functionalization of periodic mesoporous organosilica with ureidopropyl groups by a direct synthesis...
TRANSCRIPT

www.elsevier.com/locate/micromeso
Microporous and Mesoporous Materials 101 (2007) 381–387
Functionalization of periodic mesoporous organosilica withureidopropyl groups by a direct synthesis method
Qi Wei, Li Liu, Zuo-Ren Nie *, Hui-Qiao Chen, Yan-Li Wang, Qun-Yan Li, Jing-Xia Zou
College of Materials Science and Engineering, Beijing University of Technology, 100 Pingleyuan, Chaoyang District, Beijing 100022, PR China
Received 1 June 2006; received in revised form 8 September 2006; accepted 14 September 2006Available online 16 January 2007
Abstract
Ureidopropyl groups were used to functionalize the pore channels of ethane-bridged periodic mesoporous organosilica by the co-con-densation of 1,2-Bis(triethoxysilyl)ethane (BTESE) and ureidopropyltriethoxysilane (UPTES) in the presence of Poly(ethylene glycol)-B-Poly(propylene glycol)-B-Poly(ethylene glycol) (P123) surfactants under acidic conditions. The final materials were investigated in detailby means of XRD, TEM, solid-state NMR, FT-IR and N2 adsorption, in order to study the effect of ureidopropyl groups concentrationon their mesoscopic order and pore structure. The results show that bridging groups in the framework do not cleave and ureidopropylgroups are attached covalently to the pore wall of periodic mesoporous organosilica after functionalization. The mesoscopic orderdecreases with increasing amount of UPTES except for the sample with 20 mol% UPTES concentration, which exhibits a highly orderedtwo-dimensional hexagonal symmetry. The surface area and pore size decrease as the concentration of UPTES increases, but the mate-rials with 20 mol% UPTES still preserve a desirable pore structure, with a surface area of 565 m2/g, a pore volume of 1.1 cm3/g and amean pore size of 10.1 nm.� 2006 Published by Elsevier Inc.
Keywords: Periodic mesoporous organosilica; Terminal functionalization; Ureidopropyl groups; Mesoscopic order; Pore structure
1. Introduction
As a novel class of organic–inorganic nanocomposites,periodic mesoporous organosilicas (PMOs) have drawnmuch attention due to potential applications such ashost-guest inclusion, nanotechnology, chemoselectiveseparation and adsorption, chemical sensing, and catalysis[1–8]. These PMOs materials are synthesized by the supra-molecular–assembly route in the presence of structure-directing agents using a silsesquioxane of the type(EtO)3Si–R–Si(OEt)3 as the sole precursor, therefore theorganic groups R are located within the channel walls asbridges between Si centers. These homogenously distrib-uted functional groups provide PMOs materials with manyof the favorable properties associated with organic poly-mers, however, they are less reactive and accessible com-
1387-1811/$ - see front matter � 2006 Published by Elsevier Inc.
doi:10.1016/j.micromeso.2006.09.014
* Corresponding author.E-mail address: [email protected] (Z.-R. Nie).
pared to the functional terminal organic groupsprotruding into the pores due to steric and electronic differ-ences [1,2,9], which are generally incorporated to the sur-face of ordered mesoporous silica by both post-syntheticgrafting and co-condensation of trialkoxyorganosilanewith tetraethyl orthosilicate (TEOS) [10–13]. A combina-tion of both bridging organic functional moieties in theframework and terminal organic functional groups pro-truding into the pores stands for a judicious alternative,conferring to PMOs materials additional functional func-tionalities [14]. Such approach was recently explored forvarious groups on periodic mesoporous organosilicas.Asefa et al. functionalized vinyl groups into periodicmesoporous organosilicas by the co-condensation ofbis(triethoxysiyl)ethylene and triethoxyvinylsilane in thepresence of cetyltrimethylammonium bromide under basiccondition, leading to a so-called bifunctional periodicmesoporous organosilicas (BPMOs) in which the bridgingethylene plays a structural and mechanical role and the

382 Q. Wei et al. / Microporous and Mesoporous Materials 101 (2007) 381–387
terminal vinyl groups are readily accessible for chemicaltransformations [9]. Ethylene-bridged periodic mesoporousorganosilicas have been further functionalized with a vari-ety of functional groups, including amines, mercaptans,and simple aromatics in Markowitz’s group [15]. Propyl-and arene-sulfonic acid groups were used to functionalizeperiodic mesoporous ethanesilica over two different surfac-tant assemblies and it was found that their acid capacityand proton conductivity reached 1.38 meq/g and1.6 · 10�2 S/cm, respectively [16]. More recently, Wahabet al. incorporated vinyl, ethyl, glycidoxypropyl and cyano-propyl groups into ethane-bridged periodic mesoporousorganosilicas using binary surfactant mixtures under basicconditions [17]. Ureidopropyl groups contain two amineattached directly to a carbonyl and seem to be an attractiveclass of functional groups since the lone pair electron inoxygen and nitrogen may offer the functionality such aspotential activity to form chelate complex with other reac-tant [18]. It is of considerable significance if ureidopropylgroups can be functionalized into periodic mesoporousorganosilicas because they can be used in many applicationfields such as covalent coupling of protein to the surface ofsilica materials and selective adsorption of heavy metalions such as Zn2+, Cr6+ and Ni2+ [19–21]. To the best ofour knowledge, ureidopropyl groups have not yet beenused to functionlize periodic mesoporous organosilicas inpublished literature.
In the present paper, we report the functionalization ofperiodic mesoporous organosilicas with ureidopropylgroups by the co-condensation of bis(triethoxysilyl)ethaneand ureidopropyltriethoxysilane under acidic conditionsand the characterization of final materials by means ofX-ray diffraction (XRD), transmission electron microscopy(TEM), solid-state nuclear magnetic resonance (NMR) andN2 adsorption in detail. This paper aims to offer evidencesthat ureidopropyl groups have been successfully attachedon the pore surface and discuss the effect of the funcional-ization on the mesoscopic order and pore structure of peri-odic mesoporous organosilicas.
2. Experimental
2.1. Chemicals
1,2-Bis(triethoxysilyl)ethane (BTESE,96%) andpoly(ethylene glycol)-B-poly(propylene glycol)-B-Poly(eth-ylene glycol)(P123) were obtained from Aldrich. Ure-idopropyltriethoxysilane (UPTES, 97%) was purchasedfrom Alfa Aesar. Hydrochloric acid (HCl) and ethanol(C2H5OH) were produced in China. All chemicals wereused as received.
2.2. Materials synthesis
Functionalized mesoporous materials were preparedusing BTESE as the source of bridging organic groups,UPTES as the source of terminal organic groups, and
P123 as the structure-directing agent. For a typical synthe-sis, a molar ratio of BTESE, 1 minus x; UPTES x; P123,0.034; HCl, 11.7; H2O, 326 was used, where x varies from0% to 40%. In a typical procedure, a solution of P123, HCland water was prepared at 40 �C. To this solution wasadded dropwise a mixture of BTESE and UPTES. Themixture was allowed to stir for 24 h until white precipitatesformed, and then the products were moved into Teflon-lined autoclaves and aged for 72 h at 100 �C. The whiteprecipitates were recovered by filtration, washed with wateruntil reaching a pH of 6–7, and then dried at atmosphere.The surfactant template was removed from organosilicamaterials through solvent extraction. An as-synthesizedsample (0.5 g) was gently stirred for 6 h in a solution ofHCl (36 wt.%, 5 g) and ethanol (100 g) in 50 �C waterbath. This procedure was repeated several times until thesurfactants were totally removed. The powder was filtered,washed with ethanol, and air-dried at 60 �C overnight toobtain final material.
2.3. Materials characterization
Powder X-ray diffraction patterns were obtained on aBruker D8/advance diffractometer using a high powerNi-filtered CuKa radiation (1.541 A) source with a resolu-tion of 0.02� and scanning speed of 0.5�/min. Nitrogenadsorption measurements were carried out at 77 K on aMicromeritics ASAP 2020 M volumetric adsorption ana-lyzer. Before the measurements, the samples were out-gassed under vacuum at 110 �C for 5 h. The surface areawas calculated according to BET equation at the relativepressures ranging from 0.05 to 0.20 and the pore size distri-bution was obtained from the adsorption branch of iso-therms using BJH approach modified by Kruk–Jaroniec–Sayari (KJS) method. The morphology of the sampleswas observed by transmission electron microscopy (JEOLJEM-2010). The solid-state magic angle spinning 1Hnuclear magnetic resonance (MAS NMR) experimentswere performed at 9.4 T on a Varian Infinityplus-400 spec-trometer using 4 mm probes under magic-angle spinningspeed of 10 KHz. The resonance frequency is 400.1 MHzfor 1H. 1H single-pulse experiment was used. The 1H 90�pulse width was measured to be 4.0 ls with repetition timeof 4 s. The solid state cross-polarized magic angle spinning29Si and 13C nuclear magnetic resonance (CP MAS NMR)spectra were recorded on a Bruker AV300 spectrometeroperating at a frequency of 59.62 MHz for 29Si and75.47 MHz for 13C. Chemical shifts for 1H, 29Si and 13Cwere referenced to tetramethylsilane (TMS) at 0 ppm.Infrared spectra were acquired from KBr pellets with aNicolet 5700 FT-IR spectrophotometer with a resolutionof 4 cm�1 and a scan number of 32.
3. Results and discussion
Fig. 1 shows the XRD patterns of the PMOs materialsfunctionalized with different molar ratio of UPTES. The

0.5 1.0 1.5 2.0 2.5 3.0
(200)(110)
(100)
Inte
nsity
(a.
u.)
Á5
0%
40%30%
20%
10%
2 È/o
5%
Fig. 1. X-ray diffraction patterns of PMOs materials functionalized withdifferent molar ratio of UPTES. The region where (110) and (200) peaksappear is scaled 5 times for the sample with 20 mol% UPTES.
Q. Wei et al. / Microporous and Mesoporous Materials 101 (2007) 381–387 383
pure PMOs sample displays only a sharp diffractionindexed to (100) at 2h value of 0.83�, suggesting an uniformpore structure but not a highly ordered structure for thelack of the higher order diffractions. However, one verybroad peak at 2h ranging from 0.8 to 0.9 is observed onthe samples functionalized with 5 mol% UPTES, indicatinga more amorphous structure. As the UPTES concentrationfurther increases to 10 mol%, the intensity of this peakbecomes stronger than that of the corresponding peak inthe samples with 5 mol% UPTES, but the samples stillremain an amorphous structure. The samples with20 mol% UPTES exhibit a highly ordered pore system,with a distinct peak at the 2h value of 0.75� and two addi-tional well-resolved diffractions at the 2h value between 1�and 2�, which can be assigned to (100), (110) and (200)diffractions, respectively, and indexed according to a two-dimensional P6mm hexagonal symmetry. It is particularlyinteresting that the samples with 20 mol% UPTES exhibita better mesoscopic order than that of the pure PMOsmaterials, which is not expected as the presence of organos-ilane would be more or less disruptive to the structuralorder as reported previously in a similar synthetic system[15,17,22]. Such observation is further confirmed by theTEM images. As shown in Fig. 2, the pure PMOs materialdisplays predominantly a wormhole-motif pore structure(Fig. 2b), but an uniform pore channel (Fig. 2a) and asmall domain reflecting a more ordered structure inFig. 2b are also observed. However, the materials function-alized with 20 mol% UPTES show perfect hexagonallypacked pore arrangement (Fig. 2d) and straight pore chan-nels (Fig. 2c). Such observation encourages us to furtherexamine the structural order of functionalized samples pre-pared in the presence of higher concentration of UPTES.However, an absolutely opposite trend is observed withfurther increasing amount of UPTES. The presence of30 mol% UPTES leads to a more disordered structure asevidenced by the pronounced decrease of the intensity of
the (100) reflection and the disappearance of the higherorder reflections. It is also the case for the samples with40 mol% UPTES. Therefore, in order to obtain a highlyordered functionalized PMOs material, the concentrationof UPTES should be fixed to 20 mol%. The mesostructureof functionalized materials might be attributed to thebehavior of UPTES and the assembly procedure in themixture under different conditions during the sol-gel reac-tion. It is possible that under synthetic conditions mostUPTES may react with BTESE through co-hydrolysisand condensation to form silica network, especially in thecase of low UPTES concentration in the mixture, thereforeureidopropyl groups are attached to the surface of the finalmaterials. However, ureidopropyl groups might have astrong influence on the self-assembly of polymer P123,leading to the destruction of the interface between silicatesspecies and P123, which is formed by a combination of H-bonding, electrostatic, and van der Waals interactions [23],thus, resulting in the formation of a poor ordered meso-structure. However, functionalized materials have an excel-lent mesoscopic order as the concentration of UPTES isequal to 20 mol%, which seems to be incompatible to theabove-mentioned mechanism. Such surprising observationmay be related to the special nature of ureidopropyl groupsin solution. As the concentration of UPTES increases grad-ually, a fraction of UPTES tends to form the so-called oli-gomerized silicate species through the intermolecularcondensation of the hydrolysate of UPTES. The oligomer-ized silicate species, consisting of hydrophobic moieties andhydrophilic sections, have a similar behavior in solution asthe organosilicon surfactants. The hydrophobic section ofthe oligomerized silicate species inserts into the hydropho-bic block of P123 to form mixed surfactants, leading to areduction of the charge density for the polar heads ofmixed surfactants, and therefore it is more easy for themixed surfactants to form micelles and to be organized intoordered assemblies. The competition between theformation of mixed surfactants and the disruptive effectof ureidopropyl groups compromises at the UPTES con-centration of 20 mol%. The silicate species and the mixedsurfactants are assembled together via the H-bonding, elec-trostatic and van der Waals interactions to form a highlyordered mesostructure. As mentioned above, the materialsbecome structurally amorphous with further increase ofUPTES concentration. This observation is attributed tothe fact that UPTES precursors contain fewer hydrolysablegroups (3 in UPTES vs. 6 in BTESE) and provide less sites(–OR or –OH groups, where R is referred to ethoxygroups) for condensation or interacting with triblockcopolymer P123 in comparison with the BTESE precur-sors. Therefore, if large amount of UPTES substitutes forBTESE in the initial mixture, the degree of cross-linkingwill decrease and the silicate species will be poorly orga-nized around P123 surfactants to such an extent that anordered mesostructure does not form [9]. Additionally,the steric hindrance resulted from ureidopropyl groups inUPTES also prevents more or less the co-condensation of

Fig. 2. TEM images of PMOs materials with UPTES molar ratio of (a,b) 0 mol% and (c,d) 20 mol% taken perpendicular to the direction of the pore axis(a,c) and along the pore axis (b,d).
384 Q. Wei et al. / Microporous and Mesoporous Materials 101 (2007) 381–387
UPTES and BTESE, leading to a reduction of the densityand an increase of the defects for the hybrid frameworks,and furthermore resulting in a disruptive local and longrange order. From the exact positions of the (100) peaks,the d1 0 0 spacings of the samples can be calculated andthe results are listed in Table 1. It can be seen from Table1 that increasing the concentration of UPTES in the mix-ture causes an increase of d1 0 0 spacings from 10.6 nmfor the pure PMOs to 13.1 nm for the sample with30 mol% UPTES. This indicates an expansion of theframework, resulting from the lower degree of the conden-sation of UPTES and BTESE.
Solid state NMR experiments are used to determinewhether the functional groups have been attached to pore
Table 1Textural data of the functionalized PMOs with different molar ratio of UPTE
UPTES concentration (mol%) Surface area (m2 g�1) Pore volu
0 1071 1.55 1004 1.410 876 1.320 565 1.130 539 0.9
channels of PMOs and whether surfactant species havebeen removed after ethanol extraction. The 1H MASNMR spectra of the pure and functionalized samples areshown in Fig. 3. The resonance of H2 protons (as shownin the inset in Fig. 3) almost overlaps with that of H3 pro-tons, resulting in a broad peak at 7.9 ppm in the function-alized samples. A signal at 1.3 ppm is related to themethylene within the silicate framework and in theattached ureidopropyl groups (H1), whereas the intensityof the signal in functionalized materials is larger than thatin pure PMOs samples. This observation, together with thepresence of resonances related to amine proton, indicatesthat ureidopropyl groups have been functionalized intoPMOs materials. The peak at 4.8 ppm is assigned to phys-
S
me (cm3 g�1) Mean pore diameter (nm) d1 0 0 (nm)
8.2 10.610.1 10.38.3 11.3
10.1 11.712.1 13.1

15 10 5 0 -5
(b)
(a)
7.9
4.1
1.3
1.3
1.6
4.1
4.8
Chemical shift/ppm
H1
Si CH2 CH2 CH2 NH C NH2
O
Si CH2 CH2
OH O
OO
O
H1 H2 H3
H4
Si OSi CH 2 CH2
OH O
OO
O
H OHH1
H4
Fig. 3. Solid state 1H MAS NMR spectra of the surfactant-removedsamples: (a) pure PMOs and (b) PMOs functionalized with 20 mol%UPTES.
Q. Wei et al. / Microporous and Mesoporous Materials 101 (2007) 381–387 385
isorbed water while the signal at 1.6 ppm is resulted fromisolated silanol species (H4) [24]. The signal at 4.1 ppmmay be attributed to the impurities in the precursors.
Fig. 4 shows a comparison of the 13C CP MAS NMRspectra for the as-synthesized or solvent-extracted materi-als, including the pure and 20 mol% UPTES-functionalizedPMOs. A prominent peak is observed at the chemical shiftof 5.0 ppm for all samples, which is attributed to ethane-carbon atoms covalently linked to Si atoms in the frame-work [25,26]. The presence of such resonance indicates thatno Si–C bond cleavage occurs during either the synthesis orthe surfactant extraction process. For the functionalizedmaterials, the peaks occurring at ca. 22, 24, 42 and161 ppm are assigned to the C1, C2, C3 and C4 carbonatoms (as shown in the inset of Fig. 4) contained in ureido-
200 150 100 50 0
C4 C3 C2
C1
P123
P123
(d)
(c)
(b)
(a)
Chemical shift/ppm
Si CH2 CH2 CH2 NH C NH2
O
C1 C2 C3 C4
Si CH2 CH2 Si
C2H5OH
Fig. 4. Solid state 13C CP MAS NMR spectra of the samples: (a) purePMOs, before surfactant-removal; (b) pure PMOs, after surfactant-removal; (c) PMOs functionalized with 20 mol% UPTES, before surfac-tant-removal and (d) PMOs functionalized with 20 mol% UPTES, aftersurfactant-removal.
propyl groups [27,28]. It can be confirmed that ureidopro-pyl groups remain intact and have successfully beenattached to the pore channels of PMOs materials due tothe presence of resonances that are characteristic of thesemoieties. The resonance peak at ca. 60 ppm for theextracted materials is derived from ethanol, which maybe trapped during the surfactant extraction. It is shownthat surfactants have been removed by ethanol extractionsince the signals at 17 and 73 ppm assigned to remnantP123 surfactants are not observed in the extracted samples[12]. Fig. 5 depicts the 29Si CP MAS NMR spectra of thesurfactant-removed pure silica PMOs and 20 mol%UPTES-functionalized samples. Both samples display T2
and T3 resonances ascribed to Si(OSi)2OHR and Si(OSi)3Rframework silicon sites [29], respectively, with only slightdifferences in chemical shifts. The absence ofQn[Si(OSi)n(OH)4�n] Si sites indicates that the Si–C bondsremain intact under our synthesis and subsequent treat-ment conditions. Since there are no signals of –OC2H5
groups in the 1H MAS NMR and 13C CP MAS NMRspectra, we can deduce that all UPTES precursors intro-duced in the mixture have involved in the co-condensationreaction, and that all ureidopropyl groups except thosemixed with P123 have been integrated in the final materials.
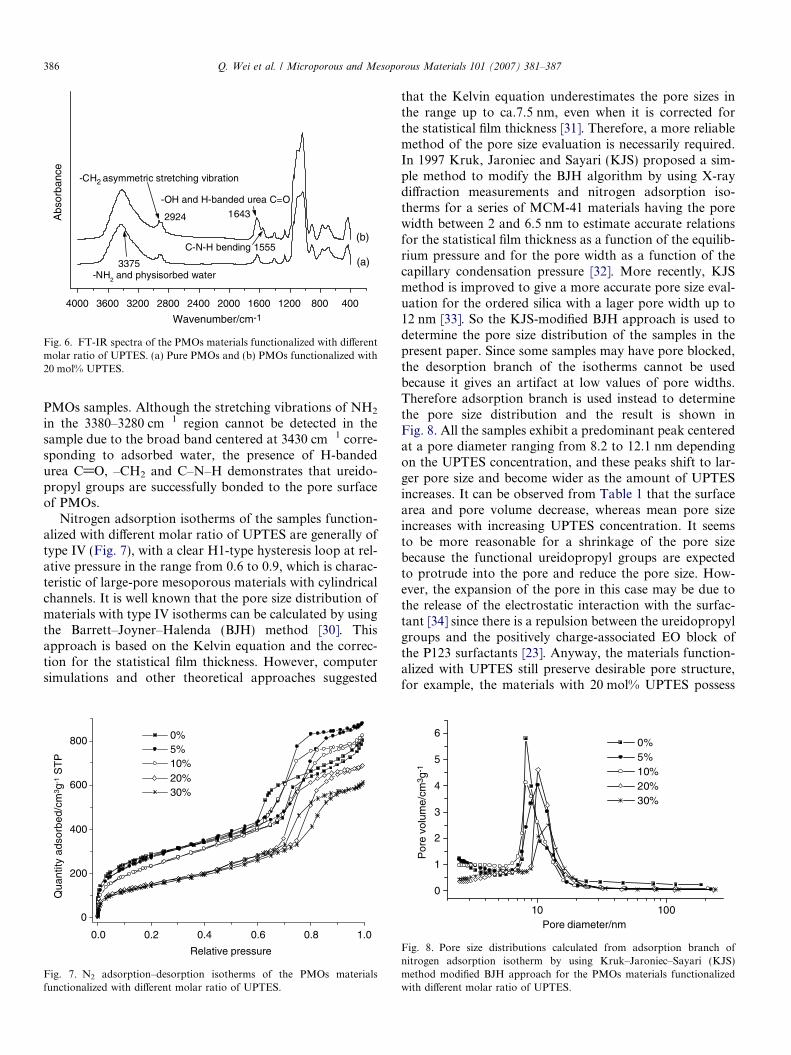
The presence of ureidopropyl groups in functionalizedsamples can also be confirmed by the FT-IR spectra inFig. 6. The absorption bands at 2924 cm�1 are attributedto the –CH2 asymmetric stretching vibration within boththe framework and ureidopropyl groups. The C–N–Hbending absorption can be found in functionalized PMOsmaterials at a wavenumber of 1550 cm�1, but this vibrationis not observed in pure POMs samples. The H-bandedC@O absorption occurs at about 1643 cm�1, overlappedwith that of silanol groups, but the presence of H-bandedurea C@O in functionalized materials can be confirmedby the observation that the 1643 cm�1 absorption in func-tionalized samples is much stronger than that of pure
0 -20 -40 -60 -80 -100 -120
T3
T2
Chemical shift/ppm
SiO Si C
OH
T2
SiO Si C
OSi
OSi
OSi
T3
(a)
(b)
Fig. 5. Solid state 29Si CP MAS NMR spectra of the surfactant-removedsamples: (a) pure PMOs and (b) PMOs functionalized with 20 mol%UPTES.
4000 3600 3200 2800 2400 2000 1600 1200 800 400
(b)
(a) 3375
1643
-CH2 asymmetric stretching vibration
2924
-NH2
and physisorbed water
-OH and H-banded urea C=O
C-N-H bending 1555
Abs
orba
nce
Wavenumber/cm-1
Fig. 6. FT-IR spectra of the PMOs materials functionalized with differentmolar ratio of UPTES. (a) Pure PMOs and (b) PMOs functionalized with20 mol% UPTES.
386 Q. Wei et al. / Microporous and Mesoporous Materials 101 (2007) 381–387
PMOs samples. Although the stretching vibrations of NH2
in the 3380–3280 cm�1 region cannot be detected in thesample due to the broad band centered at 3430 cm�1 corre-sponding to adsorbed water, the presence of H-bandedurea C@O, –CH2 and C–N–H demonstrates that ureido-propyl groups are successfully bonded to the pore surfaceof PMOs.
Nitrogen adsorption isotherms of the samples function-alized with different molar ratio of UPTES are generally oftype IV (Fig. 7), with a clear H1-type hysteresis loop at rel-ative pressure in the range from 0.6 to 0.9, which is charac-teristic of large-pore mesoporous materials with cylindricalchannels. It is well known that the pore size distribution ofmaterials with type IV isotherms can be calculated by usingthe Barrett–Joyner–Halenda (BJH) method [30]. Thisapproach is based on the Kelvin equation and the correc-tion for the statistical film thickness. However, computersimulations and other theoretical approaches suggested
0.0 0.2 0.4 0.6 0.8 1.0
0
200
400
600
800 0% 5% 10% 20% 30%
Qua
ntity
ads
orbe
d/cm
3 g-1
ST
P
Relative pressure
Fig. 7. N2 adsorption–desorption isotherms of the PMOs materialsfunctionalized with different molar ratio of UPTES.
that the Kelvin equation underestimates the pore sizes inthe range up to ca.7.5 nm, even when it is corrected forthe statistical film thickness [31]. Therefore, a more reliablemethod of the pore size evaluation is necessarily required.In 1997 Kruk, Jaroniec and Sayari (KJS) proposed a sim-ple method to modify the BJH algorithm by using X-raydiffraction measurements and nitrogen adsorption iso-therms for a series of MCM-41 materials having the porewidth between 2 and 6.5 nm to estimate accurate relationsfor the statistical film thickness as a function of the equilib-rium pressure and for the pore width as a function of thecapillary condensation pressure [32]. More recently, KJSmethod is improved to give a more accurate pore size eval-uation for the ordered silica with a lager pore width up to12 nm [33]. So the KJS-modified BJH approach is used todetermine the pore size distribution of the samples in thepresent paper. Since some samples may have pore blocked,the desorption branch of the isotherms cannot be usedbecause it gives an artifact at low values of pore widths.Therefore adsorption branch is used instead to determinethe pore size distribution and the result is shown inFig. 8. All the samples exhibit a predominant peak centeredat a pore diameter ranging from 8.2 to 12.1 nm dependingon the UPTES concentration, and these peaks shift to lar-ger pore size and become wider as the amount of UPTESincreases. It can be observed from Table 1 that the surfacearea and pore volume decrease, whereas mean pore sizeincreases with increasing UPTES concentration. It seemsto be more reasonable for a shrinkage of the pore sizebecause the functional ureidopropyl groups are expectedto protrude into the pore and reduce the pore size. How-ever, the expansion of the pore in this case may be due tothe release of the electrostatic interaction with the surfac-tant [34] since there is a repulsion between the ureidopropylgroups and the positively charge-associated EO block ofthe P123 surfactants [23]. Anyway, the materials function-alized with UPTES still preserve desirable pore structure,for example, the materials with 20 mol% UPTES possess
10 100
0
1
2
3
4
5
6 0% 5% 10% 20% 30%
Por
e vo
lum
e/cm
3 g-1
Pore diameter/nm
Fig. 8. Pore size distributions calculated from adsorption branch ofnitrogen adsorption isotherm by using Kruk–Jaroniec–Sayari (KJS)method modified BJH approach for the PMOs materials functionalizedwith different molar ratio of UPTES.

Q. Wei et al. / Microporous and Mesoporous Materials 101 (2007) 381–387 387
a surface area of 565 m2/g, a pore volume of 1.1 cm3/g anda mean pore diameter of 10.1 nm.
4. Conclusion
On the basis of the co-condensation of bis(triethoxysi-lyl)ethane and ureidopropyltriethoxysilane, ureidopropylgroups are terminally functionalized to periodic mesopor-ous organosilicas without any damage to the ethane bridg-ing groups in the framework. The functionalized materialsbecome structurally disordered with increasing of ureido-propyl group except for the sample with 20 mol% UPTES,which displays a highly ordered two-dimensional hexago-nal structure, with a better mesoscopic order than that ofpure PMOs. This material still possesses a desirable porestructure with a surface area of 565 m2/g, a pore volumeof 1.1 cm3/g, and a mean pore size of 10.1 nm, althoughit shows a trend that the surface area and pore volumedecrease with increasing amount of UPTES for functional-ized materials.
Acknowledgements
The financial support of National Natural ScienceFoundation of China (Grant Nos. 50502002, 50525413)and Scientific Research Common Program of BeijingMunicipal Commission of Education (Grant No.KM200610005016) is gratefully acknowledged.
References
[1] T. Asefa, M.J. MacLachlan, N. Coombs, G.A. Ozin, Nature 402(1999) 867–871.
[2] B.J. Melde, B.T. Holland, C.F. Blanford, A. Stein, Chem. Mater. 11(1999) 3302–3308.
[3] S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna, O. Terasaki, J. Am.Chem. Soc. 121 (1999) 9611–9614.
[4] B. Hatton, K. Landskron, W. Whitnall, D. Perovic, G.A. Ozin, Acc.Chem. Res. 38 (2005) 305–312.
[5] O. Olkhovyk, M. Jaroniec, J. Am. Chem. Soc. 127 (2005) 60–61.[6] G. Kickelbick, Angew. Chem. Int. Ed. 43 (2004) 3102–3104.[7] K. Landskron, B.D. Hatton, D.D. Perovic, G.A. Ozin, Science 302
(2003) 266–269.[8] X.Y. Bao, X.S. Zhao, J. Phys. Chem. B 109 (2005) 10727–10736.
[9] T. Asefa, M. Kruk, M.J. MacLachlan, N. Coombs, H. Grondey, M.Jaroniec, G.A. Ozin, J. Am. Chem. Soc. 123 (2001) 8520.
[10] X. Feng, G.E. Frywell, L.Q. wang, A.Y. Kim, J. Liu, K.M. Kemner,Science 276 (1997) 923–926.
[11] A.S.M. Chong, X.S. Zhao, A.T. Kustedjo, S.Z. Qiao, Microp.Mesop. Mater. 72 (2004) 33–42.
[12] Y. Wang, B. Zibrowius, C.-M. Yang, B. Spliethoff, F. Schuth, Chem.Commun. (2004) 46–47.
[13] R.J.P. Corriu, A. Mehdi, C. Reye, C. Thieuleux, Chem. Mater. 16(2004) 159–166.
[14] S. Hamoudi, S. Kaliaguine, Micropor. Mesopor. Mater. 59 (2003)195–204.
[15] M.C. Burleigh, M.A. Markowitz, M.S. Spector, B.P. Gaber, J. Phys.Chem. B 105 (2001) 9935.
[16] S. Hamoudi, S. Royer, S. Kaliaguine, Micropor. Mesopor. Mater. 71(2004) 17–25.
[17] M.A. Wahab, I. Imae, Y. Kawakami, C.-S. Ha, Chem. Mater. 17(2005) 2165–2174.
[18] Y.J. Gong, Z.H. Li, D. Wu, Y.H. Sun, F. Deng, Q. Luo, Y. Yong,Micropor. Mesopor. Mater. 49 (2001) 95–102.
[19] A.M. Liu, K. Hidajat, S. Kawi, D.Y. Zhao, Chem. Commun. (2000)1145.
[20] T. Gass, F.S. Ligler, Immobilized Biomolecules in Analysis – APractical Approach, Oxford University Press, New York, 1998.
[21] A. Subramanian, S.J. Kennel, P.I. Oden, Enzyme Microb. Technol.(1999) 24, 26.
[22] R. Corriu, A. Mehdi, C. Reye, C. Thieuleux, Chem. Commun. (2002)1382–1383.
[23] D.Y. Zhao, Q.S. Huo, J.L. Feng, B.F. Chmelka, G.D. Stucky, J. Am.Chem. Soc. 120 (1998) 6024–6036.
[24] Jirı Brus, Jirı Dybal, Macromolecules 35 (2002) 10038–10047.[25] A. Sayari, S. Hamoudi, Y. Yang, I.L. Moudrakovski, J.R. Ripme-
ester, Chem. Mater. 12 (2000) 3857.[26] W. Guo, J.-Y. Park, M.-O. Oh, H.W. Jeong, W.J. Cho, I. Kim, C.S.
Ha, Chem. Mater. 15 (2003) 2295–2298.[27] J.J. Moreau, L. Vellutini, M.W.C. Man, C. Bied, Chem. Eur. J. 9
(2003) 1594–1598.[28] D.Q. Yu, J.S. Yang, J.X. Xie, Handbook of Analytical Chemistry:
Nuclear Magnetic Resonance, Chemical Industry Press, Beijing, 1989(in Chinese) pp. 476, 542 & 548.
[29] X.Y. Bao, X.S. Zhao, X. Li, P.A. Chia, J. Li, J. Phys. Chem. B 108(2004) 4684–4689.
[30] E.P. Barrett, L.G. Joyner, P.P. Halenda, J. Am. Chem. Soc. 73 (1951)373–380.
[31] C. Lastoskie, K.E. Gubbins, N. Quirke, J. Phys. Chem. 97 (1993)4786–4796.
[32] M. Kruk, M. Jaroniec, A. Sayari, Langmuir 13 (1997) 6267–6273.[33] M. Jaroniec, L.A. Solovyov, Langmuir 22 (2006) 6757–6760.[34] T. Yokoi, H. Yoshitake, T. Tatsumi, Chem. Mater. 15 (2003) 4536–
4538.