estradiol induces functional inactivation of p53 by ... · estradiol induces functional...
TRANSCRIPT

[CANCER RESEARCH 60, 2594–2597, May 15, 2000]
Advances in Brief
Estradiol Induces Functional Inactivation of p53 by Intracellular Redistribution 1
Anna Maria Molinari, Paola Bontempo, Ettore M. Schiavone, Vincenzo Tortora, M. Antonietta Verdicchio,Massimo Napolitano, Ernesto Nola, Bruno Moncharmont,2 Nicola Medici, Vincenzo Nigro, Ignazio Armetta,Ciro Abbondanza, and Giovanni A. PucaIstituto di Patologia Generale ed Oncologia, Facolta di Medicina e Chirurgia, Seconda Universita degli Studi di Napoli, I-80138 Naples, Italy
Abstract
Estrogen treatment of MCF-7 cells grown in serum-free medium in-duced a modification of the intracellular distribution of p53 protein.Western blot analysis and immunofluorescence staining showed that p53was localized in the nucleus of untreated cell and that after 48 h ofhormone treatment, it was mostly localized in the cytoplasm. This effectwas blocked by the antiestrogen ICI182,780. Intracellular redistributionof p53 was correlated to a reduced expression of the WAF1/CIP1 geneproduct and to the presence of degradation fragments of p53 in thecytosol. Estradiol treatment prevented the growth inhibition induced byoligonucleotide transfection, simulating DNA damage. This observationindicated that the wild-type p53 gene product present in the MCF-7 cellcould be inactivated by estradiol through nuclear exclusion to permit thecyclin-dependent phosphorylation events leading to the G1-S transition. Inaddition, the estradiol-induced inactivation of p53 could be involved in thetumorigenesis of estrogen-dependent neoplasm.
Introduction
Different types of stress (e.g.,hypoxia or DNA damage) induce acellular response leading to growth arrest or to apoptosis through afunctional activation of p53 gene product (1). DNA-dependent proteinkinase (2) or the gene product mutated in ataxia telangiectasia, ATM(3, 4), are involved in alternative pathways leading to p53 activation.Activated p53 protein acts as a transcription factor, increasing theexpression of genes such asp21 (5), GADD45(6), or bax (7), whoseproducts inhibit cell cycle progression or induce apoptosis. Activationof p53 protein is limited by a short feedback loop involving theMDM2 gene product (8). p53 stimulates transcription of theMDM2gene (9), and MDM2 protein binds to activated p53 protein (10). Thisinteraction inhibits p53 transcriptional activity and promotes its exportto the cytoplasm for proteasome-mediated degradation (11). In manyhuman and animal tumors, thep53gene is functionally inactivated bydeletion or point mutations, participating, by this mechanism, in theprocess of neoplastic transformation as an antioncogene (12).
The MCF-7 breast cancer cell line responds to stimulation by aphysiological concentration of estradiol with an increase in prolifer-ation rate (13). This cell line contains a wild-typep53gene (14) whoseproduct is mostly localized in the nucleus during the G1 phase andmoves to the cytoplasm after the G1-S transition (15). In this report,we present evidence that, in MCF-7 cells, estradiol was able to inducefunctional inactivation of p53 protein by intracellular redistribution.
Materials and Methods
Cell Culture. MCF-7 breast cancer cells were grown in 75-cm2 flasks inDMEM, supplemented with 5% FCS, 100 units/ml penicillin G, 100 units/mlstreptomycin, 50mg/ml gentamicin, and 2 mM L-glutamine in a 5-% CO2 atmo-sphere. For induction experiments, 30% confluent cells were grown in phenol red-and serum-free medium for 2 days (cells were transferred to fresh medium twicea day) and then 10mg/ml insulin and 10 nM estradiol were added in the absenceor the presence of 1mM ICI182,780. All tissue culture media, sera, and reagentswere from Life Technologies, INC., Grand Island, NY.
For oligonucleotide transfection, cells were grown in 6-cm plates. Trans-fection was performed with 8mg/dish of 17-mer double-stranded oligonucleo-tides with a 59-end overhang (GATC) premixed with 30ml of DOTAP3
liposomal transfection reagent (1 mg/ml; Boehringer Mannheim, Mannheim,Germany), according to manufacturer’s instructions. In mock transfections,only DOTAP liposomal transfection reagent was added. A rough estimate ofthe cell number per well was obtained by staining the cells with crystal violetand measuring the absorbance on a densitometer (16).
Colony Formation Assay in Methylcellulose.MCF-7 cells were culturedin 50% DMEM and 50% RPMI, supplemented with 20% FCS, and treated withdextran-coated charcoal and 0.8% methylcellulose (MethoCult H4100; Terry Foxlaboratories, Vancouver BC, Canada) in 6-well plates (53 103 cells/well). Col-onies were analyzed after 7 days. Photographs were taken at3100 magnification.
Cytosol and Nuclear Extract Preparation. Cells were rinsed with coldPBS containing 1 mM EDTA, and harvested with 2 mM EDTA. The cell pellet waswashed twice with ice-cold PBS, once with ice-cold buffer A [10 mM HEPES (pH7.9), 10 mM KCl, 0.5 mM DTT, 1.5 mM MgCl2], resuspended in three volumes ofbuffer A, and homogenized in a Dounce homogenizer (10 strokes with pestle B).The homogenate was centrifuged at 33003 g for 30 min at 4°C to obtain a nuclearpellet. The supernatant was collected and centrifuged for 1 h at 100,0003 g at4°C; the supernatant referred to as cytosol. The nuclear pellet was resuspendedwith buffer B [20 mM HEPES (pH 7.9), 450 mM NaCl, 0.2 mM DTT, 1.5 mM
MgCl2, 0.5 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 25% (v/v) glyc-erol], homogenized (two to five strokes in a Dounce homogenizer, pestle B),incubated for 30 min with gentle shaking, and centrifuged for 1 h at 25,0003 gat 4°C. The supernatant, referred to as nuclear extract, was clarified and aliquoted.Complete protease inhibitor cocktail TM was added to all buffers (1 tablet/50 ml;Boehringer Mannheim).
Electrophoresis and Western Blot Analysis.SDS PAGE was performed inreducing conditions in 10 or 11% acrylamide gels (acrylamide/bisacrylamide ratio,40/1, w/w). Twenty-fivemg of protein form cytosol or nuclear extract were appliedto each lane. For Western blot analysis, proteins were electrophoretically trans-ferred to a 0.45mm nitrocellulose sheet. Membranes were then blocked by 5%nonfat milk for 1 h in TBST buffer [20 mM Tris-HCl (pH 7.5), 135 mM NaCl, and0.05% Tween 20]. After repeated washes with TBST buffer, the membranes wereincubated with the primary antibodies for 1 h at room temperature in the samebuffer. Proteins were detected using mouse monoclonal antibodies Ab-6 (1mg/ml;Calbiochem-Oncogene Research Products, Cambridge, MA) or BP53-12 (1mg/ml; Sigma Immunochemicals, St. Louis, MO) to p53, or mouse monoclonalantibody to WAF1 (Calbiochem-Oncogene Research Products). At the end of theincubation, the membrane was washed once for 15 min and three time for 5 minwith TBST buffer. Antibody reactions were revealed by incubation for 1 h, at roomtemperature with enzyme-coupled antimouse IgG (1:10000 dilution; Amersham,
Received 1/17/00; accepted 3/31/00.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby markedadvertisementin accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1 This investigation was supported by grants from the Italian Ministry for Universityand Scientific and Technological Research, and from Regione Campania (L.R. 41/90, rep.8397).
2 To whom correspondence should be addressed, at Istituto di Patologia Generale edOncologia, Facolta di Medicina e Chirurgia, Seconda Universita degli Studi di Napoli,Larghetto Sant’Aniello a Caponapoli, 2, I-80138 Naples, Italy. Phone: 39 081 5665686;fax: 39 081 5665695; E-mail: [email protected].
3 The abbreviations used are: DOTAP,N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimeth-ylammonium methylsulfate; TBST, Tris-buffered saline-Tween 20; EMSA, electro-phoretic mobility shift assay.
2594
Research. on April 14, 2017. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
Aylersbury, Bucks, United Kingdom), in TBST buffer containing a 0.5% solutionof blocking reagent, followed by a washing cycle, and development with achemiluminescent substrate (ECL; Amersham), according to manufacturer’s in-structions.
EMSA. Whole-cell extract was obtained from cells transfected with oligonu-cleotides (or mock-transfected) by freeze-thawing (three cycles) in 50 mM Tris-HCl (pH 8.0), 1 mM DTT, 0.2 mM EDTA, 0.02 mg/ml BSA, and 0.2mg/mlpoly(dI-dCzdI-dC). Ten pmol of theGADD45p53-binding site (59-GATCCTG-CAGCAGAACATGTCTAAAGCATGCTGGGCTCGAG-39) were incubatedwith 10 units of polynucleotide kinase and 50mCi of g[32P]ATP at 37°C for 30min. The forward and reverse strands were annealed, and double-stranded se-quences were purified by 12% PAGE. Each reaction mixture contained 5mg ofprotein from whole-cell extract, 50,000 cpm of labeled probe (;3000 Ci/mmol),and where indicated, 20 pmol of double-stranded oligomers containing a 59overhang. EMSA was performed as described (17).
Immunocytochemistry. Cells were grown on slides for immunofluores-cence (bioMerieux SA, Marcy l’Etoile, France), fixed with 4% (w/v) formal-dehyde in PBS for 10 min and washed three times with PBS. Slides weretreated at 4°C with methanol and 0.1% hydrogen peroxide for 10 min,permeabilized with 0.2% Triton X-100 in PBS for 10 min (where indicated),and blocked with PBS containing 1% goat serum (Life Technologies) and 5%BSA (Sigma) for 1 h. Cells were stained overnight at 4°C with antibodies top53 (clone BP53; Sigma) diluted 1:3000, washed twice with PBS, and incu-bated with FITC-conjugated goat antimouse IgG antibody (1:40 dilution;Ortho Diagnostic Systems Inc., Raritan, NJ) for 45 min. After two washes withPBS, slides were mounted in Fluo permounting medium (bioMerieux SA).
Results
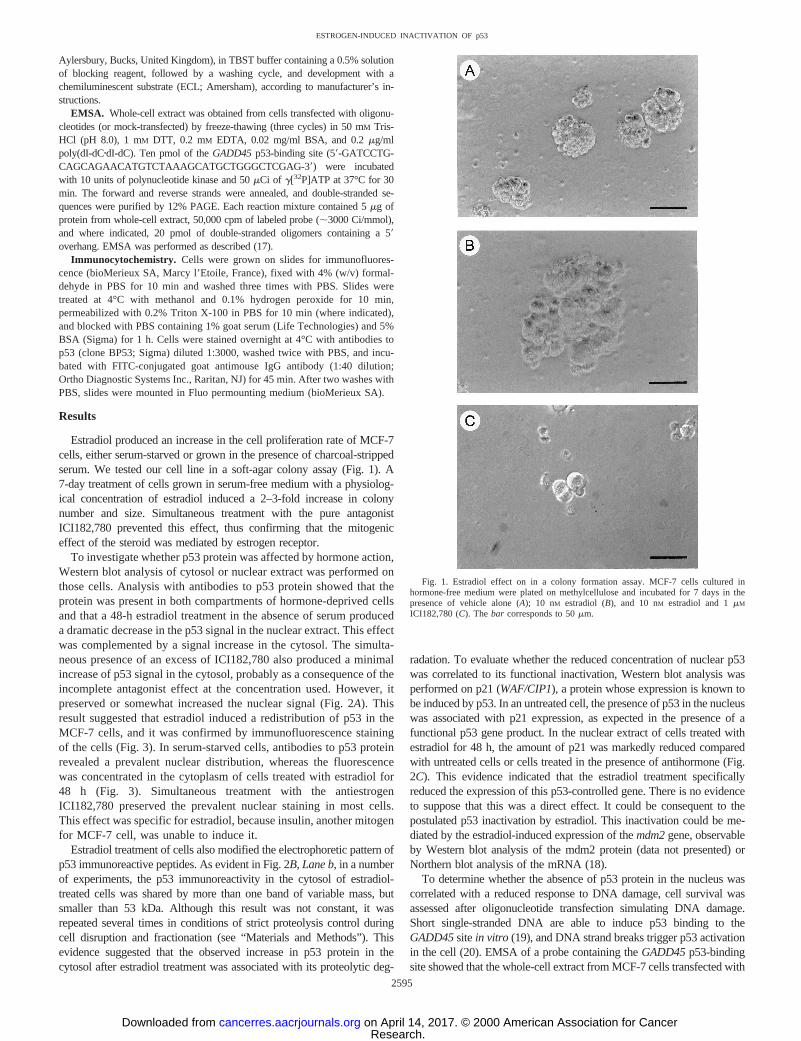
Estradiol produced an increase in the cell proliferation rate of MCF-7cells, either serum-starved or grown in the presence of charcoal-strippedserum. We tested our cell line in a soft-agar colony assay (Fig. 1). A7-day treatment of cells grown in serum-free medium with a physiolog-ical concentration of estradiol induced a 2–3-fold increase in colonynumber and size. Simultaneous treatment with the pure antagonistICI182,780 prevented this effect, thus confirming that the mitogeniceffect of the steroid was mediated by estrogen receptor.
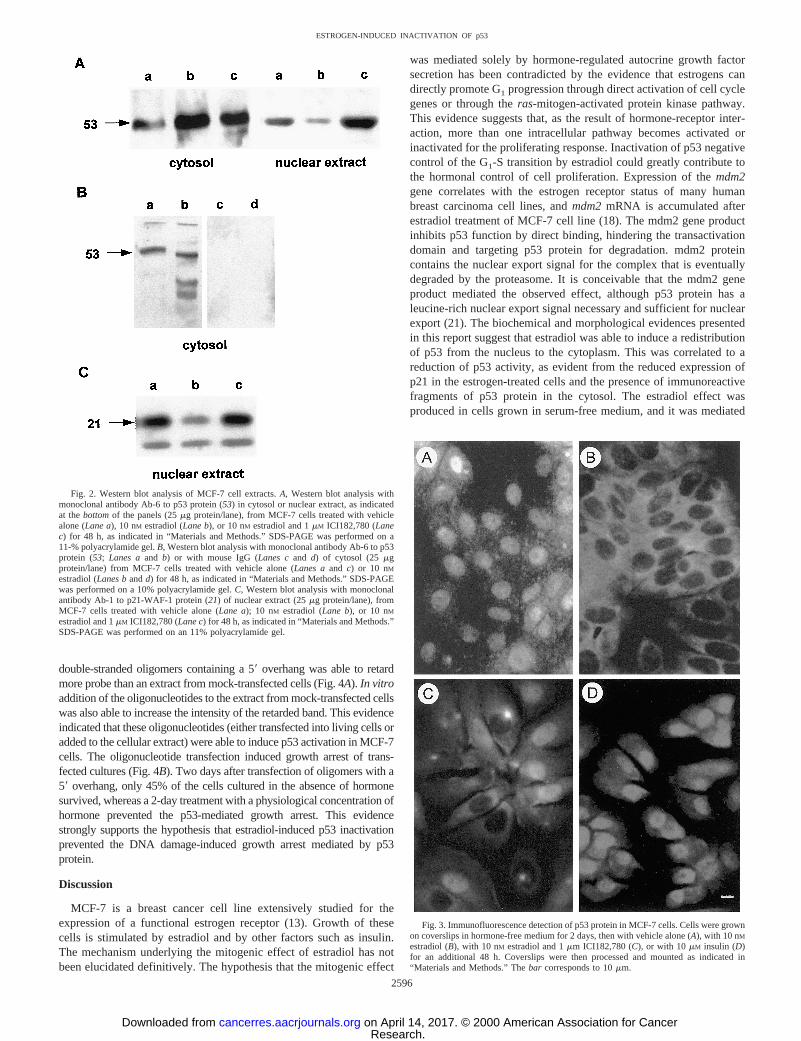
To investigate whether p53 protein was affected by hormone action,Western blot analysis of cytosol or nuclear extract was performed onthose cells. Analysis with antibodies to p53 protein showed that theprotein was present in both compartments of hormone-deprived cellsand that a 48-h estradiol treatment in the absence of serum produceda dramatic decrease in the p53 signal in the nuclear extract. This effectwas complemented by a signal increase in the cytosol. The simulta-neous presence of an excess of ICI182,780 also produced a minimalincrease of p53 signal in the cytosol, probably as a consequence of theincomplete antagonist effect at the concentration used. However, itpreserved or somewhat increased the nuclear signal (Fig. 2A). Thisresult suggested that estradiol induced a redistribution of p53 in theMCF-7 cells, and it was confirmed by immunofluorescence stainingof the cells (Fig. 3). In serum-starved cells, antibodies to p53 proteinrevealed a prevalent nuclear distribution, whereas the fluorescencewas concentrated in the cytoplasm of cells treated with estradiol for48 h (Fig. 3). Simultaneous treatment with the antiestrogenICI182,780 preserved the prevalent nuclear staining in most cells.This effect was specific for estradiol, because insulin, another mitogenfor MCF-7 cell, was unable to induce it.
Estradiol treatment of cells also modified the electrophoretic pattern ofp53 immunoreactive peptides. As evident in Fig. 2B, Lane b, in a numberof experiments, the p53 immunoreactivity in the cytosol of estradiol-treated cells was shared by more than one band of variable mass, butsmaller than 53 kDa. Although this result was not constant, it wasrepeated several times in conditions of strict proteolysis control duringcell disruption and fractionation (see “Materials and Methods”). Thisevidence suggested that the observed increase in p53 protein in thecytosol after estradiol treatment was associated with its proteolytic deg-
radation. To evaluate whether the reduced concentration of nuclear p53was correlated to its functional inactivation, Western blot analysis wasperformed on p21 (WAF/CIP1), a protein whose expression is known tobe induced by p53. In an untreated cell, the presence of p53 in the nucleuswas associated with p21 expression, as expected in the presence of afunctional p53 gene product. In the nuclear extract of cells treated withestradiol for 48 h, the amount of p21 was markedly reduced comparedwith untreated cells or cells treated in the presence of antihormone (Fig.2C). This evidence indicated that the estradiol treatment specificallyreduced the expression of this p53-controlled gene. There is no evidenceto suppose that this was a direct effect. It could be consequent to thepostulated p53 inactivation by estradiol. This inactivation could be me-diated by the estradiol-induced expression of themdm2gene, observableby Western blot analysis of the mdm2 protein (data not presented) orNorthern blot analysis of the mRNA (18).
To determine whether the absence of p53 protein in the nucleus wascorrelated with a reduced response to DNA damage, cell survival wasassessed after oligonucleotide transfection simulating DNA damage.Short single-stranded DNA are able to induce p53 binding to theGADD45sitein vitro (19), and DNA strand breaks trigger p53 activationin the cell (20). EMSA of a probe containing theGADD45p53-bindingsite showed that the whole-cell extract from MCF-7 cells transfected with
Fig. 1. Estradiol effect on in a colony formation assay. MCF-7 cells cultured inhormone-free medium were plated on methylcellulose and incubated for 7 days in thepresence of vehicle alone (A); 10 nM estradiol (B), and 10 nM estradiol and 1mM
ICI182,780 (C). Thebar corresponds to 50mm.
2595
ESTROGEN-INDUCED INACTIVATION OF p53
Research. on April 14, 2017. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
double-stranded oligomers containing a 59 overhang was able to retardmore probe than an extract from mock-transfected cells (Fig. 4A). In vitroaddition of the oligonucleotides to the extract from mock-transfected cellswas also able to increase the intensity of the retarded band. This evidenceindicated that these oligonucleotides (either transfected into living cells oradded to the cellular extract) were able to induce p53 activation in MCF-7cells. The oligonucleotide transfection induced growth arrest of trans-fected cultures (Fig. 4B). Two days after transfection of oligomers with a59 overhang, only 45% of the cells cultured in the absence of hormonesurvived, whereas a 2-day treatment with a physiological concentration ofhormone prevented the p53-mediated growth arrest. This evidencestrongly supports the hypothesis that estradiol-induced p53 inactivationprevented the DNA damage-induced growth arrest mediated by p53protein.
Discussion
MCF-7 is a breast cancer cell line extensively studied for theexpression of a functional estrogen receptor (13). Growth of thesecells is stimulated by estradiol and by other factors such as insulin.The mechanism underlying the mitogenic effect of estradiol has notbeen elucidated definitively. The hypothesis that the mitogenic effect
was mediated solely by hormone-regulated autocrine growth factorsecretion has been contradicted by the evidence that estrogens candirectly promote G1 progression through direct activation of cell cyclegenes or through theras-mitogen-activated protein kinase pathway.This evidence suggests that, as the result of hormone-receptor inter-action, more than one intracellular pathway becomes activated orinactivated for the proliferating response. Inactivation of p53 negativecontrol of the G1-S transition by estradiol could greatly contribute tothe hormonal control of cell proliferation. Expression of themdm2gene correlates with the estrogen receptor status of many humanbreast carcinoma cell lines, andmdm2mRNA is accumulated afterestradiol treatment of MCF-7 cell line (18). The mdm2 gene productinhibits p53 function by direct binding, hindering the transactivationdomain and targeting p53 protein for degradation. mdm2 proteincontains the nuclear export signal for the complex that is eventuallydegraded by the proteasome. It is conceivable that the mdm2 geneproduct mediated the observed effect, although p53 protein has aleucine-rich nuclear export signal necessary and sufficient for nuclearexport (21). The biochemical and morphological evidences presentedin this report suggest that estradiol was able to induce a redistributionof p53 from the nucleus to the cytoplasm. This was correlated to areduction of p53 activity, as evident from the reduced expression ofp21 in the estrogen-treated cells and the presence of immunoreactivefragments of p53 protein in the cytosol. The estradiol effect wasproduced in cells grown in serum-free medium, and it was mediated
Fig. 2. Western blot analysis of MCF-7 cell extracts.A, Western blot analysis withmonoclonal antibody Ab-6 to p53 protein (53) in cytosol or nuclear extract, as indicatedat thebottomof the panels (25mg protein/lane), from MCF-7 cells treated with vehiclealone (Lane a), 10 nM estradiol (Lane b), or 10 nM estradiol and 1mM ICI182,780 (Lanec) for 48 h, as indicated in “Materials and Methods.” SDS-PAGE was performed on a11-% polyacrylamide gel.B, Western blot analysis with monoclonal antibody Ab-6 to p53protein (53;Lanes aand b) or with mouse IgG (Lanes cand d) of cytosol (25mgprotein/lane) from MCF-7 cells treated with vehicle alone (Lanes aand c) or 10 nMestradiol (Lanes bandd) for 48 h, as indicated in “Materials and Methods.” SDS-PAGEwas performed on a 10% polyacrylamide gel.C, Western blot analysis with monoclonalantibody Ab-1 to p21-WAF-1 protein (21) of nuclear extract (25mg protein/lane), fromMCF-7 cells treated with vehicle alone (Lane a); 10 nM estradiol (Lane b), or 10 nMestradiol and 1mM ICI182,780 (Lane c) for 48 h, as indicated in “Materials and Methods.”SDS-PAGE was performed on an 11% polyacrylamide gel.
Fig. 3. Immunofluorescence detection of p53 protein in MCF-7 cells. Cells were grownon coverslips in hormone-free medium for 2 days, then with vehicle alone (A), with 10 nM
estradiol (B), with 10 nM estradiol and 1mm ICI182,780 (C), or with 10mM insulin (D)for an additional 48 h. Coverslips were then processed and mounted as indicated in“Materials and Methods.” Thebar corresponds to 10mm.
2596
ESTROGEN-INDUCED INACTIVATION OF p53
Research. on April 14, 2017. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from

by estrogen receptor, because the estrogen antagonist ICI182,780prevented it.
Nuclear exclusion is one of the mechanisms of p53 inactivation inbreast cancer, and cytoplasm staining for p53 of neoplastic cellsstrongly correlated with the presence of a wild-type, intact p53 proteinby sequence analysis (22). In addition, cytoplasm sequestration of p53is visible in normal lactating breast tissue (22). Our observations,therefore, strengthen the hypothesis that p53 inactivation by nuclearexclusion is a necessary step for estrogen-induced cell proliferation inthe presence of an active, wild-typep53 gene. This p53 inactivationhas a permissive role for the observed induction of cyclin-dependentkinase activity and the consequent phosphorylation of retinoblastomaprotein induced by estradiol and inhibited by coadministration ofICI182,780 in the MCF-7 cell line (23). Apparently contradictoryevidence that in the T47D breast cancer cell line estradiol is able toincrease the expression of p53 protein (24) further confirms ourhypothesis that nuclear exclusion is a nonmutational mechanism forabrogating the inhibitory function of wild-type p53 protein. This cellline, in fact, expresses an inactive p53 protein harboring a pointmutation at codon 194 (14) and responds to estradiol treatment withincreased proliferation and retinoblastoma protein phosphorylationdespite the induced expression of p53 protein (25).
These results, indicating that estradiol treatment prevented thep53-mediated growth arrest induced by the presence of intracellularDNA fragments, have allowed further speculation. Through inactiva-tion of the tumor suppressor function of p53, estradiol could play anadditional role in the multistep process of tumorigenesis of targettissues in which thep53 gene is not altered by genetic mutations.Absence of an active p53 protein by nuclear exclusion could, in fact,contribute to the genetic instability of target cells in which estradiol is
able to induce a proliferative response. A hormone-independent phe-notype usually emerges during the process of tumor progression, bothin human breast cancer and in mouse mammary tumor models, despitethe presence of a hormonal environment conferring a growth advan-tage to the hormone-dependent cells.
References
1. Maltzman, W., and Czyzyk, L. UV irradiation stimulates levels of p53 cellular tumorantigen in nontransformed mouse cells. Mol. Cell. Biol.,4: 1689–1694, 1984.
2. Woo, R. A., McLure, K. G., Lees-Miller, S. P., Rancourt, D. E., and Lee, P. W.DNA-dependent protein kinase acts upstream of p53 in response to DNA damage.Nature (Lond.),394: 700–704, 1998.
3. Waterman, M. J., Stavridi, E. S., Waterman, J. L., and Halazonetis, T. D. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3proteins. Nat. Genet.,19: 175–178 1998.
4. Canman, C. E., Lim, D. S., Cimprich, K. A., Taya, Y., Tamai, K., Sakaguchi, K.,Appella, E., Kastan, M. B., and Siliciano, J. D. Activation of the ATM kinase byionizing radiation and phosphorylation of p53. Science (Washington DC),281:1677–1679, 1998.
5. el-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R., Trent, J. M.,Lin, D., Mercer, W. E., Kinzler, K. W., and Vogelstein, B. WAF1, a potentialmediator of p53 tumor suppression. Cell,75: 817–825, 1993.
6. Kastan, M. B., Zhan, Q., el-Deiry, W. S., Carrier, F., Jacks, T., Walsh, W. V.,Plunkett, B. S., Vogelstein, B., and Fornace, A. J., Jr. A mammalian cell cyclecheckpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia.Cell, 71: 587–597, 1992.
7. Miyashita, T., and Reed, J. C. Tumor suppressor p53 is a direct transcriptionalactivator of the humanbax gene. Cell,80: 293–299, 1995.
8. Wu, X., Bayle, J. H., Olson, D., and Levine, A. J. The p53-mdm-2 autoregulatoryfeedback loop. Genes Dev.,7: 1126–1132, 1993.
9. Barak, Y., Juven, T., Haffner, R., and Oren, M. mdm2 expression is induced by wildtype p53 activity. EMBO J.,12: 461–468, 1993.
10. Momand, J., Zambetti, G. P., Olson, D. C., George, D., and Levine, A. J. The mdm-2oncogene product forms a complex with the p53 protein and inhibits p53-mediatedtransactivation. Cell,69: 1237–1345, 1992.
11. Freedman, D. A., and Levine, A. J. Nuclear export is required for degradation ofendogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol.,18:7288–7293, 1998.
12. Hollstein, M., Sidransky, D., Vogelstein, B., and Harris, C. C. p53 mutations inhuman cancers. Science (Washington DC),253: 49–53, 1991.
13. Lippman, M., Bolan, G., and Huff, K. The effects of estrogens and antiestrogens onhormone-responsive human breast cancer in long-term tissue culture. Cancer Res.,36: 4595–4601, 1976.
14. Runnebaum, I. B., Nagarajan, M., Bowman, M., Soto, D., and Sukumar, S. Mutationsin p53 as potential molecular markers for human breast cancer. Proc. Natl. Acad. Sci.USA, 88: 10657–10661, 1991.
15. David-Pfeuty, T., Chakrani, F., Ory, K., and Nouvian-Dooghe, Y. Cell cycle-depen-dent regulation of nuclear p53 traffic occurs in one subclass of human tumor cells andin untransformed cells. Cell. Growth Differ.,7: 1211–1225, 1996.
16. McKeehan, W. L., McKeehan, K. A., Hammond, S. L., and Ham, R. G. Improvedmedium for clonal growth of human diploid fibroblasts at low concentrations ofserum protein. In Vitro,13: 399–416, 1977.
17. Medici, N., Nigro, V., Abbondanza, C., Moncharmont, B., Molinari, A. M., and Puca,G. A. In vitro binding of the purified hormone-binding subunit of the estrogenreceptor to oligonucleotides containing natural or modified sequences of an estrogenresponsive element. Mol. Endocrinol.,5: 555–563, 1991.
18. Sheikh, M. S., Shao, Z. M., Hussain, A., and Fontana, J. A. The p53-binding proteinMDM2 gene is differentially expressed in human breast carcinoma. Cancer Res.,53:3226–3228, 1993.
19. Jayaraman, J., and Prives, C. Activation of p53 sequence-specific DNA binding byshort single strands of DNA requires the p53 C-terminus. Cell,81: 1021–1029, 1995.
20. Nelson, W. G., and Kastan, M. B. DNA strand breaks: the DNA template alterationsthat trigger p53-dependent DNA damage response pathways. Mol. Cell. Biol.,14:1815–1823, 1994.
21. Stommel, J. M., Marchenko, N. D., Jimenez, G. S. Moll, U. M., Hope, T., J., andWahl, G. M. A leucine-rich nuclear export signal in the p53 tetramerization domain:regulation of subcellular localization and p53 activity by NES masking. EMBO J.,18:1660–1672, 1999.
22. Moll, U. M., Riou, G., and Levine, A. J. Two distinct mechanisms alter p53 in breastcancer: mutation and nuclear exclusion. Proc. Natl. Acad. Sci. USA,89: 7262–7266,1992.
23. Foster, J. S., and Wimalasena, J. Estrogen regulates activity of cyclin-dependentkinases and retinoblastoma protein phosphorylation in breast cancer cells. Mol.Endocrinol.,10: 488–498, 1996.
24. Hurd, C., Khattree, N., Alban, P., Nag, K., Jhanwar, S. C., Dinda, S., and Moudgil,V. K. Hormonal regulation of the p53 tumor suppressor protein in T47D human breastcarcinoma cell line. J. Biol. Chem.,270: 28507–28510, 1995.
25. Hurd, C., Khattree, N., Dinda, S., Alban, P., and Moudgil, V. K. Regulation of tumorsuppressor proteins, p53 and retinoblastoma, by estrogen and antiestrogens in breastcancer cells. Oncogene,15: 991–995, 1997.
Fig. 4. Estradiol effect on MCF-7 cell survival after simulated DNA damage.A, EMSAof the probe containing theGADD45p53-binding site incubated with MCF-7 cell extracts(5 mg of protein). Cells were mock-transfected with DOTAP (Lanes aand b) or trans-fected with DOTAP premixed with oligonucleotides (Lanes candd) and incubated for anadditional 48 h in the same medium containing 10% charcoal-treated FCS. The probe wasincubated with cell extract in the absence (Lanes aandc) or presence (Lanes bandd) of20 pmol of double-stranded oligomers containing a 59overhang. Thearrow indicatesmigration of uncomplexed probe.B, cells were grown in 6-well microtiter plates to 60%confluency and incubated for 36 h in hormone-free medium before the addition of 10 nM
estradiol or vehicle. After 48 h, cells were mock-transfected with DOTAP (hatchedcolumns) or transfected with DOTAP premixed with oligonucleotides (gray columns) andincubated for an additional 48 h in the same medium containing 10% charcoal-treated FCSin the absence or in the presence of hormone. Cells were stained with crystal violet, andthe absorbance was measured at 595 nm.Bars, SD.
2597
ESTROGEN-INDUCED INACTIVATION OF p53
Research. on April 14, 2017. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from

2000;60:2594-2597. Cancer Res Anna Maria Molinari, Paola Bontempo, Ettore M. Schiavone, et al. RedistributionEstradiol Induces Functional Inactivation of p53 by Intracellular
Updated version
http://cancerres.aacrjournals.org/content/60/10/2594
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/60/10/2594.full.html#ref-list-1
This article cites 25 articles, 13 of which you can access for free at:
Citing articles
/content/60/10/2594.full.html#related-urls
This article has been cited by 10 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on April 14, 2017. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from