genetic modeling of human...
TRANSCRIPT

Genetic Modeling of Human Rhabdomyosarcoma
Corinne M. Linardic,1,2Diane L. Downie,
2Stephen Qualman,
5Rex C. Bentley,
3
and Christopher M. Counter2,4
Departments of 1Pediatrics, 2Pharmacology and Cancer Biology, 3Pathology, and 4Radiation Oncology, Duke University Medical Center, Durham,North Carolina; and 5Center for Childhood Cancer, Columbus Children’s Research Institute and Children’s Hospital, Columbus, Ohio
Abstract
Rhabdomyosarcoma, a malignancy showing features ofskeletal muscle differentiation, is the most common softtissue sarcoma of childhood. The identification of distinctclinical presentation patterns, histologic tumor types, and riskgroups suggests that rhabdomyosarcoma is a collection ofhighly related sarcomas rather than a single entity. In aneffort to understand this seemingly heterogeneous malignan-cy, we constructed a genetically defined but malleable modelof rhabdomyosarcoma by converting less differentiatedhuman skeletal muscle cell precursors (SkMC) and committedhuman skeletal muscle myoblasts (HSMM) into their malig-nant counterparts by targeting pathways altered in rhabdo-myosarcoma. Whereas the two cell types were bothtumorigenic, SkMCs gave rise to highly heterogeneous tumorsoccasionally displaying features of rhabdomyosarcoma,whereas HSMMs formed rhabdomyosarcoma-like tumors withan embryonal morphology, capable of invasion and metasta-sis. Thus, despite introducing the same panel of geneticchanges, altering the skeletal muscle cell of origin led todifferent tumor morphologies, suggesting that cell of originmay dictate rhabdomyosarcoma tumor histology. The abilityto now genetically induce human rhabdomyosarcoma-liketumors provides a representative model to dissect themolecular mechanisms underlying this cancer. (Cancer Res2005; 65(11): 4490-5)
Introduction
Malignant tumors resembling skeletal muscle, collectively knownas rhabdomyosarcomas, are the most common soft tissue sarcomaof childhood, with a 3-year failure-free survival of high-risk patientsof only 20% (1). Tumor histology plays a significant role in theprognosis of rhabdomyosarcoma (1), and combined with variationsin clinical group and stage, suggest that the etiology ofrhabdomyosarcoma is heterogeneous. There are only a few in vivoexperimental systems to study the variable molecular eventsleading to rhabdomyosarcoma. A number of human rhabdomyo-sarcoma tumor cell lines have been established, but these linesrepresent the final stages of rhabdomyosarcoma development andhence are not amenable to dissecting early events. Rhabdomyo-sarcoma tumors appear at low or variable incidence in a variety oftransgenic mouse backgrounds (2, 3), and recently, mouse modelshave been generated for both the embryonal and alveolar histologicvariants of rhabdomyosarcoma (4, 5). However, tumorigenesis canbe different between humans and rodents (6, 7); hence, there is
value in studying cancer in human cells. To elucidate the cellularmechanisms underlying human rhabdomyosarcoma, we thereforesought to define the molecular events sufficient to drive normalhuman skeletal muscle cell precursors towards a cancerous fate.Rhabdomyosarcoma shares a number of changes common to
other human malignancies. Specifically, the p53 tumor suppressorpathway is impaired in up to 50% of rhabdomyosarcoma tumorsand cell lines (2, 8, 9) and rhabdomyosarcoma can occur in childrenwith germ line inactivation of p53 (10). The RB tumor suppressorpathway also seems dysregulated in rhabdomyosarcoma throughamplification of cyclin-dependent kinase CDK4 and/or deletion ofthe tumor suppressor p16INK4A (2, 9, 11). The MYCN proto-oncogene is up-regulated in rhabdomyosarcoma (2, 12), targeting40% of genes similarly activated by c-Myc (13). Telomerestabilization and correspondingly cell immortalization is illegiti-mately restored in rhabdomyosarcoma by reactivation of thehTERT catalytic subunit or through alternative telomere lengthen-ing mechanisms (14–16). Lastly, activation of the Ras pathway (17)occurs in rhabdomyosarcoma through point mutations in Ras(2, 18, 19), activation of upstream tyrosine kinase receptors (2), orloss of the negative regulator neurofibromin (20). To recapitulatethe genetic alterations of rhabdomyosarcoma in a human modelsystem, we therefore disrupted these pathways (21) in humanprimary cells. The cell of origin of rhabdomyosarcoma is unknownbut suggested to be from cells developing at any point along theskeletal muscle cell axis (2). Hence, we chose to introduce thesealterations in human primitive fetal skeletal muscle cell precursors(SkMC) as well as human postnatal skeletal muscle myoblasts(HSMM) already committed to the skeletal muscle lineage. We usedexpression of the SV40 large T oncoprotein to inactivate the tumorsuppressors p53 and RB, small t oncoprotein to inactivate PP2A,leading to stabilization of the c-Myc oncprotein (22), hTERT toimpart immortalization (23), and oncogenic (V12G) Ras to provideself-sufficiency in growth signals (17).
Materials and Methods
Cell lines. Low-passage normal human fetal SkMCs or normal HSMMs
from a teenage donor (Clonetics Cell Systems, Cambrex Corp., EastRutherford, NJ) were sequentially infected with amphotrophic retroviruses
derived from pBABE-neo-T/t-Ag , pBABE-bleo-FLAG-H-RasV12G (7), pBABE-
hygro-FLAG-hTERT , pBABE-puro-FLAG-H-Ras V12G (21), or the
corresponding empty vectors and sequentially selected for 7 to 10 days inmedium supplemented with 0.25 Ag/mL puromycin (Sigma Chemical Co.,
St. Louis, MO), 50 Ag/mL hygromycin B, 250 Ag/mL G418, or 800 Ag/mL
Zeocin (all from Life Technologies Invitrogen, Carlsbad, CA). Cells were
verified to be of skeletal muscle lineage by expression of one or more skeletalmuscle markers: SkMCs were desmin positive, skeletal muscle–specific
actin, and myoglobin negative, whereas HSMMs were desmin and skeletal
muscle–specific actin positive and myoglobin negative (data not shown).Detection of gene products. For Western blotting, 100 Ag of whole
cell lysates were separated and immunoblotted with antibodies anti–T Ag
SC-147, anti-actin SC-8432 (Santa Cruz Biotechnology, Santa Cruz, CA),
Requests for reprints: Christopher M. Counter, Duke University Medical Center,Box 3813, Durham, NC 27710. Phone: 919-684-9890; Fax: 919-684-8958; E-mail:[email protected].
I2005 American Association for Cancer Research.
Cancer Res 2005; 65: (11). June 1, 2005 4490 www.aacrjournals.org
Priority Report
Research. on July 13, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
anti-FLAG-M2 (Sigma Chemical), anti-desmin M0760, anti–muscle-specificactin M0635, or anti-myoglobin A0324 (DakoCytomation, Carpinteria, CA),
using established protocols (7).
For immunohistochemistry, 5-Am sections of paraffin-embedded tissue
were subject to heat-induced antigen retrieval using the steam method andincubated with antibodies (DakoCytomation) against myoglobin (A0324,
1:3,000), muscle-specific actin (M0635, 1:200), desmin (M0760, 1:50),
myogenin (M3559, 1:50), and MyoD1 (M3512, 1:50) at 37jC for 45 minutes.
Biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) wasincubated for 20 minutes, and the tertiary detection system was ABCElite
Complex (Vector Laboratories), with a 3,3V-diaminobenzidine chromagen
substrate (Innovex Biosciences, Richmond, CA). Slides were lightly
counterstained with hematoxylin. Normal human skeletal muscle andisotype-specific antibodies were used as positive and negative controls,
respectively. Pathologists with experience in the evaluation of pediatric
solid tumors (S.Q. and R.C.B.) evaluated slides. Standard H&E stainedsections were also prepared.
For telomerase assays, 0.5 Ag cellular lysates were assayed for activity
using a PCR-based telomere repeat amplification assay, as described
previously (21).For reverse transcription-PCR, 2 Ag of total RNA, prepared using the
RNAzol B reagent (TEL-TEST, Friendswood, TX), was reverse transcribed
using the Ominscript RT kit (Qiagen, Valencia, CA) with OligodT primer
(Life Technologies Invitrogen), after which 4 AL of each reaction was PCRamplified using primers specific for insulin-like growth factor-II mRNA (5V-ATCGTT-GAGGAGTGCTGTTTCC-3V; 5V-ATTGCTGGCCATCTCTGG-3V) andglyceraldehyde-3-phosphate dehydrogenase (21).
For transmission electron microscopy, tumor samples were processed as
described (24) except that glutareldehyde was 4%, OsO4 buffer was 0.2 mol/L
cacodylate, stain/counterstain was uranyl acetate/lead citrate, and images
were captured on a Philips EM410 electron microscope.Transformation and tumorigenesis. Soft agar growth was assessed
after 4 weeks as described previously (7). For xenograft assays, 1 � 107 cells
were suspended in 50 AL Matrigel (BD Biosciences, San Jose, CA) and
injected s.c. into the flank of a severe combined immunodeficient/Beige
mouse as previously described (7). For orthotopic assays, 3 � 105 cellssuspended in 50 AL PBS were injected into the right gastrocnemius muscle.
For metastasis assays, 4 � 106 cells suspended in 200 AL medium were
injected into the tail vein. Each cell line was tested in quadruplicate. All
experiments were done under the Duke Institutional Animal Care and UseCommittee–approved protocols.
Results and Discussion
Genetic transformation of skeletal muscle cell precursors.Unlike adult carcinomas, which derive from the malignanttransformation of epithelial cells, the histogenesis of pediatricmesenchymal tumors such as rhabdomyosarcoma is less clear. It hasbeen suggested that rhabdomyosarcoma tumors may derive fromthe transformation of cells developing at any point along the skeletalmuscle cell axis (2), generically termed here as ‘‘skeletal muscle cellprecursors.’’ Although historically these precursors were presumedto be satellite cell myoblasts located beneath the basementmembrane of the skeletal myofiber, skeletal muscle cell precursorsare also believed to include multipotential stem cells, which derivefrom the bone marrow but reside in skeletal muscle tissue, andpossibly skeletal muscle myonuclei, which may be stimulated toreenter the cell cycle after specific cues (25). In an effort to modelrhabdomyosarcoma, we postulated that transformation of anunselected primary skeletal muscle cell population might yieldtumors akin to rhabdomyosarcoma. Therefore, a heterogeneouspopulation of skeletal muscle cell precursors derived from humanfetal muscle, termed SkMC cells, was serially infected and selectedfor antibiotic resistance with amphotrophic retroviruses encodingT/t-Ag , hTERT, and H-RasV12G, or a combination thereof, in whichone or more transgenes was substitutedwith an empty vector. Stablepolyclonal cell lines representing all possible combinations of T/t-Ag(T), and/or hTERT (H), and/or H-RasV12G (R), and/or vector (V),
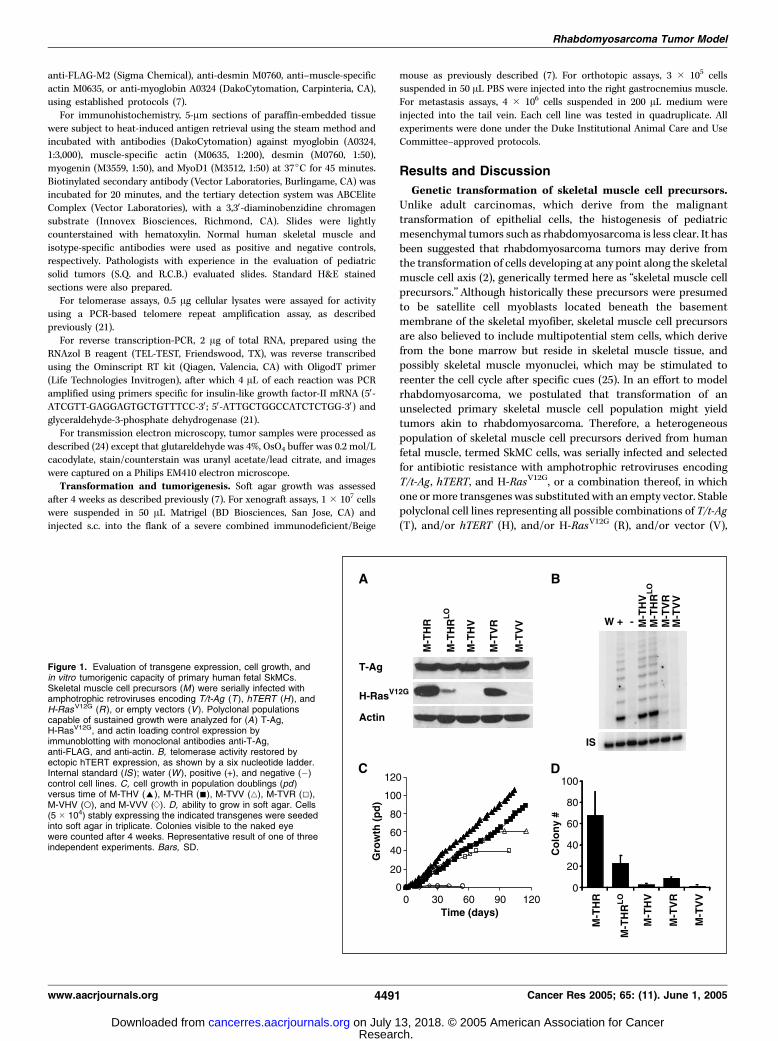
Figure 1. Evaluation of transgene expression, cell growth, andin vitro tumorigenic capacity of primary human fetal SkMCs.Skeletal muscle cell precursors (M ) were serially infected withamphotrophic retroviruses encoding T/t-Ag (T ), hTERT (H ), andH-RasV12G (R ), or empty vectors (V ). Polyclonal populationscapable of sustained growth were analyzed for (A) T-Ag,H-RasV12G, and actin loading control expression byimmunoblotting with monoclonal antibodies anti-T-Ag,anti-FLAG, and anti-actin. B, telomerase activity restored byectopic hTERT expression, as shown by a six nucleotide ladder.Internal standard (IS ); water (W ), positive (+), and negative (�)control cell lines. C, cell growth in population doublings (pd )versus time of M-THV (E), M-THR (n), M-TVV (4), M-TVR (5),M-VHV (o), and M-VVV (d). D, ability to grow in soft agar. Cells(5 � 104) stably expressing the indicated transgenes were seededinto soft agar in triplicate. Colonies visible to the naked eyewere counted after 4 weeks. Representative result of one of threeindependent experiments. Bars, SD.
Rhabdomyosarcoma Tumor Model
www.aacrjournals.org 4491 Cancer Res 2005; 65: (11). June 1, 2005
Research. on July 13, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

were generated and confirmed to appropriately express the desiredtransgenes (Fig. 1A-B). A cell line expressing 5-fold lower levels ofH-RasV12G was also generated (Fig. 1A ; M-THRLo) to address theeffect of oncogenic Ras expression on rhabdomyosarcoma tumor-igenicity, as low oncogenic Ras expression has been found to limittumor growth in human mammary tumors (26).
Tumorigenic conversion of skeletal muscle cell precursorsby expression of T/t-Ag, hTERT, and H-RasV12G. To explore theeffects of these genetic changes on the tumorigenic process, theresulting cell populations were assayed in vitro for immortalizationand anchorage-independent growth, common features of cancercells, and in vivo for tumor growth in immunocompromised mice.As regards immortalization, cells lacking T/t-Ag entered apermanent growth arrest between population doublings 13 and15 with morphologic features consistent with senescence (Fig. 1C ;uninfected, M-VVV, M-VHV), in agreement with the results ofothers (27). The stable expression of H-RasV12G alone or incombination with hTERT resulted in cell death (Fig. 1C ; M-VVR,M-VHR), presumably reflecting a cellular response against Ras up-regulation in the presence of an intact p53 pathway (28). Asexpected (29), expression of T/t-Ag alone or in the presence ofoncogenic Ras extended the proliferative life span of the cells by upto 60 population doublings, at which time the cells entered crisisand perished (Fig. 1C ; M-TVV, M-TVR). As with other cell types (21),independent of RasV12G expression, only T/t-Ag in conjunction withhTERT greatly extended cellular life span (Fig. 1C ; M-THR, M-THV).Thus, combined ectopic expression of T/t-Ag and hTERT endowshuman SkMCs with an infinite life span, in agreement with theobserved dysregulation of the p53 , RB ,Myc , and hTERT pathways inrhabdomyosarcoma tumors and cell lines (2, 14–16).As regards transformation, each of the above-described cell lines
was also assayed for growth in soft agar, one of the most stringentassays for transformation in vitro . Expression of all four transgenes,and not any less, was necessary and sufficient for anchorage-independent growth of SkMCs (Fig. 1D ; M-THR). This growthdepended upon increased oncogenic Ras expression, as both thenumber of colonies and the heterogeneity of colony sizes decreasedupon lowering oncogenic Ras expression 5-fold (Fig. 1D ; other data
Table 1. Xenograft tumor formation in immunocompro-mised mice
Parental
cell type
Cell line Injection
route
Mice developing
tumors/mice
injected
Earliest time
to palpable
tumor
SkMC M-TVV SQ 0/4 —
M-THV SQ 0/8 —M-TVR SQ 0/4 —
M-THRLo SQ 6/8 11 wks
M-THR SQ 4/4 4.5 wks
HSMM MY-THR SQ 5/5 2 wksMY-THR IV 4/5 NA
MY-THR IM 4/4 8 wks
NOTE: Polyclonal cell lines derived from normal human SkMCs or
normal human myoblasts (HSMM) stably expressing SV40 T/t-Ag (T),
and/or hTERT (H), and/or H-RasV12G (R), and/or empty vector (V )
were injected s.c., i.v., or i.m. to assay for tumor growth. ‘‘Lo’’ is a cellline expressing low levels of H-RasV12G.
Abbreviations: NA, not applicable (as mice injected via tail vein
developed lung nodules); SQ, s.c.; IV, i.v.; IM, i.m.
Figure 2. S.c. tumor xenografts derived from SkMC cells stablyexpressing T/t-Ag , hTERT , and H-RasV12G show variablerhabdomyoblastic morphology and immunohistochemical staining.H&E (A ) and (B) desmin staining of the least differentiated tumorxenograft. H&E (C ), (D ) desmin, skeletal muscle–specific actin(E), and myoglobin (F ) staining of the most differentiated tumorcontaining areas of spindle-shaped cells. Immunoreactivity(brown). Elongate cell with biopolar cytoplasmic processes forminga ‘‘spindle’’ shape (open arrow ). Small foci of immunoreactivity(closed arrows ). Magnification, 400�.
Cancer Research
Cancer Res 2005; 65: (11). June 1, 2005 4492 www.aacrjournals.org
Research. on July 13, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
not shown). Thus, Ras expression is required in addition to T/t-Agand hTERT for anchorage-independent growth of SkMCs.The most telling assay of tumorigenesis is tumor growth itself.
SkMCs expressing all four transgenes, or control SkMCs expressingtwo or three of these transgenes, were injected s.c. intoimmunocompromised mice and monitored for tumor growth. Asin soft agar, expression of all four transgenes was required fortumor growth (Table 1). This growth was sensitive to Rasexpression, as a decrease in expression of this oncogene delayedor abolished tumor growth (Table 1), suggesting an important rolefor the stimulation of the Ras pathway in rhabdomyosarcoma. Insum, SkMCs minimally must undergo dysregulation of the p53 , RB ,Myc (and possibly other targets of PP2A), Ras, and hTERT pathwaysto form tumors in vivo .SkMC-derived tumors ranged from highly undifferentiated small
round blue cell tumors with large nuclei (Fig. 2A) and scant
cytoplasm to tumors with foci of spindle-shaped cells (Fig. 2C). Noclassic embryonal or alveolar histology could be identified by lightmicroscopy, nor were there cytoplasmic cross-striations to confirmskeletal muscle differentiation. Immunohistochemical staining fordesmin, muscle-specific actin, and myoglobin, markers used inclinical practice to evaluate for rhabdomyosarcoma (1), showedsome biochemical evidence of skeletal muscle differentiation.Staining for these markers ranged from undetectable (Fig. 2B) topositive for all three antigens in the tumor that showed foci ofspindle-shaped cells (Fig. 2D-F). Staining for myogenin and MyoD1,muscle-specific transcription factors expressed in the nuclei ofrhabdomyosarcomas (1), was negative (data not shown). All tumorsshowed brisk mitotic activity and areas of necrosis, consistent withhigh-grade sarcomas. Thus, introducing genetic changes charac-teristic of rhabdomyosarcoma in cultures of human SkMCs led to abroad spectrum of sarcomas, ranging from undifferentiated small
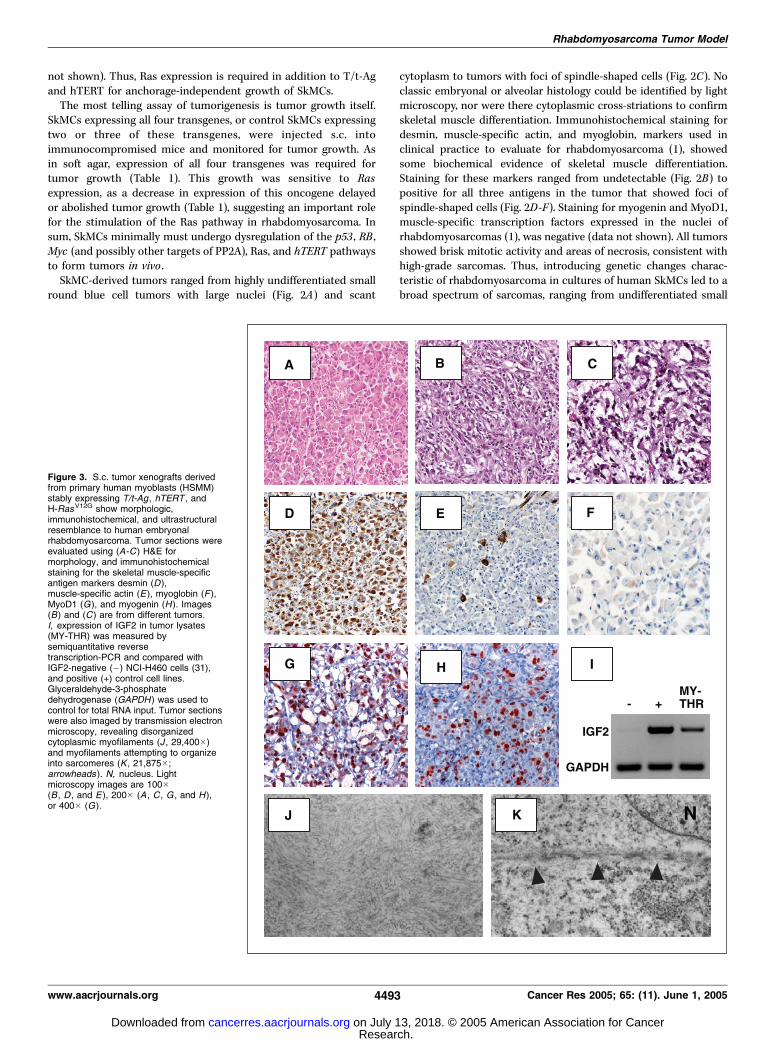
Figure 3. S.c. tumor xenografts derivedfrom primary human myoblasts (HSMM)stably expressing T/t-Ag , hTERT , andH-RasV12G show morphologic,immunohistochemical, and ultrastructuralresemblance to human embryonalrhabdomyosarcoma. Tumor sections wereevaluated using (A-C ) H&E formorphology, and immunohistochemicalstaining for the skeletal muscle-specificantigen markers desmin (D),muscle-specific actin (E ), myoglobin (F ),MyoD1 (G), and myogenin (H ). Images(B) and (C ) are from different tumors.I, expression of IGF2 in tumor lysates(MY-THR) was measured bysemiquantitative reversetranscription-PCR and compared withIGF2-negative (�) NCI-H460 cells (31),and positive (+) control cell lines.Glyceraldehyde-3-phosphatedehydrogenase (GAPDH ) was used tocontrol for total RNA input. Tumor sectionswere also imaged by transmission electronmicroscopy, revealing disorganizedcytoplasmic myofilaments (J , 29,400�)and myofilaments attempting to organizeinto sarcomeres (K , 21,875�;arrowheads ). N, nucleus. Lightmicroscopy images are 100�(B , D , and E ), 200� (A , C , G , and H ),or 400� (G).
Rhabdomyosarcoma Tumor Model
www.aacrjournals.org 4493 Cancer Res 2005; 65: (11). June 1, 2005
Research. on July 13, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

round blue cell tumors (sarcomas, not otherwise specified) totumors exhibiting some differentiation markers characteristic ofrhabdomyoblasts, but lacking frank histopathologic features ofeither embyronal or alveolar rhabdomyosarcoma.Transformation of human skeletal muscle myoblasts gen-
erates an embryonal rhabdomyosarcoma phenotype. Given thevariable tumor histology resulting from a mixed population ofSkMCs, we hypothesized that the skeletal muscle cell-of-originmight underlie differences in rhabdomyosarcoma tumor histology.As recent data suggests that embryonal rhabdomyosarcoma mightderive from satellite cell myoblasts (30), which should be present inthe SkMC population, an enriched population of HSMMs wasinfected with retroviruses encoding T/t-Ags , hTERT, and H-RasV12G,generating a new polyclonal myoblast MY-THR cell line. These cellswere tumorigenic in mice (Table 1). However, in contrast to SkMCcells, transformed HSMM cells showed rhabdomyosarcoma mor-phology, characterized by large numbers of rhabdomyoblasts withabundant, deeply eosinophilic cytoplasm (Fig. 3A), and spindle-shaped cells in a myxoid background (Fig. 3B-C). Immunohisto-chemical staining showed skeletal muscle differentiation, withdiffuse and strong desmin staining, focally positive muscle-specificactin, and diffusely positive myoglobin (Fig. 3D-F). The tumorswere focally positive for MyoD1 and myogenin (Fig. 3G-H) andexpressed IgF2 (Fig. 3I), a fetal growth factor overexpressed inrhabdomyosarcoma tumors (reviewed in ref. 2). When examined byelectron microscopy, the most rigorous test for establishing skeletalmuscle origin, these tumors had cytoplasmic myofilaments (Fig. 3J),and myofilaments with z-band material attempting to formsarcomeres (Fig. 3K). Thus, genetic changes observed in rhabdo-myosarcoma can convert HSMMs to tumors resembling humanrhabdomyosarcoma. Moreover, given the presence of embryonal
rhabdomyosarcoma-specific findings (nuclear pleomorphism, myx-oid change with spindling, focal staining of MyoD1, and myogenin)and the absence of alveolar findings (nuclear monotony, diffuseimmunostaining with MyoD1 and myogenin, collagenous septaelined by rhabdomyoblasts with associated nesting of tumor cells),these tumors are most consistent with an embryonal rhabdomyo-sarcoma histology.Genetically defined embryonal rhabdomyosarcoma tumors
are invasive and metastatic. Aside from histologic markers,human rhabdomyosarcoma tumors are characterized by theirability to invade adjacent tissue and metastasize. Therefore, thegenetically defined myoblast-derived tumor cells (MY-THR) wereintroduced into the systemic circulation, after which four of fiveinjected mice developed clinically and anatomically apparent lungmetastases by 8 weeks (Table 1), with almost complete obliterationof normal pulmonary alveolar architecture (Fig. 4A). Tumor cellstended to localize around pulmonary arterioles, often encasingpulmonary bronchioles (Fig. 4B). Histologic evaluation was againconsistent with an embryonal rhabdomyosarcoma morphology(Fig. 4C). Although the tail vein assay evaluates many steps inmetastasis, it does not assay the capacity of tumor cells to bemotile or invasive. In this regard, nodules located at the lungperiphery were noted to invade locally, eroding through the visceraland parietal lung pleura, and infiltrated through basementmembrane into the adjacent chest wall skeletal muscle and ribs(Fig. 4D). Moreover, MY-THR cells injected orthotopically into thegastrocnemius muscle of four mice developed as tumors (Table 1)that displaced the normal tissue (Fig. 4E) and invaded into thesurrounding native skeletal muscle (Fig. 4F), although not asextensively as the lung nodules. Intriguingly, local invasion was notobserved in s.c. xenografts (data not shown), suggesting that the
Figure 4. Primary human myoblasts stably expressing T/t-Ag ,hTERT , and H-RasV12G show metastatic and invasive behaviorwhen introduced systemically through the tail vein or orthotopicallyinto native skeletal muscle. A, low-power view of mouse lunglargely replaced by tumor nodules (10�). Dashed box, areamagnified for (B). B, single tumor nodule encasing pulmonarybronchus and vein (100�). Asterisks, areas of remaining intactalveoli. C, magnification of metastatic nodule showing embryonalmorphology. D, invasion of adjacent chest wall by peripheralpulmonary nodules (10�). Closed arrows, area of local invasion.Rib is cut transversely for orientation. E, low-power view ofgastrocnemius muscle largely replaced by tumor xenograft (10�).Hind limb is cut transversely for orientation. Dashed box, regionmagnified for (F ). F, tumor cells (open arrows ) streaming intoadjacent native skeletal muscle (200�).
Cancer Research
Cancer Res 2005; 65: (11). June 1, 2005 4494 www.aacrjournals.org
Research. on July 13, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

surrounding microenvironment provides factors necessary tosupport this more aggressive phenotype. Thus, expression of T/t-Ags , hTERT, and H-RasV12G endowed human myoblasts with theinvasive and metastatic phenotypes characteristic of rhabdomyo-sarcoma tumors.
Summary
The presentation of rhabdomyosarcoma is heterogeneous,arguing that treatments may need to be tailored to tumor type.To explore rhabdomyosarcoma etiology, we found that heteroge-neous populations of SkMCs could be converted to a broadspectrum of tumors via the corruption of the p53 , RB , Myc ,telomerase, and Ras pathways, whereas by limiting these changesto committed myoblasts (HSMMs), embryonal-like rhabdomyosar-coma tumors were generated. We therefore argue that cell typemay play a key role in rhabdomyosarcoma, and now with the
described ability to genetically model rhabdomyosarcoma, itshould be possible to dissect the histogenesis and molecularmechanisms underlying this disease using normal primary humancells.
Acknowledgments
Received 9/2/2004; revised 3/29/2005; accepted 4/6/2005.Grant support: NIH grants 5K12-HD043494-03 (C.M. Linardic) and CA94184 (C.M.
Counter), Duke Children’s Miracle Network (C.M. Linardic), North CarolinaCooperative Extension (C.M. Linardic), and Leukemia and Lymphoma Society (C.M.Counter).
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank members of the laboratories of Chris Counter, Xiao-Fan Wang, andAnthony Means, especially Stacey Adam, Kian-Huat Lim, Chaoyu Ma, and Tom Ribarfor assistance with mouse experiments; Philip Breitfeld and Darrell Yamashiro forreview of the report; and Henry Estrada and Sara Miller for assistance with electronmicroscopy.
References
1. Meyer WH, Spunt SL. Soft tissue sarcomas ofchildhood. Cancer Treat Rev 2004;30:269–80.
2. Merlino G, Helman LJ. Rhabdomyosarcoma: workingout the pathways. Oncogene 1999;18:5340–8.
3. Nanni P, Nicoletti G, De Giovanni C, et al. Develop-ment of rhabdomyosarcoma in HER-2/neu transgenicp53 mutant mice. Cancer Res 2003;63:2728–32.
4. Sharp R, Recio JA, Jhappan C, et al. Synergismbetween INK4a/ARF inactivation and aberrant HGF/SFsignaling in rhabdomyosarcomagenesis. Nat Med 2002;8:1276–80.
5. Keller C, Arenkiel BR, Coffin CM, El Bardeesy N,DePinho RA, Capecchi MR. Alveolar rhabdomyosarco-mas in conditional Pax3:Fkhr mice: cooperativity ofInk4a/ARF and Trp53 loss of function. Genes Dev 2004;18:2614–26.
6. Ghebranious N, Donehower LA. Mouse models intumor suppression. Oncogene 1998;17:3385–400.
7. Hamad NM, Elconin JH, Karnoub AE, et al. Distinctrequirements for Ras oncogenesis in human versusmouse cells. Genes Dev 2002;16:2045–57.
8. Takahashi Y, Oda Y, Kawaguchi K, et al. Alteredexpression and molecular abnormalities of cell-cycle-regulatory proteins in rhabdomyosarcoma. Mod Pathol2004;17:660–9.
9. Gordon AT, Brinkschmidt C, Anderson J, et al. A noveland consistent amplicon at 13q31 associated withalveolar rhabdomyosarcoma. Genes ChromosomesCancer 2000;28:220–6.
10. Birch JM, Hartley AL, Blair V, et al. Cancer in thefamilies of children with soft tissue sarcoma. Cancer1990;66:2239–48.
11. Maelandsmo GM, Berner JM, Florenes VA, et al.Homozygous deletion frequency and expression levelsof the CDKN2 gene in human sarcomas: relationship to
amplification and mRNA levels of CDK4 and CCND1. BrJ Cancer 1995;72:393–8.
12. Williamson D, Lu YJ, Gordon T, et al. Relationshipbetween MYCN copy number and expression in rhab-domyosarcomas and correlation with adverse prognosisin the alveolar subtype. J Clin Oncol 2005;23:880–8.
13. Boon K, Caron HN, van Asperen R, et al. N-mycenhances the expression of a large set of genesfunctioning in ribosome biogenesis and protein syn-thesis. EMBO J 2001;20:1383–93.
14. Kleideiter E, Schwab M, Friedrich U, Koscielniak E,Schafer BW, Klotz U. Telomerase activity in cell linesof pediatric soft tissue sarcomas. Pediatr Res 2003;54:718–23.
15. Terasaki T, Kyo S, Takakura M, et al. Analysis oftelomerase activity and telomere length in bone andsoft tissue tumors. Oncol Rep 2004;11:1307–11.
16. Henson JD, Hannay JA, McCarthy SW, et al. A robustassay for alternative lengthening of telomeres in tumorsshows the significance of alternative lengthening oftelomeres in sarcomas and astrocytomas. Clin CancerRes 2005;11:217–25.
17. Bos JL. Ras oncogenes in human cancer: a review.Cancer Res 1989;49:4682–9.
18. Wilke W, Maillet M, Robinson R. H-ras-1 pointmutations in soft tissue sarcomas. Mod Pathol 1993;6:129–32.
19. Yoo J, Robinson RA. H-ras and K-ras mutations in softtissue sarcoma: comparative studies of sarcomas fromKorean and American patients. Cancer 1999;86:58–63.
20. Reed N, Gutmann DH. Tumorigenesis in neurofibro-matosis: new insights and potential therapies. TrendsMol Med 2001;7:157–62.
21. Hahn WC, Counter CM, Lundberg AS, BeijersbergenRL, Brooks MW, Weinberg RA. Creation of humantumour cells with defined genetic elements. Nature 1999;400:464–8.
22. Yeh E, Cunningham M, Arnold H, et al. A signallingpathway controlling c-Myc degradation that impactsoncogenic transformation of human cells. Nat Cell Biol2004;6:308–18.
23. Collins K, Mitchell JR. Telomerase in the humanorganism. Oncogene 2002;21:564–79.
24. Linardic CM, Jayadev S, Hannun YA. Activation of thesphingomyelin cycle by brefeldin A: effects of brefeldinA on differentiation and implications for a role forceramide in regulation of protein trafficking. CellGrowth Differ 1996;7:765–74.
25. Grounds MD, White JD, Rosenthal N, BogoyevitchMA. The role of stem cells in skeletal and cardiacmuscle repair. J Histochem Cytochem 2002;50:589–610.
26. Elenbaas B, Spirio L, Koerner F, et al. Human breastcancer cells generated by oncogenic transformation ofprimary mammary epithelial cells. Genes Dev 2001;15:50–65.
27. Seigneurin-Venin S, Bernard V, Moisset PA, et al.Transplantation of normal and DMD myoblastsexpressing the telomerase gene in SCID mice. BiochemBiophys Res Commun 2000;272:362–9.
28. Bringold F, Serrano M. Tumor suppressors andoncogenes in cellular senescence. Exp Gerontol 2000;35:317–29.
29. Mouly V, Edom F, Decary S, Vicart P, Barbert JP,Butler-Browne GS. SV40 large T antigen interferes withadult myosin heavy chain expression, but not withdifferentiation of human satellite cells. Exp Cell Res1996;225:268–76.
30. Tiffin N, Williams RD, Shipley J, Pritchard-Jones K.PAX7 expression in embryonal rhabdomyosarcomasuggests an origin in muscle satellite cells. Br J Cancer2003;89:327–32.
31. Quinn KA, Treston AM, Unsworth EJ, et al. Insulin-like growth factor expression in human cancer celllines. J Biol Chem 1996;271:11477–83.
Rhabdomyosarcoma Tumor Model
www.aacrjournals.org 4495 Cancer Res 2005; 65: (11). June 1, 2005
Research. on July 13, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

2005;65:4490-4495. Cancer Res Corinne M. Linardic, Diane L. Downie, Stephen Qualman, et al. Genetic Modeling of Human Rhabdomyosarcoma
Updated version
http://cancerres.aacrjournals.org/content/65/11/4490
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/65/11/4490.full#ref-list-1
This article cites 31 articles, 10 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/65/11/4490.full#related-urls
This article has been cited by 13 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/65/11/4490To request permission to re-use all or part of this article, use this link
Research. on July 13, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from