forensic stain identification by rt-pcr analysis (updated)
TRANSCRIPT

The author(s) shown below used Federal funds provided by the U.S. Department of Justice and prepared the following final report: Document Title: Forensic Stain Identification By RT-PCR
Analysis (Updated) Author: Trisha Conti, Ph.D., Eric Buel, Ph.D. Document No.: 236537
Date Received: November 2011 Award Number: 2007-DN-BX-K149 This report has not been published by the U.S. Department of Justice. To provide better customer service, NCJRS has made this Federally-funded grant final report available electronically in addition to traditional paper copies.
Opinions or points of view expressed are those
of the author(s) and do not necessarily reflect the official position or policies of the U.S.
Department of Justice.

FORENSIC STAIN IDENTIFICATION BY RT-PCR ANALYSIS
Award Number: 2007-DN-BX-K149
Trisha Conti, PhD and Eric Buel, PhD
ABSTRACT
With the advent of innovative molecular biological techniques becoming the norm in the forensic
laboratory, it is plausible to imagine the eventual replacement of traditional serological testing
methods used to identify questioned stains with molecular biological techniques. New tests that
are tissue-specific and designed to be multiplexed would yield rapid results on a minimal amount
of sample. Such testing could employ mRNA as the tissue-specific determinant by testing for
the appropriate tissue-specific mRNAs. Analyses can also be performed to demonstrate that
mRNA is relatively stable and can thus be of great use in a wide variety of forensic cases. The
nature of this research was to identify mRNA transcripts that will definitively identify the tissue
of origin, determine if such transcripts survive the typical environmental insults that forensic
samples may encounter, and to develop rapid multiplex assays to assess these molecules using
small amounts of sample. A crucial prerequisite to these analyses is the development of a
DNA/RNA co-extraction method to minimize sample requirements and eliminate the need for
two separate extractions. Through collaboration with Promega, a RNA/DNA co-isolation
technique was developed which effectively extracts both nucleic acids of sufficient quality and
quantity for downstream real-time PCR and STR analyses. The stability of RNA over time was
established using real-time PCR assays. Two separate technologies were used to multiplex
assays once candidates were shown to be tissue-specific. The Plexor® One-Step qRT-PCR
1

System was used to design a semen-saliva multiplex assay in collaboration with Promega.
Unfortunately, Promega has decided to discontinue development of this assay. Lastly,
homebrew TaqMan® assays were developed for semen-sperm identification as well as a brain
screening assay.
2

TABLE OF CONTENTS
ITEM PAGE(S)
ABSTRACT 1-2
EXECUTIVE SUMMARY 4-16
MAIN BODY 17-87
• Introduction 17-25
o Statement of the Problem 17-18
o Literature Citations and Review 18-23
o Rationale for the Research 23-25
• Methods 25-34
• Results 35-66
o Statement of Results 35-52
o Tables 53-63
o Figures 64-66
• Conclusions 67-79
o Discussion of Findings 67-74
o Implications for Policy and Practice 75-76
o Implications for Further Research 76-79
• References 80-85
• Dissemination of Research Findings 86-87
3

EXECUTIVE SUMMARY
Identification of the tissue origin of the suspect DNA is often an important issue in forensic
casework, which may also aid in predicting the success of DNA analysis. Therefore, it is
paramount to the criminal justice system that suspect stains be identified definitively and
accurately. Currently, the serological approaches for stain identification involve enzymatic or
immunologic tests. While these tests have improved in selectivity over the years, several
problems still exist such as the possibility of cross-reactivity with other species and the lack of
specificity for particular tissues. In addition, there are several tissues for which no such tests are
available.
New tests that are fluid/tissue-specific and designed to be multiplexed would yield rapid results
on a minimal amount of sample. Such testing could employ mRNA as the specific determinant.
While the DNA of all tissues from an individual is essentially identical, the mRNA spectrum
made by the different cells in each tissue is very different. Each tissue or cell type makes a
unique constellation of mRNAs, some specific for only that tissue or cell type. Some body
fluids, such as blood, contain cells as part of their function while other fluids, such as urine,
contain cells that have been shed from their tissue of origin. Therefore, analysis of the “RNA
profile” in a sample can uniquely identify the fluid or tissue of origin.
As the demand for sample analysis increases, forensic laboratories continue to balance
manpower and cost issues versus the value of the analysis when evaluating new techniques. In
order for laboratories to invest in RNA technology, they will require a straight-forward
extraction procedure. A method that could co-purify RNA and DNA from a single sample with a
4

minimal number of steps would be attractive to those seeking new technologies. A preferred
extraction method would demonstrate the best ability to co-isolate DNA and RNA in terms of
yield and amplifiability while remaining simple, efficient, and ideally, involve non-hazardous
reagents.
The thrust of this research was to identify mRNA transcripts that will definitively identify the
tissue of origin and to develop rapid multiplex assays to assess these molecules using small
amounts of sample. The proposal had the following goals: 1) to select the best method of
DNA/RNA co-extraction from a wide variety of stain types, 2) to find 2-3 genes specific for
numerous stain and tissue types, 3) to develop multiplex assays for the identification of tissue
and stain types, 4) to validate these assays for forensic casework, and 5) to disseminate these
results to the forensic community.
Our first goal was to identify the best method to co-extract DNA and RNA from a variety of
stain types. By utilizing one extraction step, a DNA sample would be ready and waiting for STR
profiling if the RNA screening assay deemed it worthy of such analysis. In addition, obtaining
RNA and DNA from a single stain would prevent the possibility of conclusions being drawn
regarding the identity of one stain which may not hold true for a nearby stain. Therefore, a
significant amount of time was spent optimizing a procedure which would co-extract the two
nucleic acids so that they were of sufficient quality and quantity for downstream analyses.
Based on preliminary experiments, the TRIzol® method was identified as an efficient and
straight-forward procedure for the isolation of DNA and RNA. Despite the success of the
TRIzol® extractions, there are several disadvantages which led us to actively seek an alternative
5

isolation procedure. Although the TRIzol® reagent has the capacity to isolate RNA and DNA,
this requires essentially two extraction pathways following the first initial steps. Therefore, we
sought procedures which would be less labor-intensive and yield sufficient amounts of high-
quality nucleic acids. Through collaboration with Promega we developed a homebrew extraction
method that would work for RNA and DNA. The optimization experiments for this Tris-based
phenol method were detailed in the Final Report for 2004-DN-BX-K002. Although this method
involves numerous hands-on steps, it is faster than the TRIzol® method, and produces
significantly better DNA yields. Under the current award, we performed further studies to
compare the DNA and RNA yields generated from the co-isolation method with the gold-
standard organic method used by our laboratory for casework samples. The yields were fairly
comparable and more than sufficient to perform downstream STR amplification. Further studies
are required to assess the quality of the isolated DNA by STR analysis.
Lastly, we sought to determine whether the Tris-based phenol process could be modified to
handle mixtures of male and female DNA. Currently, most labs use a differential lysis procedure
to achieve this separation. Because the Tris-based phenol protocol as previously described
provides a single extract containing all DNA, we adapted the Tris-based phenol process to
produce epithelial and sperm extracts. The results demonstrate that RNA survives the initial 2-
hour incubation with female extraction buffer and the DNA yields were sufficient to perform
downstream analysis (i.e. STRs). Together these results indicate that the adapted Tris-based
phenol procedure may be a viable extraction method for samples containing both male and
female components. Further research is necessary to compare the quality and quantity of the
nucleic acids isolated via the adapted method with the conventional procedures for DNA and
RNA extraction (i.e. differential method, Tris-based phenol).
6

The third goal of this project was to identify gene candidates which were specific for each tissue
of interest. The candidates used throughout the course of the grant were identified through
surveys of literature (PubMed), Gene and other databases. Initially we identified several genes
that appeared to be specific for each tissue/fluid (i.e. brain, semen, sperm). These screening
studies were performed using TaqMan® primer/probe sets from Applied Biosystems because
they were pre-designed, inexpensive and thoroughly tested for specificity. We tested the
specificity and sensitivity of each assay by analysis of mRNA isolated from the body fluid of
interest (seminal fluid) or control RNA (brain). Our diverse sample bank was used to assess the
specificity of the candidate tissue-specific genes. These samples included blood, semen, saliva,
menstrual blood, vaginal secretions, kidney, colon, adipose, skin, and control human RNAs
(brain, heart, liver, kidney, intestine).
To test the specificity of the TaqMan® sets (only the control B2M and tissue-specific sets should
give amplification), mRNA was isolated from dried blood, semen, saliva, menstrual blood or
vaginal secretions using TRIzol®. Human tissues were extracted using the Absolutely RNA® Kit
and control human RNAs from Ambion were diluted to 100 ng/µl. The RNA samples were then
reverse transcribed and PCR performed with each of the TaqMan® sets. There was some cross-
reactivity with the seminal fluid candidates. Some minor amplification occurred with vaginal
secretions (SEMG1 and ACPP) and blood (ACPP). Interestingly, MSMB amplified from a
control intestine sample, but not with human colon tissues. ACPP and CRISP1, although ideal
candidates based on literature searches, were not suitable assays for the specific detection of
semen. The sperm marker PRM2 amplified from a seminal fluid sample, but nothing else that
7

was tested. The control B2M was detected in each of the fluids and tissues. Based on these
results, TGM4 and PRM2 have demonstrated their specificity for semen/sperm.
Furthermore, even when some candidates appeared to be specific based on the results using the
control RNAs, differing results were seen with actual human tissue samples. Therefore, before
any claims can be made regarding the specificity of the tissues candidates, experiments using
actual human tissues (like what was done for kidney and colon/intestine) need to be performed.
We were not been able to obtain human heart, brain or liver samples through our collaborators at
FAHC.
The specificity studies using the tissue assays for brain yielded mixed results. All three of the
brain assays amplified from the control brain sample as expected, but also had varying degrees of
cross-reactivity with other control tissues. DRD1 amplified from liver and intestine, GPM6A
amplified from liver and ADCY1 amplified from heart and kidney, respectively. The Cts for
ADCY1 and GPM6A were the lowest for brain samples out of the three assays which indicates
that they are expressed to a higher extent than DRD1.
The sensitivity of the candidate assays for semen (TGM4) and sperm (PRM2) was evaluated
using a range of semen volumes spotted onto cotton cloth. Both TGM4 and PRM2 expression
was detected in the lowest sample volumes tested (i.e. 1 µl for TGM4 and 0.01 µl for PRM2)
indicating that they are very sensitive assays. But, since the two lowest volumes, namely 0.01 µl
and 0.1 µl, were not analyzed with the TGM4 assay, the lower limits are unknown, but assumed
to be lower than 1 µl based on the Cts.
8

The sensitivities of the candidate assays for brain were evaluated using a range of control brain
RNA (1 pg/µl to 1 µg/µl). The various concentrations of RNA were reverse transcribed and the
resulting cDNA was amplified using the TaqMan® assays. ADCY1 was the most sensitive of the
three assays tested being detected in 10 pg of brain RNA; similar to the control B2M target.
Alternatively, GPM6A expression was detected in 100 pg of brain RNA and DRD1 expression
was detected in 1 ng of RNA.
A major aim of stain identification using mRNA expression profiling, and the fourth goal of the
project, is working towards multiplexing the real-time PCR assays once mRNAs are identified
that clearly define specific types of stains. Since these assays are designed to function more
qualitatively than quantitatively, a test of a single stain should typically give amplification with
only one candidate. It is possible that a mixture could give several amplifications. However, it
is of greater concern to know that a mixture does exist, than to know the exact amount of each
fluid present. The Plexor® system from Promega was one technology we employed to develop
multiplex assays. Depending on the dye-capability of the real-time instrument that is utilized,
this system allows up to six mRNAs to be multiplexed in one assay, thus reducing the amount of
sample needed and time of analysis. The Plexor® qRT-PCR system takes advantage of the
specific interaction between two modified nucleotides to achieve quantitative PCR analysis.
Promega’s Plexor® design software allows the generation of primers and probes which span an
intron by designating a certain base to include in the primer/probe design.
In our previous grant (2004-DN-BX-K002) we described an ongoing collaboration with Promega
to develop stain identification assays using their Plexor® platform. The combination of
Promega’s extensive knowledge of the Plexor® platform with our sample inventory and insight
9

into the needs of the forensic laboratory forged a strong partnership. The goal was to generate
Plexor® Tissue Typing Systems; multiplexed qRT-PCR systems for determining the source and
quantity of a variety of human stains and/or tissues. The systems would include Plexor® primers
for the detection of tissue-specific mRNA transcripts associated with semen, sperm, blood,
menstrual blood, saliva, etc. By limiting the initial system to two-color detection (i.e. FAM and
HEX detection), it would be compatible with the majority of real-time thermal cyclers in forensic
laboratories. We sought to explore the potential to include controls (e.g. a housekeeping gene) or
multiple, tissue-specific targets using the ability of the Plexor® Data Analysis Software to
distinguish two different amplicons in the same dye channel, based upon thermal melt properties.
The Stain ID assay which we decided to first focus on was for detection of semen and saliva
using the targets TGM4 (semen), HTN3 (saliva) and GAPDH (housekeeper). The preliminary
experiments were not performed using sample types and sizes which are reflective of forensic-
type samples. Although these studies gave an idea as to the relative sensitivities of the assay, it
was important to assess how the assay would perform with typical forensic samples (i.e. various
volumes of unknown RNA yields). Once the draft Technical Manual was completed by
Promega, we alpha-tested the assay using a large panel of in-house samples including: blood,
menstrual blood, saliva, semen, urine, vaginal secretions, buccal swabs on FTA®, kidney,
adipose, colon and skin. In addition, mixtures of blood/saliva, blood/semen, and saliva/semen
were tested and multiple samples were used that ranged in age and size. The results
demonstrated that the assay was indeed specific and sensitive. Amplification of the HTN3 and
TGM4 targets only occurred in samples composed of saliva and semen, respectively, whereas all
of the samples (with the exception of the negative controls) showed amplification of GAPDH.
The only exception was a lack of GAPDH amplification in the 1 µl urine samples which is likely
10

due to the minute amount of sample remaining on the substrate at the time of extraction. Non-
specific amplification wasn’t detected in any of the samples.
Promega invited a number of forensic labs from across the county to participate in an alpha test
of the prototype kit. As part of this process, Promega included a series of blind swabs which
were provided to all of the test sites. There were a total of eight swabs; four different swabs
prepared in duplicate. Based solely on the software calls, the results were as expected with the
exception of two samples. However, by looking at the raw data (i.e. Cts), both samples show
strong positives for semen and GAPDH, but the Tms drifted slightly below the control melting
curve and thus, were not called “yes” by the software. Scientists at Promega have seen this
happen on occasion, particularly with swabs that have a lot of sample material dried onto them.
Their theory is that perhaps something is being carried over from the extraction process in those
samples and slightly affects the melt temperature. A protocol for removal of these impurities is
included in the manual accompanying the Stain ID kit.
The usefulness of the Stain ID assay was tested using additional mock and actual casework
samples. Following serological confirmatory testing (i.e. microscopic sperm search), residual
extracts (some containing the original cuttings) were saved at -20 ˚C until nucleic acid isolation
was performed using the Tris-buffered phenol protocol. In additional, vaginal swabs (with and
without seminal fluid contributions) and a skin swab from a female donor following male
salivary deposition were included in the experiment. Several of the casework samples which
were found to contain sperm tested negative for TGM4 in the Stain ID Assay. The cause for
these false negatives warrants further examination. In addition to examining the RNA
component isolated from these samples, the co-isolated DNA was amplified for STR analysis to
11

determine if amplifiable DNA is produced using this procedure. The results indicate that the co-
isolation method is capable of producing DNA of sufficient quantity and quality for downstream
STR analysis.
The next step was to produce data for a validation paper using Stain ID materials that were made
and quality controlled by the manufacturing department at Promega. Furthermore, these samples
would also be evaluated for DNA yields and generation of STR profiles. However, as we were
anxiously awaiting the next steps, Promega put a hold on this project based on feedback from the
alpha testers. There were questions regarding the extraction method, requests for additional
markers in the assay, and general concerns as to the marketability of the assay. To date, no
further work has been performed on the Stain ID assay here at the VFL.
A major disadvantage to using the Gene Expression TaqMan® assays from Applied Biosystems
is the inability to multiplex the assays since they are all labeled with the same dye. Therefore,
we set out to design our own TaqMan® probes and primers in hopes of developing several
specific multiplex assays. Work on these assays began with Grant 2004-DN-BX-K002 and was
described in the Final Report. The first multiplex we designed was a semen-sperm detection
assay using the TGM4 and PRM2 markers in addition to the housekeeper B2M. Often, it is
important to determine if semen is present even if the male is sterile or has had a vasectomy (i.e.
no sperm). In other cases, it is important to know if sperm are present. An assay that could
determine whether the stain 1) is seminal fluid and 2) contains sperm could alleviate the need to
perform extensive microscopy for the identification of sperm.
12

Once experiments were carried out to optimize the amount of primers and probe for each
candidate, the optimal amounts of primers and probes were combined into a single multiplex
reaction and compared to singleplex reactions. Preliminary data reported in Table 23 of the Final
Report for 2004-DN-BX-K002 shows that while each primer/probe set amplified from semen
when alone in the reaction, the combination into a multiplex reaction caused the dropout of
TGM4 and B2M amplification. Alternatively, the amplification using the PRM2 primer/probe
set was unchanged regardless of the reaction conditions. Since PRM2 is highly expressed in
seminal fluid samples, it may be competing against the other sets for reaction components.
Therefore, we decreased the primer and probe conditions for this target. The results showed that
changing the amount of PRM2 primers and probes was successful in preventing the loss of
TGM4 and B2M amplification in the multiplex reaction. In an attempt to decrease the Ct for
B2M amplification, the primer and probe concentrations were altered, which only had a minor
effect on the results.
In other studies we have seen that changing the master mix used in the reaction can have an
impact on the quality of the results. We compared eight commercially available master mixes to
determine whether amplification could be improved for the multiplex assay. We also decided to
add BSA to the reactions to see if it would have a beneficial effect on amplification. The
addition of BSA did increase amplification in some of the reactions (i.e. lower the Cts).
However, the best overall amplification was when the Sigma JumpStart Taq ReadyMix was used
and the presence of BSA had no additional effect on the results.
The optimized semen-sperm assay was tested for its sensitivity using three year old seminal fluid
samples ranging from 0.01 – 20 µl. Amplification of both seminal fluid markers occurred in the
13

lowest volume sample tested which demonstrates the sensitivity of this assay since the control
target B2M was only detected down to the 0.1 µl sample. The specificity of the assay was
assessed using a panel of cDNAs prepared from a variety of RNAs extracted from body fluids
(blood, semen, saliva, vaginal secretions, menstrual blood), tissues (kidney, colon, adipose, skin)
and control RNAs (brain, heart, liver, kidney, intestine). Unfortunately, there was nonspecific
amplification of PRM2 in the saliva, vaginal secretions, menstrual blood, kidney and colon
samples. At this point, a decision was made to combine the “homebrew” TGM4 and B2M
primer/probe sets with the Gene Expression TaqMan® PRM2 assays from Applied Biosystems.
In order to test the new triplex containing the commercial PRM2 assay, we compared the original
“homebrew” multiplex assay with the new modified triplex and the PRM2 stand-alone assay
from Applied Biosystems. Amplification of TGM4 and B2M was lost with the modified
homebrew assay. A titration of the amount of PRM2 assay added to the modified triplex led to
an increase in amplification of TGM4. However, amplification of B2M in the modified
multiplex was still decreased compared to the original homebrew assay. One last ditch effort
was made to save this modified homebrew assay; three master mixes were tried in order to
improve the amplification of B2M. The best result, as determined by amplification of all three
targets, was with samples run using the QuantiTech Multiplex PCR Kit (Qiagen).
The sensitivity of the modified semen-sperm assay was evaluated using a range of semen
volumes (0.01 – 20 µl) spotted onto cotton cloth. A dCt was determined for each of the samples
amplified with the semen-sperm assay by subtracting the body fluid gene (PRM2 or TGM4)
from the housekeeping gene (B2M) Ct value. Based on the dCt values, PRM2 expression was
positively identified at the lowest sample volume tested, whereas the presence of TGM4 was
14

detected in the 0.1 µl semen sample. To test the specificity of the modified semen-sperm assay,
a panel of RNAs isolated from various body fluids (blood, semen, saliva, vaginal secretions,
menstrual blood), tissues (kidney, colon, adipose, skin) and control RNAs (brain, heart, liver,
kidney, intestine) were reverse transcribed and PCR performed. The amplification results show
that the only positive dCt values were for the semen sample. Every other body fluid or tissue
tested had a negative dCt and was therefore negative for the presence of semen/sperm. Based on
these results, the semen-sperm assay appears to be sensitive and specific for seminal fluid.
The second TaqMan®-based multiplex assay we have developed is for the identification of brain
tissue using ADCY1, GPM6A and the housekeeper B2M. Preliminary data published in the
Final Report for Grant 2004-DN-BX-K002 (Table 24) showed that each primer/probe set
amplified from brain both when alone in the reaction and in combination with the other
primer/probe sets. Furthermore, there was no significant decrease in the degree of amplification
when the sets were alone or combined into the multiplex. There was some minor amplification
in the no template control sample when the ADCY1 primers/probe were alone and when the
GPM6A set was used. This experiment was repeated using less of the GPM6A primers and
probes in the reaction. As a result, there was a significant decrease in the nonspecific
amplification although there was still some bleed through into the HEX/GPM6A dye channel.
However, this multiplex looks very promising for a brain screening assay. Additional studies
using different master mixes may further improve the data and experiments using human brain
samples would aid in the evaluation of this multiplex as a viable screening assay.
The final goal of this project was to disseminate our results to the forensic community. To this
end we have published a chapter outlining our work to identify specific gene targets for the
15

purpose of multiplexing, are in the process of compiling two additional manuscripts, presented
our work at a number of scientific conferences, and hope our work to develop Stain ID assays in
collaboration with Promega will regain interest.
16

MAIN BODY
I. Introduction
1. Statement of the Problem
In an age of scientific advances in molecular biology, DNA profiling has proven itself an
invaluable tool in solving crimes. However, the potential exists for the tissue origin of the
suspect DNA to be called into question. For example, in a court of law, a semen stain containing
suspect DNA can have far more serious consequences than a saliva stain. Furthermore, it is
paramount to the criminal justice system that suspect stains be identified definitively and
accurately. Traditional serological approaches for stain identification often involve a
presumptive color test, followed by a confirmatory test that typically employs a specific antibody
designed to complex with a known protein; such as hemoglobin for blood and P30 or prostatic
antigen for seminal fluid. In the past decade, unparalleled progress has occurred in the design of
tests necessary to identify and individualize crime scene samples. While these testing methods
have improved in simplicity and selectivity over the years, several problems still exist such as the
possibility of cross-reactivity with other species and the lack of specificity for particular tissues.
For example, the traditional test for saliva involves detecting the presence of the enzyme
amylase. While this enzyme is found in relatively high concentrations in saliva, it is also present
at lower levels in other fluids. Therefore, definitively determining the presence of saliva is not
possible by this method.
17

In order to develop more robust assays, we explored the possibility of using mRNA as a
determinant of tissue specificity. Each tissue or cell type makes a unique constellation of
mRNAs, some specific for only that tissue or cell type. An assay to detect these specific mRNAs
will be indicative of the tissue of origin. Some body fluids, such as blood, contain cells as part of
their function while other fluids, such as urine, contain cells that have been shed from their tissue
of origin. In our Final Report for Grant 2004-DN-BX-K002 we described the use of real-time
PCR to indicate that stains can be identified by determining which mRNAs they contain.
Furthermore, mRNAs extracted from stains ranging from 1 day to 3 years of age were
successfully amplified which demonstrates the stability of mRNA over time. Assays using a
real-time format can yield a quick and accurate identification of an unknown tissue or stain.
The longer-term goal of this research was to develop simple mRNA extraction and analysis
methods to allow the quick, unequivocal identification of body fluid stains and tissues. The
specific aims of this grant were 1) to select the best method of DNA/RNA co-extraction from a
wide variety of stain types, 2) to find 2-3 genes specific for numerous stain and tissue types, 3) to
develop multiplex assays for the identification of tissue and stain types, 4) to validate these
assays for forensic casework, and 5) to disseminate these results to the forensic community.
2. Literature Citations and Review
Although the DNA of all tissues from an individual is essentially identical, the RNA spectrum
made by the different cells in each tissue is very different. Therefore, analysis of the RNA in a
sample, especially identification of certain “tissue specific” mRNA transcripts, can provide an
“RNA profile” which will uniquely identify the tissue of origin. A major interest in cancer
18

research today is to determine the spectra of mRNAs made in different cell types in hopes of
elucidating which changes turn normal cells into cancerous cells. For these studies, it is crucial
to be able to distinguish the mRNA produced by normal cells compared to malignant cells.
Thus, a body of knowledge is being developed regarding the distribution of various mRNAs in
human tissues. Databases of information regarding the amounts of each mRNA present in every
tissue have been and are being created. Examples include: NHGRI Tissue Microarray Project,
Gene Expression Omnibus, BODYMAP and HugeIndex.
When molecular biologists began isolating mRNA for experiments, it was thought that mRNA
was very ephemeral and that tissues needed to be processed rapidly in separate rooms with
dedicated instruments and often with hazardous chemicals. However, due to the development of
new techniques and the recent increase in knowledge concerning mRNA, it has been shown to be
relatively stable. mRNA can be isolated from such items as formalin-preserved tissues and
Guthrie blood spots (Liu et al., 2002; Fritsch et al., 2003; Macabeo-Ong et al., 2002; Matsubara
et al., 1992; Cao and Cousins, 2000; Krafft et al., 1997; Lewis et al., 2001; Tetali et al., 2001; Pai
et al., 1994; Cassol et al., 1997; Abe et al., 1998; Spielberg et al., 2000; Katz et al., 2002). For
example, Tetali et al. (2001) typed individuals for the CCR5 32bp deletion using RT-PCR of
blood spots that had been dried up to 12 months. Others have isolated mRNA from decade old
samples (Mizuno et al., 1998; Li et al., 1997) and mRNA has also been isolated from dried blood
smears (Schoch et al., 2001; Crisan and Anstett, 1995).
A major obstacle which needs to be overcome in order for laboratories to invest in RNA
technology is the development of a straight-forward extraction procedure. A method that could
co-purify RNA and DNA from a single sample with a minimal number of steps would be
19

attractive to those seeking new technologies. A number of methods describing the simultaneous
isolation of DNA and RNA have been reported (Alvarez et al., 2004; Bauer and Patzelt, 2003;
Chomczynski, 1993). However, most of these have not been optimized to deal with the reduced
quantity or quality of samples encountered in forensic casework. Alternatively, the co-isolation
reports using forensically relevant samples (Alvarez et al., 2004; Bauer and Patzelt, 2003)
require numerous time-consuming steps that would not benefit fast and simple stain
identification assays.
Many companies now sell kits for quick and easy isolation of DNA or RNA from a variety of
sample types. However, despite the availability of simple and convenient commercial kits, little
has been done in the forensic field to combine DNA and RNA extraction into a convenient dual
extraction; specifically, to detect bodily fluid or tissue-specific mRNAs in crime scene samples.
The standard commercial method of DNA/RNA co-extraction utilizes the TRIzol® Reagent from
Invitrogen. Despite the quality of nucleic acids isolated using this method, a major disadvantage
is the number or steps required making the process very time-consuming. A promising
commercial extraction procedure is the AllPrep DNA/RNA Mini Kit from Qiagen. This kit is
designed for purifying both DNA and RNA from a single sample in as little as 30 minutes with
no need to divide the original sample into two for separate purification procedures. Although the
potential for DNA/RNA extraction from animal cells and tissue homogenates using the AllPrep
kit had been demonstrated, its application to forensically relevant samples is widely unknown.
Groups have isolated mRNAs from blood, semen and saliva for research and diagnostic
purposes. For blood, groups have isolated mRNA from dried blood spots for RT-PCR and
restriction or quantitation (Matsubara et al., 1992; Cao and Cousins, 2000; Zhang and McCabe,
20

1992; Watanapokasin et al., 2000). For semen, a number of groups have developed methods to
detect hepatitis C viral mRNA or HIV mRNA in seminal fluid (Bourlet et al., 2002; Dulioust et
al., 1998). Researchers have also isolated nuclear mRNAs such as calcium channel subunit
mRNAs from the sperm in semen (Goodwin et al, 2000). In fact, mRNAs of a number of genes
have been found in human spermatozoa (Miller, 2000; Richter et al., 1999). In addition, beta-
hCG mRNA has been isolated from the prostate cells in human ejaculate (Daja et al., 2000). For
saliva, mRNA of viruses such as measles has been detected (Jin et al., 2002). In terms of
forensic analysis, Bauer et al. (1999) have detected mRNAs specific for epithelial (endometrial)
cells in menstrual blood samples. They found that they could isolate mRNA after 6 months of
room temperature storage and detect a number of mRNAs species. Further study (Bauer and
Patzelt, 2002) found that matrix metalloproteinase was a good marker for menstrual blood.
Bauer et al. (2003) have a more recent paper where they studied 106 bloodstains stored up to 15
years. They found that mRNA levels as measured by laser-induced fluorescence capillary
electrophoresis correlates with the age of the sample and that “mRNA suitable for RT-PCR can
be isolated from samples stored up to 15 years”.
Juusola and Ballantyne (2003) have isolated mRNA from blood, semen and saliva stains and
used it for RT-PCR of the control genes S15, beta-actin and GAPDH. In addition, they studied
the saliva specific genes statherin, histatin 3, PRB1, PRB2 and PRB3. mRNA for these latter
genes was found only in the saliva stains.
Technology in the field of multiplexing gene expression assays is rapidly improving. The
Plexor® One-Step qRT-PCR System takes advantage of the specific interaction between two
modified nucleotides to achieve quantitative PCR analysis. It is possible to design assays to
21

quantify multiple targets within the same reaction using primer pairs with a different fluorophore
for each target sequence. Therefore, you are limited by the dye capability of your real-time PCR
instrument. However, since DNA quantitation is moving towards a molecular biological
approach which utilizes three- or four-dye real-time PCR instruments, most labs could
implement this technology without purchasing new equipment.
A new technology that may offer great promise to the forensic community is the Bio-Plex™
system which uses the multiplexing technology of Luminex Corp. to enable the simultaneous
quantitation of up to 100 analytes. This technology uses polystyrene beads (xMAP® beads)
internally dyed with differing ratios of two spectrally distinct fluorophores. Each fluorophore
can have any of 10 possible levels of fluorescent intensity, thereby creating a family of 100
spectrally addressed bead sets. Multiplex assays can be created by mixing bead sets to
simultaneously test for many analytes in one sample. This valuable technique could be used in
routine testing and could assess many samples in an automated fashion.
Another adaptation to the Luminex-bead technology is the QuantiGene® Plex Reagent System
offered by Panomics. This system combines branched DNA with the multi-analyte profiling
beads. Together they enable simultaneous detection of multiple RNA targets directly from
purified RNA. Branched DNA technology is a sandwich nucleic acid hybridization assay that
amplifies the reporter signal rather than the sequence. Groups have been able to utilize this
technology to measure gene expression from blood (Zheng et al., 2006), formalin-fixed, paraffin-
embedded tissues (Yang et al., 2006), and directly from cell lysates and tissue homogenates
without the need for RNA purification (Zhang et al., 2005; Flagella et al., 2006).
22

Altogether these studies and commercially available techniques indicate that DNA and RNA can
be co-extracted and the RNA fraction used in multiplexed real-time PCR assays. We proposed
to develop real-time PCR assays to detect tissue-specific transcripts for human fluids and tissues.
These assays would ultimately be multiplexed for faster determination of tissue origin. A major
advantage to these assays is that a single sample extract will be used to classify the sample as
blood, semen, vaginal secretions, brain, heart, etc. which will drastically reduce the number of
identification tests performed prior to DNA profiling (if required).
3. Rationale for the Research
The implementation of DNA analysis within the forensic laboratory has been a tremendous
benefit to the criminal justice community. However, as technologies progress, there are more
cases and items per case requiring DNA analysis and, unfortunately, the manpower and
resources needed to work the cases have not kept up with demand. The research into stain
identification using real-time PCR seeks to find faster and more efficient methods to work DNA
cases. The goal is to develop methods that broaden real-time PCR applications and to investigate
a new technology that could radically change the analysis of biological stains and tissues. The
biochemical approaches currently used in tissue identification are limited in scope and often
imply, but not truly identify, the source fluid/tissue. Many believe that the positive identification
of fluids and tissues can be performed quickly and efficiently, and that tissues not routinely
evaluated could be easily assessed by all laboratories so that an equality of testing could be
realized across the country. The evaluation of mRNA through real-time PCR will be a technique
that can offer a level of confidence and expand our knowledge of the materials we routinely
23

examine. This research into new technologies will demonstrate the power of multiplexing for
forensic analysis.
As the demand for sample analysis increases, forensic laboratories continue to balance
manpower and cost issues versus the value of the analysis when evaluating new techniques. In
order for laboratories to invest in RNA technology, they will require a straight-forward
extraction procedure. Furthermore, a crucial prerequisite to these analyses is the development of
a DNA/RNA co-extraction method to minimize sample requirements and eliminate the need for
two separate extractions. A method that could co-purify RNA and DNA from a single sample
with a minimal number of steps would be attractive to those seeking new technologies. A
number of methods describing the simultaneous isolation of DNA and RNA have been reported,
however, most of these have not been optimized to deal with the reduced quality of samples
encountered in forensic casework. Alternatively, the co-isolation reports using forensically
relevant samples require numerous time-consuming steps that would not benefit a fast and
simple stain identification assay. A preferred extraction method would demonstrate the best
ability to co-isolate DNA and RNA in terms of yield and amplifiability while remaining simple,
efficient, and ideally, involve non-hazardous reagents.
The purpose of this RNA-based stain identification research is to develop a simple mRNA
extraction and analysis method to allow the quick, unequivocal identification of body fluid stains
and tissues. The proposed assays will be designed so that a single stain will amplify with only
the corresponding mRNA(s) of its fluid/tissue type; a mixture, however, should amplify
representing various fluids/tissues. The assays are designed to function more qualitatively than
quantitatively, so although they may indicate which fluids/tissues are present, they may not give
24

the exact ratio of each fluid present. However, it is of greater concern to know what kind of
mixture exists, than to know the exact amount of each fluid present. Furthermore, a major
advantage to these assays is that a single test will be used to classify the sample as blood, semen,
vaginal secretions, brain, heart, etc. which will drastically reduce the number of identification
analyses performed prior to DNA profiling (if required).
II. Methods
Sample Preparation
Blood, seminal fluid, saliva and urine samples were collected over the course of the grant by
pipetting known amounts of fresh fluid onto swatches of cotton cloth and allowing the spots to
dry at room temperature. Vaginal swabs were collected on cotton swabs, whereas menstrual
blood was collected on tampons and allowed to dry at room temperature. Each sample was
stored in a glassine envelope, which was kept at room temperature in a box designated for the
particular fluid at the Vermont Forensic Laboratory. Human tissues (kidney, colon, adipose,
skin) were collected from the Autopsy Service and Surgical Pathology Suite at Fletcher Allen
Health Care and frozen at -20˚C until use. Human control RNAs (brain, heart, liver, kidney,
intestine) were purchased from Ambion (Austin, TX) for use in several experiments. These
RNAs were supplied as 1 µg/µl and diluted as noted for experiments.
25

RNA/DNA Extraction
Various commercially available RNA/DNA isolation kits, as well as several homebrew methods
were used over the course of this grant including:
Absolutely RNA® Miniprep Kit (Stratagene, La Jolla, CA) This method employs a spin cup
with a silica-based fiber matrix that binds RNA in the presence of chaotropic salt while a series
of washes removes contaminants. The lysis buffer contains guanidine thiocyanate to lyse the
cells and to prevent RNA degradation by RNases. Following cell lysis, the sample is prefiltered
in a spin cup to remove particles and to reduce the amount of DNA. The filtrate is then
transferred to a spin cup with a silica-based fiber matrix which binds the RNA. Treatment with a
low-salt wash buffer and digestion with DNase removes the remaining DNA. A series of washes
removes the DNase and other proteins. Highly pure RNA is eluted from the fiber matrix with a
small volume of RNase-free water and captured in the microcentrifuge tube.
Differential Method (Vermont Forensic Laboratory, Waterbury, VT) Epithelial cells are
preferentially lysed in a female extraction buffer containing detergent for two hours at 37 ˚C.
Following centrifugation to pellet the sperm cells, the supernatant is removed to a new tube
(female fraction). The sperm pellet (male fraction) is washed three times with the female
extraction buffer before incubation with male extraction buffer containing detergent and DTT for
two hours at 37 °C. Phenol:chloroform:isoamyl alcohol is added to the female and male
fractions and following centrifugation, the DNA aqueous phase is separated from the organic
solvent. The samples are purified using Microcon 100 Concentrators (Millipore Corporation,
Bedford, MA) and the DNA is eluted in TE-4.
26

Organic Method (Vermont Forensic Laboratory, Waterbury, VT) Samples are lysed in stain
extraction buffer containing detergents and proteinase K overnight at 56°C.
Phenol:chloroform:isoamyl alcohol is added to the lysed samples and following centrifugation,
the DNA aqueous phase is separated from the organic solvent. The samples are purified using
Microcon 100 Concentrators (Millipore Corporation) and the DNA is eluted in TE-4.
PureYieldTM RNA Midiprep System (Promega, Madison, WI) This system is designed to
quickly and easily isolate high yields of pure total RNA while eliminating the co-purification of
DNA. The protocol uses the PureYieldTM silica-membrane technology to isolate intact RNA.
The PureYieldTM RNA Midiprep System provides many unique features to purify total RNA
without using DNase treatment, phenol:chloroform extractions, protease digestion or alcohol
precipitations. The PureYieldTM RNA Midiprep System avoids the problems routinely involved
with DNA contamination and its subsequent removal by selectively eliminating DNA prior to
total RNA isolation, using the PureYieldTM Clearing Agent, which preferentially binds DNA
leaving the RNA virtually free of DNA.
RNAgents® Total RNA Isolation System (Promega) This procedure utilizes the RNAgents®
Denaturing Solution to lyse cells or tissue under conditions that rapidly inhibit ribonucleases
using two potent inhibitors of RNase, guanidine thiocyanate and β-mercaptoethanol. The
Solution is designed to be used in concert with acidic phenol:chloroform and alcohol
(isopropanol) for purification of total RNA.
27

Tris-Buffered Phenol Method (Stain ID extraction) (Promega and Vermont Forensic
Laboratory) This protocol is optimized for the simultaneous extraction of both DNA and RNA
from a sample. The resulting total nucleic acid is suitable for analysis using both the Stain ID
system as well as Promega’s STR systems. The procedure uses a guanidine thiocyanate-based
denaturing solution in combination with a Proteinase K treatment step to provide rapid sample
lysis and protein precipitation, effectively protecting the RNA during sample lysis. A Tris-
buffered phenol extraction step efficiently extracts both DNA and RNA from the lysed sample.
Alcohol precipitation serves to wash the total nucleic acids of residual salts.
TRIzol® Reagent (Invitrogen, Carlsbad, CA) This reagent is a monophasic solution of phenol
and guanidine isothiocyanate suitable for isolating total RNA, DNA, and proteins. During
sample lysis, TRIzol® Reagent maintains the integrity of the RNA, while disrupting cells and
dissolving cell components. Addition of chloroform followed by centrifugation separates the
solution into an aqueous phase and an organic phase. RNA remains exclusively in the aqueous
phase. After transfer of the aqueous phase, the RNA is recovered by precipitation with isopropyl
alcohol. After removal of the aqueous phase, the DNA in the sample can be recovered by
precipitation with ethanol to yield DNA from the interphase.
cDNA Synthesis
For the reverse transcription (RT) reaction, 6 µl of total RNA template was combined with 2 µl
of random decamers (50 µM) (Applied Biosystems, Foster City, CA) and 4 µl of nuclease-free
water (final volume of 12 µl) and heated at 80 ˚C for 3 minutes. To the reaction, 2 µl of 10X
RT-PCR buffer (Ambion, Austin TX), 4 µl of dNTP mix (10 mM) (Ambion), 1 µl of RNase
28

inhibitor (40 U/µl) (Applied Biosystems) and 1 µl of MMLV-RT (100 U/µl) (Applied
Biosystems) were added to yield a final reaction volume of 20 µl. This reaction mixture was
incubated at 43 ˚C for 1 hour and then at 92 ˚C for 10 minutes to inactivate the RT.
Real-time PCR
TaqMan® Analysis. Singleplex assays using real-time PCR on cDNA were performed using
Assays-on-Demand™ Gene Expression Products (Applied Biosystems) (Table 1). These are a
comprehensive collection of pre-designed and tested primer and probe sets that allow researchers
to perform quantitative gene expression studies on any human gene. They are designed against
GenBank transcripts, transcripts from the Mammalian Gene Collection, and human Celera
transcripts. Each assay is built on 5’ nuclease chemistry and consists of two unlabeled PCR
primers and a FAM™ dye-labeled TaqMan® minor groove binder (MGB) probe. The
components are formulated into a single 20X mix and designed to run under universal conditions
for reverse transcription and PCR. Assays with “m1” in the ID code indicate an assay whose
probe spans an exon junction and therefore is designed to amplify only target cDNA without
amplifying genomic DNA. This is the result of targeting primer sites that span regions of mRNA
in which introns have been removed, making the mRNA different from the DNA that it
originated from. Those with “g1” in the ID code may possibly amplify genomic DNA.
The TaqMan® probe consists of two types of fluorophores, which are the fluorescent parts of
reporter proteins (Figure 1). While the probe is attached or unattached to the template DNA and
before the polymerase acts, the quencher (Q) fluorophore reduces the fluorescence from the
reporter (R) fluorophore. It does this by the use of fluorescence resonance energy transfer
29

(FRET), which is the inhibition of one dye caused by another without emission of a photon. The
reporter dye is found on the 5’ end of the probe and the quencher at the 3’ end. Once the
TaqMan® probe has bound to its specific piece of the template DNA after denaturation and the
reaction cools, the primers anneal to the DNA. Taq polymerase then adds nucleotides and
removes the TaqMan® probe from the template DNA. This separates the quencher from the
reporter, and allows the reporter to give off its energy which is quantified using a computer. The
more times the denaturation and annealing takes place, the more opportunities there are for the
TaqMan® probe to bind and, in turn, the more emitted light is detected.
In addition to the Assays-on-DemandTM gene expression products purchased from Applied
Biosystems, we designed our own in-house TaqMan® assays for seminal fluid/sperm and brain.
Primers and TaqMan® probes were designed using the Beacon Designer program (PREMIER
Biosoft International, Palo Alto, CA). Primers were made for PRM2 (F,
AGTCACCTGCCCAAGAAACAC; R, ACTTTTGCTCGTTTCACTCAGATC), TGM4 (F,
CTGGATGAAGCGACCGGATC; R, ATGTCACCTTTGCGGATGGC), B2M-Semen (F,
TCCTGAAGCTGACAGCATTCG; R, GGATGACGTGAGTAAACCTGAATC), ADCY1 (F,
CCTGCTGTCAACCTCTCCTC; R, CTAGTGGAAAGGGGACCATAAGG), GPM6A (F,
CACCTCACTGCCAGTTTACATG; R, TCACAATTCCAAACTGACGAAGG) and B2M-
brain (F, CATTCGGGCCGAGATGTCTC; R, TGCTGGATGACGTGAGTAAACC) and
purchased from Biosearch Technologies (Novato, CA). TaqMan® probes (PRM2,
CTTCTCGGCGGCAACTCAGGGCT; TGM4, CCCAAGGGCTACGACGGCTGGC; B2M-
Semen, TGTCTCGCTCCGTGGCCTTAGCTG; ADCY1,
CTGCCTTGTCCCTGCTCCTGTGCT; GPM6A, TGTGGACCATCTGCCGGAACACCA;
B2M-Brain, TGGCCTTAGCTGTGCTCGCGCT) were labeled with FAM (PRM2, ADCY1),
30

CAL Orange 560 (TGM4, GPM6A) or CAL Red 610 (B2M-Semen, B2M-Brain) on the 5’end
and were also purchased from Biosearch Technologies. Real-time PCR primer pairs were
designed to span at least one exon-exon boundary (with the exception of PRM2) and be 20-24
bases in length, but needed to function with high annealing temperatures (58-60 ˚C) and short
mRNA/cDNA amplicons (100-150 bp). The real-time PCR probe for each gene was designed to
anneal at the exon-exon boundary enclosed by the primers, but be 20-25 bases in length and have
an annealing temperature approximately 10 ˚C higher than the respective primer pair (~68 ˚C).
Real-time PCR was performed on a Qiagen Rotor-Gene Q (Valencia, CA). Two microliters of
the RT-reaction were amplified in a total reaction volume of 10 µl. Each standard singleplex
reaction included 5 µl of TaqMan® Universal PCR Master Mix, No AmpErase® UNG, 0.5µl of
Assay-on-Demand primer/probe set and 2.5µl of nuclease-free water. For the homebrew assays,
optimal primer and probe concentrations were determined experimentally. The optimal master
mix was determined by comparing amplification results using the following: QuantiTect
Multiplex PCR NoROX Kit (Qiagen), ABsoluteTM QPCR Mix (Thermo Fisher Scientific,
Rockford, IL), JumpStartTM Taq DNA Polymerase (Sigma, St. Louis, MO), Brilliant Multiplex
QPCR Master Mix (Stratagene), HotMaster Mix (Eppendorf, Hauppauge, NY), Full Velocity
QPCR Master Mix (Stratagene), QPCR MasterMix Plus (Eurogentec, Fremont, CA) and
Universal PCR Master Mix (Applied Biosystems). Real-time PCR cycling conditions consisted
of a denaturation step (95˚C for 10 min) followed by 50 cycles (95˚C, 15 sec and 60˚C for 60
sec) with acquiring to FAM, VIC/JOE and ROX. For data analysis, the threshold was manually
set to 0.030. A delta Ct (dCt) was determined by subtracting the body fluid gene (i.e. PRM2 or
TGM4) Ct value from the housekeeping gene (B2M) Ct value. A positive dCt value indicates
that a body fluid gene was present at a higher level than the housekeeping gene and that the body
31

fluid gene was present. A negative dCt value indicates that the body fluid gene was present at a
lower level than the housekeeping gene or not detected at all, and therefore the body fluid was
not detected. In instances when no Ct value was obtained, a Ct value equal to the highest cycle
number used in the assay (i.e. 50) was substituted into the calculation.
Plexor® Analysis. Eragen Biosciences developed and synthesized a series of new DNA base
pairs (Figure 2). The Plexor® technology is based on one of these new base pairings: isoG and
isoC. IsoC and IsoG nucleotides are incorporated by DNA polymerase; however, neither isoC
nor isoG can base pair with any of the other conventional bases. These two novel bases only
interact and base pair with one another and are not found in nature. Although similar to the
conventional G-C pair, you can see the hydrogen bonding pattern is much different.
The Plexor® assay uses two primers that are specific for the target of interest. One of the primers
contains a 5’ modified nucleotide (iso-dCTP) linked to a fluorescent label. The second primer is
unlabeled. The fluorescently labeled C residue only pairs with iso-dG, not regular G residues.
The Plexor® primer and the normal downstream primer begin the process of replicating the DNA
sequence of interest into new double-stranded template (Figure 3). The process is fed by
conventional dNTPs, as with any amplification. At the end of the amplimer the polymerase is
confronted with the isoC base. The Plexor® System master mix includes iso-dGTP bound to the
quencher dabcyl. In subsequent rounds of PCR, iso-dG is incorporated into the new DNA strand
opposite from iso-dC, bringing the quencher into close proximity with the fluorescent dye,
resulting in very efficient quenching of the fluorescent reporter. The fluorescent signal decreases
in direct proportion to the amount of PCR product made. The number of cycles required to reach
a significant decrease in fluorescence, the cycle threshold (Ct), is dependent on the amount of
32

template DNA present. The amount of template DNA can be quantitated by comparison to a
standard curve generated from known amounts of target DNA. As a quality check, the Plexor®
Systems allow you to measure the melting temperature of the PCR products. Homogeneous
product creates a well defined melting curve. The Plexor® methodology lends itself to multiplex
real-time amplification. A single Plexor® reaction can contain multiple Plexor® primer sets; each
primer pair is specific to a different target sequence, and labeled with different fluors. The
dabcyl-iso-dGTP in the Plexor® master mix will quench the fluorescence of all the dyes present
in the reaction.
Real-time qRT-PCR was performed on a Qiagen Rotor-Gene Q. Five microliters of RNA was
amplified in a total reaction volume of 20 µl. Each Stain ID reaction included 10 µl of Stain ID
2X Master Mix, 0.4µl of RNasin RNase Inhibitor, 0.16 µl of ImProm-II RT, 1 µl of Stain ID
20X Primer Mix and 3.44 µl of nuclease-free water. Real-time RT-PCR cycling conditions
consisted of 45 ˚C for 5 min, 95 ˚C for 2 min, followed by 38 cycles of 95 ˚C for 5 sec and 60˚C
for 35 sec (acquiring to FAM, VIC/JOE and ROX). This was followed by 95 ˚C for 5 sec, a 50 –
95 ˚C ramp (acquiring to FAM, VIC/JOE and ROX) and 50 ˚C for 30 sec. Data analysis was
performed using Plexor Analysis Software.
DNA Quantitation
The TaqMan® duplex human/Y DNA quantitation assay was used to compare the DNA isolation
methods. This technique was developed by Nicklas and Buel (2006) and is based on PCR
amplification of the Alu sequence and the Y chromosome-specific DYZ5 sequence. A dilution
series of human genomic DNA ranging from 64 ng/µl to 0.0039 ng/µl, along with a negative
33

control is run with each assay. The RotorGene instrument uses the Ct values of the dilution
series to generate a standard curve from which the concentrations of the unknown samples are
automatically determined. Primers (Alu-F, Alu-R, DYZ-F, DYZ-R) and MGB probe (VIC-Alu,
FAM-DYZ5) were obtained from Applied Biosystems. All reactions were performed in a Rotor-
Gene Q (Qiagen) using 10 μl volumes and included 1X Absolute QPCR Mix (ABgene,
Rochester, NY), 100 uM Alu-F, 200 uM Alu-R, 200 uM DYZ5-F, 100 uM DYZ5-R, 200 uM
probe, and 160 ng/ul BSA. The PCR reaction was initiated with a 15 minute denaturation at 95
ºC followed by 40 cycles at 95 ºC for 30 seconds and 60 ºC for 1 minute.
STR Analysis
DNA was amplified using the PowerPlex® 16 HS Amplification System from Promega
(Madison, WI) in a GeneAmp PCR System 9700 (Applied Biosystems). The PowerPlex® 16 HS
kit amplifies 15 loci (D3S1358, THO1, D21S11, D18S51, Penta E, D5S818, D13S317, D7S820,
D16S539, CSF1PO, Penta D, vWA, D8S1179, TPOX, FGA) and amelogenin. PCR reactions
were performed as per manufacturer’s protocol, using 7.5 µl of sterile dH2O per reaction and 10
µl of normalized sample (~0.8 ng total DNA) in a 25 ul reaction volume using a 10/20 cycling
parameter. One µl of each amplification product was analyzed on an ABI 3130 Genetic
Analyzer (Applied Biosystems) as per manufacturer’s protocol, using software 3130 Data
Collection v3.0 (Applied Biosystems) and GeneMapper® ID v3.2 Software (Applied
Biosystems) to analyze the data.
34

III. Results
1. Statement of Results
Aim #1: To select the best method of DNA/RNA co-extraction from a wide variety of stain types.
Our first goal was to identify the best method to co-extract DNA and RNA from a variety of
stain types. Through the course of our previous grant (2004-DN-BX-K002), we tried several
commercial extraction kits and homebrew methods which had various degrees of success. A
major selling point of RNA-based stain identification assays will need to be the development of a
co-isolation method for RNA and DNA extraction. By utilizing one extraction step, a DNA
sample would be ready and waiting for STR profiling if the RNA screening assay deemed it
worthy of such analysis. In addition, obtaining RNA and DNA from a single stain would prevent
the possibility of conclusions being drawn regarding the identity of one stain which may not hold
true for a nearby stain. Therefore, a significant amount of time was spent optimizing a procedure
which would co-extract the two nucleic acids so that they were of sufficient quality and quantity
for downstream analyses.
Based on preliminary experiments, the TRIzol® method was identified as an efficient and
straight-forward procedure for the isolation of DNA and RNA. However, despite the success of
the TRIzol® extractions, there are several disadvantages which led us to actively seek an
alternative isolation procedure. The TRIzol® reagent has the capacity to isolate RNA and DNA;
but this requires essentially two extraction pathways following the first initial steps. Therefore,
35

we sought a procedure which would be less labor-intensive and yield sufficient amounts of high-
quality nucleic acids.
Through collaboration with Promega we developed a homebrew extraction method that would
work for RNA and DNA. The optimization experiments for this Tris-based phenol method were
detailed in the Final Report for 2004-DN-BX-K002. The final protocol which yields sufficient
quality and quantity DNA and RNA for downstream analyses is detailed below:
To the sample, 100 µl of RNagents denaturation solution, 90 µl of 1X PBS and 10 µl of 20
mg/ml proteinase K is added. The sample is vortexed and incubated for 10 minutes at 56°C.
The sample is transferred to a spin basket and centrifuged at max speed for 2 minutes. To the
flow-through, 20 µl of 2M sodium acetate is added and mixed followed by the addition and
mixing of 12 µl of 1M Tris (pH 9.0). A volume of 230 µl phenol:chloroform:isoamyl alcohol is
added and the sample vortexed and centrifuged at max speed for 5 minutes. The aqueous phase
is removed into a new tube and 220 µl of chloroform:isoamyl alcohol is added and vortexed.
The sample is centrifuged at max speed for 5 minutes and the aqueous phase is removed into a
new 1.5 mL tube. Four µl of 5 µg/µl glycogen and 210 µl of isopropanol is added and vortexed.
The sample is centrifuged at max speed for 10 minutes and following removal of the supernatant,
the sample is washed with 1 mL of ice-cold 95% ethanol. Centrifuge at max speed for 5 minutes
and remove supernatant and wash with 200 µl of 75% ethanol. Centrifuge at max speed for 5
minutes and remove supernatant and allow pellet to air dry for 1 minute. The pellet is
resuspended in 30 µl of TE-4 and stored at -20°C.
36

Since the majority of the preliminary experiments involved comparing RNA yields to alternative
methods, we wanted to compare the DNA yields generated from the co-isolation method with the
gold-standard organic method used by our laboratory for casework samples. Five different
samples prepared in duplicate were extracted using the Tris-based phenol and organic methods
and the resulting extracts were quantitated for total human DNA using an Alu-based real-time
PCR assay. As shown in Table 2, with the exception of the blood sample, the yields were fairly
comparable and more than sufficient to perform downstream STR amplification. It appears as
though extraction of the blood sample using the organic method didn’t work and would need to
be repeated before conclusions can be drawn for that comparison. In addition, although the
quantity of DNA generated from the two methods is comparable, the quality needs to be assessed
by STR analysis.
Evidence from sexual assault cases often involves swabs or clothing containing a mixture of
female and male contributions (i.e. mixtures). In these instances, it is helpful to separate the
female and male DNA prior to STR analysis. Currently, most labs use a differential lysis
procedure to achieve this separation. Because the Tris-based phenol protocol as previously
described provides a single extract containing all DNA, we adapted the Tris-based phenol
process to produce epithelial and sperm extracts. To achieve this, we added an up front 2-hour
incubation with a female extraction buffer. Following the incubation, the samples were
centrifuged and the supernatant (containing lysed epithelial cells) was removed. In this
experiment, the sperm pellet was not extracted further, but could easily be if needed. Instead,
DNA in the supernatant was further isolated using the Tris-based phenol process. Real-time
PCR was performed on the resulting extracts to determine the DNA yield and whether RNA
could be detected. Interestingly, Table 3 demonstrates that RNA survives the initial 2-hour
37

incubation since PRM2 was detected in the samples containing seminal fluid. Furthermore, the
DNA yields were sufficient to perform downstream analysis (i.e. STRs). Together these results
indicate that the adapted Tris-based phenol procedure may be a viable extraction method for
samples containing both male and female components. Further research is necessary to compare
the quality and quantity of the nucleic acids isolated via the adapted method with the
conventional procedures for DNA and RNA extraction (i.e. differential method, Tris-based
phenol).
Aim #2: To find 2-3 genes specific for numerous stain and tissue types.
Survey of literature including PubMed, Gene and other databases have allowed for the selection
of several genes that appear to be specific for the various fluids and tissues assessed during the
course of this grant (i.e. brain, semen, sperm). For semen, we are interested in mRNAs specific
for the sperm as well as for the prostatic components. In a number of cases, it is important to
determine if semen is present even if the male is sterile or has had a vasectomy. It other cases, it
is important to know if sperm are likely to be present. The following alphabetical list is a brief
description of each gene target utilized over the course of the project:
ACPP (acid phosphatase, prostate) - enzyme which catalyzes the conversion of orthophosphoric monoester to alcohol and orthophosphate; secreted by the epithelial cells of the prostrate gland ADCY1 (adenylate cyclase 1, brain) - encodes a form of adenylate cyclase expressed in brain B2M (beta 2 microglobulin) - serum protein found in association with the major histocompatibility complex class I heavy chain on the surface of nearly all nucleated cells CRISP1 (cysteine-rich secretory protein 1) - expressed in the epididymis and plays a role at fertilization in sperm-egg fusion
38

DRD1 (dopamine receptor 1) - G-protein coupled receptor stimulates adenylyl cyclase and activates cyclic AMP-dependent protein kinases; regulate neuronal growth and development, mediate some behavioral responses, and modulate dopamine receptor D2-mediated events GPM6A (glycoprotein M6A) - stress-responsive gene involved in hippocampal formation MSMB (microseminoprotein, beta) - synthesized by the epithelial cells of the prostate gland and secreted into the seminal plasma PRM2 (protamine 2) - major DNA-binding protein in the nucleus of sperm and packages DNA PSA (kallikrein 3) - protease present in seminal plasma involved in liquefaction of seminal coagulum SEMG1 (semenogelin I) - involved in formation of gel matrix that encases ejaculated spermatozoa TGM4 (transglutaminase 4) - catalyzes the cross linking of proteins and the conjugation of polyamines to specific proteins in the seminal tract
Applied Biosystems has created TaqMan® (Figure 1) primer/probe sets for many human genes
(Assay-on-DemandTM products). These have been carefully designed and thoroughly tested for
specificity. Most of these sets are mRNA specific (cross exon-exon boundaries and have no
cross-reaction with pseudogenes) although some do react with genomic DNA. Since these sets
are available, optimized and cost only $150 each, we decided to use these pre-designed sets for
our initial studies rather than expend the time and resources necessary to design our own. Table
1 lists the Assay-on-DemandTM numbers for each of the genes of interest. The mRNA specific
assays (probe spans an exon/exon junction) end in “_m1”. Those ending in “_g1” are not
guaranteed to be mRNA specific.
In order for a candidate to be implemented into a stain identification assay, it needs to be
detectable for years following deposition of the sample. One of the earliest experiments we
performed was to see whether amplifiable RNA could be detected from an aged semen sample.
One or twenty µl seminal fluid stains aged for 2 weeks to 2 years were easily detected using the
39

PRM2 assay (Table 4) showing that expression of this target is maintained in samples aged over
2 years.
We tested the specificity and sensitivity of each assay by analysis of mRNA isolated from the
body fluid of interest. In addition to the target of interest, we tested the assay on mRNA isolated
from other fluids and tissues to demonstrate that the assay is specific. We obtained anonymous
samples of a variety of tissues (kidney, colon, adipose, skin) from the Surgical Pathology Service
at Fletcher Allen Health Care. Lastly, control RNAs (liver, kidney, brain, heart, intestine) were
purchased from Ambion to ensure that our assays are specific only for the intended tissue.
To test the specificity of the TaqMan® sets (only the control B2M and tissue-specific sets should
give amplification), mRNA was isolated from dried blood, semen, saliva, menstrual blood or
vaginal secretions using TRIzol®. Human tissues were extracted using the Absolutely RNA® Kit
and control human RNAs from Ambion were diluted to 100 ng/µl. The RNA samples were then
reverse transcribed and PCR performed with each of the TaqMan® sets. The amplification
results for the semen and sperm assays are depicted in Table 5. For the 6 semen assays there
were mixed results. PSA, SEMG1, MSMB and TGM4 all appeared to be specific for semen
when tested against blood, semen and saliva. But upon further investigation, SEMG1 cross-
reacted with vaginal secretions and kidney. Interestingly, MSMB amplified from a control
intestine sample, but not with human colon tissues. ACPP and CRISP1, although ideal
candidates based on literature searches, were not suitable assays for the specific detection of
semen. The sperm marker PRM2 amplified from a seminal fluid sample, but nothing else that
was tested. The control B2M was detected in each of the fluids and tissues. Based on these
results, TGM4 and PRM2 have demonstrated their specificity for semen/sperm.
40

The sensitivity of the candidate assays for semen (TGM4) and sperm (PRM2) was evaluated
using a range of semen volumes spotted onto cotton cloth. In the first experiment (TGM4),
between 1 µl and 20 µl of semen was allowed to air dry at room temperature for 41 days prior to
extraction using TRIzol®. In a subsequent experiment (PRM2), between 0.01 µl and 20 µl of
semen was allowed to air dry at room temperature for 8 days prior to extraction using TRIzol®.
cDNA was amplified using the TaqMan® assays. Both TGM4 and PRM2 expression was
detected in the lowest sample volumes tested (i.e. 1 µl for TGM4 and 0.01 µl for PRM2)
indicating that they are very sensitive assays (Table 6). But, since the two lowest volumes,
namely 0.01 µl and 0.1 µl, were not analyzed with the TGM4 assay, the lower limits are
unknown, but assumed to be lower than 1 µl based on the Cts.
The specificity studies using the tissue assays for brain yielded mixed results (Table 7). All three
of the brain assays amplified from the control brain sample as expected, but also had varying
degrees of cross-reactivity with other control tissues. DRD1 had a Ct value of 24.90 with brain,
but Cts of 28.62 and 29.86 with liver and intestine, respectively. GPM6A had a Ct value of
19.89 with brain, but a Ct of 24.76 with liver. ADCY1 had a Ct value of 19.59 with brain, but
Cts of 23.39 and 23.78 with heart and kidney, respectively. The Cts for ADCY1 and GPM6A
were the lowest for brain samples out of the three assays which indicates that they are expressed
to a higher extent than DRD1.
The sensitivities of the candidate assays for brain were evaluated using a range of control brain
RNA (1 pg/µl to 1 µg/µl). The various concentrations of RNA were reverse transcribed and the
resulting cDNA was amplified using the TaqMan® assays. ADCY1 was the most sensitive of the
41

three assays tested being detected in 10 pg of brain RNA; similar to the control B2M target
(Table 8). Alternatively, GPM6A expression was detected in 100 pg of brain RNA and DRD1
expression was detected in 1 ng of RNA.
Aims #3 and #4: To develop multiplex assays for the identification of tissue and stain types & To
validate these assays for forensic casework.
A major aim of stain identification using mRNA expression profiling is working towards
multiplexing the real-time PCR assays once mRNAs are identified that clearly define specific
types of stains. Since these assays are designed to function more qualitatively than
quantitatively, a test of a single stain should typically give amplification with only one candidate.
It is possible that a mixture could give several amplifications. However, it is of greater concern
to know that a mixture does exist, than to know the exact amount of each fluid present.
Plexor® Analysis
One methodology to achieve this goal is the Plexor® system from Promega. Depending on the
dye-capability of the real-time instrument that is utilized, this system allows up to six mRNAs to
be multiplexed in one assay, thus reducing the amount of sample needed and time of analysis.
The Plexor® qRT-PCR system takes advantage of the specific interaction between two modified
nucleotides to achieve quantitative PCR analysis (Figure 2). Promega’s Plexor® design software
allows the generation of primers and probes which span an intron by designating a certain base to
include in the primer/probe design.
42

In our previous grant (2004-DN-BX-K002) we described an ongoing collaboration with Promega
to develop stain identification assays using their Plexor® platform. The combination of
Promega’s extensive knowledge of the Plexor® platform with our sample inventory and insight
into the needs of the forensic laboratory forged a strong partnership. The goal was to generate
Plexor® Tissue Typing Systems; multiplexed qRT-PCR systems for determining the source and
quantity of a variety of human stains and/or tissues. The systems would include Plexor® primers
for the detection of tissue-specific mRNA transcripts associated with semen, sperm, blood,
menstrual blood, saliva, etc. By limiting the initial system to two-color detection (i.e. FAM and
HEX detection), it would be compatible with the majority of real-time thermal cyclers in forensic
laboratories. We sought to explore the potential to include controls (e.g. a housekeeping gene) or
multiple, tissue-specific targets using the ability of the Plexor® Data Analysis Software to
distinguish two different amplicons in the same dye channel, based upon thermal melt properties.
The Stain ID assay which we decided to first focus on was for detection of semen and saliva.
Plexor® primers were designed and optimized to amplify the targets TGM4 (semen), HTN3
(saliva) and GAPDH (housekeeper). The preliminary experiments were not performed using
sample types and sizes which are reflective of forensic-type samples. The RNA samples which
were extracted in-house included vaginal secretions, whole blood, semen and saliva. In all cases,
RNA was extracted from extremely large sample sizes (i.e. 2 mL of semen, 10 mL of saliva, 5
mL of whole blood) and the samples were all fresh. These were obviously not reflective of
actual casework samples, but intended to isolate a large pool of RNA that experiments could be
performed with. The semen, saliva and whole blood RNA samples were extracted with phenol-
chloroform and the vaginal secretion was extracted using Promega’s PureYield RNA midiprep
kit. After extraction, the RNA was quantitated and 100 ng of RNA was used per reaction.
43

Titration experiments were performed and indicated that semen had a detection threshold of
approximately 10 pg of RNA whereas the threshold for saliva was approximately 1 ng of RNA.
Although these studies gave an idea as to the relative sensitivities of the assay, it was important
to assess how the assay would perform with typical forensic samples (i.e. various volumes of
unknown RNA yields).
At the time we were writing the Final Repot for Grant 2004-DN-BX-K002, Promega was in the
process of generating a draft Technical Manual that would accompany the Stain ID kit. In
conjunction with the Stain ID assay, Promega developed analysis software based on the expected
target melt temperatures of the three targets. Following the real-time PCR run the analyst sets
the expected target melt temperature and range for each of the three dye channels. This range is
instrument dependent however Promega provides an average range for each of the targets. The
melt threshold is the level of signal that must be reached for the software to “call” the melt
results. A “yes” or “no” indicates whether a sample Tm is within the expected target melt
temperature range. A “no call” indicates that the melt curve displays the expected target melt
temperature, but there is insufficient amplification product to cause the melt curve to cross the
melt threshold.
Once the draft Technical Manual was completed, we alpha-tested the assay using a large panel of
in-house samples including: blood, menstrual blood, saliva, semen, urine, vaginal secretions,
buccal swabs on FTA®, kidney, adipose, colon and skin. In addition, mixtures of blood/saliva,
blood/semen, and saliva/semen were tested and multiple samples were used that ranged in age
and size. The samples were extracted using the Tris-buffered phenol protocol outlined above.
The results for this study can be found in Table 9. The results demonstrated that the assay was
44

indeed specific and sensitive. Amplification of the HTN3 and TGM4 targets only occurred in
samples composed of saliva and semen, respectively, whereas all of the samples (with the
exception of the negative controls) showed amplification of GAPDH. The only exception was a
lack of GAPDH amplification in the 1 µl urine samples which is likely due to the minute amount
of sample remaining on the substrate at the time of extraction. Non-specific amplification wasn’t
detected in any of the samples.
Promega invited a number of forensic labs from across the county to participate in an alpha test
of the prototype kit. As part of this process, Promega included a series of blind swabs which
were provided to all of the test sites. There were a total of eight swabs; four different swabs
prepared in duplicate. The swabs were extracted using the Tris-buffered phenol protocol and the
Stain ID results can be found in Table 10. Swabs A1/A2 contained 10 µl of semen, Swabs
B1/B2 contained 10 µl of saliva, Swabs C1/C2 contained 1 µl of blood and Swabs D1/D2 were
vaginal swabs spiked with 10 µl of semen. Based solely on the software calls, the results were as
expected with the exception of samples D1 and D2. By looking at the raw data (i.e. Cts), both
D1 and D2 show strong positives for semen and GAPDH, but the Tms drifted slightly below the
control melting curve and thus, were not called “yes” by the software. Scientists at Promega
have seen this happen on occasion, particularly with swabs that have a lot of sample material
dried onto them. Their theory is that perhaps something is being carried over from the extraction
process in those samples and slightly affects the melt temperature. A protocol for removal of
these impurities is included in the manual accompanying the Stain ID kit.
The usefulness of the Stain ID assay was tested using additional mock and actual casework
samples. Following serological confirmatory testing (i.e. microscopic sperm search), residual
45

extracts (some containing the original cuttings) were saved at -20 ˚C until nucleic acid isolation
was performed using the Tris-buffered phenol protocol. In additional, vaginal swabs (with and
without seminal fluid contributions) and a skin swab from a female donor following male
salivary deposition were included in the experiment. Table 11 shows a comparison of the
serological and Stain ID results. Several of the casework samples which were found to contain
sperm tested negative for TGM4 in the Stain ID Assay (#2, #3, #4, #7). The cause for these false
negatives warrants further examination. In addition to examining the RNA component isolated
from these samples, the co-isolated DNA was amplified for STR analysis to determine if
amplifiable DNA is produced using this procedure. The STR data following amplification with
the PowerPlex 16 kit is summarized in Table 11. The results indicate that the co-isolation
method is capable of producing DNA of sufficient quantity and quality for downstream STR
analysis.
The next step was to produce data for a validation paper using Stain ID materials that were made
and QC’d by the manufacturing department at Promega. Furthermore, these samples would also
be evaluated for DNA yields and generation of STR profiles. However, as we were anxiously
awaiting the next steps, Promega put a hold on this project based on feedback from the alpha
testers. There were questions regarding the extraction method, requests for additional markers in
the assay, and general concerns as to the marketability of the assay. To date, no further work has
been performed on the Stain ID assay here at the VFL.
46

TaqMan® Analysis
As stated previously, one of the disadvantages to using the Gene Expression TaqMan® assays
from Applied Biosystems is the inability to multiplex the assays since they are all labeled with
the same dye. Therefore, we set out to design our own TaqMan® probes and primers in hopes of
developing several specific multiplex assays. Work on these assays began with Grant 2004-DN-
BX-K002 and was described in the Final Report. The first multiplex we designed was a semen-
sperm detection assay. Often, it is important to determine if semen is present even if the male is
sterile or has had a vasectomy (i.e. no sperm). In other cases, it is important to know if sperm are
present. An assay that could determine whether the stain 1) is seminal fluid and 2) contains
sperm could alleviate the need to perform extensive microscopy for the identification of sperm.
The semen-sperm assay uses the TGM4 (CAL Orange 560; HEX) and PRM2 (FAM) markers in
addition to the housekeeper B2M (CAL Red 610; ROX). Using the Beacon Designer program
we designed a set of probes and primers which were ordered from Biosearch Technologies.
Initially, experiments were conducted to optimize the amount of probe and primer for each gene
of interest. In monoplex reactions, 100 nM, 200 nM, 300 nM, 400 nM or 500 nM of primers
were combined with 200 nM of the corresponding probe and tested using semen cDNA. Once
the ideal primer concentrations were determined, the amount of probe in the reaction was
optimized using 100 nM, 200 nM and 300 nM.
Once these experiments were carried out for all three candidates, the optimized amounts of
primers and probes were combined into a single multiplex reaction and compared to singleplex
reactions. Preliminary data reported in Table 23 of the Final Report for 2004-DN-BX-K002
47

shows that while each primer/probe set amplified from semen when alone in the reaction, the
combination into a multiplex reaction caused the dropout of TGM4 and B2M amplification.
Alternatively, the amplification using the PRM2 primer/probe set was unchanged regardless of
the reaction conditions. Since PRM2 is highly expressed in seminal fluid samples, it may be
competing against the other sets for reaction components. Therefore, we decreased the primer
and probe conditions for this target. As shown in Table 12, changing the amount of PRM2
primers and probes was successful in preventing the loss of TGM4 and B2M amplification in the
multiplex reaction. In an attempt to decrease the Ct for B2M amplification, the primer and probe
concentrations were altered, which only had a minor effect on the results (Table 13).
In other studies we have seen that changing the master mix used in this reaction can have an
impact on the quality of the results. To date, we had been using the Universal PCR Master Mix
from Applied Biosystems for our TaqMan studies. We compared this master mix to 7 other
commercially available master mixes to determine whether amplification could be improved for
the multiplex assay. Table 14 summarizes this study. Use of three of the master mixes caused
near or complete loss of amplification (Eppendorf HotMaster Mix, ABgene Absolute QPCR
Mix, Statagene Full Velocity QPCR Master Mix). We decided to add BSA to the reactions to
see if it would have a beneficial effect on amplification. Multiplex reactions were performed
using five of the master mixes which showed potential in the previous experiment. Findings
from the BSA study can be found in Table 15. The addition of BSA did increase amplification
in some of the reactions (i.e. lower the Cts). The best overall amplification was when the Sigma
JumpStart Taq ReadyMix was used. The addition of BSA to this mix had no additional effect on
the results and was not included in the assay.
48

The optimized semen-sperm assay was tested for its sensitivity and specificity. First, three year
old seminal fluid samples ranging from 0.01 – 20 µl were extracted using TRIzol. cDNA was
generated by reverse transcription and real-time PCR performed in multiplex reactions using the
Sigma mastermix. Amplification of both seminal fluid markers occurred in the lowest volume
sample tested which demonstrates the sensitivity of this assay since the control target B2M was
only detected down to the 0.1 µl sample (Table 16). The specificity of the assay was assessed
using a panel of cDNAs prepared from a variety of RNAs extracted from body fluids (blood,
semen, saliva, vaginal secretions, menstrual blood), tissues (kidney, colon, adipose, skin) and
control RNAs (brain, heart, liver, kidney, intestine). Unfortunately, there was nonspecific
amplification of PRM2 in the saliva, vaginal secretions, menstrual blood, kidney and colon
samples (Table 17). To be thorough, this experiment was repeated, but the outcome was the
same (data not shown). At this point, a decision was made to combine the “homebrew” TGM4
and B2M primer/probe sets with the Gene Expression TaqMan® PRM2 assays from Applied
Biosystems.
In order to test the new triplex containing the commercial PRM2 assay, we compared the original
“homebrew” multiplex assay with the new modified triplex and the PRM2 stand-alone assay
from Applied Biosystems. As shown in Table 18, amplification of TGM4 and B2M was lost
with the modified homebrew assay. We decided to perform a titration of the amount of PRM2
assay added to the modified triplex in the hopes that the other targets would amplify with less
PRM2 present in the reaction. Indeed, decreasing the amount of PRM2 assay added to the
reaction led to an increase in amplification of TGM4 (Table 19). However, amplification of
B2M in the modified mutiplex was still decreased compared to the original homebrew assay.
When this last experiment was performed, the gains on the Rotor Gene instrument were not
49

changed to the settings typically used for these reactions. Therefore, we repeated part of this
experiment using the 0.2 µl of PRM2 assay in the modified multiplex with the notion that
correcting the gains would improve the data. Unfortunately, correcting the gains on the
instrument didn’t improve amplification of B2M (Table 20). One last ditch effort was made to
save this modified homebrew assay; three master mixes were tried in order to improve the
amplification of B2M. The best result, as determined by amplification of all three targets, was
with samples run using the QuantiTech Multiplex PCR Kit (Qiagen) (Table 21).
The sensitivity of the modified semen-sperm assay was evaluated using a range of semen
volumes (0.01 – 20 µl) spotted onto cotton cloth. The samples were extracted using TRIzol and
the resulting RNA was reverse transcribed and amplified with the modified triplex assay. A dCt
was determined for each of the samples amplified with the semen-sperm assay by subtracting the
body fluid gene (PRM2 or TGM4) from the housekeeping gene (B2M) Ct value. Based on the
dCt values, PRM2 expression was positively identified at the lowest sample volume tested,
whereas the presence of TGM4 was detected in the 0.1 µl semen sample (Table 22). To test the
specificity of the modified semen-sperm assay, a panel of RNAs isolated from various body
fluids (blood, semen, saliva, vaginal secretions, menstrual blood), tissues (kidney, colon,
adipose, skin) and control RNAs (brain, heart, liver, kidney, intestine) were reverse transcribed
and PCR performed. The amplification results are depicted in Table 23 and show that the only
positive dCt values were for the semen sample. Every other body fluid or tissue tested had a
negative dCt and was therefore negative for the presence of semen/sperm. Based on these
results, the semen-sperm assay appears to be sensitive and specific for seminal fluid.
50

One TaqMan®-based multiplex assay which we sought to develop was for the identification of
brain tissue. Based on our preliminary studies using the Gene Expression Assays from Applied
Biosystems, we identified the genes ADCY1 and GPM6A as potential tissue-specific candidates.
Again using the Beacon Designer program, we designed a set of probes and primers for ADCY1
(FAM), GPM6A (CAL Orange 560; HEX) and the housekeeper B2M (CAL Red 610; ROX)
which were ordered from Biosearch Technologies. Preliminary experiments were conducted to
optimize the amount of probe and primer for each gene of interest. In monoplex reactions, 100
nM, 200 nM, 300 nM, 400 nM or 500 nM of primers were combined with 200 nM of the
corresponding probe and tested using cDNA generated from control brain RNA (Ambion). Once
the ideal primer concentrations were determined, the amount of probe in the reaction was
optimized using 100 nM, 200 nM and 300 nM.
Once these experiments were carried out for all three candidates, the optimized amounts of
primers and probes were combined into a single multiplex reaction and compared to singleplex
reactions. Data published in the Final Report for Grant 2004-DN-BX-K002 (Table 24) showed
that each primer/probe set amplified from brain both when alone in the reaction and in
combination with the other primer/probe sets. Furthermore, there was no significant decrease in
the degree of amplification when the sets were alone or combined into the multiplex. There was
some minor amplification in the no template control sample when the ADCY1 primers/probe
were alone and when the GPM6A set was used. This experiment has since been repeated using
less of the GPM6A primers and probes in the reaction in order to alleviate some of this
amplification. Table 24 shows a significant decrease in the nonspecific amplification when less
of the GPM6A primers and probes are used. Although there is still some bleed through into the
HEX/GPM6A dye channel, this issue may be resolved by changing the gains (here were FAM 7;
51

HEX 9; ROX 9) on the instrument or use of a different master mix. This experiment was run a
third time using the improved primer/probe conditions, but the gains were changed (FAM 5;
HEX 9.33; ROX 9). Unfortunately, the change in gains did not resolve the bleed through issue
(Table 25). However, this multiplex looks very promising for a brain screening assay.
Additional studies using different master mixes may further improve the data and experiments
using human brain samples would aid in the evaluation of this multiplex as a viable screening
assay.
52

2. Tables
Table 1. Assays-on-Demand™ Gene Expression products used in the grant.
TISSUEsemenogelin I [SEMG1]
(Hs00268141_m1)kallikrein 3 [PSA] (Hs00426859_g1)
acid phosphatase, prostate [ACPP] (Hs00173475_m1)
cysteine-rich secretory protein 1[CRISP1] (Hs00538261_m1)
microseminoprotein, beta- [MSMB] (Hs00159303_m1)
transglutaminase 4 [TGM4] (Hs00162710_m1)
Spermprotamine 2 [PRM2] (Hs00172518_m1)
Braindopamine receptor 1 [DRD1]
(Hs00377719_g1)glycoprotein M6A [GPM6A]
(Hs00245530_m1)adenylate cyclase 1, brain [ADCY1]
(Hs00299832_m1)
Controlbeta 2 microglobulin [B2M]
(Hs99999907_m1)
Assays on Demand (ABI)
Semen
Table 2. Comparison of DNA yields from Organic and Tris-Based Phenol Methods.
SAMPLE ng/ul total ng ng/ul total ng
blood - 10ul 0.003 0.150 8.971 269.130semen - 1ul 0.710 35.500 6.293 188.790semen - 10ul 2.456 122.800 2.622 78.660saliva - 1ul 0.072 3.600 0.296 8.880
saliva - 10ul 1.297 64.850 5.491 164.730
ORGANIC TRIS-BASED PHENOL
DNA Results RNA Results
SAMPLE total DNA (ng/ul) male DNA (ng/ul) TaqMan-based PRM2 Assay (Ct)
semen - 5ul 0.343 0.276 26.91 vaginal swab 46.663 0.000 -
vaginal swab + semen - 5ul 74.127 0.187 33.11
Table 3. DNA and RNA yields following the Differential Extraction Procedure.
53
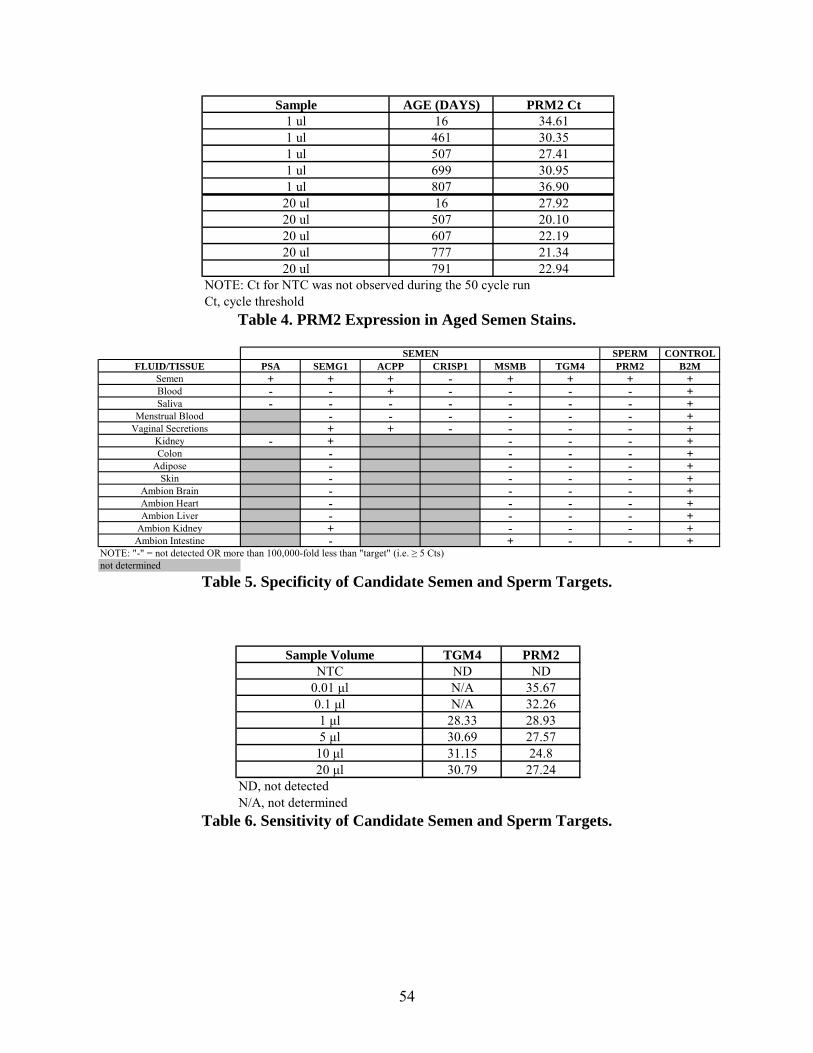
Table 4. PRM2 Expression in Aged Semen Stains.
Sample AGE (DAYS) PRM2 Ct1 ul 16 34.611 ul 461 30.351 ul 507 27.411 ul 699 30.951 ul 807 36.90
20 ul 16 27.9220 ul 507 20.1020 ul 607 22.1920 ul 777 21.3420 ul 791 22.94
NOTE: Ct for NTC was not observed during the 50 cycle runCt, cycle threshold
Table 5. Specificity of Candidate Semen and Sperm Targets.
SPERM CONTROLFLUID/TISSUE PSA SEMG1 ACPP CRISP1 MSMB TGM4 PRM2 B2M
Semen + + + - + + + +Blood - - + - - - - +Saliva - - - - - - - +
Menstrual Blood - - - - - - +Vaginal Secretions + + - - - - +
Kidney - + - - - +Colon - - - - +
Adipose - - - - +Skin - - - - +
Ambion Brain - - - - +Ambion Heart - - - - +Ambion Liver - - - - +
Ambion Kidney + - - - +Ambion Intestine - + - -
NOTE: "-" = not detected OR more than 100,000-fold less than "target" (i.e. ≥ 5 Cts)not determined
SEMEN
+
Table 6. Sensitivity of Candidate Semen and Sperm Targets.
Sample Volume TGM4 PRM2NTC ND ND
0.01 μl N/A 35.670.1 μl N/A 32.261 μl 28.33 28.935 μl 30.69 27.57
10 μl 31.15 24.820 μl 30.79 27.24
ND, not detectedN/A, not determined
54
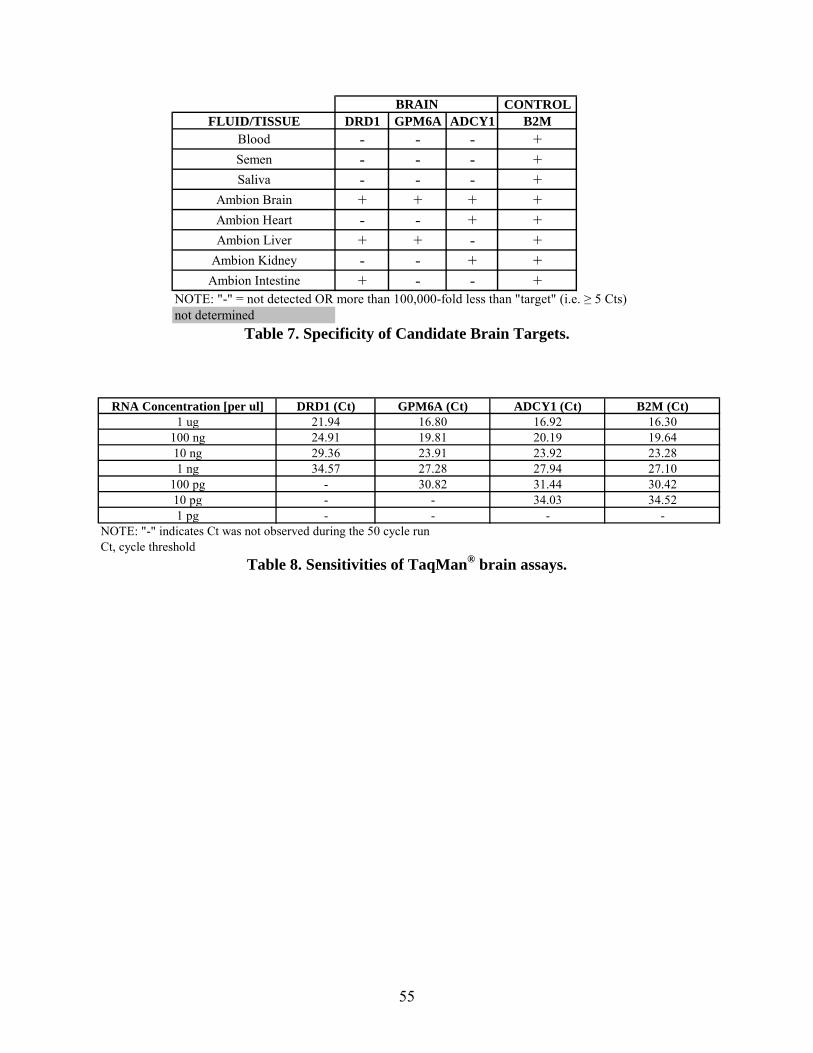
Table 7. Specificity of Candidate Brain Targets.
CONTROLFLUID/TISSUE DRD1 GPM6A ADCY1 B2M
Blood - - - +Semen - - - +Saliva - - - +
Ambion Brain + + + +Ambion Heart - - + +Ambion Liver + + - +
Ambion Kidney - - + +Ambion Intestine + - - +
NOTE: "-" = not detected OR more than 100,000-fold less than "target" (i.e. ≥ 5 Cts)not determined
BRAIN
Table 8. Sensitivities of TaqMan® brain assays.
RNA Concentration [per ul] DRD1 (Ct) GPM6A (Ct) ADCY1 (Ct) B2M (Ct)1 ug 21.94 16.80 16.92 16.30
100 ng 24.91 19.81 20.19 19.6410 ng 29.36 23.91 23.92 23.281 ng 34.57 27.28 27.94 27.10
100 pg - 30.82 31.44 30.4210 pg - - 34.03 34.521 pg - - - -
NOTE: "-" indicates Ct was not observed during the 50 cycle runCt, cycle threshold
55

Table 9. Detection of semen and saliva using a Plexor®-based Stain ID assay.
SAMPLE AGE HTN3 TGM4 B2MNTC - No No NoNTC - No No No
Positive Control - Yes Yes YesPositive Control - Yes Yes YesNegative Control - No No NoNegative Control - No No No
Blood (1ul) ~ 2 years No No YesBlood (1ul) ~ 2 years No No Yes
Blood (10ul) ~ 2 years No No YesBlood (10ul) ~ 2 years No No Yes
Blood/Saliva (5ul ea) ~ 3 years Yes No YesBlood/Saliva (5ul ea) ~ 3 years Yes No YesBlood/Semen (5ul ea) ~ 2 years No Yes YesBlood/Semen (5ul ea) ~ 2 years No Yes Yes
Buccal Swab ~ 2 years No No YesBuccal Swab ~ 2 years No No Yes
Menstrual Blood ~ 2 years No No YesMenstrual Blood ~ 2 years No No Yes
Saliva (1ul) ~ 3 years Yes No YesSaliva (1ul) ~ 3 years Yes No Yes
Saliva (10ul) ~ 3 years Yes No YesSaliva (10ul) ~ 3 years Yes No Yes
Saliva/Semen (5ul ea) ~ 3 years Yes Yes YesSaliva/Semen (5ul ea) ~ 3 years Yes Yes Yes
Semen (1ul) ~ 1 year No Yes YesSemen (1ul) ~ 1 year No Yes Yes
Semen (10ul) ~ 1 year No Yes YesSemen (10ul) ~ 1 year No Yes YesUrine (1ul) ~ 3 years No No No CallUrine (1ul) ~ 3 years No No No Call
Urine (10ul) ~ 3 years No No YesUrine (10ul) ~ 3 years No No Yes
Vaginal Swab ~ 3 years No No YesVaginal Swab ~ 3 years No No Yes
Kidney ~ 1 year No No YesKidney ~ 1 year No No YesAdipose ~ 1 year No No YesAdipose ~ 1 year No No YesColon ~ 1 year No No YesColon ~ 1 year No No Call YesSkin ~ 1 year No No YesSkin ~ 1 year No No Yes
.
56
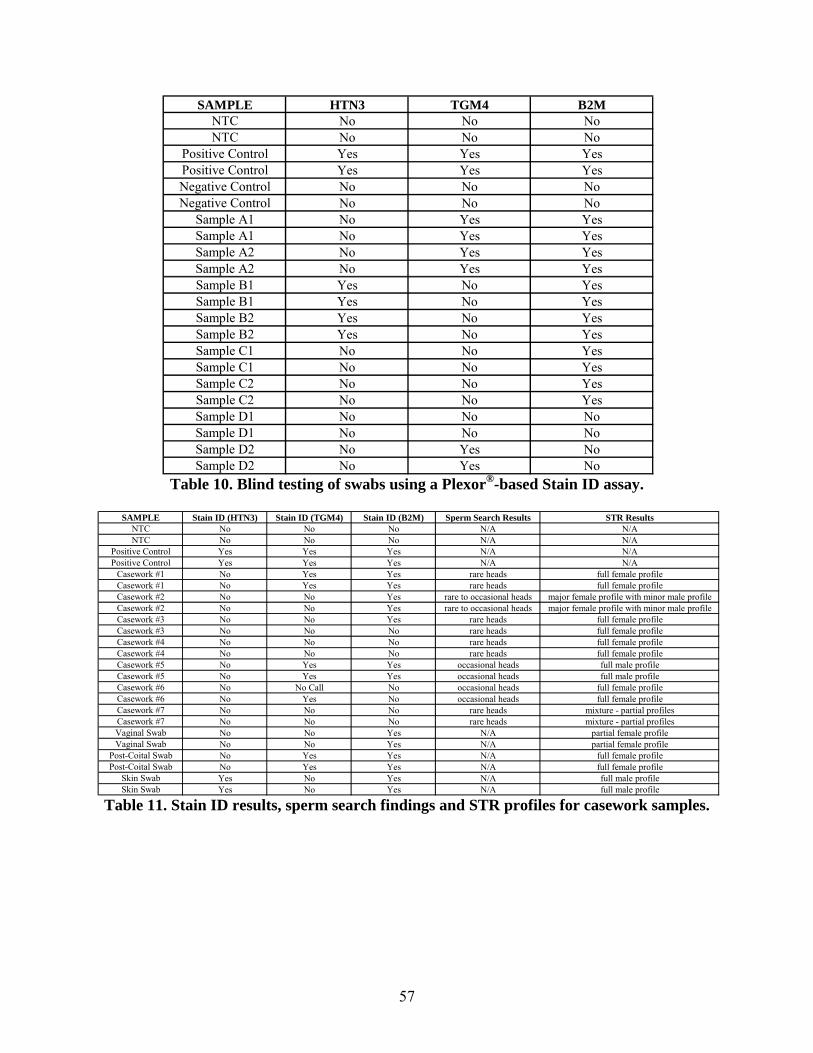
Table 10. Blind testing of swabs using a Plexor®-based Stain ID assay.
SAMPLE HTN3 TGM4 B2MNTC No No NoNTC No No No
Positive Control Yes Yes YesPositive Control Yes Yes YesNegative Control No No NoNegative Control No No No
Sample A1 No Yes YesSample A1 No Yes YesSample A2 No Yes YesSample A2 No Yes YesSample B1 Yes No YesSample B1 Yes No YesSample B2 Yes No YesSample B2 Yes No YesSample C1 No No YesSample C1 No No YesSample C2 No No YesSample C2 No No YesSample D1 No No NoSample D1 No No NoSample D2 No Yes NoSample D2 No Yes No
Table 11. Stain ID results, sperm search findings and STR profiles for casework samples.
SAMPLE Stain ID (HTN3) Stain ID (TGM4) Stain ID (B2M) Sperm Search Results STR ResultsNTC No No No N/A N/ANTC No No No N/A N/A
Positive Control Yes Yes Yes N/A N/APositive Control Yes Yes Yes N/A N/A
Casework #1 No Yes Yes rare heads full female profileCasework #1 No Yes Yes rare heads full female profileCasework #2 No No Yes rare to occasional heads major female profile with minor male profileCasework #2 No No Yes rare to occasional heads major female profile with minor male profileCasework #3 No No Yes rare heads full female profileCasework #3 No No No rare heads full female profileCasework #4 No No No rare heads full female profileCasework #4 No No No rare heads full female profileCasework #5 No Yes Yes occasional heads full male profileCasework #5 No Yes Yes occasional heads full male profileCasework #6 No No Call No occasional heads full female profileCasework #6 No Yes No occasional heads full female profileCasework #7 No No No rare heads mixture - partial profilesCasework #7 No No No rare heads mixture - partial profilesVaginal Swab No No Yes N/A partial female profileVaginal Swab No No Yes N/A partial female profile
Post-Coital Swab No Yes Yes N/A full female profilePost-Coital Swab No Yes Yes N/A full female profile
Skin Swab Yes No Yes N/A full male profileSkin Swab Yes No Yes N/A full male profile
57

Table 12. Detection of semen using a multiplex TaqMan® assay.
SAMPLE (PRIMER/PROBE SET USED)
FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
NTC (PRM2) ND ND NDSemen (PRM2) 21.70 ND NDNTC (TGM4) ND ND ND
Semen (TGM4) ND 26.45 NDNTC (B2M) ND ND ND
Semen (B2M) ND ND 31.73NTC (Multiplex) ND ND ND
Semen (Multiplex) 21.90 26.42 38.12ND = Not Detected
Table 13. Detection of semen using a multiplex TaqMan® assay.
SAMPLE (PRIMER/PROBE SET USED)
FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
NTC (PRM2) ND ND NDSemen (PRM2) 21.57 ND NDNTC (TGM4) ND ND ND
Semen (TGM4) ND 26.80 NDNTC (B2M) ND ND ND
Semen (B2M) ND ND 31.71NTC (Multiplex) ND ND ND
Semen (Multiplex) 22.49 26.65 35.65ND = Not Detected
Table 14. Detection of semen using a multiplex TaqMan® assay – master mix comparison.
SAMPLE (MASTERMIX USED) FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
NTC (Universal PCR Master Mix) ND ND NDSemen (Universal PCR Master Mix) 23.61 27.54 38.43
NTC (HotMaster Mix) ND ND NDSemen (HotMaster Mix) ND ND ND
NTC (ABsolute QPCR Mix) ND ND NDSemen (ABsolute QPCR Mix) 22.92 ND ND
NTC (Brilliant Multiplex QPCR Master Mix) ND ND NDSemen (Brilliant Multiplex QPCR Master Mix) 21.42 27.11 30.68
NTC (QPCR Mastermix Plus) ND ND NDSemen (QPCR Mastermix Plus) 21.66 28.22 ND
NTC (Full Velocity QPCR Master Mix) ND ND NDSemen (Full Velocity QPCR Master Mix) ND ND 35.65
NTC (QuantiTech Multiplex PCR Kit) ND ND NDSemen (QuantiTech Multiplex PCR Kit) 22.11 28.32 33.37
NTC (JumpStart Taq ReadyMix) ND ND NDSemen (JumpStart Taq ReadyMix) 22.38 26.64 30.82
ND = Not Detected
58
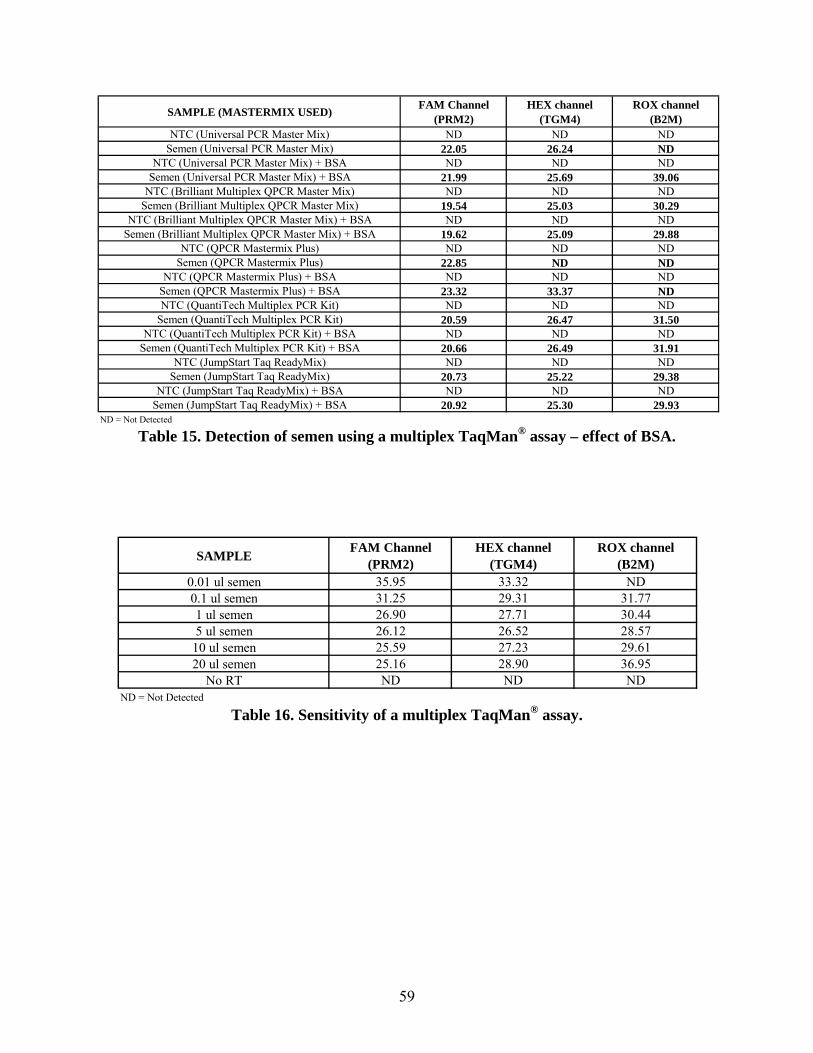
Table 15. Detection of semen using a multiplex TaqMan® assay – effect of BSA.
SAMPLE (MASTERMIX USED) FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
NTC (Universal PCR Master Mix) ND ND NDSemen (Universal PCR Master Mix) 22.05 26.24 ND
NTC (Universal PCR Master Mix) + BSA ND ND NDSemen (Universal PCR Master Mix) + BSA 21.99 25.69 39.06
NTC (Brilliant Multiplex QPCR Master Mix) ND ND NDSemen (Brilliant Multiplex QPCR Master Mix) 19.54 25.03 30.29
NTC (Brilliant Multiplex QPCR Master Mix) + BSA ND ND NDSemen (Brilliant Multiplex QPCR Master Mix) + BSA 19.62 25.09 29.88
NTC (QPCR Mastermix Plus) ND ND NDSemen (QPCR Mastermix Plus) 22.85 ND ND
NTC (QPCR Mastermix Plus) + BSA ND ND NDSemen (QPCR Mastermix Plus) + BSA 23.32 33.37 NDNTC (QuantiTech Multiplex PCR Kit) ND ND ND
Semen (QuantiTech Multiplex PCR Kit) 20.59 26.47 31.50NTC (QuantiTech Multiplex PCR Kit) + BSA ND ND ND
Semen (QuantiTech Multiplex PCR Kit) + BSA 20.66 26.49 31.91NTC (JumpStart Taq ReadyMix) ND ND ND
Semen (JumpStart Taq ReadyMix) 20.73 25.22 29.38NTC (JumpStart Taq ReadyMix) + BSA ND ND ND
Semen (JumpStart Taq ReadyMix) + BSA 20.92 25.30 29.93ND = Not Detected
Table 16. Sensitivity of a multiplex TaqMan® assay.
SAMPLE FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
0.01 ul semen 35.95 33.32 ND0.1 ul semen 31.25 29.31 31.771 ul semen 26.90 27.71 30.445 ul semen 26.12 26.52 28.57
10 ul semen 25.59 27.23 29.6120 ul semen 25.16 28.90 36.95
No RT ND ND NDND = Not Detected
59

Table 17. Specificity of a multiplex TaqMan® assay.
SAMPLE FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
Blood - 20 ul ND ND 17.97Semen - 20 ul 26.81 28.01 28.80Saliva - 20 ul 28.65 ND 25.82
Vaginal Secretions 26.07 ND 14.02Menstrual Blood 35.98 ND 28.67
Kidney 33.58 ND 16.71Colon 33.84 ND 15.84
Adipose ND ND 17.13Skin ND ND 15.98
Control Brain ND ND 18.03Control Heart ND ND 16.12Control Liver ND ND 16.10
Control Kidney ND ND 16.18Control Intestine ND ND 14.85
ND = Not Detected
Table 18. Detection of semen using a homebrew and modified homebrew TaqMan®-based
multiplex assay.
SAMPLE (PRIMER/PROBE SET USED)
FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
NTC (Original Homebrew) ND ND NDSemen (Original Homebrew) 21.75 25.75 30.62NTC (Modified Homebrew) ND ND ND
Semen (Modified Homebrew) 21.28 ND NDNTC (PRM2) ND ND ND
Semen (PRM2) 21.07 ND NDND = Not Detected
60

Table 19. Optimization of modified homebrew TaqMan®-based assay.
SAMPLE (PRIMER/PROBE SET USED)
FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
NTC (Original Homebrew) ND ND NDSemen (Original Homebrew) 20.88 25.32 32.18
NTC (Modified Homebrew - 0.5ul PRM2) ND ND NDSemen (Modified Homebrew - 0.5ul PRM2) 20.62 ND NDNTC (Modified Homebrew - 0.4ul PRM2) ND ND ND
Semen (Modified Homebrew - 0.4ul PRM2) 21.41 25.38 NDNTC (Modified Homebrew - 0.3ul PRM2) ND ND ND
Semen (Modified Homebrew - 0.3ul PRM2) 21.51 25.43 NDNTC (Modified Homebrew - 0.2ul PRM2) ND ND ND
Semen (Modified Homebrew - 0.2ul PRM2) 21.42 25.55 NDNTC (Modified Homebrew - 0.1ul PRM2) ND ND ND
Semen (Modified Homebrew - 0.1ul PRM2) ND 25.93 37.26NTC (PRM2 - 0.5ul) ND ND ND
Semen (PRM2 - 0.5ul) 21.45 ND NDND = Not Detected
Table 20. Further optimization of modified homebrew TaqMan®-based assay.
SAMPLE (PRIMER/PROBE SET USED)
FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
NTC (Original Homebrew) ND ND NDSemen (Original Homebrew) 21.07 25.60 30.32
NTC (Modified Homebrew - 0.2ul PRM2) ND ND NDSemen (Modified Homebrew - 0.2ul PRM2) 21.93 25.40 ND
NTC (PRM2 - 0.2ul) ND ND NDSemen (PRM2 - 0.2ul) 21.78 ND ND
ND = Not Detected
Table 21. Continued optimization of modified homebrew TaqMan®-based assay – master
mix comparison.
PRIMER/PROBE SET USED (MASTER MIX)
FAM Channel (PRM2)
HEX channel (TGM4)
ROX channel (B2M)
Original Homebrew (JumpStart Taq Ready Mix) 21.32 25.84 30.18Modified Homebrew (JumpStart Taq Ready Mix) 22.59 25.72 ND
PRM2 (JumpStart Taq Ready Mix) 22.05 ND NDOriginal Homebrew (ABsolute QPCR Mix) 20.11 25.41 31.24Modified Homebrew (ABsolute QPCR Mix) 20.46 25.80 ND
PRM2 (ABsolute QPCR Mix) 20.04 ND NDOriginal Homebrew (QuantiTech Multiplex PCR Kit) 20.07 26.65 31.12Modified Homebrew (QuantiTech Multiplex PCR Kit) 21.50 27.02 32.36
PRM2 (QuantiTech Multiplex PCR Kit) 20.90 ND NDND = Not Detected
61
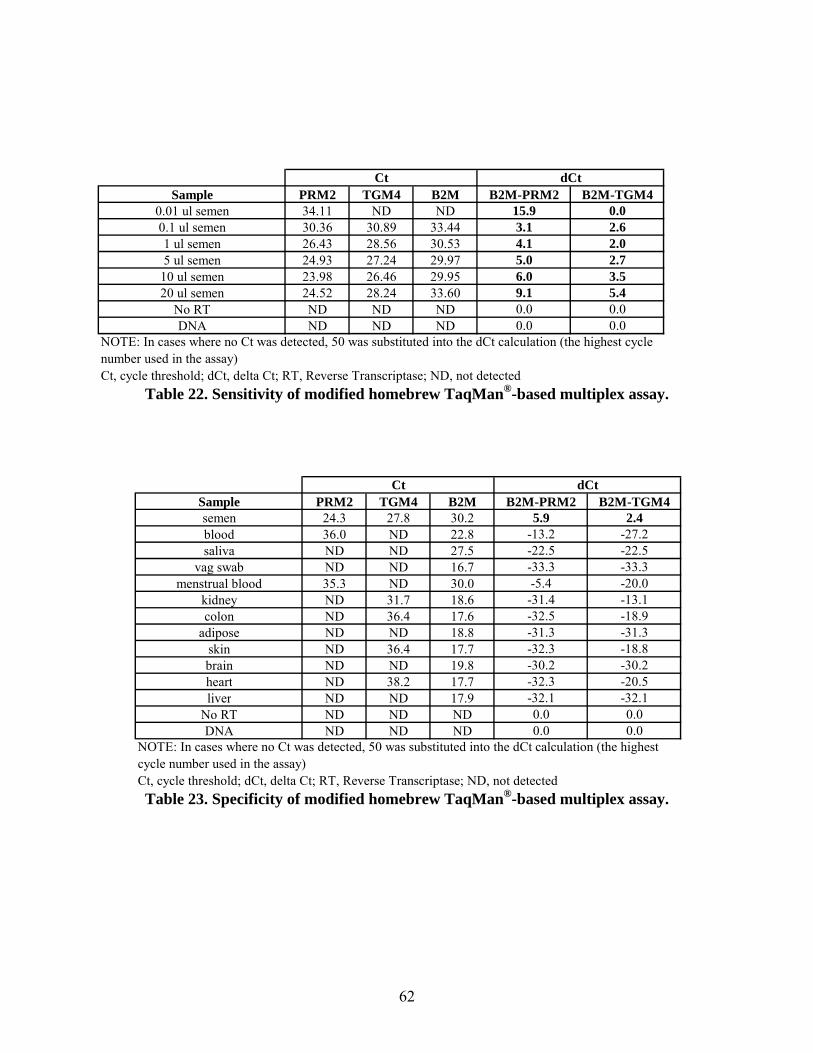
Table 22. Sensitivity of modified homebrew TaqMan®-based multiplex assay.
Sample PRM2 TGM4 B2M B2M-PRM2 B2M-TGM40.01 ul semen 34.11 ND ND 15.9 0.00.1 ul semen 30.36 30.89 33.44 3.1 2.61 ul semen 26.43 28.56 30.53 4.1 2.05 ul semen 24.93 27.24 29.97 5.0 2.7
10 ul semen 23.98 26.46 29.95 6.0 3.520 ul semen 24.52 28.24 33.60 9.1 5.4
No RT ND ND ND 0.0 0.0DNA ND ND ND 0.0 0.0
Ct, cycle threshold; dCt, delta Ct; RT, Reverse Transcriptase; ND, not detected
NOTE: In cases where no Ct was detected, 50 was substituted into the dCt calculation (the highest cycle number used in the assay)
Ct dCt
Table 23. Specificity of modified homebrew TaqMan®-based multiplex assay.
Sample PRM2 TGM4 B2M B2M-PRM2 B2M-TGM4semen 24.3 27.8 30.2 5.9 2.4blood 36.0 ND 22.8 -13.2 -27.2saliva ND ND 27.5 -22.5 -22.5
vag swab ND ND 16.7 -33.3 -33.3menstrual blood 35.3 ND 30.0 -5.4 -20.0
kidney ND 31.7 18.6 -31.4 -13.1colon ND 36.4 17.6 -32.5 -18.9
adipose ND ND 18.8 -31.3 -31.3skin ND 36.4 17.7 -32.3 -18.8brain ND ND 19.8 -30.2 -30.2heart ND 38.2 17.7 -32.3 -20.5liver ND ND 17.9 -32.1 -32.1
No RT ND ND ND 0.0 0.0DNA ND ND ND 0.0 0.0
Ct, cycle threshold; dCt, delta Ct; RT, Reverse Transcriptase; ND, not detected
NOTE: In cases where no Ct was detected, 50 was substituted into the dCt calculation (the highest cycle number used in the assay)
Ct dCt
62

Table 24. Detection of brain using a multiplex TaqMan® assay.
SAMPLE (PRIMER/PROBE SET USED)
FAM Channel (ADCY1)
HEX channel (GPM6A)
ROX channel (B2M)
NTC (ADCY1) ND ND NDBrain (ADCY1) 21.40 23.68 NDNTC (GPM6A) ND ND NDBrain (GPM6A) ND 16.91 ND
NTC (B2M) ND ND NDBrain (B2M) ND ND 19.14
NTC (Multiplex) ND ND NDBrain (Multiplex) 21.53 17.29 19.14
ND = Not Detected
Table 25. Detection of brain using a multiplex TaqMan® assay – Gain modifications.
SAMPLE (PRIMER/PROBE SET USED)
FAM Channel (ADCY1)
HEX channel (GPM6A)
ROX channel (B2M)
NTC (ADCY1) ND ND NDBrain (ADCY1) 22.00 23.86 NDNTC (GPM6A) ND ND NDBrain (GPM6A) ND 17.89 ND
NTC (B2M) ND ND NDBrain (B2M) ND ND 19.32
NTC (Multiplex) ND ND NDBrain (Multiplex) 21.80 18.53 18.99
ND = Not Detected
63

3. Figures
Figure 1. Schematic of TaqMan® based real-time PCR analysis. Each reaction contains a gene specific primer and a fluorescently labeled TaqMan® probe. The probe contains a 5’ reporter dye and a 3’ quencher dye. The probe is designed to anneal to the target sequence between the forward and reverse PCR primers. While the probe is intact, the quencher suppresses the fluorescence of the reporter dye. During amplification, Taq DNA polymerase cleaves the probe and displaces it from the target, allowing extension to continue. Cleavage of the probe separates the reporter dye from the quencher dye, resulting in an increase in fluorescence. The increased fluorescence only occurs if the target sequence is amplified and is complimentary to the probe, thus preventing detection of non-specific amplification.
64

Figure 2. Schematic of synthetic isoC and isoG bases developed by Eragen Biosciences. Eragen Biosciences developed and synthesized a series of new DNA base pairs. The Plexor® technology is based on one of these new base pairings: isoG and isoC. IsoC and IsoG nucleotides are incorporated by DNA Polymerase; however, neither isoC nor isoG can base pair with any of the other conventional bases. These two novel bases only interact and base pair with one another and are not found in nature. Although similar to the conventional G-C pair, you can see the hydrogen bonding pattern is much different.
G
C
isoG
isoC
G
C
isoG
isoC
65

Figure 3. Schematic of Plexor® chemistry. Plexor® takes a conventional PCR primer and adds the isoC base to the 5’ end of the primer. To this is attached a fluorescent reporter, by conventional 5’ end labeling methods. The Plexor® Primer and a normal downstream PCR primer begin the process of replicating the DNA sequence of interest. This process is fed by conventional dNTPs, as with any amplification. At the end of the amplimer, the polymerase is confronted with the isoC base. The Plexor® Master Mix contains isoG bases that are labeled with the quencher dabcyl. The pairing of the isoC and isoG bases brings the dabcyl quencher within close proximity of the fluor, resulting in very efficient quenching of the fluorescent reporter.
66

IV. Conclusions
1. Discussion of Findings
The first goal of this project was to identify the best method to simultaneously extract DNA and
RNA from a wide variety of stain types. This was a major objective since the purpose of the
stain identification assays is to determine whether a sample is worthy of STR analysis.
Therefore, it is critical that the same sample extract be profiled since nearby stains may be of
different origins. Furthermore, for small, limited samples, there must be sufficient sample
remaining to obtain a profile or the RNA analysis may prove to be a waste of time.
In the previous grant we assessed several methods for their ability to extract RNA and DNA.
The most reliable RNA extraction method for all stain types tested was the TRIzol® reagent.
This straight-forward procedure is cost-effective and provides high yields of quality RNA.
However, there are disadvantages to using this method; it is labor-intensive (involves separate
RNA and DNA steps) and provides poor DNA yields. Therefore TRIzol® is not recommended
for limited samples. The Absolutely RNA® kit was used to extract RNA from human tissues.
However, a major disadvantage of the Absolutely RNA® kit is its inability to co-isolate DNA.
A major outcome of the Stain ID assay development with Promega was the generation of a co-
isolation method for RNA and DNA extraction. A combination of the RNagents Total RNA
Isolation System (Promega) with a Tris buffered phenol protocol was optimized for simultaneous
extraction of the nucleic acids. The final extraction method was shown to extract sufficient
quantities of quality RNA and DNA from 1 and 10 µl of semen and saliva demonstrating its
67

utility as a dual extraction technique. Although this method involves numerous hands-on steps,
it is faster than the TRIzol® method, and produces significantly better DNA yields. To date, it’s
the best co-isolation method we’ve tested in terms of yields and amplifiability.
Since all labs routinely examine cases involving samples which contain a mixture of female and
male contributions (i.e. mixtures), we attempted to adapt the Tris-based phenol process to
produce epithelial and sperm extracts. The findings demonstrate that RNA survives the initial 2-
hour incubation with female extraction buffer since PRM2 was detected in the samples
containing seminal fluid. Furthermore, the DNA yields were sufficient to perform downstream
analysis (i.e. STRs). Together these results indicate that the adapted Tris-based phenol
procedure may be a viable extraction method for samples containing both male and female
components. Further research is necessary to compare the quality and quantity of the nucleic
acids isolated via the adapted method with the conventional procedures for DNA and RNA
extraction (i.e. differential method, Tris-based phenol).
Additional experiments were conducted to compare the DNA yields generated from the co-
isolation method with the “gold-standard” organic method used by our laboratory for casework
samples. Results suggest that the yields were fairly comparable and more than sufficient to
perform downstream STR amplification. Future studies need to assess the quality of the DNA by
STR analysis.
The second goal of this project was to identify gene candidates which were specific for each
tissue of interest. The gene candidates utilized through the course of this grant and the previous
grant were identified through surveys of the literature including PubMed, Gene and other
68

databases. Initially we identified 2-3 genes that appeared to be specific for each tissue. These
screening studies were performed using TaqMan® primer/probe sets from Applied Biosystems
because they were pre-designed, inexpensive and already optimized for their intended target.
For each target, the sensitivity and specificity for the body fluid of interest were assessed. Once
the assay was shown to be robust, we tested it on mRNA isolated from other fluids and tissues to
demonstrate that the assay is specific.
In order for a candidate to be implemented into a stain identification assay, it needs to be
detectable for years following deposition of the sample. One of the earliest experiments we
performed was to see whether amplifiable RNA could be detected from an aged semen sample.
Stability of the PRM2 target in particular was demonstrated by its amplification in stains aged
over 2 years.
Sensitivity studies of the semen/sperm candidate assays demonstrated how minute volumes of
seminal fluid could be detected using mRNA profiling. Amplification using the TGM4 and
PRM2 TaqMan® sets occurred with 1 µl and 0.01 µl of semen, respectively. The lower detection
limit for TGM4 was not determined, but assumed to be lower than the 1 µl tested based on the Ct
since it was far from nearing Cts typically observed in negative samples. The sensitivities for
brain candidate assays were evaluated using a range of control brain RNA. As a result we have a
general idea about which assays are more or less sensitive than the others, but conclusions
regarding sensitivity can’t be made until human brain tissue samples are used.
Our diverse sample bank was used to assess the specificity of the candidate tissue-specific genes.
These samples included blood, semen, saliva, menstrual blood, vaginal secretions, kidney, colon,
69

adipose, skin, and control RNAs (brain, heart, liver, kidney, intestine). The semen results
showed varying degrees of cross-reactivity. Some minor amplification occurred with vaginal
secretions (SEMG1 and ACPP), blood (ACPP), kidney (SEMG1) and intestine (MSMB). But,
for the most part, the assays only amplified from semen stains. Based on these studies PSA,
TGM4 and PRM2 were the most specific.
On the other hand, the specificity studies using brain assays weren’t as straight forward. The
three brain assays amplified from the control brain sample as expected, but also had varying
degrees of cross-reactivity with other control tissues (DRD1 with liver and intestine, GPM6A
with liver, ADCY1 with heart and kidney). As mentioned previously, before any definitive
conclusions can be drawn regarding the brain assays, analysis using actual human tissues must
be performed.
A major aim of this grant was developing multiplex real-time PCR assays once mRNAs are
identified that clearly define specific types of stains. Since these assays are designed to function
more qualitatively than quantitatively, a test of a single stain should typically give amplification
with only one candidate. It is possible that a mixture could give several amplifications.
However, it is of greater concern to know that a mixture does exist, than to know the exact
amount of each fluid present.
One methodology to achieve this goal is the Plexor® system from Promega. Depending on the
dye-capability of the real-time instrument that is utilized, this system allows up to six mRNAs to
be multiplexed in one assay, thus reducing the amount of sample needed and time of analysis. In
our previous grant (2004-DN-BX-K002) we described an ongoing collaboration with Promega to
70

develop stain identification assays using their Plexor® platform. The combination of Promega’s
extensive knowledge of the Plexor® platform with our sample inventory and insight into the
needs of the forensic laboratory forged a strong partnership. The goal was to generate Plexor®
Tissue Typing Systems; multiplexed qRT-PCR systems for determining the source and quantity
of a variety of human stains and/or tissues. The systems would include Plexor® primers for the
detection of tissue-specific mRNA transcripts associated with semen, sperm, blood, menstrual
blood, saliva, etc. By limiting the initial system to two-color detection (i.e. FAM and HEX
detection), it would be compatible with the majority of real-time thermal cyclers in forensic
laboratories. We sought to explore the potential to include controls (e.g. a housekeeping gene) or
multiple, tissue-specific targets using the ability of the Plexor® Data Analysis Software to
distinguish two different amplicons in the same dye channel, based upon thermal melt properties.
The Stain ID assay which we have developed to date detects semen and saliva. Plexor® primers
were designed to amplify the targets TGM4 (semen), HTN3 (saliva) and GAPDH (housekeeper).
Through numerous optimization studies, we generated saliva primers that show specific
reactivity with saliva RNA and no cross-reactivity with other RNA samples or with genomic
DNA. The semen primers (TGM4) show specific reactivity with semen RNA and no cross-
reactivity with other RNA samples or with genomic DNA. At the time we were writing the Final
Repot for Grant 2004-DN-BX-K002, Promega was in the process of generating a draft Technical
Manual that would accompany the Stain ID kit. In conjunction with the Stain ID assay, Promega
developed analysis software based on the expected target melt temperatures of the three targets.
Once the draft Technical Manual was completed, we tested the assay using a large panel of in-
house samples representative of typical forensic samples. The results demonstrated that the
71

assay was indeed specific and sensitive. Amplification of the HTN3 and TGM4 targets only
occurred in samples composed of saliva and semen, respectively, whereas all of the samples
(with the exception of the negative controls) showed amplification of GAPDH. We tested the
usefulness of the Stain ID assay using additional mock and actual casework samples. Several of
the casework samples which were found to contain sperm tested negative for TGM4 in the Stain
ID Assay (#2, #3, #4, #7). The cause for these false negatives warrants further examination. In
addition to examining the RNA component isolated from these samples, the co-isolated DNA
was amplified for STR analysis to determine if amplifiable DNA is produced using this
procedure. The results indicate that the co-isolation method is capable of producing DNA of
sufficient quantity and quality for downstream STR analysis. Promega invited a number of
forensic labs from across the county to participate in an alpha test of the prototype kit, but the
results from that study are confidential. However, we do know that based on the feedback from
the alpha testers, Promega put a hold on this project. There were questions regarding the
extraction method, requests for additional markers in the assay, and general concerns as to the
marketability of the assay. To date, no further work has been performed on the Stain ID assay
here at the VFL.
A second strategy for multiplexing tissue-specific assays was with the TaqMan® technology.
Since the Gene Expression TaqMan® assays from Applied Biosystems are incapable of
multiplexing, as they are all labeled with the same dye, we set out to design our own TaqMan®
probes and primers in hopes of developing several specific multiplex assays. Work on these
assays began with Grant 2004-DN-BX-K002 and was described in the Final Report. The first
multiplex we designed was a semen-sperm detection assay using the TGM4 and PRM2 markers
in addition to the housekeeper B2M.
72

Experiments were carried out to optimize the amount of primers and probes in the reaction, the
components were combined into a single multiplex reaction. While each primer/probe set
amplified from semen when alone in the reaction, the combination into a single multiplex
reaction caused the dropout of TGM4 and B2M amplification. However, by decreasing the
primer and probe conditions for PRM2, we were able to prevent the loss of TGM4 and B2M
amplification in the multiplex reaction. Further optimization was conducted which included
changing the master mix and adding BSA. However, although the optimized semen-sperm assay
was sensitive, we saw non-specific PRM2 amplification in saliva, vaginal secretions, menstrual
blood, kidney and colon samples. Therefore, we the semen-sperm assay was modified to include
the Gene Expression TaqMan® PRM2 assay in place of the homebrew PRM2 primers and probe.
The modified assay was optimized for the amount of PRM2 assay to be included, the instrument
gains, and master mix. Once the preliminary experiments were finished, the sensitivity and
specificity of the modified semen-sperm assay were evaluated. Based on the dCt values, the
assay proved to be sensitive and specific for seminal fluid.
The second TaqMan®-based multiplex assay we developed was for the identification of brain
tissue using ADCY1, GPM6A and the housekeeper B2M. In our previous grant, we reported
that each primer/probe set amplified from brain both when alone in the reaction, and in
combination with the other primer/probe sets with no significant decrease in the degree of
amplification when the sets were alone or combined into the multiplex. However, there was
some minor amplification in the no template controls. Under the current grant, we lowered the
amount of GPM6A primers and probes in the reaction which led to a significant decrease in the
nonspecific amplification. Further studies are required to rectify bleed through into the
73

HEX/GPM6A dye channel by such means as using a different master mix. Importantly, future
experiments using human brain samples would aid in the evaluation of this multiplex as a viable
screening assay.
A third technology that we’d hoped to use for the purpose of multiplexing was the Bio-Plex™
system which uses the multiplexing technology of Luminex Corp. to enable the simultaneous
quantitation of up to 100 analytes. In our previous grant we had used the QuantiGene® Plex
Reagent System as one platform that can be coupled with the Luminex instrument to enable the
simultaneous detection of multiple RNA targets directly from purified RNA. We planed to
design additional QuantiGene® Plex assays based on the needs of the community, but time
constraints did not allow us to expand on this line of study.
The work conducted here and by others in the field shows that the analysis of mRNA could
provide a viable alternative to existing methods to identify forensically relevant stains. Studies
have shown that mRNA survives in dried stains for long periods of time and the extraction and
subsequent steps to identify mRNA are amenable to the identification of this nucleic acid. The
studies presented here detail options for extraction and analysis that could be further studied and
refined to allow the implementation of mRNA analysis in the forensic laboratory. Perhaps in a
few years, the co-extraction of mRNA and DNA will become routine such that downstream
analysis will provide an identification of the tissue in combination with the profile of the donor.
74

2. Implications for Policy and Practice
The biochemical approach currently used in tissue identification has undergone some changes in
the past few years but essentially still relies upon the same technology where selective antibodies
detect antigens to a particular source. These approaches have been simplified to save analyst
time but are limited in scope; many laboratories limit tissue identifications to blood and seminal
fluid only. Other tissue sources such as saliva or vaginal fluid are implied but not truly identified.
We believe that the positive identification of these and other tissues can be performed in a quick
and efficient manner which would allow analysts to provide a better service, more efficiently.
Also with this technique, we believe tissues that are currently not routinely evaluated could be
easily assessed by all laboratories so that an equality of testing could be realized across the
country. Presently, some biochemical or “serological” tests are only performed in a select few
laboratories. Through the development of a universal approach to tissue identification, one could
imagine a multitude of tests that could be carried out by anyone qualified to do any one of the
tests. As such, a wide variety of tissues could be assessed and evaluated. As time progresses, the
courts and the forensic community itself will demand tests that truly identify a tissue and allow
for a better understanding of the material composing a STR pattern. We believe that the
evaluation of mRNA through real-time PCR will be a technique that can offer that level of
confidence and expand our knowledge of the materials we routinely examine. An example of
this need was realized recently in our laboratory. A small stain was detected on the muzzle of a
revolver used in an apparent suicide; the weapon, however, was found in a separate room from
the victim. While the STR profile of the stain matched the victim, it would have been very
valuable to know if the stain contained brain tissue or was simply the victim’s blood perhaps
from an older, previous, unrelated cut.
75

We believe that in the next few years a transition from a conventional biochemical approach to a
molecular biological approach will be realized which will replace routine tissue identification.
Tests that are tissue specific and designed to be multiplexed could yield rapid results on minimal
sample. Such testing could employ mRNA as the tissue-specific determinant. Research that
moves this line of testing forward will be important to the forensic community and also to the
criminal justice community in general.
3. Implications for Further Research
To date, over the course of this grant and the previous one, we performed experiments to assess
the specificity, sensitivity and discriminatory limits of real-time assays, as well as the stability of
mRNA over time using various fluid-specific genes for blood and semen. Experiments to assess
the stability of mRNA over time showed that mRNA isolated from blood or semen spots stored
at room temperature for various amounts of time (up to 4 years) was amplified using the blood or
semen assays, respectively. In order to multiplex the real-time PCR assays, we designed Plexor®
primers and TaqMan® probes/primers to identify whether various tissues are present in a stain.
Lastly, in the search for a dual DNA/RNA extraction method we optimized a procedure that
yields quantities of DNA comparable to the current extraction methods of our lab.
While we have made significant progress during the course of the original grant, more time and
effort is required to develop and validate full multiplex assays using the aforementioned
technologies. A shorter-term goal is to further our efforts to find the best DNA/RNA co-
purification method that yields sufficient material for both mRNA analysis and DNA profiling.
76

Great strides have been made to this end through the development of a tris-buffered phenol
extraction procedure. However, further studies are needed to optimize this method for multiple
fluid and tissue samples as well as for mock casework samples. The extraction methods will
need to be evaluated for different analysis platforms to ensure that the nucleic acids produced
can successfully be analyzed. Additionally, the continued evaluation of appropriate genes for
other fluids/tissues (urine, vaginal secretions, menstrual blood, skin, adipose, muscle, etc.) is
necessary, as well as studies to assess mRNA stability, selectivity and suitability for forensic
stain identification. As the cycle continues, once genes are identified for further fluids/tissues,
multiplex assays for those genes must be developed.
Importantly, the tissue-specific genes must be evaluated using human tissue samples. The brain
studies presented in this report were performed with control RNAs. Although this provides a
strong background for preliminary determination of specificity, it is not an accurate
representation of how actual heterogeneous tissue samples will react with the assays. Through
collaboration with local hospitals, it should be possible to obtain human samples.
To date, almost all of the samples we have worked with were spotted onto clean, cotton cloth.
Although samples have been collected on different substrates (cardboard, carpet, vinyl, metal,
etc.), they have yet to be analyzed to determine the stability of the RNA on various substrates.
Furthermore, the effect of storage conditions on RNA stability was only superficially addressed
in the previous project. Studies were conducted to assess the differences between room
temperature storage and storage at 37˚C, but other environments encountered by forensic stains
(sunlight, rain, soil, etc.) are yet untested.
77

If Promega were to re-initiate work on the Stain ID assays there are several improvements that
could be made. Future kits may contain additional primer pairs, facilitating the detection and
quantification of multiple sample types. For example, it may be possible to detect eight unique
transcripts/targets using a real-time thermal cycler capable of four-color detection. This is
possible by designing the assay to detect two different amplicons in each dye channel. Each
amplicon could be discriminated based upon the thermal melt characteristics of the amplicon.
Other potential features include the inclusion of primers for genomic DNA targets and/or internal
amplification controls. The included features will be dictated by the degree of multiplexing
supported by the real-time thermal cyclers. The inclusion of primers for genomic targets would
allow the user to assess the probability of generating a useful genotype in subsequent STR typing
experiments (this may also be possible using the RNA targets). The inclusion of internal
amplification controls would allow the end-user to assess the presence of amplification
inhibitors. As previously noted, primers targeting ubiquitously-expressed transcripts could serve
as controls for RNA integrity.
Altogether these studies indicate that DNA and RNA can be co-extracted and the RNA fraction
used in multiplexed real-time PCR assays. The development of real-time PCR assays to detect
tissue-specific transcripts for human fluids and tissues is the focus of many laboratories. These
assays can ultimately be multiplexed for faster determination of tissue origin. A major
advantage to these assays is that a single sample extract will be used to classify the sample as
blood, semen, vaginal secretions, brain, heart, etc. which will drastically reduce the number of
identification tests performed prior to DNA profiling (if required). The realization of RNA-
based profiling is in the immediate future, yet a large body of work remains to optimize these
78

techniques for the vast number of sample types and conditions which are routinely encountered
in the forensic laboratory.
79

V. References
Abe K, Konomi N (1998) Hepatitis C virus RNA in dried serum spotted onto filter paper is
stable at room temperature. J Clin Microbiol 36:3070-3072.
Alvarez M, Juusola J, Ballantyne J (2004) An mRNA and DNA co-isolation method for forensic
casework samples. Anal Biochem 335(2):289-298.
Bauer M, Kraus A, Patzelt D (1999) Detection of epithelial cells in dried blood stains by reverse
transcriptase-polymerase chain reaction. J Forensic Sci 44:1232-1236.
Bauer M, Patzelt D (2002) Evaluation of mRNA markers for the identification of menstrual
blood. J Forensic Sci 47:1278-1282.
Bauer M, Patzelt D (2003) A method for simultaneous RNA and DNA isolation from dried blood
and semen stains. Forensic Sci Int 136:76-78.
Bauer M, Polzin S, Patzelt D (2003) Quantification of RNA degradation by semi-quantitative
duplex and competitive RT-PCR: a possible indicator of the age of bloodstains? Forensic Sci Int
138:94-103.
Bourlet T, Berthelot P, Grattard F, Genin C, Lucht FR, Pozzetto B (2002) Detection of GB virus
C/hepatitis G virus in semen and saliva of HIV type-1 infected men. Clin Microbiol Infect 8:352-
357.
80

Cao J, Cousins RJ (2000) Metallothionein mRNA in monocytes and peripheral blood on nuclear
cells and in cells from dried blood spots increases after zinc supplementation of men. J Nutr
130:2180-2187.
Cassol S, Gill MJ, Pilon R, Cormier M, Voigt RF, Willoughby B, Forbes J (1997) Quantification
of human immunodeficiency virus type 1 RNA from dried plasma spots collected on filter paper.
J Clin Microbiol 35:2795-2801.
Chomczynski P (1993) A reagent for the single-step simultaneous isolation of RNA, DNA, and
proteins from cell and tissue samples. Biotechniques 15(3):532-537.
Conti T, Buel E (May 2009) Forensic Stain Identification by RT-PCR Analysis. NIJ Grant
Report (2004-DN-BX-K002), U.S. Department of Justice.
Crisan D, Anstett MJ (1995) Myeloperoxidase mRNA detection for lineage determination of
leukemic blasts: retrospective analysis. Leukemia 9:1264-1275.
Daja MM, Aghmesheh M, Ow KT, Rohde PR, Barrow KD, Russell PJ (2000) Beta-human
chorionic gonadotropin in semen: a marker for early detection of prostate cancer?. Mol Urol
4:421-427.
81

Dulioust E, Tachet A, De Almeida M, Finkielsztejn L, Rivalland S, Salmon D, Sicard D,
Rouzioux C, Jouannet P (1998) Detection of HIV-1 in seminal plasma and seminal cells of HIV-
1 seropositive men. J Reprod Immunol 41:27-40.
Flagella M, Bui S, Zheng Z, Nguyen CT, Zhang A, Pastor L, Ma Y, Yang W, Crawford KL,
McMaster GK, Witney F, Luo Y (2006) A multiplex branched DNA assay for parallel
quantitative gene expression profiling. Anal Biochem 352(1):50-60.
Fritsch MK, Bridge JA, Schuster AE, Perlman EJ, Argani P (2003) Performance Characteristics
of a Reverse Transcriptase-Polymerase Chain Reaction Assay for the Detection of Tumor-
specific Fusion Transcripts from Archival Tissue. Pediatr Dev Pathol 6(1):43-53.
Goodwin LO, Karabinus DS, Pergolizzi RG, Benoff S (2000) L-type voltage-dependent calcium
channel alpha-1C subunit mRNA is present in ejaculated human spermatozoa. Mol Hum Reprod
6:127-136.
Jin L, Vyse A, Brown DW (2002) The role of RT-PCR assay of oral fluid for diagnosis and
surveillance of measles, mumps and rubella. Bull World Health Organ 80:76-77.
Juusola J, Ballantyne J (2003) Messenger RNA profiling: a prototype method to supplant
conventional methods for body fluid identification. Forensic Sci Int 135:85-96.
Katz RS, Premenko-Lanier M, McChesney MB, Rota PA, Bellini WJ (2002) Detection of
measles virus RNA in whole blood stored on filter paper. J Med Virol 67:596-602.
82

Krafft AE, Duncan BW, Bijwaard KE, Taubenberger JK, Lichy JH (1997) Optimization of the
Isolation and Amplification of RNA From Formalin-fixed, Paraffin-embedded Tissue: The
Armed Forces Institute of Pathology Experience and Literature Review. Mol Diagn 2:217-230.
Lewis F, Maughan NJ, Smith V, Hillan K, Quirke P (2001) Unlocking the archive--gene
expression in paraffin-embedded tissue. J Pathol 195:66-71.
Li J, Wirtz RA, McCutchan TF (1997) Analysis of malaria parasite RNA from decade-old
Giemsa-stained blood smears and dried mosquitoes. Am J Trop Med Hyg 57:727-731.
Liu H, Huang X, Zhang Y, Ye H, Hamidi AE, Kocjan G, Dogan A, Isaacson PG, Du MQ (2002)
Archival fixed histologic and cytologic specimens including stained and unstained materials are
amenable to rt-PCR. Diagn Mol Pathol 11:222-227.
Macabeo-Ong M, Ginzinger DG, Dekker N, McMillan A, Regezi JA, Wong DT, Jordan RC
(2002) Effect of duration of fixation on quantitative reverse transcription polymerase chain
reaction analyses. Mod Pathol 15:979-87.
Matsubara Y, Ikeda H, Endo H, Narisawa K (1992) Dried blood spot on filter paper as a source
of mRNA. Nucleic Acids Res 20(8):1998.
Miller D (2000) Analysis and significance of messenger RNA in human ejaculated spermatozoa.
Mol Reprod Dev 56:259-264.
83

Mizuno T, Nagamura H, Iwamoto KS, Ito T, Fukuhara T, Tokunaga M, Tokuoka S, Mabuchi K,
Seyama T (1998) RNA from decades-old archival tissue blocks for retrospective studies. Diagn
Mol Pathol 7:202-208.
Nicklas J, Buel E (2006) Simultaneous determination of total human and male DNA using a
duplex real-time PCR assay. J Forensic Sci 51(5):1005-1015.
Pai JT, Tsai SF, Horng CJ, Chiu PC, Cheng MY, Hsiao KJ, Wuu KD (1994) Absence of FMR-1
gene expression can be detected with RNA extracted from dried blood specimens. Hum Genet
93:488-493.
Richter W, Dettmer D, Glander H (1999) Detection of mRNA transcripts of cyclic nucleotide
phosphodiesterase subtypes in ejaculated human spermatozoa. Mol Hum Reprod 5:732-736.
Schoch R, Pitako J, Schafhausen P, Jenisch S, Haferlach T, Kneba M, Suttorp M (2001)
Semiquantitative reverse transcription polymerase chain reaction analysis for detection of bcr/abl
rearrangement using RNA extracts from bone marrow aspirates compared with glass slide
smears after 0, 2 and 4 d of storage. Br J Haematol 115:583-587.
Spielberg F, Critchlow C, Vittinghoff E, Coletti AS, Sheppard H, Mayer KH, Metzgerg D,
Judson FN, Buchbinder S, Chesney M, Gross M (2000) Home collection for frequent HIV
testing: acceptability of oral fluids, dried blood spots and telephone results. HIV Early Detection
Study Group. Aids 14:1819-1828.
84

Tetali S, Lee EM, Kaplan MH, Romano JW, Ginocchio CC (2001) Chemokine receptor CCR5
Delta 32 genetic analysis using multiple specimen types and the NucliSens Basic Kit. Clin Diagn
Lab Immunol 8:965-971.
Watanapokasin Y, Winichagoon P, Fuchareon S, Wilairat P (2000) Relative quantitation of
mRNA in beta-thalassemia/Hb E using real-time polymerase chain reaction. Hemoglobin
24:105-116.
Yang W, Maqsodi B, Ma Y, Bui S, Crawford KL, McMaster GK, Witney F, Luo Y (2006)
Direct quantitation of gene expression in homogenates of formalin-fixed, paraffin-embedded
tissues. Biotechniques 40(4):481-486.
Zhang A, Pastor L, Nguyen Q, Luo Y, Yang W, Flagella M, Chavli R, Bui S, Nguyen CT, Zheng
Z, He W, McMaster G, Witney F (2005) Small interfering RNA and gene expression analysis
using a multiplex branched DNA assay without RNA purification. J Biomol Screen 10(6):549-
556.
Zhang YH, McCabe ER (1992) RNA analysis from newborn screening dried blood specimens.
Hum Genet 89:311-314.
Zheng Z, Luo Y, McMaster GK (2006) Sensitive and quantitative measurement of gene
expression directly from a small amount of whole blood. Clin Chem 52(7):1294-1302.
85

VI. Dissemination of Research Findings
While the stain identification assays we sought to develop would be useful in our own
laboratory, a major goal of this grant was to distribute the information to the forensic community
at large to improve criminal justice in the United States. To this end, we took a multi-tiered
approach at disseminating our work. The first step was talking about our progress at scientific
meetings which included the NIJ Conferences and the Promega Meeting:
Presentations at Scientific Conferences:
The National Institute of Justice Conference, Arlington, VA, July 2008; “Development of RNA-
Based Screening Assays for Forensic Stain Identification”, Poster Presentation
19th International Symposium on Human Identification, Hollywood, CA, October 2008;
“Development of an RNA-Based Screening Assay for Forensic Stain Identification”, Oral
Presentation
The National Institute of Justice Conference, Arlington, VA, June 2009; “Forensic Stain
Identification by Real-Time PCR Analysis”, Poster Presentation
The National Institute of Justice Conference, Arlington, VA, June 2010; “Forensic Stain
Identification by Real-Time PCR Analysis”, Poster Presentation
86

87
One approach to disseminating our results was to work with a company to produce our assays as
kit(s) for sale to the forensic community. We forged a relationship with Promega and although
they were extremely interested in making stain identification screening kits based on their
proprietary Plexor® method, have since decided to put a hold on the project.
The last way to disseminate our work was to publish our results in forensic journals. To date, we
have published one chapter and we anticipate submitting at least one or two other manuscripts
based on the results we have compiled during the course of this project.
Citations:
Noreault-Conti, T., Buel, E., “Bringing tissue identification into the 21st century: mRNA analysis
as the next molecular biology revolution in forensic science?”, F. Gonzalez-Andrade Ed.,
Forensic Genetics Research Progress, 2010.