european infant neuroblastoma study* - chped.it · 2 1 premesse 1.1 età ed estensione della...
TRANSCRIPT

Versione definitiva al 17 dicembre 1999
Approvato dal Comitato Etico dell'Istituto Giannina Gaslini Alla stesura di questo protocollo hanno partecipato rappresentanti di: Austria, Belgio, Danimarca, Francia (Societé Francaise d’Oncologie Pédiatrique, SFOP), Italia (Associazione Italiana Ematologia-Oncologia Pediatrica, AIEOP), Norvegia, Regno Unito (United Kingdom Children's Cancer Study Group, UKCCSG) Spagna (Grupo de Neuroblastoma, SEOP), Svezia, Svizzera. CONTRIBUTI PER LE VARIE COMPETENZE: Oncologia: Jonas Abrahamson, Maja Nenadov-Beck, Bruno De Bernardi, Peppy Brock, Adela Canete, Annabel Foot, Mary Gerrard, Ruth Ladenstein, Genevieve Laureys, Ingeborg Mathieson, Jean Michon, Andrew Pearson, Dominique Plantaz, Catherine Rechnitzer, Hervé Rubie. Chirurgia: Giovanni Cecchetto, Claudio Granata, Maurizio Guglielmi, Pierre Helardot, Keith Holmes, Antonino Rizzo, Roly Squire. Anatomia-patologica: Emanuele D’Amore, Claudio Gambini, Samuel Navarro, Michel Peuchmaur, Borghild Roald, Dick Variend. Biologia: Peter Ambros, Gian Paolo Tonini. Radioterapia: Salvina Barra, Ann Barrett, Mark Gaze. Statistica: Paolo Bruzzi, Yann De Rycke, John Imeson, Veronique Mosseri. Gestione del protocollo in Italia: Andrea Di Cataldo, coordinatore. Claudio Gambini, anatomia patologica. Claudio Granata, chirurgia. Gian Michele Magnano, radiologia. Salvina Barra, radioterapia. Giampiero Villavecchia, medicina nucleare. Gian Paolo Tonini, biologia. Luca Boni, statistica. *Traduzione italiana a cura di Maria Serena Lo Piccolo e Bruno De Bernardi
EUROPEAN INFANT NEUROBLASTOMA STUDY*

1
INDICE DEL CONTENUTO
1 PREMESSE ................................................................................................................................................. 2
2 L'ESPERIENZA ITALIANA..................................................................................................................... 5
3 OBIETTIVI DELLO STUDIO .................................................................................................................. 6
4 SCHEMI DI TRATTAMENTO................................................................................................................. 8
5 ELEGGIBILITA’ ALLO STUDIO ......................................................................................................... 11
6 INDAGINI PRE-TRATTAMENTO........................................................................................................ 15
7. LINEE GUIDA ANATOMO-PATOLOGICHE..................................................................................... 17
8 LINEE GUIDA BIOLOGICHE............................................................................................................... 28
9 CHIRURGIA............................................................................................................................................. 41
10 TRATTAMENTO: TUMORI SENZA AMPLIFICAZIONE DI MYCN.............................................. 43
11 TRATTAMENTO: TUMORI CON AMPLIFICAZIONE DI MYCN.................................................. 52
12 LEUCOAFERESI ..................................................................................................................................... 53
13 CHEMIOTERAPIA.................................................................................................................................. 54
14 DIAGNOSI E TERAPIA DELLA VOD.................................................................................................. 58
15 LINEE GUIDA RADIOTERAPICHE .................................................................................................... 60
16 VALUTAZIONE DELL'ESTENSIONE DI MALATTIA E DELLA RISPOSTA .............................. 62
17 MODIFICAZIONI DELLA TERAPIA IN RAPPORTO ALLA TOSSICITA' .................................. 63
18 VALUTAZIONE DEL PAZIENTE DURANTE IL TRATTAMENTO............................................... 64
19 VALUTAZIONE DOPO LA FINE DELLA TERAPIA ........................................................................ 65
20 CONSIDERAZIONI STATISTICHE ..................................................................................................... 66
21 BIBLIOGRAFIA....................................................................................................................................... 78

2
1 PREMESSE 1.1 Età ed estensione della malattia alla diagnosi costituiscono i fattori
prognostici più importanti del neuroblastoma1. Tuttavia alcune caratteristiche biologiche, come l'amplificazione di MYCN, il livello plasmatico di LDH ed il contenuto nucleare di DNA si sono dimostrati prognosticamente influenti2.
1.2 Dal punto di vista clinico si osservano due popolazioni distinte: una,
costituita da pazienti che per lo più non sopravvivono alla malattia, nonostante una terapia aggressiva; l'altra, cui appartengono i casi con malattia localizzata operabile e quelli di stadio 4s, che possono guarire grazie a un trattamento non aggressivo o addirittura senza terapia3-4. Evitare la somministrazione di chemioterapia o di radioterapia quando non sono necessarie è un obiettivo importante, specie per i bambini più piccoli, maggiormente suscettibili di sviluppare effetti tossici a distanza.
1.3 La SFOP ha descritto 316 casi con neuroblastoma localizzato5, di cui 152 sotto l'anno di età. L’ amplificazione di MYCN è risultata il fattore prognostico più importante: dei 6 infant con tale alterazione (su 115 testati), uno è deceduto dopo l’intervento, 4 sono recidivati e poi deceduti per progressione della neoplasia.
1.4 Una analisi di Evans6 su 119 casi di malattia localizzata, distinti in 1 gruppo di 21 pazienti trattati con chemioterapia, ed un altro di 98 non trattati, riporta rispettivamente nei due gruppi una EFS di 79% e 89%, ed una OS di 85% e 98%.
1.5 Un precedente studio di Castleberry7 aveva concluso che gli infant con infiltrazione dei linfonodi regionali hanno buona prognosi dopo "limitata" chemioterapia postoperatoria.
1.6 Si segnala ancora l'esperienza monocentrica di Kushner8 che ha descritto un gruppo di 31 pazienti con neuroblastoma di stadio 1, 2, 3, e 4s senza amplificazione di MYCN. Sei pazienti hanno presentato progressione di malattia: di essi solo uno ha ricevuto chemioterapia (per la presenza di delezione 1p), mentre gli altri hanno raggiunto la remissione spontaneamente o con il solo intervento chirurgico.
1.7 Infine, Yamamoto9 ha recentemente descritto un gruppo di infant con neuroblastoma diagnosticato grazie allo screening urinario, e regredito senza alcun trattamento.

3
1.8 Nella maggior parte degli infant con neuroblastoma stadio 4s, la neoplasia
tende alla regressione spontanea. Alcuni casi tuttavia, per lo più diagnosticati nei primi 2 mesi di vita, sviluppano sintomi gravi per i quali è indicata la somministrazione di chemioterapia.
1.9 Per gli infant di stadio 4s, Hsu10 ha recentemente proposto uno schema per la
valutazione dei sintomi, allo scopo di individuare i casi per i quali è indicato il trattamento chemioterapico.
1.10 Ancora in questo gruppo di pazienti, per quanto concerne il ruolo
dell'asportazione chirurgica del "tumore primitivo", Martinez11 la considera vantaggiosa, mentre un gruppo italiano12 ed uno studio del POG13 non trovano differenza in termini di outcome tra i pazienti operati o meno.
1.11 Il neuroblastoma negli infant ha una prognosi migliore rispetto a quella dei
bambini più grandi, indipendentemente dal trattamento eseguito. Tuttavia alcuni di essi possono sviluppare progressioni o recidive. La possibilità di identificare tali pazienti, evidenziando peculiari caratteristiche clinico-biologiche, consentirebbe di disegnare un trattamento specifico che potrebbe migliorarne la prognosi.
1.12 L'analisi dei dati (MP Gerrard, non pubblicati) riguardanti gli infant di stadio 4
e 4s, diagnosticati nel periodo 1992-1996 in Austria, Belgio, Francia, Germania, Italia, Nazioni scandinave, Regno Unito, Spagna e Svizzera (285 casi: 97 di stadio 4 e 188 di stadio 4s), indica che l'amplificazione di MYCN e la delezione di 1p sono associate a peggiore prognosi. Infatti, mentre 146 bambini senza amplificazione di MYCN hanno avuto una sopravvivenza a 2 anni dell'88%, nei 27 con amplificazione la sopravvivenza è stata 29%. In particolare, 5 casi su 6 di stadio 4s sono deceduti. La delezione di 1p è stata studiata in 19 pazienti di stadio 4: i 10 senza delezione hanno avuto una OS di 88%, contro 50% dei 9 con delezione. Dei 24 casi di stadio 4s studiati per 1p, 17 non avevano delezione e sono tutti sopravissuti, mentre dei 7 con delezione solo il 24% sopravvive. Il dato trova conferma in un recente studio del POG 13.
1.13 Dati della SFOP relativi alla terapia ad alte dosi suggeriscono che i regimi
contenenti Busulfano sono associati a migliore prognosi, nonostante il rischio di malattia veno-occlusiva (VOD). Nello studio di Hartmann14 11 pazienti (di età inferiore a 1 anno al momento della diagnosi) hanno ricevuto Busulfano
(150 mg/m2/die x 4 giorni) in associazione a Melphalan. La VOD è stata osservata in 8 di essi, compresi 5 di età inferiore a 1 anno al momento della terapia ad alte dosi. Sette pazienti sopravvivono ad oltre 2 anni dalle alte dosi, ed è stato registrato un solo decesso per tossicità. Gli autori suggeriscono di ridurre la dose di Busulfano a 120 mg/m2/die.

4
1.14 I criteri di stadiazione INSS 15,16 sono oggi largamente usati. E' tuttavia difficile
da accettare che un infant venga definito di stadio 4 solo in quanto il tumore primitivo supera la linea mediana. Dati della SFOP17 e di Paul18 suggeriscono che gli infant con tale condizione o con infiltrazione midollare, ma senza metastasi scheletriche, hanno una prognosi migliore rispetto al vero e proprio stadio 4 con metastasi scheletriche.
1.15 Anche l'analisi dei dati europei del periodo 1992-1996 relativi agli stadi 4 e 4s
indica che i pazienti di stadio 4 senza interessamento scheletrico e senza amplificazione di MYCN hanno una prognosi eccellente (solo un decesso su 20 casi).
1.16 Questo studio prospettico europeo intende registrare sistematicamente gli
infant con neuroblastoma, basando il trattamento sullo stadio e sullo stato di MYCN.

5
2 L'ESPERIENZA ITALIANA 2.1 Neuroblastoma localizzato (Stadi 1-3). Tra il 1992 e il 1995 sono stati
osservati 72 infants con neuroblastoma localizzato. Nello stadio 1-2 il trattamento è stato esclusivamente chirurgico. Nello stadio 3 sono stati somministrati 6 cicli di CADO, seguiti, quando possibile, dall'intervento. Stadio 1, 28 casi. 2 recidive locali, 1 locale e midollare. Nessun decesso. Stadio 2, 20 casi. 9 di stadio 2A, nessun evento. Degli 11 di stadio 2B, 1 decesso per complicanze chirurgiche, mentre l'unico caso con recidiva locale sopravvive. Stadio 3, 24 casi. 3 decessi, 1 per complicanze chirurgiche e 2 per progressione di malattia. 58 pazienti sono stati valutati per amplificazione di MYCN: 1 paziente dei 3 che ne presentavano 3 copie è deceduto, come anche l'unico con 10 copie dell'oncogene. La sopravvivenza complessiva a 3 anni è stata 94% (stadio 1, 100%; stadio 2, 95%; stadio 3, 87%). La sopravvivenza libera da eventi a 3 anni è stata 89% (stadio 1-2, 89%; stadio 3, 87%).
2.2 Neuroblastoma metastatico (Stadio 4). Tra il 1992 e il 1995 sono stati osservati 16 infants di stadio 4. Quattro pazienti sono deceduti, 3 per progressione di malattia e 1 per tossicità da chemioterapia. Un paziente sopravvive dopo recidiva locale. Dei 7 pazienti con metastasi ossee, 3 sono deceduti, mentre dei 9 senza coinvolgimento osseo solo 1 è deceduto. La sopravvivenza complessiva a 3 anni è stata 73%. La sopravvivenza libera da eventi a 3 anni è stata 67%.
2.3 Neuroblastoma metastatico (Stadio 4s). Tra il 1992 e il 1995 sono stati osservati 35 infants di stadio 4s. Per essi non era previsto alcun trattamento se non in caso di sintomi gravi comportanti il rischio di decesso. Sei pazienti sono deceduti, 5 per progressione epatica di malattia e 1 per complicanze chirurgiche. Gli altri 6 che hanno presentato progressione di malattia, sopravvivono dopo trattamento. Non è stato trovato, tra i 21 pazienti studiati per MYCN, nessun caso con amplifcazione. La sopravvivenza complessiva a 3 anni è stata 84%. La sopravvivenza libera da eventi a 3 anni è stata 68%.

6
3 OBIETTIVI DELLO STUDIO 3.1 OBIETTIVI PRIMARI 3.1.1 Determinare l’outcome (sopravvivenza e morbidità) per gli infant con malattia
localizzata ma non resecabile, senza amplificazione di MYCN, utilizzando un regime chemioterapico di moderata intensità ed un approccio più conservativo possibile (Trial 99.1).
L’obiettivo è una sopravvivenza a 3 anni > 90%. 3.1.2 Confermare che la prognosi degli infant di stadio 4s, senza amplificazione di
MYCN, è favorevole, anche senza ricorso a terapie antitumorali. Queste vengono riservate ai casi che sviluppano progressione di malattia tale da compromettere la vita del paziente (Trial 99.2). Questo approccio sarà utilizzato anche per gli infant con voluminoso tumore primitivo che supera la linea mediana, e metastasi che non coinvolgono lo scheletro, la pleura, il polmone od il SNC.
L’obiettivo è una sopravvivenza a 2 anni > 85%. 3.1.3 Confermare che i pazienti di età < 1 anno, di stadio 4 per coinvolgimento
osseo, pleurico, polmonare o del SNC, ma senza amplificazione di MYCN, non richiedono un consolidamento con chemioterapia intensiva ad alte dosi (Trial 99.3).
L’obiettivo è una sopravvivenza a 2 anni > 70%. 3.1.4 Determinare la sopravvivenza degli infant con tumore di stadio superiore ad 1
ed amplificazione di MYCN, trattati con chemioterapia intensiva, che includa un ciclo di terapia ad alte dosi con infusione di cellule staminali autologhe (Trial 99.4).
L’obiettivo è una sopravvivenza a 2 anni non inferiore al 50% 3.2 OBIETTIVI SECONDARI 3.2.1 Adottare per tutti i bambini con diagnosi confermata di neuroblastoma, posta
prima dell’età di 12 mesi, un trattamento uniforme sulla base di stadio e stato di MYCN.
3.2.2 Definire il comportamento del neuroblastoma negli infant utilizzando una
stadiazione uniforme e la raccolta dei dati biologici. Verranno utilizzati i criteri INSS con una eccezione: l'inclusione nello stadio 4s verrà estesa ai casi con neoplasia primitiva che supera la linea mediana, senza metastasi ossee, riconosciute con radiografia standard (o TC del cranio).

7
3.2.3 Definire nuovi criteri prognostici utili per una migliore stratificazione terapeutica (in particolare la delezione del cromosoma 1p e il contenuto di DNA).
8
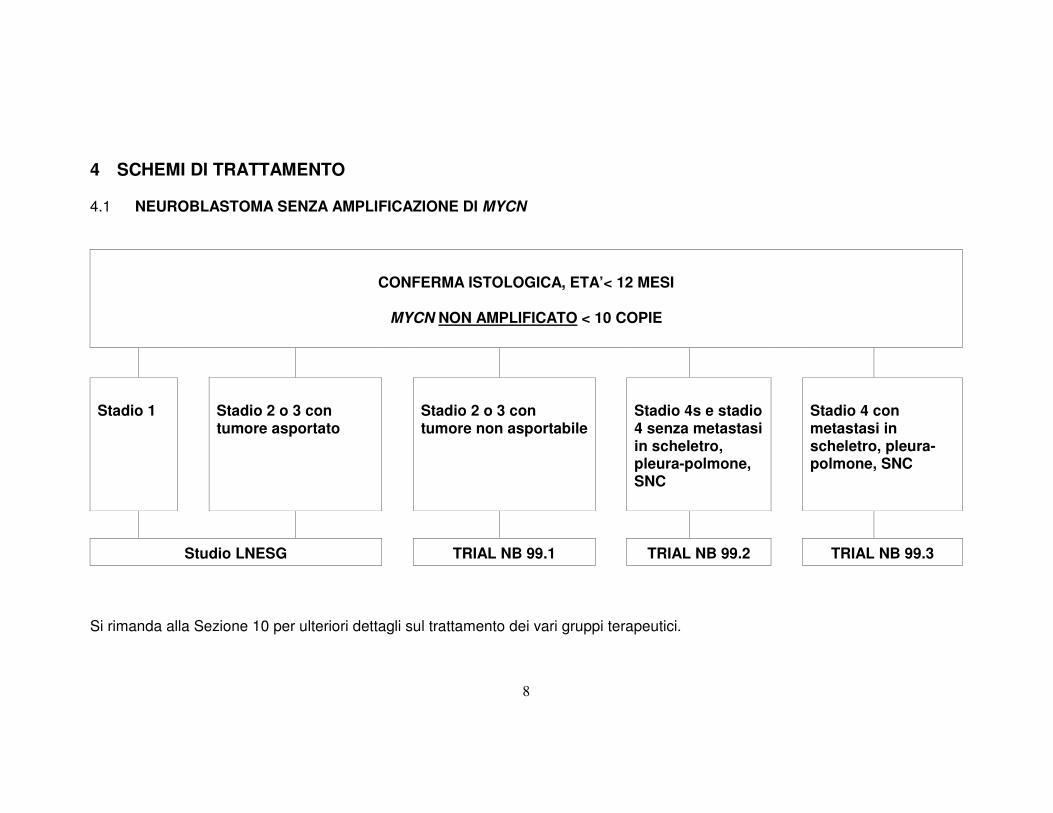
4 SCHEMI DI TRATTAMENTO 4.1 NEUROBLASTOMA SENZA AMPLIFICAZIONE DI MYCN
CONFERMA ISTOLOGICA, ETA’< 12 MESI
MYCN NON AMPLIFICATO < 10 COPIE
Stadio 1
Stadio 2 o 3 con tumore asportato
Stadio 2 o 3 con tumore non asportabile
Stadio 4s e stadio 4 senza metastasi in scheletro, pleura-polmone, SNC
Stadio 4 con metastasi in scheletro, pleura-polmone, SNC
Studio LNESG TRIAL NB 99.1 TRIAL NB 99.2 TRIAL NB 99.3
Si rimanda alla Sezione 10 per ulteriori dettagli sul trattamento dei vari gruppi terapeutici.

9
4.2 NEUROBLASTOMA CON AMPLIFICAZIONE DI MYCN
CONFERMA ISTOLOGICA, ETA’ < 12 MESI
AMPLIFICAZIONE DI MYCN > 10 COPIE
Stadio 1
Stadio 2
Stadio 3
Stadio 4s
Stadio 4
Studio LNESG
TRIAL NB 99.4
Trattamento iniziale con Carbo-VP, seguito da CADO Terapia ad alte dosi con reinfusione di cellule staminali
Radioterapia sul tumore primitivo
Si rimanda alla Sezione 11 per ulteriori dettagli sul trattamento.

10
4.3 TRATTAMENTO IN RELAZIONE A STADIO E BIOLOGIA
La scelta della terapia appropriata viene posta in base a stadio e stato di MYCN, e, per quanto riguarda lo stadio 4, all’eventuale coinvolgimento osseo. In particolare, la scelta relativa allo stadio 4 discende dalla difficoltà di distinguerlo nettamente dallo stadio 4s. In questo studio sono inclusi nello stadio 4s i casi con tumore primitivo inoperabile. La ripartizione dei pazienti nei diversi Trial in base a stadio e MYCN è mostrata nella tabella sottostante. TRIAL
STADIO
99.1 99.2 99.3 99.4 STUDIO LNESG
1 MYCN <10 copie
1 MYCN > 10 copie
2 asportato MYCN <10 copie
2 non asportato MYCN <10 copie
2 MYCN > 10 copie
3 asportato MYCN <10 copie
3 non asportato MYCN <10 copie
3 MYCN > 10 copie
4s MYCN <10 copie
4s MYCN > 10 copie
4 MYCN <10 copie (senza metastasi ossee, pleura-polmone,SNC)
4 MYCN <10 copie (con metastasi ossee, pleura-polmone,SNC)
4 MYCN > 10 copie
I pazienti di qualsiasi stadio con stato di MYCN non noto non vengono registrati in alcun trial, ma saranno seguiti ed analizzati separatamente.

11
5 ELEGGIBILITA’ ALLO STUDIO 5.1 CRITERI DI INCLUSIONE 5.1.1 Sono eleggibili tutti gli infant con nuova diagnosi di neuroblastoma o
ganglioneuroblastoma (secondo i criteri INSS, Addendum I) di età inferiore a 12 mesi al momento della diagnosi.
Data della diagnosi: corrisponde alla data della diagnosi istologica. La definizione comprende anche gli infant diagnosticati attraverso lo “screening”. Data di eleggibilità: corrisponde alla data in cui tutti i criteri di inclusione sono stati controllati dal Centro Coordinatore. Inclusione in un Trial: richiede che tutti i criteri di eleggibilità (compresi i dati biologici) siano registrati al Centro Raccolta Dati entro 6 settimane dalla diagnosi.
5.1.2 Il Trial LNESG 95.01 (solo chirurgia per malattia localizzata) sarà chiuso
contestualmente all'attivazione del presente protocollo. Tuttavia gli Infant con neoplasia localizzata operata, continueranno ad essere registrati in attesa dell'attivazione di un nuovo protocollo ad hoc. A questo fanno eccezione i casi di stadio 2 e 3 con amplificazione di MYCN, eleggibili per il trial 99.4.
5.1.3 Gli infant registrati prima dell’età di 12 mesi, che ricevono terapia dopo il
compimento del 12° mese, rimangono eleggibili per lo studio. 5.1.4 Consenso informato dei genitori o del tutore. 5.2 CRITERI DI ELEGGIBILITA’ PER I SINGOLI TRIAL Poichè lo stato del MYCN è essenziale per determinare l’eleggibilità per un
trial, il risultato deve essere disponibile al momento della inclusione nel trial stesso. L’esame deve essere effettuato in uno dei laboratori di riferimento del Trial (per l'Italia dott. Tonini - Genova CBA).
Per una corretta stadiazione e la conseguente assegnazione ad un Trial è essenziale seguire le linee-guida indicate in Sezione 6, in particolare per quanto concerne lo studio dell’infiltrazione midollare e scheletrica.
Tutti i criteri di eleggibiltà devono essere controllati ed il paziente registrato non oltre 6 settimane dopo la diagnosi. E’ prevedibile che alcuni infant saranno avviati al trattamento prima della verifica dell’eleggibilità risultando poi non eleggibili per quel particolare Trial. E’ il caso di pazienti di stadio 2 o 3 con tumore non asportabile, trattati inizialmente con CO ed in seguito riconosciuti avere MYCN amplificato. Essi verranno trattati secondo il protocollo del trial 99.4.

12
5.3 Nota bene: poichè lo studio intende acquisire informazioni sul possibile ruolo prognostico della delezione di 1p e del contenuto di DNA, è necessario che i risultati di tali indagini siano già disponibili (o in corso) al momento della registrazione.
5.4 PAZIENTI IN "STUDIO" (inclusi quelli non eleggibili per i Trial)
Gli infant non eleggibili per alcun Trial a causa di problemi inerenti la determinazione del MYCN, o non registrati in un Trial entro 6 settimane dalla diagnosi, saranno seguiti raccogliendo un numero limitato di dati da analizzarsi separatamente.

13
5.5 TRIAL NB 99.1 Criteri di inclusione: • Tumore localizzato ma inoperabile (Addendum II, linee guida
chirurgiche), confermato istologicamente. • Nei casi in cui una biopsia a cielo aperto sia considerata troppo
rischiosa in ragione della sede del tumore o delle condizioni del bambino, è consentita la diagnosi citologica, purchè si sia ottenuto materiale adeguato per identificare la natura tumorale delle cellule ed il loro stato di MYCN.
• Assenza di amplificazione di MYCN. Criteri di esclusione: • Infiltrazione tumorale midollare. • Scintigrafia con MIBG (o Tecnezio) positiva a livello scheletrico,
oppure evidenza radiologica di interessamento osseo. • Ecografia patologica a livello epatico. • Amplificazione di MYCN. 5.6 TRIAL NB 99.2 Criteri di inclusione • Tumore, confermato istologicamente, di qualsiasi stadio localizzato,
con metastasi limitate a cute, midollo, linfonodi o fegato, senza coinvolgimento di scheletro (definito radiograficamente o con TC cranica), di pleura-polmone o SNC.
• Assenza di amplificazione di MYCN. Criteri di esclusione: • Lesioni scheletriche evidenziate radiologicamente o con TC cranica . • Amplificazione di MYCN. 5.7 TRIAL NB 99.3 Criteri di inclusione • Tumore, confermato istologicamente, con metastasi scheletriche
identificabili attraverso radiografia standard (o TC cranica), o coinvolgimento pleurico, polmonare, o del SNC.
• Assenza di amplificazione di MYCN. Criteri di esclusione: • Amplificazione di MYCN. 5.8 TRIAL NB 99.4

14
Criteri di inclusione • Tumore, confermato istologicamente, con amplificazione di MYCN
(>10 copie), ad eccezione dello stadio 1. Criteri di esclusione: • Stadio 1 con MYCN amplificato.

15
6 INDAGINI PRE-TRATTAMENTO Nota E’ essenziale che il bilancio di estensione di malattia e lo studio di MYCN
siano completati prima di iniziare qualunque trattamento, a parte i casi in cui è urgente erogare terapia antitumorale. In questo caso, si cerchi di completare quanto sopra entro 7 giorni.
1] Anamnesi completa con particolare attenzione a: pallore, sudorazione,
perdita di peso, diarrea, irritabilità. Segnalare la durata di tali sintomi. 2] Esame obiettivo accurato con particolare attenzione a lesioni cutanee. Rilevazione di peso, altezza, circonferenza cranica e pressione arteriosa. Rilevare eventuale presenza di opsoclono e di segni di compressione
midollare. 3] Emocromo completo. 4] Esami bio-umorali: profilo coagulativo, funzionalità epatica e renale.
latticodeidrogenasi (LDH), enolasi neurono-specifica (NSE), ferritina. 5] Catecolamine urinarie (AVM, AOV e dopamina). 6] Diagnostica per immagini:
• Radiografia AP del torace. • Ecografia addominale. • TC o RMN del tumore primitivo (con misurazione delle 3 D). • Scintigrafia con I123 o I131-MIBG. • Scintigrafia con Tc99-MDP se MIBG non disponibile (o non valutabile, o
negativa). • Radiografia di qualsiasi area scheletrica sintomatica o positiva alle
scansioni isotopiche. Questo è essenziale in quanto determina l’ingresso nel Trial 99.4 per gli infant con malattia metastatica.
• TC del cranio.
• Per ulteriori informazioni sulle valutazioni radiologiche vedi Appendice 3 (linee guida radiologiche).
7] Aspirato midollare in quattro sedi per morfologia e studio di MYCN (possibile
in caso di infiltrazione evidente). La biopsia osteo-midollare è raccomandata ma di difficile esecuzione.
8] Istologia del tumore primitivo. Si raccomanda una biopsia del tumore
primitivo, anche in presenza di metastasi, allo scopo di ottenere informazioni sulle caratteristiche biologiche. Se tale biopsia presenta rischi importanti, ad esempio nei casi di stadio 4s con tumore primitivo di piccole dimensioni ed epatomegalia massiva, ci si può limitare ad una biopsia epatica (a cielo aperto o tru-cut ).
Una biopsia di noduli cutanei può essere diagnostica.

16
Se la chirurgia a cielo aperto è considerata troppo rischiosa a causa della sede del tumore o delle condizioni del bambino, viene accettata la diagnosi citologica, purchè sia ottenuta una quantità di materiale adeguata per l’identificazione di cellule tumorali e lo studio di MYCN.
9] Valutazione e definizione dell’ infiltrazione scheletrica.
MIBG valutabile con captazione nel tumore primitivo, o con aumentata captazione epatica*
captazione scheletrica di MIBG Assenza di captazione scheletrica di MIBG
Radiografie standard delle aree positive
(eseguire TC del cranio in caso di captazione in tale sede)
Alterazione ossea attribuibile al tumore (Rx o TC).
Eleggibile per il Trial 99.3
Se MYCN amplificato: trattare con Trial 99.4
Nessuna alterazione:
Assenza di malattia scheletrica
Eleggibile per il Trial 99.2 a meno che non vi sia malattia pleuro-polmonare o SNC, o amplificazione di MYCN.
Non ulteriori indagini (scintigrafia conTc99MDP non necessaria)
* La scintigrafia con Tc99
-MDP è obbligatoria per la valutazione dello scheletro se la scintigrafia con MIBG è negativa a livello del tumore primitivo, o non è valutabile, come nel caso in cui non vi è chiara evidenza di aumentata captazione nel fegato. Anche in questo caso le aree positive vanno studiate con radiografia standard, o con TC del cranio se la captazione è presente in quella sede.

17
7. LINEE GUIDA ANATOMO-PATOLOGICHE 7.1 CONSIDERAZIONI GENERALI E RACCOMANDAZIONI 7.1.1 E' compito fondamentale del patologo trattare il tumore secondo modalità che
consentano un corretto studio sia anatomo-patologico che biologico. 7.1.2 Per la corretta interpretazione dei risultati genetico-molecolari è necessario
determinare l'esatto contenuto cellulare del campione esaminato, cosa possibile solo se il patologo valuta i campioni speculari rispetto a quelli utilizzati per le analisi genetico-biologiche (per i dettagli vedi oltre). Il referto relativo al contenuto cellulare deve essere spedito insieme al campione.
7.1.3 Il patologo è responsabile della manipolazione del tessuto tumorale secondo
le indicazioni del presente protocollo. 7.1.4 Il tessuto tumorale va immediatamente trasferito, in condizioni di sterilità,
dalla sala operatoria al Servizio di Anatomia Patologica. Le sezioni devono essere eseguite dal patologo nel più breve tempo possibile, indicando l’intervallo trascorso dall’asportazione chirurgica al momento del campionamento istologico.
7.1.5 Si tratti di resezioni tumorali o di biopsie, è sempre necessario prelevare
materiale derivante da aree tumorali differenti (sono di particolare interesse i noduli emorragici!) per le indagini genetico-molecolari e biologiche. Il motivo di tale raccomandazione è basato sull'osservazione che l'eterogeneità del tumore dal punto di vista genetico (ad esempio per il gene MYCN e la delezione di 1p36) e/o istologico (ganglioneuroblastoma, sottotipo nodulare secondo la INPC*) ha importanti implicazioni prognostiche.
7.1.6 E' indispensabile una stretta cooperazione tra patologi e biologi. I
patologi devono informare i biologi se aree di aspetto morfologicamente sfavorevole sono presenti solo nel materiale in paraffina e assenti nei campioni selezionati per le indagini biologiche. Tali aree dovrebbero essere analizzate usando il materiale paraffinato (vedi sotto).
7.1.7 Un campione di sangue periferico del paziente (5-10 ml in EDTA o sodio
citrato) è necessario come controllo per gli studi biologico-molecolari e va inviato al laboratorio di riferimento assieme al tessuto tumorale. La mancanza del campione di sangue periferico non permetterà di eseguire l'analisi di LOH per i loci D1S80 e D1S76 sul cromosoma 1p36.
7.1.8 Il materiale prelevato per le indagini genetico-molecolari/biologiche va inviato
prima possibile al Laboratorio Biologico Nazionale di Riferimento. __________________________________________________________________ *Shimada et al. “Terminology and morphologic criteria of neuroblastic tumors” and “The international neuroblastoma pathology classification the Shimada system”. Cancer 1999; 63: 349-372.

18
7.1.9 Nel caso in cui i campioni del tumore prelevati per le indagini genetico-
molecolari non siano idonei ad ottenere risultati affidabili, è possibile determinare l'amplificazione di MYCN, la presenza di delezione di 1p36 ed il contenuto di DNA adoperando il materiale incluso in paraffina. Tuttavia l'analisi su materiale in paraffina risulta più indaginosa e sarebbe quindi da evitare.
7.2 TRATTAMENTO E CONSERVAZIONE DI MATERIALE TUMORALE IN CASO
DI TUMORI ASPORTABILI
7.2.1 Si raccomanda la seguente procedura:
Tagliare il tumore secondo il dametro maggiore e prelevare almeno due campioni da aree, se presenti, di aspetto morfologicamente differente (1x1x1 cm). E’ obbligatorio campionare eventuali aree di aspetto nodulare. Identificare i campioni con lettere maiuscole (A, B, etc.) e tagliare ognuno di essi in quattro sezioni macroscopiche seriate che saranno marcate con numeri progressivi (v. disegno). Se si sospetta dall'aspetto macroscopico un ganglioneuroma o un ganglioneuroblastoma si ricerchi con attentenzione l'eventuale presenza di noduli!
Si noti che bisogna indicare come A1 e B1 una delle sezioni centrali!
� A1 e B1: eseguire 10 apposizioni da una superficie appena tagliata. I
vetrini vanno asciugati all' aria e non fissati e, se necessario, conservati a -20° C (la conservazione a temperatura ambiente per una settimana non compromette l'esecuzione delle successive analisi). Le apposizioni vengono utilizzate per FISH e citometria statica. Fatto questo, i pezzi vengono fissati in formalina per l'esame istologico di routine che comprende la determinazione e l'indicazione del contenuto di cellule tumorali in rapporto al contenuto in cellule normali (come cellule

19
di Schwann, linfociti, etc); deve inoltre essere indicata la percentuale di necrosi. Tali informazioni sono di fondamentale importanza per l'interpretazione dei risultati di FISH e di citometria statica, come pure di citogenetica, PCR e SB (eseguiti adoperando i pezzi 2, 3 o 4!)
� A2, A3, A4; B2, B3, B4: congelare appena possibile in fiale distinte
contenenti azoto liquido o biossido di carbonio a -70°C. Le sezioni A2 ed A3 saranno usate per valutazione di MYCN e stato del cromosoma 1p36. Le sezioni A4 e B4 verranno invece utilizzate per la definizione del contenuto di DNA e per le determinazioni immunoistochimiche su materiale congelato.
NOTA: Si noti che al contrario del precedente protocollo non é piú indispensabile porre i frammenti A2 e B2 in mezzo di coltura sterile (RPMI 1640). Tale procedura resta tuttavia facoltativa, ed é anzi preferibile se il materiale puó pervenire al centro coordinatore entro la giornata di venerdí. E’ comunque indispensabile preavvertire il Servizio di Anatomia Patologica dell’Ospedale Gaslini dell’arrivo di materiale fresco, in modo che possano essere predisposte tutte le misure atte alla immediata processazione dello stesso.
CONGELARE I CAMPIONI ED INVIARLI PRIMA POSSIBILE UTILIZZANDO IL BIOCASE GIA' DISPONIBILE DAL PROTOCOLLO NB 97
7.2.2 FISSAZIONE IN PARAFFINA
Al termine di tale procedura, il resto dei campioni tumorali può essere fissato in formalina e trattato secondo le comuni linee-guida. Va inclusa l'intera sezione centrale di 4 mm del tumore, ottenuta a livello del diametro più largo, nonchè un minimo di una sezione tumorale per ogni centimetro dall'intero campione (compresa l'area centrale e quelle periferiche). I margini chirurgici devono essere marcati con inchiostro.
7.3 TRATTAMENTO E CONSERVAZIONE DI MATERIALE TUMORALE IN
CASO DI TUMORI NON ASPORTABILI 7.3.1 Si seguano le raccomandazioni generali descritte al paragrafo 7.1.
L’approccio al campione tumorale varia in rapporto alla quantità di materiale disponibile per la diagnosi istologica e gli studi di biologica molecolare. Le apposizioni devono essere eseguite prima di fissare ed includere il materiale tumorale (per biopsie tru cut e aspirati con ago sottile vedi sotto).

20
7.3.2 BIOPSIA A CIELO APERTO
In questo caso, vanno biopsiate due differenti aree del tumore. I campioni prelevati dovrebbero avere dimensioni non inferiori a 1x 1x 1cm. In accordo con le raccomandazioni fornite sopra, i campioni vanno contrassegnati con lettere maiuscole (A, B, etc.), tagliati in 4 pezzi, seguendo le indicazione date al paragrafo 7.2 In caso di campioni bioptici di dimensioni minori il materiale può essere suddiviso secondo una delle seguenti modalità: 3 frammenti: frammento 1: per apposizioni ed istologia
frammenti 2 e 3: per congelamento (o in alternativa frammento 2 in RPMI e 3 per congelamento).
oppure in: 2 frammenti: frammento 1: per apposizioni ed istologia frammento 2: per congelamento
7.3.3 BIOPSIE TRU CUT
Considerazioni generali In caso di biopsie tru cut si suggerisce di prelevare quattro campioni (almeno due in caso di tumori di piccole dimensioni) da differenti aree tumorali. Si consiglia di adoperare un ago del diametro di 18 gauge. Le biopsie tru cut sono di solito eseguite dal radiologo sotto guida ecografica. Si consiglia vivamente che il patologo assista alla biopsia, così da poter valutare immediatamente la qualità dei campioni, riducendo il rischio di dover in seguito esaminare materiale non valutabile (necrotico).
E' essenziale, in vista dello studio bio-molecolare, che il patologo quantifichi la percentuale di cellule tumorali presenti nel materiale bioptico.
Gestione delle biopsie tru cut L’esecutore della biopsia non deve rimuovere il tessuto bioptico dall'ago adoperando un bisturi o altro strumento, ma deve porre l'ago in un contenitore con RPMI 1640 sterile o in qualsiasi altro mezzo isotonico sterile e agitare delicatamente fino a che il campione non scivoli nel liquido. Tale manovra consente di evitare artefatti dovuti a schiacciamento. Inoltre le agobiopsie tendono ad aderire l’un l’altra rendendo difficile la successiva

21
separazione del materiale. Ogni biopsia va mantenuta in una singola provetta con RPMI 1640 o altra soluzione isotonica sterile.
Controllo di qualità I campioni devono essere almeno di lunghezza non inferiore al cm e di spessore non inferiore a 0.1 cm. Materiale troppo minuto o frammentato non è accettabile. Se esistono dubbi sulla qualità del materiale, si agiti la provetta con attenzione una o due volte. La frammentazione in più parti del campione suggerisce che il materiale può essere necrotico, in questo caso il patologo deve chiedere di ripetere la biopsia. Poichè non si può giudicare dall’aspetto macroscopico se una agobiopsia contenga cellule tumorali o prevalentemente stroma, la biopsia tru cut va preceduta da un agoaspirato "fine needle" che
a) dà informazioni sul contenuto di cellule neoplastiche dell'area prescelta in pochi minuti
b) fornisce sospensioni cellulari per la valutazione del contenuto di DNA ed un adeguato numero di campioni citologici per lo studio con FISH.
Trattamento di biopsie tru cut
4 frammenti: 2 di essi vanno trasferiti da RPMI 1640 o altra soluzione isotonica in formalina 4% tamponata, in contenitori separati ed inclusi in blocchetti di paraffina distinti, contrassegnati con le lettere A e B, per eseguire le analisi morfologiche ed immunoistochimiche.
2 frammenti vanno trasferiti con attenzione in tubi di plastica distinti a fondo piatto, accuratamente ricoperti da un composto per inclusioni a base acquosa per la preparazione di sezioni di tessuto congelato (si cerchi di mantenere il campione al fondo della provetta!), e congelate prima possibile in azoto liquido o in biossido di carbonio a -70°C per gli studi genetico-molecolari, biologici e per l'immunoistochimica. Eseguire 10 apposizioni.
2 frammenti: 1 di questi va trasferito in formalina ed incluso in paraffina,
mentre l'altro va direttamene congelato dopo aver eseguito 10 apposizioni.
Modalità di esecuzione delle apposizioni Una buona analisi FISH richiede che il materiale da esaminare (apposizioni o cytospin) contenga nuclei intatti. Poichè il materiale a fresco proveniente da agobiopsia raramente consente di ottenere un adeguato numero di cellule, si suggerisce di eseguire le apposizioni dallo stesso materiale congelato.

22
Dopo aver rimosso accuratamente il frammento con le pinze riscaldando la provetta con le mani, si tagli una sezione congelata cominciando dall'estremità inferiore, si colori con E&E e si valuti la presenza di cellule neoplastiche morfologicamente integre (rappresentatività). La superficie fredda del frammento può essere usata per eseguire le apposizioni su vetrini a temperatura ambiente. Attenzione a ricongelare il pezzo in tempo utile, poichè il completo scongelamento distrugge la morfologia e rende il materiale inappropriato per qualsiasi analisi in situ a livello di singola cellula. Tali campioni potranno essere utilizzati come fonti per l'estrazione di DNA/RNA e di proteine (SB, PCR, comparative genome hybridisation, Northern blot, Western blot). Ulteriore commento Se il materiale congelato non contiene un numero sufficiente di cellule tumorali, la FISH e l'analisi del contenuto di DNA possono essere realizzate sui nuclei estratti dal materiale bioptico incluso in paraffina (vedi sopra). Si ricorda che è auspicabile evitare questa procedura,
7.3.4 CITOLOGIA DA AGOASPIRATI CON AGO SOTTILE
Considerazioni generali
In caso di tumori non asportabili, il patologo ed il biologo si possono trovare difronte a materiale proveniente da aspirati eseguiti con ago sottile.
In generale vanno eseguiti almeno due distinti aspirati. In base alle dimensioni del tumore, l'ago deve essere mosso avanti e indietro all'interno di diverse aree mantenendo una aspirazione costante allo scopo di campionare tessuto proveniente da più zone tumorali. Si raccomanda inoltre di eseguire un aspirato prima della biopsia tru cut (ciò permette di valutare la rappresentatività dell'area tumorale esplorata). Si consiglia di adoperare aghi con diametro esterno di 0.6-0.7 mm, 22-23 gauge.
L’aspirazione va eseguita in stretta collaborazione con radiologo e citopatologo. In base alla valutazione della massa tramite esame clinico ed ecografico l’agoaspirato viene eseguito o direttamente o sotto guida ecografica.
Nota bene: attenzione al gel adoperato per l'ecografia! Quando aspirato accidentalmente può alterare la morfologia delle cellule. La cute deve essere quindi accuratamente asciugata prima dell'esecuzione dell'agoaspirato.
Trattamento del materiale aspirato Un'aspirazione adeguata conterrà spesso 104-105 cellule.

23
L'aspirato deve sempre essere utilizzato per la preparazione di vetrini e sospensioni cellulari. 1. In rapporto a quanto disponibile, si allestiscano un minimo di quattro
strisci sottili asciugati all'aria. 2. Successivamente l'ago e il resto dell'aspirato vanno lavati all'interno
con 1 ml di soluzione fisiologica sterile tamponata e la sospensione così ottenuta va conservata in una provetta Eppendorf.
3. Uno striscio da ogni aspirato va colorato con il metodo DiffQuik che richiede 3 min. per ottenere un preparato valutabile alla microscopia ottica (preferibilmente eseguita da un tecnico specializzato)
4. In rapporto alla valutazione preliminare dell'adeguatezza del campione il patologo consiglierà o meno se ripetere l’aspirato.
Raccomandazioni per analisi successive Gli strisci servono per:
• valutazione morfologica (colorazione con May-Grünwald-Giemsa)
• immunocitologia • FISH (MYCN e delezione 1p36) • Citometria statica, contenuto di DNA.
Le sospensioni cellulari servono per: • analisi della ploidia • colture cellulari (ad es. per citogenetica) • preparazione di cytospin (circa 103 cellule per
vetrino) asciugati all'aria durante la notte ed usati per gli stessi scopi degli strisci.
Valutazione di qualità Il numero di cellule tumorali contenuto in una serie di cytospin è solitamente più uniforme rispetto a quello contenuto in una serie di apposizioni o di strisci preparati individualmente. Uno striscio/cytospin di ogni serie/aspirato deve essere analizzato morfologicamente/istologicamente allo scopo di valutare il contenuto di cellule neoplastiche. Tale informazione deve essere inviata insieme al vetrino al laboratorio di riferimento. Cytospin, strisci e sospensioni cellulari possono essere congelati a –20°C o meglio a –80° C per future analisi.

24
7.3.5 ASPIRATI MIDOLLARI In presenza di una massiva infiltrazione midollare, in pazienti di stadio 4, il midollo osseo può essere utilizzato per la caratterizzazione genetica delle cellule tumorali circolanti. Se il contenuto di cellule tumorali supera il 60%, il numero di copie di MYCN può anche essere determinato con Southern Blot. In presenza di bassi livelli di infiltrazione lo studio di MYCN e di del 1p36 va eseguito con FISH.
7.4 REFERTO ISTOLOGICO/CITOLOGICO
7.4.1 Tumore asportato chirurgicamente Classificazione morfologica: il tumore deve essere classificato secondo la International Neuroblastoma Pathology Classification, INPC. Descrizione morfologica: va indicata la quantità di cellule di Schwann, la eventuale presenza di noduli, il grado di differenziazione delle cellule tumorali, l'indice mitotico-cariorettico complessivo, la possibile presenza di aree con alto indice mitotico e di calcificazioni. Margini di resezione chirurgica: il patologo può riferire la presenza di margini di resezione infiltrati senza che questo implichi una definizione di residuo micro- o macroscopico. Contenuto di cellule tumorali: deve essere definita la percentuale relativa di cellule neuroblastiche/gangliari rispetto alle cellule di Schwann o altre cellule normali, come pure la quota di necrosi e calcificazioni. Viene eseguito dal Centro di riferimento. Referto istologico relativo ai campioni A1, B1 (C1, etc): il referto deve indicare chiaramente la percentuale stimata di cellule tumorali, ad es. cellule neuroblastiche/gangliari versus cellule di Schwann ed altre cellule normali contenute nei campioni utilizzati per gli studi biologici. Una copia di tale referto va inviata al biologo molecolare. Viene eseguito dal Centro di riferimento.
7.4.2 Biopsie In caso di campioni bioptici di piccole dimensioni, è possibile che il tessuto tumorale ottenuto non sia rappresentativo dell'intera neoplasia. Ad esempio, nel caso di Ganglioneuroblastoma nodulare, la biopsia può essere ottenuta sia da un nodulo neuroblastico che da area ganglioneuromatosa. In tali casi critici si raccomanda l'uso dei seguenti termini, in accordo con la INPC: • Tumore neuroblastico, inclassificabile: tale termine si riferisce ad una neoplasia che appartiene inequivocabilmente al gruppo dei tumori

25
neuroblastici periferici, ma non può essere allocata con certezza in una delle quattro fondamentali categorie, che sono: • Neuroblastoma (Schwann cell stroma-poor) • Ganglioneuroblastoma intermixed (Schwann cell stroma-rich) • Ganglioneuroma (Schwann cell stroma-dominant) • Ganglioneuroblastoma nodulare (Schwann cell stroma-rich/-dominant and
stroma-poor). Altri termini raccomandati dalla INPC per tumori che danno origine a problemi nella classificazione sono: Neuroblastoma (Schwann cell stroma-poor), NOS: tale termine è usato per neoplasie con una categorizzazione non equivoca, ma i sottotipi, ad es. indifferenziato, poco differenziato, differenziante, non possono essere definiti a causa della scarsa qualità delle sezioni, di estese aree emorragiche, necrotiche o artefatti tecnici (vedi INPC). Ganglioneuroblastoma, NOS: definisce tumori con aspetto di stroma-rich/dominant contenenti estese aree di calcificazione che possono celare un nodulo stroma-poor.
7.4.3 Citologia da aspirato con ago sottile
I preparati citologici di tumori neuroblastici non contengono le informazioni relative all'architettura tissutale, necessarie per la classificazione istologica. Se la diagnosi è posta su materiale ottenuto con agoaspirato il referto che il patologo invierà al biologo includerà informazioni su numero delle cellule, la loro morfologia (stato di differenziazione delle cellule tumorali, delle cellule di Schwann, residui necrotici), ed il loro immunofenotipo .
7.5 MATERIALE TUMORALE OTTENUTO DOPO TERAPIA CITOTOSSICA
3.1.1 Il campionamento di tumore asportato o biopsiato dopo terapia citotossica segue le stesse linee guida valide per campioni ottenuti all’esordio (vedi 7.1, 7.2, 7.3). Si tenga conto tuttavia che il tumore trattato può aver subito trasformazioni necrotiche o calcifiche massive.
3.1.2 Sia la chemio che la radioterapia possono determinare importanti
modificazioni morfologiche, citodifferenziazione e maturazione (con sviluppo di stroma Schwannico), ma probabilmente non modificano le originali caratteristiche genetiche del tumore. In questo caso i tumori possono essere classificati secondo la INPC, ma senza l’assegnazione ai sottogruppi prognostici, sottolineando nel referto anatomo-patologico che il tumore è stato trattato prima della chirurgia.

26
3.1.3 La descrizione morfologica deve fornire informazioni sulla percentuale di cellule tumorali vitali e sugli aspetti di differenziazione (cellule gangliari, stroma ganglioneuromatoso), regressione (necrosi, calcificazione, sclerosi) e infiltrazione linfocitaria.
7.6 LINFONODI REGIONALI 7.6.1 La biopsia dei linfonodi locoregionali è fortemente raccomandata, qualora
fattibile, indipendentemente dal loro aspetto macroscopico. 7.6.2 Referto istologico. Deve contenere informazioni su:
• Sede e numero di linfonodi positivi • Tipo di infiltrazione metastatica:
Presenza di micrometastasi (<2 mm) Metastasi parcellari intranodali Metastasi intranodali massive Metastasi linfonodale con estensione extracapsulare.
• Descrizione morfologica dell'infiltrato tumorale.
7.7 IMMUNO-ISTOLOGIA/-CITOLOGIA
� Diagnosi differenziale: talvolta, ad es. in alcuni neuroblastomi sottotipo indifferenziato (secondo l'INPC), la diagnosi differenziale può essere difficoltosa. In questi casi si raccomanda di adoperare i seguenti anticorpi: CD56 (N-CAM), NB84a, (NSE monoclonale, NF, sinaptofisina, tirosina idrossilasi, pgp 9.5). Questi marker possono essere negativi in caso di neuroblastomi indifferenziati.
Commenti: NB84a può cross-reagire con cellule epiteliali ed endoteliali (vedi sotto). Sebbene il GD2 è virtualmente positivo in tutti i casi di tumore neuroblastico ed è estremamente utile nella evidenziazione di cellule neuroblastiche nel midollo osseo, colorazioni con antiGD2 non possono essere utilizzate nel caso di materiale paraffinato (colorazione di fondo molto forte). Si tenga conto che l'espressione del GD2 non è specifica soltanto dei tumori neuroblastici! MIC2 (CD99), actina, desmina, citocheratina a basso peso molecolare, antigene comune leucocitario (CD45) e vimentina risultano di solito negativi nei tumori neuroblastici.
� Diagnostica linfonodale: la definizione di infiltrazione va sostenuta dalla
immunoistochimica con anticorpi anti-CD56 e NSE. Se si utilizza l’anticorpo NB84a, va tenuta presente la sua cross-reattività con le cellule endoteliali. Per altri anticorpi vedi oltre.
� Determinazione esatta del contenuto di cellule tumorali: nei campioni
utilizzati per indagini bio-molecolari, si raccomanda l'uso della S-100, per l’identificazione delle cellule di Schwann, e dell'antigene leucocitario comune.

27
� Materiale citologico: per la diagnosi di infiltrazione midollare si usi
l’anticorpo anti-GD2. Per gli agoaspirati si utilizzino gli anticorpi anti GD2, anti CD 56 ed anti S-100. Per la diagnosi differenziale vedi oltre.
7.6 ARCHIVIO ANATOMO-PATOLOGICO E REVISIONE DEI PREPARATI
Al patologo di riferimento vanno spediti:
• All'esordio i campioni in formalina al 10% A1, B1, ecc. che corrispondono agli inclusi in paraffina dei prelievi corrispondenti ai campioni tumorali utilizzati per gli studi “biologici”.
• Successivamente 10 sezioni non colorate prelevate dai blocchetti in paraffina più rappresentativi della diagnosi effettuata dal Centro periferico (oppure se possibile i blocchi). Se per caso i campioni in formalina non sono stati centralizzati alla diagnosi, inviare successivamente i blocchetti.
• Copia del referto istologico comprendente i risultati immunoistologici. Nel caso di biopsie incisionali a cielo aperto é anche necessario inviare gli inclusi in paraffina corrispondenti ai prelievi utilizzati per gli studi biologici, o almeno 1 sezione colorata per ogni blocchetto di materiale incluso e 10 sezioni non colorate dei blocchetti piú rappresentativi. Il materiale centralizzato verrà sottoposto a revisione da parte di un Panel di Patologi.

28
8 LINEE GUIDA BIOLOGICHE
8.1 CONSIDERAZIONI GENERALI E RACCOMANDAZIONI 8.1.1 L'analisi del MYCN, del cromosoma 1p36.3 e del contenuto di DNA delle
cellule tumorali sono indagini obbligatorie, che devono essere eseguite nel Laboratorio di Riferimento Nazionale. I risultati forniti da altri laboratori non facenti parte dell'European Neuroblastoma Quality Control Assessment (ENQUA), non saranno accettati per l'assegnazione del gruppo per il trattamento.
8.1.2 Per rendere affidabile l'interpretazione dei risultati genetico-molecolari, deve
essere accuratamente determinato l'esatto contenuto di cellule tumorali dei campioni utilizzati per tali indagini. Ciò è possibile solo se il patologo valuta attentamente i campioni speculari rispetto a quelli usati per gli studi bio-molecolari (per i dettagli vedi sotto).
8.1.3 Per l'eleggibilità ai Trial è necessario che il risultato dello studio del MYCN sia
disponibile entro quattro settimane (al massimo sei settimane). 8.1.4 Il gruppo ENQUA ha adottato una comune terminologia per definire i
risultati di MYCN e di 1p36; tale terminologia è descritta in seguito ed il suo uso è fortemente raccomandato.
8.1.5 Un controllo di qualità eseguito nel 1998 ha messo in evidenza il rischio di
errore insito in ogni metodica. 8.2 STUDI DI BIOLOGIA MOLECOLARE
8.2.1 NUMERO DI COPIE DI MYCN
Metodi e considerazioni generali Il numero di copie di MYCN può essere determinato con Southern Blot (SB) o con FISH. Si consiglia di non adoperare la PCR senza la conferma di un secondo metodo. Si ricorda che usando il SB come unico metodo per la determinazione del MYCN, la possibilità di non evidenziare eterogeneità nello stato del MYCN (vedi Capitolo1) o interpretare erroneamente bassi gradi di amplificazione è maggiore (a causa dell'effetto "diluizione") rispetto alla FISH, che analizza la singola cellula. Se si utilizzano SB o PCR il contenuto di cellule tumorali deve essere superiore al 60%. Se si utilizza FISH vanno analizzate almeno 200 cellule tumorali da differenti aree sul vetrino.

29
• SB: usare pNb1 (Dr. Schwab, Heidelberg, FRG) o un probe equivalente, ed un probe per un gene a singola copia situato sul cromosoma 2 (L2.3).
• FISH: usare il probe Oncor (Vysis) per MYCN o il pNb9 o il pNb101 (Dr. Schwab, Heidelberg, FRG), ed un probe specifico per il cromosoma 2, per il centromero (D2Z, Oncor) o per il cromosoma 2pter (per evitare errate interpretazioni a causa di associazioni centromeriche).
• PCR: questa indagine è accettata soltanto quando i suoi risultati vengano confrontati con quelli di FISH o SB. Un protocollo dettagliato su tale metodica (scritto dal Dr.Olivier Delattre) verrà fatto circolare.
� Numero di copie del MYCN
A prescindere dal metodo usato, il numero di copie di MYCN deve essere in rapporto al numero di cromosomi 2.
� Definizione dello stato di MYCN e referto:
Studio con SB e PCR • Amplificazione di MYCN: aumento superiore a 4 volte della intensità della
banda in rapporto alla banda di riferimento interno (sul cromosoma 2!)
• Imbalance: aumento da 2 a 4 volte della intensità della banda in rapporto alla banda di riferimento interno (sul cromosoma 2!) Necessità di ulteriore verifica con la FISH
• Assenza di amplificazione di MYCN • Nessun risultato: risultato dubbio o non interpretabile
campioni contenenti meno del 60% di cellule tumorali mancanza di DNA mancanza di cellule tumorali nel campione

30
Studio con FISH • Amplificazione di MYCN: aumento del numero di segnali di MYCN 4 volte superiore in relazione al numero di cromosomi 2 • MYCN gain: aumento del numero di copie di MYCN da 2 a 4 volte in relazione al numero di cromosomi 2. Richiede ulteriori indagini!
• Amplificazione focale di MYCN • Assenza di amplificazione di MYCN
• Nessun risultato: risultato dubbio o non interpretabile quantità insufficiente di cellule nel campione
mancanza di cellule tumorali nel campione 8.2.2 STATO DEL CROMOSOMA 1p36.3
� Metodi e considerazioni generali
L'integrità del braccio corto del cromosoma 1 (1p36.3) può essere determinata con 3 metodi: SB, PCR e FISH.
Deve essere ben chiaro che gli studi effettuati con SB e PCR forniscono informazioni sullo stato allelico della regione cromosomica 1p36.3. Di contro, le analisi con FISH danno informazioni sullo stato del cromosoma, ad es. sul rapporto fra il numero dei centromeri e delle regioni subtelomeriche del cromosoma 1. Un rapporto 2:1 trovato con FISH (ad es. 2 centromeri contro 1 regione subtelomerica) molto probabilmente corrisponde ad una perdita di eterozigosità (LOH) riscontrata con SB o PCR. Ogni altra sproporzione fra regioni centromeriche e subtelomeriche ma con più di un segnale 1p36.3 (ad es. un rapporto 3:2, 4:3, 5:3, etc.) documentato con FISH, non riflette necessariamente la presenza di una LOH. Viceversa, l'assenza di LOH non necessariamente riflette l'assenza di aberrazioni citogenetiche a carico del cromosoma 1p36.3.
E’ quindi fortemente raccomandato l'uso di entrambi i metodi (SB/PCR e FISH) per la ricerca di aberrazioni a carico del cromosoma 1p36.3, allo scopo di poter raccogliere l'informazione completa sulle possibili mutazioni di tale importante regione cromosomica.

31
Se si utilizzano SB o PCR il contenuto di cellule tumorali deve essere superiore al 60%. Se utilizza FISH vanno analizzate almeno 200 cellule tumorali da differenti aree sul vetrino. SB: usare ad es. il probe CEB15 o altri (come D1S7, Dr. Jeffreys) all’interno la regione di consenso (i.e. 1p36.3). PCR: usare ad es. i primer specifici per D1S76 e D1S80 o altri all'interno della consensus region. FISH: usare ad es.il probe D1Z2 insieme ad un probe centromerico (DZ), o un probe situato sul braccio lungo del cromosoma 1 (per evitare errate interpretazioni dovute ad associazioni centromeriche) con tecnica FISH a doppia colorazione.
� Definizione del cromosoma 1p36.3 e referto
Studio con PCR e SB
• Perdita allelica (LOH): completa o quasi completa scomparsa di una banda
• Imbalance allelico (non conclusivo): una banda più debole rispetto all’altra.
Ciò può sia significare " squilibrio allelico " (ad es. due cromosomi paterni ed uno materno) sia LOH Questo risultato deve essere confermato da studio con FISH!
• Assenza di perdita allelica o di imbalance allelico
• Nessun risultato: dubbio o non interpretabile
omozigosità costituzionale campioni contenenti meno del 60% di cellule tumorali mancanza di DNA mancanza di cellule tumorali nel campione

32
Studio con FISH
• Delezione: presenza di un'unica regione subtelomerica del braccio corto del cr.1 (rapporto 2:1, possibilmente insieme con 4:2 nello stesso tumore, 3:1, 4:1)
• Imbalance con FISH: sproporzione del rapporto dei centromeri e delle regioni subtelomeriche del braccio corto del cr.1, con presenza di più d'una regione subtelomerica (rapporto 3: 2, 4:3, 4:2, 5:3 etc.)
• Assenza di delezione, assenza di imbalance con FISH evidenziata con i probes citati precedentemente
• Nessun risultato: risultato dubbio o non interpretabile quantità insufficiente di cellule nel campione mancanza di cellule tumorali nel campione
Raccomandazioni: se meno del 50% delle cellule tumorali mostra delezione o squilibrio con FISH, è bene analizzare ulteriori vetrini, campioni o nuclei isolati; qualora la FISH sulle apposizioni fallisca (ad es. ganglioneuroma) può essere adoperato materiale in paraffina.
� REFERTO DEGLI STUDI DI BIOLOGIA MOLECOLARE
Tale referto comprenderà due parti:
• Risultati: vengono riferiti in dettaglio, indicando i metodi utilizzati o le cause di mancanza del risultato (es. mancanza di campionamento adeguato, campione con scarso numero di neuroblasti, ecc.).

33
• Conclusioni: il risultato deve essere riportato in breve e con precisione usando la terminologia sopradescritta.
8.2.3 CONTENUTO DI DNA
� Metodi e considerazioni generali I metodi comunemente usati per la valutazione del contenuto tumorale di DNA sono Citometria a flusso e Citometria statica. Il numero di cromosomi 1 ottenuto con FISH non può essere usato per la valutazione del contenuto di DNA, poichè un tumore trisomico può essere disomico per il cr.1 e tumori diploidi possono essere trisomici per il cr.1, etc. Per la Citometria a flusso usare come cellule di riferimento le cellule del paziente stesso. Nel caso in cui il materiale tumorale non sia adeguato per la determinazione del contenuto di DNA con Citometria a flusso ed il rispettivo laboratorio di riferimento non effettui l'analisi in Citometria statica, si inviino alcune apposizioni al Dr. Per Kogner, Karolinska Institute, Stockholm, Sweden, per l'indagine con Citometria statica. � Referto dei risultati Il referto deve indicare la metodica usata e specificare il numero di cellule tumorali versus cellule normali contenute nel campione studiato. Il risultato del contenuto di DNA delle cellule tumorali deve essere espresso in numeri assoluti.
8.3 REVISIONE CENTRALIZZATA
I risultati di MYCN, cromosoma 1p36.3 e contenuto di DNA saranno rivisti centralmente in apposite riunioni coinvolgenti i Laboratori di Riferimento Nazionali.
8.4 ASPIRATI MIDOLLARI
8.4.1 Materiale necessario: 2-4 siringhe, 10-20 vetrini per gli strisci midollari, 1 vetrino coprioggetto sgrassato, anticoagulante adeguato (eparina, EDTA, ACD, in proporzione 1:5). Sono necessarie due aspirazioni: una per gli strisci e la seconda per citoimmunologia, PCR o altre tecniche.

34
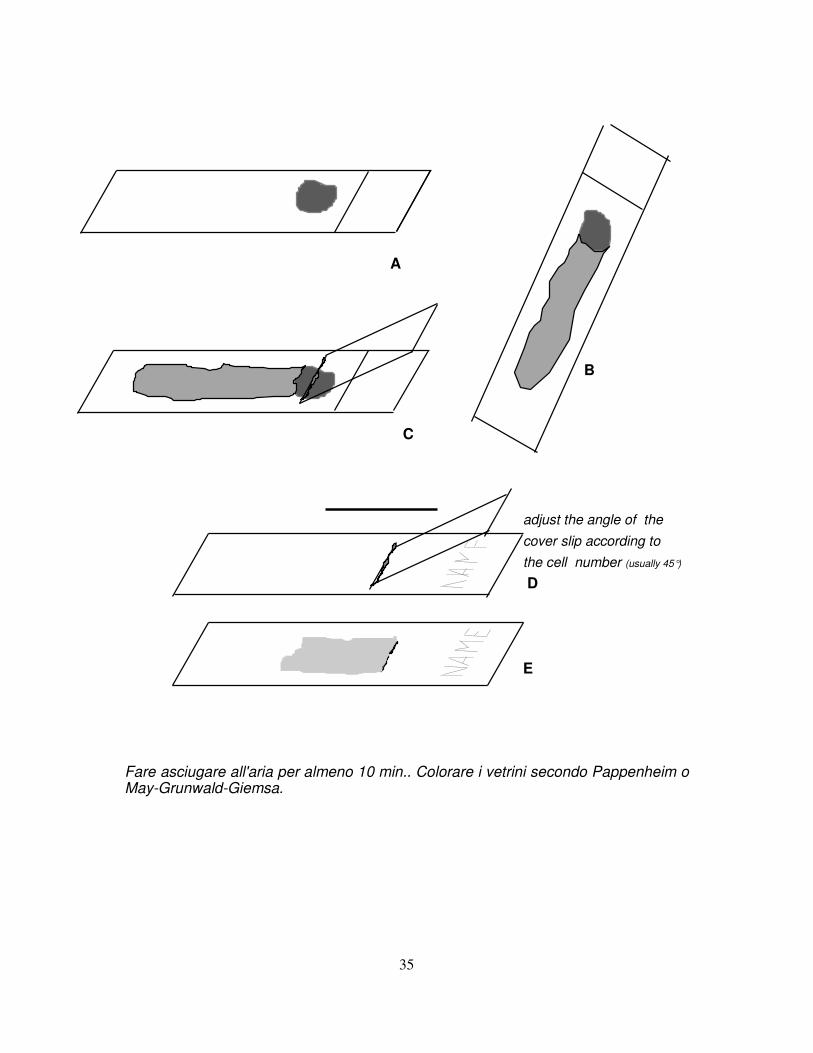
• Prima aspirazione e preparazione degli strisci midollari (vedi figura)
Si raccomanda vivamente di eseguire gli aspirati di midollo nel numero previsto dal protocollo clinico (2-4) da sedi diverse. Aspirare con decisione per ottenere 0.2-0.5 ml di midollo e porre l'intera quantità su un vetrino (A). Inclinare il vetrino (B) in modo da lasciar defluire il sangue in eccesso. Toccare la goccia di midollo con il coprioggetti (C) e strisciare il coprioggetti stesso su un nuovo vetrino (D); ripetere tale manovra 3-5 volte per ogni aspirato.
• Seconda aspirazione per immunocitologia Adoperare come anticoagulante eparina, EDTA o ACD (1:5). Aspirare la appropriata quantità di anticoagulante nella siringa. Per evitare una diluizione eccessiva con sangue periferico ogni aspirato non dovrebbe suprare i 4-5 ml, meglio se 2-3 ml. Agitare immediatamente per consentire all'anticoagulante di mescolarsi con il midollo. Ripetere tale procedura per ogni sede di puntato. Chiudere la siringa ed evitare fuoriuscita di materiale. Trasportare la siringa chiusa al laboratorio di ematologia o di immunologia a temperatura ambiente prima possibile. Può essere accettato un ritardo fino a 48 ore.
8.4.2 Gestione delle cellule midollari in laboratorio
� Separazione di cellule mononucleate, preparazione e conservazione
dei cytospin
Separare le cellule mononucleate con un Lymphoprep (Nycomed) o in Fycoll. Sarebbe ottimale separare ogni sede indipendentemente. In ogni caso procedere seguendo le indicazioni del coordinatore nazionale, indicando con chiarezza il contenuto delle provette. Dopo aver separato con cura la frazione delle cellule mononucleate, lavare in PBS o in TRIS pH 7,0, senza FCS e centrifugare lentamente (10 min a 252 g). La frazione monucleata recuperata deve essere contata e valutata per la % di cellule vitali con il test di esclusione al tripan blu. Allestire i cytospin. I vetrini dovranno contenere almeno 106 cellule mononucleate. Qualunque metodo si usi i preparati cellulari devono contenere un adeguato numero di cellule mononucleate ben separate, allo scopo di evitare falsi positivi o falsi negativi. Tale preparato di cellule mononucleate può essere ottenuto in diversi modi: con 10-20 cytospin del diametro di 5mm (centrifuga tipo Shandon) o preferibilmente 2-3 citospin larghi (17mm) utilizzando la centrifuga Hettich, con sedimentazione di cellule mononucleate su vetrini viscosi (Poly-L-Lysine; Cell-Line, Cell-Tak).
35
A
B
C
adjust the angle of the
cover slip according to
the cell number (usually 45°)
D
E Fare asciugare all'aria per almeno 10 min.. Colorare i vetrini secondo Pappenheim o May-Grunwald-Giemsa.

36
Le preparazioni cellulari vengono asciugate all'aria per 2-24 ore e fissate con paraformaldeide al 1-4% per 10 min (dato che il GD2 è un antigene lipidico devono essere evitati fissativi a base alcolica). I campioni vanno subito colorati o conservati a -20°C o a -80°C (per conservazioni prolungate) in camere di plastica a chiusura ermetica o avvolte ermeticamente in fogli di alluminio. Dopo lo scongelamento, lasciare i campioni ad asciugare (in contenitori chiusi) per almeno due ore prima di processarli.
� Immunocitologia con GD2
Attualmente, l'uso di anticorpi anti-GD2 per individuare cellule di neuroblastoma dà i risultati migliori in termini di sensibilità e specificità. L'antigene GD2 è un disialoganglioside altamente e costantemente espresso sulle cellule neuroblastomatose, mentre non lo è sulle cellule normali. Inoltre, tale metodica è relativamente semplice, economica ed universalmente applicabile. L’uso di ulteriori tecniche indipendenti e complementari (ad es. RT-PCR per trascritti specifici di NB, FISH per aberrazioni genetiche associate al NB) è in corso di studio da parte dei ricercatori della SIOP Europe Neuroblastoma. • purchè un evento positivo sia direttamente esaminato e confermato al
microscopio (esclusa la citometria a flusso) sia immunocitologia a fluorescenza che tecniche immunocitochimiche sono ugualmente accettabili. I risultati vanno archiviati in apposito registro e sottoposti a regolari controlli di qualità.
• Se il midollo presenta una contaminazione tumorale <0.1% devono essere
analizzate almeno 106 cellule mononucleate. In caso di contaminazione maggiore può essere esaminato un numero inferiore di cellule.
• Si raccomanda conservare almeno tre cytospin non colorati per controlli di
qualità. L’ anticorpo monoclonale primario da impiegare va scelto fra i seguenti:
- 14.G2A (Ralph Reisfeld, La Jolla) - 3F8 (Nai-Kong Cheung, New York)
Esso può essere impiegato nella forma purificata o come supernatante di ibridoma fino alla minima concentrazione capace ancora di colorare in modo continuo il controllo positivo senza background. Il tipo di anticorpi secondari e i sistemi di sviluppo da impiegare sono lasciati alla scelta del ricercatore purchè il sistema di rilevazione sia validato con diluizioni scalari di linee cellulari e vengano adoperati gli opportuni controlli sia positivi che negativi (vedi sotto).

37
� Protocolli di colorazione
Colorazione per Fluorescenza (Peter F. Ambros, CCRI Vienna, Tel +43-1-40470-412) La tecnica include sempre controlli positivi e negativi. Si usino o vetrini a fresco (asciugati all'aria per 12 ore) o vetrini congelati (dopo scongelamento per almeno due ore a temperatura ambiente in contenitori chiusi o fasciati). Colorare e fissare i vetrini con DAPI/paraformaldeide per 10 min.. Preparare una soluzione di lavoro di DAPI diluendo 10µl della soluzione base (2 mg/ml in PBS) in 10 ml di PBS (conservare al buio a 4°C). Usare 50µl della soluzione di lavoro, diluire in 850 µl di PBS e aggiungere 100 µl di paraformaldeide al 40%. Non colorare o fissare vetrini in una camera in quanto le cellule tumorali possono debordare e aderire ad altri vetrini così da mascherare il quadro. Idealmente tutte le procedure di colorazione e fissaggio devono essere eseguite sotto un coprioggetti. Dopo aver fissato lavare con PBS. Assicurarsi che le cellule non siano essiccate durante l'intera procedura di colorazione! • Rimuovere l'eccesso di PBSC ed incubare le cellule per 10 min. a 37°C
con l’anticorpo anti-GD2 diluito in PBS contenente BSA al 3%. Valutare la corretta concentrazione del colorante dell’anticorpo attraverso diluizioni progressive.
• Lavare con PBS contenente BSA al 3%. • Incubare per 30 min. con un anticorpo anti-mouse, etc. (ad es. rabbit anti
mouse, FITC labelled, DAKO F0313) 1:60 in PBS contenente BSA al 3%. • Lavare di nuovo come sopra. • Incubare con anticorpo di maiale anti-coniglio FITC (DAKO F205) 1:60 in
PBS contenente BSA al 3%. Lavare di nuovo con PBS, rimuovendo l'eccesso di liquido e montare il coprogetto (senza asciugare le cellule) con Glicerolo contenente un “antifade” (ad es. Citifluor AF3; UKC, Chem. Lab. Canterbury, CT2 7NH, UK in Glycerol+Citifluor+PBS, 8+1+1, or Vectashield+DAPI). Per evidenziare le cellule tumorali osservare il vetrino con microscopio a fluorescenza (lente da immersione a 25x) munito di filtro FITC, per un'analisi più dettagliata aumentare l'ingrandimento di 50-100 volte. Possibili errori: - Per evitare erronee interpretazioni di cellule positive (macrofagi o
plasmacellule, colorazione di membrana nucleare derivanti dalla rottura di

38
quella cellulare etc.) controllare la morfologia con contrasto di fase e colorazione con DAPI.
- Quando l'infiltrato cellulare tumorale è inferiore a 1 su 104 cellule mononucleate, controllare con FISH o altra tecnica se le caratteristiche genetiche di tali cellule corrispondono a quelle di cellule neoplastiche (ad es. amplificazione di MYCN, del 1p, o altre aberrazioni cromosomiche).
Colorazione enzimatica A. (Klaus Beiske, Norwegian National Hospital Oslo, fax 0047-22-868596)
• Per la dimostrazione di cellule tumorali al microscopio ottico, fissare cytospin asciugati all'aria in parafolmaldeide al 4% per 10 min. (senza soluzione DAPI) ed incubare con anticorpi anti-GD2 seguiti da anticorpi Ig policlonali di coniglio anti-mouse (DAKO D 314, Dakopatts, Copenhagen, Denmark) e di maiale anti-coniglio (DAKO D 306), entrambi coniugati con fosfatasi alcalina. • Ogni tappa di incubazione si conclude con un triplo lavaggio con buffer
TRIS-HCL 0.01M a pH 7.6. • Visualizzare il legame specifico trattando con soluzione substrato (15 mg
Naphtol AS-TR phosphate [Sigma], 20mg Fast Red ITR salt [Sigma], e 12 mg Levamisole [Sigma], in 50 ml tampone acetato Veronal Michaelis pH 7.6) per 45 min. a 37°C, dando luogo così ad un prodotto ad intensa colorazione rossa.
• Controcolorare i campioni con ematossilina, montare dall’acqua con gel di glicerina ed esaminare con microscopio ottico.
B. Metodo legante Avidina marcata con Cytospin Hettich (106 cellule mononucleate per cytospin) (Lawrence Faulkner, Ospedale Mayer -Firenze, fax 0039-55-570380)
• Risospendere le cellule mononucleate a 1-2 x 106 cellule/ml. • Preparare i cytospin (contenenti ognuno fino a 106 cellule mononucleate
su una singola area di 17 mm di diametro) su normali vetrini centrifugando 0.4-0.8 ml della sospensione di cellule mononucleate a 400g per 10’ (Centrifuga Hettich, Tuttlingen, Germany).
• Asciugare i cytospin a temperatura ambiente per 4-24 ore. • Dopo aver fissato i vetrini per 10 min. in acetone freddo, lavarli in PBS per
5 min., asciugarli per 5 min. ed infine incubarli per 30 min. a temperatura ambiente in una camera umida con l’anticorpo monoclonale primario murino anti-GD2.
• Dopo aver risciacquato, lavato in PBS per 10 min. ed asciugato per 5 min., incubare per 30 min. con un anticorpo policlonale biotinilato anti-mouse, indi risciacquare, rilavare in PBS per 10 min. ed asciugare per 5 min., incubare per la terza volta per 30 min. con un complesso streptavidina fosfatasi alcalina (LSAB2, DAKO).
• Dopo aver rilavato, incubare con un nuovo cromogeno a base di fucsina (DAKO) per 8 min. per colorare complessi legati alla fosfatasi alcalina). Controcolorare incubando per 5 min. con ematossilina.

39
• Per ogni procedura di colorazione processare un controllo negativo (sostituendo la soluzione di anticorpo primario con PBS) ed uno positivo (cytospin di linea cellulare di NB)
Nota: i campioni colorati enzimaticamente non sono appropriati per studi di citogenetica in interfase (come FISH), poichè il complesso enzima-substrato può coprire il nucleo e impedire l'ibridazione. Ulteriori cytospin devono essere disponibili per colorazioni con anticorpi marcati fluorescenti allo scopo di aumentare l'affidabilità nel rilevare infiltrazione midollare.

40
8.5 INTERPRETAZIONE DEI TEST, REFERTAZIONE E CONTROLLI DI
QUALITA'
8.5.1 Per ogni test va esaminato un controllo negativo con un numero confrontabile
di cellule mononucleate. Almeno 0.5-1 x106 cellule mononucleate vanno valutate sia per controllo negativo (in cui l'anticorpo primario viene sostituito con una soluzione salina tamponata) che per il campione vero e proprio. Per ogni procedura di colorazione va allestito un controllo positivo (con una linea cellulare di NB). Il numero di cellule può essere ridotto nei casi di infiltrazione tumorale evidente (>5%).
8.5.2 Una cellula è considerata positiva se ha un nucleo ben riconoscibile ed un
citoplasma colorato nella sua totalità. Un test è interpretabile quando il controllo negativo non ha una colorazione di fondo tale da interferire con il riconoscimento di una cellula positiva e/o non possiede cellule da classificare come positive. Il controllo positivo deve avere una colorazione brillante. In tutte le altre circostanze il test deve essere refertato come non interpretabile.
8.5.3 Nei casi dubbi o in quelli con bassa infiltrazione tumorale, è necessario
evidenziare le aberrazioni genetiche delle cellule tumorali, allo scopo di assicurarsi che le cellule colorate immunologicamente posseggano le stesse anomalie genetiche del tumore primario.
8.5.4 In alcuni casi possono essere evidenziati agglomerati di materiale amorfo
GD2 positivo (membrane cellulari? Micelle?) che vanno riportati come tali e quantificati nel miglior modo possibile. Per campioni fortamente positivi (>1 cell GD2+ per campo a 200x) il risultato può essere riportato come percentuale dopo aver contato almeno 30 campi.
8.5.5 Il numero di cellule positive va riportato insieme al numero totale di cellule
esaminate. Il ricercatore deve sapere quante cellule ha effettivamente esaminato in totale con un errore non superiore al 10-20% (la media delle cellule perse in durante la preparazione dei vetrini/cytospin e/o la colorazione vanno verificate all’inizio per ogni metodo immuno-citologico). I vetrini colorati per la miscroscopia ottica vanno conservati a temperatura ambiente (sia positivi che negativi). I vetrini per la fluorescenza vanno conservati a +4°C. I segnali fluorescenti dovuti al legame tra anticorpi e/o sonde ibridizzate vanno registrati e archiviati con microfotografie processate con mezzi ottici o digitali.
8.5.6 I Vetrini non colorati di almeno 5 diversi pazienti e diluizioni di linee cellulari
saranno fatti circolare tra i laboratori che si occupano di immunocitologia con anti-GD2. I risultati di tali test saranno discussi alle riunioni fra i vari gruppi in apposite riunioni.

41
9 CHIRURGIA 9.1 Un importante obiettivo di questo protocollo è evitare ogni intervento
chirurgico mutilante. In conseguenza, la valutazione di resecabilità, utilizzando le moderne tecniche radiologiche, riveste un ruolo decisivo. In caso di dubbia resecabilità, è preferibile limitarsi ad un intervento bioptico che consenta l’acquisizione di materiale tumorale per le indagini biologiche, dal cui risultato dipende la strategia terapeutica.
9.2 Il giudizio di radicalità implica l’assenza di fattori di rischio (per ulteriori
dettagli vedi Addendum II). 9.3 L’intervento immediato è considerato appropriato quando il chirurgo, sulla
base dei dati clinici e radiologici ed in seguito a discussione con l’oncologo, ritiene possibile effettuare una asportazione radicale del tumore. In particolare, vanno evitate manovre chirurgiche rischiose o che comportino asportazione di organi.
9.4 In caso di malattia disseminata, l'asportazione del tumore non è consigliata, a
meno che non costituisca l’unica modalità che consente l’acquisizione di tessuto tumorale per lo studio istologico e biologico.
9.5 In corso di intervento differito, si dovrà mirare all'asportazione radicale o con
residui minimi, evitando rischi ingiustificati o l’asportazione di qualunque organo (compreso il rene). Fanno eccezione i casi con amplificazione di MYCN.
9.6 Nello stadio 4s la chirurgia differita del tumore primitivo non è ritenuta
necessaria, specie se la malattia è in regressione (Guglielmi12). 9.7 FATTORI DI RISCHIO (vedi anche Addendum II: Chirurgia)
La formulazione del giudizio di operabilità di un tumore localizzato dipende da un attento esame clinico unito ad uno studio radiologico accurato.
La decisione se operare o meno è la conclusione di una valutazione multidisciplinare che coinvolge oncologo, radiologo e chirurgo. In linea di massima, la presenza di un qualunque fattore di rischio dovrebbe sconsigliare un intervento primario.
9.8 Fattori di rischio correlati alla sede della neoplasia
9.8.1 Collo
• Inglobamento dell’arteria vertebrale • Inglobamento delle radici del plesso brachiale • Superamento della linea mediana

42
9.8.2 Torace • Inglobamento della trachea o di un bronco principale • Inglobamento dell’origine o di diramazioni delle succlavie • Tumore fusiforme peri-aortico • Tumore toraco-addominale • Tumore del mediastino inferiore, infiltrante la giunzione costo-vertebrale
fra T9 e T12 9.8.3 Retroperitoneo
• Infiltrazione dell’ilo epatico • Tumore surrenalico che ingloba i rami dell’arteria mesenterica superiore a
livello della radice del mesentere • Tumore surrenalico che circonda l’origine del tronco celiaco e dell'arteria
mesenterica superiore • Invasione di uno o di entrambi i peduncoli renali • Tumore fusiforme che circonda l’aorta infrarenale • Inglobamento dei vasi iliaci • Tumore pelvico che supera la incisura ischiatica • Tumore che coinvolge i vasi ipogastrici
9.8.4 Altri fattori di rischio
• Tumore a clessidra con segni neurologici, ma senza indicazione alla laminectomia (Addendum IV)
• Dimensioni del tumore • Fragilità del tumore per presenza di aree necrotico-colliquative.

43
10 TRATTAMENTO: TUMORI SENZA AMPLIFICAZIONE DI MYCN Dettagli sulla chemioterapia, incluse dosi e modalità di somministrazione, possono essere trovati nella Sezione 13 e nell'Addendum VI. 10.1 Stadio 1, stadio 2 asportato, stadio 3 asportato 10.1.1 Il trattamento è limitato alla sola chirurgia, anche se incompleta. Non è
prevista chemioterapia precauzionale. E’ importante che la chirurgia sia effettuata nei casi in cui siano assenti fattori di rischio. E’ opportuno assicurarsi che chirurgo e oncologo abbiano studiato le direttive chirurgiche radiologiche (Addendum II e III) prima del’intervento.
10.1.2 E’ possibile che l’analisi dei dati raccolti nel LNESG porti all’identificazione di nuovi fattori di rischio non considerati in questo studio. Questo potrebbe portare modifiche all’attuale protocollo.
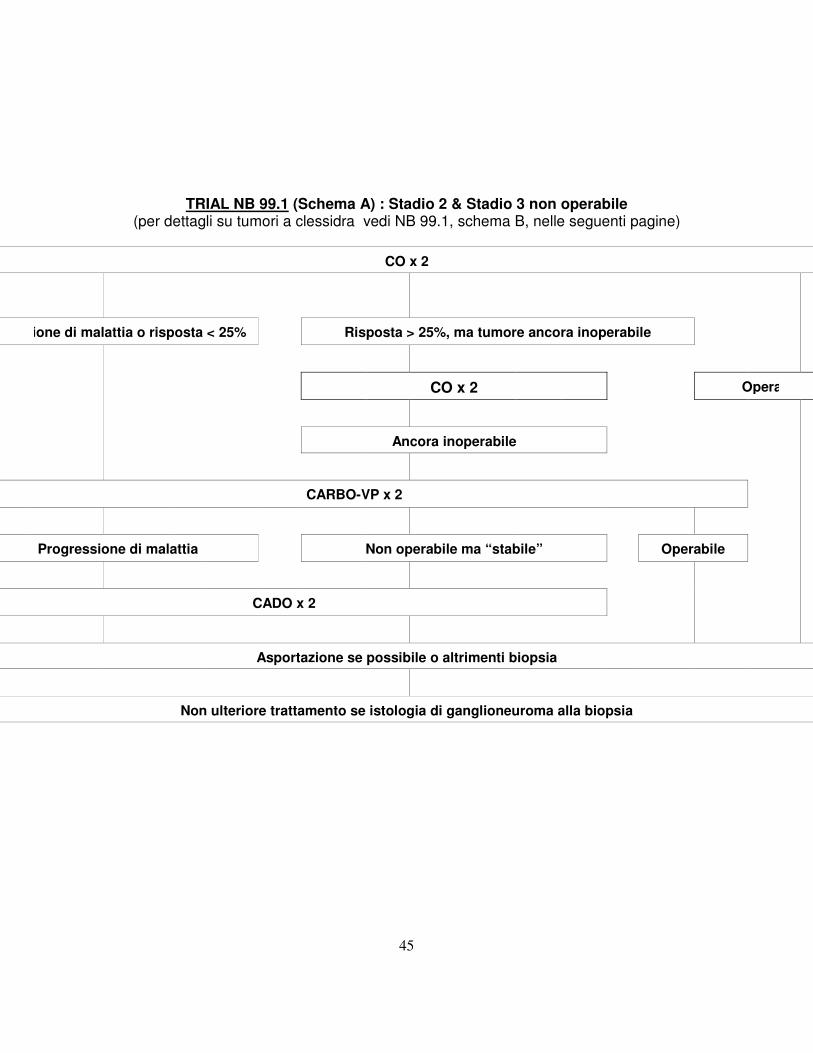
10.2 TRIAL NB 99.1 Stadio 2 e 3 non operabile Nota bene: vi sono 2 schemi di trattamento (A e B) in rapporto alla
presenza o meno di compressione midollare sintomatica. 10.2.1 Questo Trial riguarda infant con neoplasia localizzata (senza amplificazione
di MYCN), ma non operabile, sottoposti a biopsia diagnostica per conferma istologica e studio biologico. L’approccio terapeutico iniziale consiste in 2 cicli di Ciclofosfamide e Vincristina (CO), seguiti da valutazione della risposta ed intervento chirurgico, ove possibile.
10.2.2 Se dopo i 2 cicli di CO la chirurgia non è ancora possibile, ma si osserva una risposta (definita come riduzione di almeno il 25% nel prodotto dei due diametri maggiori), saranno somministrati altri 2 cicli di CO, seguiti da ulteriore rivalutazione di operabilità. Se il tumore verrà asportato non si somministrerà ulteriore chemioterapia. Nota bene: la valutazione della risposta richiede la misurazione delle 3 dimensioni quando possibile, valutando per il calcolo i due diametri maggiori.
10.2.3 Se dopo 2 cicli di CO vi è una risposta < 25% del prodotto dei due diametri
maggiori o vi è evidenza di progressione di malattia, il trattamento prosegue con Carboplatino ed Etoposide (Carbo-VP). Dopo 2 cicli si valuterà la risposta e quindi la resecabilità.
10.2.4 Se dopo 2 cicli di Carbo-VP il tumore rimane non asportabile o vi è
evidenza di progressione, il trattamento proseguirà con 2 cicli CADO

44
(Ciclofosfamide, Doxorubicina, Vincristina), a seguito dei quali il tumore verrà asportato o biopsiato. Se l'istologia mostra trattarsi di ganglioneuroma non sarà erogata ulteriore terapia.
10.2.5 Si raccomanda di discutere i casi che presentano difficoltà con il
Coordinatore Nazionale. 10.2.6 Infant con compressione midollare sintomatica sono inizialmente trattati
con Carbo-VP (si rimanda al Trial 99.1 Schema B nelle pagine seguenti) x 2 cicli prima della valutazione della risposta. Se questa è > 50% verrà asportata la componente extraspinale, se possibile. In caso contrario, si continuerà con 2 cicli CADO.
Nota bene: alcune neoplasie, pur rispondendo ai due cicli CO, rimangono inoperabili in virtù della particolare sede (esempio, neoplasia fusiforme periaortica). In questi casi si raccomanda di somministrare 4 cicli CO ed eseguire una biopsia prima di passare a Carbo-VP o CADO (discutere il caso con il Coordinatore Nazionale). Vedi Sezione 13 e Addendum VI per dettagli sulla chemioterapia. Nota bene: i dosaggi per i neonati (età < 1 mese) o infant con peso < 5 kg vanno ridotti di un terzo.
45
TRIAL NB 99.1 (Schema A) : Stadio 2 & Stadio 3 non operabile (per dettagli su tumori a clessidra vedi NB 99.1, schema B, nelle seguenti pagine)
CO x 2
Progressione di malattia o risposta < 25% Risposta > 25%, ma tumore ancora inoperabile
CO x 2 Operabile
Ancora inoperabile
CARBO-VP x 2
Progressione di malattia Non operabile ma “stabile” Operabile
CADO x 2
Asportazione se possibile o altrimenti biopsia
Non ulteriore trattamento se istologia di ganglioneuroma alla biopsia

46
TRIAL 99.1 (Schema B) tumori a clessidra
Non segni di compressione midollare Sintomi di compressione spinale
Trattare secondo Schema A (vedi pagine precedenti)
CO x 2, se risposta continuare CO x 2 (o chirurgia se possibile) Se progressione o la componente extraspinale
rimane inoperabile dopo CO x 4, passare a Carbo-VP
Se progressione o se la componente extraspinale rimane non operabile dopo Carbo-VP, passare a
ADO x 2.
Carbo-VP x 2 cicli
(vedi nota 10.2.6 per tumori inoperabili per la sede particolare)
Miglioramento dei sintomi e tumore extraspinale
operabile.
Non miglioramento nei sintomi, o tumore extraspinale ancora non operabile * se deterioramento neurologico vedi Nota 3, in seguito.
CADO x 2
Asportazione chirurgica (tumore EXTRASPINALE) se possibile, biopsia se non operabile
Non ulteriore trattamento se istologia di ganglioneuroma alla biopsia (discutere con il Coordinatore del Trial in caso di tumore vitale)
Note: L’asportazione chirurgica è rivolta esclusivamente alla componente extraspinale.
Se vi è progressione dei sintomi neurologici durante chemioterapia la laminectomia/laminotomia va considerata (Addendum IV)

47
10.3 TRIAL NB 99.2 Stadio 4s e stadio 4 (senza metastasi ossee, pleuro-
polmonari o del SNC) senza amplificazione di MYCN 10.3.1 Questo Trial riguarda lo stadio 4s (senza amplificazione di MYCN) che
richiede trattamento, e lo stadio 4 senza metastasi ossee (o pleuro-polmonari o del SNC). Sono inclusi alcuni infant che rientrerebbero nello stadio 4 secondo i criteri INSS, solo in quanto il tumore primitivo oltrepassa la linea mediana. Nota Bene: sono eleggibili gli infant con infiltrazione midollare, purchè senza amplificazione di MYCN.
10.3.2 Per quanto concerne lo stadio 4s, si raccomanda di trattare soltanto i
pazienti con sintomi gravi. Una biopsia per l’istologia e gli studi biologici DEVE essere eseguita in tutti i pazienti. Se la biopsia del tumore primitivo è considerata rischiosa, può essere sufficiente biopsiare una lesione epatica o un nodulo cutaneo.
10.3.3 Il ”Philadelphia Scoring System” descritto da Hsu et al (Addendum V)
intende guidare il giudizio sulla opportunità di trattamento in base all'assegnazione di un punteggio. Detto punteggio riflette la compromissione d’organo, in particolare polmoni, reni, apparato gastrointestinale e fegato.
10.3.4 Il trattamento consta di Carbo-VP. Poichè una regressione può avvenire
precocemente (anche dopo un solo ciclo), la decisione di continuare il trattamento sarà presa valutando il paziente dopo ogni ciclo. Infant con un punteggio > 2 (o > 1 per i neonati) riceveranno un ulteriore ciclo. In caso di infiltrazione midollare che persista dopo 2 cicli, si suggerisce la discussione del caso con il Coordinatore Nazionale.
10.3.5 Se non si osserva risposta dopo 2 cicli di Carbo-VP, si passa a CADO
(massimo 4 cicli).
Vedi Sezione 13 ed Addendum VI per i dettagli sulla chemioterapia. Nota Bene: per i neonati (età < 1 mese) o per gli infant di peso < 5 kg, i dosaggi vanno ridotti di un terzo
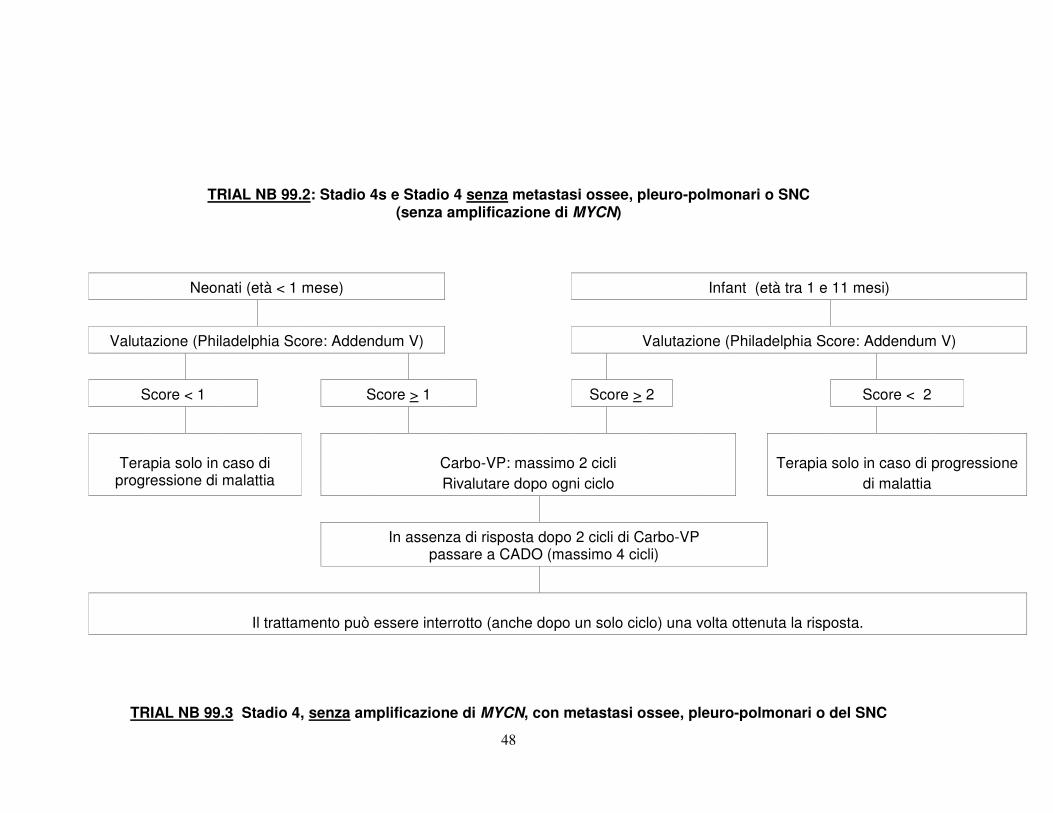
48
TRIAL NB 99.2: Stadio 4s e Stadio 4 senza metastasi ossee, pleuro-polmonari o SNC (senza amplificazione di MYCN)
Neonati (età < 1 mese) Infant (età tra 1 e 11 mesi)
Valutazione (Philadelphia Score: Addendum V) Valutazione (Philadelphia Score: Addendum V)
Score < 1 Score > 1 Score > 2 Score < 2
Terapia solo in caso di progressione di malattia
Carbo-VP: massimo 2 cicli Rivalutare dopo ogni ciclo
Terapia solo in caso di progressione
di malattia
In assenza di risposta dopo 2 cicli di Carbo-VP passare a CADO (massimo 4 cicli)
Il trattamento può essere interrotto (anche dopo un solo ciclo) una volta ottenuta la risposta.
TRIAL NB 99.3 Stadio 4, senza amplificazione di MYCN, con metastasi ossee, pleuro-polmonari o del SNC
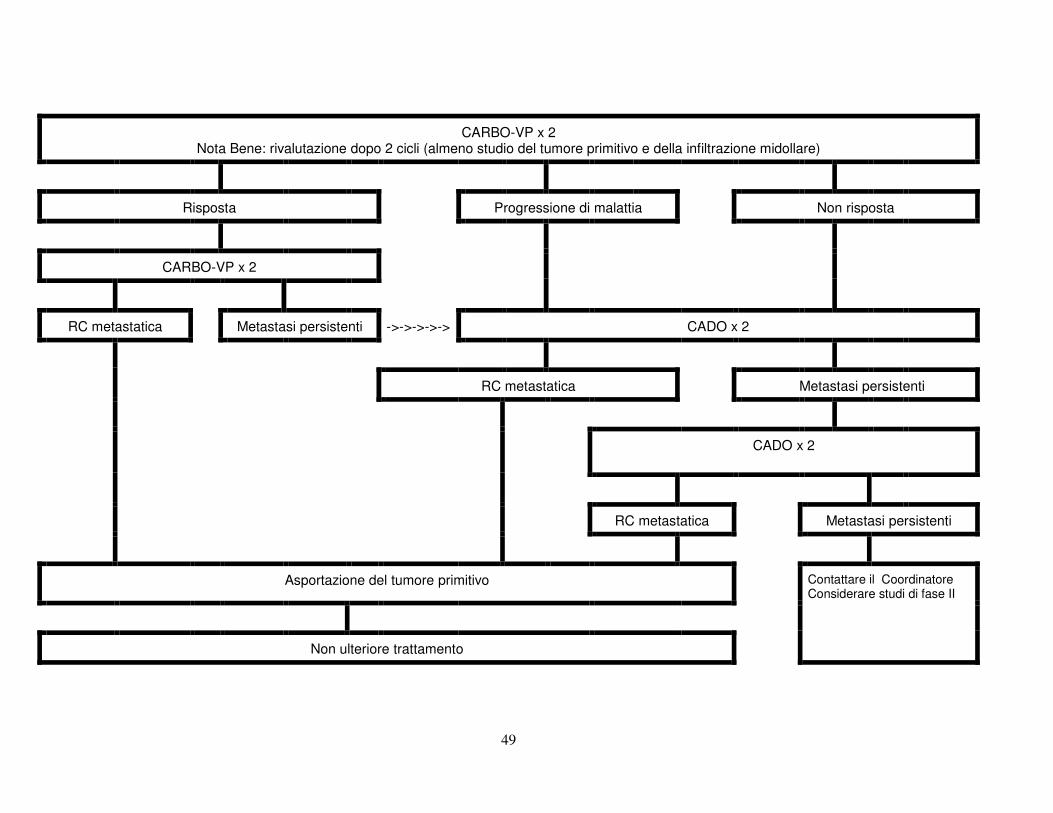
49
CARBO-VP x 2 Nota Bene: rivalutazione dopo 2 cicli (almeno studio del tumore primitivo e della infiltrazione midollare)
Risposta Progressione di malattia Non risposta
CARBO-VP x 2
RC metastatica Metastasi persistenti ->->->->-> CADO x 2
RC metastatica Metastasi persistenti
CADO x 2
RC metastatica Metastasi persistenti
Asportazione del tumore primitivo Contattare il Coordinatore Considerare studi di fase II
Non ulteriore trattamento

50
10.4 TRIAL NB 99.3 Stadio 4 (metastasi ossee, pleuriche, polmonari o del SNC) senza amplificazione di MYCN
10.4.1 Questo Trial riguarda gli infant con metastasi scheletriche documentate con
radiografia standard (o TC) e/o coinvolgimento pleuro-polmonare e/o del SNC, ma senza amplificazione di MYCN.
10.4.2 Questi infant ricevono 2 cicli di Carbo-VP, seguiti, in assenza di
progressione, da altri due. Si eseguirà poi rivalutazione completa di tutte le sedi coinvolte: in caso di RC metastatica, sarà eseguita asportazione chirurgica del tumore primario, ove possibile. In assenza di risposta, o in presenza di progressione dopo 2 cicli di Carbo-VP, si passerà a CADO (vedi paragrafo successivo).
10.4.3 Se dopo 4 cicli di Carbo-VP non è stata ancora raggiunta la RC
metastatica, si passerà a CADO (massimo 4 cicli). La risposta verrà valutata dopo 2 cicli CADO: in caso di RC metastatica, verrà asportato il tumore primario, ove possibile; in caso di persistenza di metastasi si somministrano altri 2 cicli di CADO, rivalutando poi lo stato di malattia.
10.4.4 Se dopo un totale di 4 cicli CADO il paziente presenta ancora evidenza di
malattia metastatica, si raccomanda di discutere l'ulteriore scelta terapeutica (inclusi studi di fase II) con il Coordinatore Nazionale.
10.4.5 Non è previsto alcun trattamento dopo la chirurgia, a meno che non vi sia
progressione di malattia. In particolare, non si raccomanda di somministrare radioterapia su un eventuale tumore residuo.
10.4.6 Alla luce di pubblicazioni che riportano un’ottima prognosi di infant con
tumore stadio 4 MYCN non-amplificato, trattati con chemioterapia moderatamente intensiva (Paul18; Matthay19), non viene somministrata a questo gruppo di pazienti terapia ad alte dosi.
Vedi Sezione 13 ed Addendum VI per dettagli sulla chemioterapia. Nota Bene: per i neonati (età < 1 mese) o per gli infant di peso < 5 kg, i dosaggi vanno ridotti di un terzo

TRIAL NB 99.4 Stadi 2, 3, 4, 4s con amplificazione di MYCN (Lo stadio 1 è trattato con sola chirurgia)
Carbo-VP x 2 seguiti da CADO x 2
Valutazione completa
Raccolta di cellule staminali periferiche (o midollo osseo purchè non infiltrato)
Chirurgia, se non è stata già eseguita una resezione completa.
Nello stadio 4, in assenza di RC metastatica, ulteriore terapia previo contatto con il Coordinatore Nazionale
Post-chirurgia: Carbo-VP x 1 seguito da CADO x 1
Terapia ad alte dosi (Bu-Mel) + infusione di cellule staminali periferiche (PBSC) o midollari
Radioterapia sul tumore primitivo (non prima di 2 mesi dalla terapia ad alte dosi)

11 TRATTAMENTO: TUMORI CON AMPLIFICAZIONE DI MYCN 11.1 TRIAL NB 99.4 Tumore con amplificazione di MYCN: riguarda infant di
stadio 2, 3, 4, 4s, con tumore MYCN-amplificato (> 10 copie). 11.2 E’ essenziale che il risultato di MYCN sia disponibile il più rapidamente
possibile, e comunque non oltre 6 settimane dalla diagnosi. Sulla base del risultato di MYCN:
• gli infant con malattia metastatica e amplificazione di MYCN
continuano la terapia con CADO, • gli infant con malattia metastatica senza amplificazione di MYCN
continuano terapia secondo Trial 99.2 o 99.3 in rapporto alle sedi metastatiche interessate,
• gli infant con malattia localizzata e amplificazione di MYCN sono trattati secondo il Trial 99.4, ma saranno eleggibili solo quelli non precedentemente trattati con CO,
• gli infant con malattia localizzata e amplificazione di MYCN trattati con CO (terapia iniziata prima dell’informazione sullo stato di MYCN) sono registrati, seguiti ed analizzati separatamente.
11.3 Tutti gli infant con amplificazione di MYCN (eccetto stadio 1) vengono
trattati nella stessa maniera, sulla base del fatto che anche nei casi con malattia localizzata questa anomalia genetica implica una pessima prognosi (Rubie5). La terapia consiste in 2 cicli di Carbo-VP seguiti da 2 cicli di CADO e quindi chirurgia (se non eseguita in precedenza).
11.4 I pazienti di stadio 4, non in RC dopo Carbo-VP e CADO escono dallo
studio, e il loro successivo trattamento verrà discusso con i Coordinatori Nazionali.
11.5 Ad adeguato intervallo dalla chirurgia viene ripresa la chemioterapia per
complessivi 2 cicli (Carbo-VP x 1, CADO x 1). 11.6 Dopo almeno 3 settimane dall’ultimo ciclo somministrato (CADO) il paziente
riceve chemioterapia ad alte dosi (con Bu-Mel) seguito da infusione di cellule staminali. Vedi Sezione 12 per dettagli sulla procedura leucoaferetica.
11.7 Il protocollo prevede la radioterapia sul letto tumorale a distanza di almeno
2 mesi dalla terapia ad alte dosi (Sezione 15: Radioterapia).

53
12 LEUCOAFERESI 12.1 La raccolta di cellule staminali (da sangue periferico o, se ciò non fosse
possibile, da midollo osseo) va eseguita dopo il 4° ciclo di terapia, quando il midollo è privo di infiltrazione neoplastica (morfologia ed anti GD2).
12.2 La raccolta da sangue periferico viene eseguita nella fase di recupero
ematologico, con somministrazione e.v. o sotto cute di G-CSF, alla dose di 10 µg/kg per 5 giorni consecutivi, con monitoraggio quotidiano del livello di CD34+ circolanti/µl, iniziando da valori di GB >1000/µl. Il giorno di inizio della stimolazione coincide con il nadir ematologico. La raccolta va eseguita il giorno stesso o il giorno successivo alla determinazione di un livello di CD34+ circolanti >20/µl. Se fattibile in base al livello di CD34+ circolanti la raccolta va ripetuta il giorno successivo. In ogni caso la procedura deve essere ripetuta dopo il ciclo di chemioterapia successivo se le cellule CD34+ raccolte sono <5x106/kg.
12.3 Per infant di peso inferiore a 10 kg, si raccomanda che il separatore cellulare
sia tarato con globuli bianchi depleti CMV-negativi, globuli rossi risospesi in albumina al 5% e diluiti con soluzione fisiologica in modo da ottenere valori in linea con l’ematocrito del paziente.20
12.4 Non sono previsti tentativi di “purificare” le cellule raccolte. Le cellule raccolte
vanno congelate e reinfuse dopo la chemioterapia ad alte dosi (Bu-Mel). 12.5 Per quanto concerne la logistica relativa alla terapia ad alte dosi valgono le
indicazioni date nell’ambito del protocollo AIEOP NB97. Vedi Sezione 13 ed Addendum VI per dettagli sulla chemioterapia. Nota Bene: per neonati (età < 1 mese) o per gli infant di peso < 5 kg, i dosaggi vanno ridotti di un terzo.

54
13 CHEMIOTERAPIA Per dettagli sui farmaci fare riferimento all’Addendum VI. Le dosi dei farmaci sono espresse in mg/kg. Per i neonati (età < 1 mese) e per gli infant di peso < 5 kg le dosi vanno ridotte di un terzo. 13.1 CICLOFOSFAMIDE E VINCRISTINA (CO) I cicli di CO sono somministrati ad intervalli di 2 settimane. FARMACO Dose (mg/kg) Somministrazione Ciclofosfamide 5 e.v.lenta x 5 giorni Vincristina 0.05 e.v.lenta x 1 giorno
FARMACO GIORNO
1 2 3 4 5
Vincristina •
Ciclofosfamide • • • • •
13.2 CARBOPLATINO ED ETOPOSIDE (CARBO-VP)
I cicli Carbo-VP sono somministrati, via catetere venoso centrale, ad intervalli di tre settimane, ai giorni 1-3 di ogni ciclo. Per quanto concerne idratazione e prevenzione del vomito, si rimanda al protocollo AIEOP NB 97.
FARMACO Dose (mg/kg) Somministrazione Carboplatino 6.6 in SG 5% (5 ml/kg) in 1 ora x 3 giorni consecutivi Etoposide (VP16) 5 in SF (12.5 ml/kg) in 2 ore x 3 giorni consecutivi FARMACO GIORNO
1 2 3
Carboplatino • • •
Etoposide • • •

55
13.3 CICLOFOSFAMIDE, DOXORUBICINA E VINCRISTINA (CADO)
I cicli CADO vanno somministrati via catetere venoso centrale, ad intervalli di tre setttimane. FARMACO Dose (mg/kg) Somministrazione Ciclofosfamide 10 in SG 5% (5 ml/kg) in 1 ora x 5 giorni consecutivi Doxorubicina 1 in SF in 6 ore ai giorni 4 e 5 Vincristina 0.05 in bolo ai giorni 1 e 5 FARMACO GIORNO
1 2 3 4 5
Ciclofosfamide • • • • •
Doxorubicina • • Vincristina • •
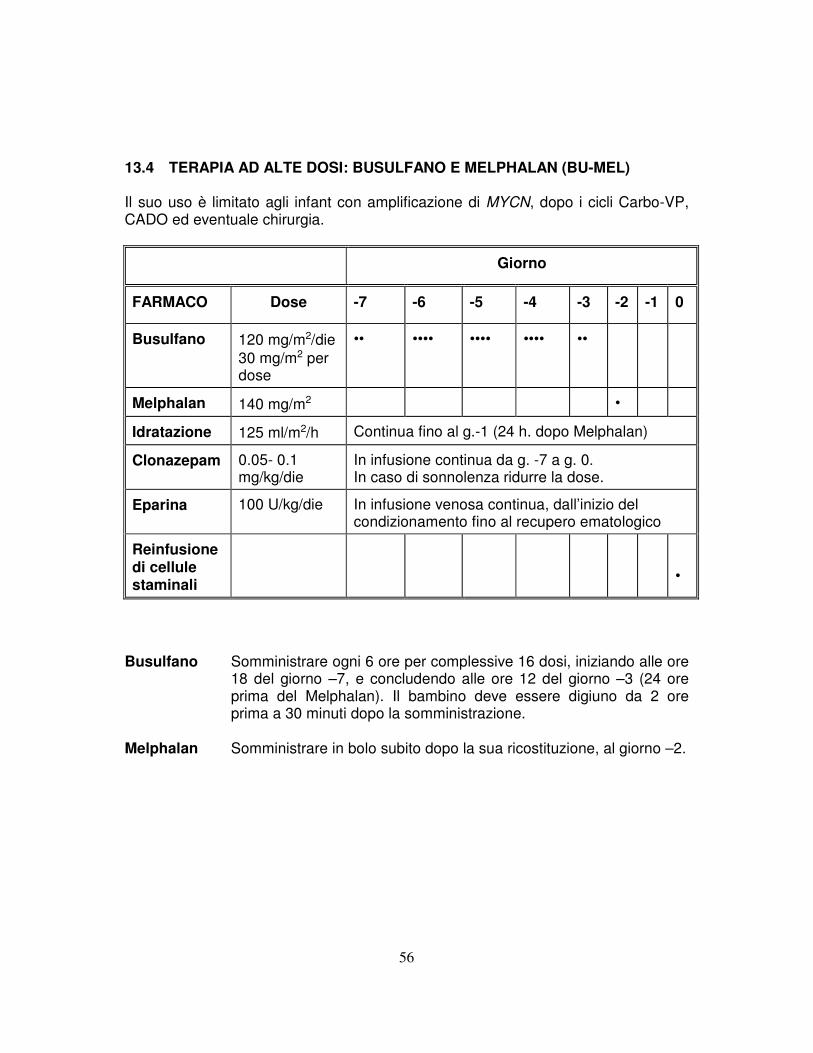
56
13.4 TERAPIA AD ALTE DOSI: BUSULFANO E MELPHALAN (BU-MEL) Il suo uso è limitato agli infant con amplificazione di MYCN, dopo i cicli Carbo-VP, CADO ed eventuale chirurgia.
Giorno
FARMACO Dose -7 -6 -5 -4 -3 -2 -1 0
Busulfano 120 mg/m2/die 30 mg/m2 per dose
•• •••• •••• •••• ••
Melphalan 140 mg/m2 •
Idratazione 125 ml/m2/h Continua fino al g.-1 (24 h. dopo Melphalan)
Clonazepam 0.05- 0.1 mg/kg/die
In infusione continua da g. -7 a g. 0. In caso di sonnolenza ridurre la dose.
Eparina 100 U/kg/die In infusione venosa continua, dall’inizio del condizionamento fino al recupero ematologico
Reinfusione di cellule staminali
•
Busulfano Somministrare ogni 6 ore per complessive 16 dosi, iniziando alle ore
18 del giorno –7, e concludendo alle ore 12 del giorno –3 (24 ore prima del Melphalan). Il bambino deve essere digiuno da 2 ore prima a 30 minuti dopo la somministrazione.
Melphalan Somministrare in bolo subito dopo la sua ricostituzione, al giorno –2.

57
13.5 TERAPIA DI SUPPORTO DURANTE LA TERAPIA AD ALTE DOSI 13.5.1 Attenzione
Questo ciclo va somministrato in Centri esperti nella gestione di bambini sottoposti a terapia ad alte dosi. Valutare, se necessario, l’eventuale trasferimento del bambino in un altro Centro per questa parte della terapia.
13.5.2 Idratazione
Liquidi endovena (SG 5% + elettroliti) alla velocità di 125 ml/m2/h (equivalenti a 3000 ml/m2/die) dovrebbero essere somministrati, attraverso una vena centrale, dal g. –8 (il giorno prima dell’inizio del Busulfano) al g.-1 (giorno seguente la somministrazione di Melphalan). Se necessario somministrare diuretici per mantenere un adeguato bilancio urinario. Valutare la necessità di aumentare la concentrazione del glucosio o di somministrare nutrizione parenterale totale in caso di diminuzione dell'apporto calorico per os.
13.5.3 Terapia anticonvulsivante. Si suggerisce l'uso del Clonazepam in infusione
continua (da 0.05 a 0.1 mg/kg/die) dal g. -7 (primo giorno di Busulfano) al g. 0 (giorno della infusione di cellule staminali periferiche). In caso di sonnolenza considerare l’uso di una preparazione orale.
13.5.4 Profilassi della malattia veno-occlusiva epatica (VOD)
• Eparina, 100 U/kg/die in infusione venosa continua dall’inizio del condizionamento fino al recupero ematologico. Controllare settimanalmente il profilo coagulativo; oppure
• Acido ursidesossicolico, 300 mg/m2/die in 2-3 somministrazioni, dal giorno –7 a giorno +80. Il farmaco (nome commerciale Ursodiol) è disponibile in compresse da 300 mg.
13.5.5 Cotrimossazolo (“Bactrim” o simili: sulfametossazolo + trimethoprim)
Va somministrato per via orale, alla dose di 25 mg/kg di sulfametossazolo tre volte alla settimana, dall’inizio del trattamento fino a 6 mesi dopo la infusione delle cellule staminali. Può essere sospeso se la neutropenia persiste oltre il g.+20 dalla infusione. In caso di allergia il cotrimossazolo può essere sostituito con pentamidina nebulizzata.
13.5.6 Antibiotici. La decisione di somministrare profilassi antibiotica è lasciata alla discrezione dei singoli Centri.
13.5.7 Profilassi antifungina. La profilassi con ketoconazolo, itraconazolo o fluconazolo, a causa dell’aumentato rischio di VOD con tali farmaci, va evitata a meno che non siano state utilizzate cellule staminali midollari, situazione nella quale il rischio di infezioni micotiche è superiore.

58
14 DIAGNOSI E TERAPIA DELLA VOD 14.1 Quadro clinico La VOD è definita clinicamente dalla presenza di almeno due dei seguenti
criteri21: • Epatomegalia e/o dolore riferito all’ipocondrio destro. • Ittero • Ascite • Aumento di peso (> 2.5% rispetto al peso iniziale) non altrimenti spiegato.
Può coesistere versamento pleurico, con conseguente distress respiratorio. La febbre è frequente, ma va valutata la possibilità che sia di origine infettiva.
14.2 Alterazioni bio-umorali In più del 90% dei casi si osserva alterazione della funzionalità epatica, con
iperbilirubinemia. Ipertransaminasemia è osservata nel 60-70% dei casi. Meno frequenti, anche in assenza di segni clinici, sono le alterazioni della coagulazione, in particolare a carico dei Fattori VIII e X.
Una diminuzione dell’escrezione urinaria di sodio (< 10 mmol/L) è un segno
precoce e costante, e la sua normalizzazione è frequentemente il primo segno del recupero clinico. Sono a volte presenti alterazioni della funzione renale, che possono tuttavia riflettere la restrizione dell’apporto di liquidi.
La richiesta piastrinica può essere aumentata.
14.3 Ecografia L’indagine dimostra ingrandimento del fegato e della milza, ascite,
ispessimento della parete della colecisti, riduzione del diametro delle vene epatiche, aumento del diametro della vena porta e visualizzazione della vena paraombelicale. L’eco-doppler è utile ai fini diagnostici e nel controllo dell’evoluzione clinica23.
14.4 Aspetti istologici La biopsia epatica va eseguita solo in caso di dubbio diagnostico.
Istologicamente, la VOD corrisponde ad un ispessimento concentrico subendoteliale e ad un restringimento del lume delle venule epatiche terminali o delle sovralobulari, ad opera di reticolo edematoso o collagene24.
14.5 Prevenzione Esistono due studi randomizzati che dimostrano una certa efficacia sia
dell’eparina 25 che dell’acido ursodesossicolico21.

59
14.6 Terapia I bambini che sviluppano VOD richiedono una specifica terapia di supporto:
• Restrizione dei liquidi (60 ml/kg/die), corretta in base alla funzione renale. • Restrizione dell’apporto di sodio, ricordando che le trasfusioni piastriniche
ne contengono in quantità considerevole. • Spironolattone (5 mg/kg/die), 5 giorni la settimana, con intervallo di due
giorni in rapporto alla lunga emivita del farmaco. • La furosemide è di solito inefficace e talvolta aggrava l’insufficienza renale. • Il livello piastrinico va mantenuto al di sopra di 20 x 109/l. • Il dolore addominale può essere intenso e richiedere trattamento
analgesico con oppiacei. • Il ricorso alla paracentesi va strettamente limitato ai casi di insufficienza
respiratoria. • La somministrazione di albumina è consigliata solo quando la sua
concentrazione scende al di sotto di 15 g/l. • Farmaci come rt-PA, PGE o defibrotide, pur non entrati nell’uso corrente,
possono essere utilizzati in rapporto all'esperienza dei singoli Centri.

60
15 LINEE GUIDA RADIOTERAPICHE 15.1 La radioterapia non ha fatto parte finora del trattamento di prima linea degli
infant con neuroblastoma. 15.2 Essa è a volte utilizzata:
• nello stadio 4s in progressione non responsiva alla chemioterapia; • negli stadi 3 e 4 con malattia residua o progressione dopo
chemioterapia e chirurgia. 15.3 Nel presente protocollo tutti i bambini con amplificazione di MYCN, eccetto lo
Stadio 1, sono candidati a ricevere radioterapia sulla sede del tumore primitivo (dopo avere concluso il programma di chemioterapia senza progressione), dopo il recupero ematologico dalla terapia ad alte dosi, prevedibilmente non prima di 2 mesi dall'infusione di cellule staminali periferiche. La funzione epatica deve essere nella norma.
15.4 La radioterapia deve essere effettuata utilizzando un acceleratore lineare. Il
piano di cura radioterapico viene disegnato sulla base delle immagini TC o RMN. In caso di residuo a livello della sede del tumore primitivo, il campo di irradiazione deve comprendere il GTV (Gross Tumor Volume) con 0,5-1 cm di margine per eventuali aree di malattia macroscopica e 0,5-1 cm per riposizionamento e respirazione. In totale il PTV (Planning Tumor Volume) deve corrispondere al GTV con 1 o 2 cm di margine. Si raccomanda l'uso di più campi personalizzati con variazioni di dose comprese tra +/- 5%.
15.5 Se la radioterapia è somministrata dopo asportazione completa del tumore
primitivo il letto tumorale va trattato in base alle immagini pre-operatorie, alla dose di 21 Gy in frazioni giornaliere di 1.5 Gy. Un boost di 9 Gy, in 6 frazioni (fino a raggiungere la dose totale di 30 Gy, in 20 frazioni complessive) va erogato su eventuali residui persistenti dopo la terapia ad alte dosi. La dose deve essere specificata in accordo con le raccomandazioni ICRU.
15.6 Alcuni pazienti non sono eleggibili al trattamento radioterapico sopra
formulato a causa della sede e/o del volume del tumore primitivo. Per alcuni organi esiste infatti una limitata tolleranza alla radioterapia, della quale la formulazione del piano di cura deve tener conto (in queste situazioni si suggerisce di discutere il caso con il radioterapista e con il Coordinatore del Trial).
Fegato: in caso di tumori dell’emiaddome destro la radioterapia non deve interessare più di metà del volume epatico, e la dose erogata all'organo non deve superare i 20 Gy.

61
Midollo spinale: il tratto di midollo incluso nel campo di irradiazione non deve superare i 10 cm, e la dose totale i 20 Gy ad eccezione di una piccola porzione che può ricevere fino ad un massimo di 30 Gy. Reni: la dose erogata al singolo rene non deve superare i 15 Gy. Quella erogata a parte di un rene non deve superare i 20 Gy Osso: è inevitabile un effetto "tossico" su vertebre situate nel campo di irradiazione per dosi superiori a 15 Gy. Attenzione va rivolta al mantenimento della simmetria del campo di irradiazione sull’intera vertebra. Altri organi: è improbabile che altri organi vengano irradiati con dosi superiori a quella tollerata. In alcuni casi tuttavia, come per voluminose neoplasie mediastiniche, può essere necessario includere una parte rilevante di cuore e/o polmone nel campo di trattamento.

62
16 VALUTAZIONE DELL’ESTENSIONE DI MALATTIA E DELLA RISPOSTA (secondo i criteri INSS, Addendum I)
Sede tumore
Valutazione consigliata
Tumore primitivo
TC e/o RMN con misurazione dei 3 diametri (*) Scintigrafia con MIBG, se possibile Scintigrafia con Tc99 MDP, se MIBG non disponibile
Metastasi
Midollo osseo
Aspirati delle due creste iliache posteriori e biopsie osteo-midollari (devono contenere almeno 1 cm di midollo, cartilagine esclusa)
Scheletro Scintigrafia con MIBG Scintigrafia con Tc99 MDP, se MIBG non disponibile o negativa sul tumore primitivo
Linfonodi
Esame clinico (linfonodi palpabili). TC dei linfonodi non palpabili (con misurazione delle 3 D)
Addome
TC o RMN con misurazione dei 3 diametri di eventuali masse.
Torace Radiografia standard.
TC o RMN se la radiografia risulta positiva, o se la massa o i linfonodi addominali si estendono fino al torace.
(*) L’esame ecografico non è incluso in quanto non sempre consente accurate misurazioni in bambini sotto l‘anno d’età.

63
17 MODIFICAZIONI DELLA TERAPIA IN RAPPORTO ALLA TOSSICITA’
17.1 Prima di ogni ciclo di chemioterapia la conta dei neutrofili deve essere
>1.000/mmc e quella delle piastrine >100.000/mmc. In caso di valori inferiori la terapia va rinviata, fino all’ottenimento di quelli sopraindicati.
17.2 Ridurre del 20% le dosi di Carboplatino ed Etoposide nei casi in cui il nadir
dei neutrofili scenda sotto i 500/mmc, e/o si siano verificati importanti problemi infettivi.
17.3 Si consideri la possibilità che la somministrazione di Etoposide può provocare
reazioni allergiche, anche gravi. In tal caso si sostituisca l’Etoposide con la Teniposide (VM26).
17.4 Si consiglia di eseguire un ecocardiogramma prima della somministrazione di
Doxorubicina e dopo ogni 2 cicli. La frazione di eiezione non deve scendere sotto il 30%.
17.5 La funzionalità renale va valutata con GFR prima del trattamento con alte
dosi. In caso di valori inferiori alla norma per l’età (Adendum VIII), contattare il Coordinatore del protocollo.
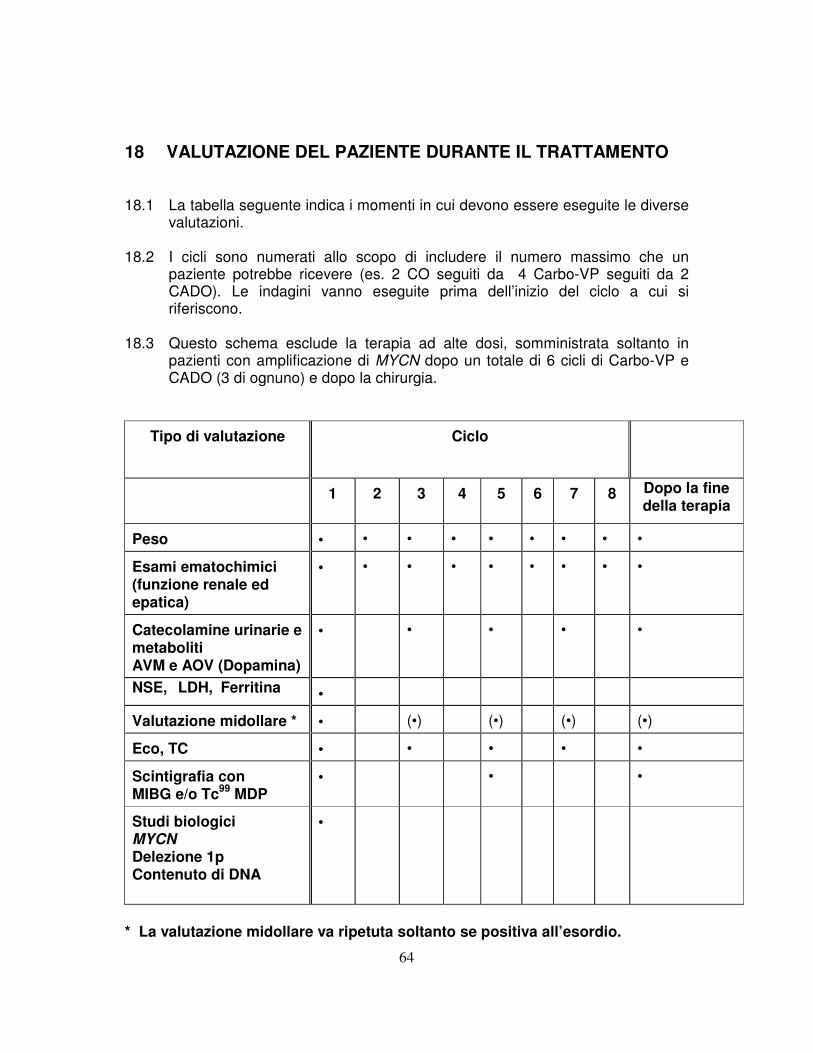
64
18 VALUTAZIONE DEL PAZIENTE DURANTE IL TRATTAMENTO 18.1 La tabella seguente indica i momenti in cui devono essere eseguite le diverse
valutazioni. 18.2 I cicli sono numerati allo scopo di includere il numero massimo che un
paziente potrebbe ricevere (es. 2 CO seguiti da 4 Carbo-VP seguiti da 2 CADO). Le indagini vanno eseguite prima dell’inizio del ciclo a cui si riferiscono.
18.3 Questo schema esclude la terapia ad alte dosi, somministrata soltanto in
pazienti con amplificazione di MYCN dopo un totale di 6 cicli di Carbo-VP e CADO (3 di ognuno) e dopo la chirurgia.
Tipo di valutazione Ciclo
1
2
3
4
5
6
7 8 Dopo la fine della terapia
Peso • • • • • • • • •
Esami ematochimici (funzione renale ed epatica)
• • • • • • • • •
Catecolamine urinarie e metaboliti AVM e AOV (Dopamina)
• • • • •
NSE, LDH, Ferritina •
Valutazione midollare * • (•) (•) (•) (•)
Eco, TC • • • • •
Scintigrafia con MIBG e/o Tc
99 MDP
• • •
Studi biologici MYCN Delezione 1p Contenuto di DNA
•
* La valutazione midollare va ripetuta soltanto se positiva all’esordio.

65
19 VALUTAZIONE DOPO LA FINE DELLA TERAPIA 19.1 Infant trattati con sola chirurgia
1. Studio radiologico (ecografia, TC e scintigrafia con MIBG se positiva all’esordio) ogni 3 mesi nel primo anno, ogni sei nel secondo e terzo anno, indi annualmente.
2. Dosaggio amine simpatiche urinarie e marcatori plasmatici a discrezione del clinico responsabile.
19.2 Infant trattati con chemioterapia
Il follow up dovrebbe comprendere valutazioni volte a determinare eventuale tossicità a lungo termine.
19.3 Carboplatino: follow up della funzione renale Una valutazione del GFR deve essere eseguita alla fine del trattamento e, se
alterato, ripetuta annualmente. Nei bambini in cui è possibile ottenere una raccolta urine delle 24 ore, è sufficiente la valutazione della clearance della creatinina. Quando ciò non è possibile è preferibile valutare il GFR con DTPA o Cr-EDTA.
19.4 Carboplatino: follow up della funzione uditiva Un controllo audiometrico dovrebbe essere eseguito alla fine del trattamento
e poi annualmente, fino all’età di 4-5 anni. 19.5 Doxorubicina: follow up della funzione cardiaca. Un ecocardiogramma dovrebbe essere eseguito entro 6 mesi dalla fine del
trattamento ed in seguito ogni 2 anni. In questo protocollo, la dose massima cumulativa di antracicline non dovrebbe superare i 180 mg/m2. Se la dose cumulativa supera i 300 mg/mq, si consiglia di seguire il paziente per tutta la vita.
19.6 Etoposide: rischio di seconda neoplasia E’ fondamentale che qualsiasi seconda neoplasia comparsa fra i pazienti
trattati con chemioterapia sia registrata urgentemente al centro raccolta dati.

66
20 CONSIDERAZIONI STATISTICHE 20.1 INTRODUZIONE
Sulla base dell’incidenza nota nei vari paesi potenzialmente coinvolti nello studio, si considera un arruolamento annuo di circa 150 pazienti. Si stima che il 90% sarà eleggibile e reclutato nello studio (90% di compliance) per un reclutamento annuo complessivo di 135 infant. In questa sezione sono definite considerazioni statistiche e stopping rules per i vari gruppi terapeutici.
• Trial 99.1
Si assume che il 35% degli infant eleggibili per lo studio sarà di stadio 2 o 3 (15% stadio 2, 20% stadio 3); fra questi circa un terzo dei tumori di stadio 2 ed il 75% di stadio 3 potrebbe essere non asportabile all’esordio e meno del 5% dovrebbe presentare amplificazione di MYCN. In accordo con questi assunti, il reclutamento annuo stimato dovrebbe essere di circa 25 pazienti. Con una durata di arruolamento di 4 anni, in questo studio di fase II non randomizzato potrebbero entrare 100 pazienti.
• Trial 99.2 Si assume che il 25% degli infant eleggibili per lo studio sia di stadio 4S o stadio 4 senza metastasi ossee e che meno del 5% di questi abbia tumori con amplificazione di MYCN. Dunque, il reclutamento annuo stimato dovrebbe essere intorno a 32-33 pazienti. Con una durata di arruolamento di 4 anni, in questo studio di fase II non randomizzato potrebbero entrare 130 pazienti.
• Trial 99.3
Si assume che il 15% degli infant eleggibili per lo studio sia di stadio 4 (con metastasi ossee, pleuro-polmonari o SNC) e che circa il 20% di questi presenti amplificazione di MYCN. In accordo con questi assunti, il reclutamento annuo stimato dovrebbe essere intorno a 15 pazienti. Con una durata di arruolamento di 4 anni, in questo studio di fase II non randomizzato potrebbero entrare 60 pazienti.
• Trial 99.4
Si assume che il 25% degli infant eleggibili per lo studio sia di stadio 4S o stadio 4 non osseo, il 15% stadio 4 di altro tipo, il 35% stadio 2 o 3 e che il 15% degli stadi 4 o 4S presenti amplificazione di MYCN, contro il 5% degli stadi 2 o 3. In accordo con questi assunti, il reclutamento annuo stimato di casi con MYCN amplificato dovrebbe essere di circa 10 pazienti. Con una durata di arruolamento di 4 anni, in questo studio di fase II non randomizzato potrebbero entrare 40 pazienti.

67
20.2 ENDPOINTS 20.2.1 Per ogni Trial viene definito un endpoint basato sulla sopravvivenza a lungo
termine. Inoltre, come garanzia di sicurezza, allo scopo di evitare un eccesso di eventi aspettando 2-3 anni e l'arruolamento di molti pazienti, prima dell'analisi finale, viene proposta per ogni Trial una stopping rule basata su criteri a breve termine.
20.2.2 Le stopping rules sono basate sulla procedura multi stage di Fleming28.
Saranno eseguite 4 analisi ad interim, quando il 25%, 50%, 75% e 100% dei pazienti avranno raggiunto il minimo follow up necessario. Se un limite più alto è superato durante un’analisi ad interim, la stopping rule sarà stata raggiunta.
20.2.3 La decisione se chiudere o meno un Trial è essenzialmente medica, e le
stopping rules statistiche rappresentano semplicemente delle linee guida. E’ possibile stabilire dei limiti basati su precedenti Trial, tuttavia ogni decisione relativa alla chiusura dello studio è una decisione clinica.
20.2.4 Se durante l’analisi ad interim non tutti i pazienti hanno il follow up minimo
richiesto (poiché alcuni vengono persi di vista), la percentuale di eventi o di decessi verrà calcolata con il metodo di Kaplan-Meier29. Il risultato sarà paragonato al limite superiore di rifiuto di Fleming. Se risulterà uguale o superiore al corrispondente limite, allora le stopping rules statistiche saranno state raggiunte.
20.2.5 Ciò significa che l’arruolamento continuerà prima della valutazione della
risposta nel gruppo precedente di pazienti. E’ chiaro che al momento dell’esecuzione della terza analisi (effettuata sul 75% dei pazienti seguiti per il tempo richiesto), la maggior parte dei pazienti sarà già stata inclusa nel Trial.
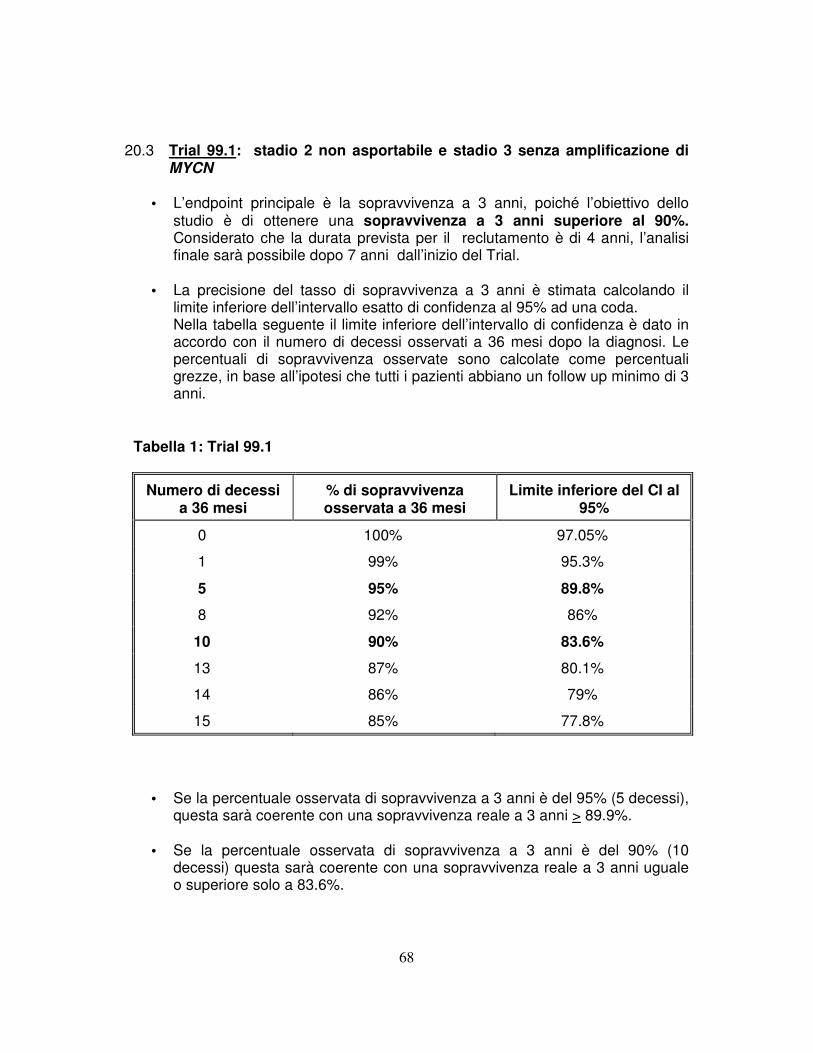
68
20.3 Trial 99.1: stadio 2 non asportabile e stadio 3 senza amplificazione di
MYCN
• L’endpoint principale è la sopravvivenza a 3 anni, poiché l’obiettivo dello studio è di ottenere una sopravvivenza a 3 anni superiore al 90%. Considerato che la durata prevista per il reclutamento è di 4 anni, l’analisi finale sarà possibile dopo 7 anni dall’inizio del Trial.
• La precisione del tasso di sopravvivenza a 3 anni è stimata calcolando il
limite inferiore dell’intervallo esatto di confidenza al 95% ad una coda. Nella tabella seguente il limite inferiore dell’intervallo di confidenza è dato in
accordo con il numero di decessi osservati a 36 mesi dopo la diagnosi. Le percentuali di sopravvivenza osservate sono calcolate come percentuali grezze, in base all’ipotesi che tutti i pazienti abbiano un follow up minimo di 3 anni.
Tabella 1: Trial 99.1
Numero di decessi a 36 mesi
% di sopravvivenza osservata a 36 mesi
Limite inferiore del CI al 95%
0 100% 97.05%
1 99% 95.3%
5 95% 89.8%
8 92% 86%
10 90% 83.6%
13 87% 80.1%
14 86% 79%
15 85% 77.8%
• Se la percentuale osservata di sopravvivenza a 3 anni è del 95% (5 decessi), questa sarà coerente con una sopravvivenza reale a 3 anni > 89.9%.
• Se la percentuale osservata di sopravvivenza a 3 anni è del 90% (10
decessi) questa sarà coerente con una sopravvivenza reale a 3 anni uguale o superiore solo a 83.6%.

69
STOPPING RULE BASATA SULLA PERCENTUALE DI EVENTI A 12 MESI
• La stopping rule è basata sulla percentuale di sopravvivenza libera da eventi
a 12 mesi, allo scopo di interrompere il Trial se l'incidenza di eventi è considerata troppo alta.
• Ogni decesso, compresi quelli dovuti alla tossicità (chirurgica o
chemioterapica) o a progressione di malattia, verificatosi entro i primi 12 mesi dalla diagnosi, sarà considerato un evento.
• Il calcolo dei limiti è stato effettuato assumendo come accettabile una
percentuale di eventi a 12 mesi <6%, e controllando la potenza allo scopo di riconoscere nel 90% dei casi una percentuale di eventi a 12 mesi > 15% (rischio di errore β del 10% di una accettazione scorretta del nuovo protocollo, se la percentuale di eventi reale è > 15%). Il rischio di errore α di chiudere erroneamente il protocollo è limitato allo 0.05 (probabilità di interrompere il Trial e rifiutare il protocollo, con una percentuale di eventi a 12 mesi reale < 6%).
• I primi 25 pazienti inclusi nello studio saranno analizzati quando sarà stato
raggiunto un follow up minimo di 12 mesi. La stopping rule sarà raggiunta se fra questi 25 pazienti un numero > 6 eventi viene osservato durante i primi 12 mesi (percentuale di eventi osservati =24%).
• Se il numero di eventi osservati è < 5, il reclutamento potrà continuare fino
alla analisi successiva, eseguita non appena un altro gruppo di 25 pazienti sarà stato seguito per almeno 12 mesi. La stopping rule verrà raggiunta qualora sia stato osservato fra questi 50 pazienti un numero > 8 eventi durante i primi 12 mesi (percentuale di eventi osservati =16%).
• Una terza analisi sarà effettuata quando altri 25 pazienti arruolati saranno stati seguiti per almeno 12 mesi. La stopping rule sarà raggiunta se fra questi 75 pazienti un numero > 9 eventi sarà stato osservato durante i primi 12 mesi (percentuale di eventi osservati = 12%).
• Se sarà stato osservato un numero di eventi < 8, l’ultima analisi sarà eseguita
sui 100 pazienti, dopo un follow up di almeno 12 mesi. Se, durante i primi 12 mesi, sarà stato osservato un numero di eventi > 11 (percentuale di eventi osservati =11%) il limite superiore sarà oltrepassato in favore di una percentuale di eventi eccessivamente alta (percentuale a 12 mesi > 6%).

70
20.4 Trial 99.2: stadio 4S e stadio 4 senza metastasi ossee (MYCN non
amplificato)
• L’endpoint principale è la sopravvivenza a 2 anni, e lo scopo dello studio è di ottenere una sopravvivenza a 2 anni superiore all’ 85%. Con una durata prestabilita per il reclutamento di 4 anni, l’analisi finale sarà possibile 6 anni dopo l’inizio del Trial.
• La precisione del tasso di sopravvivenza a 2 anni è stimata calcolando il
limite inferiore dell’intervallo esatto di confidenza al 95%. Nella tabella seguente, il limite inferiore dell’intervallo suddetto è calcolato in base al numero di decessi osservati a 24 mesi dalla diagnosi. Le percentuali di sopravvivenza osservate sono calcolate come dato grezzo, assumendo che tutti i pazienti abbiano un follow up minimo di 2 anni.
Tabella 2: Trial 99.2
Numero di decessi a 24 mesi
% di sopravvivenza osservata a 24 mesi
Limite inferiore del CI al
95%
0 100% 97.7%
5 96.1% 92.1%
10 92.3% 87.3%
12 90.8% 85.5%
13 90% 84.6%
14 89.2% 83.7%
19 85.4% 79.3%
20 84.6% 78.4%
21 83.8% 77.6%
• Se la percentuale di sopravvivenza a 2 anni osservata è del 90.8% (12
decessi), questa sarà coerente con una sopravvivenza reale a 2 anni > 85.5%.
• Se la percentuale di sopravvivenza a 2 anni osservata è del 90% (13
decessi) questa sarà coerente con una sopravvivenza reale a 2 anni > 84.6%.
• Comunque, se la percentuale di sopravvivenza a 2 anni è all’incirca 85% (19
o 20 decessi), questa sarà coerente con una percentuale reale soltanto del 78.4 (79.3 se i decessi sono 19).

71
STOPPING RULE BASATA SULLA PERCENTUALE DI DECESSI A 6 MESI
• La stopping rule è basata sulla percentuale di sopravvivenza a 6 mesi, allo scopo di interrompere il Trial se il numero di decessi a 6 mesi è considerato troppo alto.
• Qualsiasi decesso verificatosi durante i primi 6 mesi dopo la diagnosi sarà
considerato un evento. • Il calcolo dei limiti è stato effettuato considerando accettabile una
percentuale di eventi a 6 mesi <5%, e controllando la potenza allo scopo di riconoscere, nel 99% dei casi una percentuale di eventi a 6 mesi >15% (rischio di errore β del 2% di una accettazione scorretta del nuovo protocollo, se la percentuale di eventi reale è >15%). Il rischio di errore α di rifiutare erroneamente il protocollo è limitato allo 0.04 (probabilità di interrompere il Trial e rifiutare il protocollo, con una percentuale di decessi a 6 mesi reale <5%).
• I primi 32 pazienti inclusi nello studio saranno analizzati quando sarà stato
raggiunto un follow up minimo di 6 mesi. La stopping rule sarà raggiunta se, fra questi 32 pazienti, viene osservato durante i primi 6 mesi un numero > 7 decessi (percentuale di decessi osservata =21.9%).
• Se il numero di eventi osservati è < 6, il reclutamento potrà continuare fino
alla seconda analisi, che verrà eseguita non appena un altro gruppo di 32 pazienti sarà stato seguito per almeno 6 mesi. La stopping rule verrà raggiunta qualora sia osservato fra questi 64 pazienti durante i primi 6 mesi un numero > 8 decessi (percentuale di eventi osservati =12%).
• Una terza analisi sarà effettuata quando altri 32 pazienti arruolati saranno stati seguiti per almeno 6 mesi. La stopping rule sarà raggiunta se fra questi 96 pazienti un numero > 10 decessi sarà stato osservato durante primi 6 mesi (percentuale di eventi osservati = 10.4%).
• Se sarà stato osservato un numero di decessi < 9, l’ultima analisi sarà
eseguita sui 130 pazienti, dopo un follow up di almeno 6 mesi. Se sarà stato osservato un numero di decessi > 12 durante i primi 6 mesi (percentuale di eventi osservati =9.2%) il limite superiore sarà oltrepassato in favore di una percentuale di eventi eccessivamente alta (percentuale a 6 mesi > 5%).
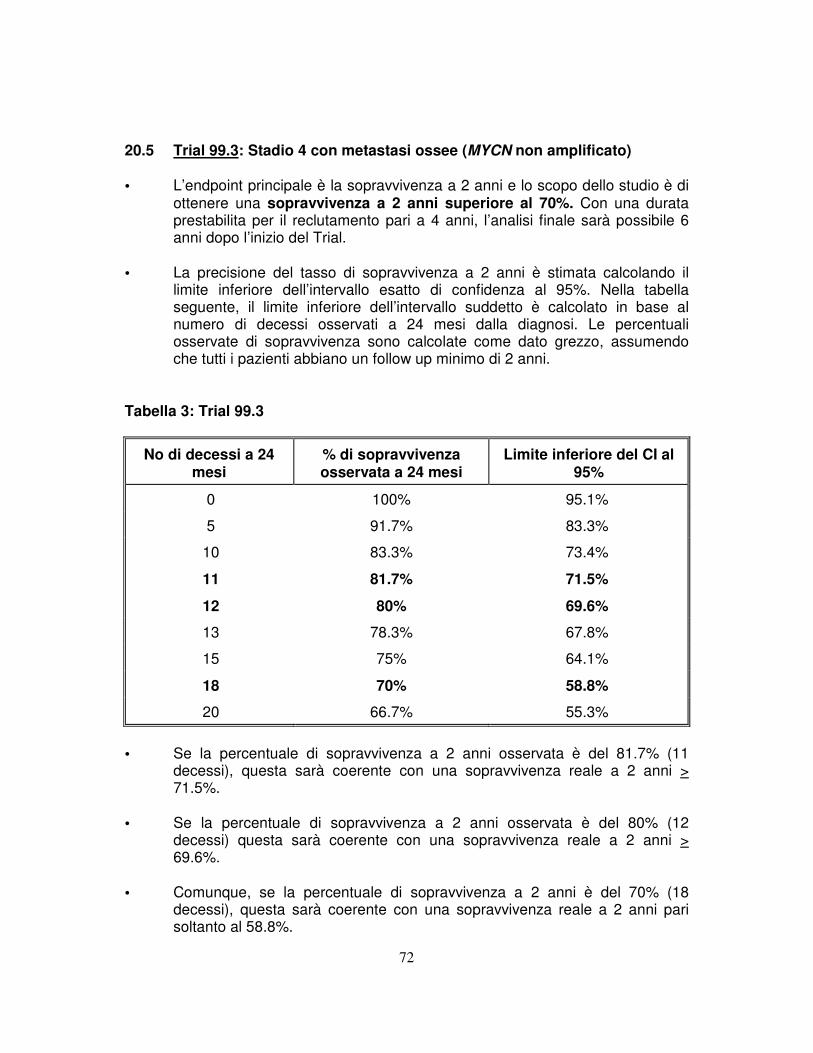
72
20.5 Trial 99.3: Stadio 4 con metastasi ossee (MYCN non amplificato) • L’endpoint principale è la sopravvivenza a 2 anni e lo scopo dello studio è di
ottenere una sopravvivenza a 2 anni superiore al 70%. Con una durata prestabilita per il reclutamento pari a 4 anni, l’analisi finale sarà possibile 6 anni dopo l’inizio del Trial.
• La precisione del tasso di sopravvivenza a 2 anni è stimata calcolando il
limite inferiore dell’intervallo esatto di confidenza al 95%. Nella tabella seguente, il limite inferiore dell’intervallo suddetto è calcolato in base al numero di decessi osservati a 24 mesi dalla diagnosi. Le percentuali osservate di sopravvivenza sono calcolate come dato grezzo, assumendo che tutti i pazienti abbiano un follow up minimo di 2 anni.
Tabella 3: Trial 99.3
No di decessi a 24 mesi
% di sopravvivenza osservata a 24 mesi
Limite inferiore del CI al 95%
0 100% 95.1%
5 91.7% 83.3%
10 83.3% 73.4%
11 81.7% 71.5%
12 80% 69.6%
13 78.3% 67.8%
15 75% 64.1%
18 70% 58.8%
20 66.7% 55.3%
• Se la percentuale di sopravvivenza a 2 anni osservata è del 81.7% (11
decessi), questa sarà coerente con una sopravvivenza reale a 2 anni > 71.5%.
• Se la percentuale di sopravvivenza a 2 anni osservata è del 80% (12
decessi) questa sarà coerente con una sopravvivenza reale a 2 anni > 69.6%.
• Comunque, se la percentuale di sopravvivenza a 2 anni è del 70% (18
decessi), questa sarà coerente con una sopravvivenza reale a 2 anni pari soltanto al 58.8%.

73
STOPPING RULE BASATA SULLA PERCENTUALE DI EVENTI A 12 MESI • La stopping rule è basata sulla percentuale di sopravvivenza libera da eventi
a 12 mesi, allo scopo di interrompere il Trial se il numero di eventi a 12 mesi è considerato troppo alto.
• Qualsiasi decesso (inclusi quelli per tossicità) verificatosi durante i primi 12
mesi dopo la diagnosi sarà considerato un evento. • Il calcolo dei limiti è stato effettuato considerando accettabile una
percentuale di eventi a 12 mesi <10%, e controllando la potenza allo scopo di riconoscere, nel 91% dei casi una percentuale di eventi a 12 mesi > 25% (rischio di errore β del 9% di una accettazione scorretta del nuovo protocollo, se la percentuale di eventi reale è >25%). Il rischio di errore α di rifiutare erroneamente il protocollo è limitato allo 0.05 (probabilità di interrompere il Trial e rifiutare il protocollo, con una percentuale reale di eventi a 12 mesi <10%).
• I primi 15 pazienti inclusi nello studio saranno analizzati quando sarà stato
raggiunto un follow up minimo di 12 mesi. La stopping rule sarà raggiunta se fra questi 15 pazienti un numero > 6 eventi viene osservato durante i primi 12 mesi (percentuale osservata di eventi =40%).
• Se il numero di eventi osservati è < 5, il reclutamento potrà continuare fino
alla seconda analisi eseguita non appena un altro gruppo di 15 pazienti sarà stato seguito per almeno 12 mesi. La stopping rule verrà raggiunta qualora fra questi 30 pazienti sia stato osservato un numero > 8 eventi durante i primi 12 mesi (percentuale di eventi osservati =26.7%).
• Una terza analisi sarà effettuata quando altri 15 pazienti arruolati saranno stati seguiti per almeno 12 mesi. La stopping rule sarà raggiunta se fra questi 45 pazienti un numero > 9 eventi sara stato osservato durante primi 12 mesi (percentuale di eventi osservati = 20%).
• Se sarà stato osservato un numero di decessi < 8, l’ultima analisi sarà
eseguita sui 60 pazienti, dopo un follow up di almeno 12 mesi. Se sarà stato osservato un numero di eventi > 11 durante i primi 12 mesi (percentuale di eventi osservati =18.3%) il limite superiore sarà oltrepassato in favore di una percentuale di eventi eccessivamente alta (percentuale a 12 mesi > 10%).

74
20.6 Trial 99.4: tutti gli stadi (eccetto stadio 1) con amplificazione di MYCN • L'endopoint principale è la sopravvivenza a 2 anni, e lo scopo è di
raggiungere una sopravvivenza a 2 anni di circa il 50%. Con una durata prestabilita per il reclutamento pari a 4 anni, l’analisi finale sarà possibile 6 anni dopo l’inizio del Trial.
• La precisione del tasso di sopravvivenza a 2 anni è stimata calcolando il
limite inferiore dell’intervallo esatto di confidenza al 95%. Nella tabella seguente, il limite inferiore dell’intervallo suddetto è calcolato in accordo con il numero di decessi osservati a 24 mesi dalla diagnosi. Le percentuali osservate di sopravvivenza sono calcolate come dato grezzo, assumendo che tutti i pazienti abbiano un minimo follow up di 2 anni.
Table 4: Trial 99.4
No di decessi a 24 mesi
% di sopravvivenza osservata a 24 m.
Limite inferiore del CI al 95%
0 100% 92.8%
10 75% 61.3%
13 67.5% 53.4%
14 65% 50.8%
15 62.5% 48.3%
18 55% 40.9%
19 52.5% 38.5%
20 50% 36.1%
23 42.5% 29.2
25 37.5% 24.7%
• Se la percentuale di sopravvivenza a 2 anni osservata è del 65% (14
decessi), questa sarà coerente con una sopravvivenza reale a 2 anni > 50.8%.
• Se la percentuale di sopravvivenza a 2 anni osservata è del 62.5% (15
decessi) questa sarà coerente con una sopravvivenza reale a 2 anni >48.3%. Entrambi i casi sono compatibili con una percentuale reale di sopravvivenza di circa il 50%.

75
• Se, comunque, la percentuale di sopravvivenza a 2 anni è del 50% (20 decessi), questa sarà coerente con una soprevvivenza reale a 2 anni pari soltanto al 36.1%.

76
STOPPING RULE BASATA SULLA PERCENTUALE DI EVENTI A 12 MESI • La stopping rule è basata sulla percentuale di sopravvivenza libera da eventi
a 12 mesi, allo scopo di interrompere il Trial se il numero di eventi a 12 mesi è considerato troppo alto.
• Qualsiasi decesso (inclusi quelli per tossicità) verificatosi durante i primi 12
mesi dopo la diagnosi sarà considerato un evento. • Il calcolo dei limiti è stato effettuato considerando accettabile una percentuale
di eventi a 12 mesi <25%, e controllando la potenza allo scopo di riconoscere, nel 78% dei casi una percentuale di eventi a 12 mesi > 45% (rischio di errore β del 22% di una accettazione scorretta del nuovo protocollo, se la percentuale di eventi reale è >45%). Il rischio di errore α di chiudere il protocollo è limitato allo 0.035 (probabilità di interrompere il Trial e rifiutare il protocollo, con una % di eventi a 12 mesi reale <25%).
• I primi 10 pazienti inclusi nello studio saranno analizzati non appena sarà
stato raggiunto un follow up minimo di 12 mesi. La stopping rule sarà raggiunta se fra questi 10 pazienti un numero > 8 eventi viene osservato durante i primi 12 mesi (percentuale di eventi osservata =80%).
• Se il numero di eventi osservati è < 7, il reclutamento potrà continuare fino
alla seconda analisi, che verrà eseguita non appena un altro gruppo di 10 pazienti arruolati sarà stato seguito per almeno 12 mesi. La stopping rule verrà raggiunta qualora fra questi 20 pazienti sia stato osservato un numero > 11 eventi durante i primi 12 mesi (percentuale di eventi osservati =55%).
• Una terza analisi sarà effettuata quando altri 10 pazienti arruolati saranno stati seguiti per almeno 12 mesi. La stopping rule sarà raggiunta se fra questi 30 pazienti un numero > 13 eventi sara stato osservato durante primi 12 mesi (percentuale di eventi osservati = 43.3%).
• Se sarà stato osservato un numero di eventi < 12, l’ultima analisi sarà
eseguita su tutti i pazienti dopo un follow up di almeno 12 mesi. Se sarà stato osservato un numero di eventi > 16 durante i primi 12 mesi (percentuale di eventi osservati =40%) il limite superiore sarà stato oltrepassato in favore di una percentuale di eventi eccessivamente alta (percentuale a 12 mesi > 25%).
STOPPING RULE BASATA SULLA PERCENTUALE DI MORTI TOSSICHE • Tutti i decessi dovuti a tossicità devono essere considerati come eventi se si
verificano durante i primi 8 mesi seguenti la diagnosi, a prescindere dal fatto che si verifichino dopo la chemioterapia ad alte dosi. I decessi per tossicità

77
occorsi durante tale periodo vanno tenuti in considerazione secondo le stopping rule, senza escludere i pazienti usciti dallo studio (analisi del “intent to treat”). I decessi verificatisi durante tale periodo, non correlati al trattamento, non saranno considerati.
• A causa dell’alta percentuale di decessi non dovuti a tossicità previsti durante
il primo anno, è difficile proporre un procedimento statistico come una stopping rule, poiché i decessi per malattia non possono essere considerati nell’ambito delle stopping rules relative alla tossicità.
• Si suggerisce di stimare la percentuale di decessi per tossicità attraverso il
metodo di Kaplan-Meier, e se possibile con il competing risks method, durante ognuna delle 4 analisi ad interim previste, e calcolare il suo intervallo di confidenza alla scopo di valutare se la percentuale è troppo alta. Una percentuale del 5% è da considerare accettabile.

78
21 BIBLIOGRAFIA 1] Prognostic Factors in Neuroblastoma AE Evans et al Cancer 59;1853-1859 (1987) 2] Clinical relevance of tumour cell ploidy and N myc amplification in childhood
neuroblastoma: a Paediatric Oncology Group study. AT Look et al J Clin Oncol 9;581-591 (1991) 3] Locally resectable neuroblastoma: results of the second study of the Italian
Cooperative Group for neuroblastoma. De Bernardi et al J Clin Oncol 13;884-893 (1995) 4] Disseminated neuroblastoma (stage IV and IV-S) in the first year of life. De Bernardi et al Cancer 70;1625-1633 (1992) 5] Survival from locally invasive or widespread neuroblastoma without cytotoxic
therapy. BH Kushner et al J Clin Oncol 14;373-381 (1996) 6] Successful management of low stage neuroblastoma without adjuvant
therapies: a comparison of two decades, 1972-1981 and 1982-1992 in a single institution.
AE Evans et al J Clin Oncol 14;2405-2410 (1996) 7] N Myc amplification is a major prognostic factor in localised neuroblastoma:
results of the French NBL 90 study. H Rubie et al J Clin Oncol 15;1171-1182 (1997) 8] Infants with neuroblastoma and regional lymph node metastases have a
favourable outlook after limited postoperative chemotherapy: A POG study. RP Castleberry et al J Clin Oncol 10;1299-1304 (1992) 9] Spontaneous regression of localised neuroblastoma detected by mass
screening. K Yamamoto et al J Clin Oncol 16;1265-1269 (1998)

79
10] Hepatomegaly in neuroblastoma stage 4S: criteria for treatment of the vulnerable neonate.
LL Hsu et al Med Ped Oncol 27;521-528 (1996) 11] Resection of the primary tumour is appropriate for children with stage IV-S
neuroblastoma: an analysis of 37 cases. DA Martinez et al J Pediatr Surg 27 1016-1021 (1992) 12] Resection of the primary tumour at diagnosis in stage IV-S neuroblastoma:
does it affect the clinical course? M Guglielmi et al J Clin Oncol 14;1537-1544 (1996) 13] Prognostic significance of age, MYCN oncogene amplification, tumour cell
ploidy and histology in 110 infants with stage D(S) neuroblastoma: the Paediatric Oncology Group experience.
HM Katzenstein et al J Clin Oncol 16;2007-2017 (1998) 14] Stage IV neuroblastoma over 1 year of age autografted: the combination used
in the conditioning regimen is the major prognostic factor. O Hartmann et al Med Ped Oncol 29;320 (1997) 15] International Criteria for diagnosis, staging and response to treatment in
patients with neuroblastoma. GM Brodeur et al J Clin Oncol 6;1874-18881 (1988) 16] Revisions in the international criteria for neuroblastoma diagnosis, staging, and
response to treatment. GM Brodeur et al J Clin Oncol 11;1466-1477 (1993) 17] Stage 4 neuroblastoma: adverse outcome of infants with N Myc amplification. O Hartmann et al Advances in Neuroblastoma Research abstracts (1998) 18] Stage IV neuroblastoma in infants. SR Paul et al Cancer 67;1493-1497 (1991) 19] Prognosis for stage IV neuroblastoma less than 1 year at diagnosis: a
prospective Children's Cancer Group study. K Matthay et al

80
ASCO proceedings 14; 446 (1995) 20] Peripheral blood stem cell collection in extremely low weight infants. W Nussbaumer et al Bone Marrow Transplantation 18;15-17 (1996) 21] Ursodiol prophylaxis against hepatic complications of allogeneic bone marrow transplantation. A randomised, double blind, placebo controlled trial. JH Essell et al Ann Int Med 128;975-981 (1998) 22] Veno-occlusive disease of the liver after bone marrow transplantation:
diagnosis, incidence and predisposing factors. GB McDonald et al Hepatology 4;16 (1984) 23] Complications hepatique apres chimotherapie lourde et autogreffe de moelle
dans les tumeurs solide de l'enfant. Brugieres L et al Presse Med 17;1305-1308 (1988) 24] Hepatic veno-occlusive disease after myeloablative treatment and bone
marrow transplantation: value of gray scale and Doppler ultrasound in 100 patients.
Lassau N et al Radiology 204;545-552 (1997) 25] An analysis of hepatic veno-occlusive disease and centrilobular hepatic
degeneration following bone marrow transplantation. Schulman HM et al Gastroenterology 79;1178-1191 (1980) 26] Prevention of hepatic veno-occlusive disease after bone marrow
transplantation by continuous infusion of low dose heparin. A prospective randomised trial.
Attal M et al Blood 79;2834-2840 (1992) 27] Cisplatin ototoxicity in children: a practical grading system. PR Brock et al Med Ped Oncol 19;295-300 (1991) 28] 24 hour endogenous creatinine clearance in infants and children without renal
disease. J Winberg Acta Pediatr 48; 443-52 (1959)

81
29] One sample multiple testing procedure for Phase II clinical trials. TR Fleming Biometrics 38;143-151 (1982) 30] Nonparametric estimation from incomplete observations. EL Kaplan, P Meier et al J Am Stat Assoc 53; 457-481 (1958)

ADDENDA
ADDENDUM 1 INTERNATIONAL CRITERIA FOR DIAGNOSIS, STAGING AND
RESPONSE TO TREATMENT............................................... 83
ADDENDUM 2 SURGICAL GUIDELINES ............................................................. 87
ADDENDUM 3 RADIOLOGICAL GUIDELINES ...................................................... 1
ADDENDUM 4 NEUROBLASTOMA WITH INTRASPINAL EXTENSION................ 8
ADDENDUM 5 PHILADELPHIA SCORE FOR STAGE 4S DISEASE ................... 13
ADDENDUM 6 DRUG INFORMATION.................................................................. 15
ADDENDUM 7 CLASSIFICATION OF CISPLATIN-INDUCED BILATERAL
FREQUENCY HEARING LOSS.............................................. 18
ADDENDUM 8 GLOMERULAR FILTRATION RATE ........................................... 19
ADDENDUM 9 RECOMMENDATIONS FOR GRADING OF TOXIC EFFECTS..... 20
ADDENDUM 10 PARENT INFORMATION SHEET/INFORMED ONSENT.............. 22
ADDENDUM 11 INTERNATIONAL STUDY CO-ORDINATION.............................. 25
ADDENDUM 12 DECLARATION OF HELSINKI .................................................... 31

83
ADDENDUM 1: INTERNATIONAL CRITERIA FOR DIAGNOSIS, STAGING AND RESPONSE TO TREATMENT
These definitions are taken from the Revised Guidelines published in 1993 (Brodeur et al Journal of Clinical Oncology volume 11 pp 1466-1477) Table 1 DIAGNOSIS OF NEUROBLASTOMA A diagnosis of neuroblastoma is established if : 1. An unequivocal pathological diagnosis is made from tumour tissue by light
microscopy (with or without immunohistology, electron microscopy, increased urine or serum catecholamines or metabolites )Ψ
OR 2. Bone marrow contains unequivocal tumour cells Ψ (eg syncytia or
immunocytologically positive clumps of cells) and increased urine or serum catecholamines or metabolites *.
Notes Ψ If histology is equivocal, karyotypic patterns or abnormalities in tumour cells
characteristic of other tumours [eg t(11;22)], then exclude a diagnosis of neuroblastoma, whereas genetic features characteristic of neuroblastoma (1p deletion, N-myc amplification) would support this diagnosis.
* Catecholamines and metabolites include dopamine, HVA and/or VMA; levels
must be > 3 SD above the mean for age (measured in �mol per mmol creatinine)# to be considered increased, and at least two of these metabolites must be measured.
# Timed urine collections are difficult in young children so normalisation per mmol
creatinine does away with the necessity for a timed collection and also avoids the problem of false negatives due to dilute urine.

84
Table 2 ASSESSMENT OF EXTENT OF DISEASE
Tumour Site
Recommended Tests
Primary Tumour
CT and/or MRI scan* with 3D measurements. MIBG scan if available†.
Metastatic Sites
Bone marrow Bilateral posterior iliac crest marrow aspirates and trephine (core) bone marrow biopsies required to exclude marrow involvement. A single positive site documents marrow involvement. Core biopsies must contain at least 1 cm of marrow (excluding cartilage) to be considered adequate
Bone MIBG† scan; 99Tc scan is required if MIBG scan negative or unavailable, and plain radiographs of positive lesions are recommended.
Lymph nodes Clinical examination (palpable nodes), confirmed histologically. CT scan for non-palpable nodes (3D measurements)
Abdomen/liver CT and/or MRI scan* with 3D measurements
Chest AP and lateral chest radiographs. CT/MRI necessary if chest radiograph positive, or if abdominal mass/nodes extend into the chest
Notes * Ultrasound examination considered suboptimal for accurate 3D measurements † The MIBG scan is applicable to all sites of disease.

85
Table 3 INSS STAGING Stage 1 Localised tumour with complete gross excision, with or without
microscopic residual disease; representative ipsilateral lymph nodes negative for tumour microscopically (nodes attached to and removed with the primary tumour may be positive).
Stage 2A Localised tumour with incomplete gross excision; representative
ipsilateral non-adherent lymph nodes negative for tumour microscopically.
Stage 2B Localised tumour with or without complete gross excision, with ipsilateral
non-adherent lymph nodes positive for tumour. Enlarged contralateral lymph nodes must be negative microscopically.
Stage 3 Unresectable unilateral tumour, infiltrating across the midline*, with or
without regional lymph node involvement; or localised unilateral tumour with contralateral regional lymph node involvement; or midline tumour with bilateral extension by infiltration or lymph node involvement.
Stage 4 Any primary tumour with dissemination to distant lymph nodes, bone,
bone marrow, liver and/or other organs. (except as defined for Stage 4S). Stage 4S Localised primary tumour (as defined for Stage 1, 2A or 2B), with
dissemination limited to liver, skin, and/or bone marrow †. (limited to infants < 1 year of age) Notes Multi-focal primary tumours (eg bilateral adrenal primary tumours) should be
staged according to the greatest extent of disease, as defined above, and be followed by a subscript "M" (eg Stage 3M).
* The midline is defined as the vertebral column. Tumours originating on one
side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column.
† Marrow involvement in Stage 4S should be minimal, ie less than 10% nucleated
cells on bone marrow biopsy or quantitative assessment of nucleated cells on marrow aspirate. More extensive marrow involvement should be considered Stage 4. The MIBG scan (if done) should be negative in the marrow for Stage 4S.
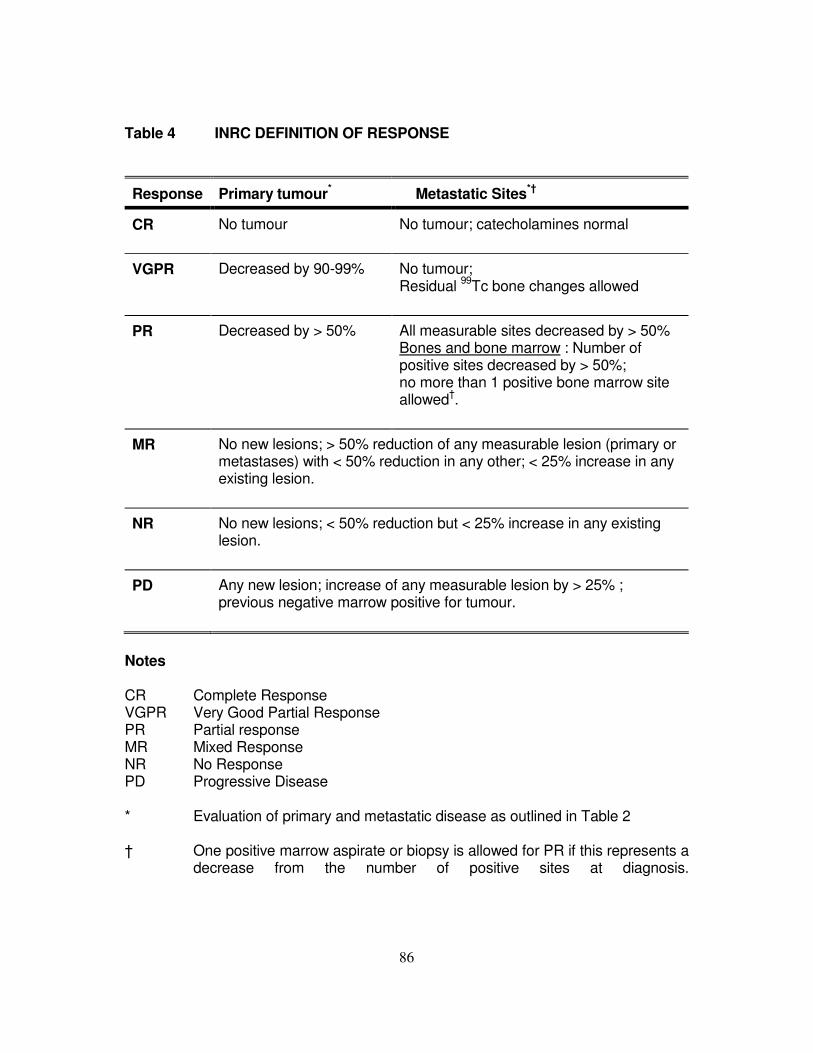
86
Table 4 INRC DEFINITION OF RESPONSE
Response Primary tumour* Metastatic Sites
*†
CR No tumour No tumour; catecholamines normal
VGPR Decreased by 90-99% No tumour; Residual 99Tc bone changes allowed
PR Decreased by > 50% All measurable sites decreased by > 50% Bones and bone marrow : Number of positive sites decreased by > 50%; no more than 1 positive bone marrow site allowed†.
MR No new lesions; > 50% reduction of any measurable lesion (primary or metastases) with < 50% reduction in any other; < 25% increase in any existing lesion.
NR No new lesions; < 50% reduction but < 25% increase in any existing lesion.
PD Any new lesion; increase of any measurable lesion by > 25% ; previous negative marrow positive for tumour.
Notes CR Complete Response VGPR Very Good Partial Response PR Partial response MR Mixed Response NR No Response PD Progressive Disease * Evaluation of primary and metastatic disease as outlined in Table 2 † One positive marrow aspirate or biopsy is allowed for PR if this represents a
decrease from the number of positive sites at diagnosis.

87
ADDENDUM 2 SURGICAL GUIDELINES
1 DEFINITIONS
1.1 Primary surgery 1.11 The definition of primary surgery as outlined here aims to distinguish initial surgery from
intentional biopsy. Primary surgery is defined as any surgery of the primary tumour performed before any other treatment. It can lead to radical or partial tumour excision, or be limited to a simple biopsy.
1.12 It is a primary intention of this protocol to avoid mutilating surgery in infants with neuroblastoma,
so every effort should be made to pre-operatively assess resectability using MR or CT imaging. If in doubt, initial biopsy is preferred to immediate radical resection. As biological investigations are essential for selecting treatment strategy in this study, biopsy at least should be carried out on all patients (see section 1.2).
1.2 Biopsy 1.21 Biopsy should be used for all patients with tumours that are evaluated as unresectable, and is
usually appropriate for infants with metastatic disease. 1.22 Biopsy aims to obtain the amount of tumour tissue required for cyto or histological diagnosis and
the biological investigations indicated in the protocol. In particular it is essential to that sufficient material is obtained for the accurate determination of N myc status.
1.23 Needle core biopsy can be considered if the local laboratory facilities allow full biological
investigation to be carried out on that tissue specimen, or arrangements are in place for needle core specimens to be assessed for biological investigations at a reference laboratory.
1.3 Radical excision 1.31 Radical excision should only be considered if a localised (non-metastatic) tumour appears
resectable on pre operative imaging. This implies the absence of risk factors. See section 6 of this appendix for further details.
1.32 This term defines the macroscopic extirpation of the tumour, including the removal of
macroscopically abnormal lymph nodes and sampling of normal ones. The surgical report must state that the excision has been radical.
1.33 The pathologic examination of the tumour mass and biopsies performed on tumour bed tissues
adds the information that the excision has been radical with or without microscopic residual tumour.
1.4 Excision with macroscopic residue 1.41 The tumour is thought pre-operatively to be resectable, but there is postoperative macroscopic
residue. The amount of the macroscopic residue must be stated as clearly as possible and should be evaluated by postoperative imaging.

88
2 PREOPERATIVE CRITERIA OF OPERABILITY 2.1 Age of the patient: It is not justified to adopt a more aggressive surgical approach in
infants with the aim of avoiding chemotherapy. 2.2 Definition of resectability In primary surgery the tumour is stated to be operable when its radical excision appears feasible
without risk to the patient, does not require any mutilation, and will not lead to important sequelae.
For further details of risk factors related to localisation of the tumour see Section 6 of Appendix. 2.3 Evaluation of the feasibility of radical excision The imaging techniques presently available should allow evaluation in most cases as to whether
the tumour is resectable. When MR or CT imaging suggest that the radical excision is unlikely, primary surgery should be avoided. However, there may be occasions where macroscopic residue remains when an excision which was expected to be complete turns out to be incomplete.
2.4 Neuroblastoma with intraspinal extension (See also Appendix 4) Children with dumbbell tumours in general are not candidates for primary radical resection, and
should be treated according to protocol (Trial 98.1). 2.5 Nephrectomy If preoperative MR or CT imaging has shown that radical surgery is feasible only by performing a
nephrectomy, primary surgery should be avoided. 2.6 Tumour spillage by dissection or rupture When preoperative imaging suggests that a radical tumour resection will be possible through the
dissection or the opening of the tumour itself, the surgeon is allowed to operate. At this time, he/she may face two different situations:
1) the tumour appears friable. The opening of the tumour with consequent tumour cell
spillage should be avoided. 2) the tumour appears firm and compact (this situation is most likely to occur in the thoracic
region). The surgeon may proceed and try to remove the tumour even though its section may be necessary.
2.7 Tumour relation with great vessels In order to obtain a further insight in the accuracy of data derived from the imaging techniques,
the surgeon should accurately describe the intra-operative findings and the technical difficulties encountered in the points of contact between tumour and vessels as documented in the MR or CT imaging. The lack of a fat layer between tumour and vessels does not contraindicate a primary surgery.

89
3 SURGICAL PROCEDURES 3.1 This section of the guidelines applies to situations where resection of the tumour is being
attempted: either as a primary procedure, or as a delayed procedure after initial chemotherapy. 3.2 Surgery of neuroblastoma aims not only to radically remove the primary tumour, but also to
evaluate its extension (staging) and provide adequate tumour tissue for histology and biological studies. Therefore, it includes the following steps:
a) biopsy of regional lymph nodes; b) exploration of the abdominal and/or pleural cavity (tumour bed), with biopsy of any
suspected lesion. 3.3 Surgical approach 3.31 In case of retroperitoneal neuroblastoma, a transverse laparotomy (at times extended to a
thoraco-phreno-laparotomy) is recommended. In cases of pelvic site of the primary tumour, a vertical laparotomy is recommended.
3.32 A double thoracotomy at a distance of 2-3 intercostal spaces, using the same incision, is
indicated in case of very large thoracic neuroblastoma, in order to obtain good access to the tumour.
3.33 In some instances, a combined surgical approach can be indicated. This may sometimes by
performed during the same operative session (other times, two subsequent sessions could be required).
The following points indicate some instances of combined surgical approach: a) approach through cervicotomy and thoracotomy in cervico-mediastinal or upper-
mediastinal neuroblastoma (single session); b) laparotomy and posterior sacral - coccygeal approach in some cases of pelvic
neuroblastoma (single session); c) approach through thoracotomy in some cases of mediastinal neuroblastoma extending
beyond the midline (two subsequent session with 3-4 weeks interval between them); d) laminectomy associated with thoracic or abdominal operation. 3.4 Use of clips The use of clips may affect interpretation of CT scan and MR examinations and is therefore not
recommended. 3.5 Lymph node evaluation As the available evidence is that infants with non-metastatic disease plus/minus nodal
involvement is good, and extensive surgical examination of nodes can be damaging it is not recommended that an aggressive search and sampling of nodes is undertaken. (RP Castleberry JCO 1992) According to the site of the primary tumour, lymph nodes from the following regions should be examined and biopsied, if they appear abnormal:
a) latero-cervical region: jugular chain and supraclavicular area; b) chest: mediastinal lymph nodes above and below the tumour; c) abdomen: infra-diaphragmatic are (coeliac nodes); mid aortic area (at the level of the
renal vessels); iliac region (bilaterally). Note: In some instances it may be difficult or even impossible to identify any lymph nodes. This
condition is defined as "absence of lymph nodes".

90
4 TIME AND MODE OF OPERATION 4.1 Immediate resection is considered appropriate when the surgeon, on the basis of clinical and
radiologic data, and after discussion with the oncologist, thinks it possible to perform a radical resection of the tumour. In other words, all localised (non-metastatic) tumours likely to be successfully operated should undergo immediate resection. However as stated above an initial surgical resection should only be undertaken if this can be complete and achieved without undue risk. No difficult vascular dissection or organ resection should be undertaken.
4.2 In cases of disseminated disease, it is inappropriate to carry out immediate resection unless
removal of the primary tumour is also considered the most satisfactory way of obtaining tissue for analysis.
4.3 Immediate surgery is not recommended when the surgeon, on the basis of clinical and
radiological data, thinks it is not possible to carry out a complete resection (or leave only minimal residue) in a neuroblastoma without metastases. A biopsy should be performed for diagnostic purposes and the attempt at radical surgery be delayed.
4.4 Where the surgeon realises it is impossible to obtain a complete excision during immediate
surgery, it is recommended that a biopsy is carried out and decision regarding removal of the tumour during a subsequent operation, can be made with the knowledge of the histology and biological data.
4.5 During delayed or second-look operations, the surgeon should aim to obtain a radical tumour
resection or a resection with minimal residual disease, without taking risks and/or provoking mutilation such as a nephrectomy.
4.6 Second-look surgery should be avoided in the following instances: a) complete resection or resection with minimal residue has been obtained during a
previous operation and no sign of relapse is evident; b) during treatment before second-look surgery, clear tumour progression or appearance
of metastases has occurred. 4.7 Second-look or delayed surgery can be avoided when chemotherapy has induced a complete
regression of the primary tumour, documented by imaging techniques (CT, MRI, US and MIBG scan).
4.8 Second-look surgery is to be considered in all cases of incomplete tumour resection or simple
biopsy during a previous operation and there has been a response to the subsequent therapy. Second-look surgery is mandatory even after complete excision or excision with minimal residue in case of signs of local relapse. As previously mentioned, second-look surgery is associated with accurate exploration of the tumour site and lymph node biopsies. If second-look surgery involves an incomplete resection or consists of biopsy only, subsequent re-operation is left to the surgeon's discretion.
4.9 It is recommended that the oncologist and pathologist attend any operation. One of their tasks is
to subdivide tumour tissue for histology, molecular studies, tumour tissue bank (see section s 7 & 8 in the main protocol). A detailed surgical report should be written down during the operation or immediately after.

91
5 SURGICAL MANAGEMENT IN STAGE 4S TUMOURS 5.1 Resection of the primary tumour is not required, in infants with stage 4S disease, unless this is
removed as part of the initial biopsy procedure. The available evidence does not demonstrate a benefit in terms of reducing recurrence if the
primary tumour is removed. (Guglielmi et al JCO 1996) 6 SURGICAL RISK FACTORS 6.1 Close attention to clinical examination and imaging studies can assist the surgeon (in
consultation with the oncologist and radiologist) in deciding whether a localised tumour is amenable to primary surgical resection. The presence of any of the risk factors listed below should lead to discussion as to whether primary surgery is the most appropriate therapy, or whether this should include an initial biopsy followed by management informed by the biological characteristics of the tumour.
6.2 RISK FACTORS RELATED TO LOCALISATION 6.21 Neck • Tumour encasing vertebral artery • Tumour encasing brachial plexus roots • Tumour crossing the midline 6.22 Thorax • Tumour encasing the trachea or principal bronchus • Tumour encasing the origin and branches of the subclavian vessels • Thoraco-abdominal tumour, peri-aortic fusiform tumour • Lower left mediastinal tumour, infiltrating the costo-vertebral junction between T9 and
T12 6.23 Retroperitoneal space • Adrenal tumour infiltrating the porta hepatis • Suprarenal tumour infiltrating the branches of the superior mesenteric artery at the
mesenteric root • Suprarenal tumour surrounding the origin of the coeliac axis, and of the superior
mesenteric artery • Tumour invading one or both renal peduncles • Fusiform tumour surrounding the infrarenal aorta • Tumour encasing the iliac vessels • Pelvic tumour crossing the sciatic notch • Tumour invading the branches of the hypogastric vessels 6.3 OTHER RISK FACTORS • Dumbbell tumour with neurological signs and no indication for emergency laminectomy
(see Appendix 4: Intraspinal neuroblastoma) • Risk factors related to tumour size • Risk factors related to tumour fragility (necrotic and liquid filled areas on tumour surface)

1
ADDENDUM 3 RADIOLOGICAL GUIDELINES
1 INTRODUCTION 1.1 The aim of these guidelines is to define standard procedures that will enable
accurate evaluation of the resectability of a localised neuroblastoma. 1.2 The different sections of the notification forms are intended to answer specific
questions. They should be completed according to the results of the whole imaging work-up.
1.3 It is not necessary to describe which investigation (ie US or CT) has provided a
given piece of information. 2 METHODS OF INVESTIGATION 2.1 The radiologist should select the most convenient imaging methods according
to the available equipment in his/her institution but should follow the INSS guidelines. [Appendix 1]
- Thoracic MRI. If not available, CT with IV contrast - Abdominal CT or MRI plus US - Cervical MRI. If not available CT with IV contrast - Pelvic CT or MRI 2.2 The relationship between the tumour and major adjacent vessels is of critical
importance for defining the resectability of a localised neuroblastoma. 2.3 Different techniques can provide this information eg US or CT after injection of
contrast medium. For MRI the choice of vertical planes parallel to the great vessels is indicated.
2.3 Pre-operative MRI sequences should be chosen to give maximum anatomical
detail. Contrast medium is not mandatory with MRI unless there are doubts about the tumour limits.

2
3 RADIOLOGICAL FORMS 3.1 Choice of the form 3.1.1 There is a specific form for each major anatomic region: cervical, thoracic,
abdominal, pelvic. 3.1.2 If a tumour extends across one compartment (cervico-thoracic,
thoraco-abdominal, abdomino-pelvic) its centre will define the primary tumour site. In this case a second form should be completed for the second compartment involved by the tumour.
3.1.3 For example: an abdominal tumour extending across the pelvic brim will be
evaluated by completing the abdominal form, while the part of the tumour extending in the pelvic area will be evaluated by completing the form for pelvic neuroblastoma.
3.2 Tumour size 3.2.1 In case of a round and well defined tumour the size will be defined by the
diameter. 3.2.2 If the tumour cannot be distinguished from adherent lymph nodes (in which
case the term “mass” or “tumour mass” will be preferred) the measurements should be approximated to the nearest half-centimetre and should be taken in the 3 orthogonal axes (x/y/z).
3.2.3 If the tumour has an irregular shape, tangents to the boundaries should define
the three dimensions. 4 DEFINITIONS OF TERMS TO BE USED WHEN DESCRIBING TUMOUR
EXTENT 4.1 TUMOUR and MASS 4.1.1 The term “tumour” indicates a well defined neoplasm, whereas the term “mass”
or “tumour mass” should be used when there are lymph nodes indistinguishable from the tumour because of its irregular shape. This should be clearly indicated when completing the forms.
4.2 LOCATION 4.2.1 For cervical, thoracic or pelvic neuroblastomas the vertebrae are identified by
two boxes: the first indicates the cervical, thoracic, lumbar or sacral level; the second indicates the number of the involved vertebra.

3
4.2.2 For abdominal neuroblastomas the definition of midline or lateral is given by the centre of the tumour. The limits of the midline are defined by INSS criteria ie a tumour is crossing over the midline if it infiltrates beyond the opposite side of the vertebral column. At a level of the sacrum the lateral limits are defined by a tangent to the lateral border of L5.
4.2.3 Midline tumours are classified according to their relationship with the great
vessels. 4.2.4 For example, a perivascular tumour encompasses the aorta while a lateral
tumour is described as “intervascular-renal” if it is located between one of the great vessels (IVC or aorta) and the kidney (which is usually displaced).
4.3 LOCAL EXTENSION OF THE PRIMARY TUMOUR 4.3.1 Extension to intervertebral foramina and spinal canal This defines the tumour involvement by continuity of one or more intervertebral
foramina with or without tumour extension into the spinal canal. In most cases it is difficult to measure the intraspinal tumour proportion due to
its half-moon shape. For practical purposes we may describe it by the percentage of its maximum size in relation to the diameter of the spinal canal: <25%, between 25 and 50%, >50%.
An abdominal neuroblastoma has a pelvic extension if the tumour passes over
the pelvic brim. 4.3.2 Relationship between tumour and vessels. The use of the following three terms is suggested: separated, in contact and
encased. - separated describes the situation where a clear-delineated-fat plane is found
between tumour and vessels. Its thickness has little relevance. - in contact describes the situation where no clear-delineated-fat plane is
found between tumour and vessels. - encased means that one or more vessels are within the tumour mass.

4
4.3.3 Relationship between tumour and contiguous organs. The use of the following two terms is suggested: separated or in contact. - separated describes a situation where either a clear-delineated-fat plane is
found between the tumour and contiguous organs or where a relative mobility is seen on dynamic US.
- in contact describes the situation where no clear-delineated-fat plane is
found between the tumour and contiguous organs. 5 INFORMATION DERIVED BY THE IMAGING STUDIES THAT MAY HELP
THE SURGEON TO DEFINE THE RESECTABILITY OF A NEUROBLASTOMA
5.1 THORACIC NEUROBLASTOMA 5.1.1 Relevant information includes: a) crossing of the midline, b) extension into
contiguous compartments (neck or abdomen), c) intraspinal extension, d) relation to the airways.
There is no practical benefit in localising the oesophagus. 5.1.2 Superior thoracic neuroblastoma The surgical access by thoracotomy does not usually allow a good view of the
upper part of the tumour. Therefore the pre-surgical evaluation should concentrate on: a) the possible cervical extension of the tumour, b) the involvement of the subclavian vessels and of their branches, c) the involvement of the roots of the brachial plexus.
5.1.3 Inferior thoracic neuroblastoma of the left side The medullary artery originates in 85% of the cases between T9 and T12 on
the left side. However, in 15% of the cases it originates on the right side and is usually associated with a supplementary circulatory circuit. Tumour growth within this area induces the formation of collaterals.
Therefore arteriography of the medullary artery is not mandatory, since the pre-
operative risk is minimal, although it should be mentioned to the parents. On the other hand an invasive procedure such as arteriography in very young children is risky.
Important elements to evaluate risk are: a) the precise tumour extension
towards the vertebrae T9-T12, b) the infiltration of the intervertebral foramina, and c) the infiltration of the posterior wall of the descending aorta.

5
5.2 ABDOMINAL NEUROBLASTOMA 5.2.1 Vascular investigation: the relationship of tumour to the veins is important to
define as it allows a better assessment of surgical risk. In fact, arterial injuries are easier and quicker to repair than injuries to veins.
5.2.2 Porta hepatis: the invasion can be detected if the distance between the
structures is enlarged. The relationship of the tumour with this region is crucial at the spleno-mesenteric confluence duodeno-pancreatic block).
5.2.3 Inferior mesenteric artery: visualisation is of limited interest, except if the
coeliac axis and the superior mesenteric artery are involved. 5.2.4 Renal vessels: evaluation is essential to assess the possibility and ease or
otherwise of performing a nephrectomy. To this purpose it is especially important to define the relationship between the tumour and both renal artery and vein (separated, in contact or encasing). If the tumour is encasing the vessels, it should be specified if this is also true for the hilar region, defined by a tangent to the upper and lower lip of the hilus (in axial plane). Stretching of the vessels by the tumour is considered as a pre-operative risk factor.
5.2.4 Contiguous organs: definition of the relation of the tumour to the contiguous
organs relies on the evaluation of the dividing planes which are often better seen by real time ultrasound. However, the lack of demonstrable dividing planes (in contact) does not allow prediction of surgical difficulties. In perspective, the study of the information derived by the pre-operative diagnostic forms and the surgical report could help to clarify this aspect.
5.2.5 Duodeno-pancreatic region: this is a difficult area to examine but in most
patients ultrasound with CT or MRI with IV contrast medium will provide adequate assessment. Imaging studies of the gastro-intestinal tract add no practical information.
5.2.6 Renal function: the course of the ureter should be evaluated and this may
require an abdominal x-ray at the end of investigation by CT scan. If the examination is performed by MRI an intravenous pyelogram should also be done. It may be helpful to define the borders between the tumour and the adrenal gland.
5.3 PELVIC NEUROBLASTOMA 5.3.1 The tumour extension at the level of the crossing of the iliac vessels is
important. It is especially relevant to know the relationship of the tumour with the iliac veins and their branches. The involvement of the sciatic notch is considered as a risk factor.

6
6 EVALUATION AND DEFINITION OF BONY DISEASE (See adjoining schema) 6.1 An MIBG scan should be carried out on all infants using I123. 6.1.1 If the result is positive: ie those infants who have MIBG uptake in the primary
tumour (or who have enhanced liver uptake) an assessment of the MIBG uptake in the skeleton is essential. If there is no MIBG uptake in the skeleton further investigation is not required (this includes MDP Tc99 scan).
6.1.2 Those infants who have uptake of MIBG in the skeleton should have plain
radiographs of the involved areas (plus a CT scan of the skull if the skull takes up MIBG). Those infants with abnormal cortical bones (or CT scan of skull) are eligible for Trial 98.3 (as long as no MYCN amplification).
6.1.3 Those infants who have positive MIBG scans in the skeleton, but no cortical
bone abnormalities do not have bone disease and should be treated on the 98.2 Trial (except in the presence of pleural or CNS disease).
6.2 If the MIBG scan is negative (or non-evaluable, because no clear evidence of
enhanced MIBG liver uptake) a scan with MDP Tc99 is mandatory, in order to detect scintigraphic abnormalities. Any positive areas should be checked by plain radiographs and/or CT scan of the skull. In these cases the definition of bone disease is the same as for infants with positive MIBG: that is there must be visible abnormalities of the cortical bones on X ray or skull CT scan of sites that are positive on Tc scintigraphy.
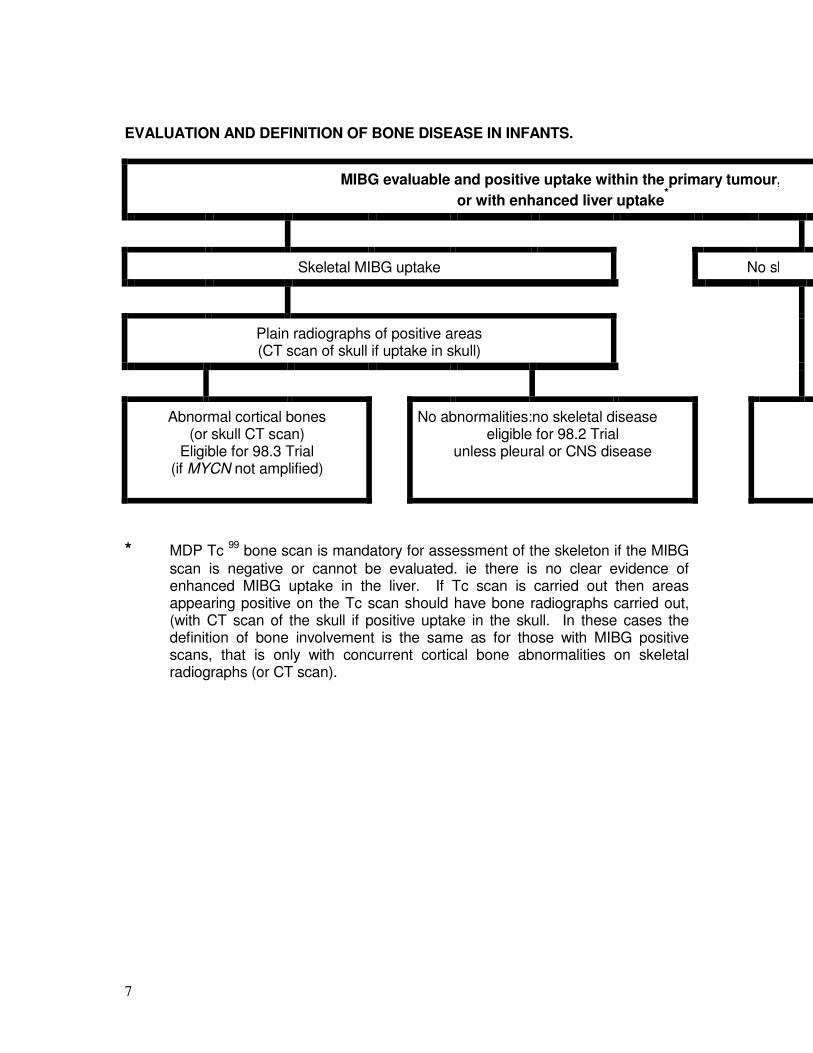
7
EVALUATION AND DEFINITION OF BONE DISEASE IN INFANTS.
MIBG evaluable and positive uptake within the primary tumour,
or with enhanced liver uptake*
Skeletal MIBG uptake No skeletal MIBG uptake
Plain radiographs of positive areas (CT scan of skull if uptake in skull)
Abnormal cortical bones (or skull CT scan) Eligible for 98.3 Trial (if MYCN not amplified)
No abnormalities:no skeletal disease eligible for 98.2 Trial unless pleural or CNS disease
* MDP Tc 99 bone scan is mandatory for assessment of the skeleton if the MIBG
scan is negative or cannot be evaluated. ie there is no clear evidence of enhanced MIBG uptake in the liver. If Tc scan is carried out then areas appearing positive on the Tc scan should have bone radiographs carried out, (with CT scan of the skull if positive uptake in the skull. In these cases the definition of bone involvement is the same as for those with MIBG positive scans, that is only with concurrent cortical bone abnormalities on skeletal radiographs (or CT scan).

ADDENDUM 4 NEUROBLASTOMA WITH INTRASPINAL
EXTENSION
1 Background 1.1 The natural connections that exist between the sympathetic nervous system
and the spinal cord account for the ability of neuroblastoma to infiltrate the intervertebral foramina with occasional involvement of the spinal canal. Despite the fact that tumour growth almost invariably remains extradural, it may still cause spinal cord compression which can progress to irreversible paraplegia 1,2,3,4. Early diagnosis and prompt treatment of spinal cord compression in neuroblastoma is therefore of critical importance. 5
1.2 Dumbbell neuroblastoma is significantly more frequent amongst children with
localised disease, in comparison to the incidence amongst children with metastatic disease. 4, 6. Children with spinal involvement tend to present at a younger age 2,4,5,6,7. Dumbbell neuroblastoma is more commonly associated with intra-thoracic disease 2, 4, 7, 8
. 1.3 Evaluation of the intraspinal extension of neuroblastoma has been greatly
improved by modern diagnostic imaging (CT scan and MRI) (9). MRI has proved especially effective in demonstrating in close detail infiltration of the intervertebral foramina and invasion of the spinal canal 10. By these means 10-15% of children with neuroblastoma have documentation of spinal cord involvement. However, only 60% present with signs of spinal cord compression, a minority present with complete paraplegia 7.
1.4 Treatment of symptomatic spinal cord compression has evolved over recent
years. Until the mid-80s decompressive laminectomy was performed as a rule with the intent of avoiding progression towards paraplegia. Surgery was frequently carried out even when paraplegia had developed 4,5,11. Radiotherapy was considered less effective, although some reports suggested this could be as effective as surgery in metastatic disease 12, 13.
1.5 Both laminectomy and radiotherapy have potential disadvantages. The former
requires experienced hands and may carry the risk of late spine deformities (4. Radiation reduces and alters the growth of irradiated vertebrae and increases the danger of a late second malignancy.
1.6 The use of chemotherapy as an effective alternative to laminectomy or
radiotherapy was first reported in 1984 15. Since then several authors have confirmed that chemotherapy can successfully be used instead of surgery or radiotherapy, without compromising the chance of neurologic recovery. The concern that chemotherapy could not work quickly enough to prevent

permanent neurologic damage has been contradicted by the evidence of many reported cases 7,16,17,18.
1.7 In conclusion, chemotherapy in association with dexamethasone
19 may represent adequate treatment for children with localised neuroblastoma who present with signs of spinal cord compression, with the possible exception of cases who have rapid neurologic deterioration, where laminectomy, or laminotomy is still considered the treatment of choice.
2 Diagnosis and Evaluation of Spinal Cord Compression 2.1 Diagnosis: Early detection of spinal cord compression can be difficult
especially in younger children, unless one thinks of this possibility in any child with a tumour. The most common symptoms are back pain, reduced mobility of legs and/or arms, sensory and sphincteric dysfunction.
The presence of motor deficit is particularly important since children who develop complete motor loss usually experience little or no recovery. Infants with congenital dumbbell tumours have a particularly poor outcome with regard to neurologic recovery 20.
2.2 The extent of motor loss can be graded as follows; 0 Capable of unassisted ambulation, may have pain and/or difficulty with
micturition. 1 Can walk with assistance only. 2 Antigravity strength only. 3 Presence of trace movement only. 4 Complete motor and sensory loss. 2.3.1 Evaluation: As previously stated, MRI is the best method to detect the
infiltration of the intervertebral foramina and the invasion of the spinal canal by neuroblastoma. Although a CT scan may be an adequate study in many cases it is recommended that MRI be used wherever possible, particularly in the diagnostic workup of infants with cervical, thoracic or pelvic disease.
2.3.2 It may be difficult to accurately assess the volume of the intraspinal tumour may
be difficult due to the half-moon configuration commonly assumed by the tumour in that particular site. As indicated in the Radiologic Guidelines (Addendum 3) it is suggested that the maximum diameter of the tumour is expressed as a percentage in relation to the diameter of the spinal canal: < 25%, between 25-50%, > 50%.
2.3.3 Myelography and lumbar puncture are of no diagnostic use, and are
absolutely contraindicated.

3 Treatment of spinal cord compression
3.1 Spinal cord compression without symptoms 3.1.1 The regular use of MRI has increased the incidence of children with documented infiltration of
foramina (with or without invasion of the spinal canal). However, in the majority of cases, especially when the intraspinal component is modest (less the 50% of the diameter), there are no neurological symptoms.
3.1.2 There is very little, if any, evidence that an asymptomatic intraspinal tumour may grow further after the extraspinal component of the tumour has been resected. Information related to the few well documented cases suggests that the intraspinal neuroblastoma in patients with no neurological symptoms tend to remain stable or even regress without specific treatment.
3.1.3 In patients with an unresectable tumour and asymptomatic intraspinal extension the
chemotherapy given to reduce the tumour is likely to be effective even in treating the intraspinal component. No specific additional treatment is required unless neurologic symptoms develop.
3.2 Spinal cord compression with neurologic signs 3.2.1 Patients with localised neuroblastoma who present with signs of spinal cord compression do
require specific treatment. If neurological deficits are present and/or progress rapidly therapeutic decisions must be taken in a matter of hours or at maximum within 1-2 days. Laminectomy, or laminotomy is preferred only in those infants where neurologic deterioration has occurred very rapidly. This situation occurs infrequently. For infants with partial compromise or a stable neurological deficit, the decision to use chemotherapy should be discussed as a matter of urgency between the oncologist and the neurosurgeon.
3.2.2 If it is decided that chemotherapy should be given, there is no urgent indication to remove the
extraspinal tumour (which is likely to be unresectable, and surgery runs the risk of causing a worsening of the neurological deficit). The tumour should be biopsied (by Tru cut or fine needle, or open biopsy), it may be necessary to delay this for a few days, to avoid postponing initial chemotherapy.
The medical treatment is as follows: Dexamethasone 0.5 mg/kg IV bolus followed by 0.2 mg/kg/day IV in 3 divided daily doses.
Chemotherapy is given using "Carbo-VP16". A second course should be given 21 days after the beginning of the first course. (See section 10 of protocol for further details about chemotherapy doses and administration).
A further MRI scan should be obtained following the first course of chemotherapy. If there is no
improvement/or deterioration in the neurological signs and there is no response of the intraspinal component, laminotomy and excision of the intraspinal component should be considered.
If the tumour remains unresectable after 2 courses of VP-Carbo, 2 courses of CADO should be
given, and assessment carried out with MRI as before. 4 Asymptomatic intraspinal residual tumour following chemotherapy: It is not necessary to remove asymptomatic intraspinal residual tumour after chemotherapy. The
removal of any residual extraspinal component should be undertaken only after a careful assessment of the intraspinal residue.

REFERENCES 1. Boogerd W. Van der Sande: Diagnosis and treatment of spinal cord compression in malignant
disease. Cancer Treat Rep 19: 129-150, 1993. 2 Lepeintre J. Schweisguth O. Labrune M. Lemerle J: Le neuroblastome en sablier. Etude de 22
cas. Arch Fr Pediatr. 1969; 26;829-47 3. Lewis DW. Packer RJ. Raney B. et al: Incidence, presentation and outcome of spinal cord
disease in children with systemic cancer. Pediatrics 78: 438-443, 1986. 4. Punt J. Pritchard J. Pincott JR: Neuroblastoma. A review of 21 cases presenting with spinal
cord compression. Cancer 45: 3095-3101, 1980. 5. Traggis DG. Filler R. Druckman H et al: Prognosis of children with neuroblastoma presenting
with paralysis. J Pediatr Surg 12: 419-425, 1976. 6. Plantaz D. Hartmann O. Kalifa C. et al: Neuroblastomes en sablier. Arch Fr Pediatr 48: 529-533, 1991. 7. Plantaz D. Rubie H. Michon J et al. The treatment of neuroblastoma with intra-spinal extension
with chemotherapy followed by the surgical removal of residual disease: a prospective study of 42 cases. Cancer 1996; 78, 311-319
8 King D. Goodman J. Hawk T. Boles ET. Sayers MP. Dumbbell neuroblastoma in children. Arc
Surg 110; 881-91 1975 9. Miller JH. Sato JK: Adrenal origin tumours. In: Imaging in paediatric oncology. Williams R
Wilkins. Baltimore/London, 1985, pp 305-340. 10 Dietrich RB. Kangarloo H. Lenarsky C. Feig SA. Neuroblastoma: the role of MR imaging. Am J
Roentgenol 1987; 148:937-42 11 Plantaz D. Hartmann O. Kalifa C. Sainte Rose C. et al: Localised dumbbell neuroblastoma: a
study of 25 cases treated between 1982 and 1987, using the same protocol. Med Ped Oncol. 21: 249-253, 1993
12. Leviov M. Dale J. Stein M: The management of metastatic spinal cord compression: a
radiotherapeutic success ceiling. Int J Radiation Oncology Biol 27: 231-234, 1993. 13. Klein SL. Sandford RA. Muhlbauer MS: Paediatric spinal epidural metastases. J Neurosurg 74:
70-75, 1991 14. Mayfield JK. Riseborough EJ. Jaffe N. et al: Spinal deformity in children treated for
neuroblastoma. J Bone Joint Surg 63: 183-193, 1981. 15. Hayes FA. Thompson EI. Huizdala E: Chemotherapy as an alternative to laminectomy and
radiation in the management of epidural tumour. Journal of Pediatrics 2: 221-224, 1984. 16. Hayes FA, Green AA, O'Connor DM. Chemotherapeutic management of epidural
neuroblastoma . Med Ped Oncol 1989; 17:6-8 17 De Bernardi B. Pianca C. Mancini A. Casale F. De Laurentis C. Bagnulo S: Spinal cord
compression in neuroblastoma at diagnosis. Treatment and outcome with 70 patients. Proc in SIOP XXVI meeting Paris 1994, Abstract #14 Med Ped Oncol 1994;23:173
18. Sanderson IR. Pritchard J. Marsh HT: Chemotherapy as the initial treatment of spinal cord
compression due to disseminated neuroblastoma. J Neurosurg 70: 688-690, 1989. 19. Posner JB. Howieson J. Cvitkovic E: Disappearing spinal cord compression : oncolytic effects of
glucocorticoids (and other chemotherapeutic agents) on epidural metastases. Ann Neurol 2: 409, 1977.

20. Sainte-Rose C. Roux FX. Pierre Kahn A. et al. Les neuroblastomes intra-rachidiens congénitaux. A propos de 7 cas opérés. Neurochirugie 28;409-15 1982
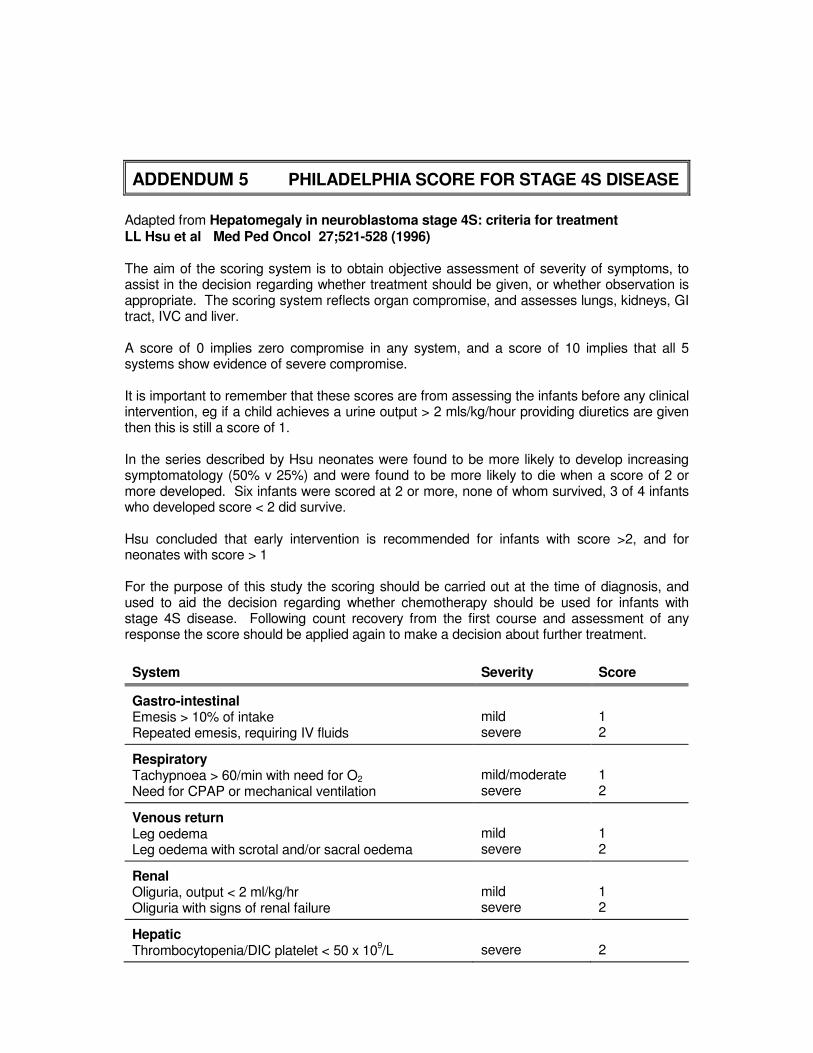
ADDENDUM 5 PHILADELPHIA SCORE FOR STAGE 4S DISEASE
Adapted from Hepatomegaly in neuroblastoma stage 4S: criteria for treatment LL Hsu et al Med Ped Oncol 27;521-528 (1996) The aim of the scoring system is to obtain objective assessment of severity of symptoms, to assist in the decision regarding whether treatment should be given, or whether observation is appropriate. The scoring system reflects organ compromise, and assesses lungs, kidneys, GI tract, IVC and liver. A score of 0 implies zero compromise in any system, and a score of 10 implies that all 5 systems show evidence of severe compromise. It is important to remember that these scores are from assessing the infants before any clinical intervention, eg if a child achieves a urine output > 2 mls/kg/hour providing diuretics are given then this is still a score of 1. In the series described by Hsu neonates were found to be more likely to develop increasing symptomatology (50% v 25%) and were found to be more likely to die when a score of 2 or more developed. Six infants were scored at 2 or more, none of whom survived, 3 of 4 infants who developed score < 2 did survive. Hsu concluded that early intervention is recommended for infants with score >2, and for neonates with score > 1 For the purpose of this study the scoring should be carried out at the time of diagnosis, and used to aid the decision regarding whether chemotherapy should be used for infants with stage 4S disease. Following count recovery from the first course and assessment of any response the score should be applied again to make a decision about further treatment.
System Severity Score
Gastro-intestinal Emesis > 10% of intake Repeated emesis, requiring IV fluids
mild severe
1 2
Respiratory Tachypnoea > 60/min with need for O2 Need for CPAP or mechanical ventilation
mild/moderate severe
1 2
Venous return Leg oedema Leg oedema with scrotal and/or sacral oedema
mild severe
1 2
Renal Oliguria, output < 2 ml/kg/hr Oliguria with signs of renal failure
mild severe
1 2
Hepatic Thrombocytopenia/DIC platelet < 50 x 109/L
severe
2


ADDENDUM 6 DRUG INFORMATION
CARBOPLATIN Formulation Vials containing 50 mg/5 mls, 150 mg/15 mls or 450 mg/45 mls. Storage At room temperature. Reconstitution Dilute in water for injection BP or 5% dextrose to a minimum strength of 500 �gms
per ml. Stability When reconstituted with water for injection or glucose 5%, 8 hours at room
temperature, 24 hours in refrigerator. After dilution in glucose 5% infusion stable for 24 hours in refrigerator.
Administration In this protocol as 1 hour intravenous infusion. Toxicity Myelosuppression, especially thrombocytopenia, nausea and vomiting, low
incidence of nephrotoxicity, ototoxicity, neurotoxicity and abnormalities of liver function. May cause urinary loss of serum magnesium, potassium and calcium, so levels of these should be monitored.
CYCLOPHOSPHAMIDE Formulation 100 mg, 200 mg, 500 mg and 1 G vials for reconstitution. Storage At room temperature. Stability Unreconstituted vials stable for 5 years at room temperature. A solution of
cyclophosphamide appears to be chemically stable for at least 28 days when stored at 4° C. Reconstituted solution (20 mg/ml) should be used within 3 hours when stored at room temperature, unless prepared under strict aseptic conditions, when it is may be used within 8 hours.
Administration Mesna is not required but hydration is given (see individual course details for more
information). Toxicity Myelosuppression, nausea, vomiting, alopecia, haemorrhagic cystitis, sterility,
second malignancies including leukaemia or bladder cancer. May exacerbate the cardiotoxic effects of anthracycline therapy.

DRUG INFORMATION DOXORUBICIN Formulation Vials containing 10 mg or 50 mg in solution (2 mg/ml) Vials containing 10 mg, 50 mg as powder. Storage Solution at 2� - 8�C in refrigerator Powder can be stored at room temperature. Reconstitution Each 10 mg should be reconstituted with 5 mls water for injection. Solution should
be further diluted in 0.9% saline or 5% dextrose. Stability Powder 4 years at room temperature. Solution 2 years (2-8�C in refrigerator) or 1
month at room temperature. Reconstituted solution (100 �g/ml) in 5% dextrose or 0.9% saline is stable for 28 days when stored in refrigerator. Solutions should be protected from light during storage and administration unless concentration is > 500 �g/ml and freshly prepared. Substantial photodegradation may occur at concentrations below 100 �g/ml if exposed to light.
Administration Mix with 0.9% saline and given over 6 or 48 hours. Toxicity Local necrosis if extravasation occurs. Cardiotoxicity. Bone marrow suppression,
mucosal ulceration, nausea, vomiting, alopecia. ETOPOSIDE (VP16) Formulation Vials containing 100 mg etoposide in 5 ml. Storage At room temperature. Reconstitution Ideally dilute to a concentration of 0.25-0.4 mg/ml in 0.9% sodium chloride or 5%
dextrose. Stability Vials are stable for 5 years at room temperature. At concentrations of 0.4 mg/ml in
0.9% saline solutions are stable for 96 hours at room temperature in normal fluorescent lighting, in PVA containers. Solution in PVC infusion bags should be used immediately, to avoid leaching out of potential carcinogenic plasticisers.
Administration By intravenous infusion over 1 - 2 hours - protected from light. Caution:
Anaphylactic reaction usually manifested as severe hypotension may occur if infusion given too rapidly. Avoid extravasation.
Toxicity Bone marrow suppression. Alopecia, headache, fever, hypotension, nausea,
vomiting, anaphylactic reactions, second malignancies including leukaemia.

DRUG INFORMATION VINCRISTINE Formulation 1 mg, 2 mg, 5 mg vials with 10 mls diluent. 1 ml, 2 ml, 5 ml vials of solution 1
mg/ml. Also available in pre-filled syringes containing 1 mg in 1 ml, 2 mg in 2 mls
unpreserved. Storage At 2-8�C in refrigerator. Stability Depends on formulation : Lyophilised powder (0-6�C) 3 years. Solution (0-6�C) 2
years. Reconstituted injection is stable for 14 days (2-8�C). Diluted infusion (in 0.9% saline, 5% dextrose or Ringer's lactate) is stable for 24 hours at 20 �g/ml.
Administration By bolus intravenous injection. Ensure that needle is well into the vein to avoid
extravasation. It is strongly recommended that all vincristine injections are labelled "FOR INTRAVENOUS USE ONLY".
Toxicity Local necrosis if extravasated. Jaw pain, paresis, constipation, neurotoxicity and
alopecia, alopecia, paralytic ileus and SIADH. BUSULPHAN Formulation 0.5 and 2 mg coated tablets Storage 0.5 mg tablets - keep dry and store at 2-8�C 2 mg tablets - keep dry and store < 25�C Stability Three years from manufacture. If 0.5 mg tablets are stored between 8-25�C they
are stable for 6 months. Administration For oral administration No product licence (in UK) for neuroblastoma Toxicity Myelosuppresion, thrombocytopenia, mucositis, nausea, vomiting, diarrhoea,
anorexia, hyperpigmentation, interstitial pulmonary fibrosis. MELPHALAN Formulation 20 mg vials containing 50 mg anhydrous melphalan hydrochloride with 10 ml
buffered diluent. Storage Below 30�C protect from light Reconstitution Add 10 ml diluent to 50 mg vial, shake vigorously until dissolution complete. This
gives a solution of 5 mg/ml which should be used immediately, or diluted further in 0.9% saline and infused within 2 hours. Dextrose solutions are incompatible with melphalan.
Administration As IV bolus into fast running infusion via a central venous line, or as an infusion
over 1-2 hours. Toxicity Myelosuppresion, irreversible bone marrow aplasia, amenorrhoea, sterility,
nausea, vomiting, stomatitis, diarrhoea, haematological malignancies

ADDENDUM 7 CLASSIFICATION OF CISPLATIN-INDUCED BILATERAL FREQUENCY HEARING LOSS.
Taken from Brock et al "Cisplatin ototoxicity in children : a practical grading system." Medical and Pediatric Oncology (1991) 19; 295 - 300
Bilateral hearing loss
1 Grade
< 40 dB at all frequencies2
0
> 40 dB at 8000 Hz only2
1
> 40 dB at 4000 Hz and above2
2
> 40 dB at 2000 Hz and above2
3
> 40 dB at 1000 Hz and above2
4
Notes 1 The results are obtained by pure-tone audiometry, from the "better" ear. 2 < 40 dB at lower frequencies
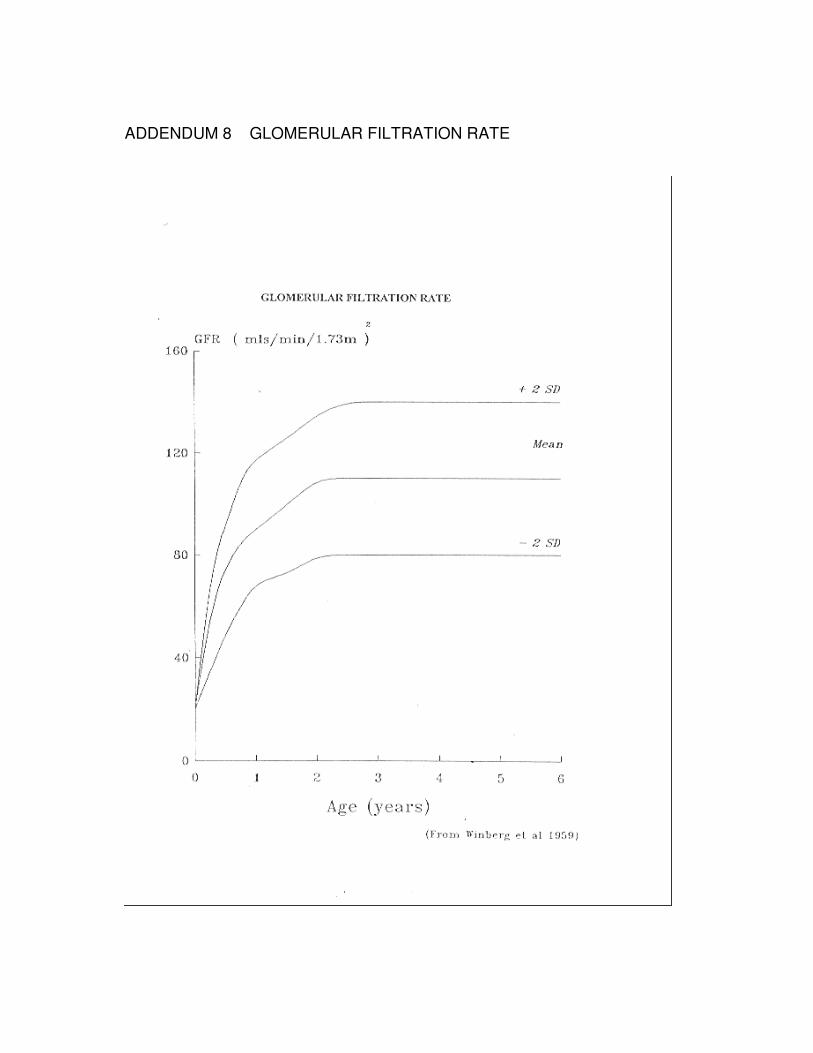
ADDENDUM 8 GLOMERULAR FILTRATION RATE

ADDENDUM 9
RECOMMENDATIONS FOR GRADING OF TOXIC EFFECTS
Taken from CTC-NCIC criteria
Site Grade 0 Grade 1 Grade 2 Grade 3
H Haematological
H1 Haemoglobin (g/100 ml) WNL > 10.0 8.0-9.9 6.5-7.9 H2 Leucocytes (1000/mm3) <4.0 3.0-3.9 2.0-2.9 1.0-1.9 H3 Granulocytes (1000/mm3) < 2.0 1.5-1.9 1.0-1.4 0.5-0.9 H4 Platelets (100`0/mm3) WNL > 75 50-75 25-50 H5 Haemorrhage None Petechiae Mild blood loss Needing blood transfusion
or fundal haemorrhages
D Digestive
D1 Bilirubin WNL < 1.5 x N 1.5 - 3 x N
D2 Transaminases
(SGOT/SGPT) < 1.25 x N
1.26-2.5 x N
2.6 -5 x N 5.1 -20 x N
D3 Alkaline phosphatase < 1.25 x N 1.26-2.5 x N 2.6 -5 x N 5.1 -20 x N
D4 Amylase < 1.25 x N < 1.5 x N 1.6-2 x N 2.1 - 5 x N
D5 Stomatitis No change Mild soreness, erythema Painful erythema, oedema, ulcers but can eat solids
Ulcerated lesions, requiring liquid
D6 Nausea/vomiting None Nausea Transient vomiting < 5 episodes in 24 hrs
Transient vomiting > 5 episodes in 24 hrs
D7 Diarrhoea None Transient < 2 days Tolerable but > 2 days Intolerable requiring therapy
D8 Constipation None or no change Mild Moderate
Severe, abdominal distension
M Metabolic/Renal
M Blood creatinine WNL < 1.5 x N 1.5 - 3 x N 3.1 - 6 x N
M Proteinuria No change 1+ or < 3g/l 2-3 + or 3-10 g/l 4+ or > 10 g/l
M3 Haematuria No change Microscopic Gross no clots Gross + clots
M4 Na + mmol/L 135-145 146-149/130-134 150-155/125/129 156-164/116/124
M5 K+ mmol/lL 3.5-5.4 5.5-5.9/3.1-3.4 6-6.4/2.6-3 6.5-6.9/2.1-2.5
M6 Ca + mmol/L 2.15-2.59 2.6-2.89/1.9-2.1 2.9-3.09/1.7-1.89 3.1-3.3/1.5-1.69
M7 Mg++ mmol/l 1.5-2.0 1.2-1.4 0.9-1.1 0.6-0.8
P Pulmonary
P1 PA O2 > 90 80-89 65-79 50-64
P2 DL CO 100-75% 74-65% 64-55% 54-40%
P3 CV 100-75% 74-65% 64-55% 54-40%
P4 Function No change Mild symptoms Exertional dyspnoea Dyspnoea at normal levels of exertion
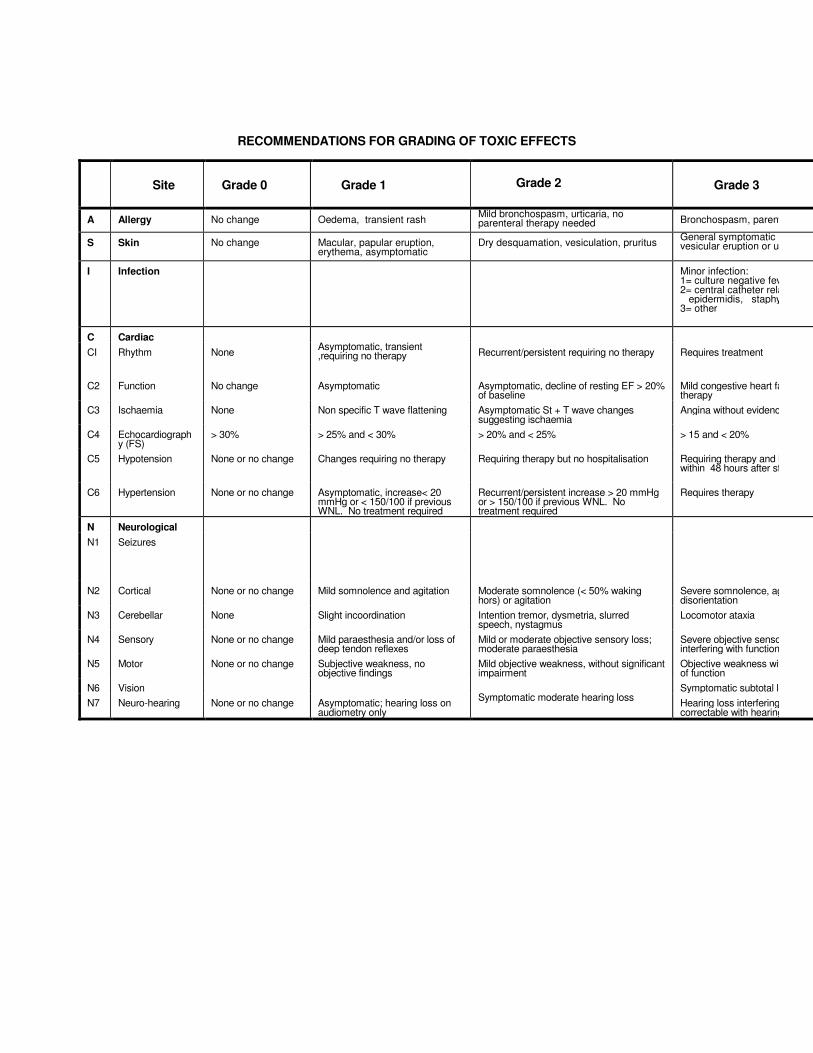
RECOMMENDATIONS FOR GRADING OF TOXIC EFFECTS
Site
Grade 0
Grade 1
Grade 2 Grade 3
A Allergy No change Oedema, transient rash Mild bronchospasm, urticaria, no parenteral therapy needed Bronchospasm, parenteral therapy required
S Skin No change Macular, papular eruption, erythema, asymptomatic
Dry desquamation, vesiculation, pruritus General symptomatic macular, papular or vesicular eruption or ulceration
I Infection
Minor infection: 1= culture negative fever without septic shock 2= central catheter related bacteraemia epidermidis, staphylococcus 3= other
C Cardiac
CI Rhythm None Asymptomatic, transient ,requiring no therapy
Recurrent/persistent requiring no therapy Requires treatment
C2 Function No change Asymptomatic Asymptomatic, decline of resting EF > 20% of baseline
Mild congestive heart failure responsive to therapy
C3 Ischaemia None Non specific T wave flattening Asymptomatic St + T wave changes suggesting ischaemia
Angina without evidence of infection
C4 Echocardiography (FS)
> 30% > 25% and < 30% > 20% and < 25% > 15 and < 20%
C5 Hypotension None or no change Changes requiring no therapy Requiring therapy but no hospitalisation Requiring therapy and hospitalisation. resolves within 48 hours after stopping the agent
C6 Hypertension None or no change Asymptomatic, increase< 20 mmHg or < 150/100 if previous WNL. No treatment required
Recurrent/persistent increase > 20 mmHg or > 150/100 if previous WNL. No treatment required
Requires therapy
N Neurological
N1 Seizures
N2 Cortical None or no change Mild somnolence and agitation Moderate somnolence (< 50% waking hors) or agitation
Severe somnolence, agitation, confusion, disorientation
N3 Cerebellar None Slight incoordination Intention tremor, dysmetria, slurred speech, nystagmus
Locomotor ataxia
N4 Sensory None or no change Mild paraesthesia and/or loss of deep tendon reflexes
Mild or moderate objective sensory loss; moderate paraesthesia
Severe objective sensory loss, or paraesthesia interfering with function
N5 Motor None or no change Subjective weakness, no objective findings
Mild objective weakness, without significant impairment
Objective weakness withof function
N6 Vision Symptomatic subtotal loss of vision
N7 Neuro-hearing None or no change Asymptomatic; hearing loss on audiometry only
Symptomatic moderate hearing loss Hearing loss interfering with funcorrectable with hearing aid

ADDENDUM X INFORMAZIONI PER I GENITORI E MODULO PER
IL CONSENSO
Cari genitori, vi è stato comunicato che il vostro bambino ha un neuroblastoma. Si tratta di una malattia tumorale molto particolare, della quale conosciamo bene alcuni aspetti, mentre altri sono in fase di studio. Cosa è ormai chiaro del neuroblastoma ?
• i bambini con neuroblastoma di età inferiore ad un anno hanno una probabilità di guarire superiore rispetto a quella dei bambini diagnosticati in età successive;
• l'intervento chirurgico non è sempre necessario, in quanto alcune volte il tumore smette di crescere e tende a ridursi fino a scomparire grazie ad un trattamento poco aggressivo o, talora, anche senza alcuna terapia, e questo succede soprattutto in questa fascia di età;
• i bambini con neuroblastoma localizzato, soprattutto se operabile, guariscono più facilmente dei bambini con neuroblastoma con metastasi;
• le caratteristiche delle cellule del tumore possono influenzare la possibilità di guarigione del bambino;
• la più importante di queste caratteristiche biologiche che finora conosciamo si chiama MYCN, un gene presente anche nelle cellule normali, ma che nelle cellule di alcuni neurobIastomi è presente con un numero di copie elevato. Se il neuroblastoma presenta molte copie di MYCN, la possibilità di raggiungere la guarigione è più bassa che negli altri casi;
• è difficile che un bambino di età inferiore a 1 anno con neuroblastoma possa avere molte copie di MYCN, ma quando questo succede è necessario un trattamento particolarmente aggressivo, con chemioterapia a dosi elevate;
• per i bambini di età inferiore a 1 anno in cui il neuroblastoma presenta un numero di copie di MYCN normale è possibile ridurre l’intensità del trattamento allo scopo di evitare i gravi effetti tossici legati alla cura, senza per questo ridurne le possibilità di guarigione.
Quindi se il vostro bambino presenta un neuroblastoma localizzato il trattamento iniziale è l’intervento chirurgico. Il tumore viene analizzato per quanto riguarda le sue caratteristiche biologiche, e solo se sono presenti molte copie di MYCN viene eseguita una chemioterapia allo scopo di aumentare le possibilità di guarigione.

Se il tumore non è operabile viene effettuata una biopsia, cioè un intervento che permette di asportare una piccola parte del tumore, così da potere effettuare ugualmente gli studi biologici, e sulla base di questi viene stabilito il trattamento da eseguire. Cosa non è ancora chiaro del neuroblastoma ?
• oltre a MYCN sono state individuate altre caratteristiche biologiche del neuroblastoma, ma quanto dipende da queste la possibilità di guarigione del bambino ?
Proprio per dare una risposta a questo interrogativo è necessario raccogliere informazioni su molti casi . Ma dal momento che il neuroblastoma è una malattia poco frequente, i singoli Centri seguono un numero ristretto di bambini e pertanto non sono in grado di raccogliere un numero sufficiente di dati per fare gli studi necessari a migliorare le conoscenze sul neuroblastoma. Ecco che allora pediatri oncologi, chirurghi, biologi, radiologi e radioterapisti esperti di neuroblastoma nelle più importanti nazioni europee, compresa l’Italia, hanno lavorato insieme per preparare questo protocollo, con l’obiettivo di studiare e trattare in modo chiaro e sistematico tutti I bambini di età inferiore a 1 anno affetti da neuroblastoma, e di raccogliere più dati possibile relativi a questi pazienti. Speriamo così di scoprire ciò che di questa malattia conosciamo ancora male, e di individuare il miglior trattamento possibile in ogni singolo caso. Il programma di cura utilizzato in questo protocollo, che noi crediamo essere altrettanto o più efficace di quelli utilizzati in altre nazioni del mondo, tiene conto di tutto quanto già è assodato sul comportamento clinico del neuroblastoma. Se voi siete favorevoli a che il vostro bambino venga trattato secondo questo protocollo, i dati relativi al tumore e all’andamento clinico del vostro bambino saranno raccolti e conservati sia in Italia che nel centro europeo di raccolta dati situato a Parigi. Tutte le informazioni relative al vostro bambino saranno verificate nel più stretto riserbo ed eventuali pubblicazioni di dati sui risultati di questo studio non permetteranno mai di identificare i singoli bambini. Anche se acconsentite ad inserire vostro figlio in questo protocollo, potrete in qualsiasi momento modificare questa decisione, senza che questo privi il bambino della possibilità di essere curato nel modo più efficace possibile. Chiedete qualsiasi altra informazione ai medici che si occupano del vostro bambino.

EUROPEAN INFANT NEUROBLASTOMA STUDY 1999 CONSENSO INFORMATO
I genitori devono compilare per intero questo foglio
Avete letto la copia del foglio informativo che vi è stata consegnata ?
SI NO
Avete avuto la possibilità di chiedere chiarimenti e di discutere con I medici sul protocollo ?
SI NO
Sono state date risposte esaurienti alle vostre domande ?
SI NO
Avete compreso che se siete d’accordo ad inserire il vostro bambino in questo protocollo, il trattamento dipenderà da quanto il neuroblastoma è esteso e dalle sue caratteristiche biologiche (numero di copie di MYCN)?
SI NO
Chi vi ha illustrato il protocollo ? Dr .................................................. Avete compreso che vi sarà possibile modificare la vostra decisione escludendo il bambino dal protocollo: • in qualsiasi momento • senza dover dare alcuna spiegazione • senza compromettere il futuro trattamento di vostro figlio
SI NO
Siete dunque d'accordo ad inserire il bambino in questo protocollo ?
SI NO
Nome del bambino .............................................................. Nome e firma dei genitori .............................................................. .............................................................. Data ..............................................................
Nome e firma del Medico ................................................................ Data .................................................................

ADDENDUM 10 INTERNATIONAL STUDY CO-ORDINATION
The conduct of this study will be according to the following agreed procedures: 1 Status of Study This is a collaborative study between a number of participating national groups.
The coordinators for each group are listed below. There are four Trials included in the study, the coordinators for each Trial are listed below.
2 The Protocol 2.1 A common protocol will be used for the International study by all national groups.
The finalised master protocol in the English language will be held at the UKCCSG Data Centre, and also at the international data centre in Paris. Other national groups will be responsible for producing a literal translation in their own language.
2.2 Each national coordinator will be responsible for distribution of protocols to centres
within their national group via their national data centre as appropriate. 2.3 Addenda may be added independently by any of the national groups to address
local needs, provided they have no bearing on the essential aims of the international protocol. However no change will be allowed to the eligibility criteria or the treatment procedures for each Trial.
2.4 Subsequent to finalisation, any amendments to the protocol must be agreed by the
Trial coordinators and the national coordinators. Any future amendments to the protocol will be issued from the UKCCSG in English to be translated as appropriate.
3 Study Forms 3.1 One common set of forms will be used. The master version (in English) of the
study forms will be held at the International Data Centre in Paris. 3.2 Each national coordinator will be responsible for distribution of forms to centres
within his/her country (using their national data centre as appropriate). 3.3 Additional forms may be produced independently by any national group for the
collection of data additional to that required for the International study. 3.4 Subsequent to finalisation, amendments to the forms must be agreed by all
national groups. The International Data Centre will be responsible for the issue of amended forms.

4 Inclusion procedure for new patients New patients will be registered by each participating centre to its national co-
ordinating centre, which will then forward the Trial entry form to the International Data Centre in Paris. The eligibility forms must be completed by the investigator, and checked by the national co-ordinator (ascertaining that biological data concerning MYCN results are available from the national reference laboratory, and that additional biological material is available for 1p studies). This information must be transmitted to the International data Centre in Paris within 6 weeks of the date of diagnosis (defined as the date at which material was obtained for studies).
5 Data Collection 5.1 Each national coordinator will be responsible for submitting the data to the
International Data Centre in Paris. It is expected that national groups may wish to hold the master database for its own patients, and shall be responsible for data quality according to local practice.
5.2 The content of the database shall be identical to the data collected on the study
core data forms. 5.3 The master database for the entire study will be held at the International Data
Centre in Paris. 5.4 A complete data set will be transferred by computer disk at least 6-monthly to the
master database at the International Data Centre in Paris. 5.5 Forms returned from the treating institutions shall be stored at the national data
centre for each national group. 6 Confidentiality of patient data The use of names as patient identifiers on paper forms and on national databases
will be according to national practice. An abbreviated patient identifier will be used for data transfer and for the master database.
7 Data Quality Control 7.1 On receipt of forms at the national data centre, common range and logical checks,
agreed by the Trial coordinators, will be carried out on data prior to transfer to the master database. Any amendment to the checking programmes shall require mutual agreement of the Trial coordinators.
7.2 Data entry verification shall be carried out according to current national practice.
Cross checks of data entry will be carried out occasionally, between national centres, on a sample of forms.
7.3 Data amendments shall only be carried out at the national data centre on the
national database. Errors noted on the master database, after receipt of the group database, shall be reported back to the national centre.

7.4 Data audit of study forms against the patient record forms at the treating institution
shall be performed to satisfy national requirements. 8 Data analysis and monitoring 8.1 Data will be released 6 monthly from the International Database to the Trial
coordinator responsible for interim analysis of the appropriate group of patients. 8.2 Results of the interim analysis of outcome and toxicity shall be reported to an
independent international data monitoring committee (DMC) as scheduled by protocol. The DMC may recommend early stopping, continuation or extension of the study to the international study committee.
8.3 Reports prepared from the national databases on overall outcome may be
circulated locally subject to approval for release by the DMC. No other publication will be allowed prior to the completion of the study.
8.4 The Trial coordinators (and national coordinators) shall meet as appropriate to
consider patient treatment, eligibility and outcome to ensure the smooth running of the study.
9 Adverse Events 9.1 Any adverse event (death, relapse or Grade 4 non-haematological life threatening
toxicity) shall be reported immediately by the treating institution to the national centre and relayed to the Trial coordinators and the International Data Centre for further reporting according to local practice.
9.2 The Toxicity criteria used in completion of the Data forms will be the same for all
participating groups and appear in Appendix 7. 10 Chemotherapy Review Chemotherapy forms shall be reviewed by the national group co-ordinator(s) and
protocol deviations noted on the database. The review information shall be reported to the Trial coordinators.
11 Pathology Review Pathology review concerning eligibility shall be rapidly reviewed by national group
reviewer(s). National group pathology data shall be discussed at the meeting of the international study committee.
12 Follow up data All registered patients shall be followed up by the national group data centre during
and after completion of treatment according to current protocol.

13 Study approval The study shall open simultaneously in each of the participating countries. 14 Institutional/Local ethical approval and patient consent Institutional/local ethical approval shall follow accepted national practice. National procedures for patient consent as documented shall be used 15 Data Monitoring Committee (DMC) 15.1 An independent Data Monitoring Committee composed of 4 international
experts will monitor the progress of the four trials on ethical and scientific grounds. The role of the DMC will be:
To review accrual rate To examine interim analysis. 15.2 Each interim analysis will be reported to the DMC. These interim analyses will remain confidential. On the basis of these analyses, the DMC will recommend whether the study can continue, or whether it should be changed or terminated prematurely. 15.3 To monitor toxicity.
Every 6 months the statistician for the trial will circulate a report to the members of the DMC about toxicity. The DMC will review these interim toxicity data which will also be forwarded to each Trial coordinator. This biannual procedure prevents against problems of major toxicity.
15.4 To examine other trials.
The DMC will review reports of related studies performed by other groups or organisations to determine whether such information materially affects the aims or preliminary findings of the trial.
15.5 Other.
The DMC will be asked to review any major modification to the study proposed by the study committee prior to its implementation.

16 TRIAL COORDINATORS
TRIAL 98.1 Dr Herve Rubie Phone +33 534 558609 Hôpital Des Enfants Fax +33 534 558612 330 avenue de Grande-Bretagne E mail [email protected] B.P. 3119-31026 Toulouse Cedex 3 France
TRIAL 98.2 Dr Mary Gerrard Phone +44 114 2717366 Sheffield Children's Hospital Fax +44 114 2762289 Western Bank E mail [email protected] Sheffield, S10 2TH United Kingdom
TRIAL 98.3 Dr Bruno De Bernardi Phone +39 010 5636464 Dept Hematology/Oncology Fax +39 010 3762322 G Gaslini Children's Hospital E mail brunodebernardi@ospedale-
gaslini.ge.it Largo Gerolamo Gaslini 5 16148 Genova Italy
TRIAL 98.4 Dr Adela Canete Phone +34 96 3862789 Hospital Infantil "La-Fe" Fax +34 96 3868700 Avenida Campanar 21 E mail [email protected] 46009 Valencia Spain

16 INFANT NEUROBLASTOMA STUDY: NATIONAL COORDINATORS
AUSTRIA Dr Ruth Ladenstein St Anna's Children's Hospital Kinderspitalgasse 6 1090 VIENNA Austria Tel + 43 1 40470 Fax + 43 1 40170 759
E mail [email protected]
PORTUGAL
Dr Ana Forjaz de Lacerda Instituto de Oncologia R Prof Lima Basco 1093 Lisboa Codex PORTUGAL Tel + 35 11 7200429 Fax + 35 11 7200417
BELGIUM
Dr Genevieve Laureys Universital Hospital Gent De Pintelaan 135 9000 GENT Belgium Tel + 32 9 2402111 Fax + 32 9 2403875
E mail [email protected]
SPAIN Dr Adele Cañete Hospital Infantil "La-Fe" Avenida Campanar 21 46009 VALENCIA Spain Tel + 34 96 3862789 Fax + 34 96 3868700
E mail [email protected]
DENMARK Dr Catherine Rechnitzer Dept of Pediatrics Rigshospitalet DK-2100 COPENHAGEN Denmark Tel + 45 35 454756 Fax + 45 35 454673
E mail [email protected]
SWEDEN Dr Jonas Abrahamson Dept of Pediatrics SU/Östra sjukhuset S-14685 GÖTEBORG Sweden Tel + 46 31 374000 Fax + 46 31 843010 E mail
FRANCE Dr Herve Rubie Hôpital Des Enfants 330 avenue de Grande-Bretagne B.P. 3119-31026 TOULOUSE, Cedex 3, France
Tel + 33 5 34 558609
Fax + 33 5 34 558612
E mail [email protected]
SWITZERLAND Dr Maja Nenadov Beck University Hospital(chuv) Rue du Bugnon 1011 LAUSANNE Switzerland Tel + 41 21 3143567 Fax + 41 21 3143558
E mail [email protected]
ITALY Dr Bruno De Bernardi Dept Hematology/Oncology G Gaslini Children's Hospital Largo Gerolamo Gaslini 5 16148 GENOVA Italy Tel + 39 010 5636464 Fax + 39 010 3762322
E mail [email protected]
UNITED KINGDOM Dr Annabel Foot Royal Hospital for Sick Children St Michael's Hill BRISTOL BS2 8BJ Tel + 117 9215411 Fax + 117 9285682
E mail [email protected]
NORWAY Dr Ingebjörg S-Mathieson University Hospital Dept Paediatrics 0027 OSLO Norway Tel + 47 22 869067 Fax + 47 22 422822
E mail
INTERNATIONAL DATA BASE
Clinical Coordinator Dr Jean Michon Section Medicale et Hospitaliere
Statistician Dr Veronique Mosseri Institut Curie

Institut Curie, 26, Rue D'Ulm 75231 PARIS CEDEX 5 France Tel + 33 14 4324558 Fax + 33 14 4324005
E mail [email protected]
Unite de Biostaistiques 75231 PARIS CEDEX 5 France Tel + 33 14 44324665 Fax + 33 14 44324078
E mail [email protected]
ADDENDUM 11 DECLARATION OF HELSINKI
Recommendations guiding physicians in biomedical research involving human subjects
Adopted by the 18th World Medical Assembly
Helsinki, Finland, June 1964
amended by the 29th World Medical Assembly
Tokyo, Japan, October 1975 and
the 35th World Medical Assembly Venice, Italy, October 1983
and the 41st World Medical Assembly
Hong Kong, September 1989 and
the 48th General Assembly Somerset West, Republic of South Africa, October 1996
INTRODUCTION It is the mission of the physician to safeguard the health of the people. His or her knowledge and conscience are dedicated to the fulfilment of this mission. The Declaration of Geneva of the World Medical Association binds the physician with the words, "The health of my patient will be my first consideration," and the International Code of Medical Ethics declares that, "A physician shall act only in the patient's interest when providing medical care which might have the effect of weakening the physical and mental condition of the patient." The purpose of biomedical research involving human subjects must be to improve diagnostic, therapeutic and prophylactic procedures and the understanding of the aetiology and pathogenesis of disease. In current medical practice most diagnostic, therapeutic or prophylactic procedures involve hazards. This applies especially to biomedical research.

Medical progress is based on research which ultimately must rest in part on experimentation involving human subjects. In the field of biomedical research a fundamental distinction must be recognised between medical research in which the aim is essentially diagnostic or therapeutic for a patient, and medical research, the essential object of which is purely scientific and without the implying direct diagnostic or therapeutic value to the person subjected to the research. Special caution must be exercised in the conduct of research which may effect the environment, and the welfare of animals used for research must be respected. Because it is essential that the results of laboratory experiments be applied to human beings to further scientific knowledge and to help suffering humanity, the World Medical Association has prepared the following recommendations as a guide to every physician in biomedical research involving human subjects. They should be kept under review in the future. It must be stressed that the standards as drafted are only a guide to physicians all over the world. Physicians are not relieved from criminal, civil and ethical responsibilities under the laws of their own countries. I. BASIC PRINCIPLES 1. Biomedical research involving human subjects must conform to generally
accepted scientific principles and should be based on adequately performed laboratory and animal experimentation and on a thorough knowledge of the scientific literature.
2. The design and performance of each experimental procedure involving
human subjects should be clearly formulated in an experimental protocol which should be transmitted for consideration, comment and guidance to a specially appointed committee independent of the investigator and the sponsor provided that this independent committee is in conformity with the laws and regulations of the country in which the research experiment is performed.
3. Biomedical research involving human subjects should be conducted only by
scientifically qualified persons and under the supervision of a clinically competent medical person. The responsibility for the human subject must always rest with a medically qualified person and never rest on the subject of the research, even though the subject has given his or her consent.
4. Biomedical research involving human subjects cannot legitimately be carried
out unless the importance of the objective is in proportion to the inherent risk to the subject.
5. Every biomedical research project involving human subjects should be
preceded by careful assessment of predictable risks in comparison with

foreseeable benefits to the subject or to others. Concern for the interests of the subject must always prevail over the interests of science and society.
6. The right of the research subject to safeguard his or her integrity must always
be respected. Every precaution should be taken to respect the privacy of the subject and to minimise the impact of the study on the subject's physical and mental integrity and on the personality of the subject.
7. Physicians should abstain from engaging in research projects involving human subjects unless they are satisfied that the hazards involved are believed to be predictable. Physicians should cease any investigation if the hazards are found to outweigh the potential benefits.
8. In publication of the results of his or her research, the physician is obliged to
preserve the accuracy of the results. Reports of experimentation not in accordance with the principles laid down in this Declaration should not be accepted for publication.
9. In any research on human beings, each potential subject must be adequately
informed of the aims, methods, anticipated benefits and potential hazards of the study and the discomfort it may entail. He or she should be informed that he or she is at liberty to abstain from participation in the study and that he or she is free to withdraw his or her consent to participation at any time. The physician should then obtain the subject's freely-given informed consent, preferably in writing.
10. When obtaining informed consent for the research project the physician
should be particularly cautious if the subject is in a dependent relationship to him or her or may consent under duress. In that case the informed consent should be obtained by a physician who is not engaged in the investigation and who is completely independent of this official relationship.
11. It case of legal incompetence, informed consent should be obtained from the
legal guardian in accordance with national legislation. Where physical or mental incapacity makes it impossible to obtain informed consent, or when the subject is a minor, permission from the responsible relative replaces that of the subject in accordance with national legislation.
Whenever the minor child is in fact able to give consent, the minor's consent
must be obtained in addition to the consent of the minor's legal guardian. 12. The research protocol should always contain a statement of the ethical
considerations involved and should indicate that the principles enunciated in the present Declaration are complied with.

II. MEDICAL RESEARCH COMBINED WITH PROFESSIONAL CARE (Clinical Research) 1. In the treatment of the sick person, the physician must be free to use a new
diagnostic and therapeutic measure, if in his or her judgement it offers hope of saving life, re-establishing health or alleviating suffering.
2. The potential benefits, hazards and discomfort of a new method should be
weighed against the advantages of the best current diagnostic and therapeutic methods.
3. In any medical study, every patient - including those of a control group, if any
- should be assured of the best proven diagnostic and therapeutic method. This does ot exclude the use of inert placebo in studies where no proven diagnostic or therapeutic method exists.
4. The refusal of the patient to participate in a study must never interfere with
the physician-patient relationship. 5. If the physician considers it essential not to obtain informed consent, the
specific reasons for this proposal should be stated in the experimental protocol for transmission to the independent committee (I, 2).
6. The physician can combine medical research with professional care, the
objective being the acquisition of new medical knowledge, only to the extent that medical research is justified by its potential diagnostic or therapeutic value for the patient.
III. NON-THERAPEUTIC BIOMEDICAL RESEARCH INVOLVING HUMAN
SUBJECTS (Non-Clinical Biomedical Research) 1. In the purely scientific application of medical research carried out on a human
being, it is the duty of the physician to remain the protector of the life and health of that person on whom biomedical research is being carried out.
2. The subjects should be volunteers - either healthy persons or patients for
whom the experimental design is not related to the patient's illness. 3. The investigator or the investigating team should discontinue the research if
in his/her or their judgement it may, if continued, be harmful to the individual. 4. In research on man, the interest of science and society should never take
precedence over considerations related to the well-being of the subject.
