early changes in immune, coagulation, and organ function
TRANSCRIPT

Institute of Clinical and Experimental Trauma Immunology,
Ulm University Hospital
Prof. Dr. med. Markus Huber-Lang
Early changes in immune, coagulation, and organ function after
clinical and experimental polytrauma
Dissertation submitted in partial fulfillment of the requirements for the degree
of ”Doctor rerum naturalium” (Dr. rer. nat.) of the International Graduate
School in Molecular Medicine Ulm
Rebecca Halbgebauer
Born in Ludwigsburg
2018

Current Dean of the Medical Faculty: Univ.-Prof. Dr. Thomas Wirth
Current Chairman of the Graduate School: Univ.-Prof. Dr. Michael Kühl
Thesis Advisory Committee:
- First supervisor: Univ.-Prof. Dr. Markus Huber-Lang
- Second supervisor: Univ.-Prof. Dr. Anita Ignatius
- Third supervisor: Univ.-Prof. Dr. Martijn van Griensven
External reviewer: Univ.-Prof. Dr. Michael Kirschfink
Day doctorate awarded: November 30, 2018


Results gained in my thesis have previously been published in the following publication:
Halbgebauer R, Braun CK, Denk S, Mayer B, Cinelli P, Radermacher P, Wanner GA,
Simmen HP, Gebhard F, Rittirsch D, Huber-Lang M: Hemorrhagic shock drives glycocalyx,
barrier and organ dysfunction early after polytrauma. J Crit Care 44: 229-237 (2017)
Permission for reuse in this dissertation, including modification of figures, has been obtained
from the publisher.

I
Table of contents
1 Introduction .................................................................................................................. 1
1.1 Acute posttraumatic inflammatory response ........................................................... 1
1.2 Mechanisms of acute trauma-induced coagulopathy .............................................. 3
1.3 Systemic response to severe blood loss ................................................................... 4
1.4 Development of trauma-induced organ dysfunction and failure ............................. 5
1.5 Lack of reliable immune and organ monitoring in trauma care .............................. 6
1.6 Aims of the study .................................................................................................... 7
2 Materials and methods ................................................................................................. 8
2.1 Consumables ........................................................................................................... 8
2.2 Buffers and media ................................................................................................... 8
2.3 Reagents and chemicals........................................................................................... 9
2.4 Primers................................................................................................................... 10
2.5 Antibodies and isotype controls ............................................................................ 10
2.6 Immunoassays ....................................................................................................... 11
2.7 Devices .................................................................................................................. 13
2.8 Software................................................................................................................. 13
2.9 PT study design at Ulm University Hospital ......................................................... 14
2.10 Plasma and serum collection ................................................................................. 14
2.11 Flow cytometric analysis of whole blood.............................................................. 14
2.12 TruCulture® assay ................................................................................................. 15
2.13 PAI-1 plasma detection ......................................................................................... 16
2.14 Neutrophil isolation ............................................................................................... 16
2.15 PAI-1 surface detection on stimulated neutrophils ............................................... 16
2.16 Gene expression analysis in neutrophils ............................................................... 16
2.17 Detection of surface PAI-1 and microvesicles after whole blood stimulation ...... 17
2.18 PT study design at Zurich University Hospital ..................................................... 18

II
2.19 Rotational thromboelastometry analysis ............................................................... 19
2.20 Testing of endothelial glycocalyx and intestinal mucus components in rotational
thromboelastometry ............................................................................................... 20
2.21 Murine model of PT and HS ................................................................................. 20
2.22 Statistical analyses ................................................................................................. 23
3 Results ........................................................................................................................ 24
3.1 Alterations in leukocyte surface molecules after trauma and correlation to shock
parameters ............................................................................................................. 24
3.2 PAI-1 in PT plasma and ex vivo on the granulocyte surface ................................. 34
3.3 Gene expression of PAI-1 and urokinase receptor in stimulated neutrophils ....... 38
3.4 Thromboelastic alterations in blood after PT ........................................................ 39
3.5 Cytokine response to bacterial stimulus in an ex-vivo whole blood model........... 43
3.6 Impact of HS on barrier disturbance and organ dysfunction after PT ................... 49
4 Discussion .................................................................................................................. 65
4.1 Leukocyte surface profiling in polytraumatized patients ...................................... 66
4.2 Coagulopathy in polytraumatized patients ............................................................ 68
4.3 Functional immune monitoring in trauma ............................................................. 69
4.4 HS as a major driver of barrier and organ dysfunction after PT ........................... 70
4.5 Conclusion ............................................................................................................. 73
5 Summary .................................................................................................................... 74
6 List of references ........................................................................................................ 76

III
List of abbreviations
AIS Abbreviated Injury Score
AKI Acute kidney injury
aPTT Abbreviated partial thromboplastin time
ANOVA Analysis of variance
BSA Bovine serum albumin
C3a Complement activation product 3a
C5a Complement activation product 5a
C5aR Complement activation product 5a receptor
CC16 Clara cell secretory protein
CD Cluster of differentiation
cDNA Complementary DNA
CRP C-reactive protein
Ctrl Control
DAMP Damage-associated molecular pattern
DNase Deoxyribonuclease
EDTA Ethylenediaminetetraacetic acid
ELISA Enzyme-linked immunosorbent assay
FGF basic Basic fibroblast growth factor
FITC Fluorescein isothiocyanate
G-CSF Granulocyte colony-stimulating factor
GCS Glasgow Coma Scale
GM-CSF Granulocyte-macrophage colony-stimulating factor
Healthy Healthy volunteers
HR Heart rate
HMGB1 High mobility group box 1
HS Hemorrhagic shock

IV
I-FABP Intestinal fatty acid-binding protein
ICU Intensive care unit
IL Interleukin
IL1-RA Interleukin-1 receptor antagonist
IL-12 (p70) Interleukin -12 heterodimer
IP-10 Interferon gamma-induced protein 10
ISS Injury Severity Score
L-FABP Liver-type fatty acid-binding protein
LPS Lipopolysaccharide
MAP Mean arterial pressure
MCP-1 Monocyte chemoattractant protein 1
MDSC Myeloid-derived suppressor cell
MFI Mean fluorescence intensity
MIP-1 Macrophage inflammatory protein-1
MMP Matrix metalloproteinase
MODS Multiple organ dysfunction syndrome
MOF Multiple organ failure
MP Microparticle
mRNA Messenger RNA
MV Microvesicle
NET Neutrophil extracellular trap
NGAL Neutrophil gelatinase-associated lipocalin
PAI-1 Plasminogen-activator inhibitor-1
PAMP Pathogen-associated molecular pattern
PBS Phosphate-buffered saline
PCT Procalcitonin
PDGF-bb Platelet-derived growth factor subunit B

V
PICS Persistent low-grade inflammation, immunosuppression, and
high protein catabolism syndrome
PMA Phorbol 12-myristate 13-acetate
PRR Pattern recognition receptor
PT Polytrauma
PTC Polytrauma cocktail
qPCR (Real-time) quantitative polymerase chain reaction
R Pearson correlation coefficient
RAGE Receptor for advanced glycation end products
RBC Red blood cell
RISC Revised Injury Severity Classification
ROTEM® Rotational thromboelastometry
RPMI Roswell Park Memorial Institute medium
ROS Reactive oxygen species
SDHA Succinate dehydrogenase complex, subunit A, flavoprotein
variant
SIRS Systemic inflammatory response syndrome
SOFA score Sepsis-related or Sequential Organ Failure Assessment score
Sphingosine-1-P Sphingosine-1-phosphate
TASH score Trauma Associated Severe Hemorrhage score
TBI Traumatic brain injury
TCC Terminal complement complex
TIC Trauma-induced coagulopathy
TLR Toll-like receptor
TM Thrombomodulin
TNF Tumor necrosis factor
TREM Triggering receptor expressed on myeloid cells
TXT Thoracic trauma

VI
uPA Urokinase (plasminogen activator)
uPAR Urokinase receptor
VE-cadherin Vascular endothelial cadherin
VEGF Vascular endothelial growth factor

Introduction
1
1 Introduction
Physical injuries cause more than 5 million deaths worldwide every year, representing the
major cause of death in the population below the age of 44 years and accounting for 10% of
global mortality [89,123]. Despite improved public safety measures, rescue chains, as well
as emergency and intensive care, trauma remains a major burden of disease in all societies
[58]. In 2016, 33,000 people sustained injuries that required primary care in the emergency
room and a subsequent stay at the intensive care unit (ICU) in Germany alone. Patients were
predominantly male, mostly suffered from blunt trauma and stayed in hospital an average of
16 d. Furthermore, 20% developed multiple organ failure and 11.3% died in the hospital
[56]. Polytrauma (PT), defined by a complex injury pattern affecting several physical regions
or organ systems and including the severity of the injuries so that at least one injury or the
combination of several injuries are life-threatening (Injury Severity Score (ISS) ≥ 16)
[3,130,131,169], was present in 55% of patients [56].
1.1 Acute posttraumatic inflammatory response
After severe injury, an immediate systemic response occurs to the traumatic insult.
Breakdown of cellular structures and barriers results in the concomitant release of damage-,
but also pathogen-associated molecular patterns (DAMPs and PAMPs, respectively). These
molecules trigger the activation of different humoral serine protease systems, including the
coagulation and the complement system, with the aim of sealing off injured areas to stop
bleeding and recognizing invading microbes to stop their growth by transmitting the danger
signal to phagocytic leukocytes [27,35,53,85]. Leukocytes can also directly recognize
DAMPs and PAMPs via pattern recognition receptors [5,24,120]; complement and
leukocyte activation induces the secretion of pro-inflammatory cytokines and initiates a
systemic inflammatory response which can culminate in development of the systemic
inflammatory response syndrome (SIRS). Besides clinical parameters [8], SIRS is
characterized by local and systemic production and release of acute-phase proteins,
components of the contact phase and coagulation systems, complement factors, pro- and
anti-inflammatory cytokines, as well as an accumulation of immune cells at the site of tissue
damage [54,69,81]. An excessive immune reaction can reinforce dysfunction of cellular
barriers [86], allowing the entry and generation of more PAMPs and DAMPs, thereby
amplifying a vicious cycle of tissue injury and harmful immune processes [5,54,81] (Fig. 1).
However, in parallel to the pro-inflammatory response, there is also a marked induction of
suppressive processes immediately after injury which may render patients susceptible to

Introduction
2
infection [62,118,166]. It is therefore considered essential that the initial immune reaction
contains both pro- and anti-inflammatory aspects to establish an immune balance facilitating
clearance of the damaged tissues and induction of an effective regeneration [69] (Fig. 1).
Fig. 1: Posttraumatic immune response. Injury induces the release of molecular danger signals, so-called
pathogen-associated and damage-associated molecular patterns (PAMPs and DAMPs, respectively) after cell
damage and destruction of local barriers. These are sensed by activation of the complement and the coagulation
systems which may be reinforced by local hypoxia and acidosis. The danger signal is transmitted to leukocytes
via pattern recognition receptors (PRRs), inducing chemotaxis towards the inflammatory stimulus, release of
cytokines, reactive oxygen species (ROS), and microparticles (MPs), neutrophil extracellular trap (NET)
generation, and phagocytosis of opsonized particles. Effective immune control can aid to establish a balanced
reaction, allowing timely and effective clearance of damaged tissue, repair and regeneration. Adapted and
personalized trauma care may support the avoidance of an escalation of the danger response due to hemorrhage,
extended surgical interventions or nosocomial infection. Influenced by individual risk factors, the innate
immune response can become dysbalanced, with patients developing coagulopathy and barrier dysfunction,
resulting in edema formation, invasion of microorganisms, development of sepsis and multiple organ
dysfunction syndrome (MODS). From Huber-Lang et al. [69].
Apart from pre-existing physical conditions, exogenous factors such as the severity of the
injury itself (“first hit”) and surgical interventions (“second hit”) play an important role in
SIRS development and posttraumatic outcome [149]. Therefore, in the clinical setting,
additional tissue injury should be minimized during life-saving procedures and primary

Introduction
3
surgery immediately after trauma; secondary reconstructive surgery is recommended to be
delayed until after a stabilization phase ranging from a few days up to three weeks [4,54].
1.2 Mechanisms of acute trauma-induced coagulopathy
Tissue damage is almost always associated with rupture of blood vessels, and several studies
have reported that approximately 25% of PT patients arrive at the emergency department
with hemodynamic instability and acute traumatic coagulopathy, more recently termed
trauma-induced coagulopathy (TIC) [16,97-99]. About 40% of deaths after injury are caused
by bleeding [80]. Disintegration of macro- (e.g. the skin) and microbarriers such as
endothelial cell membranes immediately activates multiple hemostatic pathways of the
coagulation system, including fibrinolysis, with rapid consumption of coagulation factors
and thrombocytes [21]. This hypocoagulable state is mainly caused by tissue hypoperfusion
and predominantly involves protein C activation [13]. Moreover, platelets have been shown
to become hyporesponsive to pro-coagulatory stimuli with impaired clot assembly and
stability as early as 30 min after injury [90,180]. The risk of bleeding is reinforced by
hypoxia, acidosis, and hypothermia, also termed the “lethal triad” [106]. Furthermore,
increased secretion of catecholamines contributes to the activation of endothelial cells and
results in downregulation of thrombomodulin and secretion of plasminogen-activator
inhibitor-1 (PAI-1) [40,70,105,183]. Increased adrenaline levels and hypoperfusion also lead
to shedding of endothelial glycocalyx components and thereby amplify endothelial
dysfunction and coagulopathy [76]. Additionally, hyperfibrinolysis was demonstrated to be
a central risk factor for posttraumatic mortality [73,79,153]. Conversely, other studies
indicated that shutdown of fibrinolysis may be even more prominent than hyperfibrinolysis,
especially in severely injured patients, in regard to incidence and mortality [113,114]. As a
further driver of coagulation disturbances, fluid resuscitation especially with crystalloids
seems to increase the severity of TIC since the replaced volume was demonstrated to be
proportional to the degree of coagulopathy [98]. In other studies, however, TIC was also
present in patients who had received little or no fluids [13,14,34,51], suggesting that the
injury pattern and severity may also play a significant role. Currently, apart from addressing
hypoxia, acidosis, and hypothermia, treatment strategies such as balanced resuscitation,
antifibrinolytics, patient-tailored recombinant coagulation factors as well as fibrinogen
concentrates are employed. Guided by conventional coagulation assays or viscoelastic
functional testing, all of these can significantly improve the outcome [27].

Introduction
4
1.3 Systemic response to severe blood loss
Deaths due to bleeding occur for different reasons, ranging from delayed admission to
surgery to detrimental effects of prolonged shock periods despite successful control of
bleeding [15]. Upon arrival at the emergency room, 31% of patients with severe trauma as
defined by Paffrath et al. [130] and Pape et al. [131] present with shock (systolic blood
pressure ≤ 90 mmHg) and 24% with acidosis (base excess ≤ –6 mmol/l) as characteristics of
significant hemorrhage. Of note, in the presence of shock, mortality was increased to 37%
[56].
Severe blood loss and hypovolemia due to blunt or penetrating injury initiate a compensatory
response via sympathetic reflexes, inducing peripheral vasoconstriction in order to maintain
blood pressure. Blood flow in internal organs such as the liver, pancreas, kidney, and
gastrointestinal tract can be reduced (centralized) to such an extent that patients remain
normotensive despite severe shock [115]. Only after the loss of more than 30% of the blood
volume, decreases in cardiac output, blood pressure, and organ perfusion may become
evident [179]. The reduction in oxygen supply and transport of metabolites quickly results
in tissue hypoxia and an increase in lactate levels. In the following, sustained mitochondrial
anaerobic metabolization of glucose can lead to cellular destruction and metabolic acidosis
[19]. If rapid and effective restoration of normotension and removal of the source of bleeding
is not achieved, the accumulation of cell damage leads to disseminated intravascular
coagulopathy, microvascular damage, and SIRS with a mostly unfavorable outcome [115].
Recent studies have furthermore suggested hemorrhagic shock (HS) as a major driver of
systemic inflammation and disintegration of the blood-organ barrier. In rat models of
hemorrhage, a loss in intestinal tight-junction proteins and increased amounts of
mitochondrial DAMPs in the mesenteric lymph were detected, suggesting cellular
breakdown and a deterioration in barrier function [165,187]. These processes seem to be
mainly dependent on complement activation [49]. Shock has also been demonstrated to be a
potent inducer of mitochondrial DAMP release which in turn increases secretion of pro-
inflammatory mediators in neutrophils and other tissues [192]. Furthermore, posttraumatic
bleeding is also considered to play a central role in development of coagulopathy and
complementopathy [18,50,51,74] as well as organ failure [78]. These are presumed to be
especially driven by pathological activation of the endothelium, termed endotheliopathy. In
this regard, disturbances in the endothelial glycocalyx layer after trauma may depend on
shock severity: in severely injured patients, higher catecholamine levels were shown to lead

Introduction
5
to an increase in glycocalyx shedding from the endothelial surface [75,125,126] which is
likely to advance barrier failure and organ damage.
1.4 Development of trauma-induced organ dysfunction and failure
In all patients admitted to emergency departments in Germany, early mortality during the
first 24 h accounted for 5.1% of deaths while another 5.3% of patients died later during the
first 30 d after injury. On the ICU, 34% developed organ failure which culminated in
multiple organ failure (MOF) in 20% of patients [56]. After injury, the aforementioned
activation of serine protease cascades (such as the complement system) and release of
cytokines and other pro-inflammatory mediators from immune cells and tissues, especially
from the gut, prime and mobilize neutrophil granulocytes and other leukocytes
[100,112,122,155]. Neutrophil priming can be augmented by ischemia/reperfusion
conditions. Further iatrogenic contributors are represented by blood products which are
deemed to be immunoactive and contain considerable amounts of pro-inflammatory
coagulation and complement factors as well as cytokines and lipids [41,63]. The risk of MOF
is additionally increased in patients after severe hemorrhage due to tissue hypoperfusion
during the initial shock phase [167,175,178]. Early neutrophilia often turns into neutropenia,
suggesting organ sequestration based on increased adhesion molecules on neutrophils and
endothelium and transmigration into damaged tissues [12,26]. This is accompanied by
increased endothelial and epithelial permeability facilitating electrolyte and protein shifts
and subsequent edema formation. Degranulation of neutrophils in inflamed tissues releases
further pro-inflammatory cytokines, but also proteases and reactive oxygen species which
can damage host tissues and promote bystander injury [163,170]. These processes are
reinforced by ischemia and reperfusion; when substantial amounts of oxygen reenter the
tissue, accumulation of superoxide anions, hydrogen peroxide, and hydroxyl radicals causes
the peroxidation of cell membranes, inducing apoptosis and necrosis and stimulating the
local generation of inflammatory mediators [144,151,172]. In the context of the proceeding
compensatory anti-inflammatory response in parallel to SIRS development, Gentile et al.
established the concept of the persistent low-grade inflammation, immunosuppression, and
high protein catabolism syndrome (PICS). PICS is characterized by defects in innate and
adaptive immunity with decreased activity of dendritic cells, macrophages, and T effector
cells mediated by myeloid-derived suppressor cells (MDSCs), culminating in an increased
risk of late MOF [55].

Introduction
6
1.5 Lack of reliable immune and organ monitoring in trauma care
Despite considerable advances in trauma care in the last decades, some issues remain which
continue to complicate treatment and may impede further improvements in patient
management. There are several well-established techniques in order to monitor organ
function on the ICU, including clinical parameters and organ performance scores such as the
Sepsis-related or Sequential Organ Failure Assessment (SOFA) score [177]. Considering the
deleterious effects of extensive surgical interventions with substantial DAMP and PAMP
generation when the patient is currently at high risk of overwhelming inflammation or
immunosuppression, there is an obvious need for parallel monitoring of the immune function
during the time course after injury. Nevertheless, to date, no tests exist to provide the
clinician with reliable data on the remaining functional capacity of the immune system and
support decision-making in order to optimize timing of necessary interventions or definitive
surgical care. In this context, there is also a lack of information on the alterations in
peripheral blood leukocytes regarding their responsiveness to inflammatory or pathogenic
stimuli in order to implement an immune reaction after injury. Furthermore, not much is
known on how leukocytes may be involved in disturbed coagulation after trauma and
hemorrhage.
Although bleeding is a frequent concomitant condition in trauma patients, there is a lack of
studies assessing the influence of HS on the posttraumatic inflammatory response and
especially its impact on organ damage. For this purpose, reliable and specific organ damage
markers might provide better insight into pathophysiological processes particularly before
the onset of organ dysfunction. Given the central role that a loss in integrity of the endothelial
glycocalyx layer seems to play in posttraumatic coagulopathy and mortality, assessing the
extent of damage inflicted on the glycocalyx after traumatic hemorrhage could also support
an early recognition of barrier breakdown preceding loss of function with improved and
personalized treatment options for the severely injured. However, immune and organ
monitoring during the early response after severe trauma is not yet established neither
scientifically nor clinically.

Introduction
7
1.6 Aims of the study
The following hypotheses were tested in the course of this thesis:
Leukocyte surface molecules interacting with complement, DAMPs, and coagulation are
altered during the time course after injury.
Using a clinically applicable approach, the cellular immune response to an inflammatory
stimulus can be monitored.
The posttraumatic development of coagulopathy can be observed reliably using
functional thromboelastometry.
The organ injury after severe trauma is aggravated by an additional HS, and this damage
can be assessed by specific serum markers.
HS plays a significant role in the loss in endothelial glycocalyx and barrier function after
PT.

Materials and methods
8
2 Materials and methods
2.1 Consumables
BD FACS Clean BD Biosciences, Heidelberg, Germany
BD FACS Flow BD Biosciences, Heidelberg, Germany
BD FACS Shutdown Solution BD Biosciences, Heidelberg, Germany
Cannula, 25 G B. Braun Melsungen AG, Melsungen,
Germany
Centrifugation tube, 15 ml and 50 ml BD Biosciences, Heidelberg, Germany
Cup & Pin mini tem, Munich, Germany
Filter (0.2 µm pore size) GE Healthycare, Solingen, Germany
Nunc™ MicroWell™ 96-Well plate NUNC A/S, Roskilde, Denmark
Nunc™ MaxiSorp™ 96-Well ELISA plate NUNC A/S, Roskilde, Denmark
Mylar® polyester film, 50 µm Du Pont de Nemur, Bad Homburg,
Germany
Polyethylene catheter (0.62 mm) Föhr Medical Instruments GmbH,
Seeheim/Ober-Beerbach, Germany
Polystyrene tube, round bottom, 5 ml BD Biosciences, Heidelberg, Germany
Reaction tube, 0.5 ml, 1.5 ml and 2 ml Eppendorf, Hamburg, Germany
TruCulture® tube (Null, LPS) HOT Screen GmbH, Germany
S-Monovette® 7.5 ml, Clotting Activator/
Serum
Sarstedt, Nümbrecht, Germany
S-Monovette® 9 ml K3E (EDTA) Sarstedt, Nümbrecht, Germany
S-Monovette® 10 ml 9NC, Citrate 3.2%
(1:10)
Sarstedt, Nümbrecht, Germany
Safety Multifly cannula, 18 G Sarstedt, Nümbrecht, Germany
Silkam® 5/0 suture B. Braun Melsungen AG, Melsungen,
Germany
Syringe, 1 ml BD Biosciences, Heidelberg, Germany
2.2 Buffers and media
Aqua dest. Fresenius Kabi, Bad Homburg, Germany

Materials and methods
9
DPBS-/- (Dulbecco’s Phosphate-Buffered
Saline without Ca2+/Mg2+)
Gibco BRL, Grand Island, NY, USA
HBSS+/+ (Hank´s Balanced Salt Solution
with Ca2+/Mg2+)
Gibco BRL, Grand Island, NY, USA
Jonosteril Fresenius Kabi, Bad Homburg, Germany
Sodium chloride solution (0.9%) Fresenius Kabi, Bad Homburg, Germany
RPMI 1640 (Roswell Park Memorial
Institute-Medium)
Gibco BRL, Grand Island, NY, USA
2.3 Reagents and chemicals
AffinityScript QPCR cDNA Synthesis Kit Agilent, Santa Clara, CA, USA
Annexin V-Alexa Fluor 647 Invitrogen, Carlsbad, CA, USA
Annexin V binding buffer (10x) BD Biosciences, Heidelberg, Germany
Bovine serum albumin Sigma-Aldrich, Steinheim, Germany
Brilliant III Ultra-Fast SYBR® Green QPCR
Master Mix
Agilent, Santa Clara, CA, USA
Buprenorphine (Temgesic®) Boehringer, Mannheim, Germany
C5a from human serum Complement Technology, Inc., Tyler, TX,
USA
CellFIX (10x, concentrate) BD Biosciences, Heidelberg, Germany
Chloroform (≥ 99%) Sigma-Aldrich, Steinheim, Germany
CountBright™ Absolute Counting Beads Invitrogen, Carlsbad, CA, USA
DEPC-treated water (RNase-free) Invitrogen, Carlsbad, CA, USA
DNase I Invitrogen, Carlsbad, CA, USA
Ethanol (pure) Sigma-Aldrich, Steinheim, Germany
Ethylenediaminetetraacetic acid (EDTA) Sigma-Aldrich, Steinheim, Germany
Ex-tem Tem, Munich, Germany
FACS Lysing Solution BD Biosciences, Heidelberg, Germany
Fib-tem Tem, Munich, Germany
Ficoll-Paque GE Healthcare, Freiburg, Germany

Materials and methods
10
Heparan sulfate Amsbio, Abingdon, UK
Human recombinant C3a Calbiochem Inc., San Diego, USA
Human recombinant interleukin-1 PeproTech, Rocky Hill, NJ, USA
Human recombinant interleukin-6 Biomol, Hamburg, Germany
Human recombinant interleukin-8 Biomol, Hamburg, Germany
Human recombinant syndecan-1 R&D, Wiesbaden-Nordenstadt, Germany
In-tem Tem, Munich, Germany
Isopropanol Sigma-Aldrich, Steinheim, Germany
Latex beads, amine-modified polystyrene,
fluorescent red (1.0 μm)
Sigma-Aldrich, Steinheim, Germany
Latex beads, carboxylate-modified
polystyrene, fluorescent red, (0.5 μm)
Sigma-Aldrich, Steinheim, Germany
Latex beads, polystyrene (0.3 µm) Sigma-Aldrich, Steinheim, Germany
Lipopolysaccharides from Escherichia coli
O55:B5
Sigma-Aldrich, Steinheim, Germany
Mucin from porcine stomach Type II Sigma-Aldrich, Steinheim, Germany
Norepinephrine Sanofi, Frankfurt am Main, Germany
Phorbol 12-myristate 13-acetate Sigma-Aldrich, Steinheim, Germany
Sevofluran (SevoraneTM) Abbott, Wiesbaden, Germany
TRIzol™ Reagent Thermo Fisher Scientific, Waltham, MA,
USA
2.4 Primers
Human PLAUR QuantiTect® Primer Assay QIAGEN, Hilden, Germany
Human SDHA QuantiTect® Primer Assay QIAGEN, Hilden, Germany
Human SERPINE1 QuantiTect® Primer
Assay
QIAGEN, Hilden, Germany
Human TBP QuantiTect® Primer Assay QIAGEN, Hilden, Germany
2.5 Antibodies and isotype controls
Mouse α-human C3aR, PE BioRad, Kidlington, UK

Materials and methods
11
Mouse α-human TREM-1, PE BioRad, Kidlington, UK
Mouse IgG1 Negative Control, PE BioRad, Kidlington, UK
Mouse α-human C5L2, PE BioLegend, San Diego, CA, USA
Mouse IgG2a, κ Isotype Control, PE BioLegend, San Diego, CA, USA
Mouse α-human CD66b, FITC BD Biosciences, Heidelberg, Germany
Mouse IgM, κ Isotype Control, FITC BD Biosciences, Heidelberg, Germany
Mouse α-human CD88, FITC BioRad, Kidlington, UK
Mouse IgG2a Negative Control, FITC BioRad, Kidlington, UK
Mouse α-human CD142/Tissue factor, FITC BioRad, Kidlington, UK
Mouse IgG1 Negative Control, FITC BioRad, Kidlington, UK
Mouse α-human CD282 (TLR2), PE BioRad, Kidlington, UK
Mouse α-human CD284 (TLR4), PE BioRad, Kidlington, UK
Mouse IgG2a Negative Control, PE BioRad, Kidlington, UK
Mouse α-human PAR-2, PE Santa Cruz Biotechnology, Heidelberg,
Germany
Mouse IgG2a κ Isotype Control, PE Santa Cruz Biotechnology, Heidelberg,
Germany
Mouse α-human TCC, FITC Hycultec, Beutelsbach, Germany
Mouse IgG2a Isotype control, FITC Hycultec, Beutelsbach, Germany
Mouse α-human Thrombomodulin, FITC Abcam, Cambridge, UK
Mouse IgG1 Isotype Control, FITC Abcam, Cambridge, UK
Rabbit α-human PAI-1, FITC Bioss, Woburn, MA, USA
Rabbit α-human RAGE/AGER, FITC Bioss, Woburn, MA, USA
Rabbit IgG Isotype Control, FITC Bioss, Woburn, MA, USA
2.6 Immunoassays
2.6.1 Multiplex immunoassay
Bio-Plex Pro™ Human Cytokine 27-plex
Assay
BioRad, Hercules, CA, USA

Materials and methods
12
2.6.2 ELISA kits for human proteins
ADM / Adrenomedullin ELISA Kit LifeSpan BioSciences, Seattle, WA, USA
Angiopoietin-2 DuoSet ELISA R&D, Wiesbaden-Nordenstadt, Germany
C-Reactive Protein/CRP Quantikine ELISA
Kit
R&D, Wiesbaden-Nordenstadt, Germany
CC16 (Clara Cell 16kD protein) ELISA Kit Elabscience, Bethesda, MD, USA
Claudin-5(CLDN5) ELISA kit Cusabio, College Park, MD, USA
FABP2/I-FABP DuoSet ELISA R&D, Wiesbaden-Nordenstadt, Germany
HS (Heparan Sulfate) ELISA Kit Elabscience Biotechnology Co., Houston,
TX, USA
IL-6 ELISA Set BD Biosciences, Heidelberg, Germany
L-FABP ELISA kit Hycultec, Beutelsbach, Germany
Lipocalin-2/NGAL DuoSet ELISA R&D, Wiesbaden-Nordenstadt, Germany
MUC2 ELISA Kit (Human) Aviva Systems Biology, San Diego, CA,
USA
Serpin E1/PAI-1 DuoSet ELISA R&D, Wiesbaden-Nordenstadt, Germany
Serum Albumin DuoSet ELISA R&D, Wiesbaden-Nordenstadt, Germany
Sphingosine 1 Phosphate ELISA Kit MyBiosource, San Diego, CA, USA
Syndecan-1 DuoSet ELISA R&D, Wiesbaden-Nordenstadt, Germany
Total MMP-9 DuoSet ELISA R&D, Wiesbaden-Nordenstadt, Germany
Total MMP-13 DuoSet ELISA R&D, Wiesbaden-Nordenstadt, Germany
2.6.3 ELISA kits for murine proteins
ALB/Serum Albumin ELISA Kit LifeSpan BioSciences, Seattle, WA, USA
Claudin-5 (CLDN5) ELISA kit Cusabio, College Park, MD, USA
Hspg2 (Basement membrane-specific
heparan sulfate proteoglycan core protein)
ELISA Kit
Amsbio, Abingdon, UK
MMP9/Gelatinase B ELISA Kit LifeSpan BioSciences, Seattle, WA, USA
MMP13 ELISA Kit LifeSpan BioSciences, Seattle, WA, USA

Materials and methods
13
SDC1/Syndecan 1/CD138 ELISA Kit LifeSpan BioSciences, Seattle, WA, USA
THBD/CD141/Thrombomodulin ELISA Kit LifeSpan BioSciences, Seattle, WA, USA
2.7 Devices
Anesthesia device Vapor Drägerwerk AG, Lübeck,
Germany
Bio-Plex® 200 platform BioRad, Hercules, CA, USA
Blood pressure analyzer Data Sciences International, New
Brighton, MN, USA
FACSCanto II BD Biosciences, Heidelberg, Germany
Mx3000P QPCR System Agilent, Santa Clara, CA, USA
Vial Program 1 syringe pump BD Medical, Heidelberg, Germany
Qubit 2.0 Thermo Fisher Scientific, Waltham, MA,
USA
ROTEM® delta Tem, Munich, Germany
Tecan Sunrise Reader Tecan, Crailsheim, Germany
Temperature plate Föhr Medical Instruments, Seeheim/Ober-
Beerbach, Germany
Test tube rotator Snijders, Tilburg, Netherlands
ThermoMixer C Eppendorf, Hamburg, Germany
2.8 Software
BD FACSDivaTM Software (Version 6.1.2) Becton Dickinson, Heidelberg, Gemany
FlowJo (Version 10.2) FlowJo, LLC, Ashland, OR, USA
Ponemah (Version 5.0) Data Sciences International, New
Brighton, MN, USA
SAS (Version 9.3) SAS, Cary, NC, USA
SigmaPlot (Version 11.0) Systat Software, Erkrath, Germany
SigmaStat (Version 3.5) Systat Software, Erkrath, Germany
XFluor4 (Version 4.51) Tecan, Crailsheim, Germany

Materials and methods
14
2.9 PT study design at Ulm University Hospital
A prospective clinical study was conducted in 12 patients after severe PT (ISS ≥ 32) that
were admitted to the University Hospital Ulm between December 2013 and May 2015. The
study protocol was approved by the Independent Local Ethics Committee of the University
of Ulm (approval numbers 244/11 and 94/14). The study was registered on
ClinicalTrials.gov, identifiers NCT00710411 and NCT02682550, and was performed in
accordance with the Declaration of Helsinki and its recent modifications. Exclusion criteria
were age < 18 years, pregnancy, infection with the human immunodeficiency virus,
cardiogenic shock as the primary underlying disease, underlying hematologic disease,
cytotoxic therapy given within the previous 6 months, and the presence of rapidly
progressing underlying disease anticipating death within the next 24 h. Whole venous blood
was drawn at the time of admission (0 h) and 4 h, 12 h, 24 h, 48 h, 5 d and 10 d after trauma
with a maximum divergence from time points of ± 10%. Seven healthy volunteers served as
a control group. Before inclusion, written informed consent was obtained from all patients
and volunteers; if the patient was incapable of making decisions because of intubation,
sedation or altered mental status, informed consent was obtained directly after recovery or
from the next of kin.
2.10 Plasma and serum collection
At all seven time points after injury, blood was drawn into serum, ethylenediaminetetraacetic
acid (EDTA) and sodium citrate tubes and transported on ice. Tubes with EDTA-chelated
and citrated blood were centrifuged immediately at 800 x g and 4°C for 10 min; supernatants
were centrifuged again at 16,000 x g at 4°C for 2 min to remove platelets. For serum
collection, blood was allowed to coagulate at 4°C for 30–120 min; tubes were centrifuged at
1,560 x g at 4°C for 10 min. All supernatants were aliquoted on ice and stored at –80°C until
further analysis.
2.11 Flow cytometric analysis of whole blood
To assess changes in surface molecules related to complement activation, modulation of
coagulatory processes and sensing of DAMPs and PAMPs on leukocytes, leukocytes in
whole blood were analyzed by flow cytometry during the course after trauma and compared
to healthy volunteers. 100 µl of EDTA-anticoagulated blood were stained for the cellular
receptors for the complement component 5a (C5a, C5aR1 and C5aR2), the terminal
complement complex (TCC), plasminogen activator inhibitor-1 (PAI-1), thrombomodulin,
receptor for advanced glycation end products (RAGE), toll-like receptors 2 and 4

Materials and methods
15
(TLR-2/-4), and triggering receptor expressed on myeloid cells (TREM) using fluorescence-
labeled antibodies. Respective IgG isotype controls were used to correct for unspecific
binding. After staining at room temperature in the dark for 20 min, erythrocyte lysis was
performed for 12 min using FACS Lysing Solution. Cells were pelleted at 340 x g for 5 min,
washed with phosphate-buffered saline (PBS), resuspended in 100 µl CellFix and stored at
4°C for no more than 8 h until the analysis by flow cytometry. Light scatter characteristics
were used to distinguish granulocytes, lymphocytes, and monocytes, and at least 2,000 cells
of each population were recorded. The mean fluorescence intensity (MFI) for the cell
populations was analyzed using FlowJo (version 10.2, FlowJo, LLC, Ashland, OR, USA).
2.12 TruCulture® assay
Whole blood ex vivo stimulation was performed using TruCulture® tubes with blood taken
4 h, 24 h and 5 d after injury. Tubes were prefilled by the manufacturer under standardized
conditions with 2 ml culture medium, and contained unfractionated heparin as an
anticoagulant at a final concentration of 50 IU/ml. To assess the immune reaction to a
bacterial stimulus, tubes with or without 100 ng/ml lipopolysaccharide (LPS, from
Escherichia coli, O55:B5, or Null) were used. The TruCulture® system minimizes the risk
of contamination, but also reduces intra-individual variation by direct blood withdrawal
without additional pipetting steps [117]. Tubes were stored at –20°C and thawed in a water
bath at 37°C immediately before use. 1 ml of blood was drawn into blood collection tubes
from healthy volunteers and PT patients at indicated time points. After incubation at 37°C
for 24 h, sedimented cells were separated from the supernatant using a valve, and
supernatants were stored at –80°C until analysis. In order to increase clinical applicability,
a second set of tubes ± LPS was taken at the second time point (24 h after trauma), and
incubation at 37°C was reduced to 4 h before supernatant collection. Samples were analyzed
using a multiplexed sandwich immunoassay on a multiplex platform according to the
manufacturer’s recommendations. Since many samples were out of range for the pro-
inflammatory marker interleukin (IL)-6, they were re-assessed using the IL-6 Quantikine kit
(BD Biosciences, Germany). Concentrations of cytokines after LPS stimulus in supernatants
from PT patients were compared with those from healthy volunteers. To evaluate the shorter
incubation period, the differences in released mediators in stimulated and control samples
after 4 h incubation were compared to those after 24 h incubation.

Materials and methods
16
2.13 PAI-1 plasma detection
PAI-1 concentrations in EDTA-plasma of PT patients and healthy volunteers were measured
using a commercially available enzyme-linked immunosorbent assay (ELISA) kit strictly in
accordance to the manufacturer’s protocol (R&D Systems and BD Biosciences,
respectively).
2.14 Neutrophil isolation
Venous blood from healthy volunteers was drawn into tubes containing 3.2% sodium citrate
from the antecubital vein. After mixing the blood with an equal volume of isotonic saline
(0.9% NaCl), 20 ml were layered carefully over 10 ml of Ficoll in a 50 ml centrifugation
tube and cells were separated at 340 x g for 30 min with slow brakes. The supernatant
containing plasma, mononuclear cells and Ficoll was removed, and the remaining pellet was
mixed with dextran in 0.9% NaCl at a final concentration of 1%. After sedimentation of
erythrocytes for 30 min, the supernatant was collected and neutrophils were pelleted by
centrifugation at 340 x g for 5 min. The remaining erythrocytes were lysed with distilled
water, followed by addition of 2.7% NaCl to restore isotonic conditions. Neutrophils were
pelleted and resuspended in medium at 5–10*106/ml.
2.15 PAI-1 surface detection on stimulated neutrophils
For surface staining, freshly isolated neutrophils were resuspended in RPMI 1640 + 0.1%
bovine serum albumin (BSA) at 5*106/ml. After addition of LPS (5 µg/ml) or the so-called
polytrauma cocktail (PTC) [65], containing complement component 3a (C3a, 500 ng/ml),
C5a (10 ng/ml), IL-1 (200 pg/ml), IL-6 (500 pg/ml), and IL-8 (150 pg/ml), cells were
incubated at 37°C for 1 h while rotating at 80 rpm to simulate blood circulation. After
centrifugation, 5*105 cells were stained with anti-PAI-1 antibody or the isotype control at
4°C for 20 min, followed by washing and fixation. Measurement of PAI-1 surface binding
on at least 10,000 cells was performed by flow cytometry and analyzed using FlowJo.
2.16 Gene expression analysis in neutrophils
For messenger RNA (mRNA) isolation, neutrophils isolated from citrated blood were
adjusted to 107/ml in Roswell Park Memorial Institute (RPMI) medium + 0.1% BSA and
stimulated as described in 2.15. After pelleting at 800 x g for 5 min at 4°C, 2*107/ml cells
per sample were lysed using Trizol, and mRNA was isolated following the manufacturer’s
protocol. In brief, the cell lysate was mixed with chloroform and centrifuged to separate the
aqueous phase containing RNA from the interphase and the phenol-chlorophorm phase
containing DNA and cellular proteins. RNA was precipitated with isopropanol, pelleted,

Materials and methods
17
washed with 75% ethanol, dried and solubilized in ribonuclease-free water by incubation at
60°C for 10 min. Until further analysis, RNA was stored at –80°C.
RNA concentrations were determined using the Qubit fluorometer and the respective
quantitation assay, employing a dye that only produces a fluorescent signal when bound to
RNA. Genomic DNA was removed by deoxyribonuclease (DNase) I digestion of 1 µg RNA
per sample. RNA was mixed with 10x DNase I Reaction Buffer, DNase and diethyl
pyrocarbonate water and incubated for 15 min at room temperature. To inactivate the DNase,
EDTA was added to a concentration of 2.5 mM before heating the sample at 65°C for 10 min.
For reverse transcription of 550 ng RNA per sample, the AffinityScript QPCR cDNA
Synthesis Kit was used. After mixing RNA, first strand master mix, oligo(DT) primers and
AffinityScript RT/RNase Block enzyme mixture, samples were incubated at 25°C for 5 min
to allow primer annealing. Complementary DNA (cDNA) synthesis was performed at 42°C
for 15 min and the reaction was terminated at 95°C for 5 min. cDNA was diluted 1:5 with
water and stored at –20°C until further analysis.
Gene expression was measured using the Brilliant III Ultra-Fast SYBR® Green QPCR
Master Mix and commercially available primers in reaction volumes of 20 µl in duplicates
on the Mx3000P real-time quantitative PCR (qPCR) system. Expression was compared to
the neutrophil house-keeping genes succinate dehydrogenase complex, subunit A,
flavoprotein variant (SDHA) and TATA-binding protein [91] employing the 2–ΔΔCT method
[95]. For final analyses, SDHA was used as a reference.
2.17 Detection of surface PAI-1 and microvesicles after whole blood stimulation
In order to analyze membrane-bound molecules on leukocytes in a more physiological
setting, EDTA-anticoagulated blood was drawn from healthy volunteers. Blood was
stimulated with PTC, 5 µg/ml LPS or 20 ng/ml of the protein kinase C activator phorbol 12-
myristate 13-acetate (PMA) [23] at 37°C and rotating at 80 rpm to simulate blood circulation.
Whole blood was stained for PAI-1 for 20 min at room temperature. Following erythrocyte
lysis, cells were washed and fixed and fluorescence was detected by flow cytometry.
To assess microvesicles released by neutrophils during stimulation, plasma was obtained by
centrifugation at 2,200 x g for 15 min, and stored at –20°C. Plasma was mixed with Alexa
Fluor 647-labeled Annexin V, fluorescein isothiocyanate (FITC)-labeled anti-cluster of
differentiation (CD)66b antibody and CountBright™ Absolute Counting Beads in
Annexin V binding buffer (filtered with 0.2 µm pore size). 20 mM EDTA was added as a
control to inhibit Annexin V binding. After incubation for 15 min in the dark, samples were

Materials and methods
18
washed with binding buffer and measured using the FACSCanto II flow cytometer. For
compensation, single stain controls were used. Gates were set using latex beads with defined
sizes (0.3 µm, 0.5 µm and 1 µm) and Annexin V-positive microvesicles with a diameter of
up to 1 µm were analyzed. The gating strategy is shown in Fig. 2. For volume control,
measurement was stopped after detecting 1000 counting beads corresponding to 2 µl of
plasma.
Fig. 2: Representative gating strategy for detection of microvesicles in plasma after whole-blood
stimulation. After gating for size according to light scatter characteristics (side scatter area, SSC-A, and
forward scatter area, FSC-A), annexin-V-positive vesicles were selected and mean fluorescence intensity as
well as percentage of fluorescein isothiocyanate (FITC)-positive microvesicles were analyzed. Counting beads
were used for volume control. Here, samples after control (Ctrl) or phorbol 12-myristate 13-acetate (PMA)
stimulus are shown.
2.18 PT study design at Zurich University Hospital
Since the low number of patients in the study Ulm PT group did not allow stratification for
the presence or absence of HS, a randomly selected subcohort of 30 patients from a mono-
centered, observational, prospective study at the University Hospital Zurich (Trauma Level
I Center) including a total of 104 patients (ClinicalTrials.gov identifier: NCT02508272,

Materials and methods
19
[146,147]) was used. The study was approved by the Cantonal Ethic Commission Zurich
(StV 26–2007) and performed in accordance with local and international guidelines. Patients
with an ISS ≥ 18, age ≥ 18 y, and time passed since the injury < 6 h were included under
informed consent. Blood was drawn upon arrival at the emergency room (day 0) and daily
during the next 21 d.
The subcohort of 30 patients was stratified for signs of manifest HS [88,108,190] on day 0;
the presence of HS was assumed when at least one of the following criteria was fulfilled: a
Trauma Associated Severe Hemorrhage (TASH) score ≥ 10, base excess < −6 mmol/l, lactate
≥ 2.5 mmol/l, and/or red blood cell (RBC) transfusion during the first 24 h > 2 U. Venous
whole blood was drawn into serum or sodium citrate collection tubes and centrifuged at 2000
x g at 4°C for 10 min. Serum concentrations of IL-6, the organ damage markers clara cell
secretory protein (CC16), neutrophil gelatinase-associated lipocalin (NGAL), intestinal fatty
acid-binding protein (I-FABP), and liver-type fatty acid-binding protein (L-FABP), albumin,
sphingosine-1-phosphate, thrombomodulin, angiopoietin-2, matrix metalloproteinases
(MMP)-9 and -13, claudin-5, vascular endothelial (VE-) cadherin, syndecan-1, heparan
sulfate and mucin-2 were determined serially using commercially available sandwich ELISA
kits. After determining optimal dilution conditions, samples were incubated on ELISA plates
over night at 4°C; afterwards, samples were transferred to the next plates and ELISAs were
performed according to the manufacturers’ recommendations, allowing measurement of up
to 4 parameters in one sample. According to a previous publication, serial ELISA
measurements produce valid and reliable data [127]. Concentrations of fibrinogen, D-dimer,
soluble TCC and C5a in citrated plasma were determined by respective commercial
sandwich ELISA kits. To correct for dilution effects after mass transfusion, absolute serum
protein was determined using a bicinchonic acid assay (Pierce). Measurements in patients
with signs of HS were compared to those without shock; six healthy volunteers served as
controls.
2.19 Rotational thromboelastometry analysis
Due to the low sample volume available from PT patients and a required volume of 310 µl
for common rotational thromboelastometry (ROTEM®) analyses, small-volume ROTEM®
was established in collaboration with Prof. van Griensven, Munich; employing smaller cups
and pins allowed a reduction of the sample volume to 105 µl per analysis. To avoid variations
in measurements due to long time periods between patient inclusions, citrate plasma from
patients included in the Ulm PT study was stored at –80°C until analysis. For analysis,
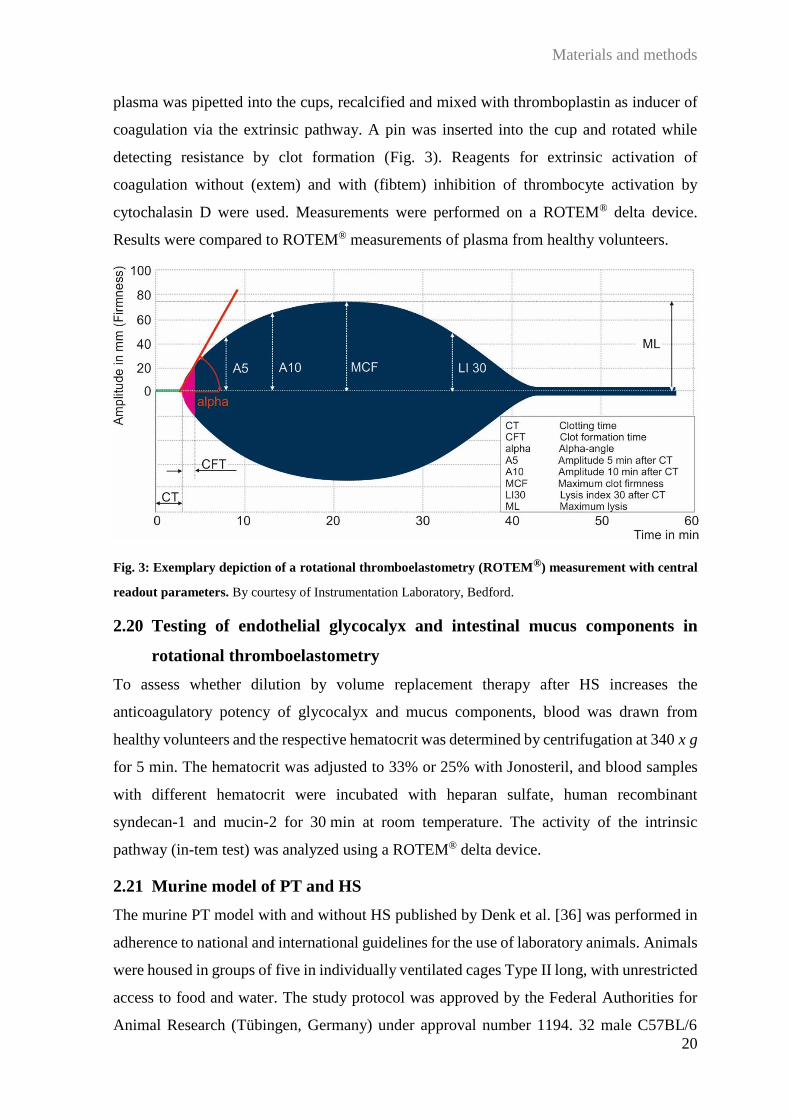
Materials and methods
20
plasma was pipetted into the cups, recalcified and mixed with thromboplastin as inducer of
coagulation via the extrinsic pathway. A pin was inserted into the cup and rotated while
detecting resistance by clot formation (Fig. 3). Reagents for extrinsic activation of
coagulation without (extem) and with (fibtem) inhibition of thrombocyte activation by
cytochalasin D were used. Measurements were performed on a ROTEM® delta device.
Results were compared to ROTEM® measurements of plasma from healthy volunteers.
Fig. 3: Exemplary depiction of a rotational thromboelastometry (ROTEM®) measurement with central
readout parameters. By courtesy of Instrumentation Laboratory, Bedford.
2.20 Testing of endothelial glycocalyx and intestinal mucus components in
rotational thromboelastometry
To assess whether dilution by volume replacement therapy after HS increases the
anticoagulatory potency of glycocalyx and mucus components, blood was drawn from
healthy volunteers and the respective hematocrit was determined by centrifugation at 340 x g
for 5 min. The hematocrit was adjusted to 33% or 25% with Jonosteril, and blood samples
with different hematocrit were incubated with heparan sulfate, human recombinant
syndecan-1 and mucin-2 for 30 min at room temperature. The activity of the intrinsic
pathway (in-tem test) was analyzed using a ROTEM® delta device.
2.21 Murine model of PT and HS
The murine PT model with and without HS published by Denk et al. [36] was performed in
adherence to national and international guidelines for the use of laboratory animals. Animals
were housed in groups of five in individually ventilated cages Type II long, with unrestricted
access to food and water. The study protocol was approved by the Federal Authorities for
Animal Research (Tübingen, Germany) under approval number 1194. 32 male C57BL/6

Materials and methods
21
mice at the age of 8–9 weeks (Charles River WIGA GmbH, Sulzfeld, Germany) with a mean
body weight of 25 g (± 2.5 g) were randomly assigned to sham treatment, PT, HS, or
combined PT with HS. Anesthesia was induced by inhalation of 2.5% sevoflurane in oxygen
in a tube for narcosis induction and maintained during the experiment after transferring
animals to a face mask. Mice received 0.03 mg/kg body weight buprenorphine
subcutaneously for analgesia. Animals were placed on a feed-back loop temperature control
device with connected rectal probe to maintain a core temperature of 37°C. Chest and upper
abdomen, legs and head were shaved. Sham animals were catheterized and monitored, but
did not undergo trauma or HS. An overview of the study protocol is shown in Fig. 4.
Fig. 4: Overview of the study protocol. Animals in deep anesthesia underwent polytrauma including thoracic
trauma (TXT), traumatic brain injury (TBI), a closed fracture and soft tissue injury. After instrumentation, HS
was applied to achieve a mean arterial pressure (MAP) of 30 mmHg. MAP, heart rate (HR) and temperature
were monitored until termination of the experiment 240 min after trauma. From Denk et al. [36].
2.21.1 Trauma application
PT was induced by combining blunt bilateral chest trauma, traumatic brain injury and femur
fracture/soft tissue trauma. Animals were fixated in a supine position, and blunt chest trauma
was induced using a blast wave generator as established by Knöferl et al. [84]. Mice were
positioned 1.6 cm below the generator’s nozzle with its lower edge in alignment with the
costal margin. The upper section of the nozzle served as pressure reservoir which was
connected to a storage tank of compressed air and separated from the lower opening by a 50-
µm polyester film. The film would burst when the pressure in the upper part, created by
opening the storage tank, exceeded 17 bar, and generate a reproducible single blast wave
with a duration of 1−2 ms. The described protocol induces local and systemic inflammation,
but does not cause injuries of the bony thorax or of abdominal organs [83,84,94,133,134].

Materials and methods
22
For induction of traumatic brain injury, animals were positioned on the abdomen and fixated.
The left galea aponeurotica frontoparietal was incised with a scalpel and the scalp was
exposed. A weight of 333 g was dropped on the cranial vault of the left hemisphere from a
height of 2.0 cm, inducing a contusion of the underlying brain tissue without injuring the
bony lamina interna [157].
A closed fracture was applied on the center of the right femur using a weight-drop device
modified from Bonnarens and Einhorn [10]. After positioning of the limb on the device, a
weight of 50 g was dropped from 120 cm. The resulting dynamic impulse induced a dynamic
three-point bending, leading to a transverse diaphyseal fracture and injury of the surrounding
soft tissue.
2.21.2 Induction of pressure-controlled HS
After trauma application, mice were instrumented aseptically by insertion of an arterial
polyethylene catheter (diameter 0.62 mm) in the distal left femoral artery, using a minimally
invasive dissection technique. The arterial catheter was connected to a device for blood
pressure monitoring. The same technique was employed to insert a catheter into the left
jugular vein for volume replacement and continuous infusion of norepinephrine regulated
by a perfusor. Hemorrhage was induced by controlled bleeding via the arterial catheter until
a mean arterial pressure of 30 mmHg ± 5 mmHg was reached; this was maintained over a
period of 60 min. Afterwards, animals were reperfused over the venous catheter with a bolus
of 400 µl Jonosteril over 5 min and per slower infusion of additional Jonosteril until the
fourfold volume of the blood drawn during hemorrhage had been given. Animals in the sham
or PT groups only received the bolus of 400 µl. After HS, mice were monitored for 120 min
after a standardized protocol, maintaining a mean arterial pressure of 50 mmHg by reducing
the depth of anesthesia or by norepinephrine support (0.01−0.12 µg/kg per min) via the
venous catheter. 240 min after PT or sham treatment, animals were exsanguinated by
thoracotomy and cardiac puncture. Blood was collected in EDTA tubes; plasma was
obtained by centrifugation at 800 x g and 4°C for 5 min; supernatants were centrifuged at
13,000 x g and 4°C for 2 min to remove platelets and stored at −80°C for further analyses.
2.21.3 Plasma analysis
Mouse plasma was analyzed using commercially available sandwich ELISA kits for serum
albumin, claudin-5, heparan sulfate proteoglycan core protein, MMP-9 and -13, syndecan-1
and thrombomodulin following the manufacturers’ recommendations.

Materials and methods
23
2.22 Statistical analyses
Experimental results were compared using paired t-test for two groups or one-way analysis
of variance (ANOVA) followed by Student-Newman-Keuls post-hoc test for three or more
groups. Correlation analyses were performed using Pearson Product Moment Correlation.
Differences in leukocyte cytokine secretion after endotoxin stimulus were compared by
Student’s t-test. Clinical parameters of patients with and without HS were compared using
the Chi-square test in case of categorical variables and Student’s t-test in case of continuous
parameters. These statistical analyses were performed using SigmaPlot. To evaluate whether
the presence or absence of HS significantly changed overall plasma values of barrier and
organ molecules during the time course after trauma, repeated-measures ANOVA was used
employing SAS. For all ANOVA testing, no formal statistical test on normality was applied
due to its limited validity regarding the available sample size [140]. Results are presented as
mean ± standard error of the mean. A p-value < 0.05 was considered statistically significant.
Results
24
3 Results
3.1 Alterations in leukocyte surface molecules after trauma and correlation to
shock parameters
In order to analyze whether a severe trauma was reflected by changes in the surface
expression patterns of central receptors and molecules interacting with DAMPs/PAMPs,
complement, and coagulation, whole blood was stained during the course after injury and
the surface expression was measured by flow cytometry. Expression levels were then
correlated to clinical parameters with focus on hemorrhagic shock.
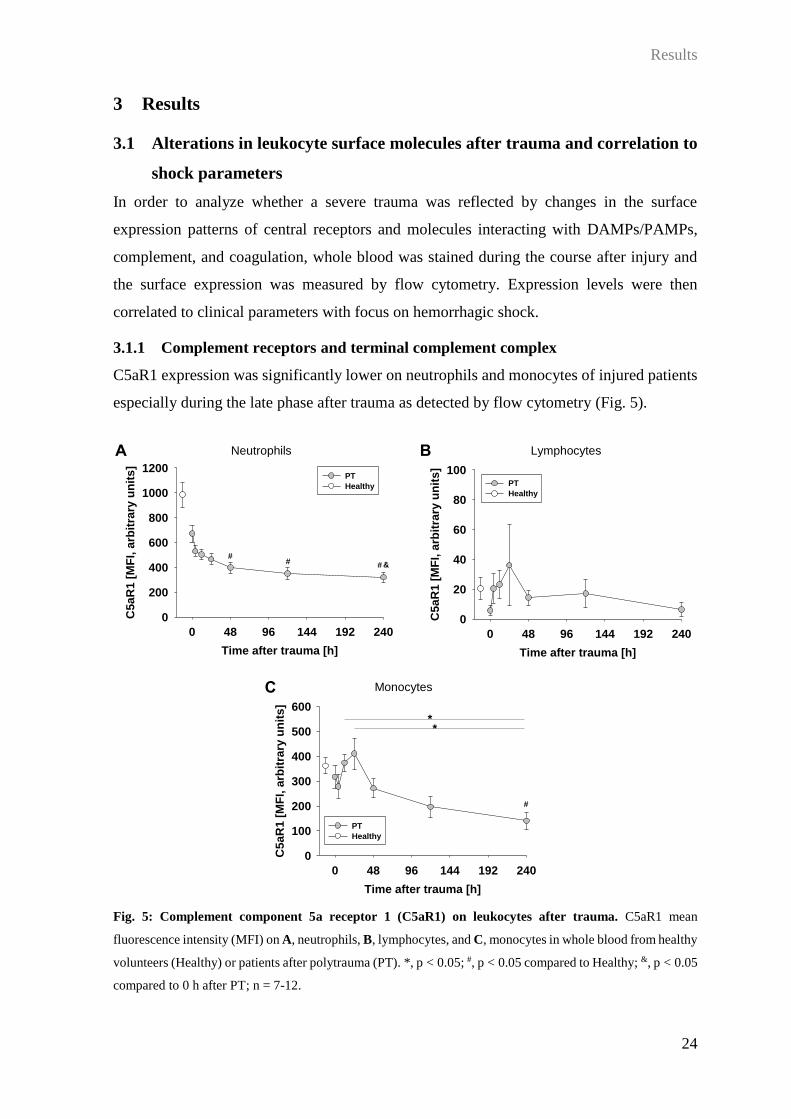
3.1.1 Complement receptors and terminal complement complex
C5aR1 expression was significantly lower on neutrophils and monocytes of injured patients
especially during the late phase after trauma as detected by flow cytometry (Fig. 5).
Fig. 5: Complement component 5a receptor 1 (C5aR1) on leukocytes after trauma. C5aR1 mean
fluorescence intensity (MFI) on A, neutrophils, B, lymphocytes, and C, monocytes in whole blood from healthy
volunteers (Healthy) or patients after polytrauma (PT). *, p < 0.05; #, p < 0.05 compared to Healthy; &, p < 0.05
compared to 0 h after PT; n = 7-12.
A
Time after trauma [h]
0 48 96 144 192 240
C5
aR
1 [
MF
I, a
rbit
rary
un
its
]
0
200
400
600
800
1000
1200PT
Healthy
##
# &
Time after trauma [h]
0 48 96 144 192 240
C5
aR
1 [
MF
I, a
rbit
rary
un
its
]
0
20
40
60
80
100PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
C5
aR
1 [
MF
I, a
rbit
rary
un
its
]
0
100
200
300
400
500
600
PT
Healthy
**
#
A B
C
Neutrophils Lymphocytes
Monocytes

Results
25
Similarly, C5aR2 was reduced on neutrophils 10 d after injury (Fig. 6 A), and demonstrated
a significant decrease already after 4 h on monocytes when compared to controls (Fig. 6 B).
Of note, expression of both C5a receptors was considerably lower on lymphocytes compared
to granulocytes and monocytes (Fig. 6 C).
Fig. 6: Complement component 5a receptor 2 (C5aR2) on leukocytes after trauma. C5aR2 mean
fluorescence intensity (MFI) on A, neutrophils, B, lymphocytes, and C, monocytes in whole blood from healthy
volunteers (Healthy) or patients after polytrauma (PT). #, p < 0.05 compared to Healthy; &, p < 0.05 compared
to 0 h after PT; n = 4-7.
Time after trauma [h]
0 48 96 144 192 240
C5a
R2 [
MF
I, a
rbit
rary
un
its]
0
500
1000
1500
2000PT
Healthy
&
Time after trauma [h]
0 48 96 144 192 240
C5a
R2 [
MF
I, a
rbit
rary
un
its]
0
100
200
300
400PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
C5a
R2 [
MF
I, a
rbit
rary
un
its]
0
500
1000
1500
2000
PT
Healthy
#
A B
C
Neutrophils Lymphocytes
Monocytes

Results
26
On neutrophils and lymphocytes, TCC could not be detected on the cell surface neither in
healthy volunteers nor in trauma patients. In contrast, on monocytes, considerable membrane
formation of the TCC was measured, which, after an early slight reduction, increased
significantly in PT patients 24 h and 48 h after injury, and returned to baseline levels on days
5 and 10 (Fig. 7).
Fig. 7: Terminal complement complex (TCC) on monocytes. Mean fluorescence intensity (MFI) on
monocytes in whole blood from healthy volunteers (Healthy) or patients after polytrauma (PT). *, p < 0.05; #,
p < 0.05 compared to Healthy; &, p < 0.05 compared to 0 h after PT; $, p < 0.05 compared to 240 h after PT; n
= 7-12.
The TCC formation at 4 h after injury was significantly associated with the number of
applied erythrocyte concentrates during the first day (Fig. 8).
Fig. 8: Pearson correlation between transfused blood products and terminal complement complex (TCC)
expression on monocytes. Number of applied erythrocyte concentrates during the first day in correlation with
TCC on monocytes as determined by mean fluorescence intensity (MFI). R, Pearson correlation coefficient.
Time after trauma [h]
0 48 96 144 192 240
TC
C [
MF
I, a
rbit
rary
un
its]
0
200
400
600
800
1000
1200
1400PT
Healthy
& &$ $
**
Erythrocyte concentrates [units]
0 2 4 6 8 10
TC
C [
MF
I, a
rbit
rary
un
its]
0
100
200
300
400
500
600
700
r = 0.87p = 0.005

Results
27
3.1.2 DAMP and PAMP receptors
While RAGE expression was detectable, but not significantly increased on neutrophils and
monocytes in patients during the later course after PT, there were no alterations in its surface
levels on lymphocytes (Fig. 9).
Fig. 9: Receptor for advanced glycation end products (RAGE) on peripheral leukocytes after trauma.
RAGE mean fluorescence intensity (MFI) on A, neutrophils, B, lymphocytes, and C, monocytes in whole
blood from healthy volunteers (Healthy) or patients after polytrauma (PT). N = 7-11.
Time after trauma [h]
0 48 96 144 192 240
RA
GE
[M
FI, a
rbit
rary
un
its]
0
100
200
300
400
PT
Healthy
Time after trauma [h]
0 48 96 144 192 240R
AG
E [
MF
I, a
rbit
rary
un
its]
0
50
100
150
200
250
300PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
RA
GE
[M
FI, a
rbit
rary
un
its]
0
100
200
300
400
500PT
Healthy
A B
C
Neutrophils Lymphocytes
Monocytes

Results
28
However, RAGE expression on lymphocytes measured at the early time points after injury
correlated significantly with the base excess and the number of transfused erythrocyte
concentrates (Fig. 10 A-C). Furthermore, expression on monocytes 48 h and 5 d post trauma
correlated negatively with blood lactate at admission and positively with erythrocyte
concentrates given during the first day (Fig. 10 D, E).
Fig. 10: Pearson correlation between receptor for advanced glycation end products (RAGE) expression
on leukocytes and clinical parameters. Shown are correlations between initial base excess, the number of
applied erythrocyte concentrates during the first day, and blood lactate concentrations upon admission and
RAGE expression on lymphocytes 0 h (A), 4 h (B), and 12 h (C) after trauma and on monocytes 48 h (D) and
5 d (E) after injury as determined by mean fluorescence intensity (MFI). R, Pearson correlation coefficient.
Base excess [mmol/l]
-6 -4 -2 0 2 4RA
GE
MF
I [a
rbit
rary
un
its]
0
50
100
150
200
250
300
r = 0.64p = 0.04
Base excess [mmol/l]
-6 -4 -2 0 2 4RA
GE
MF
I [a
rbit
rary
un
its]
0
200
400
600
800
r = 0.92p = 0.001
Erythrocyte concentrates [units]
0 2 4 6 8 10RA
GE
MF
I [a
rbit
rary
un
its]
0
100
200
300
400
r = 0.82p = 0.007
Erythrocyte concentrates [units]
0 2 4 6 8 10RA
GE
MF
I [a
rbit
rary
un
its]
0
200
400
600
800
1000r = 0.72p = 0.04
Lactate [mmol/l]
0 1 2 3 4RA
GE
MF
I [a
rbit
rary
un
its]
0
100
200
300
400
500
600
r = 0.74
p = 0.03
A B
C D
E

Results
29
The expression of TLR-2 and TLR-4 was notably decreased on all leukocyte types to around
50% especially during the early time course after trauma, but alterations were not significant
compared to healthy volunteers (Fig. 11).
Fig. 11: Toll-like receptor (TLR) expression on leukocytes after trauma. Mean fluorescence intensity
(MFI) of TLR-2 (A, C, E) and TLR-4 (B, D, F) on granulocytes (A, B), lymphocytes (C, D) and monocytes
(E, F) of healthy volunteers (Healthy) and polytrauma patients (PT). N = 4-12.
Time after trauma [h]
0 48 96 144 192 240
TL
R-4
[M
FI, a
rbit
rary
un
its]
0
20
40
60
80
100
120
140
160PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
TL
R-4
[M
FI, a
rbit
rary
un
its]
0
20
40
60
80
100
120
140
160PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
TL
R-4
[M
FI, a
rbit
rary
un
its]
0
100
200
300
400PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
TL
R-2
[M
FI, a
rbit
rary
un
its]
0
20
40
60
80
100PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
TL
R-2
[M
FI, a
rbit
rary
un
its]
0
10
20
30
40
50
60PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
TL
R-2
[M
FI, a
rbit
rary
un
its]
0
100
200
300
400
500PT
Healthy
A B
C D
E F
Neutrophils
Lymphocytes
Monocytes
Neutrophils
Lymphocytes
Monocytes

Results
30
Nevertheless, TLR-2 on monocytes 4 h and 12 h after injury revealed a significant positive
correlation with the patients’ TASH score upon admission (Fig. 12 A, B). Similarly, TLR-4
on monocytes 12 h after trauma correlated positively with the TASH score (Fig. 12 C).
Several negative correlations were found for TLR-4 expression: at 24 h on granulocytes and
monocytes with base excess, and on lymphocytes with erythrocyte concentrates (Fig. 12
D-F).
Fig. 12: Toll-like receptor (TLR) expression in correlation to clinical parameters. Pearson correlation
between Trauma Associated Severe Hemorrhage (TASH) score, base excess, and the number of applied
erythrocyte concentrates during the first day with TLR-2 (A, B) and TLR-4 (C-F) on monocytes 4 h (A) and
12 h (B, C) after trauma, and on granulocytes (D), monocytes (E), and lymphocytes (F) 24 h after injury as
determined by mean fluorescence intensity (MFI). R, Pearson correlation coefficient.
TASH score [points]
0 2 4 6 8 10 12 14TL
R-2
[M
FI,
arb
itra
ry u
nit
s]
0
100
200
300
400
500
600r = 0.71p = 0.03
TASH score [points]
0 2 4 6 8 10 12 14TL
R-2
[M
FI, a
rbit
rary
un
its]
0
100
200
300
400
500 r = 0.66p = 0.04
Base excess [mmol/l]
-6 -4 -2 0 2 4TL
R-4
[M
FI,
arb
itra
ry u
nit
s]
0
50
100
150
200r = -0.68p = 0.03
Base excess [mmol/l]
-6 -4 -2 0 2 4TL
R-4
[M
FI, a
rbit
rary
un
its]
0
100
200
300
400
500r = -0.7p = 0.03
Erythrocyte concentrates [units]
0 2 4 6 8 10TL
R-4
[M
FI, a
rbit
rary
un
its]
0
20
40
60
80r = -0.72p = 0.02
TASH score [points]
0 2 4 6 8 10 12 14TL
R-4
[M
FI, a
rbit
rary
un
its]
0
100
200
300
400r = 0.68p = 0.03
A B
C D
E F

Results
31
While TREM-1 expression on granulocytes and lymphocytes was unaltered over the time
course after PT (Fig. 13 A, B), monocytes demonstrated some increase in surface TREM-1
already at the time of admission which was significant after 12 h compared to healthy
controls. After 10 d, TREM-1 values had returned to baseline levels (Fig. 13 C).
Fig. 13: Triggering receptor expressed on myeloid cells-1 (TREM-1) expression on peripheral leukocytes
during the time course after trauma. TREM-1 detected as mean fluorescence intensity (MFI) on A,
neutrophils, B, lymphocytes, and C, monocytes in whole blood from healthy volunteers (Healthy) or patients
after polytrauma (PT). *, p < 0.05; #, p < 0.05 compared to Healthy; &, p < 0.05 compared to 0 h after PT; n =
4-12.
A B
C
Neutrophils Lymphocytes
Monocytes
Time after trauma [h]
0 48 96 144 192 240
TR
EM
-1 [
MF
I, a
rbit
rary
un
its]
0
1000
2000
3000
4000PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
TR
EM
-1[M
FI, a
rbit
rary
un
its
]
0
20
40
60
80PT
Healthy
Time after trauma [h]
0 48 96 144 192 240
TR
EM
-1 [
MF
I, a
rbit
rary
un
its
]
0
500
1000
1500
2000
2500
3000
PT
Healthy
#&
**

Results
32
In line with these results, monocyte TREM-1 expression at 4 h post injury correlated
significantly with lactate concentration and the number of transfused erythrocyte
concentrates at admission (Fig. 14).
Fig. 14: Triggering receptor expressed on myeloid cells-1 (TREM-1) correlated to clinical parameters.
Pearson correlation between initial lactate concentration (A) and the number of applied erythrocyte
concentrates during the first day (B) with TREM-1 expression as detected by mean fluorescence intensity
(MFI) on monocytes 4 h after trauma. R, Pearson correlation coefficient.
3.1.3 Molecules interacting with the coagulation system
Thrombomodulin was detectable only on monocytes, and surface expression was
significantly increased 24 h and 48 h after trauma compared to healthy volunteers and values
upon admission (Fig. 15). There were no significant correlations between thrombomodulin
and clinical parameters of shock.
Time after trauma [h]
0 48 96 144 192 240
TM
[M
FI, a
rbit
rary
un
its]
0
50
100
150
200
250PT
Healthy#&#&
Fig. 15: Thrombomodulin (TM) expression on monocytes after trauma. TM mean fluorescence intensity
(MFI) on monocytes in whole blood from healthy volunteers (Healthy) or patients after polytrauma (PT). #,
p < 0.05 compared to Healthy; &, p < 0.05 compared to 0 h after PT; n = 7-12.
Lactate [mmol/l]
0 1 2 3 4TR
EM
[M
FI,
arb
itra
ry u
nit
s]
0
500
1000
1500
2000
2500
3000
r = 0.73p = 0.04
Erythrocyte concentrates [units]
0 2 4 6 8 10TR
EM
[M
FI,
arb
itra
ry u
nit
s]
0
500
1000
1500
2000
2500
3000
r = 0.73
p = 0.04
A B

Results
33
On the surface of all leukocytes, PAI-1 was detected in increased amounts after trauma
compared to healthy volunteers. While neutrophils and monocytes demonstrated a
significant increase only 5 d after trauma (Fig. 16 A, C), binding on lymphocytes was
significantly increased 24 h and 48 h compared to emergency room values, and days 5 and
10 only showed some increase compared to controls (Fig. 16 B).
Fig. 16: Surface plasminogen activator inhibitor-1 (PAI-1) on leukocytes during the time course after
trauma. PAI-1 mean fluorescence intensity (MFI) on A, neutrophils, B, lymphocytes, and C, monocytes in
whole blood from healthy volunteers (Healthy) or patients after polytrauma (PT). *, p < 0.05; #, p < 0.05
compared to Healthy; &, p < 0.05 compared to 0 h after PT; n = 7-12.
Time after trauma [h]
0 48 96 144 192 240
PA
I-1
[M
FI,
arb
itra
ry u
nit
s]
0
100
200
300
400
500
PT
Healthy
#
***
&
Time after trauma [h]
0 48 96 144 192 240P
AI-
1 [
MF
I, a
rbit
rary
un
its
]
0
10
20
30
40
50
60PT
Healthy
#
#
*
*
Time after trauma [h]
0 48 96 144 192 240
PA
I-1
[M
FI,
arb
itra
ry u
nit
s]
0
100
200
300
400PT
Healthy&
A B
C
Neutrophils Lymphocytes
Monocytes

Results
34
PAI-1 on granulocytes (Fig. 17 A) and on lymphocytes (Fig. 17 B, C) correlated significantly
with the initial number of needed erythrocyte concentrates.
Fig. 17: Plasminogen activator inhibitor-1 (PAI-1) in correlation to clinical parameters. Pearson
correlation between the number of applied erythrocyte concentrates during the first day with PAI-1 expression
as detected by mean fluorescence intensity (MFI) on granulocytes 4 h (A) and lymphocytes 4 h (B) and 12 h
(C) after injury. R, Pearson correlation coefficient.
3.2 PAI-1 in PT plasma and ex vivo on the granulocyte surface
Since PAI-1 was detected in increased amounts on all leukocytes especially on day 5 after
injury, soluble PAI-1 was measured in plasma samples to check whether higher plasma
concentrations could induce an increased surface binding. However, plasma PAI-1 was only
increased during the initial days post trauma (Fig. 18), therefore presumably not accounting
for increased surface binding on leukocytes.
Erythrocyte concentrates [units]
0 2 4 6 8 10PA
I-1 [
MF
I, a
rbit
rary
un
its]
0
10
20
30
40
50
60
70r = 0.72p = 0.04
Erythrocyte concentrates [units]
0 2 4 6 8 10PA
I-1 [
MF
I, a
rbit
rary
un
its]
0
50
100
150
200
250
300
350
r = 0.87p = 0.01
Erythrocyte concentrates [units]
0 2 4 6 8 10PA
I-1 [
MF
I, a
rbit
rary
un
its]
0
10
20
30
40
50
60
r = 0.8p = 0.03
A B
C
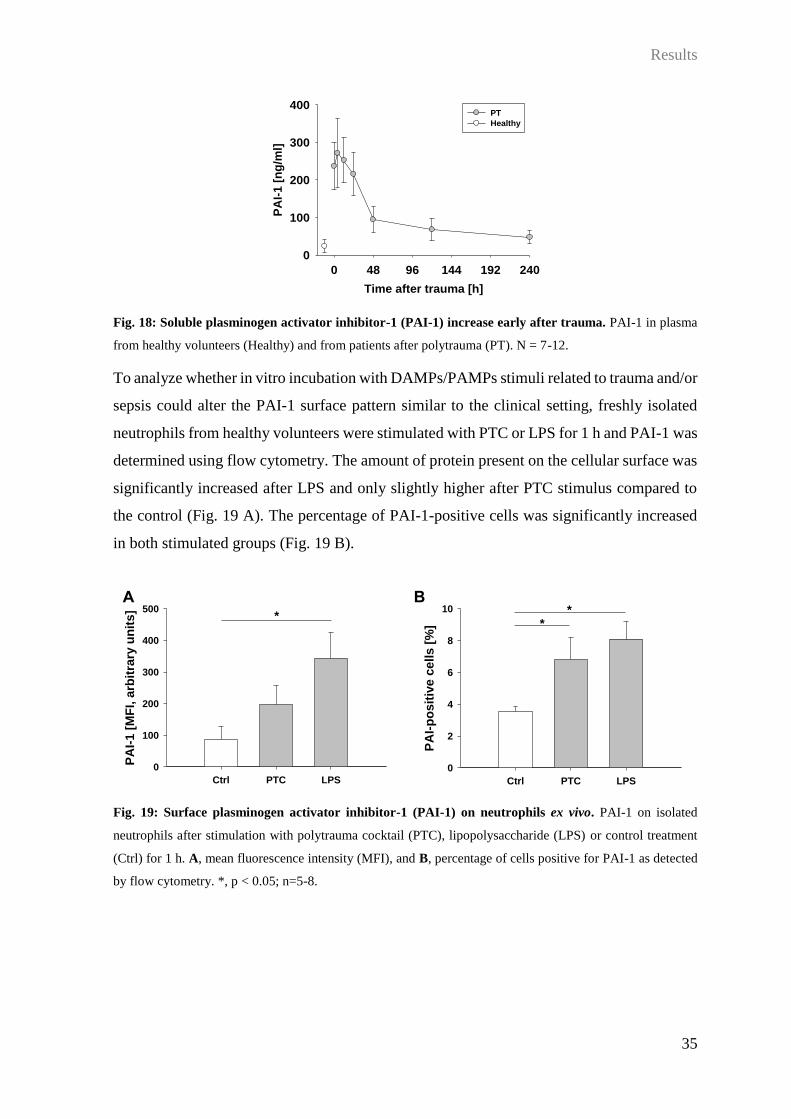
Results
35
Time after trauma [h]
0 48 96 144 192 240P
AI-
1 [
ng
/ml]
0
100
200
300
400PT
Healthy
Fig. 18: Soluble plasminogen activator inhibitor-1 (PAI-1) increase early after trauma. PAI-1 in plasma
from healthy volunteers (Healthy) and from patients after polytrauma (PT). N = 7-12.
To analyze whether in vitro incubation with DAMPs/PAMPs stimuli related to trauma and/or
sepsis could alter the PAI-1 surface pattern similar to the clinical setting, freshly isolated
neutrophils from healthy volunteers were stimulated with PTC or LPS for 1 h and PAI-1 was
determined using flow cytometry. The amount of protein present on the cellular surface was
significantly increased after LPS and only slightly higher after PTC stimulus compared to
the control (Fig. 19 A). The percentage of PAI-1-positive cells was significantly increased
in both stimulated groups (Fig. 19 B).
Fig. 19: Surface plasminogen activator inhibitor-1 (PAI-1) on neutrophils ex vivo. PAI-1 on isolated
neutrophils after stimulation with polytrauma cocktail (PTC), lipopolysaccharide (LPS) or control treatment
(Ctrl) for 1 h. A, mean fluorescence intensity (MFI), and B, percentage of cells positive for PAI-1 as detected
by flow cytometry. *, p < 0.05; n=5-8.
Ctrl PTC LPS
PA
I-1
[M
FI,
arb
itra
ry u
nit
s]
0
100
200
300
400
500*
Ctrl PTC LPS
PA
I-p
os
itiv
e c
ell
s [
%]
0
2
4
6
8
10 **
A B

Results
36
Since isolated neutrophils demonstrate a considerable apoptosis rate after longer incubation
periods (own unpublished data), whole blood anticoagulated with EDTA was used for a
longer incubation protocol. As a positive control for cell stimulation, the protein kinase C
activator PMA was used. In contrast to the results for isolated neutrophils, stimulation of
whole blood by LPS or PTC resulted in no increase in surface PAI-1 expression. Only PMA
was able to induce a significant increase, and this was true for both 1 h (Fig. 20 A, B) and
the prolonged 24 h incubation time (Fig. 20 C, D).
Fig. 20: Surface plasminogen activator inhibitor-1 (PAI-1) on neutrophils after whole-blood stimulation.
Blood was incubated with polytrauma cocktail (PTC), lipopolysaccharide (LPS), phorbol 12-myristate 13-
acetate (PMA) or without stimulant (Ctrl) for 1 h (A, B) and 24 h (C, D). A, C, mean fluorescence intensity
(MFI) on neutrophils and B, D, percentage of neutrophils positive for PAI-1 as assessed by flow cytometry.
***, p < 0.001; n = 3.
To analyze whether this increase in surface PAI-1 on neutrophils stimulated in whole blood
was due to a reduced shedding of microvesicles from the cell membrane, microvesicle
number and origin in plasma were determined using flow cytometry. After 1 h, there were
no differences in the total microvesicle number (Fig. 21 A). Similarly, nor the absolute count
nor the percentage of CD66b-positive microvesicles was altered after either stimulus (Fig.
21 C, E). After 24 h, there was some increase in total microvesicle numbers (Fig. 21 B) and
Ctrl PTC LPS PMA
PA
I-1 [
MF
I, a
rbit
rary
un
its]
0
50
100
150
200
250 **
**
A B
C D
Ctrl PTC LPS PMA
PA
I-1
[M
FI, a
rbit
rary
un
its
]
0
200
400
600
800
1000
1200
1400
1600 ******
***
Ctrl PTC LPS PMA
PA
I-p
osit
ive c
ells [
%]
0
20
40
60
80
100*
***
Ctrl PTC LPS PMA
PA
I-p
osit
ive c
ells [
%]
0
20
40
60
80
100
*****
***

Results
37
a significant elevation in total and relative numbers of CD66b-positive microvesicles after
PMA incubation, but again, there were no changes in samples with PTC or LPS (Fig. 21 D,
F).
Fig. 21: Microvesicles (MV) in supernatant after whole-blood stimulation. Blood was incubated with
polytrauma cocktail (PTC), lipopolysaccharide (LPS), phorbol 12-myristate 13-acetate (PMA) or without
stimulant (Ctrl) for 1 h (A, C, E) and 24 h (B, D, F). A, B, total microvesicles per µl plasma, C, D, number of
CD66b+ MV per µl plasma, and E, F, percentage of CD66b+ MV as assessed by flow cytometry. ***, p < 0.001;
n = 3.
Ctrl PTC LPS PMA
MV
[n
/µl p
las
ma
]
0
2000
4000
6000
8000
10000
Ctrl PTC LPS PMA
MV
[n
/µl p
las
ma
]
0
20000
40000
60000
80000
Ctrl PTC LPS PMA
CD
66
b+
MV
[n
/µl
pla
sm
a]
0
100
200
300
400
Ctrl PTC LPS PMA
CD
66
b+
MV
[n
/µl
pla
sm
a]
0
2000
4000
6000
8000***
******
Ctrl PTC LPS PMA
CD
66
b+
MV
[%
]
0
2
4
6
8
10
12 ******
***
Ctrl PTC LPS PMA
CD
66
b+
MV
[%
]
0
1
2
3
4
5
6
A B
C D
E F
1 h 24 h

Results
38
3.3 Gene expression of PAI-1 and urokinase receptor in stimulated neutrophils
To test the hypothesis that the increased PAI-1 detected on isolated neutrophils after
inflammatory stimuli was due to an increase in gene transcription, mRNA was isolated from
stimulated isolated neutrophils and expression of SERPINE1 (coding for PAI-1) and
PLAUR (coding for the urokinase receptor, uPAR) was analyzed using qPCR. There were
no changes in PAI-1 expression in neutrophils after PTC stimulation for 1 h and 4 h; in
contrast to the surface protein findings, incubation with LPS for 4 h lead to a significant
downregulation of PAI-1 expression (Fig. 22 A). When analyzing the cellular anchor for
PAI-1, uPAR, which is a receptor for urokinase (uPA) and thus facilitates PAI-1 binding, a
slight, but nonsignificant increase in uPAR expression was detected after 1 h in PTC and
LPS samples compared to unstimulated controls. However, the expression was
downregulated in cells after 4 h incubation (Fig. 22 B).
Fig. 22: Expression of plasminogen-activator inhibitor-1 (PAI-1) and urokinase receptor (uPAR) in
isolated neutrophils. A, SERPINE1 (coding for Pai-1) and B, PLAUR (coding for uPAR) in neutrophils 1 h
and 4 h after polytrauma cocktail (PTC) or lipopolysaccharide (LPS) stimulation as fold-expression compared
to control treatment (Ctrl) and normalized to the housekeeping gene. **, p = 0.001; ***, p < 0.001; n=2-8.
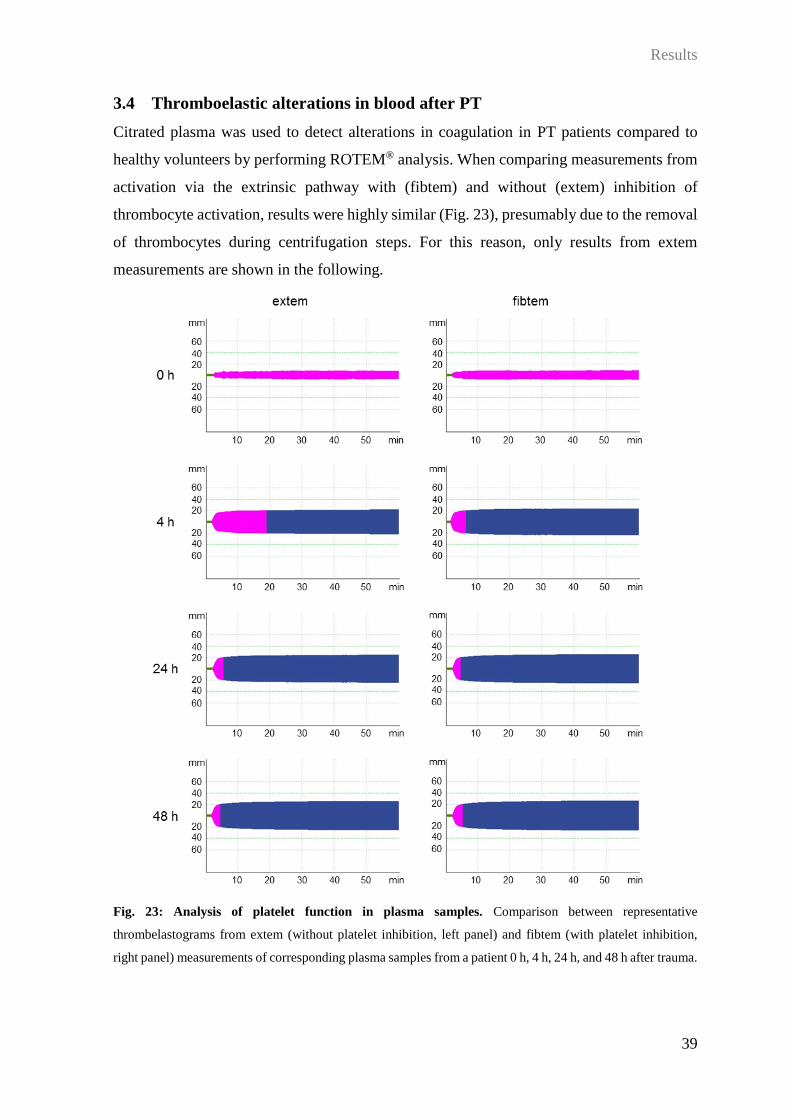
Results
39
3.4 Thromboelastic alterations in blood after PT
Citrated plasma was used to detect alterations in coagulation in PT patients compared to
healthy volunteers by performing ROTEM® analysis. When comparing measurements from
activation via the extrinsic pathway with (fibtem) and without (extem) inhibition of
thrombocyte activation, results were highly similar (Fig. 23), presumably due to the removal
of thrombocytes during centrifugation steps. For this reason, only results from extem
measurements are shown in the following.
Fig. 23: Analysis of platelet function in plasma samples. Comparison between representative
thrombelastograms from extem (without platelet inhibition, left panel) and fibtem (with platelet inhibition,
right panel) measurements of corresponding plasma samples from a patient 0 h, 4 h, 24 h, and 48 h after trauma.

Results
40
3.4.1 Coagulation activation and clot polymerization
During the first 10 d after PT, clotting time in patients was significantly higher than in
healthy controls, reaching a peak 48 h post injury (Fig. 24 A). Clot formation time was higher
in tendency, but comparison of results was difficult with a partly low n-size due to several
samples not reaching a 20 mm amplitude (Fig. 24 B). The -angle as the angle between the
baseline and the tangent to the clotting curve through the 2-mm point was significantly
reduced in trauma patients during the first 48 h, but then normalized on days 5 and 10 (Fig.
24 C). In line, the clot formation rate as the angle between the baseline and the tangent at
maximum slope was significantly lower at the early time point after trauma and returned to
normal values after 5 d (Fig. 24 D).
Fig. 24: Coagulation activation and clot polymerization after severe injury. Clotting time (A), clot
formation time (B), -angle (C), and clot formation rate (D) in citrated plasma from healthy volunteers (white
bars) and polytrauma patients (grey bars) at indicated time points after trauma. *, p < 0.05; #, p < 0.05 compared
to Healthy; §, p < 0.05 compared to 5 d; $, p < 0.05 compared to 10 d; n = 6-11 (A, C, D), n = 1-8 (B).
A B
C D
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Clo
t fo
rmati
on
rate
[°]
0
20
40
60
80
100
#§$#§#§
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
-An
gle
[°]
0
20
40
60
80
100
#§$ # §$#§$
#§$
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Clo
t fo
rmati
on
tim
e [
s]
0
200
400
600
800
1000
1200
1400
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Clo
ttin
g t
ime [
s]
0
20
40
60
80
100
120
140
160 *
#
#
## # #

Results
41
3.4.2 Clot firmness
Besides analyzing the onset of coagulation, ROTEM® also allows to determine the clot
firmness during the course of measurement. The maximal clot firmness during the 90-min
assay was significantly reduced in patients upon admission to the emergency room compared
to healthy volunteers. The reduction remained present until 48 h after injury, and disappeared
on day 5 (Fig. 25 A). This pattern was already apparent 10 min and 30 min after starting the
assay (Fig. 25 B, C). Time to maximal clot firmness, however, was only slightly increased
in patients 48 h after injury (Fig. 25 D).
Fig. 25: Parameters of clot firmness after polytrauma. Maximal clot firmness (A), clot firmness after 10
min (B) and 30 min (C), and time to maximal clot firmness (D) in citrated plasma from healthy volunteers
(white bars) and polytrauma patients (grey bars) at indicated time points after trauma. *, p < 0.05; #, p < 0.05
compared to Healthy; §, p < 0.05 compared to 5 d; $, p < 0.05 compared to 10 d; n = 6-11.
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Clo
t fi
rmn
ess [
mm
]
0
5
10
15
20
25
30
§$ §$
**
§$
#
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Clo
t fi
rmn
ess [
mm
]
0
5
10
15
20
25
30
§ $
§$
*
§
#
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Clo
t fi
rmn
ess [
mm
]
0
5
10
15
20
25
30
§$§$
**
§$
#
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Tim
e t
o m
axim
al
clo
t fi
rmn
ess [
s]
0
500
1000
1500
2000
A B
C D

Results
42
3.4.3 Clot characteristics
As could be concluded from the lysis index at 30 min, there was no detectable clot lysis in
any of the samples (Fig. 26 A). Maximum velocity as the calculated maximum of the curve’s
first derivative was reduced in patient samples at all time points compared to healthy
volunteers (Fig. 26 B). In line with the results for clot firmness, the shear elastic modulus
strength was significantly reduced as early as upon hospital admission and remained low
until 24 h after trauma, returning to baseline levels after 48 h and even surpassing healthy
values on days 5 and 10 (Fig. 26 C).
Fig. 26: Clot characteristics in polytrauma patients. Lysis index after 30 min (A), maximum velocity (B),
and shear elastic modulus strength (C) in citrated plasma from healthy volunteers (white bars) and polytrauma
patients (grey bars) at indicated time points after trauma. *, p < 0.05; #, p < 0.05 compared to Healthy; §,
p < 0.05 compared to 5 d; $, p < 0.05 compared to 10 d; n = 6-11.
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Maxim
um
velo
cit
y [
mm
/min
]
0
20
40
60
80
# ## #
#
#
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Lysis
in
dex [
%]
0
20
40
60
80
100
120
Healthy 0 h 4 h 24 h 48 h 5 d 10 d
Sh
ear
ela
sti
c m
od
ulu
s s
tren
gth
0
500
1000
1500
2000
2500
*
§$§$
§
A B
C

Results
43
3.5 Cytokine response to bacterial stimulus in an ex-vivo whole blood model
Whole blood from healthy volunteers and from patients at different time points after PT was
incubated with LPS for 24 h and cytokines in the supernatant were analyzed. For all
cytokines, only results in stimulated samples were compared; unstimulated values are shown
as reference.
IL-6 generation was lower by tendency, but not significantly, in PT patients compared to
healthy volunteers (Fig. 27 A). TNF release was significantly reduced in patients 4 h and 5
d after injury (Fig. 27 B). IL-1 production and the IL-1/IL-RA ratio were significantly
decreased in stimulated patient groups at all time points (Fig. 27 C, D).
Fig. 27: Release of pro-inflammatory cytokines in whole blood after lipopolysaccharide (LPS) stimulus.
Interleukin (IL)-6 (A), tumor necrosis factor (TNF, B), and IL-1 (C) concentrations, and ratio between IL-1
and IL-1 receptor antagonist (IL1-RA) concentrations (D) in supernatants after whole blood stimulation with
(+) and without (-) LPS for 24 h. Healthy, healthy volunteers; PT, blood samples taken from polytrauma
patients at indicated time points after trauma. *, p < 0.05 compared to Healthy for analysis of stimulated
samples; unstimulated values are shown as reference. N = 5-7.

Results
44
Fig. 28: Cytokine release in whole blood after lipopolysaccharide (LPS) stimulus. Interferon- (IFN-, A),
interleukin (IL)-10 (B), IL-12 (C), IL-17 (D), eotaxin (E), basic fibroblast growth factor (FGF basic, F),
granulocyte colony-stimulating factor (G-CSF, G), and granulocyte-macrophage colony-stimulating factor
(GM-CSF, H) in supernatants after whole blood stimulation with (+) and without (-) LPS for 24 h. Healthy,
healthy volunteers; PT, blood samples taken from polytrauma patients at indicated time points after trauma.
Analysis was performed for stimulated samples; unstimulated values are shown as reference. N = 5-7.

Results
45
Although there were some alterations in the unstimulated samples after trauma, there were
no significant differences in the production of interferon-IFN- Fig. 28 A), IL-10 (Fig. 28
B), IL-12 (Fig. 28 C) or IL-17 (Fig. 28 D). Similarly, trauma also did not induce any
significant changes in the secretion of eotaxin (Fig. 28 E), basic fibroblast growth factor
(Fig. 28 F), granulocyte-colony stimulating factor (Fig. 28 G), or granulocyte-macrophage
colony-stimulating factor (Fig. 28 H).
In contrast, 24 h after trauma, there was a significantly increased release of IL-2 (Fig. 29 A),
IL-4 (Fig. 29 B), and IL-5 (Fig. 29 C). IL-7 production after LPS stimulus was also
significantly higher in patients 24 h and 5 d after trauma compared to healthy volunteers
(Fig. 29 D). IL-9 was slightly increased in patient samples 4 h and 5 d after trauma, and
significantly higher 24 h after injury (Fig. 29 E). IL-13 was elevated in trauma patients only
by tendency (Fig. 29 F). IL-15 secretion was significantly increased in patients at all time
points compared to healthy volunteers (Fig. 29 G). Monocyte chemoattractant protein 1
release after LPS stimulus was significantly higher in blood samples from patients 4 h and
24 h after injury (Fig. 29 H).

Results
46
Fig. 29: Release of inflammatory mediators in whole blood after lipopolysaccharide (LPS) stimulus.
Interleukin (IL)-2 (A), IL-4 (B), IL-5 (C), IL-7 (D), IL-9 (E), IL-13 (F), IL-15 (G), and monocyte
chemoattractant protein 1 (MCP-1, H) in supernatants after whole blood stimulation with (+) and without (-)
LPS for 24 h. Healthy, healthy volunteers; PT, blood samples taken from polytrauma patients at indicated time
points after trauma. *, p < 0.05 compared to Healthy for analysis of stimulated samples; unstimulated values
are shown as reference. N = 5-7.

Results
47
When comparing a shortened incubation time of 4 h in samples taken 24 h after trauma, there
was a highly similar pattern of cytokine release (Fig. 30).
Fig. 30: Comparison of differences in cytokine concentrations in stimulated and unstimulated samples
after 4 h and 24 h incubation. Data are shown as mean in pg/ml. FGF-basic, basic fibroblast growth factor;
G-CSF, granulocyte-colony stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor;
IFN- interferon-; IL, interleukin; IL1-RA, IL-1 receptor antagonist; IL-12 (p70), IL-12 heterodimer; IP-10,
interferon gamma-induced protein 10; MCP-1, monocyte chemoattractant protein 1; MIP-1, macrophage
inflammatory protein-1; PDGF-bb, platelet-derived growth factor subunit B; TNF, tumor necrosis factor;
VEGF, vascular endothelial growth factor.
IL-1βIL-1RA
IL-2
IL-4
IL-5
IL-6
IL-7
IL-9
IL-10
IL-12 (p70)
IL-13
IL-15IL-17
Eotaxin
FGF-basic
G-CSF
GM-CSF
IFN-γ
IP-10
MCP-1
MIP-1α
PDGF-bb
TNF-α
VEGF4 h
24 h

Results
48
A comparison of p-values for the difference between stimulated and unstimulated samples
for the two incubation periods is shown in Table 1.
Table 1: Comparison between sample incubation for 4 h or 24 h. Shown are p-values for differences
between stimulated and unstimulated samples.
Parameter p-value LPS vs. Ctrl
4 h incubation 24 h incubation
IL-1 0.018 0.009
IL1-RA 0.002 0.003
IL-2 < 0.001 0.006
IL-4 0.001 0.056
IL-5 0.005 0.010
IL-6 0.006 0.003
IL-7 0.532 0.021
IL-9 0.001 0.011
IL-10 0.002 0.009
IL-12 < 0.001 0.078
IL-13 0.015 0.030
IL-15 0.001 0.004
IL-17 0.003 0.014
Eotaxin 0.001 0.011
FGF basic 0.002 0.040
G-CSF 0.003 0.002
GM-CSF 0.021 0.019
IFN- < 0.001 0.009
IP-10 0.001 0.010
MCP-1 0.069 0.002
MIP-1 0.083 0.012
MIP-1 0.006 0.033
PDGF-bb 0.615 0.145
TNF 0.009 0.038
VEGF 0.003 0.031
Abbreviations: FGF-basic, basic fibroblast growth factor; G-CSF, granulocyte-colony stimulating factor; GM-
CSF, granulocyte-macrophage colony-stimulating factor; IFN- interferon-; IL, interleukin; IL1-RA, IL-1
receptor antagonist; IP-10, interferon gamma-induced protein 10; MCP-1, monocyte chemoattractant protein
1; MIP-1, macrophage inflammatory protein-1; PDGF-bb, platelet-derived growth factor subunit B;
TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.

Results
49
3.6 Impact of HS on barrier disturbance and organ dysfunction after PT
Since there were several significant correlations between surface molecules on leukocytes
in our PT cohort and clinical parameters related to the presence or absence of HS (see 3.1),
the influence of an additional HS after severe trauma, especially focusing on inflammation,
vascular barrier and organ function, was analyzed. For this purpose, a PT patient cohort and
a murine model of PT and/or HS were studied.
3.6.1 Parameters of inflammation, barrier and organ dysfunction, and coagulation in
a human PT study and correlation with clinical data
A patient cohort from Zurich University Hospital was stratified for the presence or absence
of HS according to established parameters. The strategy identified 10 patients without and
20 patients with HS. The cohorts had similar injury patterns except for the prevalence of
abdominal trauma. Despite similar ISS values, patients with HS displayed a higher maximal
SOFA score during the course of observation and required a three-fold higher ICU and two-
fold increased in-hospital length of stay. Stratification criteria such as the TASH score,
numbers of packed RBC transfusion, and initial lactate levels were also significantly
different in the two groups. Further clinical parameters and a statistical comparison are
shown in Table 2.
Several inflammatory mediators and parameters related to barrier and organ (dys-)function
were analyzed over the course of the first 5 days after injury employing sandwich ELISAs.
P-values < 0.05 represent a significant difference between patients with compared to those
without HS over the 5-day period.

Results
50
Table 2: Descriptive statistics and comparison of demographic parameters in stratified groups. Statistical
comparison of stratified groups is shown as p-values. Modified from Halbgebauer et al. [60].
Polytrauma without HS
(n=10)
Polytrauma with HS
(n=20)
P-value
Stratification of
hemorrhagic shock
All criteria fulfilled:
base excess ≥ −6 mmol/l
lactate < 2.5 mmol/l
pRBC ≤ 2 units on day 0
TASH score < 10 points
One or more criteria
fulfilled:
base excess < −6 mmol/l
lactate ≥ 2.5 mmol/l
pRBC > 2 units on day 0
TASH score ≥ 10 points
Mean ± SEM Mean ± SEM
Demographics
Age [y] 44.4 ± 6.3 37.6 ± 3.8 0.45
Sex [male/total] 4/10 (40%) 17/20 (85%) 0.011
GCS 11.5 ± 1.5 12 ± 0.9 0.93
Traumatic brain injury (GCS ≤ 12) 4/10 (40%) 7/20 (35%) 0.789
AIS max. 4.6 ± 0.9 4.5 ± 0.8 0.7
AIS head/neck/cervical spine 2.9 ± 0.9 2.25 ± 0.4 0.45
AIS face 0.3 ± 0.3 0.8 ± 0.2 0.2
AIS thorax/thoracic spine 3.3 ± 0.6 3.7 ± 0.3 0.49
AIS abdomen/lumbar spine 1.1 ± 0.4 2.8 ± 0.3 0.01
AIS upper/lower extremity 2.3 ± 0.4 2.8 ± 0.2 0.23
ISS 36.2 ± 6.3 38.7 ± 2.8 0.54
SOFA score initial 4.6 ± 1.2 5.7 ± 0.7 0.4
SOFA score max. 6 ± 1.1 10.5 ± 1 0.009
Outcomes
RISC [% survival] 79.1 ± 10.2 80 ± 5.7 0.52
Survival 3/10 (30%) 4/20 (20%) 0.542
Hospital length of stay [d] 17.2 ± 3.2 34 ± 4.1 0.01
Intensive care unit length of stay [d] 5.9 ± 1.3 19.2 ± 2.8 0.001
Allogenic blood transfusions
TASH score [points] 4.6 ± 0.9 10.5 ± 1.2 0.004
Initial (d0) RBC transfusion [units] 1.1 ± 0.4 7.9 ± 1.5 0.002
Total RBC transfusion [units] 3.8 ± 0.5 18.2 ± 3.5 < 0.001
Massive transfusion rate 0/10 6/20 (30%) 0.053
Infectious complications
Nosocomial infections 4/10 (40%) 15/20 (75%) 0.872
Sepsis 0/10 6/20 (20%) 0.053
Hemostasis and blood gas analysis
Initial base excess [mmol/l] −2.2 ± 0.4 −3.6 ± 0.7 0.38
Initial lactate [mmol/l] 1.5 ± 0.2 2.7 ± 0.3 0.007
Initial Quick [%] 73.5 ± 5.7 53.4 ± 4.1 0.008
Initial aPTT [s] 29.7 ± 1.5 46.1 ± 6.4 0.04
Initial hematocrit [%] 33.5 ± 1.5 26.6 ± 1.7 0.013
Initial temperature [C] 35.1 ± 0.2 35.4 ± 0.3 0.58
P-values < 0.05 are bold.
Abbreviations: AIS, Abbreviated Injury Score; aPTT, abbreviated partial thromboplastin time; GCS, Glasgow
Coma Scale; HS, hemorrhagic shock; ISS, Injury Severity Score; RBC, red blood cells; PT, polytrauma; RISC,
Revised Injury Severity Classification; SOFA score, Sequential Organ Failure Assessment score; TASH score,
trauma associated severe hemorrhage score.
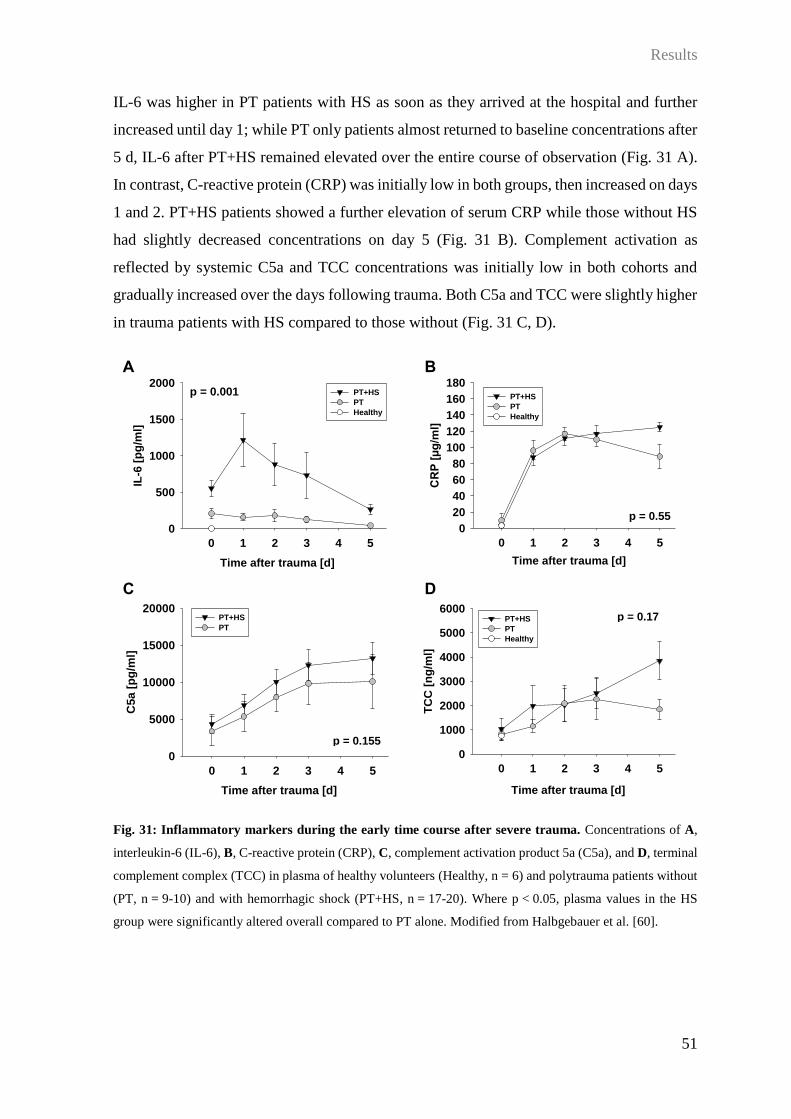
Results
51
IL-6 was higher in PT patients with HS as soon as they arrived at the hospital and further
increased until day 1; while PT only patients almost returned to baseline concentrations after
5 d, IL-6 after PT+HS remained elevated over the entire course of observation (Fig. 31 A).
In contrast, C-reactive protein (CRP) was initially low in both groups, then increased on days
1 and 2. PT+HS patients showed a further elevation of serum CRP while those without HS
had slightly decreased concentrations on day 5 (Fig. 31 B). Complement activation as
reflected by systemic C5a and TCC concentrations was initially low in both cohorts and
gradually increased over the days following trauma. Both C5a and TCC were slightly higher
in trauma patients with HS compared to those without (Fig. 31 C, D).
Fig. 31: Inflammatory markers during the early time course after severe trauma. Concentrations of A,
interleukin-6 (IL-6), B, C-reactive protein (CRP), C, complement activation product 5a (C5a), and D, terminal
complement complex (TCC) in plasma of healthy volunteers (Healthy, n = 6) and polytrauma patients without
(PT, n = 9-10) and with hemorrhagic shock (PT+HS, n = 17-20). Where p < 0.05, plasma values in the HS
group were significantly altered overall compared to PT alone. Modified from Halbgebauer et al. [60].
Time after trauma [d]
0 1 2 3 4 5
IL-6
[p
g/m
l]
0
500
1000
1500
2000PT+HS
PT
Healthy
p = 0.001
Time after trauma [d]
0 1 2 3 4 5
CR
P [
µg
/ml]
0
20
40
60
80
100
120
140
160
180PT+HS
PT
Healthy
p = 0.55
Time after trauma [d]
0 1 2 3 4 5
C5a
[p
g/m
l]
0
5000
10000
15000
20000PT+HS
PT
p = 0.155
Time after trauma [d]
0 1 2 3 4 5
TC
C [
ng
/ml]
0
1000
2000
3000
4000
5000
6000PT+HS
PT
Healthy
p = 0.17
A B
C D

Results
52
To assess the extent of organ dysfunction, markers specific for damage of lungs (CC16),
kidneys (NGAL), intestine (I-FABP) and liver (L-FABP) were analyzed. All showed
significantly higher concentrations in PT+HS patients compared to those with PT only, but
with a very distinct temporal pattern. CC16 was increased starting on day 1 after trauma and
gradually returned to healthy values until day 5 while PT patients demonstrated slightly
elevated concentrations only on day 0 (Fig. 32 A). In contrast, NGAL was initially low in
both groups, but then increased strongly in the PT+HS cohort over the course of observation
and remained high until day 5 (Fig. 32 B). I-FABP was initially high in PT+HS patients and
only slightly elevated in PT only; during the course of observation, the values approached
baseline which was reached on day 3 (Fig. 32 C). L-FABP was initially increased in both
groups; while after PT alone, concentrations returned to levels found in healthy volunteers
already on day 2, they remained elevated until day 5 in PT+HS (Fig. 32 D).
Fig. 32: Organ-damage markers during the early time course after severe trauma. Concentrations of A,
clara cell secretory protein (CC16), B, neutrophil gelatinase-associated lipocalin (NGAL), C, intestinal fatty
acid-binding protein (I-FABP), and D, liver-type fatty acid-binding protein (L-FABP) in plasma of healthy
volunteers (Healthy, n = 6) and polytrauma patients without (PT, n = 9-10) and with hemorrhagic shock
(PT+HS, n = 17-20). When p < 0.05, overall plasma values in the HS group were significantly altered
compared to PT alone. Modified from Halbgebauer et al. [60].
Time after trauma [d]
0 1 2 3 4 5
CC
16 [
pg
/ml]
0
200
400
600
800
1000
1200
1400PT+HS
PT
Healthy
p = 0.049
Time after trauma [d]
0 1 2 3 4 5
NG
AL
[p
g/m
l]
0
105
2x105
3x105
4x105
PT+HS
PT
Healthy
p = 0.0003
Time after trauma [d]
0 1 2 3 4 5
I-F
AB
P [
pg
/ml]
0
2000
4000
6000
8000
10000
12000
14000PT+HS
PT
Healthy
p = 0.02
Time after trauma [d]
0 1 2 3 4 5
L-F
AB
P [
pg
/ml]
0
2000
4000
6000
8000
10000
12000
14000 PT+HS
PT
Healthy
p = 0.004
A B
C D

Results
53
In order to determine whether certain clinical parameters revealed an influence on organ
damage, correlation analyses were performed between damage markers and clinical data.
CC16 on day 0 significantly correlated with the total number of transfused erythrocyte
concentrates during the first 21 d (Fig. 33 A), and on day 1, higher plasma concentrations
were associated with a higher fluid balance on day 2 (Fig. 33 B). NGAL on day 1 correlated
significantly with the AIS of the abdomen (Fig. 33 C), and on day 3 with concurrent
procalcitonin concentrations (Fig. 33 D). Upon arrival, I-FABP serum concentrations
correlated with lactate (Fig. 33 E) and with the fluid balance (Fig. 33 F). L-FABP on day 1
correlated negatively with Quick values (Fig. 33 G), and positively with procalcitonin two
days later (Fig. 33 H).

Results
54
Fig. 33: Pearson Product Moment Correlation of plasma concentrations of organ damage markers to
clinical parameters. Correlation analyses were performed for polytrauma patients without (PT) and with
hemorrhagic shock (PT+HS) on indicated days after injury. Significant correlations were found for clara cell
secretory protein (CC16) with A, total applied red blood cell (RBC) concentrates and B, the fluid balance,
neutrophil gelatinase-associated lipocalin (NGAL) with C, the abbreviated injury score (AIS) of the abdomen
and D, procalcitonin (PCT). Intestinal fatty acid-binding protein (I-FABP) concentrations correlated with E,
CC16 d0 [pg/ml]
0 103 2x103 3x103 4x103 5x103
RB
C t
ota
l [u
nit
s]
0
10
20
30
40
50
60
70PT+HS
PT
r = 0.62p = 0.001
NGAL d1 [pg/ml]
105 2x105 3x105 4x105 5x105
AIS
ab
do
men
0
1
2
3
4
5
6
PT+HS
PT
r = 0.66p < 0.001
NGAL d3 [pg/ml]
105 2x105 3x105 4x105 5x105
PC
T d
3 [
µg
/l]
0
2
4
6
8
10PT+HS
PT
r = 0.85p < 0.001
I-FABP d0 [pg/ml]
0 104 2x104 3x104 4x104
Lacta
te d
0 [
mm
ol/l]
0
2
4
6
8
PT+HS
PT
r = 0.71p < 0.001
I-FABP d0 [pg/ml]
0 104 2x104 3x104 4x104
Flu
id b
ala
nce d
0 [
ml]
-2000
0
2000
4000
6000
8000
10000
12000
PT+HS
PT
r = 0.51p = 0.008
L-FABP d1 [pg/ml]
5x103 104 2x104 2x104
Qu
ick d
1 [
%]
0
20
40
60
80
100
120PT+HS
PT
r = -0.45p = 0.01
L-FABP d1 [pg/ml]
5x103 104 2x104 2x104
PC
T d
3 [
µg
/l]
0
5
10
15
20PT+HS
PT
r = 0.80p < 0.001
CC16 d1 [pg/ml]
0 1000 2000 3000 4000
Flu
id b
ala
nce d
2 [
ml]
-2000
0
2000
4000
6000
8000
PT+HS
PT
r = 0.65p < 0.001
A B
C D
E F
G H

Results
55
blood lactate and F, the fluid balance; liver-type fatty acid-binding protein (L-FABP) was significantly
correlated with G, Quick values and H, PCT concentrations. R, Pearson correlation coefficient. Modified from
Halbgebauer et al. [60].
As could be expected from hemodilution conditions by resuscitation protocols, albumin
concentrations were lower in all patients compared to healthy volunteers, but there was no
significant difference between the patient cohorts (Fig. 34 A). Thrombomodulin as
endothelial damage marker was initially lower in patients, but then increased until day 5
especially in PT+HS patients (Fig. 34 B). The lipid mediator sphingosine-1-phosphate was
decreased in all patients compared to controls, but there was no significant difference
between the cohorts (Fig. 34 C). After lower concentrations in patients upon hospital
admission compared to healthy individuals, the vascular growth factor angiopoietin-2
normalized in both groups on day 1. Starting on day 2, there was an increase in serum
concentrations in PT+HS patients, and concentrations remained elevated until day 5 (Fig. 34
D). The vasodilator adrenomedullin was lower in PT only patients compared to controls
during the course of observation. In contrast, PT+HS patients revealed slightly increased
serum concentrations on day 0 which further increased until highest levels were reached on
day 2, then slowly decreasing until day 5 (Fig. 34 E).

Results
56
Fig. 34: Mediators of vascular permeability during the early time course after severe trauma.
Concentrations of A, albumin, B, thrombomodulin, C, sphingosine-1-phosphate, D, angiopoietin-2, and E,
adrenomedullin in plasma of healthy volunteers (Healthy, n = 6) and polytrauma patients without (PT, n = 9-
10) and with hemorrhagic shock (PT+HS, n = 17-20). Where p < 0.05, overall plasma values in the HS group
were significantly altered compared to PT alone. Modified from Halbgebauer et al. [60].
Time after trauma [d]
0 1 2 3 4 5
Alb
um
in [
mg
/ml]
0
10
20
30
40
50
60PT+HS
PT
Healthy
p = 0.3
Time after trauma [d]
0 1 2 3 4 5
Th
rom
bo
mo
du
lin
[p
g/m
l]
0
1000
2000
3000
4000
5000
6000PT+HS
PT
Healthy
Time after trauma [d]
0 1 2 3 4 5
An
gio
po
ieti
n-2
[p
g/m
l]
0
1000
2000
3000
4000PT+HS
PT
Healthy
p = 0.004
Time after trauma [d]
0 1 2 3 4 5Sp
hin
go
sin
e-1
-ph
osp
hate
[p
g/m
l]
0
2000
4000
6000
8000
10000
12000PT+HS
PT
Healthy
p = 0.03
Time after trauma [d]
0 1 2 3 4 5
Ad
ren
om
ed
ullin
[p
g/m
l]
0
100
200
300
400PT+HS
PT
Healthy
p = 0.047
p = 0.0005
A B
C D
E

Results
57
In both patient cohorts, MMP-9 was strongly increased upon arrival at the emergency room;
concentrations dropped already on day 1, but remained elevated until day 5 compared to
healthy volunteers (Fig. 35 A). MMP-13, however, showed a very different pattern; PT
patients had concentrations comparable to those in controls which only increased slightly on
day 5; levels in PT+HS patients were strongly increased on day 0 and slowly decreased after
that (Fig. 35 B). Claudin-5 as an important tight-junction molecule was initially increased in
both patient cohorts, although more pronounced in PT+HS patients; while concentrations
returned to healthy values in PT patients on day 5, they remained high in PT+HS patients
(Fig. 35 C). VE-cadherin was decreased in both patient cohorts over the observation period
compared to controls, but there was no difference between the cohorts (Fig. 35 D).
Fig. 35: Mediators and components of endothelial intercellular junctions during the early time course
after trauma. Concentrations of A, matrix metalloproteinase (MMP)-9, B, MMP-13, C, claudin-5, and D,
vascular endothelial (VE-) cadherin in plasma of healthy volunteers (Healthy, n = 6) and polytrauma patients
without (PT, n = 9-10) and with hemorrhagic shock (PT+HS, n = 17-20). Where p < 0.05, overall plasma
values in the HS group were significantly altered compared to PT alone. Modified from Halbgebauer et al.
[60].
p = 0.72
Time after trauma [d]
0 1 2 3 4 5
VE
-Ca
dh
eri
n [
ng
/ml]
0
50
100
150
200
250
PT+HS
PT
Healthy
Time after trauma [d]
0 1 2 3 4 5
MM
P-1
3 [
pg
/ml]
0
500
1000
1500
2000PT+HS
PT
Healthy
p = 0.001
Time after trauma [d]
0 1 2 3 4 5
Cla
ud
in-5
[p
g/m
l]
0
200
400
600
800PT+HS
PT
Healthy
p = 0.049
Time after trauma [d]
0 1 2 3 4 5
MM
P-9
[n
g/m
l]
0
20
40
60
80PT+HS
PT
Healthy
A B
C D
p = 0.58
p = 0.72
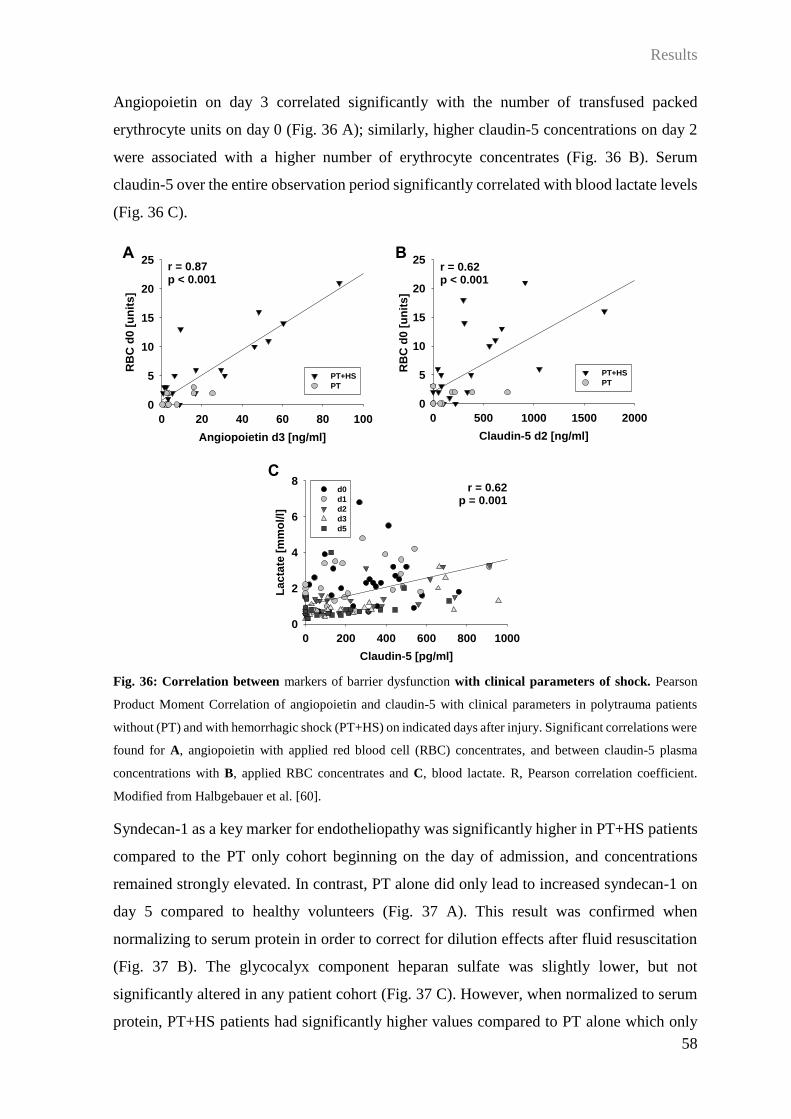
Results
58
Angiopoietin on day 3 correlated significantly with the number of transfused packed
erythrocyte units on day 0 (Fig. 36 A); similarly, higher claudin-5 concentrations on day 2
were associated with a higher number of erythrocyte concentrates (Fig. 36 B). Serum
claudin-5 over the entire observation period significantly correlated with blood lactate levels
(Fig. 36 C).
Fig. 36: Correlation between markers of barrier dysfunction with clinical parameters of shock. Pearson
Product Moment Correlation of angiopoietin and claudin-5 with clinical parameters in polytrauma patients
without (PT) and with hemorrhagic shock (PT+HS) on indicated days after injury. Significant correlations were
found for A, angiopoietin with applied red blood cell (RBC) concentrates, and between claudin-5 plasma
concentrations with B, applied RBC concentrates and C, blood lactate. R, Pearson correlation coefficient.
Modified from Halbgebauer et al. [60].
Syndecan-1 as a key marker for endotheliopathy was significantly higher in PT+HS patients
compared to the PT only cohort beginning on the day of admission, and concentrations
remained strongly elevated. In contrast, PT alone did only lead to increased syndecan-1 on
day 5 compared to healthy volunteers (Fig. 37 A). This result was confirmed when
normalizing to serum protein in order to correct for dilution effects after fluid resuscitation
(Fig. 37 B). The glycocalyx component heparan sulfate was slightly lower, but not
significantly altered in any patient cohort (Fig. 37 C). However, when normalized to serum
protein, PT+HS patients had significantly higher values compared to PT alone which only
Claudin-5 [pg/ml]
0 200 400 600 800 1000
La
cta
te [
mm
ol/l]
0
2
4
6
8d0
d1
d2
d3
d5
r = 0.62p = 0.001
Claudin-5 d2 [ng/ml]
0 500 1000 1500 2000
RB
C d
0 [
un
its]
0
5
10
15
20
25r = 0.62p < 0.001
PT+HS
PT
Angiopoietin d3 [ng/ml]
0 20 40 60 80 100
RB
C d
0 [
un
its
]
0
5
10
15
20
25r = 0.87p < 0.001
PT+HS
PT
A B
C

Results
59
slowly normalized until day 5 (Fig. 37 D). Mucin-2 as major component of the intestinal
mucosa was slightly, but not significantly lower in the PT cohort compared to PT+HS (Fig.
37 E); in contrast, normalization to total protein revealed significantly higher mucin-2
concentrations in PT+HS although levels were similar on day 5 (Fig. 37 F).
Fig. 37: Components of the endothelial glycocalyx and intestinal mucus after trauma. Plasma
concentrations of A, syndecan-1, C, heparan sulfate, and E, mucin-2 in plasma of healthy volunteers (Healthy,
n = 6) and polytrauma patients without (PT, n = 9-10) and with hemorrhagic shock (PT+HS, n = 17-20). B,
syndecan-1, D, heparan sulfate, and F, mucin-2 concentrations after normalization to total plasma protein. For
p < 0.05, overall plasma values in the HS group were significantly altered compared to PT alone. Modified
from Halbgebauer et al. [60].
Time after trauma [d]
0 1 2 3 4 5
Syn
de
ca
n-1
[p
g/m
l]
0
1000
2000
3000
4000
PT+HS
PT
Healthy
p < 0.0001
Time after trauma [d]
0 1 2 3 4 5
Syn
decan
-1 [
pg
/mg
]
0
20
40
60
80
100
120
140
PT+HS
PT
Healthy
p < 0.0001
Time after trauma [d]
0 1 2 3 4 5
He
pa
ran
su
lfa
te [
ng
/ml]
0
200
400
600
800
1000
1200PT+HS
PT
Healthy
p = 0.1
Time after trauma [d]
0 1 2 3 4 5
He
pa
ran
su
lfa
te [
ng
/mg
]
0
5
10
15
20
25
30PT+HS
PT
Healthy
p = 0.003
Time after trauma [d]
0 1 2 3 4 5
Mu
cin
-2 [
pg
/ml]
0
1000
2000
3000
4000
PT+HS
PT
Healthy
p = 0.07
Time after trauma [d]
0 1 2 3 4 5
Mu
cin
-2 [
pg
/mg
]
0
20
40
60
80
100
120
PT+HS
PT
Healthy
p = 0.0005
A B
C D
E F

Results
60
On day 1, higher MMP-13 concentrations correlated significantly with serum heparan sulfate
(Fig. 38 A), and syndecan-1 was significantly associated with the measured maximal SOFA
score (Fig. 38 B). Furthermore, over the entire observation period, syndecan-1 serum
concentrations correlated significantly with L-FABP (Fig. 38 C), NGAL concentrations
(Fig. 38 D), and the abbreviated prothrombin time (Fig. 38 E).
Fig. 38: Correlation analysis of markers of endothelial activation and clinical parameters. Pearson
Product Moment Correlation in polytrauma patients without (PT) and with hemorrhagic shock (PT+HS) on
indicated days after injury. A, correlation of matrix metalloproteinase 13 (MMP-13) with heparan sulfate;
syndecan plasma concentrations correlated to B, the maximal sequential organ failure score (SOFA max.)
during the course of observation, C, liver-type fatty acid-binding protein (L-FAPB), D, neutrophil gelatinase-
associated lipocalin (NGAL), and E, the abbreviated prothrombin time (aPTT). R, Pearson correlation
coefficient. Modified from Halbgebauer et al. [60].
MMP-13 d1 [pg/ml]
0 500 1000 1500 2000 2500
He
pa
ran
su
lfa
te d
1 [
ng
/ml]
0
200
400
600
800
1000
1200
1400
PT+HS
PT
r = 0.53p = 0.003
Syndecan [pg/ml]
0 2x103 4x103 6x103 8x103 104
aP
TT
[s]
0
20
40
60
80
100
120
140d0
d1
d2
d3
d5
r = 0.62p < 0.001
Syndecan d1 [pg/ml]
0 2x103 4x103 6x103 8x103
SO
FA
ma
x.
0
2
4
6
8
10
12
14
16
18
20
PT+HS
PT
r = 0.56p = 0.001
Syndecan [pg/ml]
0 2x103 4x103 6x103 8x103 104
L-F
AB
P [
pg
/ml]
0
104
2x104
3x104
4x104
d0
d1
d2
d3
d5
r = 0.39p < 0.001
Syndecan [pg/ml]
0 2000 4000 6000 8000
NG
AL
[p
g/m
l]
0
2x105
4x105
6x105
8x105
106
1x106
r = 0.4p < 0.001
d0
d1
d2
d3
d5
A B
C D
E

Results
61
Fibrinogen was significantly lower in PT+HS patients compared to the PT cohort, but
concentrations increased in both groups during the observation period (Fig. 39 A). The fibrin
degradation product D-dimer was lower in all patients compared to healthy controls, and
concentrations were significantly lower after HS compared to PT alone (Fig. 39 B). In an
experiment simulating hemodilution in patients after extensive fluid resuscitation,
adjustment of the hematocrit in the blood from healthy volunteers lead to a reduction in D-
dimer concentrations (Fig. 39 C).
Fig. 39: Fibrinogen and fibrin degradation after severe trauma and hemodilution. Plasma concentrations
of A, fibrinogen and B, D-dimer in plasma of healthy volunteers (Healthy, n = 6) and polytrauma patients
without (PT, n = 9-10) and with hemorrhagic shock (PT+HS, n = 13-16). P < 0.05 indicates a significant
alteration of overall plasma values in the HS group compared to polytrauma alone. C, D-dimer concentrations
in plasma from healthy volunteers after hematocrit adjustment.
Time after trauma [d]
0 1 2 3 4 5
0.0
0.5
1.0
1.5
2.0
2.5
PT+HS
PT
HealthyD
-dim
er
[µg
/ml]
Time after trauma [d]
0 1 2 3 4 5
Fib
rin
og
en
[µ
g/m
l]
0
2000
4000
6000
8000
PT+HS
PT
Healthy
Adjusted hematocrit [%]
45 33 25
D-d
ime
r [t
urb
ido
me
tric
un
its
]
0.0
0.2
0.4
0.6
0.8
A B
C
p = 0.0004 p = 0.02

Results
62
In a similar experiment, dilution of whole blood from healthy volunteers induced a
significant increase in clotting time (Fig. 40 A) and clot formation time (Fig. 40 B).
Incubation of blood with normal hematocrit with heparan sulfate concentrations comparable
to those detected in patients lead to a significant delay in coagulation. This was also visible,
but not significant, in samples with lower hematocrit (Fig. 40 A). Addition of mucin-2 did
not alter clot formation time in blood with a normal hematocrit, but had an even stronger
effect on clot formation time in samples with the lowest hematocrit (Fig. 40 B).
Fig. 40: Influence of glycocalyx components on coagulation. Clotting time (A) and clot formation time (B)
in rotational thromboelastometry measurements of citrated blood from healthy volunteers with normal (45%,
n = 3-8) or adjusted (33% and 25%, n = 3-5) hematocrit after incubation with heparan sulfate and mucin-2 at
the indicated concentrations; *, p < 0.05 vs. hematocrit 45% without heparan sulfate/mucin-2; #, p=0.005 vs.
hematocrit 25% without mucin-2. Modified from Halbgebauer et al. [60].
Heparan sulfate [ng/ml]
0 100 500 1000
Clo
ttin
g t
ime
[s
]
180
200
220
240
260
280Hematocrit 45%
Hematocrit 33%
Hematocrit 25%
*
$
Mucin-2 [pg/ml]
0 100 1000 5000
Clo
t fo
rmati
on
tim
e [
s]
0
50
100
150
200
250Hematocrit 45%
Hematocrit 33%
Hematocrit 25%
#
$
A B
*
*

Results
63
3.6.2 Barrier disturbances in a murine model of traumatic HS
In order to investigate the impact of HS in trauma in a highly standardized and clinically
relevant model, mice underwent PT or HS or the combination of both; healthy animals
served as controls. Plasma was analyzed 4 h after trauma.
Plasma albumin was increased only in HS animals, but was significantly lower after
additional PT (Fig. 41 A). Thrombomodulin was significantly decreased in both groups
which were exposed to HS (Fig. 41 B).
Fig. 41: Modulators of endothelial permeability in a murine trauma model. Plasma albumin (A) and
thrombomodulin (B) in mice after polytrauma (PT), hemorrhagic shock (HS), combined PT+HS, or in control
(Ctrl) animals. *, p < 0.05; n = 5-8.
MMP-9 concentrations in plasma were significantly higher in PT+HS animals compared to
controls, but also increased to some extent after HS or PT alone (Fig. 42 A). MMP-13
showed the same pattern, but results were not significantly different (Fig. 42 B). Claudin-5
concentrations were similar to healthy values in HS animals and higher by tendency in both
PT groups (Fig. 42 C). Heparan sulfate was slightly, but not significantly increased in all
trauma groups compared to controls (Fig. 42 D). Syndecan-1 was elevated after HS or PT
alone, but only the combination lead to a significant increase compared to healthy animals
(Fig. 42 E).
Ctrl HS PT PT+HS
Alb
um
in [
mg
/ml]
0
10
20
30
40
50
60
*
Ctrl HS PT PT+HS
Th
rom
bo
mo
du
lin
[p
g/m
l]
0
5000
10000
15000
20000
25000
30000
* **
*
A B

Results
64
Fig. 42: Mediators and components of endothelial intercellular junctions in a murine model of severe
trauma. Plasma matrix metalloproteinase (MMP)-9 (A), MMP-13 (B), claudin-5 (C), heparan sulfate (D), and
syndecan (E) in mice after polytrauma (PT), hemorrhagic shock (HS), combined PT+HS, or in control (Ctrl)
animals. *, p < 0.05; n = 5-8.
Ctrl HS PT PT+HS
Cla
ud
in-5
[p
g/m
l]
0
50
100
150
200
250
300
Ctrl HS PT PT+HS
MM
P-9
[n
g/m
l]
0
10
20
30
40*
Ctrl HS PT PT+HS
MM
P-1
3 [
ng
/ml]
0
500
1000
1500
2000
2500
3000
Ctrl HS PT PT+HS
Syn
de
ca
n [
ng
/ml]
0
10
20
30
40*
A B
C D
E
Ctrl HS PT PT+HS
Hep
ara
n s
ulf
ate
[n
g/m
l]
0.0
0.4
0.8
1.2
1.6

Discussion
65
4 Discussion
In the present thesis, the influence of a severe injury on the molecular and cellular “first line”
danger response in peripheral blood and development of coagulopathy in PT patients was
evaluated. Furthermore, the role of an additional HS on endothelial and organ barrier
dysfunction after PT was investigated in a larger patient cohort and in a standardized murine
model of multiple injury. Main results found during the course of this thesis are summarized
in Fig. 43.
Fig. 43: Summary of main findings. Severe trauma leads to early changes in the surface expression of
molecules interacting with coagulation, receptors for complement activation products as well as endogenous
and exogenous danger signals on peripheral leukocytes, resulting in alterations in the intracellular response to
danger signals. Increased inflammation, augmented by an additional hemorrhagic shock, induces the
breakdown of tight junctions and of the endothelial glycocalyx, water efflux into extravascular tissues and
causes organ damage with a distinct spatial and temporal onset pattern which can be monitored using plasma
markers. Heparin-like structures released during cleavage of glycocalyx components can hinder coagulation,
resulting in trauma-induced coagulopathy. C5a, complement component 5 a; C5aR, C5a receptor; CC16, clara
cell secretory protein; IL-6, interleukin-6; I-FABP, intestinal fatty acid-binding protein; L-FABP, liver-type
fatty acid-binding protein; MMP, matrix metalloproteinase; NGAL, neutrophil gelatinase-associated lipocalin;
PAI-1, plasminogen activator inhibitor-1; sphingosine-1-P, sphingosine-1-phosphate, TCC, terminal
complement complex; TLR, toll-like receptor; TM, thrombomodulin; TREM, triggering receptor expressed on
myeloid cells; uPA, urokinase plasminogen activator; uPAR, uPA receptor. Modified from Halbgebauer et al.
[60].

Discussion
66
4.1 Leukocyte surface profiling in polytraumatized patients
In the first part of this study, the role of leukocytes in the molecular danger response after
severe trauma was investigated. Confirming a previous study in polytraumatized patients
[1], there were significant reductions in the expression of complement receptors on
leukocytes from PT patients, possibly due to microvesicle shedding [171], cleavage from the
cell surface by neutrophil proteases [174], or internalization and subsequent degradation or
recycling processes [160]. Furthermore, TLR-2 and TLR-4 as the main receptors for
bacterial and fungal PAMPs [135,161], but also for endogenous DAMPs such as
extracellular histones and high mobility group box 1 (HMGB-1) [46,186] were rather
decreased on most leukocytes post trauma, possibly causing a reduced responsiveness to
microbial or DAMP stimulus. TREM-1, an amplifier of TLR activation by microbial
components [28], but also involved in sterile inflammation with potential ligands such as
HMGB-1 and heat-shock protein-70 [43,162] was only significantly higher on monocytes,
but mostly unaltered on granulocytes and lymphocytes in trauma patients. Strikingly, there
were several significant correlations of expression levels of RAGE, a multi-ligand receptor
for several endogenous molecules including advanced glycation end products [52,92], TLR-
2, TLR-4, and TREM-1 with parameters related to the presence of a HS, but not with injury
severity (data not shown), prompting the assumption that presence or absence of HS might
alter receptor expression in peripheral leukocytes. However, the relatively small number of
patients prevented us from a more detailed analysis in this regard.
All blood leukocytes were demonstrated to express complement regulatory proteins,
including CD59 as an inhibitor of TCC formation on healthy cells, and to increase their
surface levels in response to PT [1]. This may be the reason for a lack of detectable TCC on
neutrophils and lymphocytes. However, despite considerable amounts of CD59, TCC was
detectable on monocytes and there was a significant increase in patients compared to early
time points. Furthermore, early TCC surface levels correlated strongly with the number of
erythrocyte concentrates transfused during the first day after admittance, suggesting
increased complement activation after severe blood loss and extensive reperfusion therapy.
Sublytic amounts of TCC have been demonstrated to activate the inflammasome in
endothelial cells [168]. Further in vitro experiments will clarify whether this mechanism is
similar in monocytes and may induce a pro-inflammatory cellular phenotype.
Thrombomodulin is mainly expressed by endothelial cells; binding of thrombin strongly
reduces cleavage of fibrinogen and thereby clot formation, inhibits activation of endothelial

Discussion
67
thrombin receptors which may decrease vascular leakage, and enhances activation of
protein C with several anti-inflammatory and anti-coagulatory features [45,72,173].
Detection of thrombomodulin on monocytes and especially the strongly increased surface
expression in traumatized patients indicate a modulatory function of monocytes during
excessive inflammation and coagulation.
PAI-1 as the most potent inhibitor of fibrinolysis was bound to the surface of all leukocytes
and levels increased significantly especially during the later course of injury, suggesting that
peripheral leukocytes might be involved in altered coagulation and an increased risk of
thrombosis in severely injured patients [21]. In line, higher PAI-1 surface levels on
granulocytes and lymphocytes correlated with the numbers of given erythrocyte
concentrates, suggesting that patients with a higher blood loss showed higher binding of
PAI-1 on the leukocyte surface. In order to evaluate several possible causes for the
aforementioned increase in membrane-bound PAI-1 around day 5 after trauma,
concentrations in plasma from patients and healthy volunteers were measured. There was a
strong increase in circulating PAI-1 likely due to an increased expression in the liver [136]
during the early phase, but not in the later course after trauma, therefore presumably not
accounting for the increased surface detection on leukocytes. In a clinical study, a decrease
in active PAI-1 upon admission was detected in the 10% of patients after trauma with signs
of severe hyperfibrinolysis [22], which did not seem to be the case in our cohort. A more
recent study suggests that excessive tissue plasminogen activator release and complexation
of PAI-1 may be responsible for initial hyperfibrinolysis and trauma-induced coagulopathy
[25]. Of note, these studies only analyzed clotting disturbances in the very early phase and
did not monitor coagulation during the later time course after injury. As a next step, isolated
neutrophils were stimulated with LPS or a combination of inflammatory mediators (C3a,
C5a, IL-1, IL-6, IL-8) in trauma-relevant concentrations [65] in order to simulate the pro-
inflammatory conditions in severely injured patients, resulting in a fast increase in PAI-1
detectable on the cell surface. However, after stimulating cells in a more complex setting
using whole blood, this result could only be confirmed after stimulation with a potent
activator of the protein kinase C [23], but not with LPS or trauma-relevant mediators.
Similarly, alterations in microvesicles shed from the neutrophil surface were only detectable
after protein kinase C activation. As an additional approach, gene expression levels of PAI-
1 and its cellular anchor uPAR [6] were analyzed by qPCR. However, PAI-1 expression
levels were not increased after short-term exposure to inflammatory stimuli in contrast to
previous studies [145]. UPAR gene expression was by tendency higher after a short

Discussion
68
incubation with inflammatory cytokines or LPS, suggesting that a modest increase in uPAR
surface expression may be partly causative for elevated detection of PAI-1 on the cell
surface. Considering the observed changes especially in PAI-1, it is tempting to speculate
that blood leukocytes are involved in regulation of coagulation and may partially contribute
to thrombotic events, including venous thromboembolism, in patients after trauma [21] since
several leukocytes have been demonstrated to switch to pro-coagulatory phenotypes in
inflammatory (micro)environments [103,145,182]. Furthermore, several neutrophil
components, such as their cellular surface, shed microvesicles, granule proteins, or NETs,
have been established to be significantly involved in the modulation of coagulatory
processes [33,71,150] and can be presumed to play a role in coagulopathy after trauma.
Therefore, future experiments need to characterize in detail which factors in patient blood
might be responsible for changes in the expression of coagulation factors on peripheral
leukocytes. In this regard, a promising approach would also be the stimulation with
endogenous DAMPs such as HMGB-1, histones, or mitochondrial DNA which are able to
substantially activate pro-inflammatory processes in leukocytes.
4.2 Coagulopathy in polytraumatized patients
Disturbances in processes of coagulation represent a central risk factor for mortality in
trauma patients [16,97] and optimized transfusion strategies seem to be essential in order to
improve outcome [32,42,67]. To assess whether there was apparent coagulopathy in our PT
cohort, thromboelastometric analyses were performed in stored plasma samples. Patients
revealed impaired clotting as evident in delayed clot formation and a reduced clot stability
especially during the early time course after trauma. As summarized recently [15,176],
abnormal clot amplitude or MCF are indicators of coagulopathy and can be valuable tools
in the prediction of needed blood transfusion and mortality. The data in the present study
suggest substantial disturbances predominantly in initial activation of coagulation, although,
surprisingly, no hyperfibrinolysis as typically present after trauma [79,153] was detected in
this cohort. This may be due to a difficulty in diagnosing moderate fibrinolysis employing
thromboelastometric approaches by which the percentage of patients with fibrinolytic events
may be underestimated [139]. Since a differential analysis of the contribution of the cellular
blood components, including, but not limited to thrombocytes, was not possible due to
analysis of plasma samples, future coagulation analyses in whole blood from patients during
the time course after injury and corresponding correlations with flow cytometric data could
also shed more light on the earlier discussed role of leukocyte surface molecules in the
regulation of coagulatory and fibrinolytic processes.

Discussion
69
4.3 Functional immune monitoring in trauma
In a translational pilot study using a highly standardized ex vivo whole blood model, blood
from healthy volunteers and from polytraumatized patients was stimulated with LPS in order
to develop a tool for the diagnosis of remaining functional capacity of a patient’s immune
system. This experiment was performed with two aims: defining the changes in the
immediate immune reaction to a pathogenic stimulus after severe injury, and shortening the
incubation time in order to render this application of functional immune monitoring more
suitable for clinical use. Several studies have been performed focusing on the effects of
trauma, shock, or sepsis on peripheral blood cells and the alterations in their response to
bacterial stimuli [17,39,44,61,62,64,87,102,142,156,159]. However, those studies analyzed
isolated cells, not including effects of soluble plasma mediators or intercellular actions, or
used protocols with low standardization potential which are not suitable for testing in clinical
routine.
In the present study, when whole blood was stimulated with LPS, the secretion of
predominantly monocyte- and macrophage-, but also lymphocyte-derived cytokines was
significantly reduced during the time course after trauma in line with the literature
[11,68,81,129,156,158], although this effect was less distinct 24 h after injury. In contrast,
T cell function as reflected by the secretion of mediators mainly involved in T cell activation
after endotoxin incubation was not compromised, indicating that the observed alterations in
PAMP/DAMP surface receptors did not influence the lymphocytic capability to react to a
defined PAMP stimulus. As a major limitation, the small sample size, also due to the high
cost/sample ratio, and large interindividual variation [117] prevented a broad and definitive
assessment of immune functionality in this approach. Validation in a larger patient cohort
might also facilitate the detection of more subtle changes in the immune response as e.g.
mediated by MDSCs [31,119]. Furthermore, for some of the assessed cytokines (e.g. IL-9
and IL-12), there were no alterations in the secreted amounts upon stimulus since
concentrations in unstimulated samples were already higher. Nevertheless, this approach
provided a rather comprehensive analysis of the complex immune response in peripheral
blood. In addition, highly significant differences in stimulated and unstimulated samples
despite the markedly shortened incubation time demonstrated that this methodology could
provide a highly useful diagnostic tool for the immune surveillance of trauma patients and
may aid to optimize timing of surgical procedures during the posttraumatic immune response
[55,185]. Moreover, shortening stimulation time even further and analyzing only a few

Discussion
70
selected parameters relevant for the current functional immune status in clinical laboratories
might enable a valid and reliable immune monitoring and maximize clinical usefulness.
4.4 HS as a major driver of barrier and organ dysfunction after PT
In the past years, there has been accumulating evidence that severe blood loss may be one
of the central causes for posttraumatic barrier damage and organ dysfunction and failure
[49,78,154,188]. In order to assess whether the presence or absence of a HS was predictive
for organ damage during the early time course after injury, a larger PT patient cohort from a
clinical study at the University Hospital Zurich was stratified into those patients with and
those without HS according to established clinical parameters [88,108,190]. Despite equal
severity of injury in the two cohorts as reflected by the ISS and similar values in most
assessed inflammatory mediators except for IL-6, HS induced a spatially and temporally
specific injury of the lung, kidney, the intestine, and the liver as reflected by distinct serum
organ damage markers [7,143,184].
The present data did not confirm that CC16 correlates with the contused lung tissue volume
after PT [184] since there was no association with the AIS of the thorax; however, the
correlation of serum CC16 concentrations with transfused volume and positive fluid balance
suggest an increase in the permeability of the air-blood barrier [66], possibly leading to fluid
accumulation in the lung. A similar result was recently published in our murine trauma
model where only a combination of PT and HS induced a significant increase in plasma
CC16 [36].
The kidneys represent central organs in the pathophysiology after PT and HS as evidenced
by a report from a cohort of more than 2000 injured patients [181]; of those patients with
early acute kidney injury (AKI), 78% developed multiple organ failure and 27% died; both
rates are higher than those in patients with early lung, heart, or liver injury. NGAL has been
described to be produced by a number of tissues and cells, including the kidneys, neutrophils,
lungs, and liver [30,82,107,110]. HS is a strong inducer of renal hypoperfusion and hypoxia
[189] and release of NGAL from renal tubular cells was shown in response to ischemia-
reperfusion injury [116], concurrent with an early increase in serum NGAL in the HS patient
cohort in this study. This is indicative of an early remote kidney injury induced by prerenal
shock pathophysiology. Interestingly, NGAL values were associated with the initial severity
of abdominal injury although the kidneys are rarely affected by the trauma hit, suggesting
that HS significantly increases the risk of trauma-caused AKI and multiple organ failure.
This was also shown in our recent study in murine PT and HS [36]. Furthermore, correlation

Discussion
71
of serum NGAL with procalcitonin may be due to infectious complications since septic AKI
is often induced by high amounts of trauma-induced release of DAMPs and PAMPs. It is
noteworthy that NGAL was established as a marker for kidney damage with promising
results [7,109], but its specificity has ever since been discussed controversially [59,104].
Nevertheless, its validity in diagnosing AKI especially in combination with other markers
or in the urine remains high [2].
Abdominal injury was assessed using L-FABP, expressed in liver, intestine, kidney and
pancreas [38,93,96] as a reliable marker of abdominal and especially liver damage
[20,111,143], and I-FABP which is specifically expressed in the small intestine [132]. L-
FABP was higher in HS patients and correlated with coagulation parameters as well as
procalcitonin, likely due to the high blood loss in patients with abdominal injury, also
reflected by the association of HS with higher AIS of the abdomen, and a well-established
connection between abdominal trauma and procalcitonin [101,152]. A disruption of the
intestinal barrier was indicated by an immediate increase in I-FABP serum concentrations
in HS patients, and levels correlated with lactate and the fluid balance, again suggesting a
connection with the shock-induced loss in intraluminal blood volume. A recent study in burn
patients with multiple organ dysfunction demonstrated a highly similar serum I-FABP
profile, most likely due to injury-induced intestinal ischemia and barrier breakdown [128].
It can therefore be presumed that HS is detrimental in the course of posttraumatic abdominal
ischemia and organ damage.
A breakdown in endothelial barrier function is likely to play a significant role in the
development of posttraumatic organ pathology. Of note, the different organ damage markers
showed very distinct time patterns, possible due to differential alterations in the respective
blood-organ barriers. Barrier permeability can be regulated by a large number of molecules.
The lipid mediator sphingosine-1-phosphate, besides its involvement in activation of the
immune system, seems to play a significant role in this regulation especially when stabilized
by binding to plasma albumin [124,191]. Therapeutically, a functional sphingosine-1-
phosphate analogue has already been applied in the context of trauma/HS [9]. Lower serum
albumin upon admission has recently been linked to endotheliopathy in severely injured
patients [148]. Concentrations of both sphingosine-1-phosphate and albumin clearly
decreased early in patients after trauma, although HS only seemed to play a minor role here.
Interestingly, HS alone in the murine trauma setting rather induced an increase in serum
albumin, while a reduction was only visible after combined PT+HS. Furthermore, a decrease

Discussion
72
in plasma thrombomodulin, detected immediately after injury in the PT+HS cohort and in
both mouse groups which had undergone HS, can indicate the activation of endothelial cells
[70]. These data are suggestive of an initiation of endotheliopathy with disturbances in
anticoagulatory surface properties and barrier function as soon as patients arrive at the
emergency room. Angiopoietin-2 as a mediator involved in pro-inflammatory processes and
vascular leakage [57], but also adrenomedullin with rather opposite effects [164] were
increased only in the HS patient cohort and rather at the later time points after trauma. These
findings and the correlation of angiopoietin with the number of initially transfused
erythrocyte concentrates point to a strong activation of pathways regulating endothelial
permeability exclusively in patients after HS. This is also supported by initial increases in
MMP-9 (albeit in both patient cohorts) and MMP-13, matrix proteases which have been
shown to be involved in cleaving components of the apical glycocalyx layer off endothelial
cells [138,191]. In line, increased levels of syndecan-1 and heparan sulfate were detected
especially in the HS cohort; similar results were obtained in the murine PT setting where an
isolated HS already induced a clear enhancement of MMP-9, -13, and syndecan-1 plasma
concentrations compared to controls, but animals with combined PT+HS had highest
corresponding concentrations. Shedding of glycocalyx components is likely to be
responsible for increased vascular permeability after trauma and HS [137]. In this context,
syndecan-1 and its fragment heparan sulfate have been increasingly used as markers of
endothelial glycocalyx degradation especially by the group around P.I. Johansson
[75,76,125,148] and may induce a so-called “autoheparinization” effect due to their
anticoagulatory features [126,141]. This was assessed in an ex vivo experiment in blood
samples with artificially adjusted hematocrits similar to those in healthy volunteers (45%),
PT (33%), and PT+HS (25%) patients where heparan sulfate significantly delayed clotting.
Furthermore, correlations of syndecan-1 with serum markers of organ dysfunction indicate
a causal relationship between endothelial damage and organ failure. Mechanistically, release
of (nor-)epinephrine induced by sympathoadrenal activation may be a central driver of
endotheliopathy [77,125]. As a limitation of the present study, catecholamine levels were
not measured in this patient cohort. However, in addition to endogenous production, PT
patients especially after HS are likely to have received catecholamine support during the
initial phase after trauma as part of the standard emergency care protocols. Plasma fibrinogen
was lower in HS patients throughout the entire observation period; D-dimers were initially
decreased in both cohorts, but an additional effect of HS was not detectable, presumably due
to plasma dilution after extensive fluid replacement with crystalloids.

Discussion
73
Increased endothelial permeability was furthermore assessed by measurement of circulating
intercellular tight-junction and adherens-junction molecules. As recently published, we
found an early increase in plasma JAM-1, a central component of tight junctions, in humans
and mice after PT [37]. Claudin-5 as another molecule involved in regulation of tight-
junction permeability, especially of the blood-brain barrier, but also in other organs and
tissues [29,47,121], was increased in PT+HS patients and correlated with markers of HS,
suggesting that barrier disturbances after HS not only include the apical glycocalyx, but also
intercellular junctions. Unexpectedly, we also detected increased amounts of mucin-2, a
central component of intestinal mucus, in the circulation, indicating a disturbed blood-gut
barrier possible due to a disruption of intestinal mucus layers by pancreatic enzymes post
trauma [48]. Similarly to heparan sulfate, mucin-2 in concentrations measured in our patient
cohorts (up to 6000 pg/ml) prolonged coagulation in blood samples with low hematocrit,
suggesting that components of the intestine released by a direct or indirect gut barrier damage
may interfere with coagulation and could be partly responsible for coagulopathy in severely
injured patients.
4.5 Conclusion
Severe trauma induces several alterations in leukocyte surface molecules and is associated
with cell type-specific changes in the immune response to bacterial stimuli. This study
demonstrated that coagulation is significantly impaired predominantly during the early phase
after trauma, and leukocyte surface molecules may play a detrimental role. Furthermore, a
clinically relevant testing system was established to allow functional bed-side monitoring of
the patient’s remaining immune capacity which revealed a deficit especially in monocyte
function. Lastly, the differential analysis of a larger patient cohort revealed HS as a major
driver of posttraumatic organ damage. In addition, several molecules associated with barrier
(dys-)function were significantly altered by posttraumatic HS with very distinct time
patterns, underlining the importance of effective management of bleeding and reliable
monitoring of barrier function in order to treat latent and evident organ damage before onset
of leakage syndrome and organ failure.

Summary
74
5 Summary
Despite the considerable progress made in public safety measures, emergency and intensive
care in the last decades, trauma remains a major medical issue and is responsible for a large
number of deaths worldwide particularly in young people. In regard to the posttraumatic
immune response, development of disturbances in coagulation, and detection and monitoring
of end organ damage, several issues remain unresolved. Therefore, this thesis focused on the
alterations in peripheral blood leukocytes as “first line of defense” after severe injury
employing static and functional analyses. Furthermore, disturbances in coagulation were
observed over a 10-day time course after trauma. In a larger patient cohort, the impact of an
additional hemorrhagic shock on inflammation, organ damage, and breakdown of the
endothelial glycocalyx was assessed. These findings were re-translationally evaluated in a
murine model of isolated and combined trauma and hemorrhage.
In patients after severe trauma, peripheral leukocytes rapidly changed their surface
expression profile of several receptors for complement activation products as well as
damage- and pathogen-associated molecular patterns, involved in regulating the immune
response, but also modulators of coagulation. In a pilot study, a highly standardized ex vivo
testing system revealed defects in production of early pro-inflammatory cytokines such as
tumor-necrosis factor and interleukin-1 upon incubation with microbial endotoxin in
monocytes, but not in the lymphocyte function after severe injury. A shortened standardized
exposure time (4 h) resulted in a similar immune response compared to the established
incubation protocol. After further validation and optimization, this method might offer a
useful tool to monitor the immune system’s remaining capacity to react to inflammatory or
pathogenic stimuli. In a similar approach, functional monitoring of coagulation over the time
course after injury detected striking defects in coagulation immediately after trauma which
partly lasted until 10 days later, but no hyperfibrinolysis was found.
In a larger patient cohort, hemorrhagic shock was demonstrated to significantly contribute
to posttraumatic inflammation and organ damage. Despite comparable injury severity,
patients after blood loss had increased serum concentrations of specific damage markers of
the lung (Clara cell secretory protein), liver (liver-type fatty acid-binding protein), kidneys
(neutrophil gelatinase-associated lipocalin) and the intestine (intestinal fatty acid-binding
protein) in distinct spatial and temporal patterns. Furthermore, presumably by increased
generation of matrix metalloproteinases-9 and -13, there was an elevation of circulating
components of the glycocalyx such as syndecan-1 and heparan sulfate, suggesting

Summary
75
endothelial damage and increased permeability. Interestingly, increased concentrations of
elements of the intestinal mucus (mucin-2) were detected after trauma/hemorrhage, implying
a deterioration in intestinal barrier function. In vitro experiments revealed that the detected
amounts of glycocalyx as well as intestinal mucus components were able to interfere with
coagulation, indicating that the posttraumatic barrier breakdown might also impair clotting
and increase the risk of bleeding. These findings were mostly confirmed in a murine model
of PT and hemorrhage, where shock alone already demonstrated marked impact on barrier
dysfunction and glycocalyx breakdown. The data is indicative of an early development of
barrier and organ damage, presumably hours to days before manifestation of barrier
breakdown and organ dysfunction. Monitoring the posttraumatic course using novel organ
damage markers in addition to traditional functional scores may aid to detect subpar
tendencies before onset of (multiple) organ dysfunction.
In conclusion, severe trauma leads to significant alterations in leukocyte and immune
function, coagulation, and considerable barrier and organ dysfunction which is worsened in
the presence of a hemorrhagic shock. Reliable monitoring of the clinical course using valid
functional markers and assays as well as rapid implementation into clinical routine and
advanced treatment strategies may be decisive for improved outcome.

List of references
76
6 List of references
1. Amara U, Kalbitz M, Perl M, Flierl MA, Rittirsch D, Weiss M, Schneider M, Gebhard
F, Huber-Lang M: Early expression changes of complement regulatory proteins and
C5A receptor (CD88) on leukocytes after multiple injury in humans. Shock 33: 568-
575 (2010)
2. Asada T, Isshiki R, Hayase N, Sumida M, Inokuchi R, Noiri E, Nangaku M, Yahagi
N, Doi K: Impact of clinical context on acute kidney injury biomarker performances:
differences between neutrophil gelatinase-associated lipocalin and L-type fatty acid-
binding protein. Sci Rep 6: 33077 (2016)
3. Baker SP, O'Neill B, Haddon W, Jr., Long WB: The injury severity score: a method
for describing patients with multiple injuries and evaluating emergency care. J Trauma
14: 187-196 (1974)
4. Bhandari M, Tornetta P, III, Sprague S, Najibi S, Petrisor B, Griffith L, Guyatt GH:
Predictors of reoperation following operative management of fractures of the tibial
shaft. J Orthop Trauma 17: 353-361 (2003)
5. Billiar TR, Vodovotz Y: Time for trauma immunology. PLoS Med 14: e1002342
(2017)
6. Binder BR, Mihaly J, Prager GW: uPAR-uPA-PAI-1 interactions and signaling: a
vascular biologist's view. Thromb Haemost 97: 336-342 (2007)
7. Bolignano D, Donato V, Coppolino G, Campo S, Buemi A, Lacquaniti A, Buemi M:
Neutrophil gelatinase-associated lipocalin (NGAL) as a marker of kidney damage. Am
J Kidney Dis 52: 595-605 (2008)
8. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM,
Sibbald WJ: Definitions for sepsis and organ failure and guidelines for the use of
innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee.
American College of Chest Physicians/Society of Critical Care Medicine. Chest 101:
1644-1655 (1992)
9. Bonitz JA, Son JY, Chandler B, Tomaio JN, Qin Y, Prescott LM, Feketeova E, Deitch
EA: A sphingosine-1 phosphate agonist (FTY720) limits trauma/hemorrhagic shock-
induced multiple organ dysfunction syndrome. Shock 42: 448-455 (2014)

List of references
77
10. Bonnarens F, Einhorn TA: Production of a standard closed fracture in laboratory
animal bone. J Orthop Res 2: 97-101 (1984)
11. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman
SD, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS:
Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA
306: 2594-2605 (2011)
12. Botha AJ, Moore FA, Moore EE, Sauaia A, Banerjee A, Peterson VM: Early
neutrophil sequestration after injury: a pathogenic mechanism for multiple organ
failure. J Trauma 39: 411-417 (1995)
13. Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF: Acute
traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C
pathway? Ann Surg 245: 812-818 (2007)
14. Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, Pittet JF: Acute
coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and
hyperfibrinolysis. J Trauma 64: 1211-1217 (2008)
15. Brohi K, Eaglestone S: Traumatic coagulopathy and massive transfusion: improving
outcomes and saving blood. Programme Grants for Applied Research 5: (2017)
16. Brohi K, Singh J, Heron M, Coats T: Acute traumatic coagulopathy. J Trauma 54:
1127-1130 (2003)
17. Bulger EM, Cuschieri J, Warner K, Maier RV: Hypertonic resuscitation modulates the
inflammatory response in patients with traumatic hemorrhagic shock. Ann Surg 245:
635-641 (2007)
18. Burk AM, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, Bruckner U, Stahl
GL, Blom AM, Perl M, Gebhard F, Huber-Lang M: Early complementopathy after
multiple injuries in humans. Shock 37: 348-354 (2012)
19. Cairns CB: Rude unhinging of the machinery of life: metabolic approaches to
hemorrhagic shock. Curr Opin Crit Care 7: 437-443 (2001)
20. Cakir OO, Toker A, Ataseven H, Demir A, Polat H: The Importance of Liver-Fatty
Acid Binding Protein in Diagnosis of Liver Damage in Patients with Acute Hepatitis.
J Clin Diagn Res 11: OC17-OC21 (2017)

List of references
78
21. Cap A, Hunt B: Acute traumatic coagulopathy. Curr Opin Crit Care 20: 638-645
(2014)
22. Cardenas JC, Matijevic N, Baer LA, Holcomb JB, Cotton BA, Wade CE: Elevated
tissue plasminogen activator and reduced plasminogen activator inhibitor promote
hyperfibrinolysis in trauma patients. Shock 41: 514-521 (2014)
23. Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y: Direct activation
of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting
phorbol esters. J Biol Chem 257: 7847-7851 (1982)
24. Cekic C, Linden J: Purinergic regulation of the immune system. Nat Rev Immunol 16:
177-192 (2016)
25. Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Chandler JG, Mitra S,
Ghasabyan A, Chin TL, Sauaia A, Banerjee A, Silliman CC: Overwhelming tPA
release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured
trauma patients. J Trauma Acute Care Surg 80: 16-23 (2016)
26. Ciesla DJ, Moore EE, Johnson JL, Burch JM, Cothren CC, Sauaia A: A 12-year
prospective study of postinjury multiple organ failure: has anything changed? Arch
Surg 140: 432-438 (2005)
27. Cohen MJ, Christie SA: Coagulopathy of Trauma. Crit Care Clin 33: 101-118 (2017)
28. Colonna M, Facchetti F: TREM-1 (triggering receptor expressed on myeloid cells): a
new player in acute inflammatory responses. J Infect Dis 187 Suppl 2: S397-S401
(2003)
29. Cooper I, Cohen-Kashi-Malina K, Teichberg VI: Claudin-5 expression in in vitro
models of the blood-brain barrier. Methods Mol Biol 762: 347-354 (2011)
30. Cowland JB, Borregaard N: Molecular characterization and pattern of tissue
expression of the gene for neutrophil gelatinase-associated lipocalin from humans.
Genomics 45: 17-23 (1997)
31. Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM,
Heyworth PG, Efron PA, Moldawer LL: A paradoxical role for myeloid-derived
suppressor cells in sepsis and trauma. Mol Med 17: 281-292 (2011)
32. Curry NS, Davenport RA, Hunt BJ, Stanworth SJ: Transfusion strategies for traumatic
coagulopathy. Blood Rev 26: 223-232 (2012)

List of references
79
33. Darbousset R, Thomas GM, Mezouar S, Frere C, Bonier R, Mackman N, Renne T,
Dignat-George F, Dubois C, Panicot-Dubois L: Tissue factor-positive neutrophils bind
to injured endothelial wall and initiate thrombus formation. Blood 120: 2133-2143
(2012)
34. Davenport R, Manson J, De'Ath H, Platton S, Coates A, Allard S, Hart D, Pearse R,
Pasi KJ, MacCallum P, Stanworth S, Brohi K: Functional definition and
characterization of acute traumatic coagulopathy. Crit Care Med 39: 2652-2658 (2011)
35. Degn SE, Thiel S: Humoral pattern recognition and the complement system. Scand J
Immunol 78: 181-193 (2013)
36. Denk S, Weckbach S, Eisele P, Braun CK, Wiegner R, Ohmann JJ, Wrba L, Hoenes
FM, Kellermann P, Radermacher P, Wachter U, Hafner S, McCook O, Schultze A,
Palmer A, Braumuller S, Gebhard F, Huber-Lang M: Role of Hemorrhagic Shock in
Experimental Polytrauma. Shock 49: 154-163 (2018)
37. Denk S, Wiegner R, Hones FM, Messerer DA, Radermacher P, Weiss M, Kalbitz M,
Ehrnthaller C, Braumuller S, McCook O, Gebhard F, Weckbach S, Huber-Lang M:
Early Detection of Junctional Adhesion Molecule-1 (JAM-1) in the Circulation after
Experimental and Clinical Polytrauma. Mediators Inflamm 2015: 463950 (2015)
38. Derikx JP, Poeze M, van Bijnen AA, Buurman WA, Heineman E: Evidence for
intestinal and liver epithelial cell injury in the early phase of sepsis. Shock 28: 544-
548 (2007)
39. Dimofte G, Alexander A, Carlson G, Little R, Irving M: TNF alpha and IL-6
involvement in surgical trauma. II. In vitro cytokine production. Rev Med Chir Soc
Med Nat Iasi 105: 493-498 (2001)
40. Dirkmann D, Radu-Berlemann J, Gorlinger K, Peters J: Recombinant tissue-type
plasminogen activator-evoked hyperfibrinolysis is enhanced by acidosis and inhibited
by hypothermia but still can be blocked by tranexamic acid. J Trauma Acute Care Surg
74: 482-488 (2013)
41. Dunne JR, Malone DL, Tracy JK, Napolitano LM: Allogenic blood transfusion in the
first 24 hours after trauma is associated with increased systemic inflammatory
response syndrome (SIRS) and death. Surg Infect (Larchmt ) 5: 395-404 (2004)

List of references
80
42. Eastridge BJ, Mabry RL, Seguin P, Cantrell J, Tops T, Uribe P, Mallett O, Zubko T,
Oetjen-Gerdes L, Rasmussen TE, Butler FK, Kotwal RS, Holcomb JB, Wade C,
Champion H, Lawnick M, Moores L, Blackbourne LH: Death on the battlefield (2001-
2011): implications for the future of combat casualty care. J Trauma Acute Care Surg
73: S431-S437 (2012)
43. El MR, El GM, Seeds MC, McCall CE, Dreskin SC, Nicolls MR: Endogenous signals
released from necrotic cells augment inflammatory responses to bacterial endotoxin.
Immunol Lett 111: 36-44 (2007)
44. Ertel W, Kremer JP, Kenney J, Steckholzer U, Jarrar D, Trentz O, Schildberg FW:
Downregulation of proinflammatory cytokine release in whole blood from septic
patients. Blood 85: 1341-1347 (1995)
45. Esmon CT: Protein C anticoagulant system--anti-inflammatory effects. Semin
Immunopathol 34: 127-132 (2012)
46. Fan J, Li Y, Levy RM, Fan JJ, Hackam DJ, Vodovotz Y, Yang H, Tracey KJ, Billiar
TR, Wilson MA: Hemorrhagic shock induces NAD(P)H oxidase activation in
neutrophils: role of HMGB1-TLR4 signaling. J Immunol 178: 6573-6580 (2007)
47. Fernandez AL, Koval M, Fan X, Guidot DM: Chronic alcohol ingestion alters claudin
expression in the alveolar epithelium of rats. Alcohol 41: 371-379 (2007)
48. Fishman JE, Sheth SU, Levy G, Alli V, Lu Q, Xu D, Qin Y, Qin X, Deitch EA:
Intraluminal nonbacterial intestinal components control gut and lung injury after
trauma hemorrhagic shock. Ann Surg 260: 1112-1120 (2014)
49. Fleming SD, Phillips LM, Lambris JD, Tsokos GC: Complement component C5a
mediates hemorrhage-induced intestinal damage. J Surg Res 150: 196-203 (2008)
50. Frith D, Davenport R, Brohi K: Acute traumatic coagulopathy. Curr Opin Anaesthesiol
25: 229-234 (2012)
51. Frith D, Goslings JC, Gaarder C, Maegele M, Cohen MJ, Allard S, Johansson PI,
Stanworth S, Thiemermann C, Brohi K: Definition and drivers of acute traumatic
coagulopathy: clinical and experimental investigations. J Thromb Haemost 8: 1919-
1925 (2010)
52. Fritz G: RAGE: a single receptor fits multiple ligands. Trends Biochem Sci 36: 625-
632 (2011)

List of references
81
53. Ganter MT, Brohi K, Cohen MJ, Shaffer LA, Walsh MC, Stahl GL, Pittet JF: Role of
the alternative pathway in the early complement activation following major trauma.
Shock 28: 29-34 (2007)
54. Gebhard F, Huber-Lang M: Polytrauma--pathophysiology and management
principles. Langenbecks Arch Surg 393: 825-831 (2008)
55. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL,
Moore FA: Persistent inflammation and immunosuppression: a common syndrome
and new horizon for surgical intensive care. J Trauma Acute Care Surg 72: 1491-1501
(2012)
56. German Trauma Society (DGU), Committee on Emergency Medicine ICaTMSN,
AUC - Academy for Trauma Surgery: TraumaRegister DGU® - Annual Report 2017.
<[11] Journal> <[12] Volume>: (9-15-2017)
57. Giamarellos-Bourboulis EJ, Kanellakopoulou K, Pelekanou A, Tsaganos T,
Kotzampassi K: Kinetics of angiopoietin-2 in serum of multi-trauma patients:
correlation with patient severity. Cytokine 44: 310-313 (2008)
58. Haagsma JA, Graetz N, Bolliger I, Naghavi M, Higashi H, Mullany EC, Abera SF,
Abraham JP, Adofo K, Alsharif U, Ameh EA, Ammar W, Antonio CA, Barrero LH,
Bekele T, Bose D, Brazinova A, Catala-Lopez F, Dandona L, Dandona R, Dargan PI,
De LD, Degenhardt L, Derrett S, Dharmaratne SD, Driscoll TR, Duan L, Petrovich
ES, Farzadfar F, Feigin VL, Franklin RC, Gabbe B, Gosselin RA, Hafezi-Nejad N,
Hamadeh RR, Hijar M, Hu G, Jayaraman SP, Jiang G, Khader YS, Khan EA,
Krishnaswami S, Kulkarni C, Lecky FE, Leung R, Lunevicius R, Lyons RA, Majdan
M, Mason-Jones AJ, Matzopoulos R, Meaney PA, Mekonnen W, Miller TR, Mock
CN, Norman RE, Orozco R, Polinder S, Pourmalek F, Rahimi-Movaghar V, Refaat A,
Rojas-Rueda D, Roy N, Schwebel DC, Shaheen A, Shahraz S, Skirbekk V, Soreide K,
Soshnikov S, Stein DJ, Sykes BL, Tabb KM, Temesgen AM, Tenkorang EY, Theadom
AM, Tran BX, Vasankari TJ, Vavilala MS, Vlassov VV, Woldeyohannes SM, Yip P,
Yonemoto N, Younis MZ, Yu C, Murray CJ, Vos T: The global burden of injury:
incidence, mortality, disability-adjusted life years and time trends from the Global
Burden of Disease study 2013. Inj Prev 22: 3-18 (2016)
59. Haase M, Bellomo R, Devarajan P, Schlattmann P, Haase-Fielitz A: Accuracy of
neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute

List of references
82
kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 54: 1012-1024
(2009)
60. Halbgebauer R, Braun CK, Denk S, Mayer B, Cinelli P, Radermacher P, Wanner GA,
Simmen HP, Gebhard F, Rittirsch D, Huber-Lang M: Hemorrhagic shock drives
glycocalyx, barrier and organ dysfunction early after polytrauma. J Crit Care 44: 229-
237 (2017)
61. Hassett S, Moynagh P, Reen D: TNF-alpha is a mediator of the anti-inflammatory
response in a human neonatal model of the non-septic shock syndrome. Pediatr Surg
Int 22: 24-30 (2006)
62. Hazeldine J, Naumann DN, Toman E, Davies D, Bishop JRB, Su Z, Hampson P,
Dinsdale RJ, Crombie N, Duggal NA, Harrison P, Belli A, Lord JM: Prehospital
immune responses and development of multiple organ dysfunction syndrome
following traumatic injury: A prospective cohort study. PLoS Med 14: e1002338
(2017)
63. Hebert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, Tweeddale
M, Schweitzer I, Yetisir E: A multicenter, randomized, controlled clinical trial of
transfusion requirements in critical care. Transfusion Requirements in Critical Care
Investigators, Canadian Critical Care Trials Group. N Engl J Med 340: 409-417 (1999)
64. Heftrig D, Sturm R, Oppermann E, Kontradowitz K, Jurida K, Schimunek L, Woschek
M, Marzi I, Relja B: Impaired Surface Expression of HLA-DR, TLR2, TLR4, and
TLR9 in Ex Vivo-In Vitro Stimulated Monocytes from Severely Injured Trauma
Patients. Mediators Inflamm 2017: 2608349 (2017)
65. Hengartner NE, Fiedler J, Schrezenmeier H, Huber-Lang M, Brenner RE: Crucial role
of IL1beta and C3a in the in vitro-response of multipotent mesenchymal stromal cells
to inflammatory mediators of polytrauma. PLoS One 10: e0116772 (2015)
66. Hermans C, Knoops B, Wiedig M, Arsalane K, Toubeau G, Falmagne P, Bernard A:
Clara cell protein as a marker of Clara cell damage and bronchoalveolar blood barrier
permeability. Eur Respir J 13: 1014-1021 (1999)
67. Holcomb JB, McMullin NR, Pearse L, Caruso J, Wade CE, Oetjen-Gerdes L,
Champion HR, Lawnick M, Farr W, Rodriguez S, Butler FK: Causes of death in U.S.
Special Operations Forces in the global war on terrorism: 2001-2004. Ann Surg 245:
986-991 (2007)

List of references
83
68. Hotchkiss RS, Karl IE: The pathophysiology and treatment of sepsis. N Engl J Med
348: 138-150 (2003)
69. Huber-Lang M, Lambris JD, Ward PA: Innate immune responses to trauma. Nat
Immunol (2018)
70. Hunt BJ, Jurd KM: Endothelial cell activation. A central pathophysiological process.
BMJ 316: 1328-1329 (1998)
71. Iba T, Miki T, Hashiguchi N, Tabe Y, Nagaoka I: Is the neutrophil a 'prima donna' in
the procoagulant process during sepsis? Crit Care 18: 230 (2014)
72. Ito T, Kakihana Y, Maruyama I: Thrombomodulin as an intravascular safeguard
against inflammatory and thrombotic diseases. Expert Opin Ther Targets 20: 151-158
(2016)
73. Ives C, Inaba K, Branco BC, Okoye O, Schochl H, Talving P, Lam L, Shulman I,
Nelson J, Demetriades D: Hyperfibrinolysis elicited via thromboelastography predicts
mortality in trauma. J Am Coll Surg 215: 496-502 (2012)
74. Jenkins DH, Rappold JF, Badloe JF, Berseus O, Blackbourne L, Brohi KH, Butler FK,
Cap AP, Cohen MJ, Davenport R, DePasquale M, Doughty H, Glassberg E, Hervig T,
Hooper TJ, Kozar R, Maegele M, Moore EE, Murdock A, Ness PM, Pati S, Rasmussen
T, Sailliol A, Schreiber MA, Sunde GA, van de Watering LM, Ward KR, Weiskopf
RB, White NJ, Strandenes G, Spinella PC: Trauma hemostasis and oxygenation
research position paper on remote damage control resuscitation: definitions, current
practice, and knowledge gaps. Shock 41 Suppl 1: 3-12 (2014)
75. Johansson PI, Henriksen HH, Stensballe J, Gybel-Brask M, Cardenas JC, Baer LA,
Cotton BA, Holcomb JB, Wade CE, Ostrowski SR: Traumatic Endotheliopathy: A
Prospective Observational Study of 424 Severely Injured Patients. Ann Surg 265: 597-
603 (2017)
76. Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR: A high admission syndecan-
1 level, a marker of endothelial glycocalyx degradation, is associated with
inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma
patients. Ann Surg 254: 194-200 (2011)

List of references
84
77. Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR: High circulating adrenaline
levels at admission predict increased mortality after trauma. J Trauma Acute Care Surg
72: 428-436 (2012)
78. Johnson JL, Moore EE, Kashuk JL, Banerjee A, Cothren CC, Biffl WL, Sauaia A:
Effect of blood products transfusion on the development of postinjury multiple organ
failure. Arch Surg 145: 973-977 (2010)
79. Kashuk JL, Moore EE, Sawyer M, Wohlauer M, Pezold M, Barnett C, Biffl WL,
Burlew CC, Johnson JL, Sauaia A: Primary fibrinolysis is integral in the pathogenesis
of the acute coagulopathy of trauma. Ann Surg 252: 434-442 (2010)
80. Kauvar DS, Wade CE: The epidemiology and modern management of traumatic
hemorrhage: US and international perspectives. Crit Care 9 Suppl 5: S1-S9 (2005)
81. Keel M, Trentz O: Pathophysiology of polytrauma. Injury 36: 691-709 (2005)
82. Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N: Isolation and primary structure
of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem
268: 10425-10432 (1993)
83. Knoferl MW, Liener UC, Perl M, Bruckner UB, Kinzl L, Gebhard F: Blunt chest
trauma induces delayed splenic immunosuppression. Shock 22: 51-56 (2004)
84. Knoferl MW, Liener UC, Seitz DH, Perl M, Bruckner UB, Kinzl L, Gebhard F:
Cardiopulmonary, histological, and inflammatory alterations after lung contusion in a
novel mouse model of blunt chest trauma. Shock 19: 519-525 (2003)
85. Kohl J: The role of complement in danger sensing and transmission. Immunol Res 34:
157-176 (2006)
86. Kottke MA, Walters TJ: Where's the Leak in Vascular Barriers? A Review. Shock 46:
20-36 (2016)
87. Kremer JP, Jarrar D, Steckholzer U, Ertel W: Interleukin-1, -6 and tumor necrosis
factor-alpha release is down-regulated in whole blood from septic patients. Acta
Haematol 95: 268-273 (1996)
88. Kruse O, Grunnet N, Barfod C: Blood lactate as a predictor for in-hospital mortality
in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc
Emerg Med 19: 74 (2011)

List of references
85
89. Kung HC, Hoyert DL, Xu J, Murphy SL: Deaths: final data for 2005. Natl Vital Stat
Rep 56: 1-120 (2008)
90. Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM,
Nelson MF, Cohen MJ: Characterization of platelet dysfunction after trauma. J Trauma
Acute Care Surg 73: 13-19 (2012)
91. Ledderose C, Heyn J, Limbeck E, Kreth S: Selection of reliable reference genes for
quantitative real-time PCR in human T cells and neutrophils. BMC Res Notes 4: 427
(2011)
92. Lee EJ, Park JH: Receptor for Advanced Glycation Endproducts (RAGE), Its Ligands,
and Soluble RAGE: Potential Biomarkers for Diagnosis and Therapeutic Targets for
Human Renal Diseases. Genomics Inform 11: 224-229 (2013)
93. Lieberman JM, Sacchettini J, Marks C, Marks WH: Human intestinal fatty acid
binding protein: report of an assay with studies in normal volunteers and intestinal
ischemia. Surgery 121: 335-342 (1997)
94. Liener UC, Bruckner UB, Knoferl MW, Steinbach G, Kinzl L, Gebhard F: Chemokine
activation within 24 hours after blunt accident trauma. Shock 17: 169-172 (2002)
95. Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time
quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408 (2001)
96. Lowe JB, Boguski MS, Sweetser DA, Elshourbagy NA, Taylor JM, Gordon JI: Human
liver fatty acid binding protein. Isolation of a full length cDNA and comparative
sequence analyses of orthologous and paralogous proteins. J Biol Chem 260: 3413-
3417 (1985)
97. MacLeod JB, Lynn M, McKenney MG, Cohn SM, Murtha M: Early coagulopathy
predicts mortality in trauma. J Trauma 55: 39-44 (2003)
98. Maegele M, Lefering R, Yucel N, Tjardes T, Rixen D, Paffrath T, Simanski C,
Neugebauer E, Bouillon B: Early coagulopathy in multiple injury: an analysis from
the German Trauma Registry on 8724 patients. Injury 38: 298-304 (2007)
99. Maegele M, Schochl H, Cohen MJ: An update on the coagulopathy of trauma. Shock
41 Suppl 1: 21-25 (2014)
100. Maier B, Lefering R, Lehnert M, Laurer HL, Steudel WI, Neugebauer EA, Marzi I:
Early versus late onset of multiple organ failure is associated with differing patterns of

List of references
86
plasma cytokine biomarker expression and outcome after severe trauma. Shock 28:
668-674 (2007)
101. Maier M, Wutzler S, Lehnert M, Szermutzky M, Wyen H, Bingold T, Henrich D,
Walcher F, Marzi I: Serum procalcitonin levels in patients with multiple injuries
including visceral trauma. J Trauma 66: 243-249 (2009)
102. Majetschak M, Krehmeier U, Ostroverkh L, Blomeke B, Schafer M: Alterations in
leukocyte function following surgical trauma: differentiation of distinct reaction types
and association with tumor necrosis factor gene polymorphisms. Clin Diagn Lab
Immunol 12: 296-303 (2005)
103. Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD: Complement and
coagulation: strangers or partners in crime? Trends Immunol 28: 184-192 (2007)
104. Martensson J, Bellomo R: The rise and fall of NGAL in acute kidney injury. Blood
Purif 37: 304-310 (2014)
105. Martini WZ, Pusateri AE, Uscilowicz JM, Delgado AV, Holcomb JB: Independent
contributions of hypothermia and acidosis to coagulopathy in swine. J Trauma 58:
1002-1009 (2005)
106. Mikhail J: The trauma triad of death: hypothermia, acidosis, and coagulopathy. AACN
Clin Issues 10: 85-94 (1999)
107. Minami K, Saito T, Narahara M, Tomita H, Kato H, Sugiyama H, Katoh M, Nakajima
M, Yokoi T: Relationship between hepatic gene expression profiles and hepatotoxicity
in five typical hepatotoxicant-administered rats. Toxicol Sci 87: 296-305 (2005)
108. Minei JP, Cuschieri J, Sperry J, Moore EE, West MA, Harbrecht BG, O'Keefe GE,
Cohen MJ, Moldawer LL, Tompkins RG, Maier RV: The changing pattern and
implications of multiple organ failure after blunt injury with hemorrhagic shock. Crit
Care Med 40: 1129-1135 (2012)
109. Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, Ruff SM, Zahedi K,
Shao M, Bean J, Mori K, Barasch J, Devarajan P: Neutrophil gelatinase-associated
lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet
365: 1231-1238 (2005)

List of references
87
110. Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P:
Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary
biomarker for ischemic renal injury. J Am Soc Nephrol 14: 2534-2543 (2003)
111. Monbaliu D, de VB, Crabbe T, van HE, Verwaest C, Roskams T, Fevery J, Pirenne J,
Buurman WA: Liver fatty acid-binding protein: an early and sensitive plasma marker
of hepatocellular damage and a reliable predictor of graft viability after liver
transplantation from non-heart-beating donors. Transplant Proc 37: 413-416 (2005)
112. Moore EE, Johnson JL, Cheng AM, Masuno T, Banerjee A: Insights from studies of
blood substitutes in trauma. Shock 24: 197-205 (2005)
113. Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin TL, Silliman CC, Banerjee
A, Sauaia A: Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown:
the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J
Trauma Acute Care Surg 77: 811-817 (2014)
114. Moore HB, Moore EE, Liras IN, Gonzalez E, Harvin JA, Holcomb JB, Sauaia A,
Cotton BA: Acute Fibrinolysis Shutdown after Injury Occurs Frequently and Increases
Mortality: A Multicenter Evaluation of 2,540 Severely Injured Patients. J Am Coll
Surg 222: 347-355 (2016)
115. Moore K: The physiological response to hemorrhagic shock. J Emerg Nurs 40: 629-
631 (2014)
116. Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen
X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K,
Mukoyama M, Kunis C, D'Agati V, Devarajan P, Barasch J: Endocytic delivery of
lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion
injury. J Clin Invest 115: 610-621 (2005)
117. Mueller SC, Marz R, Schmolz M, Drewelow B: Intraindividual long term stability and
response corridors of cytokines in healthy volunteers detected by a standardized
whole-blood culture system for bed-side application. BMC Med Res Methodol 12: 112
(2012)
118. Munford RS, Pugin J: Normal responses to injury prevent systemic inflammation and
can be immunosuppressive. Am J Respir Crit Care Med 163: 316-321 (2001)

List of references
88
119. Nacionales DC, Szpila B, Ungaro R, Lopez MC, Zhang J, Gentile LF, Cuenca AL,
Vanzant E, Mathias B, Jyot J, Westerveld D, Bihorac A, Joseph A, Mohr A,
Duckworth LV, Moore FA, Baker HV, Leeuwenburgh C, Moldawer LL, Brakenridge
S, Efron PA: A Detailed Characterization of the Dysfunctional Immunity and
Abnormal Myelopoiesis Induced by Severe Shock and Trauma in the Aged. J Immunol
195: 2396-2407 (2015)
120. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-
Bourboulis EJ, Golenbock D, Gresnigt MS, Heneka MT, Hoffman HM, Hotchkiss R,
Joosten LAB, Kastner DL, Korte M, Latz E, Libby P, Mandrup-Poulsen T, Mantovani
A, Mills KHG, Nowak KL, O'Neill LA, Pickkers P, van der Poll T, Ridker PM,
Schalkwijk J, Schwartz DA, Siegmund B, Steer CJ, Tilg H, van der Meer JWM, van
de Veerdonk FL, Dinarello CA: A guiding map for inflammation. Nat Immunol 18:
826-831 (2017)
121. Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S: Size-
selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol
161: 653-660 (2003)
122. Niu CY, Li JC, Zhao ZG, Zhang J, Shao XH: Effect of intestinal lymphatic circulation
blockage in two-hit rats. World J Gastroenterol 12: 5805-5812 (2006)
123. Norton R, Kobusingye O: Injuries. N Engl J Med 368: 1723-1730 (2013)
124. Obinata H, Hla T: Sphingosine 1-phosphate in coagulation and inflammation. Semin
Immunopathol 34: 73-91 (2012)
125. Ostrowski SR, Henriksen HH, Stensballe J, Gybel-Brask M, Cardenas JC, Baer LA,
Cotton BA, Holcomb JB, Wade CE, Johansson PI: Sympathoadrenal activation and
endotheliopathy are drivers of hypocoagulability and hyperfibrinolysis in trauma: A
prospective observational study of 404 severely injured patients. J Trauma Acute Care
Surg 82: 293-301 (2017)
126. Ostrowski SR, Johansson PI: Endothelial glycocalyx degradation induces endogenous
heparinization in patients with severe injury and early traumatic coagulopathy. J
Trauma Acute Care Surg 73: 60-66 (2012)
127. Osuchowski MF, Remick DG: The repetitive use of samples to measure multiple
cytokines: the sequential ELISA. Methods 38: 304-311 (2006)

List of references
89
128. Osuka A, Kusuki H, Yoneda K, Matsuura H, Matsumoto H, Ogura H, Ueyama M:
Glycocalyx Shedding is Enhanced by Age and Correlates with Increased Fluid
Requirement in Patients with Major Burns. Shock (2017)
129. Osuka A, Ogura H, Ueyama M, Shimazu T, Lederer JA: Immune response to traumatic
injury: harmony and discordance of immune system homeostasis. Acute Medicine &
Surgery 1: 63-69 (2014)
130. Paffrath T, Lefering R, Flohe S: How to define severely injured patients? -- an Injury
Severity Score (ISS) based approach alone is not sufficient. Injury 45 Suppl 3: S64-
S69 (2014)
131. Pape HC, Lefering R, Butcher N, Peitzman A, Leenen L, Marzi I, Lichte P, Josten C,
Bouillon B, Schmucker U, Stahel P, Giannoudis P, Balogh Z: The definition of
polytrauma revisited: An international consensus process and proposal of the new
'Berlin definition'. J Trauma Acute Care Surg 77: 780-786 (2014)
132. Pelsers MM, Hermens WT, Glatz JF: Fatty acid-binding proteins as plasma markers
of tissue injury. Clin Chim Acta 352: 15-35 (2005)
133. Perl M, Gebhard F, Braumuller S, Tauchmann B, Bruckner UB, Kinzl L, Knoferl MW:
The pulmonary and hepatic immune microenvironment and its contribution to the early
systemic inflammation following blunt chest trauma. Crit Care Med 34: 1152-1159
(2006)
134. Perl M, Gebhard F, Bruckner UB, Ayala A, Braumuller S, Buttner C, Kinzl L, Knoferl
MW: Pulmonary contusion causes impairment of macrophage and lymphocyte
immune functions and increases mortality associated with a subsequent septic
challenge. Crit Care Med 33: 1351-1358 (2005)
135. Poltorak A, He X, Smirnova I, Liu MY, Van HC, Du X, Birdwell D, Alejos E, Silva
M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B: Defective
LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science
282: 2085-2088 (1998)
136. Raeven P, Salibasic A, Drechsler S, Weixelbaumer KM, Jafarmadar M, van GM,
Bahrami S, Osuchowski MF: A non-lethal traumatic/hemorrhagic insult strongly
modulates the compartment-specific PAI-1 response in the subsequent polymicrobial
sepsis. PLoS One 8: e55467 (2013)

List of references
90
137. Rahbar E, Cardenas JC, Baimukanova G, Usadi B, Bruhn R, Pati S, Ostrowski SR,
Johansson PI, Holcomb JB, Wade CE: Endothelial glycocalyx shedding and vascular
permeability in severely injured trauma patients. J Transl Med 13: 117 (2015)
138. Ramnath R, Foster RR, Qiu Y, Cope G, Butler MJ, Salmon AH, Mathieson PW,
Coward RJ, Welsh GI, Satchell SC: Matrix metalloproteinase 9-mediated shedding of
syndecan 4 in response to tumor necrosis factor alpha: a contributor to endothelial cell
glycocalyx dysfunction. FASEB J 28: 4686-4699 (2014)
139. Raza I, Davenport R, Rourke C, Platton S, Manson J, Spoors C, Khan S, De'Ath HD,
Allard S, Hart DP, Pasi KJ, Hunt BJ, Stanworth S, MacCallum PK, Brohi K: The
incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb
Haemost 11: 307-314 (2013)
140. Razali NM, Wah YB: Power comparisons of shapiro-wilk, kolmogorov-smirnov,
lilliefors and anderson-darling tests. Journal of statistical modeling and analytics 2:
21-33 (2011)
141. Rehm M, Bruegger D, Christ F, Conzen P, Thiel M, Jacob M, Chappell D,
Stoeckelhuber M, Welsch U, Reichart B, Peter K, Becker BF: Shedding of the
endothelial glycocalyx in patients undergoing major vascular surgery with global and
regional ischemia. Circulation 116: 1896-1906 (2007)
142. Reikeras O, Sun J, Wang JE, Foster SJ, Aasen AO: Differences in LPS and PepG
induced release of inflammatory cytokines in orthopedic trauma. J Invest Surg 21: 255-
260 (2008)
143. Relja B, Szermutzky M, Henrich D, Maier M, de Haan JJ, Lubbers T, Buurman WA,
Marzi I: Intestinal-FABP and liver-FABP: Novel markers for severe abdominal injury.
Acad Emerg Med 17: 729-735 (2010)
144. Remick DG, Villarete L: Regulation of cytokine gene expression by reactive oxygen
and reactive nitrogen intermediates. J Leukoc Biol 59: 471-475 (1996)
145. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, Rafail S, Kartalis
G, Sideras P, Lambris JD: A novel C5a receptor-tissue factor cross-talk in neutrophils
links innate immunity to coagulation pathways. J Immunol 177: 4794-4802 (2006)
146. Rittirsch D, Schoenborn V, Lindig S, Wanner E, Sprengel K, Gunkel S, Blaess M,
Schaarschmidt B, Sailer P, Marsmann S, Simmen HP, Cinelli P, Bauer M, Claus RA,

List of references
91
Wanner GA: An Integrated Clinico-transcriptomic Approach Identifies a Central Role
of the Heme Degradation Pathway for Septic Complications after Trauma. Ann Surg
264: 1125-1134 (2016)
147. Rittirsch D, Schoenborn V, Lindig S, Wanner E, Sprengel K, Gunkel S, Schaarschmidt
B, Marsmann S, Simmen HP, Cinelli P, Bauer M, Claus RA, Wanner GA:
Improvement of prognostic performance in severely injured patients by integrated
clinico-transcriptomics: a translational approach. Crit Care 19: 414 (2015)
148. Rodriguez EG, Cardenas JC, Lopez E, Cotton BA, Tomasek JS, Ostrowski SR, Baer
LA, Stensballe J, Holcomb JB, Johansson PI, Wade CE: Early Identification of the
Patient with Endotheliopathy of Trauma by Arrival Serum Albumin. Shock (2017)
149. Rotstein OD: Modeling the two-hit hypothesis for evaluating strategies to prevent
organ injury after shock/resuscitation. J Trauma 54: S203-S206 (2003)
150. Ruf W, Ruggeri ZM: Neutrophils release brakes of coagulation. Nat Med 16: 851-852
(2010)
151. Sasaki M, Joh T: Oxidative stress and ischemia-reperfusion injury in gastrointestinal
tract and antioxidant, protective agents. J Clin Biochem Nutr 40: 1-12 (2007)
152. Sauerland S, Hensler T, Bouillon B, Rixen D, Raum MR, Andermahr J, Neugebauer
EA: Plasma levels of procalcitonin and neopterin in multiple trauma patients with or
without brain injury. J Neurotrauma 20: 953-960 (2003)
153. Schochl H, Frietsch T, Pavelka M, Jambor C: Hyperfibrinolysis after major trauma:
differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J
Trauma 67: 125-131 (2009)
154. Seitz DH, Perl M, Liener UC, Tauchmann B, Braumuller ST, Bruckner UB, Gebhard
F, Knoferl MW: Inflammatory alterations in a novel combination model of blunt chest
trauma and hemorrhagic shock. J Trauma 70: 189-196 (2011)
155. Senthil M, Brown M, Xu DZ, Lu Q, Feketeova E, Deitch EA: Gut-lymph hypothesis
of systemic inflammatory response syndrome/multiple-organ dysfunction syndrome:
validating studies in a porcine model. J Trauma 60: 958-965 (2006)
156. Seshadri A, Brat GA, Yorkgitis BK, Keegan J, Dolan J, Salim A, Askari R, Lederer
JA: Phenotyping the Immune Response to Trauma: A Multiparametric Systems
Immunology Approach. Crit Care Med (2017)

List of references
92
157. Stahel PF, Shohami E, Younis FM, Kariya K, Otto VI, Lenzlinger PM, Grosjean MB,
Eugster HP, Trentz O, Kossmann T, Morganti-Kossmann MC: Experimental closed
head injury: analysis of neurological outcome, blood-brain barrier dysfunction,
intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes
for pro-inflammatory cytokines. J Cereb Blood Flow Metab 20: 369-380 (2000)
158. Stoecklein VM, Osuka A, Lederer JA: Trauma equals danger--damage control by the
immune system. J Leukoc Biol 92: 539-551 (2012)
159. Sturm R, Heftrig D, Mors K, Wagner N, Kontradowitz K, Jurida K, Marzi I, Relja B:
Phagocytizing activity of PMN from severe trauma patients in different post-traumatic
phases during the 10-days post-injury course. Immunobiology 222: 301-307 (2017)
160. Suvorova ES, Gripentrog JM, Miettinen HM: Different endocytosis pathways of the
C5a receptor and the N-formyl peptide receptor. Traffic 6: 100-115 (2005)
161. Takeuchi O, Akira S: Pattern recognition receptors and inflammation. Cell 140: 805-
820 (2010)
162. Tammaro A, Derive M, Gibot S, Leemans JC, Florquin S, Dessing MC: TREM-1 and
its potential ligands in non-infectious diseases: from biology to clinical perspectives.
Pharmacol Ther 177: 81-95 (2017)
163. Tanaka H, Sugimoto H, Yoshioka T, Sugimoto T: Role of granulocyte elastase in
tissue injury in patients with septic shock complicated by multiple-organ failure. Ann
Surg 213: 81-85 (1991)
164. Temmesfeld-Wollbruck B, Hocke AC, Suttorp N, Hippenstiel S: Adrenomedullin and
endothelial barrier function. Thromb Haemost 98: 944-951 (2007)
165. Thuijls G, de Haan JJ, Derikx JP, Daissormont I, Hadfoune M, Heineman E, Buurman
WA: Intestinal cytoskeleton degradation precedes tight junction loss following
hemorrhagic shock. Shock 31: 164-169 (2009)
166. Timmermans K, Kox M, Vaneker M, van den Berg M, John A, van LA, van der
Hoeven H, Scheffer GJ, Pickkers P: Plasma levels of danger-associated molecular
patterns are associated with immune suppression in trauma patients. Intensive Care
Med 42: 551-561 (2016)

List of references
93
167. Tran DD, Cuesta MA, van Leeuwen PA, Nauta JJ, Wesdorp RI: Risk factors for
multiple organ system failure and death in critically injured patients. Surgery 114: 21-
30 (1993)
168. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP: The complement membrane
attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome
activation. J Cell Sci 126: 2903-2913 (2013)
169. Tscherne H: [The treatment of the seriously injured at an emergency station]. Chirurg
37: 249-252 (1966)
170. Tsukamoto T, Chanthaphavong RS, Pape HC: Current theories on the pathophysiology
of multiple organ failure after trauma. Injury 41: 21-26 (2010)
171. Unnewehr H, Rittirsch D, Sarma JV, Zetoune F, Flierl MA, Perl M, Denk S, Weiss M,
Schneider ME, Monk PN, Neff T, Mihlan M, Barth H, Gebhard F, Ward PA, Huber-
Lang M: Changes and regulation of the C5a receptor on neutrophils during septic
shock in humans. J Immunol 190: 4215-4225 (2013)
172. Vajdovich P: Free radicals and antioxidants in inflammatory processes and ischemia-
reperfusion injury. Vet Clin North Am Small Anim Pract 38: 31-123, v (2008)
173. Van de Wouwer M, Collen D, Conway EM: Thrombomodulin-protein C-EPCR
system: integrated to regulate coagulation and inflammation. Arterioscler Thromb
Vasc Biol 24: 1374-1383 (2004)
174. van den Berg CW, Tambourgi DV, Clark HW, Hoong SJ, Spiller OB, McGreal EP:
Mechanism of neutrophil dysfunction: neutrophil serine proteases cleave and
inactivate the C5a receptor. J Immunol 192: 1787-1795 (2014)
175. van Wessem KJP, Leenen LPH: Reduction in Mortality Rates of Postinjury Multiple
Organ Dysfunction Syndrome: A Shifting Paradigm? A Prospective Population-Based
Cohort Study. Shock 49: 33-38 (2018)
176. Veigas PV, Callum J, Rizoli S, Nascimento B, da Luz LT: A systematic review on the
rotational thrombelastometry (ROTEM(R)) values for the diagnosis of coagulopathy,
prediction and guidance of blood transfusion and prediction of mortality in trauma
patients. Scand J Trauma Resusc Emerg Med 24: 114 (2016)
177. Vincent JL, Moreno R, Takala J, Willatts S, De MA, Bruining H, Reinhart CK, Suter
PM, Thijs LG: The SOFA (Sepsis-related Organ Failure Assessment) score to describe

List of references
94
organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related
Problems of the European Society of Intensive Care Medicine. Intensive Care Med 22:
707-710 (1996)
178. Vogel JA, Liao MM, Hopkins E, Seleno N, Byyny RL, Moore EE, Gravitz C, Haukoos
JS: Prediction of postinjury multiple-organ failure in the emergency department:
development of the Denver Emergency Department Trauma Organ Failure score. J
Trauma Acute Care Surg 76: 140-145 (2014)
179. Waechter J, Kumar A, Lapinsky SE, Marshall J, Dodek P, Arabi Y, Parrillo JE,
Dellinger RP, Garland A: Interaction between fluids and vasoactive agents on
mortality in septic shock: a multicenter, observational study. Crit Care Med 42: 2158-
2168 (2014)
180. Wohlauer MV, Moore EE, Thomas S, Sauaia A, Evans E, Harr J, Silliman CC, Ploplis
V, Castellino FJ, Walsh M: Early platelet dysfunction: an unrecognized role in the
acute coagulopathy of trauma. J Am Coll Surg 214: 739-746 (2012)
181. Wohlauer MV, Sauaia A, Moore EE, Burlew CC, Banerjee A, Johnson J: Acute kidney
injury and posttrauma multiple organ failure: the canary in the coal mine. J Trauma
Acute Care Surg 72: 373-378 (2012)
182. Wojta J, Kaun C, Zorn G, Ghannadan M, Hauswirth AW, Sperr WR, Fritsch G, Printz
D, Binder BR, Schatzl G, Zwirner J, Maurer G, Huber K, Valent P: C5a stimulates
production of plasminogen activator inhibitor-1 in human mast cells and basophils.
Blood 100: 517-523 (2002)
183. Wolberg AS, Meng ZH, Monroe DM, III, Hoffman M: A systematic evaluation of the
effect of temperature on coagulation enzyme activity and platelet function. J Trauma
56: 1221-1228 (2004)
184. Wutzler S, Lehnert T, Laurer H, Lehnert M, Becker M, Henrich D, Vogl T, Marzi I:
Circulating levels of Clara cell protein 16 but not surfactant protein D identify and
quantify lung damage in patients with multiple injuries. J Trauma 71: E31-E36 (2011)
185. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL,
Hennessy L, Moore EE, Minei JP, Bankey PE, Johnson JL, Sperry J, Nathens AB,
Billiar TR, West MA, Brownstein BH, Mason PH, Baker HV, Finnerty CC, Jeschke
MG, Lopez MC, Klein MB, Gamelli RL, Gibran NS, Arnoldo B, Xu W, Zhang Y,
Calvano SE, McDonald-Smith GP, Schoenfeld DA, Storey JD, Cobb JP, Warren HS,

List of references
95
Moldawer LL, Herndon DN, Lowry SF, Maier RV, Davis RW, Tompkins RG: A
genomic storm in critically injured humans. J Exp Med 208: 2581-2590 (2011)
186. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT: Extracellular histones are
mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol
187: 2626-2631 (2011)
187. Yi J, Slaughter A, Kotter CV, Moore EE, Hauser CJ, Itagaki K, Wohlauer M, Frank
DN, Silliman C, Banerjee A, Peltz E: A "Clean Case" of Systemic Injury: Mesenteric
Lymph After Hemorrhagic Shock Elicits A Sterile Inflammatory Response. Shock 44:
336-340 (2015)
188. Younger JG, Sasaki N, Waite MD, Murray HN, Saleh EF, Ravage ZB, Hirschl RB,
Ward PA, Till GO: Detrimental effects of complement activation in hemorrhagic
shock. J Appl Physiol (1985 ) 90: 441-446 (2001)
189. Yu L, Seguro AC, Rocha AS: Acute renal failure following hemorrhagic shock:
protective and aggravating factors. Ren Fail 14: 49-55 (1992)
190. Yucel N, Lefering R, Maegele M, Vorweg M, Tjardes T, Ruchholtz S, Neugebauer
EA, Wappler F, Bouillon B, Rixen D: Trauma Associated Severe Hemorrhage
(TASH)-Score: probability of mass transfusion as surrogate for life threatening
hemorrhage after multiple trauma. J Trauma 60: 1228-1236 (2006)
191. Zeng Y, Adamson RH, Curry FR, Tarbell JM: Sphingosine-1-phosphate protects
endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ
Physiol 306: H363-H372 (2014)
192. Zhang Q, Itagaki K, Hauser CJ: Mitochondrial DNA is released by shock and activates
neutrophils via p38 map kinase. Shock 34: 55-59 (2010)

VII
Declaration
I hereby declare that I wrote the present dissertation with the topic
Early changes in immune, coagulation, and organ function after clinical and
experimental polytrauma
independently and used no other aids that those cited. In each individual case, I have clearly
identified the source of the passages that are taken word for word or paraphrased from other
works.
I also hereby declare that I have carried out my scientific work according to the principles
of good scientific practice in accordance with the current „Satzung der Universität Ulm zur
Sicherung guter wissenschaftlicher Praxis“ [Rules of the University of Ulm for Assuring
Good Scientific Practice].
Ulm, 28.03.2018

VIII
Acknowledgements
The acknowledgements were removed for data privacy protection reasons.

IX
Curriculum vitae
Personal information
Name Rebecca Halbgebauer, née Wiegner
Year of birth 1989
Place of birth Ludwigsburg, Germany
Education
Since 04/2014 PhD student in the International Graduate School in Molecular
Medicine Ulm (IGradU)
Since 10/2013 Doctoral thesis in the Institute of Clinical and Experimental
Trauma Immunology, Ulm University Hospital
Head: Prof. Dr. med. Markus Huber-Lang
Topic: Early immune reaction after severe trauma
10/2011-07/2013 Master of Science in Molecular Medicine, University of Ulm
Master thesis at the Institute of Orthopaedic Research and
Biomechanics, University of Ulm
Head: Prof. Dr. med. vet. Anita Ignatius
Title: Role of neutrophils in fracture healing
10/2008-09/2011 Bachelor of Science in Molecular Medicine, University of
Ulm
09/1999 - 07/2008 Abitur at Gymnasien im Ellental, Bietigheim-Bissingen,
Germany

X
Oral presentations
10/2017 Halbgebauer R, Denk S, Taylor R, Weiss M, Huber-Lang M
Komplement-induzierte Veränderung der Neutrophilenmorphologie während
der Entzündung
German Congress of Orthopaedics and Trauma Surgery (DKOU), Berlin,
Germany
05/2017 Wiegner R, Denk S, Gebhard F, Huber-Lang M
Early Barrier Dysfunction in Polytrauma
Retreat of the Collaborative Research Centre 1149, Oberstdorf, Germany
02/2017 Wiegner R, Denk S, Eisele P, Weckbach S, Braun CK, Cinelli P,
Radermacher P, Wachter U, Hafner S, Palmer A, Braumüller S, Wanner GA,
Simmen HP, Gebhard F, Rittirsch D, Huber-Lang M
Hämorrhagischer Schock im Polytrauma
Gathering of the Trauma Research Network (Netzwerk Traumaforschung,
NTF), Frankfurt, Germany
10/2016 Wiegner R, Denk S, Kalbitz M, Weiß M, Gebhard F, Huber-Lang M
Leukozyten als Interaktionsplattform zwischen Komplement- und
Gerinnungssystem bei polytraumatisierten Patienten
German Congress of Orthopaedics and Trauma Surgery (DKOU), Berlin,
Germany
06/2016 Wiegner R, Denk S, Perl M, Huber-Lang M
Immune monitoring in trauma
IgradU Student Retreat 2016, Kleinwalsertal, Austria
04/2016 Wiegner R, Brenner RE, Weiss M, Lampl L, Huber-Lang M
Role of stem cells after polytrauma
Student Symposium on Molecular Medicine, Ulm, Germany
02/2016 Wiegner R, Lampl L, Weiss M, Brenner R, Huber-Lang M
Rolle von VSELs im Polytrauma
Gathering of the Trauma Research Network (Netzwerk Traumaforschung,
NTF), Frankfurt, Germany

XI
09/2015 Wiegner R, Hönes F, Kalbitz M, Weiss M, Gebhard F, Huber-Lang M,
Denk S
Leukocytes mediate the crosstalk between the complement and coagulation
system in polytrauma patients
XVI. Congress of the European Shock Society, Cologne, Germany
04/2015 Wiegner R, Denk S, Weiss M, Gebhard F, Huber-Lang M
Coagulation-complement crosstalk on leukocytes in polytrauma patients
Gathering of the Trauma Research Network (Netzwerk Traumaforschung,
NTF), Frankfurt, Germany
03/2014 Wiegner R, Kovtun A, Ignatius A
Role of neutrophils in fracture healing
German Congress of Surgery (DGCH), Berlin, Germany
Poster presentations
09/2017 Wiegner R, Braun CK, Denk S, Cinelly P, Radermacher P, Gebhard F,
Rittirsch D, Huber-Lang M
Hemorrhagic shock drives organ dysfunction in polytrauma patients
XVI. Congress of the European Shock Society, Paris, France
06/2015 Wiegner R, Hönes F, Kalbitz M, Weiss M, Braumüller S, Huber-Lang M,
Denk S
Cellular crosstalk between the complement and coagulation system in
polytrauma patients
15th European Meeting on Complement in Human Disease, Uppsala, Sweden
Awarded with a Travel Award
05/2015 Wiegner R, Denk S, Hönes F, Kalbitz M, Gebhard F, Huber-Lang M
Immunophenotyping of severely injured patients
IgradU Student Retreat 2015, Como, Italy
06/2014 Wiegner R, Perl M, Denk S, Braumüller S, Huber-Lang M
Important role of Hsp70 in complement component C5a-induced delayed
apoptosis of neutrophil granulocytes
37th Annual Conference on Shock, Charlotte, NC, USA

XII
Original articles
van Griensven M, Ricklin D, Denk S, Halbgebauer R, Braun CK, Schultze A, Hones F,
Koutsogiannaki S, Primikyri A, Reis E, Messerer D, Hafner S, Radermacher P, Biglarnia
AR, Resuello RRG, Tuplano JV, Mayer B, Nilsson K, Nilsson B, Lambris JD, Huber-Lang
M: Protective Effects of the Complement Inhibitor Compstatin CP40 in Hemorrhagic Shock.
Shock (2018). Doi: 10.1097/SHK.0000000000001127
Denk S, Weckbach S, Eisele P, Braun CK, Wiegner R, Ohmann JJ, Wrba L, Hoenes FM,
Kellermann P, Radermacher P, Wachter U, Hafner S, McCook O, Schultze A, Palmer A,
Braumuller S, Gebhard F, Huber-Lang M: Role of Hemorrhagic Shock in Experimental
Polytrauma. Shock 49: 154-163 (2018)
Halbgebauer R, Braun CK, Denk S, Mayer B, Cinelli P, Radermacher P, Wanner GA,
Simmen HP, Gebhard F, Rittirsch D, Huber-Lang M: Hemorrhagic shock drives glycocalyx,
barrier and organ dysfunction early after polytrauma. J Crit Care 44: 229-237 (2017)
Braun CK, Kalbitz M, Halbgebauer R, Eisele P, Messerer DAC, Weckbach S, Schultze A,
Braumuller S, Gebhard F, Huber-Lang MS: Early structural changes of the heart after
experimental polytrauma and hemorrhagic shock. PLoS One 12: e0187327 (2017)
Wiegner R*, Rudhart NE*, Barth E, Gebhard F, Lampl L, Huber-Lang MS, Brenner RE:
Mesenchymal stem cells in peripheral blood of severely injured patients. Eur J Trauma
Emerg Surg (2017). doi: 10.1007/s00068-017-0849-8. *, equally contributing first authors
Denk S, Taylor RP, Wiegner R, Cook EM, Lindorfer MA, Pfeiffer K, Paschke S, Eiseler T,
Weiss M, Barth E, Lambris JD, Kalbitz M, Martin T, Barth H, Messerer DAC, Gebhard F,
Huber-Lang MS: Complement C5a-Induced Changes in Neutrophil Morphology During
Inflammation. Scand J Immunol 86: 143-155 (2017)
Denk S, Neher MD, Messerer DAC, Wiegner R, Nilsson B, Rittirsch D, Nilsson-Ekdahl K,
Weckbach S, Ignatius A, Kalbitz M, Gebhard F, Weiss ME, Vogt J, Radermacher P, Kohl J,
Lambris JD, Huber-Lang MS: Complement C5a Functions as a Master Switch for the pH
Balance in Neutrophils Exerting Fundamental Immunometabolic Effects. J Immunol 198:
4846-4854 (2017)

XIII
Kovtun A, Bergdolt S, Wiegner R, Radermacher P, Huber-Lang M, Ignatius A: The crucial
role of neutrophil granulocytes in bone fracture healing. Eur Cell Mater 32: 152-162 (2016)
Denk S, Wiegner R, Hones FM, Messerer DA, Radermacher P, Weiss M, Kalbitz M,
Ehrnthaller C, Braumuller S, McCook O, Gebhard F, Weckbach S, Huber-Lang M: Early
Detection of Junctional Adhesion Molecule-1 (JAM-1) in the Circulation after Experimental
and Clinical Polytrauma. Mediators Inflamm 2015: 463950 (2015)
Review articles
Halbgebauer R, Schmidt CQ, Karsten CM, Ignatius A, Huber-Lang M: Janus face of
complement-driven neutrophil activation during sepsis. Semin Immunol (2018). doi:
10.1016/j.smim.2018.02.004
Huber-Lang M, Ekdahl KN, Wiegner R, Fromell K, Nilsson B: Auxiliary activation of the
complement system and its importance for the pathophysiology of clinical conditions. Semin
Immunopathol 40: 87-102 (2018)
Wiegner R, Chakraborty S, Huber-Lang M: Complement-coagulation crosstalk on cellular
and artificial surfaces. Immunobiology 221: 1073-1079 (2016)
Huber-Lang M, Gebhard F, Schmidt CQ, Palmer A, Denk S, Wiegner R: Complement
therapeutic strategies in trauma, hemorrhagic shock and systemic inflammation - closing
Pandora's box? Semin Immunol 28: 278-284 (2016)
Huber-Lang M, Wiegner R, Lampl L, Brenner RE: Mesenchymal Stem Cells after
Polytrauma: Actor and Target. Stem Cells Int 2016: 6289825 (2016)