ehlers danlos
TRANSCRIPT

Síndrome de ehlers Danlo
sPor:
Alejandra Barletta
José Gnecco
Luis Romero
Mayra Saumet

Así se le llama a grupo variado de
enfermedades hereditarias de
tejido conectivo.Una serie de anomalías genéticas
diferentes, que causan
problemas en la elaboración del colágeno en el
cuerpo.
¿Qué es?


Es la proteína más abundante
del tejido conectivo.
Actúa como una "goma" dándole a
los tejidos estructura, fuerza
y flexibilidad.
Colágeno
Los genes alterados afectan las propiedades mecánicas de la
piel, las articulaciones, los
ligamentos, los vasos sanguíneos
y los órganos internos, pudiendo alterar todas las
funciones corporales.

Mutación
Autosómica Dominante 50% de posibilidades de que el hijo tenga SED.
Autosómica Recesiva 25% de posibilidades de que el hijo tenga SED.
Herencia Ligada al Cromosoma XSi la mujer es la portadora: 50% de posibilidades de que su hijo herede el SED y un 50% de que su hija lo herede.

ClasificaciónNombre Número Descripción Gen(es)
Clásico Tipos 1 y 2
*Prevalencia: de 2 a 5 entre 100.000 habitantes.*Mutaciones: Colágenos tipo I y V.*Síntomas: Piel suave, aterciopelada y muy elástica que se rasga, lacera o presenta hematomas con facilidad y que cura lentamente, con tendencia a desarrollar crecimientos grasos o fibrosos benignos en áreas bajo presión. A veces, las venas se transparentan y hay una gran elasticidad en manos, sobre todo, hasta el punto de doblar los dedos por completo. El embarazo puede suponer un peligro para la vida en esta variante.
HGNCCOL5A1 ,HGNCCOL5A2 ,HGNCCOL1A1
Hipermovilidad Tipo 3
*Prevalencia: Afecta a entre 1 por 10.000 a 15.000*Autosómico-dominante*Mutaciones: En genes también implicados en la variedad vascular y en la de deficiencia de Tenascina-X.*Único tipo no diagnosticable mediante biopsia de piel o tejidos. Se emplean observaciones clínicas.*Síntomas: Puede darse piel sensible al dolor, aterciopelada; piel moderadamente hiperextensible y articulaciones inestables y laxas. Dislocaciones y subluxaciones frecuentes. Puede darse enfermedad degenerativa articular.
COL3A1,TNXB
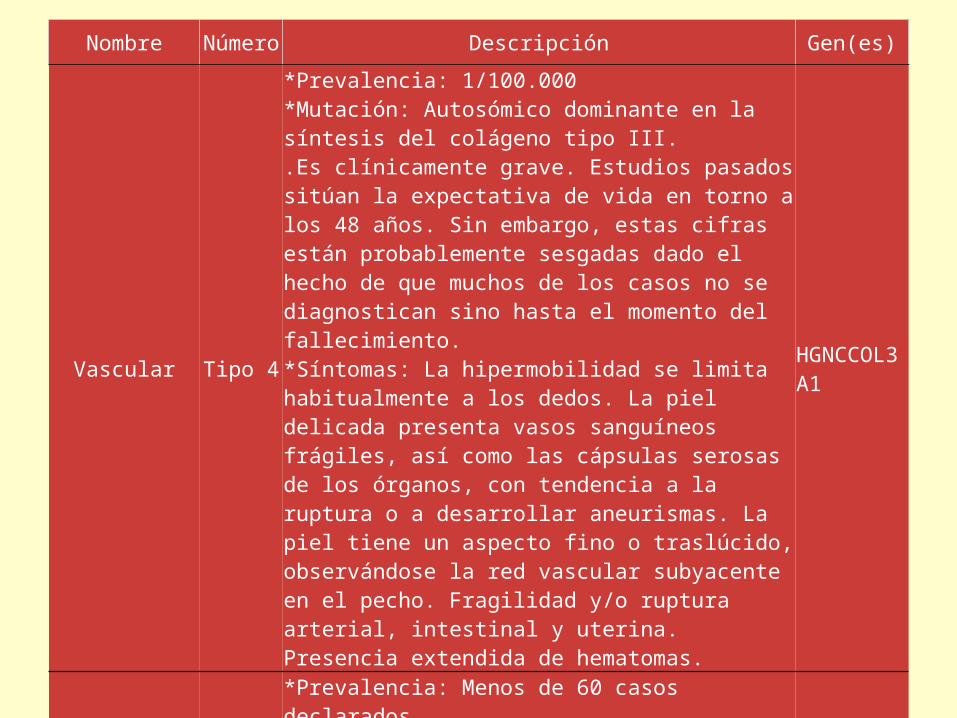
Nombre Número Descripción Gen(es)
Vascular Tipo 4
*Prevalencia: 1/100.000*Mutación: Autosómico dominante en la síntesis del colágeno tipo III..Es clínicamente grave. Estudios pasados sitúan la expectativa de vida en torno a los 48 años. Sin embargo, estas cifras están probablemente sesgadas dado el hecho de que muchos de los casos no se diagnostican sino hasta el momento del fallecimiento.*Síntomas: La hipermobilidad se limita habitualmente a los dedos. La piel delicada presenta vasos sanguíneos frágiles, así como las cápsulas serosas de los órganos, con tendencia a la ruptura o a desarrollar aneurismas. La piel tiene un aspecto fino o traslúcido, observándose la red vascular subyacente en el pecho. Fragilidad y/o ruptura arterial, intestinal y uterina. Presencia extendida de hematomas.
HGNCCOL3A1
Cifoscoliosis Tipo 6
*Prevalencia: Menos de 60 casos declarados.*Mutación: Autosómico recesiva debido a un defecto de la enzima lisil hidrolasa.*Síntomas: escoliosis progresiva, debilidad muscular progresiva y esclerótica frágil.
HGNCPLOD1
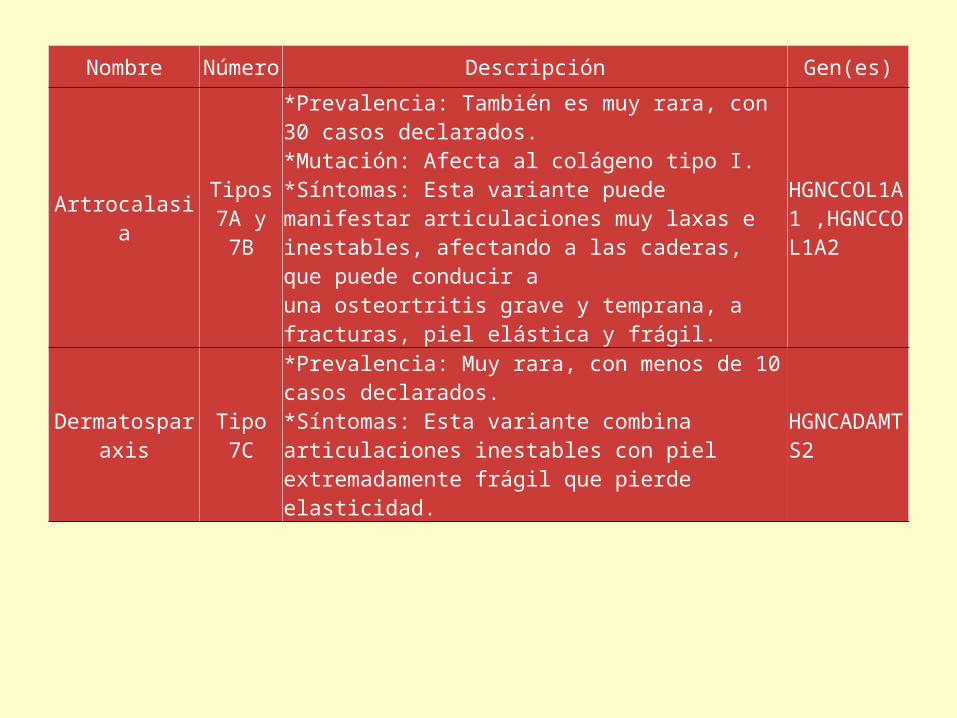
Nombre Número Descripción Gen(es)
Artrocalasia Tipos 7A y 7B
*Prevalencia: También es muy rara, con 30 casos declarados.*Mutación: Afecta al colágeno tipo I.*Síntomas: Esta variante puede manifestar articulaciones muy laxas e inestables, afectando a las caderas, que puede conducir a una osteortritis grave y temprana, a fracturas, piel elástica y frágil.
HGNCCOL1A1 ,HGNCCOL1A2
Dermatosparaxis Tipo 7C
*Prevalencia: Muy rara, con menos de 10 casos declarados.*Síntomas: Esta variante combina articulaciones inestables con piel extremadamente frágil que pierde elasticidad.
HGNCADAMTS2

Las siguientes mutaciones puede provocar el síndrome de Ehlers Danlos:
Proteínas Fibrosas: COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, y TNXB
Enzimas: ADAMTS2, PLOD1
Genética
