Download - Using and combining the different tools for predicting the pathogenicity of sequence variants

Using and combining the different tools for predicting
the pathogenicity of sequence variants
Identification and analysis of sequence variants in sequencing projects: fundamentals and tools, session 2
!Casandra Riera, VHIR
Jan, 18th 2015

Thank you!
‘Amics del VHIR’

Outline
A general view of a prediction method !Who are the players? How do they work? Looking a bit in more detail !!What can we learn from their use? Practical cases. Some tips to consider !What else can we do?

Introduction
Typical pipeline for identification of deleterious variants found in coding sequence (WES, panels, …)
Input amino acid
substitution and protein ID
SequencePhysicochemical prop.
Biochemical properties ...
Ann. from swissprot predicted scores
dbSNP, …
3D structure Secondary structure
Surface area properties B factors, …
Machine learning
approaches / theoretical
models
Output predictionsStructure
Annotation
AlignmentsConservation score
Entropy, frequencies, …

The players
SIFT MutAssessor
PolyPhen-2 Fathmm
PMut PhD-SNP
SNAP SNPs3D
…
Primary methods Consensus methods
PredictSNP Condel CADD PON-P
KGGSep Carol
…

Basics of some methods - SIFT
SIFT (“Sorting Intolerant From Tolerant”)
!MSA based on PSI-BLAST. !Features when scoring an AA variant: - Is position conserved for a single AA? - Is position conserved for AA with a
particular chemical property? - How different is mut AA from most
common AA? Nat Protoc. 2009;4(7):1073-81
Based on Alignment Using sequence homology, scores are calculated using position-specific scoring matrices.

SIFT… some details
Dataset site-directed mu-tagenesis for T4 lysozyme, HIV-1 protease, and Lac re-pressor as train. Not based on human proteins.
Gen
ome
Res.
20
01
;11
(5):8
63
-74
Output Prediction is labeled as “damaging”/“Tolerated”. Scores range from 0-1 Threshold at 0.05

Basics of some methods - PolyPhen2
PolyPhen2 (“Polymorphisms Phenotyping v2”)
It uses a Naive Bayes classifier to score variants based on 11 predictive features.
Eight Sequence/MSA Features: !PSIC score for wt AA ΔPSIC score (wt-mt) Seq id to closest homolog w/ variat. Congruency of mt allele to the MSA CpG context of transition mutations Alignment depth at mutation site ΔVolume Pfam domain annotation
Three Structural Features: !Accessibility of wt Change in hydrophobic propensity Crystallographic β-factor reflecting conformational mobility of wt !!

PolyPhen2… some details
The most informative predictive features characterize are related to the alignment:
Nature Methods 7, 248 – 249 (2010) [Suppl]!

Dataset HumVar Disease variants in UniProt + common nsSNPs w/o annotated involvement in disease. HumDiv Disease variants in UniProt + variants found in close homologs. !
Output Prediction as “Probably/possibly damaging” and “Benign” based on FPR. Scores range from 0-1 & Threshold in 0.5 + FPR correction.
PolyPhen2… Some details

PolyPhen2… Some details

Basics of some methods - Mut.Assessor
Mutation Assessor
“Predicts functional impact of AA substitutions in general and in cancer in particular… The functional impact is assessed based on evolutionary conservation of affected AA in protein homologs.” !3D structure shown in output but aren’t part of functional impact score.

MutationAssessor… some details
Calculates two scores for each AA substitution: Conservation (across entire protein family) Specificity (within subfamily, but not conserved in entire family)
Nucl.
Acid
s Res
. 39
(20
11
)
Score & Labels:
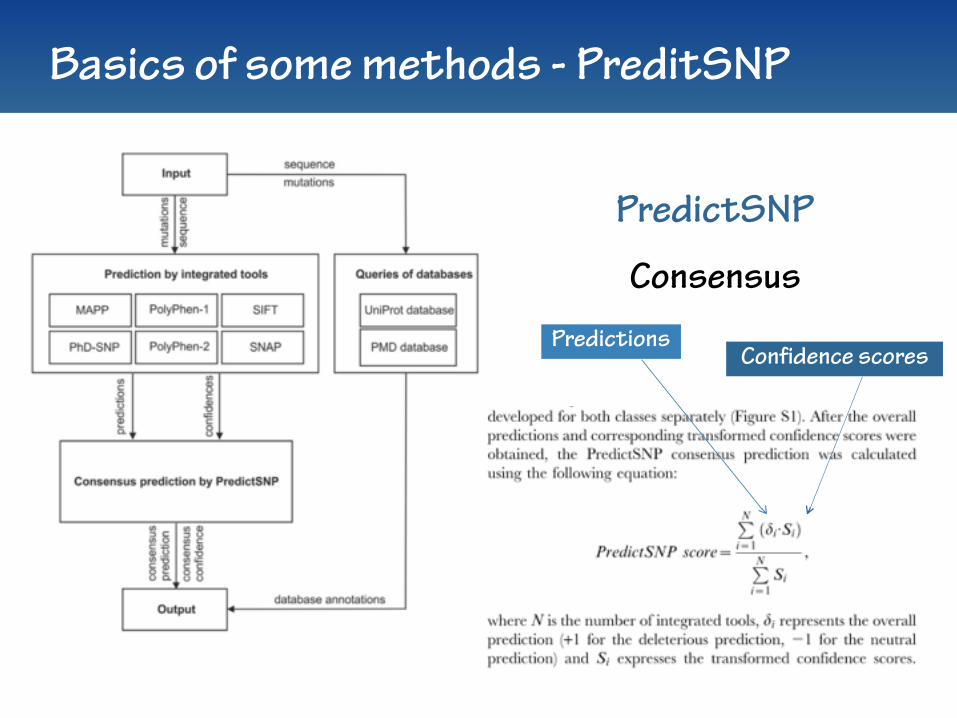
Basics of some methods - PreditSNP
PredictionsConfidence scores
PredictSNP
Consensus

PredictSNP… some details

Method Main features Further info
SIFT MSA (normalized probabilities) Ng and Henikoff, 2001
MutationAssessor MSA ( conservation in subfamilies) Reva et al., 2011
PANTHER MSA (subPSEC) Thomas et al., 2003
SNPs3D Structure (stability) // MSA (conserv. + prob) Yue et al., 2006
SNP&GO MSA (C+P) + sequence + PANTHER + GO terms Calabrese et al., 2009
CADD MSA + Regulatory info + SIFT, PPH & Grantham Kircher et al., 2014
PhD-SNP MSA (C+P) + sequence Capriotti et al., 2006
PolyPhen-2 MSA + sequence + structure Adzhubei et al., 2010
PMut MSA + sequence + structure Ferrer-Costa et al., 2005
SNAP MSA + sequence + structure + annotation Bromberg and Rost, 2007
MuD MSA + sequence + structure + annotation + SNAP Wainreb et al., 2010
CHASM MSA + sequence + structure + annotation Wong et al., 2011
FATHMM MSA + GO Shihab et al., 2012
Condel FATHHM & Mutation Assessor Gonzalez-Pérez et al, 2011
PredictSNP MAPP, PPH-1, PPH-2, Sift, PhD-SNP & SNAP Bendl et al., 2014
… … …
Summary of some of the available methods

Which method?
Riera C, Lois S, de la Cruz X. Prediction of pathological mutations in proteins. Wiley Interdiscip Rev Comput Mol Sci, 2014; 4(3):249-68.
Use 1-2 methods uniquely based on conservation (e.g. SIFT) !Use 1-2 methods including additional features, such as structure (e.g. PolyPhen, etc).

Outline
A general view of a prediction method !Who are the players? How do they work? Looking a bit in more detail !!What can we learn from their use? Practical cases. Some tips to consider !What else can we do?

Introduction
Input amino acid
substitution and protein ID
SequencePhysicochemical prop.
Biochemical properties ...
Ann. from swissprot predicted scores
dbSNP, …
3D structure Secondary structure
Surface area properties B factors, …
Machine learning
approaches / theoretical
models
Output predictionsStructure
Annotation
AlignmentsConservation score
Entropy, frequencies, …
Almost all contemporary functional prediction algorithms incorporate MSAs in some manner

Multiple Sequence alignments
" Most methods incorporate MSAs but differ in their construction and further interpretation. !
" How many sequences? " Which species should include? " Can we predict all protein families the same? " How do we quantify conservation? !Answers
MSAs are suboptimal

Conservation may not mean the same in all families
Can we predict all protein families the same?
Riera C, Lois S, de la Cruz X. Prediction of pathological mutations in proteins. Wiley Interdiscip Rev Comput Mol Sci, 2014; 4(3):249-68.

How many sequences? What species?
Example - 1

How are they aligned? Submit your own…
Example - 1

Practical experience
Submiting own aligments to PolyPhen
PolyPhen2 precomputed
alignmentPolyPhen2
Mutations to test
Predictions
Example - 2

Provide personalized alignments to calculate MSA-features
PolyPhen2 precomputed
alignmentPolyPhen2
Mutations to test
?
Own alignments
Submit own aligments to PPH2
Example - 2

Submit own aligments to PPH2
PolyPhen2 precomputed
alignmentPolyPhen2
Mutations to test
Own alignments
!PolyPhen2 only works well when using its own alignments. Otherwise, very biased predictions.
Predictions
Example - 2

Alignment depth - Mut.Assessor!MutAssessor tends to label as Neutral when there’s very few sequences at that position in the alignment. !
Example - 3

Alignment depth - Mut.Assessor!MutAssessor tends to label as Neutral when there’s very few sequences at that position in the alignment. !
Example - 3

Sequence identity /divergence - SIFT
“Confidence in a substitution predicted to be deleterious depends on the diversity of the sequences in the alignment. If the sequences used for prediction are closely related, then many positions will [wrongly] appear conserved… This leads to a high false positive error...”
SIFT therefore returns a conservation score to indicate the diversity of sequences used in the alignment.
Example - 4

!“If an alteration is a 'true' SNP, it is automatically predicted to be a polymorphism. […] We advise you not to exclude an alteration due to a dbSNP ID. Many SNPs from dbSNP are not validated and some are even known to be disease causing variant” !Reading…
Automatic Annotation
Example - 5
Mutation Taster

Introduction
Typical pipeline for identification of deleterious variants found in coding sequence (WES, panels, …)
Input amino acid
substitution and protein ID
SequencePhysicochemical prop.
Biochemical properties ...
Ann. from swissprot predicted scores
dbSNP, …
3D structure Secondary structure
Surface area properties B factors, …
Machine learning
approaches / theoretical
models
Output predictionsStructure
Annotation
AlignmentsConservation score
Entropy, frequencies, …

Understanding output scores
Score Scales in SIFT High scores usually associated to deleteriousness Thresholds at the middle of the scale (0.5)
Example - 6

Understanding output scores
Score Scales in SIFT High scores usually associated to deleteriousness Thresholds at the middle of the scale (0.5) …but SIFT threshold for damaging at <= 0.05.
Example - 6This links to…

Score
What users see…
Pathological Neutral
How much can we trust the score?

Score
It’s easy to forget about the error
Pathological Neutral
How much can we trust the score?

What else can we do?
Select best predictions Confidence score in different methods !Consensus methods Congruency methods Manual revision

Many prediction methods acompained output with an error estimate (confidence/reliabilty score). !
High confident predic-tions increase the accu-racy although it wi l l reduce coverage.
Reliability: Select best predictions

Filtering for high quality predictions
0
0,225
0,45
0,675
0,9
Abril Mayo Junio JulioAccuracy AccuracyCoverage Coverage
PolyPhen-2 for BRCA1 dataset PolyPhen-2 for BRCA2 dataset
100 %
100 %
60.0 %65.6 %
Example - 7

Filtering for high quality predictions
0
0,25
0,5
0,75
1
Abril Mayo Junio JulioAccuracy AccuracyCoverage Coverage
PolyPhen-2 for BRCA1 dataset PolyPhen-2 for BRCA2 dataset
72.4 %83.9 %
73.7 %78.2 %
Example - 7

Reliability in different methods…
Output from 0 (benign) to 1 (damaging) with threshold at 0.5: !!!!Additionally, estimates of false positive rate (FPR) and true positive rate (TPR) used to tag mutations qualitatively as benign, possibly damaging and probably damaging. ! For HumDiv uses 5% / 10% (prob < posib < benign) For HumVar uses 10% / 20% Lack of data: unknown
PolyPhen2

Reliability in SIFT
“Confidence in a prediction depends on the diversity of the sequences in the alignment. If the sequences are closely related, many positions will [wrongly] appear conserved… This leads to a high false positive error...”
SIFT

Reliability in SIFT

Reliability in MutationAssessor

Reliability in AlignGVGD
More likely to cause damage
Less likely to cause damage
BQº variation
BQ distance

Discrepancy and reliability
Mutations with low reliability incorrectly predicted !GLA protein ! M76L - Neutral variant A97V - Damaging variant
Example - 8
0.86 0.550.15* 0.91
SIFT PolyPhen-2

What else can we do?
Select best predictions Confidence score Some examples !Consensus methods Congruency methods Manual revision

Consensus methods
• Dependency of primary methods (server, updates) Example: Lack of prediction for MutAssessor for GLA protein
Example - 9All neutral variants predicted as pathological - 100% FP

Manual approach
Consensus methods
• Complemented by view/analysis at 3D/MSA.
• More training, but add info.

Take home messages
Consensus methods
• Plenty of prediction methods available !
• Common features although particularities !
• Alignment is a key element, but many solutions !
• Understanding output and reliability
!• Complementary approaches







