genómica y mejora genética · 2019. 2. 5. · genómica y mejora genética...
TRANSCRIPT

Almería, 6 de febrero 2019
Jordi Garcia-MasIRTA, Centre de Recerca en Agrigenòmica CSIC-IRTA-UAB-UB
Genómica y mejora genética

- Introducción
- Secuenciación de DNA
- Secuenciación de genomas
- Anotación de genomas
- Descubrimiento de SNPs (GBS, resecuenciación de genomas)
- Genotipado de alto rendimiento
- GWAS, Selección genómica
- Edición génica

El paso de la vida nómada al sedentarismo durante el neolítico (aprox. 10,000 AC) favoreceel desarrollo de la agricultura
Origen de la agricultura
Pintura mural 1200 AC
La domesticación de les especies cultivadas sucede en diversos centros de origen

Domesticación de las especies cultivadas
Gewin, PLoS Biology 2003
Cong et al. Nature Genetics 2008

La domesticación y la selección provocan la pérdida de variabilidad genética
The funnel effect (Tanksley i McCouch 1997)

Tipos de variabilidad
- Intraespecífica: buscamos variabilidad dentro de la misma especie- Especies cercanas compatibles (ex. tomate y género Solanum) - Creación de nueva variación:
1. Inducida: colecciones de mutantes (EMS, rayos X…)2. Transgenes, edición génica
Búsqueda y creación de variabilidad
Domestic and wild tomatoes. L to R: Solanum lycopersicum, and wild relatives S. pimpinellifolium, S. habrochaites and S. pennellii. (Brad Townsley, UC Davis)

The process of Plant Breeding: use of markers
Search and creationof variability
Selection
EvaluationMultiplication &
commercialization
Variability assessmentGenetic distancePedigree validation
Marker-assisted selection
Hybrid purityFingerprintingBreeder’s rights enforcement

CHROMOSOME
Interesting region
Gene of interest

Pathogen inoculation
S R
DNA extraction
PCR
d= 0-2 cM
M2M1 G
Selección asistida por marcadores (MAS)
Uso de marcadores de DNA, estrechamenteligados a un carácter genético, como alternativa al fenotipado del carácter.
Asumimos el fenotipo a partir del genotipo.

Bevan and Uauy 2013
Markers in the genomics era

x
BC1
BC 6-8
P1 P2
Método del retrocruzamiento:selección para un solo marcador
F1

x
BC1
BC2 BC3
P1 P2
Whole genome selection:selección para muchos marcadores
F1

DNA sequencing technologies

- Maxam and Gilbert- Sanger- Next generation sequencing (NGS) 454, SOLID, Illumina- New technologies (single-molecule sequencing)

Maxam and Gilbert sequencing (1976)
- Chemical modificationof DNA and cleavage
- Radioactive labellingof DNA at 5’
- Technically complex

Sanger sequencing (1976)
- Enzymatic method
- Di-deoxynucleotides are used
- Automated

Sanger sequencing evolution

Automatic Sanger sequencing

• Advantages Long reads of aproximatelly 900 bp are obtained
Suitable for small projects
• Disadvantages Low throughput
Expensive
Automatic Sanger sequencing


Next generation sequencing technologies

Nature 437, 376-380 (15 September 2005)
Sanger vs NGS

Shendure and Ji 2008 Nat Biotechnol
Sanger vs NGS

Metzker 2010 Nat Rev Genetics
Clonally amplified templates

Direct detection: (IonTorrent)
pH changes due to release of H+
Fluorescence: (Illumina)
Nitrogenated base labeled
phosphate labeled
Pyrosequencing: (454-Roche)
Released pyrophosphates are transformed into light signal
Sequencing by synthesis and detection

Kahvejian et al 2008 Nat Biotechnol
DNA sequencing applications

Single-molecule templates
Helicos Heliscope and PacBio are new systems that rely in the sequencing of a single molecule: no amplification

Pacific Biosciences real-time sequencing
- Single molecule real-time sequencing (SMRT)- Nanotechnology- Long reads (10 kb), high error rate

• Single molecule
• Real time sequencing
• Polymerase fixed in a plate
• bp lengths 10 kb
• Phospholinked fluorescent Nucleotides
• Well size 70-100 nm (Roche 44 µm)
• 10 million wells
• 100 Gb/h, 15x human genome in 15 min
http://www.youtube.com/watch?v=v8p4ph2MAvI


Evolution of DNA sequencing

Genome sequencing

Genome Sequencing
• Genome sequencing: obtaining the order of nucleotides across agenome… but
• Genomes are large (hundreds to thousands of Mb) and DNAsequencing methods can handle only short stretches (hundreds of bp)of DNA at once… so
• Solution: Sequence small pieces and then use computers to assemblethem. Assembly of the reads into sequences represents acomputational problem.

Genome Sequencing: some terminology
Assembly: allocating sequenced fragments of DNA into theircorrect chromosomal positions.Contig: contiguous sequence of DNA created by assemblingoverlapping sequenced fragments of a chromosome.Draft sequence: genome sequence with lower accuracy thana finished sequence.Physical map: a map of the locations of identifiable markersspaced along the chromosomes.Scaffold: a series of contigs that are in the right order but arenot necessarily connected in one continuous stretch ofsequence.Shotgun sequencing: breaking DNA into many small pieces,sequencing the pieces, and assembling the fragments.

Genome sequencing
35
Short fragments of DNA
AC..GC
TT..TCCG..CA
AC..GC
TG..GT TC..CC
GA..GCTG..AC
CT..TG
GT..GC AC..GC AC..GC
AT..ATTT..CC
AA..GC
Short DNA sequences
ACGTGACCGGTACTGGTAACGTACA
CCTACGTGACCGGTACTGGTAACGT
ACGCCTACGTGACCGGTACTGGTAA
CGTATACACGTGACCGGTACTGGTA
ACGTACACCTACGTGACCGGTACTG
GTAACGTACGCCTACGTGACCGGTA
CTGGTAACGTATACCTCT...
Sequenced genome
Genome
break sequence
assemble

Strategies for genome sequencing
- Clone-by-clone (e.g. human)
- Break genome into many long fragments and clone
- Map each long fragment onto the genome and select clones
- Sequence each selected clone with shotgun
- Walking with clone-ends (e.g. rice)
- Break genome into many long fragments and clone
- Start sequencing some clones with shotgun
- Construct map as you go
- Whole Genome Shotgun (e.g. poplar)
One large shotgun pass on the whole genome

Clone-by-clone strategy
• Create a physical map, using hibridization,SNaPshot, or WGP (whole genome profiling) usingmainly BAC libraries.
• Select a minimum tiling path
• shotgun sequence each selected BAC clone
• assemble the individual BACs
BAC clone
mapgenome


Walking strategy
- Build a redundant library of BACs and sequence clone-ends (BES)
- Sequence seed clones
- Walk from seeds with clone-ends to pick clones that extend both sides
- A complex method
genome
BAC clones


Whole-genome shotgun (WGS)strategy
• Randomly ‘shotgun’ the genome and clone in libraries of different insert size (plasmid, cosmid, phage, BAC)
• Sequence each fragment
• Reassemble the reads
• No previous order is needed

Whole Genome Shotgun Sequencing
cut many times at
random
genome
forward-reverse paired
reads
plasmids (2 – 10 Kbp)
cosmids (40 Kbp) known distance
~500 bp~500 bp

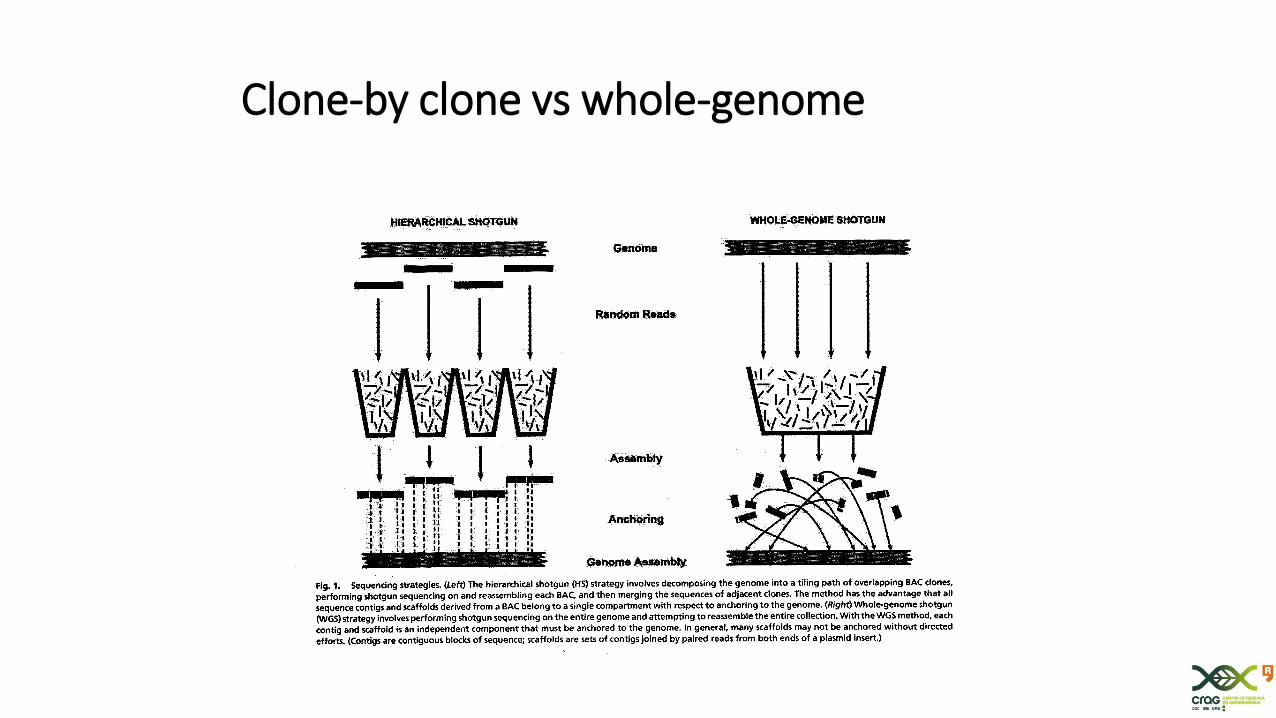
Clone-by clone vs whole-genome

Advantages and disadvantages
• Clone-by-clone and walking strategies:
easier assemblies, but a physical map is necessary. Redundancy in clone sequencing.
• Whole-genome shotgun:
No mapping and no redundant sequencing, but repeats are difficult to resolve

Fragment (or read) assembly and repeats are the main problems
• To construct the genome from the reads we find some difficulties:• Incomplete coverage. Leaves contigs separated by gaps
of unknown size.
• Sequencing errors.
• Unknown orientation of contigs
• Repeats: – Complex organisms have repetitive elements

many pieces
to assemble
High coverage:
Assembly: how much sequencing?
Low coverage:
A few pieces
to assemble
a few contigs,
a few gaps
many contigs,
many gaps
Input OutputLander and Waterman,
1988

Main steps to assemble a genome

Now we have NGS sequencingtechnologies
• We can use the above mentioned methods (clone-by clone sequencing or whole-genome shotgun) but instead of using Sanger, we can use 454, Illumina or hybrid approaches

Minimum tilling path (MTP)
BAC library
BAC pooling
Shotgun sequencingand assembly
BAC-to-BAC sequencing strategy

Shotgun sequencingand assembly
Whole genome shotgun sequencing strategy
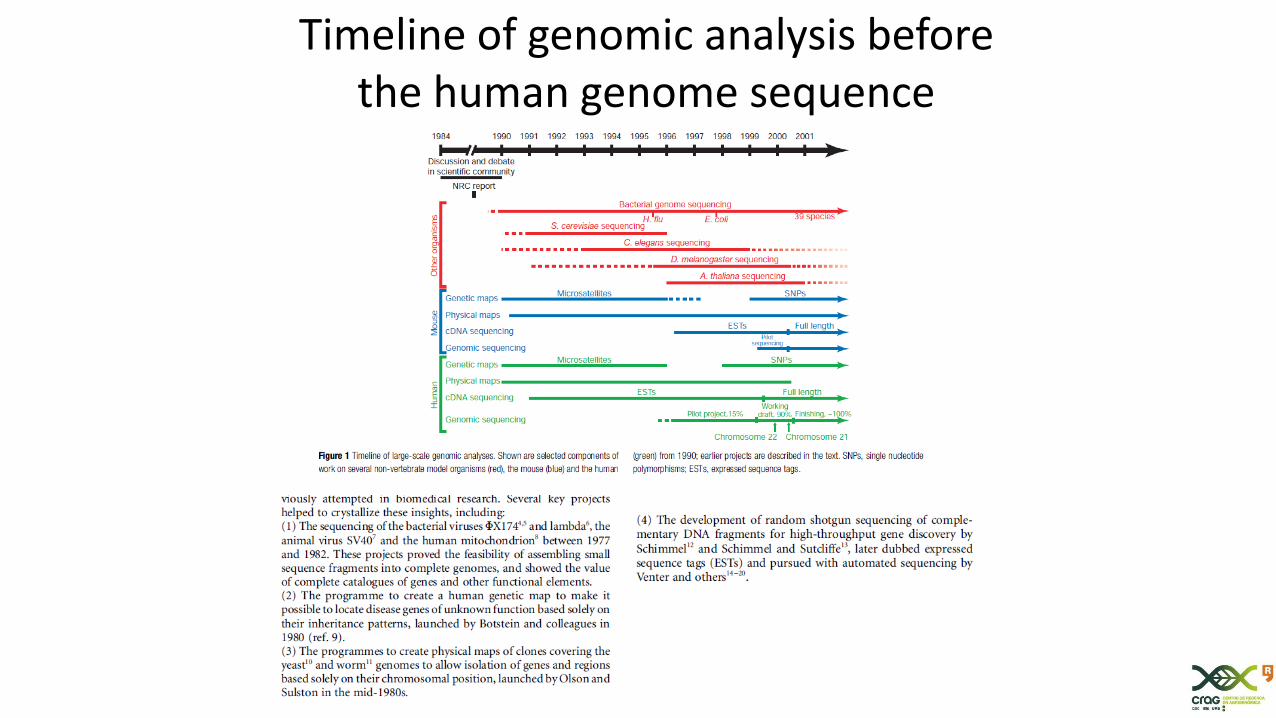
Timeline of genomic analysis beforethe human genome sequence

2009: Illumina
40-50K$
Secuenciación del genoma humano
2001: Human Genome Project
2.7G$, 11 years
2001: Celera
100M$, 3 years
2007: 454
1M$, 3 months
https://www.genome.gov/27565109/the-cost-of-sequencing-a-human-genome/

Many plant genomes available

The challenge to efficiently manage the
amount of generated data:
bioinformatics
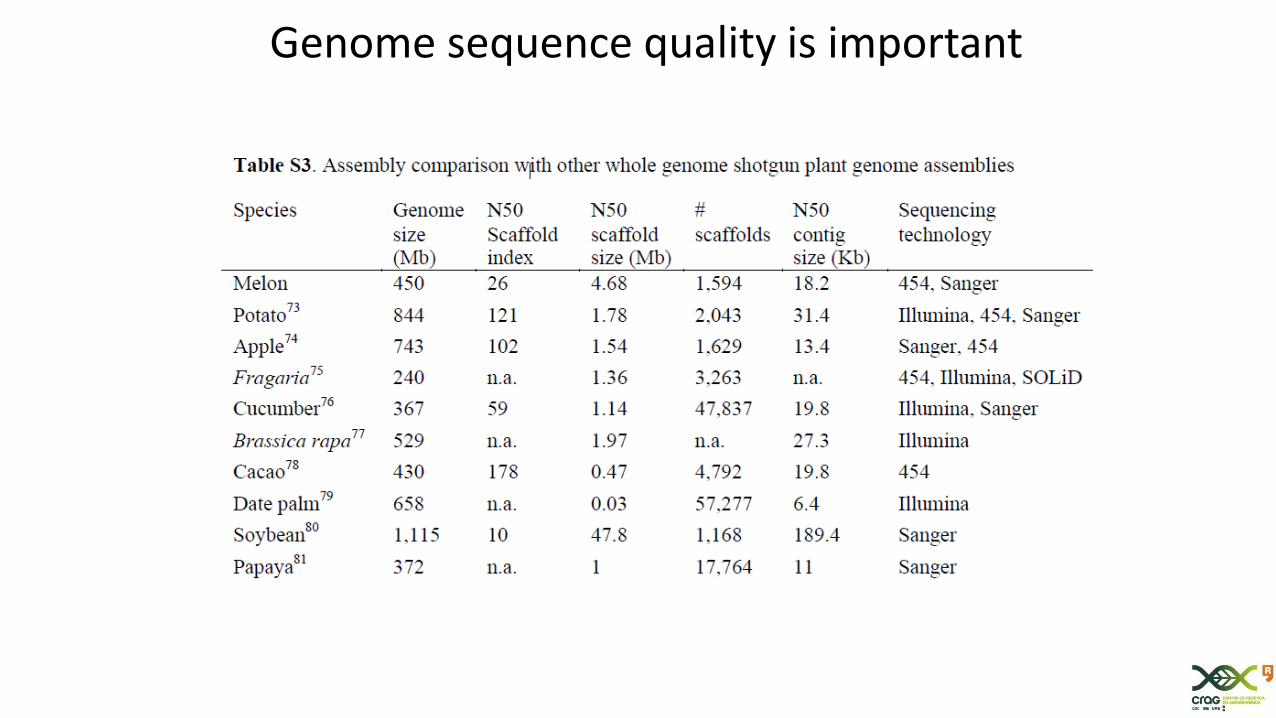
Genome sequence quality is important

Genome annotation

• Annotation: attachment of biological information to sequences
• Genome annotation
Structural: placing functional elements in the genome as ORFs, gene structures, regulatory elements
Functional: attaching biological information to these elements
• Either automated and/or manual
Genome annotation

Cruz et al. GigaScience 2016
https://www6.inra.fr/decodage/layout/set/print/TriAnnot
Pipelines for genome annotation

Sequencing the DHL92 melon genome
Shotgun Paired Ends (3, 8, 20 kb)
Assembly with Newbler 2.5
BAC-end sequences
12 x 6 x
DHL92
Ti
0.06 x (50% paired)
Etiolated plants
nuclei purification
Garcia-Mas et al. PNAS (2012)

- 19.7 % of the assembled genome corresponds to transposon sequences (probably an underestimation)
Garcia-Mas et al. PNAS (2012)www.melonomics.net
A: Physical mapB: ncRNA distribution (orange)C: predicted genes (green)D: transposable elements (blue)E: NBS-LRR genes (brown)F: duplicated blocks
0
10
20
30
40
50
60
70
0 1 2 3 4 5 6 7 8 9 10 11
nu
mb
er
of co
pie
s
million years
- The melon genome has expanded the TE content after the split of the melon/cucumber ancestor 11 My ago. This may, in part, explain the genome size difference between cucumber (350 Mb) and melon (450 Mb) - No recent WGDs were found
Insertion time of melon LTR retrotransposons
- We predicted 27,427 genes in the melon genome,encoding 34,838 transcripts and 32,487 proteins- 69 % of gene predictions are supported bytranscript or protein alignment
Sequencing the DHL92 melon genome

Genome anchoring to chromosomes: pseudomolecules
- F2 map with SNP usingIllumina GoldenGate assay
- 98.2 % of the scaffoldassembly anchored to the 12melon LGs (355 Mb)
Garcia-Mas et al. PNAS (2012); Argyris et al . BMC Genomics (2015)
Number of scaffolds
% assembly
- N90 index is 78 scaffolds.N50 scaffold size is 4.6 Mb

Assignment of chromosomes to the genome sequence
- The karyiotype of “Piel de Sapo” was obtained- FISH allowed assigning each melon chromosome with the corresponding genome assembly- Centromere-specific repetitive sequences identified the putative centromeric regions
Argyris et al. BMC Genomics (2015)

High synteny between melon and cucumber (Cucumis sativus)
Garcia-Mas et al. PNAS (2012)
- The melon genome has expanded in pericentromeric regions, probably due to TEamplification, when compared to cucumber
- Synteny exists among cucurbit genomesbut it is complex after extensive rearrangements. Dysploidy in cucumber2n=2x=14 (Yang et al Plant J 2014)
Guo et al. Nat Genet (2012)

The Tomato Genome Consortium, Nature (2012)
The tomato genome

Improved maize genome sequence
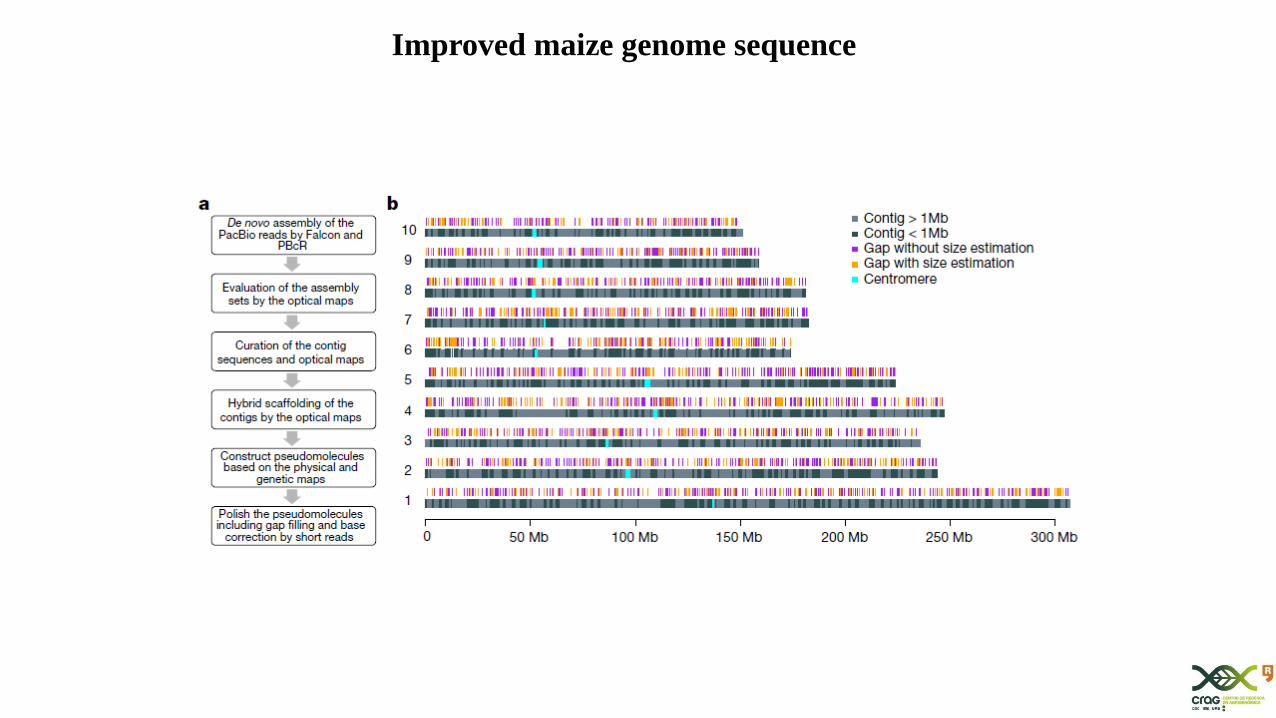
Improved maize genome sequence

Fast de novo genome sequencing

Improved melon genome sequence
(under analysis)

SNP discovery

Method for SNP identification
PrerequisitesCurrent false
discovery rate (%)Specifics, limitations
EST sequence dataLarge number of available EST-sequences
15-50
Dependent on the expression level or need for normalized libraries, difficulties in the discrimination of orthologous from paralogous sequences, low sequence quality
Array analysisUnigene sets based on EST-sequences, array technology
>20Not all SNPs identified, large genomes require complexity reduction
Amplicon resequencing
Unigene sets based on EST-sequences, amplification primers for many individual genes
<5
High reliability but costly, detailed haplotype analysis possible, many lines can be compared, allele frequency data with pools of DNA
No genomic sequence and next-generation sequencing technologies
Novel sequencing technologies, complexity reduction methods, bioinformatic tools
15-25
Generates large amounts of data, costly bioinformatics, false discovery rate for genomes without full sequence is relatively high
Genomic sequence is available either through conventional sequencing or next-generation sequencing
Reference genome, bioinformatic tools
<5-10
Small genomes can be fully sequenced and compared for SNPs, for large genomes targeted approaches will be necessary (e.g. exon capture and multiplex amplification)
Ganal et al. (2009) COPLBI,621Summary and comparison of SNP identification methods.

in silico SNPs in databases
in silico SNP
contigESTs
pSNP (for putative SNP)
Definition: sequence variation in silico that is present
in at least two ESTs between at least two varieties.

Verification of pSNP

SNP discovery in the -omics era
1. SNP discovery through transcriptome sequencing
2. SNP discovery through partial genome sequencing (GBS)
3. SNP discovery through genome resequencing

Genotyping by sequencing

Methods to reduce genome complexity
Simultaneous marker discovery and genotyping. For large genomes,
complexity has to be reduced prior to perform genotype by sequencing
We can use target enrichment (sequence capture, long range PCR,
molecular inversion probes) or Restriction enzymes
Use of sequence capture of gene regions avoids screening non-coding
regions of DNA, which may be a problem
image from Cornell University webpage

• Reduced-representation sequencing
i. reduced-representation libraries (RRLs)
ii. complexity reduction of polymorphic sequences (CRoPS)
• RAD-seq
• Low coverage genotyping
i. Multiplexed shotgun genotyping (MSG)
ii. genotyping by sequencing (GBS)
• RNA-seq
• Sequence capture
Davey et al. (2011) Nature Reviews
Types of NGS-based reduction of genome complexity

Davey et al. (2011) Nature Reviews
Platform specific

Reduced-representation libraries (RRL)
Fragments digested with frequently cutting
restriction enzymes are pooled, selected by size
and sequenced
Partial but genome-wide sequence data. Typically
used to sequence pools of DNA samples, not
allowing the detection of the sequence of each
individual. Each marker is sequence at high
coverage enabling markers to be genotyped
accurately across many individuals
Complexity reduction of polymorphic
sequences (CoRPS)
By adapting the amplification of AFLP fragments to
enable sequencing on the Roche sequencing
platform
First method to identify polymorphisms in each
individual sample by incorporating short barcode
identifier sequences into the barcodes on the
Roche platform. Each marker is sequence at high
coverage enabling markers to be genotyped
accurately across many individuals
RAD-Seq
RAD-seq sequences short regions surrounding the
restriction sites for a given restriction
endonuclease. Restriction fragments are randomly
sheared to a length suitable for the platform
chosen. Selective PCR is used to amplify for
sequencing only those fragments containing a
restriction site
Samples can be individually barcoded. Each
marker is sequence at high coverage enabling
markers to be genotyped accurately across many
individuals
Genotyping by sequencing (GBS)
Involves digestion of genomic DNA with frequent
cutter and sequencing of the ends of all resulting
restriction fragments. Adaptors containing
barcodes and common adaptors without barcodes
are mixed and used in the ligation process. Only
fragments with both types of adaptors or with
appropriate size will be sequenced. Thus, fragment
reduction occurs during the ligation process
Samples can be individually barcoded. Targeted
markers are sequenced at low coverage and not all
genotyped in all individuals. o missing data can be
inferred based on probabilistic calculations.
Suitable for mapping.
Multiplexed shotgut genotyping (MSG)Similar to GBS but with only one adaptor.
Fragments are size-selected before sequencing
Samples can be individually bar-coded. Targeted
markers are sequenced at low coverage and not all
genotyped in all individuals. Physical position of
markers known so missing data can be inferred
based on probabilistic calculations. Suitable for
mapping.

- We target a reduced representation of a genome for analysis.
- Discovery and genotyping of thousands of markers in a single step.
- Can be applied to species with no genetic information available.
- Directly applied to GWAS.
- Methods developed for high-throughput genetic marker (SNP) discovery often include:
1. Digestion of multiple samples of genomic DNA with RE
2. Selection or reduction of the resulting restriction fragments
3. NGS of the final set of fragments (<1kb size)
- Use of methylation sensitive REs reduces repetitive regions
Genotyping by sequencing

Schematic diagram of a GBS procedure
Deschamps et al (2012) Biology

GBS 2011
GBS in Buckler’s lab

image from Cornell University webpage

GBS library construction


IBM maize mapping population: parents + 276 RILs
200,000 markers mapped
Barley Oregon Wolf (OWB): parents + 43 DHLs
25,000 markers mapped
GBS original paper

Resequencing and RNA-seq

Utilització de Next Generation Sequencing (NGS)
SNP discovery from RNAseq
454 GS FLX400 Mb (1.000.000 sequences of 700 bp per run)

RNAseq of 8 tomato varieties
RNA extraction
cDNA synthesis
High throughput sequencing 454 Titanium
SNP detection
High throughput genotyping (Fluidigm, KASPar)
Unigenes total 191122 Newbler
69 MIRA
SNPs total 618445 Newber
169 MIRA
Label Reads %
Re
gio
n1
LB0502 29465 6,62
LB0417-5-2 63819 14,33
LB0757 38903 8,73
A0124-9-1DA 75474 16,95
Re
gio
n2
B0422-2-1 54026 12,13
BDU0303-11-1 57610 12,94
A908-8 55129 12,38
LB0801 49952 11,22
Unidentified 21000 4,72
Total 445378 100

- The zucchini genome is not available. Alternatively, a transcriptome is available, which was used as a reference.
- RNA from 7 zucchini breeding varietes was sequenced using Illumina Hi-Seq2000 in a Flow-cell. RNA from fruit, leaf and flower.
SQUASH LINES BARCODEREAD
LENGTHAPPLICATION
A000422R K445 2X75 pb mRNAseq
A020351R K446 2X75 pb mRNAseq
A020231R K447 2X75 pb mRNAseq
A00081 K448 2X75 pb mRNAseq
LA701 K449 2X75 pb mRNAseq
A030362VH K450 2X75 pb mRNAseq
P0220 K451 2X75 pb mRNAseq
A000422R K445 2X75 pb mRNAseq
RNAseq of 7 zucchini varieties

- Remove low quality reads- Remove adaptors- Mapping against public 49,000 zucchini unigenes- SNP calling- Filtering no intron in a 60 bp window: 24,466 - Filtering read depth, SNP quality: 4,558- Filtering 3 SNPs/unigene: 2,719
Zucchini SNP mining

A problem when discovering SNPs from transcriptome data is the presence of introns
Software is available to predict introns: Intron Finder
Or BLAST against the reference genome, if available
An experimental validation is necessary
Genes have introns...

Reduced-representation or whole-genome sequencing?
Whole-genome sequencing feasible in species with small genomes
In larger genomes, reduced-representation methods are nowadays more convenient. Many research questions can be answered with a small set of markers and the whole genome sequence is not necessary
GBS or whole genome sequencing?

- Once we have a reference genome sequence, resequencing individualsis not complex.
- Illumina is the best option, regarding cost and throughput.
- PE sequencing to low coverage is the choice (10-20 x), depending inthe number of genotypes, genome size and application.
- PE reads are between 76-150 bp.
- Reads are mapped to the reference genome, using BWA, Bowtie orothers, generating BAM/SAM files.
- We can visualize the read alignments with different software, as IGV.
Resequencing

Mapped read visualization with IGV

- Variants (SNPs, short indels and structuralvariations (SVs)) between individuals
- SV (> 50bp) include CNV (copy-numbervariation), indels and duplications.
Genetic variation observedafter resequencing

Variant browser

Lai et al 2010 Nat Genet
- 6 maize elite inbred lines sequenced.- 1 milion SNP, 30,000 indel, hundreds ofpresence/absence genes

Lai et al 2010 Nat Genet
Pedigree analysis of maize inbred lines

Chia et al 2012 Nat Genet
- 103 lines of pre-domesticated and domesticated maize- Relative genus Tripsacum is included- 55 milion SNPs

Jiao et al 2012 Nat Genet
- 278 maize inbreds- Modern breeding afected thousands of genes and non-genicregions. Reduction in nucleotide diversity and increase of rarealleles.

Hufford et al 2012 Nat Genet
- 75 wild, landraces and improved maize lines- Domestication is studied and genes with signals of selectionare identified

SNP discovery for cucurbit breeding: cucumber
Cs CUCUMBER LINES TYPE LANEMULTIPLEX
INDEX
READ
LENGTHAPPLICATION
Cs1 A 202-3-1 Holandés 1 1 2x100 WG-Seq
Cs2 A 801-7 Holandés 1 2 2x100 WG-Seq
Cs3 P 501 Francés 1 3 2x100 WG-Seq
Cs4 A 0313-8-2-1 Mini verano 1 4 2x100 WG-Seq
Cs5 A 0318-7-4-1 Mini verano 2 5 2x100 WG-Seq
Cs6 A 808-1-1 Mini invierno 2 6 2x100 WG-Seq
Cs7 A 910-3-2 Mini invierno 2 7 2x100 WG-Seq
Cs8 A 919-2-4 Cornichon 2 8 2x100 WG-Seq
- There is a need of SNPs for MAS that are polymorphic in the narrow germplasm used in breeding- As the cucumber genome is available (Huang et al 2009), 8 cucumber breeding varietes were re-sequenced using Illumina Hi-Seq2000 at 25X (2 Flow-cell lanes, MIDS used)

- Remove low quality reads- Remove adaptors- Mapping against reference genome- SNP calling- Filtering SNPs Phred>20, heterozygous, SNP in all lines: 557,110- Filtering at least 8 reads in each line: 407,753- Filtering no SNPs in 50 bp windows: 20,501
Cucumber SNP mining

Cucumber SNP validation
483 SNPs evenly distributed in the genome were used for validation using Kaspar chemistry
93% of the SNPs were experimentally validated
SNPs now used in breeding programs routinely

- Resequencing of 7 representative varieties with Illumina paired-end sequencing (35x106 paired reads/sample, read
length 150 bp, 22x coverage, 10 Gb/sample)
- 4,556,377 SNPs and 718,832 small insertion/deletions (DIPs ) were called
- Structural variation (SV) and transposable element (TE) polymorphisms were also called
- TE polymorphism analysed with Jitterbug
Exploring natural variation using genome resequencing
Trigonous (India)
C-836 (Cabo Verde)
PI 124112 (India)
C-1012 (Irak)
PI 161375 (Korea)
Védrantais (France)
Piel de sapo (Spain)
Sanseverino et al. 2015

Non-uniform distribution of diversity across the genome
Sanseverino et al. 2015

SV and TE polymorphism
Sanseverino et al. 2015

SNP discovery
PI 161375 Piel de sapo T111
Sanseverino et al. 2015

SNP genotyping
technologies

• CAPS• Denaturing HPLC (DHPLC)• Pyrosequencing• SNaPshot• Taqman• Molecular beacons• High resolution melting (HRM)• SNPlex• Illumina GoldenGate and Infinium• Kaspar• Fluidigm • Microarray Single Feature Polymorphisms (SFPs)
… and many others
SNP• SNP detection systems, many available

• Single-base extension (SBE)
– Sequenom (MS), SNaPshot
• Allele-Specific Extension
– Illumina
• Primer Extension
– Pyrosequencing
• Differential Hybridization
– Taqman, Affymetrix/GeneChip
SNP detection methods: type of detection

Taqman
- 5’-3’ nuclease assay: activity of the enzyme Taq-polymerase and fluorophore-based detection.-Two specific primers targeting the region flanking the SNP and two TaqMan fluorescent probes with a Minor Groove Binder (MGB) (quencher).- Single SNP assay

High resolution melting (HRM)
- High Resolution Melting Analysis is based on PCR melting
(dissociation) curve techniques.
- High Resolution Melting Analysis (HRM) is a post PCR method.
- The region of interest is first amplified using PCR. During this process, special
saturation dyes are added to the reaction, that fluoresce only in the
presence of double stranded DNA. Such dyes are known as Intercalating dyes
(eg SYBR green)
- When melting dsDNA, fluorescence fades away

High resolution melting (HRM)

BioMark™ (Fluidigm)
Each 96.96 Dynamic Array is capable of producing 9,216
real-time qPCR data points using just 1/200th the amount of
reagents.
- SNPtype assays available- Compatible with other chemistries as Taqman or Kaspar

Fluidigm SNPtype assays
- Allele specific PCR- Suitable for multiplexing medium number of SNPs (48-96)

Illumina genotyping
http://www.youtube.com/watch?v=lVG04dAAyvY

• Two competitive, allele-specific, tailed forward
primers and one common (reverse) primer
• Proprietary FRET cassette for fluorescent (FAM, VIC)
signal generation
• Uses passive reference dye (ROX)
• Two components: a SNP-specific assay mix (primers)
and reaction mix (everything else)
• Includes specially-developed KTaq polymerase
• Uses same equipment as TaqMan®
KASP chemistry
https://www.youtube.com/watch?v=_S0m2PrwPdE

End-point Analyser
KBioscience-LIMS
KBioscience Assay Dispenser
96/384 DNA stamping
KBioscience Laser Sealer KBioscience waterbath PCR
Projects/routine
SNPline components

Genome-wide association
studies (GWAS) and
Genomic selection

Definition of GWAS
A genome-wide association study (GWAS) is defined as any study of genetic variation across the entire genome that is designed to identify genetic associations with observable traits.
• Whole genome information combined with phenotypic data has the potential for understanding of basic biological processes.
• High-throughput, cost-effective methods for genotyping are providing powerful research tools for identifying genetic variants that contribute to interesting agronomic traits.
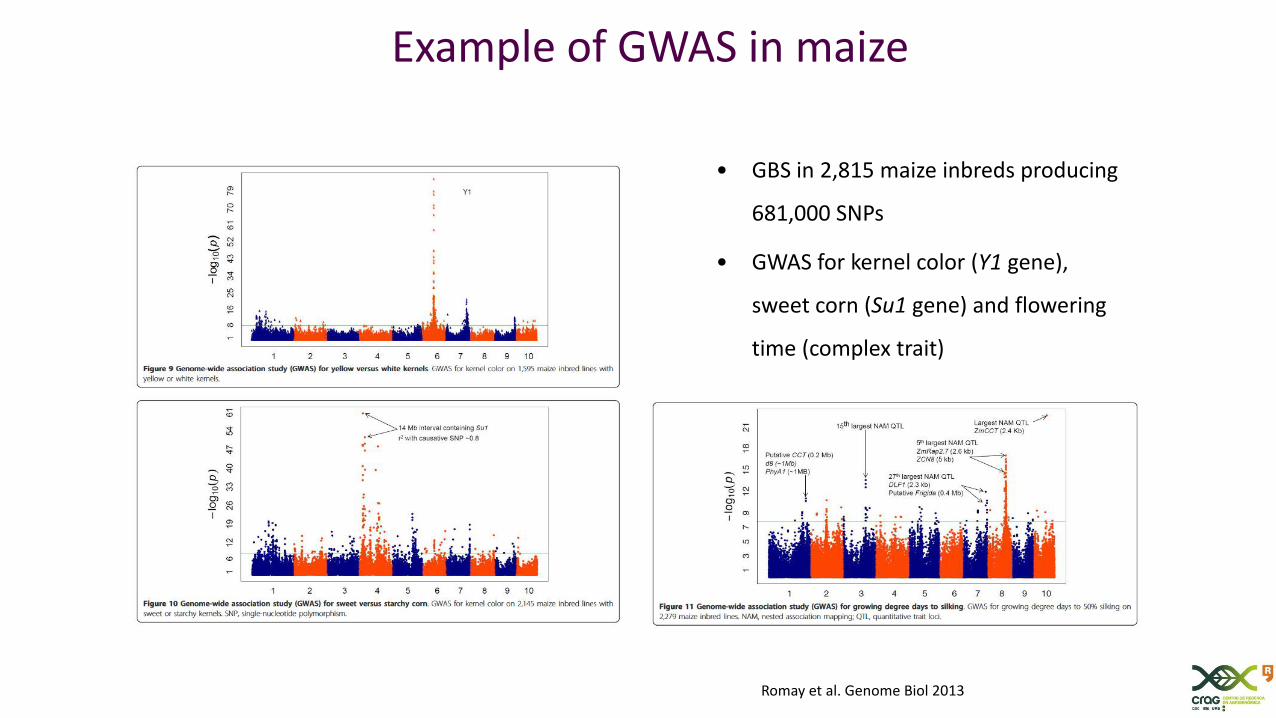
• GBS in 2,815 maize inbreds producing
681,000 SNPs
• GWAS for kernel color (Y1 gene),
sweet corn (Su1 gene) and flowering
time (complex trait)
Example of GWAS in maize
Romay et al. Genome Biol 2013

Example of GWAS in tomato
• Resequencing of 360
accessions
• GWAS for pink color (Y gene)
using 231 accessions
• Association with a SNP 8 kb
upstream of the SLMYB12 gene
Lin et al. Nat Genet 2014
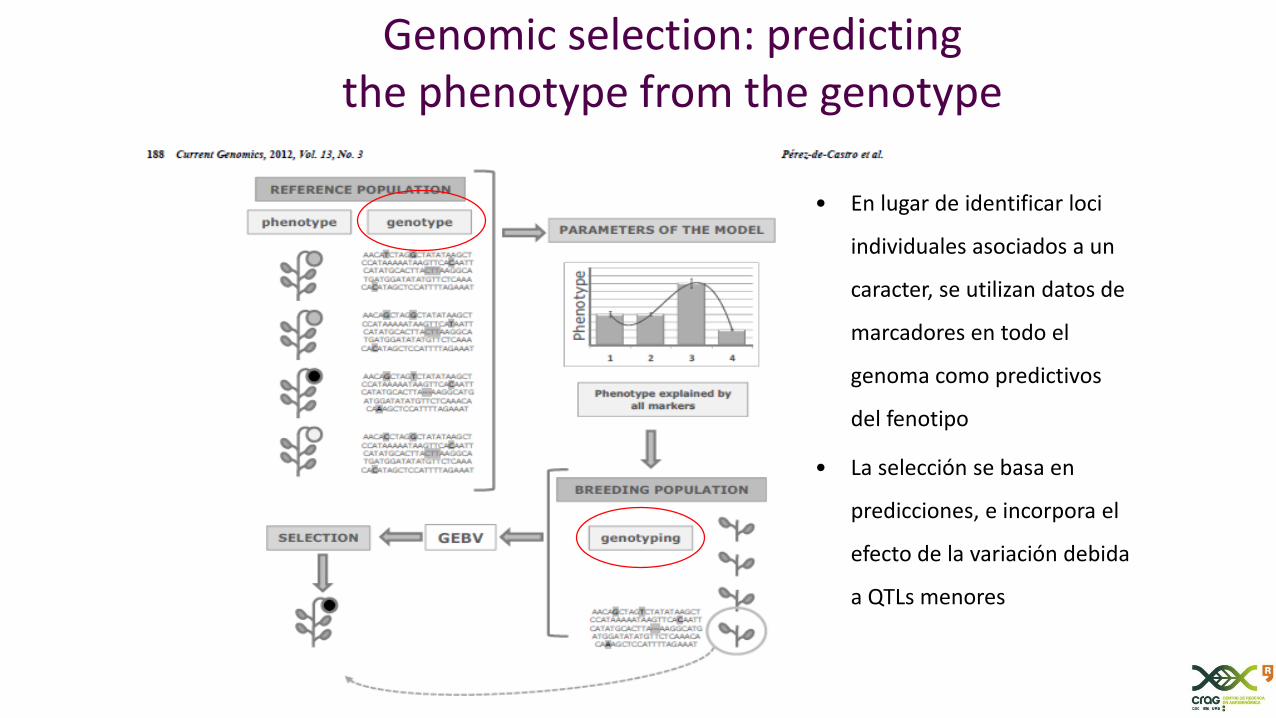
Genomic selection: predicting the phenotype from the genotype
• En lugar de identificar loci
individuales asociados a un
caracter, se utilizan datos de
marcadores en todo el
genoma como predictivos
del fenotipo
• La selección se basa en
predicciones, e incorpora el
efecto de la variación debida
a QTLs menores

Genomic selection pipeline

Gene editing

Gene editing
Precision removal or change of a few nucleotides
- Oligo directed mutagenesis (ODM)- Site-directed nucleases (SDN)
- Zinc finger mucleases- Meganucleases- TALENs- CRISPR/Cas9

Hsu et al. Cell 2014
The endogenous DNA repair mechanism

Natural mechanisms of microbial CRISPR

Belhaj et al. Curr Op Biotechnol 2015
Engineering the CRISPR editing tool

Ding et al. Front Plant Sci 2016
Cas9/gRNA genome editing

Belhaj et al. Curr Op Biotechnol 2015
Pipeline for CRISPR/Cas9 editing in plants

CRISPR/Cas9 genome editing ofcucumber eIF4E

Some examples

Virus disease resistance in cucumber
2) BSA + NGS : resequence of 15 R and 15 S homozygous families from F3 population
Strategies for the development of linked markers
1 ) SNP mining from resequenced parentals lines, mapping resistance in F2 population; gene mapped in chr5
Resistant parental x Susceptible parental
F1
F2 population (135 pl)
F3 population (135 families) pathology test(1 dominant gene)

Bulk segregant analysis combined with a next generation sequencing strategy
- Selection of 15 R homozygous (bulk R)+ 15 S homozygous (bulk S) DNA extraction
- Resequencing of parental lines + 30 individuals tagged (12 x coverage)
- Bioinformatic analysis and SNP mining between R and S bulks
15 R 15 S
2 parentals
Mapping reads against cucumber genome and filtering
~1,400,000 SNPs
Quality and coverage
1,214,344 SNPs
Not against ref genome & 30 individuals genotyped
129,301 SNPs
1,133 SNPs
SNPs bulk R against bulk S
All SNPs in Ch5 in a 794 Kbinterval (containing 64 genes)
Selection of 48 SNPs in the interval and genotyping of 135 F3 families. A singlecandidate gene identified.
A set of markers for MAS is available

- eth3.5 and eth6.3 induce climacteric ripening
independently.
- Both QTLs interact in SC3-5-1 to induce precocity in fruit
ripening.
- SC alleles in the PS background partially recover the
capacity to produce ethylene.
8M35 8M318M40
PS SC
PS and SC are non-climacteric, but two climacteric NILs were found
Moreno et al. TAG (2008); Vegas et al. TAG (2013)
eth3.5 + eth6.3
eth3.5eth6.3
PS

Mapping of eth6.3
Vegas et al TAG (2013)
≈ 2,8 Mbp
eth6.3 lies in the centromeric region of LGVI, a region with low recombination rate

SNP1 SNP2
SNP1 SNP2 Recombinants
B H 10
A H 6
H B 6
H A 5
Total 27
Fine mapping of eth6.3
- Recombinant search in 1,400 F2 individuals withflanking markers: 27 recombinants

eth6.3: NAC domain-containing protein
≈ 2,8 Mbp
Fine mapping of eth6.3 resulted in the identification of a candidate gene
139 kb interval containing 5 annotated genes
Gene
mRNA
SNP1
SNP3 SNP2
SNP2SNP1 SNP3

Validation of eth6.3
- In a climacteric genetic background TILLING population (URGV, France), 6,200 families were screened. 17 mutants identified: 5 non-coding regions, 4 silent, 8 non-synonymous.
Phenotyping in 2014 and 2015 revealed an increased time period from pollination to dehiscence and to change of external fruit color in mutated fruits in two families, confirming the role of this gene in climacteric ripening.
