genetic variation and natural selection detection · pdf filegenetic variation and natural...
TRANSCRIPT

Genetic Variation and Natural Selection Detection
CAS-MPG Partner Institute for
Computational Biology (PICB)
Shuhua Xu
Otto Warburg International Summer School and Research Symposium 2013

• Genetic Variation is variation in alleles of genes,
occurs both within and among populations.
– Mutation
– Polymorphism
Genetic variation

• Polymorphism is often defined as the presence
of more than one genetically distinct type in a
single population.
• Rare variations are not classified as
polymorphisms; and mutations by themselves do
not constitute polymorphisms.
Polymorphism

• RFLP (Restriction Fragment Length Polymorphism)
• AFLP (Amplified Fragment Length Polymorphism)
• RAPD (Random Amplification of Polymorphic DNA)
• VNTR (Variable Number Tandem Repeat, or Minisatellite)
• STR (Short Tandem Repeat, or Microsatellite)
• SNP (Single Nucleotide Polymorphism)
• SFP (Single Feature Polymorphism)
• CNV (Copy Number Variation)
DNA polymorphism

Information from NGS
The 1000 Genomes Project • Full sequence data
• Polymorphisms
• Rare mutations
• CNVs
• Small indels
• Recombination

• Number of alleles
– More alleles, larger diversity;
• Minor allele frequency (MAF)
– is the frequency of the less (or least) frequent allele in a given locus and a given population.
Intuitive statistics
Mutation: MAF ≤1% Polymorphism: MAF >1%

Heterozygosity
• The fraction of individuals in a population that are heterozygous for a particular locus.
• It can also refer to the fraction of loci within an individual that are heterozygous.
where n is the number of individuals in the population, and ai1, ai2
are the alleles of individual i at the target locus.
Observed
where m is the number of alleles at the target locus, and fi is the
allele frequency of the ith allele at the target locus.
Expected

• Heterozygosity and HWD
– Comparison of Ho and He
• Gene diversity
Heterozygosity related issues

Population Mutation Rate (q )
• Under mutation-drift equilibrium:
– q = 4Nem for autosome
– q = Nem for Y and mtDNA
– q = 3Nem for X chromosome
qautosome > qX > qY

• Number of segregating sites (θK);
• Average pairwise differences (θ∏);
• Number of alleles (θE);
• Mean number of mutations since the MRCA (θΩ);
• Singleton
• ……
Estimators of θ

Number of segregating sites (K)
►Under the infinite site model, K is equal to the number of mutations since the most recent common ancestor of the sequences in the sample.
►Therefore, K has a clear biological meaning.
►However, K depends on the sample size.

Normalized K
K
n
K
aq
1 11
2 1na
n

Variance of θK
KE q q
2
2
nK
n n
bVar
a a
qqq
2
1 11
4 1nb
n
► Under the neutral Wright-Fisher model with
constant effective population size,

• N diploid individuals. Generations are non-overlapping. At each generation, each chromosome inherits its genetic material from a uniformly chosen chromosome from the previous generation, independently from all other chromosomes.
• In its most basic form, the Wright-Fisher model overlooks many important details: – 1. Mutation – 2. Recombination – 3. Sexes – 4. Non-overlapping generations – 5. Population size changes – 6. Family size distribution – 7. Population structure – 8. Selection
Wright-Fisher model

• θK is independent of sample size.
• However, the usefulness of θK is not clear
under other population genetic models,
such as those with natural selection.
• θK is sensitive to the number of rare alleles,
or mutants of low frequency.
The properties of θK

How many common SNPs in human genome?
Common SNPs: minor allele frequency (MAF) >0.05; Suppose we have 50 samples of African, European, Asian
respectively; Theta=1.2/kb for African population; Theta=0.8/kb for European and Asian population; Autosome length (L)=2.68 billion bp;
► We expect 9.8 million common SNPs in 50 African samples;
► We expect 6.5 million common SNPs in 50 European samples;
► We expect 6.5 million common SNPs in 50 Asian samples
1
1
1
1
1
n
i
iK n
i i
q
104
5%
6
1MAF K
i
E S L iq
where

θK =1.2/kb (African)

θK =0.8/kb (non-African)

• Also known as
– sequence diversity
– mean number of nucleotide differences
between two sequences.
Average pairwise differences (∏)
2
1ij
i j
dn n
2
2
,
2 31
3 1 9 1
E
n nnVar
n n n
q
q q

• ∏ as a measure of genetic variation has clear biological meanings which do not depend on the underlying evolutionary process.
• In comparison to θK, it is insensitive to the rare alleles, or mutants of low frequency.
• ∏ is an useful measure of persistent genetic variation, and neutral genetic variation when purifying selection is operating.
• However, because its variance is considerably larger than that of θK, it is not as good as θK for neutral locus.
The properties of ∏

Number of alleles
• Ewens (1972) shows that under the infinite allele model
11 1
E kn
q q
q q
• An estimate of θ can be obtained by resolving the above
equation for θ with E(k) replaced by k.
• The estimate is known as Ewens’s estimator θE.

• Under the infinite allele model, θE is about the
best estimator one can devise.
• However, θE is slightly upward biased estimator
particularly when θ is large.
The properties of θE

• The mean number Ω of mutations since the most recent common ancestor (MRCA) of a sample is another intuitive summary statistic, but seldom used in practice.
• This is probably partly due to that its use requires knowing for each segregating site the ancestral nucleotide, and partly because its because its statistical properties are not well understood.
Mean number of mutations since the MRCA (Ω)

Mean number of mutations since the MRCA (Ω)
• Let ωl be the number of mutations in sequence l
since MRCA.
• Then the average is given by
1
1 n
l
l
An
• Note that a mutation of size i is counted as one
mutation in i of n sequences, we therefore have
1
1 n
i
l
A in

• It follows that
Mean number of mutations since the MRCA (Ω)

Singleton mutations
• The number ξi of mutations of size 1 in a sample is of special interest because it captures mostly the recent mutations in a sample.
• According to Fu and Li (1993),

Classify the above summary statistics
• ∏0,0 =θ K
• ∏1,1 =θ∏

Weight of ∏k,l statistics

• Individuals with favorable traits are more likely to leave more offspring better suited for their environment.
• Also known as “Differential Reproduction”
• Also called “Survival of the fittest"
Natural selection

• The selective breeding of domesticated plants and animals by man.
Artificial Selection

Range of values at time 1
Nu
mb
er
of
ind
ivid
uals
Range of values at time 2 N
um
ber
of
ind
ivid
uals
Range of values at time 3
Nu
mb
er
of
ind
ivid
ua
ls
Favors the intermediate variants
Selects against the extreme phenotypes
e.g. Human birth weight
Stabilizing Selection

Range of values at time 1
Nu
mb
er
of
ind
ivid
uals
Range of values at time 3
Nu
mb
er
of
ind
ivid
uals
Range of values at time 2 N
um
ber
of
ind
ivid
uals
Favors variants of opposite extremes
e.g. London's peppered moths
Disruptive Selection

Range of values at time 3
Nu
mb
er
of
ind
ivid
uals
Range of values at time 2
Nu
mb
er
of
ind
ivid
uals
Range of values at time 1
Nu
mb
er
of
ind
ivid
uals
Directional Selection
Favors one extreme phenotype or other extreme
e.g. beak length of the Galapagos finches

1. Population has variations.
2. Some variations are favorable.
3. More offspring are produced than
survive
4. Those that survive have favorable traits.
5. A population will change over time.
Darwin’s 5 points

• Balancing selection refers to a number of selective processes by which multiple alleles are actively maintained in the gene pool of a population at frequencies above that of gene mutation.
• heterozygote advantage
• frequency-dependent selection
Balancing selection

• Negative selection or purifying selection is the selective removal of alleles that are deleterious.
• Positive selection is selection on a particular trait and the increased frequency of an allele in a population
Negative selection and positive selection

• Loss of genetic diversity
• Screwed allele frequency spectrum
• Unexpected substitution ratio
• Extended haplotype homozygosity
• Elevated linkage disequilibrium
Footprints of natural selection in genomes

Gene Trees and Evolutionary Hypotheses
Neutral Balancing Selection Selective Sweep
Gene Trees and Evolutionary Hypotheses

Are most substitutions due to drift or natural selection?
“Neutralist” vs. “selectionist”
Agree that:
Most mutations are deleterious and are removed.
Some mutations are favourable and are fixed.
Dispute:
Are most replacement mutations that fix beneficial or neutral?
Is observed polymorphism due to selection or drift?
Neutralist vs. selectionist view

Whether a locus has been evolving under natural selection is often of interest if the locus represent a gene or linked to one.
As typical in many branches of sciences, a simpler explanation of phenomenon is often preferred unless there is strong evidence to suggest otherwise.
In population genetics study, the neutral hypothesis of evolution is arguably simpler than any other hypotheses and is much better understood statistically.
As a result, it is now generally used as the null model for analyzing polymorphism.
A significant deviation from the null model may signal the presence of forces that are absent or factors that are over-simplified in the null model.
Neutral hypothesis as the null model

• There are several ways statistical tests can be constructed to see if the null model is adequate for explaining the observed amount and pattern of polymorphism.
• Many summary statistics (estimators of θ) have quite different expectation when the null model is violated, this offer an opportunity of testing by considering the difference between two measures of polymorphism.
Statistical tests using estimators of θ

Statistical tests using estimators of θ
Suppose L1 and L2 are two different summary statistics such that E(L1) =E(L2) under the hypothesis of strict neutrality.
Then one way to test the null hypothesis of strict neutrality is to use the normalized difference
as test statistic.
Normalization is intended to minimize the effect of unknown
parameter(s) so that the resulting test is more rigorous.
Note that V ar(L1−L2) is a function of θ so its value needs
to be estimated.

Although every pair of statistics L1 and L2 can be used to construct a test as long as E(L1) = E(L2) and V ar(L1−L2) can be computed, such a test is useful only if the values of L1 and L2 are likely different when the locus under study depart from neutrality.
Unfortunately the distribution of a test of the form above is not well approximated by any standard distribution, so that obtaining critical values from a large number of simulated samples is commonly used, which means that the best way to apply such tests is to use a computer package that implement the test.
Therefore, we will focus on discussing the rational of several tests rather than detail of their computations.
Statistical tests using estimators of θ

Tajima test
• the parameter θ required for computing the variance is estimated by K/an.
/
/
n
n
K aD
Var K a

• Since K ignores the frequency of mutants, it is
strongly affected by the existence of deleterious
alleles, which are usually kept in low frequencies.
• In contrast, ∏ is not much affected by the
existence of deleterious alleles because it takes
the frequency of mutants into consideration.
• Therefore, a D value that is significantly different
from 0 suggests that the null hypothesis should
be rejected.
Rational of Tajima test

• When a population has been under selective sweeps (and population growth), K/an will likely be larger than ∏, resulting in negative value of D.
• When a population has been under balance selection (or population structure with sampling from many populations), K/an will likely be smaller than ∏, resulting in positive value of D.
Indication of Tajima’s D

• Neutrality: D=0
• Balancing Selection: D>0
– Divergence of alleles (π) increases • Purifying or Positive Selection: D<0
– Divergence of alleles decreases
• Also
– Bottleneck, D>0 (S decreases)
– Population expansion: D<0 (Divergence of alleles decreases: many low frequency alleles)
Tajima’s D Expectations

Tajima’s D Expectations
selective sweep
balancing selection
neutral
K
D = 0
K
Many low frequency variants and singletons,
D negative
q
Pairwise differences (k) increase faster than S
D positive

Distribution of Tajima’s D with θ = 5 and n = 100
Distribution of Tajima’s D

Fu and Li test
where the parameter required for computing variances is also estimated by K/an.

• Test D is preferred over D* whenever the size of singleton can be resolved, for example, by using an outgroup sequence or by the help of phylogeny reconstruction.
• The reasons for focusing on external or singleton mutations are as follows. – In the presence of natural selection, deleterious mutations are likely to
be eliminated from a population quickly or present in low frequencies. – In other words, deleterious mutations are usually recent mutations
and they are most likely to be found in the external branches of the sample genealogy, i.e., they are most likely external mutations or singletons.
– In contrast, mutations found in the internal branches are not as young, they are more likely to be neutral and their frequency is less affected by the presence of selection.
– Therefore, contrast between external and internal mutations, or contrast between singletons and non-singletons can be used to detect the presence of natural selection.
Rational of Fu and Li test

• Negative values of D and D* indicates an excess
of recent mutations or rare alleles (positive
selection and/or population expansion).
• Positive values indicates an excess of common
alleles (balance selection and/or population
structure).
Indication of Fu and Li D

Distribution of Fu and Li D and D*
Distribution of Fu and Li D and D* with θ = 5 and n = 100

Fay and Wu test
• Fay and Wu(2000) proposed a test which in our notation is
• ∏0,0 =θ K
• ∏1,1 =θ∏
• ∏1,0 =θΩ

Fay and Wu test

Fay and Wu test

Distribution of H’

• For proteins, two major categories of changes
are synonymous (KS) and non-synonymous (KA)
• The likelihood of synonymous vs. non-
synonymous change depends upon the
nucleotide codon position (first, second or third)
KS and KA
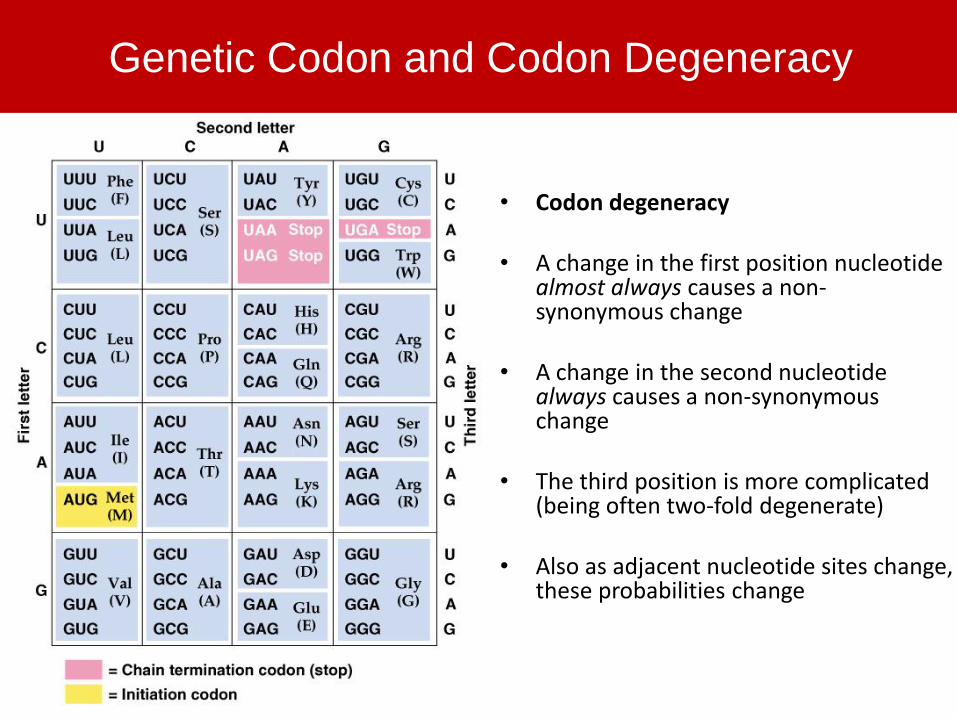
Genetic Codon and Codon Degeneracy
• Codon degeneracy
• A change in the first position nucleotide almost always causes a non-synonymous change
• A change in the second nucleotide always causes a non-synonymous change
• The third position is more complicated (being often two-fold degenerate)
• Also as adjacent nucleotide sites change, these probabilities change

• To get at these rates, need to classify nucleotide sites as
synonymous vs. non-synonymous
• KS = The number of synonymous changes divided by
the number of synonymous sites
• KA = The number of non-synonymous changes divided
by the number of non-synonymous sites
KS, KA

KA/KS test
►Neutral theory prediction if a non-syn. substitution is neutral.
►Neutral theory prediction if a non-syn. substitution is under purifying selection
►Selection theory prediction if a non-syn. substitution is under positive selection
1
1
1
N A
S S
N A
S S
N A
S S
d K
d K
d K
d K
d K
d K

► Tracks synonymous versus nonsynonymous substitutions
Fixed between species
►Non-synonymous NF
►Synonymous SF
Polymorphic within species (pairwise comparisons)
►Non-synonymous NP
►Synonymous SP
► NF/SF=NP/SP under neutrality ► More sensitive to detection of positive selection
McDonald-Kreitman Test

►Silent sites - always neutral - fix slowly - contribute to polymorphism
►Replacement sites – mainly unfavourable – if neutral, fix at same rate as silent and contribute to
polymorphism – proportion of replacement mutations that are neutral
determines dN / dS for polymorphism – if favourable, fix quickly and do not contribute to
polymorphism: higher dN / dS for fixed differences, lower rate for polymorphism
McDonald-Kreitman logic

Time to fixation: favorable and neutral

H0: All mutations are neutral.
Then, dN / dS for polymorphic sites should equal
dN / dS for fixed differences
H1: replacements are favoured. Favoured
mutations fix rapidly, so dN / dS for polymorphic
< dN / dS fixed
McDonald-Kreitman hypotheses

McDonald-Kreitman test
‘coding’ ‘non-coding’

Example of MK test: ADH in Drosophilia
Compare sequences of D. simulans and D. yakuba for ADH (alcohol dehydrogenase)
Fixed differences
Polymorphic sites
Replacement 7 2
Silent 17 42
% fixed 7 / 24 = 29% 2 / 44 = 5%
Significance? Use χ2 test for independence

Neutral polymorphism and divergence
D = 2m (low m )
t (in generations)
m m
Divergence
Polymorphism q = 4Nm low m, N
Polymorphism q = 4Nm high m N
D = 2m (high m)
Ratio D/q 2mt /4Nm
t/2N

The HKA test (Hudson-Kreitman-Aquade)
• Compares the level of polymorphism within species with the level of divergence between species – Expected level of polymorphism is estimated from the
level of divergence
– Ratio of polymorphism to divergence should be the same for all neutral loci and is set by the mutation rate for a locus
– Level of neutral divergence should be unaffected by occasional selective sweeps

The HKA test (Hudson-Kreitman-Aquade)
In the HKA test, the levels of polymorphism and
divergence in two or more loci are considered:
Locus 1 Locus 2
Polymorphisms P1 P2
Substitutions S1 S2
2
1 1
22
2
)(ˆ
)(ˆ
)(ˆ
)(ˆ
i
k
i i
ii
i
ii
SV
SES
PV
PEPX

The HKA test (Hudson-Kreitman-Aquade)
Divergence Between Species at Locus X
Variation Within Species at Locus X
Frequency-Dependent Selection Balancing Selection
Adaptive Divergence Selective Sweep
A Prediction of the Neutral Theory of Molecular Evolution
And Departures from Neutrality
Neutral Zone
Fixed Poly
Locus 1 50 5
330
HKA Test
Locus 2
Hudson Kreitman Aguadé test

Population Genomics
Population Genetics

Local adaptation (positive selection)
Functional restriction (negative selection)
Disease (negative selection)

Easy to distinguish the selection signals from demographic events.
Need not a prior knowledge of the gene function.
Population genomics approach
Joshua M. Akey
Genome Res. 2009 19: 711-722
A typical population genomics study design for detecting positive selection

Problem
We do not fully know the shape of the neutral
distribution and how it’s affected by other factors
such as demographic history.
Finding selective sweeps in
genomic (NGS) data
However, the best we
can do:
• use statistic based on
simulations
• apply it to empirical
genome-wide data sets
• Identify the loci in the
extreme tail
Most likely
candidate
of
selection

Old alleles:
• low or high frequency
• short-range LD
Positive Selection
Test based on the relationship between
allele frequency and extent of linkage disequilibrium
Young alleles:
• low frequency
• long-range LD (long haplotypes)
No Selection
Young alleles:
• high frequency
• long-range LD

The signal of selection
frequency
Lin
ka
ge
Dis
eq
uili
briu
m
(Ho
mo
zygosity)
Neutrality
Positive Selection

Long-range multi-SNP haplotypes
100%
Decay of homozygosity
(probability, at any distance, that any two haplotypes that start out the same have all the same SNP genotypes)
18%
gene
C/T A/G A/G C/T C/T C/T
Core markers
Long-range markers
G G
C
C
C
C
T
T
T
T
C
T
75% 35%
T T C
C
A G
Slide by: David Reich, Broad Institute

iHS: Measures the extent of haplotypes along alleles at a given SNP
EH
H
Genetic Distance
Ancestral Allele
Derived Allele
0.05
iHHA : iHH with respect to Ancestral core allele.
iHHD : iHH with respect to Derived core allele.

iHS Score
• Useful for variants that have not yet reached fixation.
• Large negative iHS: derived allele has swept up in frequency
• Large positive iHS: an ancestral alleles hitchhike with the selected sites.
• Hence, both cases are considered interesting!

• Footprints of natural selection could be detected by examining allele frequency spectrum and LD pattern
Exome sequencing data:
• KA/KS
• HKA
• MK
Whole genome sequencing data:
• Tajima’D
• LD-based or EHH-based approaches
Summary