genespring12 - tokushima u · genespring12 で解析できるマイクロアレイデータ...
TRANSCRIPT

GeneSpring12.5 ベーシックトレーニング資料
2013/01/16
GeneSpringベーシックトレーニング向け日本語資料

2
内容 はじめに ................................................................................................................................................5
GeneSpringについての各種情報 ......................................................................................................5
用語解説 ............................................................................................................................................6
GeneSpring12 で解析できるマイクロアレイデータ .....................................................................7
インターフェイスの説明 .................................................................................................................8
データの準備 ...................................................................................................................................... 11
シグナルデータの準備 .................................................................................................................. 11
テクノロジーの作成 ...................................................................................................................... 12
テクノロジーのダウンロード .................................................................................................. 12
テクノロジーの更新 .................................................................................................................. 12
エクスペリメントの作成 .................................................................................................................. 13
Agilent .......................................................................................................................................... 13
Affymetrix .................................................................................................................................... 15
Illumina ........................................................................................................................................ 21
ノーマライズとベースラインオプション .................................................................................. 23
ノーマライズの影響 .................................................................................................................. 23
ベースラインオプションの影響 .............................................................................................. 24
Baseline to median of Control samplesの設定方法 ................................................................... 25
基本的なグラフ .............................................................................................................................. 26
参考資料 Raw Signal valueとNormalized Signal value ................................................................ 28
グループの設定(Experiment Grouping) ................................................................................... 29
パラメーターをファイルからインポートする(必要なときのみ) .................................. 30
ブラウザでの表示順を変更する(必要なときのみ) .......................................................... 31
Quality control on sample のPCAや 2way実験の表示順を変更(必要なときのみ) ........... 31
インタープリテーションの作成(Create Interpretation) ....................................................... 32
クオリティコントロール .................................................................................................................. 34
サンプルの品質管理(Quality control on samples) .................................................................. 34
3D PCA Scores .............................................................................................................................. 34
Correlation plot ............................................................................................................................ 34

3
Hybridization Controls .................................................................................................................. 35
サンプルの除外 .......................................................................................................................... 35
発現量によるフィルタリング(Filter Probesets by Expression) .............................................. 36
フラグによるフィルタリング(Filter Probesets by Flags) ....................................................... 37
シグナルデータのフィルタリング(Filter Probesets on Data files) ........................................ 38
再現性によるフィルタリング(Filter Probesets by Error) ....................................................... 39
解析 1 遺伝子リストの絞込み ....................................................................................................... 40
統計検定(Statistical Analysis) .................................................................................................... 40
実験デザインと利用する検定の対応 ...................................................................................... 40
t-testによる検定 ......................................................................................................................... 41
統計検定(t-test)の操作手順 ................................................................................................. 42
ANOVAによる検定 ...................................................................................................................... 44
統計検定(ANOVA)の操作手順 .............................................................................................. 45
Fold change(Fold change) ........................................................................................................... 47
解析 2 クラスタリングなど ........................................................................................................... 49
クラスタリング(Clustering) ...................................................................................................... 49
k-means ........................................................................................................................................ 50
Hierarchical ................................................................................................................................... 51
Self-Organizing Map(SOM) ........................................................................................................... 53
発現パターンの似ているエンティティの検索(Find Similar Entities) .................................. 54
数値パラメーターと相関するエンティティの検索(Filter on Parameters) ......................... 55
Principal Component Analysis(主成分分析) .............................................................................. 56
結果の解釈 .......................................................................................................................................... 59
GO解析(GO Analysis) .................................................................................................................. 59
GO Analysisから、ほしい遺伝子だけをピックアップする ................................................... 60
GSEA解析(GSEA) ......................................................................................................................... 62
データの準備 .............................................................................................................................. 62
GSEAの実行 ................................................................................................................................. 64
パスウェイ解析 .................................................................................................................................. 66
データの準備 .................................................................................................................................. 66

4
Single Experiment Analysis .............................................................................................................. 66
ゲノムブラウザ .................................................................................................................................. 68
データの準備 .................................................................................................................................. 68
使い方 ............................................................................................................................................. 68
参考資料 ............................................................................................................................................. 71
便利な機能 ...................................................................................................................................... 71
Translate機能 .............................................................................................................................. 71
Export as Image ............................................................................................................................ 73
Export List .................................................................................................................................... 74
セレクションモードとズームモードの切り替え .................................................................. 75
アノテーションの閲覧 .............................................................................................................. 77
アノテーションからのエンティティの検索 .......................................................................... 78
グラフの見方 .................................................................................................................................. 79
GX7 とGX9 以降の検定の対応 ...................................................................................................... 84
1sample T-test(主に 2color実験の場合に使用可能) ........................................................... 85
Similarity Metrics ............................................................................................................................. 87
GeneSpringの保守・管理 ................................................................................................................... 89
プロジェクト単位のエクスポート .............................................................................................. 89
データのバックアップと復元 ...................................................................................................... 90
GeneSpringのバージョンアップ ................................................................................................... 90
お問い合わせ .................................................................................................................................. 91

5
はじめに
GeneSpringについての各種情報 GeneSpringについて情報を収集する場合は下記をご利用ください。
GeneSpringのヘルプページ
GeneSpringのHelpメニューからDocumentation Indexで英文の機能解説があります。
GeneSpringのサポートページ
http://genespring-support.com/
最新のインストーラーがダウンロードできます。※要ユーザー登録
Agilent日本のGeneSpringGXページ
http://www.chem.agilent.com/scripts/pds.asp?lpage=27881
日本語マニュアルやデモデータなどを閲覧できます。
AgilentのFAQページ
http://www.chem-agilent.com/contents.php?id=1001256
アジレントテクノロジーが作成している日本語FAQページです
Tomy digital biologyのFAQページ
http://www.digital-biology.co.jp/j_faq
トミーデジタルバイオロジーが作成している日本語FAQページです。

6
用語解説 ここでは、GeneSpringGXで使用する用語を解説します。
プロジェクト(Project)
ユーザーがデータを格納するワークスペースです。GeneSpringGXを複数人で共有する場合な
ど、データを分けることができます。用途の異なるデータは混乱を招くため異なるプロジ
ェクトにしたほうが無難です。
エクスペリメント(Experiment)
エクスペリメントはデータの解析単位になります。比較したいシグナルデータのインポー
トを行うとエクスペリメントが 1 つできます。
サンプル(Sample)
アレイから得られたシグナルデータです。シグナルデータのファイル名がSample名です。
ノーマリゼーション(Normalization)
アレイ間の誤差を修正し、発現量を比較するための処理です。複数の方法が搭載されてい
ます。ノーマライズはエクスペリメント単位で決定され変更することはできません。
パラメーター(Parameter)
データを解析する上での条件の名称です(例 Timeなど)
コンディション(Condition)
設定されたパラメーター内にある条件です(例 1 時間、2 時間、3 時間など)
インタープリテーション(Interpretation)
グルーピングと表示方法の設定です(例 時系列での表示、性別での表示など)
エンティティ(Entity)
マイクロアレイで扱われる 1 分子を指します。DNAマイクロアレイはほとんどの場合遺伝子
を指しますがmiRNAアレイやExonアレイを使った場合はそれぞれの概念に従ってください。
テクノロジー(Technology)
アレイに搭載されているエンティティの機能や公共DBのIDを含んだアノテーションセット
です。解析結果の解釈を得るためには十分なアノテーションが必要になります。

7
GeneSpring12 で解析できるマイクロアレイデータ GeneSpring12 のGXモジュールでは、下記のプラットフォームについて解析を行うことがで
きます。下記以外の対応していないプラットフォームでも、テキストファイルがあればカ
スタムデータとして登録することができます。
プラットフォーム GeneSpring 内の表記 備考
アジレント
Agilent Expression Single Color 遺伝子発現 1 色法 Agilent Expression Two Color 遺伝子発現 2 色法 Agilent Exon Single Color Exon アレイ 1 色法 Agilent Exon Two Color Exon アレイ 2 色法 Agilent miRNA microRNA アレイ
アフィメトリクス
Affymetrix Association Analysis SNP ジェノタイピング解析 Affymetrix Copy number コピーナンバー解析 Affymetrix Exon Expression GeneST/ExonST を使った遺
伝子発現解析 Affymetrix Exon Splicing GeneST/ExonST を使ったス
プライシング解析 Affymetrix Expression 3’IVT 遺伝子発現
カスタム Generic Single Color カスタムテクノロジー 1 色法 Generic Two Color カスタムテクノロジー 2 色法
イルミナ Illumina Association Analysis SNP ジェノタイピング解析 Illumina Copy Number コピーナンバー解析 Illumina Single Color 遺伝子発現
※カスタムデータの取り扱いに関しては、AgilentのFAQページ( http://www.chem-agilent.com/contents.php?id=1001256 )にあるGenespirng12 カスタムテクノロジーの作
成.pdfをご覧ください。

8
インターフェイスの説明
メニュー群:各操作を行うためのメニューです
アイコン群:よく使う操作はアイコン群になっています
プロジェクトナビゲーター:自分の閲覧したいデータを選択します
ブラウザ:プロジェクトナビゲーターで選んだデータが表示されます
ワークフロー:データ解析に関するメニューが並んでいます
レジェンド: ブラウザの情報について補足がある場合、情報が表示されます。
インスペクター機能
エクスペリメントやエンティティリストを右クリックし、Inspect~を選択すると、対象のイ
ンスペクターが開きます。オブジェクトにもよりますが作成日や作成条件、解析結果など
の情報が書き込まれています、よくわからないデータがあった場合は、ダブルクリックす
るか、右クリックしてください。
メニュー群
アイコン群
プロジェクト
ナビゲーター ブラウザ
ワークフロー
レジェンド

9
プロジェクトを開く
ProjectメニューからOpen Projectまたは、 アイコンですでに作成済みのプロジェクトを
選択して開くことができます。
エクスペリメントを開く
プロジェクトナビゲーターのExperimentsフォルダ以下にあるアイコンをダブルクリックす
ると既存のエクスペリメントが開き、ブラウザにデータが表示され、データ解析可能な状
態になります。※プロジェクトにエクスペリメントが 1 つしかない場合、自動的にそのエ
クスペリメントが開きます。
ダブルクリ
ックで開く

10
エクスペリメントの切り替え
エクスペリメントを複数開いている場合は、プロジェクトナビゲーターのエクスペリメン
ト名かブラウザ上部のタブをクリックで切り替えることが可能です。
複数のエクスペリメントを開くと、エンテ
ィティリストを共有することができます。
グラフ画面の切り替え
複数のグラフの切り替えはグラフ下部のタブを切り替えて行います。必要のないタブは右
クリックからCloseを選択して閉じることも可能です。
Windowの並べ替え
WindowsメニューからTile>Bothと選ぶと、複数のグラフを一画面に並べて表示することが
できます。一度に閲覧できる情報量が多くなり、選択操作などが連動します。

11
データの準備 シグナルデータとテクノロジーの作成について紹介します。
シグナルデータの準備
Agilent: Feature Extractionのアウトプットテキストファイル・1 色法・2 色法どちらも可能
Affymetrix:CELファイル、CHPファイル、テキストファイル
※ Affyのデータは日本語が含まれるフォルダに保存しないでください。
Illumina:GenomeStudioでbgx manifest filesを使って作成されたテキストファイル
Illuminaのテキストデータ例(エクセルで表示しています)
実際使用するカラムはProbeID・Average Signal・Detection p-valueです。
自動認識で取り込めない場合は、カスタムテクノロジーで対応してください。
その他: GPR形式、行列形式のテキストファイルなど
※その他のデータは、別資料カスタムテクノロジーの作成方法をご参照ください。
Array Content が GeneSpring の
Technology と同名である必要があります

12
テクノロジーの作成 テクノロジー(アレイごとのデータフォーマットや遺伝子等のアノテーション)は解析結
果を解釈するために必要な情報です。Affymetrix、Agilent、Illuminaの市販されているアレイ
のテクノロジーはGeneSpring GXを使い、インターネット経由でダウンロードすることがで
きます。
それ以外の製品をお使いの方は、カスタムテクノロジーを作成する必要があります。
テクノロジーのダウンロード 1. AnnotationsメニューのCreate technologyからFrom Agilent serverを選択する 2. 新しく開いたWindowで、お使いのアレイ名にチェックを入れてUpdateボタンをクリック 3. ダウンロード容量が表示されるので、HDDの空き容量を確認して、了解をクリック 4. ダウンロードが成功すると終了します
※ ステップ 2 でダウンロードするテクノロジーの一覧が表示されない場合は、イ
ンターネット接続が適切ではない場合があります、下記を確認してください。
・Toolsメニュー/options/Miscellaneous/Network settingsからProxyの設定を見直す
・セキュリティソフトを停止して試す
・所属の機関で下記のURLにアクセスできるかを管理者に確認する
http://lcosgens.cos.agilent.com
テクノロジーの更新 すでにGeneSpringにダウンロードしてあるテクノロジーの更新版がリリースされると
GeneSpringの起動時に更新を促すメッセージが表示されます。ダウンロードと同じ手順で更
新することができます。
※ テクノロジーを更新すると、遺伝子の情報等が以前と変わる場合があるのでご注意ください。

13
エクスペリメントの作成 解析したいシグナルデータセットをインポートすると、エクスペリメントが 1 つ作成され
ます。アレイプラットフォームよりって少々項目が異なりますが、エクスペリメント作成
時にはアレイ間のノーマライズやベースライン補正などが行われます。
Agilent 1. ProjectメニューからNew projectをクリックし、Project名を入力します
※すでにProjectを作成してある場合は、ProjectメニューからNew experimentをクリック
2. Create new experimentを選択
※すでに作成済みのエクスペリメントを開くにはOpen existing experimentを選択
3. 解析条件を入力する
4. Choose Filesボタンからテキストファイルを指定してNext
Single color か Two color を選択
Data Import Wizard を選択

14
5. Flagの設定を行う(デフォルト推奨)
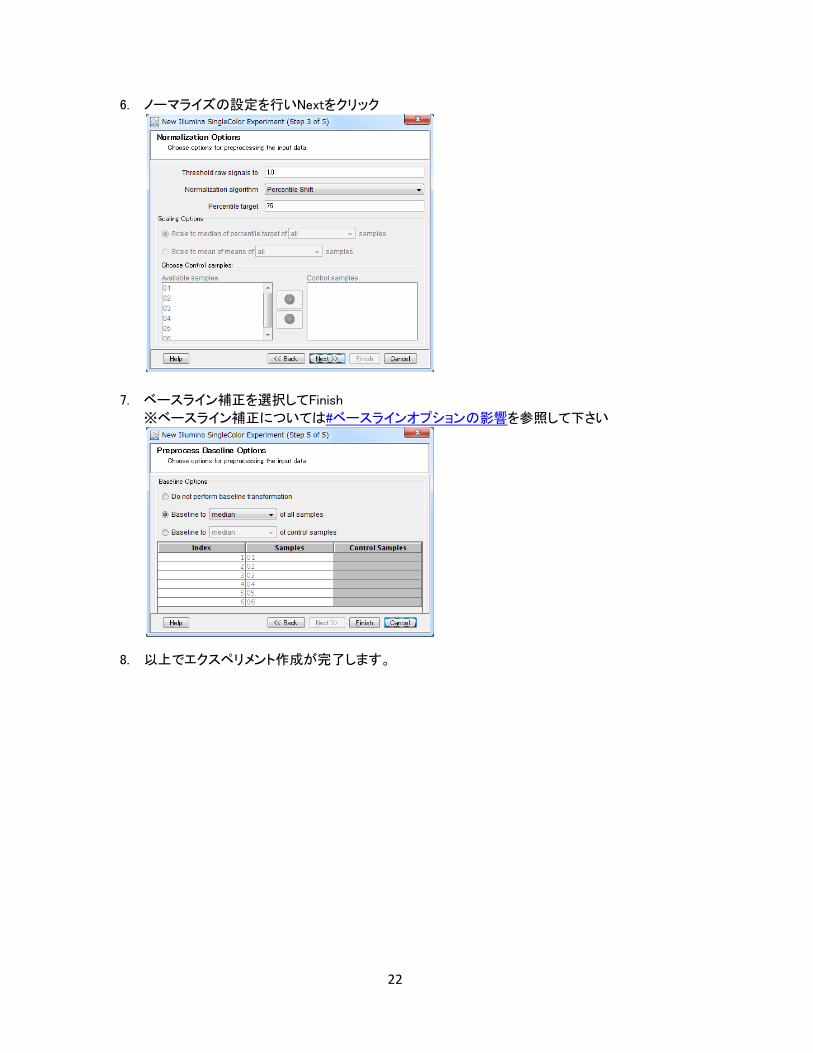
6. ノーマライズの設定を行い、Nextをクリック
7. ベースライン補正の設定を行いFinishボタンをクリック
※ベースライン補正については#ベースラインオプションの影響を参照して下さい
8. 以上でエクスペリメント作成が完了します
Percentile Shift で Percentile target75 を推奨

15
Affymetrix Affymetrixのデータは、CEL・CHP・テキストファイルをインポート可能です。
CELファイルをインポートする場合
※ CELファイルはパスに日本語を含まない場所に保存してください。
1. ProjectメニューからNew projectをクリックし、Project名を入力します
※すでにProjectを作成してある場合は、ProjectメニューからNew experimentをクリック
2. Create new experimentを選択
※すでに作成済みのエクスペリメントを開くにはOpen existing experimentを選択
3. 解析条件を入力する
Analysis type:HG-U133 などの 3’IVTはExpression、GeneST/ExonSTはExon。
Experiment type:3’IVTはAffymetrixExpression、GeneST/ExonSTの遺伝子レベル解析はExon expression、GeneST/ExonSTのExonレベル解析はExon splicingを選択。
4. Choose FilesボタンからCELファイルを指定してNext
Data Import wizard を選択

16
5. Attributeファイルがある場合は指定する、無い場合はそのままNext
6. ノーマライズとベースライン補正を選択してFinish
※ベースライン補正については#ベースラインオプションの影響を参照して下さい
ベーシックトレーニングではMAS5 を利用します
7. 以上でエクスペリメント作成が完了します。

17
CHPファイルをインポートする場合
※ CHPファイルはパスに日本語を含まない場所に保存してください。 1. ProjectメニューからNew projectをクリックし、Project名を入力します
※すでにProjectを作成してある場合は、ProjectメニューからNew experimentをクリック
2. Create new experimentを選択
※すでに作成済みのエクスペリメントを開くにはOpen existing experimentを選択
3. 解析条件を入力する
Analysis type:HG-U133 などの 3’IVTはExpression、GeneST/ExonSTはExon。
Experiment type:3’IVTはAffymetrix Expression、GeneST/ExonSTの遺伝子レベル解析はExon expression
4. Choose FilesボタンからCHPファイルを指定してNext
Data Import wizard を選択

18
5. Attributeファイルがある場合は指定する、無い場合はそのままNext
6. ノーマライズの設定を行いNextをクリック
7. ベースライン補正を選択してFinish
※ベースライン補正については#ベースラインオプションの影響を参照して下さい
8. 以上でエクスペリメント作成が完了します。

19
テキストファイルをインポートする場合
1. ProjectメニューからNew projectをクリックし、Project名を入力します
※すでにProjectを作成してある場合は、ProjectメニューからNew experimentをクリック
2. Create new experimentを選択
※すでに作成済みのエクスペリメントを開くにはOpen existing experimentを選択
3. 解析条件を入力する
Analysis type:HG-U133 などの 3’IVTはExpression
Experiment type:3’IVTはAffymetrix Expression
4. Choose Filesボタンからテキストファイルを指定してNext
Data Import wizard を選択

20
5. インポートするGene Chipのテクノロジーを選択する
※Expression console以外のテキストファイルの場合はChoose templateでCustomを選んでシ
グナルデータやFlag情報を指定してください。
6. Attributeファイルがある場合は指定する、無い場合はそのままNext
7. ノーマライズの設定を行いNextをクリック
8. ベースライン補正を選択してFinish
※ベースライン補正については#ベースラインオプションの影響を参照して下さい
9. 以上でエクスペリメント作成が完了します。

21
Illumina 1. ProjectメニューからNew projectをクリックし、Project名を入力します
※すでにProjectを作成してある場合は、ProjectメニューからNew experimentをクリック
2. Create new experimentを選択
※すでに作成済みのエクスペリメントを開くにはOpen existing experimentを選択
3. 解析条件を入力する
4. Choose Filesボタンからテキストファイルを指定してNext
5. Detection p-valueをFlagに変換する設定を行う
Data Import wizard を選択
22
6. ノーマライズの設定を行いNextをクリック
7. ベースライン補正を選択してFinish
※ベースライン補正については#ベースラインオプションの影響を参照して下さい
8. 以上でエクスペリメント作成が完了します。

23
ノーマライズとベースラインオプション GeneSpringには異なるアレイ間の誤差を補正するノーマライズと、ノーマライズ後のデータ
をパターン化して視覚的に見やすくするためのベースラインオプションがあります。ここ
ではそれぞれの影響について紹介します。※AffymetrixのCELファイルをインポートする場合
はSummarizationの際にノーマライズが行われます。
ノーマライズの影響
GeneSpringは上記の様なグローバル補正のほかに、特定の遺伝子を使用して補正する
Normalize to Control genesや、サンプル毎に一定の数値で補正するNormalize to external valueがあります。これらは事前に補正するための情報を必要としますが、mRNAの総量が大きく
変わるサンプルや、網羅的ではない、ある機能に特化した遺伝子のみを集めたアレイ等を
利用する場合に有効です。
ノーマライズなし
Percentile shift 75%
Quantile
75%
50%
25%
各サンプルの
75%を 0 に補正
実験誤差と考えられるばらつき
各サンプルの
全域を補正

24
ベースラインオプションの影響 Percentile shift 75%でノーマライズした後の、各ベースラインオプションの影響について紹
介します。ベースラインオプションは条件が多い場合にデータを単純なパターンにして解
析を行うことができ、クラスタリングの際などに有効です。
ベースライン補正なし
Baseline to median of all samples
Baseline to median of control samples
発現量の情報は保持
されているが、視覚
的にパターンを見つ
けにくい
発現量の情報は失わ
れるが、サンプル間
の発現比は保持され
る。パターンを見つ
けやすい
Control sample の発現
量の情報は失われる
が、control sample か
らの比になりパター
ンを見つけやすい
Control sample

25
Baseline to median of Control samplesの設定方法 ベースラインオプションでコントロールサンプルを設定する際の操作方法を紹介します。
Baseline to median of Control samplesを使いたい場合は、エクスペリメント作成の際に下記の
操作を行ってください。
1. Baseline to median of control samplesにチェックを入れる
2. 補正したいサンプルをドラッグしてハイライトしてからAssign valueをクリック
3. コントロールに指定するサンプルを右側のウィンドウに移動してOKをクリック
4. 以上でコントロールサンプルが指定されます。
2 群のデータで 2 種類のコントロールがある場合は、2-3 の操作を繰り返してください。
ドラッグ

26
基本的なグラフ ここでは頻繁に使用する 4 つのグラフについて紹介します。巻末の参考資料にほかのグラ
フの説明をのせてあります。
Box Whisker Plot
Box Whisker Plotは複数サンプルの分布を 1 画面で閲覧することができます。中央値の揃い方
やデータの対称性などをみることで、ノーマライズの妥当性を判断することができます。
青い四角の中央の線が中央値、上端が 75%値、下端 25%値になります。BoxWhiskerPlotの形
が大きく異なる場合は、ばらつきの原因を検討してください。
・右クリックメニューのpropertyから
Columnタブで表示するサンプルを選択でき
ます。
・Normalizeを評価する場合は、Baseline optionを使用せずにExperimentを作ります。
Scatter plot
横軸と縦軸にそれぞれ任意のコンディションを設定すると、2 つの条件の再現性や発現量の
違いを見ることができます。数値はすべてlog2 のNormalize値の表示になります。
※ViewメニューからPlot Log10/Linear valueで、log10 やLinearのグラフを描画できます。
※2 色法の場合はアイコン右のプルダウンメニューからCy3/Cy5 plotを選択できます。
・Plotされた点をダブルクリックすると
アノテーションを閲覧。
・画面をドラッグするとエンティティを選択。
・Shift+ドラッグでフリーハンドで選択。
・選択したエンティティはエンティティリストで保存。
・右クリックメニューからSave Entitiesで 2 倍以上・2倍以下のEntitiesを保存。

27
Profile Plot
Profile Plotは縦軸にNormalized Intensity、横軸にコンディションが設定され、エンティティ
ごとに折線グラフが描かれます。Profile Plotは複数条件の発現量変化を表示することができ
ます。GX11.5 以降でnumeric parameterを作成すると、パラメータをスケールに合わせて表
示できるようになりました。後述のExperiment grouping機能でnumeric parameterを設定する
必要があります。
・線をダブルクリックするとアノテ
ーションを閲覧。
・右クリックメニューから
Properties>VisualizationでColor Byを選
択することで、任意のコンディショ
ンの発現量で色を変えられます。
Venn diagram
Venn Diagramは最大 4 つのエンティティリストのAND、OR、NOTを表示することでリストの
比較をすることができます。また、図を表示した後でもドラッグ&ドロップでエンティテ
ィリストを変更することができます。
興味のあるエリアをクリックしてハイライトしてから(ハイライトすると黄色い枠が表示
されます)、左上の アイコン(Fig赤枠)をクリックすると、ハイライトしたエリアのエン
ティティリストを保存できます。 Venn図からエンティティリストを作成する場合、必ずこ
のアイコンから保存してください。
・右クリックメニューのpropertiesから
VisualizationタブからSelect Entity listsを選
択することで、比較するエンティティリ
ストの数を変更可能。
・ をクリックすると重なり具合を
考慮した図になります。

28
参考資料 Raw Signal valueとNormalized Signal value GeneSpringにデータをインポートすると、Raw Signal valueとNormalized Signal value の 2 つの
数値データが得られます。大まかに言って、補正前の値がRaw Signal value で補正後の値が
Normalized Signal value です、アレイプラットフォームによる違いを下記にまとめました。
Agilent 1colorの場合(IlluminaやGeneric Single colorを含む)
Raw Signal value:ノーマライズとベースライン補正前のシグナル値・整数
Normalized Signal value :ノーマライズとベースライン補正後のシグナル値・底 2 の対数
Agilent 2colorの場合
Raw Signal value:Cy3 とCy5 のシグナル値・整数
Normalized Signal value :Cy3/Cy5 の比(Cy5/Cy3 に変更可)・底 2 の対数
Generic 2colorの場合
Raw Signal value:Cy3 とCy5 のシグナル値・整数
Normalized Signal value :ノーマライズしたCy3/Cy5 の比(Cy5/Cy3 に変更可)・底 2 の対数
AffymetrixのCELファイルの場合
Raw Signal value:MAS5 やRMA等のSummarization後のシグナル値・整数
Normalized Signal value :ベースライン補正後のシグナル値・底 2 の対数
※CHPやテキストをインポートした場合はAgilent 1colorの場合と同等です

29
グループの設定(Experiment Grouping) インポートが終わったサンプルに実験条件を入力します。同じ処理群のサンプルは同じパ
ラメーターを入力しておくと、インタープリテーションを作成する際に平均することがで
きます。
1. ワークフローのExperiment setupからExperiment Groupingをクリック
2. Add Parameterをクリック
3. Parameter nameとParameter Valuesを入力
Parameter name:任意の名称(TimeやDisease type等) Parameter value:実験条件の名称
Parameter type:Normal, Diseaseなどのカテゴリカルな値はNon-numericで設定します。 時系列等のデータはNumericに設定し、数値のみを入力します。
4. 必要なパラメーターの入力が全て終わったらOKボタンで終了します。もし変更が必要な場
合は、パラメーターを選択してからEdit parameterボタンで編集してください。
3-1 サンプル選択
3-2 Assign value 選択
3-3 値を入力

30
パラメーターをファイルからインポートする(必要なときのみ) サンプル数やコンディションが膨大な場合、ファイルからパラメーターを取り込むことが
できます。サンプルとコンディションの対応表をタブ区切りのテキスト(.txt)で作ってお
くと、ウィンドウ左上のアイコン「Load experiment parameter from file」から取り込むこと
ができます。
例:下記のように情報をエクセル等で作成し、タブ区切りテキストで保存する。
Import parameter from fileアイコンから上記のテキストファイルを指定する。

31
ブラウザでの表示順を変更する(必要なときのみ) パラメーターのブラウザ上の表示順を変更したい場合は、並べ替えたいパラメーターを選
択し、Re-order parameter valuesをクリックしてください。ポップアップしたウィンドウで
表示する順番を変更することができます。上から順にブラウザでは左から並びます。
Quality control on sample のPCAや 2way実験の表示順を変更(必要なときのみ) 複数のパラメーターがある場合、Quality control on sample のPCAの表示や、2way実験での表
示はExperiment Groupingでの左右の順番に依存します。
左側にあるパラメーターから利用していくので、Move parameter leftまたはrightで利用した
いパラメーターを左に寄せておくと使いやすくなります。

32
インタープリテーションの作成(Create Interpretation) 先ほど作成したExperiment Groupingの設定を使用し、インタープリテーションを作成します。
これによりこの後の解析の比較解析を行うグループが決まります。
1. ワークフローのExperiment setupからCreate Interpretationをクリック 2. インタープリテーションを作成したいパラメーターにチェックしてNextをクリック
3. NumericalかCategoricalを設定する
Numerical:Profile plot(線グラフ)の線をつないで表示 Categorical:Profile plot(線グラフ)の線をつなげない
4. 作成されるコンディションを確認、解析したくないコンディションがある場合、チェックを外す
※Use measurements flaggedのチェックをはずすと任意のFlagのエンティティを除去できま
すが、欠損値が出現するため検定やクラスタリングできないエンティティが出てきます。
もし除外したい条件があれ
ば、チェックをはずす
Averaged:ブラウザ上の表示の際に平均する
Non-Averaged:平均しない
Both:上記の両方を作る

33
5. 作成されるインタープリテーションの名前を確認してFinishをクリック
6. Interpretationsフォルダの下にインタープリテーションが作成される。
例 1:Numericalで 4 回繰り返した実験を平均した場合の表示
例 2:Numericalで 4 回繰り返した実験を平均しない場合(Non-averaged)の表示
平均したインタープリテーション
平均してないインタープリテーション

34
クオリティコントロール
サンプルの品質管理(Quality control on samples) ワークフローのQuality controlからQuality control on samplesをクリックします。
3D PCA Scores ノーマライズする前の値でのPCAの結果を表示です、各サンプルの点がコンディション別に
分かれるかどうかを確認します。X・Y・Zの 3 軸がありX軸で分かれたサンプルの要因が一
番影響が大きくなります。Ctrlキーを押しながらドラッグすると角度を変えることができ、
Shiftキーを押しながらドラッグすると、拡大縮小をすることができます。※ノーマライズ後
の値でPCAをする場合はAnalysisにあるPrincipal Component Analysisを使用します。
Correlation plot それぞれのSample間の相関係数をグラフで表示しています。相関係数が高くなるほど赤く
表示されます。レプリケイト間の相関が高いか、コンディション別で相関に変化があるか
などをチェックしてください。相関係数は絶対的な指標ではなく、他のサンプルと比べる
場合などの相対的な指標として使用してください。
Color rangeは自動的に設定されますが、rangeを任意に設定したい場合はCorrelation plot上で
右クリックするとpropertyが選択できるので、Visualizationタブから設定してください。

35
Hybridization Controls アレイを作成した各メーカーが使用するコントロールプローブの値を表示します。それぞ
れのサンプルが同じようなパターンを示していれば、均一な実験をしていると考えられま
す。使用するアレイのメーカーにより表示される情報が異なります。
Agilentの場合
Affymetrixの場合
サンプルの除外 今までの情報を基に除外したいsampleがある場合は、グラフ上で該当するサンプルを選択し
てからAdd/Remove Samplesで除外すると、指定したサンプルが除外されたExperimentが再構
築されます。元に戻したい場合もAdd/Remove Samplesで元に戻せます。
注意:ある程度解析を進めてからこの操作を行うと、Experimentを再構築の際に発現量も再
計算されるために再構築前のデータと異なる値になる場合があります。

36
発現量によるフィルタリング(Filter Probesets by Expression) 発現量の低いエンティティは再現性が低いため、ある程度除去します。
1. ワークフローのQuality controlからFilter probe sets by Expressionをクリック
2. フィルターをかけるエンティティリストとインタープリテーションを選択します
3. フィルターをかける範囲を設定してNext
Retain entities in which:指定した範囲で抽出する際の条件を設定します。 上段:インタープリテーションで定義した各コンディションから通過する%を設定
下段:すべてのサンプルから通過させる数を選ぶ
値を変更した後は、必ずEnterキーを押して設定を反映させてください。
4. 結果が表示されます、設定を変えたい場合はBack保存する場合はNext
5. 名前を確認してFinishで保存します。
Raw Data:Normalize 前の値 Raw 値を用います。
Normalized Data:Normalized 値を用います。
Filter by value:数値で範囲を設定
Filter by Percentile:パーセンタイルで範囲を設定
Upper cutoff:最大値を設定
Lower cutoff:最小値を設定

37
フラグによるフィルタリング(Filter Probesets by Flags) Flagはエンティティごとに付加される品質情報です、Flag情報は解析するアレイのプラット
フォームで内容が異なります。
AgilentのFlagの種類
Detected:シグナルが検出されたSpot
Not-Detected:シグナルが検出されなかったSpot
Compromised:Spotの品質が低い(SaturationやNon-uniformなど)
AffymetrixのFlagの種類(3’IVT GenechipをMAS5 でSummarizeしたときのみ利用可)
P:シグナルがPM特異的に検出されたプローブセット
M: PかAを判別できないプローブセット
A:シグナルが検出されない、またはクロスハイブリの可能性のあるプローブセット
1. Quality controlからFilter probesets by flagを選択する 2. エンティティリストとインタープリテーションを選択する
3. フィルター条件を設定してNext
Retain entities in which:指定した範囲で抽出する際の条件を設定します。
上段:インタープリテーションで定義した各コンディションから通過する%を設定
下段:すべてのサンプルから通過させる数を選ぶ
値を変更した後は、必ずEnterキーを押して設定を反映させてください。
4. 結果が表示されます、設定を変えたい場合はBack保存する場合はNext 5. 名前を確認してFinishでエンティティリストを保存します。

38
シグナルデータのフィルタリング(Filter Probesets on Data files) インポートしたシグナルデータがテキストファイルの場合は、Filter Probesets on Data filesを使うとテキストファイルに存在する情報についてフィルターをかけることができます。
1. ワークフローのQuality controlからFilter Probesets on Data filesをクリック
2. フィルターをかけるエンティティリストとインタープリテーションを選択してNextをクリック
3. フィルターをかけるカラムを選択
Retain entities in which:指定した範囲で抽出する際の条件を設定します。 上段:インタープリテーションで定義した各コンディションから通過する%を設定 下段:すべてのサンプルから通過させる数を選ぶ
4. 結果が表示されます、設定を変えたい場合はBack保存する場合はNext 5. 名前を確認してFinishボタンで保存します。
テキストデータのプレビュー
検索対象のカラムを選択
=や>などの条件を設定
検索に使用する値を入力

39
再現性によるフィルタリング(Filter Probesets by Error) 再現性の低いデータをCV値やSD値をもとにフィルターすることができます。
1. ワークフローのQuality controlからFilter probesets by Errorをクリック
2. フィルターをかけるエンティティリストとインタープリテーションを選択して
Nextをクリック
※インタープリテーションは平均化されているものを選んでください。
3. フィルターの設定を行います。
CV値(0-100)かSD値(0-∞)にチェックを入れ、数値を入力します。どちらも 0 に近
い値がより再現性が高くなります。
Retain entities in which:指定した範囲で抽出する際の条件を設定します。
上段:インタープリテーションで定義した各コンディションから通過する%を設定
下段:すべてのサンプルから通過させる数を選ぶ
値を変更した後は、必ずEnterキーを押して設定を反映させてください。
4. 結果が表示されます、設定を変えたい場合はBack保存する場合はNext
5. 名前を確認してFinishボタンで保存します。

40
解析 1 遺伝子リストの絞込み
統計検定(Statistical Analysis) 統計検定は、繰り返し実験から得られた様々な統計量を基に確率的な有意差で発現変動し
ている遺伝子を抽出することができます。
実験デザインと利用する検定の対応 統計検定は 2 群比較、多群比較などで利用する手法が異なります。下記に実験デザインと
利用する検定の対応表を示しました。
Unpaired test 例)薬剤効果を比較するグループ間で個人が一致しないとき
1 パラメータ
2 条件
1 パラメータ
3 条件
2 パラメータ
2 条件以上
パラメトリックテスト (等分散)
T-test unpaired
Moderated t-test ANOVA 2-way ANOVA
パラメトリックテスト (不等分散)
T-test unpaired unequal variance
ANOVA unequal variance
-N/A
ノンパラメトリックテ
スト Mann Whitney
unpaired Kruskal Wallis -N/A
Paired test 例)薬剤効果を比較するグループ間で個人が一致しているとき
1 パラメータ
2 条件
1 パラメータ
3 条件以上
パラメトリックテスト T-test paired Repeated measures
ノンパラメトリックテ
スト Mann Whitney
paired Friedman

41
t-testによる検定 t-testは繰り返し実験を行った 2 条件の遺伝子の発現差を検定する手法です。有意差の指標
であるp-valueが低いほど 2 条件で発現量が同じ確率が低くなります。p-valueが低くなる要
因は主に下記の 2 つになります。
1. 検定する遺伝子の 2 条件間の平均値が大きく離れている 2. 検定する遺伝子の条件内の再現性が良い
例:ある遺伝子のBox plot
t-testのオプション
統計検定はインプットした遺伝子(エンティティ)の数だけ繰り返されます、これにより
統計検定の回数が増えると偽陽性が検出される確率もその分増えることになります。偽陽
性の増加を制限するために、GeneSpringには多重検定の補正(Multiple testing correction)オ
プションが選択可能です。デフォルトではFDRに設定されています。
t-testとFold changeを組み合わせると有効
繰り返し実験の数が少ない場合や、非常に再現性が高い場合などは平均値の差が 1.1 倍でも
検出する場合があります。この場合は後述のFold changeと組み合わせます。
※t-testとFold changeを同時に行うFilter on Volcano plot機能も搭載されています。
P=0.91 P=0.01
遺伝子 A(発現差なしと判断) 遺伝子 B(発現差ありと判断)

42
統計検定(t-test)の操作手順 1. ワークフローのAnalysisからStatistical Analysisを選択
2. 検定するエンティティリストとインタープリテーションを選択
3. 比較する条件と検定の種類を設定
※注意:結果で表示されるFold changeの計算は、Condition1/Condtion2 です、Condition2 にコ
ントロールに該当するコンディションを指定すると結果がわかりやすくなります。
4. 多重検定の補正を設定する

43
5. 結果を閲覧する
値を変更した後は、必ずEnterキーを押して設定を反映させてください。
Result Summary(左上) p-valueとFC(Fold change)で段階的にフィルターした結果を表示しています。 たとえば、P < 0.05 とFC > 2.0 に該当するセルを選択すると、右側にあるVolcano plotに該
当するエンティティが緑でハイライトされます。ハイライトしたエンティティはSave custom listで保存することができます。 ※検定の際Multiple testing correctionを使用した場合はCorrected p-valueになります。 ※Expected by chanceは予測される偽陽性(Type I Error)の数です。 p-values(左下) 検定を通過したエンティティです Volcano Plot(右) 縦軸に有意差(-log10 p-value)、横軸に発現差(log2 Fold change)をプロットしたグラフで
す。縦軸はp-valueを-log10 に変換しているため、縦軸が高いほうがp-valueが低いエンテ
ィティになります。
6. 名前(変更可能です)を確認してFinish
7. 入力したエンティティリストの下層に検定済みの新しいリストが保存されます。
p-value の Cutoff を変更可能

44
ANOVAによる検定 ANOVAは 3 条件またはそれ以上の比較で、いずれかの条件で発現差がある遺伝子を検定す
る手法です。1way-ANOVAや 1 元配置の分散分析と同義です。有意差の指標であるp-valueが低くなる要因は主に下記の 2 つになります。
1. 検定する遺伝子の条件内のばらつきが小さい
2. 検定する遺伝子のサンプル全体の平均値から各条件の平均値のばらつきが大きい
ANOVAは条件内のばらつきよりも、サンプル全体の平均値から各条件の平均値のばらつき
が十分大きい遺伝子が得られます。
ANOVAのオプション 1
統計検定はインプットした遺伝子の数だけ繰り返されます、これにより統計検定の回数が
増えると偽陽性が検出される確率もその分増えることになります。偽陽性の増加を制限す
るために、GeneSpringには多重検定の補正(Multiple testing correction)オプションが選択可
能です。デフォルトではFDRに設定されています。
ANOVAのオプション 2
ANOVAは複数の条件を同時に比較することができ、どこか 1 条件でも変動していると発現
差ありとみなされます。どの条件で変動しているかを確認するには、ANOVAを通過した後
の遺伝子リストに対してPost Hoc testを行うことで確認できます。
ANOVAとFold changeを組み合わせると有効
繰り返し実験の数が少ない場合や、非常に再現性が高い場合などは平均値の差が 1.1 倍でも
検出する場合があります。この場合は後述のFold changeと組み合わせます。
P=0.45 P=0.008
遺伝子 A(発現差なしと判断) 遺伝子 B(発現差ありと判断)

45
統計検定(ANOVA)の操作手順 1. ワークフローからStatistical Analysisを選択
2. 検定を行うエンティティリストとインタープリテーションを選択
3. 検定の種類を選択する
4. Post Hoc testを選択する
各コンディションでいくつのエンティティに違いがあるかを検定することができます。TukeyHSDが良いでしょう
5. 多重検定の補正方法を選択
6. ANOVAの結果に指定した条件からのFold changeを付加する事ができます
※ここではFold changeでのカットオフはかかりません。

46
7. 結果を確認
値を変更した後は、必ずEnterキーを押して設定を反映させてください。
Result Summary(左上) 段階的にp-valueでフィルターした結果を表示しています。 ※検定の際Multiple testing correctionを使用した場合はCorrected p-valueになります。 ※Expected by chanceは予測される偽陽性(Type I Error)の数です。 p-values(左下) 検定を通過したエンティティです。 Post-hoc Analysis Report(右)※Post Hoc testを選択した時のみ表示されます。 ANOVAを通過したエンティティについて、Post Hoc testを行った結果を表示します。 各コンディションをすべて比較し、発現差があるエンティティ数を青いセル、発現差がない
エンティティを赤いセルで表示します。興味のあるセルを選択してからSave custom listをク
リックするとエンティティリストとして保存されます。複数のセルを選択すると、Unionでマー
ジしたエンティティリスト、Intersectionで重複部分のみのエンティティリストが保存されます。
8. 名前(変更可能です)を確認してFinish
9. インプットしたエンティティリストの下層に結果が保存されます
p-value の Cutoff を変更可能

47
Fold change(Fold change) Fold changeは発現量の変化が大きいエンティティを抽出することができます。
1. ワークフローのAnalysisからFold changeをクリックする
2. エンティティリストとインタープリテーションを指定する
3. 比較するコンディションを指定する
Select pairing optionで比較方法を 2 種類選択可能です
All against single conditionの場合は特定のコントロールを選択する。
Pairs of conditionsの場合は比較するペアを選択する。
※注意:Fold changeの計算は、Condition1/Condtion2 です、Condition2 にコントロールに該当
するコンディションを指定すると結果がわかりやすくなります。
また、画像赤枠のボタンでCondition1⇔Condition2 を切り替えることができます。

48
4. 結果のプレビューが表示される
5. 名前を確認してFinish
6. 比較したペアごとにUPとDOWNのエンティティリストが保存されます
発現差のカットオフは
変更可能
複数ペアがある場合は
カットオフ可能
Fold change 後のエンテ
ィティ数を表示

49
解析 2 クラスタリングなど
クラスタリング(Clustering) 膨大なデータの中からエンティティやコンディションの発現パターンが似ているものを分
類します。コンディションが少ないものはScatter plotやProfile plotでも変動をつかめますが、
コンディションが多くなるとそれぞれのエンティティの発現変動を理解しづらくなるので、
クラスタリングでパターン別に分けて、それぞれのクラスターに、後述のGO Analysisなどで
意味付けしていきます。
クラスタリングする際の注意点1:Baseline Transformationを利用する
Baseline transformationは発現値を同じスケールに置き換えパターンに変換します。 この補
正は個々の遺伝子の条件変化による変動を見つけやすくなります。
クラスタリングする際の注意点 2:はずれ値を除去する
クラスタリングでは、極端な値を持つはずれ値や変動していないエンティティの影響を受
けます。事前にQuality controlを行い、信頼性の高い、かつ変動したエンティティを絞り込
むことが重要です。

50
k-means K-meansは、エンティティまたはコンディションをユーザーが指定したK個のクラスターに
分けるアルゴリズムです。例えば、「ある病態に 3 つのサブタイプが存在する」という仮
説がある場合など、コンディションを 3 つのクラスターに分けるK-meansを実行する価値が
あります。初期のK個のクラスターはランダムに配置されるので、結果が安定しない場合が
あります。
メモリ使用量が低く非常に処理が早いアルゴリズムですが、エンティティ同士の関連やク
ラスター同士の関連情報は得られません。
1. ワークフローのAnalysisからClusteringを選択する 2. エンティティリスト、インタープリテーションを選択し、K-meansを選択してNext
3. Parametersを設定する
Cluster on:クラスタリングする対象を選択、EntitiesかCondition、または両方 Distance metric:距離の定義を設定します Number of clusters:クラスター数を指定します Number of iterations:クラスターを探す工程を繰り返す回数(大きいほど再現性が良くなる)
4. Previewボタンで結果が表示されます。保存する場合はOKをクリックします。
5. 名前を確認してFinish
6. 結果がプロジェクトナビゲーターに保存されます。
7. 結果を右クリックしてExpand as Entity listをクリックするとクラスター毎のリストが保存され
ます。
各クラスターに後述のGO解析やパスウェイ解析をお試しください
エンティティリストを指定
インタープリテーションを指定
K-means を指定

51
Hierarchical Hierarchicalは階層的クラスタリングです。エンティティとコンディションの距離をツリーで
表現します。枝が近いほど似ていると解釈します。全ての関連を表示することができます
が、クラスターの明確な境界が無いためその点はユーザーが判断する必要があります。
1. ワークフローのAnalysisからClusteringを選択する
2. エンティティリスト、インタープリテーションを選択し、Hierarchicalを選択してNext
3. Parametersの設定 Normalized intensity values:Normalized intensityを利用します Associated values:P-valueやFold change等エンティティリストに付加された値を利用。 Cluster on:クラスタリングする対象を選択、EntitiesかCondition、またはBoth(両方) Distance metric:距離の定義を設定します Linkage rule:クラスター間を比較するためのアルゴリズムを選択します Single:クラスター間の最近のノードの距離を比較(最近隣距離法) Complete:クラスター間の最遠のノードの距離を比較(最遠距離法) Average:クラスター間のそれぞれのノードの平均距離を比較(群平均距離法) Centroid:クラスター内の重心(平均)同士の距離を比較(重心距離法)
Ward’s:ANOVAに似たアプローチで、クラスター内の誤差の平均値を計算し全体
の誤差の平均値から差が小さいものを新しいクラスターに結合します。
4. Previewをクリックすると、結果が表示されます、保存する場合はOKをクリックします
5. 名前を確認してFinish
エンティティリストを指定
インタープリテーションを指定
Hierarchical を指定

52
6. 結果がプロジェクトナビゲーターに保存されます。
7. 保存後は、発現量に応じた階層的クラスタリングの図が表示されます。
画面左側の任意の位置のツリーをクリックすると、そのツリー以下に存在する遺伝子が選
択されます。 アイコンで、選択した遺伝子をエンティティリストとして保存可能です。
全体図
拡大図
サンプルまたは
コンディション
Gene
発現量のカラ
ーチャート

53
Self-Organizing Map(SOM) SOMはユーザーが行と列を指定したグリッドに対して、近いグリッドが近い値を持つよう
に値を繰り返し更新して、クラスターを作ります。グリッドの隣り合ったクラスターはパ
ターンが似ている傾向があります。
1. ワークフローのAnalysisからClusteringを選択する
2. エンティティリスト、インタープリテーションを選択し、Self-Organizing Mapを選択する
3. Parametersの設定 Cluster on:クラスタリングする対象を選択、EntitiesかCondition、または両方 Distance metric:距離の定義を設定します Maximum number of iterations:クラスターの更新の回数を設定 Number of grid rows:グリッドの列の数を指定 Number of grid columns:グリッドの行の数を指定 ※作成されるクラスター数=上記で指定した 列×行 になります。
4. Previewボタンをクリックすると結果が表示されます、保存する場合はOKをクリックします SOMの結果は、隣り合うクラスターはパターンが似ているという特徴があります。
5. 名前を決めて、Finishボタンをクリックすると、SOMの結果がプロジェクトナビゲーターに保
存されます。
6. SOMの結果を右クリックしてExpand as Entity listをクリックすると各クラスター毎のリストが
保存されます。
それぞれのクラスターに後述のGO解析やパスウェイ解析等をお試しください
エンティティリストを指定
インタープリテーションを指定
Self-Organizing Map を指定

54
発現パターンの似ているエンティティの検索(Find Similar Entities) 注目すべき発現パターンのエンティティが既にある場合、そのエンティティと似た相関ま
たは逆相関する発現パターンを示すエンティティを検索します。
1. AnalysisからFind Similar Entitiesを選びます。 2. エンティティリスト、インタープリテーション、Query Entity、Similarly metricを選択します。
Query Entityは注目しているエンティティをSelectボタンから検索して設定してください。
ピアソン相関係数は線形の相関解析です。非線形の相関を解析したい場合はスピア
マン順位相関係数を使用します。
3. 検索結果が表示されます。
Cutoff値のレンジは 1 に近いと相関、0 で相関なし、-1 に近いと逆相関になります。
4. エンティティリストの名前を確認してFinishボタンでエンティティリストを保存します。

55
数値パラメーターと相関するエンティティの検索(Filter on Parameters) タイムコース実験や薬剤の投与量など量的なパラメーターがある場合、パラメーターと発
現量について相関、逆相関するエンティティを検索します。時間依存で変動するエンティ
ティや薬剤の量特異的に変動するエンティティなどの抽出が可能です。
事前にExperiment GroupingでParameter typeがNumericのパラメーターを作成します。
1. AnalysisからFilter on parameterを選択します。 2. エンティティリスト、インタープリテーション、相関を探すパラメータ、Similarity Metricを選択
ピアソン相関係数は線形の相関解析なので、非線形の相関を解析したい場合はスピ
アマン順位相関係数を使用します。
3. 結果を閲覧する
Cutoff値のレンジは 1 に近いと相関、0 で相関なし、-1 に近いと逆相関になります。
4. 名前を確認してFinishボタンでエンティティリストを保存します。

56
Principal Component Analysis(主成分分析) PCAはアレイデータをベクトルデータとみなし、空間にプロットすることでその分布から新
たな軸(主成分)を作ります。これによりエンティティやコンディションを視覚的に分類
することができます。PCA on EntityとPCA on Conditionのどちらかを選べます。
Quality control on samplesのPCAとは違い、ノーマライズ後のデータを使い、使用するエンテ
ィティリストやインタープリテーションも選択可能です。
初めに
Toolsメニュー/Options/Data Analysis Algorithmsフォルダを選択し、Principal Component Analysisの 3-D Scoresにチェックを入れてからApplyしてください。
1. AnalysisからPrincipal component Analysisを選択 2. エンティティリストとインタープリテーションを選択
3. 各条件を設定してNext
PCA on:EntityかConditionを選択する(ここではConditionを選択) Number of principal components:入力した数字だけ主成分を作成します

57
4. 結果を閲覧する
PCA Scores:X軸(PCA Component1)とY軸(PCA Component 2)の 2 次元のスキャ
ッタープロットです。
3D PCA Scores:X軸(PCA Component1)とY軸(PCA Component 2)Z軸(PCA Component3)のスキャッタープロットです、デフォルトでは影響の大きいX>Y>Zの順に並びます。
PCA Loadings:PCA on Conditionを選んだ場合、各Entityが各PCA Componentに対して
持っている負荷量をプロットすることができます。PCA終了後にエンティティリス
トに付加される値と同じものです。
C-C Plot:共分散と相関をプロットしたグラフです。
Eigen values:この値は各PCA Componentがデータの持つ変動を全体からどの程度説
明しているかを表す寄与率です。青は累積の寄与率になります。累積寄与率が
80%程度までのPCA Componentを見れば、十分な情報を捉えていることいえるでし
ょう。

58
5. Entity listの保存 NEXTをクリックするとPCAの結果をEntity listで保存できます。この時、Entity listに付加され
るAssociated valueとしてPCA loading factorが付加されます。これは各PCA Componentに対する個々のEntityのインパクトを表します。
6. PCAの結果はプロジェクトナビゲーターに保存され、ダブルクリックで結果を見ることができ
ます。
7. PCAの結果を右クリックしてExpand Entity listをクリックすると、エンティティリストとして結果
が保存されます。 ※ViewメニューのPlot list associated valueを使うと、PCAのLoading factorをヒストグラム等
で表示することができます。

59
結果の解釈
GO解析(GO Analysis) GO(Gene Ontology project:http://www.geneontology.org/)は生物学的な語彙とそれに関連
する遺伝子を収集し、遺伝子の機能や、局在、プロセスごとにカテゴライズしています
GeneSpringのGO Analysisは自分の解析で得たエンティティリストがGOで分類された遺伝子の
リストと偶然一致する確率を計算します。偶然一致する確率が低い(p-valueが低い)≒関
連が高いと解釈します。
1. ワークフローのResults InterpretationからGO Analysisを選択します
2. エンティティリストを選択してNextをクリックします。
3. 結果の閲覧 4. Spreadsheetの見方
基本的にはCorrected p-valueの値が低い順に見ていきます。
Change cutoffボタンをクリックすることで、p-value(実際にはFDR)のカットオフを変えること
ができます。カットオフ値を低くすると、検定はより厳しくなります。 GO ACCESSION:GO TermのID GO Term:GOによって定義されたカテゴリ名 P-value:Fisher’s exact testのp-value Corrected p-value:Multiple testing correction後のp-value。この値でCutoffします。 Count in selection:自分のエンティティリストとGOカテゴリで重複した遺伝子数 %Count in selection:自分のエンティティリストでそのGOカテゴリに該当し割合 Count in total: All entitiesから、そのGO Termが含まれる遺伝子の数 %count in total:All entitiesから、そのGO Termが含まれる遺伝子の割合

60
5. FinishでProject Navigatorにエンティティリストに保存されます。
GO Analysisから、ほしい遺伝子だけをピックアップする 自分のエンティティリストから興味のある遺伝子だけを検索する場合は、下記の方法を使
用します。
1. 通常通りGO Analysisを行います。
2. 次にCut offを 1 に変更します。
3. GO Treeタブを選択します。

61
4. 検索単語を入力してFindをクリックします。
※複数のGO Termがヒットする場合は、Selectをクリックすると、すべて選択されます。
5. Save custom listボタンで保存します。 ※注意 :Cutoffを 1 にしたまま、Finishボタンをクリックしないでください、すべてのGO Termが保存さ
れるため、大幅に時間がかかります。 終了する際は、Cancelすることをお勧めします。

62
GSEA解析(GSEA) GSEA (Gene Set Enrichment Analysis)はユーザーが指定したエンティティリストと発現量を
もとに、独自に定義されたGeneset毎にEnrichment scoreを算出し、そのEnrichment Scoreが偶
然に発生する確率を計算します。これによりユーザーが指定したエンティティリストに
GSEAで定義されたGene setがどの程度含まれるかを検索します。
GSEAは個々の遺伝子の発現量変化というよりも、遺伝子群としての発現量変化を重視する
のでFold changeや検定をする前のリストに対して有効です。
BROAD Instituteが配布しているGene sets
C1 Cytogenetic sets:染色体位置情報セット C2 Functional sets: 既知のパスウェイやバイオマーカー遺伝子セット C3 Regulatory-Motif sets:microRNAや転写因子の結合配列モチーフセット C4 Neighborhood sets:癌関連遺伝子セット C5 Gene Ontology sets:GOで定義された遺伝子セット ※Genesetの定義の詳細は下記をご覧ください http://www.broadinstitute.org/gsea/msigdb/collections.jsp#C1
データの準備 BROAD Instituteが管理しているGenesetをインポートします。Genesetをインポートすること
でGeneSpringでGSEAを使うことができるようになります。ダウンロードにはGSEAのWEBページでのユーザー登録が必要になるので、各自で行ってください。
1. http://www.broad.mit.edu/gsea/index.jspにアクセスし、Downloadsをクリックします
2. 登録したE-mailアドレスでログインします。

63
3. GSEA解析に使いたいGene Setをダウンロードします。(例:C2 の既知のパスウェイの場合) ※必ずダウンロードする各Genesetは ~symbols.gmt をダウンロードしてください。
4. GeneSpringGXのToolsメニューからImport BROAD GSEA Gene setsをクリックします
5. ダウンロードしたGene setを指定し、インポートしてください。 ※ファイルタイプは*gmtを選択するとファイルが表示されます
6. 該当するGenesetのカテゴリーを選択してOKをクリックするとインポートを開始します。

64
GSEAの実行 1. ワークフローのResults InterpretationからGSEAを選択します
2. エンティティリストとインタープリテーションを選択します
インタープリテーションは比較したいグループで作成したものを選択してください
3. 発現量を比較する条件を選択する
4. 解析したいカテゴリーを選択して、Nextをクリックする
Minimum number of marking Genes:指定値以上の遺伝子がhitしたGene Setを出力 Maximum number of permutations:ランダムシャッフリングの回数(100 回程度で十分) Simple Search:BROAD Gene setsに対する解析を行う Advanced Search:自分で準備したエンティティリストに対して解析を行う

65
5. 解析結果の閲覧
基本的にq-valueをソートして値の小さい順に確認します
Gene Sets: 解析を通過したGene sets Details: Gene setに対する注釈 Total Genes: gene setの遺伝子総数. Genes Found: 自分のエンティティリストがGene setにヒットした 数 P value: p-value Q value: 検定の総数で補正したp-value ES value: エンリッチメントスコア NES value: エンリッチメントスコアの最大値で補正されたされたスコア
6. Finishボタンをクリックするとエンティティリストとして保存されます 各Gene Setの定義の詳細は、http://www.broadinstitute.org/gsea/msigdb/index.jsp で確
認してください。 ※全てのGenesetは、GeneSpring内にはエンティティリストとして登録されています、内容を
確認したい場合や、削除したい場合などSearchメニューからEntityで検索してください。

66
パスウェイ解析 GeneSpring12 ではWikiPathwayに対応したことにより、自分のデータを簡単に既知のパスウ
ェイにマップすることができるようになりました。
データの準備 インタラクションDBのダウンロード
1. AnnotationsメニューのUpdate pathway interaction/From serverを選択
2. 初めてこの機能を使う場合は基本データのダウンロードが始まります
3. Interactiondbのリストが表示されたら目的の生物種を選択してUpdateをクリック
4. しばらくするとインポートが完了します
※Humanなどはファイルサイズが 1GB程度あるのでかなり時間がかかります
BridgeDbのダウンロード
1. AnnotationsメニューからUpdate BridgeDb/From Agilent Serverを選択
2. 目的の生物種がある場合はチェックを入れUpdateをクリック
WikiPathwayのダウンロードとインポート
1. http://wikipathways.org/index.php/Download_Pathways にブラウザでアクセスして目的の生物
種のPathwayをダウンロードしする。
2. ダウンロードしたファイルを解凍する
3. GeneSpringのToolsメニューからImport pathway from file>GPMLを選択
4. 新しく開いたImport pathway画面のChoose file(s)をクリックし、3 で解凍したファイルを指定
5. Import pathway画面でOKをクリック
6. 生物種を確認してOKをクリック
7. しばらくするとインポートが完了します。
Single Experiment Analysis Single experiment analysisは自分の解析したエクスペリメント 1 つと既知のパスウェイデータ
との関連を調べることができます。マイクロアレイ解析では主にこちらを使います。また、
Multi-Omic Analysisは、ほぼ同じ操作で 2 つのExperimentから遺伝子リストを選び既知のパス
ウェイデータとの関連を調べることができます。
1. ワークフローのPathway AnalysisからSingle Experiment Analysisをクリック
2. 対象生物と解析対象のパスウェイを選択します

67
3. 解析するインタープリテーションとエンティティリスト、利用するIDを選択
4. 結果が表示されるのでNext
5. Save pathway listが開くので、名前を決めてFinish
6. ブラウザで結果を閲覧
パスウェイリストは、p-valueでソートして小さい順に見てください。
パスウェイリスト
ブラウザ
遺伝子リスト
フィルタ

68
ゲノムブラウザ Technologyにゲノムの位置情報があればゲノムブラウザ上にエクスペリメントを表示するこ
とができます。遺伝子発現だけでなく、CGHのようなゲノム構造、メチル化や転写因子とい
った発現制御の情報なども同時にゲノムブラウザで閲覧することができます。
データの準備 1. AnnotationsメニューからAnnotations managerをクリック
2. Annotations managerが開いたら、左上のListからAnnotation serverを選択
3. 生物種一覧が表示されるので、必要な生物種にチェックを入れる
4. Updateボタンをクリックするとダウンロードが始まります。
5. ダウンロードが終わると完了です。
使い方
1. アイコンでゲノムブラウザを起動する
2. ゲノムビルドが複数ある場合は、使用するゲノムビルドを選択する

69
3. データを表示させたいエクスペリメントをブラウザにドラッグ&ドロップする
エンティティリストもドラッグ&ドロップでゲノムブラウザに表示させることができます。
4. アノテーションを表示させたい場合は右側のAnnotationタブからダブルクリックで表示する
※ライセンスによっては表示されないアノテーションもあります
5. ゲノムブラウザの表示はScatter plotですが、右クリックメニューのEdit track propertyでグラフタ
イプや軸の設定などを変更することができます。
表示したいインタープリテ
ーションとサンプルを選ぶ

70
ゲノムブラウザ操作アイコンの説明
A:ゲノムブラウザの全画面表示
B:トラックのマージ:Ctrlキーを押しながら 2 つトラックを選択してからマージします
C:トラックの自動サイズ調整
D:トラックのフルズームアウト
E:トラックのズームアウト
F:トラックのズームイン※Shiftキー+ドラッグでも拡大できます。
G:トラックの方向切り替え
H:トラックの表示順変更
I:画像ファイルのエクスポート
J:染色体の切り替え
K:閲覧する染色体位置の指定、またはGene Symbolを入力して遺伝子の検索
A B C D E F G H I J K

71
参考資料
便利な機能
Translate機能 異なるエクスペリメント間でエンティティリストをコピーしたい場合に使用します。アレ
イのプラットフォームや生物種が異なっても自動的に対応する遺伝子リストに変換する機
能があります。アレイのプラットフォームが異なる場合はアノテーションのEntrez GeneIDをキーに変換し、生物種が異なる場合はEntrez GeneIDをキーにNCBIのHomologeneの情報を参
照して生物間の遺伝子を決定しています。
Translateの操作手順(例:AエクスペリメントからBエクスペリメントにTranslate)
1. エンティティリストをTranslateする先のBのエクスペリメントを選択
2. Aのエクスペリメントのエンティティリストを右クリックし、Translate Listを選択
3. IDと一緒にTranslateする値を選択
Import list associated valuesにチェックを入れると、エンティティリストにp-valueやFold changeな
どのassociated valueがある場合は一緒にTranslateできます。
Import signal values:選択したインタープリテーションのNormalized値かRaw値を一緒に
Translateできます。
A
B

72
4. Translation mappingが表示される
Percentage of entities translated ~ で変換されたパーセンテージを確認できます。
この対応表を保存したい場合、図上で右クリックメニューのExport As textを使用してください。
5. 名前を確認してFinishをクリック
6. BのエクスペリメントにTranslate結果が保存される
Translateの注意点
・アノテーションにEntrez GeneIDが無い場合、利用できません。
・Homologene(http://www.ncbi.nlm.nih.gov/homologene)で定義されていない生物種は利用
できません。
・ProbeIDとEntrez GeneIDが 1 対 1 ではない場合(ほとんどの場合 1 対 1 ではない)
Translate前後でエンティティリストの数が変化します
A

73
Export as Image ブラウザ上の画像をファイルでエクスポートすることができます。
1. エクスポートしたい画面上を右クリックでExport as Imageをクリック
2. ファイル名の決定、画像の大きさ、解像度を指定してOKをクリックすると保存されます。

74
Export List Entity Listにアノテーションを付加した情報をテキストファイルでエクスポートすることが
できます。論文作成や他のソフトウェアへのデータ排出に使用します。
1. Entity Listを右クリックでExport List
2. エクスポートしたいデータにチェックを入れます、アノテーションは選択して矢印ボタンで移動す
ることができます。Selected itemsに含まれるアノテーションがエクスポートされます。
3. ファイル名とファイルタイプを指定して保存します。

75
セレクションモードとズームモードの切り替え ブラウザ上で右クリックすることで、セレクションモードとズームモードを選択できます。
セレクションモード
スキャッタープロットの興味深い分布を範囲選択することで後述の機能でエンティ
ティリストを作ることができます。
ズームモード
見えにくい部分を拡大する場合、ズームモードに切り替えて範囲選択することで選
択した部分を拡大することができます。
Create Entity List from selection
Scatter plotやProfile plotのブラウザで選択したエンティティをエンティティリストとして保
存できます。
1. セレクションモードで興味のあるエンティティを選択する 2. Create Entity list from selectionをクリック 3. 名前をつけて保存する。

76
Import Entity List from File
論文や参考書にある外部の遺伝子リストをGeneSpringGXにインポートします。
1. インポートしたい遺伝子リストをタブ区切りテキストで準備する 準備するリストにはGeneSpringGX内のデータとマッチするIDが必要です。お使いのチップ
のプローブIDやEntrez GeneID等を準備してください。
2. UtilitiesからImport entity list from fileをクリックする
Choose file:テキストファイルを指定 Choose column to match:テキストファイル内のIDに相当するものを選択 Choose technology column:テキスト内のIDとマッチさせるGX内のIDを選択
Choose columns to import:テキストファイル内にID以外にインポートしたい
情報があれば、Available ItemsからSelected Itemsに移動させる
3. OKボタンをクリックして取り込み完了
※取り込めない場合は、マッチさせるテキスト内のIDとGX内のアノテーションが同じタイプ
のIDかどうかを確認してください。

77
アノテーションの閲覧 自分が解析しているTechnologyにはどんなアノテーションが含まれるかどうかを確認します。
1. SearchメニューからTechnologyを選択、何も入力せずにNext
2. インポート済みのTechnology一覧が表示されるのでアノテーションを閲覧したい
Technologyを選択してInspectボタンをクリック
3. アノテーションを閲覧する
Configure columnボタンで表示するアノテーションを選べます。

78
アノテーションからのエンティティの検索 Technologyに含まれるアノテーションから興味のあるエンティティを検索できます。ブラウ
ザ上でCtrlキー+Fでも検索ウィンドウを開くことができます。
1. SearchメニューからEntitiesを選択 2. 検索条件を決定してNext
Search for:検索するテキストを入力
検索対象のアノテーションを左から選んで右に移動します。
3. 検索結果を確認してNext
4. 名前を確認して、Finishで保存します。

79
グラフの見方 GeneSpringGXは解析データの解釈を助けるため多くのViewを持っています。
Scatter plot
横軸と縦軸にそれぞれ任意のコンディションを設定すると、2 つの条件の再現性や発現量の
違いを見ることができます。数値はすべてlog2 のNormalize値の表示になります。
※ViewメニューからPlot Log10/Linear valueで、log10 やLinearのグラフを描画できます。
※2 色法の場合はアイコン右のプルダウンメニューからCy3/Cy5 plotを選択できます。
・Plotされた点をダブルクリックすると
アノテーションを閲覧。
・画面をドラッグするとエンティティを選択。
・Shift+ドラッグでフリーハンドで選択。
・選択したエンティティはエンティティリストで保存。
・右クリックメニューからSave Entitiesで 2 倍以上・2倍以下のEntitiesを保存。
MvA plot
図の下のプルダウンメニューから、横軸と縦軸にそれぞれ任意のコンディションを設定す
ると、横軸に 2 条件の平均値、縦軸に 2 条件の発現差(Fold change)をプロットした図で
す。
※2 色法実験の場合は、Cy5 とCy3 を比較することができます。
・Plotされた点をダブルクリックすると
アノテーションを閲覧。
・画面をドラッグするとエンティティを選択。
・Shift+ドラッグでフリーハンドで選択。

80
Profile Plot
Profile Plotは縦軸にNormalized Intensity、横軸にコンディションが設定され、エンティティ
ごとに折線グラフが描かれます。Profile Plotは複数条件の発現量変化を表示することができ
ます。GX11.5 以降でnumeric parameterを作成すると、パラメータをスケールに合わせて表
示できるようになりました。後述のExperiment grouping機能でnumeric parameterを設定する
必要があります。
・線をダブルクリックするとアノテ
ーションを閲覧。
・右クリックメニューから
Properties>VisualizationでColor Byを選
択することで、任意のコンディショ
ンの発現量で色を変えられます。
Histogram
Histogramは縦軸に積み上げのエンティティ数、横軸にIntensityの範囲を表示することで、サ
ンプル内のIntensityの分布をみることができます。SingleとMultipleがあり、Multipleを選択す
ると、複数サンプルのヒストグラムを見ることができます。
・Samplesで表示するサンプルの選択。
・右クリックメニューのProperties/Datasetタブ
でRaw とNormalize を切り替え。
・VisualizationタブでUse explicit binningにチェ
ックを入れ、Number of binを変更することでグ
ラフの解像度を変更。

81
Matrix plot
Matrix plotはExperimentに含まれるSampleを総当たりでScatter plot表示します。
どこのSample間で相関しているか、逆相関しているかを見つけることができます。
・右クリックメニューのRenderingタブのPageから
1 ページに表示する数を指定できます、また
ColumnsタブからMatrix plotを作成するサンプル数
を変更。
Venn diagram
Venn Diagramは最大 4 つのエンティティリストのAND、OR、NOTを表示することができ、リ
ストの比較をすることができます。また、図を表示した後でもドラッグ&ドロップでエン
ティティリストを変更することができます。
興味深いエリアをクリックしてハイライトしてから(ハイライトすると黄色く表示されま
す)、左上の アイコン(Fig赤枠)をクリックすると、ハイライトしたエリアのエンティテ
ィリストを保存できます。 Venn図からエンティティリストを作成する場合、必ずこのアイ
コンから保存してください。
・右クリックメニューのpropertiesから
VisualizationタブからSelect Entity listsを選
択することで、比較するエンティティリ
ストの数を変更可能。
・ をクリックすると重なり具合を
考慮した図になります。

82
Box Whisker Plot
Box Whisker Plotは複数サンプルの分布を 1 画面で閲覧することができます。中央値の揃い方
やデータの対称性などをみることで、Normalizeの妥当性を判断することができます。青い
四角の中央の線が中央値、上端が 75%値、下端 25%値になります。BoxWhiskerPlotの形が大
きく異なる場合は、ばらつきの原因を検討してください。
・右クリックメニューのpropertyから
Columnタブで表示するサンプルを選択でき
ます。
・Normalizeを評価する場合は、Baseline optionを使用せずにExperimentを作ります。
Heatmap
Heatmapは縦軸にエンティティ横軸にコンディションを配置し、発現量を色で表示していま
す。
これにより発現の強弱を視覚的にとらえることができます。エンティティはエンティティ
リストの順番で並んでいます。発現パターンが似ているものを集めるにはクラスタリング
を行う必要があります。

83
Spreadsheet
SpreadsheetはID、Normalized Intensity、Annotationをテキストで閲覧することができます。
エクスポートする場合は右クリックメニューからExport as textでテキストファイルに保存で
きます。
Summary Statistics
各サンプルの統計量を閲覧することができます。

84
GX7 とGX9 以降の検定の対応 GX7 とGX9 以降について統計検定機能の対応表です
T-test
ANOVA
2-way ANOVA
※2color実験で使う、GXのT-test against ZeroはGX7 のFilter on Confidenceに相当します。

85
1sample T-test(主に 2color実験の場合に使用可能) 2color実験に使用します。それぞれのEntityについて、2 色のDyeの比が 1(log2 表示だと 0)である可能性を検定します。結果はp-valueで表され、p-valueが低いほど 2 色のDyeの比が 1である可能性が低い、つまり変動している可能性があるといいかえられます。
1. 2color実験でレプリケイトのあるInterpretationを作る
2. AnalysisからStatistical Analysisを選択
3. エンティティリストとインタープリテーションを選択してNext
4. T-test against Zeroを選択

86
5. P-value computationとMultiple testing correctionを選択してNext
6. 結果を閲覧してNext
p-valueのCutoff値を変更するにはChange cutoffボタンをクリックして値を変更します。 結果の見方 Result Summary(左上) p-valueとFC(Fold change)で段階的にフィルターした結果を表示しています。たとえば、P < 0.05 とFC > 2.0 に該当するセルを選択すると、右側にあるVolcano plotに該当するEntityがハイライトされます。ハイライトしたEntityはSave custom listで保存することができます。 p-values(左下):検定を通過したEntityのリストです。 Volcano Plot(右) 縦軸に有意差、横軸に変動差をプロットしたグラフです。 縦軸はp-valueを-log10 に変換しているため、縦軸が高いほうがp-valueが低くなります。横
軸は 2 コンディションの平均値を比較したときの値です。
7. 名前を確認してFinishボタンで保存

87
Similarity Metrics クラスタリングやFind similar entitiesなどで利用される、エンティティやコンディションの
距離の定義について性質を解説します。
距離系の定義
Euclidian: Standard sum of squared distance (L2-norm) between 2 entities. Takes into account the direction and magnitude of vectors.
Squared Euclidian: This accentuates the distance between entities and tends to give more weight to outliers because of the lack of the squared root.
Manhattan: Manhattan distance is greater than Euclidian. Data using this method might appear less compact.
Chebychev: The absolute value of the maximum difference in any dimension (L-Infinity-norm). This will simply pick the largest distance between 2 corresponding genes. This is resilient to noise.
.

88
Differential: The difference in slopes between the expression profiles of two entities and computing the Euclidean norm of the resulting vector. Useful in time series analyses, where changes in the expression values over time are of interest, rather than absolute values at different times.
Correlation系の定義
Pearson Absolute: The absolute value of the Pearson Correlation Coefficient between two entities positively and negatively correlated entities are close to 1 and -1, respectively. Unrelated ones are close to 0.
Pearson Centered: The 1-centered variation of the Pearson Correlation Coefficient. Positively correlated entities are close to 1; negatively correlated are close to 0, and unrelated entities close to 0.5.
Pearson Uncentered: Entities are not mean-centered. Positively and negatively correlated entities are close to 1 and -1, respectively. Unrelated ones are close to 0.

89
GeneSpringの保守・管理
プロジェクト単位のエクスポート 現在開いているプロジェクトをエクスポートすることで、部分的なデータのバックアップ
や、他のGeneSpringへのデータ移行が可能になります。
1. エクスポートしたいプロジェクトを開く
2. ProjectメニューからExport Projectを選択
3. Choose Experiments画面で、Exportしたいエクスペリメントにチェックを入れる
4. テクノロジー(アノテーション)も同時にExportする場合はチェックを入れる
5. 名前をつけて保存先を指定する(日本語パスを含めないように)
6. 拡張子が.tarのファイルが保存されると完了

90
データのバックアップと復元 この手順はGeneSpringに含まれる全てのデータをバックアップします。おおむねデータ容量
が大きくなるため、かなり時間がかかります。さらに古いWindowsなどで利用している
FAT32 ファイルシステムの場合は 4GB以上のファイルを作成できないので、かならずNTFSでフォーマットされた十分に空き容量のあるディスクを保存先にしてください。
またバックアップしたデータの復元は、同じバージョンのGeneSpringで復元してください。
バックアップ方法
1. ToolsメニューからBackup Repositoryを選択
2. 保存先を指定する
3. バックアップが完了するまで待つ。
デフォルトで、backup.tarが作成されます。これを外付けHDDなどに保存してください。
復元方法
1. ToolsメニューからRestore backupを選択
2. バックアップで作成したbackup.tarを指定する
3. 復元が終わるまで待つ。
GeneSpringのバージョンアップ HelpメニューからUpdate product/From Agilent serverでインターネット経由でGeneSpringをバ
ージョンアップ可能です。
バージョンアップはファイルサイズの大きいデータのダウンロードが必要になるため、ネ
ットワーク環境によりダウンロードできない場合は必要なデータをCDなどで取り寄せてか
ら、Update product/From fileでファイルからバージョンアップすることが可能です。

91
お問い合わせ GeneSpringのサポートは、大まかに企業ユーザーはアジレントテクノロジー、アカデミック
ユーザーはトミーデジタルバイオロジーと分かれています。ここにはそれぞれの会社のサ
ポートと連絡先を記述しましたので、ご不明な点がある場合は下記までご連絡ください。
事前にGeneSpringGXのバージョンやお使いのアレイの名称を準備いただけるとスムーズに対
応できますのでお手数ですがよろしくお願い致します。
アジレントテクノロジー株式会社
〒192-8510 東京都八王子市高倉町 9-1
電話:0120-477-111
E-mail : [email protected]
担当 石井
トミーデジタルバイオロジー株式会社
〒110-0008 東京都台東区池之端 2-9-1
電話:03-5834-0810
E-mail : [email protected]
担当 田中・高木