fact sheets print
TRANSCRIPT

1 | P a g e Ist edition by dr.amir abdelazim ahmed
Rapid
Notes Information for DOCTORS about the
Disorders included in the Kuwait’ Newborn Screening Panel
By Dr.Amir Abdelazim Ahmed
Clinical pathology specialist
Kuwait newborn screening laboratories

2 | P a g e Ist edition by dr.amir abdelazim ahmed
Content :
Subject
1 Panel of newborn screening program in Kuwait
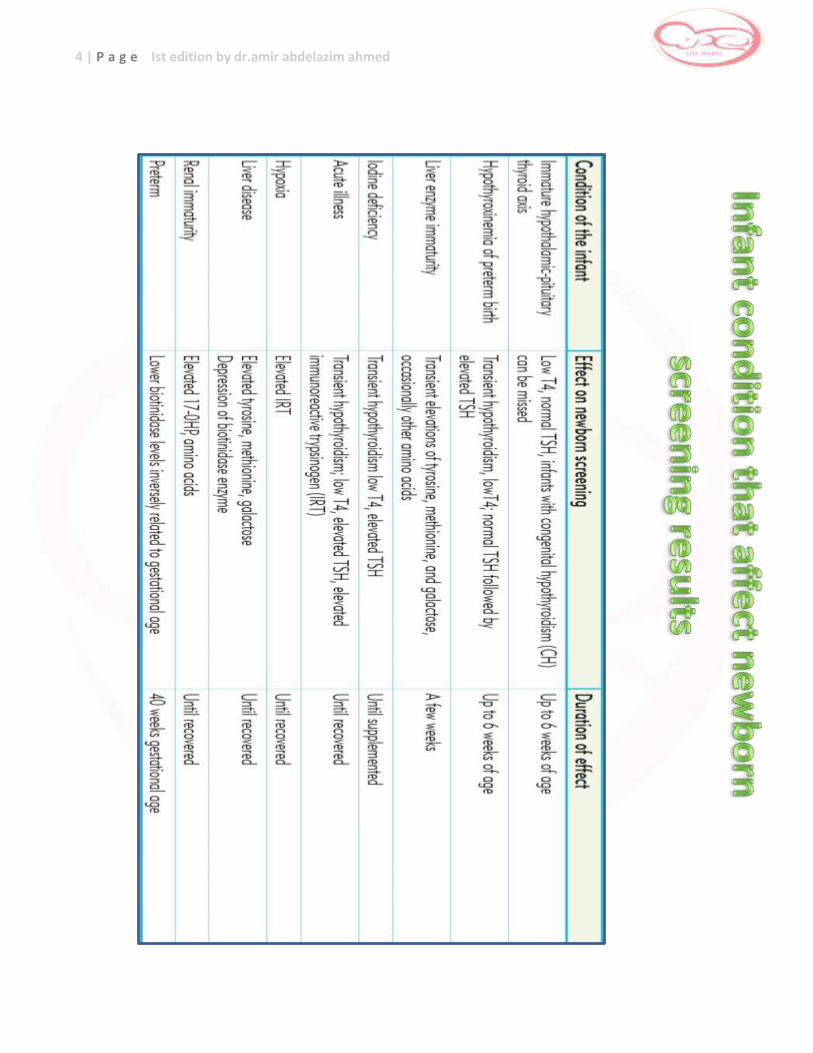
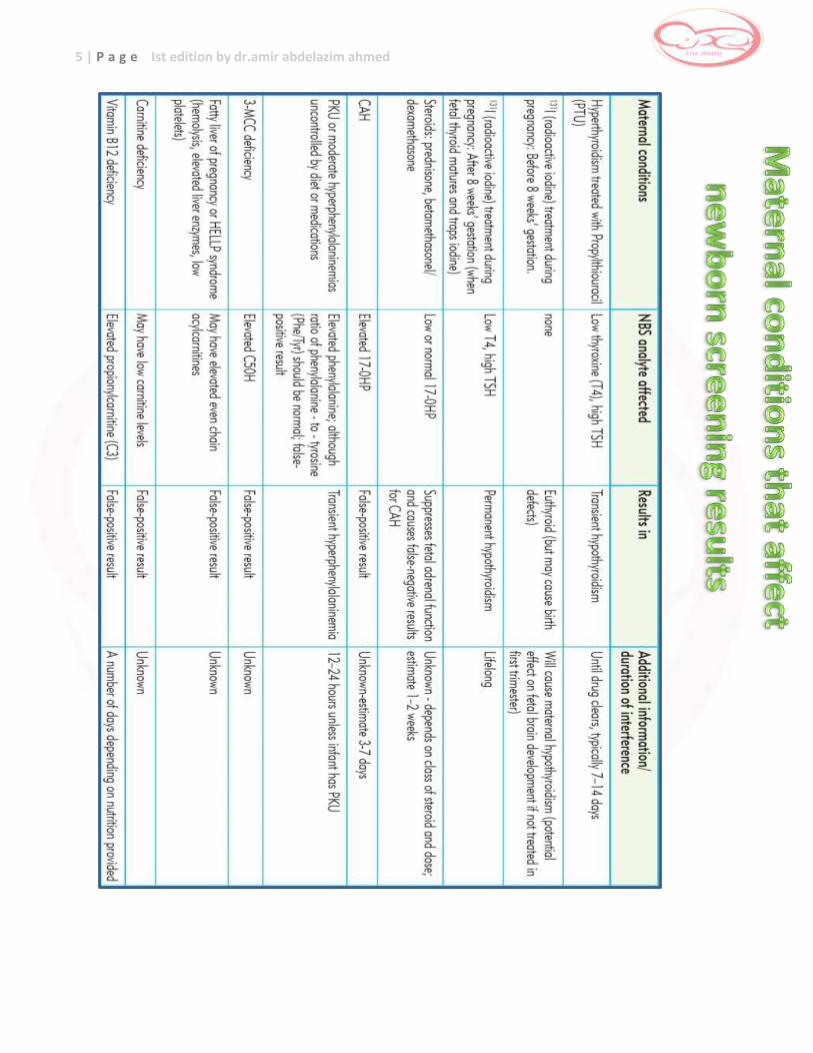
2 Summary for conditions affect newborn screening results
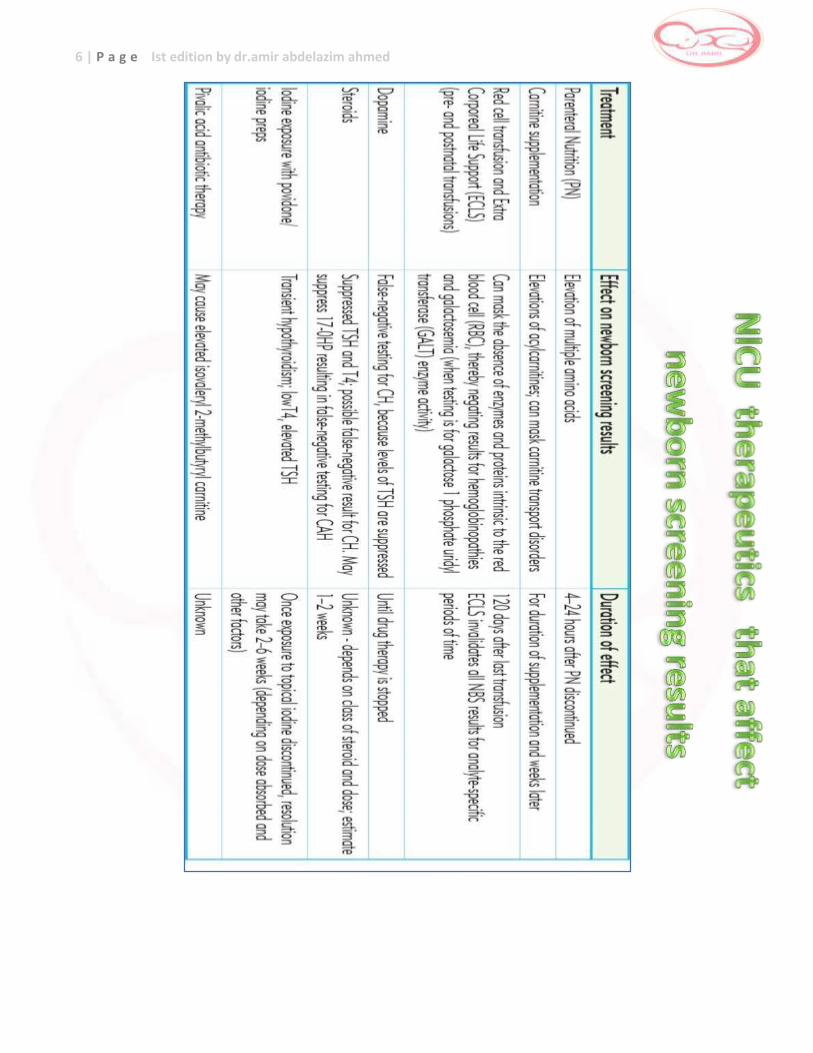
3 Table for notes in clinical and therapeutic principles
4 Amino acid disorders 5 Fatty acid disorders 6 Organic acid disorders 7 Endocrine disorders 8 Galactosemia
3 | P a g e Ist edition by dr.amir abdelazim ahmed
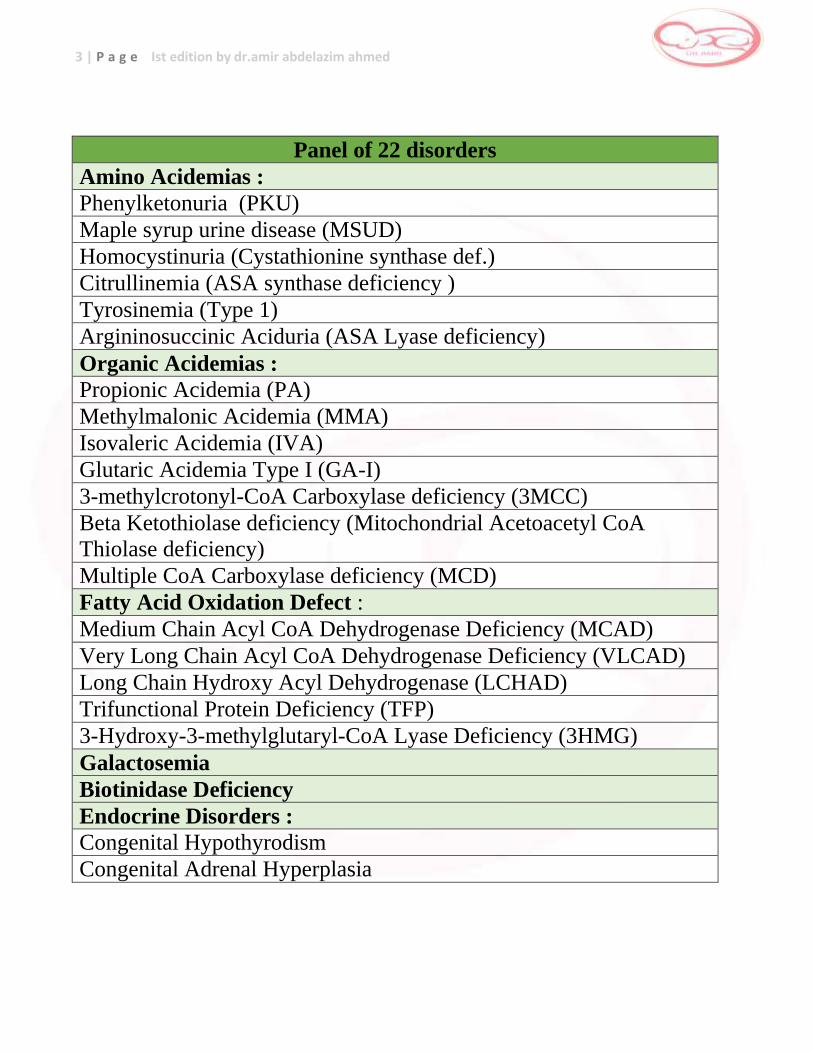
Panel of 22 disorders
Amino Acidemias :
Phenylketonuria (PKU)
Maple syrup urine disease (MSUD)
Homocystinuria (Cystathionine synthase def.)
Citrullinemia (ASA synthase deficiency )
Tyrosinemia (Type 1)
Argininosuccinic Aciduria (ASA Lyase deficiency)
Organic Acidemias :
Propionic Acidemia (PA)
Methylmalonic Acidemia (MMA)
Isovaleric Acidemia (IVA)
Glutaric Acidemia Type I (GA-I)
3-methylcrotonyl-CoA Carboxylase deficiency (3MCC)
Beta Ketothiolase deficiency (Mitochondrial Acetoacetyl CoA
Thiolase deficiency)
Multiple CoA Carboxylase deficiency (MCD)
Fatty Acid Oxidation Defect :
Medium Chain Acyl CoA Dehydrogenase Deficiency (MCAD)
Very Long Chain Acyl CoA Dehydrogenase Deficiency (VLCAD)
Long Chain Hydroxy Acyl Dehydrogenase (LCHAD)
Trifunctional Protein Deficiency (TFP)
3-Hydroxy-3-methylglutaryl-CoA Lyase Deficiency (3HMG)
Galactosemia
Biotinidase Deficiency
Endocrine Disorders :
Congenital Hypothyrodism
Congenital Adrenal Hyperplasia
4 | P a g e Ist edition by dr.amir abdelazim ahmed
5 | P a g e Ist edition by dr.amir abdelazim ahmed
6 | P a g e Ist edition by dr.amir abdelazim ahmed
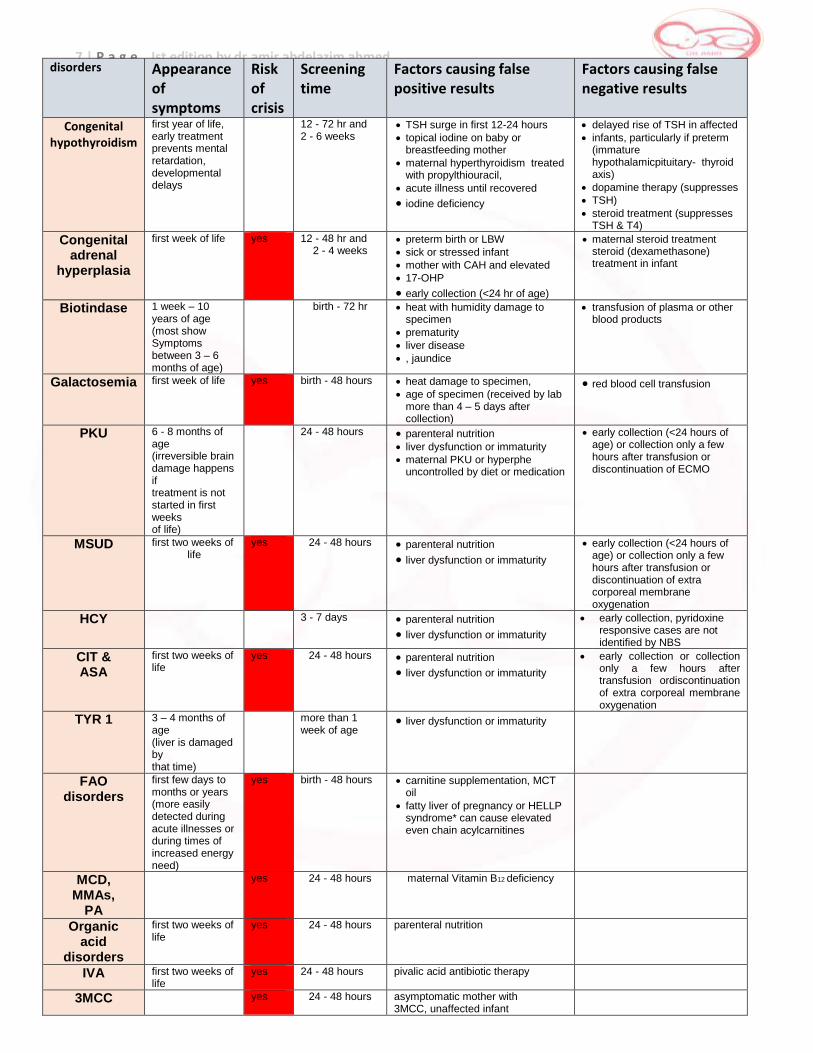
7 | P a g e Ist edition by dr.amir abdelazim ahmed disorders Appearance
of symptoms
Risk of crisis
Screening time
Factors causing false positive results
Factors causing false negative results
Congenital hypothyroidism
first year of life, early treatment prevents mental retardation, developmental delays
12 - 72 hr and 2 - 6 weeks
TSH surge in first 12-24 hours
topical iodine on baby or breastfeeding mother
maternal hyperthyroidism treated with propylthiouracil,
acute illness until recovered
iodine deficiency
delayed rise of TSH in affected
infants, particularly if preterm (immature hypothalamicpituitary- thyroid axis)
dopamine therapy (suppresses
TSH)
steroid treatment (suppresses TSH & T4)
Congenital adrenal
hyperplasia
first week of life yes 12 - 48 hr and 2 - 4 weeks
preterm birth or LBW
sick or stressed infant
mother with CAH and elevated
17-OHP
early collection (<24 hr of age)
maternal steroid treatment steroid (dexamethasone) treatment in infant
Biotindase 1 week – 10 years of age (most show Symptoms between 3 – 6 months of age)
birth - 72 hr heat with humidity damage to specimen
prematurity
liver disease
, jaundice
transfusion of plasma or other blood products
Galactosemia first week of life yes birth - 48 hours heat damage to specimen,
age of specimen (received by lab more than 4 – 5 days after collection)
red blood cell transfusion
PKU 6 - 8 months of age (irreversible brain damage happens if treatment is not started in first weeks of life)
24 - 48 hours parenteral nutrition liver dysfunction or immaturity
maternal PKU or hyperphe uncontrolled by diet or medication
early collection (<24 hours of age) or collection only a few hours after transfusion or discontinuation of ECMO
MSUD first two weeks of life
yes 24 - 48 hours parenteral nutrition
liver dysfunction or immaturity
early collection (<24 hours of age) or collection only a few hours after transfusion or discontinuation of extra corporeal membrane oxygenation
HCY 3 - 7 days parenteral nutrition
liver dysfunction or immaturity
early collection, pyridoxine responsive cases are not identified by NBS
CIT & ASA
first two weeks of life
yes 24 - 48 hours parenteral nutrition
liver dysfunction or immaturity
early collection or collection only a few hours after transfusion ordiscontinuation of extra corporeal membrane oxygenation
TYR 1 3 – 4 months of age (liver is damaged by that time)
more than 1 week of age
liver dysfunction or immaturity
FAO disorders
first few days to months or years (more easily detected during acute illnesses or during times of increased energy need)
yes
birth - 48 hours carnitine supplementation, MCT oil
fatty liver of pregnancy or HELLP syndrome* can cause elevated even chain acylcarnitines
MCD, MMAs,
PA
yes 24 - 48 hours maternal Vitamin B12 deficiency
Organic acid
disorders
first two weeks of life
yes 24 - 48 hours parenteral nutrition
IVA first two weeks of life
yes 24 - 48 hours pivalic acid antibiotic therapy
3MCC yes 24 - 48 hours asymptomatic mother with 3MCC, unaffected infant
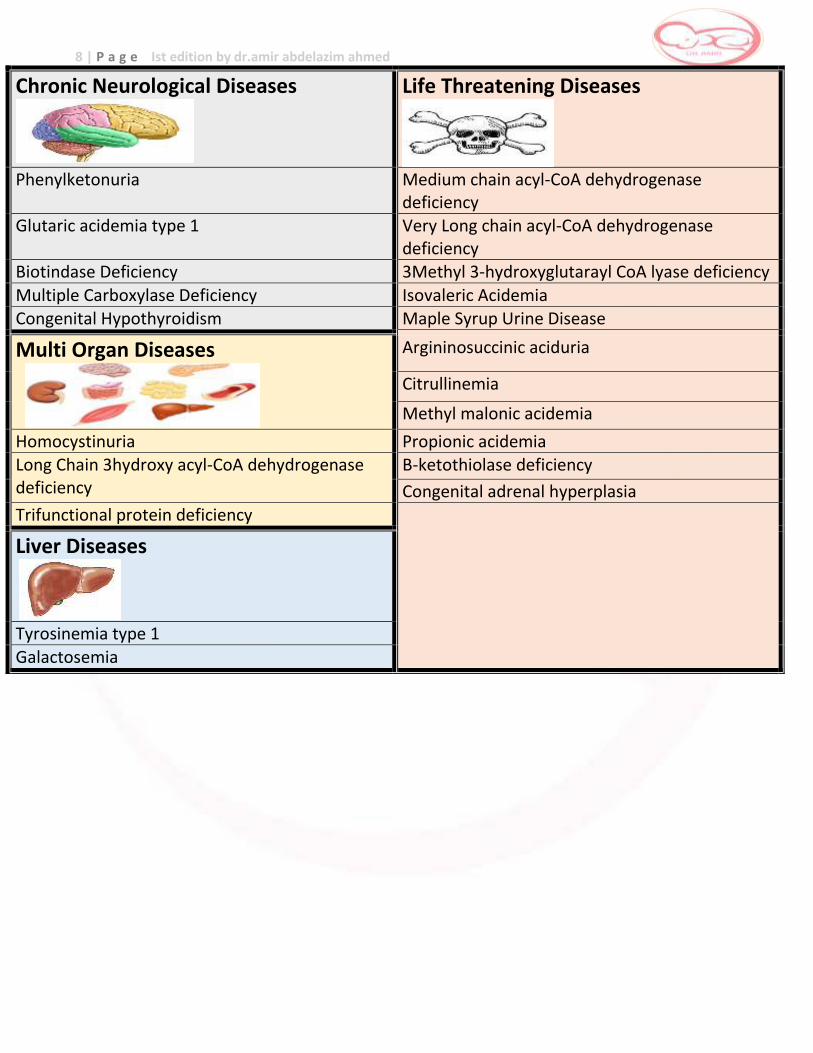
8 | P a g e Ist edition by dr.amir abdelazim ahmed
Chronic Neurological Diseases
Life Threatening Diseases
Phenylketonuria Medium chain acyl-CoA dehydrogenase
deficiency Glutaric acidemia type 1 Very Long chain acyl-CoA dehydrogenase
deficiency Biotindase Deficiency 3Methyl 3-hydroxyglutarayl CoA lyase deficiency
Multiple Carboxylase Deficiency Isovaleric Acidemia Congenital Hypothyroidism Maple Syrup Urine Disease
Multi Organ Diseases
Argininosuccinic aciduria
Citrullinemia
Methyl malonic acidemia
Homocystinuria Propionic acidemia Long Chain 3hydroxy acyl-CoA dehydrogenase deficiency
B-ketothiolase deficiency
Congenital adrenal hyperplasia
Trifunctional protein deficiency
Liver Diseases
Tyrosinemia type 1
Galactosemia
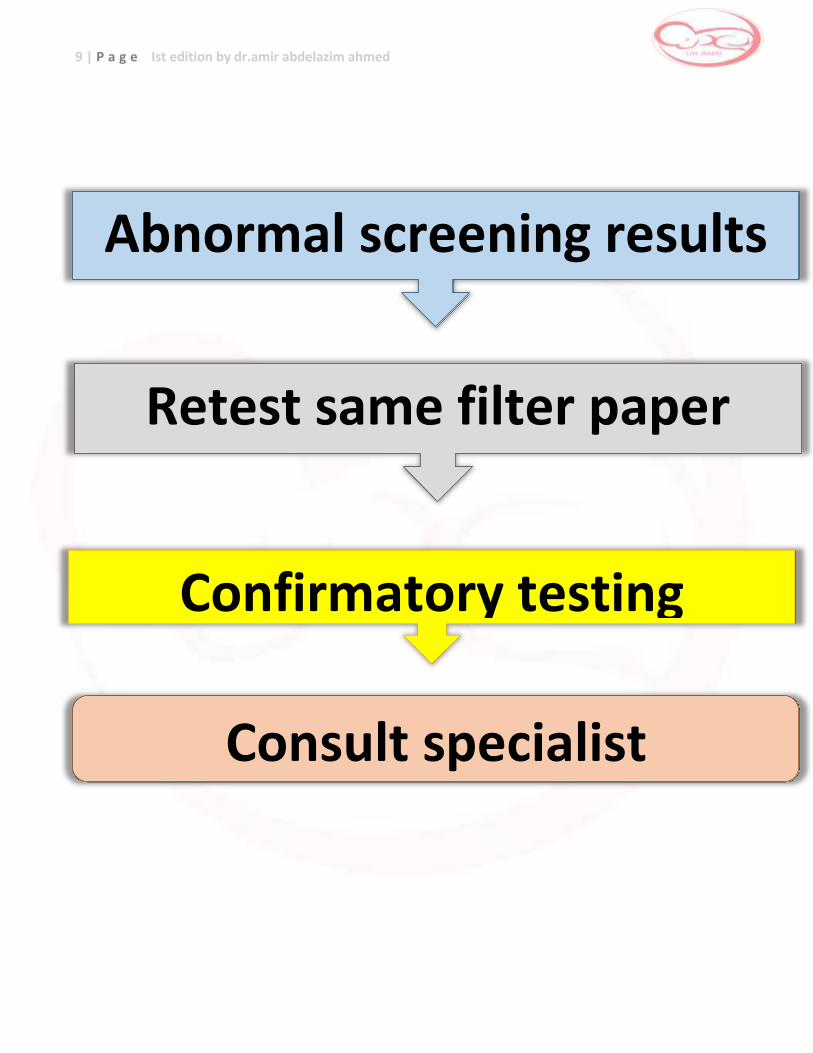
9 | P a g e Ist edition by dr.amir abdelazim ahmed
Abnormal screening results
Retest same filter paper
Confirmatory testing
Consult specialist
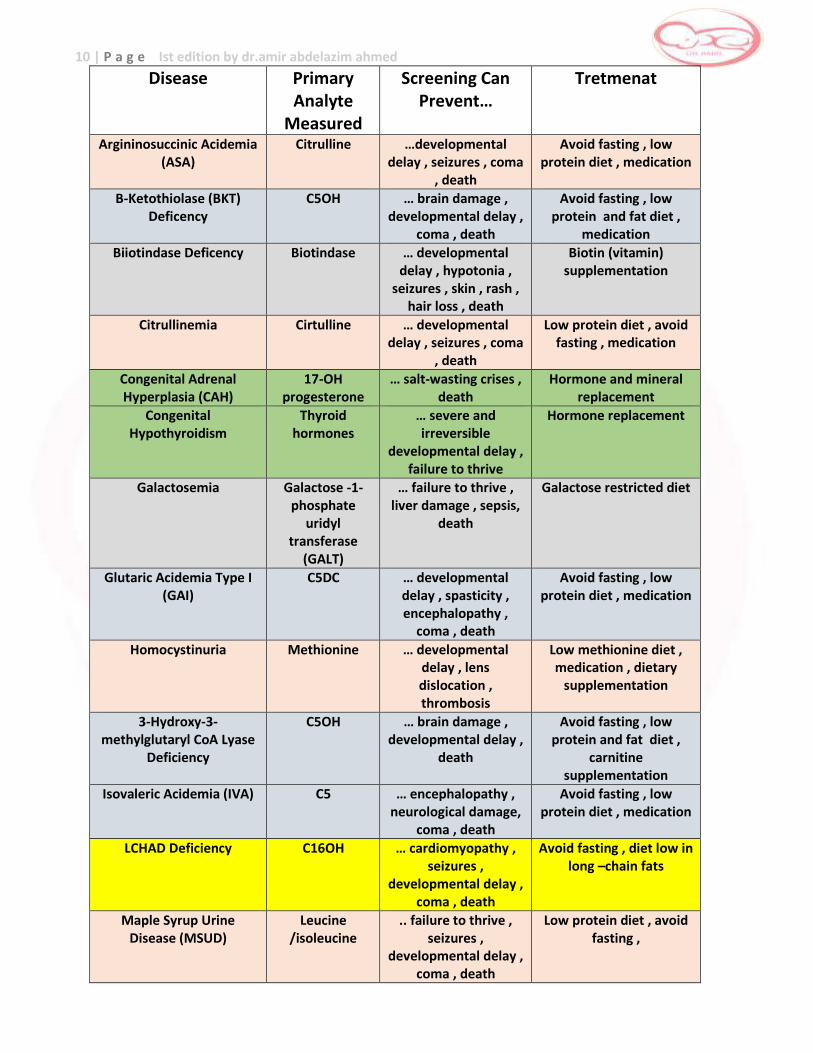
10 | P a g e Ist edition by dr.amir abdelazim ahmed
Disease Primary Analyte
Measured
Screening Can Prevent…
Tretmenat
Argininosuccinic Acidemia (ASA)
Citrulline …developmental delay , seizures , coma
, death
Avoid fasting , low protein diet , medication
Β-Ketothiolase (BKT) Deficency
C5OH … brain damage , developmental delay ,
coma , death
Avoid fasting , low protein and fat diet ,
medication
Biiotindase Deficency Biotindase … developmental delay , hypotonia ,
seizures , skin , rash , hair loss , death
Biotin (vitamin) supplementation
Citrullinemia Cirtulline … developmental delay , seizures , coma
, death
Low protein diet , avoid fasting , medication
Congenital Adrenal Hyperplasia (CAH)
17-OH progesterone
… salt-wasting crises , death
Hormone and mineral replacement
Congenital Hypothyroidism
Thyroid hormones
… severe and irreversible
developmental delay , failure to thrive
Hormone replacement
Galactosemia Galactose -1-phosphate
uridyl transferase
(GALT)
… failure to thrive , liver damage , sepsis,
death
Galactose restricted diet
Glutaric Acidemia Type I (GAI)
C5DC … developmental delay , spasticity , encephalopathy ,
coma , death
Avoid fasting , low protein diet , medication
Homocystinuria Methionine … developmental delay , lens dislocation , thrombosis
Low methionine diet , medication , dietary
supplementation
3-Hydroxy-3-methylglutaryl CoA Lyase
Deficiency
C5OH … brain damage , developmental delay ,
death
Avoid fasting , low protein and fat diet ,
carnitine supplementation
Isovaleric Acidemia (IVA) C5 … encephalopathy , neurological damage,
coma , death
Avoid fasting , low protein diet , medication
LCHAD Deficiency C16OH … cardiomyopathy , seizures ,
developmental delay , coma , death
Avoid fasting , diet low in long –chain fats
Maple Syrup Urine Disease (MSUD)
Leucine /isoleucine
.. failure to thrive , seizures ,
developmental delay , coma , death
Low protein diet , avoid fasting ,
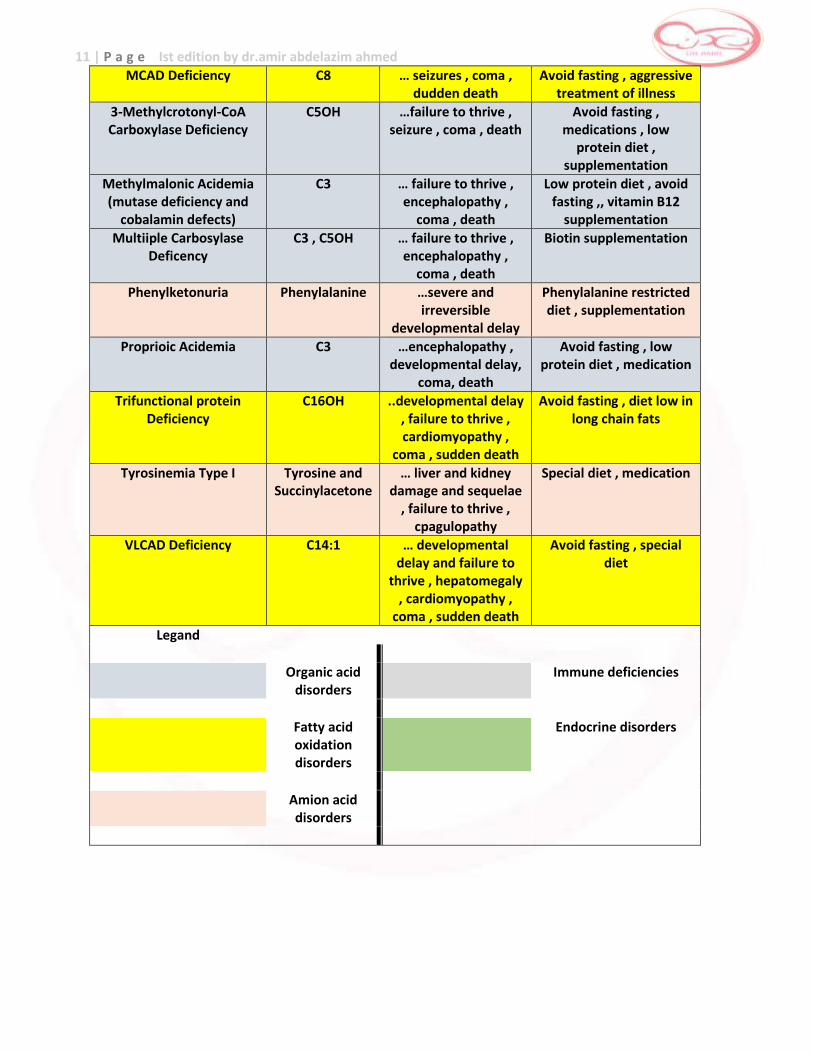
11 | P a g e Ist edition by dr.amir abdelazim ahmed
MCAD Deficiency C8 … seizures , coma , dudden death
Avoid fasting , aggressive treatment of illness
3-Methylcrotonyl-CoA Carboxylase Deficiency
C5OH …failure to thrive , seizure , coma , death
Avoid fasting , medications , low
protein diet , supplementation
Methylmalonic Acidemia (mutase deficiency and
cobalamin defects)
C3 … failure to thrive , encephalopathy ,
coma , death
Low protein diet , avoid fasting ,, vitamin B12
supplementation
Multiiple Carbosylase Deficency
C3 , C5OH … failure to thrive , encephalopathy ,
coma , death
Biotin supplementation
Phenylketonuria Phenylalanine …severe and irreversible
developmental delay
Phenylalanine restricted diet , supplementation
Proprioic Acidemia C3 …encephalopathy , developmental delay,
coma, death
Avoid fasting , low protein diet , medication
Trifunctional protein Deficiency
C16OH ..developmental delay , failure to thrive , cardiomyopathy ,
coma , sudden death
Avoid fasting , diet low in long chain fats
Tyrosinemia Type I Tyrosine and Succinylacetone
… liver and kidney damage and sequelae
, failure to thrive , cpagulopathy
Special diet , medication
VLCAD Deficiency C14:1 … developmental delay and failure to
thrive , hepatomegaly , cardiomyopathy ,
coma , sudden death
Avoid fasting , special diet
Legand
Organic acid disorders
Immune deficiencies
Fatty acid oxidation disorders
Endocrine disorders
Amion acid disorders
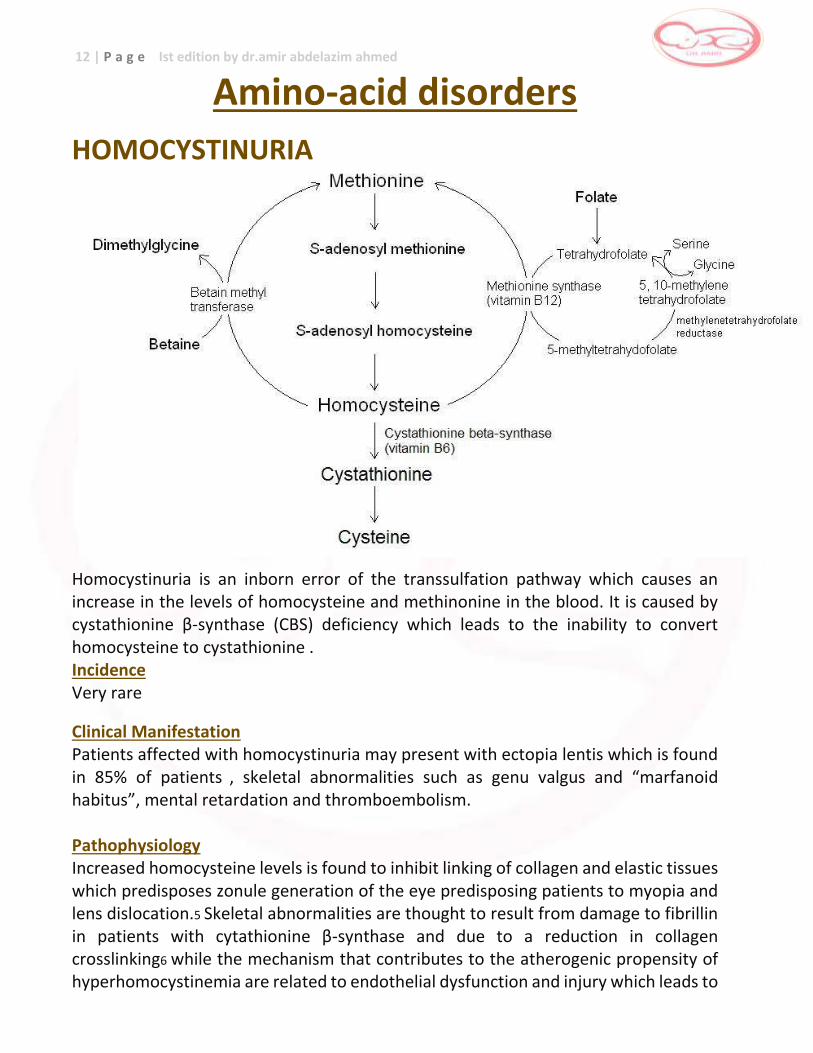
12 | P a g e Ist edition by dr.amir abdelazim ahmed
Amino-acid disorders
HOMOCYSTINURIA
Homocystinuria is an inborn error of the transsulfation pathway which causes an increase in the levels of homocysteine and methinonine in the blood. It is caused by cystathionine β-synthase (CBS) deficiency which leads to the inability to convert homocysteine to cystathionine . Incidence Very rare
Clinical Manifestation Patients affected with homocystinuria may present with ectopia lentis which is found in 85% of patients , skeletal abnormalities such as genu valgus and “marfanoid habitus”, mental retardation and thromboembolism. Pathophysiology Increased homocysteine levels is found to inhibit linking of collagen and elastic tissues which predisposes zonule generation of the eye predisposing patients to myopia and lens dislocation.5 Skeletal abnormalities are thought to result from damage to fibrillin in patients with cytathionine β-synthase and due to a reduction in collagen crosslinking6 while the mechanism that contributes to the atherogenic propensity of hyperhomocystinemia are related to endothelial dysfunction and injury which leads to

13 | P a g e Ist edition by dr.amir abdelazim ahmed
platelet aggregation and thrombus formation.7 Chemical abnormalities and the repeated thromboemolic strokes may contribute to the mental retardation. Inheritance autosomal recessive Screening: increased methionine on MSMS
Confirmatory Testing Total homocysteine in plasma. Amino acids in plasma, methylmalonic acid in urine and enzyme study in fibroblasts may be used to confirm the diagnosis. Prognosis Early diagnosis and treatment can prevent thromboembolic events and reduce the complications brought about by increased levels of homocysteine
Treatment of HCY Treatment is through the dietary restriction of protein and the supplementation of formula lacking methionine. Vitamin B6, folic acid and betaine are also given.
Preliminary / Initial Management During Metabolic Crisis Metabolic crises may be caused by illness, prolonged fasting or stressful situations such as SURGERY and severe infection. The goal of treatment is to reverse the catabolic state and prevent essential amino acid deficiency.

14 | P a g e Ist edition by dr.amir abdelazim ahmed
Long Term Management
The aim of treatment is to reduce plasma total homocysteine levels to as close to normal as possible while maintaining normal growth rate. This can be done in the following ways: Supplementation of Vitamins
Pyridoxine (Vitamin B6)- may start with 50-100mg/day. May progress to 500-1000mg/day guided by plasma homocysteine and methionine monitoring. About half of patients with CBS deficiency respond often only partially to large doses of pyridoxine. But since high doses of pyridoxine has been associated with sensoryneuropathy, it should then be kept at the lowest dose that is able to achieve a good metabolic control. Doses higher than 250mg/day should be avoided in newborns and young infants. If patients do not respond to pyridoxine, a low methionine, high cystine diet must be introduced and continued throughout life. Folic acid – may start at 5-10 mg/day as response to pyridoxine may also be influenced by folate depletion Vitamin C supplementation has been shown to ameliorate endothelial dysfunction in CBS patients suggesting its possible value in reducing the long term risk of atherothrombotic complications. One may give it at 100mg/ day Diet
_ Low Methionine Diet- synthetic methionine free amino acid mixtures for infants _ Supplements of essential fatty acids and carbohydrates are also required _ After infancy, foods containing proteins low in methionine can be introduced.
What to Do: If unwell and cannot tolerate oral intake: a. Nothing per orem b. Ensure patient’s airway is secure c. Insert IV access. Collect samples for methionine and homocystine levels (contact the Biochemical Genetic Laboratory NIH). May request for other investigations (i.e. CBC, Blood gas) as needed. May give fluid boluses if patient requires. d. Start D12.5% 0.3 NaCl at full maintenance. Assess patient clinically, if there is need to increase fluid, may do so up to 1.2 or 1.5X the maintenance especially if the patient will undergo surgery. e. Make sure that the patient is well hydrated. Monitor input and output strictly (q6 hours) f. Start betaine, folic acid and vitamin B6 If unwell but is able to tolerate oral intake: a. Insert oro- or nasogastric tube and start continuous feeding with HCY formula to run at maintenance rate b. Insert IV access. Collect samples for methionine and homocystine level (contact the Biochemical Genetics Laboratory, NIH). May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. c. Start D12.5% 0.3 NaCl at 5-10 cc/hr. Make sure that the patient is well hydrated especially if he will undergo surgery. Monitor input and output strictly (6 hours) d. Start betaine, folic acid and vitamin B6 *Children should not be protein restricted for longer than necessary (24-48 hours). * Inform metabolic doctor on call for further guidance regarding on-going management.

15 | P a g e Ist edition by dr.amir abdelazim ahmed
Betaine
Betaine is a homocysteine lowering agent (remethylates homocysteine to methionine) that is especially useful when compliance to the diet is unsatisfactory. One can start at 100mg/kg/day with a maximum dose of 6-9 grams in adults. Monitoring of plasma homocysteine and methionine levels
Plasma monitoring of methionine, cysteine, cysteine:homocysteine disulfide and homocysteine should be done every 3 months. The goal is a plasma homocysteine level of <60umol/L.
Key metabolite : Methionine , elevated Emergency key : Low
Action : Referral to a metabolic center
Confirmation analysis :
Total homocysteine in plasma Amino acids in plasma Organic(mwthylmalonic)acids in urine Mutation analysis
Therapy :
Diet restricted in methionine Betaine Pyridoxine in responsive patients Vitamene B12 Folic acid
Signs and symptoms :
Mental retardation Dislocation of the lenses Marfanoid habitus Osteoporosis Thromboembolism
Prognosis : Good References 1 Schulze A, Matern D, Hoffmann GF. Chapter 2: Newborn screening in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 17-32. 2 Yap S. Homocystinuria due to cystathionine β-synthase deficiency. Orphanet 2005. http://www.orpha.net/data/photo/GBuk-CbS.pdf Accessed Feb. 16, 2012. 3 Chapter 22 Homocystinuria. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 146-151. 4 Cruysburg JR, Boers GHJ, Trijbels FMJ et al. Delay in diagnosis of homocystinuria: retrospective study of consecutive patients. BMJ 1996;313:1037-1040. 5 Burke JP, O’Keefe M, Bowell R and Naughten ER. Ocular Complications in Homocystinuria – Early and Late Treated. Br J Ophthalmol. 1989 June; 73 (6):427-431. 6 Mudd SH, Levy HL, Skovby F. Disorders of transsulfuration. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. Vol 2. New York: McGraw-Hill, 2001:2007-2056.

16 | P a g e Ist edition by dr.amir abdelazim ahmed
MAPLE SYRUP URINE DISEASE [MSUD]
Maple syrup urine disease (MSUD) is due to a defect or deficiency of the branched chain ketoacid dehydrogenase (BCKD) enzyme complex leading to the elevated quantities of leucine, isoleucine, valine and their corresponding oxoacids in body fluids.1 Accumulation of the latter amino acids will result in life threatening encephalopathy if not adequately treated. Incidence Very rare Clinical Manifestation There are different classifications of MSUD based on the enzyme activity and these include: classical, intermediate, intermittent, thiamine responsive and E-3 deficient MSUD. Classical MSUD (residual enzyme <2%) is the most severe and common form with symptoms of poor suck, lethargy, hypo and hypertonia, opisthotonic posturing, seizures and coma developing 4-7 days after birth.1 The characteristic odor of maple syrup may be detected as soon as neurological symptoms develop. Patients with intermediate MSUD (residual enzyme 3-30%) have gradual neurologic problems resulting in mental retardation.1 Intermittent form of MSUD go into metabolic crisis when there is a stressful situation such as infection or after surgery. Thiamine-responsive MSUD’s clinical symptomatology and metabolic disturbance is ameliorated once pharmacologic dose of thiamine has been given. E-3 deficient MSUD present with symptoms similar to those of intermediate MSUD but they also have lactic acidosis. Pathophysiology Due to mutations in the gene coding for the branched chain keto-acid dehydrogenase enzyme, the levels of leucine, valine and isoleucine increase in blood. The increase in leucine may cause competitive inhibition with other precursors of neurotransmitters causing the neurologic manifestations. Inheritance: autosomal recessive
Screening: leucine + isoleucine, valine, (leucine + isloeucine)/phe ratio
Confirmatory Testing Diagnosis is confirmed by detection of the highly increased branched-chain amino acid levels via quantitative amino acid analysis and/or by increased urinary excretion of α-

17 | P a g e Ist edition by dr.amir abdelazim ahmed
keto and hydroxyl acids and branched chain amino acids using gas chromatography-mass spectrometry (GC-MS) and quantitative amino acid analysis.2 Prognosis Patients with MSUD are now expected to survive, they are generally healthy between episodes of metabolic imbalance and some attend regular school. However, the average intellectual performance is clearly below those of normal subjects.
Treatment of MSUD Treatment is through the dietary restriction of protein and the supplementation of formula lacking leucine, valine and isoleucine.
Preliminary / Initial Management During Metabolic Crisis Metabolic crises may be caused by illness, prolonged fasting or stressful situations such as surgery and severe infection. The goal of treatment is to lower down the levels of leucine, isoleucine and valine, reverse the catabolic state and prevent essential amino acid deficiency.
What to Do: If unwell and cannot tolerate oral intake: a. Nothing per orem b. Ensure patient’s airway is secure c. Insert IV access. Collect samples for leucine level, plasma amino acids, blood glucose and urine ketones. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. d. Start D12.5% 0.3 NaCl at full maintenance. Assess patient clinically, if there is need to increase fluid, may do so up to 1.2 or 1.5X the maintenance. e. Start intralipid at 1g/kg/24 hours. f. Monitor input and output strictly (q6 hours) If unwell but is able to tolerate oral intake: a. Insert oro- or nasogastric tube and start continuous feeding with BCAD formula to run at maintenance rate b. May give valine at 50mg/kg/day divided into 6 doses and isoleucine 30mg/kg/day divided into 6 doses c. Insert IV access. Collect samples for leucine level, plasma amino acids, blood glucose and urine ketones. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. d. Start D12.5% 0.3 NaCl at 5-10 cc/hr. e. Monitor input and output strictly (q6 hours) *Children should not be protein restricted for longer than necessary (24-48 hours). *If patient does not improve with the initial management (within 12 hours), hemodialysis may be indicated. Monitor patient clinically, the necessity of hemodialysis will depend on patient’s clinical status. * Inform metabolic doctor on call for further guidance regarding on-going management.

18 | P a g e Ist edition by dr.amir abdelazim ahmed
Long term Management
The aim of life long maintenance therapy is to maintain the branched chain amino acid levels at near normalconcentrations. Regular evaluation of nutritional status, metabolic control, growth percentiles as well as developmental progress are imperative for a good clinical and cognitive outcome. Diet
The major component of the diet is a special formula that do not contain any leucine, isoleucine or valine but are otherwise nutritionally complete. They contain all the necessary vitamins, minerals, calories and the other amino acids needed for growth. They will also be given a formula supplemented with carefully controlled amounts of a protein-based formula. The protein-based formula provides the infant with the limited amount of branched chain amino acids needed to grow and develop normally. As children with MSUD grow, they continue taking the special formula. They are allowed other foods which are weighed or measured in the home to supply the prescribed amount of leucine each day. Typically the MSUD diet does not include any high protein foods such as meat, nuts, eggs, and most dairy products. Children gradually learn to accept the responsibility for controlling their diets and generally being on low protein at all times. Frequent determination of leucine levels are likewise encouraged so that proper dietary adjustments be done for effective management of the condition. Special supplements
Occasionally, small amounts of free valine and isoleucine must be added to the amounts provided by the natural protein because the tolerance for leucine is lower than the other two. Under conditions of high leucine and low valine and isoleucine levels, a rapid fall of plasma leucine can be achieved only by combining a reduced leucine intake with a temporary supplement of leucine and isoleucine. Treatment of intercurrent decompensations
Acute intercurrent episodes are prevented by being aware of those situations that may induce protein catabolism. These include intercurrent infections, immunizations, trauma, anesthesia and surgery. Parents must have at their disposal a semi emergency diet in which natural protein intakes are reduced by half or an emergency diet in which natural proteins are suppressed. In both, energy supply is reinforced using carbohydrates and lipids. Solutions containing a mixture of glucose polymer and lipids can be used. Timely evaluation and intensive treatment of minor illnesses at any age is essential, as late death attributed to recurrence of metabolic crises with infections has occurred.

19 | P a g e Ist edition by dr.amir abdelazim ahmed
Emergency Protocol for Maple Syrup Urine Disease Important points to be relayed to the parents over the phone:
1. Avoid delay and bring the child to the hospital at once
2. Bring formula (if known MSUD patient)
3. Bring isoleucine and valine tablets (if known MSUD patient)
4. Ask for child’s current weight
5. Ask about an estimated time of arrival at the ER
Alert Emergency Department of the patient’s arrival
1. Talk to the Admitting Officer and Nursing Team Leader
2. Ask them to do an urgent clinical assessment (history and physical examination)
3. *Prepare 12.5% dextrose (maintenance)
4. *Prepare Intralipid 2g/kg/day
5. Collect blood for **plasma amino acids or on dried blood spot. Check for urine ketones. Other examinations as required.
6. Contact the Physician on call once patient arrives at ER ————————— * Please prescribe for weight before the patient arrives. ** Collect in green top tube. Transport immediately to Biochemical Genetics Laboratory
Principles of Management Reversion of catabolism
Start IV infusion using 12.5% dextrose -maintenance + %dehydration (add potassium if serum K is not high). If the patient is encephalopathic, additional sodium may be required (up to 6 mmols/kg/day). If there is a concern about cerebral edema (focal neurologic signs or fluctuating level of consciousness) fluids may need to be restricted. _ Stop natural protein. _ Intralipid at 2g/kg/day. This can be infused in the same line peripherally. _ The patient may also have an enteral emergency sick day regimen, which can be administered continuously via a nasogastric feeding tube. _ Treat underlying cause. Treat dehydration, electrolyte imbalance, infection and acidosis _ Consider dialysis if with acute deterioration of cerebral function consider the
following
_ Maintain plasma concentrations of isoleucine and valine more than 200 umol/L
20 | P a g e Ist edition by dr.amir abdelazim ahmed
Key metabolite : Leucine + isoleucine , valine , elevated
Emergency key : High
Action : Immediate referral to metabolic specialist
Confirmation analysis :
Amino acids in plasma Organic acid in urine Enzyme activity in lymphocytes Mutation analysis
Therapy :
Acute management : Discontinue natural protein Provide large amount of calories ,fluids and electrolytes Enteral therapy : Special formula that contains all required amino acids but is free of leucine , valine and isoleucine
Signs and symptoms :
Progressive encephalopathy Maple syrup smell of urine Mental retardation
Prognosis : Moderate , often mild mental impairment even in well treated children
References 1 Chapter 24 Maple syrup urine disease. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd
ed. Great Britain:Oxford University Press, 2005 pp 159-164 2 Hoffman GF and Schulze A. Chapter 7: Organic Acidurias in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 93-94. 3 Schulze A, Matern D, Hoffmann GF. Chapter 2: Newborn screening in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 17-32. 4 Wendel U and de Baulny H. branched chain organic acidurias/acidemias. Inborn Metabolic Diseases Chapter 19 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp246-256

21 | P a g e Ist edition by dr.amir abdelazim ahmed
PHENYLKETONURIA [PKU]
Phenylketonuria is a disorder of aromatic amino acid metabolism in which phenylalanine cannot be converted to tyrosine due to a deficiency or absence of the enzyme phenylalanine hydroxylase. Phenylalanine hydroxylase requires the co-factor 6-pyruvoyltetrahydropterin or BH4 for activity in the hydroxylation to tyrosine, absence of this co-factor may present with an increase in plasma phenylalanine similar to phenylketonuria but is considered a separate disorder. Incidence 1:15,000 worldwide
Clinical Manifestation Patients affected with PKU appear normal at birth.2,4 The most important and sometimes the only manifestation of PKU is mental retardation.2 Patients may present with constitutional, intellectual and neurologic abnormalities and signs as well as hypopigmentation of the skin and hair and iris rapidly develop due to impaired metabolism of melanin.4 Seizures occur in a fourth of patients. The odor of the phenylketonuric patient is that of phenylacetic acid described as mousy, barny, or musty.
Pathophysiology PKU results from a deficiency of activity of a liver enzyme, phenylalanine hydroxylase leading to increased concentrations of phenylalanine in the blood and other tissues.4
Elevated phenylalanine interfere with myelination, synaptic sprouting and dendritic pruning; and in addition, it competitively inhibits the uptake of neutral amino acids in the blood-brain barrier causing reduced tyrosine and tryptophan concentrations thereby limiting the production of neurotransmitters.4 Inheritance autosomal recessive Screening increased phenylalanine levels on MSMS
Confirmatory Testing The demonstration of decreased enzyme activity is confirmatory. However, in the presence of increased phenylalanine levels, it is important to differentiate phenylketonuria from a BH4 deficiency. This is accomplished through administration of tetrahydrobiopterin (doses of 2mg/kg intravenously and 7.5-20mg/kd orally) which leads to a prompt decrease to normal in the concentration of phenylalanine. Pterin
22 | P a g e Ist edition by dr.amir abdelazim ahmed
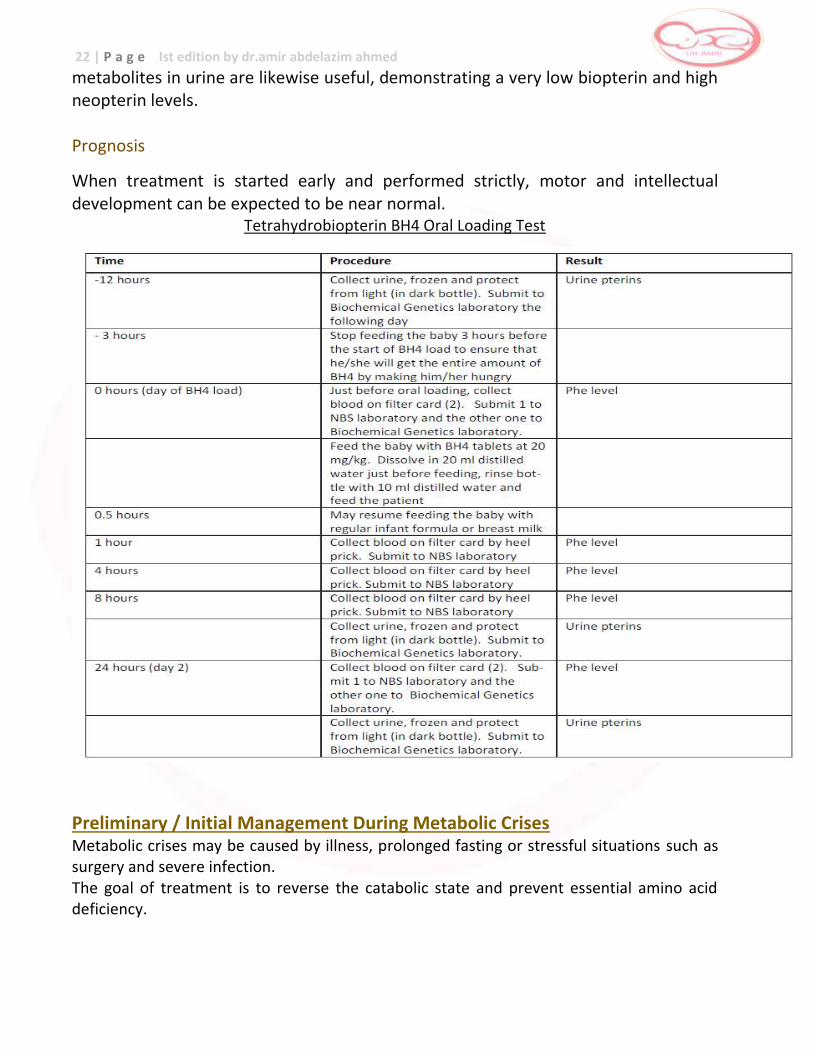
metabolites in urine are likewise useful, demonstrating a very low biopterin and high neopterin levels. Prognosis
When treatment is started early and performed strictly, motor and intellectual development can be expected to be near normal.
Tetrahydrobiopterin BH4 Oral Loading Test
Preliminary / Initial Management During Metabolic Crises Metabolic crises may be caused by illness, prolonged fasting or stressful situations such as surgery and severe infection. The goal of treatment is to reverse the catabolic state and prevent essential amino acid deficiency.

23 | P a g e Ist edition by dr.amir abdelazim ahmed
Long Term Management Diet
Dietary management is the key to treatment. The diet of patients has four components: _ complete avoidance of food containing high amounts of phenylalanine; _ calculated intake of low protein/phenylalanine natural food _ sufficient intake of fat and carbohydrates to fulfill the energy requirements of the patient and; _ calculated intake of phenylalanine free amino acid mixture supplemented with vitamins, minerals and trace elements as the main source of protein. In young children
At the start of treatment in infants with blood phenylalanine levels above 1200 umol/L, a period (usually 24-48 hrs) of phenylalanine free milk brings levels down at a rate of 400 umol/l per day. As levels approach the therapeutic range (120-360umol/L), phenylalanine is then added (around 1-1.5g/kg/day). Infants with lesser degrees of phenylalanine accumulation need less rigorous restriction and smooth control is easier to achieve. The prescription of phenylalanine is adjusted until serial blood levels have stabilized.
What to Do: If unwell and cannot tolerate oral intake: a. Nothing per orem b. Ensure patient’s airway is secure c. Insert IV access. Collect samples for phenylalnine levels. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. d. Start D12.5% 0.3 NaCl at full maintenance. Assess patient clinically, if there is need to increase fluid, may do so up to 1.2 or 1.5X the maintenance. e. Start Intralipid at 1g/kg/day f. Monitor input and output strictly (q6 hours)
If unwell but is able to tolerate oral intake: a. Insert oro- or nasogastric tube and start continuous feeding with PKU formula to run at maintenance rate b. Insert IV access. Collect samples for phenylalanine levels. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. c. Start D12.5% 0.3 NaCl at 5-10 cc/hr. d. Monitor input and output strictly (q6 hours) *Children should not be protein restricted for longer than necessary (24-48 hours). * Inform metabolic doctor on call for further guidance regarding on-going management

24 | P a g e Ist edition by dr.amir abdelazim ahmed
In older children, adolescents and adults
Given the practical difficulties involved in sustaining a strict low phenylalanine diet, a relaxation of the diet at some point before adolescence is allowed. It is recommended that older children be offered the opportunity to remain on a diet that keep blood phenylalanine concentrations ar or below 700umol/L after mid-childhood and into adulthood. Phenylalanine levels rise in response to minor events such as intercurrent illness, decline in energy intake or in growth rate, reduction in the amount of protein substitute and rise in phenylalanine intake, thus diet should be adjusted as needed. Managing illness
During illness, children cannot take their prescribed diet. High energy fluids with or without fat emulsion will help reduce catabolism and are more acceptable to children during time of illness. As anabolism takes over, it is important to reintroduce phenylalanine allowance to avoid phenylalanine deficiency as diet is re-established. Monitoring of phenylalanine levels and growth and development
Regular monitoring of phenylalanine levels (at least monthly or more frequent depending on the clinical status of patient) should be done religiously. There is evidence that raising blood phenylalanine concentrations is associated with reversible impairments in neuropsychological performance, thus assessment of mental development should likewise be enforced. The risk of maternal phenylketonuria in adolescent girls and women of reproductive age should also be emphasized as this risk increases linearly in proportion to maternal phenylalanine concentrations. Defects of Biopterin Metabolism (i.e. 6 Pyruvoyltetrahydrobiopterin synthase deficiency)
There is no diet restriction in these types of disorders. The following medications should be given: _ Tetrahydrobiopterin: 5-10 mg/kg/day
L-Dopa 8-12 mg/kg/day (neonates 1-3mg/kg/day, infants 4-7 mg/kg/day) _ 5-OH-tryptophan (max 6-9mg/kg/day)
Key metabolite : Phe , elevated Emergency key : Moderate
Action : Refer to metabolic specialist Confirmation analysis : Amion acid in plasma
Pterin analysis in urine DHPR-activity in DBS
Therapy : Phe restricted diet Signs and symptoms : Severe mental retardation –seizures
Prognosis : Excellent , normal development
25 | P a g e Ist edition by dr.amir abdelazim ahmed
References 1Chapter 20: Phenylketonuria. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 127-133. 2Chapter 21 Hyperphenylalaninemia and defective metabolism of tetrahydrobiopterin. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 136-145 3Burgard P, Lui X, Hoffmann GF. Chapter 13: Phenylketonuria in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 163-168. 4Kaye CI and the Committee on Genetics. Newborn screening fact sheets. Pediatrics 2006;118:934-963. 5 Walter JH, Lee P, Burgard P, Hyperphenylalaninemia. Inborn Metabolic Diseases Chapter 17 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp224-226 6 Zschocke J and Hoffman G. Vademecum Metabolicum (Diagnosis and Treatment of Inborn Errors of
Metabolism) 3rd edition pp 153.
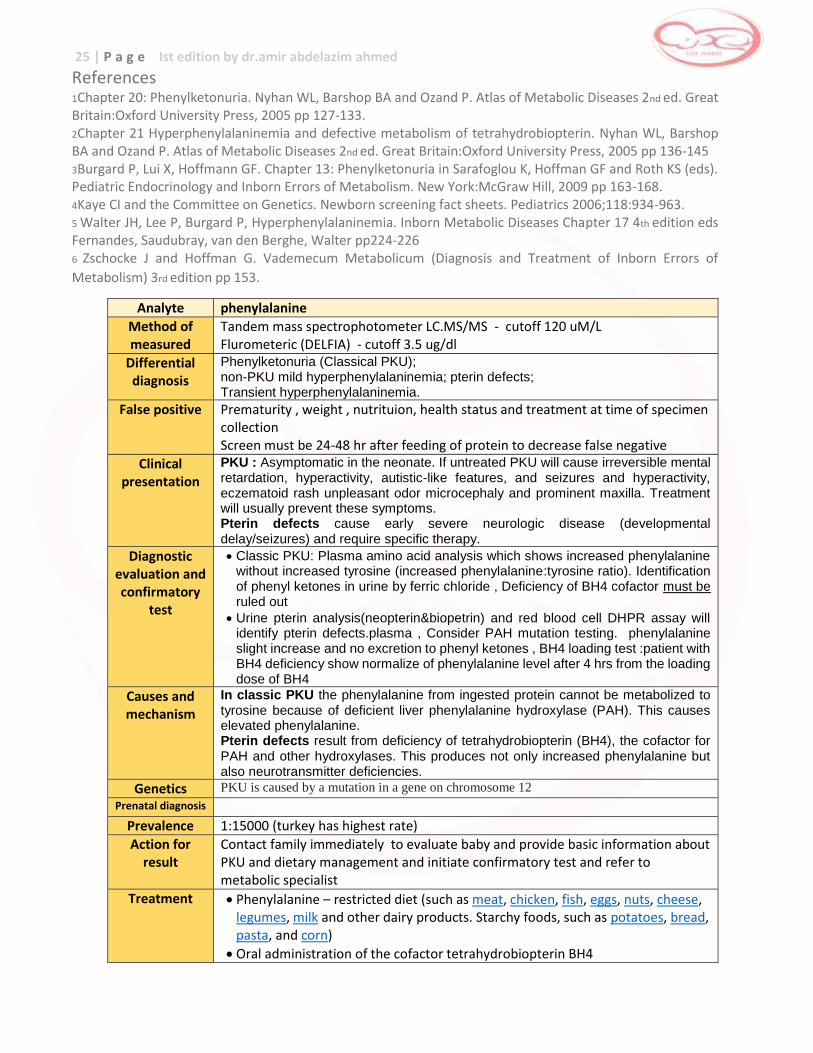
Analyte phenylalanine
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 120 uM/L Flurometeric (DELFIA) - cutoff 3.5 ug/dl
Differential diagnosis
Phenylketonuria (Classical PKU); non-PKU mild hyperphenylalaninemia; pterin defects; Transient hyperphenylalaninemia.
False positive Prematurity , weight , nutrituion, health status and treatment at time of specimen collection Screen must be 24-48 hr after feeding of protein to decrease false negative
Clinical presentation
PKU : Asymptomatic in the neonate. If untreated PKU will cause irreversible mental retardation, hyperactivity, autistic-like features, and seizures and hyperactivity, eczematoid rash unpleasant odor microcephaly and prominent maxilla. Treatment will usually prevent these symptoms. Pterin defects cause early severe neurologic disease (developmental delay/seizures) and require specific therapy.
Diagnostic evaluation and confirmatory
test
Classic PKU: Plasma amino acid analysis which shows increased phenylalanine without increased tyrosine (increased phenylalanine:tyrosine ratio). Identification of phenyl ketones in urine by ferric chloride , Deficiency of BH4 cofactor must be ruled out
Urine pterin analysis(neopterin&biopetrin) and red blood cell DHPR assay will identify pterin defects.plasma , Consider PAH mutation testing. phenylalanine slight increase and no excretion to phenyl ketones , BH4 loading test :patient with BH4 deficiency show normalize of phenylalanine level after 4 hrs from the loading dose of BH4
Causes and mechanism
In classic PKU the phenylalanine from ingested protein cannot be metabolized to tyrosine because of deficient liver phenylalanine hydroxylase (PAH). This causes elevated phenylalanine. Pterin defects result from deficiency of tetrahydrobiopterin (BH4), the cofactor for PAH and other hydroxylases. This produces not only increased phenylalanine but also neurotransmitter deficiencies.
Genetics PKU is caused by a mutation in a gene on chromosome 12
Prenatal diagnosis
Prevalence 1:15000 (turkey has highest rate)
Action for result
Contact family immediately to evaluate baby and provide basic information about PKU and dietary management and initiate confirmatory test and refer to metabolic specialist
Treatment Phenylalanine – restricted diet (such as meat, chicken, fish, eggs, nuts, cheese, legumes, milk and other dairy products. Starchy foods, such as potatoes, bread, pasta, and corn)
Oral administration of the cofactor tetrahydrobiopterin BH4

26 | P a g e Ist edition by dr.amir abdelazim ahmed
TYROSINEMIA
There are 2 clinically recognized types of tyrosinemia. Type I (hepatorenal) is characterized by liver toxicity from increased concentrations of tyrosine. There is anssociated renal tubular defects and peripheral neuropathy. There is also a high risk for hepatocellular carcinoma. The deficient enzyme is fumarylacetoacetase.
Type II (oculocutaneous) tyrosinemia exhibits with corneal lesions and hyperkeratosis of palms and soles. It is caused by the deficiency of the enzyme, tyrosine aminotransferase. Incidence Very rare Clinical Manifestation Tyrosine-I is usually asymptomatic in newborns, but if left untreated it affects liver, kidney, bone, and peripheral nerves. Two patterns are reported: an acute or chronic form. The acute form presents with acute hepatic decompensation where infants are noted to have jaundice, abdominal distention, failure to thrive, ascites and hepatomegaly, renal disease is also prominent and a “boiled cabbage” odor in urine is observed; the chronic liver disease feature is that of hepatic cirrhosis. Tyrosinemia type II is a distinctive oculocutaneous syndrome. Eye findings can be limited to lacrimation, photophobia, and redness. Cutaneous lesions includepainful nonpruritic blisters or erosions that crust and become hyperkeratotic. Mental retardation is also an infrequent finding. Pathophysiology In type I, the deficient enzyme, fumarylacetoacetase catalyzed the last step in tyrosine degradation. The increased concentrations of tyrosine and its metabolites is postulated to inhibit many transport functions and enzymatic activities. In type II, deficiency of the rate limiting enzyme tyrosine transaminase in tyrosine catabolism leads to accumulation of tyrosine, phenolic acids, tyramine in the blood ad urine.1 Inheritance autosomal recessive Screening increased tyrosine and succinylacetone for type I; increased tyrosine for type II
Confirmatory Testing Confirmation can be done through plasma amino acid levels (increased tyrosine) and urine metabolic screening (increased succinylacetone).

27 | P a g e Ist edition by dr.amir abdelazim ahmed
Prognosis If untreated, death from liver failure may occur in the first year of life for hepatorenal
tyrosinemia.
Treatment of Tyrosinemia Treatment is through the dietary restriction of protein and the supplementation of formula lacking tyrosine. Patients are also given nitisinone (NTBC) which is an inhibitor of p-hydroxyphenylpyruvate dioxygenase as maintenance medication.
Preliminary / Initial Management During Metabolic Crisis Metabolic crises may be caused by illness, high consumption of protein, prolonged fasting or stressful situations such as surgery and severe infection. The goal of treatment is to control level of tyrosine, correct bleeding parameters, reverse the catabolic state and prevent essential amino acid deficiency.
What to Do: If unwell and cannot tolerate oral intake: a. Nothing per orem except medications b. Ensure patient’s airway is secure c. Insert IV access. Collect samples for blood glucose, plasma amino acids, liver function tests, coagulation studies and urine succinylacetone. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. d. Start D12.5% 0.3 NaCl at full maintenance. Assess patient clinically, if there is need to increase fluid, may do so up to 1.2 or 1.5X the maintenance. e. Start nitisinone (2mg/kg) per orem. f. Monitor input and output strictly (6 hours) If unwell but is able to tolerate oral intake: a. Insert oro- or nasogastric tube and start continuous feeding with tyrosine free formula to run at maintenance rate b. Start nitisinone (2mg/kg) per NGT c. Insert IV access. Collect samples for blood glucose, plasma amino acids, liver function tests, coagulation studies and urine succinylacetone. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. d. Start D12.5% 0.3 NaCl at 5-10 cc/hr. e. Monitor input and output strictly (q6 hours) *Children should not be protein restricted for longer than necessary (24-48 hours). * Inform metabolic doctor on call for further guidance regarding on-going management.

28 | P a g e Ist edition by dr.amir abdelazim ahmed
Long Term Management
Tyrosinemia type I
Treatment options for tyrosinemia I include dietary therapy (restriction of phenylalanine and tyrosine), liver transplantation and use of the pharmacologic agent 2(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione or NTBC. NTBC
The rationale for the use of NTBC is to block tyrosine degradation at an early step so as to prevent production of toxic down stream metabolites such as fumarylacetoacetate, maleylacetoacetate and succinylacetone. It is recommended at an initial dose of 1 mg/kg/day. The risk of hepatocellular carcinoma appears to be much reduced in patients started early on NTBC treatment (before 6 months of age). Diet
Dietary restriction of phenylalanine and tyrosine is necessary to prevent the known adverse effects of hypertyrosinemia. Tyrosine levels are aimed between 200-400 umol/L using a combination of a protein restricted diet and phenylalanine and tyrosine free amino acid mixtures. Supportive therapy
In the acutely ill patient, supportive treatment is essential. Clotting factors, albumin, electrolytes and acid/base balance should be closely monitored and corrected as necessary. Tyrosine and phenylalanine intake should be kept to a minimum during acute decompensation. Addition of vitamin D may be required to treat rickets. Infections should be treated aggressively. Monitoring of patients on NTBS should include regular blood tests for liver function, blood counts, clotting studies, alpha feto protein, tests of renal and tubular function, hepatic imaging and plasma amino acid profile. Blood levels of phenylalanine and tyrosine should be checked every 3 months and the diet should be supervised regularly. Tyrosinemia type II Diet
Treatment consists of phenylalanine and tyrosine restricted diet and the skin and eye symptoms resolve within weeks of treatment. In general, skin and eye symptoms do not occur at tyrosine levels <800umol/L, however, as hypertyrosinemia may be involved in the pathogenesis of neurodevelopmental symptoms, it may be beneficial to maintain much lower levels. Growth and nutritional status should be regularly monitored.
29 | P a g e Ist edition by dr.amir abdelazim ahmed
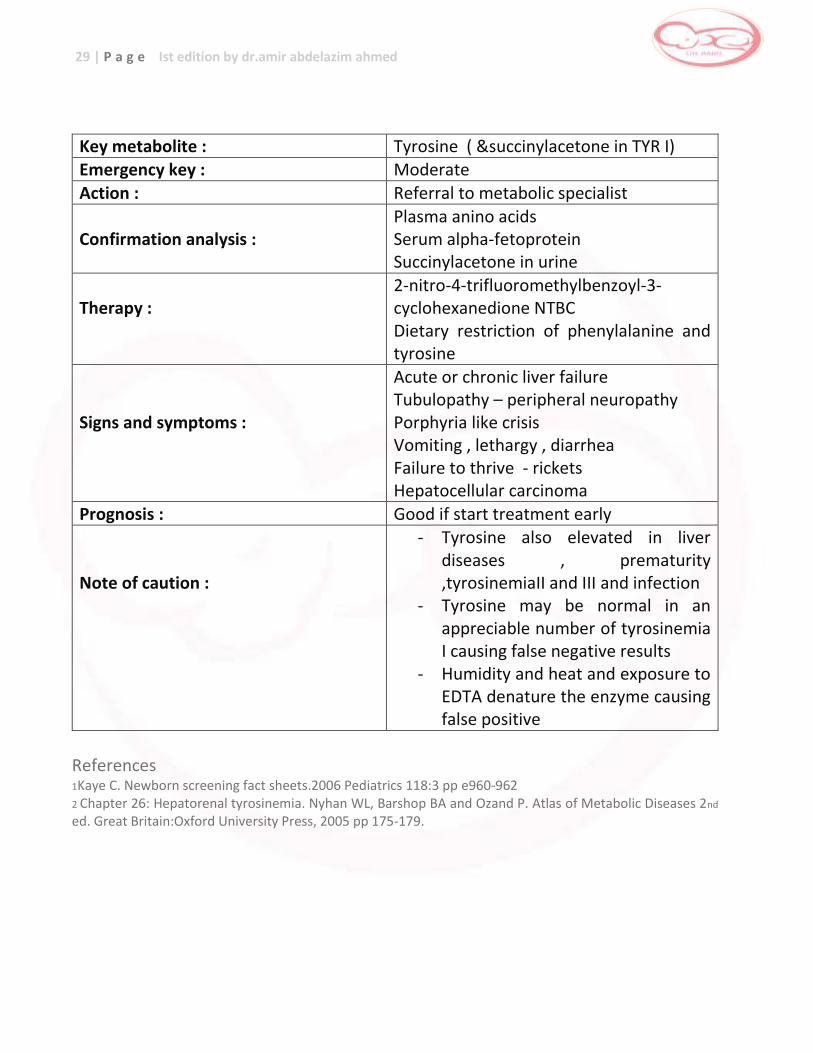
Key metabolite : Tyrosine ( &succinylacetone in TYR I) Emergency key : Moderate
Action : Referral to metabolic specialist
Confirmation analysis :
Plasma anino acids Serum alpha-fetoprotein Succinylacetone in urine
Therapy :
2-nitro-4-trifluoromethylbenzoyl-3-cyclohexanedione NTBC Dietary restriction of phenylalanine and tyrosine
Signs and symptoms :
Acute or chronic liver failure Tubulopathy – peripheral neuropathy Porphyria like crisis Vomiting , lethargy , diarrhea Failure to thrive - rickets Hepatocellular carcinoma
Prognosis : Good if start treatment early Note of caution :
- Tyrosine also elevated in liver diseases , prematurity ,tyrosinemiaII and III and infection
- Tyrosine may be normal in an appreciable number of tyrosinemia I causing false negative results
- Humidity and heat and exposure to EDTA denature the enzyme causing false positive
References 1Kaye C. Newborn screening fact sheets.2006 Pediatrics 118:3 pp e960-962 2 Chapter 26: Hepatorenal tyrosinemia. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd
ed. Great Britain:Oxford University Press, 2005 pp 175-179.
30 | P a g e Ist edition by dr.amir abdelazim ahmed
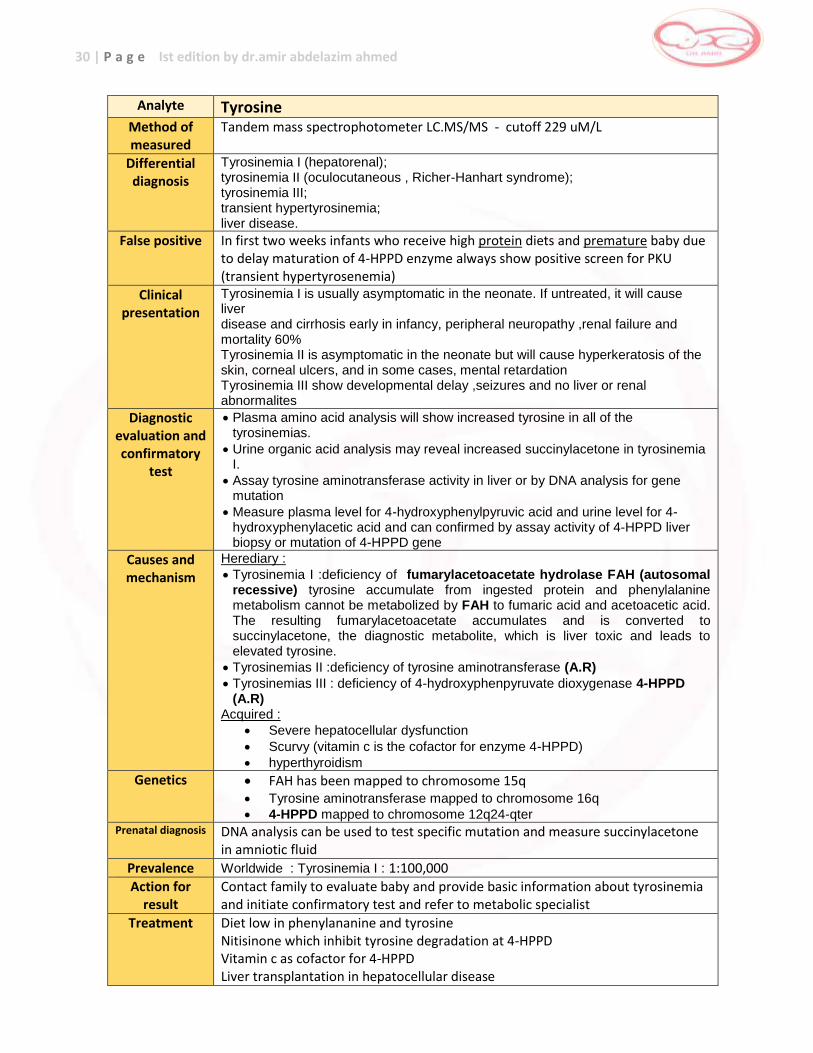
Analyte Tyrosine Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 229 uM/L
Differential diagnosis
Tyrosinemia I (hepatorenal); tyrosinemia II (oculocutaneous , Richer-Hanhart syndrome); tyrosinemia III; transient hypertyrosinemia; liver disease.
False positive In first two weeks infants who receive high protein diets and premature baby due to delay maturation of 4-HPPD enzyme always show positive screen for PKU (transient hypertyrosenemia)
Clinical presentation
Tyrosinemia I is usually asymptomatic in the neonate. If untreated, it will cause liver disease and cirrhosis early in infancy, peripheral neuropathy ,renal failure and mortality 60% Tyrosinemia II is asymptomatic in the neonate but will cause hyperkeratosis of the skin, corneal ulcers, and in some cases, mental retardation Tyrosinemia III show developmental delay ,seizures and no liver or renal abnormalites
Diagnostic evaluation and confirmatory
test
Plasma amino acid analysis will show increased tyrosine in all of the tyrosinemias.
Urine organic acid analysis may reveal increased succinylacetone in tyrosinemia I.
Assay tyrosine aminotransferase activity in liver or by DNA analysis for gene mutation
Measure plasma level for 4-hydroxyphenylpyruvic acid and urine level for 4-hydroxyphenylacetic acid and can confirmed by assay activity of 4-HPPD liver biopsy or mutation of 4-HPPD gene
Causes and mechanism
Herediary :
Tyrosinemia I :deficiency of fumarylacetoacetate hydrolase FAH (autosomal recessive) tyrosine accumulate from ingested protein and phenylalanine metabolism cannot be metabolized by FAH to fumaric acid and acetoacetic acid. The resulting fumarylacetoacetate accumulates and is converted to succinylacetone, the diagnostic metabolite, which is liver toxic and leads to elevated tyrosine.
Tyrosinemias II :deficiency of tyrosine aminotransferase (A.R)
Tyrosinemias III : deficiency of 4-hydroxyphenpyruvate dioxygenase 4-HPPD (A.R)
Acquired :
Severe hepatocellular dysfunction
Scurvy (vitamin c is the cofactor for enzyme 4-HPPD)
hyperthyroidism
Genetics FAH has been mapped to chromosome 15q Tyrosine aminotransferase mapped to chromosome 16q
4-HPPD mapped to chromosome 12q24-qter Prenatal diagnosis DNA analysis can be used to test specific mutation and measure succinylacetone
in amniotic fluid
Prevalence Worldwide : Tyrosinemia I : 1:100,000
Action for result
Contact family to evaluate baby and provide basic information about tyrosinemia and initiate confirmatory test and refer to metabolic specialist
Treatment Diet low in phenylananine and tyrosine Nitisinone which inhibit tyrosine degradation at 4-HPPD Vitamin c as cofactor for 4-HPPD Liver transplantation in hepatocellular disease

31 | P a g e Ist edition by dr.amir abdelazim ahmed
UREA CYCLE DEFECTS
CITRULLINEMIA
Citrullinemia is an inborn error of metabolism resulting from the deficiency of argininosuccinate synthetase, an enzyme present in all tissues but the level of which is highest in the liver where it functions in the urea cycle. Incidence Very rare Clinical Manifestation Following a brief hiatus in which the newborn appears normal, anorexia, vomiting and lethargy develop followed rapidly by progression to deep coma. The symptoms mimic that of sepsis and affected newborns present with severe lethargy, poor feeding to respiratory distress, jitteriness and seizures. A late onset form may occur as late as 20 years old and present as symptoms such as slurred speech, irritability, insomnia or delirium. Pathophysiology Argininosuccinate synthetase is an enzyme that converts citrulline to argininosuccinate, the absence of which causes an increase in plasma citrulline and ammonia levels.3
32 | P a g e Ist edition by dr.amir abdelazim ahmed
Inheritance autosomal recessive Screening increased citrulline and low arginine on MSMS Confirmatory Testing Confirmatory testing may be done through the demonstration of amino acids in plasma (decreased arginine and high citrulline), presence of orotic acid in urine and increased levels of ammonia in blood. Prognosis Prognosis for intellectual development depends on the nature of the initial hyperammonemia especially its duration or those of recurrent episodes.
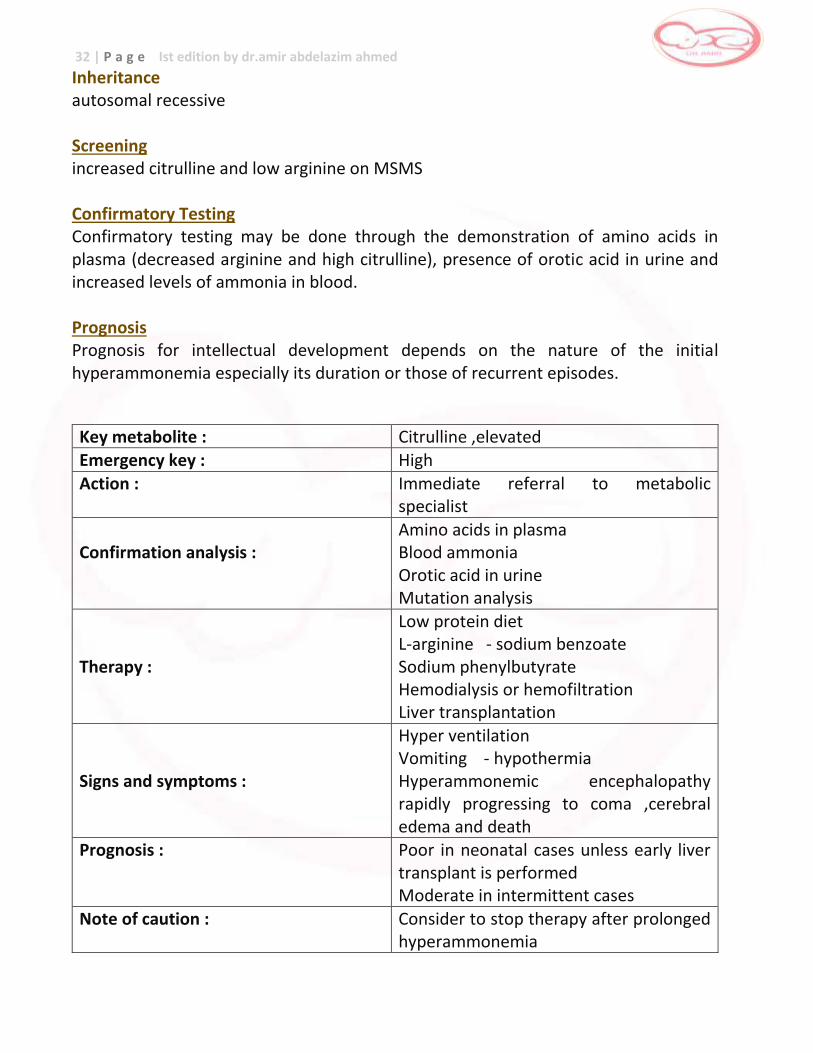
Key metabolite : Citrulline ,elevated Emergency key : High
Action : Immediate referral to metabolic specialist
Confirmation analysis :
Amino acids in plasma Blood ammonia Orotic acid in urine Mutation analysis
Therapy :
Low protein diet L-arginine - sodium benzoate Sodium phenylbutyrate Hemodialysis or hemofiltration Liver transplantation
Signs and symptoms :
Hyper ventilation Vomiting - hypothermia Hyperammonemic encephalopathy rapidly progressing to coma ,cerebral edema and death
Prognosis : Poor in neonatal cases unless early liver transplant is performed Moderate in intermittent cases
Note of caution : Consider to stop therapy after prolonged hyperammonemia

33 | P a g e Ist edition by dr.amir abdelazim ahmed
ARGININOSUCCINIC ACIDEMIA
Argninosuccinate lyase or argininosuccinase catalyzes the conversion of the argininosuccinate formed from citrulline and aspartate to fumarate and arginine.5 Incidence rare Clinical Manifestation Neonatal onset disease presents with severe hyperammonemic coma within the first few days of life with an overwhelming illness that rapidly progresses from poor feeding, vomiting, lethargy or irritability and tachypnea to seizures, coma and respiratory arrest; late onset disease are less acute and more subtle often precipitated by stress such as infection and anesthesia. A unique finding in patients is the presence of trichorrhexis nodosa where hair is very friable and breaks off easily. Pathophysiology Argininosuccinate lyase deficiency causes the accumulation of citrulline and decreasethe levels of arginine, the last compound of the urea cycle prior to the splitting off of urea.6 This causes the increased ammonia levels in blood that is responsible for the signs and symptoms observed. Inheritance: autosomal recessive Screening elevated citrulline, low arginine on MSMS
Confirmatory Testing Confirmation may be done through amino acids (elevated citrulline, low arginine, high argininosuccinate) in plasma , increased ammonia in blood, increased orotic acid in urine and enzyme studies in erythrocytes or fibroblasts. Prognosis Prognosis for intellectual development depends on the nature of initial hyperammonemia, especially its duration or the nature of recurrent episodes.
34 | P a g e Ist edition by dr.amir abdelazim ahmed
Key metabolite : Citrulline ,elevated
Emergency key : High
Action : Immediate referral to metabolic specialist
Confirmation analysis :
Amino acids in plasma Blood ammonia Orotic acid in urine Enzyme activity in erythrocytes
Therapy :
Low protein diet L-arginine (high dose ) - sodium benzoate Sodium phenylbutyrate Hemodialysis or hemofiltration Liver transplantation
Signs and symptoms :
Lethargy - hyperventilation Vomiting - hypothermia Hyperammonemic encephalopathy progressing to coma ,cerebral edema and death
Prognosis : Moderate : hyperammonemia easy to control but mental retardation will develop in most cases
Treatment of UCDs Treatment is through the dietary restriction of protein and the supplementation of a protein free formula. Sodium benzoate, an ammonia scavenger, is given as well as arginine supplementation.
Preliminary / Initial Management During Metabolic Crises Metabolic crises may be caused by an excess intake of protein, illness, prolonged fasting or stressful situations such as surgery and severe infection. The goal of treatment is to reverse the catabolic state and prevent essential amino acid deficiency.

35 | P a g e Ist edition by dr.amir abdelazim ahmed
Long Term Management Diet
Most patients require a low protein diet. Many suggest severe protein restriction but in early infancy, patients may need > 2 g/kg/day during phases of rapid growth. The protein intake usually decreases to approximately 1.2-1.5 g/kg/day during pre-school years and 0.8-1 g/kg/day in late childhood. After puberty, the quantity of natural protein may be less than 0.5 g/kg/day. However, it should be emphasized that there is considerable variation in the needs of individual patients. Some patients may not take their full protein allowance and some may not achieve good nutrition with restriction of natural protein, thus replacement with an essential amino acid mixture, giving up to 0.7 g/kg/day be added to the dietary regimen. Alternative pathways for nitrogen excretion
The effect of giving the following drugs is that nitrogen will be excreted in compounds other than urea, thus the load of the urea cycle is reduced. _ Sodium Benzoate 250-500 mg/kg/day (elimination of 1 mol NH3 per mol of glycine) _ Phenylbutyrate 250-500 mg/kg/day (elimination of 2 mol NH3 per mol of glutamine)
What to Do: If unwell and cannot tolerate oral intake: a. Nothing per orem b. Ensure patient’s airway is secure c. Insert IV access. Collect samples for serum ammonia. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. d. Start D12.5% 0.3 NaCl at full maintenance. Assess patient clinically, if there is need to increase fluid, may do so up to 1.2 or 1.5X the maintenance. e. Start IV sodium benzoate loading dose (250mg/kg) to run for four hours f. Start IV arginine loading dose (250mg/kg) to run for four hours g. Monitor input and output strictly (6 hours) If unwell but is able to tolerate oral intake: a. Insert oro- or nasogastric tube and start continuous feeding with protein free formula to run at maintenance rate b. Insert IV access. Collect samples for serum ammonia. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. c. Start D12.5% 0.3 NaCl at 5-10 cc/hr. d. Start IV sodium benzoate loading dose (250mg/kg) to run for four hours e. Start IV arginine loading dose (250mg/kg) to run for four hours f. Monitor input and output strictly (q6 hours) *Children should not be protein restricted for longer than necessary (24-48 hours). *If patient does not improve with the initial management (within 12 hours), hemodialysis may be indicated. Monitor patient clinically, the necessity of hemodialysis will depend on patient’s clinical status. * Inform metabolic doctor on call for further guidance regarding on-going management.

36 | P a g e Ist edition by dr.amir abdelazim ahmed
Replacement of deficient nutrients
Arginine is normally a nonessential amino acid, because it is synthesized within the urea cycle. For this reason, all patients with urea cycle disorders are likely to need a supplement of arginine to replace what is not synthesized. The aim should be to maintain plasma arginine concentrations between 50-200 umol/L. Monitoring
All treatments must be monitored with regular quantitative estimation of plasma ammonia and amino acids, paying particular attention to the concentration of glutamine and essential amino acids. The aim is to keep plasma ammonia levels below 80 umol/L and plasma glutamine levels below 800 umol/L. All diets must be nutritionally complete and must meet requirements for growth and development. EMERGENCY MANAGEMENT OF INTERCURRENT HYPERAMMONEMIA IN PATIENTS WITH UREA CYCLE DISORDERS Early Diagnosis and Therapy This is the most important aspect of intercurrent hyperammonemia. Delays are disastrous. A plasma ammonium level should be done as an emergency procedure on any child with these diseases who exhibits lethargy or vomiting of any degree, and the metabolic on-call physician should be alerted. Secure IV access needs to be established without delay. NB Ammonium needs to be collected in a Lithium Heparin tube, min 0.5 mls and transported IMMEDIATELY to the laboratory on ICE. Inform laboratory that the specimen is coming. If the ammonium level approaches three times the upper limits of normal, the ammonium level should be repeated and plasma obtained for electrolytes, blood gas and quantitative amino acids and urine for metabolic screening tests. Without waiting for the repeat ammonium value, the regimen described below should be followed as an emergency procedure. All dietary and intravenous protein intake should be discontinued. Because reduction of body protein breakdown is desirable a high parenteral caloric intake should be provided from 12.5% glucose and Intralipid. Intralipid (20%) should be commenced at a dose of 2gm/kg/day, grading up to 3-4gms/kg/day over the next 24 hours. Other fluids should be calculated to provide maintenance fluid as indicated by the child’s condition. Do not delay commencing priming infusion whilst organising maintenance fluids. If there are signs of cerebral edema this needs to be managed appropriately. Enteral feeding should be recommenced as soon as the patient is able to tolerate it. This needs to be done in consultation with the metabolic team. _ Give sodium benzoate up to 500 mg/kg/day-orally or intravenously. If the patient has not received any medication, give a priming dose of 250 mg/kg in 2-4 hours then 250 mg/kg in the next 20-24 hours _ Give L-arginine orally or intravenously: _ Up to 700 mg/kg/day in citrullinemia na argininosuccinic aciduria _ Up to 150 mg/kg/day in ornithine transcarbamylase deficiency and carbamoyl phosphate synthase deficiency

37 | P a g e Ist edition by dr.amir abdelazim ahmed
_ Plasma levels of ammonium, electrolytes, blood gas should be measured four hours after the completion of the priming infusion and every eight hours thereafter until plasma ammonium levels are normal or near normal, or as otherwise directed by the metabolic physician. These drugs may cause urine potassium loss; the serum potassium level should be monitored and treated as needed. _ The drugs may cause one or two vomiting episodes, usually towards the end of the 2-3 hour treatment period. Respiratory alkalosis may occur or be exacerbated during therapy with these drugs. _ If plasma ammonium level does not decrease within 8 hours urgently discuss the child with the metabolic physician. It is likely that the child will need hemodialysis. _ If intracranial pressure is elevated, conventional osmotherapy with mannitol should begin. Corticosteroids may be contraindicated because they induce negative nitrogen balance. _ When the ammonium level is stable at normal or near normal levels oral medication may be gradually added as the intravenous medication is gradually reduced. This should be done in consultation with the metabolic physician. References 1Su TS, Bock HGO, Beaudet AL et al. Molecular analysis of argininosuccinate syntehtase deficiency in human fibroblasts. J Clin Invest 1982:70:1334-1339. 2Chapter 31: Citrullinemia. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 210- 213. 3Wasant P, Viprakasit V, Srisomsap C et al. Argininosuccinate synthetase deficiency: mutation analysis in 3 Thai patients. Southeast Asian J Trop Med Pub Health 2005;36(3):757-761. 4 Leonard J. Disorders of the urea cycle and related enzymes. Inborn Metabolic Diseases Chapter 18,4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 269-271 5Chapter 32: Argininosuccinic aciduria. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 216-219. 6Chen BC, Ngu LH and Zabedah MY. Argininosuccinic aciduria: clinical and biochemical phenotype findings in Malaysian children. Malaysian J Pathol 2010;32(2):87-95. 7 Zschocke J and Hoffman G. Vademecum Metabolicum (Diagnosis and Treatment of Inborn Errors of Metabolism) 3rd
edition pp 153.
38 | P a g e Ist edition by dr.amir abdelazim ahmed
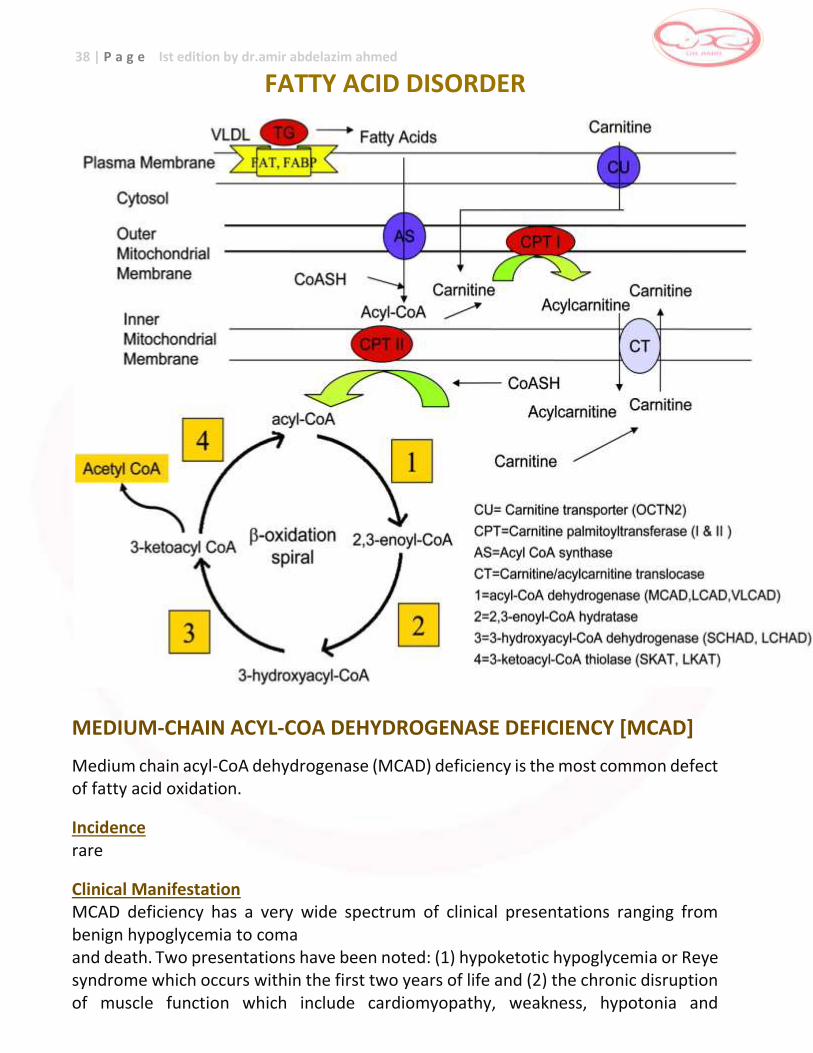
FATTY ACID DISORDER
MEDIUM-CHAIN ACYL-COA DEHYDROGENASE DEFICIENCY [MCAD]
Medium chain acyl-CoA dehydrogenase (MCAD) deficiency is the most common defect of fatty acid oxidation.
Incidence rare
Clinical Manifestation MCAD deficiency has a very wide spectrum of clinical presentations ranging from benign hypoglycemia to coma and death. Two presentations have been noted: (1) hypoketotic hypoglycemia or Reye syndrome which occurs within the first two years of life and (2) the chronic disruption of muscle function which include cardiomyopathy, weakness, hypotonia and

39 | P a g e Ist edition by dr.amir abdelazim ahmed
arrhythmia. In addition, MCAD deficiency has been shown to be associated with sudden infant death syndrome (SIDS).4 A “metabolic stress” such as prolonged fasting often in connection with viral infections is usually required to precipitate disease manifestations but patients are completely asymptomatic between episodes. Pathophysiology MCAD catalyzes the initial step in the β-oxidation of C12-C6 straight chain acyl-CoAs and MCAD deficiency results in a lack of production of energy from β-oxidation of medium chain fatty acids and hepatic ketogenesis and gluconeogenesis. Inheritance autosomal recessive
Screening increased octanoylcarnitine on MSMS and a high C10/carnitine ratio
Confirmatory Testing Urine organic acid profile will show medium chain dicarboxylic aciduria.4
Measurement of the specific MCAD enzyme activity in disrupted cultures skin fibroblasts, lymphocytes, or tissue biopsies from muscle can confirm the diagnosis.
Prognosis
Most authors report a mortality rate of 20-25% during the initial decompensation.4
Although the majority of children survive their initial episode, a significant amount of children who survived and perhaps children who have experienced clinically unrecognized episodes, suffer from long term sequelae and about 40% are judged to have developmental delay.2 Long term outcome remains dependent on constant monitoring for early signs of illness and rapid medical intervention to prevent complications Long term management Avoidance of fasting
It is essential to prevent any period of fasting which would be sufficient to require the use of fatty acids as fuel. This can be done by simply ensuring that patients have adequate carbohydrate feeding at bedtime and do not fast for more than 12 hours overnight. For young babies they should be fed every 3–4 hours with a late night feed continuing until about 9 months of age and they should not fast for longer than 6 - 8 hours. During inter- current illness (when child has poor appetite, low energy or excessive sleepiness, vomiting, diarrhea, infection or fever), care should be taken to give extra feedings of carbohydrate during
40 | P a g e Ist edition by dr.amir abdelazim ahmed
the night and inform the doctor for the “sick day regimen” which mainly consists of high energy drink. In a few patients with severe defects in fatty acid oxidation who had developed weakness and/or cardiomyopathy, addition of continuous intragastric feedings such as the use of uncooked cornstarch at bedtime might be considered as a slowly released form of glucose. Diet
Dietary fat restriction is not routine in patients with MCAD deficiency.
Emergency management of patients with MCAD deficiency
When patients with fatty acid oxidation disorders become ill, treatment with intravenous glucose should be given immediately. Delay may result on sudden death or permanent brain damage. The goal is to provide sufficient glucose to stimulate insulin secretion to levels that will only suppress fatty acid oxidation in liver and muscle, but also block adipose tissue lipolysis. Solutions of 10%dextrose should be used at infusion rates of 10 mg/kg per min or greater to maintain high to normal levels of plasma glucose, above 100mg/dl. Do not give intravenous lipids
Key metabolite : C8 (octanoyl carnitine ) , elevated Emergency key : Moderate
Action : Contact family to ascertain clinical condition and referral to metabolic specialist
Confirmation analysis :
Acylcarnitine profile in DBS/plasma Carnitine status in plasma/serum Organic acids in urine Enzyme activity fibroblasts Mutation analysis
Therapy : Avoid fasting (L-carnitine supplementation)
Signs and symptoms :
Hypoketotic hypoglycemia Reye-like syndrome Lethargy , nausea , vomiting, coma, seizures, cardiac arrest
Prognosis : excellent
Note of caution : Neonatal manifestation in rare cases References: 1Strauss AW, Andersen BS and Bennett MJ. Chapter 5: Mitochondrial Fatty Acid Oxidation Defects in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 60-62. 2 Hsu HW, Zytkovicz TH, Comeau AM et al. Spectrum of Medium chain acyl-coA dehydrogenase deficiency detected by newborn screening. Pediatrics 2008;121:e1108-e1114.
41 | P a g e Ist edition by dr.amir abdelazim ahmed 3 Chapter 40: Medium chain acyl-CoA dehydrogenase deficiency. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 260-265. 4 Wilson CJ, Champion MP, Collins JE et al. Outcome of medium chain acyl-CoA dehydrogenase deficiency after diagnosis. Arch Dis Child 1999;80:459-462. 5 Stanley C, Bennett M, Mayatepek E. Disorders of mitochondrial fatty acid oxidation and related metabolic pathways. Inborn Metabolic Diseases Chapter 23 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 184
Analyte Octanoylcarnitine (C8) (always associated with C6 and C10)
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 0.200 uM/L
Differential diagnosis
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency.
False positive
The specificity of MS/MS to identify MCAD deficiency appears to be 100%, with a few false negative results having been reported as a result of inappropriate cut-off selection False postive may be as marker “octanoylcarnitine” is not specific for MCAD deficiency and is expected to be elevated in other disorders (i.e., glutaric acidemia type II, and possibly medium-chain 3-keto acyl-CoA thiolase deficiency) and in newborns treated with valproate or fed a diet rich in medium-chain triglycerides
Clinical presentation
MCAD deficiency is usually asymptomatic in the newborn although it can present acutely in the neonate with hypoglycemia, metabolic acidosis, hyperammonemia, and hepatomegaly. MCAD deficiency is associated with high mortality unless treated promptly; milder variants exist. Hallmark features include vomiting, lethargy, and hypoketotic hypoglycemia. Untreated MCAD deficiency is a significant cause of sudden death. Prognosis : 25% sudden death in the first attack of illness Permanent brain injury occur in some patients during attack Prognosis for survivors without brain damage more than 60%
Diagnostic evaluation
and confirmatory
test
Plasma acylcarnitine analysis will show increase C8 ,C10 consistent with MCADD.
Urine organic acid analysis may also show low ketones and high medium chain dicarboxylic acids (adipic ,suberic and sebacic acids) that derive from microsomal and perioxisomal omega oxidation of fatty acid
Increase urinary acylglycines (hexanoyl-,suberyl-,3phenylpropionyl glycines)
Diagnosis can be confirmed by mutation analysis of the MCAD gene and determination of fatty acid B-oxidation in fibroblast and measure MCAD enzyme activity in fibroblast.
In acute attack show hypoketotic hypoglycemia (no metabolic acidemia)
Liver function :elevated ALT,AST and prolonged PT , PTT
Liver biopsy show micro or macro-vesicular steatosis due to triglyceride accumulation
Causes and mechanism
MCAD deficiency is a fatty acid oxidation (FAO) disorder. Fatty acid oxidation occurs mainly during prolonged fasting and/or periods of increased energy demands (fever, stress), when energy production relies increasingly on fat metabolism. In an FAO disorder, fatty acids and potentially toxic derivatives accumulate because of a deficiency in one of the mitochondrial FAO enzymes.
Genetics Diagnosis can be confirmed by finding the common A985G mutation Second common mutation T199C has been detected in infants with characteristic acylcarnitines in newborn screening test
Prenatal diagnosis
Test of sibling of affected patients important to detect asymptomatic family members as many as 50% of affected patients have never had an episode
Prevalence 1:5000 to 1:17000
Action for result
Contact family , evaluate baby for poor feeding , lethargy , hypotonia and hepatomegaly , start confirmatory investigation, educate family to avoid fasting , refer to metabolic specialist
Treatment Acute : 10% dextrose to treat hypoglycemia and suppress lipolysis Chronic: avoid fasting - restricting dietary fat or treatment with carnitine is controversial

42 | P a g e Ist edition by dr.amir abdelazim ahmed
LONG-CHAIN L-3-HYDROXYACYL-CoA DEHYDROGENASE [LCHAD]
Long chain L-3 hydroxyacyl-CoA dehydrogenase (LCHAD) is a component of trifunctional protein. Isolated LCHAD deficiency catalyzes the third step in the fatty acid oxidation spiral, converting long chain 3-hydroxyacyl- CoA esters into long chain 3-keto-CoA species by using NAD as a cofactor. Incidence Very rare Clinical Manifestation Patients exhibit moderate or severe multiorgan involvement either neonatally or during the first two years of life.They may present in the first year of life with hypoketotic hypoglycemia and liver dysfunction, Reye syndrome- like symptoms, seizures, coma and death.2 By adolescence, ophthalmologic abnormalities including loss of visual acuity, chorioretinal atrophy, progressive retinitis pigmentosa and peripheral sensorimotor polyneuropathy may be observed.2,3,4, Up to 40% of symptomatic patients may have tachycardic arrhythmias, apneic episodes, cardiopulmonary arrest and unexplained death.2 A strong association has been demonstrated with heterozygous mothers developing acute fatty liver or pregnancy or hemolysis, elevated liver enzymes and low platelet count (HELLP) syndrome when carrying an affected fetus.
Pathophysiology Since the enzyme LCHAD is part of the fatty acid oxidation, a deficiency causes a problem in the energy utilization of the body which causes the presentation of signs and symptoms as listed above. Inheritance autosomal recessive Screening elevated C16 (palmitoylcarnitine), 3-hydroxypalmitoylcarnitine, C18, 3-hydroxy-C18-carnitines and C18:1- hydroxycarnitine 2,3 Confirmatory Testing Confirmatory testing is done through enzyme assays performed in cultured cells such as skin fibroblasts. The common mutation G1528C has been identified in affected individuals and may be used for confirmation.

43 | P a g e Ist edition by dr.amir abdelazim ahmed
Prognosis Patients with LCHAD deficiency who present symptomatically often die during the acute episode or suffer from sudden, unexplained death and mortality occurs in approximately 38%. Long term management Primary goal of treatment is to avoid metabolic stress brought about by infection and long periods of fasting. Patients should be given frequent feedings, supplementation with medium chain triglycerides (MCT formula) and an overnight infusion of cornstarch. Treatment with L-carnitine remains controversial. Avoidance of fasting
Patients must be ensured to have adequate carbohydrate feeding at bedtime and do not fast for more than 12 hours overnight. For young babies they should be fed every 3–4 hours with a late night feed continuing until about 9 months of age and they should not fast for longer than 6 - 8 hours. During intercurrent illness, when appetite is diminished, care should be taken to give extra feedings of carbohydrate during the night. A” sick day regimen” containing high glucose drinks should be given. In a few patients with severe defects in fatty acid oxidation who had developed weakness and/or cardiomyopathy, addition of continuous intragastric feedings such as the use of uncooked cornstarch at bedtime might be considered as a slowly released form of glucose. Diet
Sometimes a low fat, high carbohydrate diet is recommended. Food plan is recommended. Carbohydrates give the body may types of sugar that can be used as energy. In fact, for children needing this treatment, most food in the diet should be carbohydrates (bread, pasta, fruit, etc.) and protein (lean meat and low-fat dairy foods). Any diet changes should be made under the guidance of an experienced dietitian. People with LCHADD cannot use certain building blocks of fat called “long chain fatty acids”. The dietitian can help create a food plan low in these fats. Much of the rest of fat in the diet may be in the form of medium chain fatty acids. Medium Chain Triglyceride oil (MCT oil) is often used as part of the food plan for people with LCHADD. This special oil has medium chain fatty acids that can be used in small amounts for energy. In addition to the above supplements, some doctors suggest taking DHA (docosahexanoic acid) which may help prevent loss of eyesight.

44 | P a g e Ist edition by dr.amir abdelazim ahmed
Avoid prolonged exercise
Long periods of exercise can also trigger symptoms. Problems occurring during or after exercise can include: muscle aches, weakness, cramps and reddish-brown color to the urine. It is advised to have high carbohydrate intake prior to exercise to prevent lipolysis and to restrict physical activity to levels that are not likely to precipitate an attack of rhabdomyolysis. Intercurrent illness
Advise parents to refer the child to the doctor if he/she has any of the following: _ poor appetite _ low energy or excessive sleepiness _ vomiting _ diarrhea _ an infection _ a fever _ persistent muscle pain, weakness, or reddish-brown color to the urine Children with LCHADD need to eat extra starchy food and drink more fluids during any illness - even if they may not feel hungry – or they could develop hypoglycemia or a metabolic crisis. When they become sick, children with LCHADD often need to be treated in the hospital to prevent serious health problems. Emergency management of patients with LCHAD deficiency When patients with fatty acid oxidation disorders become ill, treatment with intravenous glucose should be given immediately. Delay may result on sudden death or permanent brain damage. The goal is to provide sufficient glucose to stimulate insulin secretion to levels that will only suppress fatty acid oxidation in liver and muscle, but also block adipose tissue lipolysis. Solutions of 10%dextrose should be used at infusion rates of 10 mg/kg per min or greater to maintain high to normal levels of plasma glucose, above 100mg/dl. Do not give intravenous lipids!
45 | P a g e Ist edition by dr.amir abdelazim ahmed
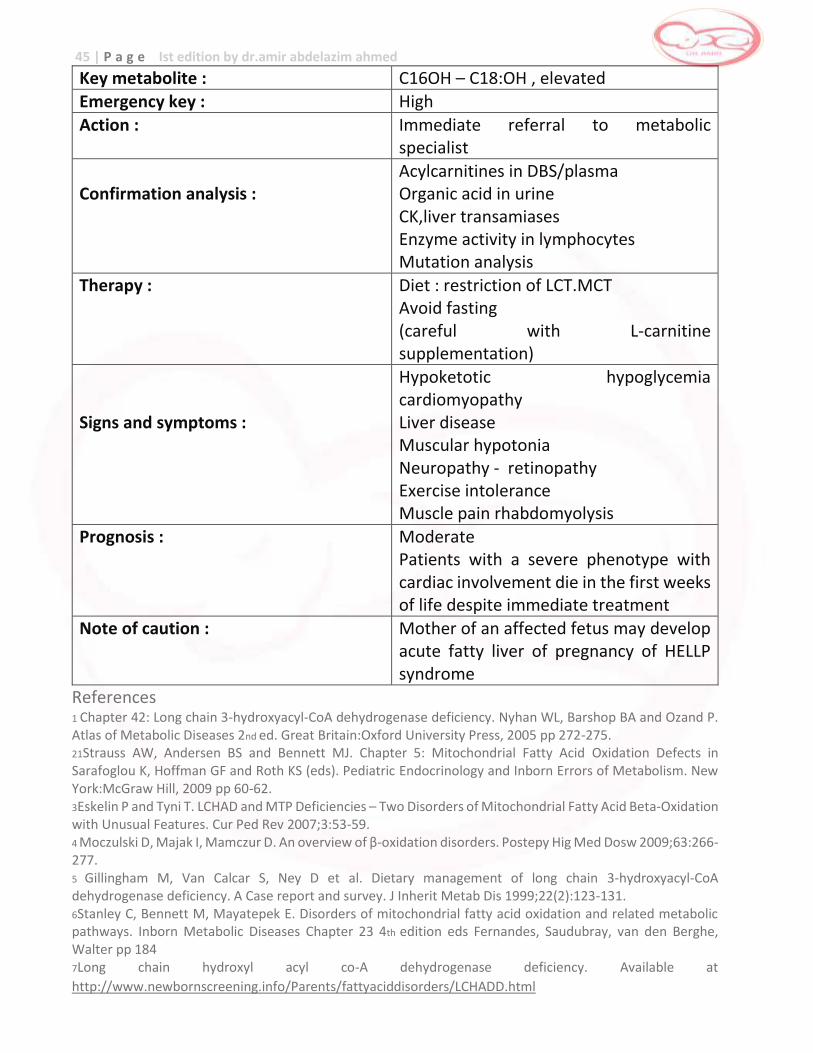
Key metabolite : C16OH – C18:OH , elevated Emergency key : High
Action : Immediate referral to metabolic specialist
Confirmation analysis :
Acylcarnitines in DBS/plasma Organic acid in urine CK,liver transamiases Enzyme activity in lymphocytes Mutation analysis
Therapy : Diet : restriction of LCT.MCT Avoid fasting (careful with L-carnitine supplementation)
Signs and symptoms :
Hypoketotic hypoglycemia cardiomyopathy Liver disease Muscular hypotonia Neuropathy - retinopathy Exercise intolerance Muscle pain rhabdomyolysis
Prognosis : Moderate Patients with a severe phenotype with cardiac involvement die in the first weeks of life despite immediate treatment
Note of caution : Mother of an affected fetus may develop acute fatty liver of pregnancy of HELLP syndrome
References 1 Chapter 42: Long chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 272-275. 21Strauss AW, Andersen BS and Bennett MJ. Chapter 5: Mitochondrial Fatty Acid Oxidation Defects in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 60-62. 3Eskelin P and Tyni T. LCHAD and MTP Deficiencies – Two Disorders of Mitochondrial Fatty Acid Beta-Oxidation with Unusual Features. Cur Ped Rev 2007;3:53-59. 4 Moczulski D, Majak I, Mamczur D. An overview of β-oxidation disorders. Postepy Hig Med Dosw 2009;63:266-277. 5 Gillingham M, Van Calcar S, Ney D et al. Dietary management of long chain 3-hydroxyacyl-CoA dehydrogenase deficiency. A Case report and survey. J Inherit Metab Dis 1999;22(2):123-131. 6Stanley C, Bennett M, Mayatepek E. Disorders of mitochondrial fatty acid oxidation and related metabolic pathways. Inborn Metabolic Diseases Chapter 23 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 184 7Long chain hydroxyl acyl co-A dehydrogenase deficiency. Available at
http://www.newbornscreening.info/Parents/fattyaciddisorders/LCHADD.html

46 | P a g e Ist edition by dr.amir abdelazim ahmed
VERY LONG-CHAIN ACYL-COA DEHYDROGENASE DEFICIENCY
[VLCADD]
Very long-chain acyl-CoA dehydrogenase catalyzes the dehydrogenation of C22-C12 straight chain fatty acids, and because the long chain fatty acids constitute a major proportion of the fatty acids, VLCAD deficiency is generally a more severe condition than MCAD or SCAD deficiency and multiple tissues are affected. Incidence rare
Clinical Manifestation The clinical presentation of symptomatic VLCAD deficiency is heterogenous with phenotypes of different severities. There are three forms described: (1) severe childhood form with neonatal onset and cardiomyopathy; (2) milder childhood form with delayed onset of symptoms often triggered by metabolic stress and presents as hypoketotic hypoglycemia and; (3) adult form which presents with isolated skeletal muscle involvement with recurrent episode of muscle pain, rhabdomyolysis and myoglobinuria. Pathophysiology VLCAD catalyzes the dehydrogenation of acyl CoA esters of 14-20 carbon length in the first step of mitochondrial fatty acid oxidation.3,4 VLCAD deficiency results in lack of production of energy from β-oxidation of longchain fatty acids. Because heart and muscle tissues depend heavily on energy from long chain fatty acid oxidation, a VLCAD deficiency severely affect these tissues. Inheritance autosomal recessive
Screening elevation of tetradecenoylcarnitine (C14:1) on MSMS
Confirmatory Testing The enzyme defect can be detected through culture skin fibroblasts.1 The gene for VLCAD has been clone and sequenced successfully and play a role in diagnosis of this disorder. Prognosis Fifty percent of patients die within 2 months of initial symptomatology.4 However, timely and correct diagnosis leads to dramatic recovery so that early detection could prevent the onset of arrhythmias, heart failure, metabolic insufficiency and death.

47 | P a g e Ist edition by dr.amir abdelazim ahmed
Preliminary / Initial Management During Metabolic Crisis Metabolic crises may be caused by illness, prolonged fasting or stressful situations such as surgery and severe infection. The goal of treatment is to reverse the catabolic state and prevent hypoglycemia.
Long term management Treatment of this disorder include avoidance of fasting by frequent feeding, overnight continuous feeding, reduction of amount of long chain fat in diet while supplying essential fatty acids in the form of canola, walnut oil or safflower oil and supplementation with medium chain triglycerides (MCT). Avoidance of fasting
Patients must be ensured to have adequate carbohydrate feeding at bedtime and do not fast for more than 12 hours overnight. For young babies they should be fed every 3–4 hours with a late night feed continuing until about 9 months of age and they should not fast for longer than 6 - 8 hours. During intercurrent illness, when appetite is diminished, care should be taken to give extra feedings of carbohydrate during the night. A” sick day regimen” containing high glucose drinks should be given.
What to Do: If unwell and cannot tolerate oral intake: a. Nothing per orem b. Ensure patient’s airway is secure c. Insert IV access. Monitor glucose levels. For patients with VLCAD, collect samples for serum CK. May request for other investigations (i.e. CBC, Blood gas) as needed. May give fluid boluses if patient requires. d. Start D10% 0.3 NaCl at full maintenance. Assess patient clinically, if there is need to increase fluid, may do so up to 1.2 or 1.5X the maintenance. e. Monitor input and output strictly (q6 hours). Check for the color of urine. If unwell and is able to tolerate oral intake: a. Insert oro- or nasogastric tube and start continuous feeding with a high glucose formula b. Insert IV access. Monitor glucose levels. For patients with VLCAD, collect samples for serum CK. May request for other investigations (i.e. CBC, Blood gas) as needed. May give fluid boluses if patient requires. c. Start D10% 0.3 NaCl at 5-10 cc/hr. d. Monitor input and output strictly (q6 hours). Check for the color of urine. *Patients with VLCAD may have rhabdomyolysis. Monitor CK levels and hydrate adequately. If CK levels continually rise, hemodialysis may be indicated. * Inform metabolic doctor on call for further guidance regarding on-going management.

48 | P a g e Ist edition by dr.amir abdelazim ahmed
In a few patients with severe defects in fatty acid oxidation who had developed weakness and/or cardiomyopathy, addition of continuous intragastric feedings such as the use of uncooked cornstarch at bedtime might be considered as a slowly released form of glucose. Diet
Sometimes a low fat, high carbohydrate diet is recommended. Food plan is recommended. Carbohydrates give the body may types of sugar that can be used as energy. In fact, for children needing this treatment, most food in the diet should be carbohydrates (bread, pasta, fruit, etc.) and protein (lean meat and low-fat dairy foods). Any diet changes should be made under the guidance of an experienced dietitian. People with VLCADD cannot use certain building blocks of fat called “long chain fatty acids”. The dietitian can help create a food plan low in these fats. Much of the rest of fat in the diet may be in the form of medium chain fatty acids. Medium Chain Triglyceride oil (MCT oil) is often used as part of the food plan for people with VLCADD. This special oil has medium chain fatty acids that can be used in small amounts for energy. Ask your doctor whether your child needs to have any changes in his or her diet. Avoid prolonged exercise
Long periods of exercise can also trigger symptoms. Problems occurring during or after exercise can include: muscle aches, weakness, cramps and reddish-brown color to the urine. It is advised to have high carbohydrate intake prior to exercise to prevent lipolysis and to restrict physical activity to levels that are not likely to precipitate an attack of rhabdomyolysis. Intercurrent illness
Advise parents to refer the child to the doctor if he/she has any of the following: _ poor appetite _ low energy or excessive sleepiness _ vomiting _ diarrhea _ an infection _ a fever
_persistent muscle pain, weakness, or reddish-brown color to the urine Children with VLCADD need to eat extra starchy food and drink more fluids during any illness - even if they may not feel hungry – or they could develop hypoglycemia or a metabolic crisis. When they become sick, children with VLCADD often need to be treated in the hospital to prevent serious health problems.
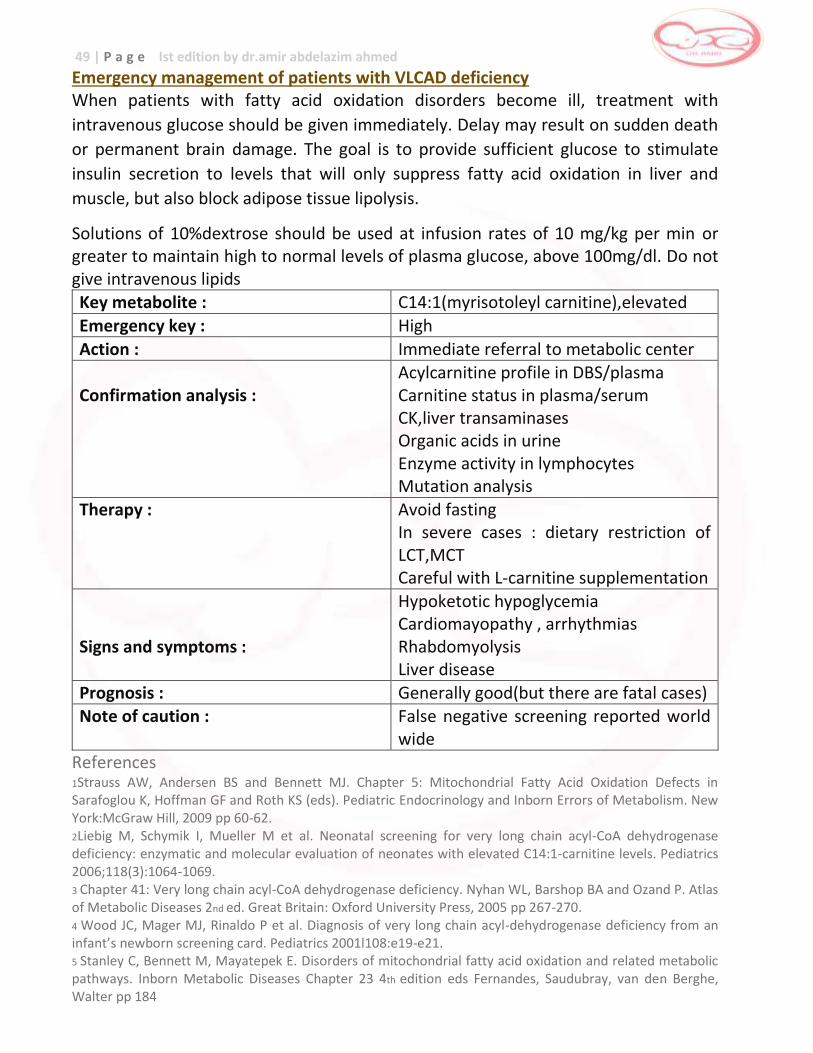
49 | P a g e Ist edition by dr.amir abdelazim ahmed
Emergency management of patients with VLCAD deficiency When patients with fatty acid oxidation disorders become ill, treatment with
intravenous glucose should be given immediately. Delay may result on sudden death
or permanent brain damage. The goal is to provide sufficient glucose to stimulate
insulin secretion to levels that will only suppress fatty acid oxidation in liver and
muscle, but also block adipose tissue lipolysis.
Solutions of 10%dextrose should be used at infusion rates of 10 mg/kg per min or greater to maintain high to normal levels of plasma glucose, above 100mg/dl. Do not give intravenous lipids Key metabolite : C14:1(myrisotoleyl carnitine),elevated
Emergency key : High
Action : Immediate referral to metabolic center Confirmation analysis :
Acylcarnitine profile in DBS/plasma Carnitine status in plasma/serum CK,liver transaminases Organic acids in urine Enzyme activity in lymphocytes Mutation analysis
Therapy : Avoid fasting In severe cases : dietary restriction of LCT,MCT Careful with L-carnitine supplementation
Signs and symptoms :
Hypoketotic hypoglycemia Cardiomayopathy , arrhythmias Rhabdomyolysis Liver disease
Prognosis : Generally good(but there are fatal cases) Note of caution : False negative screening reported world
wide References 1Strauss AW, Andersen BS and Bennett MJ. Chapter 5: Mitochondrial Fatty Acid Oxidation Defects in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 60-62. 2Liebig M, Schymik I, Mueller M et al. Neonatal screening for very long chain acyl-CoA dehydrogenase deficiency: enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics 2006;118(3):1064-1069. 3 Chapter 41: Very long chain acyl-CoA dehydrogenase deficiency. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain: Oxford University Press, 2005 pp 267-270. 4 Wood JC, Mager MJ, Rinaldo P et al. Diagnosis of very long chain acyl-dehydrogenase deficiency from an infant’s newborn screening card. Pediatrics 2001l108:e19-e21. 5 Stanley C, Bennett M, Mayatepek E. Disorders of mitochondrial fatty acid oxidation and related metabolic pathways. Inborn Metabolic Diseases Chapter 23 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 184
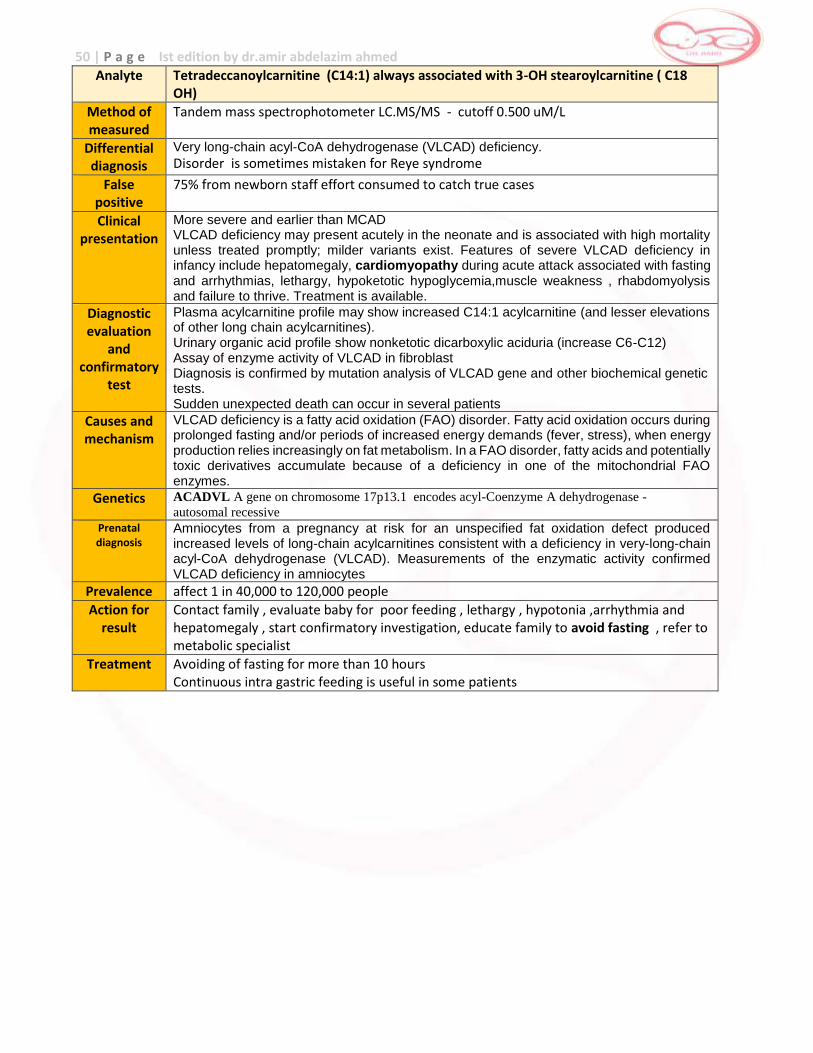
50 | P a g e Ist edition by dr.amir abdelazim ahmed
Analyte Tetradeccanoylcarnitine (C14:1) always associated with 3-OH stearoylcarnitine ( C18 OH)
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 0.500 uM/L
Differential diagnosis
Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency.
Disorder is sometimes mistaken for Reye syndrome
False positive
75% from newborn staff effort consumed to catch true cases
Clinical presentation
More severe and earlier than MCAD VLCAD deficiency may present acutely in the neonate and is associated with high mortality unless treated promptly; milder variants exist. Features of severe VLCAD deficiency in infancy include hepatomegaly, cardiomyopathy during acute attack associated with fasting and arrhythmias, lethargy, hypoketotic hypoglycemia,muscle weakness , rhabdomyolysis and failure to thrive. Treatment is available.
Diagnostic evaluation
and confirmatory
test
Plasma acylcarnitine profile may show increased C14:1 acylcarnitine (and lesser elevations of other long chain acylcarnitines). Urinary organic acid profile show nonketotic dicarboxylic aciduria (increase C6-C12) Assay of enzyme activity of VLCAD in fibroblast Diagnosis is confirmed by mutation analysis of VLCAD gene and other biochemical genetic tests. Sudden unexpected death can occur in several patients
Causes and mechanism
VLCAD deficiency is a fatty acid oxidation (FAO) disorder. Fatty acid oxidation occurs during prolonged fasting and/or periods of increased energy demands (fever, stress), when energy production relies increasingly on fat metabolism. In a FAO disorder, fatty acids and potentially toxic derivatives accumulate because of a deficiency in one of the mitochondrial FAO enzymes.
Genetics ACADVL A gene on chromosome 17p13.1 encodes acyl-Coenzyme A dehydrogenase -
autosomal recessive
Prenatal diagnosis
Amniocytes from a pregnancy at risk for an unspecified fat oxidation defect produced increased levels of long-chain acylcarnitines consistent with a deficiency in very-long-chain acyl-CoA dehydrogenase (VLCAD). Measurements of the enzymatic activity confirmed VLCAD deficiency in amniocytes
Prevalence affect 1 in 40,000 to 120,000 people
Action for result
Contact family , evaluate baby for poor feeding , lethargy , hypotonia ,arrhythmia and hepatomegaly , start confirmatory investigation, educate family to avoid fasting , refer to metabolic specialist
Treatment Avoiding of fasting for more than 10 hours Continuous intra gastric feeding is useful in some patients

51 | P a g e Ist edition by dr.amir abdelazim ahmed
TRIFUNCTIONAL PROTEIN [TFP] DEFICIENCY
The mitochondrial trifunctional protein (TFP) is a multienzyme complex of the β-oxidation cycle composed of four α-subunits harbouring long-chain enoyl-CoA hydratase and long chain L-3-hydroxyacyl-CoA dehydrogenase and four β-subunits encoding long chain 3-ketoacyl-CoA thoilase.1 General or complete TFP deficiency is defined and occurs when markedly decreased activity of all three enzymatic components, LCHAD, long chain 2,3 enoyl CoA drasate and LKAT exist. Incidence Very rare Clinical Manifestation General TFP deficiency has three phenotypes: the lethal phenotype presenting with lethal cardiac failure or sudden death due to arrhythmias, the hepatic phenotype and the neuromyopathic phenotype that has lateronset, episodic, recurrent skeletal myopathy with muscular pain and weakness often induced by exercise or exposure to cold and peripheral neuropathy. It is important to note that fetuses with complete TFP deficiency can cause maternal
liver diseases of pregnancy.
Pathophysiology Mitochondrial fatty acid β-oxidation is a major energy-producing pathway.3 Any defect in any enzyme may cause the characteristic signs and symptoms which include hypoketotic hypoglycemia. Inheritance autosomal recessive
Screening increased C16 and C18 on MSMS
Confirmatory Testing Confirmatory testing is through the demonstration of decreased enzyme activity on cultured fibroblasts.Mutations in the HADHA and HADHB gene may result in mitochondrial trifunctional protein deficiency4 and mayplay a role in confirmation. Prognosis Patients with metabolic crises do well unless the hypoglycemia and seizures are prolonged and cause developmental delay, older onset patients with rhabdomyolysis can reduce episodes significantly with dietary management and do well.
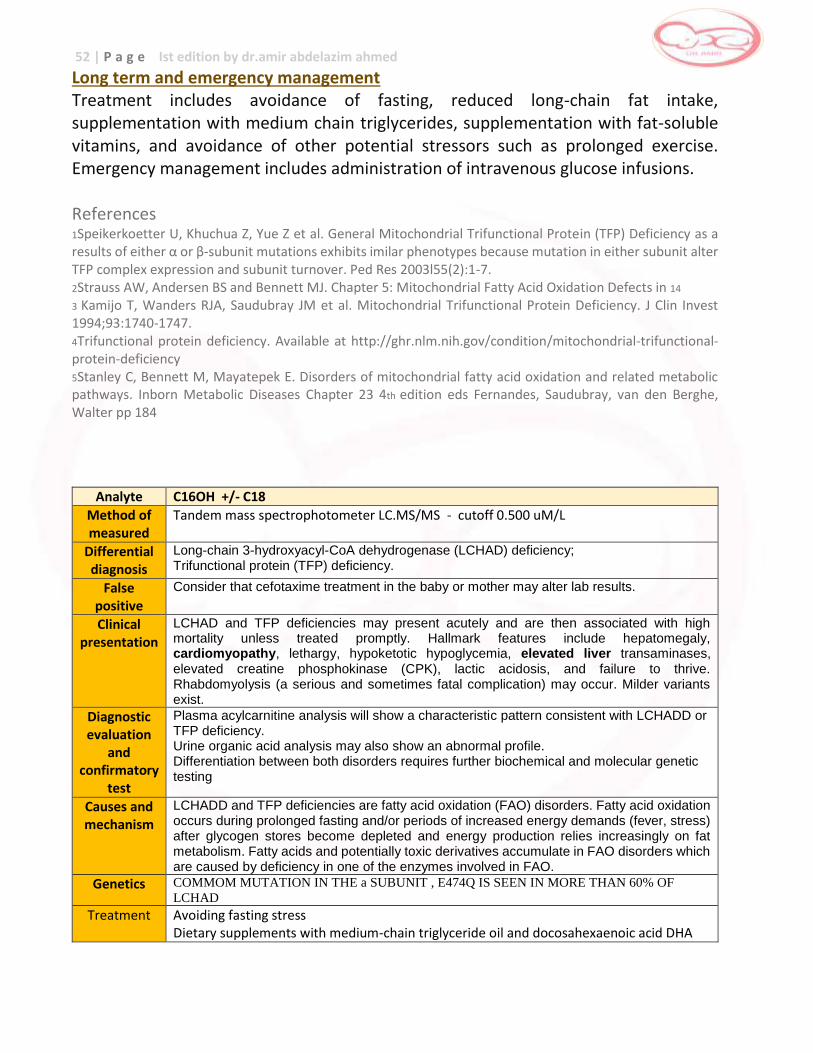
52 | P a g e Ist edition by dr.amir abdelazim ahmed
Long term and emergency management Treatment includes avoidance of fasting, reduced long-chain fat intake, supplementation with medium chain triglycerides, supplementation with fat-soluble vitamins, and avoidance of other potential stressors such as prolonged exercise. Emergency management includes administration of intravenous glucose infusions. References 1Speikerkoetter U, Khuchua Z, Yue Z et al. General Mitochondrial Trifunctional Protein (TFP) Deficiency as a results of either α or β-subunit mutations exhibits imilar phenotypes because mutation in either subunit alter TFP complex expression and subunit turnover. Ped Res 2003l55(2):1-7. 2Strauss AW, Andersen BS and Bennett MJ. Chapter 5: Mitochondrial Fatty Acid Oxidation Defects in 14
3 Kamijo T, Wanders RJA, Saudubray JM et al. Mitochondrial Trifunctional Protein Deficiency. J Clin Invest 1994;93:1740-1747. 4Trifunctional protein deficiency. Available at http://ghr.nlm.nih.gov/condition/mitochondrial-trifunctional-protein-deficiency 5Stanley C, Bennett M, Mayatepek E. Disorders of mitochondrial fatty acid oxidation and related metabolic pathways. Inborn Metabolic Diseases Chapter 23 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 184
Analyte C16OH +/- C18
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 0.500 uM/L
Differential diagnosis
Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency; Trifunctional protein (TFP) deficiency.
False positive
Consider that cefotaxime treatment in the baby or mother may alter lab results.
Clinical presentation
LCHAD and TFP deficiencies may present acutely and are then associated with high mortality unless treated promptly. Hallmark features include hepatomegaly, cardiomyopathy, lethargy, hypoketotic hypoglycemia, elevated liver transaminases, elevated creatine phosphokinase (CPK), lactic acidosis, and failure to thrive. Rhabdomyolysis (a serious and sometimes fatal complication) may occur. Milder variants exist.
Diagnostic evaluation
and confirmatory
test
Plasma acylcarnitine analysis will show a characteristic pattern consistent with LCHADD or TFP deficiency. Urine organic acid analysis may also show an abnormal profile. Differentiation between both disorders requires further biochemical and molecular genetic testing
Causes and mechanism
LCHADD and TFP deficiencies are fatty acid oxidation (FAO) disorders. Fatty acid oxidation occurs during prolonged fasting and/or periods of increased energy demands (fever, stress) after glycogen stores become depleted and energy production relies increasingly on fat metabolism. Fatty acids and potentially toxic derivatives accumulate in FAO disorders which are caused by deficiency in one of the enzymes involved in FAO.
Genetics COMMOM MUTATION IN THE a SUBUNIT , E474Q IS SEEN IN MORE THAN 60% OF
LCHAD
Treatment Avoiding fasting stress Dietary supplements with medium-chain triglyceride oil and docosahexaenoic acid DHA
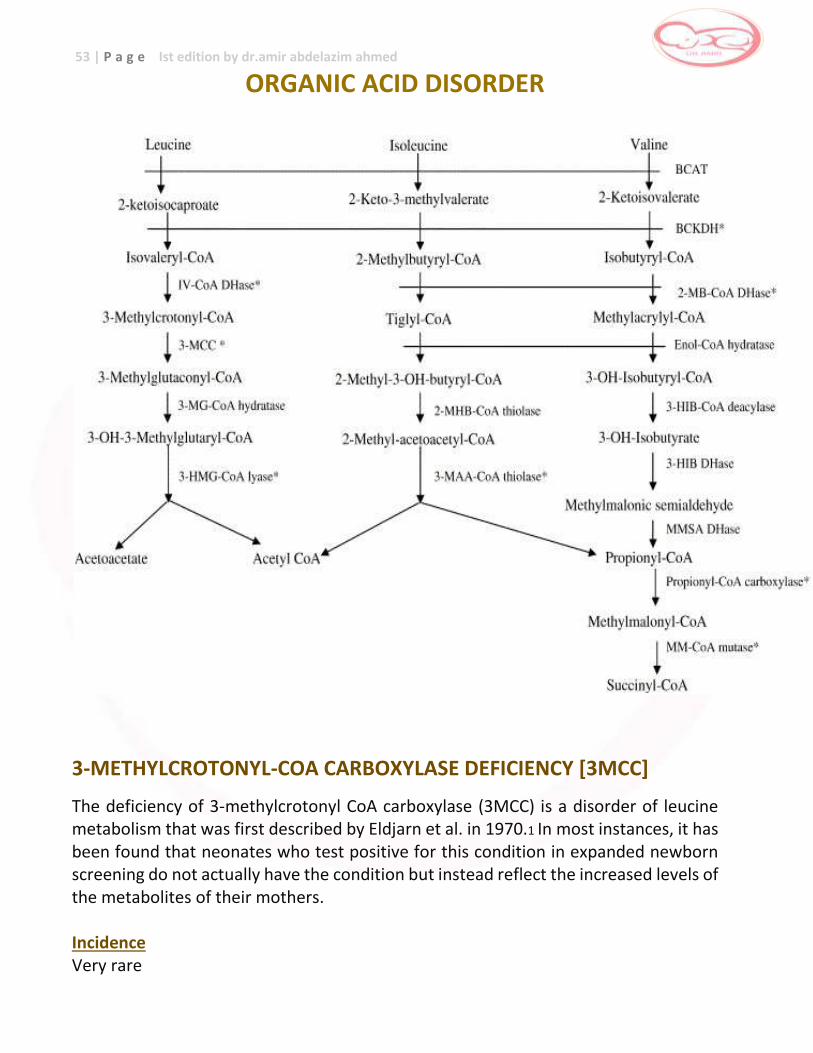
53 | P a g e Ist edition by dr.amir abdelazim ahmed
ORGANIC ACID DISORDER
3-METHYLCROTONYL-COA CARBOXYLASE DEFICIENCY [3MCC]
The deficiency of 3-methylcrotonyl CoA carboxylase (3MCC) is a disorder of leucine metabolism that was first described by Eldjarn et al. in 1970.1 In most instances, it has been found that neonates who test positive for this condition in expanded newborn screening do not actually have the condition but instead reflect the increased levels of the metabolites of their mothers. Incidence Very rare

54 | P a g e Ist edition by dr.amir abdelazim ahmed
Clinical Manifestation There is a broad spectrum of clinical presentation ranging from no symptoms to failure to thrive, hypotonia, and cardiomyopathy to severe metabolic decompensation with metabolic acidosis and hypoglycemia. Some patients may have a late presentation (1-3 years old) with an acute episode of Reye syndrome, massive ketosis, acidosis, lethary, coma leading to a fatal outcome. Pathophysiology 3-methycrotonyl CoA carboxylase is responsible for the carboxylation of 3-methylcrotonyl-CoA, the fourth step in leucine catabolism; a deficiency of which causes a disturbance in leucine catabolism. Inheritance autosomal recessive
Screening Increased 3-hydroxyisovaleryl carnitine on MSMS
Confirmatory Testing An increase in 3-hydroxyisovaleric (3 HIVA) and 3-methylcrotonyl glycine (3 MCG) are found in urine, confirmatory testing is done through the demonstration of decreased enzyme activity in cultured fibroblasts. Prognosis 3-MCC is a common, mostly benign condition; whether treatment with a low-protein diet, carnitine and glycine supplementation has the potential to change the clinical course in several affected patients remains to be elucidated.
Long term management
Long term treatment of symptomatic infants based on mildly protein restricted diet results in general improvement and reduction in the number of exacerbations. It is effective in lowering the excretion of organic acids which however, never disappears. Glycine supplementation at 175 mg/kg/day increases the excretion of 3 MCG. Carnitine supplementation at 100 mg/kg.day corrects the very low plasma carnitine levels and increases the excretion of 3 HIVA.
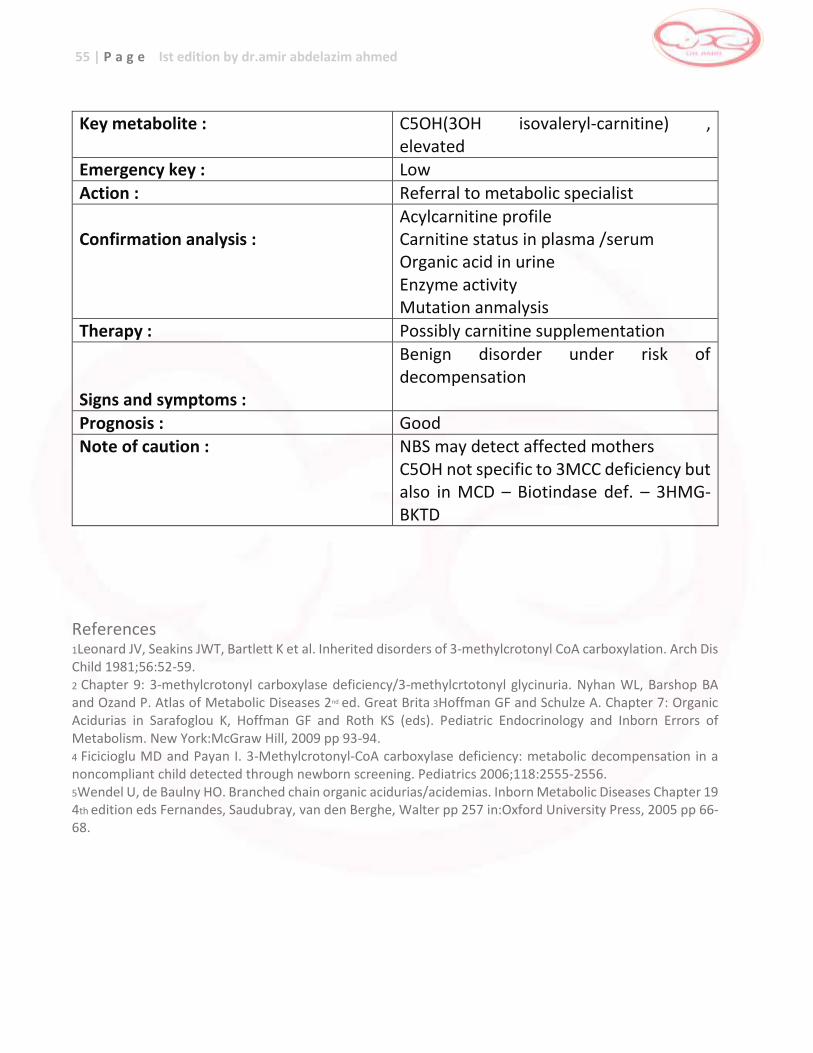
55 | P a g e Ist edition by dr.amir abdelazim ahmed
Key metabolite : C5OH(3OH isovaleryl-carnitine) ,
elevated Emergency key : Low
Action : Referral to metabolic specialist
Confirmation analysis :
Acylcarnitine profile Carnitine status in plasma /serum Organic acid in urine Enzyme activity Mutation anmalysis
Therapy : Possibly carnitine supplementation Signs and symptoms :
Benign disorder under risk of decompensation
Prognosis : Good Note of caution : NBS may detect affected mothers
C5OH not specific to 3MCC deficiency but also in MCD – Biotindase def. – 3HMG- BKTD
References 1Leonard JV, Seakins JWT, Bartlett K et al. Inherited disorders of 3-methylcrotonyl CoA carboxylation. Arch Dis Child 1981;56:52-59. 2 Chapter 9: 3-methylcrotonyl carboxylase deficiency/3-methylcrtotonyl glycinuria. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Brita 3Hoffman GF and Schulze A. Chapter 7: Organic Acidurias in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of
Metabolism. New York:McGraw Hill, 2009 pp 93-94. 4 Ficicioglu MD and Payan I. 3-Methylcrotonyl-CoA carboxylase deficiency: metabolic decompensation in a noncompliant child detected through newborn screening. Pediatrics 2006;118:2555-2556. 5Wendel U, de Baulny HO. Branched chain organic acidurias/acidemias. Inborn Metabolic Diseases Chapter 19 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 257 in:Oxford University Press, 2005 pp 66-68.
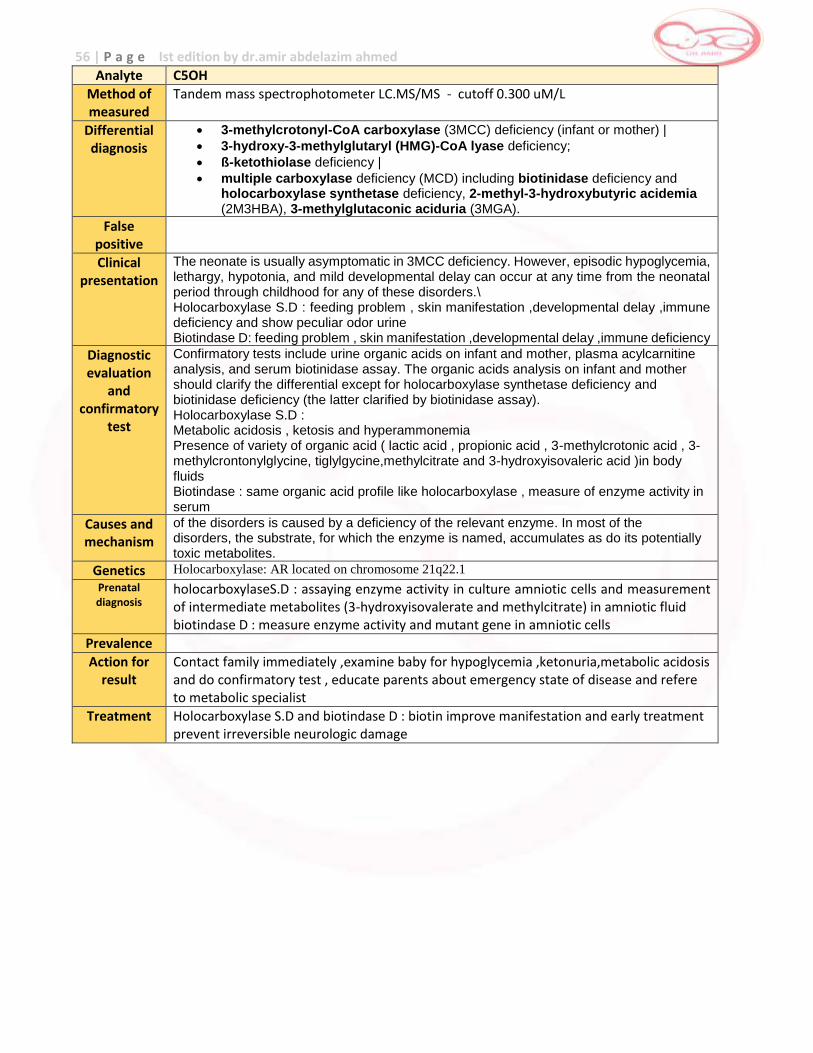
56 | P a g e Ist edition by dr.amir abdelazim ahmed
Analyte C5OH
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 0.300 uM/L
Differential diagnosis
3-methylcrotonyl-CoA carboxylase (3MCC) deficiency (infant or mother) |
3-hydroxy-3-methylglutaryl (HMG)-CoA lyase deficiency;
ß-ketothiolase deficiency |
multiple carboxylase deficiency (MCD) including biotinidase deficiency and holocarboxylase synthetase deficiency, 2-methyl-3-hydroxybutyric acidemia (2M3HBA), 3-methylglutaconic aciduria (3MGA).
False positive
Clinical presentation
The neonate is usually asymptomatic in 3MCC deficiency. However, episodic hypoglycemia, lethargy, hypotonia, and mild developmental delay can occur at any time from the neonatal period through childhood for any of these disorders.\ Holocarboxylase S.D : feeding problem , skin manifestation ,developmental delay ,immune deficiency and show peculiar odor urine Biotindase D: feeding problem , skin manifestation ,developmental delay ,immune deficiency
Diagnostic evaluation
and confirmatory
test
Confirmatory tests include urine organic acids on infant and mother, plasma acylcarnitine analysis, and serum biotinidase assay. The organic acids analysis on infant and mother should clarify the differential except for holocarboxylase synthetase deficiency and biotinidase deficiency (the latter clarified by biotinidase assay). Holocarboxylase S.D : Metabolic acidosis , ketosis and hyperammonemia Presence of variety of organic acid ( lactic acid , propionic acid , 3-methylcrotonic acid , 3-methylcrontonylglycine, tiglylgycine,methylcitrate and 3-hydroxyisovaleric acid )in body fluids Biotindase : same organic acid profile like holocarboxylase , measure of enzyme activity in serum
Causes and mechanism
of the disorders is caused by a deficiency of the relevant enzyme. In most of the disorders, the substrate, for which the enzyme is named, accumulates as do its potentially toxic metabolites.
Genetics Holocarboxylase: AR located on chromosome 21q22.1
Prenatal diagnosis
holocarboxylaseS.D : assaying enzyme activity in culture amniotic cells and measurement of intermediate metabolites (3-hydroxyisovalerate and methylcitrate) in amniotic fluid biotindase D : measure enzyme activity and mutant gene in amniotic cells
Prevalence
Action for result
Contact family immediately ,examine baby for hypoglycemia ,ketonuria,metabolic acidosis and do confirmatory test , educate parents about emergency state of disease and refere to metabolic specialist
Treatment Holocarboxylase S.D and biotindase D : biotin improve manifestation and early treatment prevent irreversible neurologic damage

57 | P a g e Ist edition by dr.amir abdelazim ahmed
B-KETOTHIOLASE DEFICIENCY
Beta ketothiolase deficiency is a defect of mitochondrial acetoacetyl-CoA thiolase involving ketone body metabolism and isoleucine catabolism. Incidence Very rare
Clinical Manifestation
This rare disorder is characterized by normal early development followed by progressive loss of mental and motor skills, it is clinically characterized by intermittent ketoacidotic episodes with no clinical symptoms in between. Some patients may present with vomiting, hypotonia, lethargy, coma, hyperventilation and dehydration.
Ketoacidotic crises may occur following a bout of infection or mild illness. Pathophysiology Mitochondrial acetoacetyl CoA thiolase is responsible for the cleavage of 2-methylacetoacetyl CoA in isoleucine metabolism, acetoacetyl CoA formation in ketogenesis and acetoacetyl CoA cleavage is ketolysis. Inheritance autosomal recessive
Screening increased tiglycarnitine and 2-methylhydroxybutylcarnitine on MSMS
Confirmatory Testing An increased excretion of 2-methyl 3-hydroxybutyric and 2-methylacetoacetic acid in urine is observed but definitive diagnosis is established by demonstrating decreased enzyme activity in cultured fibroblasts. Prognosis The frequency of ketoacidotic attacks decreases with age.3 Clinical consequences can be avoided by early diagnosis and appropriate management of ketoacidosis
Preliminary / Initial Management During Metabolic Crisis Metabolic crises may be caused by illness, prolonged fasting or stressful situations such as surgery and severe infection. The goal of treatment is to reverse the catabolic state, correct the acidosis and prevent essential amino acid deficiency.

58 | P a g e Ist edition by dr.amir abdelazim ahmed
Long term management These patients can decompensate rapidly in early childhood. To prevent this, fasting must be avoided and a high carbohydrate intake must be maintained during any metabolic stress, such surgery or infection. Drinks containing carbohydrate should be started at the first sign of illness. Hospital admission is needed if these are not tolerated or if patient develops moderate to heavy ketonuria. In hospital, patients require intravenous infusion of glucose. A moderate protein restriction is also recommended because this enzyme is also involved in isoleucine catabolism. A low fat has also been recommended. Protein and fat should certainly be avoided during illness. References 1Fukao T. Beta kethothiolase deficiency. Orphanet 2001 http://www.orpha.net/data/patho/GB/uk-T2.pdf Accessed Feb 15, 2012. 2 Chapter 17: Mitochondrial acetoacetyl CoA thiolase (3-oxothiolase) deficiency. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 102-106. 3Strauss AW, Andersen BS and Bennett MJ. Chapter 5: Mitochondrial Fatty Acid Oxidation Defects in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 60-62. 4 Morris A. Disorders of ketogenesis and ketolysis. Inborn Metabolic Diseases Chapter 14 4th edition eds
Fernandes, Saudubray, van den Berghe, Walter
What to Do: If unwell and cannot tolerate oral intake: a. Nothing per orem b. Ensure patient’s airway is secure c. Insert IV access. Monitor glucose levels. May request for investigations i.e. CBC, blood gas, urine ketones as needed. May give fluid boluses if patient requires. d. Start D10% 0.3 NaCl at full maintenance. Assess patient clinically, if there is need to increase fluid, may do so up to 1.2 or 1.5X the maintenance. e. Monitor input and output strictly (q6 hours). If unwell and is able to tolerate oral intake: a. Insert oro- or nasogastric tube and start continuous feeding with a high glucose formula b. Insert IV access. Monitor glucose levels. May request for investigations i.e. CBC, blood gas, urine ketones as needed. May give fluid boluses if patient requires. c. Start D10% 0.3 NaCl at 5-10 cc/hr. d. Monitor input and output strictly (q6 hours). * Inform metabolic doctor on call for further guidance regarding on-going management.

59 | P a g e Ist edition by dr.amir abdelazim ahmed
BIOTINIDASE DEFICIENCY
Biotinidase deficiency is a form of multiple carboxylase deficiency in which the fundamental defect is an inability to cleave biocytin for biotin recycling.1 Biotin is a water-soluble vitamin of the B complex that acts as a coenzyme in each of 4 carboxylases in humans (pyruvate carboxylase, propionyl-coenzyme A carboxylase, β- methylcrotonyl CoA caorboxylase and acetyl-CoA carboxylase). Incidence No determined Clinical Manifestation Biotinidase deficiency presents with a median age of 3 months or as late as 10 years of age, symptoms include dermatologic affectation appearing as patchy desquamation and neurological manifestations such as seizures in 70% of patients and ataxia that can interfere with walking. Some patients may also have optic atrophy and hearing loss. Individuals with partial biotinidase deficiency can present with skin manifestations and no neurologic symptoms. Pathophysiology Biotinidase deficiency results in an inability to recycle endogenous biotin which means the brain is unable to recycle biotin adequately leading to decreased pyruvate carboxylase activity in the brain and accumulation of lactate which in turn causes the neurologic symptoms. Inheritance autosomal recessive
Screening biotinidase in MSMS and presence of 3-hydroxy-isovaleric, 3-methylcrotonic, 3-hydroxypropionic, methylcitric, 3-hydroxybutyric acids, and acetoacetate in urine organic acid Confirmatory Testing Confirmatory studies are performed by determining biotinidase activity in serum.
Prognosis Once therapy is instituted, cutaneous symptoms resolve quickly as do seizures and ataxia, however other symptoms such as hearing loss and optic atrophy are less reversible
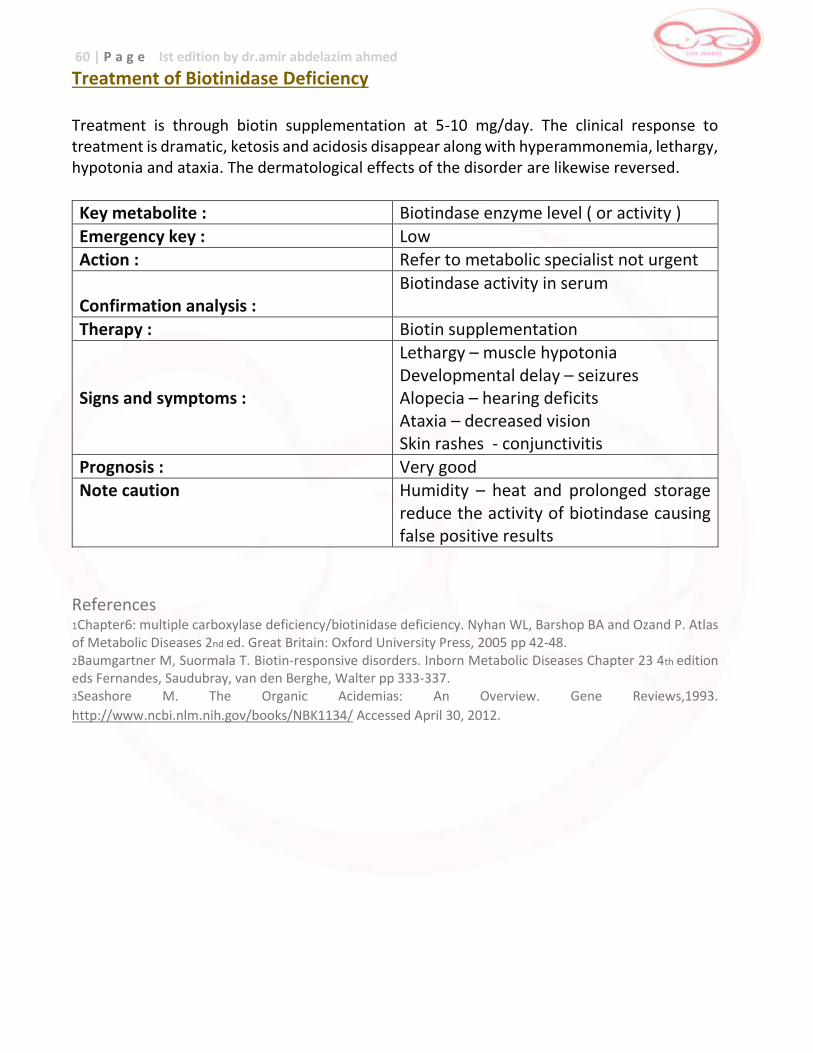
60 | P a g e Ist edition by dr.amir abdelazim ahmed
Treatment of Biotinidase Deficiency Treatment is through biotin supplementation at 5-10 mg/day. The clinical response to treatment is dramatic, ketosis and acidosis disappear along with hyperammonemia, lethargy, hypotonia and ataxia. The dermatological effects of the disorder are likewise reversed.
Key metabolite : Biotindase enzyme level ( or activity )
Emergency key : Low Action : Refer to metabolic specialist not urgent
Confirmation analysis :
Biotindase activity in serum
Therapy : Biotin supplementation Signs and symptoms :
Lethargy – muscle hypotonia Developmental delay – seizures Alopecia – hearing deficits Ataxia – decreased vision Skin rashes - conjunctivitis
Prognosis : Very good Note caution Humidity – heat and prolonged storage
reduce the activity of biotindase causing false positive results
References 1Chapter6: multiple carboxylase deficiency/biotinidase deficiency. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain: Oxford University Press, 2005 pp 42-48. 2Baumgartner M, Suormala T. Biotin-responsive disorders. Inborn Metabolic Diseases Chapter 23 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 333-337. 3Seashore M. The Organic Acidemias: An Overview. Gene Reviews,1993.
http://www.ncbi.nlm.nih.gov/books/NBK1134/ Accessed April 30, 2012.
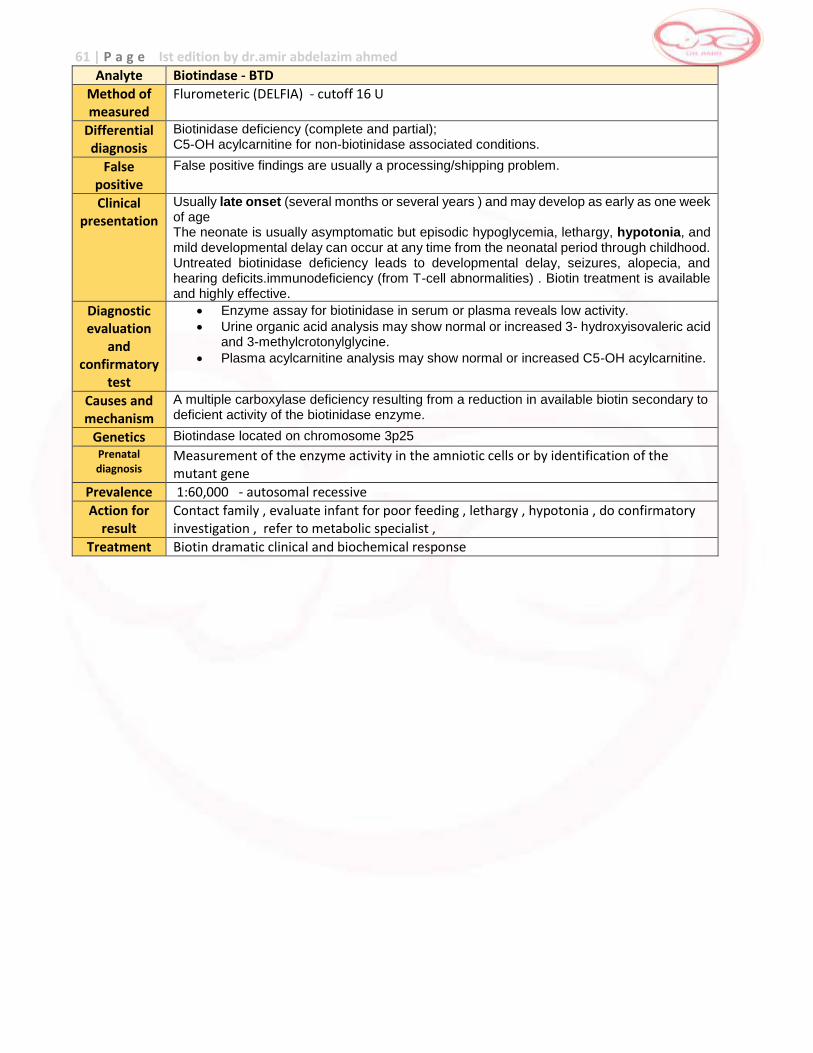
61 | P a g e Ist edition by dr.amir abdelazim ahmed
Analyte Biotindase - BTD
Method of measured
Flurometeric (DELFIA) - cutoff 16 U
Differential diagnosis
Biotinidase deficiency (complete and partial); C5-OH acylcarnitine for non-biotinidase associated conditions.
False positive
False positive findings are usually a processing/shipping problem.
Clinical presentation
Usually late onset (several months or several years ) and may develop as early as one week of age The neonate is usually asymptomatic but episodic hypoglycemia, lethargy, hypotonia, and mild developmental delay can occur at any time from the neonatal period through childhood. Untreated biotinidase deficiency leads to developmental delay, seizures, alopecia, and hearing deficits.immunodeficiency (from T-cell abnormalities) . Biotin treatment is available and highly effective.
Diagnostic evaluation
and confirmatory
test
Enzyme assay for biotinidase in serum or plasma reveals low activity.
Urine organic acid analysis may show normal or increased 3- hydroxyisovaleric acid and 3-methylcrotonylglycine.
Plasma acylcarnitine analysis may show normal or increased C5-OH acylcarnitine.
Causes and mechanism
A multiple carboxylase deficiency resulting from a reduction in available biotin secondary to deficient activity of the biotinidase enzyme.
Genetics Biotindase located on chromosome 3p25
Prenatal diagnosis
Measurement of the enzyme activity in the amniotic cells or by identification of the mutant gene
Prevalence 1:60,000 - autosomal recessive
Action for result
Contact family , evaluate infant for poor feeding , lethargy , hypotonia , do confirmatory investigation , refer to metabolic specialist ,
Treatment Biotin dramatic clinical and biochemical response

62 | P a g e Ist edition by dr.amir abdelazim ahmed
GLUTARIC ACIDURIA TYPE 1 [GA 1]
Glutaric Aciduria 1 (GA1) was first described by Goodman and colleagues in 1975.1 It is caused by a deficiency of glutaryl-CoA dehydrogenase which catalyzes the oxidative decarboxylation of glutaryl-CoA, an intermediate in the degradation of the amino acids lysine and tryptophan.2 This causes an increase in glutaric, 3- hydroxyglutaric, glutaconic and glutarylcarnitine. Incidence rare Clinical Manifestation Two subsets of patients are characterized based on the levels of glutaric acid excreted in the urine: the low (<100 mmol/mmol creatinine) and high excretors (>100 mmol/mmol creatinine).4 However, the risk of developing striatal injury resulting in neurologic dysfunction is the same regardless of excretion status. Patients with GA1 may present with hypotonia, head lag, feeding difficulties, irritability.1 Macrocephaly is seen in about 75% of infants, but this is non-specific.2 If left untreated, 90% of patients develop neurologic disease presenting as dystonic-dyskinetic posturing, athetoid movements, opisthotonus, spastic, rigidity, clenched fists, tongue thrust and profuse sweating. The encephalopathic crises precipitated by immunization, infection, surgery and fasting results in the affectation of the basal ganglia and exaggerates the neurologic manifestations which occur frequently until the 4th year of life. Patients may also be observed to have retinal hemorrhages and on MRI present with subdural hemorrhages and be mistaken to be victims of child abuse.
Pathophysiology
It was found that 3-hydroxglutaric and glutaric acid share structural similarities with glutamate which causes excitatory cell damage; futher, the accumulation of these metabolites modulate glutamatergic and GABAergic neurotransmission resulting in an imbalance of excitatory and inhibitory neurotransmitters. Inheritance autosomal recessive
Screening increased glutarylcarnitine on MSMS

63 | P a g e Ist edition by dr.amir abdelazim ahmed
Confirmatory Test Glutaric acid and 3-hydroxyglutaric acid is increased in urine organic acid analysis.1
Confirmatory testing is achieved through the demonstration of a decrease in enzyme activity in skin fibroblasts. Prognosis The early diagnosis and treatment intervention in patients with GA1 prevents striatal degeneration in 80-90% of infants.1 However, study by Beauchamp et al. (2009) showed that despite early treatment, patients with GA1 may have mild fine motor and articulation problems and raise the question of prenatal damage or subtle post-natal ongoing neurotoxic effects of glutaric and hydroxyglutaric acids or both.
Preliminary / Initial Management During Metabolic Crisis Metabolic crises may be caused by illness, prolonged fasting or stressful situations such as surgery, vaccination and severe infection. The goal of treatment is to reverse the catabolic state and prevent essential amino acid deficiency.
What to Do: If unwell and cannot tolerate oral intake: a. Nothing per orem b. Ensure patient’s airway is secure c. Insert IV access. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. d. Start D12.5% 0.3 NaCl at full maintenance. Assess patient clinically, if there is need to increase fluid, may do so up to 1.2 or 1.5X the maintenance. e. Start IV Carnitine (100mg/kg/day) q6 hours f. Monitor input and output strictly (q6 hours) If unwell but is able to tolerate oral intake: a. Insert oro- or nasogastric tube and start continuous feeding with GA1 formula or protein free formula b. Insert IV access. May request for other investigations (i.e. CBC, blood gas) as needed. May give fluid boluses if patient requires. c. Start D12.5% 0.3 NaCl at 5-10 cc/hr. d. Start IV Carnitine (100mg/kg/day) q6 hours e. Monitor input and output strictly (q6 hours) * Children should not be protein restricted for longer than necessary (24-48 hours). * Co-management with a neurologist is indicated to control the dystonia. * Inform metabolic doctor on call for further guidance regarding on-going management.

64 | P a g e Ist edition by dr.amir abdelazim ahmed
Long term management Diet
Most patient with glutaric aciduria type I are treated by restriction of natural protein in general or of lysine in particular, supplemented with a lysine free amino acid mixture. The intake of tryptophan should only be reduced. Oral supplementation with carnitine and riboflavin
Carnitine should be supplemented lifelong at 50-100mg/kg/day. If riboflavin is tried, responsiveness should be investigated by giving riboflavin in increasing doses from 50-300 mg and monitoring total glutaric acid in 24 hour urine samples. Neuropharmacologic agents
Baclofen 1-2mg/kg/day or benzodiazepines at 0.1-1 mg/kg/day reduce involuntary movements and improve motor function, probably mostly through muscle relaxation. In patients with residual motor function, anticholinergics, such as trihexyphenidyl, may improve choreoathetosis. Valproic acid is contraindicated as it effectively competes with glutaric acid for esterification with L carnitine and may promote disturbances in the mitochondrial acetyl CoA-CoA ratio. Supportive treatment
Despite the severe motor handicap in some patients, intellectual functions are preserved. Affected patients require a multidisciplinary specialist institution. The social integration of patients can be greatly improved by language computers. As involuntary movements become severe, feeding difficulties can become a major problem. Increased muscular tension and sweating require a high intake of calories and water. Percutaneous gastrostomy can lead to a dramatic improvement in nutritional status, marked decrease in psychological tension associated with feeding, reduction in the burden of care for families and even a reduction in dystonia/ dyskinesia. Neurosurgical intervention of subdural hygromas/hematomas should be avoided. Acute encephalopathic crises can be precipitated by common febrile diseases, vaccinations or surgical interventions during infancy and early childhood. If untreated, the majority of these patients manifest such crises with potentially devastating neurological sequelae. Emergency Protocol for Glutaric Aciduria type I Important points to be relayed to the parents over the phone:
1. Avoid delay and bring the child to the hospital at once
2. Bring carnitine (if known to have GAI)
3. Bring formula (Lysine free and tryptophan reduced formula)
4. Ask for child’s current weight
5. Ask about an estimated time of arrival at the ER
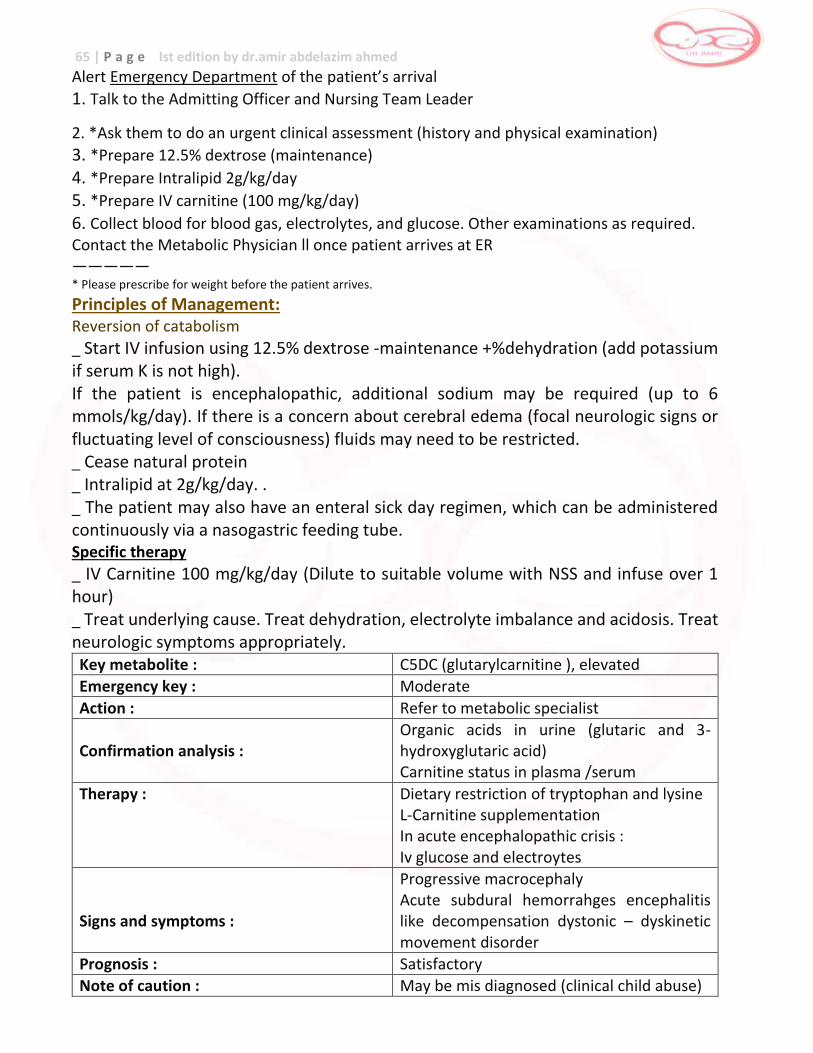
65 | P a g e Ist edition by dr.amir abdelazim ahmed
Alert Emergency Department of the patient’s arrival
1. Talk to the Admitting Officer and Nursing Team Leader
2. *Ask them to do an urgent clinical assessment (history and physical examination)
3. *Prepare 12.5% dextrose (maintenance)
4. *Prepare Intralipid 2g/kg/day
5. *Prepare IV carnitine (100 mg/kg/day)
6. Collect blood for blood gas, electrolytes, and glucose. Other examinations as required. Contact the Metabolic Physician ll once patient arrives at ER ————— * Please prescribe for weight before the patient arrives.
Principles of Management: Reversion of catabolism
_ Start IV infusion using 12.5% dextrose -maintenance +%dehydration (add potassium if serum K is not high). If the patient is encephalopathic, additional sodium may be required (up to 6 mmols/kg/day). If there is a concern about cerebral edema (focal neurologic signs or fluctuating level of consciousness) fluids may need to be restricted. _ Cease natural protein _ Intralipid at 2g/kg/day. . _ The patient may also have an enteral sick day regimen, which can be administered continuously via a nasogastric feeding tube. Specific therapy
_ IV Carnitine 100 mg/kg/day (Dilute to suitable volume with NSS and infuse over 1 hour) _ Treat underlying cause. Treat dehydration, electrolyte imbalance and acidosis. Treat neurologic symptoms appropriately. Key metabolite : C5DC (glutarylcarnitine ), elevated
Emergency key : Moderate
Action : Refer to metabolic specialist
Confirmation analysis :
Organic acids in urine (glutaric and 3-hydroxyglutaric acid) Carnitine status in plasma /serum
Therapy : Dietary restriction of tryptophan and lysine L-Carnitine supplementation In acute encephalopathic crisis : Iv glucose and electroytes
Signs and symptoms :
Progressive macrocephaly Acute subdural hemorrahges encephalitis like decompensation dystonic – dyskinetic movement disorder
Prognosis : Satisfactory
Note of caution : May be mis diagnosed (clinical child abuse)
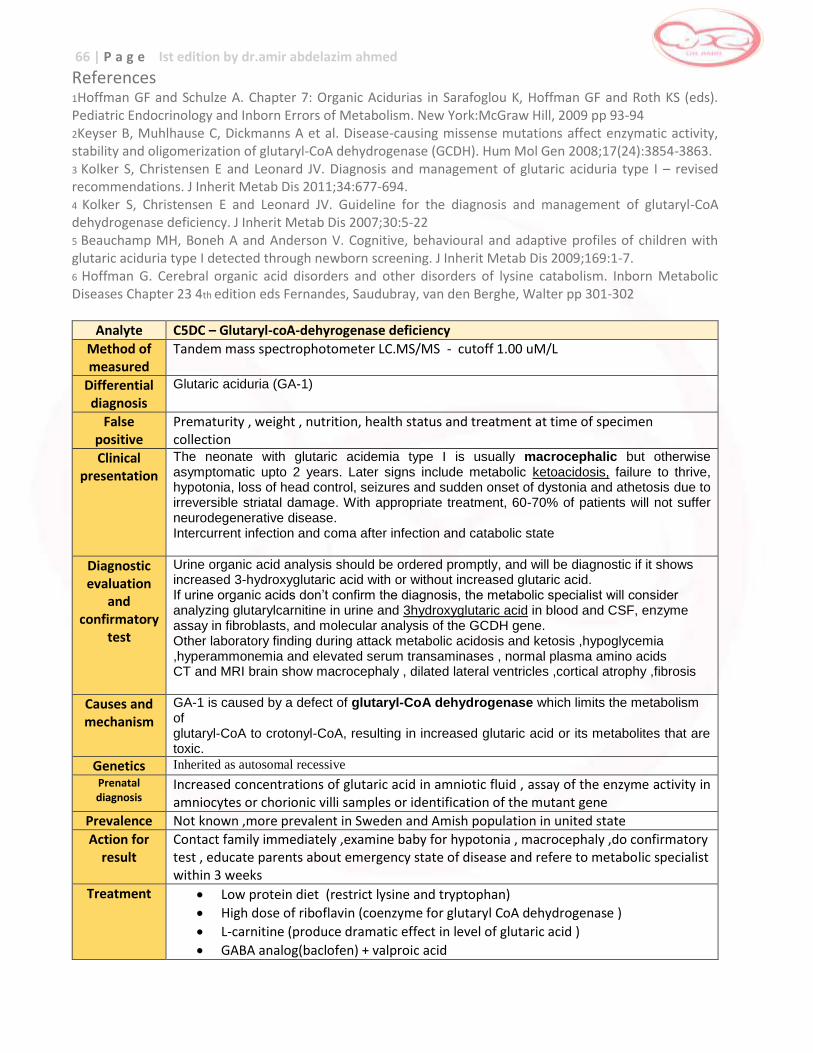
66 | P a g e Ist edition by dr.amir abdelazim ahmed
References 1Hoffman GF and Schulze A. Chapter 7: Organic Acidurias in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 93-94 2Keyser B, Muhlhause C, Dickmanns A et al. Disease-causing missense mutations affect enzymatic activity, stability and oligomerization of glutaryl-CoA dehydrogenase (GCDH). Hum Mol Gen 2008;17(24):3854-3863. 3 Kolker S, Christensen E and Leonard JV. Diagnosis and management of glutaric aciduria type I – revised recommendations. J Inherit Metab Dis 2011;34:677-694. 4 Kolker S, Christensen E and Leonard JV. Guideline for the diagnosis and management of glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 2007;30:5-22 5 Beauchamp MH, Boneh A and Anderson V. Cognitive, behavioural and adaptive profiles of children with glutaric aciduria type I detected through newborn screening. J Inherit Metab Dis 2009;169:1-7. 6 Hoffman G. Cerebral organic acid disorders and other disorders of lysine catabolism. Inborn Metabolic Diseases Chapter 23 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 301-302
Analyte C5DC – Glutaryl-coA-dehyrogenase deficiency
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 1.00 uM/L
Differential diagnosis
Glutaric aciduria (GA-1)
False positive
Prematurity , weight , nutrition, health status and treatment at time of specimen collection
Clinical presentation
The neonate with glutaric acidemia type I is usually macrocephalic but otherwise asymptomatic upto 2 years. Later signs include metabolic ketoacidosis, failure to thrive, hypotonia, loss of head control, seizures and sudden onset of dystonia and athetosis due to irreversible striatal damage. With appropriate treatment, 60-70% of patients will not suffer neurodegenerative disease. Intercurrent infection and coma after infection and catabolic state
Diagnostic evaluation
and confirmatory
test
Urine organic acid analysis should be ordered promptly, and will be diagnostic if it shows increased 3-hydroxyglutaric acid with or without increased glutaric acid. If urine organic acids don’t confirm the diagnosis, the metabolic specialist will consider analyzing glutarylcarnitine in urine and 3hydroxyglutaric acid in blood and CSF, enzyme assay in fibroblasts, and molecular analysis of the GCDH gene. Other laboratory finding during attack metabolic acidosis and ketosis ,hypoglycemia ,hyperammonemia and elevated serum transaminases , normal plasma amino acids CT and MRI brain show macrocephaly , dilated lateral ventricles ,cortical atrophy ,fibrosis
Causes and mechanism
GA-1 is caused by a defect of glutaryl-CoA dehydrogenase which limits the metabolism of glutaryl-CoA to crotonyl-CoA, resulting in increased glutaric acid or its metabolites that are toxic.
Genetics Inherited as autosomal recessive
Prenatal diagnosis
Increased concentrations of glutaric acid in amniotic fluid , assay of the enzyme activity in amniocytes or chorionic villi samples or identification of the mutant gene
Prevalence Not known ,more prevalent in Sweden and Amish population in united state
Action for result
Contact family immediately ,examine baby for hypotonia , macrocephaly ,do confirmatory test , educate parents about emergency state of disease and refere to metabolic specialist within 3 weeks
Treatment Low protein diet (restrict lysine and tryptophan)
High dose of riboflavin (coenzyme for glutaryl CoA dehydrogenase )
L-carnitine (produce dramatic effect in level of glutaric acid )
GABA analog(baclofen) + valproic acid

67 | P a g e Ist edition by dr.amir abdelazim ahmed
ISOVALERIC ACIDURIA [IVA]
Isovaleric acidemia (IVA) was the first organic acidemia to be described. It is caused by a deficiency of isovaleryl- CoA dehydrogenase, an enzyme located proximally in the catabolic pathway of the essential branched-chain amino acid leucine. Incidence rare Clinical Manifestation The clinical manifestation of IVA may be acute or chronic. An acute or neonatal presentation is characterized by non-specific findings of vomiting, lethargy, poor feeding, seizures that may progress to a comatose state. A characteristic odor in the urine described as “sweaty feet” or “dirty socks” has been reported among patients with IVA. It has also been found that in bone marrow cultures, isovaleric acid is an inhibitor of granulopoietic progenitor cell proliferation which accounts for the pancytopenia or thrombocytopenia found in patients. A chronic form may present with developmental delay or mental retardation. Both acute or chronic patients may suffer from metabolic crisis and are sometimes misdiagnosed as suffering from diabetic ketoacidosis because of the similarity in presentation: acidosis, hyperglycemia and ketosis.
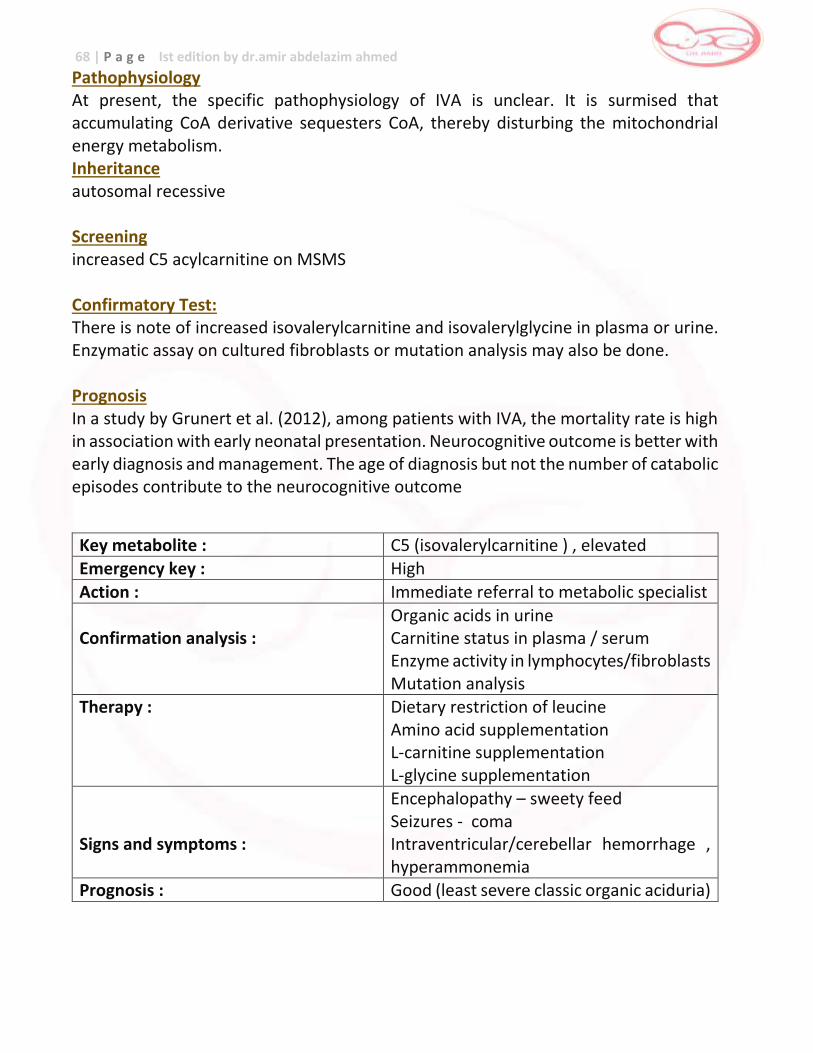
68 | P a g e Ist edition by dr.amir abdelazim ahmed
Pathophysiology At present, the specific pathophysiology of IVA is unclear. It is surmised that accumulating CoA derivative sequesters CoA, thereby disturbing the mitochondrial energy metabolism. Inheritance autosomal recessive
Screening increased C5 acylcarnitine on MSMS
Confirmatory Test: There is note of increased isovalerylcarnitine and isovalerylglycine in plasma or urine. Enzymatic assay on cultured fibroblasts or mutation analysis may also be done. Prognosis In a study by Grunert et al. (2012), among patients with IVA, the mortality rate is high in association with early neonatal presentation. Neurocognitive outcome is better with early diagnosis and management. The age of diagnosis but not the number of catabolic episodes contribute to the neurocognitive outcome
Key metabolite : C5 (isovalerylcarnitine ) , elevated Emergency key : High
Action : Immediate referral to metabolic specialist
Confirmation analysis :
Organic acids in urine Carnitine status in plasma / serum Enzyme activity in lymphocytes/fibroblasts Mutation analysis
Therapy : Dietary restriction of leucine Amino acid supplementation L-carnitine supplementation L-glycine supplementation
Signs and symptoms :
Encephalopathy – sweety feed Seizures - coma Intraventricular/cerebellar hemorrhage , hyperammonemia
Prognosis : Good (least severe classic organic aciduria)
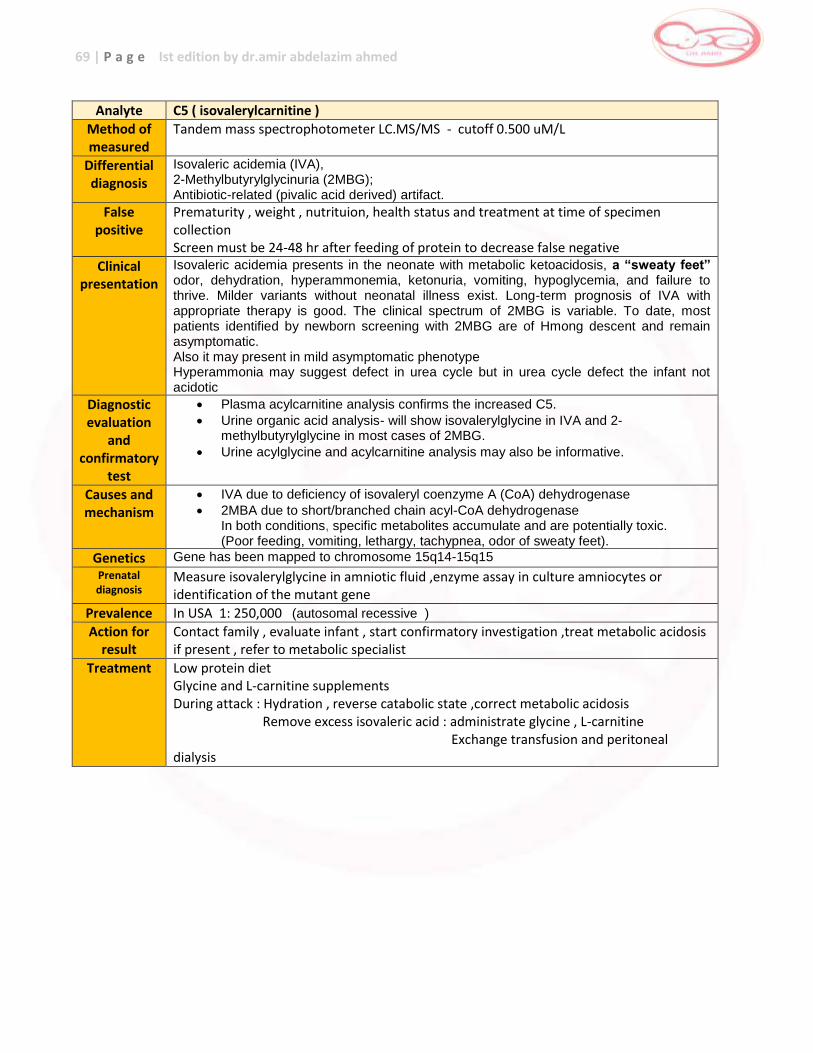
69 | P a g e Ist edition by dr.amir abdelazim ahmed
Analyte C5 ( isovalerylcarnitine )
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 0.500 uM/L
Differential diagnosis
Isovaleric acidemia (IVA), 2-Methylbutyrylglycinuria (2MBG); Antibiotic-related (pivalic acid derived) artifact.
False positive
Prematurity , weight , nutrituion, health status and treatment at time of specimen collection Screen must be 24-48 hr after feeding of protein to decrease false negative
Clinical presentation
Isovaleric acidemia presents in the neonate with metabolic ketoacidosis, a “sweaty feet” odor, dehydration, hyperammonemia, ketonuria, vomiting, hypoglycemia, and failure to thrive. Milder variants without neonatal illness exist. Long-term prognosis of IVA with appropriate therapy is good. The clinical spectrum of 2MBG is variable. To date, most patients identified by newborn screening with 2MBG are of Hmong descent and remain asymptomatic. Also it may present in mild asymptomatic phenotype Hyperammonia may suggest defect in urea cycle but in urea cycle defect the infant not acidotic
Diagnostic evaluation
and confirmatory
test
Plasma acylcarnitine analysis confirms the increased C5.
Urine organic acid analysis- will show isovalerylglycine in IVA and 2-methylbutyrylglycine in most cases of 2MBG.
Urine acylglycine and acylcarnitine analysis may also be informative.
Causes and mechanism
IVA due to deficiency of isovaleryl coenzyme A (CoA) dehydrogenase
2MBA due to short/branched chain acyl-CoA dehydrogenase In both conditions, specific metabolites accumulate and are potentially toxic.
(Poor feeding, vomiting, lethargy, tachypnea, odor of sweaty feet).
Genetics Gene has been mapped to chromosome 15q14-15q15
Prenatal diagnosis
Measure isovalerylglycine in amniotic fluid ,enzyme assay in culture amniocytes or identification of the mutant gene
Prevalence In USA 1: 250,000 (autosomal recessive )
Action for result
Contact family , evaluate infant , start confirmatory investigation ,treat metabolic acidosis if present , refer to metabolic specialist
Treatment Low protein diet Glycine and L-carnitine supplements During attack : Hydration , reverse catabolic state ,correct metabolic acidosis Remove excess isovaleric acid : administrate glycine , L-carnitine Exchange transfusion and peritoneal dialysis

70 | P a g e Ist edition by dr.amir abdelazim ahmed
METHYLMALONIC ACIDEMIA [MMA]
Methylmalonic academia (MMA) is due to a defect in methylmalonyl CoA mutase or a defect in the enzyme’s vitamin B12 derived co-factor 5’-deoxyadenosylcobalamin.5
Among patients with a defect of methylmalonyl CoA mutase, two subgroups exist: Mut0 patients have no enzyme activity while Mut- patients have a spectrum of residual activity. Incidence rare Clinical Manifestation: Patients present with severe metabolic crisis in the first months of life, progressive failure to thrive, feeding problems, recurrent vomiting, dehydration, hepatomegaly, lethargy, seizures and developmental delay. Some affected children may also have failure of linear growth, anorexia and developmental failure.5 Patients may have metabolic decompensations following bouts of acute illness or minor infections. They are prone to episodes of metabolic strokes that primarily affect the basal ganglia. Neonates affected with MMA share similar physical characteristics such as high forehead, broad nasal bridge, epicanthal folds, long smooth philtrum and triangular mouth.5 Unique to this disorder is the development of chronic renal failure in the second decade in 20-60% of patients.
Pathophysiology
Methylmalonyl CoA-mutase catalyzes the conversion of methylmalonyl CoA to succinyl CoA which can enter the tricarboxylic acid cycle. This causes the accumulation of methylmalonate in the body which may be toxic to the brain and the kidneys. Inheritance autosomal recessive
Screening increase in propionylcarnitine on MSMS
Confirmatory Testing Urine metabolic screening reveal elevated methylmalonic acid, propionylglycine, 3-hydroxypropionic acid and methylcitrate; plasma amino acids show elevated glycine, alanine and methionine.1 Definitive testing is the demonstration of decreased enzyme activity through cultured fibroblasts.
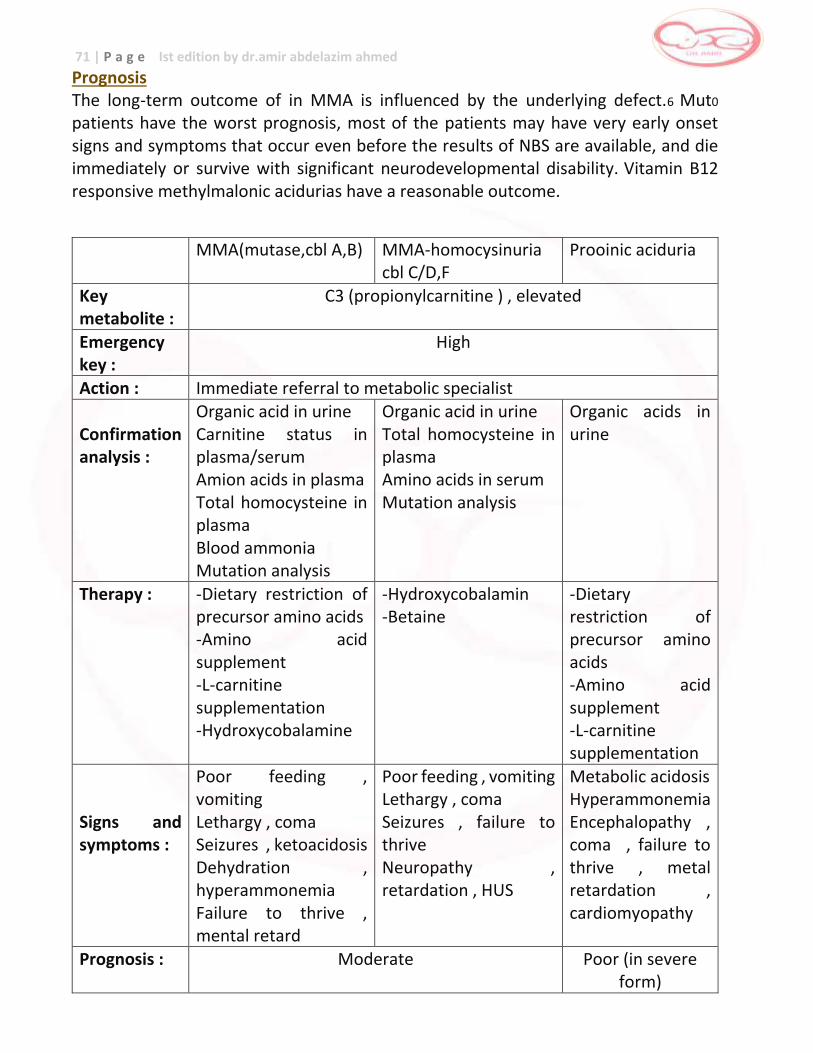
71 | P a g e Ist edition by dr.amir abdelazim ahmed
Prognosis The long-term outcome of in MMA is influenced by the underlying defect.6 Mut0
patients have the worst prognosis, most of the patients may have very early onset signs and symptoms that occur even before the results of NBS are available, and die immediately or survive with significant neurodevelopmental disability. Vitamin B12 responsive methylmalonic acidurias have a reasonable outcome.
MMA(mutase,cbl A,B) MMA-homocysinuria cbl C/D,F
Prooinic aciduria
Key metabolite :
C3 (propionylcarnitine ) , elevated
Emergency key :
High
Action : Immediate referral to metabolic specialist Confirmation analysis :
Organic acid in urine Carnitine status in plasma/serum Amion acids in plasma Total homocysteine in plasma Blood ammonia Mutation analysis
Organic acid in urine Total homocysteine in plasma Amino acids in serum Mutation analysis
Organic acids in urine
Therapy : -Dietary restriction of precursor amino acids -Amino acid supplement -L-carnitine supplementation -Hydroxycobalamine
-Hydroxycobalamin -Betaine
-Dietary restriction of precursor amino acids -Amino acid supplement -L-carnitine supplementation
Signs and symptoms :
Poor feeding , vomiting Lethargy , coma Seizures , ketoacidosis Dehydration , hyperammonemia Failure to thrive , mental retard
Poor feeding , vomiting Lethargy , coma Seizures , failure to thrive Neuropathy , retardation , HUS
Metabolic acidosis Hyperammonemia Encephalopathy , coma , failure to thrive , metal retardation , cardiomyopathy
Prognosis : Moderate Poor (in severe form)
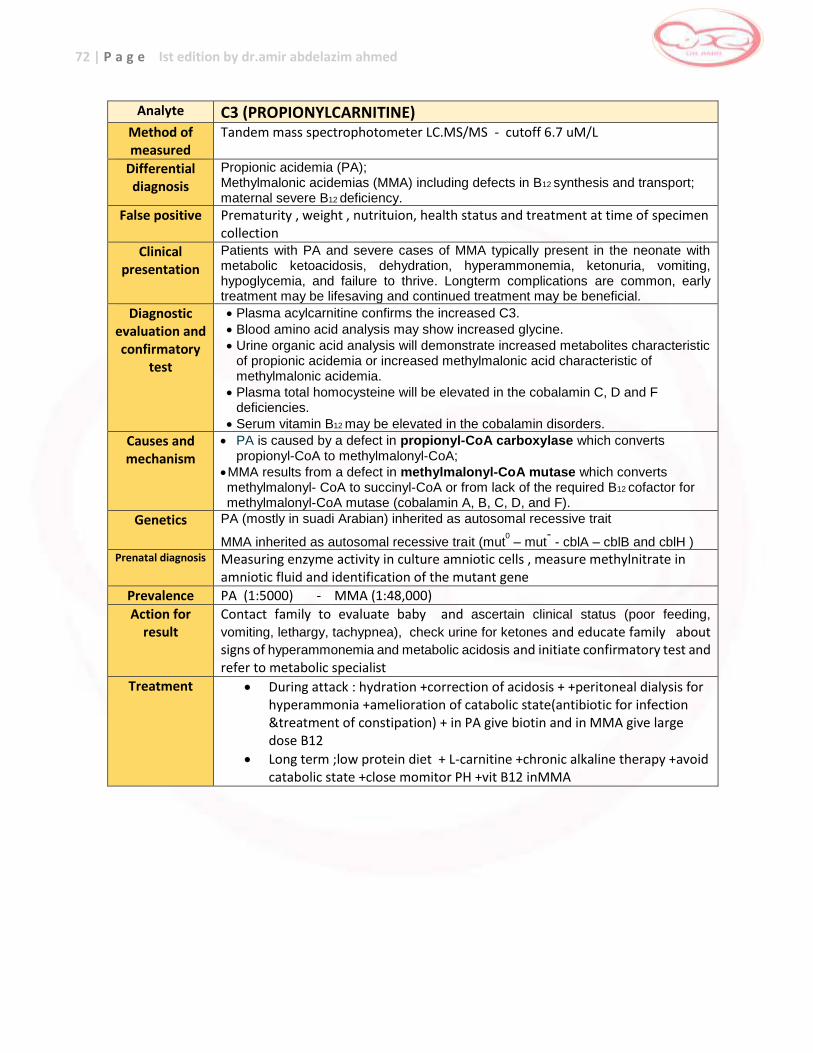
72 | P a g e Ist edition by dr.amir abdelazim ahmed
Analyte C3 (PROPIONYLCARNITINE)
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 6.7 uM/L
Differential diagnosis
Propionic acidemia (PA); Methylmalonic acidemias (MMA) including defects in B12 synthesis and transport; maternal severe B12 deficiency.
False positive Prematurity , weight , nutrituion, health status and treatment at time of specimen collection
Clinical presentation
Patients with PA and severe cases of MMA typically present in the neonate with metabolic ketoacidosis, dehydration, hyperammonemia, ketonuria, vomiting, hypoglycemia, and failure to thrive. Longterm complications are common, early treatment may be lifesaving and continued treatment may be beneficial.
Diagnostic evaluation and confirmatory
test
Plasma acylcarnitine confirms the increased C3.
Blood amino acid analysis may show increased glycine.
Urine organic acid analysis will demonstrate increased metabolites characteristic of propionic acidemia or increased methylmalonic acid characteristic of methylmalonic acidemia.
Plasma total homocysteine will be elevated in the cobalamin C, D and F deficiencies.
Serum vitamin B12 may be elevated in the cobalamin disorders.
Causes and mechanism
PA is caused by a defect in propionyl-CoA carboxylase which converts propionyl-CoA to methylmalonyl-CoA;
MMA results from a defect in methylmalonyl-CoA mutase which converts methylmalonyl- CoA to succinyl-CoA or from lack of the required B12 cofactor for methylmalonyl-CoA mutase (cobalamin A, B, C, D, and F).
Genetics PA (mostly in suadi Arabian) inherited as autosomal recessive trait
MMA inherited as autosomal recessive trait (mut0 – mut
- - cblA – cblB and cblH )
Prenatal diagnosis Measuring enzyme activity in culture amniotic cells , measure methylnitrate in amniotic fluid and identification of the mutant gene
Prevalence PA (1:5000) - MMA (1:48,000)
Action for result
Contact family to evaluate baby and ascertain clinical status (poor feeding,
vomiting, lethargy, tachypnea), check urine for ketones and educate family about signs of hyperammonemia and metabolic acidosis and initiate confirmatory test and refer to metabolic specialist
Treatment During attack : hydration +correction of acidosis + +peritoneal dialysis for hyperammonia +amelioration of catabolic state(antibiotic for infection &treatment of constipation) + in PA give biotin and in MMA give large dose B12
Long term ;low protein diet + L-carnitine +chronic alkaline therapy +avoid catabolic state +close momitor PH +vit B12 inMMA

73 | P a g e Ist edition by dr.amir abdelazim ahmed
MULTIPLE CARBOXYLASE DEFICIENCY [MCD]
Mutliple carboxylase deficiency, also known as holocarboxylase synthase leads to a failure of synthesis of all carboxylases. Incidence rare Clinical Manifestation Most patients present acutely in the first few hours of life. Patients may have dehydration, go into deep coma leading to death, ketosis, high anion gap metabolic acidosis, failure to thrive, alopecia and a characteristic erythematous eruption on the skin that can be bright, red, scaly or desquamative. Pathophysiology Holocarboxylase synthase binds biotin, an essential cofactor in gluconeogenesis, fatty acid synthesis and the catabolism of several amino acids. This in turn, leads to a failure of the synthesis of the active holocarboxylases which is the body’s main source of biotin. Inheritance autosomal recessive
Screening increased priopionyl carnitine and 3-hydroxyisovaleryl carnitine
Confirmatory Testing An increased methylcrotonylglycine and 3-hydroxyisovaleric acid in blood and urine with lactic acidosis can be observed but definitive testing is done through measurement of enzyme activity in fibroblasts. Prognosis Prognosis is good if treatment is initiated immediately and the clinical course is followed carefully by close monitoring of biochemical abnormalities. Long term management
Treatment is through giving biotin 10-20mg/day. The clinical response to treatment is dramatic, ketosis and acidosis disappear along with hyperammonemia, lethargy, hypotonia and ataxia. The dermatological effects of the disorder are likewise reversed.

74 | P a g e Ist edition by dr.amir abdelazim ahmed
Key metabolite : C5OH , (C3) , elevated
Emergency key : High Action : Immediate referral to metabolic
specialist Confirmation analysis :
Organic acid in urine Carnitine status in plasma/serum Enzyme activity fibroblasts
Therapy : Biotin Signs and symptoms :
Neurologic symptoms from neonatal decompensation , hypotonia , seizures Psychomotor retardation , skin rashes Hair loss , immune defect
Prognosis : Good
References 1 Chapter 5: Multiple carboxylase deficiency/holocarboxylase deficiency. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 36-39. 2Baumgartner M, Suormala T. Biotin-responsive disorders. Inborn Metabolic Diseases Chapter 23 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 333-337.
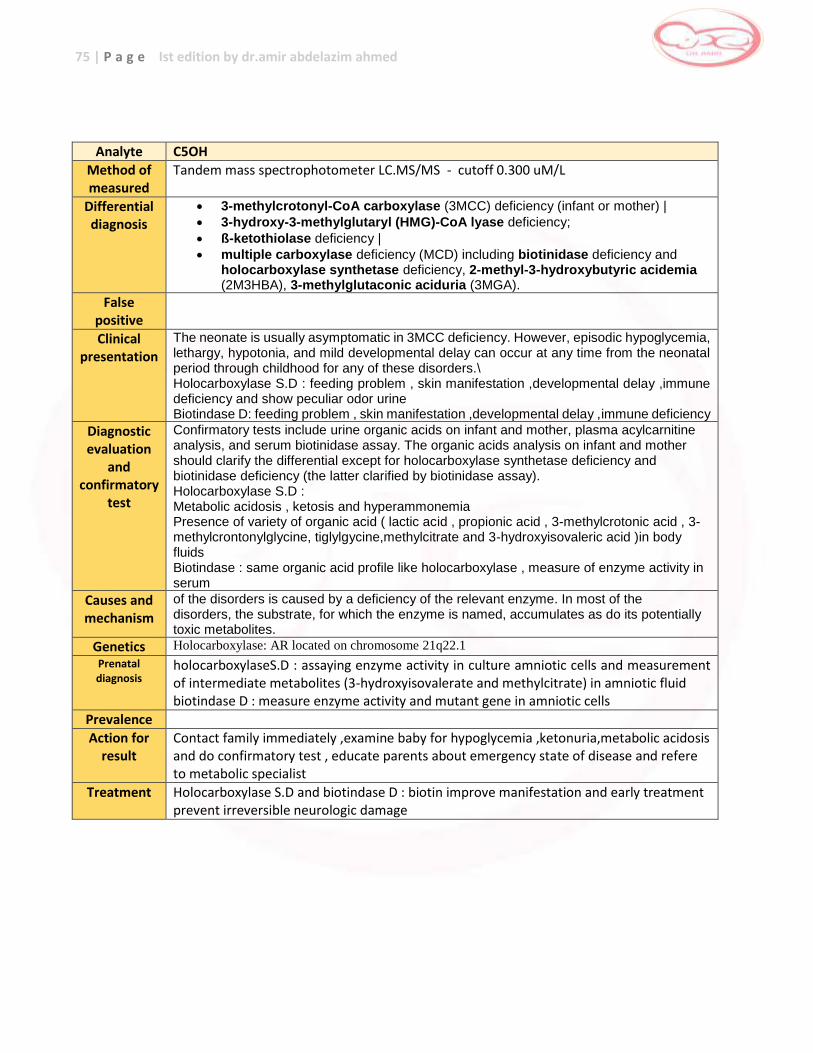
75 | P a g e Ist edition by dr.amir abdelazim ahmed
Analyte C5OH
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 0.300 uM/L
Differential diagnosis
3-methylcrotonyl-CoA carboxylase (3MCC) deficiency (infant or mother) |
3-hydroxy-3-methylglutaryl (HMG)-CoA lyase deficiency;
ß-ketothiolase deficiency |
multiple carboxylase deficiency (MCD) including biotinidase deficiency and holocarboxylase synthetase deficiency, 2-methyl-3-hydroxybutyric acidemia (2M3HBA), 3-methylglutaconic aciduria (3MGA).
False positive
Clinical presentation
The neonate is usually asymptomatic in 3MCC deficiency. However, episodic hypoglycemia, lethargy, hypotonia, and mild developmental delay can occur at any time from the neonatal period through childhood for any of these disorders.\ Holocarboxylase S.D : feeding problem , skin manifestation ,developmental delay ,immune deficiency and show peculiar odor urine Biotindase D: feeding problem , skin manifestation ,developmental delay ,immune deficiency
Diagnostic evaluation
and confirmatory
test
Confirmatory tests include urine organic acids on infant and mother, plasma acylcarnitine analysis, and serum biotinidase assay. The organic acids analysis on infant and mother should clarify the differential except for holocarboxylase synthetase deficiency and biotinidase deficiency (the latter clarified by biotinidase assay). Holocarboxylase S.D : Metabolic acidosis , ketosis and hyperammonemia Presence of variety of organic acid ( lactic acid , propionic acid , 3-methylcrotonic acid , 3-methylcrontonylglycine, tiglylgycine,methylcitrate and 3-hydroxyisovaleric acid )in body fluids Biotindase : same organic acid profile like holocarboxylase , measure of enzyme activity in serum
Causes and mechanism
of the disorders is caused by a deficiency of the relevant enzyme. In most of the disorders, the substrate, for which the enzyme is named, accumulates as do its potentially toxic metabolites.
Genetics Holocarboxylase: AR located on chromosome 21q22.1
Prenatal diagnosis
holocarboxylaseS.D : assaying enzyme activity in culture amniotic cells and measurement of intermediate metabolites (3-hydroxyisovalerate and methylcitrate) in amniotic fluid biotindase D : measure enzyme activity and mutant gene in amniotic cells
Prevalence
Action for result
Contact family immediately ,examine baby for hypoglycemia ,ketonuria,metabolic acidosis and do confirmatory test , educate parents about emergency state of disease and refere to metabolic specialist
Treatment Holocarboxylase S.D and biotindase D : biotin improve manifestation and early treatment prevent irreversible neurologic damage

76 | P a g e Ist edition by dr.amir abdelazim ahmed
PROPIONIC ACIDEMIC [PA]
Propionic acidemia (PA) is an organic acidopathy also known was propionic aciduria and ketotic hyperglycinemia. It is due to the defective activity of propionyl CoA which is the first step in the pathway of propionate metabolism in which propionyl CoA, the product of the metabolism of isoleucine, valine, theronine and methionine is converted to methylmalonyl CoA then to succinyl CoA which undergoes oxidation in the citric acid cycle. Incidence rare Clinical Manifestation Patients usually are healthy at birth but quickly develop overwhelming disease, which may be misinterpreted as sepsis or ventricular hemorrhage.1 Additional symptoms include vomiting, acidosis, dehydration, lethargy to coma, recurrent ketotic episodes, hypotonia, seizures and hyperammonemia.10 Some patients may have acute-onset neurological symptoms described as metabolic strokes, arrhythmias, cardiomyopathy and an exfoliative rash. Patients may also present with similar dysmorphic characteristics such as frontal bossing, widened nasal bridge, wide set eyes, epicanthal folds, long philtrum and upward curvature of the lips. Pathophysiology Due to an increase in propionic acid, abnormal ketogenesis occurs because propionic acid is an inhibitor of mitochondrial oxidation and succinic and alpha-ketoglutaric acid.10 Inhibition of glycine cleavage enzyme leads to hyperglycinemia and the inhibition of N-acetylglutamate synthase, an enzyme of the urea cycle, causes hyperammonemia.
Inheritance autosomal recessive
Screening elevated propionylcarnitine in MSMS
Confirmatory Testing The predominant compound found in blood and urine is 3-hydroxypropionic acid; others may include tiglic acid, tiglyglycine, butanone and propionylglycine.10 Highly elevated levels of glycine in plasma and urine can be observed but confirmatory testing is through the demonstration of low levels of enzyme on cultured fibroblasts.

77 | P a g e Ist edition by dr.amir abdelazim ahmed
Prognosis Despite early diagnosis and treatment, the neonatal onset form of PA is still complicated by early death in infancy or childhood while late onset forms reach adulthood but often are handicapped by severe extrapyramidal movement disorders and mental retardation; however, progress has been achieved in survival and prevention of neurologic sequelae in affected children with early diagnosis and treatment. Long term management for IVA, MMA and PA The goal of treatment is to reduce the production of isovaleric, methylmalonic anc
propionic acid by means of:
Diet
Patients with IVA, MMA and PA should be maintained on a low protein diet (about 1-1.5 g/kg/day). To improve the quality of this diet, it may be supplemented with a relatively small amount of synthetic amino acids free from the precursor amino acids( ie formula free of methionine, threonin, valine and isoleucine for MMA and PA). The diet must be nutritionally complete with adequate energy intake and sufficient vitamins and minerals. In children with severe forms of MMA and PA, anorexia and feeding problems can be addressed by nasogastric tube or gastrostomy tube placement. Vitamin therapy
Every patient with MMA should be tested for responsiveness to vitamin B12. Parenteral hydroxocobalamin at 1000-2000 ug/day should be tried for about 10 days during a stable metabolic condition. During this period, a 24 hour urine samples must be collected for urine oganic acid analysis. Vitamin B12 responsiveness leads to a prompt and sustained decrease of propionyl CoA by products or a drop in the urinary MMA level by more than 50%. Most B12 responsive patients need only mild protein restriction or none at all. Vitamin B12 is given orally or once per day or is administred once a week (1000-2000 ug IM). Carnitine/glycine therapy
For MMA and PA, chronic oral administration of L carnitine at 100 mg/kg/day is effective in not only preventing carnitine depletion but also in allowing urinary propionylcarnitine excretion, thus reducing propionate toxicity. For IVA, supplemental therapy with L-carnitine 50-100 mg/kgor glycine at 150-300 mg/kg/day can be used.

78 | P a g e Ist edition by dr.amir abdelazim ahmed
Metronidazole therapy
For MMA and PA, microbial propionate production can be suppressed by antibiotics. Metronidazole, an antiniotic that inhibits anaerobic colonic flora has been found to be specifically effective in reducing urinary excretion of propionate metabolites by 405 in MMA and PA patients. A dose of 10-20 mg/kg/day for ten consecutive days each month may be of significant benefit. Growth hormone
There is a place for recombinant human GH treatment as an adjuvant therapy in patients with MMA and PA, mainly in those with reduced linear growth. Biochemical Monitoring
During the course of decompensation plasma ammonia, blood gases, lactate, glucose, uric acid and ketones should bemonitored. Regular amino acid analysis is important. For MMA, levels of MMA in the plasma or urine should be controlled . The measurement of carnitine/acylcarnitine in blood may also be useful. Emergency Protocol for Propionic Aciduria/Methymalonic Aciduria Important points to be relayed to the parents over the phone:
1. Avoid delay and bring the child to the hospital at once formula ( if known patient
with MMA/PA)
2. Bring carnitine, vitamin B12 (if known patient with MMA/PA)
3. Ask for child’s current weight
4. Ask about an estimated time of arrival at the ER Alert Emergency Department of the patient’s arrival
1. Talk to the Admitting Officer and Nursing Team Leader
2. Ask them to do an urgent clinical assessment (history and physical examination)
3. *Prepare 12.5% dextrose (maintenance)
4. *Prepare Intralipid 2g/kg/day
5. *Prepare IV carnitine (100 mg/kg/day)
6. Collect blood for CBC, electrolytes, blood gas, lactate, glucose plasma methylmalonate (for MMA), plasma carnitine profile and ammonia**. Collect urine for urine organic acid c screening. Check for urine ketones. Other examinations as required.
7. Contact the Metabolic Physician once patient arrives at ER ————— * Please prescribe for weight before the patient arrives. ** Collect in green top tube. Must be on ice. Transport to lab immediately

79 | P a g e Ist edition by dr.amir abdelazim ahmed
Principles of Management: Reversion of catabolism
_ Start IV infusion using 12.5% dextrose -maintenance + %dehydration (add potassium if serum K is not high). If the patient is encephalopathic, additional sodium may be required (up to 6 mmols/kg/day). If there is a concern about cerebral edema (focal neurologic signs or fluctuating level of consciousness) fluids may need to be restricted. _ Cease natural protein _ Intralipid at 2g/kg/day. _ The patient may also have an enteral sick day regimen, which can be administered continuously via a nasogastric feeding tube. Specific therapy
_ IV Carnitine 100 mg/kg/day (Dilute to suitable volume with NS or G5W and infuse over 1 hour) _ Hydroxocobalamin 1mg/ IM ( for B12 responsive MMA only) _ Treat underlying cause. Treat infection, dehydration, electrolyte imbalance and acidosis _ Consider hemodialysis if with acute deterioration of cerebral function or if with intractable metabolic acidosis/ hyperammonemia. Also consider the following Check for amylase and lipase if suspecting pancreatitis. Be alert for signs and symptoms of cardiomyopathy _ If the patient has hyperammonemia, may give Sodium benzoate. References 1Hoffman GF and Schulze A. Chapter 7: Organic Acidurias in Sarafoglou K, Hoffman GF and Roth KS (eds). Pediatric Endocrinology and Inborn Errors of Metabolism. New York:McGraw Hill, 2009 pp 93-94. 2 Fingerhut R and Olgemoller B. Newborn screening for inborn errors of metabolism and endocrinopathies: an update. Anal Bioanal Chem. 2009; 393:1481-1497 3 Vockley J and Ensenauer R. Isovaleric academia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet 2006;142C(2):95-103. 4Gurnert SC, Wendel U, Linder M et al. Clinical and neurocognitive outcome in symptomatic isovaleric academia. Orphanet J Rar Dis 2012;7:9 5Chapter 3: Methylmalonic Acidemia. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 18-26. 6http://www.e-imd.org/rc/e-imd/htm/Article/2011/e-imd-20110728-195831-072/src/htm_fullText/fr/MethylmalonicAciduria.pdf Accessed Feb 25, 2012. 7 Cheng KH, Lie MY, Kao CH et al. Newborn screening for methylmalonic aciduria by tandem mass spectrometry: 7 years’ experience from two centers in Taiwan. J Chin Med Assoc 2010;73(6)314-319. 8Chapman KA and Summar ML. Propionic academia consensus conference summary. Mol Gen Metab 2011 article in press. 9 Chapter 2: Propionic academia. Nyhan WL, Barshop BA and Ozand P. Atlas of Metabolic Diseases 2nd ed. Great Britain:Oxford University Press, 2005 pp 8-15.
11 Pena L, Franks J, Chapman KA et al. Natural history of propionic academia. Mol Gen Metab 2011: article under press
11 Wendel U, de Baulny O. Branched chain organic acidurias/acidemias. Inborn Metabolic Diseases Chapter 19 4th edition eds Fernandes, Saudubray, van den Berghe, Walter pp 254-255
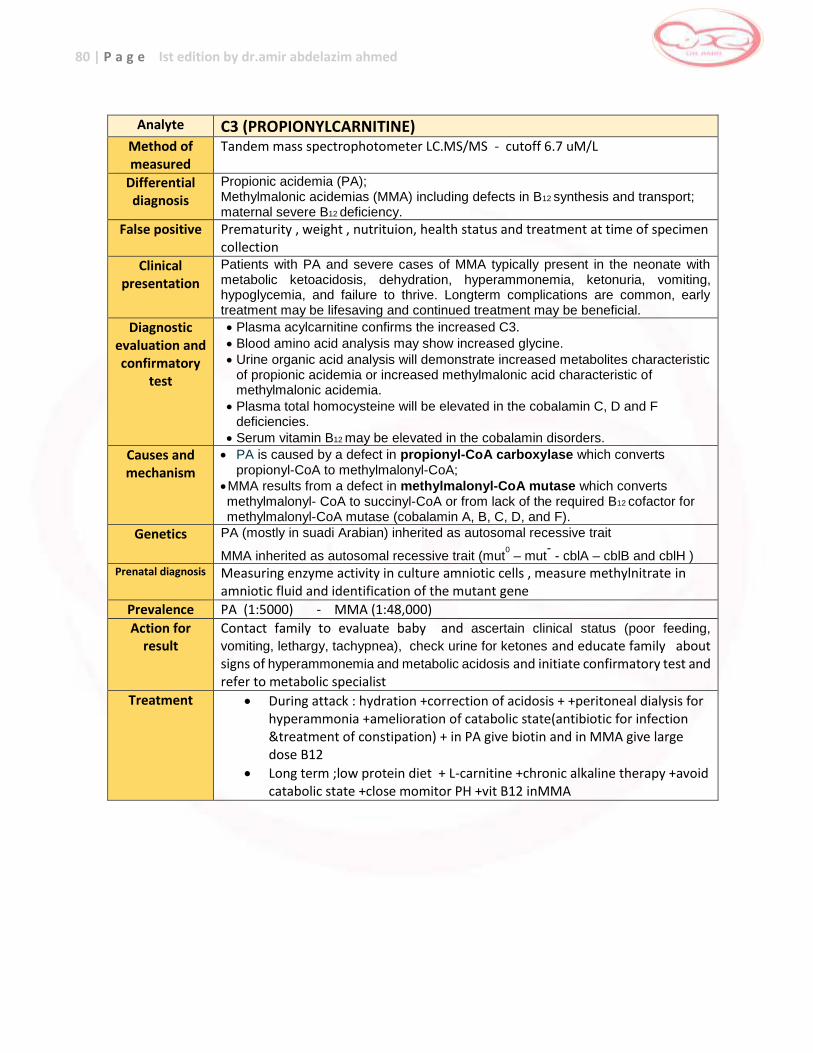
80 | P a g e Ist edition by dr.amir abdelazim ahmed
Analyte C3 (PROPIONYLCARNITINE)
Method of measured
Tandem mass spectrophotometer LC.MS/MS - cutoff 6.7 uM/L
Differential diagnosis
Propionic acidemia (PA); Methylmalonic acidemias (MMA) including defects in B12 synthesis and transport; maternal severe B12 deficiency.
False positive Prematurity , weight , nutrituion, health status and treatment at time of specimen collection
Clinical presentation
Patients with PA and severe cases of MMA typically present in the neonate with metabolic ketoacidosis, dehydration, hyperammonemia, ketonuria, vomiting, hypoglycemia, and failure to thrive. Longterm complications are common, early treatment may be lifesaving and continued treatment may be beneficial.
Diagnostic evaluation and confirmatory
test
Plasma acylcarnitine confirms the increased C3.
Blood amino acid analysis may show increased glycine.
Urine organic acid analysis will demonstrate increased metabolites characteristic of propionic acidemia or increased methylmalonic acid characteristic of methylmalonic acidemia.
Plasma total homocysteine will be elevated in the cobalamin C, D and F deficiencies.
Serum vitamin B12 may be elevated in the cobalamin disorders.
Causes and mechanism
PA is caused by a defect in propionyl-CoA carboxylase which converts propionyl-CoA to methylmalonyl-CoA;
MMA results from a defect in methylmalonyl-CoA mutase which converts methylmalonyl- CoA to succinyl-CoA or from lack of the required B12 cofactor for methylmalonyl-CoA mutase (cobalamin A, B, C, D, and F).
Genetics PA (mostly in suadi Arabian) inherited as autosomal recessive trait
MMA inherited as autosomal recessive trait (mut0 – mut
- - cblA – cblB and cblH )
Prenatal diagnosis Measuring enzyme activity in culture amniotic cells , measure methylnitrate in amniotic fluid and identification of the mutant gene
Prevalence PA (1:5000) - MMA (1:48,000)
Action for result
Contact family to evaluate baby and ascertain clinical status (poor feeding,
vomiting, lethargy, tachypnea), check urine for ketones and educate family about signs of hyperammonemia and metabolic acidosis and initiate confirmatory test and refer to metabolic specialist
Treatment During attack : hydration +correction of acidosis + +peritoneal dialysis for hyperammonia +amelioration of catabolic state(antibiotic for infection &treatment of constipation) + in PA give biotin and in MMA give large dose B12
Long term ;low protein diet + L-carnitine +chronic alkaline therapy +avoid catabolic state +close momitor PH +vit B12 inMMA

81 | P a g e Ist edition by dr.amir abdelazim ahmed
ENDOCRINE DISORDER CONGENITAL ADRENAL HYPERPLASIA [CAH]
CAH is a group of autosomal recessive disorders characterized by impaired cortisol synthesis. The most common form of CAH is caused by mutations in CYP21A2, the gene encoding the adrenal steroid 21-hydroxylase enzyme (P450c21) that accounts for approximately 95%of CAH. A cardinal feature of classic or severe virilizing CAH in newborn females is genital ambiguity. About 75% of classic CAH patients have aldosterone deficiency manifesting with salt wasting, failure to thrive, and potentially fatal hypovolemia and shock. Mild non-classic forms of CAH patients show variable degrees of postnatal androgen excess and may be asymptomatic.
Newborn Screening The screening method uses blood spot 17OHP levels by immunoassay. Figure 1 illustrates the steroidogenic pathway to demonstrate that plasma 17-OHP will elevate in CAH due to 21-hydroxylase deficiency (P450c21). Results of NBS for CAH The Attending Physician (AP) and NBS coordinator (NSF) should recall the baby within 24 hours notification of a positive CAH screen from the NSC long-term follow-up nurse.

82 | P a g e Ist edition by dr.amir abdelazim ahmed
Recommended Confirmatory Tests for CAH Serum 17-OHP (RIA) in accredited laboratories only Ancillary tests: Serum sodium and potassium Random blood glucose Chromosomal Analysis for those with ambiguous genitalia Pelvic Ultrasound for those with ambiguous genitalia Recommended Monitoring Schedule and tests (See appendix: Summary of Clinical and Biochemical Monitoring in CAH) 17-OHP Serum sodium and potassium Plasma rennin (if available) Recommended Treatment for CAH A. Medical
Acute adrenal crisis: Admit to pediatric intensive care setting (PICU or NICU) and co-manage with a pediatric endocrinologist.
1. Collect blood for confirmatory tests
2. Assess hydration and give IVF accordingly (D5 Normal Saline).
3. Monitor BP, blood glucose, plasma sodium, potassium and chloride.
4. Start IV stress dose of hydrocortisone. Neonates and infants 25 mg every 6 hours Children 50 mg every 6 hours

83 | P a g e Ist edition by dr.amir abdelazim ahmed
If not in adrenal crisis, start oral medications as follows: Hydrocortisone 10-15 mg/BSA in 2-3 divided doses Fludrocortisone 0.05- 0.1mg/day NaCl tablet: 1-2 gms/day (up to 4 gms/day) B. Surgical consultation is recommended for babies with ambiguous genitalia. C. Psychological support is recommended for affected patients and their families.
Duties of the Medical Persons Involved Attending Physician or Pediatrician
This doctor is in charge of the over-all care of the patient. He is responsible for the confirmation of CAH. He coordinates with the Pediatric endocrinologist, to ensure optimal care of the patient’s CAH. During the first patient visit (clinic or home visit), he should take the anthropometric measurements (weight, length, head circumference) and plot these on the WHO growth charts. It is important for him to assess if patient is in acute adrenal crisis (dehydrated, failing to thrive, hypotensive, low blood sugar, weak looking). If in acute crisis, admit and manage the immediate medical needs of the patient and refer to pediatric endocrinolo- gist for co-management. He should also obtain a complete history and PE. If there is any deviation from normal parameters of growth and development, further evaluation is warranted. All data should be recorded in the patient’s chart. Once the patient is stable, he schedules subsequent clinic or home visits as recommended below: 0-6 months: monthly 7-12 months: every 2 months 13-36 months: every 3 months after 3 years old: every 4-6 months It is the responsibility of the AP to make the necessary referrals to the pediatric endocrinologist and other specialists. Pediatric Endocrinologist
Oversees the comprehensive management (further diagnostics and prescriptions) of any patient referred for CAH. Interprets the confirmatory test results and makes the recommendations for treatment. Should evaluate the patient’s physical and biochemical parameters at the onset (diagnosis), and as follows: 0 -12 months: Every 3 months
12 months and onwards: Every 4-6 months
84 | P a g e Ist edition by dr.amir abdelazim ahmed
References 1. Speizer PW., Azziz R., Baskin LS., et al. Congenital Adrenal Hyperplasia due to Steroid 21 Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. JCEM 95:4133-4160, 2010. 2. Philippine Society of Pediatric Metabolism and Endocrinology (PSPME)’s Guidelines for the Management of Congenital Adrenal Hyperplasia drafted Nov 6, 2013 (unpublished).
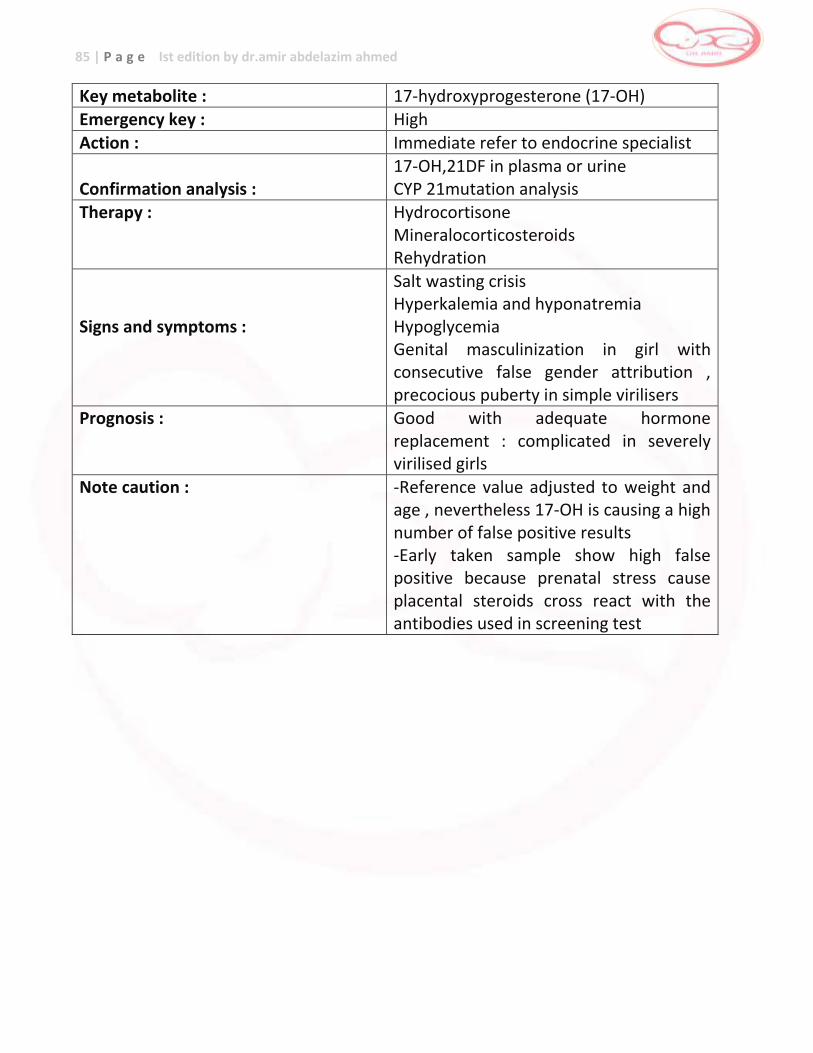
85 | P a g e Ist edition by dr.amir abdelazim ahmed
Key metabolite : 17-hydroxyprogesterone (17-OH) Emergency key : High
Action : Immediate refer to endocrine specialist Confirmation analysis :
17-OH,21DF in plasma or urine CYP 21mutation analysis
Therapy : Hydrocortisone Mineralocorticosteroids Rehydration
Signs and symptoms :
Salt wasting crisis Hyperkalemia and hyponatremia Hypoglycemia Genital masculinization in girl with consecutive false gender attribution , precocious puberty in simple virilisers
Prognosis : Good with adequate hormone replacement : complicated in severely virilised girls
Note caution : -Reference value adjusted to weight and age , nevertheless 17-OH is causing a high number of false positive results -Early taken sample show high false positive because prenatal stress cause placental steroids cross react with the antibodies used in screening test

86 | P a g e Ist edition by dr.amir abdelazim ahmed
CONGENITAL HYPOTHYROIDISM [CH]
Congenital Hypothyroidism (CH) occurs in1:3500-1:4000 screened newborns. The mental deficiency and poor physical growth in untreated CH are well recognized and this is precisely why newborn screening has been adopted to identify babies at risk for CH and to facilitate treatment before 2 weeks in order to prevent undesirable consequences. One parameter of a successful newborn screening program for CH is the mental outcome and functionality of the babies identified and treated. Hence, these recommendations were drafted as guidelines to help general practitioners ensure the comprehensive and optimal management of patients with CH. Newborn Screening primary TSH detection is the screening method. Newborn screening is best done on the 48th to 72nd hour. Before 48 hours, there is a risk of a falsely positive NBS due to the physiologic TSH surge. For premature babies, newborn screening should be done on day 7 of life or earlier if blood transfusion or exchange transfusion will be done. For term babies who require blood transfusion or exchange transfusion, newborn screening should be done before the procedure. Proper documentation of newborn screening and transport of the dried blood spot to the Newborn Screening Center (NSC) should be followed according to instructions in the National NBS Manual of Operations. Results of NBS for CH The Attending Physician (AP) and NBS coordinator (at the NSF) should recall the baby within 24 hours notification of a positive CH screen from the NSC long-term follow – up nurse. Recommended confirmatory tests for CH: Blood FT4 or T4, TSH (in accredited labs only ) Thyroid imaging: Tc 99 scan or ultrasound X-ray (AP) of the left knee and ankle for bone aging (optional) Recommended monitoring tests (biochemical parameters): Blood FT4 (preferred) or T4 and TSH Bone aging, if warranted Recommended treatment: L-thyroxine 10-15 mcg/kg/day Dose adjustments needed for the very low birth weights and premature. Please call
Pediatric Endocrinologist

87 | P a g e Ist edition by dr.amir abdelazim ahmed
Duties of the Medical Persons Involved Attending Physician or Pediatrician
This doctor is in charge of the over-all care of the patient. He is responsible for the confirmation of CH. He coordinates with the Pediatric endocrinologist, to ensure optimal care of the patient’s hypothyroidism. Dur- ing the first patient visit (clinic or home visit), he should take the anthropometric measurements (weight, length, head circumference) and plot these on the WHO growth charts. He should measure the length and width anterior fontanel. He should also obtain a complete history and PE. Any deviation deserves further evaluation. All data should be recorded in the patient’s chart. He schedules subsequent clinic or home visits as recommended below: 0-6 months: monthly 7-12 months: every 2 months 13-36 months: every 3 months after 3 years old: every 6 months It is the responsibility of the AP to make the necessary referrals to the pediatric endocrinologist and other specialists. Pediatric Endocrinologist
Oversees the comprehensive management (further diagnostics and prescriptions to include hearing evaluation at pre-school age) of any patient referred for CH. Interprets the confirmatory thyroid function results and makes the recommendations for treatment Should evaluate the patient’s physical and biochemical parameters at the onset or diagnosis (before 2 weeks), and as follows: 0 -12 months: Every 3-4 months 13-36 months: Every 6 months 36th month: Schedule re-evaluation of thyroid status. Yearly thereafter if permanent hypothyroidism is confirmed.

88 | P a g e Ist edition by dr.amir abdelazim ahmed
A. Normal FT4 and Normal TSH: No CH, No treatment B. Normal FT4 and High TSH: No treatment; Repeat FT4 and TSH after 2 weeks; correlate with clinical findings If repeat TSH elevated > 10 mU/L, treat with L-thyroxine; evaluation of thyroid function recommended at 3 years of age. If repeat TSH 6-10 mU/L, consult with Pediatric Endocrinologist. (Reference TSH at 2-6 weeks of age is 1.7-9.1 mU/L as per D. Fisher in Sperling, 2002) C. Low FT4 and High TSH: Primary CH confirmed. Treat with L-thyroxine D. Low FT4 and Normal TSH: Treat with L-thyroxine. Preterm baby and very sick neonates may also have this picture. The decision to treat will need a more extensive evaluation by the pediatric endocrinologist. E. Low FT4 and delayed elevation in TSH: Please work with a Pediatric Endocrinologist. F. Transient elevation in TSH: Treat if clinically indicated. Pediatric endocrinology consult recommended.
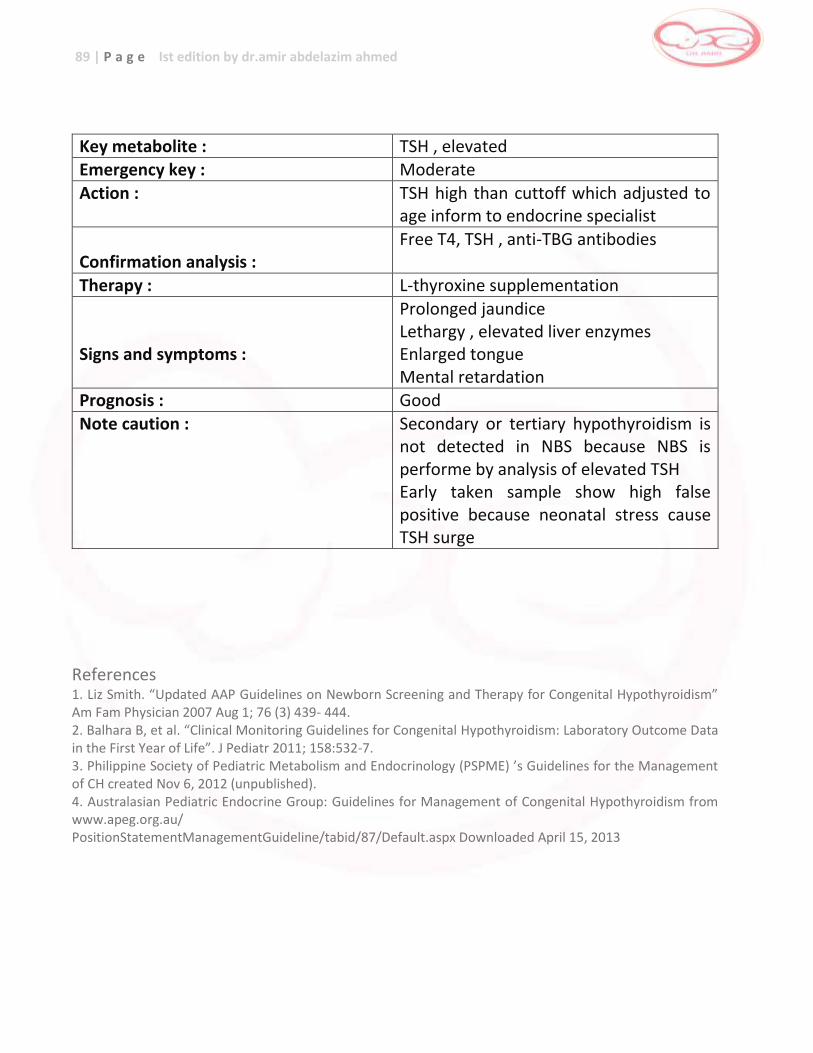
89 | P a g e Ist edition by dr.amir abdelazim ahmed
Key metabolite : TSH , elevated Emergency key : Moderate
Action : TSH high than cuttoff which adjusted to age inform to endocrine specialist
Confirmation analysis :
Free T4, TSH , anti-TBG antibodies
Therapy : L-thyroxine supplementation
Signs and symptoms :
Prolonged jaundice Lethargy , elevated liver enzymes Enlarged tongue Mental retardation
Prognosis : Good
Note caution : Secondary or tertiary hypothyroidism is not detected in NBS because NBS is performe by analysis of elevated TSH Early taken sample show high false positive because neonatal stress cause TSH surge
References 1. Liz Smith. “Updated AAP Guidelines on Newborn Screening and Therapy for Congenital Hypothyroidism” Am Fam Physician 2007 Aug 1; 76 (3) 439- 444. 2. Balhara B, et al. “Clinical Monitoring Guidelines for Congenital Hypothyroidism: Laboratory Outcome Data in the First Year of Life”. J Pediatr 2011; 158:532-7. 3. Philippine Society of Pediatric Metabolism and Endocrinology (PSPME) ’s Guidelines for the Management of CH created Nov 6, 2012 (unpublished). 4. Australasian Pediatric Endocrine Group: Guidelines for Management of Congenital Hypothyroidism from www.apeg.org.au/ PositionStatementManagementGuideline/tabid/87/Default.aspx Downloaded April 15, 2013

90 | P a g e Ist edition by dr.amir abdelazim ahmed
GALACTOSEMIA
What are Galactosemia? Galactosemia is a rare genetic metabolic disorder that is inherited in an autosomal recessive manner. It is an inborn error of carbohydrate metabolism characterized by elevated levels of galactose and its metabolites due to enzyme deficiencies involved in its metabolism. Galactose is the sugar found mainly in milk and dairy products. It is also produced by the body. Milk contains a sugar called lactose, and during digestion, lactose is broken down into the sugars glucose and galactose. Glucose can immediately be used as a source of energy by the body, but galactose needs to be further broken down before it can be utilized. Incidence : rare Pathophysiology The galactose metabolic pathway with multiple enzymatic steps is shown. The enzymes allow the subsequent conversion of galactose to galactose-1-phosphate by GALK (1); galactose-1-phosphate and uridine diphosphate glucose (UDP glucose) to glucose-1-phosphate and UDP-galactose by GALT (2) and the interconversion of UDP-glucose and UDP-galactose by GALE (3). Children with galactosemia have very little or entirely lack an enzyme that helps the body break down galactose. There are three different enzyme problems that can lead to galactosemia. In the first type or classic galactosemia, the enzyme that is reduced or missing is called galactose-1-phosphate uridyl transferase (GALT). The GALT enzyme enables the body to break down galactose into glucose. The second type of galactosemia is due to a deficiency in uridine diphosphate galactose 4- epimerase (GALE). Its severe type clinically resembles classic galactosemia. The third type, is due to a deficiency in galactokinase (GALK), and presents primarily as cataracts in untreated patients. Clinical Features Patients can present with feeding problems, failure to thrive, hepatocellular damage, bleeding, and sepsis in untreated infants. In approximately 10% of individuals, cataracts are present. Failure to thrive is the most common initial clinical symptom of classic galactosemia. Vomiting or diarrhea usually begins within a few days of milk ingestion. Jaundice of intrinsic liver disease may be accentuated by the severe hemolysis occurring in some patients. Cataracts have been observed within a few days of birth. There appears to be a high frequency of neonatal death due to E. coli sepsis in patients with classic galactosemia. The association of jaundice and hemorrhagic diathesis in the first 2 weeks of life is a clinical presentation in which galactosemia must be considered. Coagulopathy may also be present in galactosemia with little evidence of liver disease. Galactosemia also
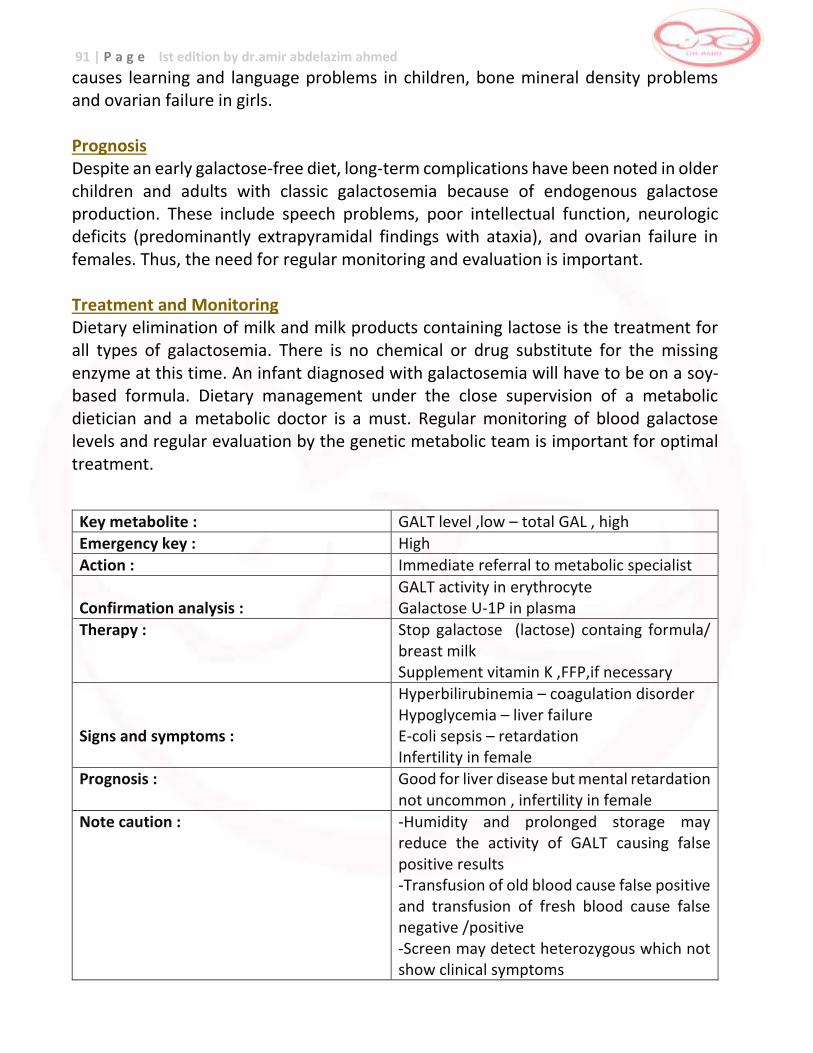
91 | P a g e Ist edition by dr.amir abdelazim ahmed
causes learning and language problems in children, bone mineral density problems and ovarian failure in girls. Prognosis Despite an early galactose-free diet, long-term complications have been noted in older children and adults with classic galactosemia because of endogenous galactose production. These include speech problems, poor intellectual function, neurologic deficits (predominantly extrapyramidal findings with ataxia), and ovarian failure in females. Thus, the need for regular monitoring and evaluation is important. Treatment and Monitoring Dietary elimination of milk and milk products containing lactose is the treatment for all types of galactosemia. There is no chemical or drug substitute for the missing enzyme at this time. An infant diagnosed with galactosemia will have to be on a soy-based formula. Dietary management under the close supervision of a metabolic dietician and a metabolic doctor is a must. Regular monitoring of blood galactose levels and regular evaluation by the genetic metabolic team is important for optimal treatment.
Key metabolite : GALT level ,low – total GAL , high
Emergency key : High
Action : Immediate referral to metabolic specialist
Confirmation analysis :
GALT activity in erythrocyte Galactose U-1P in plasma
Therapy : Stop galactose (lactose) containg formula/ breast milk Supplement vitamin K ,FFP,if necessary
Signs and symptoms :
Hyperbilirubinemia – coagulation disorder Hypoglycemia – liver failure E-coli sepsis – retardation Infertility in female
Prognosis : Good for liver disease but mental retardation not uncommon , infertility in female
Note caution : -Humidity and prolonged storage may reduce the activity of GALT causing false positive results -Transfusion of old blood cause false positive and transfusion of fresh blood cause false negative /positive -Screen may detect heterozygous which not show clinical symptoms

92 | P a g e Ist edition by dr.amir abdelazim ahmed
Diagram of invalid specimens