genetic marker family studies in familial mediterranean fever (fmf) in armenians
TRANSCRIPT

Clinical Genetics 1990: 38: 332-339
Genetic marker family studies in familial Mediterranean fever (FMF) in Armenians
T. SHOHAT', M. SHOHAT', G. M. PETERSEN', R. S. SPARKES', D. LANGFIELD', J. BICKAL',
'Medical Genetics Birth Defects Center, Departments of Medicine and Pediatrics, Cedars-Sinai Medical Center, UCLA School of Medicine, Los Angeles, the 'Division of Medical Genetics, UCLA
Center for Health Sciences, CA and the 'Division of Gastroenterology, UCLA Center for Health Sciences, CA, USA
J. R. KORENBERG', A. D. SCHWABE' A N D J. I. R o m R '
Familial Mediterranean fever is an autosomal recessive disease manifested by recurrent short episodes of fever associated with polyserositis. I t is common in a variety of Mediterranean and near Eastern populations. The biochemical defect is unknown, and there have been few studies of genetic marker associations or linkage with the disease. We have screened blood samples from members of 14 nuclear Armenian families, the population with the highest known gene frequency, for 19 different polymorphic phenotypic genetic markers. These 14 families included 31 affected and 43 unaffected family members. No association was found with any of the markers studied. Linkage could be excluded at the distance of &IS% recombination with 14 markers. Linkage could not be excluded with 5 other markers. These results exclude the F M F gene from those portions of the human gene map that are at least 0.5% recombination distance from these 14 genetic markers, and represent the first comprehensive step in the eventual localization and isolation of the FMF gene.
Received I0 October 1989. revised 2 April, accepted for publication 7 April 1990
Key words: association; familial Mediterranean fever; genetic linkage; genetic markers.
Familial Mediterranean fever (FMF) is an autosomal recessive disease characterized by recurrent, self-limited episodes of fever ac- companied by peritonitis, pleuritis, or syno- vitis (hence the alternative name recurrent polyserositis) (Sohar et al. 1967, Schwabe & Peters 1974). The most severe complication of the disease is amyloidosis leading to chronic renal failure (Sohar et al. 1967, Pras et al. 1982). Another severe complication is chronic destructive arthritis, resulting in permanent organic damage to the involved joints (Sneh et al. 1977, Loop et al. 1965).
The majority of patients are non-Ashken- azi Jews, Armenians, and Arabs (Sohar et al. 1967, Pras et al. 1982). The disease has also been described in Turks, Bulgarians,
and other ethnic groups as well (Eliakim et al. 1981). We have recently shown that the disease is recessively inherited in Armen- ians, as had been previously established for non-Ashkenazi Jews (Rogers et al. 1989, Sohar et al. 1967). This is in contrast with earlier reports which interpreted certain in- stances of vertical transmission as due to dominant inheritance of FMF (Reiman 1954). We have shown that vertical trans- mission in the Armenian population is due to a high FMF gene frequency, estimated at 8%, and consequent homozygote-hetero- zygote marriages leading to pseudodomi- nant inheritance (Rogers et al. 1989).
Even in the face of a number of immuno- logic and pathological studies, the bio-

G E N E T I C S T U D I E S t N F M F 333
chemical defect in FMF remains unknown (Eliakim et al. 1981, Shohat et al. 1989, Melamed et al. 1983). An alternative ap- proach to delineating the disorder’s funda- mental etiology is to identify linked genetic markers as an initial step in eventual identi- fication of the responsible gene. To date, no genetic markers have been reported to be linked or associated with FMF (Davrinche et al. 1985, Schlesinger et al. 1984, Chaquat et al. 1977, Chaouat et al. 1977, Pras & Gazit 1985). However, most of these studies examined only the HLA locus. We have undertaken a comprehensive effort to map the FMF -gene as a step in delineating its etiopathogenesis. Our first step in this re- gard was to investigate the relationship be- tween FMF and a panel of polymorphic phenotypic red cell and serum protein markers in patients and families of Armen- ian heritage; hence, the present study.
Material and Methods
Fourteen Armenian nuclear families living in California were ascertained through Ar- menian FMF patients seen regularly at the FMF clinic at the UCLA Center for Health Sciences. Seven families had two or more affected offspring with FMF, six had one affected offspring, and one family had an affected uncle and cousin in addition to the proband. The diagnosis of FMF in all af- fected individuals was established by the same physician (AS.), using the accepted clinical criteria for FMF (Sohar et al. 1967, Schwabe & Peters 1974). The participating patients and their first-degree relatives were asked to complete a health-related ques- tionnaire and have a blood sample taken. The study protocol was approved by the Human Subject Committee at Cedars-Sinai Medical Center. The study sample included 31 affected individuals and their 43 unaf- fected relatives, as illustrated in Fig. 1.
and screened for the 19 different polymor- phic genetic markers, as listed in Table 1. The methods utilized were the standard techniques of hemagglutination or electro- phoresis (Harris & Hopkinson 1976, Wid- man 1985).
Linkage analysis was performed by the lod score method using the computer pro- gram LINKAGE (Lathrop & Lalouel 1984). For some analyses the computer program MENDEL was used as well (Lange & Boe- hnke 1983). We considered linkage to be established when the lod score L 3 (i.e. odds for linkage 1000: 1 for a specific value of the recombination fraction (8)). Linkage can be rejected when the lod score I -2.0 (i.e. odds against linkage 1OO:l for a specific 8).
For each marker, linkage was tested and lod scores were initially calculated at speci- fied recombination fractions (0) of: 0.0001, O.OOl,O.Ol, 0.05,0.1, 0.2,0.3, and 0.4. When
Ei z x 6%; &a%
Ed% Fig. 1. FMF families studied for linkage analysis with polymoprhic markers 0 - unaffected male 0 - unaffected female
- affected male Fifteen ml of venous blood was drawn 0 - affected female

334 S H O H A T E T A L .
some degree of linkage could be rejected by this initial screen, the precise value 0 for which linkage could be excluded was esti- mated by maximizing the likelihood. Based on our previous segregation analysis and gene frequency calculations in the Armenian population (Rogers et al. 1989), the fre- quency of the FMF gene was entered as 8%, and penetrance was estimated at 100% for males and 80% for females.
Association of FMF with specific genetic markers waz tested using chi-square analysis or using Fisher's exact test (for small sample size) (Dixon & Massey 1983). Allele gene marker frequencies were determined in cases and controls. The cases were defined as: a randomly selected affected offspring from each family. The control gene frequencies were derived from parents of these affected individuals as follows. For each marker, the allele frequencies were determined in the cases, and then compared with the allele frequency of those parental alleles that were
not transmitted to the affected offspring. Although the parents are obligatory carriers of the FMF gene, at least 50% of their chromosomes reflect the normal gene fre- quency distribution among the Armenian non-FMF population. Therefore, we sub- tracted the two alleles that were transmitted to the cases from the four parental genes. The remaining alleles (those that were not transmitted to the probands) were regarded as the non-FMF control alleles. This "modi- fied" parents' gene frequency was then com- pared to the allele frequency of the cases, and significance was determined using the chi-square test. This method has been pre- viously proposed by Falk & Rubinstein (1987). If there is no association between the disease and a specific gene marker, we would expect no differences in the alleles that were passed from the parents (i.e. the allele frequency in the affected offspring) and those that were not passed to the affect- ed offspring, since the transmission of both
Table 1
Polymorphic genetic markers evaluated
Marker Symbol Possible alleles
I. Red cell antigens ABO ABO A,B,O MNSs MNSs Ms,Ms,NS,Ns Duffy N a.b,Fy Kidd JK a,b.Jk
II. Red cell enzymes Adenylate-kinase 1 AK1 1 2 6 Phosphogluconate-dehydrogenase PGD A.C Adenosine deaminase ADA 1,2 Phosphoglucomutase-1 PGMl 1,2
Acid phosphatase 1 ACP A.B.C Galactose-1-phosphate uridyltransferase GALT N,D.L,G Glutamate pyruvate transaminase GPT 1,2,3 Phosphoglycolate phosphatase PGP 1.2,3 Esterase-D ESD 1,2
Haptoglobin HP 1.2.R Transferrin TF B,Cl,C2,D Group specific component GC 1S.1F82 Complement component 3 c3 S,F Alpha-1-antitrypsin PI Ml,M2,S,Z (and other rare alleles)
(and subtypes) PGMl (sub) 1+,-1,+2.-2
111. Serum proteins

G E N E T I C S T U D I E S I N F M F 335
alleles would be random. If there is an as- sociation with the disease, then this random transmission to affected offspring would be distorted.
Results
Results of the linkage analysis are summar- ized in Table 2. More detailed results are given in Table 3. As sezii, for some of the markers, as few as two families were in- formative, whi!e for others as many as all the families were informative. Linkage (i.e. lod score 2 3) could not be established with any of the markers studied. Linkage with several markers was excluded at &15% re- combination fractions including : PGMl and PGMl subtypes, FY, ACP, GC, MNSs, GPT, ABO, AK, ESD, PI, HP, C3 and ADA. Linkage could not be excluded with PGD, TF, GALT, PGP and JK. The maximum positive lod score obtained was
0.93, for 8=0.0001 recombination for GALT.
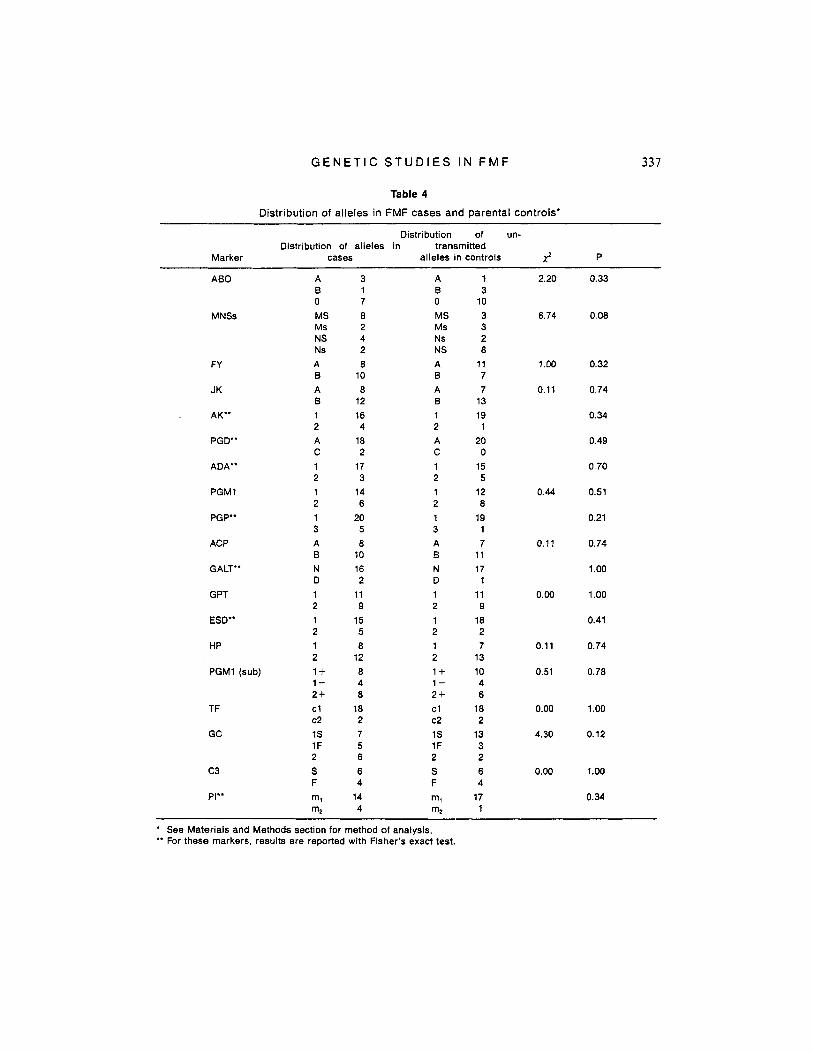
For most markers, the distribution in cases and controls was very similar, and there were no dramatic differences for any marker. Results of the association study are summarized in Table 4. No significant as- sociation was found with any of the markers studied. This includes the five loci, for which tight linkage could not be excluded: PGD, TF, GALT, PGP, and JK.
Discussion
Familial Mediterranean fever is a common inflammatory disease in Mediterranean populations. We have recently shown that FMF is an autosomal recessive disease in the Armenian population (Rogers et al. 1989), consistent with what had been pre- viously established in non-Ashkenazi Jews (Sohar et al. 1967). The observed vertical
Table 2
Linkage analysis results with polymorphic genetic markers
Mini m u m 0 Number of Chromosomal Maximum lod lod Linkage informative
Marker location score. (8) scoreb rejected families
PGMl lp22.1 - 12.5 0.050 9 PGM (sub) 1 p22.1 - 26.0 0.150 12 PGD 1 p36.2-~36.13 - 1.7 - 3 FY lq12-21 - 5.8 0.065 9
TF 3q21 0.16 (0.0001) 0.0' - 6 GC 4q12 0.01 (0.3) - 9.0 0.030 11 MNSs 4q2-31 0.03 (0.4) - 15.6 0.065 13 GPT 8q24 - 6.6 0.075 11
7 GALT 9p13 0.93 (0.0001) 0.0' - ABO 9q34 - 8.9 0.065 13 AK 9q34.1 - 4.9 0.005 8 ESD 13q 14.1 1 0.09 (0.3) - 3.85 0.001 7 PI 14q32.1 0.04 (0.4) - 2.9 0.001 3 PGP 16~13.31-~13.12 -0.1 - HP 16q22.1 - 4.6 0.010 7 JK 18ql l-q 12 -1.2 - c3 19p13.%p13.2 - 6.4 0.010 9 ADA 20q13.11 - 7.6 0.025 3
ACP 2p23 - 7.4 0.075 14
2
6
a: for markers not indicated, lod score was zero at 8=0.50. b: at 8:O.OOOl. * These lod scores were obtained at 50% recombination.

336 S H O H A T E T A L
transmission in this population has led to several reports arguing for dominant in- heritance in FMF (Reiman 1954). However, we have shown that this vertical trans- mission is due to a high gene frequency, 8% in the Armenian population, and conse- quent homozygote-heterozygote marriages leading to apparent pseudodominant in- heritance (Rogers et al. 1989). Although several immunologic abnormalities have been described during the attacks, such as increase in total complement (Ollier-Hart- mann et al. 1981), elevated fibrinogen and immunoglobulin levels (Eliakim et al. 198 l) , they are considered to be secondary to the inflammatory process. Thus, the basic de- fect is unknown. An alternative approach to the pathophysiologic one principally pur- sued thus far is a gene marker linkage ap- proach. Identification of a genetic marker linked to the disease is important for hetzro- zygote detection and for further character- ization of the gene responsible for the dis- ease.
We screened blood samples from multiple
families of Armenian patients with FMF, including their affected and unaffected rela- tives, as part of an overall effort to map the gene responsible for the disease. Linkage was rejected with 14 markers, thus excluding the FMF gene from those portions of the human gene map that is in at least tight linkage (and for most cases at greater dis- tance) from these 14 genetic markers. We could not rule out linkage with five other markers. This is most probably due to the small number of families informative for these markers. A future goal is to increase the number of informative families at these loci.
There has been a paucity of genetic marker studies of FMF. Davrinche and co- workers (1985) found no association be- tween C3, BF, and C4 protein polymor- phisms and FMF. The other reported stud- ies have examined only the HLA locus, and no evidence for association and Iinkage with FMF was observed (Schlesinger et al. 1984, Chaouat et al. 1977, Pras & Gazit 1985, Chaquat et al. 1977). No study to date has
Table 3
Linkage analysis results with polymorphic genetic markers at different lod scores
Marker Recombination fraction (8)
0.0001 0.001 0.01 0.05 0.075 0.10
PGMl - 12.47 - 5.8 - 4.76 - 2.21 - 1.63 - 1.24 PGMl (sub) - 26.00 - 19.81 12.62 - 6.53 - 4.99 - 3.93 PGD - 1.72 - 1.59 - 1.01 -0.37 - 0.34 - 0.26 FY - 5.82 - 5.00 - 3.45 - 2.56 -1.87 -1.64 ACP - 7.40 -7.13 - 5.26 -3.15 - 2.39 - 1.85 TF 0.16 0.16 0.13 0.09 0.08 0.06 GC - 9.01 - 6.36 -4.01 -1.62 - 1.07 - 0.72 MNSs - 15.64 - 14.36 - 6.64 - 2.71 -1.79 -1.2 GPT - 6.65 - 6.32 -4.36 -3.18 - 2.40 - 1.87 GALT 0.93 0.93 0.92 0.78 0.69 0.63 ABO - 8.91 -8.91 - 5.62 - 2.54 - 1.86 -1.4 AK - 4.93 - 2.84 -1.76 - 1.09 - 0.79 - 0.59 ESD -3.85 - 2.35 -1.45 - 0.44 - 0.20 - 0.07 PI - 2.86 - 2.50 - 1.57 -0.51 - 0.26 -0.11 PGP - 0.098 - 0.094 - 0.091 - 0.087 - 0.083 - 0.080 HP - 4.62 - 3.76 - 2.44 -1.32 - 0.96 - 0.71 JK - 1.25 -1.17 -0.12 -0.19 - 0.07 -0.007 c 3 - 6.42 - 4.32 - 2.39 - 1.07 - 0.77 - 0.57 ADA - 7.61 - 5.53 -3.17 -1.54 -1.13 - 0.86
G E N E T I C S T U D I E S I N F M F 337
Table 4
Distribution of alleles in FMF cases and parental controls'
Distribution of un- Distribution of alleles in transmitted
Marker cases alleles in controls J P
ABO
MNSs
FY
JK
. AK"
PGD"
ADA"
PGMl
PGP"
ACP
GALT"
G PT
ESD"
HP
PGMl (sub)
TF
GC
c3
PI"
A B 0 MS Ms NS Ns A 0 A B 1 2 A C 1 2 1 2 1 3 A B N D 1 2 1 2 1 2 1+ 1- 2 f cl c2 1s 1F 2 S F m1 m2
3 A 1 B 7 0 8 MS 2 Ms 4 Ns 2 NS 8 A
10 B 8 A
12 B 16 1 4 2
18 A 2 c
17 1 3 2
14 1 6 2 20 1 5 3 8 A
10 B 16 N 2 D
11 1 9 2
15 1 5 2 8 1
12 2 8 1+ 4 1- 8 2f
18 cl 2 c2 7 1s 5 1F 6 2 6 S 4 F
14 m, 4 m2
1 3
10 3 3 2 8
17 7 7
13 19
1 20 0
15 5
12 8
19 1 7
11 17 1
11 9
18 2 7
13 10 4 6
18 2
13 3 2 6 4 17 1
2.20
6.74
1 .oo
0.11
0.44
0.11
0.00
0.11
0.51
0.00
4.30
0.00
0.33
0.08
0.32
0.74
0.34
0.49
0.70
0.51
0.21
0.74
1 .oo
1 .oo
0.41
0.74
0.78
1.00
0.12
1.00
0.34
*
** For these markers, results are reported with Fisher's exact test. See Materials and Methods section for method of analysis.

338 S H O H A T E T A L .
examined a wider range of polymorphic markers than was done in this study.
In the association analysis of our study, we used the parents as a group from which we derived the control gene frequencies. For our comparisons we used only those par- ental alleles that were not transmitted to the affected offspring, the latter being chosen for the “cases” group. The advantage of this method is that the alleles we used for control frequencies, derived from the parents’ alle- les, reflect to a greater extent the random genes in the Armenian population than would most other control groups. Thus, this method provides a precise match for eth- nicity (Falk & Rubinstein 1987). If an as- sociation exists between a certain marker allele and FMF, this allele will be over-rep- resented in the cases when compared to the “derived” parental control frequency. No significant association was found with any of the markers studied. The lack of signifi- cance may be partially due to the relatively small number of cases and controls. How- ever, the fact that all of these could be elim- inated as tightly linked suggests that there is no strong population association, as this would likely be inferred as linkage utilizing parametric linkage methods.
Although no significant linkage or associ- ation was observed in this study, we feel that this is the first step in a comprehensive effort toward the eventual localization of the FMF gene. Future goals should include examining those loci where linkage could not be rejected, with larger samples of fam- ilies, and using molecular probes for candi- date genes such as the lipocortin gene (Shohat et al. 1989) and amyloid genes (Sack 1988) for linkage and association studies.
Acknowledgements
Supported by grant # 88-0098/1 from the US-Israel binational science foundation
(BSF). Jerusalem, Israel and the NIDDK Inflammatory Bowel Disease Center Grant (DK 35200) and a grant from the Stuart Foundations.
References
Chaouat, Y., J . P. Tormcn, P. Godeau, J. P. Ca- mus, M. F. Kahn, A. Ryckewaert, J. E. Laula, C. J. Menkes, M. Schrnid & J. Hors (1977). Marqueurs HLA chez les sujets atteints de rna- ladic pcriodiquc. Nouv. Presse Med. 6,
Chaquat, Y., J . P. Tormen & J. H. J. Dausset (1977). HLA et maladie periodique. Rev. Rhu- mat. 44, 703-708.
Davrinche, C., C. Rivat, M. P. Ollier-Hart- mann & L. Hartmann (1985). C,, BF and C, polyrnorphisms in familial Mediterranean fe- ver. Isr. J . Med. Sci. 21, 883-885.
Dixon, W. J . & F. J. Massey (1983). Enumeration statistics. In: Introduction to Statistical Analysis. New York, McGraw-Hill, Inc., pp. 228-240.
Eliakim, M., M. Levy & M. Ehrenfeld (1981). Laboratory examinations. In Recurrent Poly- serositis, M. Eliakim, M. Levy & M. Ehrenfeld (eds.). New York, Elsevier North-Holland, pp.
Falk. C. T. & P. Rubinstein (1987). Haplotype relative risks: an easy reliable way to construct a proper control sample for risk calculations. Ann. Hum. Genet. 51,227-233.
Harris, H. & D. A. Hopkinson (eds.). (1976). Handbook for Enzyme Electrophoresis in Hu- mun Genetics. Amsterdam, North Holland.
Lange, K. & M. Boehnke (1983). Extensions to pedigree analysis. Optimal calculation of Men- delian likelihoods. Hum. Hered. 31, 291-301.
Lathrop, G. M. & J. M. Lalouel (1984). Easy calculations of lod scores and genetic risks on small computers. Am. J. Hum. Genet. 36, 46Cj-465.
Loop, I. W. & D. K. Clawson (1965). Unusual arthropathy in periodic peritonitis. J . Am. Med. Assoc. 192, 11 62-1 164.
Melamed, I., Y. Shemer, V. Zakuth, N. Tzehoval, M. Pras Br Z. Spirer (1983). The immune sys- tem in familial Mediterranean fever. Clin. Exp. Immunol. 53, 659-662.
Ollier-Hartman, M. P., P. Godeau, E. Gouet & L. Hartman (1981). Le systime du complement dans la maladie pkriodique. Ann. Med. Intern. 132, 467-471.
Pras, M., N. Bronshpigel, D. Zemer & J. Gafni
2949-295 3.
87-96.

G E N E T I C S T U D I E S I N F M F 339
( I 982). Variable incidence of amyfoidosis in familial Mediterranean fever among different ethnic groups. John Hopkins Med. J. 150,
Pras, M. & E. Gazit (1985). Familial Mediter- ranean fever: no association of HLA with amy- loidosis or colchicine treatment response. Zsr. J. Med. Sci. 21, 757-758.
Reiman, H. A. (1954). Periodic peritonitis - her- edity and pathology; report of seventy-two cases. J. Am. Med. Assoc. 154, 12541259.
Rogers, D. B., M. Shohat, G. M. Petersen, J. Bickal, J. Congleton, A. Schwabe & J. I. Rotter (1989). Familial Mediterranean fever in Ar- menians: autosomal recessive inheritance with high gene frequency. Am. J. Med. Genet. 34,
Sack, G. H. (1988). Serum amyloid A (SAA) gene variations in familial Mediterranean fever. Mol. Biol. Med. 5 , 61-67.
Schles'nger, M., D. N. Ilfeld, R. Zamir & C.
no linkage with HLA. Tissue Antigens 24,
Schwabe, A. D. & R. S. Peters (1974). Familial
22-25.
168-172.
Br- 1 utbar (1984). Familial Mediterranean fever:
65-66.
Mediterranean fever in Armenians. Analysis of 100 cases. Medicine 53, 453-462.
Shohat, M., J. R. Korenberg, A. Schwabe & J. I. Rotter (1989) Familial Mediterranean fever - a genetic disorder of the lipocortin family. Am. J. Med. Genet. 34. 163-167.
Sneh, E., M. Pras, D. Michaeli, N. Shahin & J. Gafni (1977). Protracted arthritis in familial Mediterranean fever. Rheumatol. Rehabil. 16,
Sohar, E., J. Gafni, M. Pras & H. Heller (1967). Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am. J. Med.
Widmann, F. K. (1985). Technical Manual of American Association of Blood Banks. Arling- ton, American Association of Blood Banks.
102-1 06.
43, 227-253.
Address:: Jerome I . Rotter, M.D. Director, Division of Medical Genetics ASB 3 Cedars-Sinai Medical Center 8700 W Beverly Blvd. Los Angeles. CA 90048, USA