excited-st ate dynami cs of small or ganic mol ecules st...
TRANSCRIPT

E x c i t e d - s t a t e d y n a m i c s o f s m a l l o r g a n i c m o l e c u l e s
s t u d i e d b y t i m e - r e s o l v e d p h o t o e l e c t r o n s p e c t r o s c o p y
T i n g G e n g


Excited-state dynamics of small organic molecules studied by time-resolved photoelectron spectroscopy
Ting Geng

©Ting Geng, Stockholm University 2017
ISBN print 978-91-7649-758-6
ISBN PDF 978-91-7649-759-3
Printed by Universitetsservice US-AB, Stockholm 2017
Distributor: Department of Physics, Stockholm university

Abstract Ultra-violet and visible light induced processes in small organic molecules play very important roles in many fields, e.g., enviromental sciences, biology, material development, chemistry, astrophysics, and many others. Thus it is of great importance to better understand the mechanisms behind these processes. To achieve this, a bottom-up approach is most effective, where the photo-induced dynamics occurring in the simplest organic molecule (ethylene) are used as a starting point. Simple substituents and functional groups are added in a controlled manner to ethylene, and changes in the dynamics are investigated as a function of these modifications. In this manner, the dynamics occurring in more complex systems can be explored from a known base. In this thesis, the excited state dynamics of small organic molecules are studied by a combination of time-resolved photoelectron spectroscopy and various computational methods in order to determine the basic rules necessary to help understand and predict the dynamics of photo-induced processes. The dynamics occurring in ethylene involve a double bond torsion on the ππ* excited state, followed by the decay to the ground state coupled with pyramidalization and hydrogen migration. Several different routes of chemical modification are used as the basis to probe these dynamics as the molecular complexity is increased (i) When ethylene is modified by the addition of an alkoxyl group (-OCnH2n+1), a new bond cleavage reaction is observed on the πσ* state. When modified by a cyano (-CN) group, a significant change in the carbon atom involved in pyramidalization is observed. (ii) When ethylene used to build up small cyclic polyenes, it is observed that the motifs of the ethylene dynamics persist, expressed as ring puckering and ring opening. (iii) In small heteroaromatic systems, i.e., an aromatic ring containing an ethylene-like sub-structure and one or two non-carbon atoms, the type of heteroatom (N: pyrrole, pyrazole O: furan) gives rise to different bond cleavage and ring puckering channels. Furthermore, adding an aldehyde group (-C=O) onto furan, as a way to lengthen the delocalised ring electron system, opens up additional reaction channels via a nπ* state. The results presented here are used to build up a more complete picture of the dynamics that occur in small molecular systems after they are excited by a visible or UV photon, and are used as a basis to motivate further investigations. Key words: time-resolved photoelectron spectroscopy, excited-state
dynamics, organic molecules Stockholm 2017

This thesis is dedicated to my mom

List of Papers
The following papers, referred to in the text by their Roman numerals, are included in this thesis PAPER I: Cyclohexadiene revisited: a time-resolved photoelectron
spectroscopy and ab initio study. O. Schalk, T. Geng, T. Thompson, N. Baluyot, R. D. Thomas, E. Tapavicza, and T. Hansson, Journal of Physical Chemistry A, 120, 2320 (2016). PAPER II: Excited state dynamics of acrylonitrile: substituent effects at
conical intersections interrogated via time-resolved photoelectron
spectroscopy and ab initio simulation.
R. J. MacDonell, O. Schalk, T. Geng, R. D. Thomas, R. Feifel, T. Hansson, and M. S. Schuurman, Journal of Chemical Physics, 145, 114306 (2016). PAPER III: Influence of alkoxy groups on the photoinduced dynamics of
organic molecules exemplified on alkyl vinyl ethers.
O. Schalk, M. Stenrup, T. Geng, R. Lindh, R. D. Thomas, R. Feifel, and T. Hansson, Journal of Phyiscal Chemistry A, 119, 11105 (2015). PAPER IV: Substituent effects on the relaxation dynamics of furan,
furfural and β-furfural: A combined theoretical and experimental
approach.
S. Oesterling, O. Schalk, T. Geng, R. D. Thomas, T. Hansson, and R. de Vivie-Riedle, Phys. Chem. Chem. Phys, 19, 2025 (2017). PAPER V: Dynamics in higher lying excited states: Valence to Rydberg
transitions in the relaxation paths of pyrrole and methylated derivatives.
T. Geng, O. Schalk, S. P. Neville, T. Hansson, and R. D. Thomas, submitted
to Journal of Chemical Physics, January 2017.
PAPER VI: Time-resolved photoelectron spectroscopy studies on
pyrazole and its derivates.
T. Geng, I. F. Galván, O. Schalk, R. Lindh, T. Hansson, and R. D. Thomas, in manuscript.

Reprints were made with permission from the publishers.

Author’s contribution
My contributions to the papers are as follows: Paper I: Cyclohexadiene revisited: a time-resolved photoelectron
spectroscopy and ab initio study. • Participated in setting up the optics. • Participated in conducting the experiments. • Participated in the discussion of the results and the analysis
presented in the paper. PAPER II: Excited state dynamics of acrylonitrile: substituent effects at
conical intersections interrogated via time-resolved photoelectron
spectroscopy and ab initio simulation.
• Participated in setting up the optics. • Participated in conducting the experiments. • Participated in the discussion of the results and the analysis
presented in the paper.
PAPER III: Influence of alkoxy groups on the photoinduced dynamics
of organic molecules exemplified on alkyl vinyl ethers.
• Participated in setting up the optics. • Participated in conducting the experiments. • Wrote the first draft of the article and prepared the figures for the
paper. PAPER IV: Substituent effects on the relaxation dynamics of furan,
furfural and β-furfural: A combined theoretical and experimental
approach.
• Participated in setting up the optics. • Participated in conducting the experiments. • Analysed the spectra and prepared the figures for the paper.
PAPER V: Dynamics in higher lying excited states: Valence to Rydberg
transitions in the relaxation paths of pyrrole and methylated derivatives.
• Participated in setting up the optics • Participated in conducting the experiments. • Analysed the data and prepared the figures for the paper.

• Wrote the first draft of the article. PAPER VI: Time-resolved photoelectron spectroscopy studies on
pyrazole and several methylated derivatives
• Participated in setting up the optics • Participated in conducting the experiments. • Analysed the data and prepared the figures for the paper. • Wrote the first draft of the article.

Contents
Abstract V
List of Papers VII
Author’s contribution IX
Abbreviations XIII
1. Introduction ............................................................................................... 1
2. Photoinduced reactions and concepts ..................................................... 6
2.1 Electronic transition .............................................................................. 7
2.2 Franck-Condon region ........................................................................ 10
2.3 Propagation ......................................................................................... 11
2.4 Conical intersection, internal conversion and intersystem crossing ... 12
3. Excited state dynamics of molecules...................................................... 15
3.1 Excited state dynamics of ethylene ..................................................... 15
3.2 Excited state dynamics of polyenes .................................................... 17
3.2.1 Linear polyenes ............................................................................ 17
3.2.2 Cyclo-polyenes ............................................................................ 21
3.3 Excited state dynamics of molecules with heteroatoms ...................... 27
3.3.1 Acrolein........................................................................................ 27
3.3.2 Heterocycles ................................................................................ 29
4. Experimental apparatus ......................................................................... 34
4.1 Laser system ....................................................................................... 35
4.2 Optical setup- two colour experiment ................................................. 36
4.3 Interaction region and magnetic bottle spectrometer .......................... 38
5. Time-resolved photoelectron spectroscopy and data analysis ............ 41
5.1 Time-resolved photoelectron spectroscopy ........................................ 41
5.2 Time-resolved photoelectron spectrum (TRPES) and data analysis ... 42
6. Discussion of the attached papers .......................................................... 49
6.1 Dynamics of polyenes – cyclohexadiene ............................................ 50

6.2.1 Ethylene molecules with a cyano group - acrylonitrile ................ 53
6.2.2 Ethylene molecules with alkoxy group- alkyl vinyl ethers .......... 56
6.3 Dynamics of cyclopolyenes with heteroatoms .................................... 59
6.3.1 Dynamics of cyclopolyenes with oxygen - furan ......................... 59
6.3.2 Dynamics of cyclopolyenes with nitrogen - pyrrole .................... 62
6.3.2 Dynamics of cyclopolyenes with two nitrogen - pyrazole ........... 66
7. Conclusion and outlook .......................................................................... 69
Sammanfattning LXXII
Acknowledgements LXXV
References LXXVI
Attached papers LXXXI

Abbreviations
BAC Bond alternation coordinate BBO Beta-barium borate BOA Born-Oppenheimer approximation BP Bicylco [2,1,0] pentene CoIn Conical intersection CPA Chirped pulse amplification DAS Decay associated spectrum FC Franck-Condon FHG Fourth harmonic generation FWHM Full width at half-maximum HOMO Highest occupied molecular orbital IC Internal conversion ISC Intersystem crossing LUMO Lowest unoccupied molecular orbital MCP Microchannel plate SHG Second order harmonic generation TDC Time-to-digital conversion card THG Third order harmonic generation TOF Time-of-flight TP Tricyclo [2,1,0,0]pentane TRPES Time-resolved photoelectron spectroscopy Z-state Zwitterionic state

Declaration Portions of this work have been taken from my licentiate thesis. With a few modifications, chapters/sections 1, 2.4, 3.1, 5.2 are taken from my licentiate thesis. With minor modifications, chapter 4 is taken from section 4 in the licentiate thesis.

1
1. Introduction
Ultra-violet and visible light induced processing of a molecule can be described as the absorption of a photon which subsequently induces a chemical reaction in the molecule, possibly leading to a change in chemical conformation of the molecule. Such processing plays significant roles in various fields, including chemistry, physics, astrophysics, biology, and material sciences, and is exemplified by light induced photochemical smogs [1], the light induced isomerisation of retinal [2], and the operation of photoswitches [3]. In early studies, nanosecond and picosecond lasers were used to investigate these photochemical reactions, e.g. probing the lifetime of excited states and radiationless decay pathways [4, 5]. Then, with the invention of the femtosecond laser at the end of 20th century, more detailed investigations of these ultrafast chemical processes became possible, as well as giving control over the mechanisms of the chemical reaction [6]. Ultra-short laser pulses have the characteristics of a large spectral bandwidth and a high intensity: the short pulse duration allows monitoring of the molecular dynamics on the femtosecond time-scale; the large spectral bandwidth allows molecules to be excited to a number of vibrational states; the high intensity allows multiphoton absorption processes to be investigated [7]. To date, much work has been done, and a series of techniques has been developed, in order to investigate and understand the different kinds of processes and the various reaction pathways which occur in molecules upon photoexcitation [8]. Despite these advances, there are still many unresolved questions related to the various reaction mechanisms that occur after excitation. This is because even with cutting-edge tools and techniques, disentangling the important dynamics remains a non-trivial and time-consuming complex issue. This is due to i) the photon-molecule interaction itself, where molecules absorbing different photon energies can populate different excited states, e.g. pyrrole can be excited to a ππ*-state or a πσ*-state [9], and ii) the electronic environment and functionality of the molecules, where the addition of a functional group introduces states of different electronic character and opens up new and different reaction pathways, e.g. the addition of an aldehyde group to ethylene introduces the nπ* state, where the molecules can now decay to a triplet state[10], or the existence of a πσ* state in alkyl vinyl ethers, where C-O bond cleavage occurs [11].

2
In order to both obtain a broader picture and unravel the finer details, new models are needed. In this thesis I present a time-resolved photoelectron spectroscopic experimental study which is combined with various computational methods. Here, a bottom-up approach is used with the aim of working towards obtaining the information necessary to develop a consistent picture that could explain and predict the outcome of these photochemical processes. In a bottom up approach, the “recipe” to understand the excited-state dynamics is to start with a small system, and then undertake chemical modification in a controlled manner while following the effects of these modifications on the dynamics. In this context, the “starter” molecule is ethylene (H2C=CH2): the simplest unsaturated hydrocarbon. With ethylene as the “bottom” system, the dynamics may therefore be influenced/steered by the addition of extra CHx groups, e.g., making polyenes (molecules which contain one or more sequences of alternating double and single carbon–carbon bonds) or changing the topology, as in ring-structures, e.g., cycloalkenes, benzene, or by adding functional groups/heteroatoms (atoms that are not carbon or hydrogen). It has been shown that the dynamics of ethylene can be used to describe the dynamics of more complex polyenes, where two of these processes are hydrogen migration and twisting of the C=C double bond (a detailed description is given in Chapter 3) [12]. For examples, hydrogen migration happens in cyclohexene [13] and cycloheptatriene [14, 15]. However, the twist of a C=C double bond either leads to ring opening, as observed in cyclohexa-1,4-diene [13], or leads to ring puckering, as exhibited in cyclopentadiene [16, 17].
The dynamics occurring on the excited state surfaces may also be influenced by the addition of heteroatoms. Rather than introducing purely structural constraints to the molecule, heteroatoms will change the polarity and electronic energy levels, the character of states, and lead to the opening of new reaction channels, e.g. extra bond cleavage channels, charge transfer states, intersystem crossing channels, due to the addition of spin-orbit couplings, etc. With larger structures, especially rings, the location of two or more heteroatoms may also have different influences: e.g. the potential for N-N bond cleavage in pyrazole. Starting with simple molecules, coupled with inserting different functional groups at specific positions in these molecule, enables us to better understand the dynamics in increasingly complex systems, as well as to tailor functional materials used in light sensitive devices, such as molecular photoswitches [3]. Then the important questions arise. Which part of the molecule is absorbing the photon? Which parts of the molecule participate in the dynamics? Light induced processes happen due to photon-absorption by the

3
chromophore of a molecule, while the dynamics occur in a region of the molecule called the dynamophore [13]. Briefly, the chromophore is “the part of molecular entity for which the electronic transition responsible for a given spectral band is approximately localised” [18] and the dynamophore is the “part of the molecule where the dynamics are approximately localised”.
In order to localise dynamics from a delocalised wavefunction, as generated by the absorption process, we need a mixture of different states, which in turn is achieved by perturbations. Take a simple example of H2
+, illustrated in Figure 1. In H2
+, the electronic wavefunction is distributed equally among the two hydrogen nuclei. When the molecules are excited, the wavefunction is transferred from the bonding ground state to the anti-bonding excited state, where the molecule eventually dissociates forming H and H+. As the reaction progress, the wavefunction is still distributed equally at both nuclei. In order to achieve localisation in which the electronic wavefunction is localised on only one hydrogen atom, mixing of the bonding (Ψa) and antibonding orbital wavefunctions (Ψb) is required. This mixing is achieved through interaction with the environment (a heat bath, e.g. black body radiation). See reference 19 for a more detailed discussion [19].
Figure 1. Potential energy curve of H2
+ and the wavefunctions of the bonding and antibonding molecular orbitals. A more complex example is cyclohexa-1,4-diene, shown in Figure 2 [20]. The two homoconjugated double bonds create a delocalised π-system which is the chromophore of the system. After excitation, the dynamics first evolve along delocalised vibrations according to the vibrations in the ground state. Localisation occurs through mixing of different electronic states (see Figure 2b) which allows the dynamics to occur at one of the double bonds, while

4
the other acts as a “spectator”. Here, again, this localisation is obtained by interaction with the “environment” which, in this case, is provided by localised molecular vibrations.
Figure 2. (a) The chromophore in cylcohexa-1,4-diene is delocalised through the two double bonds, while the dynamophore is localised only at one of the two double bonds. (b) A linear combination forms the localisation on one of the double bonds (modified from ref [20]) Cylcohexa-1,4-diene contains a π-system, which is delocalised over the whole molecule, and, with a conjugated or homoconjugated π-system, the excitation is delocalised. However, if a molecule contains a sub-unit, for example -NH or -C=O, then an n-π* transition is often a localised excitation.
It also has been observed that even though the same chromophore is responsible for absorption of the photon, the same dynamophore is not involved in the dynamics. For example, although cyclohexa-1,3-diene also contains a delocalised conjugated system, which acts as the chromophore, the dynamophore includes the whole carbon skeleton, and so is significantly larger than the chromophore [21, 22, 23, 24]. Conversely, 1,3-butaidene, which contains a delocalised conjugated system (due to the two double carbon bonds) that acts as the chromophore, the dynamics involves only one

5
C=C bond, here a bond torsion mode, and in this case the dynamophore is smaller than the chromophore [25, 26].
This thesis is focused on the studies of the excited state dynamics of small organic molecules arising from the effects of the substituents and comparison among molecules from the point of view of dynamophore. The experimental method used is time-resolved photoelectron spectroscopy. In the following chapters, the background to photon-induced dynamics is introduced in Chapter 2. Extending these ideas from simple molecules, e.g. ethylene, to more complex ones, is illustrated in Chapter 3 to get a better picture on how to study the dynamics of complex molecules. Chapter 4 presents the experimental apparatus used in the measurements, while the data acquisition and the subsequent data analysis are described in Chapter 5. Chapter 6 summarizes the results and conclusions of the attached articles. Finally, Chapter 7 presents future perspectives.
6
2. Photoinduced reactions and concepts
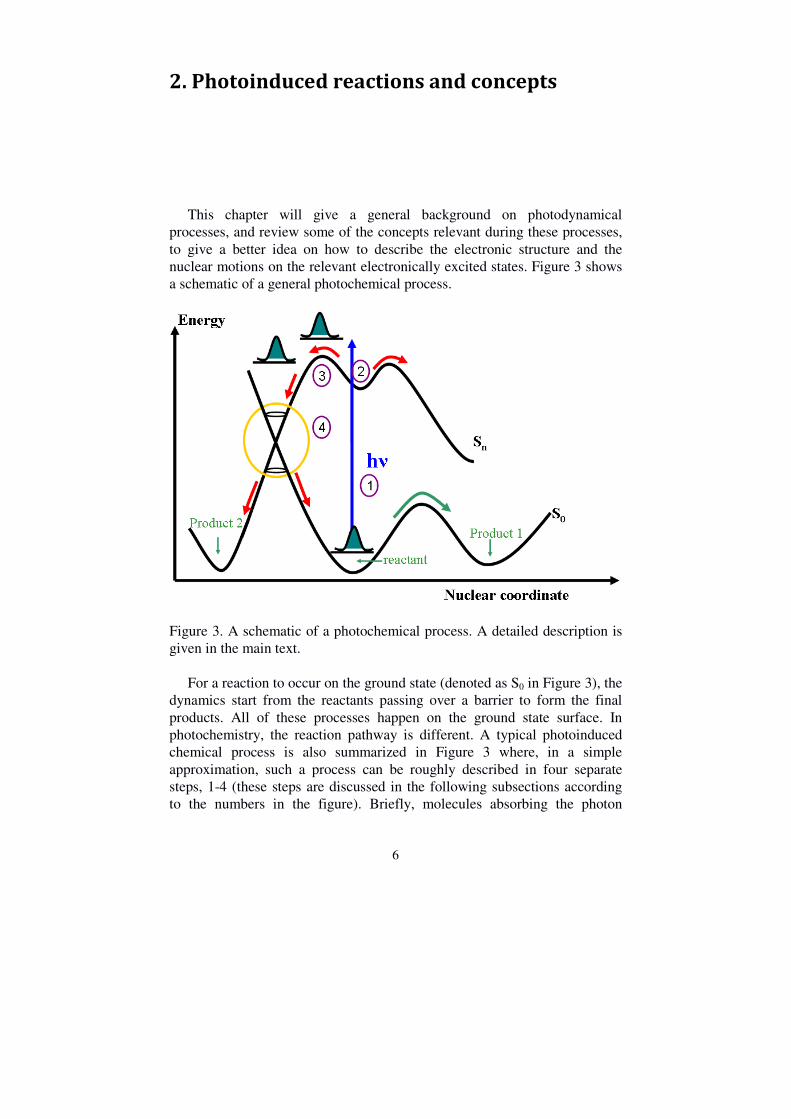
This chapter will give a general background on photodynamical processes, and review some of the concepts relevant during these processes, to give a better idea on how to describe the electronic structure and the nuclear motions on the relevant electronically excited states. Figure 3 shows a schematic of a general photochemical process.
Figure 3. A schematic of a photochemical process. A detailed description is given in the main text. For a reaction to occur on the ground state (denoted as S0 in Figure 3), the dynamics start from the reactants passing over a barrier to form the final products. All of these processes happen on the ground state surface. In photochemistry, the reaction pathway is different. A typical photoinduced chemical process is also summarized in Figure 3 where, in a simple approximation, such a process can be roughly described in four separate steps, 1-4 (these steps are discussed in the following subsections according to the numbers in the figure). Briefly, molecules absorbing the photon

7
energy are excited (1) to the Franck-Condon (FC) region of one excited state. Then molecules leave the FC region (2), flow along the excited state (3), and return to the ground state (4), e.g., through a conical intersection (CoIn).
2.1 Electronic transition In a photochemical process, the molecules, which are typically in the singlet electronic ground state (S0), are promoted to a singly electronically excited state (denoted as Sn in Figure 3) by absorbing a photon (denoted as process 1 in Figure 3).
Figure 4. (a) Formation of molecular σ and σ* bonds by overlap of atomic s or p orbitals. (b) Formation of molecular π and π* bonds by atomic p orbital overlap. (Adapted from ref [27])
The initial electronic transition in this process can be described in the molecular orbital picture. A molecular orbital [28, 29] is used to describe the wave-like behaviour of an electron in a molecule using a mathematical function. Molecular orbitals are represented as a linear combination of atomic orbitals. The efficiency of the atomic orbital interaction depends on the overlap between these orbitals. Figure 4 illustrates several examples. A constructive interaction creates a bonding molecular orbital, such as an σ

8
bond which is formed from the overlap of 1s orbitals or head-on overlapping of the (pz) atomic orbital (directed along the bond axis), or a π-bond which is formed by the overlap of the two lobes of the involved atomic (px) orbitals perpendicular to the bond-axis. A destructive interaction of atomic orbitals creates an anti-bonding molecular orbital, represented by σ* or π*. When there is no interaction between the atomic orbitals, it is termed nonbonding, and is represented by n with free electron pairs, and such molecular orbitals generally occur in molecules containing oxygen or nitrogen atoms, for example in furan, pyrrole, etc.
Figure 5. The π orbitals of the ethylene molecule (Adapted from ref [27]) Take ethylene as an example: the π and π* molecular bonds are shown in Figure 5. In the molecular ground state, two electrons are in the π orbital, which is the highest occupied molecular orbital (HOMO). The lowest unoccupied valence molecular orbital (LUMO) is the π* orbital In this example, the excited state created after a one electron transition is called a singly excited state. There are also electronic transitions from a π orbital to an anti-bonding σ* orbital, or from a non-bonding orbital to an anti-bonding π* orbital, forming the π-σ* state or the n-π* state, respectively, as observed in different molecules [9, 10, 27]. For example, in pyrrole, the existence of the π-σ* state is due to the orbital overlap integral of the σ and π orbitals allowing a π-σ* transition. The n-π* (one electron forbidden) transition in furfural (2-aldehyde furan), is an electron transition from a nonbonding oxygen p-orbital to a perpendicular carbon π orbital. These orbitals are described in Figure 6.

9
Figure 6. (a) σ* orbital in pyrrole and (b) n orbital in furfural.
If a process excites two electrons from an occupied orbital to an unoccupied orbital, this excited state is called a doubly excited state. Generally, doubly excited states are expected to require twice as large an excitation energy as the singlet excited state, due to the requirement of exciting two electrons from the HOMO to the LUMO. However, in polyenes the lowest-lying excited state often is a state with doubly excited character. This could be characterized as a HOMO2
→ LUMO2, and is explained by two reasons: one is due to a large singlet-triplet splitting, which leads to an increase in the energy of the lowest-lying singlet excited state; the other is an admixture of the singlet excited configuration with the doubly excited state [30]. According to the spin direction of the electrons, the excited state can be called a singlet state or a triplet state. If the electronic excited state has spin-paired electrons, i.e. the electrons have opposite spins, then this is called a singlet state, e.g., 1(ππ*). If an electron flips its spin direction, it is called a triplet state, e.g., 3(ππ*). However, the electronic transitions do not happen arbitrarily, they need to obey the selection rules, e.g. singlet to singlet transition is more favourable, and a singlet to triplet state transition is forbidden. Moreover, the selection rules also constrain the allowed excited state according to the number of photons absorbed. If a molecule with inversion symmetry absorbs only one photon, the excited states which the molecules can populate are called optically “bright” states, while if two photons are absorbed, the molecules will be promoted to an excited state called an optically “dark” state. The efficiency of the absorption is generally described by the oscillator strength. In addition to the above electronic transition states, there exist molecular Rydberg states. A molecular Rydberg state is a state in which an electron is excited into a molecular orbital that resembles a hydrogen-like atomic

10
orbital, and where the energy of these electronically excited states follows the Rydberg formula (shown in equation 1) as they converge to a state with an ionic core [31]. The energy of these states is described by
Ε� = −�
���� (1)
where R is the Rydberg constant, and n is the principle quantum number describing the orbital. Comparing the above formula to the standard Rydberg formula for the allowed Bohr-energies of the hydrogen atom [31], δ is called the quantum defect, which works as “screening” parameter, because the ionic core containing electrons is larger than the bare nucleus, and the quantum defect decreases both with increasing angular momentum quantum number and with increasing principle quantum number. Typical values are 0.9 for an s-, 0.5 for a p-, and 0.1 for a d-Rydberg state series.
2.2 Franck-Condon region Under normal conditions, the process of absorbing a photon is faster than the nuclear motion in the molecule, which can be considered to be static in a very good approximation. This is called the Franck-Condon approximation and, consequently, the region on the excited state to where the molecule is excited is called the Franck-Condon (FC) region (number 2 in Figure 3). On a bound excited state, the excitation induces nuclear motion through a displacement of the potential energy surfaces. These induced vibrations are called accepting modes or “Franck-Condon” active modes. These modes give rise to very fast dynamics, and are totally symmetric vibrations. Conversely, there exist the so-called promoting modes which are non-totally symmetric but vibronically active modes. These kinds of modes give an explanation to the possible dynamics which happen on the excited state. When a molecule in the vibronic ground state is excited to different vibrational modes in the excited state, it forms a so-called wavepacket which is not an eigenstate of the excited state [32]. Therefore, the wavepacket will evolve in time regardless of whether excitation occurs to a bound or non-bound state. Generally, some limited information on the electronic transition and possible dynamics can be obtained through the absorption spectrum of the molecule. The absorption spectrum consists of a series of electronic transitions which can have a superimposed vibrational structure. The spectral bandwidth of the peaks depends on various influences. It is important for this thesis that the lifetime of the excited state can be estimated from the absorption spectrum. Moreover, the existence of vibrational structure indicates that the molecule is returning to the Franck-Condon point while a structureless peak indicates a non-bound excited state.

11
When the wavepacket of a small organic molecule is lifted up to the Sn state, as illustrated in Figure 3, it will start in the Franck-Condon region as described earlier. However, it will often experience non-bound conditions, and will propagate away from the Franck-Condon region along the different gradients of the potential energy surface. Since different Franck-Condon active modes exist, the wavepacket can travel in different directions. The wavepacket can propagate with the non-Franck-Condon active modes on the excited state and move towards crossing points with lower lying excited states. These processes are indicated by the number 3 in Figure 2, and are described next.
2.3 Propagation The pathway the excited molecule takes from the initial excited state to the lower-lying state may influence the outcome of a photochemical reaction. In some cases, there may be several intermediate states but only one channel and one observed product. However, at every crossing between the different surfaces, and sometimes even on an excited surface, there can be a bifurcation, and the coupling of the wavepacket to the other surface depends on the momentum of the wavepacket and the topology of the intersection (see Chapter 2.4), e.g. allowing access to different final excited states. On the other hand, the intermediate states can also change the direction of the wavepacket. For example, in unsaturated hydrocarbon molecules, the states which play the main roles in the dynamics are the ππ* states. However, on the reaction pathway, interference from different intermediate states induces different dynamics where, e.g., low-lying Rydberg states have been studied as an intermediate state that is involved in the dynamics of ethylene [33, 34]. For unsaturated hydrocarbon molecules with heteroatoms, e.g., nitrogen, oxygen, excitation occurs mainly to the ππ* states, but the existence of close-lying πσ* states is necessary to explain the dynamics. Such a state induces bond cleavage of, e.g., N-H in pyrrole [35], or C-O in alkyl vinyl ether [11]. Furthermore, for molecules with a carbonyl group, nπ* states also become involved in the dynamics, in which it is observed that the population further decays to the nπ* or ππ* triplet states [10]. On the potential energy surface, the linking of the two minima along the reaction coordinate which has higher energy is called a barrier. Then, upon photoexcitation of molecules, the wavepacket splits on the excited state, in which some part of wavepacket tunnels through the barrier and decays to the lower-lying state. For example, in pyrrole upon excitation with low photon energies, N-H dissociation occurs on the πσ* state, and takes a long time due

12
to the existence of a barrier through which the wavepacket needs to tunnel [36].
2.4 Conical intersection, internal conversion and intersystem crossing The description of potential energy surfaces is based on the so called Born-Oppenheimer approximation (BOA). This approximation states that the electronic and nuclear motions can be separated such that the electrons adiabatically adopt any change in nuclear configuration. In this approximation, it can be shown that electronic states with the same symmetry cannot cross [37, 38]. However, when the two potential energy surfaces get closer in energy, the BOA breaks down and there is a mixing of the electronic and nuclear degrees of freedom. This is called a non-adiabatic coupling.
The two states may be coupled by a “photochemical funnel” which is also called a conical intersection (CoIn) (indicated by the number 4 in Figure 3). CoIns are not isolated molecular geometries; they are collections of geometries that form a high dimensional “seam” [39]. For nonlinear molecules, it has 3N-8 dimensions (here, N is the number of atoms, 6 degrees of freedom are for translation and vibrations and two dimensions lift the degeneracy). The potential energy surfaces intersect with each other forming a double cone, which is plotted against two special internal geometric coordinates, called “branching space coordinates”, (g) and (h), where (g) indicates the gradient difference and (h) represents the gradient of interstate coupling [40]. Motion along these coordinates lifts the degeneracy of the two excited states. This is depicted in Figure 7. As a result, in the photochemical reaction, passing through the CoIn can lead to two or more products, due to the different valleys on the ground state potential energy surface, unlike the single product obtained on the ground state in the thermal chemistry reactions.
The potential energy surface may have several minima which enable access to various CoIns that lead to the different photoproducts. As such, the nuclear motions at the conical intersections are often different. A CoIn is classified into two categories according to its topology [39, 41, 42]. One type is the “peaked” CoIn, where the nuclei have trajectories directly towards the intersection. The other type is called a “sloped” CoIn, where the potential energy surfaces have downhill slopes, and there is an increased possibility of nuclear trajectories passing from the higher excited state to the lower state and returning. At the simplest level, the “peaked” CoIn results in a high probability of transition, i.e., an ultrafast electronic transition. The “sloped” CoIn induces a low transition rate. However, Malhado et al [43]

13
assert that with the same crossing velocity (momentum), the non-adiabatic transition probability is the same for both the “peaked” and “sloped” CoIns. The higher efficiency at a “peaked” CoIn rather than at a “sloped” CoIn is due to dynamical effects, and does not depend on the topology. This is in agreement with the Landau-Zener model [44]. Figure 7. A conical intersection. The double cone is plotted against the two branching space coordinates. As the population transitions from one state to the other, if the two states are both singlet states the process is called internal conversion (IC), while if one state is a singlet state and the other is a triplet state, the process is called intersystem crossing (ISC). ISC is driven by spin-orbit coupling, which involves the coupling of an electron’s spin with orbital angular momenta. Generally, for molecules which do not contain heavy atoms, this coupling is weak, such that the ISC rate is low. However, as the El-Sayed rules assert [45, 46, 47], if the radiationless transition involves a change of molecular orbital type, the ISC rate increases significantly, e.g. the transition from a 1(ππ*) to a 3(nπ*) state or from a 1(nπ*) to 3(ππ*) in the carbonyl compounds. Furthermore, ISC is enhanced if the energy gap between the singlet and triplet states is small. As a consequence, competition between ISC and IC has also been found in benzene, which is a hydrocarbon molecule that has no obvious large spin-orbit coupling, but where this is motivated by the near degeneracy of the S1 and T2 states [48].

14
Non-radiative decay pathways have emerged as a dominant mechanism for photochemical reactions in many molecular systems. They allow for the energy transfer from the electronic excitations to the nuclear degrees of freedom of the molecules. In summary, the dynamics of small organic molecules initiated by a UV or visible photon can be described by an electronic transition from the ground state to the FC region of the excited state, which follows the standard selection rules for one-photon absorption. The molecule leaves the FC region and propagates to different final configurations through the available conical intersections via different vibrational modes. In addition, there are other factors which also need to be considered to describe the excited state dynamics of molecules. Two of these are the time taken by the molecules populated on the excited state to access to the CoIn (called the time delay), and the time taken for molecules to pass through the conical intersection to the lower-lying excited state or the ground state (called the time constant). These aspects will be discussed in more detail in Chapter 5.

15
3. Excited state dynamics of molecules
Excited state dynamics of molecules have been at the centre of interest for a long time. Here, I would like to review some important aspects from the viewpoint of the dynamophore. The chapter starts with the excited state dynamics of ethylene as the “bottom” system, using the 4-step description of the photodynamics discussed in Chapter 2. I then give an introduction to the effect of substituent groups, e.g. methylation, on the dynamics before proceeding to more complex systems, i.e., by adding more double bonds, to form linear and cyclic polyenes. Finally, I finish by explaining the effects of functional groups containing heteroatoms on the dynamics. In this way, I provide a clear picture on the change in the dynamics with controlled chemical modification of the molecules.
3.1 Excited state dynamics of ethylene Ethylene is the smallest organic molecule containing a π orbital. As the basic unit of unsaturated molecules, ethylene attracts much interest. Extensive theoretical [33, 49-55] and experimental [34, 56-60] studies have been undertaken to better understand its excited state photodynamics. Furthermore, being the “bottom”-molecule of this thesis, the ethylene dynamics serves as a model for understanding the photochemical and photobiological processes in larger systems. Ethylene is a planar molecule in the ground state. Photoexcitation to the lowest-lying ππ* state induces a rapid C=C twist motion as the molecule moves from the Franck-Condon region towards the conical intersection region, where an ultrafast internal conversion to the electronic ground state occurs [34, 60]. A sketch of the dynamics is given in Figure 8, where Φ indicates the C=C torsion angle. Upon population of the ππ* state, the molecules leave the FC region and start twisting. When the excited state potential reaches a minimum, at a 900 twist angle [52, 61, 62], the potential energy can be further lowered by a break of molecular symmetry [63, 64]. Here two different pathways are open. The first accesses a conical intersection which leads to a hydrogen-atom migration from one carbon atom to the other, forming an ethylidene-like configuration. The second

16
accesses a CoIn leading to pyramidalization of one CH2 group. The ethylidene-like and pyramidalization structures are shown in Figure 8.
Figure 8. Sketch of the excited state dynamics of ethylene. The molecules are excited to the ππ* states by the pump pulse (blue arrow) from the ground state (S0). The population on the ππ* state proceeds along the C=C bond torsion coordinate. After reaching the minimum, two reaction pathways are pyramidalization or hydrogen migration to form CH3CH. Furthermore, also on the hot ground state, hydrogen-atom elimination will occur. Φ, on the x-axis, indicates the C=C bond torsion angle. It has been identified that there is a two-electron excited zwitterionic state (π*)2 (Z state) crossing the ππ* state, and which lies slightly below it at a C=C twist angle of 900 [65-67]. Hence, the excited state experiences “sudden polarization” [67, 68], yielding a structure with a planar partially positive CH2 group and a pyramidalized partially negative CH2 group. The pyramidalization of the CH2 group means it is formally identical with a sp3- hybridization of the carbon atom which is able to stabilize the negative charge. Ethylidene (CH3CH), which can only be formed by hydrogen migration, has been detected in experiments [59]. Several investigations indicate that hydrogen elimination also occurs on the hot ground state after internal conversion [58, 69, 70].
In the context of the current discussion, the C=C group can be considered as the dynamophore, as it is the dynamics (the motion) of this bond which drives the processes along. Furthermore, along the twisting motion on the

17
excited state, it is also possible to transiently populate the π3s-Rydberg state [33, 34]. As the wavepacket progresses on the valence state, the C=C bond torsion leads to a coupling of the ππ* state and the π3s Rydberg state. As a result, there is an ultrafast population transfer from the initially excited state to the lower-lying Rydberg state. Even though the time scale is very short, it indicates the involvement of the Rydberg state in the relaxation of photoexcited ethylene. The Rydberg state dynamics following direct population of the π3s Rydberg state upon photoexcitation has also been investigated [59]. The π3s Rydberg state is lowered in energy with CC twisting, reaching a minimum at around 25-300 [71], while the ππ* state is lowered in energy by twisting to 900. The two states intersect each other, and electron population flows from the π3s Rydberg state to the ππ* state, accompanied by C=C twisting and C=C stretching. The controlled addition of substituents on a molecule may help elucidate the dynamics, where both the number of substituents and their location in the molecule could cause a change in the photodynamics. Methylation, exchanging a H-atom for a CH3-group, is one simple way to do this. The Stolow group investigated the effect of methylation on the dynamics in ethylene upon photoexcitation to the π3s Rydberg state [72]. They report that with increasing methylation the decay time of the Rydberg state increases, and that population transfer occurs on the picosecond time scale for the tetramethylethylene molecule. This is explained by an increased energy barrier to accessing the π3s/ππ* conical intersection. As a result, upon methylation, the energy barrier between the Franck-Condon region (the maximum of the torsional potential) of the π3s Rydberg state and the π3s/ππ* conical intersection increases, and causes a slowing of the C=C bond torsion mode. Methylation also affects the dynamics on the ππ* state which, in turn, affects the H-migration and pyramidalization of the CH2 group. It can be concluded that addition of the CH3-group not only affects the inertia but has other implications, as it shifts excited states in energy which can become the determining factor if they are close together. As a result, for ethylene and the methylated ethylene molecules, the chromophore and the dynamophore are the C=C double-bond.
3.2 Excited state dynamics of polyenes
3.2.1 Linear polyenes For the ethylene and methylated ethylene molecules, the chromophore and dynamophore is the C=C bond. I now increase the complexity slightly

18
by moving to linear polyenes and cyclic systems, in which we add additional C=C double bonds, i.e. the ethylene moiety, and see how these influence the dynamics. Polyenes attract much interest due to their importance in biological systems [73, 74]. Polyenes are generally considered as a family of molecules with alternating single and double carbon bonds, and constitute fundamental chromophores. Molecules containing four or more double bonds are called “longer polyenes”, while butadiene and hexatriene are “shorter polyenes”. The structure of several selected polyenes is shown in Figure 9.
Figure 9. The structure of 1,3-butadiene, 1,3,5-hextriene, and 1,3,5,7-octatetraene
Comparing the absorption spectrum of polyenes to that of ethylene shows that the lowest-lying singlet excited state is a ππ* state [75, 76]. Studies of polyenes indicate that there are two low-lying excited states involved. One is a strong singly excited state 1B (ππ*) transition, corresponding to a HOMO -> LUMO excitation, and the other is a linear combination of a doubly excited configuration with higher lying singlet excitations, the 2A state. Schuleten and Karplus [77] found the HOMO2 → LUMO2 configuration mixes with different higher single excitations, e.g. HOMO-1 → LUMO, and HOMO → LUMO+1, which brings down the energy of the 2A state and makes it lower-lying than the 1B state. The 2A state is optically “dark”, which means that it is not directly accessible via a single photon excitation.
In the photodynamics of ethylene, the 2A state, often referred to as the Z state, lies higher in energy in the Franck-Condon region than the 1B (ππ*) state compared with the longer polyenes. This is because of a decreased interaction with the singly excited states since HOMO-1 and LUMO+1 do not have π-character. Thus, when the ethylene molecules are excited to the 1B state, the 2A state does not play role until the molecule is significantly distorted. Conversely, in the shorter polyenes, like butadiene, the 2A state plays a role before the torsion distortion makes the symmetry label irrelevant. However, for the longer polyenes, the 2A state is only involved when the
19
torsion is so severe that the symmetry label becomes meaningless [25]. We can look separately at what happens to the dynamics on going from the shorter to the longer polyenes.
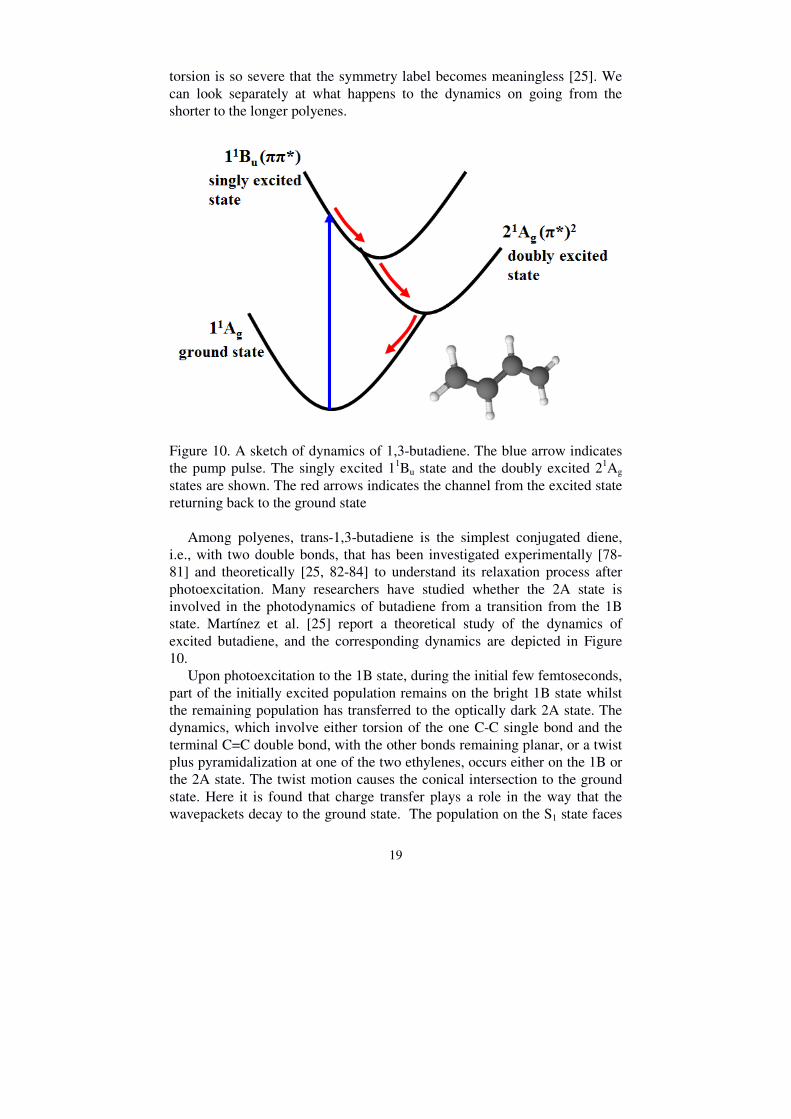
Figure 10. A sketch of dynamics of 1,3-butadiene. The blue arrow indicates the pump pulse. The singly excited 11Bu state and the doubly excited 21Ag states are shown. The red arrows indicates the channel from the excited state returning back to the ground state
Among polyenes, trans-1,3-butadiene is the simplest conjugated diene, i.e., with two double bonds, that has been investigated experimentally [78-81] and theoretically [25, 82-84] to understand its relaxation process after photoexcitation. Many researchers have studied whether the 2A state is involved in the photodynamics of butadiene from a transition from the 1B state. Martínez et al. [25] report a theoretical study of the dynamics of excited butadiene, and the corresponding dynamics are depicted in Figure 10. Upon photoexcitation to the 1B state, during the initial few femtoseconds, part of the initially excited population remains on the bright 1B state whilst the remaining population has transferred to the optically dark 2A state. The dynamics, which involve either torsion of the one C-C single bond and the terminal C=C double bond, with the other bonds remaining planar, or a twist plus pyramidalization at one of the two ethylenes, occurs either on the 1B or the 2A state. The twist motion causes the conical intersection to the ground state. Here it is found that charge transfer plays a role in the way that the wavepackets decay to the ground state. The population on the S1 state faces

20
one or two funnels to form two oppositely polarized charge transfer states, which means one twisted methylene unit is positively charged and the rest forms a negative charge. The proposed reaction channels are drawn in figure 11.
Figure 11. The proposed reaction channels happen on the excited state: pyramidalization and the charge transfer state. Excitation of butadiene to directly populate the optically dark 2A state has been investigated via two photon absorption [78, 80]. In the Franck-Condon region, the 2A state lies slightly higher in energy than the 1B state [85-87]. The C=C bond becomes weaker in the 2A excitation, being almost like a C-C single bond, and then it is broken by the twist of the terminal C=C bond.
Access to the π3s- and π3d-Rydberg states via two photon absorption has also been studied by Schalk et al. [80]. The lowest-lying 3s-Rydberg state is strongly coupled with the valence manifold, and so the decay rate of the π3s-Rydberg state is larger. Furthermore, for the π3d-Rydberg state dynamics, it is found that the different excitation wavelength does not affect the position of the Rydberg state peak, i.e., no energetic shift is found. This is due to, when the Rydberg state is ionized, the diagonality of Franck-Condon factor indicates the preservation of internal vibrational energy upon photoionization. In comparison with the dynamics of ethylene on the ππ* state, the dynamics of butadiene have a localised dynamophore, which keeps the ethylene-like torsion of the C=C bond. However, this only happens on the terminal C in one of the C=C double bonds, and the butadiene dynamics also displays delocalised charge transfer dynamics on the way from the 2A state to the ground state. Hexatriene contains one/two more double bonds than butadiene/ethylene, respectively. The dynamics of hexatriene is useful for understanding the photodynamics of cyclohexadiene, as it is a photoproduct from ring-opening in 1,3-cyclohexadiene. It is reported that the 2A state lies lower in energy than the 1B state, which reinforces the general trend for polyenes that the lowest singlet excited state has a doubly excited configuration [88]. The molecules are predominantly excited to the 1B state, which has a very short lifetime (20~40 fs) due to a rather steep slope along the double-bond

21
lengthening and single-bond shortening co-ordinate [89]. The nuclei are strongly accelerated, and leave the Franck-Condon region and undergo internal conversion to the 2A state. Quantum calculations indicate the crossing point of the 1B and 2A states is located near the Franck-Condon region, then the internal conversion proceeds along the symmetric deformation mode [90]. Several groups measured the lifetime of the 2A state and found values between 250 and 600 fs [88, 91, 92]. On the 2A state, the molecules twist around the C=C double bond and the adjacent C-C bond, and the population decays to the ground state where isomerization of the C-C single bond occurs, forming trans and cis conformers [93]. For the longer polyenes, octatetraene is taken as an example. All-trans-1,3,5,7-octatetraene contains four double bonds. Similar dynamics has been found for all trans-1,3,5,7-octatetraene as for the shorter polyenes. Upon photoexcitation, molecules predominantly populate the 1B state, and the population decays to the 2A state [93]. Calculations indicate that for all trans-1,3,5,7-octatetraene internal conversion from the 2A state to the ground state occurs due to a non-adiabatic trans-cis isomerization around the central C3 and C4 C=C double bond [94]. In comparison with 1,3,5-hexatriene, the lifetime of the 1B state is much higher in 1,3,5,7-octatetraene, which could be explained by the difference in the vibronic coupling strengths between the 1B and 2A states. Furthermore, the lifetime of the 2A state is also longer than for hexatriene, being a few nanoseconds long [93]. From butadiene to octatetraene, the photodynamics involves both the 2A and 1B states. From the point view of the dynamophore, butadiene has one localised dynamophore, due to one terminal double bond torsion, whilst the other remains planar on the 1B or 2A state and also displays a delocalised dynamophore due to the charge transfer state. For hexatriene and octatetraene, with the increased length of the double-bond chain, the cis-trans isomerization conformers obtained also indicates localised dynamics, although the dynamics are so slow that a dynamics picture of the reaction almost makes no sense.
3.2.2 Cyclo-polyenes Having reviewed the photoexcitation dynamics of linear polyenes, I now focus on ring-systems with double bonds whose dynamics include hydrogen shift, ring-puckering, and bond cleavage. Recall the dynamics of the ethylene molecule, where it undergoes a C=C double bond torsion when decaying to the ground state followed by the pyramidalization of one carbon bond. Similar dynamic processes are also observed in trans-1,3-butadiene, which displays a double bond torsion and pyramidalization of the double bond to the central single C-C bond. How will the double bond affect the

22
dynamics in a ring system? Here, cyclohexene and cyclohexa-1,4-diene will be taken as two examples to illustrate the dynamics in six-membered ring systems.
3.2.2.1 Cyclohexene and cyclohexa-1, 4-diene Cylcohexene is a six-membered ring system with only one double bond. According to early quantum calculations [95], the photochemical reactions of cyclohexene include hydrogen shift, carbene formation, bond cleavage, and ring puckering. These are depicted in Figure 12. [13, 96].
Figure 12. The photoreaction dynamics of cyclohexene. After a first arrangement on the excited state, the next reactions occur on the hot ground state (adapted from reference [13]). In the photodynamics of cyclohexene (as shown in figure 13), two excited states are involved, the ππ* state and the π3s Rydberg state [97]. The ππ* state is optically dark to one-photon absorption while the Rydberg state is weakly absorbing [98]. Upon photoexcitation to the π3s Rydberg state the molecules rapidly depart from the Franck-Condon region of the Rydberg state to the ππ* state, accompanied by a C=C bond stretch and twist. For molecules travelling along the ππ* surface, several low-lying conical intersections to the ground state can be identified: a [1,2] H-bridge, a [1,3]

23
H-shift, a [1,2] H-shift, and a bond cleavage in the α position and in the β position [99, 100]. Furthermore, recent unpublished calculations for the dynamics of cyclohexene on the ππ*-state indicate the C=C bond twisted pyramidalization is the dominant channel (95%), while the bond cleavage and hydrogen shift channels are insignificant [101].
Figure 13. Sketch of the photodynamics of cylcohexene and cyclohexa-1,4-diene upon excitation at 200 nm.
Cyclohexa-1, 4-diene can be considered as adding one more double bond to cyclohexene, where the two double bonds are separated by one –CH2– group. The photochemical pathways are summarized in Figure 14, which are similar to cyclohexene, and which include the hydrogen shift, ring puckering and bond cleavage channels.
For a comparison of the photodynamics of cylcohexene and cyclo-1,4-hexadiene, their photoelectron spectra [13] show similar features indicating similar dynamics. In cyclohexa-1,4-diene, the molecules are mainly excited to the π3s Rydberg state followed by transfer to the lower-lying ππ* state. According to recent unpublished quantum calculations on the dynamics of cyclohexa-1,4-diene on the ππ* state, the majority (~75%) of the dynamics are localised at a single double bond [101]. The molecules return from the excited state to the ground state, following the hydrogen shift or the twisted pyramidalization pathways. This indicates that the presence of the second double bond had nearly no influence on the dynamics, and the molecular orbitals also show no involvement of this second π-bond. Interestingly, there is still around 25% of the population which undergoes a twist at both double

24
bonds, meaning a twist of the entire ring to form a “chair-like” structure [101].
Figure 14. Photoreaction pathways of cyclo1,4-hexadiene. After the first arrangement upon photoexcitation, the next reactions occur on the hot ground state (modified from reference [13]). From the idea of the chromophore and the dynamophore for these two molecules, the chromophore and dynamophore of cyclohexane are both localised on one double bond, which mainly involves the twisted pyramidalization. In cyclohexa-1,4-diene, the chromophore is delocalised into the homoconjugated double bonds. A large fraction of the molecules upon photoexcitation have the same dynamophore as cyclohexene, where the large amplitude nuclear motion is localised only at single double bond, meaning that bond interaction between the two double bonds on the excited state is a minimum. However, there is still a fraction of the cyclohexa-1,4-diene molecules upon photoexcitation which have the same chromophore and dynamophore, and that the dynamics are delocalised into the whole ring system. Comparing these to the ethylene dynamics, the [1,2] hydrogen shift in a cyclo-system corresponds to the hydrogen migration in ethylene to form the ethylidene-like molecule. Moreover, in the ring system, the ethylene-like double bond twist and pyramidalization causes a similar twisted pyramidalization and, even, ring-opening in cyclohexene and cyclohexa-1,4-diene.

25
3.2.2.2 Cyclopentadiene The focus now moves to 5-membered ring systems. Cyclopentadiene is taken as an example, which is a five-membered ring system with two double bonds. This molecule is a very important precursor in organic synthesis [102]. The photochemical pathways [16, 17, 103, 104] of cyclopentadiene are described in Figure 15, and are similar to those observed in cyclohexene and cyclohexa-1,4-diene: involving a ring puckering channel, and products like bicylo[2,1,0]pentene (Bp) and tricyclo[2,1,0,0]pentane (Tp), which are highly strained structures formed via electrocyclic ring closure [103].
Figure 15. Photochemical reaction of cyclopentadiene. Tp is tricyclo[2,1,10,0] pentane and Bp is bicylco[2,1,0]pentene.
From previous studies, the optically dark 2A state has been thought to be
involved in the photodynamics [103, 104]. However, recent experimental and theoretical results show that there is only one excited state involved in the photodynamics [16, 17]. These studies indicate that after cyclopentadiene is excited to the ππ* state, the initial dynamics happen on the in-plane modes and then follows the out-of-plane modes, where the wavepacket acquires doubly excited character along these modes when accessing the conical intersection between the ππ* state and the ground state. This two-step mechanism was misunderstood as occurring separately on two different excited states. A sketch of the photodynamics of cyclopentadiene is depicted in Figure 16. The out-of plane motion causes a significant torsion in one of the double bonds, leading to the out-of-plane bend of the CH2 group and the neighbouring hydrogen, and accesses the conical intersection to the ground

26
state. Furthermore, the out-of plane motion favours the reaction pathway leading to the formation of Bp. Several methylated derivatives of cyclopentadiene are investigated by Schalk et. al to unravel the substituent effect on the dynamics [104]. The substituents clearly slow down the motion, and it could be shown that substitutions out of the ring-plane only slowed down the out of plane motion, while in-plane substitutions affected both time constants. This interpretation agrees with theoretical findings [16]. The methyl substituent has only a minor effect on the topography of the conical intersection, but adds inertia to the vibrational motion.
Figure 16. Sketch of the photodynamics of cyclopentadiene. We can make a comparison with linear polyenes and cylcopentadiene. For linear polyenes, the 2A state is the lowest-lying doubly excited state. For these molecules, upon excitation to the 1B bright state, the populations transfer to the 2A state following the C=C bond stretching and torsion. However, in cyclopentadiene, the bond torsion gives access to the conical intersection between the 1B state and the ground state, although the relative proximity of the 2A state leads to a partial doubly excited character of the potential energy surface. From the idea of the dynamophore, cyclopentadiene is similar to butadiene, cyclohexene, and a high fraction of cyclohexa-1,4-diene, upon photoexcitation, in which the dynamics is localised on one double bond and there is torsion of the H2C=CH-CH3 moiety. As mentioned earlier, there is a charge transfer state which plays a role in the dynamics of butadiene when the population decays from 2A state to the ground state. However, in

27
cyclopentadiene, the constraint of the ring structure does not allow a complete out-of-plane twist as was found in butadiene, and a smaller charge transfer was observed [16]. When considering a system with more double bonds, e.g. the cycloheptatrienes [15, 105], the bright and the dark valence states come closer together. After excitation to the ππ* state, cyclopeheptatriene reaches the dark state within a few tens of femtoseconds, and reaches the ground state via a conical intersection with a fast [1,7] hydrogen shift. This motion can be considered as a localised motion since the ring planarization coordinate seems to be decoupled from the non-adiabatic dynamics [105, 106].
3.3 Excited state dynamics of molecules with heteroatoms The photodynamics of ethylene and polyenes has been discussed which indicates that some polyenes retain the ethylene-like dynamics. Now the focus is changed to molecules with heteroatoms. These kinds of molecules are considered from two aspects; one from adding heteroatoms to ethylene, e.g. acrolein, acrylonitrile, alkyl vinyl ethers, etc. and the other is from adding the heteroatom into the ring structure, i.e. heterocycles, e.g. pyrrole and furan.
3.3.1 Acrolein The first molecules to be discussed are created by adding a heteroatom to the ethylene moiety. Acrolein is taken as one of the examples. The structure of acrolein is shown in Figure 17 (a).
Figure 17. The structure of: (a) acrolein, (b) furan and (c) pyrrole
Unlike linear polyenes, the acrolein molecule contains a –C=O functional group which introduces an additional excited state and different photochemical pathways due to the existence of nonbonding electrons from the oxygen atom. In addition to the lowest-lying ππ* state, there is another low-lying excited state, the 1nπ* state, which is due to an excitation from the

28
nonbonding lone pair orbital of the oxygen atom to the delocalised antibonding π* orbital. There are two reported major product channels from measuring acrolein in the liquid phase: one is the [1,3] Hydrogen migration channel, and the other is the α-hydrogen loss process [107].
Figure 18. The photodynamics of acrolein upon photoexcitation to the ππ* state. Upon photoexcitation to the ππ* state, the population leaves the Franck-Condon region and undergoes internal conversion to the 1nπ* state within 100 fs [108]. On the 1nπ* state, there are two possible relaxation mechanisms [109]; internal conversion to the singlet ground state, and intersystem crossing to the 3nπ* state or 3
ππ* states, of which the latter is allowed by the El-sayed’s rule. Regarding the quantum calculations [109-111], acrolein has a planar structure at room temperature, and the dynamics happens on the ππ* state, involving the terminal –CH2 group rotating out of the plane by around 900. The photodynamics of acrolein is depicted in Figure 18. Experimental studies on acrolein and its derivatives have been undertaken in both liquid phase and gas phase [10, 107, 108, 112]. Lee and co-workers [108] investigated the methylation effect on the photodynamics of acrolein. The decay time on the 1nπ* state is significantly influenced by the methylation position on the acrolein molecule. This is explained by the terminal methylene torsion on the 1nπ* state to the conical intersection between the 1nπ* state and the ground state. Furthermore, the study reported by Schalk et. al. [10] also indicates the methylation effect on the dynamics of acrolein, especially to the decay rate of the IC and ISC processes. As mentioned in Chapter 2, IC through a conical intersection has a more rapid decay rate compared to that of ISC. However, in those molecules with an aldehyde group, the rate ISC can become competitive with IC. This is both

29
because of a large spin-orbit coupling effect over an extended region of near degeneracy between the singlet and the triplet state, and that the rate of IC slows down due to a sloped conical intersection. Conversely to studies on hydrocarbons, increasing methylation does not necessarily slow down the dynamics in acroleins. For example, crotonaldehyde (γ-methyl acrolein) decays twice as fast as acrolein due to a better access to the CoIn region [108]. Acrolein displays different reaction dynamics as compared to ethylene and the linear polyenes, which now includes a 1nπ* state in the photodynamics. The factors influencing the IC rate for decay from 1nπ* state to the ground state are the twist motion of the terminal CH2 group and the velocity of the wavepacket through the conical intersection involving the two branching coordinates (the h and g vectors), i.e. the twist motion and pyramidalization at the C2 atom, respectively, where the pyramidalization dominates. Furthermore, the aldehyde group effect on the ring system, e.g. in cyclopentenone, shows dynamics similar to acrolein [10]. Here, the dynamics slows down due to an increased barrier between the 1nπ* state to the ground state. This opens the door for ISC, but the photoproduct formed after return to the ground state of cyclopentenone involves the α-cleavage reaction at the carbonyl group, and, if a γ-hydrogen is present, leading to the creation of dienole or cyclobutane [113]. Another recent study indicates that in comparison with acrolein and cyclopentenone the latter has a slower ISC rate owing to the ring-constraint, resulting in a smaller spin-orbit coupling [114]. From a study of methylated cyclopentenone, the decay rate of IC and ISC decreases with increasing methylation [10]. This is an example of the aldehyde group effect on the ethylene molecule, which changes the photodynamics but keeps the ethylene-like channel of terminal CH2 torsion. However, other molecules, like acrylonitrile, which introduces the effect of a –CN group onto ethylene, and alkyl vinyl ethers, e.g. methyl vinyl ether which adds a –OCH3 onto ethylene, have also been studied in our work, and this will be described in detail for their photodynamics and comparison with acrolein and ethylene in Chapter 6.
3.3.2 Heterocycles
Heterocycle compounds can be regarded as replacing one or more of the ring-carbon atoms with different atoms in the cyclopolyene structure, e.g. pyrrole, furan, pyrazole, oxazole, etc. These molecules play an important role in organic chemistry, pharmaceutical chemistry, and in biological systems. The dynamics of cyclopolyenes has been discussed earlier, where the general trend for molecules is the out-of-plane mode motion on the ππ* state which causes one double bond torsion and ring puckering. For

30
heterocycles, the exchanged atoms will introduce new excited states and different dynamics. For example, a new excited πσ* state participating in the pyrrole, furan, and pyrazole dynamics [115, 116], the nπ* state in the oxazole dynamics [117], and, in addition to the similar dynamics of ring-puckering, bond cleavage channels have also been observed. The detailed dynamics and comparison of heterocycles will be described below, with the following two molecules taken as an example; furan (the structure is shown in figure 17(b)), to show the effect from the oxygen atom, and pyrrole (the structure is shown in figure 17 (c)), to investigate the effect of introducing a nitrogen-hydrogen bond. Furan is a five-membered ring system, which is obtained by replacing a carbon atom in cyclopentadiene with an oxygen atom. In contrast to acrolein and cyclopentenone, the oxygen atom in the ring-system does not connect to the adjacent carbon atoms with a double bond, but forms a cyclic 6-electron π-system. Therefore, these systems are also called heteroaromatics. The interest in furan is due to it is role as a pollutant [118, 119] in atmospheric chemistry, and in combustion it has been considered as a potential biofuel [120,121]. The two lowest-lying excited states of furan are the 1B2(ππ*) state and the 1A1(ππ*) state, respectively. The lowest-lying Rydberg state is the π3s Rydberg state, whose symmetry label is 1A2, which weakly couples with the 1B2(ππ*) state. The other lower-lying Rydberg states are the 3p Rydberg states, which could split into three types: 1B1(3py),
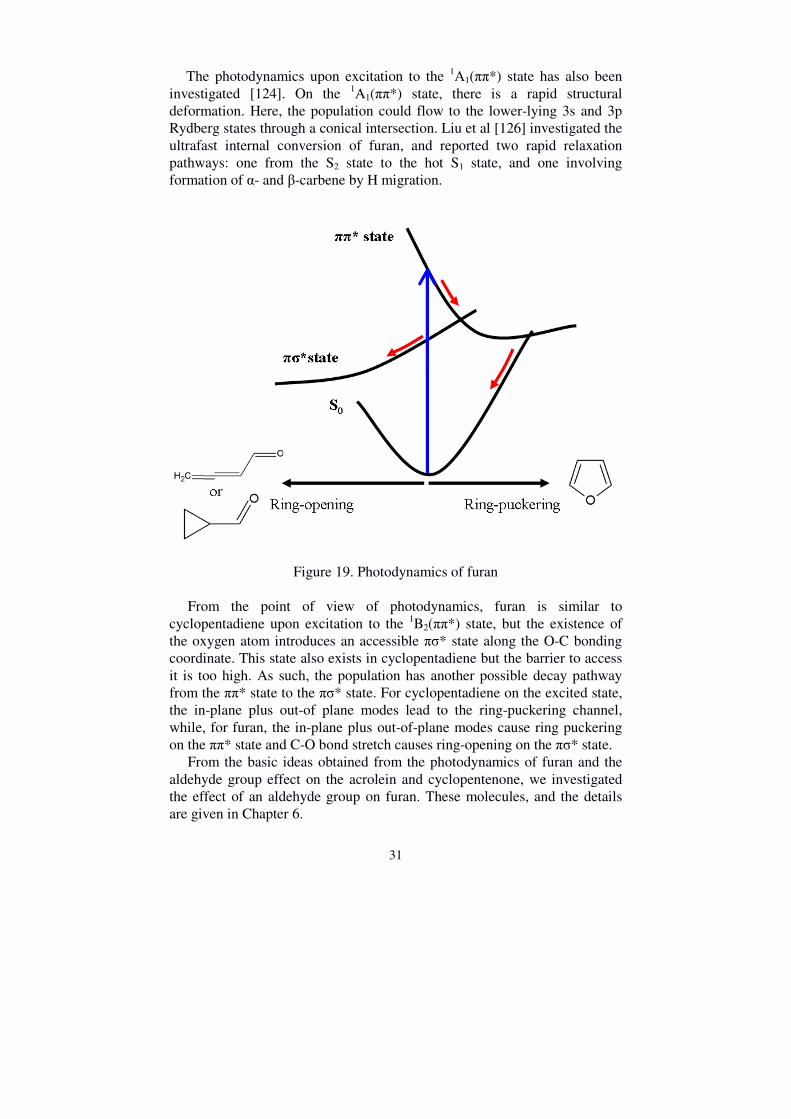
1B2(3px) and 1A2(3pz). Photoexcitation to both the 1B2(ππ*) state and the 1A2(π3s) state has been studied [4, 122-124]. On the 1B2(ππ*) state, the wavepacket motion involves the C-O stretching motion together with an out-of-plane deformation mode, and the population transfers to the ground state through a conical intersection. Quantum calculations also show [125] the two competing deactivation pathways upon photoexcitation to the ππ* state. It is found that the reduced energy of the πσ* state along the Franck-Condon active in-plane ring-breathing mode allows access the ππ* state via a conical intersection. From here, the population flows to the πσ* state, and, on the way to accessing the conical intersection with the ground state, undergoes a ring-opening, which requires a large torsion of the atomic framework. Conversely, the ring-puckered pathway is less complicated, in which relaxation along the ππ* state accesses the conical intersection with the ground state via the ring deformation. The CoIn of the ring opening channel is energetically more stable than the CoIn of the ring puckering channel; however, the barrier to access the πσ* state might cause a low quantum yield. The photodynamics of furan is described in Figure 19.
According to the photodynamics of furan, the photoproducts can be determined after excitation to the 1B2(ππ*) state, and these are also shown in Figure 19. The C-O bond-cleavage channel could form 3-butadienal and cyclopropen-3-carbaldehyde.
31
The photodynamics upon excitation to the 1A1(ππ*) state has also been investigated [124]. On the 1A1(ππ*) state, there is a rapid structural deformation. Here, the population could flow to the lower-lying 3s and 3p Rydberg states through a conical intersection. Liu et al [126] investigated the ultrafast internal conversion of furan, and reported two rapid relaxation pathways: one from the S2 state to the hot S1 state, and one involving formation of α- and β-carbene by H migration.
Figure 19. Photodynamics of furan
From the point of view of photodynamics, furan is similar to cyclopentadiene upon excitation to the 1B2(ππ*) state, but the existence of the oxygen atom introduces an accessible πσ* state along the O-C bonding coordinate. This state also exists in cyclopentadiene but the barrier to access it is too high. As such, the population has another possible decay pathway from the ππ* state to the πσ* state. For cyclopentadiene on the excited state, the in-plane plus out-of plane modes lead to the ring-puckering channel, while, for furan, the in-plane plus out-of-plane modes cause ring puckering on the ππ* state and C-O bond stretch causes ring-opening on the πσ* state. From the basic ideas obtained from the photodynamics of furan and the aldehyde group effect on the acrolein and cyclopentenone, we investigated the effect of an aldehyde group on furan. These molecules, and the details are given in Chapter 6.

32
Having introduced the effect of oxygen on a heterocycle, I now move to introducing a nitrogen atom, specifically an N-H bond. This is the pyrrole molecule. Pyrrole is a prototypical heteroatom molecule, and the most important interest in it arises from the fact that it is a significant subunit for large biological system, for examples the DNA bases, aromatic amino acids and so forth. As such, pyrrole has been measured extensively in theory and experiments [9, 35, 36, 127-136]. The absorption bands with excitation energies between 5.5 eV and 6.5 eV have been assigned by quantum calculations [137], and from low to high energy involve the A2(πσ*), B1(πσ*), π3p Rydberg, A1(ππ*), and B2(ππ*) states. The π3p Rydberg state separates into the 3pz and 3py states, and, especially for the π3py Rydberg state, the absorption structure shows a prominent, sharp feature [137, 138]. The (πσ*) states have a strong s-Rydberg admixture, and are typically respect to the bond dissociation in the dynamics. According to Barbatti [35] et al., who investigated the pyrrole dynamics theoretically, there are two main relaxation pathways: one involving the N-H bond dissociation, and the other involving ring-puckering on the excited state which accesses the conical intersection with the ground state. If the pyrrole molecules are excited initially to the B2(ππ*) state, then the population crosses the conical intersection between the B2(ππ*) state and the A2(πσ*) state, leading to N-H bond cleavage and ring puckering. This is described in Figure 20. However, if the molecules are initially excited to the A2(πσ*) state, this directly induces the N-H bond cleavage and the out-of-plane motions.
Figure 20. One kind of photodynamics in pyrrole

33
Different excitation wavelengths between 200 nm and 250 nm have been used to investigate the photodynamics of pyrrole [9]. These indicate that the longer wavelengths, e.g., 250 nm, lead to longer lifetimes on the A2(πσ*) state. Furthermore, Wu et. al. studied the dynamics on the A2(πσ*) state with different wavelengths [36]. They report that there are two pathways leading to N-H dissociation. One is a fast N-H cleavage due to the wavepacket lying above the barrier on the πσ*-state with a higher excitation energy to the dissociation channel, while the other is slow due to a lower excitation energy which puts the wavepacket below the barrier, requiring it to tunnel through the barrier to dissociate. As a result, the excitation wavelength determines the photodynamics of pyrrole and the lifetime of the dynamics.
Pyrrole also can be directly excited to the B1(πσ*) state, where there is a decay pathway for internal conversion to the A2(πσ*) state. Quantum calculations indicates that around 34% undergo N-H cleavage directly on the B1(πσ*) state, while the remainder follow the decay path to the A2(πσ*) state [36]. Methylated pyrrole, like N-methyl pyrrole, is studied with excitation to the π3p Rydberg state [139]. Here, the population transfers to the A2(πσ*) state where the ring-puckering and N-CH3 dissociation channels occur. The common point for cyclo-system molecules, like cyclopolyenes, furan, and pyrrole, is that they all undergo ring-puckering dynamics. Adding heteroatoms opens up new and different channels for different molecules, e.g. for furan, the C-O bond stretch on the excited state causes a ring-opening, and, for pyrrole, the N-H stretch leads to N-H dissociation. Conversely, comparable with ethylene and polyenes, the pyrrole and furan molecules have an additional excitation state, since there is a πσ* state where bond cleavage occurs. As the π3p Rydberg state lies lower in energy than the B2(ππ*) state, there is a possibility of transiently populating the Rydberg state upon photoexcitation to the B2(ππ*) state which can have a guiding influence on which of the NH dissociation channels is accessed. We have investigated this interaction and the details are given in Chapter 6.

34
4. Experimental apparatus
The experimental setup includes the laser system, the optical setup with the creation of the pump and probe pulses, the magnetic bottle spectrometer, and the connected electronics which are controlled by a server and which is read out by a personal computer. An overview of the setup is shown in Figure 21. The main parts of the setup are described next.
Figure 21. Sketch of the experimental apparatus. The laser beam is split into two parts, one for the pump pulse, and another for the probe pulse. Delay stage is computer controlled. The pump and probe pulses overlap and get into the reaction chamber to ionize the molecules, and the electron travels fast to the end of the magnetic bottle where an MCP-based detector is used to detect the electron and is also connected to the control computer. A pure pump only or probe only signal is obtained by using shutter.

35
4.1 Laser system A solid-state femtosecond laser system from Coherent Inc is used in all experiments. A detailed overview of the laser system is shown in Figure 22, which illustrates how the laser system is split in to an oscillator and an amplifier.
Figure 22. An overview of the laser system Firstly, the diodes in the Verdi V-5 pump laser excite a Neodymium-doped yttrium orthovandanate (Nd: YVO4) crystal. The crystal emits laser light in continuous-wave mode at 1064 nm and is intra-cavity frequency doubled to provide a doubled frequency output laser (green colour in Figure 22) with a wavelength of 532 nm and an output power of 5 W. The 532nm laser beam is used to pump the Titanium:sapphire laser (“Mira”) to produce ultra-short, broad-bandwidth, femtosecond pulses at a centre wavelength of 792 nm at a 78 MHz repetition rate and an output power of around 700 mW. This pulse is mode-locked by using the Kerr Lens mode locking method [140] which can be controlled by an aperture in the cavity. The two prisms inside the cavity are used to control the group velocity dispersion of the laser light and for tuning the position of the central wavelength of the laser. The oscillator output is coupled into a regenerative amplifier. The Ti:Sapphire crystal (in the “Legend USP-HE”), used as an amplification medium, is firstly pumped by a laser beam generated by the Evolution-30.

36
The Evolution-30 laser utilises a Nd:YLF crystal to provide high energy pulses at a low repetition rate, and is excited by diode pumping to produce 1053 nm light which is intra-cavity frequency doubled to 527 nm. The laser is Q-switched at 1 kHz, producing pulses of 250 ns duration with a power of 23 Watts. To achieve the final beam, a technique called chirped pulse amplification (CPA) [141] stretches the ultra-short pulse, amplifies the pulse, and finally compresses the pulse to its initial duration. The oscillator beam is first stretched to reduce the pulse peak intensity, which makes it successfully pass through the gain medium below its damage threshold. In the regenerative amplifier, the Pockels cells and broad-band polarizer are used for trapping and dumping the pulse. A single pulse is selected and trapped into the laser cavity, achieved by a high voltage on the Pockels cell switches, where the pulse now makes several round-trips to gain a high energy and reach saturation. At this point a second electro-optic Pockels cell is switched and the pulse is extracted from the cavity. The amplified pulse then enters into the compressor to both recover its initial duration and compensate the dispersion caused by the amplification stage. This is achieved by changing the distance of gratings in the compressor. Eventually, the final beam obtained has a wavelength of 800 nm (FWHM) with a power of 4.1 W, and a 1 kHz repetition rate with a pulse duration of less than 50 fs. The quality of the laser beam (M2-factor) is 1.5.
4.2 Optical setup- two colour experiment The optical path of a typical pump-probe experiment which is initiated from the 800 nm laser beam is shown in Figure 23. The 800 nm laser beam output from the laser system is sent through a beam splitter and split into two parts: one for the pump light, the other is the probe light. In the pump line, M1 is a concave mirror and M2 is convex mirror. The two mirrors are combined to form a telescope which reduces the beam radius. The fundamental beam is frequency doubled to provide 400 nm in the first beta-barium borate (BBO) crystal (23 degree, 600 µm), and a half-waveplate is used to rotate the polarization of the 400 nm light by π/2. The 400nm beam is then combined with the fundamental light in another BBO crystal (40 degree, 300 µm) to produce the sum frequency beam with a wavelength of 267 nm for third-order harmonic (THG) pulses. The fourth harmonic (FHG), with a wavelength of 200 nm, is produced by combining the 267 nm and 800 nm light in another BBO crystal (55 degree, 100 µm) (whose polarization is rotated by another half-wave plate). The pump beam is passed through a prism compressor, both to separate the different wavelengths and also to compress the pulses.

37
Figure 23. The optical path in two-colour light experiments. BS stands for the beam splitter. M1 and M2, M3 and M4 combine to form two telescopes. M5 is recombination mirror. M6 is a focusing mirror. The probe light, initially split beam from the fundamental, is used to obtain the third harmonic (THG) at a wavelength of 267 nm by passing through two BBO crystals with 23 (600 µm) and 40 (300µm) degrees, respectively. The probe beam is also passed through a prism compressor, and can be delayed by a computer controlled linear stage (Newport: ESP 300+M-ILS 250CCHA) with an aluminium coated hard-mounted hollow retroreflector. The linear delay stage has a 250 mm travel range, and provides 0.1 µm (0.33 fs) resolution feedback. The pulses are combined in collinear propagation using a dichroic mirror (M5 mirror), transmitting the probe and reflecting the pump. As a result, the probe light spatially overlaps with the pump light, and is focused by a concave Al mirror (M6 mirror) into the interaction region of the spectrometer. This is one optical setup for a pump-probe combination of 200-267 nm. We also use 200-400 nm (which is achieved by removing the THG crystal in the probe beam line), and 267-400 nm (achieved by removing the FHG crystal on the pump beam and one THG on the probe line) as pump-probe light for our experiments.

38
4.3 Interaction region and magnetic bottle spectrometer Injection of the sample gas into the interaction volume is controlled by a valve with a movable needle, providing fine regulation of the flow. In order to keep the interaction volume background-gas free, the systems is pumped with two turbo pumps backed by two rotary-vane fore-vacuum pumps. The base pressure for the interaction volume is around 1x10-7 mbar, and is kept below 10-5 mbar during experiments to avoid damage to the microchannel plate-base detector (MCP) which is used to detect the electrons. Liquid samples are measured by using their vapour pressure. The general back pressure for the measured samples is kept around 5.6x10-6 to 6.0x10-6 mbar.
Figure 24. (a) The Electron spectrometer. MCP is a microchannel-plate based detector. (b) The electron motion in the inhomogeneous magnetic field. Bi is the magnetic field generated by a permanent magnet, with the electron’s initial velocity vector tilted at angle θi. Bf is the magnetic field generated by solenoid, guides the electrons along the drift tube. Z is the axis of the solenoid. Time of flight (TOF) spectroscopy is used to measure the kinetic energies of the photoelectrons as they travel the fixed distance between the interaction region and the MCP detector. In this experiment, a magnetic bottle

39
spectrometer (5.5m) (as shown in figure 24(a)) is used, which was originally developed for photoelectron-photoelectron coincidence spectroscopy experiments pioneered by John Eland at Oxford University [142].
In the magnetic bottle the electrons travel through the magnetic field following a helical path as shown in Figure 24(b). This is obtained by using two different magnets, one is a permanent magnet and another is a solenoid, in the apparatus. Thus two different magnetic fields are used: one is a strong inhomogeneous field (~0.8T) in the interaction region generated by the permanent magnet, where the electrons are born and provides the initial velocity for the electrons and guides the electrons into the entrance to the spectrometer. The other one is a weak homogenous magnetic field (a few mT) to guide the electrons along the drift tube to the end of the magnetic bottle, and is produced by the solenoid. The solenoid is placed inside the flight tube and it is covered by mu-metal foil to prevent deformation of the weak magnetic field by earth’s magnetic field. In addition to the magnetic field, there is an electric potential applied between the needle (-0.3 V) and the magnet (-0.6 V) to repel the electrons by controlling the electron velocity and direct them towards the magnetic bottle. The resolution of spectrometer depends on the kinetic energy of the electrons, and the energy resolution (E/∆E) has been found to be better than ~100. The MCP-based (two-stack, metal-plate anode) detector located at the end of the spectrometer is used for detecting the electrons. The voltage over the MCP is 5.6 kV. Due to the operational requirements of the MCP at such high-voltage, there is a high-vacuum condition for which an oil-free vacuum pressure of 1x10-6 mbar or better is required. The electron signal from the MCP detector is pre-amplified and the signal is discriminated between noise and the pulse. The discriminated signals are coupled into a time-to-digital conversion card (TDC) connected to a data acquisition PC and server.
The trigger signal from the Pockel cells is also connecting into the TDC card, which provides the start signal from the laser. The TDC card is used to record the time delay from the start signal of the laser and the stop signal obtained from the MCP, and has a time resolution of 0.5 ns. The count rate of our experiment for both pump and probe pulses is generally 5~10 c/shot. The count rate from single pulses is generally 0.1~2 c/shot. The integration time for a typical experiment is 12 hours. The sever acts as an interface to another computer running a Labview-based program to control the delay stage, the pump/probe shutters and show the TOF spectrum e-kinetic energy spectrum. The pump/probe shutters are used to measure pure pump or pure probe signals, which are used for subsequent data post processing. Conversion of the time-of-flight data to the kinetic energy of the photoelectrons is calculated by equation (2)
Ε =�
���� + Ε�,Α =
�����
�, ���� = � − �� (2)

40
Here, E is the photoelectron kinetic energy, and Le is the electron flight path. E0 is a small correction due to the deviation from an ideal field-free environment. me is the electron mass, and tTOF is the time the computer measures from the TOF spectrum of the electrons generated at each laser shot. t is the time that the electron hits the detector after the trigger signal, and t0 is the actual time of ionisation with respect to the trigger signal.
Figure 25. TOF spectra of Xenon after absorption of two photons of 200 nm and, in the insert, from three photons of 200 nm.
The photoelectron spectrometer is calibrated by these factors using the photoelectrons from a known sample: here, the values of A, E0, and t0 are obtained from the photoelectron spectrum of Xenon, which has a large non-resonant ionisation feature which gives rise to well-defined and multiple sharp peaks, and the first two ionisation energies of Xe are 12.130 eV and 13.436 eV [143]. The temporal cross-correlation between the pump and probe pulse is also determined by non-resonant ionisation of Xe, which is around 150 ± 20 fs. The TOF spectrum of Xenon of two photon and three photon signal of 200 nm is shown in Figure 25. The calibration of the multiple peaks is carried out according to equation (2).

41
5. Time-resolved photoelectron spectroscopy
and data analysis
The development of time-resolved photoelectron measurements on the dynamics of excited molecules was pioneered by Carl Hayden [144], who used femtosecond pulses in its first application to non-adiabatic dynamics in polyatomic molecules. It is now a fundamental method for investigating ultrafast dynamical processes. Ultrafast processes commonly take place on the time scale of picoseconds to femtoseconds. In our experiments, the main focus is on the femtosecond time-resolved photoelectron spectroscopy measurement of gas-phase neutral polyatomic molecules. Here, very short-lived electronic excited states (~10-100 fs), and the wavepacket dynamics occurring on these states, can be followed in real-time [145]. The experiments are carried out in a pump-probe configuration. The ultrafast pump pulse initially excites the molecule into an excited state. This step is typically considered as a vertical transition, placing the ground-state wavepacket directly onto the excited electronic surface. The second ultrafast laser pulse is used as a probe to follow the evolution of the wavepacket on this excited state surface. The energy of the probe pulse is chosen to be sufficiently high to ionize the excited molecules through a photoionization process and to generate an electron and an ion.
5.1 Time-resolved photoelectron spectroscopy Time-resolved photoelectron spectroscopy [4] is a good tool to probe not only the nuclear dynamics, but also the electronic configuration. The nuclear dynamics can be mapped out through the overlap of the vibrational wavefunction between the neutral and cation state, which is described by the Franck-Condon overlap. The electronic structure can be probed according to the Koopmans’ picture, in which, during the photoionization process, the removal of the outer electron does not change the electronic configuration of the core. This is depicted in Figure 26, where S1 represents multi-configurational excited states. One of the advantages in this technique, compared with others, is that ionization is always allowed, due to relaxed selection rules. This is because the final state containing the free electron wavefunction can take on any symmetry, which allows any molecular state to be ionized [146].

42
Figure 26. Illustration of the photoionization process without a change in the electronic configuration of the core, according to Koopmans’ theorem. Another advantage in time-resolved photoelectron spectroscopy is that it is good for interpreting the existence of Rydberg states. In the dynamics, the Rydberg state is sometimes accessed directly [21, 80], e.g. molecules are initially excited to the π3s Rydberg state as in ethylene, the alkyl vinyl ethers [11] or via the two-photon absorption process in butadiene, where the molecules are excited directly to the π3s- or π3d-Rydberg states [21, 80]. Rydberg states also can play an important role as an intermediate state, e.g. the involvement of population transfer from the valence state to the Rydberg state, as mentioned in refs 33, 34 in ethylene as well as in pyrrole [147]. A molecular Rydberg state has a similar geometry to the corresponding ionic state, which means that the Franck-Condon overlap between the Rydberg state and the ionic state should be optimal due to the transition being between the same vibrational levels (i.e. the same quantum number) such that the extra vibrational energy will be stored in the ion. As such, ionisation from a Rydberg state should give rise to a sharp peak in the photoelectron spectrum because the electron kinetic energy is well-defined.
5.2 Time-resolved photoelectron spectrum (TRPES) and data analysis

43
In the photon-induced processes, the molecules absorbing one photon are excited to the Sn state (e.g. Figure 27(a)) by the pump pulse, and then ionized by the probe pulse bringing the electrons into the continuum and creating ions. The excess energy is transferred into the departing electrons as kinetic energy, and thus shows up as different peaks in the photoelectron spectrum. The position and shape of these peaks are influenced by many factors, e.g. the initial internal energy distribution of the molecule, the energy-width of the laser pulses, the shape of the potential energy surface, etc. Depending on the energy difference between the excited and the ionic states, sometimes one probe photon is not sufficient to ionise the molecule and two or more photons are required as the probe step. Generally, the features in the spectra are denoted by the number of pump (n) and probe (m’) photons required, i.e. [n,m’]. For example, [1,1’] is a one photon pump and one photon probe feature/experiment.
Figure 27. (a) Schematic overview describing the molecular dynamics of an excited state. The different colored-filled circles on the Sn excited state indicates the different time delays between the pump (blue arrow) and probe pulses (turquoise arrows). The purple circle indicates the position of conical intersection. (b) A schematic time-resolved photoelectron spectrum, with photoelectron kinetic energy (Ek) obtained at different values of the time delay between the pump and probe pulses (t). The solid black arrow on the spectrum describes the shift in the time-zero shift from higher to lower photoelectron kinetic energy. The brown arrow shows the time spent near the conical intersection. The time when pump and probe pulses overlap is called the time zero. Then, the wavepacket moves from its initial location on the excited state surface to access the conical intersection with the lower-lying excited state, and this is delayed by a period of time with respect to time-zero. This time period is often called the “induction time” [17] (expressed as a time-zero

44
shift in the fitting function). To analyze and understand the photoelectron spectrum, the most important parameters for interpreting the molecular dynamics are the time zero shift and the time constant at the different photoelectron kinetic energy regions. The time constant and their decay associated spectra (DAS) are obtained by a Levenberg-Marquart 2D global fitting routine using the following expression: [17, 104]
���, Δ� = Σ"Α"��#"�Δ�$ ⊗ &�Δ� (3) Here, Ai(E) is the DAS of the channel i. It has a time-dependent population, Pi(∆t), expressed in terms of exponential functions exp{-∆t/τi} which describes the decay with a lifetime τi (time constant). g(∆t) is the Gaussian cross-correlation function obtained from the photoelectron spectrum of Xenon. ∆t=td-t0(E) is the difference between the delay time, td, and time zero, t0(E). In the data fitting process, the time-zero can also be used as a fitting parameter in addition to Ai and τi, when the spectrum shifts for different energy regions. The experimental data are fitted either globally or for different energy slices. In the latter, the energy slices can be fitted with a time zero and τi as free fitting parameters in order to account for the temporal shift in the photoelectron spectrum and the change of the lifetime in the different regions of the potential energy surface. For different lifetimes obtained in each energy slice, only that part of dynamics is covered. For the largest value of the shift obtained, e.g. from fitting globally, this indicates that the population likely accesses the minimum of the potential energy surface where the time constant obtained here indicates the time taken for the population on the excited state to pass through the conical intersection. In order to give a clearer explanation of the time zero shift, take the schematic example shown in Figure 27. The ground-state wavepacket populates the Sn state in the Franck-Condon region, i.e. the region of the excited state potential that is accessed by the vertical transition. Probing the wavepacket here corresponds to ionizing the molecule from a position on the Sn potential energy surface given by the red circle. The steepness of the potential surface in the region induces large amplitude vibrational motions of the nuclei. As the molecules move quickly down the potential surface, they are probed on the Sn surface at the positions corresponding to the filled circles. This leads to a decrease of the photoelectron kinetic energy as a function of the pump-probe delay time. In other words, the large amplitude nuclear motions cause a rise in the ionisation potential along this motion’s co-ordinate, which also leads to a decrease in the photoelectron’s kinetic energy. The nuclear and electronic motions couple in the region of the

45
conical intersection. Here, part of the wavepacket couples to the lower lying Sn-1 state, while the other portion of the wavepacket remains on the Sn state. The kinetic energy of the photoelectron shifts as a function of the time-delay between the pump and probe laser pulses, as illustrated in Figure 27 (b). The red circle corresponds to the time-zero position, where the pump and probe pulse “overlap” in time, and produces electrons with the highest kinetic energy. With increasing pump-probe time delay, the photoelectron’s kinetic energy decreases until the point indicated by the purple circle, which is when the molecule reaches the conical intersection.
Figure 28. A typical photoelectron spectrum. Here, the spectrum of 1,3-butadiene is shown obtained from a 216 nm pump-pulse and probed by a 266 nm pulse. The x-axis is the photoelectron kinetic energy and y-axis represents the pump-probe time delay. The blue dashed line at around 1.3eV is due to the cut-off for one-photon probe ionisation. The blue dashed line at around 4eV is due to the cut-off of ionisation to the excited cation state 2Au. Red corresponds to high-intensity and blue low intensity electron signals. (modified from ref [20]) To illustrate how to interpret the photodynamics from the time-resolved photoelectron spectra, 1,3-butadiene is taken again as an example. The photodynamics of trans-1,3-butaidiene are depicted in Figure 10. A time-resolved photoelectron spectrum of trans-1,3-butaidiene obtained using a

46
216 nm (5.92 eV) pump and a 266 nm (4.65 eV) probe is shown in Figure 28 [20]. Trans-1,3-butaidiene has a vertical ionisation potential of 9.29 eV [148]. There is a strong peak in the spectrum at an energy close to the cut-off (around 1.3 eV) due to one photon probe ionisation, obtained from the calculation: Epump+Eprobe-IP. The strong peak here can be explained by the excitation to the 11Bu singlet excited state. The next cut-off, at around 3.8eV, indicates a two-photon probe ionisation (Epump+2Eprobe-IP), which is the cut-off of the ionisation to the excited 2Au cation state. The horizontal black dashed line indicates the time-zero position. The green arrows in the figure show the time-zero shift that the large amplitude motion like C=C bond twist and the torsion undergo when accessing the conical intersection to the lower lying doubly excited 21Ag state. The time-zero shift in the range of the [1, 2’] region also indicates nuclear motions like pyramidalization at the terminal -CH2 group that gives access to the conical intersection with the ground state. No more detailed pathways will be discussed here, the main idea is to introduce how to obtain information behind the molecular dynamics upon excitation from its corresponding photoelectron spectrum. The data is fitted by the convolution of the Gaussian cross-correlation function (g(∆t)) plus the time constant. Sometimes, one single exponential decay is sufficient to describe the TRPES spectra. In many cases, a second time constant, and sometimes a third, is needed to accurately but nontrivially describe the spectrum. Take a general example of fitting data with two time constants. These two time constants can represent two different kinetic models [71], one is a parallel model, the other is a sequential model. The parallel model means that the pump laser simultaneously excites the molecules on both two excited state, where these two states independently decay with two different lifetimes. The sequential model means that a singly excited state decays via a lower lying state before decaying to the ground state, where each of these steps has an associated time constant. For example, the molecules first populate the S2 state, where they then decay to the S1 state with a time constant τ1, and then subsequently decay from S1 to the ground state with a time constant τ2. Figure 29 shows the DAS for a sequential process, where the amplitude indicates the decay associated photoelectron amplitude. Here, positive amplitude represents an exponential decay of the signal, whilst negative amplitude represents an exponential growth. So, the sequential model described in Figure 29 shows the excited molecules populate the S2 state with a positive amplitude, then the molecules decay from S2 to populate S1 with an associated negative amplitude on the S2 state but a positive amplitude of S1. The time for describing this process is τ1, and subsequently the molecules decay from S1 back to the ground state with τ2. A negative amplitude in the DAS is a clear symbol for a sequential model.

47
Figure 29. Decay associated spectra for the sequential model indicating a stepwise decay mechanism with two time constants. The molecules decay from S2 to S1 with time constant τ1 and decay from S1 with decay time τ2.
Data are also fitted by cutting the 2D spectra into different energy slices, e.g. ∆E=0.05eV. The wavepackets evolution from the FC region to access to the CoIn can be described as a step-wise model of the dynamics (shown in Figure 30), and leads to the time evolution of the population density, Pi(∆t). The excited state potential is modelled by discrete intervals which show that it is populated and depopulated via single kinetic steps, e.g., |1>→|2>→|3>→…, etc [149], with a uniform transfer time constant. The lower lying part is populated with the time-decay of the former part on the excited state. This explains the increased rise time. The different numbers of steps affects the fitting quality [17].

48
Figure 30. The step model for modelling the dynamics. The decay time can be described elaborately by a discrete interval showing an increase in the rise time. It can be concluded that, at the highest photoelectron kinetic energies, the time constant one observes indicates the molecule leaving the Franck-Condon region, while at the low photoelectron kinetic energy part of the curve, the time constant is determined by the time that the population starts to pass through the conical intersection and transfer to the lower-lying state. As a result, the total time that the molecule stays on the Sn state can be described roughly by the time-zero shift plus the exponential decay time at the lowest photoelectron kinetic energy point [149].

49
6. Discussion of the attached papers
In this chapter, the results from each of the attached papers will be described following the order given in Figure 31, which shows the chemical modifications undertaken to the ethylene molecule.
Figure 31. Chemical modification from ethylene to cyclohexa-1,3-diene
(PAPER I), acrylonitrile (PAPER II), alkyl vinyl ether (PAPER III), furan
(PAPER IV), pyrrole (PAPER V), and pyrazole (PAPER VI)

50
6.1 Dynamics of polyenes – cyclohexadiene
PAPER I. Cyclohexadiene Revisited: A Time-Resolved Photoelectron Spectroscopy and ab initio Study
The dynamics upon photoexcitation of cyclohexa-1,3-diene has been investigated extensively both in theory and experiments [150, 151, 152, 153]. There are two excited states involved in the photodynamics: one of them is an optically dark 2A state, and the other one is an optically bright 2B (ππ*) state. The dynamics were previously described as follows: the molecules are initially excited to the ππ* state and the population rapidly decays to the 2A state, where the ring-opening reaction occurs. Afterwards, they pass through the conical intersection back to the ground state whereby a fraction of the molecules undergo ring opening to form hexatriene. However, as discussed in Chapter 3 on the photodynamics of cyclopentadiene, a question around the involvement of the 2A state in the excited state dynamics has arisen, in which it is asserted that only one potential energy surface is involved in the dynamics. With this in mind, it was in our interest to revisit the photodynamics of cyclohexa-1,3-diene. The experimental results obtained by time-resolved photoelectron spectroscopy using a 1 µJ pump (267 nm) and a 20 µJ probe (400 nm) are combined with calculations on the dynamics. Similarly with the dynamics observed in cyclopentadiene, we conclude that there is only one excited state potential energy surface involved in the reaction. The photodynamics we observe for cyclohexa-1,3-diene is summarized in Figure 32, and are described as follows (see Figure 32 for carbon-atom naming convention). The molecules are mainly excited to the S1 (ππ*) state. When the excited wavepacket moves toward to the conical intersection to the ground state, it is accompanied with a C5-C6 bond stretch. Once the wavepacket travels close enough to the CoIn to experience non-adiabtic coupling to the ground state, there are two main trajectories: one of them leads to ring-opening, which we call a successful trajectory; the other is where the molecules return back to the ground state with no ring opening, and which we call an unsuccessful trajectory. It is found that the successful trajectories are faster (with an incubation time 37 fs) than the unsuccessful trajectories (with an incubation time 80 fs). Both of the trajectories have direct correlations with the direction of the momentum along the bond alternation coordinate, BAC, which is defined as d(C1-C2)-d(C1-C6)+d(C3-C4)-d(C4-C5).

51
Figure 32. The photodynamics of cyclohexa-1,3-diene From these results, the successful trajectories have higher (“positive”) momenta along both the BAC and the C5-C6 bond, which explains the higher probability of the ring-opening channel (64%). In the unsuccessful trajectories the molecules do not directly return to the ground state, since if the excited molecules cannot gain enough velocity to access the ring-opening channel, and also decrease the nonadiabatic coupling, they remain in this region for an extended period time before they return to the ground state. This is supported both by a longer time constant for the unsuccessful trajectories when the successful and unsuccessful trajectories are fitted separately, and an observed wavepacket revival at 160-180 fs which is observed both in experiment and theory (as marked by the red circles in Figure 33). This figure shows a comparison between (a) the experimental and (b) the calculated time-resolved photoelectron spectrum, where the unsuccessful trajectories were selected. No revival could be observed from the successful trajectories. In addition, the overall agreement between experimental and theoretical data is excellent. Both spectra exhibit a broad band from 2.6 eV to 0.1 eV. One pump photon (267 nm = 4.65 eV) and one probe photon (400 nm = 3.10 eV) is not sufficient to ionize the molecule. So the energy cutoff of one pump photon and two probe photons, which is 4.65 eV+2x3.12 eV-8.25 eV = 2.60 eV. The vertical ionization potential of cyclohexa-1,3-diene is 8.25 eV [154]. The calculated spectrum does not show any of the detailed structure observed in the experimental spectrum

52
since intermediate Rydberg states in the two photon probe process were not considered.
Figure 33. Photoelectron spectrum from (a) experiment, and (b) calculated for unsuccessful trajectories. The green arrow indicates the time delay, and the red circles indicate the signal stretches to longer time constants. Ring opening trajectories occur mainly via a conrotatory pathway (the two double bonds rotate in the same direction) rather than disrotatory (the two double bonds rotate in a different direction). With respect to the dynamics on the hot ground state, the ring-opening causes the formation of three different conformers; trans-Z-trans (tZt), cis-Z-trans (cZt), and cis-Z-cis (cZc). The tZt conformer is the main one that the molecules access directly. Both cyclohexa-1,3-diene and cyclopentadiene have only one potential energy surface involved in the photodynamics. As described earlier, the cyclopentadiene molecules on the excited state undergo an in-plane plus out-of-plane motion along the surface to access the conical intersection to the ground state. The out-of-plane motion induces torsion about one of the double bonds. As such, the dynamics is localised on only one double bond, which is similar to that of trans-1,3-butadiene and cylcohexene. However, for cyclohexa-1,3-diene, the dynamics are delocalised around the whole system, and with the C5-C6 bond stretch and cleavage the two double bonds distort to form several different conformers. Here we can compare cyclohexa-1,3-diene and cyclohexa-1,4-diene, where both molecules have two double bonds in the ring-system but differ in their location. Firstly, the two molecules both keep the ethylene-like bond torsion. Secondly, in cyclohexa-1,3-diene, the current experimental and theoretical results show the dynamophore involving the whole molecular structure. However, in cyclohexa-1,4-diene, the calculations indicate a high fraction of molecules have a localised dynamics on one of the double bonds.

53
6.2 Dynamics of ethylenes with functional groups
6.2.1 Ethylene molecules with a cyano group - acrylonitrile
PAPER II. Excited state dynamics of acrylonitrile: Substituent effects at conical intersections interrogated via time-resolved photoelectron spectroscopy and ab initio simulation
The dynamics of ethylene molecules with the addition of a single heteroatom was discussed in Chapter 3 with the example of acrolein. Here, other kinds of molecules will be discussed: those based around acrylonitrile (Paper II), and those based around alkyl vinyl ether (Paper III). Acrylonitrile can be regarded as a molecule which is obtained by adding a –CN group onto ethylene. Many previous experimental and theoretical studies on the photodissociation dynamics of acrylonitrile have been reported, and these indicate -CN group elimination upon UV excitation [155, 156, 157]. The important point for this work is to try to understand the substituent effect (-CN group) on the ethylene molecules. Here, using a 200 nm excitation wavelength, measurements on the photodynamics of acrylonitrile are reported together with the dynamics of crotonitrile and methacrylonitrile. Combining the results from experiment and computation also shows the substituent effect on the acrylonitrile dynamics. The structures of these molecules are depicted in Figure 34.
Figure 34. The structure of acrylonitrile, crotonitrile and methacrylonitrile

54
The lowest lying electronic excited state of acrylonitrile lies between 159 nm and 213 nm, and is mainly assigned as a ππ* transition. With a 200 nm pump wavelength, the molecules are mainly excited to this state (denoted as the S2 state). There is another ππ* state which lies just below the S2 state, and this is optically dark and is denoted as the S1 state. Upon excitation of the S2 state the molecule quickly accesses the S2/S1 conical intersection following a C=C bond stretch. After internal conversion to the S1 state, the molecule undergoes a single-double bond (C=C-C) twist and a C=C-C bend accompanied by pyramidalization to give access to the conical intersection to the ground state. This step-wise mechanism can be inferred from the experimental time-resolved photoelectron spectra and from calculations, as illustrated in Figure 35(a) and (b).
Figure 35. Photoelectron spectra of (a) experiment, and (b) calculation. The green line indicates the time delay. Insets in spectra provide underlying data scaled by the given factor showing two probe photons region. It is found that the twist-pyramidalization happens at the C2 position, which is due to the partial negative charge at the nitrogen atom, and the next partial negative charge is, as expected, in the C2 position (due to alternating charges). From Figure 34, it is seen that crotonitrile and methacrylonitrile have a methyl group on the C3 and C2 positions, respectively. Analysis of the TRPES dynamics of crotonitrile and methacrylonitrile indicates that there is a methylation effect on the dynamics. It is found that the excited state decay time on the S1 state slows down in going from acrylonitrile to crotonitrile and finally to methacrylonitrile. This is explained by the effect of the pyramidalization channel. In methacrylonitrile, pyramidalization is disfavoured by the extra methyl group on C2, and, hence, it is harder to access the CoIn. This is seen in the results of the calculations, in that the

55
spawning points of methacylonitrile predominantly take place at lower degrees of pyramidalization as compared to the position of the minimum energy CoIn. This is opposite to the dynamics found in acylonitrile and crotonitrile. The photodynamics of acrylonitrile includes both the C=C bond torsion and pyramidalization, i.e. it retains the ethylene-like dynamics. One important observation is that pyramidalization happens on the central carbon position, and not the terminal carbon position as was previously expected. This is due to the influence from the cyano group, which is similar to the effect on the pyramidalization in the acrolein molecules. With respect to the effects due to methylation, similar trends are observed here: the methylation affects the decay rate, although the reason behind the much slower dynamics in acrolein is due to the tilted conical intersection on the nπ* state.

56
6.2.2 Ethylene molecules with alkoxy group- alkyl vinyl ethers
PAPER III. Influence of Alkoxy Groups on the Photoinduced Dynamics of Organic Molecules Exemplified on Alkyl Vinyl Ethers
The effect of the cyano group has been discussed in the previous section; here the akoxyl group effect on ethylene is summarized. The molecules studied are ethyl vinyl ether, propyl vinyl ether, and 2-propanol vinyl ether. The structures of these molecules are shown in Figure 36.
Figure 36. The structure of ethyl vinyl ether, propyl vinyl ether and 2-propanol vinyl ether. The simplest vinyl ether, methyl vinyl ether, has a similar structure to ethyl vinyl ether and propyl vinyl ether, and has been investigated previously [158]. Upon photoexcitation to the ππ* state, the molecule dissociates and forms the vinoxy (CH2=CHO) radical. This radical is important because it is an intermediate in combustion and photochemical smog cycles [1]. With the study of methyl vinyl ether as a reference, it is helpful to investigate the dynamics of alkyl vinyl ethers. The photodynamics obtained from the experimental and theoretical treatment is illustrated in Figure 37. With 200 nm excitation, the molecules are initially excited to the ππ* state and the π3s Rydberg state. Those molecules on the ππ* follow the double bond torsion and pyramidalization pathway to access the conical intersection to the ground state. If the molecules directly populate the π3s Rydberg state, then the population rapidly decays to either the πσ* state or the ππ* state within 20 and 60 fs, respectively. Here, similarly to the direct excitation process, the molecules on the ππ* state follow the torsion and

57
pyramidalization dynamics. However, molecules moving on the πσ* state are accompanied with a C-O bond stretch and subsequent C-O bond dissociation.
Figure 37. Photodynamics of alkyl vinyl ethers
It is concluded that the alkyl vinyl ethers have two main photochemical reaction channels: one is bond torsion and pyramidalization, and the other one is the C-O bond cleavage. From the comparison between the three molecules, the substituent effect is minor. From the dynamics, the time-scale of the process, e.g. the lifetime of the excited state, the observed rate of internal conversion also shows similar results. Another finding is that the reaction channels are dependent on the pump wavelength. In previous studies on methyl vinyl ether, using a 193 nm pump does not lead to the C-O bond dissociation pathway on the excited state. This is due to the population decaying directly from the ππ* state to the ground state [159]. However, in our experiments exciting with a 200 nm pump, this channel is observed, in which molecules that are excited to the π3s Rydberg state undergo a rapid

58
decay to the πσ* state which then forms the fast vinoxy molecules with a significant quantum yield. A comparison can be made between acrolein, acrylonitrile, and the alkyl vinyl ethers to see the functional group effect on the ethylene molecules. The ethylene-like dynamics include the double bond torsion and pyramidalization. All of these three types of molecules display the double bond torsion and pyramidalization processes, while the difference is in the position of the pyramidalization. As mentioned earlier, acrolein and acrylonitrile both have the pyramidalization occurring at the C2 position. This is because the oxygen has a partially negative charge in acrolein and the nitrogen has a partially negative charge in acrylonitrile, and where the next atom with a partial negative charge is the second carbon atom connected to them. In the alkyl vinyl ethers, the oxygen also has a partial negative charge, as does the second carbon connected to it, and, with a double bond, this is the terminal carbon. As a result, in alkyl vinyl ether, the pyramidalization happens on the terminal carbon position. From the point of view of the dynamics, the aldehyde group in acrolein introduces the nπ* state into the dynamics, where the population can decay to the ground state or to the triplet state. In acrylonitrile, there are only two ππ* states involved, and no –CN bond cleavage is observed. In the alkyl vinyl ethers, the alkoxyl group introduces the πσ* state into the dynamics, upon which the C-O bond cleavage channel occurs. The dynamics of the alkyl vinyl ethers is observed to be slower than that of the pure ethylene upon excitation to the π3s Rydberg state. This is also due to the existence of the πσ* state, where the population on the π3s Rydberg state can rapidly decay first to the πσ* state since this state can be accessed directly. Methylation on the methylene hydrogen has been found in acrolein and acrylonitrile to affect the decay rate. In conclusion, the different functional groups may introduce new reaction channels and new excited states while retaining most aspects of the “original” dynamics.

59
6.3 Dynamics of cyclopolyenes with heteroatoms
6.3.1 Dynamics of cyclopolyenes with oxygen - furan
PAPER IV. Substituent effects on the relaxation dynamics of furan, furfural and β-furfural: A combined theoretical and experimental approach
The dynamics of furan has been discussed in Chapter 3. In brief: the lowest-lying excited states of furan are the 1B2(ππ*) and 1A1(ππ*) states, respectively, and there is a coupling of the 1B2(ππ*) state and the π3s Rydberg state. The molecules are initially excited to both the 1B2(ππ*) and the π3s Rydberg states, from where the wavepacket has two main relaxation pathways; one is the C-O bond stretch, and the other is an out-of-plane motion to access the conical intersection with the ground state. In the current study, the dynamics of furan were probed with two photons of 400 nm. In addition, two derivatives were also studied, furfural and β-furfural. The structure of these two molecules is shown in Figure 38.
Figure 38. The structure of furfural and β-furfural The motivation for studying these molecules is not only to investigate the general effect of carbonyl groups on a heteraromatic system but also the role of the position. The photodynamics of furan from the current experimental and theoretical treatment is described first. Upon photoexcitation to the ππ* state, the population decays rapidly to access the conical intersection with the ground state. The two main relaxation channels of furan are ring puckering and ring opening. Previous studies have indicated that the ring puckering channel is the main relaxation pathway. However, from our theoretical calculations, we observe that the ring-opening channel is more favourable and, furthermore, it also contains πσ* character, which means that the population decays accessing the CoIn between the ππ* and πσ* states in the ring opening process. From fitting to the experimental data, the wavepackets populated on the ππ* access the CoIn within 60 fs, and the time taken for the

60
wavepackets passing through the CoIn seam connecting the ring opening and ring puckering CoIn is around 110 fs. The dynamics of furfural and β-furfural show some significant differences when compared to furan. This can be seen when comparing the photoelectron spectrum of furan and furfural, as shown in Figure 39. In Figure 39(b), the spectrum has a peak at around 0.7eV that can be explained by ionization from a nonbonding orbital of the oxygen atom in the aldehyde group. Comparing the furfural spectrum with that of furan, the former has a longer time delay. The molecules can reach the CoIn between the ππ* state and the nπ* state by rotation of the aldehyde group, and the relaxation via this CoIn needs around 140 fs. The time constant of ring opening and puckering channels is around the picosecond timescale.
Figure 39. Photoelectron spectrum of (a) furan, and (b) furfural.
The photodynamics of these two molecules is described in Figure 40. The initial excitation is again to the ππ* state, from where the wavepackets have two possible relaxation pathways: one is a return back to the ground state accompanied with a ring puckering or ring opening motion, whilst the other one is a decay to the nπ* state.
The coupling of the aldehyde group to the double bond in the ring system forms an extension of the π-orbital system, which selectively lowers the energy of the ππ* state but not the πσ* state so that the generated barrier slows down the dynamics of the furan derivatives. In addition, this extension of the conjugated π-space reduces the energy gap between the electronic states at the Franck-Condon point, which induces the less stable CoIn, which has a higher energy, compared to the Franck-Condon point in furan and its derivatives. That is to say, the aldehyde group may affect the energetic position of the CoIn, e.g., the effect on the geometry of the puckered CoIn. Here, the pyramidalization of the CoIn is highest energy in furfural, followed by that of β-furfural puckered at the α-position, furan, and finally β-furfural puckered at the δ-position.

61
Figure 40. Photodynamics of furfural and β-furfural
From the perspective of dynamics of furfural and β-furfural with that of cyclopentenone mentioned in Chapter 3, the effect of the aldehyde group on the ring system can be compared. For the three kinds of molecules, the nπ* state is found to be involved in the dynamics. However, in cyclopentenone, the dynamics on the nπ* has been described as a further decay to the triplet state or the ground state. In the furfural molecules, the intersystem crossing pathway has not been included. For the dynamics on the ππ* state, all the molecules undergo a ring puckering motion. In furan, the ring-puckering occurs at the α- and δ-positions, where it forces the oxygen atom out of the plane. With the addition of the aldehyde group to furan, the energy of the puckering mode increases, where adding the aldehyde group to the α-position has the highest energy. In cyclopentenone, there is a twist-and-pyramidalization at the C=C double bond (“bond torsion”-mode). In furfural and β-furfural, the carbon atom from the aldehyde group is a part of the extended ring system, and shows no direct effect on the bond torsion mode. However, methylation has been found to slow the dynamics when the substituent is connected to the double bond. For the ring-opening channel, C-O bond cleavage occurs in furan and its derivatives, which has a πσ* character, and looks similar to the C-O bond cleavage channel in alkyl vinyl ether, where this process occurs on the πσ* state. In conclusion, furan and its derivatives keep the ethylene-like bond torsion and pyramidalization relaxation channels, while in addition they introduce a new ring-opening channel. With respect to the influence of the aldehyde group itself, a similar influence has been observed on ethylene and the cyclic polyenes.

62
6.3.2 Dynamics of cyclopolyenes with nitrogen - pyrrole
PAPER V. Dynamics in higher lying excited states: Valence to Rydberg transitions in the relaxation paths of pyrrole and methylated derivatives
Pyrrole attracts much interest due to it being regarded as a subunit in large biological system, like DNA, or amino acids, etc. As such, the dynamics of pyrrole has been investigated previously and is summarized in Chapter 3. Briefly, pyrrole is excited to the B2(ππ*) state, where the population decays to the A2(πσ*) state with two main reaction channels: one is dissociation of the N-H bond, and the other is a ring-puckering channel. In addition to pyrrole, two additional methylated pyrrole derivatives have also been studied, N-methyl pyrrole and 2,5-dimethyl pyrrole. The structure of N-methyl pyrrole and 2,5-dimethyl pyrrole are depicted in Figure 41.
Figure 41. The structure of N-methyl pyrrole and 2,5-dimethyl pyrrole From the current results, the photodynamics of pyrrole and its methylated derivatives can be summarized by Figure 42. Upon photoexcitation with 200 nm, the molecules directly access the B2(ππ*) state. Then, during the population decay via an out-of-plane mode to access the conical intersection with the A2(πσ*) state, there is a transient population of the π3p Rydberg state, which has a short lifetime around 20 fs. When the population further decays onto the A2(πσ*) state, there are the two reaction channels available: ring puckering and N-H bond dissociation. The π3p-Rydberg state is indicated by a sharp and intense peak at around 2.4 eV as exemplified by the photoelectron spectrum of N-methyl pyrrole spectrum in Figure 43. Another sharp peak at around 1.8 eV can be explained by ionization from A2(πσ*) state which has a strong 3s Rydberg state contribution.

63
Figure 42. Photodynamics of pyrrole molecules and its derivatives.
Figure 43. Photoelectron spectrum of N-methyl pyrrole upon excitation with 200 nm. Comparison between the dynamics of pyrrole, N-methyl pyrrole and 2,5-dimethyl pyrrole, shows that all molecules transiently populate the π3p Rydberg state. This had not been considered before in theoretical treatments

64
of these molecules. However, the quantum yield of the final two reaction channels is different for the different molecules. In the pyrrole molecules, the N-H bond dissociation channel is a minor process, since there is no indication in the data for the existence of the A2(πσ*) state. This means the population probably decays from the ππ* state to the B1(πσ*) state, where the N-H dissociation happens, or it populates the ring puckering channel [35]. However, the excited state dissociation is disfavored by the short excitation wavelength [158], and so it is assumed the ring puckering is predominately accessed. In the N-methyl pyrrole, three time constants (15 fs, 100 fs, 20 ps) are obtained from the data analysis as shown in DAS spectrum in Figure 44. The second time constant indicates the lifetime of the A2(πσ*) state, the amplitude of this constant represents the timescale to access the N-CH3 bond cleavage and ring puckering channels. Due to the higher dissociation barrier observed [160, 161], access to the N-H dissociation channel is not “prompt”. In contrast, for 2,5-dimethyl pyrrole two time constants (15 fs and 120 fs) are obtained from the analysis. The amplitude of the second one is small, can be explained by a small ionization cross section from the A2(πσ*) state. Moreover, no long time constant ( >1 ps ) is observed. This can be explained by a high fraction of N-H dissociation, and a minor fraction of molecules becoming trapped in ring-puckering channel on the A2(πσ*) state. Figure 44. Normalized decay associated spectra for a) pyrrole, b) N-methyl pyrrole, and c) 2,5-dimethyl pyrrole.

65
The involvement of Rydberg states in the photodynamics has not been observed often in organic molecules. As mentioned earlier, in the dynamics of ethylene, the population on the ππ* state could populate the lower-lying π3s Rydberg state. However, the π3s Rydberg state has a high degree of valence character, and the crossing of the valence to the Rydberg states happens along the C=C bond torsion coordinate, which increases the probability of a transition. In pyrrole and its derivatives, the π3p Rydberg state does not mix with the valence state, and no modes are identified to couple the Rydberg and valence states together. As such, this is the first observation of the involvement of a close-lying Rydberg state in the photodynamics in pyrrole. Furthermore, another significance of the involvement of Rydberg state in the photodynamics is that the presence of the Rydberg state might redirect the outcome of the dynamics, e.g. from an N-H dissociation to a ring puckering intersection with the ground state, hence possibly directly affecting the yield of the fast hydrogen atom in the photodissociation of pyrrole molecules. In conclusion, interesting results have been found for the existence of a π3p Rydberg state in the photodynamics which might play a role in how the reaction pathways are accessed. As a result, the role of Rydberg states become more important when studying processes initiated on higher lying excited states. From the dynamics, for heteroatoms which are sited in the cyclopolyenes, e.g. furan and pyrrole, these molecules all keep the ethylene-like dynamics of ring puckering, while introducing new excited states and new reaction channels, i.e., C-O bond cleavage in furan and N-H dissociation in pyrrole.

66
6.3.2 Dynamics of cyclopolyenes with two nitrogen - pyrazole
PAPER VI. Time-resolved photoelectron spectroscopy studies on pyrazole and its derivatives
The photochemical dynamics of five-membered heteroaromatic compounds has been studied a lot, e.g. furan and pyrrole, which I described earlier. Here another series of five-membered heteroaromatic compounds is introduced, based on pyrazole and its methylated derivatives. These particular molecules have attracted only little previous attention, but pyrazole derivatives have many applications in the medicine [162]. The structures of the molecules we investigated are depicted in Figure 45. Compared with pyrrole, pyrazole contains an extra nitrogen atom in the 2-position. Figure 45. The structure of pyrazole, 1-methyl pyrazole, 3-methyl pyrazole and 5-methyl pyrazole. The photodynamics of all these molecules are studied with 200 nm excitation and 267 nm ionization. However, the photoelectron spectra of 1-methyl and 3-methyl pyrazole also exhibit signals toward negative time delays, indicating a reverse of the pump and probe steps. As a consequence, there is signal from 267 nm excitation mixing with signal from 200 nm excitation. As an example, the photoelectron spectra of pyrazole and 1-methyl pyrazole are shown in Figure 46. When molecules are excited with 200 nm, they can directly populate either a π3s/πσ* state or a ππ* state. When molecules are excited to the π3s/πσ* state, N-H bond dissociation is supposed to occur as concluded from previous studies [163]. However, when compared with the TRPES spectrum

67
of pyrrole obtained in our studies, a strong contribution from population of the Rydberg state would be indicated by a sharp and intense peak but here it is found to be only a relatively small in the pyrazole molecules. This indicates that direct excitation to the π3s/πσ* state is only a minor channel. Hence, molecules are mainly excited to the ππ* state from where two different decay channels are accessible: N-N bond dissociation and ring puckering. N-N bond dissociation is reached by an N-N stretch in the Franck-Condon region and subsequent access to the πσ* state. Some molecules do not have enough energy to cross to the N-N bond dissociation channel, and so will be trapped in the ππ* state leading to ring-puckering. In addition, we also find a picosecond time constant similar to the pyrrole molecules, indicating that the molecules are trapped in the ππ* state for a longer time, possibly giving rise to an intersystem crossing to the triplet state. The photodynamics can be described by Figure 19, as they are similar to the dynamics of furan.
Figure 46. Photoelectron spectrum of (a) pyrazole, and (b) 1-methyl pyrazole. A signal towards positive times denotes 200 nm excitation. When molecules are excited at 267 nm, only the bottom of the ππ*-state surface can be accessed. During the course of the experiment, no large amplitude motion is observed, and the time constant obtained is above 1ps. This possibly indicates access of the ring-puckering channel, which has a smaller barrier than the ring-opening channel (the N-H dissociation channel is energetically inaccessible). Compared with pyrazole and the methylation effect on pyrazole upon 200 nm excitation, the dynamics of pyrazole involves almost no ring-puckering, which indicates an efficient N-N bond dissociation channel. However, the methylated pyrazoles all show the ring-puckering channel in their dynamics. This can be explained by the extra inertia, which decreases the amplitude of the vibration, and hence the likelihood to enter the N-N dissociation channel. As a result, from the point of view of dynamophore, pyrazole has another reaction channel, which is N-N bond cleavage, similarly to C-O bond

68
cleavage in furan. Furthermore, it keeps the N-H bond dissociation channel like pyrrole. Finally, pyrazole keeps the dynamics common to all the five-memebered heteroaromatics compounds, which is the ring puckering channel.

69
7. Conclusion and outlook
The excited state dynamics of small organic molecules have great relevance and importance for various areas of science. In our studies to understand these dynamics, the most important aspect is to set up a basic framework by starting with a simple molecule, in a bottom-up approach, and using controlled chemical modification on these molecules to understand the dynamics of increasingly complex systems In this thesis, using the excited state dynamics of the simplest molecule, ethylene, as a template, more “complex” molecules were studied. Such molecules may involve an increased number of double bonds, e.g., to form polyenes (two double bonds), i.e., cyclohexadiene, or adding different functional groups to ethylene, e.g. to form acrylonitrile and alkyl vinyl ethers, and adding heteroatoms into cyclic systems, e.g. forming furan (-O-), pyrrole (-NH) and pyrazole (-N-N-). In addition, the aldehyde group effects on furan and methylation effects on pyrrole, pyrazole, and acrylonitrile have also been investigated, adding inertia and/or extending the delocalised electronic environment. These studies probe how the dynamics changes with respect to the molecular structure when going from linear molecules to ring-systems, and the influence on the dynamics from different functional groups, and the different locations of these groups.
To allow an interpretation of the dynamics of excited molecules, a new concept is introduced. While the part of molecule that is excited is called the chromophore, the part of molecule in which the subsequent dynamics occurs, i.e. localised to a certain part of the molecule, is called the dynamophore. The dynamophores of molecules are influenced by many factors, e.g. the structure, the location and number of substituents, etc. Though the dynamics of organic molecules are complicated, certain trends are observed from our work from comparing the investigated molecules to find the similarities and differences. These trends help to make possible prediction of the outcome after photoexcitation of more complicated molecules. Reviewing the dynamics of ethylene; this first undergoes double bond torsion on the excited state and then accesses two different conical intersections with the ground state leading to pyramidalization or hydrogen migration. In the study of the dynamics of cyclohexa-1,3-diene, the double bond torsion causes ring-puckering and ring opening to form three different conformers, and we observe that this molecule has a delocalised chromophore and dynamophore.

70
With respect to the effect of a functional group from the alkoxy and cyano groups on ethylene molecules, we find that a new bond cleavage reaction channel becomes available on the πσ* state in alkyl vinyl ether, while no bond cleavage is observed to happen in acrylonitrile. The common point they both have is that they keep the ethylene-like double bond torsion and pyramidalization processes. However, in alkyl vinyl ether, the pyramidalization happens on the terminal carbon atom, while in acrylonitrile the pyramidalization happens on the C2 atom, where the presence of a methyl-substituent is observed to slow down the dynamics. For the five-member ring molecules with O and N heteroatoms, i.e. furan pyrrole, and pyrazole, they display bond cleavage processes, C-O, N-H, or N-N, on the πσ* state, and ring puckering on the ππ* sate caused by bond torsion. Addition of an aldehyde group to furan introduces an nπ* state, and there are indications that the population probably decays from the ππ* state to the nπ* state. The other important observation is the first report in the dynamics of pyrrole of the involvement of the π3p Rydberg state, and that the Rydberg state is transiently populated from the valence state and possibly directs the outcome of the photoreaction. The excited state dynamics of molecules are obtained from analyzing the data from the experimental results of time-resolved photoelectron spectroscopy coupled with various ab initio calculations. The different photoelectron bands (their shape and position in the photoelectron spectrum), provide information on, e.g., the lifetimes of the excited states, and the time zero shifts indicates if there is large amplitude motion happening on the excited state when the molecule leaves the Franck-Condon region towards a conical intersection. The theoretical results supports the idea of the potential energy landscape of the lowest lying excited state, which kinds of channels are involved in the dynamics, and which states are accessed. A much clearer picture of molecular dynamics can be only gained by combination of experiments and theories. This thesis provides a very important bottom-up systematic approach for the excited state dynamics of organic molecules on the basis of ethylene-like dynamics. The current data, although detailed and displaying new previously unobserved processes, clearly demonstrates that more investigations on the dynamics need to be undertaken. This is due to that many factors, e.g. the excitation energy, the number of photons of pump or probe, the electro-chemical environment, etc., all influence the dynamics. In the future, for example, much higher energy probe pulses can be used to see more of the ionisation continuum, which gives more access to the product formation, and to see shifts for extending over a larger region for a better understanding the molecular dynamics on the excited state. Moreover, the role of Rydberg states in the photodynamics of molecules should be studied in more detail to see what the situation is in other molecules, and how the presence of and interplay with these Rydberg states influences the

71
reaction dynamics, especially the process initiated on higher-lying excited states.

LXXII
Sammanfattning
Processer inducerade av ultraviolett och synligt ljus i små organiska molekyler spelar en mycket viktig roll inom ett brett spektrum av områden, t.ex. miljövetenskap, biologi, materialutveckling, kemi, astrofysik och många andra. Några exempel på detta är när solljus samverkar med lättflyktiga organiska föreningar vilket leder till bildandet av fotokemisk smog, skadligt för hälsa och miljö, medan i en molekylär fotoswitch absorption av fotoenergi leder till en förändring i strukturen och switchens egenskaper. Dessutom kan fotokemiska processer användas i omvandling av solenergi vilket är av intresse för forskning kring förnybara energikällor. Dessa är bara några av många applikationer som illustrerar betydelsen av fotokemiska processer. Således är det av stor vikt att bättre förstå de mekanismer som är inblandade i dessa processer. En fotokemisk process kan beskrivas som följer. Först, en molekyl i grundtillståndet absorberar energin hos en infallande foton och lyfts upp till ett exciterat tillstånd. Den exciterade molekylen är initialt i Franck-Condon regionen för det exciterade tillståndet och lämnar därfeter denna region via en av många tillgängliga vibrationsrörelser och breder således ut sig i olika riktningar på det exciterade tillståndets potentialenergiyta. Någon av dessa ritningar kan leda till och igenom en konisk ytkorsning till ett lägre liggande exciterat tillstånd eller tillbaka till grundtillståndet. Rörelsen på det exciterade tillståndet som ger åtkomst till den koniska korsningen orsakar ibland en betydande strukturell förändring i molekylen, t.ex. bindningsvridning, bindningsklyvning, etc. Ett viktigt fokus för avhandlingen är att studera dessa typer av förändringar i små organiska molekyler, och att förstå hur de fortlöper i det exciterade tillståndet. I många av de områden där fotoprocesser är viktiga är komplexa organiska molekyler inblandade, t ex i medicin, i DNA, osv. Emellertid är fotodynamiken för komplexa molekyler svår att studera, tolka och förklara. En fundamental aspekt av min avhandling är att fastställa de grundläggande reglerna för att förstå komplexa organiska molekyler genom att utgå från en avsevärt enklare molekyl. Här är det etenmolekylen som används i ett underifrånperspektiv (”bottom-up” på engelska), där dynamiken som följer på den exciterade potentialenergiytan omfattar bindningsvridning, pyramidalisering och väteomlagring. Genom

LXXIII
att lägga till funktionella grupper, substituenter, eller att bilda cykliska system kan vi öka storleken och komplexiteten på molekylen på ett kontrollerat sätt. Utfallet av fotoabsorptionen kan då systematiskt utvärderas och användas vidare för jämförelse mellan molekyler med olika funktionella grupper på samma grundstruktur (t ex etylen) eller samma funktionella grupper på olika grundstrukturer (t ex akrolein och furfural). Tidsupplöst fotoelektronspektroskopi används för att studera fotodynamiken för molekyler. Denna teknik gör att vi kan följa dynamiken som uppstår på exciterade potentialenergiytor med en noggrannhet på ca 10 femtosekunder, som är ungefär den tid det tar för en molekyl att vibrera. De experimentella observationerna är kopplade till teoretiska studier från medarbetare och används för att tolka, stödja, och jämföra med de experimentella mätningarna för att bättre förstå denna dynamik. De molekyler som jag har undersökt i avhandlingen kan separeras i tre grupper. Den första gruppen av molekyler skapas genom att addera en funktionell grupp till eten, t ex för att bilda alkylvinyleter (-OCnH2n+1-) och akrylonitril (-CN). Vi har funnit att den dynamik som observeras i alkylvinyletrar beror på excitationsvåglängden. Med excitation vid 200 nm kan molekylerna nå π3s-Rydberg-tillståndet varifrån de går vidare till ett ππ*-tillstånd med eten-liknande dynamik eller till ett πσ* tillstånd där C-O-bindningsklyvning sker. I fotodynamiken för akrylonitril hittar vi ingen -CN-elimineringskanal. Molekylerna genomgår etenliknande bindningsvridning och pyramidalisering i det exciterade tillståndet vid passage av den koniska ytkorsningen med grundtillståndet. Den andra gruppen av molekyler skapas genom modifiering av eten till bildning av cykliska system, t.ex. cyklohexa-1,3-dien. Bara ett exciterat tillstånd observeras delta i dynamiken, och molekylerna genomgår antingen en ringöppningsreaktion eller en icke-öppningsreaktion tillbaka till grundtillståndet. Den tredje gruppen av molekyler är fem-uddiga heteroaromatiska molekyler, vilka kan betraktas som en modifikation av den cykliska cyklopentadienmolekylen. Furan (en syrehaltig heteroaromat), pyrrol (en kvävehaltig heteroaromat) och pyrazol (en dubble-kvävehaltig heteroaromat) kan följa två olika reaktionskanaler vid excitation till ππ*-tillståndet. Furan molekyler och pryazolmolekyler genomgår ringveckning genom koppling från ππ*-tillståndet till grundtillståndet eller de går vidare till πσ*-tillståndet med en C-O-/N-N-bindningsklyvning. Pyrrolmolekyler går från ett ππ* till ettπσ*-tillstånd med antingen ringveckning eller N-H-bindningsklyvning. Även inverkan av funktionella grupper undersöks för furan. Här kopplar vi en aldehydgrupp till ringen, vilket utvidgar den delokaliserade elektronstrukturen i ringen och detta har effekten att både öppna upp en ny reaktionskanal (molekylerna kan nu gå via ett np*-tillstånd) och även bromsa de befintliga kanalerna. Slutligen, i pyrrol, observerar vi

LXXIV
de första indikationerna på att ett Rydbergtillstånd som inte populeras direkt utan som ett mellantillstånd är involverat i fotodynamiken. Dessa studier bidrar till att förstå dynamiken för kol-kol-dubbelbindningar i små organiska molekyler. Det visar sig att med tämligen vitt skilda varianter på modifiering av etenmolekylerna, så bibehålls huvuddragen i etendynamiken, bindningsvridning och pyramidalisering, även om de i de cykliska systemen kommer till uttryck som en ringveckning.

LXXV
Acknowledgements
The last four years of my PhD study in Department of Fysikum has been a great experience in my life. I have met a lot of nice and friendly people who helped and supported me.. Here I would like to express my sincere gratitude to them as I would not come this far without them. My deepest gratitude to my supervisor, Richard, thanks for providing me the opportunity to start my PhD study in Stockholm University .Your patience and encouragements helped me a lot on scientific matter and others. Oliver, I am grateful for your help on the projects, not only for the experiments, but also for the data analysis. Tony, thanks for spending your time on discussing the problems and questions I got in the experiments, papers and my thesis. In addition, I would thank all the people collaborating with us and taking part in the scientific work presented in Papers I-VI. Also I would thank John for the help on the Lab-view programming which helped us for data collection. I would also like to thank Mats, thanks for being my mentor and providing me valuable advice on the scientific career. Guillermo, you are the first person working with me in the lab, thanks a lot for introducing technical stuff to me. I also would like to give my warmest gratitude to the kind colleagues and friends I met in Chemical physics division. Sifiso, I was happy to be with you in the same office when I firstly came here. You helped me a lot to get familiar with the environment here. Oleksii, it was a good time to have lunch together, sharing news and listening interesting news about your lovely daughter. Ida and Emelie, thanks for asking me go skating with you. That was a nice experience! Emelie, it was a pleasure to discuss with you on the home assignment during the quantum mechanics course. Jesper, Emelie, I am happy to have Friday lunch together with you this semester, nice sushi lunch before Christmas, although it was super expensive :D To my Chinese friends in Stockholm, Uppsala, Copenhagen, Höganäs, US and Netherlands. It was because of you I did not feel lonely. Thanks for always being there. Each trip with you is an unforgettable memory to me. To my Chinese friends in China, even though you are far away from Sweden, thank you for your supporting and concern. At last, I would like to thank my mom. 谢谢妈妈的支持, 鼓励和爱,才能让我在追逐梦想的道路上勇敢前行!

LXXVI
References [1] K. I. Barnhard, et. al., Chem. Phys. Lett. 178, 150 (1991). [2]http://www.chemistry.wustl.edu/~edudev/LabTutorials/Vision/Vision.html [3] S. Helmy, et. al., J. Am. Chem. Soc. 136, 8169 (2014). [4] A. Stolow, A. E. Bragg, and D. M. Neumark, Chem. Rev. 104(4), 1719 (2004). [5] D. M. Neumark, Annu. Rev. Phys. Chem. 52, 255 (2001). [6] E. D. Potter, et. al., Nature. 355, 66 (1992). [7] O. M. Sarkisov, Russ. Chem. Bull., Int. Ed., 57, 736 (2008). [8] D. Brinks, et.al., Chem. Soc. Rev. 43, 2476 (2014). [9] G. M. Roberts, et. al., Faraday Discuss. 163, 95 (2013). [10] O. Schalk, et.al., J. Phys. Chem. A 118, 2279 (2014). [11] O. Schalk, et.al., J. Phys. Chem. A, 119, 11105 (2015). [12] B. G. Levine, and T. J. Martínez. Annu, Rev. Phys. Chem. 58, 613 (2007). [13] O. Schalk, et. al., J. Am. Chem. Soc. 133, 16451 (2011). [14] S. A. Trushin, et. al., Phys. Chem. Chem. Phys. 1, 1431 (1999). [15] O. Schalk, et. al, J. Phys. Chem. A. 117, 10239 (2013). [16] T. S. Kuhlman, et. al., Faraday Discuss. 157, 193 (2012). [17] T. J. Wolf, et. al., Phys. Chem. Chem. Phys. 16, 11770 (2014). [18] IUPAC, Compendium of Chemical Terminology, 2nd ed.; doi: 10.1351/goldbook. C01076. [19] R. McWeeny, Farady Discuss. 135, 13 (2007). [20] O. Schalk, et. al., EPJ Web of Conference, 41, 02037 (2013). [21] K. Kosma, et. al., Phys, Chem. Chem. Phys. 11, 172 (2009). [22] Y. Dou, S. Yuan, and G. V. Lo, Appl. Surf. Sci. 253, 6404 (2007). [23] B. C. Arruda, and R. J. Session, Phys. Chem. Chem. Phys.16, 4439 (2014). [24] C. C. Bühler, et. al., J. Ato. Mol. And Opt. Phys. 2011, 637593 (2011). [25] B. G. Levine, and T. J. Martínez, J. Phys. Chem. A. 113, 12815 (2009). [26] S. Wilsey, and K. N. Houk, Photochem. Photobiol. 76, 616 (2002). [27] R. C. Evans, P. Douglas, and H. D. Burrows, Applied photochemistry. Springer Dordrecht Heidelberg New York London (2013). [28] I. Fleming, Molecular Orbitals and Organic Chemical Reactions. A John Wiley and Sons, Ltd., Publication (2010). [29] R. S. Mulliken, PhysRev. 41, 49 (1932). [30] J. H. Starcke, et. al., Chem. Phys. 329, 39 (2006). [31] R. S. Mulliken, J. Am. Chem. Soc. 86, 3183 (1964). [32] M. Gruebele, and A. H. Zewail, J. Chem. Phys. 98, 883 (1993). [33] T. Mori, et. al., J. Phys. Chem. A 116, 2808 (2012). [34] E. G. Champenois, et. al., J. Chem. Phys. 144, 014303 (2016). [35] M. Barbatti, and K. Sen, In. J, Quant. Chem. 116, 762 (2016).

LXXVII
[36] G. Wu, et. al., J. Chem. Phys. 142, 074302 (2015). [37] J. von Neumann and E.P. Wigner, Z. Physik, 30, 467 (1929). [38] F. Hund, Z. Physik, 40, 742, (1927). [39] T. J. Martínez, Nature 467, 412 (2010). [40] F. Bernadi, M. Olivucci, and M. A. Robb, Chem. Soc. Rev. 25, 321 (1996). [41] G. Atchity, S. S. Xantheas, and K. Ruedenberg, J. Chem. Phys. 95, 1862 (1991). [42] D. R. Yarkony, J. Phys. Chem. A. 105, 6277 (2001). [43] J. P. Malhado, and J. T. Hynes, J. Chem. Phys. 145, 194104 (2016). [44] C. Zener, Proc. R. Soc. London, Ser. A 137, 696 (1932). [45] M. A. EI-Sayed, J. Chem. Phys. 36, 573 (1962). [46] M. A. EI-Sayed, J. Chem. Phys. 38, 2834 (1963). [47] M. A. EI-Sayed, J. Chem. Phys. 41, 2462 (1964). [48] R. S. Minns, et. al., Phys.Chem.Chem.Phys. 12, 15607 (2010). [49] M. Ben-Nun, and T. J. Martínez, Chem. Phys. Lett. 298, 57 (1998). [50] T. J. Martínez, Acc. Chem. Res. 39, 119 (2006). [51] B. G. Levine, and T. J. Martínez, Annu. Rev. Phys. Chem. 58, 613 (2007). [52] A. J. Merer, and R. S. Mulliken, Chem. Rev. 69, 639 (1969). [53] J. Ryu, and B. S. Hudson, Chem. Phys. Lett. 245, 448 (1995). [54] K. K. Baeck, and T. J. Martínez, Chem. Phys. Lett. 375, 299 (2003). [55] B. Sellner, et. al., Mol. Phys. 111, 2439 (2013). [56] H. Tao, et.al., J. Chem. Phys. 134, 244306 (2011). [57] V. Stert, H. Lippert, H.-H. Ritze, and W. Radloff, Chem. Phys. Lett. 388, 144 (2004). [58] T. K. Allison, et. al., J. Chem. Phys. 136, 124317 (2012). [59] K. Kosma, et. al., J. Phys.Chem. A. 112, 7514 (2008). [60] T. Kobayashi, et. al., J. Phys. Chem. A . 119, 9518 (2015). [61] R. S. Mulliken, J. Chem. Phys. 71, 556 (1979). [62] M. B. Robin, Academic Press: New York, 3 (1985). [63] W. Th. A. M. Van der Lugt, and L. J. Oosterhoff, J. Am. Chem. Soc. 22, 91 (1969). [64] A. Nenov, and R. de Vivie-Riedle, J. Chem. Phys. 135, 034304 (2011). [65] A. Viel, et. al., J. Chem. Phys. 120, 11000 (2004). [66] V. Molina, et. al., Phys. Chem. Chem. Phys. 2, 2211 (2000). [67] M. Klessinger, and J. Michl, Excited States and Photochemistry of Organic Molecules, VCH, New York, NY, USA, 1995. [68] L. Salem, Science. 191, 822 (1976). [69] B. A. Balko, J. Zhang, and Y. T. Lee, J. Chem. Phys. 97, 935 (1992). [70] S. H. Lee, Y. S. Lee, and X. Yang, J. Chem. Phys. 120, 10983 (2004). [71] R. A. Rijkenberg, and W. J. Buma, J. Phys. Chem. A. 106, 3727 (2002). [72] G. Wu, et. al., J. Chem. Phys. 135, 164309 (2011). [73] C. Dugave, and L. Demange, Chem. Rev. 103, 2475 (2003).

LXXVIII
[74] M. Van der Horst, and K. Hellingwerf, Acc. Chem. Res. 37, 13 (2004). [75] D. G. Leopold, et. al., J. Chem. Phys. 81, 4218 (1984). [76] V. Vaida, Acc. Chem. Res. 19, 114 (1986). [77] K. Schulten, and M. Karplus, Chem. Phys. Lett. 14, 305 (1972) [78] A. Makida, et. al., J. Phys. Chem. Lett. 5, 1760 (2014). [79] F. Assenmacher, et. al., Phys. Chem. Chem. Phys. 3, 2981 (2001). [80] O. Schalk, A. E. Boguslavskiy and A. Stolow, J. Phys. Chem. Lett. 5, 560 (2014). [81] P. Hockett, et. al., J. Mod. Opt. 60, 1409 (2013). [82] Y. Gou, B. R. Torralva, and R. E. Allen, J. Phys. Chem. A107, 8817 (2003). [83] C. Noonenberg, S. Grimm, and I. Frank, J. Chem. Phys. 119, 11585 (2003). [84] M. Ito, I. Ohmine, J. Chem. Phys. 106, 3159 (1997). [85] R. R. Chadwick, , et.al., Chem. Phys. Lett. 115, 24 (1985). [86] J. Lappe, and R. J. Cave, J. Phys. Chem. A. 104, 2294 (2000). [87] M. A. Watson, J. Chem. Theory Comput. 8, 4013 (2012). [88] S. H. Pullen, et. al., J. Chem. Phys. 107, 4985 (1997). [89] X. Ci, A. B. Myers, J. Chem. Phys. 96, 6433 (1992). [90] M. Garavelli, et. al., J. Am. Chem. Soc. 119, 11487 (1997). [91] C. C. Hayden, and D. W. Chandler, J. Phys. Chem. 99, 7897 (1995). [92] W. Fuβ, et. al., J. Chem. Phys. 106, 2205 (1997). [93] K. Ohta, et. al., J. Phys. Chem. A. 105, 3973 (2001). [94] M. Garavelli, et. al., J. Am. Chem. Soc. 118, 11656 (1996). [95] S. Wilsey, K. N. Houk, and A. H. Zewail, J. Am. Chem. Soc. 121, 5772 (1999). [96] W. Fuβ, W. E. Schmid, and S. A. Trushin, J. Am. Chem. Soc. 123, 7101 (2001) [97] M. Merchan, et. al., J. Phys. Chem. A. 103, 5468 (1999). [98] R. McDiarmid, and J. P. Doering, J. Chem. Phys. 75, 2687 (1981). [99] S. Wilsey, and K. N. Houk, J. Am. Chem. Soc. 122, 2651 (2000). [100] S. Wilsey, and K. N. Houk, J. Am. Chem. Soc. 124, 11182 (2002). [101] M.S. Schuurman, private communication. [102] A. C. Krueger, and W. P. Malachowski, Cyclopentadiene, Jon Wiley & Sons, Ltd. [103] W. Fuβ, W. E. Schmid, and S. A. Trushin, Chemical Physics. 316, 225 (2005). [104] O. Schalk, A. E. Boguslavskiy, and A. Stolow, J. Phys. Chem. 114, 4058 (2010). [105] O. Schalk, and A.-N. Unterreiner, J. Phys. Chem. A. 111, 3231 (2007). [106] S. A. Trushin, et. al., Phys. Chem. Chem. Phys. 2, 1435 (2000). [107] W. Wu, et. al., J. Chem. Phys. 132, 124510 (2010). [108] A. M. D. Lee, et. al., J. Phys. Chem. A. 111, 11948 (2007). [109] M. Reguero, et. al., J. Am. Chem. Soc. 116, 2103 (1994).

LXXIX
[110] W.-H. Wang, J. Am. Chem. Soc. 121, 8376 (1999). [111] F. Aquilane, V. Berone, and B. O. Roos, J. Chem. Phys. 119, 12323 (2003). [112] P. Parson, et.al., J. Chem. Phys. 117, 7889 (2002). [113] F. A. Carey, and R. Sundberg, J. Organische Chemie; Wiley VCH: Weinheim, 1 (1995). [114] J. Cao, and Z. Xie, Phys. Chem. Chem. Phys. 18, 6931 (2016). [115] G. A. King, et.al., J. Chem. Phys. 132, 064305 (2010). [116] C. A. Williams, et.al., J. Phys. Chem. A. 116, 2600 (2012). [117] J. Cao, et.al., Chem. Phys. 474, 25 (2016). [118] F. Chang, et. al., Hazard. Mater. A70, 1 (1999). [119] J. Lee, and I. Tang, J. Chem. Phys. 77, 4459 (1982). [120] L. S. Tran, et. al., Energy. 43, 4 (2012). [121] Y. Roman-Leshkov, et.al., Nature 447, 982 (2007). [122] T. Suzuki, Int. Rev.Phys. Chem. 31(2), 265 (2012).f [123] D. M. Neumark, Annu, Rev. Phys. Chem. 52(1), 255 (2001). [124] R. Spesyvtsev, et. al., J. Chem. Phys. 143, 014302 (2015). [125] M. Stenrup, and Å. Larson, Chem. Phys. 379, 6 (2011). [126] Y. Liu, et. al., Chemical Physcis. 446, 142 (2015). [127] H. Köppel, E. V. Gromov, and A. B. Trofimov, Chem. Phys. 304, 35 (2004). [128] V. Vallet, et. al., Faraday Discuss. 127, 283 (2004). [129] H. Lippert, et. al., ChemPhysChem. 5, 1423 (2004). [130] B. Cronin, et. al., Phys. Chem. Chem. Phys. 8, 3440 (2006). [131] M. Patore, C. Angeli, and R. Cimiraglia, Chem. Phys. Lett. 422, 522 (2006). [132] J. Wei, et. al., Faraday Discuss. 127, 267 (2004). [133] Z. Lan, and W. Domcke, Chem. Phys. 350, 125 (2008).
[134] M. Barbatti, et. al., Chem. Phys. 375, 26 (2010). [135] R. Montero, et.al., J. Phys. Chem. Lett. 7, 2797 (2016). [136] S. P. Neville, et.al., Nat. Commu. 7, 11357 (2016). [137] S. P. Neville, and G. A. Worth, J. Chem . Phys. 140, 034317 (2014). [138] M. H. Palmer, et.al., Chem. Phys. 238, 179 (1998). [139] G. R. Wu. et.al., J. Chem. Phys.144, 014309 (2016). [140] E. Spence, P. N. Kean, and W. Sibbett, Opt. Lett. 16, 42(1991). [141] D. Strickland, and G. Mourou, Opt. Commun. 56, 219 (1985). [142] J.H.D. Eland. et al., Phys. Rev. Lett. 90, 053003 (2003). [143] C. E. Moore, Natl. Bur. Stand. Circ. 467 (1949). [144] D. R Cyr, and C. C. Hayden, J. Chem. Phys. 104, 771 (1996). [145] G. Wu, P. Hockett, and A. Stolow, Phys. Chem. Chem. Phys. 13, 18447 (2011). [146] V. Blanchet et al., Nature. 401, 52 (1999). [147] T. Geng, et. al., In manuscript. [148] D. M. Holland, et al., J. Phys. B 29, 3091 (1996).

LXXX
[149] O. Schalk, et. al., J. Phys. Chem. 118, 2279 (2014), Supplementary material. [150] M. O. Trulson, et. al., J. Am. Chem. Soc. 109, 586 (1987). [151] S. H. Pullen, et .al., J. Chem. Phys.108, 556 (1998). [152] V. S. Petrovic, et .al., J. Chem. Phys.139, 184309 (2013). [153] S.Adachi, et .al., J. Phys. Chem. Lett. 6, 343 (2015). [154] P. Bischof, and E. Heilbronner. Helv. Chim. Acta. 53, 1677 (1970). [155] W.-N. Du, C. Luo, and Z. –S. Li, J. Chem. Phys. 129, 174309 (2008). [156] C. A. Bird and D. J. Donaldson, Chem. Phys. Lett. 249, 40 (1996). [157] C. Y. Oh, et .al., J. Chem. Phys. A 107, 4333 (2003). [158] M. L. Morton, D. E. Szpunar, and L. J. Bulter, J. Chem. Phys. 115, 204 (2001). [159] B. Cronin, et .al., Phys. Chem. Chem. Phys. 6, 5031 (2004). [160] G. Piani, et .al., J. Phys. Chem. A. 113, 14554 (2009). [161] L. Blancafort, et .al., J. Phys. Chem. Lett. 7, 1231 (2016). [162] F. K. Keter, J. Darkwa.Biometals. 25, 9 (2012). [163] C. A. Willians, et .al., J. Phys. Chem. A. 116, 2600 (2012).

LXXXI
Attached papers