eliminating problems in drug - xenotech.com
TRANSCRIPT


Eliminating Problems in Drug Development – Understanding the Role of
Drug Metabolism
Andrew G. Taylor, Ph.D. Manager, Technical Support for Services
SEKISUI XenoTech
Abstract: This talk will discuss processes for the metabolism of a drug, referred to as ADME (Absorption, Distribution, Metabolism, Excretion), and the safety testing to provide requisite information to move a drug through IND and into clinical trials. Prior to marketing a new drug and actually prior to administering a candidate drug to humans in clinical trials, it is important to ensure that drug will not only be effective in the therapeutic manner prescribed, but also be safe and not bring any undo harm to patients. How a drug is metabolized is one of the most important aspects to understand. If you are involved in the development of potential new therapeutics, this discussion will help you gain necessary knowledge to inform your drug development decisions.

3
• What is ADME?• Example Case Studies of Metabolism
Based DDIs• Regulatory Requirements and in
vitro Studies to Predict Metabolism Based DDIs
• Areas of concern: Proper design & interpretation
• Timing of in vitro ADME studies
Outline

4
What is ADME?

5
What is ADME and why is it important?
It is a scientific field of study examining how compounds interact with the human body.
Used to assess and predict potential clinical safety concerns of new medications/treatments prior to
clinical trials.

6
Compounds are evaluated for ADME properties:
• Absorption – Drug Transporters, passive diffusion
• Distribution – Drug Transporters, passive diffusion
• Metabolism – Drug Metabolizing Enzymes (CYP450s,
UGTs, etc.)
• Excretion – Drug Metabolizing Enzymes and Drug
Transporters
Retrieved from https://aidsinfo.nih.gov/understanding-hiv-aids/glossary/213/drug-drug-interaction
ADME & DDI
7
Absorption (Oral Drugs)• Orally administered drugs are delivered
to the gastrointestinal (GI) tract• Compound has to be able to get into
the bloodstream • Biological barriers keep foreign
substances out of cells• E.g., skin, cell membranes
• Affected by numerous characteristics• e.g., Solubility, dissolution, ionization,
particle size, molecular size etc.
Retrieved from http://cephalicvein.com/2016/05/gi-tract/

8
Oral vs. Intravenous Administration
Oral drugs traverse the gut and liver to get into the bloodstream
Intravenous drugs go directly to the bloodstream
There is no absorption phase
Ahmad et al., Eur. J. Pharmaceut. And Biopharmaceut. 2017, 112: 234-248.

9
Distribution• Drug moves to site of action – via
bloodstream• Biological considerations
• Vascularity – where the bloodvessels go
• Transport – passive or active• Blood/placental barriers• Plasma binding proteins
Retrieved from http://7mlkbodysystems.weebly.com/circulatory-system.html

10
Metabolism• The biochemical structural modification of a drug• Often changes the chemical structure of a lipophilic compound to
make it more hydrophilic (water-soluble) for elimination• Often by DMEs in the liver; sometimes intestines, kidney etc.
Retrieved from https://pharmaxchange.info/2014/09/phase-i-metabolism-oxidative-reactions-oxidation-of-aliphatic-and-alicyclic-compounds/ibuprofen/

11
Many Enzyme Families Involved in Drug Metabolism• CYPs: Oxidation of steroids, fatty acids and many xenobiotocs
• UGTs and SULTs: Nucleophilic groups (aliphatic and aromatic hydroxyls, carboxylic acids,
amines, hydroxylamines, thiols, etc.)
• AO & XO: Heterocycles with low electron density (e.g., pteridines, quinolones, etc.)
• FMO: Soft nucleophiles (sulfur or nitrogen heteroatoms)
• CESs: Ester moieties (often for prodrugs)
• CBRs and AKRs: Ketone and quinone moieties
• MAOs: Amines (often problematic PK and other liabilities)
• Hydrolases: Often exploited for prodrug activation (e.g., esters or amides)
Foti & Dalvie. DMD. 2016; 44(8):1229-45

12
Excretion
• Irreversible loss of drug from system
• Drug-related material (drug and metabolites) has to be cleared from the organism
• Commonly occurs through• Kidneys (renal) - urine • Liver (biliary) - bile/feces• Lungs (respiratory) - breath• Other – sweat, tears, saliva

13
Example Case Studies of Metabolism Based DDIs

14
• Mibefradil is an antihypertensive Ca channel blocker approved in 1997
• After approval a serious DDI with β-blockers was observed
• Mibefradil was voluntarily withdrawn in 1998
• Turns out mibefradil was a potent TDI of CYP3A4
Example 1: Inhibition by a perpetrator drug

15
• Terfenadine is a prodrug of the active drug fexofenadine
• Terfenadine is converted to its active form predominantly by CYP3A4
• Taking terfenadine with CYP3A4 inhibitors can lead to QT prolongation
• Terfenadine was withdrawn in 1997
Example 2: Terfenadine and CYP3A4 inhibitors
Terfenadine(Seldane® - withdrawn)
Fexofenadine(Allegra®)

16
Regulatory Requirements and in vitro Studies to Predict Metabolism Based DDIs

17
Regulatory GuidanceFDA: Final January 2020 EMA: Final 2013 PMDA: Final 2019

18
Additional GuidanceFDA “MIST”: Rev 2
March 2020FDA / ICH: Final 2010

19
In vitro ADME & DDI study typesADME component Type of in vitro study
Drug Metabolism (M, E) 1. Inter-species comparative metabolism
2. Metabolite ID – Qualitative analysis of metabolite profile
3. Reaction phenotyping – Determine which CYPs are metabolizing
Drug Metabolizing Enzymes (M, E) 1. CYP Inhibition – Profile specific CYP inhibitions
2. CYP induction – Induction potential for specific CYPs
Drug Transporters (A, D, E) 1. Transporter substrate – Determine Transporter substrate profile
2. Transporter inhibition – Profile specific inhibition of major Transporters

20
Test Systems for in vitro DDI Experiments• Subcellular fractions – non-living, cell/tissue lysates that have been
fractionated to enrich for certain enzyme activities. • S9 • Microsomes • Cytosol• These subcellular fractions are derived from many relevant drug
metabolizing organs/tissues. May require co-factors.• Cryopreserved primary hepatocytes – living, isolated directly from
living liver tissue and frozen for use at a later time. From multiple small animal models and human livers.

21
Drug Metabolism Studies

22
Drug Metabolism: Inter-Species Comparative Metabolism• Design: Drug incubations with hepatocytes or subcellular fractions from various species• Typical species: Human, Rat, Mouse, Dog, Rabbit, Monkey, Pig Rat Dog
Time (min)0 60 120 180 240
Perc
ent r
emai
ning
0
20
40
60
80
100
t1/2 = > 240 min
Time (min)0 60 120 180 240
Perc
ent r
emai
ning
0
20
40
60
80
100
t1/2 = 97.6 min
Monkey Human
Time (min)0 60 120 180 240
Perc
ent r
emai
ning
0
20
40
60
80
100
t1/2 = 139 min
Time (min)0 60 120 180 240
Perc
ent r
emai
ning
0
20
40
60
80
100
t1/2 = 187 min
Metabolic Stability (Hepatocytes)
0
20
40
60
80
100
0 60 120 180 240
Perc
ent r
emai
ning
(%)
Incubation time (min)
RatHumanMonkeyDog

23
CoumarinHuman Rat
7-Hydroxycoumarin
No toxicity
Coumarin-3,4-epoxide
Hepatotoxicity
Goals: • Complete profile of metabolites• Are there human specific
metabolites? • Which other species have a similar
metabolic profile?
Drug Metabolism: Inter-Species Comparative Metabolite ID

24
Metabolite IDLC-MS/MS analysis – Qualitative identification of the metabolites
6.00 8.00 10.00 12.00 14.00 16.00
AU
0.0
1.0e-3
2.0e-3
3.0e-3
4.0e-3
5.0e-3
6.0e-3
7.0e-3
8.0e-3
9.0e-3
1.0e-2
1.1e-2
Range: 1.9.11
8.88 10.28
Repaglinide
Hydroxyrepaglinide Repaglinide dicarboxylicacid metabolite (M2)
UnknownRepaglinide glucuronide (RG)
Hydroxyrepaglinide LC-MS/MS
50 µM Repaglinide; Human hepatocytes; 60 minutes; 37°C

25
Cross-Species Metabolite IDComponent Retention time
(min) Mass shift Proposed biotransformation Mouse Rat Dog Pig Human
C1 3.43 255.9889 Sulfation + glucuronidation + + + + +
C2 3.63 354.0783 Di-glucuronidation + hydrogenation + + + + +
C3 3.78 159.9135 Di-sulfation + + + + +
C4 4.00 258.0045 Sulfation + glucuronidation + hydrogenation + + + + +
C5 4.41 161.9298 Di-sulfation + hydrogenation + + + + +
C6 4.44 194.0428 Glucuronidation + oxygenation + hydrogenation ND ND ND + +

26
• Design: Incubate drug + recombinant human CYPs or human liver microsomes or hepatocytes ± selective inhibitors
• Goal: Determine which CYPs drive the metabolism of the drug• Unique CYP metabolism is of concern High DDI potential: few enzymes involved
Follow-up studies:• Confirm with selective inhibitors• Evaluate non-CYP pathways in HLM or
hepatocytes
0102030405060708090
100
Recombinant human CYP enzyme
Rate
of m
etab
olis
m
Terfenadine(Seldane® - withdrawn)
Fexofenadine(Allegra®)
Drug Metabolism: CYP Reaction Phenotyping (Victim potential)

27
Drug Metabolizing Enzymes(Perpetrator Potential)
Enzyme Inhibition
Enzyme Induction

28
CYP Activity AssayCYP1A2 Phenacetin O-dealkylationCYP2B6 Bupropion hydroxylationCYP2C8 Amodiaquine N-dealkylationCYP2C9 Diclofenac 4´-hydroxylationCYP2C19 S-Mephenytoin 4´-hydroxylationCYP2D6 Dextromethorphan O-dealkylationCYP3A4 Testosterone 6β-hydroxylationCYP3A4 Midazolam 1´-hydroxylation
Drug Metabolizing Enzymes: CYP Inhibition
Design:Drug incubations with HLM + marker substrate ±pre-incubation
Goal:Predict clinically relevant inhibition of CYP enzymes

29
• Design: Drug incubations with HLM + marker substrate ± pre-incubation• Goal: Assess inhibition of CYP enzymes
Ketoconazole:Potent inhibitor of 3A4 precludes co-administration of other drugsMibefradilRemoved from market in 1998 due to potential for fatal DDIs
In Vitro Enzyme Inhibition

30
In Vitro CYP Induction
[Rifampin] (µM)
0.001 0.01 0.1 1 10 100
Effe
ct(F
old
Indu
ctio
n)
1
1.5
2
2.5
3
3.5
Parameter Value Std. Error
Emax 3.3378 0.1200EC50 0.1362 0.0528
[Rifampin] (µM)
0.001 0.01 0.1 1 10 100
Effe
ct(F
old
Indu
ctio
n)
2
4
6
8
Parameter Value Std. Error
Emax 6.0649 0.1981EC50 0.5795 0.1244
[Rifampin] (µM)
0.001 0.01 0.1 1 10 100
Effe
ct(F
old
Indu
ctio
n)
1
2
3
4
Parameter Value Std. Error
Emax 3.2417 0.3163EC50 0.1371 0.1427
Donor 1 Donor 2 Donor 3
• Design: Drug incubations in cultured human hepatocytes, measure mRNA (or activity) of various CYPs
• Goal: Assess induction of CYP enzymes

31
Drug Transporters
Transporter Inhibition(Perpetrator)
Substrate Potential(Victim)

32
Drug Transporter Studies
• Drug absorption, distribution, tissue-specific drug targeting, and elimination
• Drug-drug interactions• Clearance of transporter substrates (Victims) can be impacted by transporter
inhibitors or inducers (Perpetrators)• Toxicity or loss of efficacy
• Real world example - Statins• Hepatic uptake transporter (OATPs) substrates: taken up in the liver, reduce
cholesterol• Cyclosporine inhibits OATPs: up to 10-fold increase in statin exposure• Toxic side effect: rhabdomyolysis (skeletal muscles break down, cells released
into bloodstream, can lead to kidney failure and possibly death)
ADME

33
Areas of concern
for in vitro DDI studies: study
design and data
interpretation
34
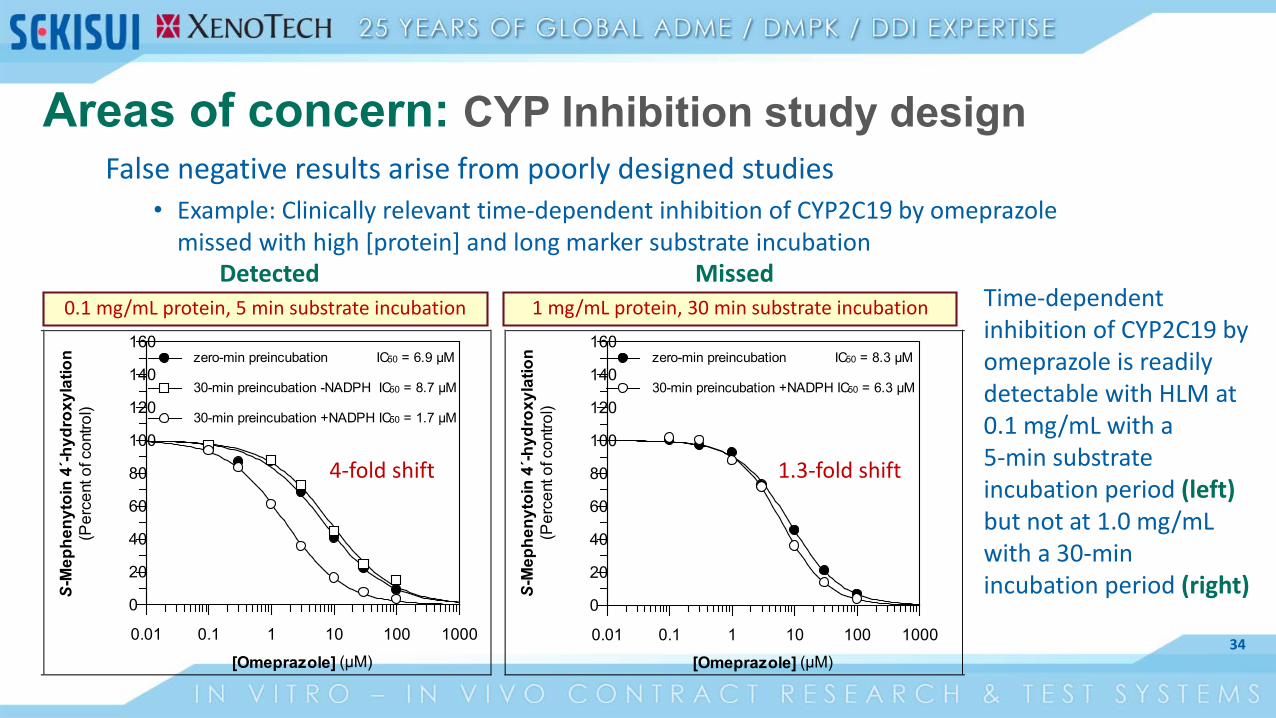
Areas of concern: CYP Inhibition study designFalse negative results arise from poorly designed studies
• Example: Clinically relevant time-dependent inhibition of CYP2C19 by omeprazole missed with high [protein] and long marker substrate incubation
[Omeprazole] (µM)0.01 0.1 1 10 100 1000
S-M
ephe
nyto
in 4
´-hyd
roxy
latio
n(P
erce
nt o
f con
trol)
0
20
40
60
80
100
120
140
160zero-min preincubation IC50 = 6.9 µM
30-min preincubation +NADPH IC50 = 1.7 µM
30-min preincubation -NADPH IC50 = 8.7 µM
[Omeprazole] (µM)0.01 0.1 1 10 100 1000
S-M
ephe
nyto
in 4
´-hyd
roxy
latio
n(P
erce
nt o
f con
trol)
0
20
40
60
80
100
120
140
160zero-min preincubation IC50 = 8.3 µM
30-min preincubation +NADPH IC50 = 6.3 µM
1 mg/mL protein, 30 min substrate incubation0.1 mg/mL protein, 5 min substrate incubation Time-dependent inhibition of CYP2C19 by omeprazole is readily detectable with HLM at 0.1 mg/mL with a 5-min substrate incubation period (left) but not at 1.0 mg/mL with a 30-min incubation period (right)
1.3-fold shift4-fold shift
Detected Missed

35
CYP induction studies: positive controls with very large induction
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
0 5 10 15 20 25 30 35 40
Rank order based on control rates
Test
oste
rone
6β-
hydr
oxyl
atio
n(p
mol
/mg/
min
)
Rifampin-treated (20 µM) hepatocytes CYP3A4 activity
Rank order of control rates
Test
oste
rone
6β-
hydr
oxyl
atio
n(p
mol
/mg/
min
)
Control hepatocytes CYP3A4 activity
0
1000
2000
3000
4000
5000
6000
7000
8000
0 5 10 15 20 25 30 35 40
0
50
100
150
200
250
300
0 5 10 15 20 25 30 35 40
Fold
indu
ctio
n
Rank order based on control rates
Fold induction
• When it comes to induction, more is not always better
• A high fold-induction (>20 fold) of CYP3A4 activity by rifampin is a sign of incomplete hepatocellular differentiation of the cultured human hepatocytes
Areas of concern: CYP Induction study design

36
Metabolism studies: Choose the right test system based on the structure• Ezetimibe is oxidized by CYP3A4 however results with HLM & NADPH alone can be
misleading.
Oxidation does not occur clinically due to rapid phenolic glucuronidation. Recombinant human UGTs or human hepatocytes would be a better test system. CYPs are not the only enzyme system.
Areas of concern: Reaction Phenotyping

37
Timing of ADME studies

38
Type of drug Lead optimization Pre-IND Phase I to NDA
Typical small molecule
1. Comparative metabolism
2. Metabolite ID3. Screening for
others
1. CYP inhibition2. CYP Induction3. Transporter
inhibition4. Limited transporter
substrate
1. Reaction phenotyping2. Additional transporter
substrate (dependent on routes of elimination)
Small molecule with orphan, breakthrough
status, etc.
1. Comparative metabolism May be able to defer
1. Metabolite ID2. CYP inhibition3. Transporter inhibition4. Reaction phenotyping5. CYP induction
Peptides, oligos, ADCs, other biologics May be able to defer May be able to defer
1. Metabolite ID2. CYP inhibition3. Transporter inhibition4. Reaction phenotyping5. CYP induction
Priority depends on strategy for each drug & need for de-risking at each stage
Drug Development Pipeline: timing in vitro DDI studies

39
“Why conduct these studies? Is this just box checking?”
No. The information in aggregate has real utility: 1. Provide deeper understanding of the molecule
• Metabolism, enzymes involved in metabolism, etc.• The information generated from DDI studies goes on the drug label• From the pharma company’s perspective these studies can inform go/no-go decisions for a drug candidate• Predictive toxicology and dose selection for certain non-clinical in vivo studies
2. Prepare for clinical studies• Prediction of FIH dose and DDI risk
3. Satisfy regulatory expectations and comply with regulatory guidance
These studies may appear deceptively simple – maximizing insight can be complex

40
Conclusions: In vitro ADME & DDI studies • Provide understanding of drug characteristics and insight concerning future
performance in in vivo systems; notably concerning predictive toxicology, dose/species selection for IND enabling studies, and FIH trial considerations.
• Satisfaction of regulatory interests is critical for prevention of delays
• Prioritization varies based on drug class and program de-risking needs
• Conduct and interpretation can be deceptively simple; they both benefit expert design and understanding
• Provide as much information of the drug as possible for appropriate guidance
41
Seeking further information? Check out our new site, detailing how we help drug developers
understand their compound’s ADME/PK & DDI potential