expression of a functional vegfr-1 in tumor cells is a ... · a major determinant of anti-plgf...
TRANSCRIPT

Expression of a functional VEGFR-1 in tumor cells isa major determinant of anti-PlGF antibodies efficacyJenny Yaoa,1, Xiumin Wua,1, Guanglei Zhuanga, Ian M. Kasmana, Tobias Vogta, Vernon Phana, Masabumi Shibuyab,Napoleone Ferraraa,2, and Carlos Baisa,2
aGenentech, Inc., South San Francisco, CA 94080; and bDepartment of Molecular Oncology, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku,Tokyo 113-8519, Japan
Contributed by Napoleone Ferrara, June 7, 2011 (sent for review April 13, 2011)
PlGF, one of the ligands for VEGFR-1, has been implicated in tumorangiogenesis. However, more recent studies indicate that geneticor pharmacological inhibition of PlGF signaling does not result inreduction of microvascular density in a variety of tumor models.Here we screened 12 human tumor cell lines and identified 3 thatare growth inhibited by anti-PlGF antibodies in vivo. We found thatefficacy of anti-PlGF treatment strongly correlates with VEGFR-1expression in tumor cells, but not with antiangiogenesis. In addi-tion, PlGF induced VEGFR-1 signaling and biological responses intumor cell lines sensitive to anti-PlGF, but not in refractory tumorcell lines or in endothelial cells. Also, genetic ablation of VEGFR-1signaling in the host did not affect the efficacy of PlGF blockade.Collectively, these findings suggest that the role of PlGF in tumor-igenesis largely consists of promoting autocrine/paracrine growthof tumor cells expressing a functional VEGFR-1 rather than stimu-lation of angiogenesis.
stroma | tyrosine kinase | placental growth factor | vascular endothelialgrowth factor | angiogenesis
The VEGF signaling pathways play important roles in angio-genesis. VEGF-A binds to two tyrosine kinase receptors,
VEGFR-1 and VEGFR-2 (1). Although both receptors areexpressed in endothelial cells, VEGFR-1 is also expressed inmonocyte/macrophages, hematopoietic stem cells, and even sometumor cells (2–4). Most of the biological effects of VEGF-A aremediated by activation of VEGFR-2 (1). VEGFR-1 has a weaktyrosine kinase activity but substantially higher binding affinity forVEGF-A than VEGFR-2 (5). The biological role of VEGFR-1 ishighly complex. Although genetic data indicate that signalingdownstream of this receptor is not required for developmental an-giogenesis (6), a role for VEGFR-1 during tumor-angiogenesis hasbeen recently suggested (7–9). PlGF is a VEGFR-1 specific ligand(10) that was identified 20 years ago (11). Under pathologicalconditions, PlGF levels are increased in various cell types, includingvascular endothelial cell, smooth muscle cells, keratinocytes, he-matopoietic cells, retinal pigment epithelial cells, and many differ-ent tumor cells (12). Plgf deficient mice are born at normalMendelian ratios and donot show anyobvious vascular defects (13).PlGF overexpression enhanced tumor growth in some models (14,15), but in others, PlGFparadoxically had an inhibitory effect, likelythrough formation of VEGF/PlGF heterodimers, which down-regulate VEGFR2 signaling (16, 17).According to Fisher et al. (7), treatment with an anti-PlGF
monoclonal antibody (Mab) reducesmicrovascular density (MVD)and inhibits primary tumor growth in a variety of murine models.However, in a subsequent study, we reported that blocking PlGFdoes not result in growth inhibition in any of the tumor modelstested (12murine and 3 human tumor cell lines) (18). Importantly,the antibodies used in these studies were able to block PlGF in vivo(18) as evidenced by their ability to inhibit metastasis of B16F10cells (7, 19, 20), wound healing (13, 21), and primary tumor growthof amurine cell line overexpressingVEGFR-1.On the other hand,it has been shown that genetic ablation of plgf results in inhibitionof tumorigenesis in somemodels, but not in others (2, 8). Becauseefficacy in these models was not associated with a reduction intumor MVD, an alternative mechanism involving vascular nor-malization has been proposed (8). In addition, it has been recently
reported that an anti-human PlGFMab inhibits growth of DangGand MDA-MB-435 xenografts (8), although the mechanismremained unknown. These observations prompted us to revisit therole of PlGF in human tumor xenograft models. This issue isparticularly timely given the ongoing evaluation of anti-PlGFtherapy in clinical trials.
ResultsEfficacy of Anti-PlGF Antibody Treatment Correlates with VEGFR-1Expression in Tumor Cells. As a first step, we sought to identify celllines that are growth inhibited by anti-PlGF treatment. To this end,we tested the ability of the validated anti-human and mouse cross-reactive anti-PlGF mAb C9.V2 (18), hereafter referred to as anti-PlGF, to inhibit growth of CALU3, H82, U87, SW480, A549,H1299, L5180, LXFL529, H460, SKUT1b, and CAKI1 tumors (SIMaterials and Methods). Consistent with previous findings (18),mostmodels evaluated (9 of 11) did not show any growth inhibition(Fig. 1 A–D, Left, and S1 A–E). However, anti-PlGF treatmentsignificantly reduced the growth of SKUT1b (Fig. 1E, Left) andCAKi1 (Fig. 1F, Left) tumors in a dose-dependent manner. Incontrast, all tumor models tested were growth inhibited by anti-VEGF-A treatment (Fig. 1 A–F and Fig. S1 A–E, Left, red line).Together, these data suggest that anti-PlGF mAb treatment doesnot result in broad inhibition of tumor-angiogenesis and that theeffects are tumor model specific. However, PlGF is expressed inboth anti-PlGF responsive and refractory tumor models (7, 8, 18)(Fig. S2). We hypothesized that VEGFR-1 expression in tumorcells (3, 4, 22) might be a potential mechanism conferring suchmodel-specific sensitivity to anti-PlGF treatment. In agreementwith this hypothesis, we found that VEGFR-1 is expressed in theanti-PlGF sensitive cell lines CAKI1 and SKUT1b (Fig. 1 E and F,Right), but it is undetectable in anti-PlGF resistant tumor cells(Fig. 1 A–D and Fig. S1, Right). Figure 1 G and H shows thatVEGFR-1 expression was detected by flow cytometry (SIMaterialsand Methods) in the positive controls [human umbilical vein en-dothelial cells (HUVECs) and HEK293-hVEGFR-1] but not inHEK293-empty vector (negative control) cells. Next, we sought todetermine whether neutralization of PlGF might be sufficient toinhibit growth of tumors known to be dependent on VEGFR-1signaling within tumor cells. To this end, we took advantage ofDU4475, aVEGFR1-positive breast carcinoma cell line previouslyshown be sensitive to anti-hVEGFR-1 mAb treatment (4). Figure1I shows that anti-PlGF mAb treatment inhibits growth of estab-lished DU4475 orthotopic tumors. Thus, PlGF blockade can in-hibit growth of xenografts dependent on VEGFR-1 signaling and,at least among the models evaluated in this study, efficacy of anti-
Author contributions: N.F. and C.B. designed research; J.Y., X.W., G.Z., I.M.K., T.V., V.P.,and C.B. performed research; V.P. and M.S. contributed new reagents/analytic tools; J.Y.,X.W., G.Z., I.M.K., T.V., and C.B. analyzed data; and N.F. and C.B. wrote the paper.
The authors declare no conflict of interest.
Freely available online through the PNAS open access option.1J.Y. and X.W. contributed equally to this work.2To whom correspondence may be addressed. E-mail: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1109029108/-/DCSupplemental.
11590–11595 | PNAS | July 12, 2011 | vol. 108 | no. 28 www.pnas.org/cgi/doi/10.1073/pnas.1109029108
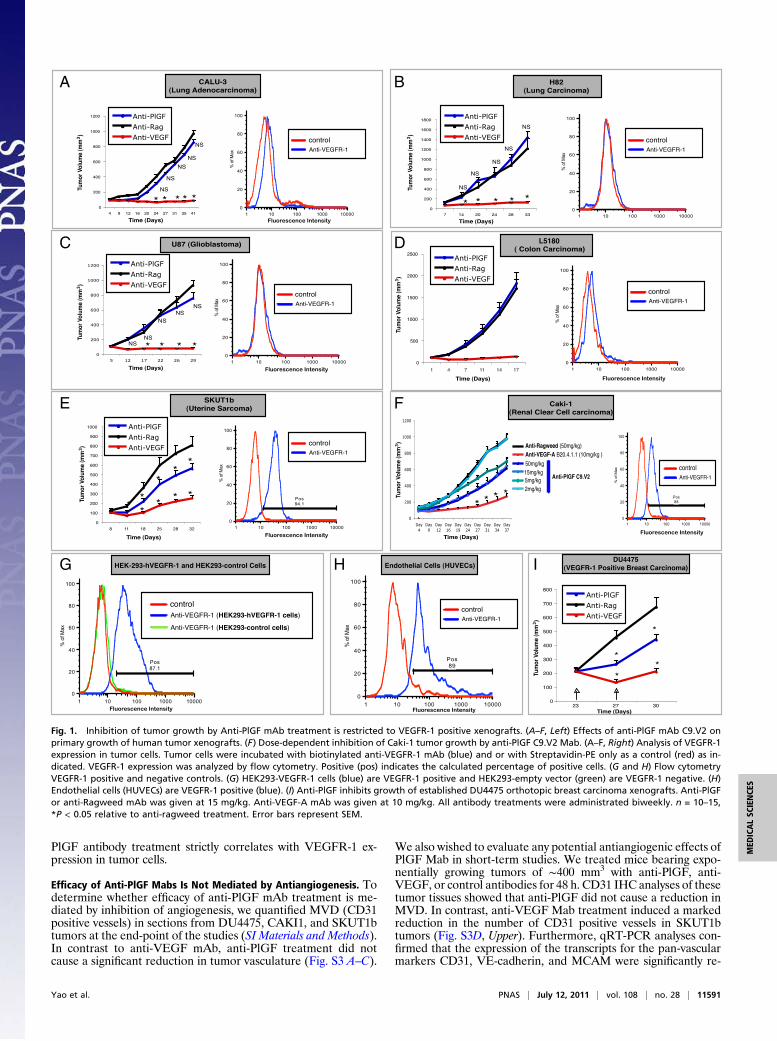
PlGF antibody treatment strictly correlates with VEGFR-1 ex-pression in tumor cells.
Efficacy of Anti-PlGF Mabs Is Not Mediated by Antiangiogenesis. Todetermine whether efficacy of anti-PlGF mAb treatment is me-diated by inhibition of angiogenesis, we quantified MVD (CD31positive vessels) in sections from DU4475, CAKI1, and SKUT1btumors at the end-point of the studies (SI Materials and Methods).In contrast to anti-VEGF mAb, anti-PlGF treatment did notcause a significant reduction in tumor vasculature (Fig. S3 A–C).
We also wished to evaluate any potential antiangiogenic effects ofPlGF Mab in short-term studies. We treated mice bearing expo-nentially growing tumors of ∼400 mm3 with anti-PlGF, anti-VEGF, or control antibodies for 48 h. CD31 IHC analyses of thesetumor tissues showed that anti-PlGF did not cause a reduction inMVD. In contrast, anti-VEGF Mab treatment induced a markedreduction in the number of CD31 positive vessels in SKUT1btumors (Fig. S3D, Upper). Furthermore, qRT-PCR analyses con-firmed that the expression of the transcripts for the pan-vascularmarkers CD31, VE-cadherin, and MCAM were significantly re-
A B
C D
E F
G H I
Fig. 1. Inhibition of tumor growth by Anti-PlGF mAb treatment is restricted to VEGFR-1 positive xenografts. (A–F, Left) Effects of anti-PlGF mAb C9.V2 onprimary growth of human tumor xenografts. (F) Dose-dependent inhibition of Caki-1 tumor growth by anti-PlGF C9.V2 Mab. (A–F, Right) Analysis of VEGFR-1expression in tumor cells. Tumor cells were incubated with biotinylated anti-VEGFR-1 mAb (blue) and or with Streptavidin-PE only as a control (red) as in-dicated. VEGFR-1 expression was analyzed by flow cytometry. Positive (pos) indicates the calculated percentage of positive cells. (G and H) Flow cytometryVEGFR-1 positive and negative controls. (G) HEK293-VEGFR-1 cells (blue) are VEGFR-1 positive and HEK293-empty vector (green) are VEGFR-1 negative. (H)Endothelial cells (HUVECs) are VEGFR-1 positive (blue). (I) Anti-PlGF inhibits growth of established DU4475 orthotopic breast carcinoma xenografts. Anti-PlGFor anti-Ragweed mAb was given at 15 mg/kg. Anti-VEGF-A mAb was given at 10 mg/kg. All antibody treatments were administrated biweekly. n = 10–15,*P < 0.05 relative to anti-ragweed treatment. Error bars represent SEM.
Yao et al. PNAS | July 12, 2011 | vol. 108 | no. 28 | 11591
MED
ICALSC
IENCE
S

duced upon VEGF blockade in SKUT1b. However, anti-PlGFtreatment did not decrease the relative mRNA expression levels inany of the vascular markers tested (Fig. S3D, Bottom).
hPlGF Induces Biological Responses in Anti-PlGF Sensitive (VEGFR-1Positive) Tumor Cells but Not in Endothelial Cells. We tested theability of anti-PlGF sensitive tumor cell lines and endothelial cellsto respond to VEGFR-1 stimulation in vitro (SI Materials andMethods). We did not observe any responses to PlGF in anti-PlGFrefractory (VEGFR-1 negative) tumor cells (Fig. S4). In contrast,anti-PlGF sensitive tumor cell lines proliferated (DU4475,SKUT1b) and migrated (CAKi1 and SKUT1b cells) in responseto hPlGF-2 (or hVEGF-A) in a dose-dependent manner (Figs. 2Aand 4D). Figure 2A also shows that anti-PlGFMab blocked PlGF-induced responses in tumor cells.We also evaluated the responsesof endothelial cells (HUVECs) to hPlGF-2 and VEGF-A. Inagreement with previous reports, HUVECs responded to VEGF-A but did not show any obvious responses to PlGF in migration(Fig. 2B,Right) and proliferation (Fig. 2B,Left) assays. It has beenpostulated that endothelial cells do not respond in vitro to exog-enous PlGF because they express high levels of endogenous PlGF(13, 23). To test this possibility, we performed PlGF knock-downin HUVECs (SI Materials and Methods). Figure 2C (Left) showsthat PlGF knock-down reduces PlGF release by more than 90%.However, HUVECs remained unresponsive to hPlGF-2 but werefully responsive to VEGF-A, bFGF, or FBS (Fig. 2C, Right).
Activation of the Mitogen-Activated Protein Kinase (MAPK) Pathway IsRequired for PlGF-Induced Biological Responses in Anti-PlGF SensitiveTumor Cells. Previous studies have shown that the (MAPK) andPI3K pathways are activated in response to ligand stimulationin some cell lines overexpressing VEGFR-1 (18, 24).
To gain further insights into PlGF/VEGFR-1 signaling in tumorcells, we first performed phospho-kinase antibody array experi-ments with cell lysates fromhPlGF-2 ormock stimulatedHEK293-VEGFR-1 cells (SIMaterials andMethods). Figure 3A (Left) showsthat p42/p44 was activated by PlGF stimulation. No significantdifferences in phosphorylation of protein kinase B (PKB/AKT) orother proteins included in this array were apparent. Nearly iden-tical results were obtained when lysates from the VEGFR-1 pos-itive uterine sarcoma cell line SKUT1b were analyzed (Fig. 3A,Right). MAPK activation by PlGF was confirmed by Western blotin both SKUT1b (Fig. 3B,Left, and Fig. S5A) and CAKI1 (Fig. 3C,Left, and Fig. S5A,Right). We next usedMAPK pathway inhibitorsto investigate whether MAPK activation is required for PlGF-in-duced migration and proliferation. Figure 3 B and C (Left) showsthat the MEK inhibitor GDC-0973/XL-518 (US patent20110086837) (25) efficiently blocks PlGF-induced MAPK phos-phorylation without affecting cell viability (Fig. 3 B and C, Right,and Fig. S5C). In addition, GDC-0973 and the RAF inhibitorGDC-0879 (26) (Fig. 3 B and C, right panels), but not Rac, JNK(SP600125), or Rho inhibitors (Fig. S5B), completely suppressedPlGF-responses. However, they only slightly reduced HGF- orFBS-induced CAKi1 and SKUT1b migration and SKUT1bsurvival/proliferation (Fig. 3 B and C, Right, and Fig. 4D). In-terestingly, the dose-dependent inhibition of PlGF-inducedMAPK phosphorylation by GDC-0973 parallels the inhibition ofmigration and proliferation induced by this agent (Fig. 3 B andC).
Inhibition of PlGF/VEGFR-1 Signaling In Tumor but Not Stromal Cells Isa Major Determinant for Anti-PlGF Efficacy. To confirm the role ofVEGFR-1 in PlGF-induced responses in anti-PlGF sensitivetumor cell lines, we knocked-down VEGFR-1 in CAKI1 andSKUT1b cells using siRNA oligonucleotides (SI Materials and
0
20000
40000
60000
80000
100000
120000
140000
160000
180000
500 0 0 0 0 0
0 50 100 2000 500 1000
hVEGF (ng/ml)
hPlGF-2 (ng/ml)
0.0
50.0
100.0
150.0
200.0
250.0
0 50 100 2000 500
500 0 0 0 0 Control SiRNA PlGF siRNASec
rete
d P
lGF
( n
g/10
00.0
00 C
ells
)
0
20
40
60
80
100
120
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
BasalMedium
hVEGF(50ng/ml)
hPlGF-2(50ng/ml)
bFGF (20ng/ml)
FBS 10%
PlGF siRNA
Control siRNA
HUVECs Proliferation HUVECs Migration PLGF KD HUVECs Migration
Cel
l Num
ber
Num
ber
of M
igra
ting
Cel
ls
Num
ber
of M
igra
ting
Cel
ls
A
B
600000
700000
hVEGF (ng/ml)hPlGF-2 (ng/ml)
100000
200000
300000
400000
500000
Anti-PlGF(C9.V2)
00 500 0 0 0 0
0 500 500 1 10
- -- - - ++
0.0
20.0
40.0
60.0
80.0
100.0
120.0
140.0
160.0
180.0
200.0
500 0 0 0 0
500 500 1 10
500 0 0 0 0
-- - - +-
0
20
40
60
80
100
120
140
160
180
200
500 500 1 10
-- - - +-
DU4475 Proliferation Caki-1 Migration SKUT1b MigrationN
umbe
r of
Mig
ratin
g C
ells
Num
ber
of M
igra
ting
Cel
ls
Cel
l Num
ber
C
Fig. 2. Anti-PlGF tumor sensitive tumor-cell lines but not endothelial cells respond to hPlGF-2 stimulation. (A) hPlGF-2 induces dose-dependent biologicaleffects in the anti-PlGF sensitive tumor cell lines DU4475 (Left), CAKI-1 (Center), and SKUT1b (Right), and these effects are blocked by anti-PlGF mAb. (B)hPlGF-2 fails to stimulate HUVEC proliferation (Left) and migration (Right) at all doses tested. (C, Left) Quantification by ELISA of hPlGF released by PlGFknock-down (KD) or control HUVECs. (C, Right) PlGF knock-down (blue bars) and siRNA control HUVECs (green bars) remain unresponsive to hPlGF-2. Thefigure shows the average values from representative experiments. Doses of ligands are indicated in the figure. Dotted lines represent basal (control) activity.Experiments were repeated at least three times with comparable results. n = 3–5. Error bars represent SD.
11592 | www.pnas.org/cgi/doi/10.1073/pnas.1109029108 Yao et al.

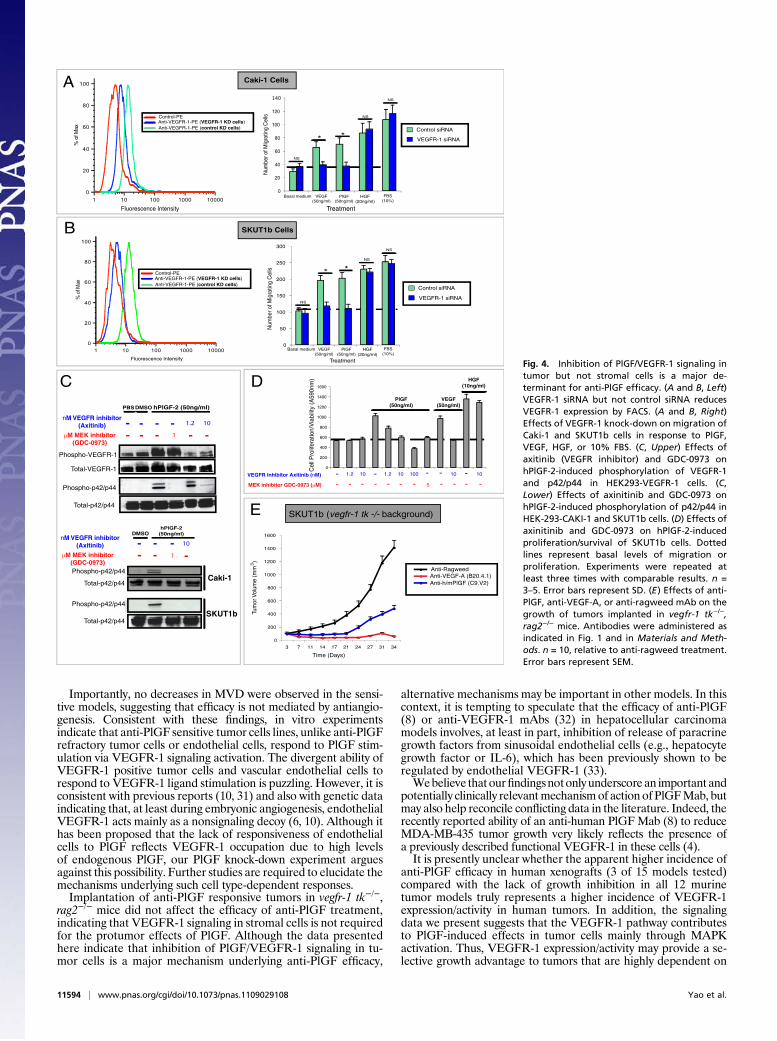
Methods). Figure 4 A and B (Left) shows that VEGFR-1 siRNAbut not control siRNA markedly decreases VEGFR-1 expressionin both cell lines. VEGFR-1 knock-down also suppressed theability of these cells to migrate in response to PlGF or VEGF-Abut did not affect their ability to respond to HGF or 10% FBS(Fig. 4 A and B, Right). Consistent with these findings, VEGFR-1depletion with a different siRNA oligonucleotide sequence(VEGFR-1 SiRNA no. 2; Fig. S6A) also specifically inhibitedVEGF- and PlGF-induced responses. We found that althoughPlGF strongly induced tyrosine phosphorylation in HEK293 cellsoverexpressing hVEGFR-1 (Fig. 4C), it barely affectedVEGFR-1phosphorylation in CAKI1 or SKUT1b (Fig. S5A). This result wasnot unexpected, because ligand-dependent tyrosine phosphory-lation of VEGFR-1 is known to be very low (or undetectable) incells endogenously expressing this receptor (27–29). To test thepotential relevance of tyrosine phosphorylation in the activationof PlGF/VEGFR-1 downstream signaling, we used the VEGFRtyrosine kinase inhibitor axitinib (30). Figure 4C shows that theMEK inhibitor GDC-0973 specifically inhibits MAPK but notVEGFR-1 phosphorylation in HEK293-VEGFR-1 cells. How-ever, axitinib inhibited both PlGF-induced phosphorylation ofVEGFR-1 and downstreamMAPKactivation in a dose-dependentmanner. Similar to anti-PlGFmAb (18) (Fig. 2A and Fig. S6C) andMEK inhibitors (Fig. 3 B and C and Fig. S6B), axitinib inhibitedhPlGF-induced signal transduction (Fig. 4C), SKUT1b cell sur-vival/proliferation (Fig. 4D) and migration of CAKI1 and SKUT1bcells (Fig. S6B). These findings indicate that VEGFR-1 expressionand phosphorylation are required for PlGF-induced biologicalresponses in anti-PlGF sensitive tumor cells.It has been postulated that anti-PlGF efficacy, in the absence of
MVD changes, is due to normalization of the vasculature asa consequence of reduced infiltration ofVEGFR-1 positive tumor-associated macrophages (TAMs) (8). To probe whether tumorgrowth inhibition by anti-PlGF indeed requires inhibition ofVEGFR-1 signaling in TAMs, hematopoietic stem cells, or otherstromal cells, we implanted SKUT1B anti-PlGF sensitive tumorcells in vegfr-1 tk −/−, rag2−/− mice (6). Because these mice expressa VEGFR-1 mutant that lacks most of its intracellular domain(including the tyrosine kinase domain), PlGF should be unable to
activate VEGFR-1 signaling in host (murine) cells. Figure 4Eshows that implantation of SKUT1b cells in vegfr-1 tk−/− does notimpair the ability of anti-PlGF to inhibit tumor growth. Similarly,Fig. S6D shows that anti-PlGF treatment has comparable effectson Caki-1 tumor growth in rag2−/− or vegfr-1 tk−/− vs. rag2−/−, vegfr-1+/+mice. These data indicate that anti-PlGF efficacy is mediatedby blockade of PlGF/hVEGFR-1 signaling in the tumor cells butnot by inhibition of VEGFR-1 signaling in host cells.
DiscussionAnti-PlGF therapy is currently being evaluated in clinical trials.Nevertheless, the significance of PlGF as a therapeutic targetremains incompletely understood.Recent studies suggest that PlGF inhibition reduces tumor
growth and angiogenesis by decreasing recruitment of macro-phages in tumor tissue (7). However, subsequent reports revealedthat inhibition of PlGF-induced signaling does not necessarilyinhibit tumor growth, nor does it correlate with pruning of tumorvessels (8). It has been also hypothesized that the efficacy of PlGFinhibition, in the absence of a significant reduction in tumorMVD, is mediated by vascular normalization following reducedTAM infiltration (8, 18). However, this hypothesis does not fullyexplain the lack of broad antitumor efficacy and the model-dependent efficacy of PlGF inhibition.AlthoughVEGFR-1 has previously been shown to be expressed
in some tumor cells (2–4), the possibility that VEGFR-1 expres-sion may confer sensitivity to PlGF inhibition was not previouslyinvestigated. It is interesting to note that of the 12 murine tumormodels we recently evaluated (18), inhibition of primary tumorgrowth by anti-PlGF treatment was restricted to a cell line engi-neered to overexpress VEGFR-1. Here, we identified three un-transfected human tumor cell lines (CAKI1, SKUT1b, andDU4475) sensitive to PlGF neutralization. Remarkably, all anti-PlGF sensitive tumor cell lines identified in the present study werefound to be VEGFR-1 positive. Conversely, all anti-PlGF re-sistant cell lines were VEGFR-1 negative. These data suggest thatblockade of PlGF/VEGFR-1 signaling in tumor cells may be re-quired for anti-PlGF mAb efficacy.
1M MEK inhibitor (GDC-0973)
hPlGF-2
0.1 1 5DMSO
Phospho-p42/p44
- -VEGF
-
Total-p42/p44
M MEK inhibitor (GDC-0973)
hPlGF-2 (50ng/ml)
0.1 1 5
DMSO
Phospho-p42/p44
- -VEGF
- 1
Total-p42/p44
0
50
100
150
200
250
300
350
MEK inhibitor GDC-0973 ( M)
Raf Inhibitor GDC-0879 ( M)
PlGF (50ng/ml) VEGF
(50ng/ml)
HGF(10ng/ml)
FBS 10%
- 51 0.1 1 5 1 1 1- - -- -- - - - - - - - - - - - -1
Num
ber
of
Mig
ratin
g C
ells
Caki-1 Cell MigrationCaki-1 MAPK phosphorylation
p42/44(T202/Y204, T185/Y187)
Phospho-array HEK293-VEGFR-1 Cells
hPlGF-2 PBS PBS
Phospho-array SKUT1b Cells
hPlGF-2
0
50
100
150
200
250
300
350
MEK inhibitor GDC-0973 ( M)
Raf Inhibitor GDC-0879 ( M)
- 51 0.1 1 5 1 1 1- - -- -- - - - - - - - - - - - -1
Num
ber
of M
igra
ting C
ells
PlGF (50ng/ml)
VEGF (50ng/ml)
HGF(10ng/ml)
FBS 10%
SKUT1b MAPK phosphorylation SKUT1b Cell Migration
A
B
C
Fig. 3. hPlGF-2-induced responsesin anti-PlGF sensitive cell lines re-quire MAPK activation. (A) Phospho-antibody array analyses of hPlGF ormock-stimulated HEK293-VEGFR-1(Left) and SKUT1b (Right) cells.The figure shows only a relevantsection of phopho-array membrane.(B, Left) Effects of MEK inhibitorGDC-0973 on PlGF-induced MAPKphosphorylation in SKUT1b cells.(B, Right) Effects of GDC-0973 orRAF inhibitor (GDC-0879) on PlGF-induced SKUT1b cell migration. (C,Left) Effect of MEK inhibitor onPlGF-induced MAPK phosphoryla-tion in CAKI-1 cells. (C, Right) Effectsof MEK inhibitor and RAF inhibitoron PlGF-induced Caki-1 cell migra-tion. Dotted lines represent basal(control) activity. Experiments wererepeated at least three times withcomparable results. n = 3–5. Errorbars represent SD.
Yao et al. PNAS | July 12, 2011 | vol. 108 | no. 28 | 11593
MED
ICALSC
IENCE
S
Importantly, no decreases in MVD were observed in the sensi-tive models, suggesting that efficacy is not mediated by antiangio-genesis. Consistent with these findings, in vitro experimentsindicate that anti-PlGF sensitive tumor cells lines, unlike anti-PlGFrefractory tumor cells or endothelial cells, respond to PlGF stim-ulation via VEGFR-1 signaling activation. The divergent ability ofVEGFR-1 positive tumor cells and vascular endothelial cells torespond to VEGFR-1 ligand stimulation is puzzling. However, it isconsistent with previous reports (10, 31) and also with genetic dataindicating that, at least during embryonic angiogenesis, endothelialVEGFR-1 acts mainly as a nonsignaling decoy (6, 10). Although ithas been proposed that the lack of responsiveness of endothelialcells to PlGF reflects VEGFR-1 occupation due to high levelsof endogenous PlGF, our PlGF knock-down experiment arguesagainst this possibility. Further studies are required to elucidate themechanisms underlying such cell type-dependent responses.Implantation of anti-PlGF responsive tumors in vegfr-1 tk−/−,
rag2−/− mice did not affect the efficacy of anti-PlGF treatment,indicating that VEGFR-1 signaling in stromal cells is not requiredfor the protumor effects of PlGF. Although the data presentedhere indicate that inhibition of PlGF/VEGFR-1 signaling in tu-mor cells is a major mechanism underlying anti-PlGF efficacy,
alternative mechanisms may be important in other models. In thiscontext, it is tempting to speculate that the efficacy of anti-PlGF(8) or anti-VEGFR-1 mAbs (32) in hepatocellular carcinomamodels involves, at least in part, inhibition of release of paracrinegrowth factors from sinusoidal endothelial cells (e.g., hepatocytegrowth factor or IL-6), which has been previously shown to beregulated by endothelial VEGFR-1 (33).Webelieve thatourfindingsnotonly underscore an important and
potentially clinically relevantmechanismof action of PlGFMab, butmay also help reconcile conflicting data in the literature. Indeed, therecently reported ability of an anti-human PlGFMab (8) to reduceMDA-MB-435 tumor growth very likely reflects the presence ofa previously described functional VEGFR-1 in these cells (4).It is presently unclear whether the apparent higher incidence of
anti-PlGF efficacy in human xenografts (3 of 15 models tested)compared with the lack of growth inhibition in all 12 murinetumor models truly represents a higher incidence of VEGFR-1expression/activity in human tumors. In addition, the signalingdata we present suggests that the VEGFR-1 pathway contributesto PlGF-induced effects in tumor cells mainly through MAPKactivation. Thus, VEGFR-1 expression/activity may provide a se-lective growth advantage to tumors that are highly dependent on
Fluorescence Intensity Treatment1 10 100 1000 10000
0
20
40
60
80
100
%of
Max
VEGFR-1 siRNA
Control siRNA
Control-PE Anti-VEGFR-1-PE (VEGFR-1 KD cells)Anti-VEGFR-1-PE (control KD cells)
Caki-1 Cells
VEGF(50ng/ml)
PlGF (50ng/ml)
HGF(20ng/ml)
FBS(10%)
Basal medium0
20
40
60
80
100
120
140
Num
ber o
f Mig
ratin
g C
ells
* *
NS
NS
NS
1 10 100 1000 100000
20
40
60
80
100
%of
Max
Control-PE Anti-VEGFR-1-PE (VEGFR-1 KD cells)Anti-VEGFR-1-PE (control KD cells)
0
50
100
150
200
250
300
VEGFR-1 siRNA
Control siRNA
Num
ber o
f Mig
ratin
g C
ells
VEGF(50ng/ml)
PlGF (50ng/ml)
HGF(20ng/ml)
FBS(10%)
Treatment
Basal medium
Fluorescence Intensity
* *
NS
NS
NS
SKUT1b Cells
A
B
C
M MEK inhibitor (GDC-0973)
nM VEGFR inhibitor (Axitinib)
- - - 1 - -- - - - 1.2 10
hPlGF-2 (50ng/ml)DMSOPBS
Phospho-VEGFR-1
Phospho-p42/p44
Total-VEGFR-1
Total-p42/p44
DVEGF
(50ng/ml)
HGF(10ng/ml)
Cel
l Pro
lifer
atio
n/V
iabi
lity
(A59
0nm
)
PlGF (50ng/ml)
0
200
400
600
800
1000
1200
1400
1600
MEK inhibitor GDC-0973 ( M)
- 101.2 1.2 10 100 10 10- - --- - - - - - - - - - -5
VEGFR Inhibitor Axitinib (nM)
0
200
400
600
800
1000
1200
1400
1600
3 7 11 14 17 21 24 27 31 34
Anti-RagweedAnti-VEGF-A (B20.4.1)Anti-h/mPlGF (C9.V2)
Tum
or V
olum
e (m
m )3
Time (Days)
SKUT1b (vegfr-1 tk -/- background)
Phospho-p42/p44
Total-p42/p44
M MEK inhibitor (GDC-0973)
nM VEGFR inhibitor (Axitinib)
- - 1 -- - - 10
hPlGF-2 (50ng/ml)DMSO
Phospho-p42/p44
Total-p42/p44
Caki-1
SKUT1b
E
Fig. 4. Inhibition of PlGF/VEGFR-1 signaling intumor but not stromal cells is a major de-terminant for anti-PlGF efficacy. (A and B, Left)VEGFR-1 siRNA but not control siRNA reducesVEGFR-1 expression by FACS. (A and B, Right)Effects of VEGFR-1 knock-down on migration ofCaki-1 and SKUT1b cells in response to PlGF,VEGF, HGF, or 10% FBS. (C, Upper) Effects ofaxitinib (VEGFR inhibitor) and GDC-0973 onhPlGF-2-induced phosphorylation of VEGFR-1and p42/p44 in HEK293-VEGFR-1 cells. (C,Lower) Effects of axinitinib and GDC-0973 onhPlGF-2-induced phosphorylation of p42/p44 inHEK-293-CAKI-1 and SKUT1b cells. (D) Effects ofaxinitinib and GDC-0973 on hPlGF-2-inducedproliferation/survival of SKUT1b cells. Dottedlines represent basal levels of migration orproliferation. Experiments were repeated atleast three times with comparable results. n =3–5. Error bars represent SD. (E) Effects of anti-PlGF, anti-VEGF-A, or anti-ragweed mAb on thegrowth of tumors implanted in vegfr-1 tk−/−,rag2−/− mice. Antibodies were administered asindicated in Fig. 1 and in Materials and Meth-ods. n = 10, relative to anti-ragweed treatment.Error bars represent SEM.
11594 | www.pnas.org/cgi/doi/10.1073/pnas.1109029108 Yao et al.

Ras/Raf/MAPK signaling. In this context it is interesting thatVEGFR-1 signaling within tumor cells previously has been shownto modulate growth and survival of several Ras/MAPK pathway-driven mouse tumor models and cell lines (3, 34). Growing evi-dence also supports a possible role for VEGFR-1 signaling incertain human cancers. In vitro studies suggested a role forVEGFR-1 signaling in survival of colorectal and pancreatic cancercell lines during epithelial to mesenchymal transition (22, 35–37).Also, VEGFR-1 signaling is required for growth of patient-derivedmalignant melanoma-initiating human cells in mice (38), and anti-hVEGFR-1 mAb treatment increases the survival of mice injectedwith acute lymphoblastic leukemia cells (39) and also inhibits tu-mor growth of VEGFR-1 positive breast carcinoma and mela-noma xenografts (4). Furthermore, expression of VEGFR-1 intumor cells has been observed in human biopsies (3, 40, 41). Fi-nally, mutations in VEGFR-1 have been found in human cancers,including ∼10% of melanomas (42).In conclusion, we show that, among the models we tested, effi-
cacy of anti-PlGF mAb treatment is limited to VEGFR-1expressing tumors, because it requires inhibition of PlGF/VEGFR-1 signaling within tumor cells. These findings may berelevant in the context of ongoing clinical evaluation of anti-PlGF(43), anti-VEGFR-1 (44) Mabs, VEGF-Trap (45), and other
VEGFR inhibitor therapies. It is tempting to speculate thatVEGFR-1 expression/activity may be a biomarker to selectpatients and indications likely to benefit from anti-PlGF therapies.
Materials and MethodsAnimals and Cell Lines. Female Beige nude and BALB/c nude mice wereobtained from Charles River. RAG2−/− mice were from Jackson Laboratories.flt1 tk−/− mice were generated as described (6). flt-1 tk, rag-2 double ko micewere generated by crossing Flt-1 tk −/− with with rag2−/− mice.
Tumor cell lines were obtained from theATCC. Tumor cells weremaintainedin RPMI-1640 containing 10% FBS (Sigma, Sigma-Aldrich), penicillin (100 units/mL), streptomycin (100 μg/mL), and L-glutamine (2 mmol/L). Hek293 cells werecultured in DMEM supplemented with 10% FBS (Sigma, Sigma-Aldrich), L-glu-tamine (2 mmol/L), and puromycin (1 μg/mL). Primary HUVEC were purchasedfrom Lonza and maintained in EGM-2 medium (Lonza). Only low-passageHUVECs were used in our experiments. All cells were cultured at 37 °C in a hu-midified incubator containing 5% CO2. Hek293-hVEGFR-1 and HEK293-controlcell lines were generated by transfection followed by puromycin selection.
ACKNOWLEDGMENTS. We thank the Genentech animal facility and theprotein purification and antibody technology groups. We also thankH. Koeppen for histopathological analysis and L. Gilmour, R. Neupane, andC.P. Poon from the FACS lab for excellent support.
1. Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. NatMed 9:669–676.
2. Fischer C, Mazzone M, Jonckx B, Carmeliet P (2008) FLT1 and its ligands VEGFB andPlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer 8:942–956.
3. Lichtenberger BM, et al. (2010) Autocrine VEGF signaling synergizes with EGFR intumor cells to promote epithelial cancer development. Cell 140:268–279.
4. Wu Y, et al. (2006) The vascular endothelial growth factor receptor (VEGFR-1)supports growth and survival of human breast carcinoma. Int J Cancer 119:1519–1529.
5. Sawano A, Takahashi T, Yamaguchi S, Aonuma M, Shibuya M (1996) Flt-1 but notKDR/Flk-1 tyrosine kinase is a receptor for placenta growth factor, which is related tovascular endothelial growth factor. Cell Growth Differ 7:213–221.
6. Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M (1998) Flt-1 lacking the tyrosinekinase domain is sufficient for normal development and angiogenesis in mice. ProcNatl Acad Sci USA 95:9349–9354.
7. Fischer C, et al. (2007) Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumorswithout affecting healthy vessels. Cell 131:463–475.
8. Van de Veire S, et al. (2010) Further pharmacological and genetic evidence for theefficacy of PlGF inhibition in cancer and eye disease. Cell 141:178–190.
9. Muramatsu M, Yamamoto S, Osawa T, Shibuya M (2010) Vascular endothelial growthfactor receptor-1 signaling promotes mobilization of macrophage lineage cells frombone marrow and stimulates solid tumor growth. Cancer Res 70:8211–8221.
10. Park JE, Chen HH, Winer J, Houck KA, Ferrara N (1994) Placenta growth factor.Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo,and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem 269:25646–25654.
11. Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG (1991) Isolation of ahuman placenta cDNA coding for a protein related to the vascular permeabilityfactor. Proc Natl Acad Sci USA 88:9267–9271.
12. Cao Y (2009) Positive and negative modulation of angiogenesis by VEGFR1 ligands. SciSignal 2:re1.
13. Carmeliet P, et al. (2001) Synergism between vascular endothelial growth factor andplacental growth factor contributes to angiogenesis and plasma extravasation inpathological conditions. Nat Med 7:575–583.
14. Hiratsuka S, et al. (2001) Involvement of Flt-1 tyrosine kinase (vascular endothelialgrowth factor receptor-1) in pathological angiogenesis. Cancer Res 61:1207–1213.
15. Marcellini M, et al. (2006) Increased melanoma growth and metastasis spreading inmice overexpressing placenta growth factor. Am J Pathol 169:643–654.
16. Schomber T, et al. (2007) Placental growth factor-1 attenuates vascular endothelialgrowth factor-A-dependent tumor angiogenesis during beta cell carcinogenesis.Cancer Res 67:10840–10848.
17. ErikssonA,etal. (2002)Placentagrowthfactor-1antagonizesVEGF-inducedangiogenesisand tumor growth by the formation of functionally inactive PlGF-1/VEGF heterodimers.Cancer Cell 1:99–108.
18. Bais C, et al. (2010) PlGF blockade does not inhibit angiogenesis during primary tumorgrowth. Cell 141:166–177.
19. Hiratsuka S, et al. (2002) MMP9 induction by vascular endothelial growth factorreceptor-1 is involved in lung-specific metastasis. Cancer Cell 2:289–300.
20. Kaplan RN, et al. (2005) VEGFR1-positive haematopoietic bone marrow progenitorsinitiate the pre-metastatic niche. Nature 438:820–827.
21. Cianfarani F, et al. (2006) Placenta growth factor in diabetic wound healing: alteredexpression and therapeutic potential. Am J Pathol 169:1167–1182.
22. Bates RC, et al. (2003) Flt-1-dependent survival characterizes the epithelial-mesenchymaltransition of colonic organoids. Curr Biol 13:1721–1727.
23. Autiero M, Luttun A, Tjwa M, Carmeliet P (2003) Placental growth factor and itsreceptor, vascular endothelial growth factor receptor-1: novel targets for stimulationof ischemic tissue revascularization and inhibition of angiogenic and inflammatorydisorders. J Thromb Haemost 1:1356–1370.
24. Wang F, et al.: RACK1 regulates VEGF/Flt1-mediated cell migration via activation ofa PI3-K/Akt pathway, J Biol Chem 2011.
25. Frémin C, Meloche S (2010) From basic research to clinical development of MEK1/2inhibitors for cancer therapy. J Hematol Oncol 3:8.
26. Hoeflich KP, et al. (2009) Antitumor efficacy of the novel RAF inhibitor GDC-0879 ispredicted by BRAFV600E mutational status and sustained extracellular signal-regulatedkinase/mitogen-activated protein kinasepathway suppression.Cancer Res 69:3042–3051.
27. Seetharam L, et al. (1995) A unique signal transduction from FLT tyrosine kinase,a receptor for vascular endothelial growth factor VEGF. Oncogene 10:135–147.
28. Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH (1994) Differentsignal transduction properties of KDR and Flt1, two receptors for vascular endothelialgrowth factor. J Biol Chem 269:26988–26995.
29. Gille H, et al. (2001) Analysis of biological effects and signaling properties of Flt-1(VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascularendothelial growth factor mutants. J Biol Chem 276:3222–3230.
30. Kindler HL, et al. (2011) Axitinib plus gemcitabine versus placebo plus gemcitabine inpatients with advanced pancreatic adenocarcinoma: a double-blind randomisedphase 3 study. Lancet Oncol 12:256–262.
31. Cao Y, et al. (1996) Heterodimers of placenta growth factor/vascular endothelialgrowth factor. Endothelial activity, tumor cell expression, and high affinity binding toFlk-1/KDR. J Biol Chem 271:3154–3162.
32. Yoshiji H, et al. (2004)Halting the interactionbetween vascular endothelial growth factorand its receptors attenuates liver carcinogenesis in mice. Hepatology 39:1517–1524.
33. LeCouter J, et al. (2003) Angiogenesis-independent endothelial protection of liver:role of VEGFR-1. Science 299:890–893.
34. Bianco R, et al. (2008) Vascular endothelial growth factor receptor-1 contributes toresistance to anti-epidermal growth factor receptor drugs in human cancer cells. ClinCancer Res 14:5069–5080.
35. Fan F, et al. (2005) Expression and function of vascular endothelial growth factorreceptor-1 on human colorectal cancer cells. Oncogene 24:2647–2653.
36. Lesslie DP, et al. (2006) Vascular endothelial growth factor receptor-1 mediatesmigration of human colorectal carcinoma cells by activation of Src family kinases. Br JCancer 94:1710–1717.
37. Wey JS, et al. (2005) Vascular endothelial growth factor receptor-1 promotesmigration and invasion in pancreatic carcinoma cell lines. Cancer 104:427–438.
38. Frank NY, et al.: VEGFR-1 expressed by malignant melanoma initiating cells isrequired for tumor growth, Cancer Res 2011.
39. Fragoso R, et al. (2006) VEGFR-1 (FLT-1) activation modulates acute lymphoblasticleukemia localization and survival within the bone marrow, determining the onset ofextramedullary disease. Blood 107:1608–1616.
40. Chung GG, et al. (2006) Vascular endothelial growth factor, FLT-1, and FLK-1 analysisin a pancreatic cancer tissue microarray. Cancer 106:1677–1684.
41. Ghosh S, et al. (2008) High levels of vascular endothelial growth factor and itsreceptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome inbreast cancer. Hum Pathol 39:1835–1843.
42. Prickett TD, et al. (2009) Analysis of the tyrosine kinome in melanoma revealsrecurrent mutations in ERBB4. Nat Genet 41:1127–1132.
43. Lassen U, et al. (2009) A phase I dose escalation study of TB-403, a monoclonal antibodydirected against PlGF, in patients with solid tumors.Mol Cancer Ther 8 (12 Suppl):A111.
44. Schwartz JD, Rowinsky EK, Youssoufian H, Pytowski B, Wu Y (2010) Vascularendothelial growth factor receptor-1 in human cancer: concise review and rationalefor development of IMC-18F1 (Human antibody targeting vascular endothelialgrowth factor receptor-1). Cancer 116 (4 Suppl):1027–1032.
45. LockhartAC, et al. (2010) Phase I studyof intravenous vascular endothelial growth factortrap, aflibercept, in patients with advanced solid tumors. J Clin Oncol 28:207–214.
Yao et al. PNAS | July 12, 2011 | vol. 108 | no. 28 | 11595
MED
ICALSC
IENCE
S