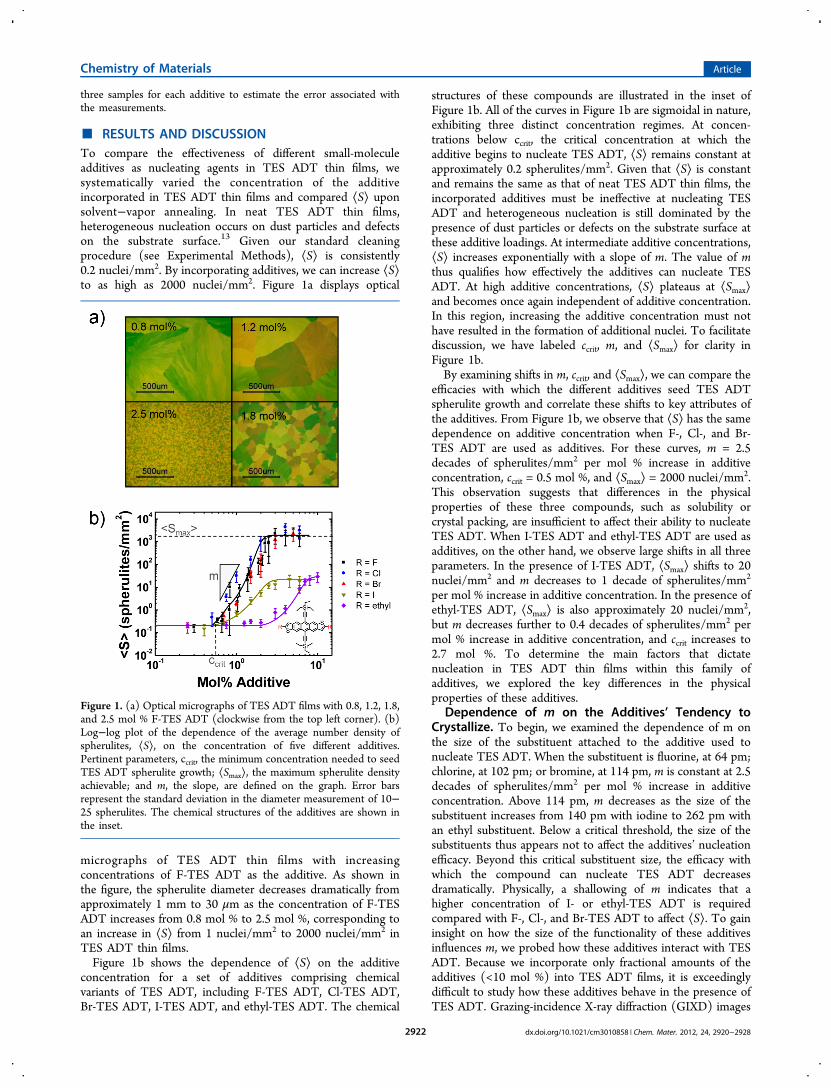
■ RESULTS AND DISCUSSIONTo compare the effectiveness of different small-moleculeadditives as nucleating agents in TES ADT thin films, wesystematically varied the concentration of the additiveincorporated in TES ADT thin films and compared ⟨S⟩ uponsolvent−vapor annealing. In neat TES ADT thin films,heterogeneous nucleation occurs on dust particles and defectson the substrate surface.13 Given our standard cleaningprocedure (see Experimental Methods), ⟨S⟩ is consistently0.2 nuclei/mm2. By incorporating additives, we can increase ⟨S⟩to as high as 2000 nuclei/mm2. Figure 1a displays optical
micrographs of TES ADT thin films with increasingconcentrations of F-TES ADT as the additive. As shown inthe figure, the spherulite diameter decreases dramatically fromapproximately 1 mm to 30 μm as the concentration of F-TESADT increases from 0.8 mol % to 2.5 mol %, corresponding toan increase in ⟨S⟩ from 1 nuclei/mm2 to 2000 nuclei/mm2 inTES ADT thin films.Figure 1b shows the dependence of ⟨S⟩ on the additive
concentration for a set of additives comprising chemicalvariants of TES ADT, including F-TES ADT, Cl-TES ADT,Br-TES ADT, I-TES ADT, and ethyl-TES ADT. The chemical
structures of these compounds are illustrated in the inset ofFigure 1b. All of the curves in Figure 1b are sigmoidal in nature,exhibiting three distinct concentration regimes. At concen-trations below ccrit, the critical concentration at which theadditive begins to nucleate TES ADT, ⟨S⟩ remains constant atapproximately 0.2 spherulites/mm2. Given that ⟨S⟩ is constantand remains the same as that of neat TES ADT thin films, theincorporated additives must be ineffective at nucleating TESADT and heterogeneous nucleation is still dominated by thepresence of dust particles or defects on the substrate surface atthese additive loadings. At intermediate additive concentrations,⟨S⟩ increases exponentially with a slope of m. The value of mthus qualifies how effectively the additives can nucleate TESADT. At high additive concentrations, ⟨S⟩ plateaus at ⟨Smax⟩and becomes once again independent of additive concentration.In this region, increasing the additive concentration must nothave resulted in the formation of additional nuclei. To facilitatediscussion, we have labeled ccrit, m, and ⟨Smax⟩ for clarity inFigure 1b.By examining shifts in m, ccrit, and ⟨Smax⟩, we can compare the
efficacies with which the different additives seed TES ADTspherulite growth and correlate these shifts to key attributes ofthe additives. From Figure 1b, we observe that ⟨S⟩ has the samedependence on additive concentration when F-, Cl-, and Br-TES ADT are used as additives. For these curves, m = 2.5decades of spherulites/mm2 per mol % increase in additiveconcentration, ccrit = 0.5 mol %, and ⟨Smax⟩ = 2000 nuclei/mm2.This observation suggests that differences in the physicalproperties of these three compounds, such as solubility orcrystal packing, are insufficient to affect their ability to nucleateTES ADT. When I-TES ADT and ethyl-TES ADT are used asadditives, on the other hand, we observe large shifts in all threeparameters. In the presence of I-TES ADT, ⟨Smax⟩ shifts to 20nuclei/mm2 and m decreases to 1 decade of spherulites/mm2
per mol % increase in additive concentration. In the presence ofethyl-TES ADT, ⟨Smax⟩ is also approximately 20 nuclei/mm2,but m decreases further to 0.4 decades of spherulites/mm2 permol % increase in additive concentration, and ccrit increases to2.7 mol %. To determine the main factors that dictatenucleation in TES ADT thin films within this family ofadditives, we explored the key differences in the physicalproperties of these additives.
Dependence of m on the Additives’ Tendency toCrystallize. To begin, we examined the dependence of m onthe size of the substituent attached to the additive used tonucleate TES ADT. When the substituent is fluorine, at 64 pm;chlorine, at 102 pm; or bromine, at 114 pm, m is constant at 2.5decades of spherulites/mm2 per mol % increase in additiveconcentration. Above 114 pm, m decreases as the size of thesubstituent increases from 140 pm with iodine to 262 pm withan ethyl substituent. Below a critical threshold, the size of thesubstituents thus appears not to affect the additives’ nucleationefficacy. Beyond this critical substituent size, the efficacy withwhich the compound can nucleate TES ADT decreasesdramatically. Physically, a shallowing of m indicates that ahigher concentration of I- or ethyl-TES ADT is requiredcompared with F-, Cl-, and Br-TES ADT to affect ⟨S⟩. To gaininsight on how the size of the functionality of these additivesinfluences m, we probed how these additives interact with TESADT. Because we incorporate only fractional amounts of theadditives (<10 mol %) into TES ADT films, it is exceedinglydifficult to study how these additives behave in the presence ofTES ADT. Grazing-incidence X-ray diffraction (GIXD) images
Figure 1. (a) Optical micrographs of TES ADT films with 0.8, 1.2, 1.8,and 2.5 mol % F-TES ADT (clockwise from the top left corner). (b)Log−log plot of the dependence of the average number density ofspherulites, ⟨S⟩, on the concentration of five different additives.Pertinent parameters, ccrit, the minimum concentration needed to seedTES ADT spherulite growth; ⟨Smax⟩, the maximum spherulite densityachievable; and m, the slope, are defined on the graph. Error barsrepresent the standard deviation in the diameter measurement of 10−25 spherulites. The chemical structures of the additives are shown inthe inset.
Chemistry of Materials Article
dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−29282922

collected on solvent-vapor annealed TES ADT films with up to6 mol % F-TES ADT, for example, exhibit reflections associatedwith TES ADT but do not reveal any reflections associated withthe crystal structure of F-TES ADT (refer to Figure S1,Supporting Information). To increase the contrast betweenTES ADT and the additives, we have chosen to study film spuncast from solutions comprising 50/50 mol % of TES ADT andthe additives. We employed atomic force microscopy (AFM) tostudy the surface topography and GIXD to examine the crystalstructure and assess the relative crystallinities of these as-spunblends. Figure 2 displays AFM images and correspondingGIXD patterns of such films. The AFM height images shown inFigure 2a reveal the surface topography of the five TES ADTfilms comprising the different additives. For films containing 50mol % F-, Cl-, and Br-TES ADT, we observe submicrometer- tomicrometer-sized features; these are absent in the AFM heightimages of films comprising 50 mol % I-TES ADT and ethyl-TES ADT. To elucidate the structure of these features, wecollected two-dimensional GIXD images on these same films.The GIXD images in Figure 2b were collected at an X-rayincident angle of 0.16° so incident X-rays can penetrate theentire depth of the 100 nm-thick films. For all GIXDexperiments, the film thickness and acquisition time wereheld constant, and the diffraction patterns were normalized bythe intensity of the incident X-ray beam. In the case of the filmwith 50 mol % F-TES ADT, we observe strong reflections,providing evidence that the features observed in the AFMmicrograph are indeed semicrystalline. The reflections in theGIXD pattern correspond to those predicted by the single-crystal structure of the high-temperature polymorph of F-TESADT,33 ruling out the possibility that cocrystals of TES ADTand F-TES ADT are present. The anisotropy in azimuthalintensity is consistent with the π-plane of F-TES ADTpreferentially oriented out-of-plane. Interestingly, we do notobserve any reflections associated with TES ADT despite thefact that we previously found the incorporation of fractionalamounts of additives (<6 mol %) to promote nucleation of TESADT.13 This enhanced nucleation in the presence of additives,however, is accompanied by a gradual decrease in the radialgrowth of the nucleated spherulites (refer to Figure S2,Supporting Information). There thus appears to be a trade-offbetween nucleation and growth of TES ADT with additiveconcentration. Extrapolating this growth rate dependence to
higher additive concentrations, we found that growth of TESADT is completely suppressed at 10.4 mol %. It is thus notsurprising that we do not observe any evidence of TES ADTcrystallinity in films comprising 50 mol % loadings of additives,with or without solvent-vapor annealing. In fact, suchsuppressed crystallization due to the presence of unlike specieshas been previously observed in films comprising comparablequantities of the two components.20
For films comprising 50 mol % Cl-TES ADT and Br-TESADT, we also do not observe reflections associated with TESADT, suggesting that the presence of these additives in highconcentrations also suppresses TES ADT crystallization.Instead, the reflections match those seen in the diffractionpattern of the film comprising a blend of F-TES ADT and TESADT, indicating that Cl-TES ADT and Br-TES ADT formpolymorphs that are similar to that of F-TES ADT in thepresence of TES ADT, and the anisotropic azimuthal intensityis consistent with the additives having their π-planespreferentially oriented perpendicular to the substrate. Theintensities of these diffraction patterns are an order ofmagnitude lower than those observed in neat films of theadditives, indicating that the films comprising 50 mol % F-, Cl-,and Br-TES ADT are only semicrystalline. Furthermore, theintensities of the diffraction patterns of TES ADT blended withF-, Cl-, and Br-TES ADT are comparable, suggesting that theadditives crystallize to comparable extents. These resultsindicate that while the substituent size increases from 64 pmwith fluorine, to 102 pm with chlorine, to 114 pm withbromine, these size differences are insufficient to affect thecrystallization behavior of the additives in the presence of TESADT. Consequently, the dependence of ⟨S⟩ on additiveconcentration is the same whether F-, Cl-, or Br-TES ADT isemployed to seed the crystallization of TES ADT. For filmscomprising 50/50 mol % blends of TES ADT and I-TES ADT,the reflections are also similar to those predicted by the high-temperature polymorph of F-TES ADT, but these reflectionsare azimuthally isotropic in their intensities. This observationindicates that crystallization of I-TES ADT occurs with nopreferential orientation in the presence of TES ADT.Furthermore, the reflections are less intense than those of thediffraction patterns of blends comprising TES ADT with F-,Cl-, and Br-TES ADT. The weaker intensities of the reflectionssuggest that the relative degree of crystallinity of films with I-
Figure 2. (a) AFM images and (b) corresponding GIXD patterns for 50/50 mol % blends of TES ADT with the labeled TES ADT additives.
Chemistry of Materials Article
dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−29282923

TES ADT is lower than that in films with F-, Cl-, and Br-TESADT. For films with 50 mol % ethyl-TES ADT, the GIXDpattern is featureless, consistent with the film being amorphous.That F-, Cl-, Br-, and I-TES ADT do in fact crystallize in the
presence of TES ADT suggests that their driving force tocrystallize is greater than that of TES ADT. We can relate therelative driving force for crystallization by measuring the meltenthalpies of the additives with that of TES ADT.34,35 WhenTES ADT is heated through its melting point of 153 °C, itexhibits a melt enthalpy of 20 J/g. The melt enthalpies of F-and Cl-TES ADT, on the other hand, are approximately 50 J/g(refer to Figure S3, Supporting Information, for differentialcalorimetry scans; Br-TES ADT and I-TES ADT degradebefore their melting point so we cannot report melt enthalpiesfor these compounds). Compared to quintessential organicsemiconductors, such as tetracene, which exhibits a meltenthalpy of 695.6 J/g,36 the driving force of crystallization inTES ADT and its additives is an order of magnitude smaller.Still, because the F- and Cl-TES ADT have larger meltenthalpies, and thus stronger tendencies to crystallize comparedto TES ADT, we expect them to crystallize more readily thanTES ADT in 50/50 mol % blends. Further evidence that thedriving force for the additives to crystallize is strongercompared to that of TES ADT is the observation that bothF-TES ADT37 and Cl-TES ADT crystallize readily upon solventevaporation during spin coating, whereas TES ADT is largelyamorphous as-spun and requires additional solvent−vaporannealing to induce crystallization.25 On the other hand, themelt enthalpy of ethyl-TES ADT is only slightly larger than thatof TES ADT, at a value of 34 J/g. Because the melt enthalpy ofethyl-TES ADT is comparable to that of TES ADT, we believethey can disrupt each other’s native crystallization habit in 50/50 mol % blends.Since the ability to seed TES ADT spherulitic growth hinges
on the ability of the additives to form nuclei, we expect theirtendency to crystallize in the presence of 50 mol % TES ADTto be directly related to their ability to aggregate into nucleiwhen present in smaller concentrations. In 50/50 mol % blendswith TES ADT, I-TES ADT and ethyl-TES ADT crystallize to alesser extent compared to F-, Cl-, and Br-TES ADT. At lowconcentrations, I-TES ADT and ethyl-TES ADT thus likelyform correspondingly fewer active nuclei. As such, higherconcentrations of I- and ethyl-TES ADT are required to achievethe same ⟨S⟩ compared to when F-, Cl-, and Br-TES ADT areused as additives. This trend is consistent with the progressiveshallowing of the slope, m, in Figure 1b, where the dependenceof ⟨S⟩ on additive concentration is strongest when F-, Cl-, andBr-TES ADT are used as additives; the dependence of ⟨S⟩ onadditive concentration becomes progressively weaker as wereplace easily crystallizable F-, Cl-, and Br-TES ADT additiveswith I- and ethyl-TES ADT.Dependence of ccrit on the Additives’ Solubility in
Toluene. Returning to Figure 1b, we observe that all theadditives begin seeding TES ADT at ccrit = 0.5 mol %, except forethyl-TES ADT. When ethyl-TES ADT is used as the additive,we do not observe significant changes in ⟨S⟩ until an additiveconcentration of 2.7 mol %. The most obvious difference in themolecular structures among the derivatives is that ethyl-TESADT has an alkyl instead of a halogen substituent. We thusspeculate that the shift in ccrit is related to differences in thesolubility of ethyl-TES ADT and those of the halogenated-TESADTs. It is well-known that adding alkyl chains to a moleculecan significantly improve the solubility of the parent compound
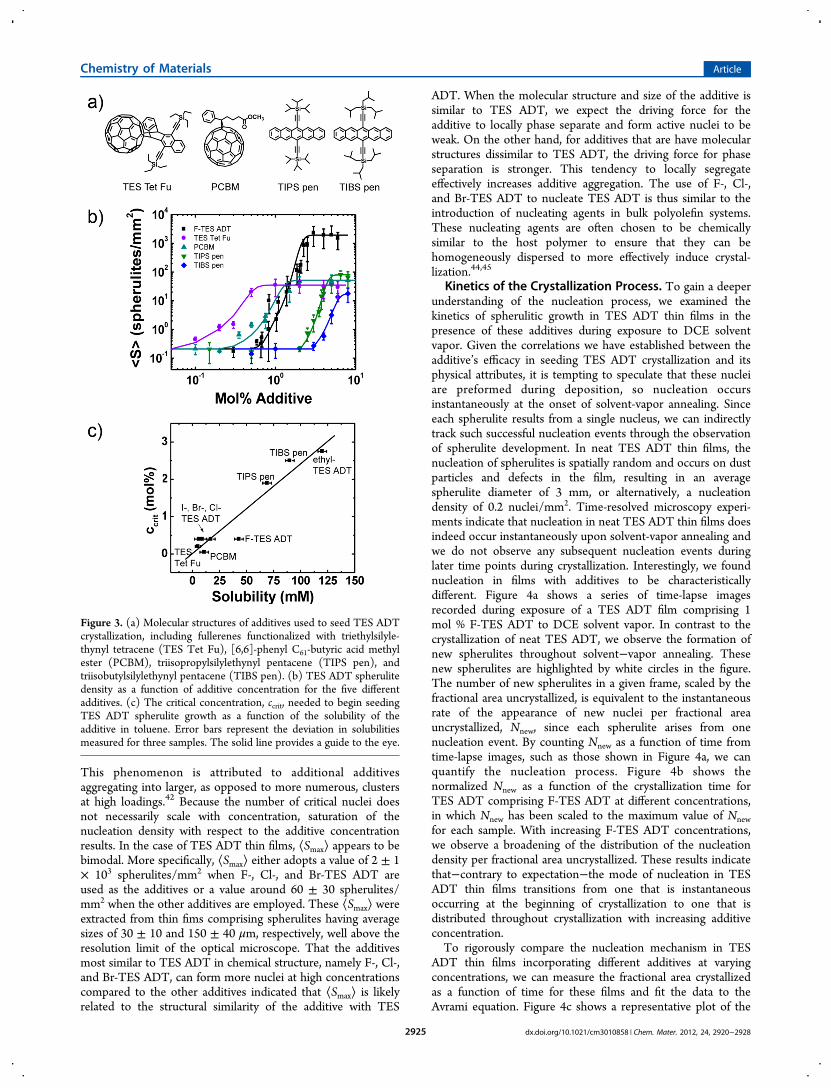
in organic solvents.38 Indeed, we estimate the solubility ofethyl-TES ADT in toluene, the solvent from which the films arecast, to be approximately 120 mM. In contrast, the solubilitiesof the F-, Cl-, Br-, and I-TES ADT in toluene are 43, 17, 9, and6 mM, respectively (see Experimental Methods). While weobserve a systematic decrease in the solubility of these additiveswith increasing halogen size, these solubilities are allsubstantially lower than that of ethyl-TES ADT in toluene.Given this correlation, we attribute the shift in ccrit in TES ADTfilms incorporating ethyl-TES ADT as the additive to itsincreased solubility compared to the halogenated-TES ADTderivatives in toluene. Since the additives must form aggregateswhose sizes are above the critical size in order for nucleation tooccur,39 additives that are more soluble in the casting solventare less likely to aggregate to form the critical nuclei required toinitiate TES ADT crystallization. As such, additives that aremore soluble will require a higher ccrit to seed crystallization ofTES ADT. Such solubility-dependent aggregation has beenobserved in P3HT-PCBM blends, in which fractional quantitiesof a poor solvent for P3HT are incorporated into the solutionprior to deposition.22,40 The poor solvent has a higher boilingpoint than the casting solvent; the presence of the poor solventafter the casting solvent evaporates thus induces P3HT topreferentially aggregate. In our case, because toluene is a goodsolvent for ethyl-TES ADT, residual toluene that is presentafter spin coating actually retards ethyl-TES ADT aggregationinto active nuclei having the critical dimensions to seed TESADT crystallization.To further verify the impact of the solubility of the additive
in the casting solvent on its ability to nucleate TES ADTcrystallization, we also explored a second set of additives whosecore structures differ substantially from that of TES ADT. Thechemical structures of these compounds are illustrated in Figure3a. Such chemical differences have allowed us to access a widerrange of solubilities. TES Tet Fu exhibits a solubility of 5 mMin toluene, while the common fullerene derivative PCBMexhibits a solubility of 11 mM in toluene; TIPS pen29 exhibits asolubility of 69 mM in toluene; and TIBS pen exhibits asolubility of 90 mM in toluene. Figure 3b displays how ⟨S⟩depends on the additive concentration for these fourcompounds; a similar graph comprising the dependence of⟨S⟩ on all of the additives in this study is depicted in Figure S4,Supporting Information. As expected, we observe a large shift inccrit depending on the solubility of the additive. Figure 3cquantifies this relationship, in which ccrit for all nine additivesexplored in this study is plotted against the solubility of theadditives in toluene. When the poorly soluble fullerenederivatives, TES Tet Fu and PCBM, are used as additives,the ccrit’s are 0.2 and 0.1 mol %, respectively. In contrast, whensoluble TIPS pen and TIBS pen are used as additives, the ccrit’sare 2.0 and 2.5 mol %, respectively. As can be seen in Figure 3c,we observe a strong and positive correlation between ccrit andthe solubility of the additive in toluene that is consistent withour hypothesis.
Dependence of Smax on the Additives’ Similarity toTES ADT. Finally, we turn our attention to ⟨Smax⟩, themaximum number density of spherulites achievable in TESADT thin films. For all the additives explored, ⟨S⟩ becomesindependent of the additive concentration beyond a criticalconcentration, plateauing at ⟨Smax⟩. This sigmoidal dependencewherein the nucleation density saturates at high additiveloadings has also been observed in the crystallization of metalalloys41,42 and polymer films43 incorporating nucleating agents.
Chemistry of Materials Article
dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−29282924
This phenomenon is attributed to additional additivesaggregating into larger, as opposed to more numerous, clustersat high loadings.42 Because the number of critical nuclei doesnot necessarily scale with concentration, saturation of thenucleation density with respect to the additive concentrationresults. In the case of TES ADT thin films, ⟨Smax⟩ appears to bebimodal. More specifically, ⟨Smax⟩ either adopts a value of 2 ± 1× 103 spherulites/mm2 when F-, Cl-, and Br-TES ADT areused as the additives or a value around 60 ± 30 spherulites/mm2 when the other additives are employed. These ⟨Smax⟩ wereextracted from thin fims comprising spherulites having averagesizes of 30 ± 10 and 150 ± 40 μm, respectively, well above theresolution limit of the optical microscope. That the additivesmost similar to TES ADT in chemical structure, namely F-, Cl-,and Br-TES ADT, can form more nuclei at high concentrationscompared to the other additives indicated that ⟨Smax⟩ is likelyrelated to the structural similarity of the additive with TES
ADT. When the molecular structure and size of the additive issimilar to TES ADT, we expect the driving force for theadditive to locally phase separate and form active nuclei to beweak. On the other hand, for additives that are have molecularstructures dissimilar to TES ADT, the driving force for phaseseparation is stronger. This tendency to locally segregateeffectively increases additive aggregation. The use of F-, Cl-,and Br-TES ADT to nucleate TES ADT is thus similar to theintroduction of nucleating agents in bulk polyolefin systems.These nucleating agents are often chosen to be chemicallysimilar to the host polymer to ensure that they can behomogeneously dispersed to more effectively induce crystal-lization.44,45
Kinetics of the Crystallization Process. To gain a deeperunderstanding of the nucleation process, we examined thekinetics of spherulitic growth in TES ADT thin films in thepresence of these additives during exposure to DCE solventvapor. Given the correlations we have established between theadditive’s efficacy in seeding TES ADT crystallization and itsphysical attributes, it is tempting to speculate that these nucleiare preformed during deposition, so nucleation occursinstantaneously at the onset of solvent-vapor annealing. Sinceeach spherulite results from a single nucleus, we can indirectlytrack such successful nucleation events through the observationof spherulite development. In neat TES ADT thin films, thenucleation of spherulites is spatially random and occurs on dustparticles and defects in the film, resulting in an averagespherulite diameter of 3 mm, or alternatively, a nucleationdensity of 0.2 nuclei/mm2. Time-resolved microscopy experi-ments indicate that nucleation in neat TES ADT thin films doesindeed occur instantaneously upon solvent-vapor annealing andwe do not observe any subsequent nucleation events duringlater time points during crystallization. Interestingly, we foundnucleation in films with additives to be characteristicallydifferent. Figure 4a shows a series of time-lapse imagesrecorded during exposure of a TES ADT film comprising 1mol % F-TES ADT to DCE solvent vapor. In contrast to thecrystallization of neat TES ADT, we observe the formation ofnew spherulites throughout solvent−vapor annealing. Thesenew spherulites are highlighted by white circles in the figure.The number of new spherulites in a given frame, scaled by thefractional area uncrystallized, is equivalent to the instantaneousrate of the appearance of new nuclei per fractional areauncrystallized, Nnew, since each spherulite arises from onenucleation event. By counting Nnew as a function of time fromtime-lapse images, such as those shown in Figure 4a, we canquantify the nucleation process. Figure 4b shows thenormalized Nnew as a function of the crystallization time forTES ADT comprising F-TES ADT at different concentrations,in which Nnew has been scaled to the maximum value of Nnewfor each sample. With increasing F-TES ADT concentrations,we observe a broadening of the distribution of the nucleationdensity per fractional area uncrystallized. These results indicatethat−contrary to expectation−the mode of nucleation in TESADT thin films transitions from one that is instantaneousoccurring at the beginning of crystallization to one that isdistributed throughout crystallization with increasing additiveconcentration.To rigorously compare the nucleation mechanism in TES
ADT thin films incorporating different additives at varyingconcentrations, we can measure the fractional area crystallizedas a function of time for these films and fit the data to theAvrami equation. Figure 4c shows a representative plot of the
Figure 3. (a) Molecular structures of additives used to seed TES ADTcrystallization, including fullerenes functionalized with triethylsilyle-thynyl tetracene (TES Tet Fu), [6,6]-phenyl C61-butyric acid methylester (PCBM), triisopropylsilylethynyl pentacene (TIPS pen), andtriisobutylsilylethynyl pentacene (TIBS pen). (b) TES ADT spherulitedensity as a function of additive concentration for the five differentadditives. (c) The critical concentration, ccrit, needed to begin seedingTES ADT spherulite growth as a function of the solubility of theadditive in toluene. Error bars represent the deviation in solubilitiesmeasured for three samples. The solid line provides a guide to the eye.
Chemistry of Materials Article
dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−29282925
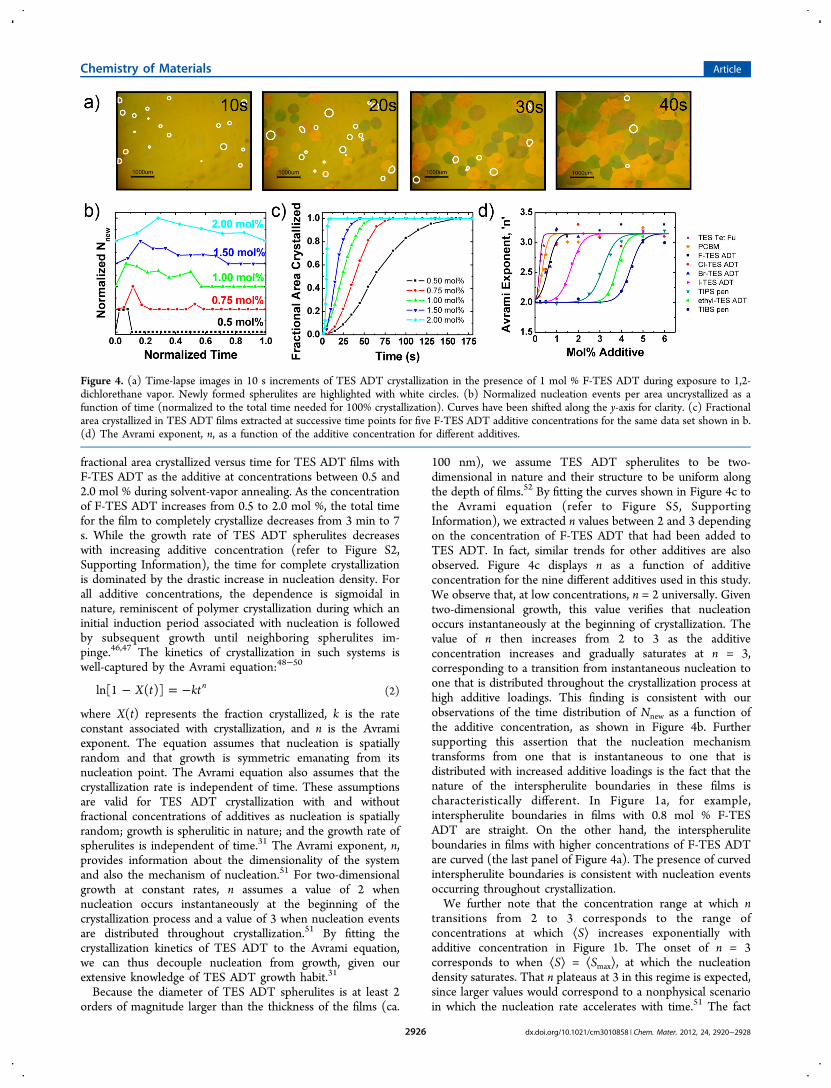
fractional area crystallized versus time for TES ADT films withF-TES ADT as the additive at concentrations between 0.5 and2.0 mol % during solvent-vapor annealing. As the concentrationof F-TES ADT increases from 0.5 to 2.0 mol %, the total timefor the film to completely crystallize decreases from 3 min to 7s. While the growth rate of TES ADT spherulites decreaseswith increasing additive concentration (refer to Figure S2,Supporting Information), the time for complete crystallizationis dominated by the drastic increase in nucleation density. Forall additive concentrations, the dependence is sigmoidal innature, reminiscent of polymer crystallization during which aninitial induction period associated with nucleation is followedby subsequent growth until neighboring spherulites im-pinge.46,47 The kinetics of crystallization in such systems iswell-captured by the Avrami equation:48−50
− = −X t ktln[1 ( )] n(2)
where X(t) represents the fraction crystallized, k is the rateconstant associated with crystallization, and n is the Avramiexponent. The equation assumes that nucleation is spatiallyrandom and that growth is symmetric emanating from itsnucleation point. The Avrami equation also assumes that thecrystallization rate is independent of time. These assumptionsare valid for TES ADT crystallization with and withoutfractional concentrations of additives as nucleation is spatiallyrandom; growth is spherulitic in nature; and the growth rate ofspherulites is independent of time.31 The Avrami exponent, n,provides information about the dimensionality of the systemand also the mechanism of nucleation.51 For two-dimensionalgrowth at constant rates, n assumes a value of 2 whennucleation occurs instantaneously at the beginning of thecrystallization process and a value of 3 when nucleation eventsare distributed throughout crystallization.51 By fitting thecrystallization kinetics of TES ADT to the Avrami equation,we can thus decouple nucleation from growth, given ourextensive knowledge of TES ADT growth habit.31
Because the diameter of TES ADT spherulites is at least 2orders of magnitude larger than the thickness of the films (ca.
100 nm), we assume TES ADT spherulites to be two-dimensional in nature and their structure to be uniform alongthe depth of films.52 By fitting the curves shown in Figure 4c tothe Avrami equation (refer to Figure S5, SupportingInformation), we extracted n values between 2 and 3 dependingon the concentration of F-TES ADT that had been added toTES ADT. In fact, similar trends for other additives are alsoobserved. Figure 4c displays n as a function of additiveconcentration for the nine different additives used in this study.We observe that, at low concentrations, n = 2 universally. Giventwo-dimensional growth, this value verifies that nucleationoccurs instantaneously at the beginning of crystallization. Thevalue of n then increases from 2 to 3 as the additiveconcentration increases and gradually saturates at n = 3,corresponding to a transition from instantaneous nucleation toone that is distributed throughout the crystallization process athigh additive loadings. This finding is consistent with ourobservations of the time distribution of Nnew as a function ofthe additive concentration, as shown in Figure 4b. Furthersupporting this assertion that the nucleation mechanismtransforms from one that is instantaneous to one that isdistributed with increased additive loadings is the fact that thenature of the interspherulite boundaries in these films ischaracteristically different. In Figure 1a, for example,interspherulite boundaries in films with 0.8 mol % F-TESADT are straight. On the other hand, the interspheruliteboundaries in films with higher concentrations of F-TES ADTare curved (the last panel of Figure 4a). The presence of curvedinterspherulite boundaries is consistent with nucleation eventsoccurring throughout crystallization.We further note that the concentration range at which n
transitions from 2 to 3 corresponds to the range ofconcentrations at which ⟨S⟩ increases exponentially withadditive concentration in Figure 1b. The onset of n = 3corresponds to when ⟨S⟩ = ⟨Smax⟩, at which the nucleationdensity saturates. That n plateaus at 3 in this regime is expected,since larger values would correspond to a nonphysical scenarioin which the nucleation rate accelerates with time.51 The fact
Figure 4. (a) Time-lapse images in 10 s increments of TES ADT crystallization in the presence of 1 mol % F-TES ADT during exposure to 1,2-dichlorethane vapor. Newly formed spherulites are highlighted with white circles. (b) Normalized nucleation events per area uncrystallized as afunction of time (normalized to the total time needed for 100% crystallization). Curves have been shifted along the y-axis for clarity. (c) Fractionalarea crystallized in TES ADT films extracted at successive time points for five F-TES ADT additive concentrations for the same data set shown in b.(d) The Avrami exponent, n, as a function of the additive concentration for different additives.
Chemistry of Materials Article
dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−29282926

that the incorporation of additives, irrespective of chemistry,increases n from 2 to 3 in qualitatively the same mannersuggests that the fundamental mechanism of additiveaggregation and subsequent nucleation of TES ADT spherulitesis independent of the physical attributes of the additives, butthe extent to which this process occurs is chemistry-specific anddepends strongly on the additives’ driving force to aggregate,their solubility in toluene, and their chemical compatibility withTES ADT.
■ CONCLUSIONSIdentifying key attributes affecting the nucleation efficacy ofsmall-molecule additives in solution-processed, organic semi-conductor thin films has strong implications in the field oforganic electronics. In systems incorporating dopants toimprove device mobilities3,4 in the case of OTFTs andconductivities5,6 and luminescence7−9 in the case of OLEDs,there exists a competition between maximizing the dopant levelto improve electronic and optical properties while minimizingdisruption to the overall film morphology because grainboundaries in organic semiconductor thin films often act asbarriers to charge transport.13−15 By designing the dopants tohave high solubilities in the casting solvent, one could minimizethe nucleation density and, thus, the density of grainboundaries, in the active layer while maximizing the dopantconcentration. Likewise, in the active layers of OLEDs, large-scale aggregation of emissive dopants can lead to a decrease inoverall light emission due to self-quenching of the dopants.8,9
In our study, we have found the degree of local phaseseparation between the guest and host compounds to be relatedto their chemical compatibility. It is thus advantageous todesign the dopant to be chemically similar to the host organicsemiconductor in order to minimize phase separation. Suchdesign parameters will aid future research in the prediction ofthe final film morphologies based on the chemical structures ofthe constituent organic semiconductor compounds, particularlyin complex multicomponent thin films.
■ ASSOCIATED CONTENT*S Supporting InformationSynthesis of TES Tet Fu. GIXD patterns of TES ADT filmswith fractional amounts of additives. Spherulitic growth ratedependence on additive concentration. DSC thermograms ofadditives. Log-log plots of the fractional area crystallized versustime. This material is available free of charge via the Internet athttp://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe acknowledge support from the NSF MRSEC programthrough the Princeton Center for Complex Materials (GrantDMR-0819860), which also provided access to the PRISMImaging and Analysis Center. This research was also supportedby the SOLAR Initiative at the NSF (DMR-1035217 and1035257). Part of this research was conducted at the CornellHigh Energy Synchrotron Source (CHESS), which issupported by the NSF and the National Institute of Health/
National Institute of General Medical Sciences (DMR-0936384). S.S.L. is supported by a National Defense Scienceand Engineering Graduate fellowship. We also thank Profs. Chi-Sing Man and Michel Jabbour of the Mathematics Departmentat the University of Kentucky for helpful discussions.
■ REFERENCES(1) Smith, J.; Hamilton, R.; McCulloch, I.; Stingelin-Stutzmann, N.;Heeney, M.; Bradley, D. D. C.; Anthopoulos, T. D. J. Mater. Chem.2010, 20, 2562.(2) Thompson, B. C.; Frechet, J. M. J. Angew. Chem., Int. Ed. 2008,47, 58.(3) Lim, E.; Jung, B.-J.; Chikamatsu, M.; Azumi, R.; Yoshida, Y.; Yase,K.; Do, L.-M.; Shim, H.-K. J. Mater. Chem. 2007, 17, 1416.(4) Ma, L.; Lee, W. H.; Park, Y. D.; Kim, J. S.; Lee, H. S.; Cho, K.Appl. Phys. Lett. 2008, 92, 063310.(5) Pfeiffer, M.; Leo, K.; Zhou, X.; Huang, J. S.; Hofmann, M.;Werner, A.; Blochwitz-Nimoth, J. Org. Electron. 2003, 4, 89.(6) Zhou, X.; Pfeiffer, M.; Blochwitz, J.; Werner, A.; Nollau, A.; Fritz,T.; Leo, K. Appl. Phys. Lett. 2001, 78, 410.(7) Tang, C. W.; VanSlyke, S. A.; Chen, C. H. J. Appl. Phys. 1989, 65,3610.(8) Adamovich, V.; Brooks, J.; Tamayo, A.; Alexander, A. M.;Djurovich, P. I.; D’Andrade, B. W.; Adachi, C.; Forrest, S. R.;Thompson, M. E. New J. Chem. 2002, 26, 1171.(9) Mi, B. X.; Gao, Z. Q.; Lee, C. S.; Lee, S. T.; Kwong, H. L.; Wong,N. B. Appl. Phys. Lett. 1999, 75, 4055.(10) Lee, S. S.; Loo, Y.-L. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 59.(11) Wei, Z.; Xi, H.; Dong, H.; Wang, L.; Xu, W.; Hu, W.; Zhu, D. J.Mater. Chem. 2010, 20, 1203.(12) Meyer zu Heringdorf, F. J.; Reuter, M. C.; Tromp, R. M. Nature2001, 412, 517.(13) Lee, S. S.; Kim, C. S.; Gomez, E. D.; Purushothaman, B.; Toney,M. F.; Wang, C.; Hexemer, A.; Anthony, J. E.; Loo, Y.-L. Adv. Mater.2009, 21, 3605.(14) Horowitz, G.; Hajlaoui., M. E. Adv. Mater. 2000, 12, 1046.(15) Horowitz, G.; Hajlaoui., M. E. Synth. Met. 2001, 122, 185.(16) Campoy-Quiles, M.; Ferenczi, T.; Agostinelli, T.; Etchegoin, P.G.; Kim, Y.; Anthopoulos, T. D.; Stavrinou, P. N.; Bradley, D. D. C.;Nelson, J. Nat. Mater. 2008, 7, 158.(17) Hamilton, R.; Smith, J.; Ogier, S.; Heeney, M.; Anthony, J. E.;McCulloch, I.; Veres, J.; Bradley, D. D. C.; Anthopoulos, T. D. Adv.Mater. 2009, 21, 1166.(18) Wang, H.; Gomez, E. D.; Kim, J.; Guan, Z.; Jaye, C.; Fischer, D.A.; Kahn, A.; Loo, Y.-L. Chem. Mater. 2011, 23, 2020.(19) Chung, Y. S.; Shin, N.; Kang, J.; Jo, Y.; Prabhu, V. M.; Satija, S.K.; Kline, R. J.; DeLongchamp, D. M.; Toney, M. F.; Loth, M. A.;Purushothaman, B.; Anthony, J. E.; Yoon, D. Y. J. Am. Chem. Soc.2011, 133, 412.(20) Zhao, J.; Swinnen, A.; Van Assche, G.; Manca, J.; Vanderzande,D.; Mele, B. V. J. Phys. Chem. B 2009, 113, 1587.(21) Gomez, E. D.; Barteau, K. P.; Wang, H.; Toney, M. F.; Loo, Y.-L. Chem. Commun. 2011, 47, 436.(22) Peet, J.; Kim, J. Y.; Coates, N. E.; Ma, W. L.; Moses, D.; Heeger,A. J.; Bazan, G. C. Nat. Mater. 2007, 6, 497.(23) Halls, J. J. M.; Pichler, K.; Friend, R. H.; Moratti, S. C.; Holmes,A. B. Appl. Phys. Lett. 1996, 68, 3120.(24) Payne, M. M.; Odon, S. A.; Parkin, S. R.; Anthony, J. E. Org.Lett. 2004, 6, 3325.(25) Dickey, K. C.; Anthony, J. E.; Loo, Y.-L. Adv. Mater. 2006, 18,1721.(26) Subramanian, S.; Park, S. K.; Parkin, S. R.; Podzorov, V.;Jackson, T. N.; Anthony, J. E. J. Am. Chem. Soc. 2008, 130, 2706.(27) Li, Z.; Lim, Y.-F.; Kim, J. B.; Parkin, S. R.; Loo, Y.-L.; Malliaras,G. G.; Anthony, J. E. Chem. Commun. 2011, 47, 7617.(28) Lloyd, M. T.; Mayer, A. C.; Subramanian, S.; Mourey, D. A.;Herman, D. J.; Bapat, A. V.; Anthony, J. E.; Malliaras, G. G. J. Am.Chem. Soc. 2007, 129, 9144.
Chemistry of Materials Article
dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−29282927

(29) Anthony, J. E.; Brooks, J. S.; Eaton, D. L.; Parkin, S. R. J. Am.Chem. Soc. 2001, 123, 9482.(30) Anthony, J. E.; Eaton, D. L.; Parkin, S. R. Org. Lett. 2001, 4, 15.(31) Lee, S. S.; Tang, S. T.; Smilgies, D.-M.; Woll, A. R.; Loth, M. A.;Mativetsky, J. M.; Anthony, J. E.; Loo, Y.-L. Adv. Mater. 2012, 24,2692.(32) Smilgies, D.-M.; Blasini, D. R.; Hotta, S.; Yanagi, H. J.Synchrotron Radiat. 2005, 12, 807.(33) Jurchescu, O. D.; Mourey, D. A.; Subramanian, S.; Parkin, S. R.;Vogel, B. M.; Anthony, J. E.; Jackson, T. N.; Gundlach, D. J. Phys. Rev.B 2009, 80, 085201.(34) Hoffman, J. D. J. Chem. Phys. 1958, 29, 1192.(35) Turnbull, D. J. Appl. Phys. 1950, 21, 1022.(36) NIST Standard Reference Database, no. 69. http://webbook.nist.gov/chemistry/ (accessed December 2011).(37) Gundlach, D. J.; Royer, J. E.; Park, S. K.; Subramanian, S.;Jurchescu, O. D.; Hamadani, B. H.; Moad, A. J.; Kline, R. J.; Teague, L.C.; Kirillov, O.; et al. Nat. Mater. 2008, 7, 216.(38) Anthony, J. E. Chem. Rev. 2006, 106, 5028.(39) Frenkel, J. Kinetic Theory of Liquids; Oxford University Press:Oxford, 1946.(40) Lee, J. K.; Ma, W. L.; Brabec, C. J.; Yuen, J.; Moon, J. S.; Kim, J.Y.; Lee, K.; Bazan, G. C.; Heeger, A. J. J. Am. Chem. Soc. 2008, 130,3619.(41) Easton, M. A.; St. John, D. H. Acta Mater. 2001, 49, 1867.(42) Greer, A. L.; Bunn, A. M.; Tronche, A.; Evans, P. V.; Bristow, D.J. Acta Metall. 2000, 48, 2823.(43) Binsbergen, F. L.; de Lange, B. G. M. Polymer 1970, 11, 309.(44) Thierry, A.; Fillon, B.; Straupe, C.; Lotz, B.; Wittmann, J. Prog.Colloid Polym. Sci. 1992, 87, 28.(45) Wilder, E. A.; Spontak, R. J.; Hall, C. K. Mol. Phys. 2003, 101,3017.(46) Lopez, L. C.; Wilkes, G. L. Polymer 1988, 29, 106.(47) Keith, H. D. Colloid Polym. Sci. 1969, 231, 421.(48) Avrami, M. J. Chem. Phys. 1939, 7, 1103.(49) Avrami, M. J. Chem. Phys. 1940, 8, 212.(50) Avrami, M. J. Chem. Phys. 1941, 9, 177.(51) Christian, J. The Theory of Transformations in Metals and Alloys.Part 1. Equilibrium and General Kinetic Theory; Pergamon Press: NewYork, 1975.(52) Lee, S. S.; Loth, M. A.; Anthony, J. E.; Loo, Y.-L. J. Am. Chem.Soc. 2012, 134, 5436.
■ NOTE ADDED AFTER ASAP PUBLICATIONThe Acknowledgments were modified in the version of thispaper published July 19, 2012. The corrected version publishedJuly 20, 2012.
Chemistry of Materials Article
dx.doi.org/10.1021/cm3010858 | Chem. Mater. 2012, 24, 2920−29282928



![Heterogeneous ice nucleation on particles composed of ...mel.xmu.edu.cn/upload_paper/2016428104154-hhj3B4.pdf · et al., 2010]. This ice nucleation apparatus allows the exposure of](https://cdn.vdocuments.net/doc/165x107/5b430b277f8b9ad23b8bac77/heterogeneous-ice-nucleation-on-particles-composed-of-melxmueducnuploadpaper2016428104154-.jpg)



