enumeration of tn5 mutant bacteria in soil by using a most...
TRANSCRIPT

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Feb. 1988, p. 446453 Vol. 54, No. 20099-2240/88/020446-08$02.00/0Copyright © 1988, American Society for Microbiology
Enumeration of Tn5 Mutant Bacteria in Soil by Using aMost-Probable-Number-DNA Hybridization Procedure and
Antibiotic ResistanceJ. K. FREDRICKSON,'* D. F. BEZDICEK,2 F. J. BROCKMAN,2 AND S. W. LI1
Pacific Northwest Laboratory, Richland, Washington 99352,1 and Department ofAgronomy and Soils,Washington State University, Pullman, Washington 991642
Received 18 May 1987/Accepted 16 November 1987
Investigations were made into the utility of DNA hybridization in conjunction with a microdilutionmost-probable-number procedure for the enumeration of Rhizobium spp. and Pseudomonas putida in soil.Isolates of Rhizobium spp. and P. putida carrying the transposon TnS were added to sterile and nonsterileBurbank sandy loam soil and enumerated over time. Soil populations of rhizobia were enumerated by colonyhybridization, most-probable-number-DNA hybridization procedure, plate counts, plant infectivity mostprobable number, and fluorescent antibody counts. Population values compared well for all methods at 5 and30 days after the addition of cells, although the fluorescent antibody method tended to overestimate the viablepopulation. In nonsterile soil, most-probable-number-DNA hybridization procedure enumerated as few as 10P. putida TnS cells g of soil-' and 100 R. leguminosarum bv. phaseoli TnS cells g of soil-' and should haveutility for following the fate of genetically engineered microorganisms released to the environment. Among theKmr isolates containing TnS, approximately 5% gave a dark, more intense autoradiograph when probed with32P-labeled pGS9 DNA, which facilitated their detection in soil. Hybridization with a pCU101 probe (pGS9without Tn5) indicated that donor plasmid sequences were being maintained in the bacterial chromosome.Transposon-associated antibiotic resistance was also utilized as a phenotypic marker. Tn5 vector-integratemutants were successfully enumerated at low populations (10 to 100 cells g of soil-') in soil by both phenotypic(Kmr) and genotypic (DNA probe) analysis. However, determination of the stability of Tn5 or Tn5 and vectorsequences in the bacteria is necessary.
The application of genetically engineered microorganismsfor waste treatment, agronomic crop production, and min-eral and energy-product recovery will likely require therelease of the altered organisms into the environment. Nu-merous concerns have been raised over protential hazards tothe ecosystem from the release of genetically engineeredmicroorganisms (6, 9, 16, 28). Assessing the risks involved inthe release of genetically engineered microorganisms re-quires tracking the environmental fate of the organisms todetermine their survival, growth, and dissemination (2).However, technologies to improve the detection and enu-meration of microorganisms in the environment have beenslow to develop.A variety of standard methods are used to detect and
enumerate microorganisms in the environment, includingdirect plating, enrichment cultures, identification of charac-teristic cell components (e.g., fatty acids, proteins, rRNAs),direct microscopy, serology, immunofluorescence, and im-munoradiography. The advantages and disadvantages ofeach have been the subject of much discussion and werereviewed by McCormick (25). One of the more recentdevelopments in the environmental detection and enumera-tion of specific microorganisms is based on the expression ofcloned Escherichia coli lac Y and lacZ genes in Pseudomo-nasfluorescens (10). One disadvantage to this method is thatit is limited to microorganisms without the intrinsic ability toutilize lactose. DNA probes have been used to detectSalmonella spp. (12) and Yersinia spp. (18) in foods and todetect specific catabolic genes in sediments (31). The advan-
* Corresponding author.
tages of DNA probes are high specificity (17) and thepotential for detection of gene or plasmid transmission.Spontaneous antibiotic-resistant mutants have been used
extensively for following populations of microorganisms insoils and the rhizosphere (13, 37, 39) and will likely continueto have utility as selective markers in microbial ecology.However, the environmental competitiveness of the organ-isms may be altered by biochemical impairment associatedwith the mutation (35). Transposable elements are recog-nized as valuable tools for genetic manipulations and char-acterizations (21), but only recently has transposon-associ-ated antibiotic resistance been used as a selective marker inmicrobial ecology experiments (36). Transposon TnS inser-tions have several advantages over spontaneous mutants inthat the mutation can be well characterized with respect tolocation in the bacterial chromosome, multiple insertionsoccur at low frequencies, insertions occur at a large numberof sites (increasing the likelihood of obtaining a mutantwhich is not biochemically impaired with respect to theparent), and a region of homology for DNA hybridization isavailable. Although the Kmr phenotype associated with theTnS insertion has been shown to be stable after growth forover 50 generations on nonselective medium (36), it is notknown to what extent transposons are stable in bacteriagrowing in soil.
In this paper we compare several methods for detectingbacteria in soil and assesses the utility of the transposableelement TnS as a phenotypic marker in microbial ecologyexperiments. In addition, a quantitative DNA hybridizationprocedure for detection of unique genotypes in soils by usingmost-probable-number (MPN) analysis is presented, whichmay be applicable to the detection and enumeration of
446
on July 8, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from

ENUMERATION OF TN5 MUTANT BACTERIA IN SOIL
genetically engineered microorganisms intended for releaseinto the environment.
MATERIALS AND METHODSBacterial strains and plasmids. Bacterial recipients for
transposon mutagenesis were Pseudomonas putida ATCC23973, harboring the TOL biodegradative plasmid, obtainedfrom G. Sayler (University of Tennessee), Rhizobium legu-minosarum bv. viceae (strain IP), and R. leguminosarum bv.phaseoli (strain Kim-5). The isolate used as a conjugativetransposon donor and the source of DNA for 32P-labeledprobes was E. coli MV12 (Leu- Thr- Thi- Thy- Trp-)transformed with the suicide plasmid vector pGS9 (CmrKmr) containing the transposon TnS (33), from V. Iyer(Carlton University). Plasmid pCU101 (Cmr), which is iden-tical to pGS9 but without TnS sequences, was maintained inE. coli HB101.
Media, culture conditions, and matings. Stock cultures ofall bacterial isolates were maintained in a glycerol-mediummixture (1:1) frozen at -80°C. P. putida was routinelycultured on Kings medium B (KB) broth or agar (20),Rhizobium strains were cultured on yeast extract-mannitol(YEM) broth or agar (38), and Et coli was cultured onLuria-Bertani (LB) medium (23). P. putida and the Rhizo-bium strains were grown at 27°C, and E. coli was grown at37°C. For bacterial matings, log-phase cultures of donor andrecipient cells were centrifuged in an Eppendorf microfugefor 5 min and washed twice in sterile deionized water beforesuspension to obtain 109 cells ml-'. Donor and recipientcells (108 0.1 ml-' each) were mixed and placed on sterile0.45-,um-pore-size nitrocellulose membrane filters on LBagar plates and incubated for 24 h at 27°C. Selection fortransconjugants was made on either Pseudomonas minimalsalts (PMS) agar (14) or Rhizobium minimal salts (RMS)containing the following (per liter): 15 g of agar, 10 g ofmannitol, 1.0 g of K2HPO4, 1 g of KH2PO4, 0.5 g of NH4CI,0.25 g of Na2SO4, 0.25 g of MgCl2 7H20, 0.1 g ofCaCl2- 2H20, 0.01 g of CaCO3, 10 mg of FeCl3 6H20, and1 mg each of biotin and thiamine with 50 or 100 jig ofkanamycin ml-' for Pseudomonas and Rhizobium cultures,respectively.
Plasmid DNA and probe preparation. E. coli MV12 wascultured in LB broth with 50,ug each of chloramphenicol andkanamycin (Sigma Chemical Co., St. Louis, Mo.) ml-1',which selected for the plasmid pGS9 and maintenance of thetransposon TnS. Plasmid DNA preparations were performedby the method of Maniatis et al. (23) by alkaline lysis with anadditional chloroform extraction followed by isopropanolprecipitation of DNA. Purified plasmid DNA for use asprobes was prepared by ultracentrifugation of plasmid prep-arations on CsCl (1.0 g ml-') density gradients with 0.5 mgof ethidium bromide ml-' at 55,000 rpm for 17 h at 20°Cfollowed by 1 h at 45,000 rpm at 20°C. Plasmid bands wereremoved and extracted four times with isopropanol-5 MNaCl (1:1, vol/vol) in 10 mM Tris (pH 8.0) buffer to remove,ethidium bromide. DNA was recovered by ethanol precipi-tation. 32P-labeled DNA probes were made with a nick-translation kit (Bethesda Research Laboratories, Gaithers-burg, Md.) by following the suggested protocol. [32P]dCTP(3,000 Ci mmol-P; New England Nuclear Corp., Boston,Mass.) was used as the labeling nucleotide. The labeledprobe was separated from unincorporated nucleotides byusing a spun-column procedure (23), and the activity wasquantified by liquid scintillation counting.Rapid visualization of plasmid DNA was accomplished by
the method of Eckhart (11) with the following modifications.
After centrifugation of cells and removal of medium, thepellet was suspended in 0.1% Sarcosyl (N-lauroylsarcosine;Sigma) in 50 mM Tris-25 mM EDTA (pH 8.8) and incubatedat room temperature for 10 min in 1.5-ml microfuge tubes.Cell suspensions were pelleted and suspended in lysozymemixture and incubated at room temperature for 10 to 15 min,followed by gentle mixing with the sodium dodecyl sulfatemixture and incubation for 10 min. Proteinase K (5 ,u, 5 mgml-'; International Biotechnologies Inc., New Haven,Conn.) was added to the microfuge tubes and mixed gently.The suspension was then transferred to the wells of a 0.7%agarose gel and sealed with molten agarose. DNA waselectrophoresed for 1 h at 35 V and then for 6 h at 100 V.Transfer of DNAs visualized by this procedure onto Gene-Screen Plus (New England Nuclear) was performed accord-ing to the manufacturer's suggested protocol.Enumeration of soil populations. Soils used for TnS mutant
recovery experiments were a Burbank sandy loam and aPalouse silt loam. Where sterile soil was used, soil wasautoclaved (121°C, 18 lb/in2) on 3 consecutive days for 30min. For enumeration of TnS mutants from soil using anti-biotic resistance as a selective marker, cultures of R. legu-minosarum and P. putida containing TnS were enumeratedwith a hemacytometer and added to 5.0 g of air-driedBurbank and Palouse soils at 106, 104, 102, and 0 cells g ofsoil-1 in 25-ml centrifuge tubes. Sterile deionized water wasadded to bring the water potential of both soils to -0.03MPa. Soils with cells were thoroughly mixed by vortexingfor 2 min and incubated at 4°C for 1 h. Sterile broth wasadded, and the contents were serially diluted and plated intriplicate. For Rhizobium cultures, samples of 100 p.1 fromeach dilution were spread onto YEM agar containing 100 ,ugeach of kanamycin sulfate, cycloheximide, and pimaricin(Sigma) ml-'. Samples from the P. putida treatments werespread onto agar plates selective for fluorescent pseudomo-nads (30) amended with 100 p.g of Km ml-'. Plates wereincubated at 27°C for 1 to 4 days before enumeration. Forenumerations by DNA colony hybridization, colony blotsfrom appropriate dilutions were performed with Whatmanno. 541 paper by the procedure of Gergen et al. (15). Filterswere dried completely at room temperature before hybrid-ization.Enumeration of R. leguminosarum and P. putida Tn5
mutants added to 5.0 g of Palouse and Burbank soils at levelsranging from 10 to 106 cells g of soil-1 were made by theMPN procedure with DNA hybridization (MPN-DNA hy-bridization). After soils and cells were mixed and incubatedas described above, 24.0 ml of sterile YEM broth containingthe antibiotics listed above or unamended KB broth wasadded to 5 g of soil to provide an initial soil/solution ratio of1:5 (wt/vol). Spontaneous mutants of R. leguminosarum bv.phaseoli (Kim-5), resistant to 100 p.g of rifampin (Sigma)ml-', were selected to provide an additional marker. Five277.5-,ul samples were removed and added to five wells in asterile microdilution plate. A serial 1:10 dilution was madeby pipetting 27.5 p.1 of the initial samples to the next set offive wells containing 250,ul of sterile broth with antibiotics toobtain a total volume of 277.5 p.1. Dilutions were made toextinction. Microdilution plates were incubated at 27°C for 3to 4 days. The entire contents of each well (250 p.l) was
quantitatively transferred to individual wells of a micro-sample filtration manifold (Schleicher & Schuell Co., Keene,N.H.), taking care to avoid disturbing and transferringsedimented soil particles, and filtered onto GeneScreen Plus.Cells were lysed, DNA was denatured and fixed by passingtwo volumes (250 p.l each) of 0.5 N NaOH through the
VOL. 54, 1988 447
on July 8, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from

448 FREDRICKSON ET AL.
individual wells followed by identical treatment with 1 MTris (pH 7.5), and filters were air dried. After DNA-DNAhybridization and autoradiography, positive blots indicatingthe presence of target DNA were scored for each dilution,and the MPN value for a five-replicate 10-fold dilution serieswas calculated (1). Uninoculated soils served as controls todiscern any background homology or nonspecific binding ofthe probe.DNA-DNA hybridization. GeneScreen Plus filters (New
England Nuclear) were placed back to back in a heat-sealable plastic bag with the manufacturer's suggestedprehybridization solution and incubated for 6 to 8 h at 65°C.Whatman 541 filter colony blots were saturated with 6x SSC(lx SSC is 0.15 M NaCl plus 0.015 M sodium citrate) andwashed for 1 to 2 h in a solution containing 50 mM Tris (pH8)-i M NaCI-1 mM EDTA-0.1% sodium dodecyl sulfate at37°C with constant agitation to remove cell debris. Prehy-bridization was done as described above for GeneScreenPlus with 5 x SSC substituting for the prehybridizationsolution. Salmon sperm carrier DNA and 32P-labeled DNAprobe were denatured by heating in boiling water for 10 minand added to plastic bags containing filters and solutions at150 ,ug and 105 dpm ml of hybridization solution-', respec-tively. Plastic bags were resealed and incubated at 65°C for12 to 14 h with agitation.
Filters were removed from bags and washed according tothe manufacturer's directions for GeneScreen Plus filters.Whatman 541 colony blots were washed three times in 2xSSC at 65°C (20 min each), followed by one wash in 0.2xSSC at 55°C for 20 min. Autoradiography was carried out at-70°C using Kodak XAR-5 X-ray film.
Stability of TnS mutants in soil. The stability of P. putidaand R. leguminosarum TnS mutants was tested in sterileBurbank soil. Bacteria were added to 10 g of sterile soil in50-ml centrifuge tubes to obtain a density of approximately200 cells g of dry weight-'. Soil water potential was adjustedto approximately -0.03 MPa with sterile water, and thetubes were incubated at 22°C. After 7 days bacterial popu-lations were enumerated by plate counts on KB and YEMfor P. putida and R. leguminosarum, respectively, samplesof 0.1 g of soil were transferred to new tubes containingsterile Burbank soil, and the cycle was repeated twice.Individual colonies were picked randomly from plates usedto enumerate populations and grown in nonselective me-dium, (no kanamycin) in microdilution plates. Susceptibilityto kanamycin was tested by replica plating from the micro-dilution plates to agar medium containing 50 jxg of kanamy-cin ml-1. The presence of donor plasmid sequences wasdetermined by spot-blot DNA hybridization with 32P-labeledpCU101 (PGS9 without Tn5) (see above) of individual colo-nies grown in microdilution plates.FA counting. Procedures for preparation of fluorescent
antibodies (FAs) and fluorescein isothiocyanate conjugateswere described previously (4, 32). Soil (1:10 dilution) wasdispersed for 5 min by shaking with 1 drop each of Tween 80and antifoam AF71 and then flocculated with 1% CaCl2 for 1h. Supernatant was diluted, when necessary, and passedthrough a 25-mm-diameter, 0.22-,um-pore-size Nucleporefilter previously stained with Irgalan black (4). Filters weretreated with a gelatin-rhodamine conjugate (4) and then withthe specific fluorescein isothiocyanate conjugate to Kim-5.Positive colonies from 20 to 30 fields were counted with aZeiss epifluorescence microscope.
Plant infectivity. MPN plant infectivity enumerations forR. leguminosarum bv. phaseoli were conducted on soildilutions with surface sterilized seeds of Phaseolis vulgaris
A B C D E F G
FIG. 1. Eckhardt-type agarose gel (left) and autoradiograph ofSouthern transfer of the gel probed with 32P-labeled pGS9 DNA(right) of R. leguminosarum bv. viceae IP Tn5 isolates (lanes A andB) and R. leguminosarum bv. viceae IP wild type (lane C) withindigenous plasmids, E. coli WA803 with pGS9 (lane D), P. putidawild type with TOL plasmid (lane E), and P. putida Tn5 isolates(lanes F and G).
cv, Viva Pink pregerminated for 5 days in sterile growthpouches (38). MPN values were determined by the presenceor absence of nodules in five replications per soil dilution.
RESULTS
Isolation and characterization of Tn5 mutants. In matingsbetween the E. coli MV12 donor and P. putida, Kmrtransconjugants were obtained at a frequency of about 10-5per recipient cell, whereas for R. legiuninosarum strains thefrequency was approximately 10-6. No attempt was made toselect or quantify auxotrophs to avoid the use of mutantscrippled in biosynthetic pathways in recovery experiments.Colony blots of Kmr mutants, probed with 32P-labeled pGS9DNA to confirm the presence of TnS, revealed signals of twointensities when autoradiographed. Those isolates with themore intense (dark) signal comprised approximately 5% ofthe total Kmr mutants for both Rhizobium spp. and P.putida. Light and dark isolates were subjected to agarose gelelectrophoresis, which revealed that pGS9 was not beingmaintained extrachromosomally (Fig. 1). Southern analysisof this gel (Fig. 1) with 32P-labeled pGS9 DNA as a probeshows that TnS is located in the chromosome of both lightand dark isolates and not on the TOL plasmid in P. putida orindigenous plasmids in Rhizobium spp.
Dot-blot analysis of P. piutida Tn5 isolates was conductedwith 20 ,ul of culture containing approximately 109 cells ml-'to obtain about the same amount of DNA for each dot.Hybridization of the blot with pCU101 probe revealed thatP. putida mutants 1 and 3 contained plasmid vector se-quences as evidenced by the relatively intense autoradio-graph (Fig. 2). Mutants 2 and 3 showed slight hybridizationdue to the small amount of Tn5 DNA present in pCU101.
Stability of Tn5 mutants in soil. The stability of Kmrmutants containing pGS9 sequences (vector integrates) wereexamined by screening individual colonies for both Tn5 anddonor plasmid sequences after growth of the bacteria insterile Burbank soil. These TnS vector-integrate mutants(Tn5 VI) were chosen for soil recovery studies because ofthe increased sensitivity of their detection by DNA hybrid-ization over those isolates containing TnS sequences alone.After growth for 33 generations none of 184 colonies of R.leguminosarum showed susceptibility to 100 ,ug of kanamy-cin ml-'. However, 22 of 184 colonies no longer hybridizedwith 32P-labeled pCU101 DNA. Of the 181 P. piutida colo-
APPL. ENVIRON. MICROBIOL.
on July 8, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from
ENUMERATION OF TNS MUTANT BACTERIA IN SOIL
'a _ 9 co
nl n. nv. n. n'
FIG. 2. Dot blot of 20 j±l of P. putida wild-type (WT) and mutantcultures probed with 32P-labeled pCU101 DNA. P. putida mutants 1and 2 correspond to those in lanes G and F, respectively, in Fig. 1.
nies, one was found to be susceptible to 100 ,ug of kanamycinml-' after 34 generations in soil, and only two failed tohybridize with pCU101 probe. The colony that was Kms alsofailed to hybridize with the pCU101 probe, indicating loss ofboth Tn5 and suicide plasmid sequences.
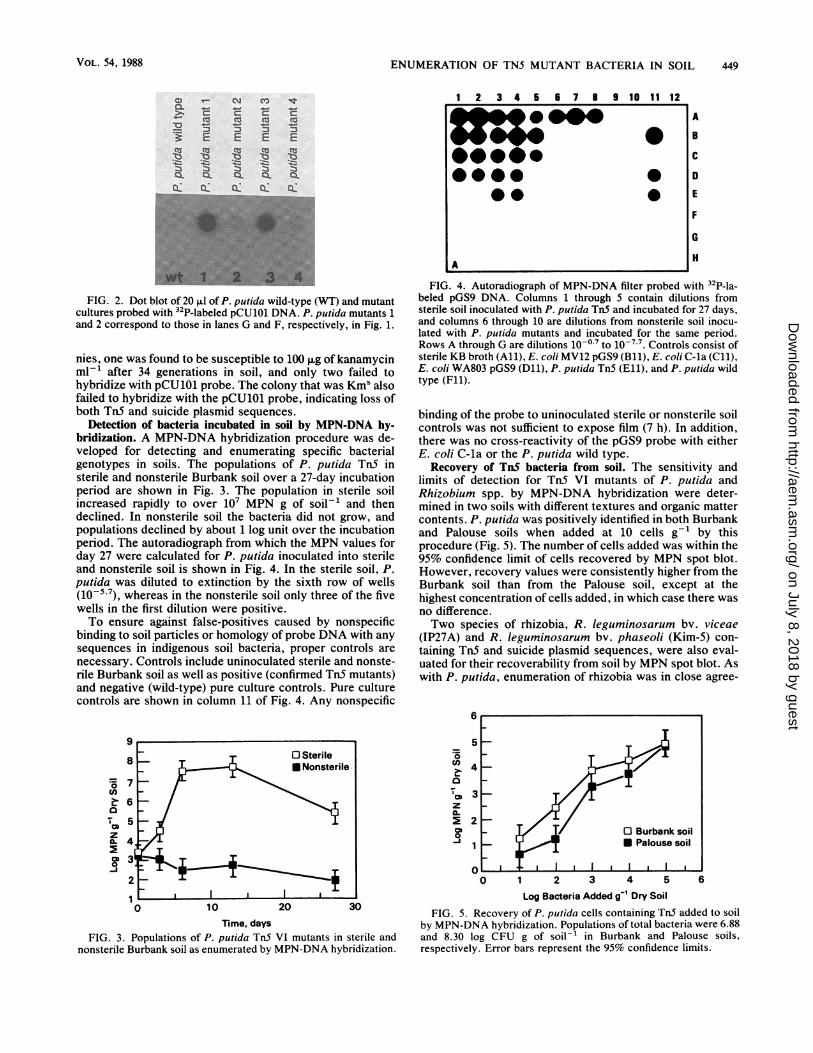
Detection of bacteria incubated in soil by MPN-DNA hy-bridization. A MPN-DNA hybridization procedure was de-veloped for detecting and enumerating specific bacterialgenotypes in soils. The populations of P. putida TnS insterile and nonsterile Burbank soil over a 27-day incubationperiod are shown in Fig. 3. The population in sterile soilincreased rapidly to over 107 MPN g of soil-' and thendeclined. In nonsterile soil the bacteria did not grow, andpopulations declined by about 1 log unit over the incubationperiod. The autoradiograph from which the MPN values forday 27 were calculated for P. putida inoculated into sterileand nonsterile soil is shown in Fig. 4. In the sterile soil, P.putida was diluted to extinction by the sixth row of wells(10-5 7), whereas in the nonsterile soil only three of the fivewells in the first dilution were positive.To ensure against false-positives caused by nonspecific
binding to soil particles or homology of probe DNA with anysequences in indigenous soil bacteria, proper controls arenecessary. Controls include uninoculated sterile and nonste-rile Burbank soil as well as positive (confirmed Tn5 mutants)and negative (wild-type) pure culture controls. Pure culturecontrols are shown in column 11 of Fig. 4. Any nonspecific
72Ca
T5
z
0-1
2
0 10 20 30
Time, daysFIG. 3. Populations of P. putida TnS VI mutants in sterile and
nonsterile Burbank soil as enumerated by MPN-DNA hybridization.
1 2 3 4 5 6 7 8 9 10 11 12
0..@
.0 ~0A O
AI
A
B
C
0
E
F
6
H
FIG. 4. Autoradiograph of MPN-DNA filter probed with 32P-la-beled pGS9 DNA. Columns 1 through 5 contain dilutions fromsterile soil inoculated with P. putida TnS and incubated for 27 days,and columns 6 through 10 are dilutions from nonsterile soil inocu-lated with P. putida mutants and incubated for the same period.Rows A through G are dilutions 10-0.7 to 10-7-7. Controls consist ofsterile KB broth (All), E. coli MV12 pGS9 (Bli), E. coli C-la (Cll),E. coli WA803 pGS9 (Dll), P. putida Tn5 (Ell), and P. putida wildtype (Fll).
binding of the probe to uninoculated sterile or nonsterile soilcontrols was not sufficient to expose film (7 h). In addition,there was no cross-reactivity of the pGS9 probe with eitherE. coli C-la or the P. putida wild type.
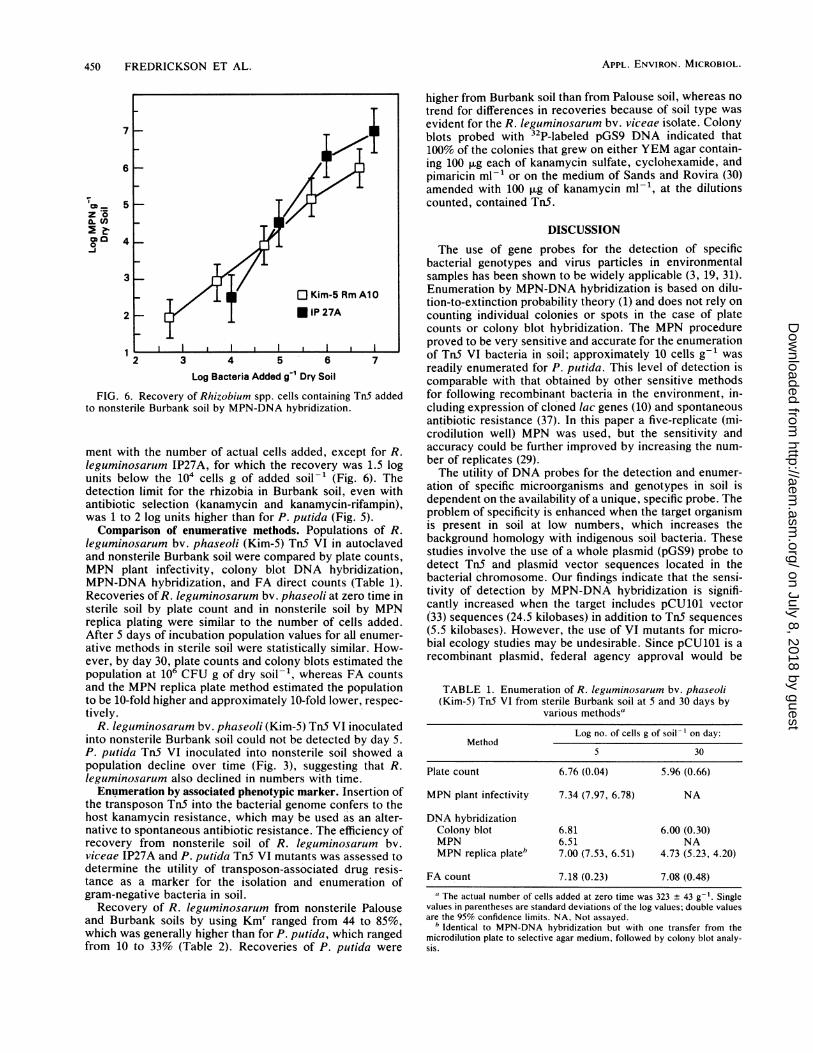
Recovery of TnS bacteria from soil. The sensitivity andlimits of detection for TnS VI mutants of P. putida andRhizobium spp. by MPN-DNA hybridization were deter-mined in two soils with different textures and organic mattercontents. P. putida was positively identified in both Burbankand Palouse soils when added at 10 cells g-1 by thisprocedure (Fig. 5). The number of cells added was within the95% confidence limit of cells recovered by MPN spot blot.However, recovery values were consistently higher from theBurbank soil than from the Palouse soil, except at thehighest concentration of cells added, in which case there wasno difference.Two species of rhizobia, R. leguminosarum bv. viceae
(IP27A) and R. leguminosarum bv. phaseoli (Kim-5) con-taining Tn5 and suicide plasmid sequences, were also eval-uated for their recoverability from soil by MPN spot blot. Aswith P. putida, enumeration of rhizobia was in close agree-
0~~~~~~
C3z
22<0~
Oll l l l 0 Bubnloil_j1 U Palouse soil
0 1 2 3 4 5 6
Log Bacteria Added g-' Dry Soil
FIG. 5. Recovery of P. putida cells containing TnS added to soilby MPN-DNA hybridization. Populations of total bacteria were 6.88and 8.30 log CFU g of soil-' in Burbank and Palouse soils,respectively. Error bars represent the 95% confidence limits.
VOL. 54, 1988 449
on July 8, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from
450 FREDRICKSON ET AL.
7
6
cz o
cm O0-j
5
4
3
2
I I' 0 Kim-5 Rm Al
I/ *IP 27A
I ,3 4 5 6
Log Bacteria Added g-' Dry Soil
10
7
FIG. 6. Recovery of Rhizobium spp. cells containing Tn5 addedto nonsterile Burbank soil by MPN-DNA hybridization.
ment with the number of actual cells added, except for R.leguminosarum IP27A, for which the recovery was 1.5 logunits below the 104 cells g of added soil-' (Fig. 6). Thedetection limit for the rhizobia in Burbank soil, even withantibiotic selection (kanamycin and kanamycin-rifampin),was 1 to 2 log units higher than for P. putida (Fig. 5).Comparison of enumerative methods. Populations of R.
leguminosarum bv. phaseoli (Kim-5) TnS VI in autoclavedand nonsterile Burbank soil were compared by plate counts,MPN plant infectivity, colony blot DNA hybridization,MPN-DNA hybridization, and FA direct counts (Table 1).Recoveries of R. leguminosarum bv. phaseoli at zero time insterile soil by plate count and in nonsterile soil by MPNreplica plating were similar to the number of cells added.After 5 days of incubation population values for aLl enumer-
ative methods in sterile soil were statistically similar. How-ever, by day 30, plate counts and colony blots estimated thepopulation at 106 CFU g of dry soil-', whereas FA countsand the MPN replica plate method estimated the populationto be 10-fold higher and approximately 10-fold lower, respec-
tively.R. leguminosarum bv. phaseoli (Kim-5) TnS VI inoculated
into nonsterile Burbank soil could not be detected by day 5.
P. putida Tn5 VI inoculated into nonsterile soil showed a
population decline over time (Fig. 3), suggesting that R.leguminosarum also declined in numbers with time.Enumeration by associated phenotypic marker. Insertion of
the transposon Tn5 into the bacterial genome confers to thehost kanamycin resistance, which may be used as an alter-native to spontaneous antibiotic resistance. The efficiency ofrecovery from nonsterile soil of R. leguminosarum bv.viceae IP27A and P. putida Tn5 VI mutants was assessed todetermine the utility of transposon-associated drug resis-tance as a marker for the isolation and enumeration ofgram-negative bacteria in soil.Recovery of R. leguminosarum from nonsterile Palouse
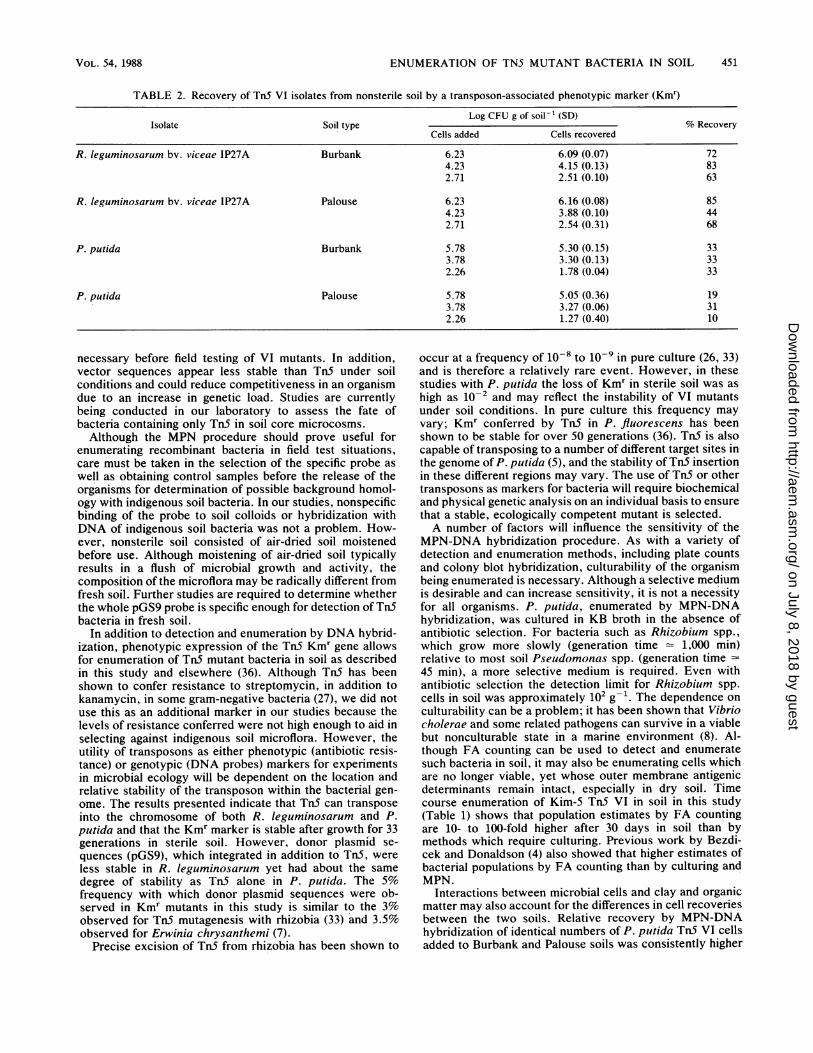
and Burbank soils by using Kmr ranged from 44 to 85%,which was generally higher than for P. putida, which rangedfrom 10 to 33% (Table 2). Recoveries of P. putida were
higher from Burbank soil than from Palouse soil, whereas notrend for differences in recoveries because of soil type wasevident for the R. leguminosarum bv. viceae isolate. Colonyblots probed with 32P-labeled pGS9 DNA indicated that100% of the colonies that grew on either YEM agar contain-ing 100 ,ug each of kanamycin sulfate, cyclohexamide, andpimaricin ml-1 or on the medium of Sands and Rovira (30)amended with 100 1Lg of kanamycin ml-', at the dilutionscounted, contained TnS.
DISCUSSION
The use of gene probes for the detection of specificbacterial genotypes and virus particles in environmentalsamples has been shown to be widely applicable (3, 19, 31).Enumeration by MPN-DNA hybridization is based on dilu-tion-to-extinction probability theory (1) and does not rely oncounting individual colonies or spots in the case of platecounts or colony blot hybridization. The MPN procedureproved to be very sensitive and accurate for the enumerationof TnS VI bacteria in soil; approximately 10 cells g-' wasreadily enumerated for P. piutida. This level of detection iscomparable with that obtained by other sensitive methodsfor following recombinant bacteria in the environment, in-cluding expression of cloned lac genes (10) and spontaneousantibiotic resistance (37). In this paper a five-replicate (mi-crodilution well) MPN was used, but the sensitivity andaccuracy could be further improved by increasing the num-
ber of replicates (29).The utility of DNA probes for the detection and enumer-
ation of specific microorganisms and genotypes in soil isdependent on the availability of a unique, specific probe. Theproblem of specificity is enhanced when the target organismis present in soil at low numbers, which increases thebackground homology with indigenous soil bacteria. Thesestudies involve the use of a whole plasmid (pGS9) probe todetect TnS and plasmid vector sequences located in thebacterial chromosome. Our findings indicate that the sensi-tivity of detection by MPN-DNA hybridization is signifi-cantly increased when the target includes pCU101 vector(33) sequences (24.5 kilobases) in addition to TnS sequences(5.5 kilobases). However, the use of VI mutants for micro-bial ecology studies may be undesirable. Since pCU101 is a
recombinant plasmid, federal agency approval would be
TABLE 1. Enumeration of R. leguminosarum bv. phaseoli(Kim-5) Tn5 VI from sterile Burbank soil at 5 and 30 days by
various methodsa
Log no. of cells g of soil-' on day:Method
5 30
Plate count 6.76 (0.04) 5.96 (0.66)
MPN plant infectivity 7.34 (7.97, 6.78) NA
DNA hybridizationColony blot 6.81 6.00 (0.30)MPN 6.51 NAMPN replica plate' 7.00 (7.53, 6.51) 4.73 (5.23, 4.20)
FA count 7.18 (0.23) 7.08 (0.48)
aThe actual number of cells added at zero time was 323 + 43 g-'. Singlevalues in parentheses are standard deviations of the log values; double valuesare the 95% confidence limits. NA, Not assayed.
b Identical to MPN-DNA hybridization but with one transfer from themicrodilution plate to selective agar medium, followed by colony blot analy-sis.
APPL. ENVIRON. MICROBIOL.
on July 8, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from
ENUMERATION OF TN5 MUTANT BACTERIA IN SOIL
TABLE 2. Recovery of Tn5 VI isolates from nonsterile soil by a transposon-associated phenotypic marker (Kmr)
Log CFU g of soil-' (SD)Isolate Soil type % Recovery
Cells added Cells recovered
R. leguminosarum bv. viceae IP27A Burbank 6.23 6.09 (0.07) 724.23 4.15 (0.13) 832.71 2.51 (0.10) 63
R. leguminosarum bV. viceae IP27A Palouse 6.23 6.16 (0.08) 854.23 3.88 (0.10) 442.71 2.54 (0.31) 68
P. putida Burbank 5.78 5.30 (0.15) 333.78 3.30 (0.13) 332.26 1.78 (0.04) 33
P. putida Palouse 5.78 5.05 (0.36) 193.78 3.27 (0.06) 312.26 1.27 (0.40) 10
necessary before field testing of VI mutants. In addition,vector sequences appear less stable than Tn5 under soilconditions and could reduce competitiveness in an organismdue to an increase in genetic load. Studies are currentlybeing conducted in our laboratory to assess the fate ofbacteria containing only TnS in soil core microcosms.Although the MPN procedure should prove useful for
enumerating recombinant bacteria in field test situations,care must be taken in the selection of the specific probe as
well as obtaining control samples before the release of theorganisms for determination of possible background homol-ogy with indigenous soil bacteria. In our studies, nonspecificbinding of the probe to soil colloids or hybridization withDNA of indigenous soil bacteria was not a problem. How-ever, nonsterile soil consisted of air-dried soil moistenedbefore use. Although moistening of air-dried soil typicallyresults in a flush of microbial growth and activity, thecomposition of the microflora may be radically different fromfresh soil. Further studies are required to determine whetherthe whole pGS9 probe is specific enough for detection of TnSbacteria in fresh soil.
In addition to detection and enumeration by DNA hybrid-ization, phenotypic expression of the TnS Kmr gene allowsfor enumeration of TnS mutant bacteria in soil as describedin this study and elsewhere (36). Although TnS has beenshown to confer resistance to streptomycin, in addition tokanamycin, in some gram-negative bacteria (27), we did notuse this as an additional marker in our studies because thelevels of resistance conferred were not high enough to aid inselecting against indigenous soil microflora. However, theutility of transposons as either phenotypic (antibiotic resis-tance) or genotypic (DNA probes) markers for experimentsin microbial ecology will be dependent on the location andrelative stability of the transposon within the bacterial gen-ome. The results presented indicate that Tn5 can transposeinto the chromosome of both R. leguminosarum and P.putida and that the Kmr marker is stable after growth for 33generations in sterile soil. However, donor plasmid se-
quences (pGS9), which integrated in addition to TnS, were
less stable in R. leguminosarum yet had about the same
degree of stability as TnS alone in P. putida. The 5%frequency with which donor plasmid sequences were ob-served in Kmr mutants in this study is similar to the 3%observed for TnS mutagenesis with rhizobia (33) and 3.5%observed for Erwinia chrysanthemi (7).
Precise excision of TnS from rhizobia has been shown to
occur at a frequency of 10-8 to l0o- in pure culture (26, 33)and is therefore a relatively rare event. However, in thesestudies with P. putida the loss of Kmr in sterile soil was ashigh as 10-2 and may reflect the instability of VI mutantsunder soil conditions. In pure culture this frequency mayvary; Kmr conferred by TnS in P. fluorescens has beenshown to be stable for over 50 generations (36). TnS is alsocapable of transposing to a number of different target sites inthe genome of P. putida (5), and the stability of TnS insertionin these different regions may vary. The use of Tn5 or othertransposons as markers for bacteria will require biochemicaland physical genetic analysis on an individual basis to ensurethat a stable, ecologically competent mutant is selected.A number of factors will influence the sensitivity of the
MPN-DNA hybridization procedure. As with a variety ofdetection and enumeration methods, including plate countsand colony blot hybridization, culturability of the organismbeing enumerated is necessary. Although a selective mediumis desirable and can increase sensitivity, it is not a necessityfor all organisms. P. putida, enumerated by MPN-DNAhybridization, was cultured in KB broth in the absence ofantibiotic selection. For bacteria such as Rhizobium spp.,which grow more slowly (generation time = 1,000 min)relative to most soil Pseudomonas spp. (generation time'=45 min), a more selective medium is required. Even withantibiotic selection the detection limit for Rhizobium spp.cells in soil was approximately 102 g-'. The dependence onculturability can be a problem; it has been shown that Vibriocholerae and some related pathogens can survive in a viablebut nonculturable state in a marine environment (8). Al-though FA counting can be used to detect and enumeratesuch bacteria in soil, it may also be enumerating cells whichare no longer viable, yet whose outer membrane antigenicdeterminants remain intact, especially in dry soil. Timecourse enumeration of Kim-S TnS VI in soil in this study(Table 1) shows that population estimates by FA countingare 10- to 100-fold higher after 30 days in soil than bymethods which require culturing. Previous work by Bezdi-cek and Donaldson (4) also showed that higher estimates ofbacterial populations by FA counting than by culturing andMPN.
Interactions between microbial cells and clay and organicmatter may also account for the differences in cell recoveriesbetween the two soils. Relative recovery by MPN-DNAhybridization of identical numbers of P. putida Tn5 VI cellsadded to Burbank and Palouse soils was consistently higher
VOL. 54, 1988 451
on July 8, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from

452 FREDRICKSON ET AL.
from the Burbank soil than from the Palouse soil (Fig. 5).The Palouse soil has a significantly higher percentage of bothclay and organic matter than the Burbank soil, which sug-gests that binding of cells to soil colloidal particles is limitingrecovery. These observations are in agreement with the dataof Stotzky and Burns (34), who cite empirical observationssuch as lack of leaching of microbes through the soil profileduring periods of heavy rain, failure to wash substantialnumbers of cells from soil in perfusion or leaching experi-ments, and increased release of microbes from soil uponsonication or treatment with surfactants as strong evidencefor the binding of microorganisms to soil particles. Bacterialinteractions with soil colloidal particles may also account forthe relative differences in recovery on selective medium(containing kanamycin) between R. leguminosarum and P.putida (Table 2) and for the lack of recovery values thatapproach 100% for either soil. Bacterial isolates vary both intheir electrokinetic potential, because of chemical differ-ences in cell wall components, and in their ability to producesubstances which increase their adherence to surfaces (24).The differences observed for growth and survival of both
P. putida (Fig. 3) and R. leguminosarum bv. phaseoli Kim-STn5 VI (Table 2) in sterile and nonsterile soil emphasize theneed for studies on the growth and survival and recombinantmicroorganisms to be conducted under environmental con-ditions which simulate (as closely as possible) the actualenvironment into which they will be released. Microbialgrowth and survival in an ecosystem are dependent onabiotic factors (e.g., pH, Eh, water potential, nutrient avail-ability) and biotic factors (e.g., predation, bacteriostasis).Although microbial survival studies in sterile soil are condu-cive to recovery and enumeration of the test organism,differences in survival patterns (22) do not allow extrapola-tion of results to predict survival in the environment.The use of DNA probes was the focal point of this work;
however, a variety of standard cultural methods as well as
new developments in detection technology will be requiredfor sensitive environmental detection and enumeration ofbacteria. A major advantage in using gene probes, in con-
juction with other methods, is that they are specific for a
gene regardless of the host and can be used for detectioneven if no selective phenotypic marker is available (orexpressed).
ACKNOWLEDGMENTS
This work was supported by the U.S. Department of Energyunder Contract DE-AC06-76RLO 1830.We thank B. Denovan, W. Trumble, and J. West for helpful
discussion and technical assistance.
LITERATURE CITED1. Alexander, M. 1982. Most probable number method for micro-
bial populations, p. 815-820. In A. L. Page, R. H. Miller, andD. R. Keeney (ed.), Methods of soil analysis, part 2, chemicaland microbiological properties, 2nd ed. American Society ofAgronomy, Madison, Wis.
2. Alexander, M. 1985. Spread of organisms with novel genotypes,p. 115-136. In A. H. Teich, M. A. Levin, and J. H. Pace (ed.),Biotechnology and the environment: risk and regulation. Amer-ican Association for the Advancement of Science, Washington,D.C.
3. Barkay, T., D. L. Fouts, and B. H. Olson. 1985. Preparation ofa DNA gene probe for detection of mercury resistance genes ingram-negative bacterial communities. Appl. Environ. Micro-biol. 49:686-692.
4. Bezdicek, D. F., and M. D. Donaldson. 1980. Flocculation ofRhizobium from soil colloids for enumeration by immunofluo-
rescence, p. 247-309. In R. C. Barkley, J. M. Lynch, J. Melling,P. R. Rutter, and B. Vincent (ed.), Microbial adhesion tosurfaces. Ellis Harwood Ltd., London.
5. Boulnois, G. J., J. M. Varley, G. S. Sharpe, and F. C. H.Franklin. 1985. Transposon donor plasmids, based on ColIb-P9,for use in Pseudomonas putida and a variety of other gram-negative bacteria. Mol. Gen. Genet. 200:65-67.
6. Brill, W. J. 1985. Safety concerns and genetic engineering inagriculture. Science 227:381-384.
7. Chatterjee, A. K., K. K. Thurn, and D. A. Feese. 1983. TnS-induced mutations in the enterobacterial phytopathogen Erwiniachrysanthemi. Appl. Environ. Microbiol. 45:644-650.
8. Colwell, R. R., P. R. Brayton, D. J. Grimes, D. B. Roszak, S. A.Huq, and L. M. Palmer. 1985. Viable but non-culturable Vibriocholerae and related pathogens in the environment: implicationsfor release of genetically engineered microorganisms. Biotech-nology 3:817-820.
9. Curtiss, R. 1976. Genetic manipulation of microorganisms:potential benefits and hazards. Annu. Rev. Microbiol. 30:507-533.
10. Drahos, D. J., B. C. Hemming, and S. McPherson. 1986.Tracking recombinant organisms in the environment: ,B-galacto-sidase as a selectable non-antibiotic marker for fluorescentpseudomonads. Biotechnology 4:439-444.
11. Eckhart, T. 1978. A rapid method for the identification ofplasmid deoxyribonucleic acid in bacteria. Plasmid 12:584-588.
12. Fitts, R., M. Diamond, C. Hamilton, and M. Neri. 1983. DNA-DNA hybridization assay for the detection of Salmonella spp. infoods. Appl. Environ. Microbiol. 46:1146-1151.
13. Fredrickson, J. K., and L. F. Elliott. 1985. Colonization ofwinter wheat roots by inhibitory rhizobacteria. Soil Sci. Soc.Am. J. 49:1173-1177.
14. Gasson, M. J. 1980. Indicator technique for antimetabolic toxinproduction by phytopathogenic species of Pseudomonas. Appl.Environ. Microbiol. 39:25-29.
15. Gergen, J. P., R. H. Stern, and P. C. Wensink. 1979. Filterreplicas and permanent collections of recombinant DNA plas-mids. Nucleic Acids Res. 7:2115-2137.
16. Gillett, J. W., S. A. Levin, M. A. Harwell, D. A. Andow, M.Alexander, and A. M. Stern. 1984. Potential impacts of environ-mental release of biotechnology products: assessment, regula-tion and research needs. Ecosystems Research Center, CornellUniversity, Ithaca, N.Y.
17. Grunstein, M., and D. S. Hogness. 1975. Colony hybridization: amethod for the isolation of cloned DNAs that contain a specificgene. Proc. Natl. Acad. Sci. USA 72:3961-3965.
18. Hill, W. E., W. L. Payne, and C. C. G. Aulisio. 1983. Detectionand enumeration of virulent Yersinia enterocolitica in food byDNA colony hybridization. Appl. Environ. Microbiol. 46:636-641.
19. Jiang, X., M. K. Estes, T. G. Metcalf, and J. L. Melnick. 1986.Detection of hepatitis A virus in seeded estuarine samples byhybridization with cDNA probes. Appl. Environ. Microbiol.52:711-717.
20. King, E. O., M. K. Ward, and D. E. Raney. 1954. Two simplemedia for the demonstration of pyocyanin and fluorescein. J.Lab. Clin. Med. 44:301-307.
21. Kleckner, N., J. Roth, and D. Botstein. 1977. Genetic engineer-ing in vivo using translocatable drug-resistant elements. J. Mol.Biol. 116:125-159.
22. Liang, L. N., J. L. Sinclair, L. M. Mallory, and M. Alexander.1982. Fate in model ecosystems of microbial species of potentialuse in genetic engineering. Appl. Environ. Microbiol. 44:708-714.
23. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
24. Marshall, K. C. 1976. Interfaces in microbial ecology. HarvardUniversity Press, Cambridge, Mass.
25. McCormick, D. 1986. Detection technology: the key to environ-mental biotechnology. Biotechnology 4:419-422.
26. Meade, H. M., S. H. Long, G. B. Ruvkun, S. E. Brown, andF. M. Ausubel. 1982. Physical and genetic characterization of
APPL. ENVIRON. MICROBIOL.
on July 8, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from

ENUMERATION OF TN5 MUTANT BACTERIA IN SOIL 453
symbiotic and auxotrophic mutants of Rhizobiumn meliloti in-duced by transposon Tn5 mutagenesis. J. Bacteriol. 149:114-122.
27. Putnoky, P., G. B. Kiss, I. Ott, and A. Kondorosi. 1983. Tn5carries a streptomycin resistance determinant downstream fromthe Kanamycin resistance gene. Mol. Gen. Genet. 191:288-294.
28. Rissler, J. F. 1984. Research needs for biotic environmentaleffects of genetically-engineered microorganisms. Recomb.DNA Tech. Bull. 7:20-30.
29. Rowe, R., R. Todd, and J. Waide. 1977. Microtechnique formost-probable-number analysis. Appl. Environ. Microbiol.33:675-680.
30. Sands, D. C., and A. D. Rovira. 1970. Isolation of fluorescentpseudomonads with a selective medium. Appl. Microbiol. 20:513-514.
31. Sayler, G. S., M. S. Shields, E. T. Tedford, A. Breen, S. W.Hooper, K. M. Sirotkin, and J. W. Davis. 1985. Application ofDNA-DNA colony hybridization to the detection of catabolicgenotypes in environmental samples. Appl. Environ. Microbiol.49:1295-1303.
32. Schmidt, E. L. 1974. Quantitative autecological study of micro-organisms in soil by immunofluorescence. Soil Sci. 118:141-149.
33. Selvaraj, G., and V. N. Iyer. 1983. Suicide plasmid vehicles forinsertion mutagenesis in Rhizobium meliloti and related bacte-ria. J. Bacteriol. 156:1292-1300.
34. Stotzky, G., and R. G. Burns. 1982. The soil environment:clay-humus-microbe interactions, p. 105-133. In R. G. Burns
and J. H. Slater (ed.), Experimental microbial ecology. Black-well Scientific Publications, London.
35. Turco, R. F., T. B. Moorman, and D. F. Bezdicek. 1986.Effectiveness and competitiveness of spontaneous antibiotic-resistant mutants of Rhizobium leguminosarum and Rhizobiumjaponicum. Soil Biol. Biochem. 18:259-262.
36. Van Elsas, J. D., A. F. Dijkstra, J. M. Govaert, and J. A. vanVeen. 1986. Survival of Pseiudomonas fluorescens and Bacillussubtilis introduced into two soils of different texture in fieldmicroplots. Microb. Ecol. 38:151-160.
37. Watrud, L. S., F. J. Perlak, M.-T. Tran, K. Kusano, E. J.Mayer, M. A. Miller-Wideman, M. G. Obukowicz, D. R. Nelson,J. P. Kreitinger, and R. J. Kaufman. 1985. Cloning of theBacillus thuringiensis subsp. kurstaki delta-endotoxin gene intoPseudomonasfluorescens: molecular biology and ecology of anengineered microbial pesticide, p. 40-46. In H. 0. Halvorson,D. Pramer, and M. Rogul (ed.), Engineered organisms in theenvironment: scientific issues. American Society for Microbiol-ogy, Washington, D.C.
38. Weaver, R. W., and L. R. Frederick. 1982. Rhizobium, p.1043-1070. In A. L. Page, R. H. Miller, and D. R. Keeney (ed.),Methods of soil analysis, part 2, chemical and microbiologicalproperties, 2nd ed. American Society of Agronomy, Madison,Wis.
39. Weller, D. M., and R. J. Cook. 1983. Suppression of take-all ofwheat by seed treatments with fluorescent pseudomonads.Phytopathology 73:463-469.
VOL. 54, 1988
on July 8, 2018 by guesthttp://aem
.asm.org/
Dow
nloaded from