drug product registration processes - dxc...
TRANSCRIPT

LEADING EDGE FORUM CSC GRANTS Copyright © 2013 Computer Sciences Corporation. All rights reserved.
Drug Product Registration
Processes
ABSTRACT Regulations in the US and Europe require technologies for registration of medicinal products in a structured format to facilitate inspections, safety tracking and licensing. The US Food and Drug Administration (FDA) system is based on a technology called “Structured Product Labeling” and is used in Drug Registration and Establishment Listings (DLER). The European Medicines Agency (EMA) has built a system primarily for pharmacovigilance, called Extended EudraVigilance Medicinal Product Dictionary (XEVMPD), which was instituted in 2012 based on an earlier, simpler format.
Both of these provide a bridge to a 2012 ISO standard called Identification of Medicinal Products, or IDMP, which is expected to go into production use in 2015 or 2016. While there are features in common to all three, there will be a significant effort required to provide a system that can migrate the required data to the international standard.
All biopharmaceutical companies will need to update the existing databases to the new standard. For the current standards, companies have been driven to create one-off registration documents in the specific formats, requiring long-term relationships with vendors familiar with the standards. However, that process does not build the knowledge base within the company.
This paper addresses the requirements of the two current and one future standard, as well as the processes used by pharmaceutical companies to maintain such information, and suggests best practices for maintenance of such databases within a company.
A key outcome is a solution architecture and set of processes for knowledge structures that would permit creation of SPL/DLER, XEVMPD and IDMP product registrations from a common set of data. This impacts a variety of roles throughout a company including obtaining, verifying and maintaining data.
Joel Finkle
CSC
CSC Grants
March 2013

ii
DRUG PRODUCT REGISTRATION PROCESSES
TABLE OF CONTENTS 1 BACKGROUND .............................................................................................................................. 1
1.1 Business Case for Drug Product Registration ......................................................................... 1 1.2 Product Terminology ................................................................................................................ 2 1.3 Regulation and Implementation ............................................................................................... 2 1.4 CSC Current Offerings ............................................................................................................. 3
2 DATA FORMATS FOR DRUG REGISTRATION ........................................................................... 4
2.1 SPL Data Listings and Establishment Registration (DLER) .................................................... 4 2.2 Extended Eudravigilance Medicinal Product Dictionary .......................................................... 5 2.3 Identification of Medicinal Products (IDMP) ............................................................................. 7 2.4 Commonalities and Differences ............................................................................................. 25
3 CHALLENGES FOR IDMP IMPLEMENTATION ......................................................................... 27
3.1 Substance Registration .......................................................................................................... 28 3.2 Data Ownership ..................................................................................................................... 28 3.3 Data Maintenance .................................................................................................................. 29
4 DRUG SPONSOR COMPANY PROCESSES .............................................................................. 30
4.1 Regulations ............................................................................................................................ 30 4.2 Data Maintenance Processes ................................................................................................ 30 4.3 Message Authoring ................................................................................................................ 31 4.4 Registration Process .............................................................................................................. 31
5 DRUG SPONSOR REQUIREMENTS ........................................................................................... 33
5.1 Consulting and Other Strategic Services ............................................................................... 33 5.2 Conversion and Authoring Services and Tools ...................................................................... 34 5.3 TRS Tracker and other Total Regulatory Solution software .................................................. 34
6 CONCLUSIONS ............................................................................................................................ 36
7 BIBLIOGRAPHY ........................................................................................................................... 38
LIST OF FIGURES Figure 1 – IDMP Data Model ............................................................................................................ 14 Figure 2 – Comparison of IDMP with DLER and XEVMPD .............................................................. 25

iii
DRUG PRODUCT REGISTRATION PROCESSES
LIST OF APPENDICES APPENDIX A : COMPARISON OF DATA ELEMENTS ...................................................................... 41
Appendix A-1 : Medicinal Product (ISO 11615) ................................................................................ 42 Appendix A-2 : Pharmaceutical Product ID (ISO 11616) .................................................................. 52 Appendix A-3 : Structured Substance (ISO 11238) .......................................................................... 55
APPENDIX B : ENTITY RELATIONSHIP MODELS ........................................................................... 68
Appendix B-1 : TRS Tracker Data Model ......................................................................................... 68 Appendix B-2 : IDMP Data Model ..................................................................................................... 70

1
DRUG PRODUCT REGISTRATION PROCESSES
1 BACKGROUND 1.1 Business Case for Drug Product Registration Current regulations in the US and Europe require substantially different technologies, terminologies and vocabularies for registration of medicinal products that are approved for use in human medical treatment, or for clinical trials. Registration of medicinal products is part of the licensing process, and is a paper activity in most countries. Europe and the US are ahead of the game by having electronic file formats specific to the drug product registration.
By providing an electronic medical products “dictionary,” the regulatory agencies are able to swiftly follow up on cases of counterfeiting, illegal import, contamination, patient safety, and supply shortages, as well as maintain the inspection audits of establishments involved in the manufacture, quality testing, packaging, import and distribution of medicines.
It is also worth noting that one of the major industry is for monitoring of drugs across classes and geographies rather than as discrete marketed products. This means that not only do market authorisation holders have to provide internally consistent data but also they need to provide data that can be sensibly aggregated with data from other companies for other products in other markets.
1.1.1 TECHNICAL CONSIDERATIONS It is important to note that the message formats used by the electronic standards are designed for consumption by an agency, and do not necessarily reflect the processes used by pharmaceutical companies for maintaining that information.
In the US, the FDA requires a “Drug Registration” and an “Establishment Listing”, both of which are based on the HL7-developed standard “Structured Product Labeling” (SPL). These describe the product, and the facilities in which it is manufactured, imported, and otherwise processed.
In Europe, the EMA initiated a new standard in 2011 called Extended Eudravigilance Medicinal Product Dictionary (XEVMPD), with a requirement that all marketed products be registered with the agency by July 2012 for the purposes of rapid access to a database of products in the case of safety issues.
The two systems have many features in common, and a number of items that are unique to each.
1.1.2 INTERNATIONAL APPROACH A third system, IDMP (IDentification of Medicinal Products) was approved in November of 2012 as an ISO standard (actually five standards, see below). IDMP was developed as an outgrowth of the Common Product Model (CPM) at HL7, so it has many features in common with SPL. The European XEVMPD standard has been stated as having a sunset date of when IDMP becomes implemented. This is estimated at 2015 or more likely 2016.
All biopharmaceutical companies will need to update existing databases to the new standard. If previous regulatory agency behavior is followed, a short (less than one year) period to adapt to the new standards will be granted for all companies to register all existing products.
For previous standards initiatives, companies have been driven to create one-off registration documents in the specific formats (SPL and XEVMPD), often requiring long-term relationships with vendors familiar with the standards, but this does not build the knowledge base within the company.
Development of a process for maintaining and messaging the information would improve the value of the information within customers’ enterprise knowledge warehouses, and improve their bottom line through the use of fewer contracted services to create and maintain such registration data.

2
DRUG PRODUCT REGISTRATION PROCESSES
Among the cases used to justify the system that will improve patient health and safety include:
• Tracking abused drugs – Realization of the level of Oxycontin abuse led to changes in its formulation to make it less appealing when a crushed tablet is inhaled.
• Product Labeling and Packaging – the accidental overdose of heparin in actor Dennis Quaid’s children has led to changes in how products are packaged and labeled (different dosage strengths now have different color labels on the bottles)
• Avoidance and mitigation of drug shortages (e.g. temporary importation from another country of the same product with a different approval number)
• Tracking drugs used in clinical trials may permit mining trial data for new uses (such as previous side effect), and tie efficacy and safety to genotypes
• Tracking generic drug usage, since generic drugs are now 80% of medicines used (but only 20% of the dollars spent), but having the same name with different manufacturers makes it difficult to gather the sum of any substance’s usage.
1.2 Product Terminology The data standards used for drug product registration include a number of similar terms which can be difficult to distinguish. They build on top of one another, and occasionally loop back into recursive structures.
The key thing to understand is the set of layers of the product definition. The product registration systems at various agencies are concerned with tracking a product for such reasons as ensuring supply, ensuring that supply is genuine and uncontaminated, and tracking any issues with using that product. For that reason, it is necessary to distinguish between what is administered to a patient, as a different thing from what is sold by the sponsor, but both have their own means of unique identification.
• MEDICINAL PRODUCT: This is a product administered to humans (or animals) for treating, preventing, diagnosing disease or restoring, correcting or modifying physiological function. The Medicinal Product is the form of a dose of the product as sold. It may have multiple components or ‘manufactured items’ and one or more ‘pharmaceutical products.’Invalid source specified.
• MANUFACTURED ITEM: This is the description of a product contained in the packaging of the Medicinal Product. Invalid source specified.. The manufactured items may include administration devices such as syringes.
• PHARMACEUTICAL PRODUCT: This is the version of the product as administered. The ISO standards described below define this as “qualitative and quantitative composition of a Medicinal Product in the dose form approved for administration in line with the regulated product information.’Invalid source specified.. In many cases, this may be the same as the Medicinal Product (e.g. a tablet), in others (e.g. a powder and solution which must be combined), a process is needed before it can be administered. In a small number of cases, there may be multiple pharmaceutical products in a medicinal product package: an antibiotic tablet packaged with a topical cream is two pharmaceutical products.
• PACKAGED MEDICINAL PRODUCT: This is the product as sold in its outermost container, e.g. a bottle of tablets, or a carton containing vials. The packaging is typically marked with an identifier of what batch it came from, to assist in tracking quality or safety issues in the product.
• PACKAGE ITEM: A Packaged Medicinal Product may contain multiple package items, such as vials or blister packs, which have their own containers. This may have several nested layers (e.g., a vial in a pouch in a carton). At the bottom level, these will contain Manufactured Items.
1.3 Regulation and Implementation Currently, only the US and the European Union members of the European Medicines Agency have a standard electronic data format for drug listings. These formats are rather different from each other,

3
DRUG PRODUCT REGISTRATION PROCESSES
partly due to differences in how the data is used, and the timing of the standards: the US has a focus on inspection of manufacturing and processing facilitiesInvalid source specified.), the EMA format was designed as a stopgap until the IDMP standard became available.
The Identification of Medicinal Product (IDMP) standard will be adopted by all three regions participating in the International Conference on Harmonisation (ICH) – adding Japan to the above US and EU countries. In addition, because it is an ISO standard, other jurisdiction are more likely to adopt an existing standard than design one from scratch. Countries that have followed ICH guidance for other electronic standards include Canada, Switzerland, and Australia for marketing applications, with influence also seen on the standards used in Russia, China, Southeast Asia (ASEAN countries), South America and the Gulf Cooperation Council. The ISO ICSR standard which is being implemented through ICH group E2B is will be used in Australia as well as the ICH partner nations.
It is expected that the US and EU will move to the IDMP standard within about two years after the development of implementation guides. Since the ICH guidance is expected in March of 2013, with regional guidance following shortly after, this should be in 2015 or 2016. It is unknown at what speed sponsors will be required to meet the new standard, but past experience has shown that agencies may be insensitive to the costs of rapidly implementing a new standard: The initial implementation of SPL for the Content of Labeling in 2005, and the update to the Physician’s Labeling Rule format in 2006 provided less than a year between distribution of the final guidance and required use. Similarly, the XEVMPD standard in the EU was announced in September 2011 for implementation by July of 2012, but the final version of the standard was not made available until March of 2012 – a mere four months prior to the deadline.
The short timeframes between final issuance of guidance and the requirement to comply mean that service and software companies will need to prepare services, software and related training and implementation services prior to the final guidance and implementation guides, with anticipation of last-minute changes to those products.
1.4 CSC Current Offerings The Life Sciences groups within CSC offers the following services and software related to drug registration dictionaries:
• SPL Conversion Services –The Regulatory Services group provides translation from Word and other formats into SPL for Content of Labeling and DLER.
• Regulatory Information Management Consulting – the author has been assigned to contracts involved in implementing Regulatory Information Management systems for clients. Other consultants have been involved in implementation of TRS Tracker, XEVMPD implementation, and data gathering in Europe.
• TRS Tracker Software and Implementation – Version 5.2 of TRS Tracker can produce reports on the Drug Product Tracking database that can be used to create XEVMPD records in the EVWEB system at EMA (see below), and create XEVMPD records directly
TRS Tracker can also be extended in the future to deliver IDMP messages, and with small changes could be delivering SPL DLER today (although content of labeling would be a significantly different effort).

4
DRUG PRODUCT REGISTRATION PROCESSES
2 DATA FORMATS FOR DRUG REGISTRATION The following sections detail the data formats of the three registration standards, and how their concepts build on each other. While the level of detail is daunting, the introductions and summaries of each section are valuable for understanding the implementation requirements, and the scope of work that medicinal product manufacturers will need to go through to implement these requirements.
2.1 SPL Data Listings and Establishment Registration (DLER) The Structured Product Labeling format was developed in 2004 by Health Level 7 (HL7) for FDA’s use, initially for the content and format of labeling. Release 3 was the first version required for use by the public, and Release 4 (SPL R4) was developed specifically to send structured data for Drug Listings and Establishment Registrations (DLER)Invalid source specified.. Per Section 510 of the Federal Food, Drug, and Cosmetic Act (FD&C Act), it is required for “firms that manufacture, prepare, propagate, compound, or process drugs in the U.S. or that are offered for import into the U.S. to register with the FDA. These domestic or foreign firms must at the time of registration, list all drugs manufactured, prepared, propagated, compounded, or processed for commercial distribution in the U.S. Foreign establishments must identify a U.S. agent at the time of their registration.”Invalid source specified..
The Drug Listing file is designed to replace the previous paper FDA forms 2657 and 2658, and includes the following information from those forms:
• Name of establishment(s) manufacturing or processing the listed drug and the type of operation(s) performed;
• DEA schedule; • Route(s) of administration; • Inactive ingredients; • Marketing information (e.g., category, start/stop date); • Information related to the application (e.g., type and year of approval) or OTC monograph
citation number; • Package size and type.
In addition to the information required on the older paper forms, the Drug Listing SPL file also includes:
• NDC Product Code for the Source Drug Repacked or Relabeled • Unique Ingredient Identifiers (UNII) • Data Universal Numbering System (DUNS®) Number for establishments and sponsor • Confidentiality Flag • Distinctive Characteristics (flavor, color and an image of solid dosage forms)
The Drug Listing and Establishment Registration may be created using the FDA-supplied E-Submitter tool, or delivered directly through the Electronic Submission gateway.
Although the SPL format is also used for the Content of Labeling, the Drug Listing and Establishment Registration does not include the sections that include the narrative labeling document. One of the criticisms of the SPL format is that it has a lot of complexity, but each document type uses only a portion of that total complexity. While that makes each document type manageable, it makes using standard XML technologies such as Microsoft Office tools extremely difficult, as the applications can not differentiate between the document types.
Approximately 8000 compounds in 50,000 unique packages are registered in SPL.Invalid source specified. FDA has recently encouraged updating all SPL files to Release 4, and in the past year

5
DRUG PRODUCT REGISTRATION PROCESSES
has managed to reduce this from 1600 separate labels in the old format to a mere 11 on eight compounds.
The most recent version of the SPL standard, Release 5, also includes structures for Device Listings. Compared to the product models in section 1.2, it’s not as complete: The Pharmaceutical Product concept is not included as a separate concept, and there is no detail on package items or manufactured items that are subsets of the whole.
2.2 Extended Eudravigilance Medicinal Product Dictionary The XEVMPD message format is called Extended Eudravigilance Product Report Message (XEVPRM). The format’s initial specifications as part of EMA Article 57, issued in September of 2011, were extensive. With a requirement to implement by July 2012, many pharmaceutical companies complained that the requirements were beyond reasonable costs of implementation, especially the information required for the “structured substance information” (SSI), which specified detailed information about active and inactive substances in each product. Coupled with this was the problem that many of the controlled vocabularies, and details on implementation of parts of the standard, had not yet been provided. This would have greatly increased the risk that sponsor organizations would not be able to comply.
EMA, however, was limited by European Union legislation, and could not delay implementation of the Eudravigilance medicinal product dictionary. The only options available were to (a) reduce the requirements to a minimum, and (b) have a flexible attitude to enforcement of compliance. The latter is significant because EMA could have removed products from the marketplace from sponsors that had not filed the XEVPRM data by July 2, 2012, but did not.
Updated specifications were issued in March of 2012Invalid source specified. – just four months prior to the deadline for submission. These updated specifications were based mainly around a subset of the requirements of the EMA Summary of Product Characteristics (SmPC)Invalid source specified.. It should be noted that the SmPC includes preclinical safety data, shelf life and Contra-indications, none of which are required by XEVMPD.
Each approved or investigational (development) product must be submitted to each country where it is marketed, in every official language of that country. The XEVPRM standard is designed as a single message (plus an acknowledgement message format from the agency) that indicates items to be registered, or changes to existing registered items. The types of items that can be registered or changed include:
• Approved or Development Products • Approved or Development Substances • Organisations (e.g. Sponsors, Regulatory Authorities) • Attachment Files • Master File Locations • Sources for information references • Updates to controlled vocabularies: ATC Codes, Pharmaceutical Dose Forms, and
Administration Routes
Each new item registered is sent with a “local key”, and the acknowledgement message will assign an official “EV Code” which is to be used for all future references to that item. Some items such as Attachments are meant to only be submitted, never modified – instead, a new file is submitted with its own code if an attachment such as labeling or the SmPC document is sent. The use of EV Codes enables references to substances. In other words, for the item types listed above, other parts of the XEVPRM message can just use the code, and not completely specify the entire set of data each time

6
DRUG PRODUCT REGISTRATION PROCESSES
and place where it is used.a Each place where EV Codes are used also requires indicating whether it is a local or EV-issued (called the Resolution Mode), and previous codes used by the same item.
Development and Approved Substances in EVPRM specify the following items:
• A valid reference source for the substance name • The English name of the substance • The Chemical Abstracts Service (CAS) Number (optional) • The molecular formula (optional) • The class of the substance (e.g. chemical, protein, etc.) (optional) • The Chemical Biological Description (CBD) (optional) • Translations and Aliases of the substance name • International Codes used to identify the substance • Attachments such as the Printed Substance Information • Structured Substance Information (optional)
Development and Approved Products in EVPRM specify the following items:
• Sponsor or Market Authorisation Holder information • Reference to the Qualified Person for Pharmacovigilance (QPPV) location • Contact Information • Market Authorisation, which consists of
• Country • Procedure • Status • Number • Authorisation and Withdrawal Dates • MRP Number • EU Number • Orphan Drug Status • Intensive Monitoring Status
• Presentation Name • References to ATC Codes • Indications • Attachments • Pharmaceutical Product information, consisting of
• Reference to the Pharmaceutical Form Codes • Administration Routes • Active Ingredients • Excipients (optional) • Adjuvants (optional) • Medical Devices (optional and rare, such as cell scaffolding)
The EVPRM model aligns closely with the product model in section 1.2. It includes the Pharmaceutical Product and Medicinal Product concepts, although its packaging information is scant.
a It is unfortunate that the same approach was not also applied to the XEVMPD documentation, as repeated sections such as references are described in identical detail every place they are used, increasing the size and tedium of the specification documentation.

7
DRUG PRODUCT REGISTRATION PROCESSES
As of September of 2012, EMA had received 229,401 XEVPRM messages, but had not yet acknowledged 20% of the total messages. This covered 236,827 products – much less than the 450,000 expected. Some of this is based on backlog of registration of applicants.
The EVPRM messages are sent to EMA by one of two methods: if the Market Authorisation Holder/Sponsor has capabilities to create the XML, files may be submitted via the EMA Gateway. This provides for greater efficiency, but requires more technical expertise on the part of the drug’s owner. Otherwise, the EVPRM messages may be created – and eventually maintained – through the EVWEB interface, a web-based application that permits entry of all information in the EVPRM data structure. This can be tedious for creating multiple records but requires little technical expertise. EMA offers an online course for using EVWEB as a series of videos on YouTube, and in fact requires the course to be taken before certifying staff for use of the system.Invalid source specified. An area which has made penetration of the service market for EVPRM difficult is that software and service vendors are not granted access to the production or test EVWEB or Gateway systems except on behalf of a market authorisation holder. As of the most recent Article 57 EMA meeting, EMA has stated that they will reconsider this position.
One of the challenges of the XEVMPD system is that EMA has spent a lot of time and effort getting compliance with the initial submission, but as of this writing has not yet completed guidance on updates to drug product information. For instance, they have stated that most updates should be done within 30 days of a change to the approved product (close of the procedure). However, they have not stated if it is necessary to send all changes or only those that require prior approval or waiting period processes, or also those that use the “do and tell” process. Those changes that are ordinarily immediately implemented and communicated to the agency within twelve months may end up requiring a more-immediate XEVMPD submission.Invalid source specified. Formal guidance on maintenance of data is not expected until March or April of 2013.
2.3 Identification of Medicinal Products (IDMP) The IDMP standard began in 2003, with a primary goal to align it as a set of identifiers for use with the ICSR (Individual Case Safety Report) standard for safety reporting. It was approved as a set of ISO standards in November of 2012:
• ISO 11238:2012: Health informatics — Identification of medicinal products — Data elements and structures for the unique identification and exchange of regulated information on substances Invalid source specified.
• ISO 11239:2012: Health informatics — Identification of medicinal products — Data elements and structures for the unique identification and exchange of regulated information on pharmaceutical dose forms, units of presentation, routes of administration and packagingInvalid source specified.
• ISO 11240 2012: Health informatics — Identification of medicinal products — Data elements and structures for the unique identification and exchange of units of measurement Invalid source specified.
• ISO 11615:2012: Health informatics — Identification of medicinal products — Data elements and structures for the unique identification and exchange of regulated medicinal product informationInvalid source specified.
• ISO 11616:2012: Health informatics — Identification of medicinal products — Data elements and structures for unique identification and exchange of regulated pharmaceutical product information Invalid source specified.
For the sake of brevity, these will be referred to throughout this document without the “2012” suffix, which only indicates the year of approval.

8
DRUG PRODUCT REGISTRATION PROCESSES
The standards were developed as an expansion of the Common Product Model at Health Level Seven (HL7). This builds on the technology and experience of the SPL standard, and the Common Product Model is used at the heart of many health standards to describe medicinal products.
The five standards build from basic concepts up through the full Medicinal Product definition. ISO 11239 is essential for understanding the concepts of dosage forms, units of presentation, routes of administration, and packaging, which are used in the ISO 11615 standard. ISO 11240 specifies the methods of communicating units of measurement, used across the rest of the standards in the set. ISO 11238 is a detailed definition of how to describe a substance (active or inactive) that is used in making up a medicinal product, from its chemical structure to its methods of manufacture, and is referenced in ISO 11615 and 11616. ISO 11616 is relatively brief: it covers only a set of identifiers used to describe the pharmaceutical product, which is the administrable form of the medicinal product. Lastly, ISO 11615 is the over-arching specification for the medicinal product, which references all of the other standards.
The primary purpose of IDMP is to create the authoritative identifiers for a medicinal product within a given regulatory authority’s jurisdiction. The specific case it was created for is the ICSR standard created by HL7, approved as ISO 27953-2:2011Invalid source specified. and implemented by ICHInvalid source specified.. By having a definitive identifier for a packaged product within each participating country, it should be possible to trace patterns in adverse drug experiences and gather statistics on where labeling should be changed, or products withdrawn. Because ICSR was approved before IDMP was completed, ICSR was implemented without requiring an IDMP identifier in their messages. After IDMP is implemented, ICSR will likely change to mandate the use of the Medicinal Product Identifier (MPID – see below).Invalid source specified.
2.3.1 UNITS OF MEASUREMENT (ISO 11240) ISO 11240 builds on the earlier ISO Guide 99:2007, which describes the SI (International System) of units, and creates data structures for communicating values and units for electronic messaging.Invalid source specified. Distinct in the ISO 11240 methodology is splitting the unit type (e.g. “grams”) from the scale (e.g. “milli”), so instead of just measuring in “mg”, the standard specifies “M” and “G” as separate terms.
There are three additional concepts crucial to measurements for medicinal products:
1. Ratios of measurements. This is typically in the form of a numerator quantity per denominator units, e.g. 5 mg/ml, which should technically be 5 mg per 1 ml.
2. Units of Presentation. For many dosage forms, the form itself is a single object, which means that the strength of a product might be expressed as “per tablet” or “per spray.” This is the presentation unit, and is an used instead of the denominator unit mentioned above.
3. Reference measurements. Some products are formulated as, for instance, a salt of a drug substance. In cases such as this, the measurement may be expressed both as the absolute (e.g. 300mg of Lithium carbonate), and its reference substance (12.2 mmol lithium). There are other reference measurements such as a particular measurement procedure.
These concepts are critical underpinnings of the Substance (11238) and Medicinal Product (11615) portions of the standard. The EVMPD uses a very similar data structure for measurements, but does not include the reference measurements as part of the standard data structure. The SPL R4 Drug Listings do not use the above means of specifying units or reference measurements.
2.3.2 ADMINISTRATION AND DOSAGE FORM TERMINOLOGY (ISO 11239) The Product definitions describe the packaged and consumed items, but additional information is needed to describe how a product is prepared for use, and how it is actually used. This is as simple

9
DRUG PRODUCT REGISTRATION PROCESSES
as a tablet, or as complicated as a powder and solution which must be combined, placed in a syringe, and then injected in a particular location.
The primary dose form information is a coded value, which will be supplied as a controlled vocabulary as part of the ICH Implementation Guide. The dictionary for dose forms is defined in ISO 11239 to further classify each value with a Basic Dose Form (e.g. “Tablet” versus “Extended Release Tablet”), Release Characteristics (the “Extended Release” portion), Transformation (e.g. dissolution of the powder in a solvent for a “Powder and Solvent for Injection”), Intended Site (e.g. “intramuscular” for an injection dose form), and Administration Method (e.g. “injection”). Not all dose forms will use all five modifiers. Because existing standards of SPL and XEVMPD have different dictionaries for dosage form and administration method, these modifiers will assist in mapping one IDMP to many XEVMPD or SPL forms, or one SPL or XEVMPD to many IDMP forms.
In addition, there are combined dose forms, where multiple dose forms listed above are transformed into a single administrable form For example, a powder may be packaged with a vial of solvent, to be combined as a “powder and solvent for solution for injection.”
It also defines the structures for the packaging class, which includes a container, its closure, and administration devices. At a minimum, there is a Packaging Category (a coded value), but it can be subdefined by one or more of the closure (“screw-cap”, “stopper”), container (“box”, “ampoule”) and administration device (“needle”, “oral syringe”) categories.
ISO 11239 also defines the controlled vocabulary that will be created for units of presentation, and the Route of Administration controlled vocabulary structure. Invalid source specified.
2.3.3 SUBSTANCE DEFINITION (ISO 11238) Unique in the ISO IDMP standards, 11238 does not have an approved message format that is already in use: There is no existing standard for substance specification. The XEVPRM format originally included one, but it was made optional: not only was it removed from the most recent guidance, to this author’s knowledge, it has not been implemented by drug sponsors. The US drug listing sections require nothing more than the identification of the substances, not a description of them, using the Unique Ingredient Identifier (UNII). In the US, the existing Substance Registration System consists of data entered by National Library of Medicine staff, based on drug submissions – it’s a system designed for use by experts to exactly match their specificationsInvalid source specified., which can then be downloaded from the NLM website.Invalid source specified. From this system are issued the Unique Ingredient Identifiers.
It is anticipated that the NLM system will become the basis for the international Substance Registration System used by IDMP, with the difference that the data will be supplied by the drug sponsor. It is not clear who is responsible for substances off patent or for common, inactive substances (water, corn starch and silicon dioxide, for instance). The current systems at the US National Library of Medicine used for maintaining the UNII identifiers are proprietary, and would do not provide information in publicly available formats. The UNII provides a unique, easy to validate identifier, and is likely to remain the ISO 11238 identifierb – although its name may change. A key benefit of the centralization of substance IDs will be that companies will have an alternative to the expensive commercial registration of a CAS Number.
Currently, countries may maintain their own substance systems. Germany has a system with 41,000 unique substances and 182,000 names. The US system is of a similar size (50,000 compounds), but not all items are the same, due to the substances which are regulated, and local pharmacopeias. b It is interesting to note that Wikipedia has chosen the UNII for tagging medicinal substances.

10
DRUG PRODUCT REGISTRATION PROCESSES
ISO 11238 specifies the following information be provided for each active or inactive substance. Invalid source specified.
• For all substances
• A Substance ID will always be listed. Without an implementation guide, it is unknown how new substances will get identified. It is likely a mechanism similar to the EVMPD “local key” will be used.
• Some substances may be subclassed into Specified Substances, indicating substances not easily otherwise defined, or specific versions of a substanceInvalid source specified.. Specified substances may be specific to a regional authority, such as a country’s pharmacopaeia’s registry. Note that Specified Substances are not yet part of the US SRS.
• Group-1: Multiple-substance materials, such as allergenic and herbal extracts • Group-2: Manufacturer and minimal manufacturing information • Group-3: Graded materials (US Pharmacopeia, European Pharmacopeia, technical,
standardized substances) • Group-4: Detailed manufacturing information
If a substance is described as a Specified Substance, it will have a Parent Substance ID – Group 4 can have a parent of any of the others including a non-specified substance, Groups 2 and 3 can have a parent of Group 1 or a non-specified substance, and Group 1’s parent would only be a non-specified substance. In some cases, the use of a specific specified substance is critical: The source of a starch, for instance corn versus potato, could affect the allergenicity of a product. Product manufacturers should be encouraged to use as specific a substance as possible.
• Detailed Name information is provided, including the official names specific to each jurisdiction (in other words translations), and reference sources for each name (e.g. US Pharmacopaeia)
• Structure information, including optical activity, molecular formula, attachments further describing the structure, and isotope information for radiolabeled compounds indicating what atoms are substituted with which nuclides.
• Where needed, Modification information, which describes how a substance is changed from a basic form. This can include any number of modifications, which may be physical, structural, or agent modification.
• Where needed, Reference information which includes classification of the substance, gene or gene elements that are the source of the material, and the target gene to be affected, and relationships with other substances. Each of these items may have a reference source.
• Source Materials, optionally indicating the organism (and specific parts or fractions) from which the material is obtained
• For Chemical Substances
• Stoichiometric Information • Comments • Properties (type, name, ID, reference substance name and amount) • Moieties (Role, ID, Name, and Amount) • Reference Sources
• For Protein Substances
• Protein Sequence Type • Subunits (sequence, length, terminal modifications) • Comments • Disulfide Linkages

11
DRUG PRODUCT REGISTRATION PROCESSES
• Modifications, if any • Molecular weight (may be multiple if different methods are used) • Properties • Glycosylations (of N, O and C type) • Reference Sources
• For Nucleic Acid Substances (DNA, RNA)
• Nucleic Acid Sequence Type • Subunits (sequence, length, terminal modifications) • Comments • Molecular weight (again, may have multiple if different methods are used) • Modifications, if any • Properties • Reference Sources
• Polymer Substances
• Polymer Class (e.g. homopolymer, copolymer) • Polymer Geometry (e.g. linear, branched, network) • Sequence type (random, block, alternating) • Comments • Monomers (ID, name, amount) • Structural Repeats (orientation of polymerization, degree of polymerization) • Molecular Weights • Source Materials, optionally • Modifications, if any • Reference Sources • Properties
• Structurally Diverse Substances (e.g. extracts from biological sources that cannot be enumerated, such as orange oil, cells and tissues)
• Comments • Molecular Weights • Source Materials • Modifications, if any • Glycosylations • Properties • Reference Sources
• Mixture Substances – These are essentially just lists of other substances
• Two or more Constituents (ID, Name, Requirement) • Mixture Type • Source Materials • Modifications
• Specified Substances Group 1
• Constituents (Substance ID, Name, Role, Amount) • Properties • Physical Form (State, Form-Type, Amount) • Reference Sources
• Specified Substances Group 2
• Parent Substance

12
DRUG PRODUCT REGISTRATION PROCESSES
• Manufacturing (Manufacturer ID and Name, Type, Production Method and System, Critical Process Version Number, Reference Sources)
• Specified Substances Group 3
• Parent Substance • Grade (Type, Name, Reference Source)
• Specified Substances Group 4
• Parent Substances • Grade (Type, Name, Reference Source) • Analytical Data (Specifications, jurisdictions for a specification, analytical method,
values, reference sources) • Constituents (Substance ID, Name, Role, Amount) • Properties • Physical Forms (State, Form-type, Amount) • Manufacturing (more detailed than Group 3, includes resultant materials, processing
materials, starting materials, processing equipment) • Reference Sources
How that substance is used is specified in the product: for example, Dextrose may be an inactive ingredient in a tablet, but the active substance in intravenous nutrition.
2.3.4 PHARMACEUTICAL PRODUCT ID SETS (ISO 11616) ISO 11616 is relatively brief, defining a sub-structure that is part of the ISO 11615 Medicinal Product. It weaves the concepts of the substances and specified substances of 11238 plus the dosage forms of 11239 and the units of measurement and reference strength of 11240. This set of identifiers is used to describe the administrable product form, after any reconstitution or transformation of the Medicinal Product.
A single Pharmaceutical Product ID Set (PhPID Set) will be described in terms of a set of active ingredients: either substances or specified substances, with an optional device (e.g. a cell substrate framework), and sub-IDs for the product with strength and unit of presentation, the administrable dose form, and both the dose form and strength. Each of the sub-IDs includes a set of substances, each with a role (active, inactive, adjuvant), and a reference strength.
The PhPIDs are generic for a given clinically equivalent product: product alone, product and dosage form; product and strength; and product, dosage form and strength. This enables tracking safety issues for, for instance, 325 mg aspirin tablets, aspirin of unknown dose, or aspirin of 325 mg of unknown dosage form, etc., all regardless of brand.Invalid source specified. It would be expected that all aspirin tablets would include two common PhPID in their sets (aspirin, aspirin tablet); all 325 mg dosage forms would have two common IDs (aspirin, aspirin 325 mg regardless of whether it’s a tablet, a capsule, or a gelcap) and so on. A similar concept exists in previous standards: The SNOMED Clinical Terms document includes a “Non-Proprietary Medicinal Preparation”Invalid source specified., and The US National Library of Medicine’s RxNorm has the concepts of Semantic Clinical Drug (SCD), Semantic Clinical Drug Form and Semantic Clinical Drug Component.Invalid source specified.
It is still not clear why each of these PhPIDs must carry their own sets of ingredients – it would be just as effective to reference a common set of ingredients for the common Pharmaceutical Product.
2.3.5 MEDICINAL PRODUCT DEFINITION (ISO 11615) The overall medicinal product in ISO 11615 combines all the concepts of the other four standards documents. Corresponding to the product model concepts presented in section 1.2, an approved

13
DRUG PRODUCT REGISTRATION PROCESSES
medicinal product (or investigational product approved for clinical trials) would be assigned a Medicinal Product Identifier (MPID) or Investigational Medicinal Product Identifier (IMPID)c. Each packaging type (e.g. a 20-capsule bottle versus a 50-capsule bottle) would receive a unique (Investigational) Medical Product Package Identifier PCID (or IPICID).
Each batch of the outermost packaging, and each batch of any intermediate packaging would receive Medicinal Product Batch Identifiers (BAID_1 and BAID_2 respectively, with IBAID_1 and IBAID_2 for investigational batches). From any of those identifiers, the product can be immediately traced to determine its name, its marketing status, which Regulatory Authority authorized it, its composition (manufacturing, ingredients, dosage form, etc.), adverse experiences and Contra-indications, and other documentation.Invalid source specified. Currently, only biological products marketed in the US have batch IDs submitted to the FDA. It is unclear whether this would be expanded beyond biologicals.
The product information is organized hierarchically in two ways: For packaged items, the data is organized from the outermost to innermost contents, down to their ingredients. For the pharmaceutical product that is actually consumed, data is organized from the administrable form down to the items that make up that form – those will be the same items as those specified in packages, although no linkage is provided between the two sets of of structures..
For the identification of a medicinal product, a change to any of the following items would constitute a different identifier:
• Product Name, Brand and Description • Formulation (active ingredients) • Strength • Dosage or Usage • Net Quantity (weight, volume or other dimension in a package) • Packaging Configuration, including sub-packagingInvalid source specified.
The MPID must indicate the ISO country code, market authorisation holder and the medicinal product code. It is expected that existing local identifiers will continue, so the MPID for a product approved in the US would have a code of US + the market authorisation holder’s DUNS number + the product’s National Drug Code (NDC) number. Other jurisdictions have their own IDs that will be repurposed for the MPID including the RVG in the Netherlands and the EU Number for European centrally-approved products. New numbers would be assigned for situations such as changes to the strain used in each season’s flu vaccine, for a change to “over-the-counter” drug sales, in addition to any physical changes to the drug or its packaging.
The rest of the identifiers will be prefixed with the MPID, then append a package code, and for batch IDs, a specific batch identifier.
The overall data structure is indicated in Figure 1. This shows the relationship between the Medicinal Product itself, the names it has, the versions (this and any previous), manufacturing information, clinical particulars (Indications, Contra-indications and Adverse Events), pharmaceutical products, packaging, and market authorisations for the product and the packaged product. The detailed structure can be found in Appendix B-2.
c It’s unknown why ISO and HL7 chose a completely separate structure for Investigational Medicinal Products, as there are few differences between them except for a few fields required in one but not the other. Note that EMA followed that lead with the EVPRM model also.

14
DRUG PRODUCT REGISTRATION PROCESSES
Figure 1 – IDMP Data Model Source: ISO Standard 11615:2012Invalid source specified.
The following subsections describe each of the boxes in detail, starting with the Medicinal Product itself and moving clockwise from the Medicinal Product Name.
2.3.5.1 Medicinal Product The root object of the standard, the Medicinal Product, provides the broadest identification of the product:
MPID (IMPID) This is the identifier for the product or investigational product. The standard specifies that the ID should always be provided, but does not specify what should be done on an initial filing, before the ID has been issued. For the IMPID, the number may include a regulator- or sponsor-provided code, depending on the jurisdiction.
Combined Pharmaceutical Dose Form
This is an optional field used only for combined products, such as “Powder and solvent for solution for injection.”
IMPID/MPID Cross Reference
The IMPID Cross Reference is used in non-investigational products, to refer to a previous investigational ID for the product. An Investigational product may have an MPID Cross Reference if being investigated in a different formulation, packaging or indication than the authorized product.
Sponsor Product Code This is found only in Investigational products. Either this or the Regulatory Product Code will be specified
Regulatory Product Code This is found only in Investigational products. Either this or the Sponsor Product Code will be specified.

15
DRUG PRODUCT REGISTRATION PROCESSES
Paediatric Use Indicator This is found only in Investigational products, indicating whether this is a specific paediatric formulation.
Additional Monitoring Indicator
This is a safety monitoring term used in the EU, such as “Black Triangle”
Special Measures This is an optional field used to describe any additional steps that are still required as part of approval, such as “Requirement to conduct post-authorization studies.” This is not found in Investigational Products
Medicinal Product Classification
One or more classification terms may be provided, listing both a code system and value, such as ATC (World Health Organization’s Anatomical Therapeutic Chemical system) DEA Schedule, etc. It is expected that ATC code will be required.
The Medicinal Product should not provide much difficulty to assemble. Except for the “chicken-and-egg” problem of getting the initial MPID, this all seems clear. The implementation guide is likely to specify a system similar to XEVMPD, where a ‘local key’ is provided and flagged as such, until the official number is issued, but the standard does not indicate any means of linking to an earlier local value the way XEVPRM does. This will likely be specified in the ICH implementation guide.
Note that many portions of the Medicinal Product structure have a near-duplicate structure for Investigational Medicinal Products. In implementation in a database, it may be more effective to have a single structure that can accommodate both the Investigational and Authorized Medicinal Products, with record-level validation to specify which fields are valid for Investigational versus Authorized products.
2.3.5.2 Medicinal Product Name The Medicinal Product Name object permits multiple names to be listed for the product. There must be at least one. Each name may be associated with one or more countries and languages.
Name This is the full name of the product as it is labeled. This may include a number of components, as described below. The rest of the terms are optional “Parts” of the whole approved name, used to assist in positively identifying variations on a product. Examples are given below. For an investigational product, a code may be present instead of a name.
Code This is only found in Investigational Products if the product is only referred to as a code instead of a name.
Invented Name Part The brand or trade name. Typically either this or the scientific name will always exist
Scientific Name Part The generic name, typically the INN. Strength Part Example: “Extra-Strength” Pharmaceutical Dose Form Part
Example: “Easy-Swallow Gelcaps”
Formulation Part Example: “Sugar-Free” Intended Use Part Example: “Arthritis Pain Formula” Target Population Part Example: “Children” (as in “Cough Syrup for Children”) Container or Pack Part Example: “Multidose Vial” Device Part Example: “Inhalatron” for an inhaler

16
DRUG PRODUCT REGISTRATION PROCESSES
Trademark or Company Name
The company name if it appears in the labeled name
Time / Period Part Example: “2012-2013” (as in ”Myvaccine Flu season 2012-2013”
Flavour Part Example: “Cherry” Country / Language Includes the ISO codes for country, sub-country jurisdictions,
and language, for example Canada / Quebec / French. Again, this should not be a difficult set of information to manage. EVMPD uses a similar, though less extensive structure.
2.3.5.3 Version One or more version sub-objects can be specified that list the date of changes to the IDMP message and one or more regulated documents associated with that version. Jurisdictions may use this to provide the approval letter, Summary of Product Characteristics, etc. A key factor is that a new version must not be a change that would cause the creation of a new Medicinal Product, e.g. the marketing authorization, legal status of supply, name, dose form, ingredients, strength, device provided and indications.
Version Date Date at which the change occurred Version Identifier A text identifier Regulated Document
Document Type Document Identifier Regulated Document Document Effective Date
Associated documents, such as SmPC, Package Leaflet, Approval Letter, Content of Labeling, etc. The information provided includes the type, an identifier for the document, the document itself as an attachment, and the time period when the document became effective.
It will be interesting to see how much Version data must be provided for a product which has been on the market for some time, or if the regulatory authorities will only require the current version with the first IDMP submission. Another question will be whether documents need to be provided with each update to the IDMP message, or if they can be sent and registered once and kept “on file.” The XEVMPD solves this by having the documents be a separate category of objects with their own identifiers, and the product parts of the message only refer to the identifier of the document.
2.3.5.4 Manufacturer The manufacturer information corresponds relatively closely to the US Establishment Registration, specifying locations, the operations they perform, and how those establishments were authorized for those operations. This information is optional for Investigational products.

17
DRUG PRODUCT REGISTRATION PROCESSES
Manufacturer / Establishment
Identifier Name Address Confidentiality Indicator Contact Person
Name Telecom Role Confidentiality Indicator
Other Locations Location Address Location Role Identifier
This includes an identifier (which may be an international ID such as DUNS), name, address, confidentiality, contact names and telecommunications codes (email, phone), and other addresses and their roles (e.g. Batch Release, Qualified Person for Pharmacovigilance [QPPV]).
Manufacturing Operation Operation Type Manufacturing Authorisation Reference Number Effective Date Confidentiality Indicator Medicines Regulatory Agency
One or more operations at the particular establishment. Operation Type may be items such as Batch Release, Re-Packaging, Re-Labelling, Manufacture, Testing. The Authorisation Reference Number, Date and Agency reflect the authorisation to perform the operation.
This information should provide little stress to drug sponsors: It’s essentially similar to what is provided for the US Drug Listing / Establishment Registration, although this information is not required for XEVMPD.
2.3.5.5 Marketing or Clinical Trial Authorisation The Marketing Authorisation is a more complex structure than the items above, containing sub-objects for periodic reporting, marketing status, and marketing authorisation procedures and applications. For Investigational Products, this is a clinical authorisation with slightly different information.
Marketing Authorisation Number
The ID number of the authorisation, which becomes part of the MPID. This may be a licensing number if there is no authorisation number (e.g. a grandfathered product)
Country One or more countries where the authorisation applies. Legal Status of Supply This indicates the type of marketability, e.g. prescription or
over-the-counter. Only for authorized products. Authorisation Status Status of the product, e.g. Active/Valid, Expired, Renewed,
Not Renewed, Withdrawn. Only for authorized products. Authorisation Status Date The date at which the status changed Validity Period Start and end date of the authorisation. Marketing Authorisation Holder
This is the information about the organization that is marketing the product, or in some cases the distributor. This is the same information provided for the Manufacturer/Establishment above.

18
DRUG PRODUCT REGISTRATION PROCESSES
Medicines Regulatory Agency
This is the information about the organization that has authorized the product. The same organizational information for the Manufacturer/Establishment applies here too.
Marketing Status Country Jurisdiction Marketing Authorisation Number Marketing Date (Start/Stop) Jurisidictional Legal Stats of Supply
This structure, while similar to the Marketing Authorisation, reports on when the product is actually marketed in each jurisdiction within the authorizing country if needed.
Marketing Authorisation Procedure
Procedure Identifier Procedure Type Procedure Date Marketing Authorisation Application
Application Identifier Application Type Application Date
If procedures are used (such as EU Centralized, Mutual Recognition and Decentralized Procedures), include the information about it here. It seems likely this may be used for other health authorities, as the Marketing Authorisation Application structure is within it, and would otherwise not be possible to add for a US application without this structure.
Periodic Safety Update Report Submission
Frequency of Submission of Safety Report Dates of Submission International Birth Date Reference Date Data Lock Point
This information specifies when Safety Update and Product Benefit-Risk Evaluation Reports must be filed. The frequency may be complex, e.g. every six months during the first two years and once a year for the next two years.
The information for a Clinical Trial Authorisation is somewhat different:
Registration Number The ID number of the clinical trial registration, which becomes part of the IMPID.
Investigation Code This is the code for a particular trial. Country One or more countries where the authorisation applies. Protocol Number The number assigned to the clinical trial protocol. Authorisation Date The date at which the clinical trial authorisation was granted Anticipated End Date The date when the clinical trial is anticipated to be completed Sponsor This is the information about the organization that is the
sponsor to which the authorisation has been granted. This is the same information as a Manufacturer/Establishment above.
Medicines Regulatory Agency
This is the information about the organization that has authorized the clinical trial. The same organizational information for the Manufacturer/Establishment applies here too.

19
DRUG PRODUCT REGISTRATION PROCESSES
Local Clinical Trial Authorisation
Local Clinical Trial Registration Number Local Investigation Code Jurisdiction Local Authorisation Date Local Anticipated End Date
This information is used for countries within a jurisdiction, such as countries within the European Union, and provides equivalent information to the overall Clinical Trial Authorisation.
Drug Safety Update Report Submission
Frequency of Submission of DSUR Dates of Submission Development International Birth Date Reference Date Data Lock Point
This information is essentially the same as the authorized product’s Periodic Safety Update Report Submission information, but for pre-authorization safety update reports.
Although the information may be complex, it should be possible to compile this information without extensive effort. The only factors making this difficult is that such information previously would be maintained by local market companies, and not necessarily provided to a central regulatory database.
2.3.5.6 Packaged Medicinal Product The Packaged Medicinal Product is something of a new concept compared to previous standards, which were more concerned with the sum of the parts than the individual pieces. Here, the product is described from outer package to inner, including their components, as it is sold or supplied. The packaged product has batch identifiers listing what has shipped, and containers which may have sub-containers. This information is optional for investigational products.
PCID / IPCID This is the overall identifier for the product as packaged. Each different package (quantity, packaging material) will have its own ID. The IPCID is for Investigational Product Packages
Package Description Text Description Marketing Authorisation Same structure as the authorisation for the product – this
may only be needed in situations where different packages have different authorisation requirements.
Batch Identifier BAID_1 / IBAID_1 BAID_2 / IBAID_2
This is the list of batch IDs for the innermost and outer packaging. The “I” versions are for batches of Investigational Products
Package Item A container of either other containers or manufactured products. See below for the full structure.
The Package Item is a complex structure in its own right. This can either be a single item, a package of different items, or a set of the same item. Each contaner may have subcontainers, and at the bottom level, manufactured items. In addition, each level may have its own storage conditions, its

20
DRUG PRODUCT REGISTRATION PROCESSES
own identifiers, packaging materials, and an application or integrated device. The manufactured items are where the individual ingredients are listed.
Package Item Type This is a coded item indicating values such as box, vial, pre-filled syringe, etc.
Package Item Quantity This is the number of this kind of item in the package enclosing it. The top-level Package Item always has a quantity of 1.
Material This is the substance that makes up the package, using an ISO 11238-registered material (e.g. cardboard).
Alternate Material If multiple materials are permitted for the package (e.g. plastic for a blister pack), they should also be listed.
Physical Characteristics Height, Width, Depth, Weight, Nominal Volume, External Diameter, Shape, Colour, Imprint, Image. Images are provided as attachments.
Other Characteristics Lists a code system and value for any other recorded characteristics
Shelf Life / Storage Shelf Life Type Shelf Life Time Period Special Precautions for Storage
Multiple shelf life periods may be listed for types of “Unopened”, “After first opening”, “After dissolution” etc. The special precautions are coded information including temperature, light and humidity descriptions. The time period will include a number and units (e.g. 3 years).
Data Carrier Identifier Code System Value
These are additional identifiers that may be on this particular level of product, such as a GTIN barcode.
Package (Component) Component Type Component Material Component Alternate Material Physical Characteristics
This is used for components of the packaging itself, not components of the product. Examples include closures, vials, etc.

21
DRUG PRODUCT REGISTRATION PROCESSES
Device Device Type Device Trade Name Device Quantity Device Listing Number Model Number Device Material Device Alternate Material Allergenicity Indicator Sterility Indicator Sterilisation Requirement Indicator Device Usage Device Nomenclature
Nomenclature System Nomenclature Value
Device Batch Identification
Batch Number Expiration Date
Physical Characteristics Other Characteristics Shelf Life / Storage Manufacturer
Devices will either be Administration (e.g. droppers or oral syringes), Integrated (pre-filled syringe), Medical Device (cell scaffold) or other (e.g. swabs) typeInvalid source specified.. If there is a trade name, it should be listed. The quantity is always listed, even if 1. Listing and model numbers should be listed where applicable. Materials, like package materials, should be coded as ISO 11238 substances. Allergenicity would be listed when it is a material of concern such as latex. The Sterility Indicator will specify both whether the device is provided as sterile, and whether it must be sterilized before use. Usage indicates the number of times the device may be used, e.g. “For single use only” Nomenclature may include any means of identifying the device, but should include the Global Medical Device Nomenclature (GMDN), which is ISO 15225, and the Unique Device Identification System (UDI). Device Batches may be listed where applicable, including their expiration date and shelf life. Devices may also have physical and other characteristics, and have a manufacturer similar to that of the product itself.
Manufactured Item Manufactured Dose Form Unit of Presentation Manufactured Item Quantity Physical Characteristics Other Characteristics Ingredient
This is the actual manufactured drug product, or one of them in the case of a combination product such as a pill plus external use ointment, powder plus solvent, or multiple antibiotics in one blister, etc. How they are combined is indicated in the overall pharmaceutical dose form, and in the Pharmaceutical Product structure below. Each item has its own manufactured dose form. For items not specified by weight or volume (in other words vials, powders, etc.), a unit of presentation (tablet, spray, etc.) should be listed. The quantity is the number of items of this type in the container. Physical and other characteristics may be specified here also. The ingredient structure is described below,
The ingredient consists of either a substance or a specified substance, strength and optionally a reference strength. Its structure is used in the same way for both Manufactured Items and the Pharmaceutical Product (which is the combined set from all Manufactured Items).
Ingredient Role The role indicates whether the ingredient is active, inactive or adjuvant.

22
DRUG PRODUCT REGISTRATION PROCESSES
Substance or Specified Substance
Substance Specified Substance Specified Substance Group Confidentiality Indicator Strength
Strength Range Presentation Strength Range Concentration Measurement Point Country Reference Strength
Substance Specified Substance Strength Range Measurement Point Country
The substance or specified substance will be listed as per ISO 11238 substance identifiers, 11239 units of presentation and 11240 units of measurement. The substance group and confidentiality indicator only apply to Specified Substances. Strength can be expressed as a range if needed (high, low and average), and a numerator and denominator each with its own units is included (for example, 5mg/ml is really 5 mg per 1 ml). Strength of presentation is the strength as sold/supplied. Strength concentration is as administered. So if a 10mg envelope of powder is to be dissolved in 20ml of solvent, it would be expressed as 10mg/20 ml for presentation, and .5 ml / 1 ml for concentration. Concentration strength is only needed if the denominator of the presentation is non-unary – thus it is not needed for solid dosage forms. The measurement point may be needed for items such as distance from an inhaler, per certain jurisdictions. The country is used only with a measurement point. The reference strength may be needed if a medicinal product is measured per another product. The example given in the standard is 300mg Lithium Carbonate, which is equivalent to 12.2 mmol of lithium.Invalid source specified.. The same features apply to the reference strength as to the strength.
It is anticipated that the level of detail for packaged items will be very complex to represent, report and compile for submission to agencies. In particular, currently the only jurisdiction that requires reporting of batch identifiers is Germany’s BfARM, and only for biological medicines – this may prove to be a large burden for manufacturers.
Strength is currently only required for the active ingredient. FDA is suggesting that the strength (quantity) of an inactive ingredient be supplied, but would be marked confidential for public databases.
2.3.5.7 Pharmaceutical Product The pharmaceutical product contains the sum of the ingredients in all the manufactured items in each manufactured product (note that if there are multiple pharmaceutical products such as a tablet plus topical cream, each has its own ingredient list), Routes of Administration, PhPID sets, Characteristics, and optionally Devices – similar to the Device model in the Packaged Product, differing only in the lack of Shelf Life / Storage and Manufacturer information.
Administrable Dose Form This is the dosage form as consumed/applied to the patient or subject. It is frequently the same as the medicinal product, except where a transformation (e.g. dissolution of a powder) is specified
Unit of Presentation This is used when the product is not a measured weight or volume. Examples include tablet, spray, actuation, etc. For a vial or bottle, it is the container, not necessarily the single dose volume or weight.
Pharmaceutical Product Quantity
Expressed as a value and a unit, this is typically “1 unit” for solid dose forms. For items administered by weight or volume, this would be the amount and units, e.g. “10 ml”.

23
DRUG PRODUCT REGISTRATION PROCESSES
(Dosing and) Route of Administration
Route of Administration First dose in humans Maximum single dose Maximum dose per day Maximum dose per treatment period Maximunm treatment period
One or more terms for how the medicinal product is applied. For authorized products, this is only the route of administration. For investigational products it includes the full set of information about how the trial can be conducted
PhPID Set One or more PhPIDs as described in ISO 11616. There can be multiple sets, but it is not clear why one Pharmaceutical Product would have multiple PhPID sets.
Pharmaceutical Product Characteristics
These are code systems and values for the pharmaceutical product, as required by the regulatory authority (not specified in the standard)
Ingredients Described as in the previous section Device Described as in the previous section, but the Shelf
Life/Storage and Manufacturer sections are not included. For a Pharmaceutical Product, devices are only included where the device is part of the applied/consumed product – essentially an ingredient – and not the means of delivering it (like a syringe). The device most frequently described for this situation is cell scaffolding for a biologic product.
Except for the rare cases of the device, this is relatively straightforward. There are some annoyances to maintaining the ingredients both for the manufactured items and the pharmaceutical product, but again this is only significant if there are differences between the manufactured and pharmaceutical products – combination packs or items that must be transformed in some way. Most products should be straightforward to construct.
It is curious why the dosing information is not required for authorized products, since it would still be part of the package label in most countries.
What may be missing from this standard is a hard linkage between the manufactured products and the pharmaceutical product it makes up. While it’s pretty clear to a human being that a “powder and solvent for solution for injection” contains a powder and a vial of solvent, a (theoretical) package that has two solvents and two powders isn’t clearly identified as which one goes with which. In any case, figuring out which manufactured products are the result of the pharmaceutical product may be a non-trivial task for a computerized system to calculate.
2.3.5.8 Clinical Particulars Clinical particulars are the reasons why or why not to be using this product, and any expected adverse experiences and interactions. It is a more detailed structure than the XEVMPD requirements, and XEVMPD only details indications, not adverse experiences, interactions or Contra-indications. The DLER does not include clinical particulars, but they are included in the SPL Content of Labeling. The standard indicates that all of these categories of information may be rendered as optional by a given jurisdiction. The Clinical Particulars consist of the following information:

24
DRUG PRODUCT REGISTRATION PROCESSES
Therapeutic Indication Indication Text Indication Disease Status Co-morbitity Intended Effect Timing/Duration Population Specifics
Age Age Range Gender Race Health Status
Other Therapy Specifics Relationship Type Medication
This should list the authorized indications for use of the product. A textual description, as well as a disease/symptom/procedure coded by controlled terms should be provided. The Disease Status (e.g. “chronic” or “acute”), Co-morbidity (other disease present at the same time, e.g. “due to cystic fibrosis”), Intended Effect (“prophylaxis”, “maintenance treatment” etc.) and Timing/duration (“for less than six months”) are optional fields used as needed. Population Specifics describe for whom an indication permits or a contra-indication prohibits the use of a product. The Age is a controlled term (e.g. “elderly” or “adolescents”) while the Age Range is a numeric range. Gender is a coded value, as is race and health status. The Other Therapy Specifics specify how a product would be used in combination with other medicinal products or therapies, e.g. “prior to treatment with DMARDs” or “second line treatment with steroids”. The Relationship type is the “prior to” and “second line” portions, and the medication is specified as a substance, medicinal product, or class of products.
Contra-indication Contra-indication Text Contra-indications Disease Status Co-Morbitity Population Specifics Other Therapy Specifics
The Contra-indication fields are identical to those in the Indication except that they indicate the conditions which would prohibit the product from being used, e.g. “Contra-indicated in the presence of severe renal impairment.” Optional for investigational products.
Undesirable Effects Undesirable Effect Text Undesirable Effect Symptom/Condtion/Effect Classification Frequency of Occurrence
These are the adverse experiences that may be expected when using the product. Typically, the serious and common events are listed. Just as with the indications, both a text and coded symptom/condition/effect are provided. In addition, there may be classifications of the effect such as the body system involved. The frequency of occurrence is a controlled vocabulary indicating how often the event is expected. Optional for investigational products.
Interactions Interaction Text Interaction Type Interaction Effect Interaction Incidence Management Actions Interactant
Code System Value
This section describes how the product could interact with other drugs, food, etc. Optional for investigational products. The Text would be a description of the interaction. The Type is a controlled term including “Drug-Drug”, “Drug-Food”, and “Drug-Laboratory Test” The Incidence is a controlled term indicating “no interaction” through “observed interaction” The Management Actions is also controlled vocabulary indicating such things as “caution”, “dose reduction”, or “not recommended in that condition”. The Interactant indicates exactly what is interacted with, such as an ATC code for a medication, a substance ID (ISO 11238) or a MedDRA code.
Although this information seems complex, it should be relatively straightforward to implement. Whether jurisdictions will require this information is as-yet unknown.

25
DRUG PRODUCT REGISTRATION PROCESSES
The population and other therapy specifics for the Indication and Contra-indication allow for more specificity than XEVMPD or SPL currently permit. As it appears that IDMP will follow XEVMPD and use MedDRA for those clinical particulars, those specifics may help alleviate some of the difficulties that companies have had in getting MedDRA to exactly match the approved indications for a drug product. However, there is concern that the process of IDMP compliance will require the expertise of a MedDRA coder.
2.4 Commonalities and Differences The XEVPRM message format was designed in part as a stopgap until IDMP becomes available. In many areas, the two formats are very similar – if not in exact schema, then at least in the information required. The Structured Substance sections (made optional in the current version) are nearly identical in the content carried to those of the IDMP standard described below. Other areas are somewhat different, particularly in areas that are optional in IDMP, or are designed for purposes other than the Pharmacovigilance concepts of the Eudravigilance organization. For instance, Confidentiality flags, used to indicate the confidential portions of the information supplied as part of a Drug Master File (US) are set as confidential to prevent those portions from being released to the public. Another major difference is that while XEVPRM includes a Pharmaceutical Product concept, it is not a separate object with its own identifiers and no concept similar to the ISO 11616 ID Set to associate with a generic product exists.
The SPL R5 format used for Drug and Device Listings is also compatible with the IDMP standard described below, with the exception of the Substance definitions, but Release 5 is backward compatible between IDMP and SPL DLER only in that the sparse data in the DLER can be stowed in the same places as in R4 – equivalent data in IDMP is not compiled in the same places.
The following chart compares the levels of completeness of the DLER and XEVMPD standards with the IDMP requirements.
Figure 2 – Comparison of IDMP with DLER and XEVMPD See Appendix A for tables showing the detailed differences between SPL DLER and Content of Labeling, XEVMPD, IDMP and the TRS Tracker Database.
0 20 40 60 80 100
Medicinal Product / Name
Manufacturing
Market Authorisation
Packaging
Pharmaceutical Product
Clinical Particulars
Substances
IDMP
XEVMPD
SPL/DLER

26
DRUG PRODUCT REGISTRATION PROCESSES
2.4.1 COMPARING SPL DLER AND IDMP The SPL Drug Listing and Establishment Registration is, compared to IDMP, just a series of broad strokes without any of the detail. The DLER does not include any specifications for substance at all (although it does reference the US UNII codes for substances). However, the US Substance Registration System currently carries a fair portion of the 11238 detail – it’s difficult to tell how much, since it is not a message format, only a web search and only relevant information on the substances looked up appears.
DLER does not include a concept that compares to the Pharmaceutical Product ID Set in ISO 11616 – since this is a new concept for IDMP, this is not surprising. This does mean that DLER does not have a good means of immediately comparing similar generics, except by items such as the ATC code.
In comparing the Medicinal Product message in ISO 11615, DLER covers most of the major topics, but none of them to the same level of detail. It does include manufacturer/establishment information that is not present in the XEVMPD specification. There is a small amount of detail on package containers, but not to the depth of IDMP, and batch IDs are not part of the process. Areas such as adverse experiences and indications which are part of the main SPL Content of Drug Labeling are not also included in the DLER.
Moving from DLER to IDMP will be challenging: A lot more information may need to be gathered, depending on what fields of IDMP are listed as required by FDA guidance.
2.4.2 COMPARING XEVPRM AND IDMP As the XEVMPD guidance for the XEVPRM record was created as a stop-gap for IDMP, it makes sense that there is more similarity between XEVPRM and the IDMP standards. The closest alignment is between the XEVPRM “Structured Substance Information” and ISO 11238, but this was dropped as an XEVMPD requirement in March of 2012. XEVPRM provides more implementation guidance, including the message and acknowledgement for creating and updating the controlled terminology, substances, products, organizations, etc.
There are areas where IDMP is more detailed or has no equivalent in XEVPRM:
• The PhPID Set is unique to IDMP, although XEVPRM includes the administrable dose form, device identification and list of ingredients present in the Pharmaceutical Product.
• The detailed “parts” of a product name do not cover as many items in XEVPRM (Formulation Part and Intended Use Part, for instance, are not present).
• Manufacturing Steps are not present except for the overall product • Periodic Reporting is not included • Only a single field is used to describe packaging in XEVMPD • Ingredient strengths do not include reference strengths, however, the February 2013
Frequently Asked Questions document specifies that the reference strength should be specified instead of the salt, ester or pro-drugInvalid source specified.
• Clinical Particulars only include indications, without the level of detail of IDMP and there is no information on interactions, contra-indications or undesirable effects.
The only area where XEVMPD exceeds IDMP capabilities is the CBD (Chemical Biological Description), which has no direct equivalent in IDMP.
Because of the closer alignment, migration to IDMP should be less challenging than for DLER to IDMP. There is still a large number of fields added, but there is a strong likelihood that many of those will be listed by EMA as not required or optional, at least in the initial implementation.

27
DRUG PRODUCT REGISTRATION PROCESSES
3 CHALLENGES FOR IDMP IMPLEMENTATION The IDMP data standard conforms closely with the data model in section 1.2, which should be obvious as that section was drawn from terminology in ISO 11615. The hierarchy of medicinal product having packaging, each with manufactured elements, and which combine to form pharmaceutical products is a key part of the structure in the HL7 Common Product Model which is the basis for IDMP.
The overall IDMP model is significantly more descriptive and flexible than the DLER and XEVMPD models: medicines such as “Z-pack” antibiotics (two different tablets in one blister pack to be taken at once), transformed items (powders and solvents to be combined), and packages containing devices can be completely described. With very little change, a medical device could also be described using the same structures.d
However, it is not clear what sections will be required by ICH, as the implementation guide has not yet been issued. On top of that, some jurisdictions may decide to make additional sections optional, essentially dumbing IDMP down to the level of the information currently provided in those countries.
It is important to note that IDMP is designed to be a per-country identifier: the Medicinal Product ID (MPID) and those for packaging and batches (PCID, BAID_1 and BAID_2) are specific to the market authorization number in a particular country. That is logical, as the majority of drug use that would require regulatory action such as a serious adverse event, counterfeiting, or black-market sales would be local to the country. There would be value in having such registrations posted to other nations, or at least accessible to other nations, in the cases of smuggling across borders or counterfeit medication carrying another country’s MPIDs. However, filing the entirety of the IMPD messages for one country with another is not a practical thing to do.
There are portions which should be standardized internationally. The controlled vocabularies for substances make sense to be registered just once, as they are entirely descriptive and do not cover the authorisation for use (see the section below for differences with XEVMPD). The PhPID Set described in ISO 11616 is another area that makes sense to be a single international registry, as it again is not relevant to individual countries, and similar products will share one or more identifiers in the PhPID set (for instance, one for all Aspirin products, one for all Aspirin tablets regardless of dosage, one for all Aspirin 325mg regardless of dose form, and one for all Aspirin 325mg tablets).
The European Union has stated a goal of full implementation by 2016. FDA has no official implementation date, but expects to be close to 2016, possibly sooner, possibly later.
The substance registration system needed for ISO 11238 is being developed by the US National Library of Medicine and is expected to be available as free and public software by early 2014. At that point, it should contain those substances already in the current US Substance Registration System as public information, including non-11238 information such as metabolites (which could be represented in the clinical particulars) and impurities.
There is speculation that because of the time and effort required in order to approve an ISO standard, that many things were put into the IDMP standard set with the expectation that they were “aspirational” rather than “operational.” Although effort is already underway to create the Substance Registration System to cover the international substance IDs, there is as-yet no registry being
d That’s assuming any changes are needed at all: The HL7 common product model was designed with devices in mind.

28
DRUG PRODUCT REGISTRATION PROCESSES
developed for the Pharmaceutical Product ID sets; and items such as Batch IDs may not be required in any jurisdiction for years to come.
If portions of the standard are not going to be implemented initially, it leaves a dilemma for vendors: Should their systems be developed to support the entire standard, knowing that it greatly increases cost of development and testing, and the difficulty in using the system, or should minimal systems to support just the union of features specified across all participating countries be created to ease use and development?
3.1 Substance Registration Currently, no regulatory jurisdiction requires the level of detail of ISO 11238 for registration of substances. The XEVMPD standard requires filing for all active, inactive and adjuvant substances in drug products, but does not go into the level of detail that ISO 11238 does (the early version of the XEVPRM guidance matched the ISO standard almost to the letter, but was scaled back in the current version). The ICH implementation guide for ISO 11238 is anticipated to specify a variation on the SPL message to exchange such information between sponsors and agencies.
In the US, the Substance Registration System is managed by experts at the US Pharmacopoeia (USP) and FDA. Data entry is not done by the sponsors, as it may require significant chemical knowledge not usually part of regulatory expertise. Of course the chemical expertise must exist somewhere within the market authorisation holder or the substance wouldn’t get manufactured at all – but it’s not typical of the data that a regulatory operations knowledge worker would have as easy access to.
The IDMP implementation for substances is expected to expand upon the US Substance Registration System. At this time, it is expected that the information will be supplied by the market authorisation holder for new chemical entities, but existing data will be adapted from the current registration systems. ISO 11238 will have a message based on the relevant portions of the HL7 Common Product Model using the SPL message format. FDA and USP are planning a publicly-funded and publicly-available tool that will be capable of authoring, compiling and searching for substance registrations. This application will permit drug companies and substance manufacturers to register the specifications of their manufactured substances, so that identifiers can be issued for use with other parts of the IDMP system. It is expected that the expansion of the US system may be used by any countries participating in the IDMP standard.
The system that the US is developing is expected to be available in early 2014. This should provide a tool for entering and searching for substance information – even beyond what is present in ISO 11238, including searching by chemical structure, linkages to product information etc. The current version of the application does not include generation of an ISO 11238 SPL file, but this is under discussion.
3.2 Data Ownership The XEVMPD process has revealed some issues in implementation, particularly around who is permitted to change data. Most of the data is clearly owned: a Medicinal Product, its Packaging and Batches, are specific to the Market Authorisation Holder (or Sponsor, for Investigational Products). Similarly, organizations such as the Market Authorisation Holder, Manufacturing Establishments and so on, are clearly owned. Those items can be transferred to other organizations, at which point the new organization would be authorized to make changes. XEVMPD also permits data to be changed for some controlled vocabularies, specifically ATC Codes, Routes of Administration and Dosage Forms. The latter two seldom change, and ATC codes may be assigned to new products but should cause little conflict.

29
DRUG PRODUCT REGISTRATION PROCESSES
The difficult part is mainly in ownership of substances. There may be multiple companies marketing a product containing inactive substances and off-patent active ingredients. The XEVMPD process has been plagued with changes to generic drugs that affect multiple companies, such as changes or removal of substance names and translations. To solve this, EMA has recently changed the process to prevent changes to existing names once entered (a legitimate change will most likely require a request outside of the XEVMPD system).
This issue will hopefully also be addressed by the ICH implementation guide for IDMP
3.3 Data Maintenance The struggles that companies have had in implementing and maintaining XEVMPD should be an indicator of what is in store for IDMP. Updates to drug listing information for SPL and XEVMPD are primarily items that reflect changes to the product registration and follow a procedure of either prior approval submission, or a “do-and-tell” change (for more minor changes) where the change is filed with an annual report (in some jurisdictions, within one year, not necessarily at the anniversary date).
IDMP supplies an additional level of complexity in the form of the Batch IDs, on both immediate packaging and exterior packaging (as previously described: the vial a solution is provided in, plus the carton of 20 vials). This implies that every manufacturing batch that is created will require an update to the IDMP message – this could be a significant effort, or it is possible that the applications may permit something as simple as an abbreviated message that states, “For MPID A’s Packaged Medicinal Product A-B, add Batch ID A-B-C, and the regulatory authorities can automatically generate an updated full IDMP record. As currently only biologic medications require batch ID submission, it is probably that it will only be required in IDMP for biologics, which will also reduce the overall effort required.
Without the ICH or regional implementation guides, only speculation is possible. Given the experience with XEVMPD, it is likely that a similar process will be required, where product updates that require prior authorization must be filed within 30 days of such authorization, and others will be required within a year (or on annual report date in the US). Scheduling batch updates is an unknown. As described in the following section, maintenance of product registration information is a key part of company processes, and automatic generation of IDMP messages should be considered a critical piece of those processes.

30
DRUG PRODUCT REGISTRATION PROCESSES
4 DRUG SPONSOR COMPANY PROCESSES All three of the existing electronic registration processes require that updates to the product which affect the information require an updated electronic message. This can be something as simple as a change to the name of a supplier, to a new inactive ingredient being included or new packaging.
4.1 Regulations While the number of details provided in IDMP is much larger than in DLER, and somewhat larger than XEVPRM, it is not expected that the number of updates will be significantly greater, as the categories of changes are essentially the same, and detail provided is merely finer on those categories. However, if there is a change to those finer details, a new version of the product information would be generated in most cases anyway.
Most regulatory agencies have a category of updates which can be called “do and tell” – minor changes to the product which do not require an immediate notification of or approval by the agency. In Europe, these are Type IA (one-a) variations. These types of changes are typically provided at the time of the annual update or periodic update in the US, and must come within 12 months of making the change in Europe. These include changes to suppliers and manufacturing sites, testing sites, minor changes to manufacturing processes, changes in batch size, tightening of specifications and testing limits, etc.Invalid source specified..
In the US, the FDA requires that Drug Listings be updated every June and December, but encourage updates to be submitted when labeling updates are made. Establishment Registration must be updated annually, but again, more frequent updates are encouraged.Invalid source specified.
4.2 Data Maintenance Processes New and changed drug products will require drug registration messages to be sent to the US and EU markets where it will be sold. The initial compilation of the data for a drug registration is not difficult (although the IDMP and XEVMPD processes are indeed much more complex than for the United States’ DLER), and the information will be fresh in the Regulatory department’s minds when the drug is being approved.
Maintaining the data adds a level of difficulty: information will be coming from a wide variety of sources: pharmacovigilance for changes in adverse experience reporting, manufacturing for changes in processing or establishments, marketing for changes in packaging or countries where it is being sold. Locating where the information is coming from and where it must be filed can be painstaking. The registration processes may be taking place centrally, whereas the information on manufacturing may be specific to a market company in another country.
The general steps for this sort of maintenance are as follows:
1. Change is identified 2. Countries affected are identified 3. Submission requirement identified (pre-authorisation versus “do and tell”) 4. Documentation gathered for the submission (either for immediate use or “stacked up” for an
annual or 6-month filing) 5. Compile changes into drug registration message (DLER, XEVPRM, and/or IDMP)
If the submission process is independent from the drug product information management, compilation of drug registration can be slow and difficult, and a great deal of re-work may be performed if it is not simple to merely modify the existing registration message.
Partly due to the complexities of product maintenance, Regulatory Information Management (RIM) systems have become more popular in recent years, with an emphasis on centralizing the data

31
DRUG PRODUCT REGISTRATION PROCESSES
management process. The RIM system should be able to assist in (a) identifying the impact of a change to product information in terms of where and when a filing is required, and (b) assisting in the creation of the registration messages.
An additional challenge common to any RIM implementation is compliance with data entry requirements. Since the registration processes involve coordination of information provided centrally (base product information, manufacturing) and local to each registered country (registration information, specific label changes, translations of labels and other documentation), there must be procedures set in place to ensure consistent, reliable data.
This would include SOPs on what data is changed for each role, e.g. global product leader versus data coordinator versus local product owner. Control over standard terms such as product names and active ingredients are critical to maintain consistency. Timeliness is also critical for centrally-managed electronic submissions of registration messages. If a local market company does not update registration changes, it is not possible for the central regulatory group to file the changes as required by FDA or EMA.
4.3 Message Authoring As described in section 5.2, there are numerous vendors providing DLER and XEVMPD tools and conversion services. At this stage, SPL authoring including DLER is a commodity, with inexpensive services offered by a number of vendors. XEVMPD is considered to be a more difficult task, because of the need to supply messages for each of the 27 EMA member nations, as opposed to the single agency that accepts SPL DLER.
Message authoring for IDMP will be similar in effort to XEVMPD. It will not be a matter of copying one message for all agencies accepting the IDMP format. While there will be some common data, region-specific information including trade names, language-specific packaging and labeling, and manufacturing differences between sites that supply different parts of the world will require that each country receives different data. However, because the format will be the same in all participating nations, the value of software to manage the messages will be leveraged across a greater amount of work, making it easier to justify the costs.
4.4 Registration Process The actual process of filing the registrations with IDMP will not change significantly from today’s XEVMPD and SPL DLER processes. Currently, US filings require the use of the Electronic Submissions Gateway (ESG), which has been in use for a number of years for both product authorization filings (e.g. New Drug Applications) and Drug Listings and Establishment Registration. To use this gateway, a Web Trader account is used, or an integrated application which communicates directly with the gateway. Both methods require a company to register with the FDA, including acquisition of a DUNS number from Dun and Bradstreet.Invalid source specified. Service providers are able to access these applications on behalf of their customers.
The Drug Listing and Establishment Registration filings do not create an automatic registration record, but the files will be validated for technical compliance, and an immediate notification provided of their validity.
As part of the XEVMPD process, EMA created an internet application called EVWEB which permits entry and maintenance of XEVMPD information in an interactive format. In addition, the EMA Gateway provides functionality similar to the US ESG: permitting submissions in a number of formats to be directly submitted. Like the US, EMA requires registration of users. A major difference is that service providers are not permitted their own accounts, and may only access the EVWEB and EMA Gateway using a customer’s account.

32
DRUG PRODUCT REGISTRATION PROCESSES
In addition to the technical compliance, EMA filings include a review of the content for correctness, resulting in the return of an Acknowledgment Message indicating which data filed with EMA has been accepted, and any errors.

33
DRUG PRODUCT REGISTRATION PROCESSES
5 DRUG SPONSOR REQUIREMENTS Every company marketing medicinal products (drugs or biological agents) is required to file information about those products, and maintain updated information, throughout the life of the product. Many companies rely on a large number of unrelated databases, often residing at international subsidiaries, to register this information.
CSC can provide services and software to ease this transition.
5.1 Consulting and Other Strategic Services CSC has performed implementation and other planning services for TRS Tracker for clients. Through analysis of customer data and processes used, and the sources of data determined during implementation engagements, CSC will develop a Best Practices environment for maintenance of product registration information for implementation of IDMP, once ICH and regional implementation guides have been issued. This will include reconciliation of disparate taxonomies and vocabularies used between the three standards.
Analysis of the various customer data sets is combined with the deconstruction of the SPL, XEVMPD and IDMP standards begun with this white paper, to identify areas of commonality, and areas where data will need to be copied into similar but not identical records to ensure unambiguous, accurate coverage of each of the standards. When it is necessary to provide two versions of similar information, it will be made clear to customers the reasons for such duplications. This will avoid inaccurate representation of information that would appear common, but is used for different purposes. As an example, the SPL standard uses 150 terms for dosage form (tablet, solution for injection, etc.), and XEVMPD has 450, with only about 40 in common. The vocabulary for IDMP is not yet finalized, but the standard has stated intentions of having mappings from both multiple legacy values to one IDMP value, and from single legacy values to multiple IDMP values.Invalid source specified.
The result of this analysis would be a Best Practices scenario with “a place for everything, and everything in its place.” This means that not only would the data be maintained in a structure usable for international product registrations, but also for maximized value to the enterprises that are maintaining the data, for understanding where their products are registered, what effort must be undertaken to continue marketing their products, and where products should be marketed next.
CSC Life Sciences consulting can then provide the following services to companies planning drug product registration and migration to IDMP:
• Assessment of existing state. This would include examination of current data management and warehousing, identification of gaps, and recommendations for best practices for Regulatory Information Management
• Implementation of Regulatory Information Management. This would include design of the TRS Tracker or third-party RIM system to best fit business practices, data migration planning, and training for use.
• Regulatory Operations staffing, including use of a Regulatory Information Management system as part of a Total Regulatory Practice.
• Authoring regulatory content as part of Total Regulatory Practice, that might result in changes to drug registrations
• Compilation of changes to drug product registrations as part of Total Regulatory Practice during assembly of regulatory submissions.
A key part of the preparation for Regulatory Information Management is a change to the corporate reguatory environment that will expect centralization of storage of regulatory information, even if the

34
DRUG PRODUCT REGISTRATION PROCESSES
sources of that information are distributed worldwide. Most multinational pharmaceutical companies rely on local management of licensing and registration. While this is the most efficient method of managing the product life cycle, it can leave gaps in the central management of product licensing and marketing, unless changes to registration and marketing status can rapidly and accurately be communicated to a central database.
5.2 Conversion and Authoring Services and Tools CSC Life Sciences currently provides SPL-based registration services, and XEVMPD registration services
The SPL format anticipated to be used for IDMP is sort of a carry-all package that is used for HL7-based messages to FDA. Currently it is used for Drug and Device Listings and Establishment Registrations, as well as the Content of Labeling. The Content of Labeling in particular requires a lot of narrative text to be authored, but also includes data components – some shared with the Drug Listing and Establishment Registration, some not.
The creation of IDMP records does not require much text authoring: its sources should be in Regulatory Information Management systems, and are data elements, versus narrative text.
Currently CSC is not offering a product for authoring SPL content, but provides services for conversion of content of labeling, and these services can also provide the DLER structure. It is anticipated that these IDMP services will be provided similarly, again after implementation guides have been issued.
Because of the data-oriented nature of IDMP, the maintenance of all of the detail in IDMP in a message format is inefficient: it would be far better to extract such information from TRS Tracker using an export module that outputs the SPL format for IDMP, and an import module that can receive the responses from the agencies. This will be developed as the implementation of the standard is finalized.
5.3 TRS Tracker and other Total Regulatory Solution software The CSC Life Sciences product, “TRS Tracker,” is designed as a regulatory information management system. Its primary functions are around management of regulatory submissions, product life cycle, national registration and licensing status and similar applications .The version available at the time of this writing is 5.2. In that version, the data needed for Drug Listing and Establishment Registration, and for XEVMPD, can be entered and managed, and direct submission of XEVPRM messages to the EMA gateway is supported, as well as the ability to compare “current-state” XEVPRM messages with the TRS Tracker database to determine what updates are required.
With IDMP implementation two to three years away, there is not a customer expectation that the current version will support IDMP. The extent of changes needed to the data model to support IDMP will not be known until the ICH and (at least) the US and EMA implementation guides have been issued. At that point, the mandatory and optional data should be known, and the architecture for changes to the data model can be designed.
To be useful, it is expected that the data model will be integrated into TRS Tracker approximately one year prior to any required date for submission to agencies, with modules for IDMP submission following, at least a few months before those deadlines. That is assuming that the agencies provide a sufficient lead time.

35
DRUG PRODUCT REGISTRATION PROCESSES
5.3.1 TRS TRACKER EXTENSION FOR IDMP Implementation of the full database to support IDMP initially appears daunting: There are many object types in the IDMP structure, but a closer investigation reveals that many of them have equivalent concepts, just a different name placed atop them: The various “Property” concepts are all just name-value pairs. Even the “standard” properties (height, weight, color, etc.) could be expressed as such name-value pairs, as opposed to distinct fields. This methodology follows third-order normalization concepts, and can actually result in some simplification, as fewer overall fields will need to be configured, and many products may leave a large number of such fields empty (for example, injectable pharmaceutical products do not have height, length, width, imprinting, flavor, etc.).
Additional work will still be needed to create and deliver the IDMP messages, and receive and interpret the acknowledgement messages from the agencies, but this should just be an upgrade from what is being built for XEVMPD. TRS Tracker does not currently have SPL/DLER messaging as part of its plan, but adding FDA gateway interfaces for the US version of IDMP should be of a similar level of effort as that for the XEVMPD gateway.

36
DRUG PRODUCT REGISTRATION PROCESSES
6 CONCLUSIONS Recent years have seen a flurry of changes in the field of electronic drug product registration. Starting with FDA’s simple Drug Listing and Establishment Registration (DLER), built as a stripped-down version of their Structured Product Language (SPL) content of labeling standard, Health Level Seven built a Common Product Model designed to be able to describe the contents, usage and safety issues of any health product.
The IDMP ISO standard was expanded from this to be a message for registering products and the substances contained within them, with the Medicinal Product ID, Packaging ID and Batch ID being used to track the products in the cases of adverse experiences, illegal import, counterfeiting and other issues of pharmacovigilance.
Because of the long implementation cycle of ISO standards, Europe “jumped the gun” and created the XEVMPD standard, in order to start gathering the drug product information that would build their pharmacovigilance system. The XEVMPD standard from EMA takes a greater step than FDA’s SPL/DLER, asking that not only do new products need to be registered, but that the backfile of every product to which the public could be exposed must also be provided in the electronic form.
The IDMP standards though, were created with an “everything for everyone” philosophy: there’s a huge amount of information that can be specified, and it supports both the new product registration tasks of SPL/DLER as well as the pharmacovigilance goals of XEVMPD. Speculation is that this standard will not all be implemented — especially in the initial stages — but that the difficulties of approving an ISO standard led the team to create an all-inclusive buffet of data that could be filled in as appropriate, as opposed to trying to amend it later. It’s good to see a standard with some flexibility that was originally conceived merely as a way of standardizing the identification of medications specified in the ICSR ISO standard for reporting adverse drug experiences that has been adopted for several years.
The difficulty in managing such a complex dataset as IDMP has already been seen in the somewhat simpler XEVMPD submission process: there are pain points in determining which of many vendors of a particular substance is permitted to modify its database entries. The complexity of completely specifying the active and inactive ingredients is also expected to be a challenge, but one that appears to be alleviated by the US Pharmacopoeia and National Institutes of Health developing an open-source application for creating and maintaining the substance data.
The Health Level Seven structures behind IDMP will result in the drug information becoming readily available for other uses. Insurance, hospital and other environments should be able to share the same identifiers for drugs, including the Pharmaceutical Product IDs which correspond to generic classifications of the drug products. In addition, the indications, contra-indications, adverse events and interactions of these products will be specified in a common, structured format. Use of these data will lead to improvements in public health through reductions in prescribing errors and unexpected drug interactions.
Unlike the preceding SPL-based Drug Listing and Establishment Registration, this will not be a simple compilation of readily-available generalized information for a single country. With DLER, the effort was small enough that a company could have a workflow of just editing the existing text, or compiling information on a new drug. XEVMPD raised the bar significantly by requiring information be sent in specific form for each country and language in the EU. It would be unexpected for the first implementation of IDMP to go much further than XEVMPD’s requirements, but this still underscores a need to manage product data in a Regulatory Information Management system: no one company location will have all the knowledge needed to assemble the IDMP messages, and those messages

37
DRUG PRODUCT REGISTRATION PROCESSES
will need to be updated to match any approved and notified changes to a product. By implementing enterprise-wide RIM systems, generation of XEVMPD and IDMP messages becomes almost a free side effect, and the value increases when the changes made to a product are reflexively collected between submissions and product registration tracking systems.

38
DRUG PRODUCT REGISTRATION PROCESSES
7 BIBLIOGRAPHY There are no sources in the current document.
About the Author
Joel Finkle is an active participant of the HL7 Regulated Product Submissions (RPS) team, developing the next generation drug product approval messaging standard. He has also participated in the Regulatory/Clinical Research Information Management (RCRIM) leadership team within HL7, which directs both the RPS as well as the IDMP standard. He is also a member of the DIA Electronic Document Management Reference Model development team. He has presented on Regulatory Information Management and the RPS standard at DIA conferences consistently over the ten years he has been part of the CSC and former ISI organizations. He makes a killer tomatillo salsa.
.

39
DRUG PRODUCT REGISTRATION PROCESSES
APPENDIX A: COMPARISON OF DATA ELEMENTS In the tables below, colored items indicate repeating sections that are used in other portions of the standard. The reused items will appear in the same color, with red or blue text color to indicate that they are the reference to the repeating section, rather than the original. The three tables below for medicinal products, pharmaceutical product ID sets, and substances do not share the colored sections listed, but the Medicinal Product section references the other two items by identification number (substances) and inclusion of the entire data structure (pharmaceutical product ID sets).
The “Q” field indicates the quantity of the item that appears in the data structure. “Regular Expression” notation was chosen rather than the typical UDL notation to save space. The meaning of this column is as follows:
Q Column Cardinality 1 [1..1] ? [0..1] * [0..n] + [1..n] 2+ [2..n]
ab (not a regular expression)
Select one or the other nearby item
The other columns are as follows:
• Information – The field name in the standards • SPL/DLER – Structured Product Language / Drug Listing Establishment Registration, the US drug listing
standard. Contains R4 and R5 indicating the version at which it is present. • SmPC – Summary of Product Characteristics, an EU labeling document (not an electronic format) • XEVMPD – Extended Eudravigilance Medical Product Dictionary, the EU drug listing standard • IDMP – Identification of Medicinal Products, the international standard for drug listing information • MP – IDMP Medicinal Product section • IMP – IDMP Investigational Medicinal Product section • Tracker – Version of TRS Tracker in which the features will be present.

40
DRUG PRODUCT REGISTRATION PROCESSES
APPENDIX A-1: MEDICINAL PRODUCT (ISO 11615)
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Medicinal Product Y Y Y Y XEVMPD lists orphan
drug indicator MPID/IMPID 1 DLER R5 Y Y Y Combined Pharm Dose Form ? DLER R4 Y Y Y IMPID Cross Reference * Y Y Y Sponsor Product Code ? N N Y Regulator Product Code ? N N Y Paediatric Use Indicator ? N N Y Additional Monitoring Indicator ? Y Y N Special Measures * N Y N Medicinal Product Name + DLER R4 Y Y 1 Only 1 name permitted
for IMPID Name 1 DLER R4 Y Y Y ? Optional, use either
Name or Code Code 1 Y N ? Optional, use either
Name or Code Invented Name Part ? Y Y Y Y Scientific Name Part ? DLER R4 Y Y Y Y SPL: genericMedicine? Strength Part ? Y Y Y Y Pharmaceutical Dose Form Part ? Y Y Y Y Formulation Part ? N Y Y Intended Use Part ? N Y Y Target Population Part ? N Y Y Container or Pack Part ? N Y Y Device Part ? N Y Y Trademark or Company Name Part ? Y Y Y Time/Period Part ? N Y Y Flavour Part ? N Y Y

41
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Country / Language + Y Y Y Considered
"translations" in XEVMPD
Country / Language 1 Y Y Y Jurisdiction ? Y Y Y Language 1 Y Y Y
Version + Y Y Y N Version Date 1 Y Y Y N Version Identifier ? Y Y Y N Regulated Document + Y Y Y N
Document Type 1 Y Y Y N Document Identifier 1 Y Y Y N Attached Doc Here Regulated Document 1 Y Y Y N Document Effective Date 1 Y Y Y N
Manufacturer/Establishment (Org) + DLER R4 Y Y N SPL uses Author, EVMPD incl. Type, comments
Identifier 1 DLER R4 Y Y N Name 1 DLER R4 Y Y N Address 1 DLER R4 Y Y N Confidentiality Indicator ? DLER R4 N Y N Contact Person + N Y N
Name 1 N Y N Telecom 1 Y Y N EVMPD has specifics for
Pharm Prod and Med Prod
Role 1 N Y N EVMPD: Role applies to entire org
Confidentiality Indicator ? N Y N Other Locations + Y Y N Used e.g. PV system
master file location
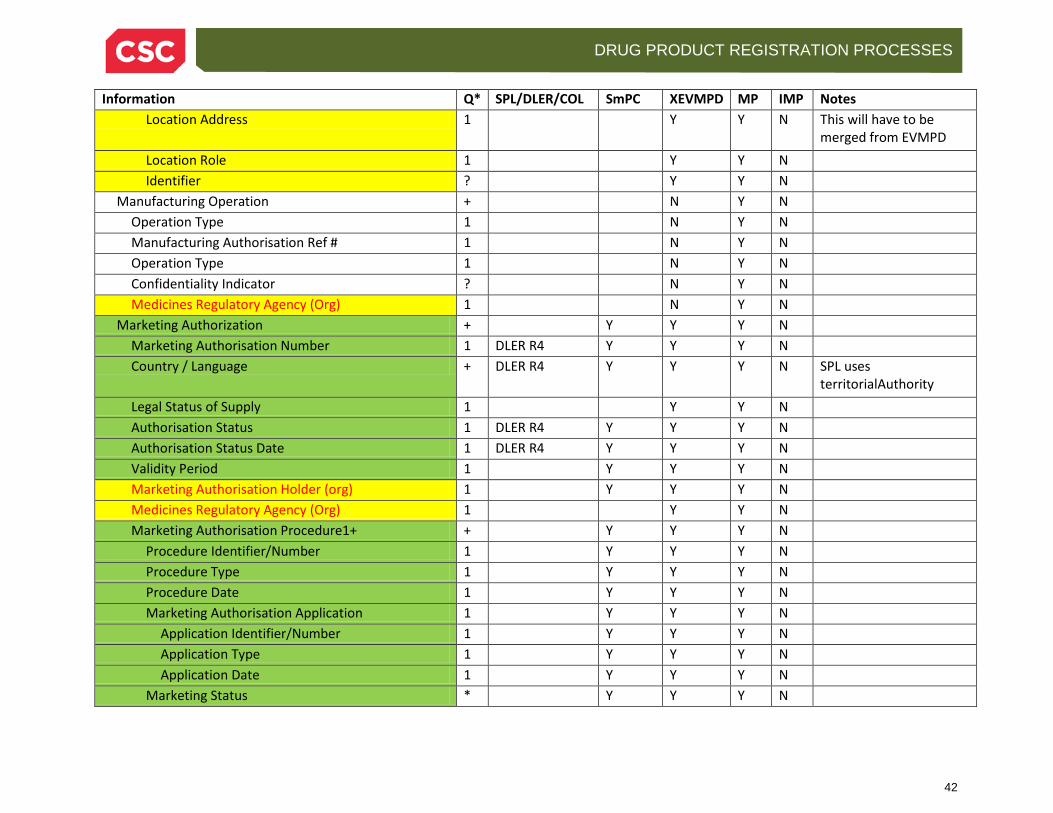
42
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Location Address 1 Y Y N This will have to be
merged from EVMPD Location Role 1 Y Y N Identifier ? Y Y N
Manufacturing Operation + N Y N Operation Type 1 N Y N Manufacturing Authorisation Ref # 1 N Y N Operation Type 1 N Y N Confidentiality Indicator ? N Y N Medicines Regulatory Agency (Org) 1 N Y N
Marketing Authorization + Y Y Y N Marketing Authorisation Number 1 DLER R4 Y Y Y N Country / Language + DLER R4 Y Y Y N SPL uses
territorialAuthority Legal Status of Supply 1 Y Y N Authorisation Status 1 DLER R4 Y Y Y N Authorisation Status Date 1 DLER R4 Y Y Y N Validity Period 1 Y Y Y N Marketing Authorisation Holder (org) 1 Y Y Y N Medicines Regulatory Agency (Org) 1 Y Y N Marketing Authorisation Procedure1+ + Y Y Y N
Procedure Identifier/Number 1 Y Y Y N Procedure Type 1 Y Y Y N Procedure Date 1 Y Y Y N Marketing Authorisation Application 1 Y Y Y N
Application Identifier/Number 1 Y Y Y N Application Type 1 Y Y Y N Application Date 1 Y Y Y N
Marketing Status * Y Y Y N

43
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Country 1 Y Y Y N Jurisdiction ? Y Y Y N Marketing Authorisation Number 1 Y Y Y N Marketing Date (start/stop) 1 Y Y Y N Jurisidictional Legal Status of Supply 1 Y Y N
Periodic Safety Update Report Submission 1 N Y N Frequency of Submission of SR 1 N Y N Dates of Submission + N Y N International Birth Date 1 N Y N Reference Date 1 N Y N Data Lock Point 1 N Y N
Clinical Trial Authorisation 1 N N Y Registration Number 1 N N Y Investigation Code 1 N N Y Country + N N Y Protocol Number 1 N N Y Authorisation Date 1 N N Y Anticipated End Date 1 N N Y Medicines Regulatory Agency (Org) 1 N N Y Sponsor (Org) 1 N N Y Local Clinical Trial Authorisation * N N Y
Local Clinical Trial Registration Number 1 N N Y Local Investigation Code 1 N N Y Jurisdiction + N N Y Local Authorisation Date 1 N N Y Local Anticipated End Date 1 N N Y
Drug Safety Update Report Submission1 1 N N Y Essemtially same as PSUR section above
Frequency of Submission of DSUR 1 N N Y

44
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Dates of Submission + N N Y Development International Birth Date 1 N N Y Reference Date 1 N N Y Data Lock Point 1 N N Y
(Inv) Packaged Medicinal Product + N Y * Optional for Investigational
PCID/PICID 1 N Y Y Package Description 1 Y Y Y Y Marketing Authorization ? N Y Y Package Item (Container) + DLER R4 N Y Y SmPC is not detailed
Package Item Type 1 DLER R4 N Y Y Package Item Quantity 1 DLER R4 N Y Y Material + N Y Y Alternate Material * N Y Y Data Carrier Identifier * N Y Y
Code System 1 N Y Y Value 1 N Y Y
Batch Identifier * N Y Y BAID1 1 N Y Y BAID2 ? N Y Y
Package (Component) * N Y Y Component Type 1 N Y Y Component Material * N Y Y Component Alternate Material * N Y Y Physical Characteristics * N Y Y
Shelf Life / Storage + N Y Y Shelf Life Type 1 N Y Y Shelf Life time Period 1 N Y Y Special Precautions for Storage ? N Y Y

45
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Device * Y Y Y
Device Type 1 N Y Y Device Trade Name ? Y Y Y Device Quantity 1 N Y Y Device Listing Number ? N Y Y Model Number ? N Y Y Device Material + N Y Y Device Alternate Material * N Y Y Allergenicity Indicator * N Y Y Sterility Indicator * N Y Y Sterilisation Requirement Indicator * N Y Y Device Usage * N Y Y Device Nomenclature * Y Y Y
Nomenclature System 1 Y Y Y Nomenclature Value 1 Y Y Y
Device Batch Identification * N Y Y Batch Number 1 N Y Y Expiration Date 1 N Y Y
Manufacturer/Establishment (Org) 1 N Y Y Physical Characteristics * N Y Y Other Characteristics * N Y Y Shelf Life / Storage * N Y Y
Manufactured Item * N Y Y Manufactured Dose Form ? N Y Y Unit of Presentation ? N Y Y Manufactured Item Quantity ? N Y Y Physical Characteristics ? N Y Y Other Characteristics ? N Y Y

46
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Ingredient + DLER R4 Y Y Y Y XEVMPD separates by
role (active, etc.) Substance ab DLER R4 Y Y Y Y
Substance 1 DLER R4 Y Y Y Y Strength * DLER R4 Y Y Y Y
Specified Substance ab Y Y Y Y Specified Substance 1 Y Y Y Y Specified Substance Group ? Y Y Y Y Confidentiality Indicator ? Y Y Y Y Strength * Y Y Y Y
Strength Range (Presentation) 1 Y Y Y Y Strength Range (Concentration ? Y Y Y Y Measurement Point ? Y Y Y Y Country ? Y Y Y Reference Strength * Y N Y Y SPL Uses Active Moiety
Reference Substance ? Y N Y Y Reference Specified Substance ? Y N Y Y Reference Strength Range 1 Y N Y Y Reference Strength Meas. Point ? Y N Y Y Reference Strength Country ? N Y Y
Physical Characteristics ? Y N Y Y Height ? Y N Y Y Width ? Y N Y Y Depth ? Y N Y Y Weight * Y N Y Y Nominal Volume 1 Y N Y Y External Diameter 1 Y N Y Y Shape * Y N Y Y Colour 1 DLER R4 Y N Y Y

47
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Imprint ? DLER R4 Y N Y Y Image 1 DLER R4 Y N Y Y
Other Characteristics * DLER R4 N Y Y Code System 1 DLER R4 N Y Y SPL uses Shape, Score,
Size, Flavor Value 1 DLER R4 N Y Y
Package Item (Container) * N Y Y (Inv) Pharmaceutical Product + Y Y Y
Administrable Dose Form 1 Y Y Y Y Unit of Presentation ? Y N Y Y Pharmaceutical Product Quantity 1 N Y Y (Dosing And) Route of Administration + Y Y Y Y
Route of Administration 1 DLER R4 Y Y Y Y First Dose in Humans ? N N Y Maximum Single Dose ? Y N N Y Maximum Dose Per Day ? Y N N Y Maximum Dose Per Treatment Period ? Y N N Y Maximum Treatment Period ? Y N N Y
PhPID Set + N N Y Y Pharmaceutical Product Characteristics * N N Y Y
Code System 1 N N Y Y Value 1 N N Y Y
Device * N Y Y Y Not including manufacturer, Shelf Life
PhPID Identifier Set 1 N N Y Y Ingredients + Y Y Y Y Part of Pharm Prod in
XEVMPD, Packaged or MP here
Therapeutic Indication + COL Y Y Y Y

48
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes indication Text 1 COL Y N Y Y Indication as "Disease/symptom/procedure" 1 COL Y Y Y Y XEVMPD uses MEDDRA Disease Status ? Y N Y Y Co-morbitidy ? Y N Y Y Intended Effect ? Y N Y Y Timing/Duration ? Y N Y Y Population Specifics * Y N Y Y
Age ? Y N Y Y Age Range ? Y N Y Y Gender ? Y N Y Y Race ? Y N Y Y Health Status ? Y N Y Y
Other Therapy Specifics * Y N Y Y Therapy Relationship Type 1 Y N Y Y Medication 1 Y N Y Y
Contra-Indication + Y N Y * Optional for Investigational
Contra-Indications Text 1 Y N Y Y Contra-Indications as "Disease/symptom/procedure" 1 Y N Y Y Disease Status ? Y N Y Y Co-morbitidy ? Y N Y Y Population Specifics * Y N Y Y Other Therapy Specifics * Y N Y Y
Undesirable Effects + COL Y N Y * Optional for Investigational
Undesirable Effect Text 1 COL Y N Y Y Undesirable Effect as "Symptom/Condition/Effect" 1 COL Y N Y Y Symptom/Condition/Effect Classification ? COL Y N Y Y Frequency of Occurrence ? Y N Y Y

49
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER/COL SmPC XEVMPD MP IMP Notes Interactions * Y N Y Y
Interaction Text 1 Y N Y Y Interaction Type 1 Y N Y Y Interaction Effect 1 Y N Y Y Interaction Incidence 1 Y N Y Y Management Actions 1 Y N Y Y
Interactant 1 Y N Y Y Interactant Code System 1 Y N Y Y Interactant Value 1 Y N Y Y
Medicinal Product Classification * DLER R4 Y Y Y Y ATC Code goes here - not as detailed as ATC unless schema has more values for these objects
Classification System 1 DLER R4 Y Y Y Y Classification System Value 1 DLER R4 Y Y Y Y

50
DRUG PRODUCT REGISTRATION PROCESSES
APPENDIX A-2: PHARMACEUTICAL PRODUCT ID (ISO 11616)
Information Q* SPL/DLER XEVMPD IDMP Notes PhPID Set 1 N N Y XEVMPD includes Route of Admin in Pharm
Product, does not distinguish between diff. ID types or specified substances, has separate lists for active, inactive and adjuvant substances.
PhPID_SUB_L1 1 N N Y ID 1 N N Y Substance Set + N Y Y
Substance 1 Y Y Y Role 1 Y Y Y Confidentiality ? N N Y Reference Strength + N N Y
Reference Substance ID ? N N Y Reference Specified Substance ? N N Y Reference Strength Range ? N N Y Reference Strength
Measurement Point ? N N Y
Reference Strength Country ? N N Y PhPID_SUB_L2 1 N N Y
ID 1 N N Y Name 1 N N Y Strength (conc) 1 N N Y Note: Strength can be represented as text if
more than one substance Strength (presentation) 1 N N Y Unit of Presentation ? N N Y Substance Set + N N Y
PhPID_SUB_L3 1 N N Y ID 1 N N Y Name 1 N N Y

51
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER XEVMPD IDMP Notes Administrable Dose Form 1 N Y Y Substance Set + N N Y
PhPID_SUB_L4 1 N N Y This is PhPID_SUB for investigational, no 1,2,3 ID 1 N N Y Name 1 N N Y Strength (conc) 1 N N Y Strength (presentation) 1 N N Y Administrable Dose Form 1 N Y Y Unit of Presentation ? N N Y Substance Set + N N Y
PhPID_SpSUB_L1 1 N N Y ID 1 N N Y Specified Substance Set + N N Y Specified Substance 1 N N Y Role 1 N N Y Confidentiality ? N N Y Reference Strength + N N Y
PhPID_SpSUB_L2 1 N N Y ID 1 N N Y Name 1 N N Y Strength (conc) 1 N N Y Strength (presentation) 1 N N Y Unit of Presentation ? N N Y Specified Substance Set + N N Y
PhPID_SpSUB_L3 1 N N Y ID 1 N N Y Name 1 N N Y Administrable Dose Form 1 N Y Y Specified Substance Set + N N Y

52
DRUG PRODUCT REGISTRATION PROCESSES
Information Q* SPL/DLER XEVMPD IDMP Notes PhPID_SpSUB_L4 1 N N Y This is PhPID_SUB for investigational, no 1,2,3
ID 1 N N Y Name 1 N N Y Strength (conc) 1 N N Y Strength (presentation) 1 N N Y Administrable Dose Form 1 N Y Y Unit of Presentation ? N N Y Specified Substance Set + N N Y
Medical Device ? N Y Y ID 1 N Y Y Name 1 N Y Y
Version + N N Y Effective Date ? N N Y

53
DRUG PRODUCT REGISTRATION PROCESSES
APPENDIX A-3: STRUCTURED SUBSTANCE (ISO 11238)
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes Substance
Substance_ID 1 Y Y Y Y XEVMPD includes CBD which is not part of IDMP
Specified_Substance_Group1 ? N Y Y specified_substance_group1_id 1 N Y Y specified_substance_group1_name 1 N Y Y Constituent + N Y Y substance_id 1 N Y Y substance_name 1 N Y Y substance_role 1 N Y Y amount_type 1 N Y Y Amount ? N Y Y Physical_Form * N Y Y physical_state 1 N Y Y physical_form_type 1 N Y Y amount_type 1 N Y Y Amount ? N Y Y Property * N Y Y Substance_Code 1 N Y Y Reference_Information ? N Y Y Substance_Name + N Y Y
Specified_Substance_Group2 ? N Y Y specified_substance_group2_id 1 N Y Y specified_substance_group2_name 1 N Y Y parent_substance_id 1 N Y Y Manufacturing 1 N Y Y
manufacturer_id 1 N Y Y

54
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes manufacturer_name 1 N Y Y manufacturing_type 1 N Y Y production_method_type 1 N Y Y production_system_type 1 N Y Y production_system 1 N Y Y critical_process_version_number 1 N Y Y Reference_Source * N Y Y Critical_Process * N Y Y
critical_process_id 1 N Y Y critical_process_site 1 N Y Y Processing_Equipment * N Y Y
number_of_eqiupments 1 N Y Y Equipment * N Y Y
equipment_id 1 N Y Y equipment_name 1 N Y Y manufacturer_id 1 N Y Y manufacturer_name 1 N Y Y
Resultant_Material + N Y Y number_of_resultant_materials 1 N Y Y material_type 1 N Y Y Resultant_Substance + N Y Y
No._of_resultant_specified_substances 1 N Y Y Resultant_Material_Substance * N Y Y
substance_id 1 N Y Y substance_name 1 N Y Y substance_role 1 N Y Y specification_type 1 N Y Y amount_type 1 N Y Y Amount 1 N Y Y

55
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes Processing_Material ? N Y Y number_of_processing_materials 1 N Y Y material_type 1 N Y Y Processing_Substance * N Y Y
number_of_processing_substances 1 N Y Y Resultant_Material_Substance * N Y Y
Starting_Material * N Y Y number_of_starting_materials 1 N Y Y amount_type 1 N Y Y Amount 1 N Y Y Resultant_Material_Substance 1 N Y Y
Critical_Parameter * N Y Y parameter 1 N Y Y amount_type 1 N Y Y Amount 1 N Y Y
Specified_Substance_Group3 ? N Y Y specified_substance_group3_id 1 N Y Y specified_substance_group3_name 1 N Y Y parent_substance_id 1 N Y Y
Grade 1 N Y Y grade_type 1 N Y Y grade_name 1 N Y Y grade_reference_source 1 N Y Y
Reference_Source * N Y Y Specified_Substance_Group4 ? N Y Y
specified_substance_group4_id 1 N Y Y parent_substance_id 1 N Y Y Physical_Form * N Y Y Property * N Y Y

56
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes Manufacturing ? N Y Y Constituent + N Y Y Grade * N Y Y Analytical_Data * N Y Y
specification_type 1 N Y Y Specification * N Y Y
specification_description 1 N Y Y specification_value_type 1 N Y Y Specification_Values * N Y Y
id 1 N Y Y name 1 N Y Y amount_type 1 N Y Y Amount 1 N Y Y
Specification_Jurisdiction * N Y Y analysis_specification_ref._jurisdiction 1 N Y Y
Analytical_Method * N Y Y analytical_method_type 1 N Y Y analytical_method 1 N Y Y analytical_method_details 1 N Y Y analytical_method_reference_data 1 N Y Y Reference_Source * N Y Y
Reference_Source * N Y Y Version + N Y Y
version_number 1 N Y Y effective_date 1 N Y Y change_made 1 N Y Y Substance_Name + Y Y Y
substance_name 1 Y Y Y substance_name_type 1 Y Y Y

57
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes language 1 Y Y Y Reference_Source * Y Y Y
public_domain 1 ? Y Y reference_source_type 1 ? Y Y reference_source_class 1 ? Y Y reference_source_id 1 ? Y Y reference_source_citation 1 Y Y Y
Official_Name * Y Y Y official_name_type 1 N Y Y official_name_status 1 N Y Y official_name_status_change_date 1 N Y Y Official_Name_Domain * N Y Y
official_name_domain 1 N Y Y Official_Name_Jurisdiction * N Y Y
official_name_jurisdiction 1 N Y Y Reference_Information ? N Y Y
comment 1 N Y Y Substance_Classification + Y Y Y
domain 1 Y Y Y substance_classification 1 Y Y Y substance_classification_type 1 Y Y Y substance_classification_code 1 Y Y Y Subtype * ? Y Y
substance_class_subtype 1 ? Y Y Reference_Source * Y Y Y Target * N Y Y
target_gene_id 1 N Y Y target_gene_name 1 N Y Y interaction_type 1 N Y Y

58
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes target_organism_type 1 N Y Y target_type 1 N Y Y Reference_Source * N Y Y
Gene * N Y Y gene_sequence_origin 1 N Y Y gene_id 1 N Y Y gene_name 1 N Y Y Reference_Source * N Y Y
Gene_Element * N Y Y gene_element_type 1 N Y Y gene_element_id 1 N Y Y gene_element_name 1 N Y Y Reference_Source * N Y Y
Substance_Relationship * Y Y Y relationship 1 Y Y Y interaction_type 1 Y Y Y substance_id 1 Y Y Y substance_name 1 Y Y Y amount_type 1 ? Y Y Amount ? ? Y Y Reference_Source * Y Y Y
Substance_Code * Y Y Y code 1 Y Y Y code_system 1 Y Y Y code_system_id 1 Y Y Y code_system_status 1 ? Y Y code_system_change_date 1 ? Y Y comment 1 ? Y Y Reference_Source * Y Y Y

59
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes Single_Substance ? Y Y Y
Structure * Y Y Y structural_representation_type 1 Y Y Y structural_presentation 1 Y Y Y structural_representation_attachment 1 Y Y Y stereochemistry 1 N Y Y optical_activity 1 N Y Y molecular_formula 1 Y Y Y Isotope * N Y Y
nuclide_id 1 N Y Y nuclide_name 1 N Y Y substitution_type 1 N Y Y
Chemical ? N Y Y stoichiometric 1 N Y Y comment 1 N Y Y Reference_Source * N Y Y Non_Stoichiometric 1 N Y Y
number_of_moieties 1 N Y Y Moiety 2+ N Y Y
moiety_role 1 N Y Y moiety_id 1 N Y Y moiety_name 1 N Y Y amount_type 1 N Y Y Amount ? N Y Y
Property * Y Y Y property_type 1 Y Y Y property_name 1 Y Y Y substance_id 1 Y Y Y substance_name 1 Y Y Y

60
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes amount_type 1 Y Y Y Amount ? Y Y Y
Polymer ? N Y Y polymer_class 1 N Y Y polymer_geometry 1 N Y Y copolymer_sequence_type 1 N Y Y comment 1 N Y Y Momomer_Description * N Y Y
number_of_monomers 1 N Y Y monomer_amount_type 1 N Y Y Monomer * N Y Y
monomer_id 1 N Y Y monomer_name 1 N Y Y amount_type 1 N Y Y Amount ? N Y Y
Structural_Repeat * N Y Y number_of_structural_repeat_units 1 N Y Y structural_repeat_unit_amount_type 1 N Y Y Structural_Repeat_Unit + N Y Y
orientation_of_polymerization 1 N Y Y structural_repeat_unit_amount_type 1 N Y Y amount_type 1 N Y Y Amount 1 N Y Y Degree_Polymerization * N Y Y
degree_polymerization 1 N Y Y amount_type 1 N Y Y Amount ? N Y Y
Molecular_Weight * N Y Y molecular_weight_method 1 N Y Y

61
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes molecular_weight_type 1 N Y Y amount_type 1 N Y Y
Source_Material N Y Y Reference_Source N Y Y Property N Y Y Modification * N Y Y
modification_type 1 N Y Y residue_modified 1 N Y Y residue_site 1 N Y Y Structural_Modification * N Y Y
structural_modification_type 1 N Y Y Moiety * N Y Y Molecular_Fragment * N Y Y
role 1 N Y Y molecular_fragment_id 1 N Y Y molecular_fragment_name 1 N Y Y amount_type 1 N Y Y Amount ? N Y Y
Agent_Modification * N Y Y agent_modification_type 1 N Y Y role 1 N Y Y modification_agent 1 N Y Y modification_agent_id 1 N Y Y modification_process 1 N Y Y amount_type 1 N Y Y Amount ? N Y Y
Physical_Modification * N Y Y role 1 N Y Y number_of_parameters 1 N Y Y

62
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes Parameter * N Y Y
parameter 1 N Y Y amount_type 1 N Y Y Amount ? N Y Y
Protein ? N Y Y sequence_type 1 N Y Y number_of_subunits 1 N Y Y disulfide_linkage 1 N Y Y comment 1 N Y Y Modification * N Y Y Property * N Y Y Molecular_Weight * N Y Y Amount 1 N Y Y Protein_Subunit + N Y Y
subunit 1 N Y Y sequence 1 N Y Y length 1 N Y Y n_terminal_modification 1 N Y Y n_terminal_modification_id 1 N Y Y c_terminal_modification 1 N Y Y c_terminal_modification_id 1 N Y Y
Glycosylation * N Y Y glycosylation_type 1 N Y Y n_glycosylation 1 N Y Y o_glycosylation 1 N Y Y c_glycosylation 1 N Y Y
Reference_Source * Y Y Y Nucleic_Acid ? N Y Y
sequence_type 1 N Y Y
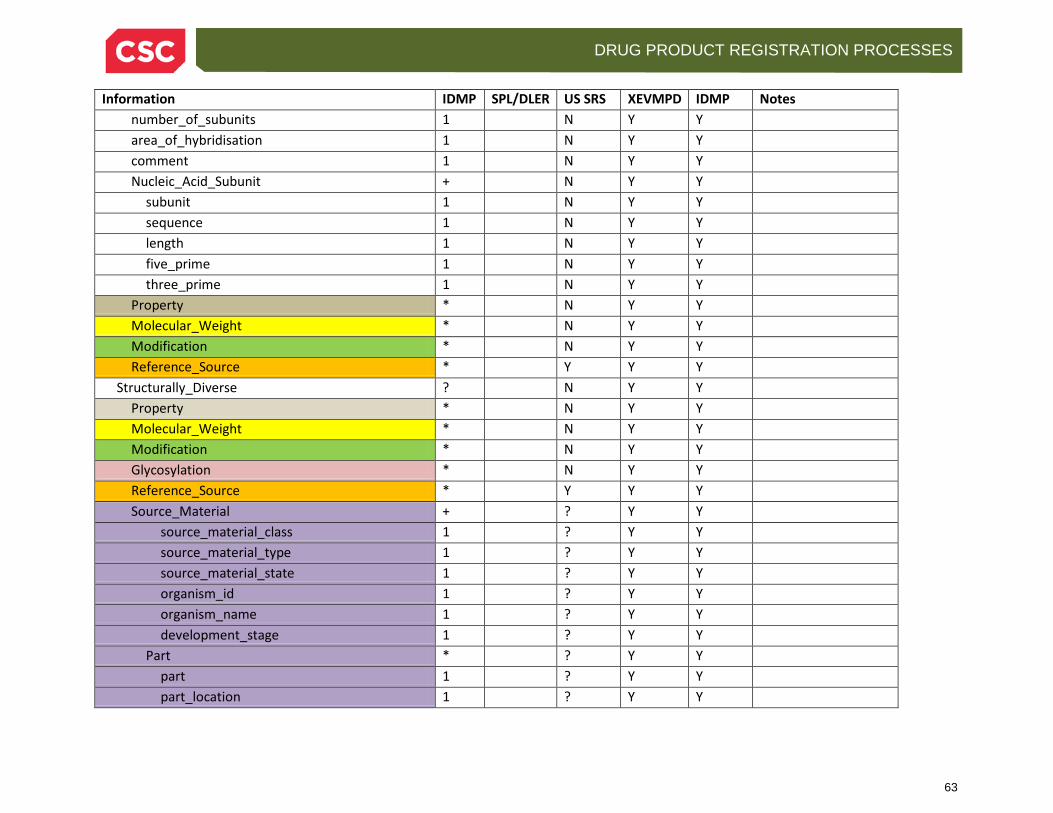
63
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes number_of_subunits 1 N Y Y area_of_hybridisation 1 N Y Y comment 1 N Y Y Nucleic_Acid_Subunit + N Y Y
subunit 1 N Y Y sequence 1 N Y Y length 1 N Y Y five_prime 1 N Y Y three_prime 1 N Y Y
Property * N Y Y Molecular_Weight * N Y Y Modification * N Y Y Reference_Source * Y Y Y
Structurally_Diverse ? N Y Y Property * N Y Y Molecular_Weight * N Y Y Modification * N Y Y Glycosylation * N Y Y Reference_Source * Y Y Y Source_Material + ? Y Y
source_material_class 1 ? Y Y source_material_type 1 ? Y Y source_material_state 1 ? Y Y organism_id 1 ? Y Y organism_name 1 ? Y Y development_stage 1 ? Y Y
Part * ? Y Y part 1 ? Y Y part_location 1 ? Y Y

64
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes Fraction * ? Y Y
material_type 1 ? Y Y fraction 1 ? Y Y
Organism * Y Y Y kingdom 1 Y Y Y phylum 1 Y Y Y class 1 Y Y Y order 1 Y Y Y family 1 Y Y Y genus 1 Y Y Y species 1 Y Y Y species_parenthetical_author 1 Y Y Y species_parenthetical_author_year 1 Y Y Y species_primary_author 1 Y Y Y species_primary_author_year 1 Y Y Y hybrid_species_materinal_organism_id 1 Y Y Y hybrid_species_paternal_organism_id 1 Y Y Y hybrid_type 1 Y Y Y infraspecific_type 1 Y Y Y infraspecific_description 1 Y Y Y infraspecific_parenthetical_author 1 Y Y Y infraspecific_parenthetical_author_year 1 Y Y Y infraspecific_primary_author 1 Y Y Y infraspecific_primary_author_year 1 Y Y Y
Mixture ? ? Y Y mixture_type 1 ? Y Y Mixture_Constituent 2+ ? Y Y
constituent_id 1 ? Y Y constituent_name 1 ? Y Y

65
DRUG PRODUCT REGISTRATION PROCESSES
Information IDMP SPL/DLER US SRS XEVMPD IDMP Notes constituent_requirement 1 ? Y Y
Source_Material * ? Y Y Modification * ? Y Y
Toxicity * Y N N Organism 1 Y N N Test Type 1 Y N N Route 1 Y N N Reported Dose (Normalized Dose) 1 Y N N effective_date 1 Y N N Source 1 Y N N

66
DRUG PRODUCT REGISTRATION PROCESSES
APPENDIX B: ENTITY RELATIONSHIP MODELS APPENDIX B-1: TRS TRACKER DATA MODEL
The following page presents an overview of the TRS Tracker 5.2 data model. Because of the complexity of the entity relationship diagram, the chart is broken into three sections, and equivalent objects on each of the three sections are represented in color in order to visualize the “overlaps.” The upper-left corner represents the product hierarchy. The upper-right corner represents the applications and submissions, and the bottom half shows the pharmaceutical product, composition, and manufacturing structures.

67
DRUG PRODUCT REGISTRATION PROCESSES

68
DRUG PRODUCT REGISTRATION PROCESSES
APPENDIX B-2: IDMP DATA MODEL
The following page includes the detailed structure of the IDMP data model. Source: ISO Standard 11615:2012Invalid source specified..
It is recommended that this page be viewed with at least 200% magnification.

69
DRUG PRODUCT REGISTRATION PROCESSES

70
DRUG PRODUCT REGISTRATION PROCESSES
Worldwide CSC Headquarters
The Americas 3170 Fairview Park Drive Falls Church, Virginia 22042 United States +1.703.876.1000
Europe, Middle East, Africa Royal Pavilion Wellesley Road Aldershot, Hampshire GU11 1PZ United Kingdom +44(0)1252.534000
Australia 26 Talavera Road Macquarie Park, NSW 2113 Australia +61(0)29034.3000
Asia 139 Cecil Street #06-00 Cecil House Singapore 069539 Republic of Singapore +65.6221.9095
About CSC The mission of CSC is to be a global leader in providing technology enabled business solutions and services.
With the broadest range of capabilities, CSC offers clients the solutions they need to manage complexity, focus on core businesses, collaborate with partners and clients, and improve operations.
CSC makes a special point of understanding its clients and provides experts with real-world experience to work with them. CSC is vendor-independent, delivering solutions that best meet each client’s unique requirements.
For 50 years, clients in industries and governments worldwide have trusted CSC with their business process and information systems outsourcing, systems integration and consulting needs.
The company trades on the New York Stock Exchange under the symbol “CSC.”
The information, views and opinions expressed in this paper constitute solely the author‘s views and opinions and do not represent in any way CSC‘s official corporate views and opinions. The author has made every attempt to ensure that the information contained in this paper has been obtained from reliable sources. CSC is not responsible for any errors or omissions or for the results obtained from the use of this information. All information in this paper is provided ―as is,‖ with no guarantee by CSC of completeness, accuracy, timeliness or the results obtained from the use of this information, and without warranty of any kind, express or implied, including but not limited to warranties of performance, merchantability and fitness for a particular purpose. In no event will CSC, its related partnerships or corporations, or the partners, agents or employees thereof be liable to you or anyone else for any decision made or action taken in reliance on the information in this paper or for any consequential, special or similar damages, even if advised of the possibility of such damages.