genomforschung und sequenzanalyse einführung in methoden ... · otu 1 cgaga c otu 2 agcga c otu 3...
TRANSCRIPT

1
1
Phylogenie IIPhylogenie II
WS 2006/2007
Genomforschung und Sequenzanalyse
Einführung in Methoden der Bioinformatik
Bernhard Lieb &Tom Hankeln
MolekulareMolekulare
2
Themen
Grundlagen und Begriffe der molekularen Phylogenie
Evolutionsmodelle
Berechnung
PrüfungStammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen

2
3
Charakter-orientierte Methoden
1. Maximum Parsimony (MP)
2. Maximum Likelihood (ML)
4
Maximum Likelihood
Zum ‘ersten’ mal von Cavalli-Sforza and Edwards (1967) für ‘gene frequency data’ benutzt, später (1981) von Felsenstein für DNA Sequenzen
L = P(D|H)
Wahrscheinlichkeit der Daten, gegeben eine Hypothese
Die Hypothee ist eine Baumtopologie, dessen Astlängen und ein Evolutionsmodell unter dessen
‘Präsenz’ die Daten evolvierten
““The explanation that makes the observed outcome the most likelyThe explanation that makes the observed outcome the most likely””

3
5
Probability and likelihood
Die Wahrscheinlichkeit Wahrscheinlichkeit liegt immer zwischenNIE (0) NIE (0) und SICHER (1).SICHER (1).
Die bedingtebedingte WahrscheinlichkeitWahrscheinlichkeit ist die Wahrscheinlichkeit eines Ereignisses A, gegeben das
Ereignis B: P(A|B)P(A|B).
“...if ‘probabilityprobability’ allows us to predict unknown outcomes based on known parameters P(H|D)P(H|D), then
‘likelihoodlikelihood’ allows us to determine unknown parameters based on known outcomes P(D|H)P(D|H)…”.
6
Likelihood
Daten: KKZKZKKZZZ
• Ereignisse sind unabhängig
• Alle ‚Kopfwürfe‘ besitzen gleiche ‚unbekannte‘ Kopfwahrscheinlichkeit p
Hypothese
=> Likelihood L(D|H) = pp(1-p)(1-p)p(1-p)pp(1-p)(1-p)(1-p)
keine Verteilung, Plot der selben Daten (KKZKZKKZZZ) gegen verschiedene Werte von p (Hypothese)
alles wird getestet (gerechnet)
=> Mit welcher Kopfwahrscheinlichkeit p bekomme ich am ehesten diese Daten?
L = P(L = P(DatenDaten | | Hypothese Hypothese ))
0,0 0,2 0,4 0,6 0,8 1,0[p]
Like
lihoo
d

4
7
Maximum LikelihoodL = P(data|hypothesis)
• Wahrscheinlichkeit der beobachteten Daten(Sequenzen!) im Lichte der Hypothese(Stammbaum).
• d.h, es wird der Stammbaum errechnet, der die beobachteten Daten (also die alignierten Sequenzen) am besten (unter der Annahme des Modells) erklärt.
Evolutions-
modell
Ein Baum wird generiert und man prEin Baum wird generiert und man prüüft dann ob die gegebenen Daten den Baum generieren kft dann ob die gegebenen Daten den Baum generieren köönnennnen
8
Maximum Likelihood
• benötigt ein explizites Evolutionsmodell• Parameter werden aus Daten + Modell
errechnet.• Explizite Verbindung Daten + Modell +
Stammbaum.• aber: schlechtes Modell => schlechter
Stammbaum• Alternative Stammbäume lassen sich testen
=> keine Methode extrahiert mehr Information aus den Daten; aber: sehr rechenintensiv
5
9
Maximum Likelihood
Evolutionsmodelle
Für DNA-Sequenzen:=> JC, K2P, F81, HKY, REV
Für Protein-Sequenzen:=> PAM, BLOSUM, JTT, WAG ...
10
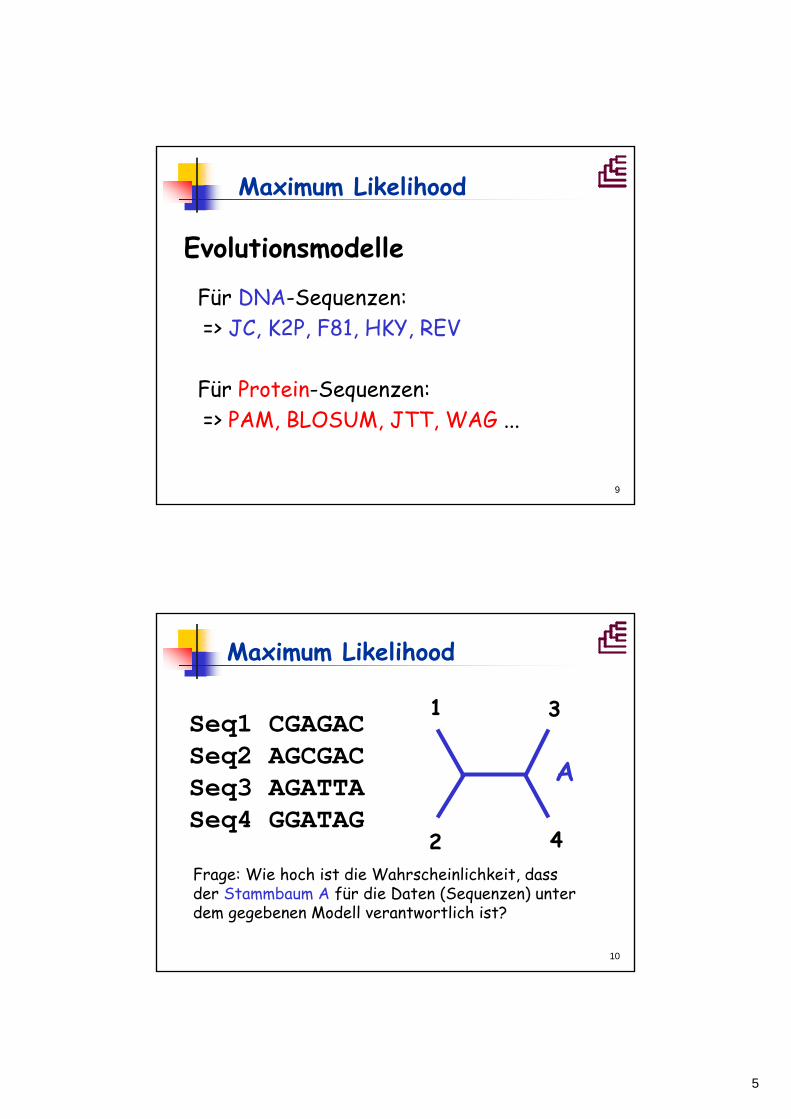
Maximum Likelihood
Seq1 CGAGACSeq2 AGCGACSeq3 AGATTASeq4 GGATAG
1
2
3
4Frage: Wie hoch ist die Wahrscheinlichkeit, dass der Stammbaum A für die Daten (Sequenzen) unter dem gegebenen Modell verantwortlich ist?
A
6
11
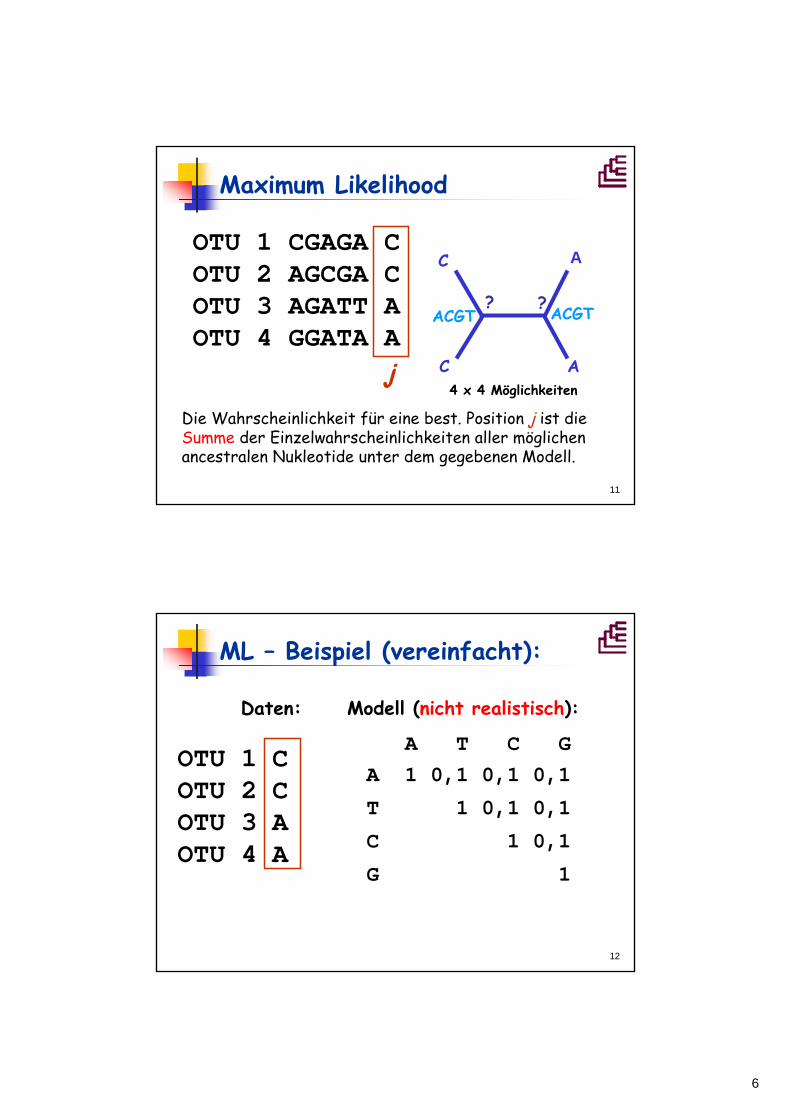
Maximum Likelihood
OTU 1 CGAGA COTU 2 AGCGA COTU 3 AGATT AOTU 4 GGATA A
j
ACGT??
C
C
A
A
ACGT
4 x 4 Möglichkeiten
Die Wahrscheinlichkeit für eine best. Position j ist die Summe der Einzelwahrscheinlichkeiten aller möglichen ancestralen Nukleotide unter dem gegebenen Modell.
12
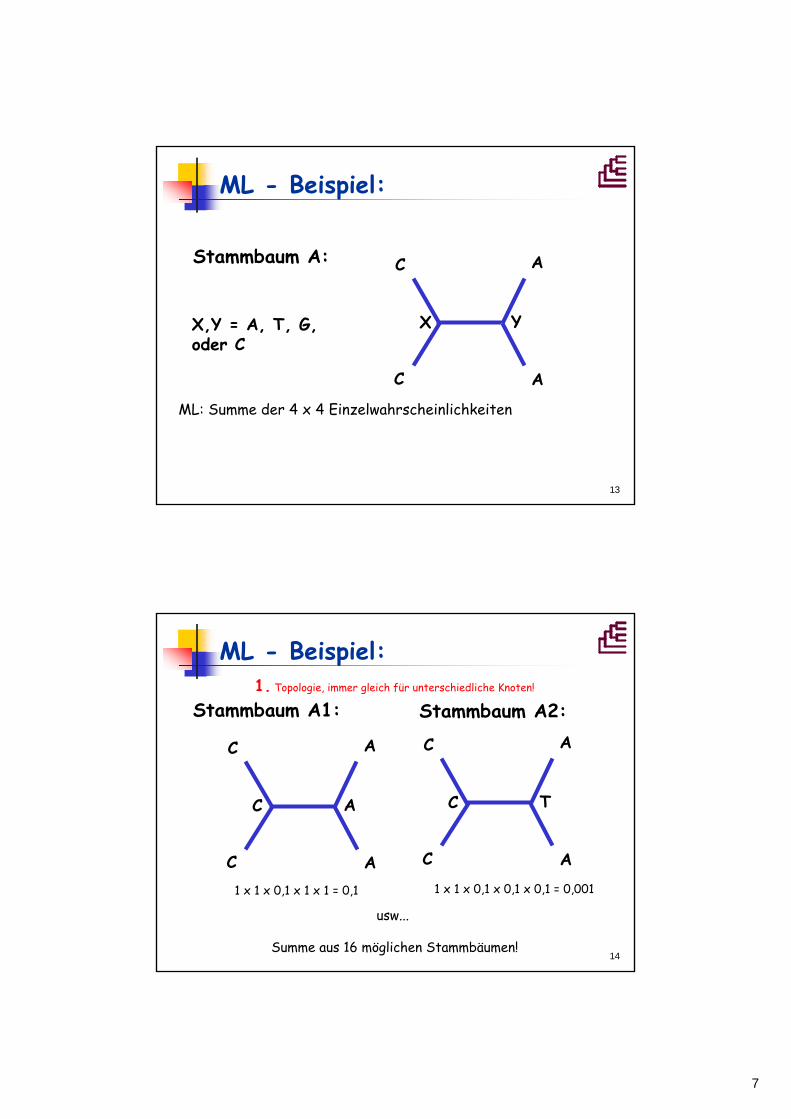
ML – Beispiel (vereinfacht):
CCAA
Daten: Modell (nicht realistisch):
A T C GA 1 0,1 0,1 0,1
T 1 0,1 0,1
C 1 0,1
G 1
OTU 1 OTU 2OTU 3OTU 4
7
13
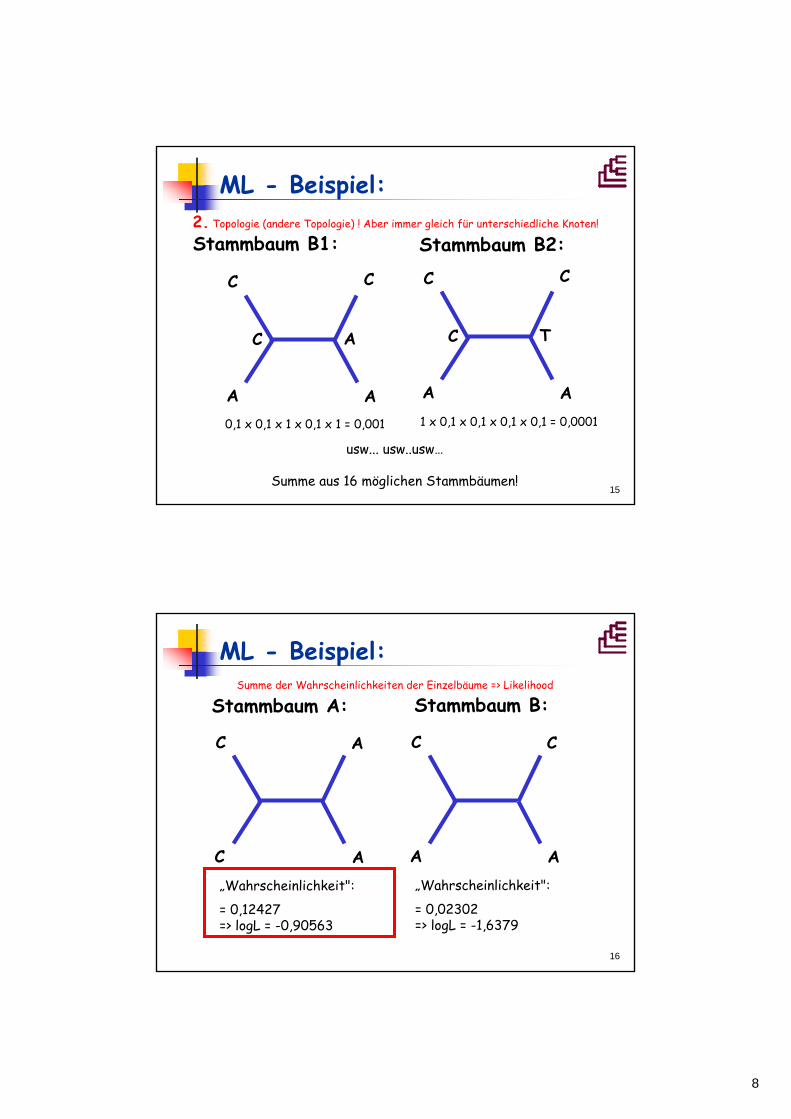
ML - Beispiel:
C
C
A
A
Stammbaum A:
X YX,Y = A, T, G, oder C
ML: Summe der 4 x 4 Einzelwahrscheinlichkeiten
14
ML - Beispiel:
Stammbaum A1:
C
C
A
A
C T
C
C
A
A
C A
1 x 1 x 0,1 x 1 x 1 = 0,1 1 x 1 x 0,1 x 0,1 x 0,1 = 0,001
usw...
Summe aus 16 möglichen Stammbäumen!
Stammbaum A2:1. Topologie, immer gleich für unterschiedliche Knoten!
8
15
ML - Beispiel:
Stammbaum B1:
C
A
C
A
C T
C
A
C
A
C A
0,1 x 0,1 x 1 x 0,1 x 1 = 0,001 1 x 0,1 x 0,1 x 0,1 x 0,1 = 0,0001
usw... usw..usw…
Summe aus 16 möglichen Stammbäumen!
Stammbaum B2:2. Topologie (andere Topologie) ! Aber immer gleich für unterschiedliche Knoten!
16
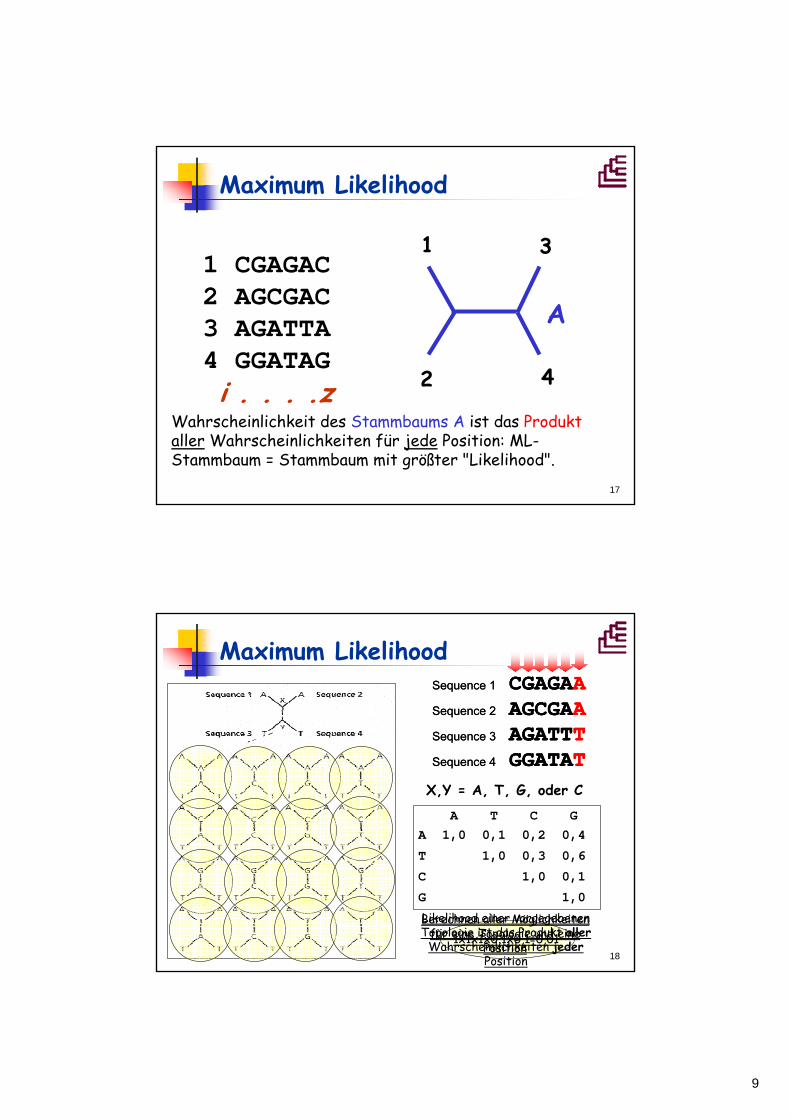
ML - Beispiel:
Stammbaum A:
C
C
A
A„Wahrscheinlichkeit":
= 0,12427=> logL = -0,90563
C
A
C
A„Wahrscheinlichkeit":
= 0,02302=> logL = -1,6379
Stammbaum B:Summe der Wahrscheinlichkeiten der Einzelbäume => Likelihood
9
17
Wahrscheinlichkeit des Stammbaums A ist das Produktaller Wahrscheinlichkeiten für jede Position: ML-Stammbaum = Stammbaum mit größter "Likelihood".
Maximum Likelihood
1 CGAGAC2 AGCGAC3 AGATTA4 GGATAGi . . . .z
1
2
3
4
A
18
X,Y = A, T, G, oder C
A T C GA 1,0 0,1 0,2 0,4
T 1,0 0,3 0,6
C 1,0 0,1
G 1,0
Sequence 1 CGAGAASequence 2 AGCGAASequence 3 AGATTTSequence 4 GGATAT
1x1x1x0,1x0,1=0,01
Sequence 1 CGAGAASequence 2 AGCGAASequence 3 AGATTTSequence 4 GGATAT
CGAGAAAGCGAAAGATTTGGATAT
Likelihood einer vorgegebenen Topologie ist das Produkt aller
Wahrscheinlichkeiten jederPosition
Berechnen aller Möglichkeiten für eine Topologie und eine
Position
Maximum Likelihood

10
19
A T C G A 1,0 0,1 0,2 0,4T 1,0 0,3 0,6C 1,0 0,1G 1,0
Sequence 1 CGAGAASequence 2 AGCGAASequence 3 AGATTTSequence 4 GGATAT
Sequence 1 CGAGAASequence 2 AGCGAASequence 3 AGATTTSequence 4 GGATAT
CGAGAAAGCGAAAGATTTGGATAT
Maximum Likelihood
Likelihood für alle anderen Topologienund
Vergleich der Likelihoods
L = P(data|hypothesis)
FFüür n=50 Sequenzen gibt es 2,84x10r n=50 Sequenzen gibt es 2,84x107676 mmöögliche Bgliche Bääume <ume <--> Atome im Universum: ~10> Atome im Universum: ~108080
20
Maximum Likelihood - Vorteile
Mathematisch gut definiertFunktioniert gut in Simulationsexperimenten Erlaubt explizite Verbindung von Evolutionsmodell und Daten (Sequenzen) "Realistische" Annahmen zur EvolutionVerschiedene Modelle und Stammbäume lassen sich testen
11
21
Maximum Likelihood – Lokale Maxima
22
Maximum Likelihood - Nachteile
Maximum likelihood ist nur konsistent (ergibt einen "wahren" Stammbaum) wenn die Evolution nach den gegebenen Modell ablief: Wie gut stimmt mein Modell mit den Daten überein? Computertechnisch nicht zu lösen wenn zu viele Taxa oder Parameter berücksichtigt werden müssen.
ffüür n=50 Sequenzen gibt es 2,84x10r n=50 Sequenzen gibt es 2,84x107676 mmöögliche Bgliche Bääumeume

12
23
Was ‚können‘ oder ‚kennen wir !?
Alignment
Distanz matrix
Evolutions-modelle
Neigbor JoiningMax.Parsimony
Max. Likelihood UPGMA
Charakter Matrix
Stammbaum
24
Viele Methoden sind Computer-technisch nicht zu lösen, insbesondere bei vielen Taxad.h., nicht alle (möglichen) Stammbäume berechenbar
Die Machbarkeit !?
L = P(data|hypothesis)
=5,5

13
25
„Intelligente Algorithmen“
- Quartet puzzling
- Bayes´sche Methode + MCMCMC
Die Machbarkeit !?
26
Quartet puzzling
Wie löst man ein großes Problem?=> Man zerlegt es in viele kleine Teilprobleme.Wenn es aber zu viele kleine Teilprobleme sind?
=> Man rechnet von der Lösung einiger Teilprobleme auf die Lösung des Gesamtproblems hoch.Quartet puzzling: Errechnung eines Gesamtstammbaums aus vielen Einzelstammbäumen mit jeweils vier Taxa => Quartetten!Programm: Tree-Puzzle (Strimmer & van Haeseler, 1996) http://www.tree-puzzle.de/
14
27
The „shell“
28
Treepuzzle
Max Likelihood
Quartet-puzzle
Majority rule consensus3.)
1.)
2.)
Drei Schritte

15
29
Quartet puzzling
OTU 1
OTU 1 OTU 2
OTU 1 OTU 2
OTU 3
OTU 1
OTU 4
OTU 2
OTU 3
OTU 1
OTU 3
OTU 2
OTU 4
OTU 1
OTU 2
OTU 3
OTU 4
3.)
4.)1.)
2.)
=> Quartett ist die minimale Einheit zur Lösung eines phylogenetischen Problems.
30
• Summe der „Likelihoods“ (eine Position, alle Möglichkeiten)
• Produkt aller „Likelihoods“(alle Positionen)
Berechnung: wie bei ML
Wahrscheinlichkeit für einen Stammbaum
∑=
n
iiL
1)(lnlnL=
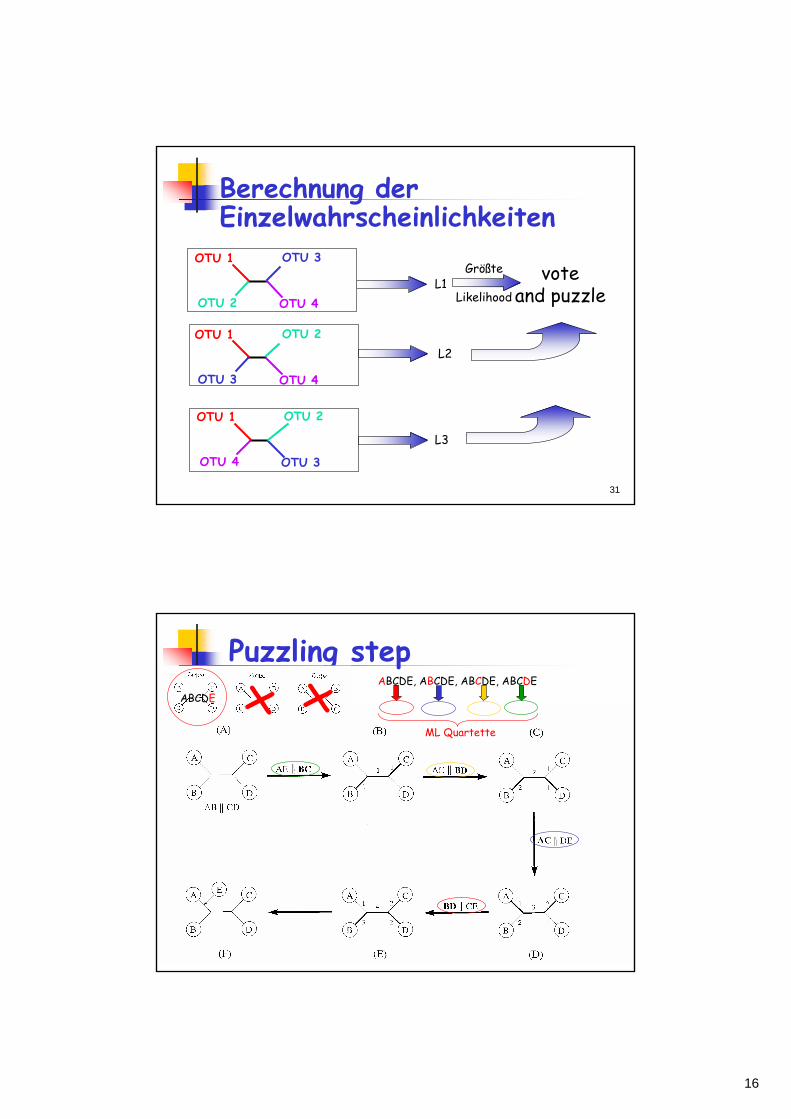
Berechnung der Einzelwahrscheinlichkeiten
16
31
Berechnung der Einzelwahrscheinlichkeiten
OTU 1
OTU 4
OTU 2
OTU 3
OTU 1
OTU 3
OTU 2
OTU 4
L1
L2
L3
voteand puzzle
Größte
Likelihood
OTU 1
OTU 2
OTU 3
OTU 4
32
Puzzling stepABCDE
ML Quartette
ABCDE, ABCDE, ABCDE, ABCDEx x
17
33
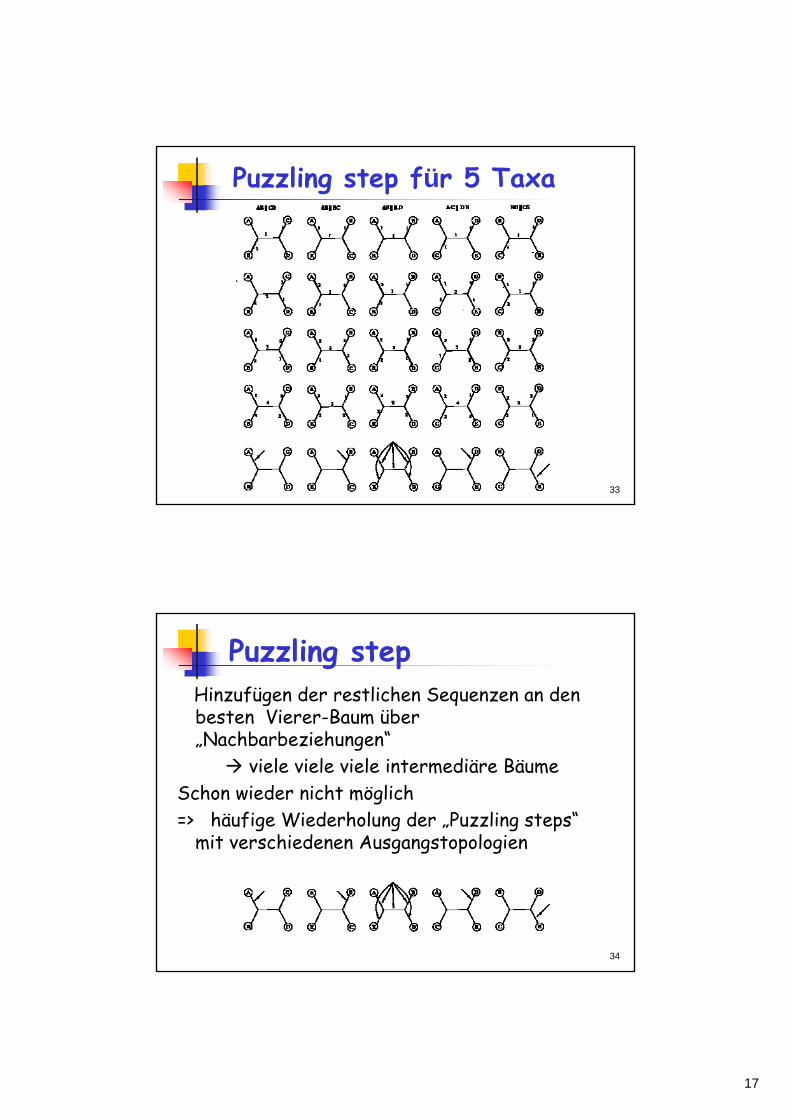
Puzzling step für 5 Taxa
34
Hinzufügen der restlichen Sequenzen an den besten Vierer-Baum über „Nachbarbeziehungen“
viele viele viele intermediäre Bäume Schon wieder nicht möglich=> häufige Wiederholung der „Puzzling steps“
mit verschiedenen Ausgangstopologien
Puzzling step
18
35
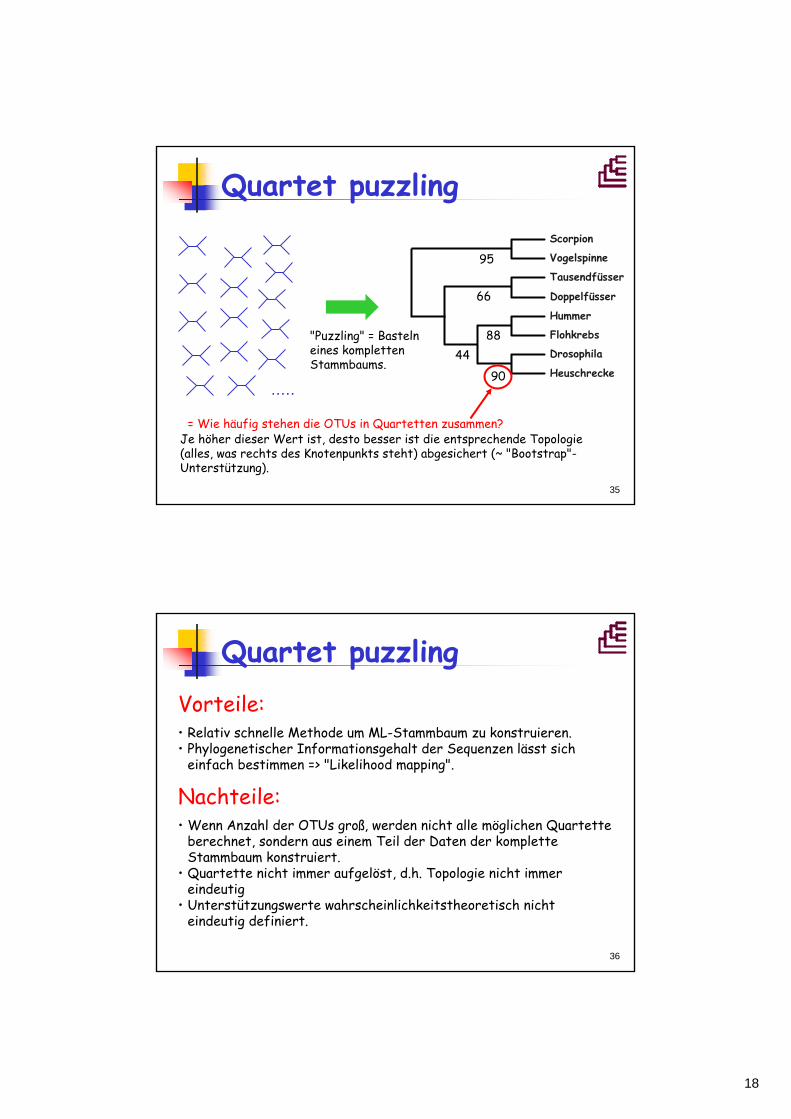
Quartet puzzling
.....
Scorpion
Vogelspinne
Tausendfüsser
Doppelfüsser
Hummer
Flohkrebs
Drosophila
Heuschrecke44
90
66
88
95
"Puzzling" = Bastelneines kompletten Stammbaums.
= Wie häufig stehen die OTUs in Quartetten zusammen?Je höher dieser Wert ist, desto besser ist die entsprechende Topologie (alles, was rechts des Knotenpunkts steht) abgesichert (~ "Bootstrap"-Unterstützung).
36
Quartet puzzlingVorteile:• Relativ schnelle Methode um ML-Stammbaum zu konstruieren. • Phylogenetischer Informationsgehalt der Sequenzen lässt sich
einfach bestimmen => "Likelihood mapping".
Nachteile:• Wenn Anzahl der OTUs groß, werden nicht alle möglichen Quartette
berechnet, sondern aus einem Teil der Daten der komplette Stammbaum konstruiert.
• Quartette nicht immer aufgelöst, d.h. Topologie nicht immer eindeutig
• Unterstützungswerte wahrscheinlichkeitstheoretisch nicht eindeutig definiert.

19
37
"Wahrscheinlichkeit" (Likelihood) der Sequenzdaten, gegeben die Topologie des Stammbaums und ein Evolutionsmodell.
Maximum Likelihood
Sequenz 5
Sequenz 3Sequenz 2Sequenz 4
Sequenz 1 Seq1 KIADKNFTYRHHNQLVSeq2 KVAEKNMTFRRFNDIISeq3 KIADKDFTYRHW-QLVSeq4 KVADKNFSYRHHNNVVSeq5 KLADKQFTFRHH-QLV
+ Modell(PAM, BLOSUM...)
Anders formuliert:Welcher Stammbaum (und welches Evolutionsmodell) Welcher Stammbaum (und welches Evolutionsmodell)
erklerkläärt am besten meine Sequenzdaten?rt am besten meine Sequenzdaten?
L = P(data|tree)
38
Wahrscheinlichkeit nach Bayes
• Reverend Thomas Bayes (1702-1761)(presbyterianischer Pfarrer)
• Wahrscheinlichkeitsrechnungen nach Bayeserst seit ein paar Jahren in der Statistik akzeptiert.
• Seit den 90er Jahren auch Anwendung in der molekularen Phylogenie.

20
39
Die Wahrscheinlichkeit ("posterior propability") beruht auf einem anfänglichen Evolutionsmodell und neuen Erkenntnissen nach einem Experiment.
Wahrscheinlichkeit nach Bayes
posterior propability P(H|D)
prior propability P(H)
40
BayesBayes´sche Statistik beruht auf dem Satz von Bayes.
AA ist die Hypothese. BB ist das beobachtete Ereignis. P(A)P(A) ist die A-Priori-Wahrscheinlichkeit von A. P(B | A)P(B | A) ist die bedingte Wahrscheinlichkeit von B, unter der Bedingung dass die HYpothese A wahr ist (als Funktion von A nennt man sie die Likelihood-Funktion):
P(B)P(B) ist die unbedingte Wahrscheinlichkeit von B.P(A | B)P(A | B) ist die A-Posteriori-Wahrscheinlichkeit von A gegeben B.
An Essay towards solving a Problem in the Doctrine of Chances. By the late Rev. Mr. Bayes, communicated by Mr. Price, in a letter to John Canton, M. A. and F. R. S.

21
41
Bayes
42
Wahrscheinlichkeit nach Bayes
⎟⎟⎠
⎞⎜⎜⎝
⎛ ×=
)()()(
)(dataP
treePtreedataPdatatreeP
Die Wahrscheinlichkeit eines Stammbaums, gegeben die Sequenzdaten, enspricht dem Produkt aus der Wahrscheinlichkeit der Daten gegeben den Stammbaum (likelihood) und der Wahrscheinlichkeit
des Stammbaums (prior probability), geteilt durch die Wahrscheinlichkeit der Daten (Randbedingungen).
PosterioreWahrscheinlichkeit für ein Ereignis A (tree) unter der
Bedingung, dass B (alignment) auftritt.
A-Priori-Wahrscheinlichkeitfür ein Ereignis A
(tree)
A-Priori-Wahrscheinlichkeit für ein Ereignis B (alignment)
Die Wahrscheinlichkeit für ein Ereignis B (alignment)unter der Bedingung, dass
A (tree) auftritt (Likelihood)

22
43
Randbedingungen
( ) )A(P)A|B(PBP ×= ∑θ
)...A(P)A|B(P)A(P)A|B(P 2211 +=
sind die Randbedingungen der Daten. D.h., es werden ALLE möglichen Werte von B berechnet. Sie geben, ebenso wie , das "Vorabwissen" an, welches man berücksichtigt.
)B(P
)A(P
)()()|()|(
BPAPABPBAP ×
=
44
Bayes
23
45
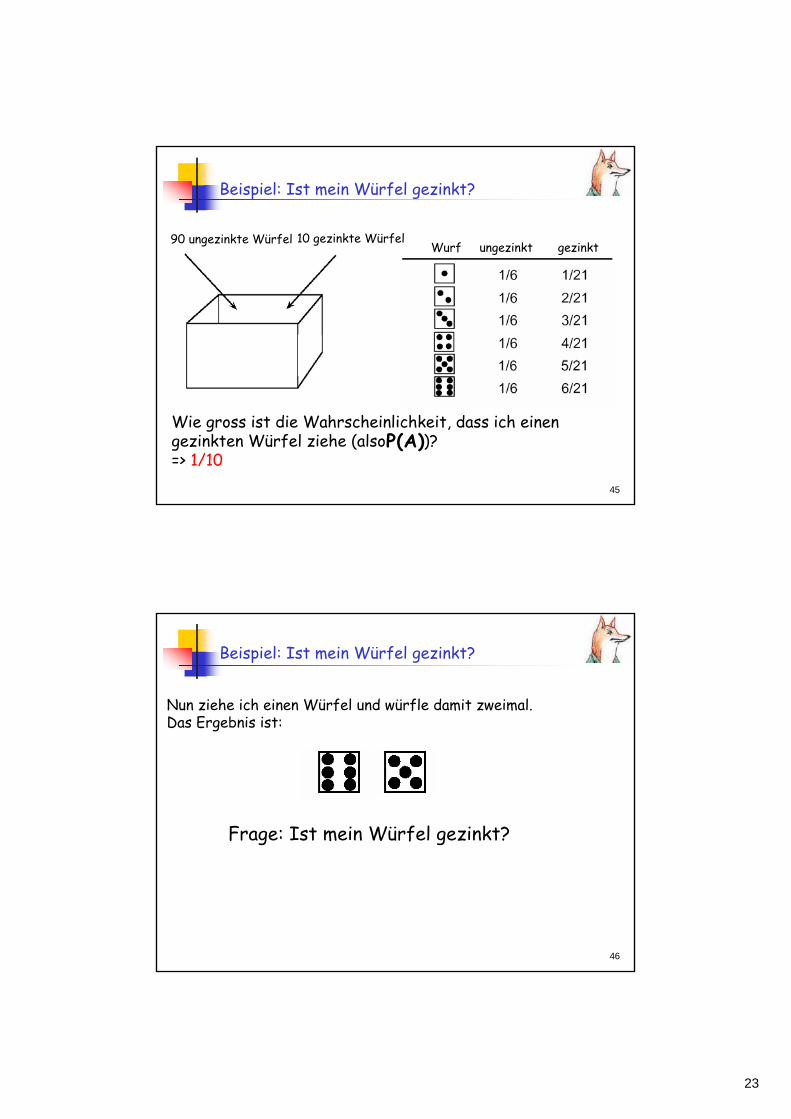
Beispiel: Ist mein Würfel gezinkt?
90 ungezinkte Würfel 10 gezinkte WürfelWurf ungezinkt gezinkt
)A(PWie gross ist die Wahrscheinlichkeit, dass ich einen gezinkten Würfel ziehe (also )? => 1/10
46
Beispiel: Ist mein Würfel gezinkt?
Nun ziehe ich einen Würfel und würfle damit zweimal.Das Ergebnis ist:
Frage: Ist mein Würfel gezinkt?

24
47
-> prior-> likelihood
Wurf ungezinkt gezinkt
-> alle Möglichkeiten
Beispiel: Ist mein Würfel gezinkt?
Bayes:
2137,0≈) |gezinkt(P
prior posterior
0,1 0,2137
9/10ungezinkt)|(1/10gezinkt)|(1/10gezinkt)|()|gezinkt(
×+××
=PP
PP
48
Mehr Daten: Ist mein Würfel gezinkt?
Wurf Häufigkeit
979,0)D|gezinkt(P =
021,0)D|ungezinkt(P =
Bayes:
Nun ziehe ich einen Würfel und würfle damit 20x. Das Ergebnis ist:
2137,0≈) |gezinkt(Pprior posterior
0,1 0,2137
vorher:

25
49
Randbedingungen
( ) )A(P)A|B(PBP ×= ∑θ
)...A(P)A|B(P)A(P)A|B(P 2211 +=
sind die Randbedingungen der Daten. D.h., es werden ALLE möglichen Werte von B berechnet. Sie geben, ebenso wie , das "Vorabwissen" an, welches man berücksichtigt.
)B(P
)A(P
)()()|()|(
BPAPABPBAP ×
=
50
Wahrscheinlichkeit nach BayesBayes sucht die Bäume mit den höchsten posterior probabilities.posterior probabilities.
( ) )A(P)A|B(PBP ×= ∑θ
)...A(P)A|B(P)A(P)A|B(P 2211 +=
)()()|()|(
BPAPABPBAP ×
=
Nenner: Summation über alle denkbaren Hypothesen (Bäume)
=> MCMCMCMCMCMC „Metropolis-Coupled Markov Chain Monte Carlo“

26
51
Metropolis-Coupled Markov Chain
Wahrscheinlichkeit nach Bayes
Zufällige Stichprobe aus der posterior probability Verteilungposterior probability Verteilung ziehenStichprobe muss groß (genug) sein
Prozentsatz mit dem ‚Clade‘ bei den Bäumen auftritt wird als Wahrscheinlichkeit interpretiert, dass ‚Clade‘ korrekt ist
Monte Carlo
MC MC 3 3 -- MCMCMCMCMCMC - „Metropolis-Coupled Markov Chain Monte Carlo“
52
Andrey (Andrei) Andreyevich MarkovRussischer Mathematiker (1856 – 1922)
Ein stochastischer Prozess besitzt die sogenannte Markov Eigenschaft, wenn:
Die bedingte Wahrscheinlichkeitsverteilung zukünftiger Zustände, bei gegebenen momentanten Status und allen vergangenen Ereignissen NUR von dem momentanen Status abhängt und NICHT von den vergangenen

27
53
Wahrscheinlichkeit nach Bayes
MC MC 3 3 -- MCMCMCMCMCMC - „Metropolis-Coupled Markov Chain Monte Carlo“
Zufällige Stichprobe aus der ‚posterior probability‘-Verteilung ziehenStichprobe muss groß (genug) sein
Prozentsatz mit dem eine Gruppierung (‚Clade‘) bei diesen zufälligen Bäumen auftritt wird als Wahrscheinlichkeit interpretiert, dass ‚Clade‘ korrekt ist
Im Falle einer Markow-Kette erster Ordnung wird hierfür sogar nur Kenntnis über den momentanen Zustand benötigt (Gedächtnislosigkeit).
Markov Chain
Ziel ist es, Wahrscheinlichkeiten für das Eintreten zukünftiger Ereignisse anzugeben
Auf lange Sicht pendelt sich Wahrscheinlichkeit auf einen Wert ein => Prozentsatz mit dem ‚Clade‘ in den Bäumen auftritt
54
Wahrscheinlichkeit nach Bayes
MC MC 3 3 -- MCMCMCMCMCMC - „Metropolis-Coupled Markov Chain Monte Carlo“
Markov Chain Monte Carlo
Zufallselement - die Bank gewinnt auf lange Sicht immer
Die Masse machts - Wahrscheinlichkeit falscher Rückschlüsse sinkt mit zunehmender Generationszahl
Kurz gesagt, was bedeutet MCMC?
Viele Zufallsproben aus der posterioren Wahrsscheinlichkeitsverteilung (Bäume) nehmen und Rückschlüsse ziehen
Zufällige Stichprobe aus der ‚posterior probability‘-Verteilung ziehenStichprobe muss groß (genug) sein
Prozentsatz mit dem eine Gruppierung (‚Clade‘) bei diesen zufälligen Bäumen auftritt wird als Wahrscheinlichkeit interpretiert, dass ‚Clade‘ korrekt ist

28
55
Wahrscheinlichkeit nach Bayes
Akzeptieren oder Verwerfen?
MetropolisMetropolis--HastingsHastings--GreenGreen--AlgorithmusAlgorithmus
Vorgegebener (Zufalls-)Baum Ti mit Topologie, Astlängen und Evolutionsmodell
Neuer Baum, neue Parameter
)()|()()|(
)()()|(
)()()|(
jj
ii
jj
ii
iesprobabilitposteriorTPTBPTPTBP
DPTPTBP
DPTPTBP
Q××
=×
×
=X
X1Q ≥ Neuen Baum akzeptieren!
1Q < Generation einer Zufallszahl (0-1) Z< Q, neuen Baum akzeptieren, sonst alter Baum
MC MC 3 3 -- MCMCMCMCMCMC - „Metropolis-Coupled Markov Chain Monte Carlo“
56
„1“
Wahrscheinlichkeit nach Bayes
MC MC 3 3 -- MCMCMCMCMCMC - „Metropolis-Coupled Markov Chain Monte Carlo“
Metropolis-Hastings-Green-Algorithmus
)()|()()|(
jj
iiiesprobabilitposterior
TPTBPTPTBPQ
××
=
priorslikelihoods
shape parameter, Astlängen..
DAS PROBLEM
),,,|()|( shapeonsubstitutilengthtreeBPTBP −= α
„Lösung“
)|()|(
ji
ij
TTPTTP
x
Änderungsvorschläge
29
57
Wahrscheinlichkeit nach Bayes
MetropolisMetropolis--HastingsHastings--GreenGreen--AlgorithmusAlgorithmus
1Q ≥ Neuen Baum akzeptieren!
1Q < Generation einer Zufallszahl (0-1) Z< Q, neuen Baum akzeptieren, sonst alter Baum
MC MC 3 3 -- MCMCMCMCMCMC - „Metropolis-Coupled Markov Chain Monte Carlo“
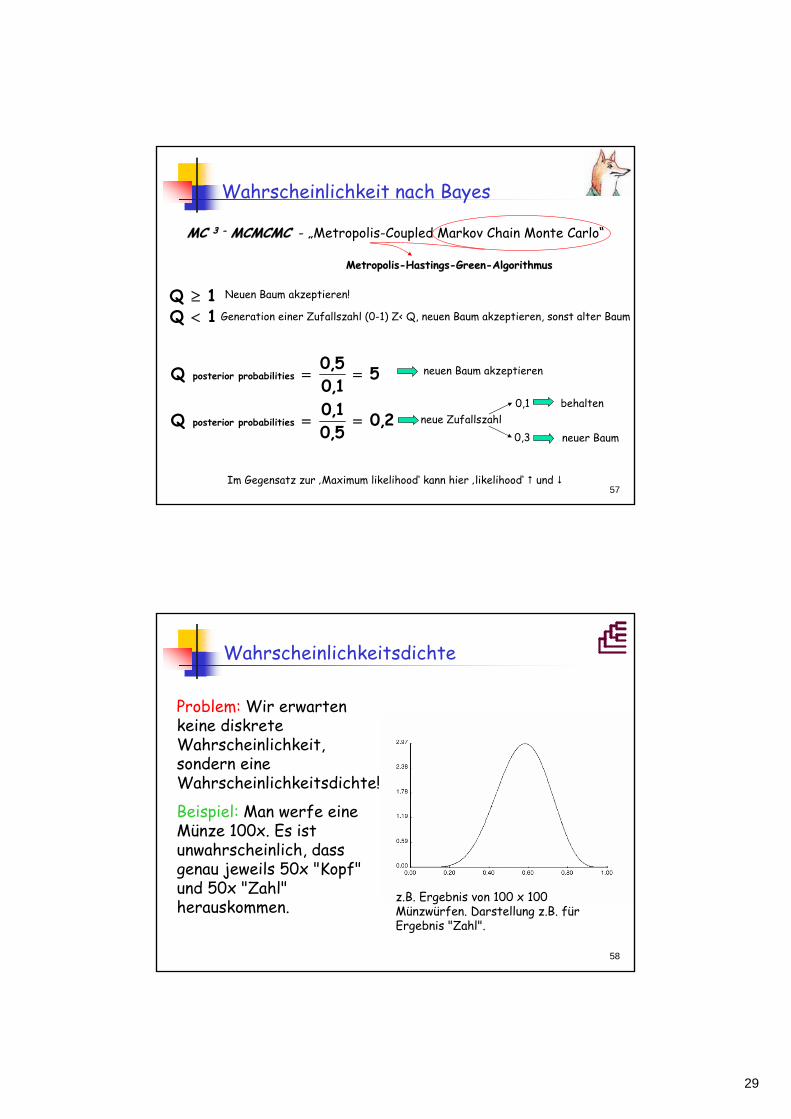
2,05,01,0Q
51,05,0Q
iesprobabilitposterior
iesprobabilitposterior
==
==
behaltenneue Zufallszahl
0,1
0,3 neuer Baum
Im Gegensatz zur ‚Maximum likelihood‘ kann hier ‚likelihood‘ und
neuen Baum akzeptieren
58
Wahrscheinlichkeitsdichte
Problem: Wir erwarten keine diskrete Wahrscheinlichkeit, sondern eine Wahrscheinlichkeitsdichte!
Beispiel: Man werfe eine Münze 100x. Es ist unwahrscheinlich, dass genau jeweils 50x "Kopf" und 50x "Zahl" herauskommen.
z.B. Ergebnis von 100 x 100 Münzwürfen. Darstellung z.B. für Ergebnis "Zahl".
30
59
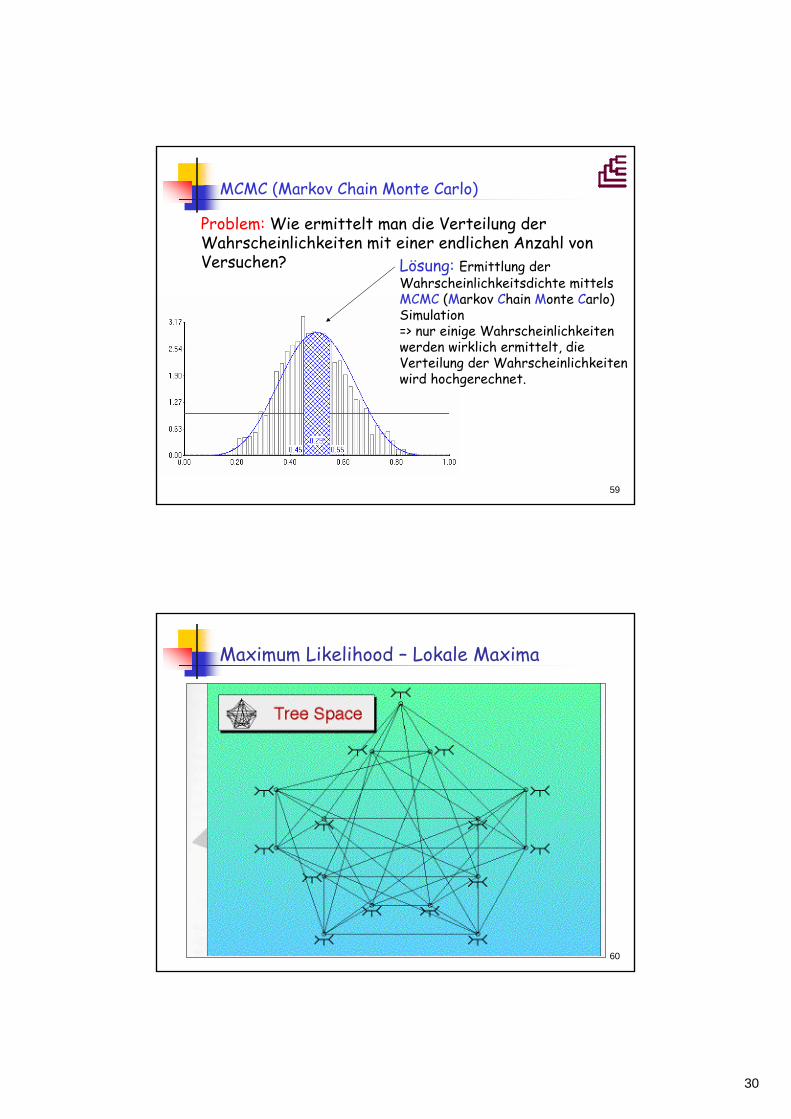
MCMC (Markov Chain Monte Carlo)
Problem: Wie ermittelt man die Verteilung der Wahrscheinlichkeiten mit einer endlichen Anzahl von Versuchen? Lösung: Ermittlung der
Wahrscheinlichkeitsdichte mittels MCMC (Markov Chain Monte Carlo) Simulation=> nur einige Wahrscheinlichkeiten werden wirklich ermittelt, die Verteilung der Wahrscheinlichkeiten wird hochgerechnet.
60
Maximum Likelihood – Lokale Maxima

31
61
MCMC (Markov Chain Monte Carlo)
62
„Start, sample and burnin“
wird verworfen
Man startet mit einen beliebigen Stammbaumdurch "burnin" werden "frühe" Ergebnisse (Stammbäume) verworfen, und über alle anderen gesamplet. => Wie gut stimmen die jeweiligen Bäume überein?Entspricht ~ Bootstrapping(kommt noch).
MrBayes

32
63
How to get up and get over?
??
64
Wahrscheinlichkeit nach Bayes
Metropolis-Coupled
=> mehrere MCMC laufen parallel und ‚kommunizieren‘
MC MC 3 3 -- MCMCMCMCMCMC - „Metropolis-Coupled Markov Chain Monte Carlo“
Zufällige Stichprobe aus der ‚posterior probability‘-Verteilung ziehenStichprobe muss groß (genug) sein
Prozentsatz mit dem eine Gruppierung (‚Clade‘) bei diesen zufälligen Bäumen auftritt wird als Wahrscheinlichkeit interpretiert, dass ‚Clade‘ korrekt ist
33
65
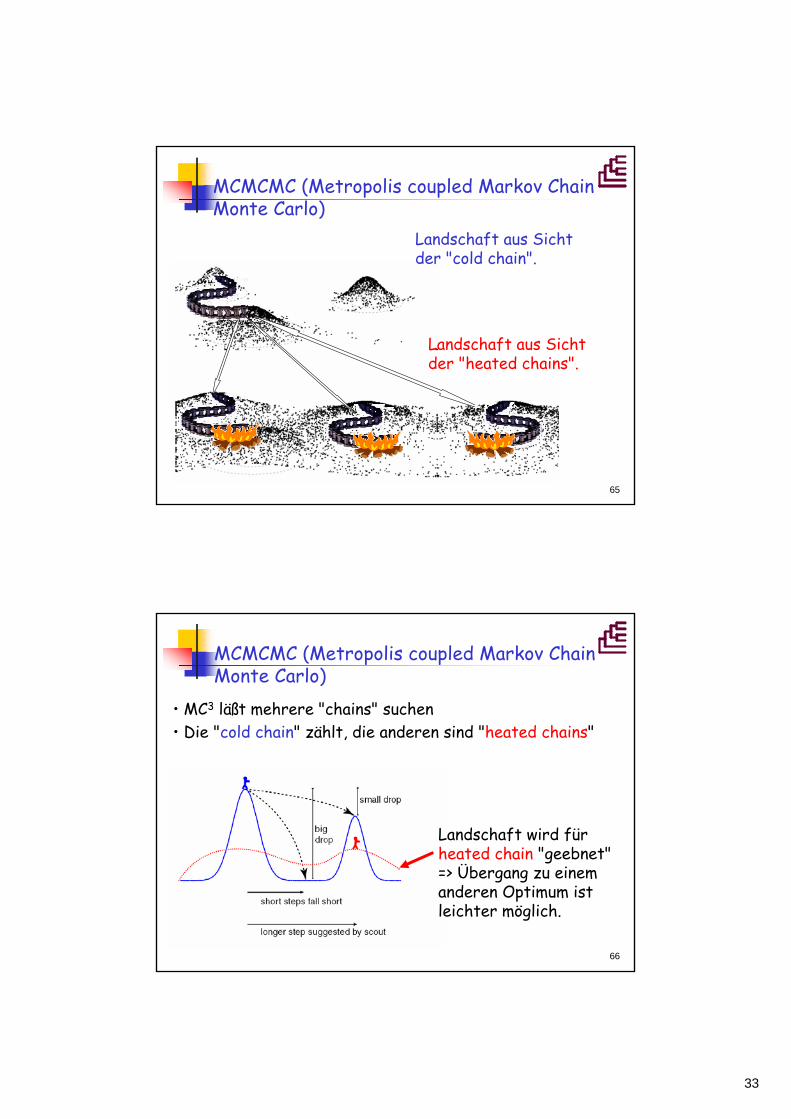
MCMCMC (Metropolis coupled Markov Chain Monte Carlo)
Landschaft aus Sicht der "cold chain".
Landschaft aus Sicht der "heated chains".
66
MCMCMC (Metropolis coupled Markov Chain Monte Carlo)
• MC3 läßt mehrere "chains" suchen• Die "cold chain" zählt, die anderen sind "heated chains"
Landschaft wird für heated chain "geebnet" => Übergang zu einem anderen Optimum ist leichter möglich.

34
67
Wahrscheinlichkeit nach Bayes
Vorteile:Vorabinformation wird berücksichtigt.Sehr schnelle ‚Lösung‘ komplexer phylogenetischer Probleme möglich!Diskrete Wahrscheinlichkeitswerte werden für jeden Ast gegeben.
Nachteile:
Vorabinformation wird berücksichtigt. Wahrscheinlichkeitstheoretisch umstritten.
68
Past and next
Molekularphylogenetische Methoden:- Quartet puzzling- Bayesian approach
Test von Stammbäumen- Bootstrapping- Likelihood tests…

35
69
Statistische Auswertung
….oder….
Wie gut ist mein Stammbaum ?
häufigste Methode ist „Bootstrapping“…
70
Bayes FaktorBayes FaktorBayes Faktor
(nach Kass & Raftery (1995) entspricht LRTLRT)Vergleich zweier Modelle Mi und Mj :
Kriterien nach Kass and Raftery (1995):Bij < 1, negative (support for Mj)1 < Bij < 3, barely worth mentioning3 < Bij < 12, positive12 < Bij < 150, strong andBij > 150, very strong.
Vorteil:‚Nested‘ und ‚non-nested data‘ kann verglichen werden
)|()|(
j
iij
MDPMDPB =

36
71
Bootstrapping Ziehen MITZurücklegen
72
D
Bootstrapping
Position Sequence 1 2 3 4 5 6 7 8 9
A A A A A G T G C A B A G C C G T G C G C A G A T A T C C A D A G A G A T C C G
OrginalsequenzenPosition
Sequence 1 2 2 4 5 5 7 8 8A A A A A G G G C CB A G G C G G C C CC A G G T A A C C CD A G G G A A C C C
Pseudosample 1
z.B. 100 WiederholungenPosition
Sequence 1 1 1 4 4 6 7 7 7A A A A A A T G G GB A A A C C T G G GC A A A T T T C C CD A A A G G T C C C
Pseudosample 2
A
D
BC
A
B
C
…100 Stammbäume
37
73
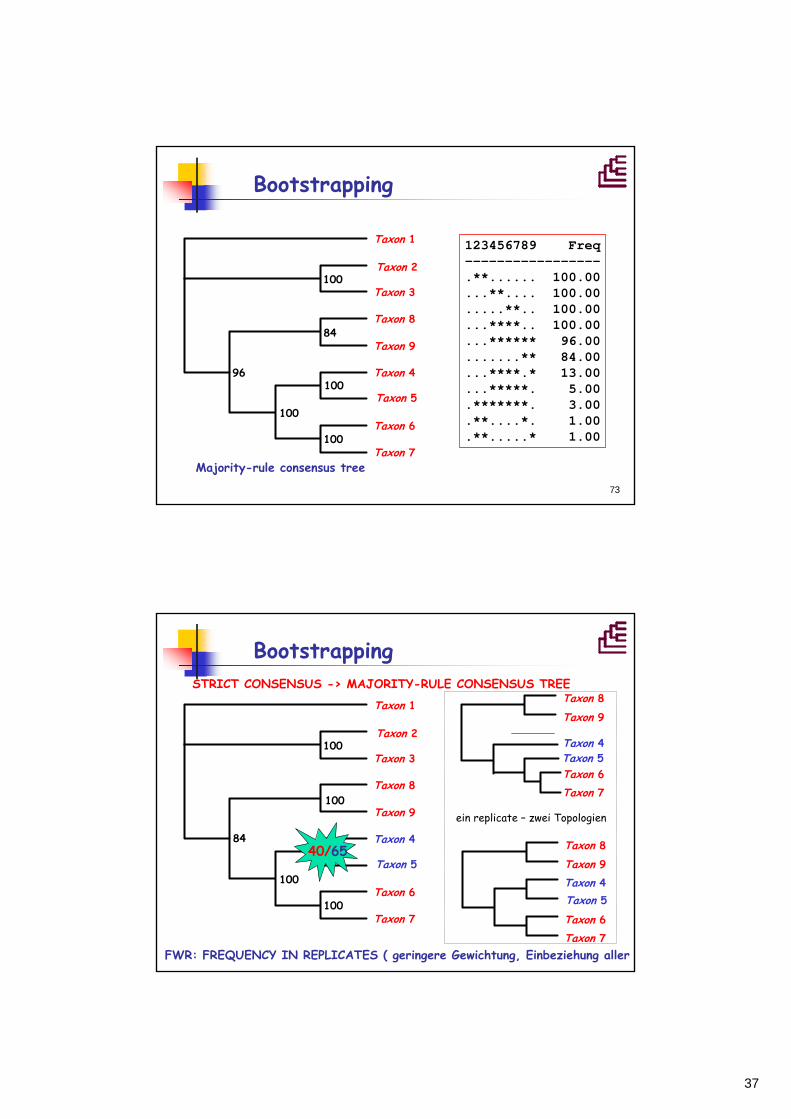
Bootstrapping
123456789 Freq-----------------.**...... 100.00...**.... 100.00.....**.. 100.00...****.. 100.00...****** 96.00.......** 84.00...****.* 13.00...*****. 5.00.*******. 3.00.**....*. 1.00.**.....* 1.00
Majority-rule consensus tree
Taxon 1
Taxon 3
Taxon 8
Taxon 9
Taxon 4
Taxon 6
Taxon 7
100
96
84
100
100
100
Taxon 2
Taxon 5
74
BootstrappingSTRICT CONSENSUS -> MAJORITY-RULE CONSENSUS TREE
Taxon 1
Taxon 3
Taxon 8
Taxon 9
Taxon 4
Taxon 6
Taxon 7
100
84
100
100
100
Taxon 2
Taxon 540/65 Taxon 8
Taxon 9Taxon 4
Taxon 6Taxon 7
Taxon 5
Taxon 8Taxon 9
Taxon 4
Taxon 6Taxon 7
Taxon 5
ein replicate – zwei Topologien
FWR: FREQUENCY IN REPLICATES ( geringere Gewichtung, Einbeziehung aller

38
75
Jackknife
• Prinzip gleich zu „bootstrap“ allerdings werden zufällig Daten gelöscht
• Unterstützungswerte ensprechen sich aber nicht
• Bootstrap-Wert in % entspricht ‚jacknife‘-Wert bei ca. 40 % gelöschte Daten
760.1
NpH
PlaxipCalloc
NuttalLepitoc
Nuttall64
Lorica StenosLepido
LoriceOnithoTonici
6062
58
AfHNotoplCrypto
6661
Likelhood mapping…Berechnung der posterior probability (Pi) aller BäumePi summiert sich auf 1 (irgendeine Topologie muss sein)3 Ecken repräsentieren die 3 TopologienJe näher P an einer der Ecke desto stärker wird dieser Baum favorisiert
28
33 30

39
77
Split Tree
78
Schwarzspitzenhai
Phylogenetic methodsEvolutionary relationships among unique mtDNA haplotypes were reconstructed using the maximummaximum--parsimony (MP)parsimony (MP) optimality criterion with all mutations weighted equally and indels treated as a fifth state. A two-nucleotide indel at positions 1045 and 1046 was treated as one event by omitting the second nucleotide from analyses. Heuristic tree Heuristic tree searchessearches were performed for all MP analyses with 1000 random-addition replications, saving a maximum of 1000 trees per replicate, and treetree--bisectionbisection––reconnectionreconnection (TBR) branch swapping. Statistical support for nodes was determined via 1000 nonparametric bootstrap replicatesbootstrap replicates(Felsenstein 1985) with 10 random-addition sequences per replicate, saving a maximum of 1000 trees per replicate, and nearest neighbour nearest neighbour interchangeinterchange (NNI) branch swapping. Haplotype trees were initially rooted using blacktip reef shark ( C. melanopterus ) and Australian blacktip shark, C. tilstoni , sequences as outgroups. Although the relationships of species within the genus Carcharhinus are not fully resolved (Lavery 1992; Naylor 1992), C. melanopterus and C. tilstoni were the closest relatives to C. limbatus for which tissue samples were available. C. melanopterus was used as the sole outgroup after C. limbatus was found to be paraphyletic to C. tilstoni in the MP analyses.
40
79
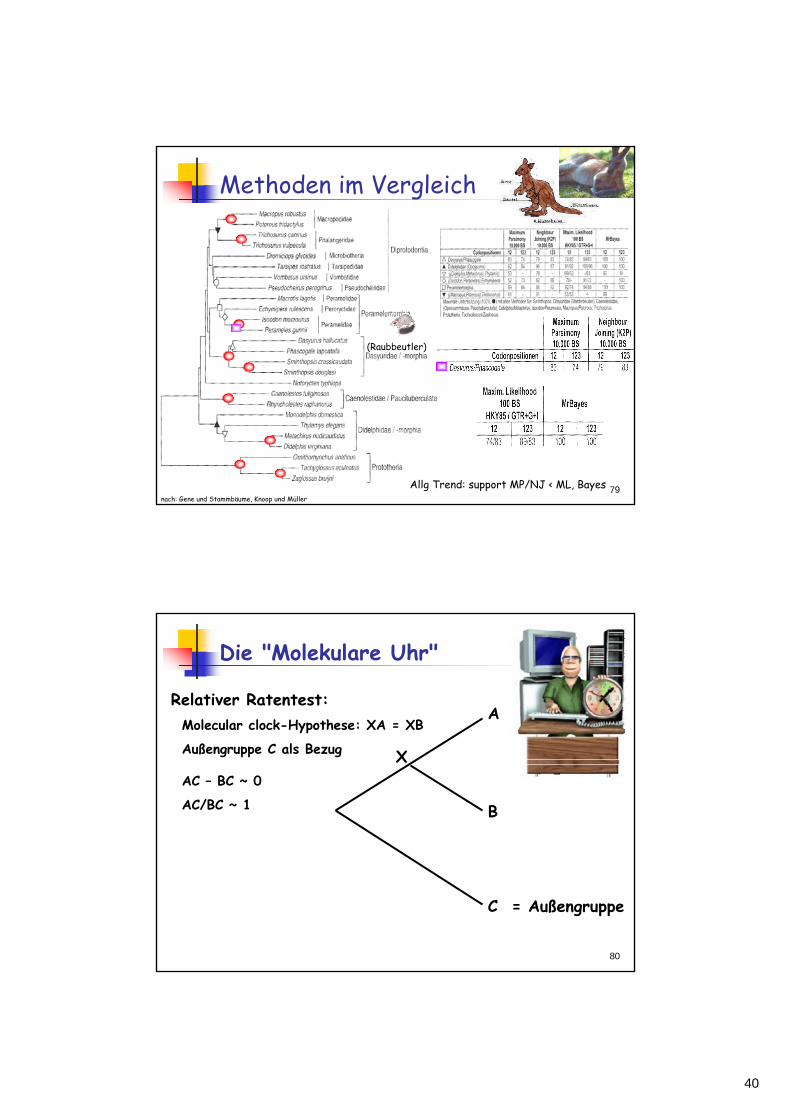
Methoden im Vergleich
maximal support
nach: Gene und Stammbäume, Knoop und Müller
(Raubbeutler)
Allg Trend: support MP/NJ < ML, Bayes
80
Die "Molekulare Uhr"
Relativer Ratentest:
X
A
B
C = Außengruppe
Molecular clock-Hypothese: XA = XB
Außengruppe C als Bezug
AC – BC ~ 0
AC/BC ~ 1

41
81
800 700 600 500 400 300 200 100 0 million years
520
420
480
220
343
370736
fossil records
Vetigastropoda
Protobranchia
Octobrachia
Decabrachia
Tetrabranchiata
Die "Molekulare Uhr"
82
Bedeutung der molekularen Phylogenie für die Systematik
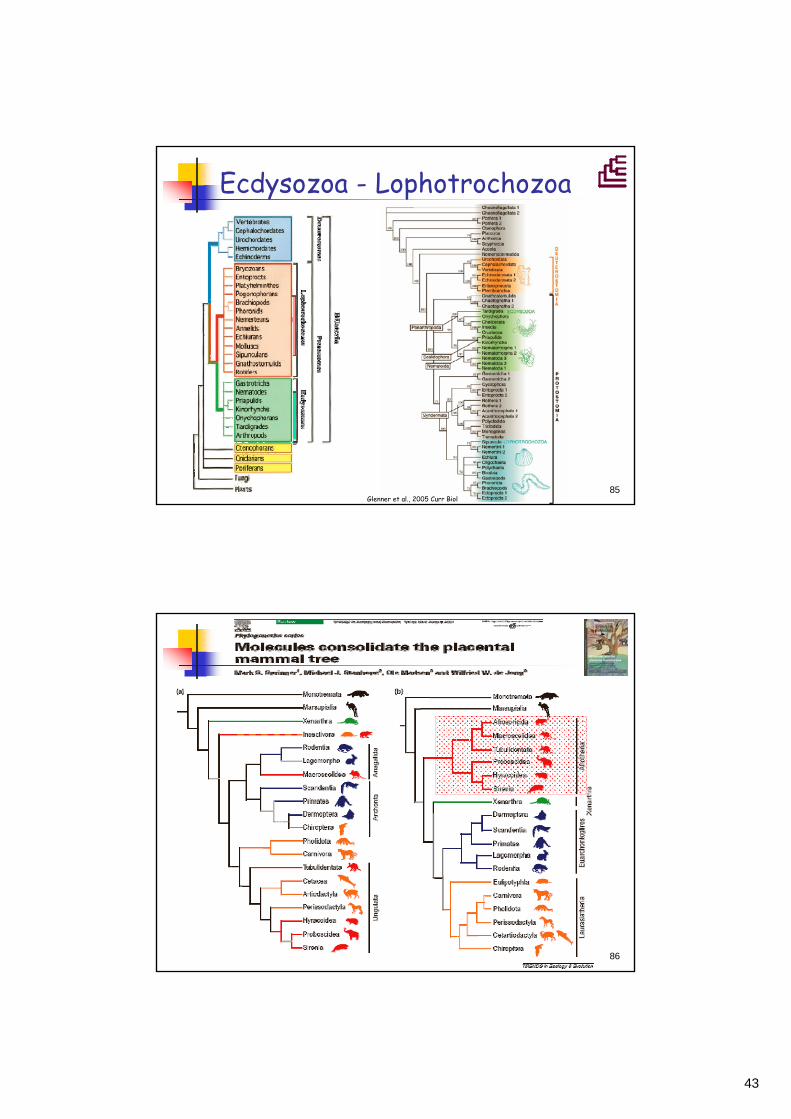
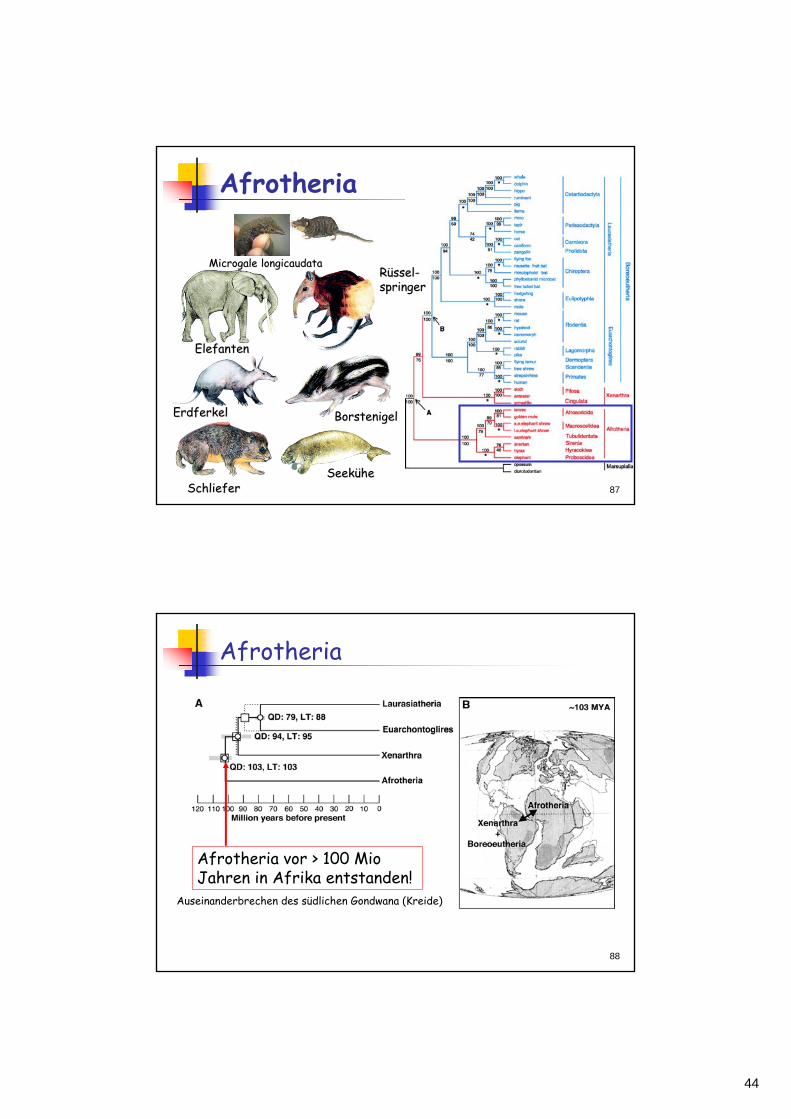
Frühere Vorstellungen zur Evolution und zu den Verwandtschaftsverhältnissen der Organismen beruhten auf morphologischen Kriterienmorphologischen KriterienDie molekulare Phylogeniemolekulare Phylogenie hat viele der früheren Vorstellungen in den letzten ~10 Jahren über den Haufen geworfen, aber die meisten Hypothesen meisten Hypothesen bestbestäätigttigt. Am interessantesten sind natürlich die "revolutionären" Vorstellungen, z.B.:- Ecdysozoa - Lophotrochozoa- Afrotheria ...

42
83
84
Ecdysozoa - Lophotrochozoa
43
85
Ecdysozoa - Lophotrochozoa
Glenner et al., 2005 Curr Biol
86
44
87
Afrotheria
RRüüsselssel--springerspringer
ElefantenElefanten
SeekSeeküühehe
ErdferkelErdferkel
SchlieferSchliefer
BorstenigelBorstenigel
MicrogaleMicrogale longicaudatalongicaudata
88
Afrotheria
Afrotheria vor > 100 MioJahren in Afrika entstanden!
Auseinanderbrechen des südlichen Gondwana (Kreide)

45
89
Konvergente Entwicklung!?
snail-like ?
90