free energies: what are they, why are they useful and how...
TRANSCRIPT

Free Energies: What are they, Why are they useful and How are they calculated?
Philip W. FowlerStructural Bioinformatics & Computational Biochemistry Unit
Department of Biochemistry, University of Oxfordhttp://sbcb.bioch.ox.ac.uk
!A = !U ! T!S

Objectives
• To give you a better appreciation of classical molecular dynamics
• To help you understand how free energies may be calculated using classical molecular dynamics
• To provide you with enough reference material to allow you to estimate whether a particular calculation is feasible or not
2

Scope
• We will
‣ discuss why knowing how the free energy changes during a process is useful
‣ relate the change in free energy to quantities that can be measured by experiment
‣ examine several theories
‣ look at several case studies
‣ only derive equations in the NVT (or canonical) ensemble
‣ finish in time for lunch
• We will not
‣ discuss thermodynamics or statistical mechanics in detail
‣ derive, or even mention all of the different approaches in the literature
‣ investigate in detail how these simulations are run3

This is NOT a lecture
• We will have periodic group discussions so that you can talk and digest the material
• If you have a question ask your neighbour, ask me, ask someone!
• Some of these concepts are hard and there is some maths
4

What is energy?
5
“ability to do work” (move or heat something)
It is a concept invented by physicists i.e it doesn’t exist
It is often used in a relative way i.e. “state A has a higher energy than state B”
has a variety of units: 1 J = 4.184 calwe often quote per mole of
substance e.g. kcal/mol

Calculating energies are central to the classical molecular dynamics approach
6
Fi = !
dE(r)
dr
Can we use all of this potential energy to do work?
Question:
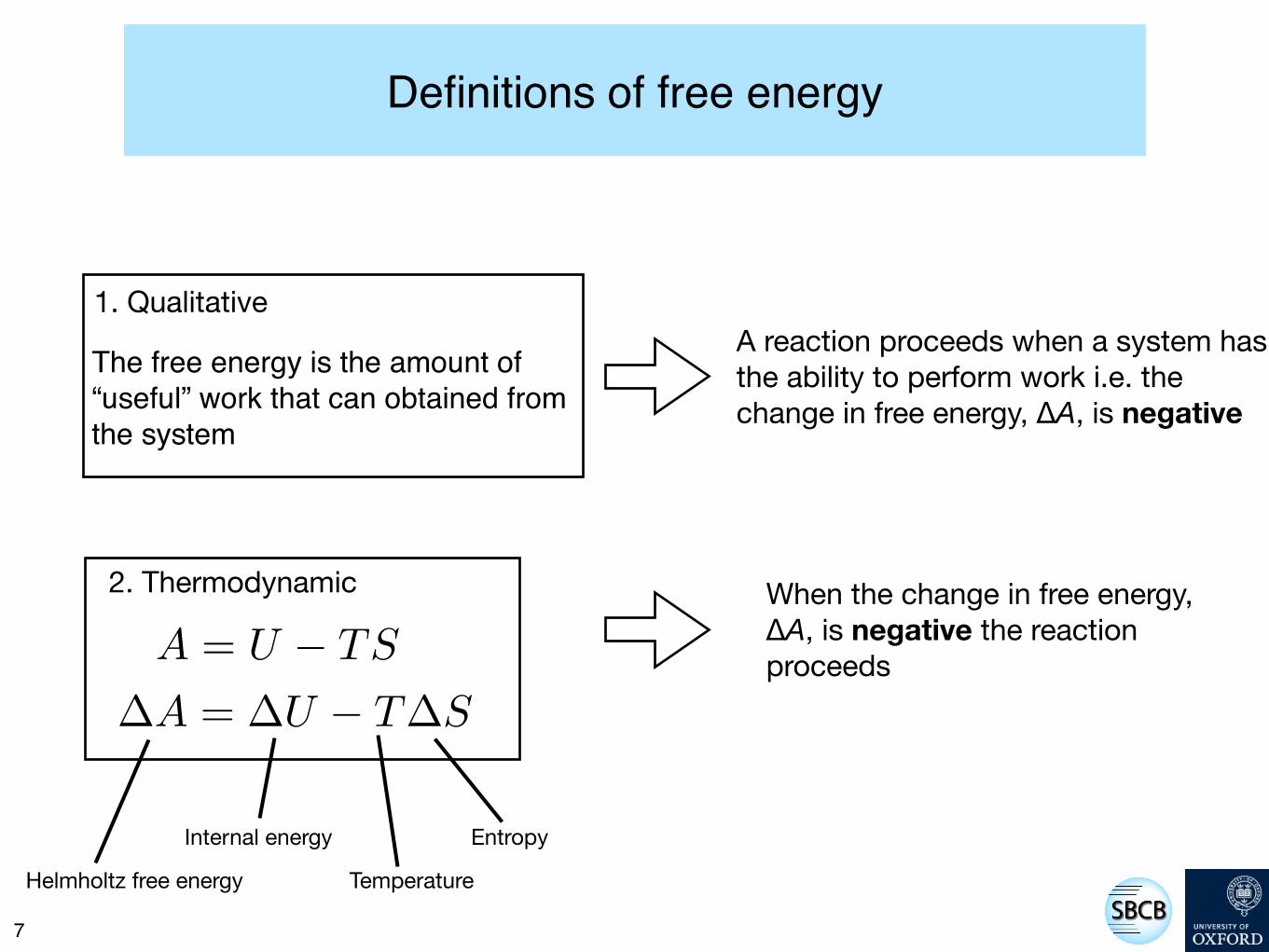
Definitions of free energy
7
The free energy is the amount of “useful” work that can obtained from the system
1. Qualitative
A = U ! TS
!A = !U ! T!S
2. Thermodynamic
A reaction proceeds when a system has the ability to perform work i.e. the change in free energy, ∆A, is negative
When the change in free energy, ∆A, is negative the reaction proceeds
Helmholtz free energy
Internal energy
Temperature
Entropy

Case study 1: protein folding
8
from V. Daggett Chem. Rev. (2006) 106 1898-1916
unfolded or extended folded or compact
The relative change in free energy between the unfolded and folded states determines where the equilibrium lies and therefore whether the reaction “proceeds” since
Gas constant
Temperature
Equilibrium constant (relate to experiment)
!A = !RT lnK

Case study 1: protein folding
9
from V. Daggett Chem. Rev. (2006) 106 1898-1916
unfolded or extended folded or compact
!A = !U ! T!S
How do ∆U and ∆S change during folding?Will the protein fold?
What chemical bonds are formed and broken as the protein folds? Does the number of states accessible to the
protein increase or decrease?
Questions:
How does the hydrophobic effect contribute to the folding of the protein?

(kJ
mol
-1)
Th Ts
∆G
∆H
T ∆S
T = 25 °C
ΔH = 0
ΔG > 0 driven by the large decrease in entropy as water orders itself around the non-polar solute
TΔS
0
1
2
0 2 4 6 8
Argon in water (25 °C)
calculated g(r)
r (Å)
g(r)
1. no interactions between solute and water
2. interactions between solute and non-polar solvent lost3. increase in the number of water hydrogen bonds
ΔH0
> 0
< 0
2 and 3 have about the same magnitude therefore: TΔS < 0
1. the water orders itself around the solute as it cannot hydrogen bond to it < 0
Caution: the thermodynamic data is for neopentane and the simulations are of Argon
The hydrophobic effect

unfolded, extended state folded, compact native statecompact molten globule
∆H
∆S
∆H
∆S
< 0
≈ 0
☹
≈ 0
native fold is less flexible than molten globule
< 0 ☺ ☺∆H
∆Sprotein
∆H
∆Swater
> 0
< 0
< 0 ☺
☹
☹
> 0 ☺ ☺
fewer protein—water hydrogen bonds
protein can adopt fewer conformations when a
molten globule
more water—water hydrogen bonds (can form
complete network)
much less local ordering of the water
clusters of non-covalent interactions form cooperatively
from the perspective of the solvent, there is little difference between the molten globule and
the folded, native state
The hydrophobic effect causes the extended polypeptide chain to collapse and form a compact but dynamic molten globule. Clusters of non-covalent interactions within the protein then form cooperatively. This leads to both secondary structure and the dense packing of the hydrophobic interior.
Protein folding

Case study 1: protein folding
12
from V. Daggett Chem. Rev. (2006) 106 1898-1916
unfolded or extended folded or compact
!A = !U ! T!Se.g. for lysozyme
523 kcal/mol- 537 kcal/mol-14 kcal/mol
The folded states of many globular proteins are marginally stable. This makes it difficult to model this process, helps explain prion-based diseases and makes it easier for proteins to be degraded
from Dobson, C. M., Sali, A. and M. Karplus Angew. Chem. Int. Ed. (1998) 37 868-893

Case study 2: the binding of small molecules to proteins
13
Questions:
If ∆U < 0 will the reaction always proceed?
If ∆U > 0 will the reaction never proceed?
How might the binding of a drug be driven purely by entropy?
Can examining only ∆U ever tell us anything useful?

Case study 2: the binding of small molecules to proteins
14
The free energy, ∆A, tells us how strongly a drug binds to a protein. It is therefore very useful if we want an efficient drug with few side effects.
The strength of binding is often quoted as the concentration required to achieve 50:50 binding e.g. “this candidate binds in the nano-molar range”
!A = !RT lnK If we know K we can calculate ∆A and vice versa

Why is computing free energies difficult?
15
Naive approach: set up a simulation with protein and drug and measure how long the drug is bound to the protein compared to how long it remains in solution.
How long might this take?Assume we estimate ∆A = - 10 kcal/mol, then K = 8 x 10-8
and if we assume that for sampling we need to observe just 1ns in the unbound state then we would need to simulate for 1.25ms.
If we had unlimited access to a supercomputer (i.e. 4ns per day) and ran this sequentially then this would take 8,500 years.
It is very difficult to calculate free energies directly: you always apply a perturbation to force the system from one state to the other

..to put it another way..
16
A = !f(U)"
you must sample the entire phase space of the protein and measure some function of the potential energy
Generate protein conformations in the correct ensemble using either classical molecular dynamics or Monte Carlo method
from V. Daggett Chem. Rev. (2006) 106 1898-1916

17
inverse temperature factor ! =1
kT
partition function
Helmholtz free energy
From thermodynamics:
multiple top and bottom by
but we can define a probability density

18
which can be rewritten as A = !f(U)"
This is similar to our earlier expression:
The final result (also know as the perturbation formula)
The configurations we generate from the MD or MC all have different energies E (or U). The likelihood of a configuration having
an energy E is proportional to the Boltzmann factor
e!!E
Conformations with higher energies are rarely generated BUT contribute significantly to the value of A.

A thermodynamic cycle
19
Discuss...
How might we use such a cycle to calculate how the free energy of binding changes as we mutate the red ligand to the blue ligand (i.e. a ∆∆A)?
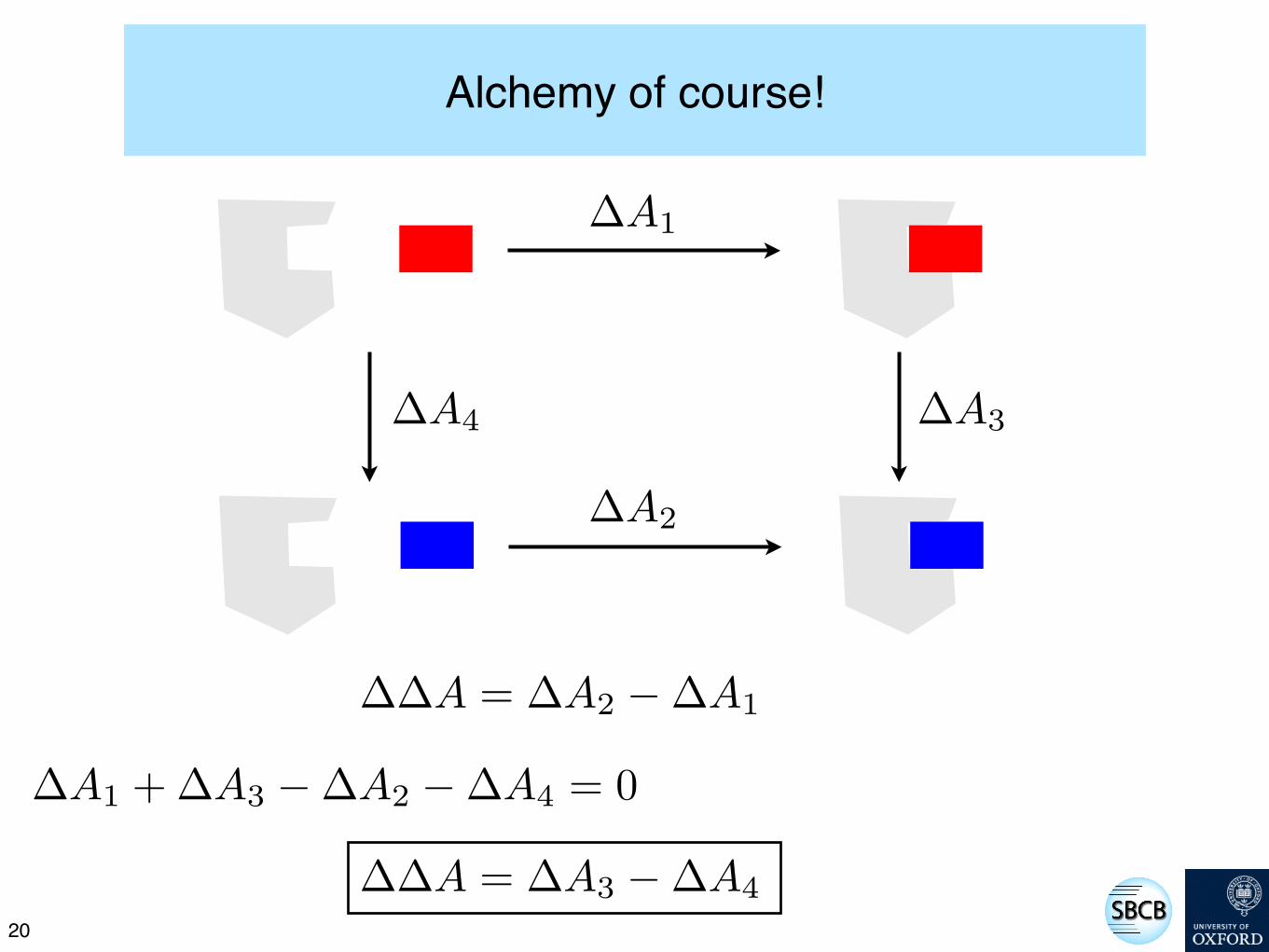
Alchemy of course!
20
!A1
!A2
!A3!A4
!A1 + !A3 ! !A2 ! !A4 = 0
!!A = !A2 ! !A1
!!A = !A3 ! !A4

Case Study 3: ∆∆Aselectivity(K+→Na+) for KcsA
21
!A1
!A2
!A3!A4
KcsA (a bacterial potassium selective ion channel)
Na+
K+
K+ bound at site S2
We shall now examine how ∆A4 is calculated using GROMACS and thermodynamic integration

Running a thermodynamic integration
22
K+
Cl-
Na+?
Cl-
λ = 0.5λ = 0 λ = 1
We shall perturb the system from λ = 0 to λ = 1 and thereby calculate ΔA
1. Assume that the Helmholtz free energy ΔA is a continuous function of λ then we can write
2. Now we substitute for A in terms of the the partition function Q
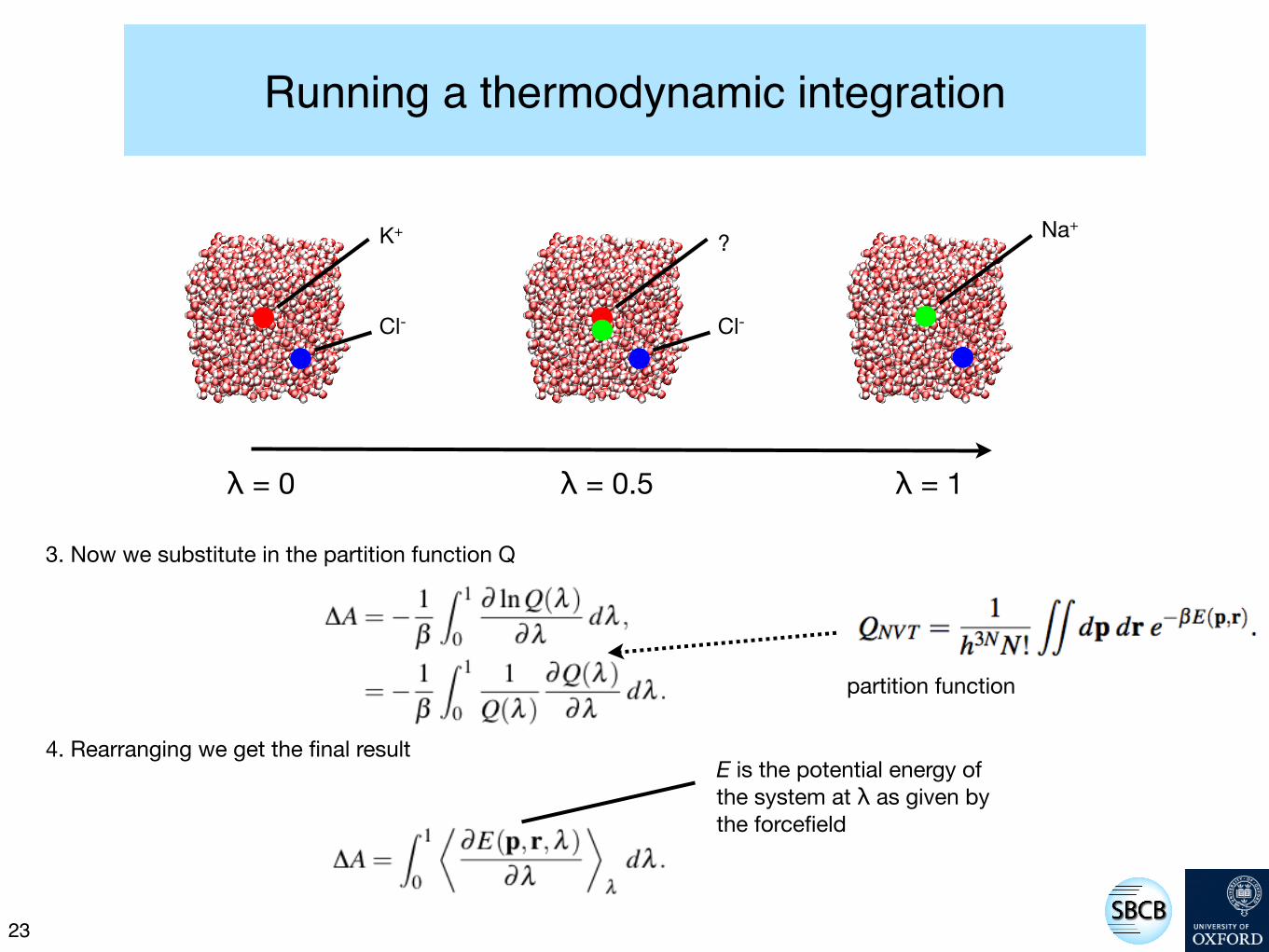
3. Now we substitute in the partition function Q
Running a thermodynamic integration
23
K+
Cl-
Na+?
Cl-
λ = 0 λ = 0.5 λ = 1
4. Rearranging we get the final result
partition function
E is the potential energy of the system at λ as given by the forcefield
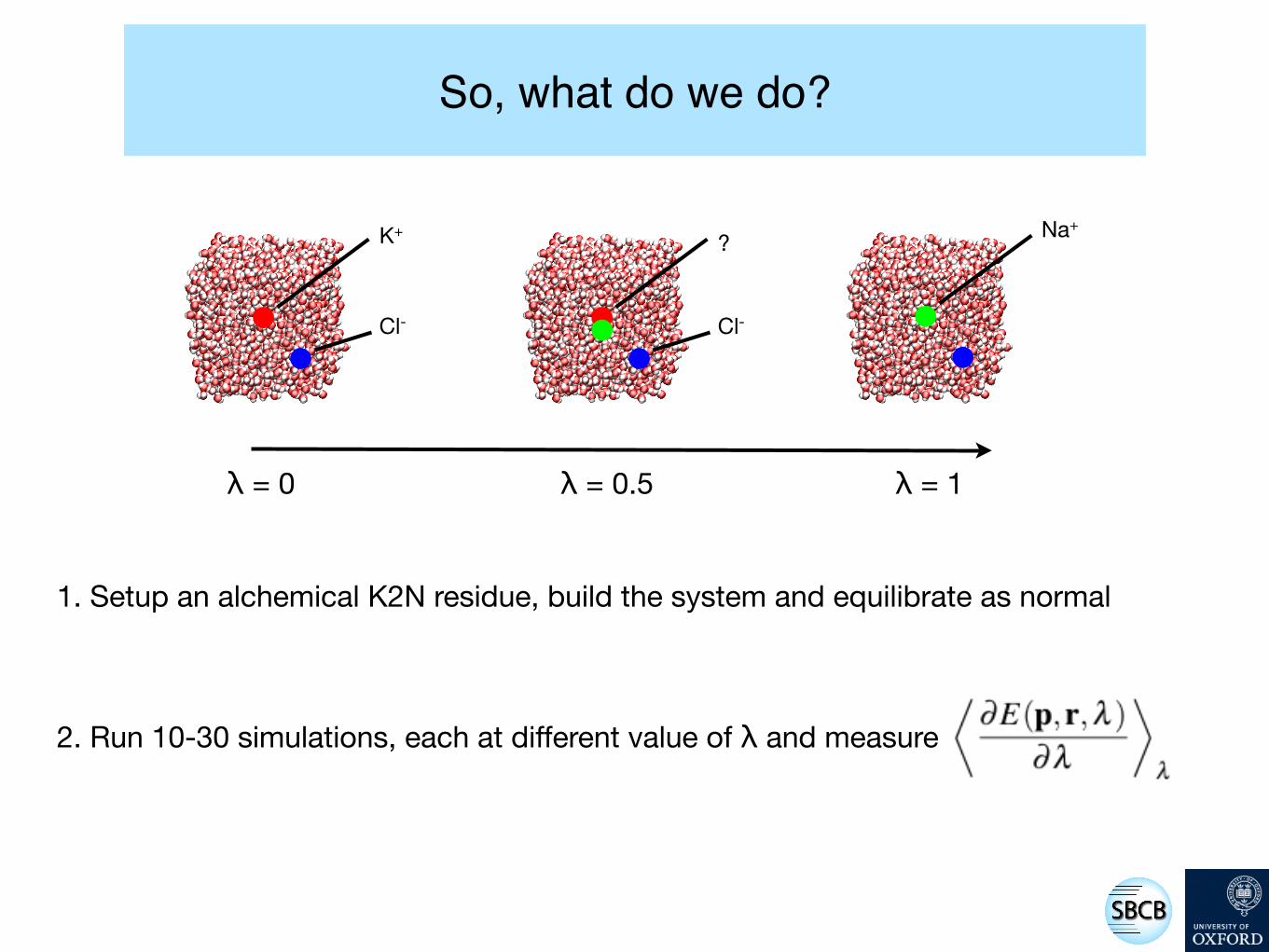
So, what do we do?
K+
Cl-
Na+?
Cl-
1. Setup an alchemical K2N residue, build the system and equilibrate as normal
2. Run 10-30 simulations, each at different value of λ and measure
λ = 0 λ = 0.5 λ = 1

So, what do we do?
25
3. Determine when each simulation has equilibrated and then plot against λ
4. Numerically integrate to get a value for ΔA
ΔAcalc = 20.6 kcal/mol
Method: NAMD2.6 with CHARMM27 forcefield, 2fs, PME, TIP3P water, 12Å cutoff
ΔAexp = 18 kcal/mol

We have ...
• defined what free energy is
• discussed why knowing it is useful
• investigated why it is difficult to calculate
• seen how applying alchemy can help make calculations tractable
• examined a single free energy calculation
26
Email: [email protected],
Practical: http://sbcb.bioch.ox.ac.uk/fowler/files/fe-practical.pdf

Sources
27
BooksMolecular Modelling. Principles and Applications. Andrew R. Leach. (2001) Second edition.Understanding Molecular Simulation. D. Frenkel and B. Smit. (2002) Second edition
Review papersT. Rodinger and R. Pomès. Curr. Opin. Struct. Biol. (2005) 15 164-170D. A. Kofke. Fluid Phase Equilib. (2005) 228-229 41-48W. L. Jorgensen Science (2004) 303 1813-1818C. Chipot and D. A. Pearlman Mol. Sim. (2002) 28 1-12
Tutorials/WebsitesGROMACS. http://www.gromacs.org. Documentation | TutorialsNAMD. http://www.ks.uiuc.edu/Research/namd/tutorial/fep/
Email: [email protected],