dr. m a maleque molla, frcp, frcpch 11 dec, 2017disorder.pdftransient hyperammonemia. ... or double...
TRANSCRIPT

Dr. M A Maleque Molla, FRCP, FRCPCH
11 Dec, 2017

Introduction Metabolism is a sequence of chemical reactions that take
place inside cells which are responsible for the breakdown of nutrients and the generation of energy in our bodies
The major building blocks which are needed to sustain life.
Carbohydrates
Amino acids
Fats (lipids)

Metabolic disorder The principal classes of metabolic disorders are:
Inborn error of metabolism
Acid-base imbalance
Metabolic brain diseases
Calcium metabolism disorders
DNA repair-deficiency disorders
Glucose metabolism disorders
Hyperlactatemia
Iron metabolism disorders
Lipid metabolism disorders
Malabsorption syndromes
Metabolic syndrome X
Mitochondrial diseases
Phosphorus metabolism disorders
Porphyrias
Proteostasis deficiency

Inborn Error of Metabolism(IEM)
Inborn errors of metabolism (IEMs) are group of rare genetic diseases that generally result from a defect in an enzyme or transport protein which results in a block in a metabolic pathway leading to clinically significant consequences.



Pathophysiology Single gene defects resulting abnormalities or defect in an
enzyme or transport protein.
• results in a block in a metabolic pathway for the synthesis or catabolism of proteins, carbohydrates, fats, or complex molecules
• Toxic accumulations of :
substrates & intermediates from alternative metabolic pathways
defects in energy production & utilization
• cause acute metabolic decompensation

Epidemiology Individual IEMs are very rare diseases, with
incidence less than 1:100,000
The incidence of IEMs, collectively, is estimated to be as high as 1 in 800 to 1:2500 live births
Male-to-female ratio is 1:1
The incidence varies within different racial and ethnic groups

Types of Inborn error of Metabolism Disorders of Protein metabolism
Amino acids (Phenylketonuria, Maple Syrup Urine Disease)
Organic acids (Methylmalonic Aciduria, Biotinidase Deficiency)
Urea cycle (Citrullinemia, Argininosuccinic Aciduria)
Disorders of Carbohydrate metabolism
Galactosemia
Fructose intolerance
Glycogen storage disease
Disorders Fatty Acid metabolism
Medium Chain Acyl CoA Dehydrogenase Deficiency(MCAD)
Very Long Chain Acyl CoA Dehydrogenase Deficiency(VLCAD)

Types of Inborn error of Metabolism
Lysosomal storage disorders, e.g., Gaucher's disease, Niemann–Pick disease
Disorders of porphyrin metabolism, e.g., acute intermittent porphyria
Disorders of purine or pyrimidine metabolism, e.g., Lesch–Nyhan syndrome
Disorders of steroid metabolism e.g., congenital adrenal hyperplasia, congenital adrenal hyperplasia
Disorders of mitochondrial function e.g., Kearns–Sayre syndrome
Disorders of peroxisomal function e.g., Zellweger syndrome

Presentation Presentation is usually in the neonatal period or infancy but
can occur at any time, even in adulthood.
Age for presentation of clinical symptoms varies for individual IEMs
Disorders of protein or carbohydrate disorders tend to present in the neonatal period or early infancy
Fatty acid oxidation defects, glycogen storage, and lysosomal storage disorders tend to present in infancy or childhood.
Disorders manifested by subtle neurologic or psychiatric features often undiagnosed until adulthood.
The onset and severity may be exacerbated by environmental factors such as diet and intercurrent illness.

Etiology
Inborn errors of metabolism is an inherited disorders.
Caused by mutations in genes coding for proteins that responsible for metabolism.
Most of the disorders are inherited as autosomal recessive, whereas autosomal dominant and X-linked disorders are also present.

Clinical manifestation The presenting features of IEM may be acute or chronic &
can affect any system.
Acute:
episodic vomiting,
dehydration or shock,
lethargy and coma,
rhabdomyolysis,
hypoglycemia associated with minor illnesses e.g. stress, or a prolonged fast.
Chronic:
growth delay/failure to thrive,
hepatomegaly, cardiomyopathy,
spastic diplegia,
developmental delay or regression.

Clinical manifestation
Can be present as Metabolic emergencies,
recurrent vomiting and dehydration,
lethargy and coma, seizures,
sudden infant death syndrome (SIDS)
apparent life-threatening event (ALTE) .

Clinical findings according to age
Neonate
Finding may be indistinguishable from those of sepsis.
Poor feeding, vomiting, diarrhea, and/or dehydration;
Temperature instability -hypo or hperthermia
Tachypnea, apnea.
Bradycardia,
Poor perfusion;
CNS: Irritability, involuntary movement, posturing, abnormal tone, seizures, and altered level of consciousness & coma.

Clinical findings according to age
Infants and young children Dysmorphic or coarse features, Recurrent vomiting, Kussmaul’s breathing suggest metabolic acidosis Poor feeding, failure to thrive, Developmental delay, occasionally with loss of milestones. Seizures, ataxia, lethargy, coma, and fulminant
hepatoencephalopathy. Skeletal abnormalities, abnormalities of the hair or skin, Dilated or hypertrophic cardiomyopathy, Hepatomegaly, jaundice, and liver dysfunction. Visual and auditory disturbances.

Clinical finding according to age
Older children, adolescents, and adults
Mild to profound mental retardation,
Autism, learning disorders, behavioral disturbances,
Hallucinations, delirium, aggressiveness, agitation, anxiety,
Panic attacks, seizures, dizziness, ataxia,
Exercise intolerance, muscle weakness, and Para paresis

Index of Suspicion for IEM Any child with unexplained following signs & symptoms, IEM should be ruled out;
Rapid deterioration in an otherwise well infant Septic appearing infant or sepsis with unusual organism
such as E. coli Unexplained metal retardation. Developmental delay or regression Neurological deterioration & Coma Motor deficit or convulsion Metabolic acidosis Hypoglycemia Inappropriate ketosis/Acidosis Hypotonia Cardiomyopathy Hepatocellular dysfunction, hepatomegaly Failure to thrive

Diagnosis History
Physical examination
Investigations

History Consanguinity, ethnicity, inbreeding
H/O neonatal deaths or fetal losses
Maternal family history:
Males - X-linked disorders
Mitochondrial disorder is maternally inherited
A positive family history may be helpful but a negative family history dose not exclude the condition

Physical examinations The physical examination, findings are nonspecific
and may be normal
Finding also varies with age and severity.
Abnormalities in general may include: dysmorphic features
failure to thrive
abnormalities of hair, skin, skeleton, or all three
abnormal odor
Organomegaly
Abnormal muscle tone

Characteristic odor & IEM Characteristic odor IEM
Musty or Mousy PKU
Boiled Cabbage Tyrosinemia or hypermethioninemia
Maple Syrup Maple syrup urine disease
Sweaty feet Isovaleric acidemia or glutaric acidemia type II
Cat urine Multiple carboxylase deficiencies (Biotin deficiency)
Rotten fish Trimethylaminuria

Investigations Specimens of blood & Urine for definitive diagnosis should be collected
while the child is acutely ill.
Complete blood count -to screen for neutropenia, anemia, and thrombocytopenia.
Serum electrolytes, bicarbonate, and blood gases levels, to detect electrolyte imbalances and to evaluate acid/base status & anion gap.
Blood glucose
Renal Function: Blood urea nitrogen and creatinine
Liver function: S. Bilirubin, transaminases, PT, aPTT, INR
Ammonia levels: <100 µmol/L in neonate & <80 µmol/L age > 1 month
Urine: pH, ketones, and reducing substances levels to evaluate for hypoglycemia.
Lactate dehydrogenase, aldolase, creatinine kinase, and urine myoglobin levels in patients with evidence of neuromyopathy

LAB interpretation & clinical clue for IEM Most IEMs can be categorized based on findings of initial laboratory evaluations with the presence of at least 1 of the following
ABG:
Anion gap metabolic acidosis-Organic acidemia
Normal anion gap metabolic acidosis-hyperglycinemia
Respiratory alkalosis+ high ammonia- Urea cycle disorder
Low BUN relative to creatinine- Urea cycle disorder
Hypoglycemia :
With hepatomegaly- Glycogen storage disease
Non-ketotic hypoglycemia- fatty acid oxidation defect
High Ammonia: >120 µmol/L in the neonate, >80 µmol/L beyond the neonatal period.
Transient hyperammonemia.
Urea cycle defects - often > 1000 mcg/dL
Ammonia in organic academia's <500 mcg/dL,
In fatty acid oxidation defects, usually le<250 mcg/dL.

Screening for IEM Newborn Screening: Heel prick Test(GUTHRIE TEST)
Congenital Hypothyroidism (CHT)
Sickle Cell disorders
Cystic Fibrosis (CF)
Inherited metabolic diseases (IMDs)
Galactocemia
Phenylketonuria (PKU)
Medium-chain acyl-CoA dehydrogenase deficiency (MCAD)
Maple syrup urine disease (MSUD)
Isovaleric acidaemia (IVA)
Glutaric aciduria type 1 (GA1) and
Homocystinuria (pyridoxine unresponsive) (HCU)
Tandem mass spectrometry – allows to screen for > 30 disorders, generally include aminoacidemias, urea cycle disorders, organic acidurias, and fatty acid oxidation disorders.

Specialized tests Quantitative plasma amino acids plasma or
serum : used to confirm the diagnosis of urea cycle disorders and other disorders of amino acid metabolism
Qualitative urine organic acids- Organic acidemia,
Serum lactate, and Pyruvate: mitochondrial disorders, glycogen storage diseases, disorders of gluconeogenesis, and disorders of pyruvate metabolism
Acylcarnitine profile: used for the diagnosis of fatty acid oxidation disorders

Specific diagnosis
Specific diagnostic tests:
Tissue biopsy or autopsy: liver, muscle, brain, bone marrow
Skin biopsy and fibroblast cultivation: for specific enzyme testing
Specific DNA testing.

DIFFERENTIAL DIAGNOSIS
In neonates: Sepsis, Congenital viral infection Duct-dependent heart disease, Drug withdrawal, Congenital adrenal hyperplasia In older children, Diabetes, Drug ingestion or intoxication, Encephalitis Adrenal insufficiency

Management Approach Goals of treatment :
Prevention of further accumulation of harmful substances,
Management of complication & correction of metabolic abnormalities
Elimination of toxic metabolites.
Treatment should be initiated as quickly as possible;
Even the apparently stable patient with mild symptoms because may deteriorate rapidly, with progression to death.
With appropriate therapy, patients may completely recover without any sequelae.

General management
In any critically ill child appropriate and aggressive treatment before confirmation of the diagnosis may be life-saving, Assessment of airway, Breathing, and Circulation. Hypoglycemia, acidosis, and hyperammonemia must be
corrected. Consider broad spectrum antibiotics in any child who may be
septic. If shock IV normal saline should be used as bolus fluid. Lactated
ringers solution should be avoided. Discontinue oral intake in patients with decreased level of
consciousness or vomiting. Correct hypoglycemia, prevent catabolism, and promote
urinary excretion of toxic metabolites. Eliminate intake or administration of potentially harmful protein
or sugars, especially galactose and fructose In case of known IEM or those with positive newborn screen
results, disease-specific offending agents should be eliminated for those with

Management of Complication Correct hypoglycemia:
IV bolus, as 10% dextrose water(DW) for neonates and 10% or 25% DW beyond the neonatal period, 0.25-1 g/kg/dose, not to exceed 25 g/dose
Bolus should followed by continuous IV administration dextrose of 8-10 mg/kg/min. Usually 10% dextrose saline as maintenance fluid.
Prevent catabolism:
For all patients suspected IEM, dextrose IV 8-10 mg/kg/min, at 1-1.5 time maintenance should be given to keep glucose level at 120-150 mg/dL, which should prevent catabolism.
Elimination of toxic metabolites: All patient with acidosis, 1.5 time maintenance fluid should be given. It will promote urinary excretion of some toxic metabolites.

Management of Complication (Cont.)
Correction of metabolic acidosis and electrolyte abnormalities:
Sodium bicarbonate should be administered to correct acidosis, 1-2 mEq/kg/hr.
For intractable acidosis, consider hemodialysis.
If the patient is hypokalemic, potassium acetate should be added.
Add maintenance concentrations of electrolytes, to correct electrolyte disturbances if present.

Management of Complication (Cont.) Correct hyperammonemia.
Significant hyperammonemia is life-threatening and must be treated immediately upon diagnosis.
Sodium phenylacetate and sodium benzoate (Ammonul); augment nitrogen excretion
Arginine HCL; should be administered in patients with urea cycle defects
L-carnitine; It is a cofactors. in primary carnitine deficiency, L carnitine should be administer.
For ammonia level greater than 500-600 mg/dL before Ammonul or greater than 300 mg/dL and rising after Ammonul, hemodialysis should be initiated.
If hemodialysis is not available, peritoneal dialysis (<10% as effective as hemodialysis) or double volume exchange transfusion (even less effective) can be performed

Management of Complication (Cont.)
Patient with seizure should be treated with IV anticonvulsant like Diazepam or phenobarbitone.
Seizures unresponsive to conventional anticonvulsants, Pyridoxine (B6) should be given to neonates as a trial.
Provide cofactor if indicated;
Vitamin B12 in case of suspected organic acidemia.
Biotin with recurrent seizures for possible biotin responsive multiple carboxylase deficiency.
Carnitine in case of supected organic acidemias
Patients should transfer to a tertiary care facility for further evaluation and treatment after stabilization

Follow up Management
Once toxic metabolites have been normalized, protein can be reintroduced using an essential amino acid solution, initially at 0.5-0.75 g/kg/day and gradually increased.
For amino and organic acidopathies & urea cycle defects, protein intake should be restricted to 40-50% of recommended daily allowance.
Lipids, 2-3 g/kg/day as 20% intralipid, can be given to increase caloric intake, but they are contraindicated for certain fatty acid oxidation defects.
Pharmacologic therapy to increase activity of abnormal cofactor-dependent enzymes e.g. thiamine [B-1], biotin PO, riboflavin [B-2], cobalamin [B-12] may be given.

Dietary Management With definitive diagnosis, specific dietary regimens,
should be initiated which can Prevent
death,
intellectual disability, or other adverse health outcomes
Diet for IEM include medical foods and dietary supplements along with dietary modifications.

Dietary Management
Two types of medical foods are used;
One type meets nutritional requirements while excluding the IEM specific nutrient that cannot be metabolized.
The second type-modified diet to be low in protein and are used in natural protein-restricted diets e.g., specially modified flour, cereals, and baked goods, meat and cheese substitutes, pasta, and rice).

Specific Management
Organ or bone marrow transplantation
Enzyme replacement therapy
Gene therapy

Specific Metabolic disorders

Phenylketonuria(PKU) Common inborn error of metabolism of essential
amino acid phenylalanine Incidence: 4-6/100,000. in USA, 10:100,000 in
Bahrain Inheritance autosomal-recessive . Due to deficiency of enzyme phenylalanine
hydroxylase an enzyme responsible for the conversion of phenylalanine to tyrosine.
In affected infants with plasma concentrations >20 mg/dL, excess phenylalanine is metabolized to phenylketones.
Elevated phenylalanine levels in the body fluid and brain negatively impact on cognitive function & brain development.

Fig. Metabolism of Amino acid Phenylalanine
Tetrahydrobiopterin(BU4)
Phenylalanine Hydroxylase
Phenylalanine Tyrosine
Dihydrobiopterin

PKU
Diagnosis:
• Tandem mass spectrometry (MS/MS) screening : Positive results for hyperphenylalaninemia.
• High Phenyl pyruvic acid.
• Diagnosis should be confirmed by quantitative measurement of plasma high phenylalanine concentration usually > 20 mg/dl
h

PKU Treatment:
• The goal of therapy is to reduce phenylalanine levels in the plasma and brain
• low-phenylalanine formula diet life long
• Tyrosine supplement
• Amino acids supplement
• Oral BH4 (Tetrahydrobiopterin).
• L – dopa and 5- hydroxytryptophan.
Prognosis: The prognosis for normal intelligence is excellent when patients have been put on a diet low in phenylalanine in the first month of life.

METHYLMALONIC ACIDEMIA
It is a type of organic acidemia due defective metabolism of specific fats and amino acids.
Defect in the conversion of methylmalonyl-coenzyme A to succinyl-CoA due to deficiencies in methylmalonyl-CoA mutase and in enzymatic synthesis of cobalamin.
Mutations leading to defects in vitamin B12 metabolism can leads to methylmalonic acidemia.
As a result there toxic level of methylmalonic acid accumulation in the blood
Inharited as an autosomal recessive disorder.
Frequency of 1 in 25,000-48,000 births
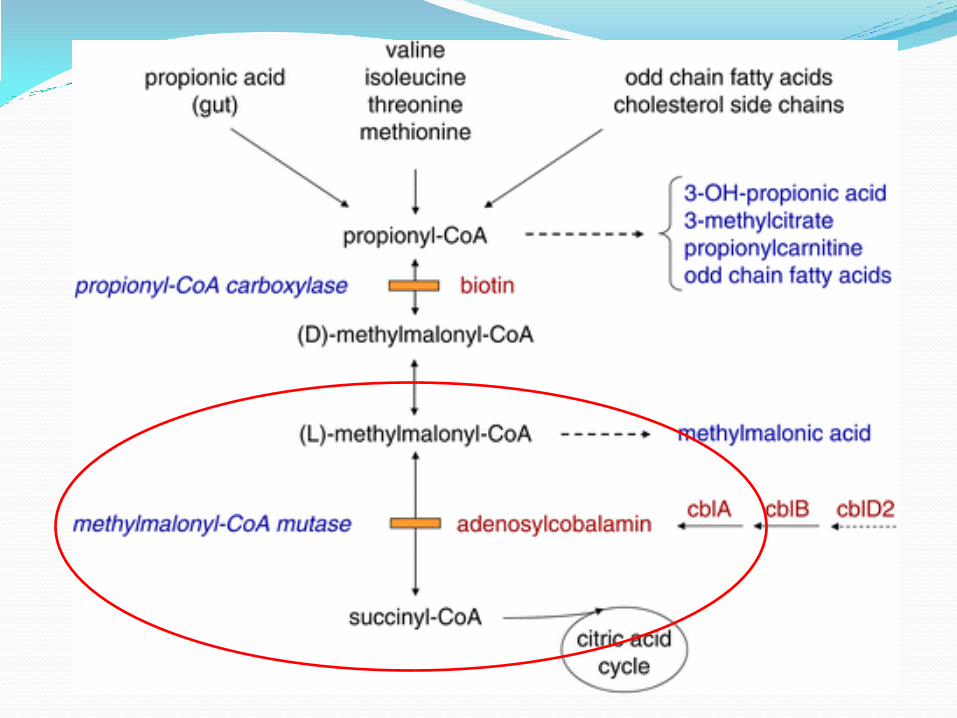

METHYLMALONIC ACIDEMIA CLINICAL MANIFESTATION
Children may be healthy at birth and develop symptoms soon after starting protein intake.
Family history may be positive for methylmalonic acidemia
In most children, present as an episode of metabolic decompensation:
Vomiting, dehydration, lethargy, seizures, recurrent infections, and progressive encephalopathy
Some children may present with strokes during a metabolic crisis.
Metabolic ketoacidosis is the clinical hallmark of methylmalonic acidemia in infants.
Hypotonia, lethargy, failure to thrive, hepatosplenomegaly, and monilial infections are some classic findings.

METHYLMALONIC ACIDEMIA
DIAGNOSIS:
Blood ammonia, glycine, and methylmalonic acid are elevated.
Serum levels of propionic acid, may also be elevated.
Urine levels of methylmalonic acid, methylcitrate, propionic acid, and 3-hydroxypropionate levels are high.
Definitive diagnosis is made after enzyme analysis of fibroblasts in search of the specific enzyme abnormality.

METHYLMALONIC ACIDEMIA MANAGEMENT
Require multispecialty care for diagnosis and treatment.
In an acute phase, treat intercurrent infections that triggered the acidotic episode.
Correct the acidosis
dialysis may be required in cases of severe ketoacidosis and hyperammonemia.
Dietary modifications must be made in a hospital setting
Implement a protein-restricted diet (0.5-1.5 g/kg/d) +
L-carnitine and cobalamin supplementation.
Cobalamin supplementation may help because cobalamin is a cofactor

GALACTOSEMIA
Most common carbohydrate metabolism disorders
There are enzymatic errors in galactose metabolism.
Most common defect is decreased activity of galactose 1-phosphate uridyltransferase (GALT). (Classic)
Incidence 1:60,000 in European population, 1:40,000 in USA
Inherited as autosomal recessive.

GALACTOSEMIA Most common disorders of carbohydrate metabolism.
Incidence 1:60,000 in European population, 1:40,000 in USA
Inherited as autosomal recessive manner.
Lactose in food is broken down to glucose and galactose by the enzyme lactase.
There are enzymatic errors in galactose metabolism.
3 enzymatic defect:
1. Galactose 1-phosphate uridyltransferase (GALT) deficiency. (Classic) most common type.
2. Galactokinase (GALK) deficiency.
3. Uridine diphosphate galactose 4-epimerase (GALE) deficiency.

Lactose
Galactose
Galactose-1-Phosphate
Glucose-1-Phosphate
Glucose Lactase
Galactokinase
Glucose-6-Phosphate
Galactose 1-phosphate uridyl transferase
Epimerase
Classic ×

GALACTOSEMIA CLINICAL FEATURES;
Infants with classic galactosemia usually present in the first few days after birth & initiation of breast milk or cow's milk feedings.
The most common findings are :
Jaundice
Vomiting, Diarrhea
Poor feeding, hypoglycemia
Hepatocellular damage-Hepatomegaly , bleeding
Failure to thrive
Lethargy
Sepsis (commonly by E. Coli)
Cataracts may be present at birth or first week of life.
Infants who survive the neonatal period and continue to ingest lactose may develop severe brain damage

GALACTOSEMIA
CLINICAL FEATURES;
Poorly treated or if untreated in older children and adults with classic galactosemia may include;
cataracts,
speech defects,
poor growth,
poor intellectual function,
neurologic deficits (predominantly extrapyramidal findings with ataxia)
Liver cirrhosis
Premature ovarian insufficiency in girl.

GALACTOSEMIA
Diagnosis
Positive reducing substances in urine.
Erythrocyte galactose-1-phosphate usually >10 mg/dL.
Confirmation by Galactose-1-PO uridyl transferase activity in RBCs.

GALACTOSEMIA
Treatment:
Immediate dietary intervention is indicated in infants whose erythrocyte GALT enzyme activity is ≤10% of control activity.
Lactose-free formula followed by dietary restriction of all lactose-containing foods later in life.
Prognosis:
Untreated infants may have severe growth failure, mental retardation, cataracts, ovarian failure, and liver cirrhosis.
Despite early and adequate intervention, some children still may develop milder signs of these clinical manifestations

Thanks for Attention