familial cholestasis: progressive familial intrahepatic cholestasis, benign recurrent intrahepatic...
TRANSCRIPT

Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553
Contents lists available at ScienceDirect
Best Practice & Research ClinicalGastroenterology
2
Familial cholestasis: Progressive familial intrahepaticcholestasis, benign recurrent intrahepatic cholestasis andintrahepatic cholestasis of pregnancy
Wendy L. van der Woerd, MD, Paediatric Resident a,b,*,Saskia W.C. van Mil, PhD, Assistant Professor b,Janneke M. Stapelbroek, MD, PhD, Paediatric Resident a,b,Leo W.J. Klomp, PhD, Associate Professor b,Stan F.J. van de Graaf, PhD, Assistant Professor b,Roderick H.J. Houwen, MD, PhD, Consultant Paediatric Gastroenterologist a
aDepartment of Paediatric Gastroenterology (KE.01.144.3), Wilhelmina Children’s Hospital, University Medical Centre Utrecht,Post-Box 85090, 3508 AB Utrecht, The NetherlandsbDepartment of Metabolic and Endocrine Diseases, University Medical Centre Utrecht, Utrecht, The Netherlands
Keywords:Progressive familial intrahepatic cholestasisPFICBenign recurrent intrahepatic cholestasisBRICIntrahepatic cholestasis of pregnancyICPATP8B1ABCB11ABCB4
* Corresponding author. Department of PaediatriMedical Centre Utrecht, Post-Box 85090, 3508 AB
E-mail addresses: [email protected] (W.Lumcutrecht.nl (J.M. Stapelbroek), [email protected]@umcutrecht.nl (R.H.J. Houwen).
1521-6918/$ – see front matter � 2010 Elsevier Ltdoi:10.1016/j.bpg.2010.07.010
Progressive familial intrahepatic cholestasis (PFIC) type1, 2 and3aredue tomutations in ATP8B1, ABCB11 and ABCB4, respectively. Each ofthese genes encodes a hepatocanalicular transporter, which isessential for the proper formation of bile. Mutations in ABCB4 canresult in progressive cholestatic disease, while mutations in ATP8B1and ABCB11 can result both in episodic cholestasis, referred to asbenign recurrent intrahepatic cholestasis (BRIC) type 1 and2, aswellas in progressive cholestatic disease. This suggests a clinicalcontinuumand these diseases are therefore preferably referred to asATP8B1 deficiency and ABCB11 deficiency. Similarly PFIC type 3 isdesignated as ABCB4 deficiency. Heterozygous mutations in each ofthese transporters can also be associated with intrahepatic chole-stasis of pregnancy. This review summarizes the pathophysiology,clinical features and current aswell as future therapeutic options forprogressive familial- and benign recurrent intrahepatic cholestasisas well as intrahepatic cholestasis of pregnancy.
� 2010 Elsevier Ltd. All rights reserved.
c Gastroenterology (KE.01.144.3), Wilhelmina Children’s Hospital, UniversityUtrecht, The Netherlands. Tel.: þ31 88 7555555; fax: þ31 88 7555342.. van der Woerd), [email protected] (S.W.C. van Mil), [email protected] (L.W.J. Klomp), [email protected] (S.F.J. van de Graaf),
d. All rights reserved.

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553542
Introduction
Familial intrahepatic cholestasis is a heterogeneous group of autosomal recessive liver disorderscharacterized by intrahepatic cholestasis, which can be divided in three main groups based onphenotypical differences: progressive familial intrahepatic cholestasis (PFIC), benign recurrentintrahepatic cholestasis (BRIC) and intrahepatic cholestasis of pregnancy (ICP). PFIC can be sub-divided in three types with slightly different clinical, biochemical and histological features, asso-ciated with mutations in ATP8B1 (PFIC1), ABCB11 (PFIC2) and ABCB4 (PFIC3) [1–3]. A smallproportion of PFIC phenotypes are not due to mutations in these three genes and therefore addi-tional genes might be involved. Mutations in ATP8B1 and ABCB11 can also result in the less severephenotype of BRIC type 1 and 2, respectively, while heterozygous mutations in all three genes areassociated with ICP. Occasionally the benign variant (BRIC) will progress to the more severe andpermanent form of intrahepatic cholestasis (PFIC), indicative of a clinical continuum, with inter-mediate phenotypes between mild and progressive disease [4–6]. Therefore these diseases arepreferably referred to as ATP8B1 deficiency and ABCB11 deficiency. Likewise, ABCB4 deficiency isused instead of PFIC3. The most prominent characteristics of these three genetic subtypes aresummarized in Table 1.
Progressive familial intrahepatic cholestasis and benign recurrent intrahepatic cholestasis
ATP8B1 deficiency
AetiologyATP8B1 deficiency is an autosomal recessive disease caused by mutations in ATP8B1 encoding
ATP8B1 (formerly designated as FIC1), a P-type ATPase. ATP8B1 is abundantly expressed in a widevariety of tissues such as the small intestine, bladder and stomach and to a lesser extent also in theliver and pancreas. It is localized on the apical membrane of epithelial cells, including the cana-licular membrane of hepatocytes [7–9]. The function of this P-type ATPase is not totally clear, butATP8B1 appears to be no bile salt transporter itself. The most widely accepted hypothesis forATP8B1 function is that of an aminophospholipid flippase, translocating phospholipids such asphosphatidylserine from the outer to the inner leaflet of the plasma membrane. In addition,a flippase-independent function of ATP8B1 in apical membrane organization was suggestedrecently [9]. Deficiency of ATP8B1 in the hepatocyte may result in the loss of asymmetric distri-bution of phospholipids in the canalicular membrane, decreasing both membrane stability andfunction of transmembrane transporters including the bile salt export pump, ABCB11 and, as such,causing cholestasis [10,11]. Extrahepatic manifestations such as the hearing loss, pancreatitis anddiarrhoea, found in patients with ATP8B1 deficiency, suggest that perturbations in cellularmembranes and/or a secondarily impaired function of transmembrane transporters can also befound in other organs [9,12–14].
To date, over 50 distinct mutations in ATP8B1 are described [15–17]. The mutations G308V found inAmish, D554N found in Inuits and I661T are amongst the most frequently detected. The severity ofATP8B1 deficiency varies from benign remitting to progressive disease. A high variability in pheno-typic presentation exists even in patients with the same mutation, so type and location of themutation correlates only partially with the severity of clinical disease. Nevertheless mutations pre-dicted to affect protein expression or function severely, such as nonsense and frameshift mutations,are more often detected in progressive disease, while missense mutations are more frequentlyidentified in benign disease, possibly as a result of residual activity of ATP8B1 [16]. Recently genotype-phenotype correlations were further clarified by investigating the effects of several ATP8B1mutationson protein expression, localization and function. In vitro, it turned out that for common missensemutations such as G308V, D554N and I661T, ATP8B1 deficiency can be regarded as a protein foldingdisease, with different degrees of retention of the mutant protein in the endoplasmic reticulum,resulting in a decreased protein expression at the plasma membrane. Incubation at a reducedtemperature could improve proper folding of some of the mutated proteins. Similarly, the
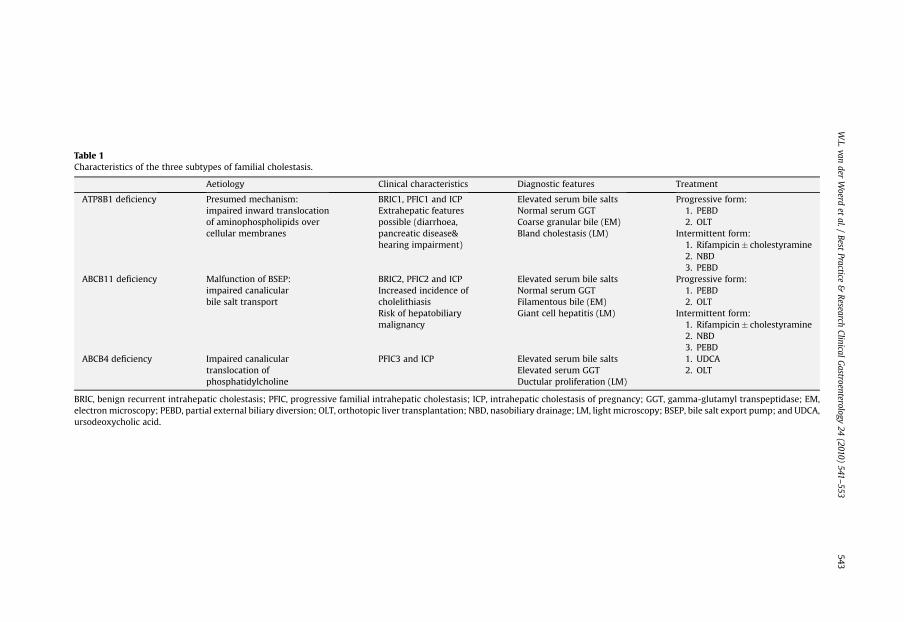
Table 1Characteristics of the three subtypes of familial cholestasis.
Aetiology Clinical characteristics Diagnostic features Treatment
ATP8B1 deficiency Presumed mechanism:impaired inward translocationof aminophospholipids overcellular membranes
BRIC1, PFIC1 and ICPExtrahepatic featurespossible (diarrhoea,pancreatic disease&hearing impairment)
Elevated serum bile saltsNormal serum GGTCoarse granular bile (EM)Bland cholestasis (LM)
Progressive form:1. PEBD2. OLT
Intermittent form:1. Rifampicin� cholestyramine2. NBD3. PEBD
ABCB11 deficiency Malfunction of BSEP:impaired canalicularbile salt transport
BRIC2, PFIC2 and ICPIncreased incidence ofcholelithiasisRisk of hepatobiliarymalignancy
Elevated serum bile saltsNormal serum GGTFilamentous bile (EM)Giant cell hepatitis (LM)
Progressive form:1. PEBD2. OLT
Intermittent form:1. Rifampicin� cholestyramine2. NBD3. PEBD
ABCB4 deficiency Impaired canaliculartranslocation ofphosphatidylcholine
PFIC3 and ICP Elevated serum bile saltsElevated serum GGTDuctular proliferation (LM)
1. UDCA2. OLT
BRIC, benign recurrent intrahepatic cholestasis; PFIC, progressive familial intrahepatic cholestasis; ICP, intrahepatic cholestasis of pregnancy; GGT, gamma-glutamyl transpeptidase; EM,electronmicroscopy; PEBD, partial external biliary diversion; OLT, orthotopic liver transplantation; NBD, nasobiliary drainage; LM, light microscopy; BSEP, bile salt export pump; and UDCA,ursodeoxycholic acid.
W.L.van
derWoerd
etal./
BestPractice
&Research
ClinicalGastroenterology
24(2010)
541–553
543

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553544
pharmacological chaperone 4-phenylbutyrate acid (4-PBA) could stabilize misfolded proteins,partially restoring cell surface expression [18,19].
Clinical characteristicsATP8B1 deficiency can present as persistent cholestasis with severe pruritus, generally in infancy
(PFIC1; formerly also Byler disease and Greenland familial cholestasis) or with episodic cholestasis atany age (BRIC1). Some BRIC1 patients show progression of the disease, though most patients continueto have intermittent cholestasis of variable duration, without evident liver damage and generally evenwith a reduction in the frequency of cholestatic attacks at an older age [4,5,12,16]. In patients withBRIC1, factors inducing a cholestatic episode are unclear, although minor infections, fever, oralcontraceptives and pregnancy are all reported by patients as triggering an attack [12,20]. In vitro, highertemperature is indeed associated with a significant decreased expression of mutant ATP8B1 at theplasma membrane, while oestrogens and progesterone metabolites seem to have the ability to inducetransinhibition of the bile salt export pump which might exacerbate the accumulation of bile salts[19,21].
Patients with the progressive form most often present with unremitting cholestasis in the first yearof life, which, without treatment, will result in cirrhosis and ultimately liver failure. Complications offat malabsorption resulting in fat-soluble vitamin deficiency can occur, such as bleeding diathesis(vitamin K), rickets (vitamin D), neuromuscular abnormalities (vitamin E) and weight-loss, althoughthese are even more often encountered in ABCB11 deficiency [3,4]. Poor growth can be persisting,despite more than adequate caloric intake. Similarly, other extrahepatic manifestations can beobserved in patients with ATP8B1 deficiency, such as diarrhoea, pancreatitis and hearing loss, allconsistent with expression of ATP8B1 in these tissues [12,14,22].
Diagnostic featuresCharacteristic for ATP8B1 deficiency but also for ABCB11 deficiency (described in the next para-
graph) is the combination of a low serum gamma-glutamyl transpeptidase (GGT) with profoundcholestasis, the latter manifestating itself as a high serum bile salt level and pruritus. Although genetictesting of the corresponding genes currently remains the only strategy to accurately discriminatebetween ATP8B1 deficiency and ABCB11 deficiency, recent studies showed some biochemical andhistological differences that can help distinguish these two entities [3,23]. For instance, serumaspartate aminotransaminase (ASAT), alanine aminotransaminase (ALAT) and bile salt levels aregenerally less elevated in patients with ATP8B1 deficiency and serum albumin levels are often lower[3,23,24]. On light microscopy hepatic architecture initially tends to be only minimally perturbed, buteventually fibrosis and cirrhosis evolves. Furthermore, intracanalicular cholestasis with coarselygranular bile on electron microscopy seems to be specific for ATP8B1 deficiency [25,26].
ABCB11 deficiency
AetiologyABCB11 deficiency is an autosomal recessive disease caused by mutations in the ABCB11 gene
encoding the bile salt export pump (BSEP), a liver-specific adenosine triphosphate (ATP)-bindingcassette transporter [27,28]. BSEP is localized solely at the canalicular membrane of hepatocytes. Itactively transports conjugated bile salts into biliary canaliculi against an extreme concentrationgradient, thereby generating bile flow. Malfunction of ABCB11 results in an isolated defect in the bilesalt excretion across the canalicular membrane, leading to cholestasis.
Over 100 mutations are now identified [29,30]. In more than half of the European families themissense mutations E297G and/or D482G are present. As in ATP8B1 deficiency, a clinical continuumfrommild to progressive phenotypes is observed. Generally missense mutations, e.g. E297G or D482G,lead to a less severe phenotype than mutations that are predicted to result in premature proteintruncation or total failure of protein production [6,23,29]. At a functional level, mutations in ABCB11 canimpair taurocholate transport function or BSEP protein processing, but can also lead to aberrant pre-messenger RNA splicing resulting in reduced levels of normal messenger RNA [31–33]. In vitro, theresidual transport function of mutant proteins correlates with the phenotypic differences between

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553 545
BRIC2 and PFIC2, with generally a diminished function in BRIC2 mutants, while complete abolishmentis more often seen in PFIC2 mutants [32].
Clinical characteristicsPatients generally present in infancy with permanent cholestasis (PFIC2; formerly sometimes
designated as Byler syndrome), which, without treatment, will result in liver cirrhosis and ultimately inliver failure. Alternatively, patients can present with episodic cholestasis (BRIC2), although the relativeproportion of BRIC patients is lower compared to ATP8B1 deficiency [6,34]. At diagnosis, the cholestasisin ABCB11 deficiency results more often in a detectable deficiency of fat-soluble vitamins [3]. Extra-hepatic symptoms are not found in ABCB11 deficiency, consistent with the specific localization of BSEPat the canalicular membrane. Another distinguishing feature from ATP8B1 deficiency is the develop-ment of cholelithiasis in approximately one third of the patients, probably due to the low bile saltconcentration in bile, secondary to impaired BSEP function, which might cause supersaturation ofcholesterol [3,6,23]. Furthermore, patients with ABCB11 deficiency are at a considerable risk for hep-atobiliary malignancy. The mechanism of carcinogenesis, specifically associated with ABCB11 defi-ciency, is unknown. Up to 15% of the patients with ABCB11 deficiency will develop hepatocellularcarcinoma or cholangiocarcinoma [3,23,29, 35,36]. Patients with two predicted protein-truncatingmutations seem to be at particular risk [29]. Close surveillance for hepatobiliary malignancy istherefore warranted in these patients.
Diagnostic featuresWhen an infant presents with low GGT cholestasis and high serum bile salts, the diagnosis of either
PFIC type 1 or 2 is likely. As mentioned earlier, only genetic investigation of the corresponding genescan confirm this diagnosis. Nevertheless at presentation some biochemical and histological featuresare more frequently seen in ABCB11 deficiency. For example ASAT, ALAT and bile salt levels tend to behigher in these patients, with ALAT usually being more than five times the upper limit of normal atpresentation in most of the patients with ABCB11 deficiency. Also serum alphafoetoprotein concen-trations are elevated in the majority of the patients in contrast to those with ATP8B1 deficiency.Histology shows a neonatal hepatitis-like patternwith giant or multinucleate cells, as opposed to blandcholestasis in ATP8B1 deficiency, while on electron microscopic investigation, canalicular bile isamorphous or finely filamentous, which can further help to distinguish ABCB11 from ATP8B1 defi-ciency [23,25,26]. Furthermore, immunohistochemical analysis for BSEP in liver tissue of patients withABCB11 deficiency will reveal absent or reduced BSEP staining in the majority of patients [29,37].However, normal BSEP expression does not exclude BSEP deficiency as some mutations only affect thebile salt transport activity.
ABCB4 deficiency
AetiologyABCB4 deficiency is an autosomal recessive disease caused by mutations in the ABCB4 gene,
encoding the class III multidrug resistance P-glycoprotein (MDR3), an ATP-binding cassette trans-porter. It is localized at the canalicular membrane of hepatocytes functioning as a phospholipidfloppase, translocating phosphatidylcholine (PC) from the inner to the outer leaflet of themembrane, where it is available for extraction into the canalicular lumen by bile salts. In thisprocess mixed micelles will be formed, which protect the membranes of the cells facing the biliarytree against the detergent properties of bile salts [38]. Consequently, diminished or absent MDR3activity causes a disbalance in the primary bile composition with a lack of PC and a surplus ofdetergent bile salts, leading to solubilization of the membranes of especially the biliary epithelium,inducing cell death and inflammation [38,39].
Over 45 disease-causing mutations in ABCB4 are now identified [39–41]. Children with missensemutations seem to have a less severe phenotype, with later onset of disease, slower progression andbetter response to treatment, as compared to patients with mutations leading to a truncated protein[1]. Possibly this is due to residual transport activity in MDR3 protein affected by missense mutations.In more than half of the patients with ABCB4 deficiency immunohistochemistry shows faint or absent

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553546
staining of canalicular MDR3 protein, as can be seen when the protein is partially retained in theendoplasmic reticulum due to a folding defect [42]. Normal MDR3 staining is observed in somepatients, indicating preserved expression and targeting, and suggesting that some mutations onlycause decreased transporter activity [37]. Heterozygous mutations in the ABCB4 gene can also cause orpredispose for a variety of other liver diseases, such as adult biliary cirrhosis, cholelithiasis, transientneonatal cholestasis, drug induced cholestasis and intrahepatic cholestasis of pregnancy [43]. Muta-tions can even lead to a cascade of several phenotypes in one patient, indicating the wide phenotypicalspectrum of ABCB4 deficiency [44].
Clinical characteristicsPatients with ABCB4 deficiency usually present with jaundice as a result of profound intrahepatic
cholestasis. Hepatomegaly, and at later stages splenomegaly, as a manifestation of portal hypertensionis often observed. Mean age at first symptoms is 34 months of age (1 month till 20 years) [1]. Withouttreatment, the persistent cholestasis usually progresses to hepatic failure before adulthood. In a fewcases, cholestasis was initially recurrent but rapidly became permanent [1,39]. Biliary lithiasis isdescribed in a few patients, but also in some parents, probably due to an increased lithogenicity of bileresulting in crystal formation. No extrahepatic features or an association with malignancies aredescribed [1].
Diagnostic featuresSerum bile salts, ALAT, ASAT and bilirubin are elevated and, unlike ATP8B1 and ABCB11 deficiency,
the serum GGT is elevated. Liver histology shows portal inflammation with strong ductular prolifer-ation at early stages, despite patency of intrahepatic and extrahepatic bile ducts [1,45]. At later stagesa typical picture of biliary cirrhosis is found. Absent or diminished immunohistochemal staining forMDR3 can help in diagnosing these patients, although it can be normal in up to half of the patients withmutations in ABCB4, limiting its diagnostic value [37,39]. Only a genetic diagnosis, i.e. finding thedisease-causing mutations, is conclusive.
Treatment and outcome
Medical therapyUnfortunately, most forms of medical therapy for ATP8B1 deficiency, ABCB11 deficiency and ABCB4
deficiency are of limited effectiveness. Nevertheless, several treatment modalities can be used inspecific patients to improve quality of life or prevent progression of disease.
Cholestyramine is a hydrophilic, water insoluble anion-exchange resin that binds bile salts, pre-venting their re-absorption in the enterohepatic circulation. In the progressive forms of familialcholestasis, relief of pruritus and normalization of biochemical parameters is only described rarelywith cholestyramine. However, in patients with intermittent cholestasis it can be helpful in shorteningepisodes [24,46,47]. Although rifampicin accelerates the hepatic detoxification and excretion ofcompounds, such as bilirubin and bile salts, it has been usedwith disappointing results in patients withprogressive cholestasis [24]. Nevertheless, in patients with intermittent episodes of cholestasis it cancompletely abort an episode [5,48].
Ursodeoxycholic acid (UDCA) is a relatively hydrophilic bile salt, which is less cytotoxic thanendogenous bile salts. Upon oral administration, it will partially replace endogenous bile salts in thebile salt pool, supposedly reducing injury of the hepatocytes during cholestasis. In ABCB4 deficiencychronic administration of UDCA normalizes liver function tests and improves clinical parameters in upto 50% of the patients [1, 49]. The therapeutic effect appears to be dependent on type of mutation, withpremature stop codons leading to a truncated protein being associated with nearly no response totherapy [1]. UDCA should therefore be first choice in the initial therapeutic management of patientswith ABCB4 deficiency, especially when a missense mutation in the corresponding gene is found.In this respect the C23-homologue of UDCA (norUDCA) might even be more potent than UDCA [50]. Inpatients with ATP8B1 or ABCB11 deficiency, the results of UDCA treatment are conflicting, ranging fromclear improvement to no effect at all [3,5,13,20,24].

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553 547
Invasive therapyIf no complete clinical or biochemical improvement is obtainedwithmedical therapy,more invasive
therapy such as biliary diversion or even liver transplantation is necessary.Biliary diversion reduces the accumulation of toxic bile salts by interruption of the enter-
ohepatic circulation, decreasing re-uptake. Partial biliary diversion (PBD) interrupts the enter-ohepatic circulation of bile salts by partially diverting bile from the gallbladder through a loop ofjejunum connecting the gallbladder to the abdominal skin (partial external biliary diversion: PEBD)or colon (partial internal biliary diversion: PIBD) [51,52]. In ileal bypass (IB) the terminal ileum isskipped by an ileocolonic anastomosis. Results of especially PEBD are promising with respect topruritus, jaundice and histology, both in patients with ATP8B1 and ABCB11 deficiency, with at leastpartial improvement in more than 75% of the patients [53]. Although this seems promising, atpresent it is unclear whether in patients responding to PEBD liver transplantation can also beavoided at long-term follow-up [54–57]. Moreover, some patients do not benefit from biliarysurgery at all. Obviously in these patients liver transplantation should be considered [56,57]. Thetype of mutation seems to be associated with the outcome of PEBD, with better prognosis indisease caused by milder mutations, especially for the ABCB11 mutations E297G and D482G [3,23].However, when severe fibrosis is already present at the moment of PEBD, prognosis is worse[54,57]. Only one patient with ABCB4 deficiency who underwent PEBD was described in literature;this patient showed no improvement [58]. No serious PEBD complications are reported, althoughproblems with the stoma (stenosis, recurrent bleeding) sometimes make a re-operation necessary.In addition excessive stomal losses can cause dehydration and electrolyte imbalance, while chol-angitis can also develop [53,55,56]. After IB symptoms may recur within one year requiringconversion to PEBD [59].
The permanent character of the PEBD makes it less suitable for patients with episodic cholestasis(BRIC). In these patients a temporary nasobiliary drain to interrupt the enterohepatic circulation(nasobiliary drainage, NBD) can be endoscopically introduced. This procedure is effective in most ofthese patients, resolving pruritus and normalising bile salts within two days [60].
If all previously described therapies fail in controlling pruritus, when severe liver cirrhosis is presentat diagnosis, or when the disease is progressive despite treatment, orthotopic liver transplantation(OLT) remains the only alternative [61,62]. Although OLT is associated with serious surgical risks andlifetime immunosuppressive therapy is necessary, it usually gives complete correction of phenotype inpatients with ABCB11 and ABCB4 deficiency. However, phenotypic recurrence of severe ABCB11 defi-ciency post-transplantation can occur as a result of the formation of autoantibodies against ABCB11[63,64]. Intensifying immunosuppressive therapy may resolve this problem. In some patients withATP8B1 deficiency, diarrhoea can become manifest or more prominent after liver transplantation. Thismight be due to an imbalance between bile salt excretion and re-absorption, since the hepatic graftexcretes a normal amount of bile salts, whereas the intestine remains functionally impaired. Theresulting increased amount of bile salts in the ileum and colon induces or worsens diarrhoea, whichmight respond to cholestyramine treatment [13,22]. Steatohepatitis post-transplantation is alsodescribed.
In conclusion, the proposed treatment strategy is to perform PEBD rapidly after diagnosis in patientswith progressive forms of ATP8B1 or ABCB11 deficiency and to consider OLT when treatment fails. Inpatients with ABCB4 deficiency UDCA treatment is the first-line therapy, if not successful followed byliver transplantation. In patients with episodic cholestasis medical treatment with rifampicin with orwithout cholestyramine can be attempted at the start of an attack. If medication is not successful inaborting the cholestatic episode NBD can be performed. In BRIC patients who progress to a morepermanent form of cholestasis, or in patients with very frequent or debilitating attacks, a PBD can beconsidered (Table 1) [65].
Future therapyAlternative therapies such as hepatocyte transplantation, the use of nuclear receptor ligands and
mutation-specific therapy are currently under investigation. Hepatocyte transplantation has beensuccessful in partially repopulating the liver, diminishing pathology in a mouse model of ABCB4deficiency, but unfortunately not yet in patients [66]. In ABCB11 deficiency it is doubtful

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553548
whether hepatocyte transplantation is a good therapeutic option since possible premalignant cells areleft in place.
Certain nuclear receptors regulate bile formation. The key nuclear receptor in bile formation is thebile salt sensor farnesoid X receptor (FXR). Activated FXR transactivates a number of genes, resulting inimproved bile salt excretion and detoxification. Targeting FXR with synthetic ligands is explored asa possible therapeutic option for cholestasis syndromes [67].
The development of mutation-specific pharmacological therapy is also making rapid progress. Forexample PTC124 (Ataluren�), that selectively induces ribosomal read through of premature termina-tion codons caused by nonsense mutations, is currently under investigation in cystic fibrosis patients[68]. Pharmacological chaperones such as 4-phenylbutyrate acid (4-PBA) have been shown to stabilizeproteins misfolded due to missense mutations, thereby preventing degradation in the endoplasmicreticulum. In vitro, 4-PBA actually enhances cell surface protein expression for some of the missensemutations found in ATP8B1 deficiency and ABCB11 deficiency [19,69]. Finally, the ability to modulateaberrant splicing is currently being investigated [33].
Intrahepatic cholestasis of pregnancy
Aetiology
Intrahepatic cholestasis of pregnancy (ICP) is a transient form of cholestasis, characterized by theonset of pruritus during pregnancy, with postnatal resolution. The aetiology of ICP is complex withendocrine, genetic and environmental components. Reproductive hormones are thought to play a rolesince ICP is more common in twin pregnancies and the disease usually starts in the third trimester,when hormone concentrations are higher. In addition, 27% of women with a history of obstetriccholestasis experience either cyclical or oral contraceptive-induced pruritus [70]. The role of repro-ductive hormones in cholestasis may be explained by their inhibiting influence on the major bile aciduptake transporter Naþ-taurocholate co-transporting polypeptide (NTCP) as well as on bile salt exportvia BSEP [21,71,72].
Furthermore, evidence of a genetic predisposition exists, since ICP was described in the heterozy-gous mothers of children with familial cholestasis. Also familial clustering and an increased incidencein some geographical regions suggest that genetic, in addition to environmental factors, are important.Recent data indeed show that mutations or polymorphisms in genes expressing hepatobiliary trans-port proteins or their nuclear regulators may contribute to the development and/or severity of ICP.Mutations in ABCB4 were the first to be described in familial intrahepatic cholestasis of pregnancy[39,73]. Since then additional ICP-associated genomic variants have been identified and a recent studyshowed that as much as 16% of Caucasian patients bear ABCB4 gene mutations [74–79]. Significantdifferences in haplotype frequencies between pregnant women with and without ICP are alsodescribed [75,77]. Variation in ATP8B1 however occurs only in a small number of ICP cases, indicatingthat it probably is not a major gene contributing to the occurrence of ICP [80,81]. Conversely, variationin ABCB11 can contribute to susceptibility of ICP and heterozygosity for common ABCB11 mutationsaccounts for at least 1% of European ICP cases [82,83]. In addition, the common BSEP polymorphismV444A also is a risk factor. Finally, ABCC2 encodingmultidrug resistance associated protein (MRP2) andNR1H4 encoding FXR are also implicated in the pathogenesis of ICP [84,85]. Genetic testing of theABCB4 gene and ongoing follow-up should be considered, at least in ICP patients who have a familyhistory of a cholestatic disorder, since heterozygous mutations in this gene can also predispose fora variety of other liver diseases [43,86].
The aetiology of the foetal complications associated with ICP is poorly understood but thought torelate to an accumulation of bile salts in the foetal compartment with different effects on various organsystems, for instance the foetal myocard, resulting in arrhythmia [87].
Clinical characteristics and diagnostic features
The main presenting symptom of ICP is pruritus, usually without clinical jaundice, becoming moresevere as the pregnancy advances and resolving after delivery. Most of the affected women present in

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553 549
the third trimester [70,88]. Characteristically serum bile salt concentration is increased, generally withsome elevation of serum ASAT, ALAT and alkaline phosphatase. In contrast, serum GGT is normal oronly slightly elevated [88]. ICP is associated with an increased incidence of foetal distress andprematurity, probably with a positive correlation between foetal complication rates and maternal bilesalt levels [70,89,90]. In addition, unexplained intrauterine death occurs more often in ICP patients,especially after 37 weeks of gestation [91].
Treatment and outcome
ICP is a condition of great discomfort and associated with serious foetal risks. Therefore, theaim of treatment is to reduce maternal symptoms and prevent foetal complications. For anextensive overview of management options we refer to a recent review of ICP [91]. Insummary, the most effective currently available pharmacological therapy seems to be UDCA.Although not officially approved for use during pregnancy, available data suggest that it is safeand effective in improving maternal symptoms and biochemical abnormalities [92,93].Furthermore UDCA might even diminish foetal problems [94]. A combination therapy of S-adenosyl-L-methionine (SAMe) and UDCA may have a synergistic effect [95]. Intensified foetalmonitoring unfortunately cannot prevent intrauterine death. Although not supported byrandomised clinical trials, current literature encourages the induction of labour between 37and 38 weeks of gestation with the aim of reducing the incidence of intrauterine death inwomen with ICP [96].
After delivery, the biochemical abnormalities normally resolve within 2–8 weeks, but recurrence ofcholestasis in subsequent pregnancies is likely and is also described on the use of oral contraceptives.Follow-up to ensure normalization of liver test is advised, and even further follow-up needs to beconsidered, as ICP is associated with the development of liver disease later in life, e.g. liver cirrhosis,gallstones and cholecystitis [97].
Summary
Familial cholestasis can be subdivided into three subtypes, formerly designated as PFIC type 1, 2and 3, each with slightly different clinical, biochemical and histological features and associated withmutations in ATP8B1, ABCB11 and ABCB4, respectively. These genes all encode hepatocanaliculartransporters, and the corresponding diseases are now generally referred to as ATP8B1 deficiency,ABCB11 deficiency and ABCB4 deficiency. The exact function of ATP8B1 remains unclear, buta function as an aminophospholipid flippase, seems likely. Deficiency of ATP8B1 may result in lossof the asymmetric phospholipid distribution in the plasma membrane, decreasing membranestability and the function of transmembrane transporters, including the bile salt export pump.Mutations in ABCB11 can result in an isolated defect in bile salt transport. MDR3, encoded by ABCB4,functions as a phospholipid floppase, translocation phosphatidylcholine. Defects result in anincreased toxicity of detergent bile salts. Mutations in these three genes can cause progressive liverdisease, but mutations in ATP8B1 and ABCB11 can result in episodic cholestasis too. Subtypes offamilial cholestasis can be distinguished by GGT concentration which is only elevated in ABCB4deficiency. Furthermore, extrahepatic symptoms are almost exclusively seen in ATP8B1 deficiency,while cholelithiasis is more often observed in ABCB11 deficiency, which is also associated withhepatobiliary malignancies. Histological data can further discriminate, as usually bland cholestasisis seen in ATP8B1 deficiency, giant cell hepatitis in ABCB11 deficiency and ductular proliferation inABCB4 deficiency. Genetic testing remains essential to discriminate between these entities. Medicaltherapy is only effective in a specific subgroup of patients and at present invasive therapy as biliarydiversion and liver transplantation is inevitable in most patients. Heterozygous mutations inATP8B1, ABCB11 and ABCB4 are also implicated in the pathogenesis of ICP, a transient form ofcholestasis with onset during the third trimester of pregnancy, which is triggered by environmentaland hormonal factors in genetically susceptible women. ICP is associated with adverse foetaloutcomes and therefore medical treatment with UDCA and induction of labour around 37 weeks ofgestation is common practice.

Research agenda
� Further elucidation of ATP8B1 function.� Developing mutation-specific pharmacological therapies.� Determine long-term prognosis after biliary diversion, especially in relation to the mutationinvolved.
� Performing randomised controlled trials to define the most effective management strategy inpreventing foetal adverse outcomes in ICP.
Clinical practice points
� Three subtypes of familial intrahepatic cholestasis are due to mutations in hepatocanaliculartransporters.
� Detection of disease-associated mutations may clarify aetiology and influence choice oftherapy.
� At present invasive therapy such as biliary diversion or liver transplantation is the onlyeffective therapy inmost patients. Medical therapy can be attempted in a specific subgroup ofpatients.
W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553550
Conflict of interest
None declared.
References
*[1] Jacquemin E, de Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, et al. The wide spectrum of multidrug resistance 3deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001;120:1448–58.
[2] van Mil SW, Houwen RH, Klomp LW. Genetics of familial intrahepatic cholestasis syndromes. J Med Genet 2005;42:449–63.
*[3] Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 childrenwith normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differencesbetween PFIC1 and PFIC2 and natural history. Hepatology 2010;51:1645–55.
[4] van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin LiverDis 2001;21:535–44.
[5] van Ooteghem NA, Klomp LW, van Berge-Henegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis pro-gressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol 2002;36:439–43.
[6] van Mil SW, van der Woerd WL, van der Brugge G, Sturm E, Jansen PL, Bull LN, et al. Benign recurrent intrahepaticcholestasis type 2 is caused by mutations in ABCB11. Gastroenterology 2004;127:379–84.
[7] Ujhazy P, Ortiz D, Misra S, Li S, Moseley J, Jones H, et al. Familial intrahepatic cholestasis 1: studies of localization andfunction. Hepatology 2001;34:768–75.
[8] van Mil SW, van Oort MM, van den Berg IT, Berger R, Houwen RH, Klomp LW. Fic1 is expressed at apical membranes ofdifferent epithelial cells in the digestive tract and is induced in the small intestine during postnatal development of mice.Pediatr Res 2004;56:981–7.
[9] Verhulst P, van der Velden L, Oorschot V, van Faassen EE, Klumperman J, Houwen RH, et al. A flippase-independentfunction of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical proteinexpression and microvillus formation in polarized epithelial cells. Hepatology 2010;51:2049–60.
[10] Cai SY, Gautam S, Nguyen T, Soroka CJ, Rahner C, Boyer JL. ATP8B1 deficiency disrupts the bile canalicular membranebilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology 2009;136:1060–9.
[11] Paulusma CC, de Waart DR, Kunne C, Mok KS, Oude Elferink RP. Activity of the bile salt export pump (ABCB11) is criticallydependent on canalicular membrane cholesterol content. J Biol Chem 2009;284:9947–54.

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553 551
[12] Tygstrup N, Steig BA, Juijn JA, Bull LN, Houwen RH. Recurrent familial intrahepatic cholestasis in the Faeroe Islands.Phenotypic heterogeneity but genetic homogeneity. Hepatology 1999;29:506–8.
[13] Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, et al. Progressive familial intrahepatic cholestasis type1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosisafter liver transplantation. J Hepatol 2003;39:447–52.
[14] Stapelbroek JM, Peters TA, van Beurden DH, Curfs JH, Joosten A, Beynon AJ, et al. ATP8B1 is essential for maintainingnormal hearing. Proc Natl Acad Sci U S A 2009;106:9709–14.
[15] Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, et al. A gene encoding a P-type ATPase mutated in twoforms of hereditary cholestasis. Nat Genet 1998;18:219–24.
*[16] Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ, et al. Characterization of mutations inATP8B1 associated with hereditary cholestasis. Hepatology 2004;40:27–38.
[17] Liu LY, Wang XH, Wang ZL, Zhu QR, Wang JS. Characterization of ATP8B1 gene mutations and a hot-linked mutation foundin Chinese children with progressive intrahepatic cholestasis and low GGT. J Pediatr Gastroenterol Nutr 2010;50:179–83.
[18] Folmer DE, van der Mark VA, Ho-Mok KS, Oude Elferink RP, Paulusma CC. Differential effects of progressive familialintrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localizationof ATP8B1. Hepatology 2009;50:1597–605.
*[19] van der Velden LM, Stapelbroek JM, Krieger E, van den Berghe PV, Berger R, Verhulst PM, et al. Folding defects in P-typeATP8B1 associated with hereditary cholestasis are ameliorated by 4-phenylbutyrate. Hepatology 2010;51:286–96.
[20] Brenard R, Geubel AP, Benhamou JP. Benign recurrent intrahepatic cholestasis. A report of 26 cases. J Clin Gastroenterol1989;11:546–51.
[21] Vallejo M, Briz O, Serrano MA, Monte MJ, Marin JJ. Potential role of trans-inhibition of the bile salt export pump byprogesterone metabolites in the etiopathogenesis of intrahepatic cholestasis of pregnancy. J Hepatol 2006;44:1150–7.
[22] Egawa H, Yorifuji T, Sumazaki R, Kimura A, Hasegawo M, Tanaka K. Intractable diarrhea after liver transplantation forByler’s disease: successful treatment with bile adsorptive resin. Liver Transpl 2002;8:714–6.
[23] Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Emerick K, Antoniou A, et al. Differences in presentation andprogression between severe FIC1 and BSEP deficiencies. J Hepatol 2010;53:170–8.
[24] Whitington PF, Freese DK, Alonso EM, Schwarzenberg SJ, Sharp HL. Clinical and biochemical findings in progressivefamilial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr 1994;18:134–41.
[25] Bull LN, Carlton VE, Stricker NL, Baharloo S, DeYoung JA, Freimer NB, et al. Genetic and morphological findings inprogressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): evidence for heterogeneity.Hepatology 1997;26:155–64.
[26] Knisely AS. Progressive familial intrahepatic cholestasis: a personal perspective. Pediatr Dev Pathol 2000;3:113–25.[27] Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. A gene encoding a liver-specific ABC transporter is
mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998;20:233–8.[28] Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, et al. The sister of P-glycoprotein represents the cana-
licular bile salt export pump of mammalian liver. J Biol Chem 1998;273:10046–50.*[29] Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerová D, Rayner A, Dutton L, et al. Severe bile salt export pump defi-
ciency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008;134:1203–14.[30] Treepongkaruna S, Gaensan A, Pienvichit P, Luksan O, Knisely AS, Sornmayura P, et al. Novel ABCB11 mutations in a Thai
infant with progressive familial intrahepatic cholestasis. World J Gastroenterol 2009;15:4339–42.[31] Wang L, Dong H, Soroka CJ, Wei N, Boyer JL, Hochstrasser M, et al. Degradation of the bile salt export pump at endo-
plasmic reticulum in progressive familial intrahepatic cholestasis type II. Hepatology 2008;48:1558–69.[32] Kagawa T, Watanabe N, Mochizuki K, Numari A, Ikeno Y, Itoh J, et al. Phenotypic differences in PFIC2 and BRIC2 correlate
with protein stability of mutant Bsep and impaired taurocholate secretion in MDCK II cells. Am J Physiol GastrointestLiver Physiol 2008;294:G58–67.
*[33] Byrne JA, Strautnieks SS, Ihrke G, Pagani F, Knisely AS, Linton KJ, et al. Missense mutations and single nuclotide poly-morphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing.Hepatology 2009;49:553–67.
[34] Lam CW, Cheung KM, Tsui MS, Yan MS, Lee CY, Tong SF. A patient with novel ABCB11 gene mutations with phenotypictransition between BRIC2 and PFIC2. J Hepatol 2006;44:240–2.
[35] Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, Portmann BC, et al. Hepatocellular carcinoma in ten childrenunder five years of age with bile salt export pump deficiency. Hepatology 2006;44:478–86.
[36] Scheimann AO, Strautnieks SS, Knisely AS, Byrne JA, Thompson RJ, Finegold MJ. Mutations in bile salt export pump(ABCB11) in two children with progressive familial cholestasis and cholangiocarcinoma. J Pediatr 2007;150:556–9.
[37] Keitel V, Burdelski M, Warskulat U, Kühlkamp T, Keppler D, Häussinger D, et al. Expression and localization of hep-atobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology 2005;41:1160–72.
[38] Oude Elferink RP, Paulusma CC. Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). PflugersArch 2007;453:601–10.
[39] de Vree JM, Jacqemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, et al. Mutations in the MDR3 gene cause progressive familialintrahepatic cholestasis. Proc Natl Acad Sci U S A 1998;95:282–7.
[40] Deleuze JF, Jacquemin E, Dubuisson C, Cresteil D, Dumont M, Erlinger S, et al. Defect of multidrug-resistance 3 geneexpression in a subtype of progressive familial intrahepatic cholestasis. Hepatology 1996;23:904–8.
[41] Degiorgio D, Colombo C, Seia M, Porcaro L, Costantino L, Zazzeron L, et al. Molecular characterization and structuralimplications of 25 new ABCB4 mutations in progressive familial intrahepatic cholestasis type 3 (PFIC3). Eur J Hum Genet2007;15:1230–8.
[42] Delaunay JL, Durand-Schneider AM, Delautier D, Rada A, Gautherot J, Jacquemin E, et al. A missense mutation in ABCB4gene involved in progressive familial intrahepatic cholestasis type 3 leads to a folding defect that can be rescued by lowtemperature. Hepatology 2009;49:1218–27.
[43] Gonzales E, Davit-Spraul A, Baussan C, Buffet C, Maurice M, Jacquemin E. Liver diseases related to MDR3 (ABCB4) genedeficiency. Front Biosci 2009;14:4242–56.

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553552
[44] Lucena JF, Herrero JI, Quiroga J, Sangro B, Garcia-Foncillas J, Zabalegui N, et al. A multidrug resistance 3 gene mutationcausing cholelithiasis, cholestasis of pregnancy, and adulthood biliary cirrhosis. Gastroenterology 2003;124:1037–42.
[45] Maggiore G, Bernard O, Hadchouel M, Lemonnier A, Alagille D. Diagnostic value of serum gamma-glutamyl trans-peptidase activity in liver diseases in children. J Pediatr Gastroenterol Nutr 1991;12:21–6.
[46] Kurbegov AC, Setchell KD, Haas JE, Mierau GW, Narkewicz M, Bancroft JD, et al. Biliary diversion for progressive familialintrahepatic cholestasis: improved liver morphology and bile acid profile. Gastroenterology 2003;125:1227–34.
[47] Uegaki S, Tanaka A, Mori Y, Kodama H, Fukusato T, Takikawa H, et al. Successful treatment with colestimide for a bout ofcholestasis in a Japanese patient with benign recurrent intrahepatic cholestasis caused by ATP8B1 mutation. Intern Med2008;47:599–602.
[48] Cancado EL, Leitao RM, Carrilho FJ, Laudanna AA. Unexpected clinical remission of cholestasis after rifampicin therapy inpatients with normal or slightly increased levels of gamma-glutamyl transpeptidase. Am J Gastroenterol 1998;93:1510–7.
[49] Jacquemin E, Hermans D, Myara A, Habes D, Debray D, Hadchouel M, et al. Ursodeoxycholic acid therapy in pediatricpatients with progressive familial intrahepatic cholestasis. Hepatology 1997;25:519–23.
[50] Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, et al. 24-norUrsodeoxycholic acid issuperior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroen-terology 2006;130:465–81.
[51] Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated withintrahepatic cholestasis. Gastroenterology 1988;95:130–6.
[52] Bustorff-Silva J, Sbraggia Neto L, Olimpio H, de Alcantara RV, Matsushima E, De Tommaso AM, et al. Partial internal biliarydiversion through a cholecystojejunocolonic anastomosis – a novel surgical approach for patients with progressivefamilial intrahepatic cholestasis: a preliminary report. J Pediatr Surg 2007;42:1337–40.
[53] Ismail H, Kalicinski P, Markiewicz M, Jankowska I, Pawlowska J, Kluge P, et al. Treatment of progressive familial intra-hepatic cholestasis: liver transplantation or partial external biliary diversion. Pediatr Transplant 1999;3:219–24.
[54] Emond JC, Whitington PF. Selective surgical management of progressive familial intrahepatic cholestasis (Byler’s disease).J Pediatr Surg 1995;30:1635–41.
[55] Ekinci S, Karnak I, Gurakan F, Yüce A, Senocak ME, Cahit Tanyel F, et al. Partial external biliary diversion for the treatmentof intractable pruritus in children with progressive familial intrahepatic cholestasis: report of two cases. Surg Today2008;38:726–30.
[56] Arnell H, Bergdahl S, Papadogiannakis N, Nemeth A, Fischler B. Preoperative observations and short-term outcome afterpartial external biliary diversion in 13 patients with progressive familial intrahepatic cholestasis. J Pediatr Surg 2008;43:1312–20.
[57] Yang H, Porte RJ, Verkade HJ, De Langen ZJ, Hulscher JB. Partial external biliary diversion in children with progressivefamilial intrahepatic cholestasis and Alagille disease. J Pediatr Gastroenterol Nutr 2009;49:216–21.
[58] Wanty C, Joomye R, van Hoorebeek N, Paul K, Otte JB, Reding R, et al. Fifteen years single center experience in themanagement of progressive familial intrahepatic cholestasis of infancy. Acta Gastroenterol Belg 2004;67:313–9.
[59] Kalicinski PJ, Ismail H, Jankowska I, Kaminski A, Pawlowska J, Drewniak T, et al. Surgical treatment of progressive familialintrahepatic cholestasis: comparison of partial external biliary diversion and ileal bypass. Eur J Pediatr Surg 2003;13:307–11.
[60] Stapelbroek JM, van Erpecum KJ, Klomp LW, Venneman NG, Schwartz TP, van Berge Henegouwen GP, et al. Nasobiliarydrainage induces long-lasting remission in benign recurrent intrahepatic cholestasis. Hepatology 2006;43:51–3.
[61] Torri E, Lucianetti A, Pinelli D, Corno V, Guizzetti M, Maldini G, et al. Orthotopic liver transplantation for Byler’s disease.Transplant Proc 2005;37:1149–50.
[62] Englert C, Grabhorn E, Richter A, Rogiers X, Burdelski M, Ganschow R. Liver transplantation in children with progressivefamilial intrahepatic cholestasis. Transplantation 2007;84:1361–3.
[63] Keitel V, Burdelski M, Vojnisek Z, Schmitt L, Häussinger D, Kubitz R. De novo bile salt transporter antibodies as a possiblecause of recurrent graft failure after liver transplantation: a novel mechanism of cholestasis. Hepatology 2009;50:510–7.
[64] Jara P, Hierro L, Martinez-Fernandez P, Alvarez-Doforno R, Yánez F, Diaz MC, et al. Recurrence of bile salt export pumpdeficiency after liver transplantation. N Engl J Med 2009;361:1359–67.
*[65] Stapelbroek JM, van Erpecum KJ, Klomp LW, Houwen RH. Liver disease associated with canalicular transport defects:current and future therapies. J Hepatol 2010;52:258–71.
[66] de Vree JM, Ottenhoff R, Bosma PJ, Smith AJ, Aten J, Oude Elferink RP. Correction of liver disease by hepatocyte trans-plantation in a mouse model of progressive familial intrahepatic cholestasis. Gastroenterology 2000;119:1720–30.
[67] Zollner G, Trauner M. Nuclear receptors as therapeutic targets in cholestatic liver diseases. Br J Pharmacol 2009;156:7–27.[68] Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by
nonsense mutations. Nature 2007;447:87–91.[69] Hayashi H, Sugiyama Y. 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type
and mutated bile salt export pumps. Hepatology 2007;45:1506–16.[70] Williamson C, Hems LM, Goulis DG, Walker I, Chambers J, Donaldson O, et al. Clinical outcome in a series of cases of
obstetric cholestasis identified via a patient support group. BJOG 2004;111:676–81.[71] Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, Meier PJ. Drug- and estrogen-induced cholestasis through inhibition of
the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology 2000;118:422–30.[72] Abu-Hayyeh S, Martinez-Becerra P, Sheik Abdul Kadir SH, Selden C, Romero MR, Rees M, et al. Inhibition of Na+-taur-
ocholate co-transporting polypeptide mediated bile acid transport by cholestatic sulphated progesterone metabolites.J Biol Chem 2010;285:16504–12.
[73] Jacquemin E, Cresteil D, Manouvrier S, Boute O, Hadchouel M. Heterozygous non-sense mutation of the MDR3 gene infamilial intrahepatic cholestasis of pregnancy. Lancet 1999;353:210–1.
[74] Dixon PH, Weerasekera N, Linton KJ, Donaldson O, Chambers J, Egginton E, et al. Heterozygous MDR3 missense mutationassociated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genet 2000;9:1209–17.
[75] Mullenbach R, Linton KJ, Wiltshire S, Weerasekera N, Chambers J, Elias E, et al. ABCB4 gene sequence variation in womenwith intrahepatic cholestasis of pregnancy. J Med Genet 2003;40:e70.

W.L. van der Woerd et al. / Best Practice & Research Clinical Gastroenterology 24 (2010) 541–553 553
[76] Pauli-Magnus C, Lang T, Meier Y, Zidan-Marin T, Jung D, Breymann C, et al. Sequence analysis of bile salt exportpump (ABCB11) and multi drug resistance p-glycoprotein 3 (ABCB4, MDR3) in patients with intrahepatic cholestasisof pregnancy. Parmacogenetics 2004;14:91–102.
[77] Wasmuth HE, Glantz A, Keppeler H, Simon E, Bartz C, RathW, et al. Intrahepatic cholestasis of pregnancy: the severe formis associated with common variants of the hepatobiliary phospholipid transporter ABCB4 gene. Gut 2007;56:265–70.
[78] Floreani A, Carderi I, Paternoster D, Soard G, Azzaroli F, Esposito W, et al. Hepatobiliary phospholipid transporter ABCB4,MDR3 gene variants in a large cohort of Italian womenwith intrahepatic cholestasis of pregnancy. Dig Liver Dis 2008;40:366–70.
[79] Bacq Y, Gendrot C, Perrotin F, Lefrou L, Chrétien S, Vie-Buret V, et al. ABCB4 gene mutations and single-nucleotidepolymorphisms in women with intrahepatic cholestasis of pregnancy. J Med Genet 2009;46:711–5.
[80] Mullenbach R, Bennett A, Tetlow N, Patel N, Hamilton G, Cheng F, et al. ATP8B1 mutations in British cases with intra-hepatic cholestasis of pregnancy. Gut 2005;54:829–34.
[81] Painter JN, Savander M, Ropponen A, Nupponen N, Riikonen S, Ylikorkala O, et al. Sequence variation in the ATP8B1 geneand intrahepatic cholestasis of pregnancy. Eur J Hum Genet 2005;13:435–9.
[82] Meier Y, Zodan T, Lang C, Zimmermann R, Kullak-Ublick GA, Meier PJ, et al. Increased susceptibility for intrahepaticcholestasis of pregnancy and contraceptive-induced cholestasis in carriers of the 1331T > C polymorphism in the bile saltexport pump. World J Gastroenterol 2008;14:38–45.
[83] Dixon PH, van Mil SW, Chambers J, Strautnieks S, Thompson RJ, Lammert F, et al. Contribution of variant alleles of ABCB11to susceptibility to intrahepatic cholestasis of pregnancy. Gut 2009;58:537–44.
[84] van Mil SW, Milona A, Dixon PH, Mullenbach R, Geenes V, Chambers J, et al. Functional variants of the bile acid sensorFXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology 2007;133:507–16.
[85] Sookoian S, Castano G, Burgueno A, Gianotti TF, Pirola CJ. Association of the multidrug-resistance-associated protein gene(ABCC2) variants with intrahepatic cholestasis of pregnancy. J Hepatol 2008;48:125–32.
[86] Hardikar W, Kansal S, Oude Elferink RP, Angus P. Intrahepatic cholestasis of pregnancy: when should you look further?World J Gastroenterol 2009;15:1126–9.
[87] Sheikh Abdul Kadir SH, Miragoli M, Abu-Hayyeh S, Moshkov AV, Xie Q, Keitel V, et al. Bile acid-induced arrhythmia ismediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS One 2010;15:e9689.
[88] Bacq Y, Sapey T, Brechot MC, Pierre F, Fignon A, Dubois F. Intrahepatic cholestasis of pregnancy: a French prospectivestudy. Hepatology 1997;26:358–64.
[89] Davies MH, da Silva RC, Jones SR, Weaver JB, Elias E. Fetal mortality associated with cholestasis of pregnancy and thepotential benefit of therapy with ursodeoxycholic acid. Gut 1995;37:580–4.
*[90] Glantz A, Marschall HU, Mattson LA. Intrahepatic cholestasis of pregnancy: relationships between bile acid levels andfetal complication rates. Hepatology 2004;40:467–74.
[91] Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol 2009;15:2049–66.[92] Diaferia A, Nicastri PL, Tartagni M, Loizzi P, Iacovizzi C, Di Leo A. Ursodeoxycholic acid therapy in pregnant women with
cholestasis. Int J Gynaecol Obstet 1996;52:133–40.[93] Palma J, Reyes H, Ribalta J, Hernández I, Sandoval L, Almuna R, et al. Ursodeoxycholic acid in the treatment of cholestasis
of pregnancy: a randomized, double-blind study controlled with placebo. J Hepatol 1997;27:1022–8.[94] Zapata R, Sandoval L, Palma J, Hernández I, Ribalta J, Reyes H, et al. Ursodeoxycholic acid in the treatment of intrahepatic
cholestasis of pregnancy. A 12-year experience. Liver Int 2005;25:548–54.[95] Nicastri PL, Diaferia A, Tartagni M, Loizzi P, Fanelli M. A randomised placebo-controlled trial of ursodeoxycholic
acid and S-adenosylmethionine in the treatment of intrahepatic cholestasis of pregnancy. Br J Obstet Gynaecol1998;105:1205–7.
[96] Mays JK. The active management of intrahepatic cholestasis of pregnancy. Curr Opin Obstet Gynecol 2010;22:100–3.*[97] Ropponen A, Sund R, Riikonen S, Ylikorkala O, Aittomäki K. Intrahepatic cholestasis of pregnancy as an indicator of liver
and biliary diseases: a population-based study. Hepatology 2006;43:723–8.