exoenzyme s of pseudomonas aeruginosa cellular targets and
TRANSCRIPT

UMEÅ UNIVERSITY MEDICAL DISSERTATIONS New Series No. 1125 ISSN: 0346-6612 ISBN: 978-91-7264-414-4
Exoenzyme S of Pseudomonas aeruginosa: cellular targets and interaction with 14-3-3
Lubna Yasmin Department of Medical Biosciences and Pathology,
Umeå University, Sweden.
November 2007

2
UMEÅ UNIVERSITY MEDICAL DISSERTATIONS New Series No. 1125 ISSN: 0346-6612 ISBN: 978-91-7264-414-4
Edited by the Dean of the Faculty of Medicine
COVER: The photo shows that HeLa cells were rounded up upon infection with Yesinia pseudotuberculosis bacteria expressing and translocating ExoS(wild type), indicated by white color rod shape. The picture has been taken on 24th of October 2003.
Copyright© 2007 by Lubna Yasmin Printed in Sweden by Arkitektkopia AB, Umeå, 2007

3
بِسْمِ اللّهِ الرَّحْمَـنِ الرَّحِيمِ.In the Name of ALLAH, the Most Beneficent, the Most Merciful.
فَبِأَيِّ آلَاء رَبِّكُمَا تُكَذِّبَانِ.Then which of the Blessings of your Lord will you deny.
Sura Ar-Rahman (The Quran)

4

TABLE OF CONTENTS
5
TABLE OF CONTENTS
1. ABSTRACT ..................................................................................................... 7 2. PAPERS IN THIS THESIS............................................................................ 8 3. ABBREVIATIONS ......................................................................................... 9 4. INTRODUCTION......................................................................................... 11
4.1 Pseudomonas aeruginosa ......................................................................... 11
4.1.1. Severity of diseases ........................................................................... 12 4.1.2. Virulence factors of Pseudomonas aeruginosa............................... 12 4.1.3. Pseudomonas aeruginosa ExoS........................................................ 15
4.1.3.1. ExoS secretion and translocation domain ............................... 16 4.1.3.2. ExoS-GAP domain ..................................................................... 16 4.1.3.3. ExoS-ADP-ribosyltransferase domain..................................... 17 4.1.3.4. ExoS-ADP-ribosyltransferase structure.................................. 18
4.2 Small GTPase ........................................................................................... 18
4.2.1. Ras small GTPases ........................................................................... 19 4.2.2. Rap small GTPases........................................................................... 20
4.3. Signaling pathways affected by ExoS ................................................... 21
4.3.1. Ras activation and downstream signaling...................................... 21 4.3.2. The Raf pathway (RAF/MEK/ERK cascade)................................ 22 4.3.3. Rap1 signaling cascade .................................................................... 24 4.3.4. The PI3-Kinase pathway ................................................................. 24 4.3.5. Regulation of apoptosis by RAF/MEK/ERK and PI3-KINASE cascades ....................................................................................................... 26
4.3.5.1. Caspases ...................................................................................... 27 4.3.5.2. Apoptosis signaling cascade ...................................................... 28
4.4. The 14-3-3 proteins ................................................................................. 29
4.4.1. The mechanisms of 14-3-3 action.................................................... 29 4.4.2. Structures of 14-3-3 dimers ............................................................. 30 4.4.3. 14-3-3/client molecule interactions ................................................. 32
4.4.3.1. The phosphorylated 14-3-3 binding motif ............................... 32 4.4.3.2. Non-phosphorylated 14-3-3 binding motifs............................. 33
4.4.4. Comparative structural analysis of the seven 14-3-3 protein isoforms ....................................................................................................... 35

TABLE OF CONTENTS
6
4.4.5. Function of 14-3-3............................................................................. 35 4.5. P. aeruginosa pathogenesis in a mouse model...................................... 36 4.6. Cell lines used for this study .................................................................. 37 4.7. ExoS constructs used for this study ...................................................... 37
5. AIMS............................................................................................................... 40 6. RESULTS AND DISCUSSION ................................................................... 41
6.1. Cells expressing activated Ras or PKB/Akt survive upon ExoS infection (Paper I)........................................................................................... 41
6.1.1. Ras is an important cellular target and determinant of cell survival ........................................................................................................ 41 6.1.2. ExoS mediates caspase-3 activity .................................................... 42
6.2. Phosphorylation independent interaction between 14-3-3/ExoS (Papers II, III, and IV) ................................................................................... 43
6.2.1. ExoS-LDL motif is important for interaction and enzymatic activity ......................................................................................................... 43 6.2.2. 14-3-3/ExoS structure (the novel binding mode IV) ..................... 45 6.2.3. Hydrophobic interaction is important for both interaction and enzymatic activity ....................................................................................... 46 6.2.4. Leu428 is an important determinant of cell death induction....... 47 6.2.5. Asp424 and Asp427 are not important for ExoS enzymatic activity ......................................................................................................... 47 6.2.6. Leu428 is critical for ExoS auto-ADP-ribosylation ...................... 47
7. CONCLUSIONS............................................................................................ 49 8. ACKNOWLEDGEMENTS.......................................................................... 50 9. REFERENCES .............................................................................................. 52 10. PAPERS I-IV................................................................................................ 67

ABSTRACT
7
1. ABSTRACT
Exoenzyme S of Pseudomonas aeruginosa: cellular targets and interaction with 14-3-3 Pseudomonas aeruginosa is an opportunistic pathogen that is a serious problem for immuno-compromised patients. Toxins such as exoenzyme (Exo) S, ExoT, ExoY and ExoU are secreted and translocated from the bacteria into the eukaryotic cell via the bacterial encoded type III secretion system. Our research focuses on ExoS, a bifunctional toxin comprising a Rho-GTPase-activating protein domain (RhoGAP) and a 14-3-3 dependent ADP-ribosyltransferase domain. In addition, ExoS contains a membrane localization domain termed MLD. In this study, cell lines expressing activated forms of various components of the Ras signaling pathway have been used to understand the functional and mechanical activation of ExoS-ADP-ribosyltransferase activity and to reveal its cellular targets in the cell. Our observations suggested that Ras GTPase is the dominant target by which ExoS mediates cell death and activated Ras is able to protect cells against cell death, regardless of whether it has been ADP-ribosylated by ExoS.
It has been reported that the 14-3-3 cofactor protein is required for ADP-ribosyltransferase activity of ExoS and a phosphorylation-independent interaction occurs between 14-3-3 and the C-terminal part of ExoS. We have undertaken a deeper analysis including structural and biological investigation of this interaction. Our results suggested that leucine-428 of ExoS is the most critical residue for ExoS enzymatic activity. Structural analysis showed that ExoS binds to 14-3-3 in a novel binding mode mostly relying on hydrophobic contacts. Our structure was supported by biochemical and cytotoxicity analyses, which revealed that the substitution of important residues of ExoS significantly weakens the ability of ExoS to modify endogenous targets such as RAS/RAP1 and to induce cell death. Further, mutation of key residues within the ExoS binding site for 14-3-3 impairs virulence in a mouse pneumonia model. Leucine residues-422, 423, 426, and 428 of ExoS are important for the interaction with the ″roof″ of the amphiphatic groove of 14-3-3.
In conclusion, we show the mechanism of cell signal transduction pathways affected upon ExoS infection and also demonstrate that the hydrophobic residues of ExoS in 14-3-3 interaction motif have a significant role for ExoS enzymatic activity.
Keywords: ExoS, ADP-ribosylation, 14-3-3 protein, Pseudomonas aeruginosa, Small GTPases, RAS, RAP

PAPERS IN THIS THEIS
8
2. PAPERS IN THIS THESIS This thesis includes the following papers referred to in the text by their roman numerals (I-IV) and is reprinted with permission of the publishers. Paper I. Yasmin, L.*, Jansson, .L.*, Downward, J., Warne, P., Palmer, R.H., and Hallberg, B. Exoenzyme S of Pseudomonas aeruginosa is not able to induce apoptosis when cells express activated proteins, such as Ras or protein kinase B/Akt. Cellular Microbiology, 8(5):815-22, May 2006 Paper II Yasmin, L.*, Jansson, A.L.*, Panahandeh, T., Palmer, R.H., Francis, M.S., and Hallberg, B. Delineation of Exoenzyme S residues that mediate the interaction with 14-3-3 and its biological activity. FEBS J. 273(3):638-46, Feb 2006 Paper III Ottmann, C., Yasmin, L., Weyand, M., Veesenmeyer, J.L., Diaz, M.H., Palmer, R.H., Francis, M.S., Hauser, A.R., Wittinghofer, A., and Hallberg, B. Phosphorylation independent interaction between 14-3-3-Exoenzyme S: from structure to pathogenesis. EMBO J. 26(3):902-13, Feb 2007 Paper IV Yasmin, L., Francis, M.S., and Hallberg, B. Electrostatic interaction is of secondary importance for the interaction between ExoS and 14-3-3. Manuscript 2007 *Equal contribution

ABBREVIATIONS
9
3. ABBREVIATIONS AANAT Arylalkylamine N-acetyltransferase ADP Adenosine diphosphate ADPRT ADP-ribosyltransferase AIDS Acquired immuno-deficiency syndrome Apaf-1 Apoptosis proteases-activating factor -1 ASK1 Apoptosis signal-regulating kinase 1 Asp Aspartate ATP Adenosine triphosphate BAD Bcl- XL /Bcl-2-associated death promoter Bak Bcl-2 homologous antagonist/killer Bax Bcl-2 -associated x protein Bcl-2 B cell leukaemia-2 cAMP Cyclic adenosine monophosphate CARD Caspase recruitment domain Caspases Cysteine aspartate-specific proteases CF Cystic fibrosis CHO Chinese Hamster Ovary CNF1 Cytotoxic necrotizing factor 1 CpA Cyclophilin A Crk CT10-regulator of kinase CRMP1 Chromosomal regional maintenance protein 1 DED Death effector domain DISC Death-inducing signalling complex EGF Epidermal growth factor EPAC Exchange protein activated by cAMP ERK Extracellular signal-regulated kinase ERM Ezrin, radixin and moesin ETA Exotoxin A ExoS Exoenzyme S F-actin Filamentous actin FADD Fas associated protein with death domain FAS Factor activating exoenzyme S FOXO Forkhead Box, class O Ftase Farnesyltransferase FTIs FTase inihibitors GAP GTPase activating proteins GDP Guanine diphosphate GEF Guanine nucleotide exchange factor GGTaseI Geranylgeranyltransferase I Grb2 Growth factor receptor-bound protein 2 GSK3 Glycogen synthase kinase 3 GTP Guanine triphosphate GTPase Guanosine triphosphatase HeLa Cell line taken from Henrietta Lacks alias Helen Lane ICMT Isoprenylcysteine carboxyl methyltransferase

ABBREVIATIONS
10
IgA Immunoglobulin A IgG Immunoglobulin G KSR Kinase suppressor of Ras LPA Lysophosphatidic acid LPS Lipopolysaccharide MAPK Mitogen-activated protein kinase Mcl-1 Myeloid cell leukaemia-1 MDCK Madin-Darby canine kidney cells MEK Mitogen-activated protein kinase/ Erk kinase MLD Membrane localization domain MP1 MEK partner 1 NAD Nicotinamide adenine dinucleotide NES Nuclear export signal NF-kB Nuclear factor-kappa Beta NGF Nerve growth factor PAK p21-associated kinase PDB Protein data bank PDGF Platelet-derived growth factor PDK PI3-K dependent Kinase PH Pleckstrin homology PI3-K Phosphoinositide 3-kinase PIP2 Phosphatidyl inositol 4,5-bisphosphate PIP3 Phosphatidyl inositol 3,4,5-triphosphate PKA Protein kinase A PKB Protein kinase B PKC Protein kinase C PTEN Phosphatase and tensin homologue deleted on chromosome ten PUMA p53 upregulated modulator of apoptosis Raf Rapidly growing fibrosarcomas RalGDS Ral GDP dissociation stimulator Rap Ras-related protein Ras Rat sarcoma Rce1 Ras converting enzyme I Rho Rhodopsin RKIP Raf kinase inhibitor protein RTK Receptor tyrosine kinase S Serine SH2 Src –homology 2 Shc Src homology 2 domain containing SOS Son-of-sevenless T Threonine Tfp Type IV pili TLR4 Toll-like receptor 4 TNF Tumor necrosis factor TPR Tetratricopeptide repeat TRAIL TNF-related apoptosis inducing ligand TTSS Type III secretion system Y Tyrosine 14-3-3 Protein eluted fraction 14 and migration position 3.3 on starch gel
electrophoresis

INTRODUCTION
11
Figure 1. Pseudomonas aeruginosa
4. INTRODUCTION
4.1 Pseudomonas aeruginosa
Pseudomonas aeruginosa is a Gram-negative, aerobic, rod-shaped bacterium (Figure 1: Yahoo image) with unipolar motility and is an opportunistic human and plants pathogen. In plants, P. aeruginosa induces symptoms of soft rot in Arabidopsis thaliana (Thale cress) and Letuca sativa (Lettuce). The associations of virulence factors are the same for plant and animal infections [1-3]. P. aeruginosa survives in almost any environment including water, soil, and plants and is known for its nutritional and ecological flexibility [4]. In humans, P. aeruginosa is one of the leading causes of nosocomical infections particularly in the settings of epithelial cell injury and immuno-compromised patients such as those with severe burns, injuries, leukaemia, acquired immuno-deficiency syndrome (AIDS) and cystic fibrosis (CF) [5] and has become one of the leading causes of hospital-acquired infections. P. aeruginosa is naturally resistant to a large range of antibiotics. The inducible AmpC chromosomal β-lactamase contributes to the resistance against ampicillin and most cephalosporins [6]. The P. aeruginosa outer membrane has low permeability and the presence of the several multidrug efflux systems contribute to its intrinsic resistance or reduced vulnerability to several antimicrobial agents. Due to the various resistance mechanisms of P. aeruginosa, the selection of effective antimicrobial agents is relatively limited making sustained infections even more difficult to treat [7, 8].
P. aeruginosa can be easily identified by a number of phenotypic markers such as production of pigments including pyocyanin (the blue pigment is used as diagnostic marker) and pyoverdin (the fluorescent pigment most often produced), denitrification ability, and growth at 41°C [9-11]. P. aeruginosa may also be able to produce several additional pigments, including the reddish pigment pyorubrin (production enhanced by the addition of glutamate to the medium) and a brown pigment, pyomelanin [12]. Colonies of P. aeruginosa are grayish, flat with irregular edges and tend to spread on the agar surface with time. Mucoid colonies frequently appear among isolates from the respiratory tract of CF patients showing the mucoid extracellular substance alginate [13].
The complete genome sequence of P. aeruginosa (published in 2000) is 6.3 Mb with 5570 predicted open reading frames and a high (G+C) content of

INTRODUCTION
12
66.6% [14]. The P. aeruginosa genome contains an extremely high number of regulatory genes that are involved in catabolism, transport, efflux of organic materials, distinct secretion, and motility. This reflects the ability of P. aeruginosa to adapt and survive in many different environments and to resist antimicrobial agents.
4.1.1. Severity of diseases
P. aeruginosa causes serious infections including bacteremia, septicemia, pneumonia, endocarditis, otitis, keratitisis, osteomyelitis, osteochondritis, meningitis, and brain abscesses in immuno-compromised patients [5]. P. aeruginosa is responsible for 16% of nosocomial pneumonia cases, 12% of hospital-acquired urinary tract infections, 8% of surgical wound infection, and 10% of bloodstream infections. In addition, neutropenic cancer and bone marrow transplant patients are particularly susceptible to opportunistic infections such as pneumonia and septicemia caused by P. aeruginosa with attributable deaths reaching 30% [5]. P. aeruginosa is also one of the most common and lethal pathogens responsible for ventilator-associated pneumonia in intubated patients with directly attributable death rates reaching 38%. P. aeruginosa infections in patients with burn injuries are associated with 60% death rates due to local deficiency of antibody medicated immunity and a decreased level of complement factors in the burn wounds [15]. In addition, P. aeruginosa bacteremia is associated with 50% of deaths in the expanding AIDS population. CF patients are characteristically susceptible to chronic infection (with approximately 70-80% incidence in the lungs) [16], leading to high rates of illness and death in this population [5, 17]. The illness causes include persistence of bacterial infection, lung damage, and gastrointestinal abnormalities leading to malabsorption and nutritional deficits [17-19]. P. aeruginosa causes severe eye infections in both healthy individuals and immuno-compromised patients such as ulcerative keratitis, a severe inflammatory infection in the cornea [4]. P. aeruginosa is also often found to be the dominant bacteria in poly microbial infections. Infection and inflammation such as microbial keratitis in contact lens wear is often associated with microbial contamination and colonization of lenses caused by several different types of microbes including P. aeruginosa [20]. All these reports demonstrate the importance of basic and functional research of P. aeruginosa for the future healthcare.
4.1.2. Virulence factors of Pseudomonas aeruginosa
To initiate infection, P. aeruginosa usually requires a substantial break in first-line defenses. Such a break can result from: (i) operation or bypass of normal cutaneous or mucosal barriers due to trauma, surgery, serious burns, or

INTRODUCTION
13
indwelling devices; (ii) disruption of the protective balance of normal mucosal flora by broad-spectrum antibiotics; or (iii) alteration of the immunologic defense mechanisms such as in chemotherapy-induced neutropenia, mucosal clearance defects from cystic fibrosis, AIDS, and diabetes mellitus. The capacity of P. aeruginosa to produce such diverse, often overwhelming infections is due to an arsenal of virulence factors [5, 21]. Many extracellular virulence factors secreted by P. aeruginosa have been shown to be controlled by a complex regulatory circuit involving cell-to-cell signaling systems that allow the bacteria to produce these factors in a coordinated, cell-density-dependent manner [22]. The most aggressive virulence factors are ADP-ribosyltransferases, elastase, lipases, phopholipase C, alkaline protease and neuraminidase expressed during acute infections [23]. Adherence to host cells/tissues is an essential step in establishing infection and this step is mediated by major adhesins such as pili, flagella, lipopolysaccharide (LPS), and a variety of other fimbrial-related molecules [24-26].
LPS, an important virulence factor for both innate and acquired host responses to infection. LPS demonstrates a fair degree of heterogeneity in its lipid A- and O-antigen structure and has two different outer-core glycoforms (one of which is ligated to the O-antigen). The lipid A can be penta-, hexa-, or hepta-acylated and these isoforms have varying potencies for host innate immunity which is activated by binding to Toll-like receptor 4 (TLR4). LPS causes inflammation and bacterial dissemination [27].
Proteases, including LasB elastase, LasA elastase and alkaline protease are assumed to play a major role during acute P. aeruginosa infection [5]. P. aeruginosa destroys the protein elastin during acute infection [28] by the action of LasB and LasA elastases that are believed to destroy elastin-containing human lung tissue and thus cause the pulmonary hemorrhages [29]. LasA is a serine protease that acts synergistically with LasB to degrade elastin. LasB is a zinc metalloprotease that acts on a number of proteins including elastin, fibrin, and collagen and inhibits human IgG , IgA, airway lysozyme, and complement components leading to tissue damage through interference with the host defense mechanism [30, 31].
Exotoxin A (ETA) is a type II secreted, 66-kDa protein toxin. Like diphtheria toxin, ETA inhibits protein synthesis in eukaryotes by irreversibly transferring the ADP-ribosyl moiety of NAD+ into elongation factor 2, leading to cell death [32, 33]. ETA is chromosomally encoded by a single copy gene that is present in 95% of P. aeruginosa strains [34] and plays a role in local tissue damage, bacterial invasion, and immuno-suppression [35, 36].

INTRODUCTION
14
Hemolysin, including phospholipase C and rhamnolipid that can act synergistically to break down lipids and lecithin and contribute to cytotoxic effects during tissue invasion [37]. The rhamnose-containing glycolipid biosurfactant rhamnolipid has a detergent like structure that is believed to solubilize the phospholipids of lung surfactant, causing the lung surfactant to be more accessible to cleavage by phospholipase C [38]. The resulting loss of lung surfactant may be responsible for the atelectasis associated with chronic and acute P. aeruginosa lung infection. Rhamnolipid also inhibits the mucociliary transport and ciliary function of human respiratory epithelium [39].
Biofilms protect bacteria from extracellular stress including antibacterial agents, host phagocytic clearance, protease defense and oxygen radicals [40]. One hypothesis for the antibiotic resistance of P. aeruginosa in the CF lung is that this bacterium exists as a biofilm that renders the cells resistant to antimicrobial treatment. Alginate slime forms the matrix of the P. aeruginosa biofilm where bacteria attach to the host surface and to each other [41].
Type IV pili (Tfp), proteinaceous retractable hair-like surface structures found in P. aeruginosa that are defined by their shared structural, biochemical, and morphological features and have a highly conserved biogenesis pathway [42] . Tfp plays a central role in motility, multicellular behavior, natural genetic transformation, and adherence. Bacterial attachment is a critical first step in both tissue colonization and biofilm formation [43]. The P. aeruginosa surface Tfp is important in biofilm development via their role in bacterial 'twitching' motility [44].
Type III secretion system (TTSS) is one of the virulence regulators of P. aeruginosa that likely plays a role to avoid innate immune clearance in mammals [45, 46]. Upon cellular contact, the P. aeruginosa TTSS is capable of delivering a combination of at least four different effector proteins, named exoenzyme S (ExoS), ExoT, ExoU, and ExoY [47, 48]. The translocated proteins ExoS and ExoU are cytotoxic to cells during infection and transfection [49]. Host cell contact or low environmental Ca2+ causes P. aeruginosa to secrete TTSS effectors either by direct injection into the host cell cytoplasm or secretion to the external environment, respectively [50].
ExoY, an adenylate cyclase activated by an unknown host cell factor that mediates rounding of cultured mammalian cells without causing loss of viability [51]. ExoY promotes cyclic AMP accumulation in vascular endothelial cells and increases vascular permeability [52]. Although the majority of P. aeruginosa isolates contain the exoY gene, little else is known about the function(s) of this effector [53]. Based on its previously described effects, it was hypothesized that ExoY would cause disruption of the epithelial cell cytoskeleton, probably

INTRODUCTION
15
leading to significant changes in actin distribution and cytoskeleton integrity [50].
ExoU, a member of the phospholipase A family of enzymes possessing phospholipase A2 activity [54]. ExoU requires either a eukaryotic specific modification or a cofactor for its activity in vitro and recently superoxide dismutase (SOD) is proposed as a cofactor for ExoU in vitro [55]. The biological effects of minimal expression of ExoU in yeast can be visualized by membrane damage to different organelles and fragmentation of the vacuole [54, 56]. In mammalian cells, the direct injection of ExoU causes irreversible damage to cellular membranes and rapid necrotic death. ExoU likely represents a unique enzyme and is the first identified phopholipase virulence factor that is translocated into the cytosol by TTSS [54]. ExoU is the major virulence factor in mice with acute pneumonia [57]. A recent study has observed that ExoU is more common in the blood from infected patients [58].
ExoT shares 76% sequence homology with ExoS [59]. Like ExoS, ExoT encodes an N terminal GTPase-activating protein (GAP) domain targeting the small GTPases Rho, Rac, and Cdc42 and a C terminal ADP-ribosyltransferase (ADPRT) domain that interacts with Crk family proteins and phosphoglycerate kinase [60-62]. ExoT only possesses 0.2-1% of 14-3-3 dependent ADP-ribosyltransferase activity in vitro and the active catalytic site E385 is homologous to E381 of ExoS [49, 59]. In contrast, the substrate specificity of the ExoT-C terminal ADPRT domain is distinct from ExoS [63]. ExoT is thought to be particularly important in the pathogenesis of P. aeruginosa infections since all strains encode this type III effector and isogenic mutants lacking ExoT showed reduced virulence in several disease models [57, 64]. Recently, the host ubiquitin ligase Cbl-b has been proposed as an interaction partner of ExoT [65].
4.1.3. Pseudomonas aeruginosa ExoS
ExoS was first described as a secreted ADP-ribosyltransferase isolated from P. aeruginosa strain 388 that possesses properties distinct from Exotoxin A [66]. ExoS ADP-ribosylated several proteins in crude extracts of wheat germ or rabbit reticulocytes without modifying elongation factor 2 which is ADP ribosylated by Exotoxin A and diphtheria toxin [66]. In addition, the ADP-ribosyltransferase activity of ExoS was not neutralized by α-Exotoxin A antibody and relative to Exotoxin A, ExoS is heat stable. Together these properties indicated ExoS to be different from the previously described Exotoxin A [66, 67].
ExoS was originally purified from the culture supernatant of P. aeruginosa strain 388 as a protein aggregates consisting of two ADP-ribosyltransferase proteins with molecular weight of 53-kDa and 49-kDa [68].

INTRODUCTION
16
Both the 53- and 49-kDa proteins had similar N-terminal amino acid sequences, immunoreactivity, and peptides upon proteolytic cleavage [69, 70] are encoded by two distinct genes exoT and exoS, respectively [71].
ExoS toxin is an important virulent factor in a mouse model of acute pneumonia and a significant contributor of increased mortality, decreased bacterial clearance from the lung, and more frequent bacterial dissemination to peripheral organs [72]. A recent study showed that secreted ExoS facilitates invasion and internalization and is associated with chronic infection in CF patients [73].
4.1.3.1. ExoS secretion and translocation domain
ExoS has a membrane localization domain (MLD) at residues 51–77, necessary for trafficking of ExoS within host cells [74]. A leucine-rich motif within the MLD contributes to intracellular localization of type III-delivered ExoS to the plasma membrane and subsequently to the perinuclear region of cultured cells [75]. Other type III cytotoxins also possess a leucine-rich motif within their respective MLDs, including ExoT, Yersinia pseudotuberculosis outer protein E (YopE), and Salmonella typhimurium type III-secreted protein (SptP) [61, 76]. It has been proposed that the presence of the MLD allows type III-delivered ExoS to ADP-ribosylate specific subsets of host proteins, including the Ras GTPases [74]. However, it has also been proposed that 14-3-3 proteins translocate ExoS toxins to the membrane [77, 78].
4.1.3.2. ExoS-GAP domain
The GAP domain (residues 96–219) of ExoS stimulates the hydrolysis of guanine triphosphate (GTP) on GTP-bound Rho GTPases and thus inactivate RhoA, Rac1, and Cdc42 both in vivo and in vitro [76, 79]. The GAP activity of ExoS abrogates the activation of RhoA, Cdc42, and Rap1 [80]. Arg146 is a catalytic residue of Rho GAP activity and functions as an arginine finger in stabilizing the transition state of the Rac-GTPase reaction [76]. Consistent with the functions of Rho GTPases as regulators of the actin cytoskeleton, the N terminus GAP of ExoS induces actin rearrangement in cultured cells. Intracellular expression of the GAP domain of ExoS causes rounding of Chinese Hamster Ovary (CHO) cells by disrupting the actin cytoskeleton [81]. The GAP domain activity is inhibited by the ADP-ribosyltransferase domain along with NAD and factor activating ExoS (FAS). In addition, auto-ADP-ribosylation of the Rho-GAP domain reduces its activity by about one order of magnitude [82]. Crystallographic studies revealed that the GAP domain of ExoS comprises seven α helices linked by two loop regions. The Rho GAP domain of ExoS stabilizes the transition state of the GTPase reaction indicating that RhoGAP is the

INTRODUCTION
17
Figure 2. ADP-ribosylation of a target host protein
Protein
ADP ribosyl - protein
Nicotinamide
Protein
Nicotinamide
ADP-ribose
NAD
Target protein i.e Ras
functional mimic of eukaryotic Rho GAPs, even though their structures in the protein data bank (PDB) are different [83, 84].
4.1.3.3. ExoS-ADP-ribosyltransferase domain
The C-terminal ADP-ribosyltransferase domain (residues 234–453) of ExoS is involved in P. aeruginosa virulence [85]. The 14-3-3-dependent ADP-ribosyltransferase domain of ExoS is multi-substrate specific and transfers ADP-ribose from NAD+ onto several substrates (Figure 2), including the small monomeric GTPases such as Ras superfamily, vimentin, and the anti-stress protein Hsp27 [80, 86]. ADP-ribosylation of Ras at Arg41 and Arg128 interferes with its ability to be reactivated in vitro by guanine nucleotide exchange factor (GEF) and it was further shown that ExoS is able to inhibit nerve growth factor (NGF) stimulated neurite formation in PC-12 cells [87]. ExoS also ADP-ribosylates Rap at position Arg41 that inhibits RapGEF to stimulate nucleotide exchange [88]. In addition, ezrin, radixin, and moesin (ERM) are also identified as high-affinity substrates for ExoS ADP-ribosyltransferase [60]. Recently it has been shown that ExoS-ADP-ribosylates and affects the function of the cytosolic protein cyclophilin A (CpA) [89].
The ADP-ribosyltransferase activity of ExoS has been shown to be dependent on the eukaryotic co-factor FAS, which has been identified as a member of the 14-3-3 family [90, 91]. Mutation in the C-terminal region of ExoS (which is responsible for its interaction with 14-3-3) reduces the ability of ExoS to ADP-ribosylate cytoplasmic proteins, both in vitro and in cell lines [78, 80]. Glu381 is a catalytic residue for the ADP-ribosyltransferase activity and a point mutation at glutamic acid 381 reduces the enzymatic activity by approximately 2000-fold [92]. Subsequent studies have identified Glu379 which is required for efficient ADP-ribosyltransferase activity [93]. Expression of ExoS in HeLa cells results in cell rounding due to expression of ADP-

INTRODUCTION
18
Secr/Transl GAP
R146 E3811 453
ADP-ribosyl transferase
Figure 3. Schematic overview of ExoS
ribosylation activity and has also been shown to be independent of the expression of Rho GAP activity [60]. In vitro, ExoS is auto-ADP-ribosylated, resulting in a reduction of GAP activity of ExoS and auto-ADP-ribosylation of ExoS in CHO cells, suggesting an intra-molecular regulation for ExoS function in intact cells [82].
4.1.3.4. ExoS-ADP-ribosyltransferase structure
There are no available crystal structures of ExoS-ADP-ribosylation domains in the PDB. However, Jianjun Sun and Joseph Barbier have compared different ADP-ribosylation proteins with ExoS [94] (Figure 3). Using protein modeling
based analysis of known structures of other bacterial ADP-ribosylation toxins they suggested that ExoS regions B (active site loop), C (ARTT motif) and E (PN loop) are necessary and sufficient to recognize target proteins. ExoS ADP-riboylation domains, similar to other ADP-ribosylating toxins, share an identical three-dimensional structural topology and a conserved NAD-binding pocket. Different electrostatic potentials in the enzyme-substrate interface determines the substrate specificity and the ExoS enzyme-substrate interface is a mixture of basic, hydrophobic and acidic clusters. ExoS region A is rich in basic charged residues (Lys243, Arg244, Lys251, and Arg255), region C contains hydrophobic (Tyr376) and basic residues (Lys377, Asn378, and Lys380), and region B contains His276. Region E is conserved between ExoS and ExoT. In addition, different electrostatic potentials within regions A, B, C and E may play a role in the ability of the members of bacterial ADP-ribosylating toxins to target specific host proteins to express their unique pathology [94].
4.2 Small GTPase
The small GTPase superfamily comprises monomeric GTP-binding proteins with molecular masses usually in the range of 20-25 kDa, are found in all eukaryotic organisms, ranging from yeast to human and consisting of more than 100 members. Members of the superfamily share approximately 30% amino acid sequence identity, mostly derived from four highly conserved domains required for the recognition of guanine diphosphate (GDP), GTP, and for

INTRODUCTION
19
Figure 4. Regulation of Small GTP binding proteins
Small GTPase GTP
Small GTPase GDP
”OFF”
”ON”
GAPGEF
Intrinsic hydrolysis
Under basal condition
Upon stimulation
Effector proteins
Small GTPase GTP
Small GTPase GDP
”OFF”
”ON”
GAPGEF
Intrinsic hydrolysis
Under basal condition
Upon stimulation
Effector proteins
GTPase activity. The small GTPase superfamily is synonymous with the Ras superfamily [95].
Small GTPases exist as monomers, which function as molecular switches in intracellular signaling to control a wide variety of cellular functions [96]. In general, the GDP-bound form is the inactive configuration of the molecular switch while the GTP-bound form is active. The switch is activated by the exchange of GDP for GTP and catalyzed by GEFs in response to a variety of upstream signals. Activated GTPases interact with one or more effector proteins, leading to activation of downstream signaling pathways. Otherwise, GAPs stimulate the intrinsic GTPase activity that results in changing the conformation back to the inactive GDP-bound form (Figure 4). The process of activation and inactivation is transient and repeated as long as the upstream stimulatory signal is present [96, 97]. Most of the small GTPase superfamily members are structurally classified into 5 distinct subfamilies: Ras, Rho/Rac, Rab, Arf/Sar1, and Ran [95, 98].
4.2.1. Ras small GTPases
The members of the Ras family (H-, K-Ras4A/4B, and N-Ras) are critical regulators of cell proliferation and differentiation [99]. Ras proteins function as a GDP/GTP-regulated switch, which is regulated by guanine nucleotide exchange factors such as RasGEFs [e.g. son of sevenless (SOS)] that promote the formation of active Ras-GTP, whereas GAPs; [e.g. neurofibromin (NF1)] stimulate GTP hydrolysis and formation of inactive Ras-GDP. In normal quiescent cells, Ras is GDP-bound and inactive. Extracellular stimuli such as epidermal growth factor (EGF) cause transient formation of the active, GTP-bound form of Ras [100].
Ras is mutationally activated in 30% of all cancers, with pancreas (90%), colon (50%), thyroid (50%), lung (30%), and melanoma (25%) having the highest prevalence. K-Ras is the more frequently mutated Ras isoform in human cancer [101]. Mutant Ras proteins are GAP-insensitive, rendering the proteins

INTRODUCTION
20
constitutively GTP-bound and activated, leading to stimulus-independent, persistent activation of downstream effectors and in particular, the Raf-MEK-ERK cascade [102].
For proper signaling, Ras proteins localize to the inner surface of the plasma membrane where they interact with upstream activators receptor tyrosine kinase complexes and downstream effectors. Ras proteins are synthesized as inactive, cytosolic proteins that quickly undergo a series of post-translational modifications that target Ras to the plasma membrane [103, 104]. This series of modifications is initiated by the enzymes cytosolic farnesyltransferase (FTase), which recognizes the Ras carboxyl-terminal CAAX motif (where a cysteine is followed by two aliphatic residues and one random amino acid) causing the covalent addition of a farnesyl isoprenoid lipid to the cysteine residue of the CAAX sequence, followed by proteolysis of the AAX peptide with the Ras converting enzyme I (Rce1). Finally, isoprenylcysteine carboxyl methyltransferase (ICMT) covalently attaches a methyl group to the farnesylated cysteine residue [105, 106]. Both Rce1 and ICMT are associated with the endoplasmic reticulum. K-Ras and N-Ras undergo alternative prenylation by a related enzyme geranylgeranyltransferase I (GGTaseI) and FTase activity is inhibited by FTase inhibitors (FTIs). Therefore, each modification is important to increase Ras membrane affinity and if the initial farnesylation step is inhibited, all subsequent steps will be inhibited [107, 108].
Ras protein controls a classical membrane localized signaling pathway known as the RAS-MEK-ERK kinase cascade that regulates proliferation and differentiation [102]. Recent studies using electron microscopy and single flurophore video tracking analysis have shown that Ras activation triggers the formation of discrete signaling nanoclusters that are critically important for Ras function. Removal of critical scaffold proteins such as Sur-8, galectin-1, or galectin-3 block the formation of Ras nanoclusters and disrupt Ras-dependent MAPK activation [109].
4.2.2. Rap small GTPases
Rap proteins (Rap1 and Rap2 in mammals) are members of the Ras family and are important regulators of fundamental cellular functions such as adhesion, secretion, and vesicle trafficking [110]. Rap1A was described in 1989 as an antagonist of Ras (Krev-1) and it was shown that over-expression of Rap1a can reverse the transformed phenotype of cells harboring an oncogenic K-Ras mutation [111]. Five isoforms of Rap protein have been identified to date: Rap1A, Rap1B, Rap2A, Rap2B, and Rap2c [112]. Like Ras, Rap1 is directed to the plasma and the endosomal membranes, via the C-terminal CAAX motif that provides sites for lipid modification [113]. Rap1 is ubiquitously expressed and is specially prevalent in hematopoietic cells [114, 115]. In its GTP-bound active

INTRODUCTION
21
form, Rap1 interacts with various effector molecules to initiate downstream signaling pathways. Initial studies showed that Rap1 interferes with Ras signaling by directly interacting with Ras effectors, but more recent results indicate that Rap1 functions in independent signaling pathways controlling diverse processes, such as cell adhesion, cell-cell junction formation and cell polarity [116]. The Rap1 small GTPase has been implicated in regulation of integrin-mediated leukocyte adhesion downstream of various chemokines and cytokines in many aspects of inflammatory and immune responses [117, 118].
It is now evident that over 30 GEFs are responsible for activating various Ras family proteins and that many of these activate Rap1 and Rap2 [119]. These GEFs share a catalytic domain consisting of a CDC25 homology domain and a Ras exchange motif. The diversity of additional regulatory domains results in many extracellular-generated signals converging on Rap1 activators. The activation of Rap1 in response to various upstream signals is mediated by GEFs such as C3G (the first identified Rap1 GEF), which associates with the adaptor protein Crk, and forms a complex with receptor and non-receptor protein tyrosine kinases in response to extracellular stimuli [120]. cAMP-dependent Rap1 activation is regulated by Epac1 (known as cAMP-GEFI) and Epac2 (known as cAMP-GEFII) that are activated by direct binding of cAMP [121].
4.3. Signaling pathways affected by ExoS
Ras is an in vivo target for ExoS-ADP-ribosyltransferase activity. ExoS modifies members of the Ras super family such as H-Ras, N-Ras, K-Ras, R-Ras, Rap1, Rap2, RalA, Rac1, Rab5, Rab8, Rab11, and cdc42 in vivo [80, 87]. This result is supported by the findings of Fraylick et al, who identified ADP-ribosylated target molecules using 2-dimentional electrophoresis [122, 123]. To understand the effect of ExoS, we have focused on the Ras mediated signaling by which we could search the putative in vivo targets of ExoS. Furthermore, it has been proposed that ExoS-ADP-ribosylation activity induces caspase-3 acitvity in infected cells [124]
4.3.1. Ras activation and downstream signaling
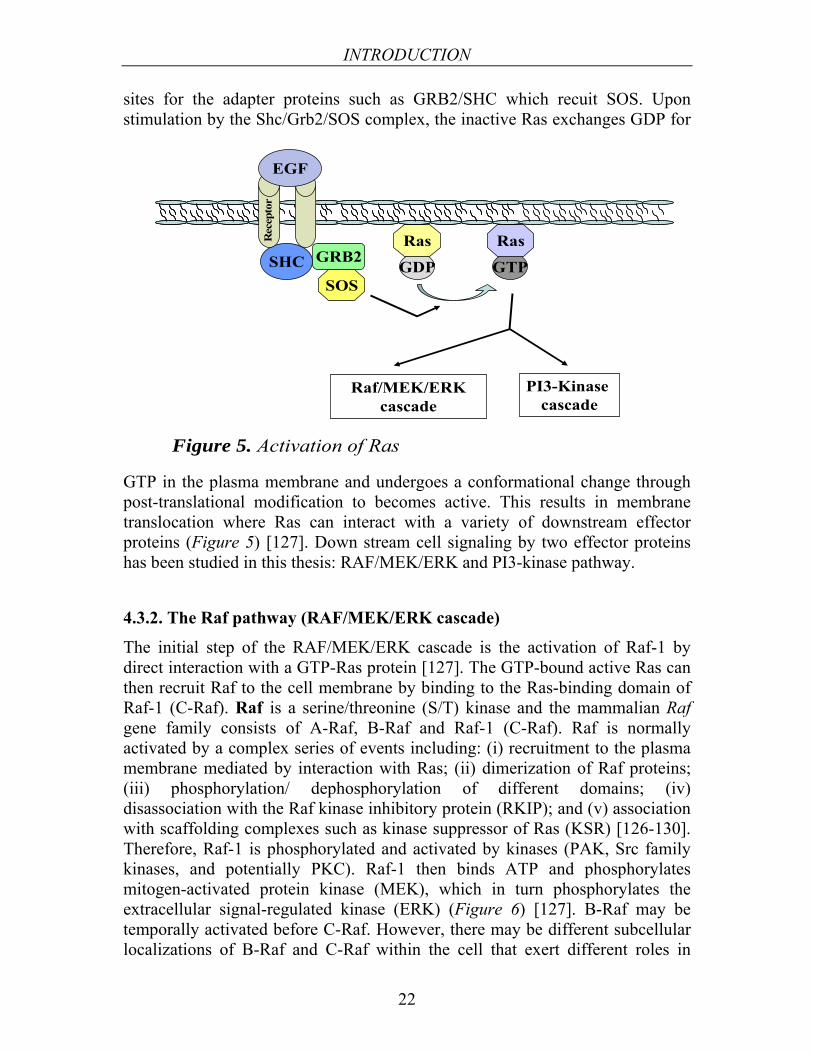
Ras is the common upstream molecule of several signaling pathways including Raf/MEK/ERK, PI3-K/Akt, and Ral-EGF signaling cascades [125]. Ras proteins show varying abilities to activate the Raf/MEK/ERK and PI3-K/Akt cascades. For example, K-Ras has been associated with the Raf/MEK/ERK pathway while H-Ras is associated with PI3-K/Akt activation [126]. Following binding and activation of EGF, growth factors or mitogens to their appropriate receptors, then activation of the coupling complex SHC/GRB2/SOS occurs in the cytosol. Activation occurs by phosphorylation of tyrosine residues that serve as docking
INTRODUCTION
22
SHC GRB2
SOSGDP GTP
Raf/MEK/ERKcascade
PI3-Kinase cascade
Ras Ras
Figure 5. Activation of Ras
EGF
Rec
epto
r
sites for the adapter proteins such as GRB2/SHC which recuit SOS. Upon stimulation by the Shc/Grb2/SOS complex, the inactive Ras exchanges GDP for
GTP in the plasma membrane and undergoes a conformational change through post-translational modification to becomes active. This results in membrane translocation where Ras can interact with a variety of downstream effector proteins (Figure 5) [127]. Down stream cell signaling by two effector proteins has been studied in this thesis: RAF/MEK/ERK and PI3-kinase pathway.
4.3.2. The Raf pathway (RAF/MEK/ERK cascade)
The initial step of the RAF/MEK/ERK cascade is the activation of Raf-1 by direct interaction with a GTP-Ras protein [127]. The GTP-bound active Ras can then recruit Raf to the cell membrane by binding to the Ras-binding domain of Raf-1 (C-Raf). Raf is a serine/threonine (S/T) kinase and the mammalian Raf gene family consists of A-Raf, B-Raf and Raf-1 (C-Raf). Raf is normally activated by a complex series of events including: (i) recruitment to the plasma membrane mediated by interaction with Ras; (ii) dimerization of Raf proteins; (iii) phosphorylation/ dephosphorylation of different domains; (iv) disassociation with the Raf kinase inhibitory protein (RKIP); and (v) association with scaffolding complexes such as kinase suppressor of Ras (KSR) [126-130]. Therefore, Raf-1 is phosphorylated and activated by kinases (PAK, Src family kinases, and potentially PKC). Raf-1 then binds ATP and phosphorylates mitogen-activated protein kinase (MEK), which in turn phosphorylates the extracellular signal-regulated kinase (ERK) (Figure 6) [127]. B-Raf may be temporally activated before C-Raf. However, there may be different subcellular localizations of B-Raf and C-Raf within the cell that exert different roles in

INTRODUCTION
23
Gene transcription
Proliferation /survival
Raf-1
MEK
ERK
GTP
Ras
Figure 6. The Raf/MEK/ERK cascade
P
P
P
Nucleus
signaling and apoptotic pathways [131]. Raf-1 is inactivated by protein dephosphorylation and binds to 14-3-3, resulting in a conformational change and translocation to the cytoplasm as an inactive form. Raf-1 also associates with RKIP leading to its inactivation. The phosphorylation/dephosphorylation events alter the configuration of Raf-1 and can result in the disassociation of the 14-3-3 protein, which unlocks the Raf-1 protein allowing it to be phosphorylated by other kinases [127].
MEK1/MEK2 are dual specificity protein kinases that target tyrosine (Y) and serine/threonine residues for phosphorylation. All three Raf family members are able to phosphorylate and activate MEK with different biochemical potencies such as B-Raf > C-Raf >>A-Raf [132]. The predominant downstream target of MEK1/MEK2 is ERK, which is an S/T kinase existing in two isoforms ERK1 and ERK 2. The activities of ERK1 and ERK2 are positively regulated by phosphorylation of both T202 and T204 in the activation loop and is mediated by MEK1 and MEK2 [133]. ERKs can directly phosphorylate many transcription factors including Ets-1, c-Jun, c-Myc, S6 kinase (p90Rsk), and NF-kB [134]. ERK2 has been positively associated with proliferation while ERK1 may inhibit the effects of ERK2 in certain cells [135]. MEK and ERK also interact with RKIP, which prevents interaction with Raf-1.

INTRODUCTION
24
Figure 7. Activation of Rap1
Rap-1
GTPRas
GTP
Hormonal activation
cAMP
Epac2
Rap-1GTP
C-Raf
B-Raf
Rac
RapL
AdhesionCellular migration
Cell differentiation
4.3.3. Rap1 signaling cascade
Rap1 is activated by distinct types of Rap1-GEFs coupled with various receptors or secondary messengers in response to diverse extra-cellular stimuli in many cell types. Activated Rap1 is down-regulated by Rap1 GAP proteins, through which Rap1 activation is controlled spatio-temporally. RapGEFs including C3G associates with the family of Crk adaptor proteins and are recruited to the perinuclear membrane where C3G is activated via phosphorylation that in turn activates Rap1 by receptor tyrosine kinase activation [136, 137]. Epac family proteins such as Epac-1 and Epac-2 (GEFs for the Rap1 and Rap2 ) are capable of binding c-AMP that results in the activation of their GEF activity via a
conformational change that leads to Rap1 activation [138]. Under the appropriate conditions, Rap1 is also a downstream target of Ras through the ability of Ras-GTP to recruit Epac2 to the plasma membrane and activate a specific pool of Rap1 [138]. Rap1-GTP specifically interacts with various effectors (including Rac, Ral, and Raf) in order to exert its biological functions (Figure 7) [116].
4.3.4. The PI3-Kinase pathway
The class IA subset of PI3-Kinases are heterodimers made up of a 110 kDa catalytic subunit (p110α, p110β, and p110δ) in a complex with one of five regulatory subunits (collectively called the ‘p85s’) [139]. p110α and p110β are found in most tissues whereas the expression of p110δ is most abundant in leukocytes [140].

INTRODUCTION
25
Survival
Figure 8. ThePI3-kinase cascade
GTP
Ras
PI3-kinase
PKB/AKT
14-3-3
PIP2PIP3
PDK1
P
P
PCytoplasm
P
Prevention of apoptotic gene transcription
Nucleus
Foxo3
Activation of the PI3-Kinase pathway can occur in response to a variety of extracellular signals through either growth factor activation or integrin receptor pathways [127]. After ligand-induced activation of specific receptors, PI3-Kinase can be activated by two mechanisms. First, a phosphorylated tyrosine residue on the receptor serves as a docking site for the p85 regulatory subunit of PI3-Kinase via its Src-homology 2 (SH2) domains and thus, recruits the p110 catalytic subunit of PI3-Kinase to this complex resulting in translocation to the membrane [141]. Alternatively, upon activation of the cytokine receptor by the appropriate ligand, the Shc protein binds to the receptor to enable the Grb-2 and SOS proteins to form a complex, which results in the activation of Ras. Ras is then able to induce the membrane translocation and activation of the p110 subunit of PI3-Kinase [133]. Activated PI3-Kinase phosphorylates phosphatidylinositol-4,5- phosphate (PIP2) at the 3' position of the inositol ring and generates phosphatidylinositol-3,4,5-phosphate (PIP3), which acts as a secondary messenger by facilitating binding sites for protein with pleckstrin homology (PH) domain. Thus, PIP3 results in localization of the phospholipid-binding domain containing proteins PDK1 via its PH domain in the membrane. Protein kinase B (Akt/ PKB) is also recruited to the lipid-rich

INTRODUCTION
26
plasma membrane by its PH domain and is phosphorylated at residues T308 in the activation loop and S473 in the C-terminal regulatory region by PDK1 and PDK2, respectively resulting in full activation of Akt/ PKB [142]. The Akt/PKB family of serine-threonine kinases consists of three members: Akt1/PKBα, Akt2/PKBβ, and Akt3/PKBγ. The Akt family has been shown to be the primary downstream mediator of PI3-Kinase and regulates a variety of cellular processes through the phosphorylation of a wide spectrum of downstream substrates [141]. Upon activation, PKB/Akt moves to the cytoplasm and nucleus, where it phosphorylates a plethora of downstream targets (Figure 8).
In addition, the activity of PI3-Kinase is negatively regulated by lipid phosphatases (including SHIP-1/2 and PTEN) that removes the 3' phosphate of PIP3 and thus, regenerate PIP2 and attenuates downstream signaling of activated PI3-Kinase [143]. The coordinated regulation of PIP3 levels in the cell provides a means of intricately controlling downstream signaling activity that is essential to maintain the fine balance required for controlled cell growth. Further study showed that activation of the c-Kit receptor in HSC-like cell lines lead to a PI3-Kinase-dependent Raf/MEK/ERK activation and PI3-Kinase inhibitors block phosphorylation of PKB, Raf-1 and Erk without affecting the activation of Ras and upstream signaling molecules such as Shc and Grb2 [144]. Ras proteins signal through direct interaction with PI3-Kinases and binding of Ras to PI-3 kinase p110α is required for ras-driven tumorigenesis in mice [145].
4.3.5. Regulation of apoptosis by RAF/MEK/ERK and PI3-KINASE cascades
Apoptosis is a distinctive form of cell death exhibiting specific morphological and biochemical characteristics including cell membrane blebbing, chromatin condensation, genomic DNA fragmentation, and exposure of specific phagocytosis signaling molecules on the cell surface [146]. Initial understanding of the molecular mechanisms that control apoptosis come from early studies in the nematode Caenorhabditis elegans and then further studies to extend the mechanisms to mammalian systems [146, 147].
A multitude of proteins and enzymes are involved in the initiation, amplification, or suppression of apoptosis. In most cells, apoptosis triggers usually lead to the activation of caspases, which ultimately mediate cell death. The Raf/MEK/ERK and PI3-Kinase pathway may also induce the phosphorylation of apoptototic regulator molecules including Bcl-2, Bad, Bim, Mcl-1, Caspase 9, and Foxo3A involved in the regulation of apoptosis [148, 149]. In conclusion, phosphorylation of these molecules is involved in regulating apoptosis by either stimulating or inhibiting transcriptional activity

INTRODUCTION
27
[133, 150]. The following molecules play a main role in the regulation of apoptosis;
AKT/PKB can be regarded as a cellular master switch to integrate incoming signals on cell growth and apoptosis. For example, activation of Akt results in a direct phosphorylation and consecutive inhibition of the pro-apoptotic factor Bad [151]. The anti-apoptotic effects of Akt may also be mediated via activation of the transcription factor NF-κB by an interaction of Akt with IκB kinase (IKK) that stimulates degradation of IκBα (a potent repressor of NF-κB) [152]. Activated Akt regulates transcription of FOXO3a target genes through modulation of FOXO3a activity by phosphorylating it at three conserved serine/threonine residues (Thr32, Ser253, and Ser315), leading to the release of FOXO3a from DNA which then translocates to cytoplasm [153].
FOXO proteins trigger apoptosis. FoxO transcription factors belong to the large Forkhead family of proteins, present in all eukaryotes. FoxO transcription factors are characterized by a conserved DNA-binding domain termed the Forkhead box (Fox). The ‘O’ subgroup FoxO contains four members FoxoO1 (FKHR), FoxoO2 (FoxO6), FoxO3 (FKHRL1), and FoxO4 (AFX) [154]. FOXO proteins play an important role in the control of cell cycle, cell death, cell metabolism, longevity, aging, cellular stress response, and development [155-157]. Both, FOXO1 and FOXO3a can induce apoptosis by stimulating the expression of the pro-apoptotic Bcl-2 interacting mediator of cell death. Foxo3A controls the expression of several genes involved in regulating apoptosis through interactions with Mcl-1 and Bax/Bak [155]. Akt-dependent phosphorylation is crucial to regulate the function of FOXO proteins. The phosphorylated FoxO transcription factors bind to 14-3-3 proteins that facilitate the translocation of FoxO from the nucleus into the cytoplasm (Figure 8). This nuclear exclusion and translocation of FoxO into the cytoplasm inhibits FoxO-dependent transcription which in turn prevents apoptosis [155, 158-160].
4.3.5.1. Caspases
Cysteine aspartate-specific proteases (caspases) are a family of cysteine proteases that normally exist as inactive precursors called procaspases, which are cleaved to give rise to the active form. The preferred cleavage site for the known caspases is at the C-terminal side of a four amino-acid motif, X-X-X-Asp (where X can be any amino acid). Activated caspases cleave various intracellular and cytoplasmic membrane substrates leading to cellular disintegration [161]. The 11 known human caspases are extremely specific with a difference in their P2–P4 specificity, cleaving their substrates exclusively after

INTRODUCTION
28
Extrinsic apoptotic signaling pathway
Intrinsic apoptotic signaling pathway
Death receptor
FADD
Pro-caspase 8
Caspase-3
DISCBH-3 molecules
Mitochondria
Cyto C
Apaf-1
Caspase-9
Apoptosome
Apoptosis
Figure 9. Shematic diagram of pathways to apoptosis
Asp residues [162]. Some caspases are implicated in apoptosis such as caspases 2, 3, 6, 7, 8, 9, and 10; while others are involved in activation of proinflammatory cytokines such as caspases 1, 4, and 5 or in keratinocyte differentiation such as caspase 14. Based on their role in apoptosis, proapoptotic caspases can be divided into the initiator caspases (2, 8, 9, and 10) and the executioner caspases (3, 6, and 7). The initiator caspases have long prodomains such as death effector domain (DED) and caspase recruitment domain (CARD), occur as inactive monomers in the cytosol and are activated via recruitment to signaling complexes. However, the executioner caspases exist as dimers in the zymogen form and require cleavage by activated initiator caspases [163, 164].
4.3.5.2. Apoptosis signaling cascade
There are two major pathways leading to apoptosis in the mammalian system: (i) an extrinsic pathway initiated by death receptors and (ii) an intrinsic pathway occurring through the mitochondria [146].
In the extrinsic pathway, ligands and receptors of the tumor necrosis factor (TNF) family are essential and they include: (i) Fas and Fas ligand FasL; (ii) "death receptors" (DR4 and DR5) and TRAIL; and (iii) TNFα and the TNF receptor (TNF-R1). Binding of the ligands to these receptors, leads to oligomerization of the death receptors. These in turn bind the adapter proteins such as fas associated protein with death domain (FADD) and thereby forming the death-inducing signaling complex (DISC). The DISC serves as an activation platform for caspases 8 and 10, which in turn activate Caspase-3 by proteolytic activation [165].

INTRODUCTION
29
The intrinsic or mitochondrial pathway is triggered by cell stress (such as growth factor withdrawal, mitochondrial stress, and DNA damage) leading to activation of proapoptotic molecules of the Bcl-2 family such as Bim, Noxa, Puma, Bid, and Bad. Activated Bax and Bak form pores in the mitochondrial membrane, leading to the recruitment of proapoptogenic factors such as cytochrome c (Cyto C). Released Cyto C can complex with apoptosis proteases-activating factor 1 (Apaf-1), resulting in a conformational change to Apaf-1 by oligomerization. In addition, procaspase-9 binds to this complex to form the apoptosome. The apoptosome then, serves as an activation platform for caspase-9, which is activated by self process and thus, activates Caspase-3 by proteolytic activation [146, 166] (Figure 9).
4.4. The 14-3-3 proteins
14-3-3 proteins are a family of highly conserved, 28-33 kDa acidic, intracellular cytosolic proteins, ubiquitously produced in all eukaryotes with no intrinsic enzymatic activity. The 14-3-3 protein was originally identified in 1967 by Moore and Perez during a systematic classification of mammalian brain proteins family [246]. In fact, the name ′14-3-3′ denotes the elution fraction (14th fraction) containing these proteins in bovine brain homogenate following DEAE-cellulose chromatography and their migration position are fractions 3.3 after subsequent starch gel electrophoresis. Further, members of the family have been renamed after discovery by other researchers based on their involvement in novel regulatory roles.
The human genome contains several distinct 14-3-3 genes, denoted beta (β), epsilon (ε), eta (η), gamma (γ), tau (τ) (also called θ), sigma (σ), and zeta (ζ) according to their respective positions in eluted HPLC fractions [167-169]. Up to 15 isoforms are present in plants and two isoforms have been identified in Yeast, Drosophila melanogaster, and Caenorhabditis elegans [170, 171]. In almost every known organism, multiple (at least two) isoforms of 14-3-3 have been observed [172] and 12 to 15 isoforms are expressed in Arabidopsis [173]. There is no novel human or mouse 14-3-3 isoforms identified in the genome sequencing projects. The majority of 14-3-3 molecules are present in the cytoplasm. Further study has shown that in the absence of bound ligands 14-3-3 locates to the nucleus [174].
4.4.1. The mechanisms of 14-3-3 action
The mechanism of 14-3-3 interaction with target proteins can be classified into several generic modes-of-action [175, 176] (Figure 10: 14-3-3 protein dimers shown in shades of grey and interacting proteins in shades of yellow and blue). The mechanisms of 14-3-3 action include the following: (I) altering the activity

INTRODUCTION
30
(I) Mode 1. Inhibition, activation or turn over
(II) Mode 2. Adaptor or scaffold
(III) Mode 3.conformational change and multi-step regulation
(IV) Mode 4. Organellar protein transport
(VI) Mode 6. Excape from ER retention
(V) Mode 5. Nuclear-cytoplasmic shuttling
Figure 10. Mode of action attributed by 14-3-3
of the target through changes to the specific activity or the half-life of an enzyme; (II) a scaffolding function, in which 14-3-3 brings together two other proteins [177]; (III) inducing conformational changes of the target protein by interactions at multiple sites on a target protein [178]; (IV) binding of 14-3-3s to cleavable signaling peptides (light-blue region) of nuclear-encoded animal mitochondria /plant chloroplast proteins that stimulate their import into these organelles [179]; (V) modulation of nuclear import and export [180]; (VI) protein transport in the endomembrane system in animal cells; and (VII) changing cellular localization of interacting partner [176].
4.4.2. Structures of 14-3-3 dimers
Previously, crystal structures of three 14-3-3 isoforms (ζ, σ, and τ) have been reported. Structural data of σ and ζ are deposited in the PDB. The ζ isoforn is deposited in its apo form [181], as peptide complexes [182-185], and in complex with serotonin N-acetyltransferase (AANAT) [186]; whereas the σ isoform is in apo- and peptide-bound states [187, 188]. In addition, the structure of τ was published although it has not been deposited in the PDB [189]. These structural data showed that the 14-3-3 proteins adopt a similar conformation in the

INTRODUCTION
31
Figure 11. Schematic representation of a monomer of 14-3-3 with side chains lining the amphipathic groove indicated. Side chains are coloured according to type: hydrophobic (green), basic (blue), acidic (red) and polar (grey). A schematic model of an alpha-helix (main chain only) docked into the groove is shown in yellow. The location of the phosphorylation site, S184, is indicated. The disordered loop formed by residues 203 to 216 is shown schematically as a thin ribbon. From: Liu: Nature, Volume 376(6536).July 13, 1995.191-194
unliganded and peptide/protein-bound structures. Therefore, the structural analysis suggests that the 14-3-3 proteins may act as molecular anvils [178].
Native 14-3-3 proteins are present in the form of homo- and hetero-dimers. The most structurally variable N- and C-termini are responsible for isoform specific protein-protein interactions and cellular localization. The first 14-3-3 (ζ and τ isoforms) crystal structures were determined at resolution 2.9 Å and 2.6 Å resolution, respectively [181, 189]. Studies by Haina Fu and Alastair Aitken showed that each 14-3-3 monomer is composed of nine antiparallel α-helices (α1-α9), organised into an N-terminal and a C-terminal domain. The helices form a palisade that clearly shows an amphipathic groove lined with residues from helices α3, α5, α7, and α9 (Figure 11; with permission from publisher). On one side of the groove, α7 and α9 present a hydrophobic surface including four exposed leucine side chains (L172, L216, L220, and L227) along with residues (V176, I217, and W228). On the other side (right side in the figure), α3 includes three basic side chains (K49, R56, and R60) and α5 includes polar and charged groups (K120, D124, Y128, and R127). The three-arginine residues (R56, R60, and R127) form a basic cluster near the top of the groove. The final 15 residues of the protein (231 to 245) form a poorly ordered loop, the end of which rests on the surface of the groove and partly obscures the basic cluster [181]. The dimer creates large negatively charged channels where the invariant residues (throughout all the isoforms) are found lining the interior of this channel, while the variable residues are located on the surface of the protein. This channel would recognize common features of target proteins and thus the specificity of interaction for specific 14-3-3 isoforms with diverse target proteins may involve the outer surface of the protein [181, 190]. The N-terminal residues of all 14-3-3 isoforms are highly variable which are important for homo- or hetero-dimer formation with a limit restriction [172, 191].

INTRODUCTION
32
4.4.3. 14-3-3/client molecule interactions
14-3-3 proteins interact via phosphorylated and non-phosphorylated binding motifs with client partners [183, 185, 192-194]. Phosphorylation-dependent and -independent binding have been shown to be targeted at the same site of the 14-3-3 proteins [182].
4.4.3.1. The phosphorylated 14-3-3 binding motif
A prominent feature of the 14-3-3 proteins is their ability to bind a very large amount of client proteins, mostly through a phosphorylated serine or threonine motif on the target proteins [192]. A novel phosphoserine containing motif with structural details of the14-3-3/peptides has been identified in Raf kinase, demonstrated by Muslin and co-workers [192] Further, the motif is refined into two subtypes Mode I and II, elucidated by Yaffe et al. [184] and Rittinger et al. [185]. Therefore, each 14-3-3 dimer with two binding pockets interacts with canonical binding motifs, defined as;
Mode I (RSXpSXP: where X is any type of residue and pS is phosphoserine) and Mode II (RXΦXpSXP: where Φ is an aromatic or aliphatic residue) [184, 185]. The crystal structure of 14-3-3 ζ(zeta) homo-dimers complexed with short peptides or native binding partners has been determined at 2 Å or 2.6 Å resolutions, where each monomer contains an amphipathic groove that acts as a ligand-binding channel [184, 185, 192] . Each dimer contains two binding pockets, which can simultaneously interact with two phosphoserine binding motifs on a single target or on separate binding partners. 14-3-3 residues (important for phospho-peptide binding) are conserved within all 14-3-3 isoforms. The binding site for the phophoserine consists of a basic pocket composed of Lys-49, Arg-56, Arg-127, Tyr-128, and Tyr-129 within the C and E helices. The proline residues are in different conformations such that the proline has a cis [184] and trans-conformation [185] in the mode I and mode II phosphopeptide, respectively. Both proline conformations result in a sharp alteration in chain direction that allows the peptide to exit the binding groove. This is also clear in the determination of co-crystal structure between 14-3-3 and arylalkylamine N-acetyltransferase (AANAT) [186]. The ligand binding groove runs in opposite directions in the stable dimer that results in an isoform specific interaction and the capacity to bind with two distinct target proteins. The involvement of the hydrophobic residues of the phosphoserine binding motif may be particularly relevant to the R18 peptide binding, whose co-crystal structure has also been characterized and 5 amino acids out of 15 bind in a similar way as mode I e.g. phospho-mimicking [183]. It is also suggested that the binding of phospho- and nonphosphorylated motifs appear to share the same binding groove [185,

INTRODUCTION
33
195]. After the initial description of the mode I and II binding motifs, target proteins such as BAD, Raf-1, and Cbl are found to have two clearly defined high-affinity 14-3-3-binding sites [184] and proteins that interact with 14-3-3 through multiple sites have been identified such as the cell-cycle regulator Cdc25B [196]. In addition, a phosphopeptide containing two motifs binds 14-3-3 with a 30-fold greater affinity than a phosphopeptide containing one motif [184]. Furthermore, a synthetic peptide with the sequence WLDLE isolated from a phage display library, bound the 14-3-3 amphipathic groove in a similar manner to a phosphorylated mode-I peptide of pS-RAF-1-259 [184].
Mode-III (pS/pT X1-2-CO2 H or SWpTX-COOH, C-terminal binding motif) has recently been demonstrated by the structural study of fungal toxin fusicoccin in activation of the plant plasma membrane H+-ATPase [197]. In this study, the crystal structure demonstrated that the 4-3-3/the C-terminus of the H+-ATPase complex is stabilized by fusicoccin and therefore, releases the auto-inhibitory activity of the regulatory domain. Based on the homology between the C-terminal 14-3-3 binding motif of the serotonin-N-acetyltransferase (AANAT, arylalkylamine-N-acetyltransferase) and H+-ATPase proteins, a so-called mode III binding motif has been proposed [197]. One further example of a mode III was revealed by the study of RKR amino acid motif endoplasmic reticulum localization signal and membrane proteins movement to the cell surface [198, 199]. In addition, interactions between 14-3-3 proteins and their target partner(s) can also occur on the 14-3-3 dimer outer surface, which probably contributes to a stable three-dimensional configuration and provides an opportunity for conformational modulation of the target [200].
4.4.3.2. Non-phosphorylated 14-3-3 binding motifs
14-3-3 also interacts in a phosphorylation-independent manner with some target proteins including human telomerase (hTERT) [201], the enteropathogenic Escherichia coli Tir protein [202], the amyloid β-protein precursor intracellular domain fragment [203], unpalmitoylated CD81 (a member of the tetraspanin superfamily of proteins) [204], histone H3 peptide (both phosphorylated and phospho-acetylated) [205] and ExoS toxin [78, 206] .
ExoS interacts with 14-3-3 in a phosphorylation-independent mechanism [77, 78] for which the 27 most C-terminal amino acids of ExoS are important for 14-3-3 binding. ExoS lacking the interaction motif is significantly less cytotoxic than ExoS wild type verifying the in vivo importance of this interaction. Although 14-3-3 clearly binds ExoS in the absence of

INTRODUCTION
34
phosphorylation, the nature of the structural interaction between these two proteins is unclear.
The best-studied example of a non-phosphorylated interaction with 14-3-3 is with the R18 artificial peptide isolated from a phage library displaying random peptides [182]. This peptide specifically binds to multiple isoforms of purified 14-3-3 proteins and is found to interact with only a single band of proteins from total cell lysates. Functionally, the R18 peptide blocks the association of 14-3-3 with Raf-1, resulting inactivation of Raf-1 [181, 183, 184, 189, 207]. In the co-crystal structure of 14-3-3ζ in complex with R18, the pentapeptide WLDLE of R18 occupies the phosphoserine recognition site in the groove [183]. Acidic residues of the pentapeptide (Asp and Glu) mimic the phosphate group of phosphoserine-containing ligands and are placed next to the basic cluster of 14-3-3ζ containing residues Lys-49, Arg-56, Arg-60, and Arg-127. Consistent with this model, a charge-reversal mutation K49E of 14-3-3ζ drastically decreases 14-3-3ζ binding to R18. Competition between R18 and Raf-1 for the same set of residues in the binding amphipathic groove of 14-3-3 may account for the inhibitory effect of R18 on the 14-3-3/Raf-1 interaction. The conserved amphipathic groove that binds R18 is also involved in binding to diverse protein ligands and R18 may also block the 14-3-3/proteins interaction. Furthermore, R18 effectively inhibits the interaction of 14-3-3ζ with unphosphorylated ExoS [206, 208]. The R18 motif (WLDLE) interacts with Lysine49 on 14-3-3 that is important for ExoS/14-3-3 interaction. Similar to the binding motif WLDLE in R18, Hallberg B et al showed that ExoS contain a DALDL sequence at position 424-428 that is required for interaction between ExoS and 14-3-3. Moreover, the ExoS-DALDL binding motif appears to be necessary for both in vivo ADP-ribosylation and cytotoxic activities [77]. For the R18 peptide, the negatively charged Asp12 and Glu14 residues within WLDLE sequence interact in the amphipathic groove of 14-3-3 and form a negative charge density in a manner similar to phosphorylated mode-I peptide of pS-RAF-1-259 [182, 183, 206, 207]. Further examples have shown that inositol polyphosphate 5-phosphatase containing the motif RSESEE binds 14-3-3 proteins via multiple negatively charged amino acids [193, 209] and Glycoprotein b-α is a 14-3-3 binding protein, which contains a reported interaction domain with the motif QDLLSTVS has a weak resemblance towards ExoS and R18 [210]. Based on these studies, it has been speculated that negatively charged ExoS residues such as Asp424 and Asp427 may be able to mimic the Raf-1 phosphorylated serine motif, which would perhaps explain the binding of 14-3-3 proteins to this motif. Therefore, based on the reported speculation, we investigated the question regarding phosphorylation-independent interactions mediated by phospho-mimicking acidic residues of ExoS (Papers II-IV).

INTRODUCTION
35
4.4.4. Comparative structural analysis of the seven 14-3-3 protein isoforms
Xiaowen Yang et al demonstrated a comparative structural analysis of the seven 14-3-3 proteins, which revealed specificity determinants for binding of phosphopeptides in a specific orientation, target domain interaction surfaces, and flexible adaptation of 14-3-3 proteins through domain movements [211]. This study provided structural details for five 14-3-3 isoforms bound to ligands and structural coverage for all isoforms of a human protein family. Specifically, the structures of the beta isoform in its apo and peptide bound forms showed that its binding site can exhibit structural flexibility to facilitate binding of its protein and peptide partners. The authors suggest that conformational flexibility in both the peptide binding site and in the dimer interface facilitates binding of 14-3-3 proteins to diverse peptides and proteins of varying sizes and sequences. The initial interaction involves conformational changes of the three C-terminal helices (αG to αI). Through these changes, the 14-3-3 protein could adapt to the type of bound peptide whether it is phosphorylated and extended or nonphosphorylated and helical. The flexibility at the dimerization interface provides a mechanism for the 14-3-3 proteins to bind proteins of varying size. This versatility is most likely enhanced by the presence of desolvation sites (S1b and S2) providing nonspecific protein-protein interaction motifs, with specificity features primarily from the variable (V1 and V2) regions. Further specificity in 14-3-3 regulation of intracellular signaling is most likely provided by its tissue, temporal distribution, and subcellular localization that allows this small protein family to be a central and specific regulator through its ability to adapt its conformation to interact with a variety of binding partners [211].
4.4.5. Function of 14-3-3
14-3-3 interacts with more than 200 partners with a broad range of functions [212]. In plants, 14-3-3 binding partners are involved in biosynthetic metabolism, transport of ions, and regulation of growth [175]. In humans, these partners play a role in physiological processes such as mitogenic signal transduction, cell proliferation, cell cycle progression, protein trafficking, rearrangement of the cytoskeleton, oncogenesis, apoptosis, and survival [212, 213].
Survival and apoptosis pathways are integrated by the dynamic 14-3-3/client protein interactions to determine cell fate. 14-3-3 targets the mitochondrial apoptotic machinery by interacting with BCL2 family members (i.e. BAD, BIM, and BAX), as well as a staggering number of signaling molecules that mediate survival and death to the mitochondrial death machinery [213, 214].

INTRODUCTION
36
Cell cycle regulation is a pivotal role of 14-3-3 proteins that act as integral components in several checkpoints and play a role during uncontrolled cell cycle progression. 14-3-3 proteins regulate the onset and timing of mitosis in cycling cells, are involved in maintaining a stable G1-arrest in non-cycling cells and are also necessary for the S-phase checkpoint after DNA-damage by UV. 14-3-3σ regulates entry into mitosis downstream of CDC25C, presumably by sequestration of CDC2-cyclin B complexes. In addition to regulating kinases and phosphatases, 14-3-3 proteins affect the cell cycle by regulating the activity of transcription factors such as p53, FOXO, MIZI etc, causing inhibition of cell cycle progression [215].
Oncogenic transformation in all four major lung cancer histologies is promoted by 14-3-3 over-expression [216]. 14-3-3 proteins bind and regulate proteins involved in each of the cancerous processes including Raf, Bcr, Bcr-Abl, polyomavirus middle tumor antigen, Bad, Bax, ASK-1, forkhead transcription factors, p53, TSC2, p27, Cdc25 A, Cdc25B, Cdc25C, Wee1, Chk1, p130 Cas, integrins β1, integrins β2, integrins β3, and Ron [217, 218]. The role of 14-3-3 proteins in cancer remains unclear as both tumor suppressing and tumor promoting activities are attributed to 14-3-3. Thus, targeting 14-3-3 may provide an effective strategy to sensitize tumor cells for therapy-induced cell death in cancer [218].
Metabolic regulation appears to be regulated by 14-3-3 proteins. In plants, 14-3-3 affects the activity of various primary metabolic enzymes and ion channels of central importance in plant biochemistry. Well-known examples are nitrate reductase and the plasma membrane H+-ATPase, which are inhibited and activated by 14-3-3, respectively [175, 219, 220].
4.5. P. aeruginosa pathogenesis in a mouse model
Shaver and Hauser have evaluated the relative contributions of ExoS, ExoT, and ExoU to virulence in the lung with a mouse model of acute pneumonia caused by P. aeruginosa [57]. In this study, genetically modified clinical bacterial strains that naturally secrete ExoU, ExoS, and ExoT were constructed to evaluate the relative roles of each effector protein to pathogenesis. By measuring mortality, bacterial persistence in the lung, and dissemination, it was elucidated that ExoU has a major effect, ExoS has an intermediate effect, and ExoT has a minor effect on virulence in acute pneumonia. It has also been demonstrated that the ExoS virulence is largely dependent on its ADP-ribosyltransferase activity and minor effect is attributed to its GAP activity [57]. Surprisingly, it has been observed that ExoS, ExoT, and ExoU are not synergistically increased when combinations of these effector proteins are secreted together [72].

INTRODUCTION
37
4.6. Cell lines used for this study
Olson et al demonstrated the functional effects of ExoS on human epithelial and murine fibroblastic cells and showed that human epithelial cells exhibit an overall increased sensitivity to the effects of translocated ExoS on cell proliferation, morphology, and re-adherence. ExoS is also found to ADP-ribosylate a greater number of low-molecular-mass G (LMMG) proteins in human epithelial cells compared to murine fibroblast [221]. The Olson study also suggested that eukaryotic cell properties of different animal origins affect the substrate targeting of ExoS-ADP-ribosyltransferase activity which in turn can influence the severity effects of ExoS on host-cell function [221]. To understand the mechanism of ExoS mediated cell death, we have used various cell lines for cytotoxicity analysis. Usually P. aeruginosa adhere lungs and other organ epithelial cells before colonizing and invasion. The Madin-Darby canine kidney (MDCK) epithelial cell line was chosen as P. aeruginosa infect kidneys and form monolayers similar to in vivo epithelial sheets (Paper I). Human epithelial cells (HeLa) were used to study actin depolymerization and cytotoxicity by infection with bacteria expressing ExoS variants (Paper II-IV). The HeLa cells were taken from a fatal cervical carcinoma with adherent cells to maintain contact inihibition. The mouse embryonic fibroblast cell line NIH3T3 was used for transfection / transformation assays (Paper I). The mouse macrophage cell line J774A.1 was chosen for cytotoxicity analysis as P. aeruginosa causes lungs infection in mice resulting in damage to mouse macrophage (Paper III).
4.7. ExoS constructs used for this study
The type III secretion system is utilized by numerous Gram-negative bacteria [46, 222]. The type III apparatus of Yersinia pseudotuberculosis is one of the most intensely studied among the Gram negative pathogens. At least 20 proteins function together to form a channel through the bacterial double envelope that allows the secretion of virulence factors in one step from the bacterial cytoplasm to the cytosol of the target cell [46]. At least three translocator proteins LcrV, YopB, and YopD that aid the effectors to cross the eukaryotic cell membrane are required for the translocation of Yop proteins into the host cell [223-225]. It has been proposed that a complex of LcrV, YopB, and YopD forms a pore in the eukaryotic cell membrane through which the virulence factors are delivered into the eukaryotic cytosol [223, 226-229]. The secreted proteins are not processed during secretion and the sequences targeting them for secretion are located in the N-terminal of the protein [230, 231]. The secreted proteins require small cytoplasmic proteins (termed chaperones) that interact with the type III system to ensure stabilization and efficient secretion [232-237]. Immediately downstream of the secretion signal resides the recognition sequence for

INTRODUCTION
38
translocation into the host cell [238]. The delivery of proteins is dependent on physical contact between the bacterium and the eukaryotic cell and the translocation process appears to be polarized [239]. The translocation apparatus of P. aeruginosa shows high sequence and functional similarity to that of Y. pseudotuberculosis at the level of the individual protein components as well as for genetic organization [59, 232, 240]. Individual selected components of the secretion system have been shown to be exchangeable between P. aeruginosa and Y. pseudotuberculosis [241]. We have used Y. pseudotuberculosis model system for ExoS infection in the Paper I-IV.
ExoS is secreted and translocated into the target cell by the Y. pseudotuberculosis type III secretion system in a similar manner for its normal cytotoxins [77, 79, 242]. Therefore, we used the type III secretion system of a genetically defined secretion and translocation mutant of Y. pseudotuberculosis to express and translocate ExoS into the eukaryotic cell (Paper I). The advantage of this approach is that it allows us to study the effect(s) of ExoS in the absence of other toxins and proteases secreted by P. aeruginosa. Moreover, Y. pseudotuberculosis expresses and delivers ExoS protein with higher efficiency than P. aeruginosa.
We used different variants of bacteria to translocate ExoS into the eukaryotic cell. To control the expression and translocation of ExoS from the bacteria into the target cell, a Y. pseudotuberculosis strain which expresses and translocates ExoS under the control of an arabinose inducible promoter was constructed [77, 243]. A controlled expression and translocation of ExoS into eukaryotic cells at a similar to the P. aeruginosa wild type was achieved by growing the bacteria in the presence of 0.1% arabinose (Paper II, III, and IV).
E.coli strains harboring the pGEX plasmid encoding for different GST-ExoS variants were used for pull down analysis in this study.
Table 1: Summary of various GST-fusion proteins constructs of ExoS used (Paper II, III, and IV)
Plasmid Substituted amino acid(s) Reference or source
1. GST alone Amersham
2. GST-ExoS(88–453), wild type S419QGLLDALDL428 [78]
3. GST-ExoS(88–453; S∆) M419AAAA428 [77]
4. GST-ExoS(LDL426–428AAA) S419QGLLDAAAA428 This study

INTRODUCTION
39
5. GST-ExoS(HDD418,424,427AAA)
S416GASQGLLAALAL428 This study
6. GST-ExoS(LLLL422,423,426,428AAAA)
S419QGAADAADA428 This study
7. GST-ExoS(DALDL424–428AAAAA)
S419QGLLAAAAA428 [77]
8. GST-ExoS(D424A; D427A) S419QGLLAALAL428 This study
9. GST-ExoS(S416A) A416GHSQGLLDALDL428 This study
10. GST-ExoS(H418A) S416GASQGLLDALDL428 This study
11. GST-ExoS(S419I) I419QGLLDALDL428 This study
12.GST-ExoS(Q420A) S419AGLLDALDL428 This study
13.GST-ExoS(G421A) S419QALLDALDL428 This study
14. GST-ExoS(L422A) S419QGALDALDL428 This study
15. GST-ExoS(L423A) S419QGLADALDL428 This study
16. GST-ExoS(D424A) S419QGLLAALDL428 This study
17. GST-ExoS(A425K) S419QGLLDKLDL428 This study
18. GST-ExoS(L426A) S419QGLLDAADL428 This study
19. GST-ExoS(D427A) S419QGLLDALAL428 This study
20. GST-ExoS(L428A) S419QGLLDALDA428 This study
21. GST-ExoS(LD426–427AA) S419QGLLDAAAL428 This study

AIMS
40
5. AIMS The general aim was to study the ExoS interaction with 14-3-3 using structural analysis, biochemical observation for survival or death, and pathogenesis experiments in mice. We also aimed to identify the cellular target molecule(s) for enzymatic activity of ExoS using a cell culture system. Specific aims were as follows;
To identify the important ExoS amino acid(s) for interaction with 14-3-3 and enzymatic activity using single amino acid mutational analysis.
To analyze the toxicity of ExoS mutants in cell culture system and in a mouse model which are able or unable to interact with 14-3-3.
To study the effects of ExoS variants in signal transduction downstream targets after infection.
To study the induction of apoptosis upon ExoS infection
To analyze the structure of ExoS/14-3-3 complex.

RESULTS AND DISCUSSION
41
6. RESULTS AND DISCUSSION
6.1. Cells expressing activated Ras or PKB/Akt survive upon ExoS infection (Paper I)
ExoS toxin is a promiscuous ADP-ribosyltransferase, able to ADP-ribosylate Ras and other small GTP-binding proteins after translocation from bacteria into the cell [80]. Specially transformed cell lines expressing oncogenic proteins important in proliferation and survival together with control cell lines were used to investigate the ExoS cellular target that induces apoptosis after ExoS translocation. The cell lines used were NIH3T3(wt), NIH3T3(K-RasV12) expressing a constitutively activated K-Ras protein, MDCK(wt), MDCK (Er-Raf) expressing a tamoxifen-inducible oncogenic Raf-1 gene, and MDCK (gag-PKB/Akt) expressing activated PKB/Akt protein. The Y. pseudotuberculosis type III secretion system was employed as a tool to express and translocate the P. aeruginosa toxin ExoS(wt) or ExoS mutant ExoS-E381A (mutated at Glu-381) proteins through the host cell membrane .
6.1.1. Ras is an important cellular target and determinant of cell survival
We showed that activated Ras is able to transmit signals to downstream targets efficiently even if ADP-ribosylation of Ras occurred by exogenous ExoS proteins (Figure 12) and ExoS is unable to downregulate activated PKB/Akt or Raf-1 proteins. This pinpointed that the cellular target of ExoS is located higher in the signal transduction cascade than the level of Raf-1 and PKB/Akt. Infection analysis with Y. pseudotuberculosis expressing ExoS(wt) and ExoS(E381A) in cell lines NIH3T3(K-RasV12), MDCK (Er-Raf), and MDCK (gag-PKB/Akt) showed that Ras proteins in all cell lines were modified efficiently by ExoS (Paper I, Fig 1D and 2D). However, Ras was still able to signal to downstream targets such as Erk 1/2 and PKB/Akt proteins, which were not inhibited upon ExoS infection (Paper I, Fig 1A, 1B and 2A, 2B). In contrast, upon ExoS infection in both NIH3T3(wt) and MDCK(wt) cell lines, Ras downstream signaling e.g. Erk 1/2 and PKB/Akt was inhibited upon stimulation with EGF (Paper I, Fig 1A, 1B and 2A, 2B).
Our results suggested that, expression of activated K-RasV12 is able to efficiently protect cells from ExoS-induced cell death (Paper I, Fig 3). Cell survival assays showed that after 5 h of infection with ExoS, approximately 85% of NIH3T3(K-RasV12) cells were viable that was similar to the non-infected NIH3T3 cells and in the control approximately 25% of NIH3T3(wt) cells were viable (Paper I, Fig 3 and 4A, 4B). The cells were rounded up and semi-detached from the Petri dish surface due to the N-terminal GAP domain activity

RESULTS AND DISCUSSION
42
of ExoS (Paper I, Fig 4A and 4B). We also suggested that activated K-Ras and PKB/Akt expressed in NIH3T3 and MDCK cells have the ability to mediate a proper survival signal even after infection and modification of cellular targets. By using a similar cell survival assay as above, we showed that approximately 80% of MDCK(gag-Akt) cells were survived and more resistant to ExoS infection. This was compared with control MDCK-wt cells which were viable at approximately 50% upon ExoS infection indicating less sensitivity to ExoS infection as compared with NIH3T3 cells (Paper I, Fig 3). MDCK(Er-Raf-1) cells showed a slight reduction in cell death with a 60% survival rate upon infection with ExoS as compared with control MDCK cells (Paper I, Fig 3). Therefore, we suggest that Ras protein is important for cell survival in response to ExoS infection, and down-regulation of Ras mediates a faster cell death upon infection with ExoS.
6.1.2. ExoS mediates caspase-3 activity
Our results suggested that activated Ras and activated PKB/Akt are more potent survival determinants than activated Raf-1. Upon infection with ExoS(wt), the caspase-3 activity(after 6h) in NIH3T3 (K-RasV12 ) cells did not increased above the basal level (Paper I, Fig 5). This was similar to non-infected controls or cells infected with the bacteria expressing ExoS(E381A). In contrast, NIH3T3-wt cells showed a significantly increased caspase-3 activity upon infection with ExoS (Paper I, Fig 5). Similar results for caspase-3 activity were obtained in
Figure 12. Mode of action attributed by ExoS upon infection into host cell
Pseudomonas aeruginosa/ Yersinia pseudotuberculosis
Type III secretion system
MDCK / NIH3T3( wt) cell
ExoS
NIH3T3(K-Rasv12) MDCK(gag-AKT/PKB), MDCK(Er-Raf) cell
14-3-3ExoS
activated
Raf-1P
activatedPKB/AKT
PP
activated
PFoxo3ERK
P
Survival
Inactive
Apoptosis
Induce Caspase-3
ERKP
PKB/AKTP
P
Ras ADP-Ribose
14-3-3ExoS
K-RasV12 ADP-Ribose

RESULTS AND DISCUSSION
43
Secr/Transl GAP
ADP-ribosyltransferase
Exoenzyme SR146 E381
1 453
14-3-3
SGHSQGLLDALDL416 429
Figure 13. ExoS Sequence important for 14-3-3 interaction
MDCK(wt) cells infected with ExoS. However, MDCK(gag-PKB/Akt) cells and MDCK(Er-Raf-1) showed reduced caspase-3 activity upon infection with ExoS(wt) in a similar manner to non-infected MDCK cells or cells infected with ExoS(E381A). Several findings [244, 245] support our evidence that activation of the Raf-1 cascade suppresses apoptosis in other experimental cell systems. Moreover, by measuring the apoptosis inducing potential FOXO3a, no inhibition of FOXO3a phosphorylation was observed in the NIH3T3(K-RasV12), MDCK(gag-PKB/Akt) and MDCK(Er-Raf-1) cells upon stimulation with EGF (Paper I, Fig 1E and 2E). In contrast, upon infection with ExoS, NIH3T3(wt) and MDCK(wt) cell showed reduced FOXO3a phosporylation (Paper I, Fig 1E and 2E). In conclusion, the observed cell death induced by ExoS in cell lines correlates with reduced phosphorylation of FOXO3a protein and increased caspase-3 activity. Expression of an activated variant of Ras or downstream mediators, such as PKB/Akt, efficiently prevents cell death, correlates with lack of caspase-3 activity, and FOXO3a protein phosphorylation (Figure 12).
6.2. Phosphorylation independent interaction between 14-3-3/ExoS (Papers II, III, and IV)
A phosphorylation-independent interaction has been reported to occur between 14-3-3 and a C-terminal domain within ExoS [78]. We investigated the effect of amino acid mutations in the C-terminal domain of ExoS on ADP-ribosyltransferase activity and interaction with 14-3-3. To address exactly which amino acid residues in the ExoS sequence were critical for 14-3-3 binding (Figure 13), a set of single substitution ExoS mutants were constructed together with some additional variants (Table 1).
6.2.1. ExoS-LDL motif is important for interaction and enzymatic activity
By employing a GST-ExoS fusion protein pull down experiement (Papers II and IV) we showed that ExoS amino acid substitutions at positions 426-428 significantly reduce the in vitro interaction potential of 14-3-3 with ExoS (Paper II, Fig 1) and both ExoS Asp424 and Asp427 are not strictly necessary

RESULTS AND DISCUSSION
44
Uninfected
ExoS-WT(88-453)
ExoS-LDL(426-428)AAA
ExoS-S(416)A
ExoS-H(418)A
ExoS-L(422)A
ExoS-L(423)A
ExoS-D(427)A
ExoS-L(428)A
ExoS-SH(416,418)AA
ExoS-DD(424,427)AA
ExoS-LLLL(422,423,426,428)AAAA
ExoS-HDD(418,424,427)AAA
ExoS-LD(426,427)AA
ExoS-D(424)A
ExoS-L(426)A
Figure 14. Cytotoxicity analysis of infection into mammalian cells with ExoS variants.Viability of HeLa cells are expressed as percentage survival rate ,24 h after intoxication with ExoS variants. HeLa cells were infected for 2 h in the presence of 0.1% arabinose with Yersinia (YPIII) carrying plasmids expressing different ExoS variants.
20
40
60
80
100
0
Surv
ival
cel
ls (%
)
for the interaction (Paper II, Fig 1). Cytotoxicity and Ras modification assays were carried out in infected HeLa cells with the Y. pseudotuberculosis arabinose inducible strains expressing ExoS wild type as well as several single, double, triple, and quadruple amino acid substitution variants. The data showed that ExoS-LDL appears to be necessary for both ADP-ribosylation and cytotoxic activities of ExoS in vivo. This was demonstrated by ExoS variants lacking the LDL motif showing no cytotoxic response (Figure 14) and no ADP ribosylation of Ras (Paper II, Fig 3A-D and Fig 4). During the course of this study, Shaver and Hauser published evidence that activation of the ADP-ribosyltransferase activity of ExoS results in cell death in an animal pneumonia model, with increased bacterial persistence in the lung and decreased host survival [57]. P. aeruginosa pathogenesis was investigated in a mouse acute pneumonia model by intranasally infecting mice with clinical isolates of P. aeruginosa expressing ExoS-wt, ExoS(LDL-AAA) and ExoS(E379A/E381A). The results showed that mutation of key residues (LDL) within the ExoS binding site for 14-3-3 impairs virulence as 50% of the mice was survived after 7 days of infection and the bacterial load in the lungs was cleared at 18h post infection (Paper III, Fig 6). In conclusion, we showed that residues L426, D427, and L428 of ExoS are critical for the virulence of ExoS. Further, we extended our study through joint collaboration with Christian Ottmann and Alfred Wittinghofer to demonstrate structural analysis and biological investigation of ExoS/14-3-3 interaction. Our

RESULTS AND DISCUSSION
45
structural analysis predicted that this phosphorylation independent interaction by L426, D427, and L428 is crucial for the function of ExoS (Paper III).
6.2.2. 14-3-3/ExoS structure (the novel binding mode IV)
In Paper III, the 14-3-3 dimer revealed the characteristic W-like shape with each monomer constituting an amphipathic groove that harbored the ExoS peptide, which was extended in a conformation occupying more than two-thirds of the 14-3-3 binding groove (Paper III, Fig. 1A and 1B). Our findings designated the novel phosphorylation-independent binding geometry as mode IV and suggested that ExoS binds to 14-3-3 in a novel binding mode mostly relying on hydrophobic contacts by L422, 423, 426, and 428. The manner by which ExoS binds 14-3-3 is also distinct from binding modes I–III of 14-3-3 with other known client partners due to following reasons:
ExoS binds to 14-3-3 mainly via hydrophobic contacts involving leucines 422, 423, 426, and 428 that form an extensive hydrophobic interaction surface with the 'roof' of the amphipathic groove by the 14-3-3 residues Pro165, Ile166, Leu216, Ile217, Leu220, and Leu172 (Paper III, Fig. 2). In addition, the peptide was coordinated by several H-bonds or electrostatic contacts involving His418, Asp424, and Asp427 of ExoS peptide and Asp213, Lys49, and Tyr 128 of 14-3-3ζ (Paper III, Fig. 2).
Based on Haina Fu, Aitken, and Rittiger findings on motifs I–III of 14-3-3 [181, 184, 185], it was suggested that the 14-3-3 residues Lys49, Arg56, and Arg127 were involved in the accommodation of a phosphorylated residue within the basic cluster of 14-3-3 [181, 184, 185]. Our crystal analysis showed that Asp427 of ExoS was too far away from the basic cluster of 14-3-3 for significant interaction and that Asp424 of ExoS only contacts Lys49 of 14-3-3 (Paper III, Fig. 3).
The binding mode of ExoS/14-3-3 differs in respect to the nature of binding forces and the general geometry of binding in that previously observed 14-3-3/client complexes were oriented from N- to C-terminus direction, which was in an opposite orientation to the ExoS/14-3-3 binding mode. The corresponding 14-3-3 binding motif from serotonin-N-acetyltransferase (AANAT) was crossed the amphipathic groove in an N- to C-terminal direction from right to left [186]. However, ExoS was orientated in an antiparallel fashion to this, starting with the N-terminal part from left to right (Paper III, Fig 4A). The 'entry point' of the AANAT N-terminus and the ExoS C-terminus were located at nearly exactly the same position and the other ends of the polypeptide chain were emerged at different locations; ExoS leaves the groove at the far end while keeping close contact to 14-3-3 whereas the AANAT peptide leaves the groove perpendicular to the groove's

RESULTS AND DISCUSSION
46
base after the proline-induced kink in its polypeptide chain (Paper III, Fig 4A and 4B).
Our structural analysis was supported by Yang et al [211]. However, in contrast to our model, Yang et al showed that the ExoS peptide is bound to the conserved binding groove with both 14-3-3 subunits (β isoform) adopting a closed conformation and the conformational change that takes place in the crystalline form results in a change of crystal symmetry. Both ExoS and R18 bind with their N to C termini in the opposite direction compared with phosphorylated peptides. In addition, the ExoS/14-3-3 complex forms a helical structure, showing a secondary structural element in the 14-3-3 peptide-binding groove [211].
6.2.3. Hydrophobic interaction is important for both interaction and enzymatic activity
In papers III and IV, we suggested that hydrophobic interactions (contributed by L422, 423, 426, and 428 of ExoS) are important for both ExoS/14-3-3 interaction and ExoS ADP-ribosyltrasferasase activity (Figure 15). Our cytotoxicity and biochemical analyses showed that cells infected with bacteria producing and translocating ExoS(L422A) and ExoS(L423A) were 59 and 54% viable respectively (Figure 14). However, over 90% of the cells survived upon infection with bacteria producing and translocating ExoS(L422:L423:L426:L428-AAAA), (Figure 14). Cytotoxicity analysis
Pseudomonas aeruginosa/ Yersinia pseudotuberculosis
Type III secretionsystem
ExoS
ExoSL428
L426L423L422
14-3-3
ExoSL428
L426L423L422
14-3-3
Ras ADP-Ribose
Eukaryotic cell
Figure 15. Schematic overview of binding mode IV

RESULTS AND DISCUSSION
47
showed <10% cell viability upon infection with ExoS variants with single, double, and triple substitutions of the residues in electrostatic contacts such as single mutants ExoS(S416A), ExoS(H418A), ExoS(D424A), and ExoS(D427A); the double mutant ExoS(S416:H418-AA); and triple mutant ExoS(H418:D424:D427-AAA) and they were as aggressive as wild-type ExoS in their ability to induce cell death (Paper IV, Fig 2 and Figure 14). By biochemical analysis, it was observed that substitution of the hydrophobic residues of ExoS (L422:L423:L426:L428-AAAA) significantly reduced its ability to modify the endogenous targets such as small GTPases RAS/RAP1 which were able to signal downstream through activation of ERK and PKB (Paper III, Fig 5 and paper IV, Fig 3). ExoS variants with electrostatic residue substitutions including S416, H418, D424, and D427 were as aggressive as ExoS-wt in their ability to modify RAS/RAP1 within the cell and impaired downstream signaling targets (Paper IV, Fig 3). In conclusion, electrostatic interaction between ExoS and 14-3-3 via Ser416, His418, Asp424, and Asp427 residues are of secondary importance as they are not sufficiently strong to significantly contribute to induce cell death after ExoS infection.
6.2.4. Leu428 is an important determinant of cell death induction
Using cytotoxicity analysis and Ras modification assay, we showed that Leu428 of ExoS is the critical residue for ExoS enzymatic activity and for modifying the eukaryotic endogenous target Ras (Paper II, Fig 3). As the GST-ExoS pull down experiments showed a strong interaction, it was evident that the substitution of Leu428 significantly weakened the ability of ExoS to mediate cell death (Figure 14) without altering ExoS/14-3-3 interaction (Paper II, Fig. 1 and Paper IV, Fig. 1).
6.2.5. Asp424 and Asp427 are not important for ExoS enzymatic activity
In papers II, III, and IV, we showed that negatively charged aspartate residues at positions 424 and 427 of ExoS do not mimic phosphorylated serine motifs for the ExoS/14-3-3 interaction as single substitutions including ExoS(D424A) or ExoS(D427A) and the double mutant ExoS(DD424:427AA) were as aggressive as wild-type ExoS in their ability to induce cell death (Figure 14).
6.2.6. Leu428 is critical for ExoS auto-ADP-ribosylation
As previously demonstrated, ExoS is auto-ADP-ribosylated [82]. Interestingly, we found that substitution in Leu428 result in loss of ExoS-ADP-ribosylation activity which may result in a lack of auto-ADP-ribosylation of the GAP domain in ExoS and resulting in faster GAP function than the ExoS-wt during infection

RESULTS AND DISCUSSION
48
Uninfected
ExoS-WT(88-4
53)
ExoS-LDL(426-428)AAA
ExoS-S(416)A
ExoS-H(418)A
ExoS-L(422)A
ExoS-L(423)A
ExoS-D(427)A
ExoS-L(428)A
ExoS-SH(416,418)AA
ExoS-DD(424,427)AA
ExoS-LLLL(422,423,426,428)AAAA
ExoS-HDD(418,424,427)AAA
ExoS-LD(426,427)AA
ExoS-D(424)A
ExoS-L(426)A0
60
120
180
240
300
100%
rou
nded
up
(min
ute)
Figure 16. Rounded up after infection of HeLa cells with Y. pseudotuberculosis expressing different ExoS variants
analysis. To test this, we measured the rounded up morphology (in percentage per minute) of cells upon infection with Y. pseudotuberculosis expressing different ExoS variants. Cells infected with bacteria expressing ExoS(L428A) were 100% rounded up by 60 minutes post infection. Cells infected with bacteria expressing ExoS-LDL(426-428)AAA and ExoS-LLLL(422,423,426,428)AAAA were rounded up by 90 minutes post infection whereas cells infected with bacteria expressing ExoS-wt were rounded up by 300 minutes. Cells infected with the rest of the mutants were rounded up by 180 minutes, respectively (L.Y. and B.H. unpublished observations).

CONCLUSIONS
49
7. CONCLUSIONS
Activated Ras is able to protect cells against cell death even if it has been ADP ribosylated by ExoS. Ras may be an important cellular target for the P. aeruginosa toxin ExoS. ExoS infection leads to cell death, which correlates with dephosphorylation of FOXO3a and increased of caspase-3activity (Paper I).
The LDL(426-428) binding motif of ExoS is important for both the interaction of 14-3-3/ExoS and ExoS ADP-ribosylation activity (Papers II and III).
Leucine428 of ExoS may be a critical residue for ExoS enzymatic activity as substitution of this leucine significantly weakens the ability of ExoS to mediate cell death. Leu428 of ExoS is also required to modify the eukaryotic endogenous target Ras (Paper II). Leucine-428 of ExoS may also be critical for ExoS auto-ADP-ribosylation activity in vivo (unpublished data).
Structural analysis of the 14-3-3/ExoS suggests a novel binding mode (IV) where the important attractive force is mainly contributed by hydrophobic contacts: Leu422, Leu423, Leu426, and Leu428 residues, extended within the ″roof″ of the 14-3-3 groove. The 14-3-3 binding motif of ExoS runs in an opposite orientation compared with the consensus peptide binding mode I-III. The 14-3-3/ExoS structural interaction analysis is supported by biological, functional and infection assays in a mouse model with acute pneumonia (Papers III and IV).
Leucine residues-422, 423, 426, and 428 are important for ExoS ADP-ribosyltrasferasase activity as ExoS with substitution of these corresponding Leucine amino acids is unabled to induce cell death or modify endogenous RAS/RAP1 (Papers III and IV).
Interaction between ExoS and 14-3-3 via electrostatic residues Serine416, Histidine418, Aspartate424 and Aspartate427 are of secondary importance. ExoS mutants with substituion in Ser416, His418, Asp424, and Asp427 are as aggressive as wild type ExoS in their ability to induce cell death and to modify endogenous ExoS targets in infected host cells (Paper IV).

ACKNOWLEDGEMENTS
50
8. ACKNOWLEDGEMENTS This work was carried out at the Department of Medical Biosciences and Pathology, University of Umeå, during the period of April 2004-November 2007.
The most important acknowledge is to my Lord Allah (SWT) (Most Merciful Most Wise) for guiding me to conceptualize, develop, and complete the thesis work. Indeed, without Allah’s help, nothing would be possible to accomplish. During this work, I met and worked with the people whom Allah (SWT) has given me to help with this project. The contributions vary but the appreciation is still large. I could not remember how many times I get depressed and YOU comfort me every single difficult moment. Allhamdullillah (all praise is due to Allah).
My warmest gratitude and thanks belong to my husband and friend Mazharul Alam and my most precious boy Ibrahim Mohammad, who share all sorts of things including good and bad, supporting me in every single situation. You guys sacrifice big parts of life and Allah make us a happy family together as we learn ″not so many things need in a short live″. Now we have to go forward and you guys always are there InshaAllah (If Allah wills).
I am grateful to my supervisor Professor Bengt Hallberg, for guiding me in the world of science; Cell Signal Transduction. Will you not miss me everyday morning with the question: Lubna what are you doing? His enthusiasm and optimistic attitude towards research inspired me during these years. Thanks to take me as a PhD student and for the opportunity to work for ExoS project. It’s a nice experience to work with you and to see you as a PROFESOR before leaving. !!Congratulation!! ″Will I survive at the next level? ″ let’s see!
I wish to thank Dr. Karin Nylander, the Head of the department of Medical Biosciences and Pathology, who provided encouraging atmosphere and specially for review of the present manuscript. Thanks Dr. Richard Palmqvist for his critical decision in ending my PhD program.
I wish to thank specially Professor Alfred Whittinghoffer and Dr. Alan Hauser for rewarding collaboration and their interest in our work.
I want to thank co-authors and collaborators: Dr. Christian Ottmann, Michael Weyand, Jeffrey L Veesenmeyers, Maureen H Diaz, Tooba Panahandeh, Patricia Warne, Dr. Julian Downward, Dr. Matt Francis, and Dr. Ruth Palmer. Contributions by all of you are greatly appreciated. All of your interests in my work have been of great importance.
I want to thank Dr. Mark Dopson for the careful revision of the language of this thesis and manuscript.
I want to thank Professor Hans Wolf-Watz, Dr. Debra Milton, Professor Victoria Shingler, Professor Maria Fällman, and Dr. Cathrine Persson for introducing knowledge on bacterial secretion machinery, signaling and parasite virulence in the Monday journal club and for critical discussions in Wednesday meeting.
Thanks Veronica Janson for the help she provided for the apoptosis assay and statistical data analysis. Dr. Stina Rudolfsson for the help she provided, especially for case study with her was really a great pleasure. Thanks to Daniel Bergemalm and Charlotta Lindahl for the case study together.
I wish to thank friends and co-workers in the professor Bengt Hallberg and Dr. Ruth palmer group: Maria, Hai-ling, Charlotte, Lovisa, Christina Loren, Theresse (painting is fun), Gaurav, Jana, Margret, Caroline, Camilla, Mattias including all others (hej guys! I am very bad at remembering names). You people get relief from bacteria in Drosophila

ACKNOWLEDGEMENTS
51
discussions. It was great to go through the selected papers from the high impact journal every Friday morning with cakes and cookies. I am going to miss it……ahh. Christina (green girl with loudest voice) good luck with your PhD and with Magnus. My dear potato friend Emma Vernerssion supported my work in countless ways. I cannot thank you enough. Wish you and Jonny a very happy couple life. Hope our friendship keep going even I leave. Anna Jansson, your departure welcome my entry in this lab and I am lucky by getting you as a very good supportive friend. Great we have two papers together.
I want to thank Åsa Lundsten for all sorts of help needed to continue the experiments in the lab and friendly co-operation for which I do not think it would be enough by telling you thank. Thanks to all of the women in bacteriology lab for the solution and media stuff which makes lab life easier. Ylva thanks a lot for the suggestions in thesis writing. Thanks to Karin Boden and Terry Persson for the administrative help, save my days. All of the people in pathology specially Bodil, Åsa, Sofie, Anna, Nina, Sofia, Oleg, Peter, Linda, Pernilla including others are great, friendly and make my department life very easy, smooth and nice. Special thanks to Per Hörstedt for fixing computer technical things. Professor Anders bergh, Professor Göran Roos, Dr. Pernilla Wikström are really inspiring. Thanks Professor Kjel Grankvist for the apoptosis lessons and Dr. Eva Lundin for inspiration. I am looking forward to work with her in future. Dr. Maria Henriksson and Dr. Bethany Van Guelpen for review of the thesis.
I want to thank Ann Dolling, Mona Bergfors, Per Wester (FUN), Ola Grimsholm (MSU) including others who stood for me and encouraged me in difficult situation.
I wish to thanks Dr. Ivor Tittawella, working with you was a nice experience to start PhD project even it was not succeeded. At least we achieved one paper together that we had been aiming for. Anyway, we had the desired results when I (Bangladeshi woman) had been experiencing snowing for the first time in the dark December-January 2002, at -31ºC.
Eva Cristine Lundstrom always provides superb company occasionally and unoccasionally, outside the world of science as a family friend. She always inspires and comforts me in all pleasant and unpleasant moment.
Finally, I want to thank my brothers Abdullah and Ashraf for being supportive by phone, email and sms. My parents and parents in laws family for making this all possible. With their blessing, it’s possible to come and continue work in foreign life. Uncle family: Mamma Prodip Kanchan, Mammi, cousin Mithun and his wife, cousin Mollika with your blessings, life is full of fun in Umeå, Alhamdullillah. Special thanks to muslim brothers and sisters in Ålidhem mosque, making our life really comfortable and pleasant in Umeå. Allhamdullillah and Jazakallahu khairan (May Allah reward you with good). I miss my family members cousins, aunts, uncles, grandparents, specially my country Bangladesh.
During this study, some of them (heart and blood related) passed away: father in Law Noor ahmed, our 2nd child Mousa, my grand father and grand mother. INNA LILLAHI WA INNA ILAYHI RAJIUN (we are from Allah and to Him are we returning).
I may forget to mention lot of people here as I am very bad at recalling name. However, all of you (not mentioned) people are included in acknowledgment.
This work was supported financially by Swedish Cancer Society, Carl Tryggers Foundation and Riksförbundet, Cystic Fibros Forskningsfond, KEMPE Fond, Swedish research council, O.E and Elsa Johanssons research fund. All these are gratefully acknowledged. Thanks Ann Germann (Arkitektkopia) for the advice and thesis printing.
-------------- SUBHANALLAH (GLORY BE TO ALLAH) ----------------

REFERENCES
52
9. REFERENCES 1. Rahme, L.G., F.M. Ausubel, H. Cao, E. Drenkard, B.C. Goumnerov, G.W. Lau, S.
Mahajan-Miklos, J. Plotnikova, M.W. Tan, J. Tsongalis, C.L. Walendziewicz, and R.G. Tompkins, Plants and animals share functionally common bacterial virulence factors. Proc Natl Acad Sci U S A, 2000. 97(16): p. 8815-21.
2. Rahme, L.G., E.J. Stevens, S.F. Wolfort, J. Shao, R.G. Tompkins, and F.M. Ausubel, Common virulence factors for bacterial pathogenicity in plants and animals. Science, 1995. 268(5219): p. 1899-902.
3. Rahme, L.G., M.W. Tan, L. Le, S.M. Wong, R.G. Tompkins, S.B. Calderwood, and F.M. Ausubel, Use of model plant hosts to identify Pseudomonas aeruginosa virulence factors. Proc Natl Acad Sci U S A, 1997. 94(24): p. 13245-50.
4. Lyczak, J.B., C.L. Cannon, and G.B. Pier, Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect, 2000. 2(9): p. 1051-60.
5. Van Delden, C. and B.H. Iglewski, Cell-to-cell signaling and Pseudomonas aeruginosa infections. Emerg Infect Dis, 1998. 4(4): p. 551-60.
6. Lambert, P.A., Mechanisms of antibiotic resistance in Pseudomonas aeruginosa. J R Soc Med, 2002. 95 Suppl 41: p. 22-6.
7. Nakae, T., Role of membrane permeability in determining antibiotic resistance in Pseudomonas aeruginosa. Microbiol Immunol, 1995. 39(4): p. 221-9.
8. Nikaido, H., Multidrug efflux pumps of gram-negative bacteria. J Bacteriol, 1996. 178(20): p. 5853-9.
9. Reyes, E.A., M.J. Bale, W.H. Cannon, and J.M. Matsen, Identification of Pseudomonas aeruginosa by pyocyanin production on Tech agar. J Clin Microbiol, 1981. 13(3): p. 456-8.
10. Mavrodi, D.V., R.F. Bonsall, S.M. Delaney, M.J. Soule, G. Phillips, and L.S. Thomashow, Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J Bacteriol, 2001. 183(21): p. 6454-65.
11. Meyer, J.M., Pyoverdines: pigments, siderophores and potential taxonomic markers of fluorescent Pseudomonas species. Arch Microbiol, 2000. 174(3): p. 135-42.
12. Honda, M., H. Seki, K. Aso, T. Tanabe, K. Araya, T. Ohta, T. Ikeno, S. Lee, and T. Muratani, [A bacteriological study of multiple-drug-resistant Pseudomonas aeruginosa isolates derived from 2 patients]. J Uoeh, 2005. 27(2): p. 209-17.
13. Hoffmann, N., B. Lee, M. Hentzer, T.B. Rasmussen, Z. Song, H.K. Johansen, M. Givskov, and N. Hoiby, Azithromycin blocks quorum sensing and alginate polymer formation and increases the sensitivity to serum and stationary growth phase killing of P. aeruginosa and attenuates chronic P. aeruginosa lung infection in Cftr -/--mice. Antimicrob Agents Chemother, 2007.
14. Stover, C.K., X.Q. Pham, A.L. Erwin, S.D. Mizoguchi, P. Warrener, M.J. Hickey, F.S. Brinkman, W.O. Hufnagle, D.J. Kowalik, M. Lagrou, R.L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L.L. Brody, S.N. Coulter, K.R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G.K. Wong, Z. Wu, I.T. Paulsen, J. Reizer, M.H. Saier, R.E. Hancock, S. Lory, and M.V. Olson, Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature, 2000. 406(6799): p. 959-64.
15. Deitch, E.A., M. Dobke, and C.R. Baxter, Failure of local immunity. A potential cause of burn wound sepsis. Arch Surg, 1985. 120(1): p. 78-84.

REFERENCES
53
16. Burns, J.L., R.L. Gibson, S. McNamara, D. Yim, J. Emerson, M. Rosenfeld, P. Hiatt, K. McCoy, R. Castile, A.L. Smith, and B.W. Ramsey, Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis, 2001. 183(3): p. 444-52.
17. Yu, H., M. Hanes, C.E. Chrisp, J.C. Boucher, and V. Deretic, Microbial pathogenesis in cystic fibrosis: pulmonary clearance of mucoid Pseudomonas aeruginosa and inflammation in a mouse model of repeated respiratory challenge. Infect Immun, 1998. 66(1): p. 280-8.
18. Govan, J.R. and V. Deretic, Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev, 1996. 60(3): p. 539-74.
19. Murray, T.S., M. Egan, and B.I. Kazmierczak, Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr Opin Pediatr, 2007. 19(1): p. 83-8.
20. Willcox, M.D., Pseudomonas aeruginosa infection and inflammation during contact lens wear: a review. Optom Vis Sci, 2007. 84(4): p. 273-8.
21. Rumbaugh, K.P., J.A. Griswold, and A.N. Hamood, The role of quorum sensing in the in vivo virulence of Pseudomonas aeruginosa. Microbes Infect, 2000. 2(14): p. 1721-31.
22. Passador, L., J.M. Cook, M.J. Gambello, L. Rust, and B.H. Iglewski, Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science, 1993. 260(5111): p. 1127-30.
23. Nicas, T.I. and B.H. Iglewski, The contribution of exoproducts to virulence of Pseudomonas aeruginosa. Can J Microbiol, 1985. 31(4): p. 387-92.
24. de Bentzmann, S., P. Roger, F. Dupuit, O. Bajolet-Laudinat, C. Fuchey, M.C. Plotkowski, and E. Puchelle, Asialo GM1 is a receptor for Pseudomonas aeruginosa adherence to regenerating respiratory epithelial cells. Infect Immun, 1996. 64(5): p. 1582-8.
25. Gilboa-Garber, N., Towards anti-Pseudomonas aeruginosa adhesion therapy. Adv Exp Med Biol, 1996. 408: p. 39-50.
26. Feldman, M., R. Bryan, S. Rajan, L. Scheffler, S. Brunnert, H. Tang, and A. Prince, Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect Immun, 1998. 66(1): p. 43-51.
27. Pier, G.B., Pseudomonas aeruginosa lipopolysaccharide: A major virulence factor, initiator of inflammation and target for effective immunity. Int J Med Microbiol, 2007.
28. Hutchison, D.C., The role of proteases and antiproteases in bronchial secretions. Eur J Respir Dis Suppl, 1987. 153: p. 78-85.
29. Kawaharajo, K., J.Y. Homma, Y. Aoyama, K. Okada, and K. Morihara, Effects of protease and elastase from Pseudomonas aeruginosa on skin. Jpn J Exp Med, 1975. 45(2): p. 79-88.
30. Galloway, D.R., Pseudomonas aeruginosa elastase and elastolysis revisited: recent developments. Mol Microbiol, 1991. 5(10): p. 2315-21.
31. Heck, L.W., P.G. Alarcon, R.M. Kulhavy, K. Morihara, M.W. Russell, and J.F. Mestecky, Degradation of IgA proteins by Pseudomonas aeruginosa elastase. J Immunol, 1990. 144(6): p. 2253-7.
32. Kessler, S.P. and D.R. Galloway, Pseudomonas aeruginosa exotoxin A interaction with eucaryotic elongation factor 2. Role of the His426 residue. J Biol Chem, 1992. 267(27): p. 19107-11.
33. Jorgensen, R., A.R. Merrill, and G.R. Andersen, The life and death of translation elongation factor 2. Biochem Soc Trans, 2006. 34(Pt 1): p. 1-6.

REFERENCES
54
34. Vasil, M.L., C. Chamberlain, and C.C. Grant, Molecular studies of Pseudomonas exotoxin A gene. Infect Immun, 1986. 52(2): p. 538-48.
35. Woods, D.E. and B.H. Iglewski, Toxins of Pseudomonas aeruginosa: new perspectives. Rev Infect Dis, 1983. 5 Suppl 4: p. S715-22.
36. Vidal, D.R., P. Garrone, and J. Banchereau, Immunosuppressive effects of Pseudomonas aeruginosa exotoxin A on human B-lymphocytes. Toxicon, 1993. 31(1): p. 27-34.
37. Woods, D.E. and P.A. Sokol, Role of Pseudomonas aeruginosa extracellular enzymes in lung disease. Clin Invest Med, 1986. 9(2): p. 108-12.
38. Liu, P.V., Extracellular toxins of Pseudomonas aeruginosa. J Infect Dis, 1974. 130 Suppl(0): p. S94-9.
39. Read, R.C., P. Roberts, N. Munro, A. Rutman, A. Hastie, T. Shryock, R. Hall, W. McDonald-Gibson, V. Lund, G. Taylor, and et al., Effect of Pseudomonas aeruginosa rhamnolipids on mucociliary transport and ciliary beating. J Appl Physiol, 1992. 72(6): p. 2271-7.
40. Hoiby, N., H. Krogh Johansen, C. Moser, Z. Song, O. Ciofu, and A. Kharazmi, Pseudomonas aeruginosa and the in vitro and in vivo biofilm mode of growth. Microbes Infect, 2001. 3(1): p. 23-35.
41. Nivens, D.E., D.E. Ohman, J. Williams, and M.J. Franklin, Role of alginate and its O acetylation in formation of Pseudomonas aeruginosa microcolonies and biofilms. J Bacteriol, 2001. 183(3): p. 1047-57.
42. Mattick, J.S., Type IV pili and twitching motility. Annu Rev Microbiol, 2002. 56: p. 289-314.
43. Winther-Larsen, H.C., M.C. Wolfgang, J.P. van Putten, N. Roos, F.E. Aas, W.M. Egge-Jacobsen, B. Maier, and M. Koomey, Pseudomonas aeruginosa Type IV pilus expression in Neisseria gonorrhoeae: effects of pilin subunit composition on function and organelle dynamics. J Bacteriol, 2007.
44. Klausen, M., A. Heydorn, P. Ragas, L. Lambertsen, A. Aaes-Jorgensen, S. Molin, and T. Tolker-Nielsen, Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol Microbiol, 2003. 48(6): p. 1511-24.
45. Lee, C.A., Type III secretion systems: machines to deliver bacterial proteins into eukaryotic cells? Trends Microbiol, 1997. 5(4): p. 148-56.
46. Hueck, C.J., Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev, 1998. 62(2): p. 379-433.
47. Blocker, A., N. Jouihri, E. Larquet, P. Gounon, F. Ebel, C. Parsot, P. Sansonetti, and A. Allaoui, Structure and composition of the Shigella flexneri "needle complex", a part of its type III secreton. Mol Microbiol, 2001. 39(3): p. 652-63.
48. Frank, D.W., The exoenzyme S regulon of Pseudomonas aeruginosa. Mol Microbiol, 1997. 26(4): p. 621-9.
49. Finck-Barbancon, V., J. Goranson, L. Zhu, T. Sawa, J.P. Wiener-Kronish, S.M. Fleiszig, C. Wu, L. Mende-Mueller, and D.W. Frank, ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol, 1997. 25(3): p. 547-57.
50. Cowell, B.A., D.J. Evans, and S.M. Fleiszig, Actin cytoskeleton disruption by ExoY and its effects on Pseudomonas aeruginosa invasion. FEMS Microbiol Lett, 2005. 250(1): p. 71-6.
51. Yahr, T.L., A.J. Vallis, M.K. Hancock, J.T. Barbieri, and D.W. Frank, ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci U S A, 1998. 95(23): p. 13899-904.

REFERENCES
55
52. Sayner, S.L., D.W. Frank, J. King, H. Chen, J. VandeWaa, and T. Stevens, Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ Res, 2004. 95(2): p. 196-203.
53. Feltman, H., G. Schulert, S. Khan, M. Jain, L. Peterson, and A.R. Hauser, Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology, 2001. 147(Pt 10): p. 2659-69.
54. Sato, H. and D.W. Frank, ExoU is a potent intracellular phospholipase. Mol Microbiol, 2004. 53(5): p. 1279-90.
55. Sato, H., J.B. Feix, and D.W. Frank, Identification of superoxide dismutase as a cofactor for the pseudomonas type III toxin, ExoU. Biochemistry, 2006. 45(34): p. 10368-75.
56. Rabin, S.D. and A.R. Hauser, Pseudomonas aeruginosa ExoU, a toxin transported by the type III secretion system, kills Saccharomyces cerevisiae. Infect Immun, 2003. 71(7): p. 4144-50.
57. Shaver, C.M. and A.R. Hauser, Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect Immun, 2004. 72(12): p. 6969-77.
58. Wareham, D.W. and M.A. Curtis, A genotypic and phenotypic comparison of type III secretion profiles of Pseudomonas aeruginosa cystic fibrosis and bacteremia isolates. Int J Med Microbiol, 2007. 297(4): p. 227-34.
59. Yahr, T.L., J. Goranson, and D.W. Frank, Exoenzyme S of Pseudomonas aeruginosa is secreted by a type III pathway. Mol Microbiol, 1996. 22(5): p. 991-1003.
60. Maresso, A.W., M.R. Baldwin, and J.T. Barbieri, Ezrin/radixin/moesin proteins are high affinity targets for ADP-ribosylation by Pseudomonas aeruginosa ExoS. J Biol Chem, 2004. 279(37): p. 38402-8.
61. Krall, R., J. Sun, K.J. Pederson, and J.T. Barbieri, In vivo rho GTPase-activating protein activity of Pseudomonas aeruginosa cytotoxin ExoS. Infect Immun, 2002. 70(1): p. 360-7.
62. Kazmierczak, B.I. and J.N. Engel, Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42. Infect Immun, 2002. 70(4): p. 2198-205.
63. Sundin, C., M.L. Henriksson, B. Hallberg, A. Forsberg, and E. Frithz-Lindsten, Exoenzyme T of Pseudomonas aeruginosa elicits cytotoxicity without interfering with Ras signal transduction. Cell Microbiol, 2001. 3(4): p. 237-46.
64. Wu, C.L., P. Domenico, D.J. Hassett, T.J. Beveridge, A.R. Hauser, and J.A. Kazzaz, Subinhibitory bismuth-thiols reduce virulence of Pseudomonas aeruginosa. Am J Respir Cell Mol Biol, 2002. 26(6): p. 731-8.
65. Balachandran, P., L. Dragone, L. Garrity-Ryan, A. Lemus, A. Weiss, and J. Engel, The ubiquitin ligase Cbl-b limits Pseudomonas aeruginosa exotoxin T-mediated virulence. J Clin Invest, 2007. 117(2): p. 419-27.
66. Iglewski, B.H., J. Sadoff, M.J. Bjorn, and E.S. Maxwell, Pseudomonas aeruginosa exoenzyme S: an adenosine diphosphate ribosyltransferase distinct from toxin A. Proc Natl Acad Sci U S A, 1978. 75(7): p. 3211-5.
67. Bjorn, M.J., O.R. Pavlovskis, M.R. Thompson, and B.H. Iglewski, Production of exoenzyme S during Pseudomonas aeruginosa infections of burned mice. Infect Immun, 1979. 24(3): p. 837-42.
68. Kulich, S.M., D.W. Frank, and J.T. Barbieri, Purification and characterization of exoenzyme S from Pseudomonas aeruginosa 388. Infect Immun, 1993. 61(1): p. 307-13.

REFERENCES
56
69. Coburn, J., Pseudomonas aeruginosa exoenzyme S. Curr Top Microbiol Immunol, 1992. 175: p. 133-43.
70. Kulich, S.M., T.L. Yahr, L.M. Mende-Mueller, J.T. Barbieri, and D.W. Frank, Cloning the structural gene for the 49-kDa form of exoenzyme S (exoS) from Pseudomonas aeruginosa strain 388. J Biol Chem, 1994. 269(14): p. 10431-7.
71. Yahr, T.L., J.T. Barbieri, and D.W. Frank, Genetic relationship between the 53- and 49-kilodalton forms of exoenzyme S from Pseudomonas aeruginosa. J Bacteriol, 1996. 178(5): p. 1412-9.
72. Shaver, C.M. and A.R. Hauser, Interactions between effector proteins of the Pseudomonas aeruginosa type III secretion system do not significantly affect several measures of disease severity in mammals. Microbiology, 2006. 152(Pt 1): p. 143-52.
73. Wareham, D.W. and M.A. Curtis, A genotypic and phenotypic comparison of type III secretion profiles of Pseudomonas aeruginosa cystic fibrosis and bacteremia isolates. Int J Med Microbiol, 2007. 297(4): p. 227-234.
74. Pederson, K.J., R. Krall, M.J. Riese, and J.T. Barbieri, Intracellular localization modulates targeting of ExoS, a type III cytotoxin, to eukaryotic signalling proteins. Mol Microbiol, 2002. 46(5): p. 1381-90.
75. Zhang, Y. and J.T. Barbieri, A leucine-rich motif targets Pseudomonas aeruginosa ExoS within mammalian cells. Infect Immun, 2005. 73(12): p. 7938-45.
76. Goehring, U.M., G. Schmidt, K.J. Pederson, K. Aktories, and J.T. Barbieri, The N-terminal domain of Pseudomonas aeruginosa exoenzyme S is a GTPase-activating protein for Rho GTPases. J Biol Chem, 1999. 274(51): p. 36369-72.
77. Henriksson, M.L., M.S. Francis, A. Peden, M. Aili, K. Stefansson, R. Palmer, A. Aitken, and B. Hallberg, A nonphosphorylated 14-3-3 binding motif on exoenzyme S that is functional in vivo. Eur J Biochem, 2002. 269(20): p. 4921-9.
78. Henriksson, M.L., U. Troller, and B. Hallberg, 14-3-3 proteins are required for the inhibition of Ras by exoenzyme S. Biochem J, 2000. 349 Pt 3: p. 697-701.
79. Frithz-Lindsten, E., Y. Du, R. Rosqvist, and A. Forsberg, Intracellular targeting of exoenzyme S of Pseudomonas aeruginosa via type III-dependent translocation induces phagocytosis resistance, cytotoxicity and disruption of actin microfilaments. Mol Microbiol, 1997. 25(6): p. 1125-39.
80. Henriksson, M.L., C. Sundin, A.L. Jansson, A. Forsberg, R.H. Palmer, and B. Hallberg, Exoenzyme S shows selective ADP-ribosylation and GTPase-activating protein (GAP) activities towards small GTPases in vivo. Biochem J, 2002. 367(Pt 3): p. 617-28.
81. Pederson, K.J., A.J. Vallis, K. Aktories, D.W. Frank, and J.T. Barbieri, The amino-terminal domain of Pseudomonas aeruginosa ExoS disrupts actin filaments via small-molecular-weight GTP-binding proteins. Mol Microbiol, 1999. 32(2): p. 393-401.
82. Riese, M.J., U.M. Goehring, M.E. Ehrmantraut, J. Moss, J.T. Barbieri, K. Aktories, and G. Schmidt, Auto-ADP-ribosylation of Pseudomonas aeruginosa ExoS. J Biol Chem, 2002. 277(14): p. 12082-8.
83. Wurtele, M., L. Renault, J.T. Barbieri, A. Wittinghofer, and E. Wolf, Structure of the ExoS GTPase activating domain. FEBS Lett, 2001. 491(1-2): p. 26-9.
84. Wurtele, M., E. Wolf, K.J. Pederson, G. Buchwald, M.R. Ahmadian, J.T. Barbieri, and A. Wittinghofer, How the Pseudomonas aeruginosa ExoS toxin downregulates Rac. Nat Struct Biol, 2001. 8(1): p. 23-6.
85. Knight, D.A., V. Finck-Barbancon, S.M. Kulich, and J.T. Barbieri, Functional domains of Pseudomonas aeruginosa exoenzyme S. Infect Immun, 1995. 63(8): p. 3182-6.

REFERENCES
57
86. Coburn, J., S.T. Dillon, B.H. Iglewski, and D.M. Gill, Exoenzyme S of Pseudomonas aeruginosa ADP-ribosylates the intermediate filament protein vimentin. Infect Immun, 1989. 57(3): p. 996-8.
87. Ganesan, A.K., D.W. Frank, R.P. Misra, G. Schmidt, and J.T. Barbieri, Pseudomonas aeruginosa exoenzyme S ADP-ribosylates Ras at multiple sites. J Biol Chem, 1998. 273(13): p. 7332-7.
88. Riese, M.J., A. Wittinghofer, and J.T. Barbieri, ADP ribosylation of Arg41 of Rap by ExoS inhibits the ability of Rap to interact with its guanine nucleotide exchange factor, C3G. Biochemistry, 2001. 40(11): p. 3289-94.
89. DiNovo, A.A., K.L. Schey, W.S. Vachon, E.M. McGuffie, J.C. Olson, and T.S. Vincent, ADP-ribosylation of cyclophilin A by Pseudomonas aeruginosa exoenzyme S. Biochemistry, 2006. 45(14): p. 4664-73.
90. Fu, H., J. Coburn, and R.J. Collier, The eukaryotic host factor that activates exoenzyme S of Pseudomonas aeruginosa is a member of the 14-3-3 protein family. Proc Natl Acad Sci U S A, 1993. 90(6): p. 2320-4.
91. Coburn, J. and D.M. Gill, ADP-ribosylation of p21ras and related proteins by Pseudomonas aeruginosa exoenzyme S. Infect Immun, 1991. 59(11): p. 4259-62.
92. Liu, S., S.M. Kulich, and J.T. Barbieri, Identification of glutamic acid 381 as a candidate active site residue of Pseudomonas aeruginosa exoenzyme S. Biochemistry, 1996. 35(8): p. 2754-8.
93. Radke, J., K.J. Pederson, and J.T. Barbieri, Pseudomonas aeruginosa exoenzyme S is a biglutamic acid ADP-ribosyltransferase. Infect Immun, 1999. 67(3): p. 1508-10.
94. Sun, J., A.W. Maresso, J.J. Kim, and J.T. Barbieri, How bacterial ADP-ribosylating toxins recognize substrates. Nat Struct Mol Biol, 2004. 11(9): p. 868-76.
95. Takai, Y., T. Sasaki, and T. Matozaki, Small GTP-binding proteins. Physiol Rev, 2001. 81(1): p. 153-208.
96. Bos, J.L., H. Rehmann, and A. Wittinghofer, GEFs and GAPs: critical elements in the control of small G proteins. Cell, 2007. 129(5): p. 865-77.
97. Vetter, I.R. and A. Wittinghofer, The guanine nucleotide-binding switch in three dimensions. Science, 2001. 294(5545): p. 1299-304.
98. Rajalingam, K., R. Schreck, U.R. Rapp, and S. Albert, Ras oncogenes and their downstream targets. Biochim Biophys Acta, 2007. 1773(8): p. 1177-95.
99. Scheele, J.S., R.E. Marks, and G.R. Boss, Signaling by small GTPases in the immune system. Immunol Rev, 2007. 218: p. 92-101.
100. Mitin, N., K.L. Rossman, and C.J. Der, Signaling interplay in Ras superfamily function. Curr Biol, 2005. 15(14): p. R563-74.
101. Downward, J., Signal transduction. Prelude to an anniversary for the RAS oncogene. Science, 2006. 314(5798): p. 433-4.
102. Roberts, P.J. and C.J. Der, Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene, 2007. 26(22): p. 3291-310.
103. Willumsen, B.M., A. Christensen, N.L. Hubbert, A.G. Papageorge, and D.R. Lowy, The p21 ras C-terminus is required for transformation and membrane association. Nature, 1984. 310(5978): p. 583-6.
104. Basso, A.D., P. Kirschmeier, and W.R. Bishop, Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res, 2006. 47(1): p. 15-31.
105. Hancock, J.F., K. Cadwallader, H. Paterson, and C.J. Marshall, A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. Embo J, 1991. 10(13): p. 4033-9.
106. Hancock, J.F., A.I. Magee, J.E. Childs, and C.J. Marshall, All ras proteins are polyisoprenylated but only some are palmitoylated. Cell, 1989. 57(7): p. 1167-77.

REFERENCES
58
107. Sebti, S.M. and C.J. Der, Opinion: Searching for the elusive targets of farnesyltransferase inhibitors. Nat Rev Cancer, 2003. 3(12): p. 945-51.
108. Wright, L.P. and M.R. Philips, Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res, 2006. 47(5): p. 883-91.
109. Tian, T., A. Harding, K. Inder, S. Plowman, R.G. Parton, and J.F. Hancock, Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol, 2007.
110. Caron, E., Cellular functions of the Rap1 GTP-binding protein: a pattern emerges. J Cell Sci, 2003. 116(Pt 3): p. 435-40.
111. Kitayama, H., Y. Sugimoto, T. Matsuzaki, Y. Ikawa, and M. Noda, A ras-related gene with transformation suppressor activity. Cell, 1989. 56(1): p. 77-84.
112. Paganini, S., G.F. Guidetti, S. Catricala, P. Trionfini, S. Panelli, C. Balduini, and M. Torti, Identification and biochemical characterization of Rap2C, a new member of the Rap family of small GTP-binding proteins. Biochimie, 2006. 88(3-4): p. 285-95.
113. Hancock, J.F., Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol, 2003. 4(5): p. 373-84.
114. Bos, J.L., J. de Rooij, and K.A. Reedquist, Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol, 2001. 2(5): p. 369-77.
115. Stork, P.J. and T.J. Dillon, Multiple roles of Rap1 in hematopoietic cells: complementary versus antagonistic functions. Blood, 2005. 106(9): p. 2952-61.
116. Kooistra, M.R., N. Dube, and J.L. Bos, Rap1: a key regulator in cell-cell junction formation. J Cell Sci, 2007. 120(Pt 1): p. 17-22.
117. Yoshikawa, Y., T. Satoh, T. Tamura, P. Wei, S.E. Bilasy, H. Edamatsu, A. Aiba, K. Katagiri, T. Kinashi, K. Nakao, and T. Kataoka, The M-Ras-RA-GEF-2-Rap1 Pathway Mediates Tumor Necrosis Factor-{alpha} dependent Regulation of Integrin Activation in Splenocytes. Mol Biol Cell, 2007. 18(8): p. 2949-2959.
118. Bos, J.L., Linking Rap to cell adhesion. Curr Opin Cell Biol, 2005. 17(2): p. 123-8. 119. Quilliam, L.A., J.F. Rebhun, and A.F. Castro, A growing family of guanine nucleotide
exchange factors is responsible for activation of Ras-family GTPases. Prog Nucleic Acid Res Mol Biol, 2002. 71: p. 391-444.
120. Hattori, S. and M. Matsuda, [Activation of Rap1, antagonist to ras, by Crk-C3G]. Gan To Kagaku Ryoho, 1997. 24(11): p. 1414-21.
121. Jia, J., Y. Wang, L. Zhou, and S. Jin, Expression of Pseudomonas aeruginosa toxin ExoS effectively induces apoptosis in host cells. Infect Immun, 2006. 74(12): p. 6557-70.
122. Fraylick, J.E., M.J. Riese, T.S. Vincent, J.T. Barbieri, and J.C. Olson, ADP-ribosylation and functional effects of Pseudomonas exoenzyme S on cellular RalA. Biochemistry, 2002. 41(30): p. 9680-7.
123. Fraylick, J.E., E.A. Rucks, D.M. Greene, T.S. Vincent, and J.C. Olson, Eukaryotic cell determination of ExoS ADP-ribosyltransferase substrate specificity. Biochem Biophys Res Commun, 2002. 291(1): p. 91-100.
124. Kaufman, M.R., J. Jia, L. Zeng, U. Ha, M. Chow, and S. Jin, Pseudomonas aeruginosa mediated apoptosis requires the ADP-ribosylating activity of exoS. Microbiology, 2000. 146 ( Pt 10): p. 2531-41.
125. Peyssonnaux, C., S. Provot, M.P. Felder-Schmittbuhl, G. Calothy, and A. Eychene, Induction of postmitotic neuroretina cell proliferation by distinct Ras downstream signaling pathways. Mol Cell Biol, 2000. 20(19): p. 7068-79.

REFERENCES
59
126. Yan, J., S. Roy, A. Apolloni, A. Lane, and J.F. Hancock, Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem, 1998. 273(37): p. 24052-6.
127. McCubrey, J.A., L.S. Steelman, W.H. Chappell, S.L. Abrams, E.W. Wong, F. Chang, B. Lehmann, D.M. Terrian, M. Milella, A. Tafuri, F. Stivala, M. Libra, J. Basecke, C. Evangelisti, A.M. Martelli, and R.A. Franklin, Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta, 2007. 1773(8): p. 1263-84.
128. Luo, Z., G. Tzivion, P.J. Belshaw, D. Vavvas, M. Marshall, and J. Avruch, Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature, 1996. 383(6596): p. 181-5.
129. Fabian, J.R., I.O. Daar, and D.K. Morrison, Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol Cell Biol, 1993. 13(11): p. 7170-9.
130. Dhillon, A.S., S. Meikle, Z. Yazici, M. Eulitz, and W. Kolch, Regulation of Raf-1 activation and signalling by dephosphorylation. Embo J, 2002. 21(1-2): p. 64-71.
131. Brummer, T., P.E. Shaw, M. Reth, and Y. Misawa, Inducible gene deletion reveals different roles for B-Raf and Raf-1 in B-cell antigen receptor signalling. Embo J, 2002. 21(21): p. 5611-22.
132. Alessi, D.R., Y. Saito, D.G. Campbell, P. Cohen, G. Sithanandam, U. Rapp, A. Ashworth, C.J. Marshall, and S. Cowley, Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. Embo J, 1994. 13(7): p. 1610-9.
133. McCubrey, J.A., L.S. Steelman, S.L. Abrams, J.T. Lee, F. Chang, F.E. Bertrand, P.M. Navolanic, D.M. Terrian, R.A. Franklin, A.B. D'Assoro, J.L. Salisbury, M.C. Mazzarino, F. Stivala, and M. Libra, Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul, 2006. 46: p. 249-79.
134. Chang, F., L.S. Steelman, J.T. Lee, J.G. Shelton, P.M. Navolanic, W.L. Blalock, R.A. Franklin, and J.A. McCubrey, Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia, 2003. 17(7): p. 1263-93.
135. Pouyssegur, J., V. Volmat, and P. Lenormand, Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol, 2002. 64(5-6): p. 755-63.
136. Wang, Z., T.J. Dillon, V. Pokala, S. Mishra, K. Labudda, B. Hunter, and P.J. Stork, Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol Cell Biol, 2006. 26(6): p. 2130-45.
137. Hattori, M. and N. Minato, Rap1 GTPase: functions, regulation, and malignancy. J Biochem (Tokyo), 2003. 134(4): p. 479-84.
138. Li, Y., S. Asuri, J.F. Rebhun, A.F. Castro, N.C. Paranavitana, and L.A. Quilliam, The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J Biol Chem, 2006. 281(5): p. 2506-14.
139. Papakonstanti, E.A., A.J. Ridley, and B. Vanhaesebroeck, The p110delta isoform of PI 3-kinase negatively controls RhoA and PTEN. Embo J, 2007. 26(13): p. 3050-61.
140. Vanhaesebroeck, B. and D.R. Alessi, The PI3K-PDK1 connection: more than just a road to PKB. Biochem J, 2000. 346 Pt 3: p. 561-76.
141. Chang, F., J.T. Lee, P.M. Navolanic, L.S. Steelman, J.G. Shelton, W.L. Blalock, R.A. Franklin, and J.A. McCubrey, Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia, 2003. 17(3): p. 590-603.

REFERENCES
60
142. Isakoff, S.J., T. Cardozo, J. Andreev, Z. Li, K.M. Ferguson, R. Abagyan, M.A. Lemmon, A. Aronheim, and E.Y. Skolnik, Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. Embo J, 1998. 17(18): p. 5374-87.
143. Sharrard, R.M. and N.J. Maitland, Regulation of protein kinase B activity by PTEN and SHIP2 in human prostate-derived cell lines. Cell Signal, 2007. 19(1): p. 129-38.
144. Wandzioch, E., C.E. Edling, R.H. Palmer, L. Carlsson, and B. Hallberg, Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood, 2004. 104(1): p. 51-7.
145. Gupta, S., A.R. Ramjaun, P. Haiko, Y. Wang, P.H. Warne, B. Nicke, E. Nye, G. Stamp, K. Alitalo, and J. Downward, Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell, 2007. 129(5): p. 957-68.
146. Xu, G. and Y. Shi, Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res, 2007.
147. Spector, M.S., S. Desnoyers, D.J. Hoeppner, and M.O. Hengartner, Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature, 1997. 385(6617): p. 653-6.
148. Zha, J., H. Harada, E. Yang, J. Jockel, and S.J. Korsmeyer, Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell, 1996. 87(4): p. 619-28.
149. Cardone, M.H., N. Roy, H.R. Stennicke, G.S. Salvesen, T.F. Franke, E. Stanbridge, S. Frisch, and J.C. Reed, Regulation of cell death protease caspase-9 by phosphorylation. Science, 1998. 282(5392): p. 1318-21.
150. Dong, S., S. Kang, T.L. Gu, S. Kardar, H. Fu, S. Lonial, H.J. Khoury, F. Khuri, and J. Chen, 14-3-3 Integrates prosurvival signals mediated by the AKT and MAPK pathways in ZNF198-FGFR1-transformed hematopoietic cells. Blood, 2007. 110(1): p. 360-9.
151. Datta, S.R., H. Dudek, X. Tao, S. Masters, H. Fu, Y. Gotoh, and M.E. Greenberg, Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell, 1997. 91(2): p. 231-41.
152. Romashkova, J.A. and S.S. Makarov, NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature, 1999. 401(6748): p. 86-90.
153. Greer, E.L. and A. Brunet, FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene, 2005. 24(50): p. 7410-25.
154. Katoh, M. and M. Katoh, Human FOX gene family (Review). Int J Oncol, 2004. 25(5): p. 1495-500.
155. Reagan-Shaw, S. and N. Ahmad, The role of Forkhead-box Class O (FoxO) transcription factors in cancer: A target for the management of cancer. Toxicol Appl Pharmacol, 2006.
156. Dijkers, P.F., R.H. Medema, C. Pals, L. Banerji, N.S. Thomas, E.W. Lam, B.M. Burgering, J.A. Raaijmakers, J.W. Lammers, L. Koenderman, and P.J. Coffer, Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol Cell Biol, 2000. 20(24): p. 9138-48.
157. Barthel, A., E.A. Ostrakhovitch, P.L. Walter, A. Kampkotter, and L.O. Klotz, Stimulation of phosphoinositide 3-kinase/Akt signaling by copper and zinc ions: Mechanisms and consequences. Arch Biochem Biophys, 2007. 463(2): p. 175-82.
158. Burgering, B.M. and G.J. Kops, Cell cycle and death control: long live Forkheads. Trends Biochem Sci, 2002. 27(7): p. 352-60.

REFERENCES
61
159. Van Der Heide, L.P., M.F. Hoekman, and M.P. Smidt, The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J, 2004. 380(Pt 2): p. 297-309.
160. van der Heide, L.P. and M.P. Smidt, Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci, 2005. 30(2): p. 81-6.
161. Nicholson, D.W., Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ, 1999. 6(11): p. 1028-42.
162. Fuentes-Prior, P. and G.S. Salvesen, The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J, 2004. 384(Pt 2): p. 201-32.
163. Turk, B. and V. Stoka, Protease signalling in cell death: caspases versus cysteine cathepsins. FEBS Lett, 2007. 581(15): p. 2761-7.
164. Kumar, S., Caspase function in programmed cell death. Cell Death Differ, 2007. 14(1): p. 32-43.
165. Riedl, S.J. and Y. Shi, Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol, 2004. 5(11): p. 897-907.
166. Li, P., D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, and X. Wang, Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell, 1997. 91(4): p. 479-89.
167. Ichimura, T., T. Isobe, T. Okuyama, N. Takahashi, K. Araki, R. Kuwano, and Y. Takahashi, Molecular cloning of cDNA coding for brain-specific 14-3-3 protein, a protein kinase-dependent activator of tyrosine and tryptophan hydroxylases. Proc Natl Acad Sci U S A, 1988. 85(19): p. 7084-8.
168. Toker, A., L.A. Sellers, B. Amess, Y. Patel, A. Harris, and A. Aitken, Multiple isoforms of a protein kinase C inhibitor (KCIP-1/14-3-3) from sheep brain. Amino acid sequence of phosphorylated forms. Eur J Biochem, 1992. 206(2): p. 453-61.
169. Aitken, A., D.B. Collinge, B.P. van Heusden, T. Isobe, P.H. Roseboom, G. Rosenfeld, and J. Soll, 14-3-3 proteins: a highly conserved, widespread family of eukaryotic proteins. Trends Biochem Sci, 1992. 17(12): p. 498-501.
170. Rosenquist, M., M. Alsterfjord, C. Larsson, and M. Sommarin, Data mining the Arabidopsis genome reveals fifteen 14-3-3 genes. Expression is demonstrated for two out of five novel genes. Plant Physiol, 2001. 127(1): p. 142-9.
171. Wang, W. and D.C. Shakes, Molecular evolution of the 14-3-3 protein family. J Mol Evol, 1996. 43(4): p. 384-98.
172. Aitken, A., Functional specificity in 14-3-3 isoform interactions through dimer formation and phosphorylation. Chromosome location of mammalian isoforms and variants. Plant Mol Biol, 2002. 50(6): p. 993-1010.
173. Rosenquist, M., P. Sehnke, R.J. Ferl, M. Sommarin, and C. Larsson, Evolution of the 14-3-3 protein family: does the large number of isoforms in multicellular organisms reflect functional specificity? J Mol Evol, 2000. 51(5): p. 446-58.
174. Brunet, A., F. Kanai, J. Stehn, J. Xu, D. Sarbassova, J.V. Frangioni, S.N. Dalal, J.A. DeCaprio, M.E. Greenberg, and M.B. Yaffe, 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol, 2002. 156(5): p. 817-28.
175. Roberts, M.R., 14-3-3 proteins find new partners in plant cell signalling. Trends Plant Sci, 2003. 8(5): p. 218-23.
176. Bridges, D. and G.B. Moorhead, 14-3-3 proteins: a number of functions for a numbered protein. Sci STKE, 2004. 2004(242): p. re10.
177. Tzivion, G., Y.H. Shen, and J. Zhu, 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene, 2001. 20(44): p. 6331-8.

REFERENCES
62
178. Yaffe, M.B., How do 14-3-3 proteins work?-- Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett, 2002. 513(1): p. 53-7.
179. May, T. and J. Soll, 14-3-3 proteins form a guidance complex with chloroplast precursor proteins in plants. Plant Cell, 2000. 12(1): p. 53-64.
180. Muslin, A.J. and H. Xing, 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal, 2000. 12(11-12): p. 703-9.
181. Liu, D., J. Bienkowska, C. Petosa, R.J. Collier, H. Fu, and R. Liddington, Crystal structure of the zeta isoform of the 14-3-3 protein. Nature, 1995. 376(6536): p. 191-4.
182. Wang, B., H. Yang, Y.C. Liu, T. Jelinek, L. Zhang, E. Ruoslahti, and H. Fu, Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry, 1999. 38(38): p. 12499-504.
183. Petosa, C., S.C. Masters, L.A. Bankston, J. Pohl, B. Wang, H. Fu, and R.C. Liddington, 14-3-3zeta binds a phosphorylated Raf peptide and an unphosphorylated peptide via its conserved amphipathic groove. J Biol Chem, 1998. 273(26): p. 16305-10.
184. Yaffe, M.B., K. Rittinger, S. Volinia, P.R. Caron, A. Aitken, H. Leffers, S.J. Gamblin, S.J. Smerdon, and L.C. Cantley, The structural basis for 14-3-3:phosphopeptide binding specificity. Cell, 1997. 91(7): p. 961-71.
185. Rittinger, K., J. Budman, J. Xu, S. Volinia, L.C. Cantley, S.J. Smerdon, S.J. Gamblin, and M.B. Yaffe, Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell, 1999. 4(2): p. 153-66.
186. Obsil, T., R. Ghirlando, D.C. Klein, S. Ganguly, and F. Dyda, Crystal structure of the 14-3-3zeta:serotonin N-acetyltransferase complex. a role for scaffolding in enzyme regulation. Cell, 2001. 105(2): p. 257-67.
187. Benzinger, A., G.M. Popowicz, J.K. Joy, S. Majumdar, T.A. Holak, and H. Hermeking, The crystal structure of the non-liganded 14-3-3sigma protein: insights into determinants of isoform specific ligand binding and dimerization. Cell Res, 2005. 15(4): p. 219-27.
188. Wilker, E.W., R.A. Grant, S.C. Artim, and M.B. Yaffe, A structural basis for 14-3-3sigma functional specificity. J Biol Chem, 2005. 280(19): p. 18891-8.
189. Xiao, B., S.J. Smerdon, D.H. Jones, G.G. Dodson, Y. Soneji, A. Aitken, and S.J. Gamblin, Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature, 1995. 376(6536): p. 188-91.
190. Aitken, A., D. Jones, Y. Soneji, and S. Howell, 14-3-3 proteins: biological function and domain structure. Biochem Soc Trans, 1995. 23(3): p. 605-11.
191. Jones, D.H., S. Ley, and A. Aitken, Isoforms of 14-3-3 protein can form homo- and heterodimers in vivo and in vitro: implications for function as adapter proteins. FEBS Lett, 1995. 368(1): p. 55-8.
192. Muslin, A.J., J.W. Tanner, P.M. Allen, and A.S. Shaw, Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell, 1996. 84(6): p. 889-97.
193. Hallberg, B., Exoenzyme S binds its cofactor 14-3-3 through a non-phosphorylated motif. Biochem Soc Trans, 2002. 30(4): p. 401-5.
194. Moorhead, G., P. Douglas, V. Cotelle, J. Harthill, N. Morrice, S. Meek, U. Deiting, M. Stitt, M. Scarabel, A. Aitken, and C. MacKintosh, Phosphorylation-dependent interactions between enzymes of plant metabolism and 14-3-3 proteins. Plant J, 1999. 18(1): p. 1-12.
195. Aitken, A., 14-3-3 and its possible role in co-ordinating multiple signalling pathways. Trends Cell Biol, 1996. 6(9): p. 341-7.

REFERENCES
63
196. Giles, N., A. Forrest, and B. Gabrielli, 14-3-3 acts as an intramolecular bridge to regulate cdc25B localization and activity. J Biol Chem, 2003. 278(31): p. 28580-7.
197. Ganguly, S., J.L. Weller, A. Ho, P. Chemineau, B. Malpaux, and D.C. Klein, Melatonin synthesis: 14-3-3-dependent activation and inhibition of arylalkylamine N-acetyltransferase mediated by phosphoserine-205. Proc Natl Acad Sci U S A, 2005. 102(4): p. 1222-7.
198. Coblitz, B., S. Shikano, M. Wu, S.B. Gabelli, L.M. Cockrell, M. Spieker, Y. Hanyu, H. Fu, L.M. Amzel, and M. Li, C-terminal recognition by 14-3-3 proteins for surface expression of membrane receptors. J Biol Chem, 2005. 280(43): p. 36263-72.
199. Shikano, S., B. Coblitz, M. Wu, and M. Li, 14-3-3 proteins: regulation of endoplasmic reticulum localization and surface expression of membrane proteins. Trends Cell Biol, 2006. 16(7): p. 370-5.
200. Coblitz, B., M. Wu, S. Shikano, and M. Li, C-terminal binding: an expanded repertoire and function of 14-3-3 proteins. FEBS Lett, 2006. 580(6): p. 1531-5.
201. Seimiya, H., H. Sawada, Y. Muramatsu, M. Shimizu, K. Ohko, K. Yamane, and T. Tsuruo, Involvement of 14-3-3 proteins in nuclear localization of telomerase. Embo J, 2000. 19(11): p. 2652-61.
202. Patel, A., N. Cummings, M. Batchelor, P.J. Hill, T. Dubois, K.H. Mellits, G. Frankel, and I. Connerton, Host protein interactions with enteropathogenic Escherichia coli (EPEC): 14-3-3tau binds Tir and has a role in EPEC-induced actin polymerization. Cell Microbiol, 2006. 8(1): p. 55-71.
203. Sumioka, A., S. Nagaishi, T. Yoshida, A. Lin, M. Miura, and T. Suzuki, Role of 14-3-3gamma in FE65-dependent gene transactivation mediated by the amyloid beta-protein precursor cytoplasmic fragment. J Biol Chem, 2005. 280(51): p. 42364-74.
204. Clark, K.L., A. Oelke, M.E. Johnson, K.D. Eilert, P.C. Simpson, and S.C. Todd, CD81 associates with 14-3-3 in a redox-regulated palmitoylation-dependent manner. J Biol Chem, 2004. 279(19): p. 19401-6.
205. Macdonald, N., J.P. Welburn, M.E. Noble, A. Nguyen, M.B. Yaffe, D. Clynes, J.G. Moggs, G. Orphanides, S. Thomson, J.W. Edmunds, A.L. Clayton, J.A. Endicott, and L.C. Mahadevan, Molecular basis for the recognition of phosphorylated and phosphoacetylated histone h3 by 14-3-3. Mol Cell, 2005. 20(2): p. 199-211.
206. Masters, S.C., K.J. Pederson, L. Zhang, J.T. Barbieri, and H. Fu, Interaction of 14-3-3 with a nonphosphorylated protein ligand, exoenzyme S of Pseudomonas aeruginosa. Biochemistry, 1999. 38(16): p. 5216-21.
207. Zhang, L., H. Wang, D. Liu, R. Liddington, and H. Fu, Raf-1 kinase and exoenzyme S interact with 14-3-3zeta through a common site involving lysine 49. J Biol Chem, 1997. 272(21): p. 13717-24.
208. Zhang, L., J. Chen, and H. Fu, Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc Natl Acad Sci U S A, 1999. 96(15): p. 8511-5.
209. Campbell, J.K., R. Gurung, S. Romero, C.J. Speed, R.K. Andrews, M.C. Berndt, and C.A. Mitchell, Activation of the 43 kDa inositol polyphosphate 5-phosphatase by 14-3-3zeta. Biochemistry, 1997. 36(49): p. 15363-70.
210. Gu, M. and X. Du, A novel ligand-binding site in the zeta-form 14-3-3 protein recognizing the platelet glycoprotein Ibalpha and distinct from the c-Raf-binding site. J Biol Chem, 1998. 273(50): p. 33465-71.
211. Yang, X., W.H. Lee, F. Sobott, E. Papagrigoriou, C.V. Robinson, J.G. Grossmann, M. Sundstrom, D.A. Doyle, and J.M. Elkins, Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc Natl Acad Sci U S A, 2006. 103(46): p. 17237-42.

REFERENCES
64
212. Mackintosh, C., Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem J, 2004. 381(Pt 2): p. 329-42.
213. Porter, G.W., F.R. Khuri, and H. Fu, Dynamic 14-3-3/client protein interactions integrate survival and apoptotic pathways. Semin Cancer Biol, 2006. 16(3): p. 193-202.
214. Datta, S.R., A. Katsov, L. Hu, A. Petros, S.W. Fesik, M.B. Yaffe, and M.E. Greenberg, 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell, 2000. 6(1): p. 41-51.
215. Hermeking, H. and A. Benzinger, 14-3-3 proteins in cell cycle regulation. Semin Cancer Biol, 2006. 16(3): p. 183-92.
216. Nakanishi, K., S. Hashizume, M. Kato, T. Honjoh, Y. Setoguchi, and K. Yasumoto, Elevated expression levels of the 14-3-3 family of proteins in lung cancer tissues. Hum Antibodies, 1997. 8(4): p. 189-94.
217. Wilker, E. and M.B. Yaffe, 14-3-3 Proteins--a focus on cancer and human disease. J Mol Cell Cardiol, 2004. 37(3): p. 633-42.
218. Tzivion, G., V.S. Gupta, L. Kaplun, and V. Balan, 14-3-3 proteins as potential oncogenes. Semin Cancer Biol, 2006. 16(3): p. 203-13.
219. Comparot, S., G. Lingiah, and T. Martin, Function and specificity of 14-3-3 proteins in the regulation of carbohydrate and nitrogen metabolism. J Exp Bot, 2003. 54(382): p. 595-604.
220. Huber, S.C., C. MacKintosh, and W.M. Kaiser, Metabolic enzymes as targets for 14-3-3 proteins. Plant Mol Biol, 2002. 50(6): p. 1053-63.
221. Rucks, E.A., J.E. Fraylick, L.M. Brandt, T.S. Vincent, and J.C. Olson, Cell line differences in bacterially translocated ExoS ADP-ribosyltransferase substrate specificity. Microbiology, 2003. 149(Pt 2): p. 319-31.
222. Galan, J.E. and H. Wolf-Watz, Protein delivery into eukaryotic cells by type III secretion machines. Nature, 2006. 444(7119): p. 567-73.
223. Rosqvist, R., A. Forsberg, and H. Wolf-Watz, Intracellular targeting of the Yersinia YopE cytotoxin in mammalian cells induces actin microfilament disruption. Infect Immun, 1991. 59(12): p. 4562-9.
224. Francis, M.S. and H. Wolf-Watz, YopD of Yersinia pseudotuberculosis is translocated into the cytosol of HeLa epithelial cells: evidence of a structural domain necessary for translocation. Mol Microbiol, 1998. 29(3): p. 799-813.
225. Boland, A., M.P. Sory, M. Iriarte, C. Kerbourch, P. Wattiau, and G.R. Cornelis, Status of YopM and YopN in the Yersinia Yop virulon: YopM of Y.enterocolitica is internalized inside the cytosol of PU5-1.8 macrophages by the YopB, D, N delivery apparatus. Embo J, 1996. 15(19): p. 5191-201.
226. Holmstrom, A., J. Olsson, P. Cherepanov, E. Maier, R. Nordfelth, J. Pettersson, R. Benz, H. Wolf-Watz, and A. Forsberg, LcrV is a channel size-determining component of the Yop effector translocon of Yersinia. Mol Microbiol, 2001. 39(3): p. 620-32.
227. Tardy, F., F. Homble, C. Neyt, R. Wattiez, G.R. Cornelis, J.M. Ruysschaert, and V. Cabiaux, Yersinia enterocolitica type III secretion-translocation system: channel formation by secreted Yops. Embo J, 1999. 18(23): p. 6793-9.
228. Neyt, C. and G.R. Cornelis, Insertion of a Yop translocation pore into the macrophage plasma membrane by Yersinia enterocolitica: requirement for translocators YopB and YopD, but not LcrG. Mol Microbiol, 1999. 33(5): p. 971-81.
229. Hakansson, S., K. Schesser, C. Persson, E.E. Galyov, R. Rosqvist, F. Homble, and H. Wolf-Watz, The YopB protein of Yersinia pseudotuberculosis is essential for the translocation of Yop effector proteins across the target cell plasma membrane and

REFERENCES
65
displays a contact-dependent membrane disrupting activity. Embo J, 1996. 15(21): p. 5812-23.
230. Anderson, D.M. and O. Schneewind, A mRNA signal for the type III secretion of Yop proteins by Yersinia enterocolitica. Science, 1997. 278(5340): p. 1140-3.
231. Anderson, D.M. and O. Schneewind, Type III machines of Gram-negative pathogens: injecting virulence factors into host cells and more. Curr Opin Microbiol, 1999. 2(1): p. 18-24.
232. Forsberg, A. and H. Wolf-Watz, Genetic analysis of the yopE region of Yersinia spp.: identification of a novel conserved locus, yerA, regulating yopE expression. J Bacteriol, 1990. 172(3): p. 1547-55.
233. Frithz-Lindsten, E., R. Rosqvist, L. Johansson, and A. Forsberg, The chaperone-like protein YerA of Yersinia pseudotuberculosis stabilizes YopE in the cytoplasm but is dispensible for targeting to the secretion loci. Mol Microbiol, 1995. 16(4): p. 635-47.
234. Wattiau, P. and G.R. Cornelis, SycE, a chaperone-like protein of Yersinia enterocolitica involved in Ohe secretion of YopE. Mol Microbiol, 1993. 8(1): p. 123-31.
235. Wattiau, P., B. Bernier, P. Deslee, T. Michiels, and G.R. Cornelis, Individual chaperones required for Yop secretion by Yersinia. Proc Natl Acad Sci U S A, 1994. 91(22): p. 10493-7.
236. Neyt, C. and G.R. Cornelis, Role of SycD, the chaperone of the Yersinia Yop translocators YopB and YopD. Mol Microbiol, 1999. 31(1): p. 143-56.
237. Day, J.B. and G.V. Plano, A complex composed of SycN and YscB functions as a specific chaperone for YopN in Yersinia pestis. Mol Microbiol, 1998. 30(4): p. 777-88.
238. Cheng, L.W., D.M. Anderson, and O. Schneewind, Two independent type III secretion mechanisms for YopE in Yersinia enterocolitica. Mol Microbiol, 1997. 24(4): p. 757-65.
239. Rosqvist, R., K.E. Magnusson, and H. Wolf-Watz, Target cell contact triggers expression and polarized transfer of Yersinia YopE cytotoxin into mammalian cells. Embo J, 1994. 13(4): p. 964-72.
240. Yahr, T.L., A.K. Hovey, S.M. Kulich, and D.W. Frank, Transcriptional analysis of the Pseudomonas aeruginosa exoenzyme S structural gene. J Bacteriol, 1995. 177(5): p. 1169-78.
241. Frithz-Lindsten, E., A. Holmstrom, L. Jacobsson, M. Soltani, J. Olsson, R. Rosqvist, and A. Forsberg, Functional conservation of the effector protein translocators PopB/YopB and PopD/YopD of Pseudomonas aeruginosa and Yersinia pseudotuberculosis. Mol Microbiol, 1998. 29(5): p. 1155-65.
242. Henriksson, M.L., R. Rosqvist, M. Telepnev, H. Wolf-Watz, and B. Hallberg, Ras effector pathway activation by epidermal growth factor is inhibited in vivo by exoenzyme S ADP-ribosylation of Ras. Biochem J, 2000. 347 Pt 1: p. 217-22.
243. Francis, M.S., S.A. Lloyd, and H. Wolf-Watz, The type III secretion chaperone LcrH co-operates with YopD to establish a negative, regulatory loop for control of Yop synthesis in Yersinia pseudotuberculosis. Mol Microbiol, 2001. 42(4): p. 1075-93.
244. Bonni, A., A. Brunet, A.E. West, S.R. Datta, M.A. Takasu, and M.E. Greenberg, Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science, 1999. 286(5443): p. 1358-62.
245. Marani, M., D. Hancock, R. Lopes, T. Tenev, J. Downward, and N.R. Lemoine, Role of Bim in the survival pathway induced by Raf in epithelial cells. Oncogene, 2004. 23(14): p. 2431-41.

REFERENCES
66
246. B.E. Moore and V.J. Perez In: F.D. Carlson, Editor, Physiological and biochemical aspects of nervous integration, Prentice-Hall, Englewood Cliffs, NJ (1967), pp. 343–359.

PAPERS
67
10. PAPERS I-IV