edqm conference new technologies & their impact on … · rtrt is attainable through a...
TRANSCRIPT

1
EDQM Conference
New technologies & their impact on the Pharmacopoeia
Workshop 2
EDQM Conference
New technologies & their impact on the Pharmacopoeia
Workshop 2
14-15 October 2010Prague, Czech Republic
©2010 EDQM, Council of Europe, All rights reserved
Workshop 2Workshop 2
Prof. Gert Ragnarsson
Mr Marc Goeller
Dr Kowid Ho
Dr Keith Pugh
©2010 EDQM, Council of Europe, All rights reserved

1
PAT, RTR, QbD, Design Space,Design of Experiments – what is behind
Gert RagnarssonProfessor and Director at Medical Products Agency, Uppsala, SwedenChairman of the EDQM PAT team
New technologies and their impact on the PharmacopoeiaWorkshop 2
EDQM, Prague October 14-15 2010
Disposition
• Some basic definitions• Current issues and challenges• Latest development of the EDQM PAT team

2
Quality by design (QbD)• systematic approach*) to development that begins withpredefined objectives and emphasises product and processunderstanding and control, based on sound science and quality risk
*) may include, for example,• incorporation of prior knowledge• results of experimental studies using design of experiments• use of quality risk management• use of knowledge management throughout the lifecycle of the product
FDA Definition of PAT (Sept 2004), alsoadopted by EMEA
Process Analytical Technology (PAT) is considered to be a system for designing, analysing and controllingmanufacturing through timely measurements (i.e. during processing of critical quality and performance attributesof raw and in-process materials and processes with the goal of ensuring final product quality)

3
Specific meaning:Process Analytical Technology i.e. as ananalytical tool with the ability to register/evaluate a process continuously (in,on,at-line) to enableadjustments/optimisation and potentially real-time release.
PAT
Variable Input
Adaptive Process & QuantitativeFormulation
Fixed Process&
Formulation
Low(er) defectrate
High(er) defectrate
Variable Input
To; Quality by design, risk and science based product development
From; Quality by control/inspection & strict compliance

4
Design Space
"the multi-dimensional combination and interaction of input variables, i.e. material attributes and process parameters, that have been demonstrated to provide adequate quality"
Design space ( the area which has been demonstrated to give adequateQuality) and control space (where theapplicant/company choose to operate)
Real Time Release Testing
The ability to evaluate and ensure the quality of in-process and/or final product based on process data, which typicallyinclude a valid combination of measured material attributesand process controls. (ICH Q8)

5
ICH Q8: Approaches to Pharmaceutical Development
Minimal approach• Empirical development• One variable at the time• Fixed manufacturing process
• Focus on reproducibility
• Off-line analysis
• Quality assurance by testing• Reactive lifecyclemanagement (correctiveactions)
Enhanced, QbD, approach*)• Systematic approach to development• Multivariate experiments, DoE• Manufacturing process (and quanti-tative formulation) adjustable within the design space
• Focus on control strategy androbustness of the process
• PAT tools used for feed forward and feed back process control
• Risk based control strategy &potentially Real Time Release
• Preventive lifecycle managementand continuous improvement
*) Optional approach. Parts may be applied.
Quality by Design - A potential win-win option andthus highly supported by regulatory authorities;• More flexible, risk-based, regulatory decisions (reviewsand inspections)
• Improved quality and less rejections• Lower production costs• Simplifies continuous process improvement (changewithin design space, fewer formal variations)
• Real Time Release may become reality (reduced or noroutine end product testing) giving faster release
• Leaner process• May facilitate/promote innovation & science
Enabled by the new ICH Q8 – Q11 guidelines

6
• the ICH guidelines are on a high level• a large number of expressions and sub-processes havebeen introduced
• lack of tools and investment costs may still be issues• monographs and pharmacopoeia texts may need to bemodified to support / facilitate the QbD approach
• different interpretations may still exist • some companies are using internal vocabulary
Current issues and challenges
A large number of expressions and sub-processes havebeen introduced;Substantial training is still needed
Enhancedunder-
standing
Designof
Experi-ments
DesignSpace
ProcessAnalytical
Technology
RealTime
Release
RegulatoryFlexibility
RiskAssess-ment
Quality System / Organisation
7
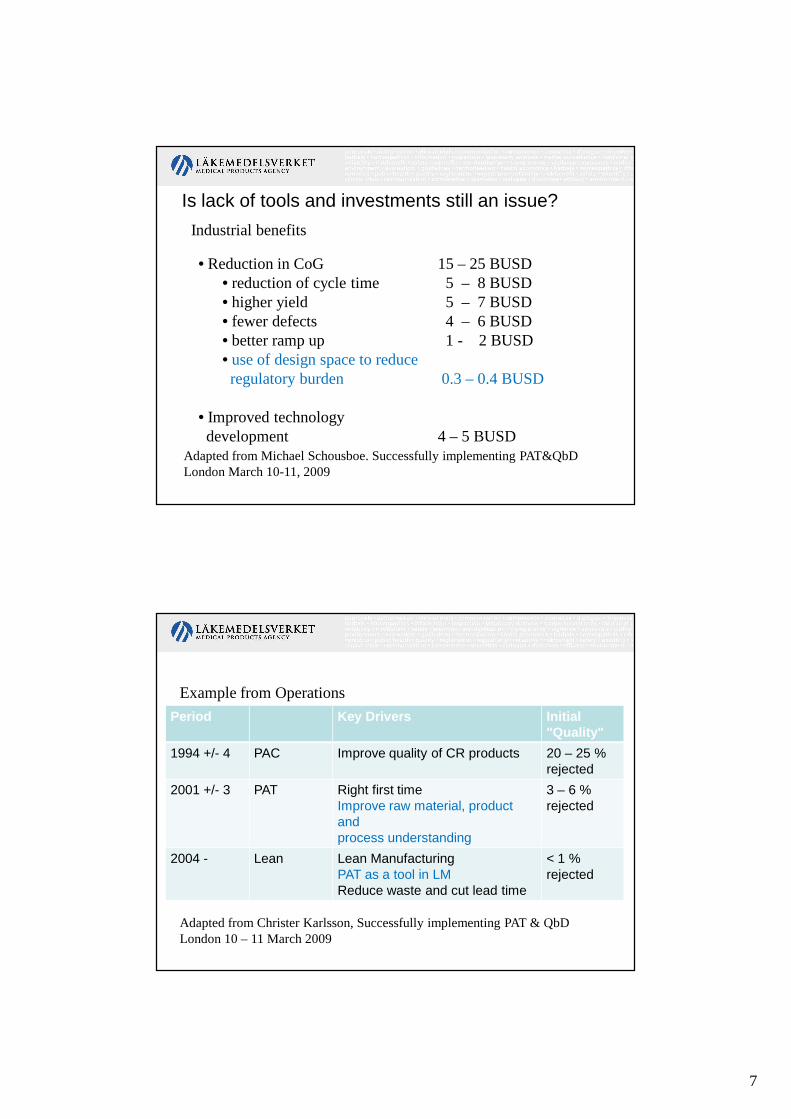
Industrial benefits
• Reduction in CoG 15 – 25 BUSD• reduction of cycle time 5 – 8 BUSD• higher yield 5 – 7 BUSD• fewer defects 4 – 6 BUSD• better ramp up 1 - 2 BUSD• use of design space to reduce regulatory burden 0.3 – 0.4 BUSD
• Improved technology development 4 – 5 BUSD
Adapted from Michael Schousboe. Successfully implementing PAT&QbDLondon March 10-11, 2009
Is lack of tools and investments still an issue?
Example from Operations
Period Key Drivers Initial "Quality"
1994 +/- 4 PAC Improve quality of CR products 20 – 25 % rejected
2001 +/- 3 PAT Right first timeImprove raw material, product andprocess understanding
3 – 6 % rejected
2004 - Lean Lean ManufacturingPAT as a tool in LMReduce waste and cut lead time
< 1 % rejected
Adapted from Christer Karlsson, Successfully implementing PAT & QbDLondon 10 – 11 March 2009
8
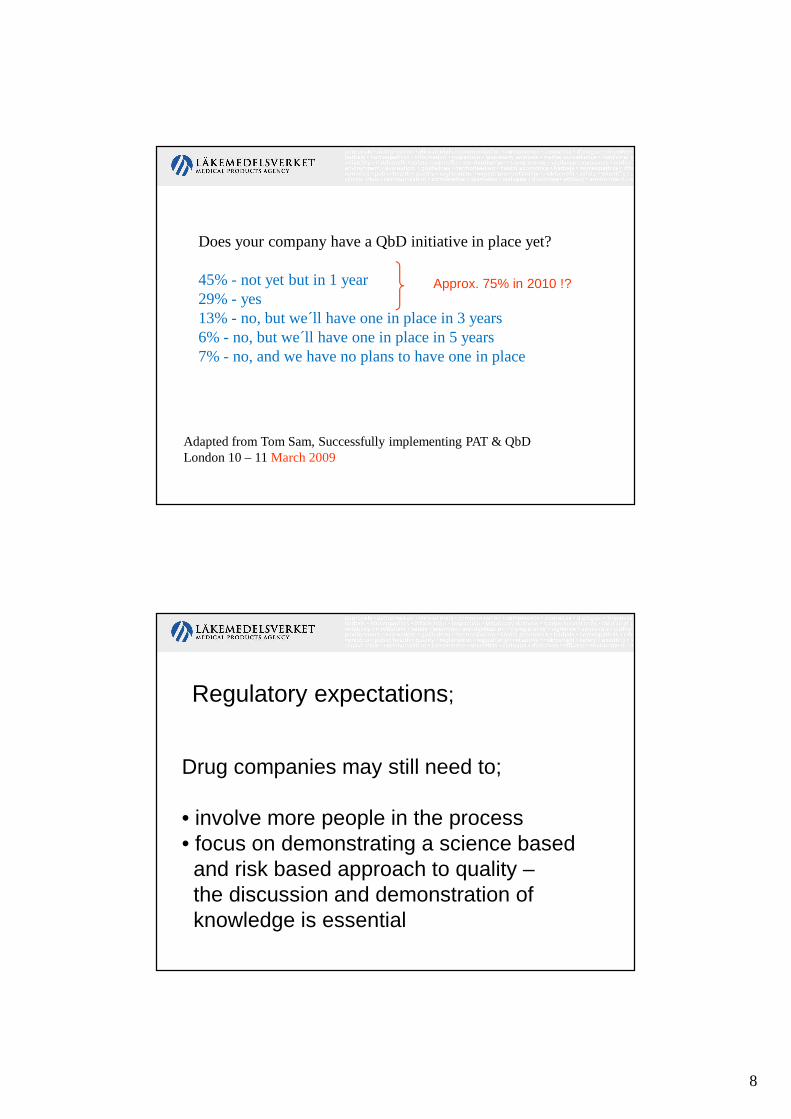
Does your company have a QbD initiative in place yet?
45% - not yet but in 1 year29% - yes13% - no, but we´ll have one in place in 3 years6% - no, but we´ll have one in place in 5 years7% - no, and we have no plans to have one in place
Adapted from Tom Sam, Successfully implementing PAT & QbDLondon 10 – 11 March 2009
Approx. 75% in 2010 !?
Drug companies may still need to;
• involve more people in the process• focus on demonstrating a science based and risk based approach to quality –the discussion and demonstration of knowledge is essential
Regulatory expectations;

9
Which departments will be involved in your company´sQbD efforts?
29 % - manufacturing28% - process development25% - quality assurance and quality control8% - discovery and research9% - analytical, engineering, regulatory affairs
Adapted from Tom Sam, Successfully implementing PAT & QbDLondon 10 – 11 March 2009
Pharmaceutical Development
To give reviewers and inspectors a comprehensive understanding
and• increased reliance • justified control strategies

10
Process Development, ICH Q8An attempt to define expectations for process understanding and quality by design
• obtain product quality and performance by design of effectivemanufacturing processes
• set product specifications based on fundamental scientific or mechanistic understanding of formulation and process factors
• form the basis for continuous improvement and”real time” assurance of quality
• identify Critical Quality Attributes (CQAs) that carry risk• form the basis for risk management principles and risk assessment of changes to be made (ICH Q9)
Guidelines are on a high level….but some suggestions how to proceed;
• Demonstrate fundamental scientific/mechanistic under-standing of formulation and process factors in relation tothe intended product profile (efficacy, safety, quality) andset specifications accordingly
• Identify Critical Quality Attributes (CQA:s) that carry risk(dissolution, drug release rate, …..)
• Identify Critical Material Attributes (CMA:s) and Critical Process Parameters (CPP:s) and assess risk

11
• Use the fundamental understanding and risk assessmentin the Design of Experiments to evaluate the impact ofCMA:s and CPP:s and the interaction between criticalparameters
• Try to stick to the vocabulary and definitions used in ICH Q8 – Q11
• Read guidelines, appendixes and Q&A:s as they become available !
What about ICH Q11?
The subject; Development and Manufacture ofDrug Substance ( chemical entities and biotechnological/biological entities)
• QbD is in principle already applicable to drug substancesaccording to Q8 – Q10
• ICH Q11 draft number 4 in progress• chemical entities and biotechnological/biological entities• what is traditional vs. QbD approaches ?• views on development and validation

12
Elements to be covered by applicants;
• Identify potential CQAs associate withdrug substancestarting materialreagentsraw materialintermediates
..so that those characteristics having an impact on productquality can be studied and controlled
• Determin CQAs of the drug substance and relevantmaterials and / or intermediates
• Definine an approriate manufacturing process• Definine a control strategy
Contribution by the EDQM PAT WP• Monographs and chapters in the European
Pharmacopoeia – View new general chapters on analytical techniques
• Relation between sample size and acceptance criteriaIt is the ambition of the team to propose an alternative to test2.9.40 (content uniformity) that is;– Independent of sample size n– Take into account difference between target and the sample mean– Considering consumers and patients risk– No re-sampling– Statistically sound– Handling different distributions– Enable online sampling– Easy to understand and apply– Link to current criteria– Get use of in process data– Optimized regarding sampling size– Lower producers and consumers risk

1
Marc GoellerNovartis Pharmaceuticals Corp.Prague - October 14-15, 2010
The application of RTR (Testing) in thePharma industry – Dream or Reality?
What is Real Time Release Testing (RTRT)?
� Real Time Release Testing (as per ICH Q8(R2)) is “the ability to evaluate and ensure the acceptable quality of in-process and/or final product based on process data, which typically includes a valid combination of measured material attributes and process controls.”
� RTRT should not be confused with “Batch Release”
2 | RTRT in Pharma: Dream or Reality?| Marc Goeller | Oct 14-15, 2010 | EDQM Prague | Business Use Only
2
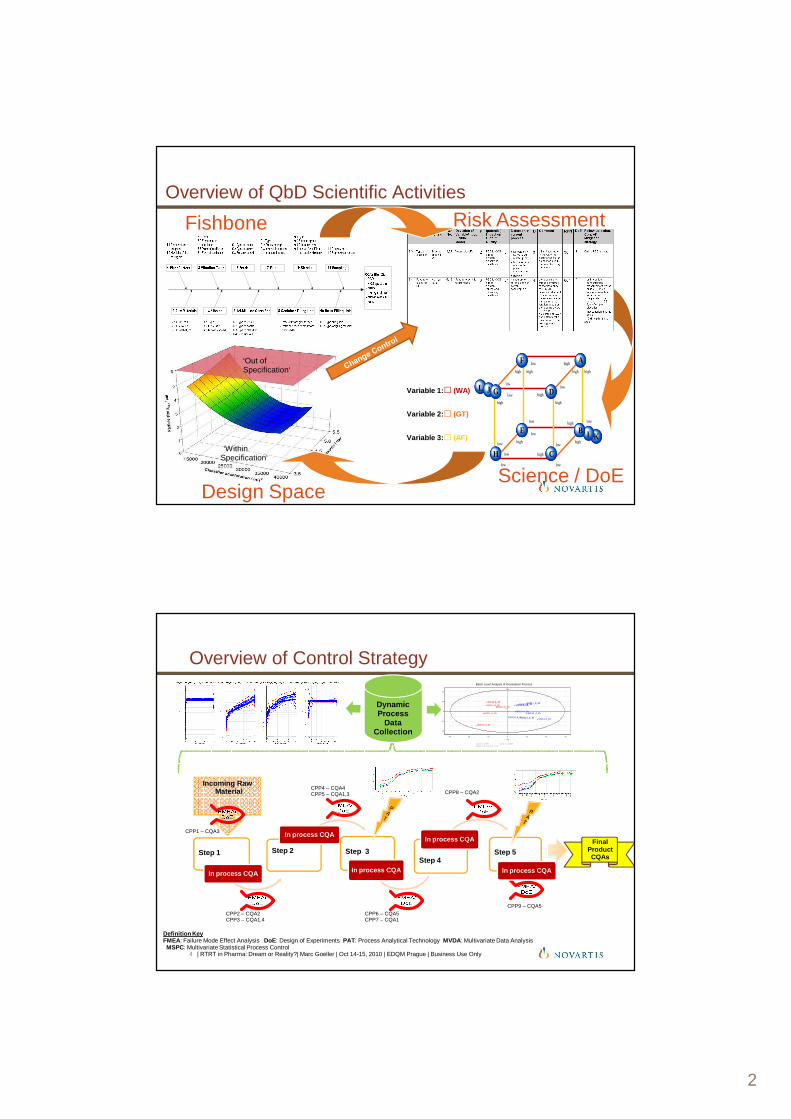
Overview of QbD Scientific Activities
•Fishbone •Risk Assessment
•Science / DoE•Design Space
high high
high
low
low
low
low
low
high
high high
low
low
high
high
high
high
low
high
high
low
low
low
low
H
A
B
C
D
E
F
G
I K
J
L •Variable 1:□ (WA)
•Variable 2:□ (GT)
•Variable 3:□ (AF)
‘Out of Specification‘
•‘Within Specification‘
Incoming Raw Material
Overview of Control Strategy
4
Dynamic Process
Data Collection
•CPP9 – CQA5
-20
-10
0
10
20
-30 -20 -10 0 10 20 30
t[2]
t[1]
Batch Level Analysis of Granulation Process
R2X[1] = 0.5656 R2X[2] = 0.298283 Ellipse: Hotelling T2 (0.95)
S0010-B_85
S0011-A_85
S0011-B_85
S0012-A_85
S0012-B_85
S0013-A_85S0013_B_85
S0014_A_85
S0014_B_85
S0015-A_85
S0015_B_85
S0016-A_85
S0016-B_85
•CPP2 – CQA2•CPP3 – CQA1,4
•CPP6 – CQA5•CPP7 – CQA1
•CPP4 – CQA4•CPP5 – CQA1,3 •CPP8 – CQA2
•CPP1 – CQA3
•Step 1
In process CQA
•Step 2
In process CQA
•Step 3
In process CQA•Step 4
In process CQA
•Step 5
In process CQA
•Definition Key•FMEA: Failure Mode Effect Analysis DoE: Design of Experiments PAT: Process Analytical Technology MVDA: Multivariate Data Analysis • MSPC: Multivariate Statistical Process Control
Final ProductCQAs
| RTRT in Pharma: Dream or Reality?| Marc Goeller | Oct 14-15, 2010 | EDQM Prague | Business Use Only

3
RTRT sought for CQAs...
�API• Specific impurity
• Polymorph
• Residual Solvent
• ....
�EMA approval received in 2009!
�FDA review ongoing
�DP• Dissolution
• CU
• Assay
• ...
5 | RTRT in Pharma: Dream or Reality?| Marc Goeller | Oct 14-15, 2010 | EDQM Prague | Business Use Only
Real Life Experience
� Quality Control:• Information received from Control Strategy tools that supports RTRT
• Operators/technicians to be trained on evaluation of new supportive data (e.g., MVDA)
• How to report RTRT on a Certificate of Analysis (CoA)?
� Quality Systems:• CoA, SOP, etc... need to be adapted
• One or several QbD variants?
• How to trend quality if you have no QC test result?
� Regulatory interactions:• Review focused on different areas between FDA and EMA
• Importance of face to face meetings with HAs
6 | RTRT in Pharma: Dream or Reality?| Marc Goeller | Oct 14-15, 2010 | EDQM Prague | Business Use Only

4
Conclusions / Outlook
� RTRT is a Reality at Novartis!
� RTRT is attainable through a proactive scientific approach that allows to establish a Design Space in combination to a sophisticated Control Strategy made of:• Predictive equations (i.e., DoE) or First Principle Understanding
• and/or MVDA
• and/or PAT
• and/or any other tool that provides the relevant detectability.
� Varying expectations and feedback from various HAs is still seen as an overall challenge for the Industry as it could lead to region-specific needs
7 | RTRT in Pharma: Dream or Reality?| Marc Goeller | Oct 14-15, 2010 | EDQM Prague | Business Use Only
Acknowledgements
� Catherine Ford, Chris Balducci, Jim Cheney and Alan Royce
� All Novartis QbD and PAT network members
� Novartis TechOps and TRD management
8 | RTRT in Pharma: Dream or Reality?| Marc Goeller | Oct 14-15, 2010 | EDQM Prague | Business Use Only

5
Thank you for your attention !
| RTRT in Pharma: Dream or Reality?| Marc Goeller | Oct 14-15, 2010 | EDQM Prague | Business Use Only

1
PAT and RTR for biologicalsIs there a need for special requirements ?
Kowid HODepartement des produits biologiques, DEMEBAfssaps
14-15 October 2010, Prague
1
Chemicals Advanced therapy
Recombinant DNAtechnology
Blood-derived
Immunologicals
AspirinMW: 0.2 kDa
IFN alfa165AA, MW: 19 kDa
IgG~1300AA,
MW: ~150 kDa
FVIII~2330AA,
MW: ~330 kDa
Virus like particleMW: ~20 000 kDa
…

2
2 K.HO – IFIS
VARIABLE REGION- Deamidation- Oxidation- N-term Pyro-Glu- Glycosylation- Glycation…
CONSTANT REGION- Deamidation- Oxidation- Acetylation- Glycation- Glycosylation (fucosylation, sialylation, galactosylation, mannosylation…)- C-term Lys- Di-sulfide bond shuffling/ cleavage- Fragmentation/clipping…
BINDING- Affinity- Avidity- Immunoreactivity / crossreactivity- Unintentional reactivity…
EFFECTOR FUNCTION- Complement interaction- FcRn, FcγR interaction- Mannan binding ligand interaction- Mannose receptor interaction…
OTHER BIOLOGICAL PROPERTIES- PK properties- Epitope / Immunogenicity- Modulatory region (Tregitope …)…
BIOLOGICAL CHARACTERISTICSPHYSICOCHEMICAL CHARACTERISTICS
3
Modes of action of Mab
Source: GB Kress, EMEA workshop on biosimilar MAB, 2009

3
4
Example: Impact of glycosylation of Mab
Source: GB Kress, EMEA workshop on biosimilar MAB, 2009
5
Peptide variants
desired product
Post-translational variants
PURITY PROFILEPURITY PROFILE
IMPURITY PROFILEIMPURITY PROFILE
??? PROFILE??? PROFILE
degradation Process related impurities
3D-structure
Product relatedimpurities
Product related substances

4
6
Quality Attributes
Controlled QA
QA « under control »
Unknown QA
DESIREDQUALITY
7
Quality Attributes

5
8
Product
Process
Control of raw and starting materials
Control of intermediates
Control of process
parameters
Control of drug substance and drug
productProcess
validation& evaluation
Good manufacturing
Practice
QUALITYQUALITY
9
Control strategy
• Control Strategy (ICH Q10) :– A planned set of controls, derived from current
product and process understanding, that assures process performance and product quality. The controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control.

6
10
CQA
non-CQA
CPP
Specification
Enhanced
IPT + PAT
Specification 1 Attribute Y
Specification XAttribute Z
End testing and/or
alternative approach
non-CPP
Process
Starting/raw material 1
Starting/raw material X
… Product
Output
Intermediate
RE
LE
ASE
TraditionalSpecification 1
Specification X
CQA
non-CQA
CPP
non-CPPSpecification
IPT + PAT
End testing and/or
alternative approach
Inputs
Design space
ENHANCED CONTROL STRATEGY
TRADITIONAL CONTROL STRATEGY
11
Control strategy elements (ICH Q6B)
• A specification:– A list of tests, references to analytical procedures, AND
appropriate acceptance criteria– Set of criteria to which a drug substance, drug product or materials
at other stages of its manufacture should conform to be considered acceptable for its intended use.
– “Conformance to specification” means that the drug substance and drug product, when tested according to the listed analytical procedures, will meet the acceptance criteria.
• Acceptance criteria: – Numerical limits, ranges, or other suitable measures for acceptance
of the results of analytical procedures which the drug substance or drug product or materials at other stages of their manufacture should meet.

7
12
Control strategy elements (ICH Q6B)
• Considerations in setting specifications:– Characterization studies
• performed in development phase• following significant process changes• Relevant and up-to-date methods• Includes:
– physicochemical properties, – biological activity, – immunochemical properties, – purity, impurities, and contaminants – quantity.
13
Control strategy elements (ICH Q6B)
• Considerations in setting specifications:– Process controls
• Process related considerations: – Adequate design of a process and knowledge of its capability– For certain impurities: testing of either the drug substance or the
drug product may not be necessary and may not need to be included in the specifications if efficient control or removal to acceptable levels is demonstrated by suitable studies. This concept may be implemented after marketing authorization.
• In-process acceptance criteria and action limits– Recorded as action limits or reported as acceptance criteria. – Performing such testing may eliminate the need for testing of the
drug substance or drug product– Internal action limits:
» to assess the consistency of the process at less critical steps» responsibility of the manufacturer, may be used to initiate
investigation or further action.

8
14
Control strategy elements (ICH Q6B)
• Justification of specifications:– Manufacturing process
• Data obtained from lots used to demonstrate manufacturing consistency• Process changes and degradation products produced during storage may differ
from those observed in the material used during preclinical and clinical development: significance to be evaluated.
– Stability of drug substance and drug product• Inherent complexity: no single stability-indicating assay or parameter that profiles
the stability characteristics.• Manufacturer: should propose a stability-indicating profile• Product-specific
– Preclinical and clinical studies• Quality of the material made at commercial scale should be representative of the
lots used in preclinical and clinical studies.
– Analytical procedures• Critical quality attributes may include items such as potency, the nature and
quantity of product-related substances, product-related impurities, and process-related impurities.
• Can be assessed by multiple analytical procedures, each yielding different results
15
Control strategy elements (Draft RTR guideline)
• Alternative to routine end-product testing– Specifications have to be established and products should
comply with them if tested (release and shelf life).
– Full testing according to the specifications may be required during a running in period
– Real Time Release• Ability to evaluate and ensure the quality of in-process and/or
final product based on process data, which typically include a valid combination of measured material attributes and process controls
• RTR testing applicable to Drug Product, Drug Substance, and Intermediates

9
16
Control strategy elements (Draft RTR guideline)
• Routine testing approach and/or a validation approach: – Routine testing approach: monitoring of the quality attribute at
appropriate step(s) of the process, in order to ensure acceptable levels in the final product.
– Validation approach: based on evidence of successful validation of the manufacturing process (e.g. establishing consistent and satisfactory impurity clearance); in such situation, process monitoring may be carried out without direct measurements of the quality attribute.
– Combination of routine testing and validation approaches (e.g. routine testing at an earlier step of the purification process and demonstrated reduction by validation in order to ensure a limit at the final product level, if tested).
17
Control systemCase study 1: Routine testing approach
…
…
Eluate Y
Column X
PP (b) PP (c)
Bioreactor
PP (a)
Eluate X
Harvest
Sufficient clearance capability NOT demonstrated
Impurity formation
ROUTINE TESTING
DS
Column Y
QA (b)
QA (c)
QA (a)
QA (d)
PP (d)
Not routinely tested
Not routinely tested
Not routinely tested

10
18
…
…
Eluate Y
Column X
PP (b) PP (c)
Bioreactor
PP (a)
Eluate X
Harvest
Sufficient clearance capability
DEMONSTRATED
Impurity formation
DS
Column Y
QA (b)
QA (c)
QA (a)
QA (d)
PP (d)
Control systemCase study 2: Validation approach
Not routinely tested
Not routinely tested
Not routinely tested
Not routinely tested
19
Control systemCase study 3: Routine testing & Validation approach
…
…
Eluate Y
Column X
PP (b) PP (c)
Bioreactor
PP (a)
Eluate X
Harvest
Moderate clearance capability
(e.g. 3 log reduction)
Impurity formation
DS
Column Y
QA (b)
QA (c)
QA (a)
QA (d)
PP (d)
Low to Moderate clearance capability
but low level (e.g. <1000ppm)
ROUTINE TESTING
Not routinely tested
Not routinely tested
Not routinely tested

11
20
Control systemCase study 4: Enhanced approach
…
…
Eluate Y
Column X
PP (b) PP (c)
Bioreactor
PP (a)
Eluate X
Harvest Impurity formation
DS
Column Y
QA (b)
QA (c)
QA (a)
QA (d)
PP (d)
PAT
Sufficient clearance capability DEMONSTRATED
when operating within DESIGN SPACE
Not routinely tested
Not routinely tested
Not routinely tested
21
PAT and RTR for biologicals
• Is there a need for special requirements ???

1
©
Safeguarding public health
RTRT and consequences for the Pharmacopoeia – a European regulatory perspective
Dr Keith Pugh MHRA
Slide 2
©
Overview of the Presentation
General• Role of the Pharmacopoeia in the regulation
of medicine (Regulatory perspective)• Regulatory Dossiers• Real Time Release Testing• Areas that potentially need Pharmacopoeial
consideration (?)

2
Slide 3
©
Pharmacopoeia - Purpose of a MonographSpecific monographs + general monographs + general texts:
• Set of requirements as quality standard for medicines that will be available to patients in the 36 Member States and beyond
• Facilitate approval procedures by giving a common basis for quality to:
- Regulatory authorities
- Official medicines control laboratories
- Pharmaceutical manufacturers
- Suppliers of the pharmaceutical industry
Slide 4
©
Role of the Pharmacopoeia
Specific monographs
• Active Substance• Finished Product
(Acceptance criterion/Test methods and limits)
General monographs
• Pharmaceutical forms• Analytical methods - NIR

3
Slide 5
©
Pharmacopoeia – General Notices
“General chapters become mandatory when referred to in monograph, unless such reference is made in a way that indicates that it is not the intention to make the text referred to mandatory but rather to cite it for information.”
“An article is not of Pharmacopoeia quality unless it complies with all the requirements stated in the monograph.”
Slide 6
©
Pharmacopoeia – General Notices
Real Time Release Testing/PAT
“This does not imply that that the performance of all the tests in the monograph is necessarily a prerequisite for a manufacturer in assessing compliance with the Pharmacopoeia before release of the product.”

4
Slide 7
©
Pharmacopoeia – General Notices
Real Time Release Testing/PAT
“The manufacturer may obtain assurance that a product is of Pharmacopoeial quality form data derived, for example, from validation studies of the manufacturing process and from in-process controls.”
Slide 8
©
Pharmacopoeia – General Notices
Alternative methods:-
“The test and assay described are the official methods upon which the standards of the Pharmacopoeia are based. With the agreement of the competent authority, alternative methods of analysis may be used for control purposes, provided the methods used enable an unequivocal decision to be made as to whether compliance with the standards of the monograph would be achieved if the official methods were used.”

5
Slide 9
©
Pharmacopoeia – General Notices
“Parametric release in circumstances deemed appropriate by the competent authority is thus not precluded by the need to comply with the Pharmacopoeia.”
Slide 10
©
Marketing Authorisation - Regulatory Dossier
Pharmacopoeia – mandatory standard
“Unless otherwise indicated in the General Notices or in the monographs, statements in monographs constitute mandatory requirements.”

6
Slide 11
©
Marketing Authorisation - Regulatory Dossier
Active substance/Finished product specification (IC HQ6A)
• Comprehensive to cover all relevant requirements• Release and shelf life aspects• Parametric release (Real Time Release Testing)• Periodic/Skip testing • Compliance only if tested
Pharmacopoeial methods – Wherever appropriate they should be used, but alternatives allowed if “comparable or superior to the official procedure”.
Slide 12
©
Marketing Authorisation - Regulatory Dossier
Active substance/Finished product specification (IC HQ6A)
Periodic/Skip testing “Performance of specified tests at release on pre-selected batches and/or at predetermined intervals, rather than on a batch to batch basis.” – approach needs to be justified and approved

7
Slide 13
©
Real Time Release Testing - Regulatory Guidance
Parametric release guideline (CPMP/QWP/3051/99) (Se p 01)
..results of in process tests and controls may constitute sufficient grounds for batch release and provide greater assurance of the finished tablet meeting certain criteria in the spe cification without the tests being repeated on a sample of the finished pr oduct...
…use of PAC, such as NIR and Raman spectroscopy, usually used in combination with multivariate analysis. Spectral data monitored on-line controlling content of active substance, polymorphism, water content, blending homogeneity, particle/powder properties or film thickness could thereby replace end-product testing like e.g. uniformity of content, tablet strength and drug dissolution...
Slide 14
©
Process Analytical Technology
Definition (ICH Q8)
“PAT is considered to be a system for designing, analyzing,and controlling manufacturing through timely measurements(i.e. during processing) of critical quality and performanceattributes of raw and in-process materials and processes withthe goal of ensuring final product quality.”

8
Slide 15
©
REAL TIME RELEASE
Real Time Release – part of overall control strategy
“Batch release is not dependent upon conventional finished product testing prior to release - Some elements are reliant on other information gained during the manufacturing process and/or knowledge of the product.”
ICH Q8(R1) -
“Real time release: The ability to evaluate and ensure the acceptable quality of in-process and/or final product based upon process data, which typically include a valid combination of assessed material attributes and process controls”.
Slide 16
©
Real Time Release Testing - Regulatory Guidance
Draft Guideline on Real Time Release Testing (EMA/CHMP/QWP/811210)/2009 Rev 1 (Consultation deadline 31 Aug 2010)
- Wider scope
- Experience, GMP

9
Slide 17
©
Parametric Release/Real Time Release Testing in regulatory submissions –
Experience to date
Limited experience – increasing due to QbD and PAT
Parametric release – sterile products (sterility)
RTRT – mainly solid dosage forms (assay, content uniformity, dissolution) – NIR
Joint assessor/inspector consideration - inspection
Slide 18
©
Real Time Release
Release is based upon some or all of the following:-
• in-process results
• Direct measurements e.g. NIR for assay• Indirect measurements e.g. process parameters for sterilisation
(parametric release)
Certificate of analysis results/Qualified Person batch release
• In process values • Predictive modelling/Algorithm prediction e.g. dissolution
Key challenge – translation of finished product specification acceptance criteria into acceptance criteria for a PAT/Real-Time Release System

10
Slide 19
©
REAL TIME RELEASE TESTING
Submission must cover the following:-
• Product and process understanding
• Appropriate controls in place throughout –
product characteristics - materials controls, …..
Validation of analytical methods (fit for purpose)
GMP – likely need for inspection
• Clear on what basis each of the finished product release criteria is based and whatever approach is used must be fully explained and appropriately justified.
• Sampling - size
Slide 20
©
Marketing Authorisation - QualityModule 2 (Quality Overall Summary) - drug substance and drug product
Module 3 Drug Substance
3.2.S Drug Substance
3.2.S.1 General Information
3.2.S.2 Manufacture3.2.S.2.2 Description of Manufacturing process and controls3.2.S.2.3 Control of Materials3.2.S.2.4 Control of critical steps and intermediates
3.2.S.3 Characterisation
3.2.S.4 Control of Drug Substance3.2.S.4.1 Specification3.2.S.4.2 Analytical procedures3.2.S.4.3 Validation of analytical procedures3.2.S.4.4 Batch Analysis3.2.S.4.5 Justification of specification
3.2.S.5 Reference Standards or Materials
3.2.S.6 Container Closure System
3.2.S.7 Stability

11
Slide 21
©
Quality aspects (continued)3.2.P Drug product
3.2.P.1 Description and Composition of the Drug Pro duct
3.2.P.2 Pharmaceutical Development
3.2.P.3 Manufacture3.2. P.3.3 Description of Manufacturing Process and Process Controls3.2. P.3.4 Controls of Critical Steps and Intermediates3.2. P.5.4 Batch Analysis3.2. P.4.5 Justification of specification
3.2.P.4 Control of Excipients
3.2.P.5 Control of Finished Product3.2.P.5.1 Specification3.2. P.5.2 Analytical procedures3.2. P.5.3 Validation of analytical procedures3.2. P.5.4 Batch Analysis3.2. P.4.5 Justification of specification
3.2.P.6 Reference Standards or Materials
3.2.P.7 Container Closure System
3.2.P.8 Stability
Slide 22
©
Pharmacopoeia –Consequences of RTRT?
RTR – potentially testing a large number of samples (large N). Slightly different proposals being made.
Need agreement - standard Pharmacopoeia acceptance criteria (?)
How should acceptance criteria be set to reflect th e increased sampling e.g. calculation of assay or consistency o f formulated products – uniformity of weight or conten t (2.9.5 and 2.9.6) or uniformity of dosage units (2.9.40)?

12
Slide 23
©
Summary
RTRT- Consequences for the Pharmacopoeia
• Principles of Parametric release and RTRT already fully acknowledged in the General Notices
• Role in developing a common standard for setting acceptance criteria for large N sampling.
Slide 24
©
Thank youfor your attention.
Any Questions?