equipment facility qualification 1
TRANSCRIPT

13Equipment andFacility Qualification
Thomas L. PeitherPECON—Peither Consulting, Schopfheim, Germany
I. INTRODUCTION
The importance of the qualification process of technical systems in the pharma-ceutical industry has been steadily increasing over the last 10 years. It has beendriven primarily by the requirements of regulatory bodies and not by the needto save money in this part of the industry. If the industry made use of the fullscope of the GMP requirements, the qualification process would be more effi-cient and the cost of qualification would drop. On the other hand, pharmaceuti-cal companies want to protect themselves from a less than perfect result duringa regulatory inspection and therefore demand 120% effort from their suppliersand service companies. New methods and tools must be implemented to reachthe goal of qualifying a technical system while minimizing effort.
Another aspect is the trend for quality assurance departments to evolvefrom being mere controllers of product quality to delivering tools and methodsto other departments, thus helping them to design a better production process.The goal is to improve overall production reliability and availability. In orderto achieve this objective, the quality assurance team must be experienced inapplying and teaching the qualification tools and methods needed. This is atrend that has not yet started in many companies. It may be seen in other indus-tries that more instruments and quality tools are necessary than those limited toqualification and validation. Qualification and validation only appear to be thebeginning of a continuous development process in the quality assurance of thepharmaceutical industry.
Copyright © 2003 Marcel Dekker, Inc.

To avoid misunderstanding, it is crucial to use the correct terms and ex-pressions during quality management. A list of definitions can be found in theappropriate section.
The following describes how the qualification of pharmaceutical equip-ment and facilities can be efficiently planned and executed.
Figure 1 depicts the most commonly used approach to the qualificationprocess as used in the pharmaceutical industry. It shows a pyramid, which isthe best way in which to plan a qualification/validation project. Investing moretime in the first phases will save time and money in later and critical phases. Ifinadequate investment is made during the start-up of a project, the later phasesof installation qualification (IQ), operational qualification (OQ), and perfor-mance qualification (PQ) will necessarily require an inordinate amount of timeand money. The project will be a pyramid again, but now it is inverted.
It must be stressed that a good engineering and project process is the bestbasis for proper qualification and validation work. It is the current opinion onqualification in the pharmaceutical industry that the later steps in the qualifica-tion process need more time and attention than the earlier steps. This may betotally different in other industry branches; they tend to spend more effort duringthe earlier stages to save time and money later on.
Figure 1 Steps of qualification.
Copyright © 2003 Marcel Dekker, Inc.

If the pharmaceutical industry adopted the lessons learned in otherbranches (e.g., aircraft industry, automotive industry) it could realize an in-creased efficiency in the qualification and validation processes. To this end ef-fort should be made to investigate statistical process control (SPC), house ofquality, Deming circuits, and so on.
At the moment the term design qualification (DQ) is the focus of somecontroversy. Performing a DQ is not a legal requirement, but it has been intro-duced to the qualification process through implementation of Annex 15 to theEC Guide on Good Manufacturing Practices for Medical Products. It is not arequirement to implement a DQ, but it seems that regulatory bodies have aninterest in promoting this element of engineering and quality management. Itshould be a requirement of a proper engineering process, and in fact although itis often a part of the engineering process, it is not declared as a separate action.Nevertheless, the activity itself should be executed in combination with an effi-cient procedure documented in a standard operating procedure (SOP). (See Sec.V.) Important aspects that should be taken into consideration before qualifica-tion aspects start are shown in Figure 2.
It is important to perform these preliminary steps conscientiously. Mostqualification projects fail because these basic activities are not performed ade-quately.
Figure 2 Preliminary steps.
Copyright © 2003 Marcel Dekker, Inc.

II. PROJECT MANAGEMENT
Good project management is the first step toward organizing the successfulqualification of a technical system. A well-structured and -planned approach toqualification is the first step toward success.
The tools and methods of project management are mainly used for largeand complex projects. It is equally important to apply these management skillsto smaller projects, however. A good project start is the best way to win thebattle.
A. Project Organization
To start with, the project organization must be defined. The different positionsmust be defined and people need to be found with the necessary knowledge tofill these positions. The most commonly required areas of expertise for a projectleader are organizational know-how, social skills, project management know-how, time management, validation know-how, and general technical know-how.A team member should have expertise in communication skills, validationknow-how, and detailed technical know-how.
B. Meeting Management
The communication structure must be defined following the definition of thebest project organization. A lot of projects waste time in meetings. Everybodyis familiar with this scenario. You find yourself sitting in a meeting thinkingthat your time is being wasted and that you might not attend another scheduledmeeting. Nobody likes to feel that his or her time is wasted, therefore thoroughplanning prior to any meeting is mandatory.
A chairperson heading the meeting must be chosen and a person desig-nated to take the minutes. Every meeting should have an agenda. People shouldbe invited based on whether or not they can help with solving the issues on theagenda. Obviously everyone attending should be well prepared. In order to facil-itate this process, meetings should be planned 6 months in advance.
C. Project Planning
After the definition of functions, responsibilities, and communication structures,the project itself must be planned. Using Gantt charts is often the best way toschedule the different tasks. This allows you to see quickly which task has tobe done when and by whom. The charts also indicate interdependencies betweendifferent tasks and show what happens if a task takes longer than planned. Dif-
Copyright © 2003 Marcel Dekker, Inc.

ferent software is available to help generate such project plans. An example fora project plan is shown in Figure 3.
People often ask for an example of a detailed project plan. Working out aspecific project plan requires in-depth knowledge of a technical system, how-ever. As each system is different, Figure 3 can only show a general overviewof a project plan.
D. Project Reporting
The next important task in the process of project management is the implemen-tation of efficient project control. A reporting system must be put into place thatdescribes the current state of the project as well as the progress of the mostimportant tasks. Additionally, the reporting system must be able to pick up andhighlight problems within the project. A functioning reporting system is thecontrolling instrument for the project manager.
E. Tools for Project Management
In order to manage a project efficiently appropriate tools must be applied. Thereare several products of project management software on the market. The deci-sion as to which system is the best suited for a given project should include thefollowing aspects:
Project focusSize of a projectNumber of team membersNumber of tasks in a projectRequired functionality
III. VALIDATION/QUALIFICATION MASTER PLAN
It is important to draw up a summarized document that describes the wholeproject. It has become common practice in the industry to develop a “validationmaster plan” (VMP). This document would usually include the qualificationaspects of a project. Alternatively, a “qualification master plan” (QMP) shouldbe drafted. In case of a large retrospective qualification project it is beneficialto write a separate QMP. The main point is to develop a document that includesthe most important information of the project and can be used like a projecthandbook.
Copyright © 2003 Marcel Dekker, Inc.
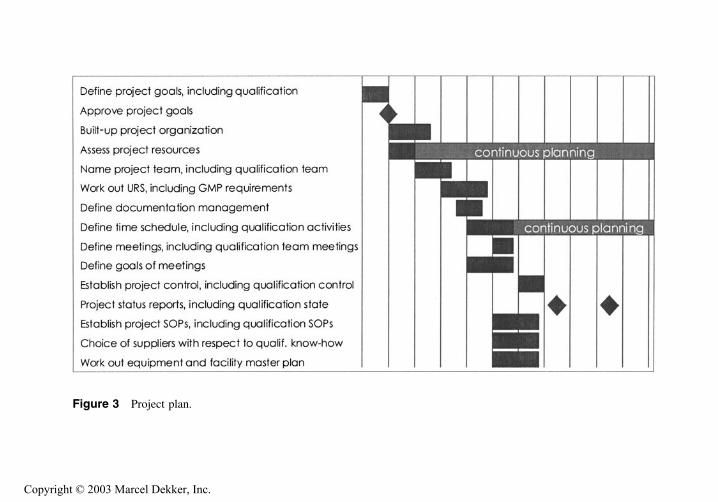
Figure 3 Project plan.
Copyright © 2003 Marcel Dekker, Inc.

Figure 4 Structure of a validation master plan.
A. Structure of a VMP or QMP
The structure of a VMP or QMP is well documented in the PIC/S document PI006 Recommendation on Validation Master Plan, Installation and OperationalQualification, Non-Sterile Process Validation and Cleaning Validation. Thisdocument is the basis for Annex 15 to the EC Guide on Good ManufacturingPractices for Medical Products. Figure 4 displays the most commonly usedtopics to be described in a VMP.
The PIC/S-document PI 006 defined the VMP as “A document providinginformation on the company’s validation work programme. It should define de-tails of and time scale for the validation work to be performed. Responsibilitiesrelating to the plan should be stated.” The EC Guide on Good ManufacturingPractices for Medical Products, Annex 15, said of the VMP: “All validationactivities should be planned. The key elements of a validation programmeshould be clearly defined and documented in a validation master plan (VMP)or equivalent documents. The VMP should be a summary document which isbrief, concise and clear.” The FDA published another interesting paper in 1995,
Copyright © 2003 Marcel Dekker, Inc.

the Guide to Inspections of Validation Documentation. This guide outlines thebasics in qualification and validation; for example, “Planning documents mayuse various formats and styles, and different descriptive terms may be used suchas master validation plan, project plans, study plans, and others. Regardless ofterminology, it is important that suitable documents denote intentions in suffi-cient detail.” It highlights the benefits of having an overall document such asVMP.
To summarize, the VMP or QMP should be a brief overview of the proj-ect, tasks, tools, resources, and methods that are going to be used during theproject. This document should be described at a very early stage of a project bythe engineering or manufacturing department of a pharmaceutical manufactureror service provider.
Standard operating procedures are an integral part of any VMP or QMP.They outline rules that have to be followed during the project and provide proj-ect members with guidelines as to which rules have to be studied before startingwork.
B. Areas of Interest
Many technical systems in a pharmaceutical production have to be validated orqualified. The requirement for a system to be validated depends on its impact onproduct quality. Whether a system is critical or not may be determined through arisk analysis. (See Design Qualification.) Following is a list of such differentsystems or clusters of systems.
1. Infrastructure and Facilities
High purity water systems (high purity water, water for injection, highlypurified water, etc.)
Clean steamGases with product contact (compressed air, nitrogen, oxygen, vacuum,
etc.)HVAC with rooms (clean area), including lighting
2. Equipment
Closures, tanks, vessels with product contactMachines with product contact (filling machine, washing machine, closing
machine, granulator, packaging lines, etc.)Machines with direct impact on product quality (autoclaves, sterilizing
units, labeling system, weighing system, production control system, fa-cility control system, etc.)
Copyright © 2003 Marcel Dekker, Inc.

IV. USER REQUIREMENT SPECIFICATION
The goal of working out user requirement specifications (URS; Fig. 5) is todocument the needs of the manufacturing department. User requirement specifi-cations are always written for a technical system that should be implemented inthe production process of a pharmaceutical product. The URS is very importantfor realizing a project, as many measurements refer to the URS. A well-preparedURS is the key to a project’s success. Projects without detailed URS have atendency to demand lots of change later on, thus increasing cost, start-up times,or both. Who should evaluate a URS? Best practice is a coordinated approachamong production, quality assurance, and engineering of the pharmaceuticalcompany. Some companies even use the services of external resources to createa URS.
A. GMP Requirements
The key aspect of any URS is to generate a document detailing all the GMPrequirements the technical system has to fulfill. The URS is an important docu-ment for the commissioning phase as well. Often the URS provides the basisfor an offer to the suppliers. A detailed URS will result in a better and morecompetitive offer for the technical system. While evaluating a supplier, it isimportant to gather as much information as possible. Without a comprehensiveURS, a pharmaceutical company cannot get a clear understanding of the supplierand may be led to make a wrong decision.
Figure 5 Content user requirement specification (URS) design qualification.
Copyright © 2003 Marcel Dekker, Inc.

B. Technical and Economic Requirements
User requirement specifications cover more aspects than only the GMP require-ment, because the URS is not written only for the validation procedure; in fact,a URS is a very important project document covering technical as well as eco-nomic requirements of the technical system. Pharmaceutical manufacturing de-partments not only check the GMP aspects of a system; additionally, followinggood engineering practice they will review the technical and economic aspectsof a technical system. Obviously, the more experience a company gains, themore comprehensive a URS become. Past experiences such as project faults,inefficient technical systems, and bad commissioning can be included in a URS.
V. DESIGN QUALIFICATION (DQ)
Design qualification is more common in Europe than in the United States. Thereis no legal requirement to perform a DQ. Sometimes this phase may not becalled DQ, but may instead be referred to as “design review,” “design assess-ment,” and so on. The intention is important in this phase. The goal is to performsomething similar to a risk analysis and to check the design documents of atechnical system to ensure that they fulfill the user requirements. For this reasona risk analysis—not yet commonly known in all companies—should be used.
A. Risk Analysis
The overall concept of all of the following tools is that of risk analysis or riskassessment. Risk analysis helps to decide whether an aspect is GMP-critical ornot. The risk analysis can be performed in a formal or more informal way.Following are two popular and import types of risk analysis. Another method,the fault tree analysis (FTA), has recently been used in the area of computervalidation. This method is not described here, as it is a complex form of riskanalysis.
B. FMEA
FMEA is a quantitative risk analysis for complex systems (Fig. 6). As thisapproach involves assessment of occurrence probabilities, detection of failures,and judgment as to the severity of a failure, it should only be chosen if somepractical experience with the technical system is available. Each of the threevalues will be assigned a number from 1 to 5. Multiplying these values resultsin the “risk priority number.” This number indicates the priority of the assessedfailure. The pure version of the FMEA is seldom practiced in the pharmaceuticalindustry.
Copyright © 2003 Marcel Dekker, Inc.

Figure 6 Failure mode and effects analysis (FMEA) overview.
Most common risk analysis forms are mutations of the fundamentalFMEA. It is easier for most companies to start initially with a more practicalway of performing a risk analysis. In the future the fundamental FMEA will bemore commonly applied, as companies will have gained confidence with varia-tions of the FMEA.
Variations are often made by cutting the detection of failures or severityof failures. Sometimes the values are decreased to a spread of 1 to 3. In othercases the risk priority number is not calculated, but the levels are noted in amatrix to see whether the point is critical or not.
C. Hazard Analysis of Critical Control Points (HACCP)
The second method is the hazard analysis of critical control points (HACCP;Fig. 7). This method is well known in the food industry. The goal of HACCPis to reduce the risk of contamination of products and to reduce the effort fortesting products during final tests. The HACCP defines critical control points(CCPs) in different grades (usually three grades). The HACCP protocols are
Copyright © 2003 Marcel Dekker, Inc.

Figure 7 Hazard analysis of critical control points (HACCP).
worked out and the results are documented in reports. Important steps are thedefinition of CCPs and their limits, the implementation of a change control sys-tem, the execution of corrective actions, and the implementation of a documenta-tion system. Equally important are regular audits of the concept and the approvalof HACCP protocols using appropriate procedures. As with the FMEA, theHACCP concept offers the opportunity to rethink all technical and organizationalaspects in an early phase of a project and to find out all critical deficiencies.
D. Documentation of DQ
The results of any risk analysis should be well documented as they become thekey input into the qualification and validation process. They are the basis fordefining tests in the IQ, OQ, and PQ phases. It is often impossible to say priorto a risk analysis what steps of qualification need to be performed. It dependson the risks and measurements defined during the risk analysis. Equally impor-tant, this procedure increases the efficiency of the qualification process. In thepast, the decision on which qualification tests to perform was outlined by writ-ing qualification protocols. These usually prompted long and expensive discus-
Copyright © 2003 Marcel Dekker, Inc.

sions. One of the challenges is to determine which parts of a system to devisetests for. It is easy to imagine that companies and people who have experiencewith risk analysis are in a better position, as they will have developed standardtests for a list of critical elements, leaving only a few additional tests to bedesigned for a particular system. The result is that the longer the qualificationsystem is in place the more effective it becomes. This is a great advantage andhelps to repay the investments of starting a risk analysis system quickly.
VI. INSTALLATION QUALIFICATION
The decision as to which system needs to be qualified should result directlyfrom the risk analysis process and should be described in the VMP or QMP. Inany case, it would be a technical system that impacts on the quality of a pharma-ceutical product. Installation qualification aims to check documentation againstreality. The result is “as-built documentation.” The other task in the IQ is toensure that the GMP requirements are fulfilled. The generally accepted way toperform an IQ is to
Develop an IQ protocol (Fig. 8)Approve the IQ protocols (by the quality assurance, production, and tech-
nical departments)Perform the IQWork out the IQ reportApprove the IQ report (by the quality assurance, production, and technical
departments)
Installation qualification is defined in the PIC/S document PI 006 as “Theperformance and documentation of tests to ensure that equipment (such as ma-chines, measuring equipment) used in a manufacturing process, are appropri-ately selected, correctly installed and work in accordance with established speci-fications.”
The OQ phase relies on valid calibration of all quality-relevant instru-ments. The best way to guarantee this is to perform the calibration at the end ofthe IQ phase. Sometimes it is performed at the beginning of the OQ. This proce-dure is acceptable as well.
The IQ phase will be executed with personnel of the supplier of a techni-cal system or with technical personnel of the pharmaceutical company. It willfollow the procedures set out in the IQ protocols. After performing the IQ, theresults are summarized and documented in an IQ report.
The qualification of the control unit of a technical system is very similarto that of the mechanical equipment of a system. This does not apply to compu-
Copyright © 2003 Marcel Dekker, Inc.

Figure 8 Example of an IQ protocol.
Copyright © 2003 Marcel Dekker, Inc.

Figure 8 Continued.
terized systems, however. These are described in detail in another part of thisbook.
The most important aspects to consider during IQ are
Provide as-built documentation (e.g., P&ID check).Check training reports.Check that documentation is complete.Check calibration reports.Identify piping and instrumentation
VII. OPERATIONAL QUALIFICATIONS
Operational qualification is defined in the PIC/S document PI 006 as “Docu-mented verification that the system or sub-system performs as intended through-out all anticipated operating ranges.”
Operational qualification tests whether or not the system works as ex-pected. The approach to a successful OQ is the same as described for IQ [de-velop OQ protocols (Fig. 9)], approve OQ protocols (by the quality assurance,production, and technical departments), perform OQ, work out OQ report, andapprove OQ report (by the quality assurance, production, and technical depart-ments).
The OQ phase normally involves personnel from the supplier of a techni-cal system or technical personnel from the pharmaceutical company. It is prefer-
Copyright © 2003 Marcel Dekker, Inc.

Figure 9 Example of an OQ protocol.
Copyright © 2003 Marcel Dekker, Inc.

able to include customer employees, as they are going to be the users of thesystem. This facilitates a better know-how transfer between supplier and cus-tomer. Again, this process follows the rules outlined in the OQ protocols. Theresults of OQ are summarized and documented in an OQ report. It is commonlyaccepted practice in the industry to produce one report for both IQ and OQresults. This saves money and time for approval.
Operational qualification of the control unit of a technical system is oneof the most important steps during the OQ phase. It tests all critical functionsand alarms of the technical system. There are no different procedures for me-chanical OQ and control unit OQ.
The result of the OQ is a documented approval that the technical systemfulfills the user requirements and all GMP-related functions of the technicalsystem.
Typical tests in the OQ include the following:
Alarm testsBehavior of the system after energy breakdownAccuracy of filling linesTransportation speed in a sterilization tunnelTemperature distribution in an autoclavePerformance of a washing machineAccuracy of a weighing system
VIII. PERFORMANCE QUALIFICATION
The PQ is the phase in which either a technical system is tested over a longperiod of time (e.g., water system), or a complex technical system is testedoverall (connected filling line). For many systems OQ is the last phase per-formed during qualification. If there are only a few performance tests needed,it might be more practical to include them during OQ or process validation.Combining OQ and PQ decreases the number of documents (less documentationwork in the future) and cuts approval time and effort. Again, the procedure forPQ is the same as for IQ and OQ ([develop PQ protocols, approve PQ protocols(by the quality assurance, production, and technical departments), perform PQ,work out the PQ report, and approve the PQ report (by the quality assurance,production, and technical departments)]. The documentation and test descriptionare identical to those in the OQ phase.
Performance qualification should be executed by customer personnel. It isa great disadvantage if it has to be performed by the supplier. Ideally this phaseallows know-how to be established at the pharmaceutical company.
The following technical systems need to be performance-tested and quali-fied:
Copyright © 2003 Marcel Dekker, Inc.

High purity water systems (monitoring of the quality parameters: pH,TOC, conductivity, CPU, temperature)
HVAC systems (temperature, pressure, humidity)Complex connected systems (e.g., filling line, BPI production line; perfor-
mance parameters)
IX. DOCUMENTATION SYSTEM
To quickly locate any given document, it is mandatory to have implemented anappropriate documentation system. In case of a fault in production or inspection,it becomes necessary to find a document within 15 to 20 min. All companiesshould test the reliability of their documentation system using internal audits.
One aspect of a working documentation system is a standardized docu-mentation structure. If every system is documented using the same documentstructure, everyone can gain access to the necessary information quickly. Figure10 shows an example of a documentation structure.
Documents do not need to be delivered to the customer in paper format.Electronic media documents such as CDs are equally acceptable. Obviously, anappropriate reading system must be in place to access the documents at a laterdate (e.g., for an inspection). Such a system must remain in place until the
Figure 10 Documentation structure.
Copyright © 2003 Marcel Dekker, Inc.

documentation is destroyed. In case of a complete electronic documentationsystem the whole system needs to be validated by computer validation.
X. CHANGE CONTROL
Change control is defined in the PIC/S document PI 006 as follows: “A formalsystem by which qualified representatives of appropriate disciplines review pro-posed or actual changes that might affect a validated status. The intent is todetermine the need for action that would ensure and document that the systemis maintained in a validated state.”
Change control is a lifetime monitoring approach. Planning for well-executed change control procedures (Fig. 11) includes the following aspects:
Figure 11 Change control procedure flowchart.
Copyright © 2003 Marcel Dekker, Inc.

Workable documentation systemDefined responsibilities and job descriptionsDefined review proceduresWell-trained staff
The implementation of a change control system is an important and neces-sary step in the validation approach for equipment and facilities. Vital to anychange control system is its efficiency in that it does not require too much timeand effort to handle changes. In order to design an efficient change controlsystem, the following aspects need to be taken into consideration:
Early categorization of a change as major or minor change (i.e., cata-logue). This should speed up the decision and approval time of achange.
Easy and logical way of document flow (production engineering, qualityassurance, production).
Easy and logical decision tree for major or minor changes or planned oremergency changes.
It is not only good practice but also essential that a requested change isonly implemented after the appropriate change control procedures and approvalshave been followed. Time and money are often wasted because a change wasnot correctly evaluated (major or minor) or personnel was not familiar with thebest practice for change control procedures. It is crucial for an efficient changecontrol process that the production, engineering, and validation departments areworking together very closely.
Clear change control procedures have to be in place for all eventualities.This must include instructions for situations in which the supervisory or man-agement personnel is not present when the problem occurs. In such a case, forexample, a change or correction might be implemented quickly by the mainte-nance or operational personnel that must then be reviewed and approved bymanagement within 24 hr.
REFERENCES
1. U.S. Food and Drug Administration. 21 CFR 210/211 Good Manufacturing Practice.Fed Reg (2001).
2. U.S. Food and Drug Administration. Guide to Inspection of Validation Documenta-tion (1995).
3. Recommendation on Validation Master Plan, Installation and Operational Qualifi-cation, Non-Sterile Process Validation and Cleaning Validation, Pharmaceutical In-spection Co-Operation Scheme PIC/S PI 006 (2002).
Copyright © 2003 Marcel Dekker, Inc.

4. EC. Guide on Good Manufacturing Practices for Medical Products. EU (2001).5. U.S. Food and Drug Administration. Guide to Inspection of Process Validation
(1987).6. The gold sheet. F-D-C Rep 34 (2000).7. Vina, B. GMP Compliance, Productivity, and Quality. Interpharm Press (1998).8. ISPE. Baseline Pharmaceutical engineering guide series: Introduction of commis-
sioning and qualification. ISPE 2000 European seminar, Amsterdam, 2000.
Copyright © 2003 Marcel Dekker, Inc.