drug discovery - جامعة نزوى · drug discovery objectives: • to ... to understand sar and...
TRANSCRIPT

10/30/2016
1
Drug Discovery
Objectives:• To understand the processes of Drug Development• To be able to develop a plan for drug discovery
Reference:• Patrick G L. An Introduction to Medicinal Chemistry. 2013
Zainab Al KharusiOffice: 33-10
Processes of Drug Development� Choose a disease
� Choose a drug target
� Identify a bioassay
� Finding a lead compound
� Isolation and purification
� Structure determination
10/30/2016 2

10/30/2016
2
Choosing a disease
� Research projects tend to focus on diseases that are important in the developed world
� Great deal on: cancer, flu, ulcers, obesity, depression and cardiovascular diseases
� Only when such disease make a sense on western societies do pharmaceutical companies sit up and take notice
� Example: when malaria spread into the southern states of USA, pharmaceutical companies started to work on diseases such as malaria, tuberculosis and dengue.
10/30/2016 3
Choosing a drug target
� Drug targets such as receptors, enzymes and nucleic acids� It is important to define the target so we know what we have to
design either agonist or antagonist� Consider target selectivity and specificity between species� Example: penicillin act on enzyme that present only in bacteria
as bacteria have a cell wall while mammalian cells do not� Also some enzymes are present in both humans and bacteria
but due to little structure differences between the two enzymes it can be selective only for the microorganism.
� Consider target selectivity and specificity within the body� Example: drugs that act on β-receptors
10/30/2016 4

10/30/2016
3
Identifying a bioassay
• The test should be quick, simple and relevant
• In vitro test: involved specific tissues, cells, enzymes or receptors. These used to identify if the drug interacts with the target.
• In vivo test: animals, transgenic animals used to identify pharmacokinetics properties.
• High throughput screening (HTS): testing large number of compounds against large number of targets
10/30/2016 5
Identifying a bioassay
� Screening by NMR• It is complement to HTS. It can screen 1000 compounds in a
day.
• It can detect weak binding interactions and can identify the binding of small molecules to different regions.
• Disadvantages, it need to purify the protein and obtain it in a significant quantity.
� Affinity screening: screening mixtures for active constituents
10/30/2016 6

10/30/2016
4
Finding a lead compound
� Screening a natural products: plants, microorganisms, marine sources and animal sources
� Screening synthetic libraries: compounds that have been synthesized and may not reach the clinical trials are stored in a libraries and are available for testing.
� Existing drugs: � ‘me better’ drugs � ‘me too’ drugs� Enhancing a side effect: sulphonamides are used as antibacterial
agents but they have a side effect of hypoglycemia and so the structure of sulphonamides have been modified to give the antidiabetic agent tolbutamide.
� Computer-aided design of lead compounds: as structure-based drug design
10/30/2016 7
Processes of Drug Development
� Finding a lead compound
• Fragment-based lead discovery: NMR spectroscopy has been used to design a lead compound. This can be done by finding an epitope or ligand that can bind to a region in the binding site. Then repeating with other ligands that can bind to a different region. This is done in the presence of the first ligand to ensure that the other ligand will bind to a different region. Then a molecule can be designed by linking both compounds.
• Properties of Lead compound by fragment-based:
� A molecular weight less than 300
� No more than 3 hydrogen bonding donors
� No more than 3 hydrogen bonding acceptors
10/30/2016 8

10/30/2016
5
Properties of lead compound
� Lipinski Rule of 5
• Designed by Christopher Lipinski and his colleagues
• Used to predict if a molecules are able to cross biological membranes and absorbed in the body
• The molecule should follow the criteria:
• Molecular Weight ˂ 500
• Log P ˂ 5
• Hydrogen Bond Donors (HBD) ˂ 5
• Hydrogen Bond Acceptor (HBA) ˂ 10
• There are limitation and cautions should be taken when using this rule
10/30/2016 9
Processes of Drug Development
� Isolation and purification
• If the lead compound or active principle is available in a mixture from a natural source or combinatorial synthesis, then it has to be isolated.
• Available techniques that are used to for isolation and purification are chromatography and freeze-drying
� Structure Determination
• X- ray crystallography
• NMR Spectroscopy
• IR Spectroscopy
• Mass Spectroscopy
10/30/2016 10

30/10/2016
1
Structure Activity Relationship (SAR)
ReferencePatrick G L. An Introduction to Medicinal Chemistry. 2013
Objectives
� To understand SAR and its importance in drug interactions and design
� To predict different interactions that a functional groups in a drug can make it with the receptor
� To identify different tests that can be used to test drug binding interactions
Zainab Alkharusi
Office: 33-10
30/10/2016 1
After drug development
• When the structure of the compound is identified, then it should be moved to study the Structure Activity Relationship (SAR), to identify parts of the molecules that are important in binding interactions
• If the structure of the target is known, then it is possible to crystallize the target with the lead compound bound to its binding site
• The binding interactions can be studied with molecular modelling software to identify important binding interactions
• If the structure of target is unknown, then it is necessary to use the traditional methods of synthesizing a selected number of compounds and then study their biological activity
30/10/2016 2

30/10/2016
2
The possible binding interactions with the target structure
• Binding Interactions with alcohols and Phenols
• Binding interactions with amines
• Binding of aromatic rings and alkenes
• Binding interactions of ketones and aldehydes
• Binding interactions of amides
• Binding interactions of quaternary ammonium salts
• Binding interactions of carboxylic acids
• Binding role of esters
30/10/2016 3
Binding interactions with alcohols and Phenols
�The oxygen can act as hydrogen bond acceptor and the Hydrogen can act as hydrogen bond donor. Analogue for alcohols and phenols could be ether and ester. Ethers can hinder hydrogen bonding as the proton of the hydroxyl group is lost. Hence, the oxygen still available can have binding interactions but to less extent. The methyl group can hinder the interactions.
�Ester analogue cannot act as HBD but still can act as HBA due to availability of the oxygen but to less extent due to hindrance from the acyl group. Also, the lone pair of electrons in the oxygen involved in resonance structure.
30/10/2016 4

30/10/2016
3
Binding interactions with amines
� The lone pair of electrons in nitrogen can act as hydrogen bond acceptor
� The N-H group in the primary and secondary amines can act as hydrogen bond donors
� Amines present in aromatic and heteroaromatic rings can act only as hydrogen donors because the pair lone of electrons interact with the rings.
� The amine may be protonated when it interacts with the binding site which mean it is ionized and cannot act as HBA but it still can form strong HBD
� To test whether an ionic or a hydrogen bonding is interacting, an amide analogue can be studied.
30/10/2016 5
Binding of aromatic rings and alkenes
� The aromatic rings are planar, hydrophobic structures mostly involved in van der Waals interactions with the flat hydrophobic region of the binding site. An analogue containing a cyclohexane ring will be less likely to bind.
� Alkenes are also, planar and hydrophobic and can interact with binding site through van der Waals interactions.
30/10/2016 6

30/10/2016
4
Binding interactions of ketones and aldehydes
The possibility that can form hydrogen bonding interactions with the binding site through the oxygen. As the oxygen can act as HBA as the two lone pairs of electrons are available on the carbonyl oxygen. Also, the carbonyl has dipole moment and so it can interact through dipole-diploe interactions.Aldehydes are less common as a drugs because they are more reactive and susceptible to metabolic oxidation to carboxylic acids.
30/10/2016 7
Binding interactions of amides
� The amides are available in peptidesand peptido-mimetics
� It can form two HBA through thecarbonyl oxygen.
� Primary and secondary amides haveN-H groups which allow them forHBD.
� The nitrogen cannot act as a HBAbecause the lone pair of electronsinteracting with neighbouring carbonylfor resonance.
� Analogues can be used to test thebinding interactions of amide.
30/10/2016 8

30/10/2016
5
Binding interactions of quaternary ammonium salts
� Can interact through ionic bonds with carboxylate groups
� Another possible interactions through induced dipole between the quaternary ammonium and any aromatic rings in the binding site.
� Example: acetylcholine has quaternary ammonium.
30/10/2016 9
Binding interactions of carboxylic acids
� Can act as HBA and HBD
� The carboxylate ion can make an ionic interactions
� To test interactions, analogues such as esters, primary amides, primary alcohols and ketones can be synthesized and tested. None of these groups can be ionized, so loss of activity imply that ionic interactions is important.
30/10/2016 10

30/10/2016
6
Binding role of esters
� Can interact as HBA only, the carbonyl oxygen is more likely to act HBA than the alkoxy oxygen as it is sterically less hindered and has greater electron density
� Esters are susceptible to metabolism in vivo by esterases, but there are a lot of drugs that have ester groups, this due to electronic factors that stabilize the ester or steric factors that protect it.
� Esters usually used to mask polar functional groups as alcohol or carboxylic acid to prevent its metabolism. Once it in the blood, it hydrolysed to release the active drug. (prodrug)
30/10/2016 11
Testing procedures
� When testing SAR for drug binding interactions, biological testing should involve
� In vitro tests: may reveal drug-target interaction
� In vivo tests: may reveal functional groups that are important in protecting or assisting the drug in its passage through the body. This is may be not a clear-cut as the drug may not reach the target.
� NMR spectroscopy
� There is a little point in designing a drug that has optimum interactions with its target if it has undesirable pharmacokinetics properties
30/10/2016 12

30/10/2016
7
Identification of a pharmacophore
� A pharmacophore summarize the important functional groups for binding and that are required for activity.
� In order to identify the 3D pharmacophore, it is important to identify the active conformation.� This can be done by:� Synthesizing rigid analogues of a flexible compound and test it for activity� Crystallize the target and the compound bound to the binding site, the x-ray crystallography
can be used to identify the structure of the complex and the active conformation�Using NMR spectroscopy to solve the active conformation of isotopically labelled molecules
bound to their binding site.� It is important to realize that the overall skeleton of the molecule is involved in the
interactions with the binding site through van der Waals and hydrophobic interactions. The 3D pharmacophore does not take this into account. Also, it does not take into account the size of the molecule and whether it will fit the binding site.
30/10/2016 13

10/30/2016
1
Drug design: optimizing access to the target
Objectives
• To understand techniques that are used to get access to the target
• To be able to predict which method can be used to get access to the target
Zainab Al KharusiOffice room: 33-10
10/30/2016 1
How to optimize access with targets
� Optimizing hydrophilic/hydrophobic properties1. Masking polar functional groups to decrease polarity2. Adding or removing polar functional groups to vary polarity3. Variation of N-alkyl substituents to vary pKa4. Varying hydrophobic substituents to vary polarity5. Bioisosteres for polar groups� Making drugs more resistant to chemical and enzymatic degradation1. Steric shield2. Electronic effects of bioisosteres3. Steric and electronic modification4. Metabolic blockers5. Group shifts� Making drugs less resistant to drug metabolism1. Introducing metabolic groups
10/30/2016 2

10/30/2016
2
Optimizing hydrophilic/hyrophobic properties
� Important because it can control the processes of ADME
� Drugs which are too polar can not cross the cell membrane easily. These drugs have polar functional groups that make them prone to protein binding, phase II metabolism and rapid excretion.
� Drugs that are too hydrophobic no better. If they are given orally, they will be dissolved in fat tissues and will be poorly absorbed. If they injected, they are poorly soluble in blood and are likely to be taken by fat tissues, resulting in low circulating levels.
10/30/2016 3
Masking polar functional groups to decrease polarity
� Converting alcohol or phenol to an ether or ester
� Converting carboxylic acid to an ester or amide
� Converting primary and secondary amines to amides or to secondary or tertiary amines
� Adding extra hydrophobic alkyl group or larger alkyl groups
10/30/2016 4

10/30/2016
3
Adding or removing polar functional groups to vary polarity
� Adding polar functional groups for non-polar and poorly soluble drugs
• Example: Ticonazole to fluconazole
� Removing polar functional groups for highly polar drugs
• Example: alkaloids or endogenous peptides
10/30/2016 5
Variation of N-alkyl substituents to vary pKa
� Drugs that have pKa outside the range 6 – 9 tend to be strongly ionized and poorly absorbed through the cell membranes
� Extra N-alkyl groups or larger N-alkyl have an increase basicity but increasing the size or number of alkyl groups increases the steric bulk around the nitrogen atom. This hinders the water molecules from solvating the ionized form and prevents stabilization of the ion. This in turn decrease the basicity of the amine.
10/30/2016 6

10/30/2016
4
Varying hydrophobic substituents to vary polarity
� To increase the polarity
• OH, NH2, CO2H
� To increase the hydrophobicity
• Replace alkyl group with larger alkyl groups
• Example: increasing the size of one alkyl group and decreasing the size of another. This called methylene shuffle.
• Addition of halogen such as chloro or fluoro
10/30/2016 7
Bioisosteres for polar groups
� Carboxylic acid is highly polar group which can ionize and hinder absorption of the drug containing it.
• One way is to make it as ester prodrug.
• Another way, is to replace it with a bioisostere which has similar physiochemical properties such as 5-substitueted tetrazole rings. It is 10 times more lipophilic than CO2H
10/30/2016 8

10/30/2016
5
Making drugs more resistant to chemical and enzymatic degradation
� Steric shields• To prevent a nucleophile or enzyme from attacking the
susceptible group
• It can be done by adding a bulky group close to the functional group
10/30/2016 9
Making drugs more resistant to chemical and enzymatic degradation
� Electronic effects of bioisosteres• Replacing the methyl group of an ethanoate with NH2
• labile ester group can be replaced by an amide group
• Pyrrole ring can be used as a bioisostere for an amide
10/30/2016 10
10/30/2016
6
Making drugs more resistant to chemical and enzymatic degradation
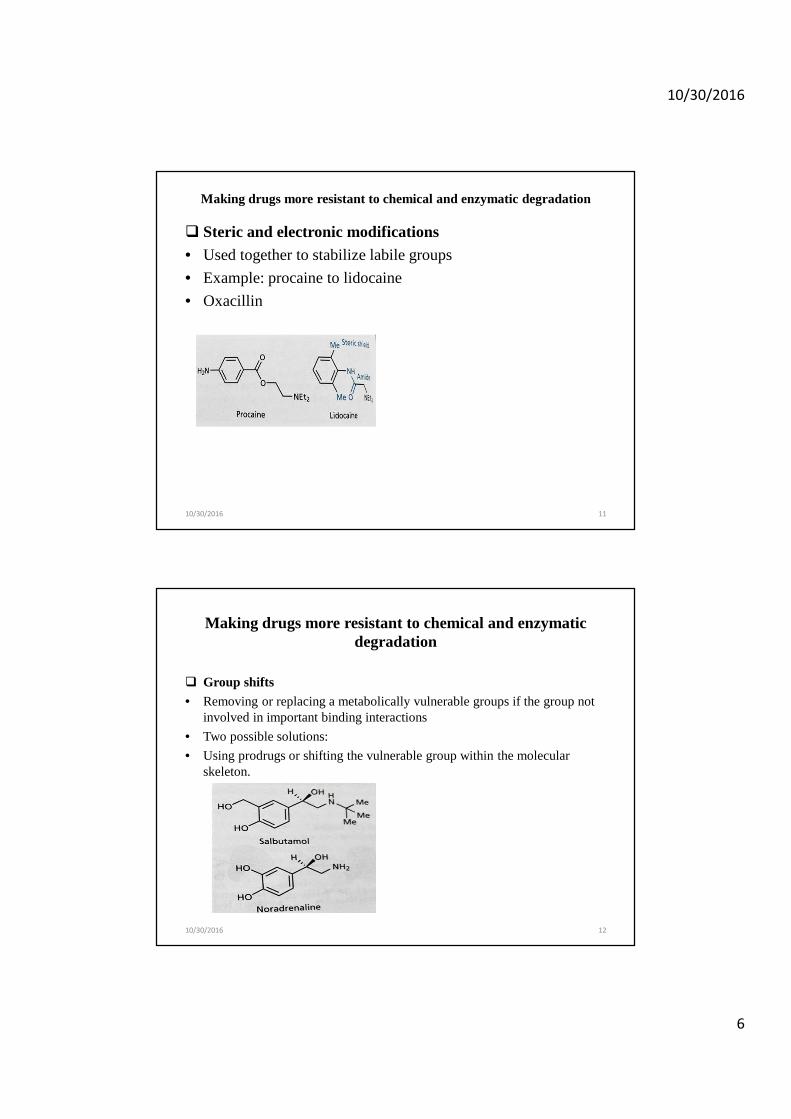
� Steric and electronic modifications• Used together to stabilize labile groups
• Example: procaine to lidocaine
• Oxacillin
10/30/2016 11
Making drugs more resistant to chemical and enzymatic degradation
� Group shifts
• Removing or replacing a metabolically vulnerable groups if the group not involved in important binding interactions
• Two possible solutions:
• Using prodrugs or shifting the vulnerable group within the molecular skeleton.
10/30/2016 12

10/30/2016
7
Making drugs more resistant to chemical and enzymatic degradation
� Metabolic blockers• Used to prolong the duration of biological activity of some drugs• Example: steroids are oxidized at position 6 of the tetracyclic ring to
introduce a polar hydroxyl group. This allows the formation of polar conjugates and so elimination from the body. By introducing a methyl group at position 6, metabolism is blocked and the activity of the steroid is prolonged.
• Flourine atom can be used in aromatic rings to block metabolism.
10/30/2016 13
Making drugs less resistant to drug metabolism
� Introducing metabolic groups
• Some drugs are stable to metabolism and are very slowly excreted as prolong activity may lead to toxicity and side effects
• This can be done by adding a methyl group that can be oxidized to polar alcohol as well to carboxylic acid
10/30/2016 14

10/30/2016
1
Quantitative Structure Activity
Relationship (QSAR)
� Objectives• To define the term of QSAR• To understand the physiochemical properties that can
affect biological properties• To understand Hansch equation and interpret it• To understand Criag Plot
• Reference• Patrick G L. An Introduction to Medicinal Chemistry.
2013
Zainab AlkharusiOffice room: 33-10
QSAR
• Quantitative Structure Activity Relationship (QSAR) attempts to identify and quantify the physiochemical properties of a drug and to see whether any of these has an effect on the drug biological activity.
• The compounds used should be related in structure, act at the same target and have the same mechanism of action.
• Each time one physiochemical properties is used to determine the biological activity.
• If the relationship is true, an equation can be drawn up which quantify the relationship and it can be say with some confidence that relationship has an important role in the pharmacokinetics or the mechanism of action of the drug.

10/30/2016
2
Graphs and Equations
• A range of compounds is synthesized in order to vary one physiochemical property and to test effects
• A graph then drawn to plot the biological activity (log 1/C) on the y-axis vs. the physiochemical features on the x-axis
• Linear regression analysis is done to obtain the best possible line.
• To measure how close the data points, a vertical lines is drawn from each point.
Graphs and Equations
• The next step is to see whether the relationship is meaningful. This can be done by regression coefficient (r2) or correlation coefficient (r)
• Correlation coefficient (r) is a measure of how well the physiochemical parameters presents in the equation explain the observed variance in the activity.
• When r = 1 the relationship is perfect. However this relationship is consider impossible and so when r ˃ 0.9 then the relationship is acceptable.
• The correlation coefficient r2 value of 0.85 is consider as good fit. If r2 multiplied by 100 it indicates the variance in biological activity accounted by the parameter
• Fisher’s F-test used to assess the significance of coefficients k for each parameter in the QSAR equation. Normally P value derived from F-test should be less than or equal to 0.05 (P ≤ 0.05).

10/30/2016
3
Physiochemical Properties
� Hydrophobicity factors
• Partition coefficient (P)
• The substituent hydrophobicity constant (π)
� Electronic factors
� Steric factors
Hydrophobicity• It determine how the drug can cross the cell membranes and how it can be
interact with the target.
� Partition coefficient (P)
• This can be tested by experiment using n-octanol/water mixture by distribution between the n-octanol layer and water layer.
• The relative distribution is known as partition coefficient (P)
• P = ����������������� ���������
����������������� �������=
� �
� �
• High P values means the compounds are hydrophobic
• Varying the substituents on the lead will lead to different analogues and then a graph can be draw by plotting log (1/C) versus log P. This will lead to straight-line graph (log P is 1-4) showing that there is a linear relationship.
• The equation will be log(1/C) = K1logP + K2

10/30/2016
4
Hydrophobicity� Partition coefficient (P)
• A straight-line relationship is observed when the range of log P is narrow (1-4). If this extended to more than this values, then the graph will be parabolic. Here the biological activity will increase until a maximum value is obtained (log P0). This will represent the optimum partition coefficient for biological activity. Beyond that point, an increase in log P results in a decrease in biological activity.
• The equation for parabolic curve is• log (1/C) = -K1(log P)2 + K2logP + k3
Hydrophobicity
� The substituent hydrophobicity constant (π)
• Log P value can be calculated theoretically to decide which compounds worth synthesizing it.
� It can be calculated by knowing the contribution that various substituents make to hydrophobicity. The contribution is known as the substituent hydrophobicity constant (π) and it is a measure of how hydrophobic a substituent relative to hydrogen
• πx = log Px – log PH
PH is the partition coefficient for the standard compound
Px is the partition coefficient for the standard compound with the substituent
• Use this equation and the table to calculate log P
• LogP = ∑ π

10/30/2016
5
Electronic effects
• The electronic effects of various substituents have an effect on a drug’s ionization or polarity which affect how easily a drug can pass through the cell membranes or how strongly it can interact with the binding site.
• Hammett substituent constant (σ), use to measure electronic effect on a substituent on an aromatic ring. This is a measure of the EW or ED ability of a substituent.
Electronic effects
• Benzoic acids containing EW substituents will have larger Kx (dissociation constant of substituent) values than benzoic acid itself KH (dissociation constant of Benzoic acid) and so the value of σx will be positive. Substituents such as Cl, CN or CF3 have a positive value.
• Benzoic acids containing ED substituents will have a smaller Kx values than benzoic acid and hence the value of σx will be negative. Substituents such as Me, Et and t-Bu will have a negative value
• Hammett substituent constant take into account both the resonance and inductive effects. Therefore, the value of σ for a substituent will depend on whether the substituent is meta or para.

10/30/2016
6
Steric Factors
� The bulk, size and shape of a drug will affect how it can interact with the binding site
� A bulky substituent may act as a shield and hinder the ideal interactions while it may help to orientate the drug into the binding site and increase the activity
� It can be measured by several methods such as
• Taft’s steric factor (Es)
• Molar refractivity (MR)
• Verloop steric parameter
Hansch equation
• In situations where only one parameter can be related, a simple equation can be drawn up.
• When more than a physiochemical parameters must be related to the biological activity, then Hansch equations can be used.
• It relate the biological activity to the most common used physiochemical properties (log P, π, σ and a steric factor)
• If the range of hydrophobicity values is limited to a small range then the equation will be linear
• Log (1/C) = k1 log P + K2σ + K3Es + K4
• If the log P values spread over a large range, then the equation will be parabolic
• Log (1/C) = k1 (log P)2 + K2Log P + K3σ + K4Es + K5

10/30/2016
7
Hansch equation
� Example 1
• The adrenergic activity was related to π and σ and did not include a steric factor. This equation tell us if the substituents have positive π and negative σthis will increase the activity. In other words the substituents should be hydrophobic and electron donating
� Example 2
• Log (1/C) = -0.015(π)2 + 0.14 πsum+ 0.27 ∑ πx + 0.40 ∑ πy + 0.65 ∑ σx + 0.88 σy + 2.34
• (n = 102, r = 0.0913, r2 = 0.834, s = 0.258)
• This the equation shows that antimalarial activity increases very slightly as the overall hydrophobicity of the molecule (πsum) increases (the constant 0.14 is low). The (πsum)2 term shows that there is an optimum overall hydrophobicity for the activity and this is found to be 4.44. Activity increases if hydrophobic substituents are present on ring X and in particular on ring Y. EWG on both rings are also beneficial to activity more in ring Y than ring X. The r2 value is 0.834 which is above the minimum acceptable value of 0.8.
Criag Plot
• There is no relationship between π and σ• You can see which groups have the
similar π values (aligned in vertical line) and that it can be interchangeable
• Groups which form a horizontal line can be identified as being iso-electronic or having similar σ
• (Positive π and positive σ) substituents are hydrophobic and EW properties
• (Negative π and negative σ) substituents are hydrophilic and electron-donating
• (Positive π and negative σ) substituents are hydrophobic and electron-donating
• (Negative π and positive σ) substituents are hydrophilic and electron-withdrawing

10/30/2016
1
Drug optimization: strategies in drug design
Objectives� To understand the strategies that are used in drug design� To be able to predict which one can be used in tackling the
problems
Reference� Patrick G L. An Introduction to Medicinal Chemistry. 2013
Zainab Al KharusiOffice room: 33-10
10/30/2016 1
Strategies
• Variation of substituents
• Extension of structure
• Chain extraction/contraction
• Ring expansion/contraction
• Isosteres and bioisosteres
• Simplification of structure
• Rigidification of the structure
10/30/2016 2
10/30/2016
2
Variation of substituents
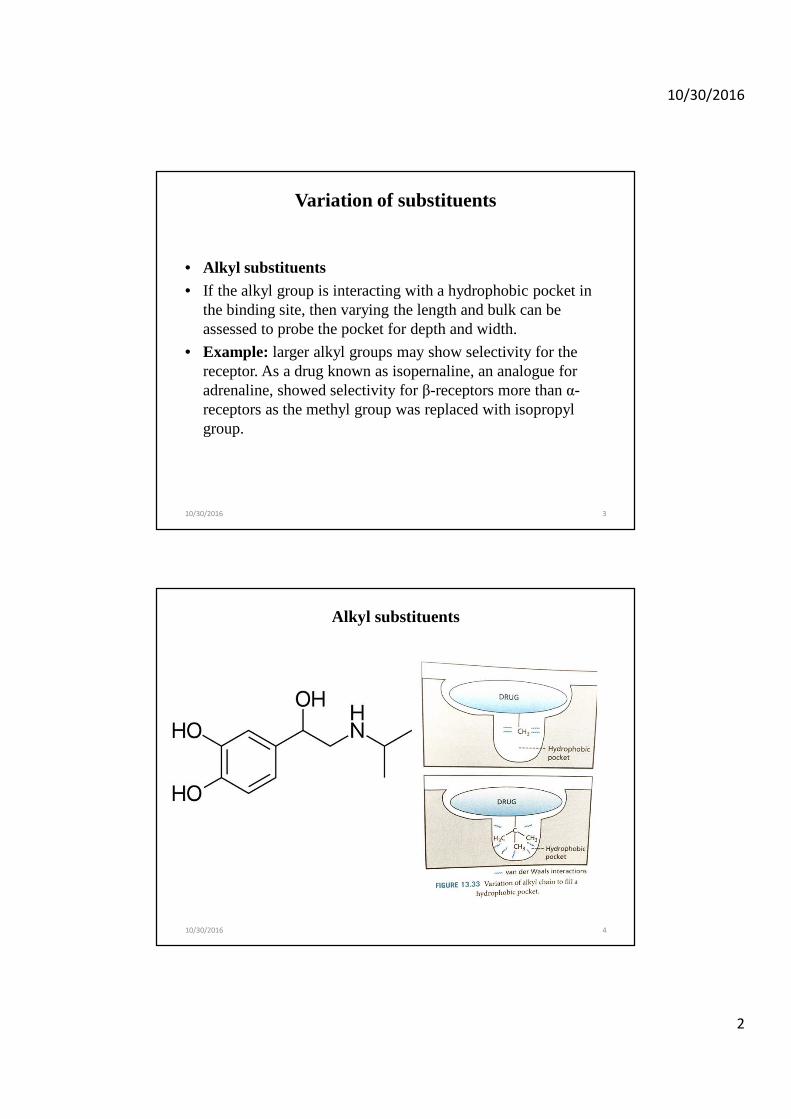
• Alkyl substituents• If the alkyl group is interacting with a hydrophobic pocket in
the binding site, then varying the length and bulk can be assessed to probe the pocket for depth and width.
• Example: larger alkyl groups may show selectivity for the receptor. As a drug known as isopernaline, an analogue for adrenaline, showed selectivity for β-receptors more than α-receptors as the methyl group was replaced with isopropyl group.
10/30/2016 3
Alkyl substituents
10/30/2016 4

10/30/2016
3
Variation of substituents
� Aromatic substituents• The position of the substituents can be varied (ortho, para or
meta).
� Example: the best anti-arythmic activity was found when the substituents sulphonamide was at position 7of the aromatic ring.
• The substituents themselves can be varied to vary hydrophobicity, steric and electronic parameters.
� Electron-withdrawing substituents may give better activity, so chloro substituents can be used.
10/30/2016 5
Aromatic substituents
10/30/2016 6

10/30/2016
4
Extension of the structure
� Involves the addition of another functional groups or substituents to the lead compound to probe for extra binding interactions with the target.
� Used to find extra hydrophobic region in the binding site by adding various alkyl or arylalkyl groups
• Example: enalaprilate design
� Used to convert agonist into antagonist
• Example: converting enzyme substrate into an inhibitor
10/30/2016 7
Extension of the structure
10/30/2016 8

10/30/2016
5
Chain extension/
contraction
• Some drugs have two important groups linked together by a chain, but the chain length is not ideal for the best interactions.
• It can be done by shortening or lengthening the chain.
10/30/2016 9
Ring expansion/contraction
• Expanding or contracting a ring may put other rings in different positions relative to each other and may lead to better interactions.
• Example: development of cilazaprilat
10/30/2016 10

10/30/2016
6
Isosteres and bioisosteres
• Isosteres used to vary the properties of the molecules such as, the size, polarity, electronic properties and bonding.
• Non-classical isosteres are groups that do not obey the steric and electronic rules that used to define classical isosteres but which have similar physical and chemical properties
• Bioisosteres are groups that can be used to replace another group while retaining the desired biological activity.
10/30/2016 11
Isosteres and bioisosteres
10/30/2016 12

10/30/2016
7
Simplification of the structure
• Used usually for lead compounds that come from natural sources.
• Start by conducting SAR and identifying the essential functional groups.
• Then non-essential groups can be discarded to simplify the structure and also to reduce side effects.
• Simplification done in small stages and then testing the biological activities each time.
• The presence of chiral centers in the compound can be reduce by the following tactics:
� Replacing the chiral carbon with nitrogen� Introducing symmetry in the original molecule
10/30/2016 13
Simplification of the structure
10/30/2016 14

10/30/2016
8
Simplification of the structure
10/30/2016 15
Rigidification of the structure
• Used to increase the activity and decrease the side effects• Flexible molecules have more conformations and only one
of them is the active conformation that will bind to a specific receptor. Other conformations may bind to other receptors which may cause another biological activities and so side effects.
• Rigidification of the molecule will decrease the number of conformations and so will decrease the side effects.
• Some of the tactics that are used to rigidify the molecule such as
� Incorporating the carbon skeleton into a ring� Incorporating a rigid functional groups such as alkene,
amide, aromatic ring
10/30/2016 16

10/30/2016
9
Rigidification of the structure
10/30/2016 17