genomic sequence alignment. overview dynamic programming & the needleman-wunsch algorithm local...
Post on 19-Dec-2015
228 views
TRANSCRIPT

Genomic Sequence Alignment

Overview
• Dynamic programming & the Needleman-Wunsch algorithm
• Local alignment—BLAST
• Fast global alignment
• Multiple sequence alignment
• Rearrangements in genomic sequences
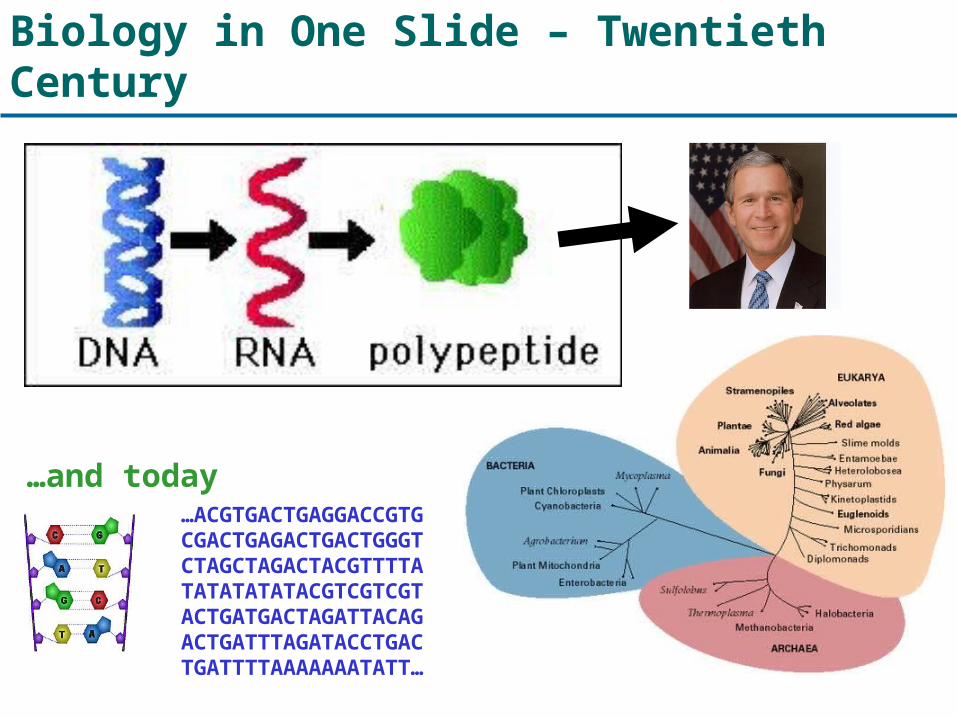
Biology in One Slide – Twentieth Century
…ACGTGACTGAGGACCGTGCGACTGAGACTGACTGGGTCTAGCTAGACTACGTTTTATATATATATACGTCGTCGTACTGATGACTAGATTACAGACTGATTTAGATACCTGACTGATTTTAAAAAAATATT…
…and today

Complete DNA Sequences
About 300 complete genomes have been
sequenced
Evolution
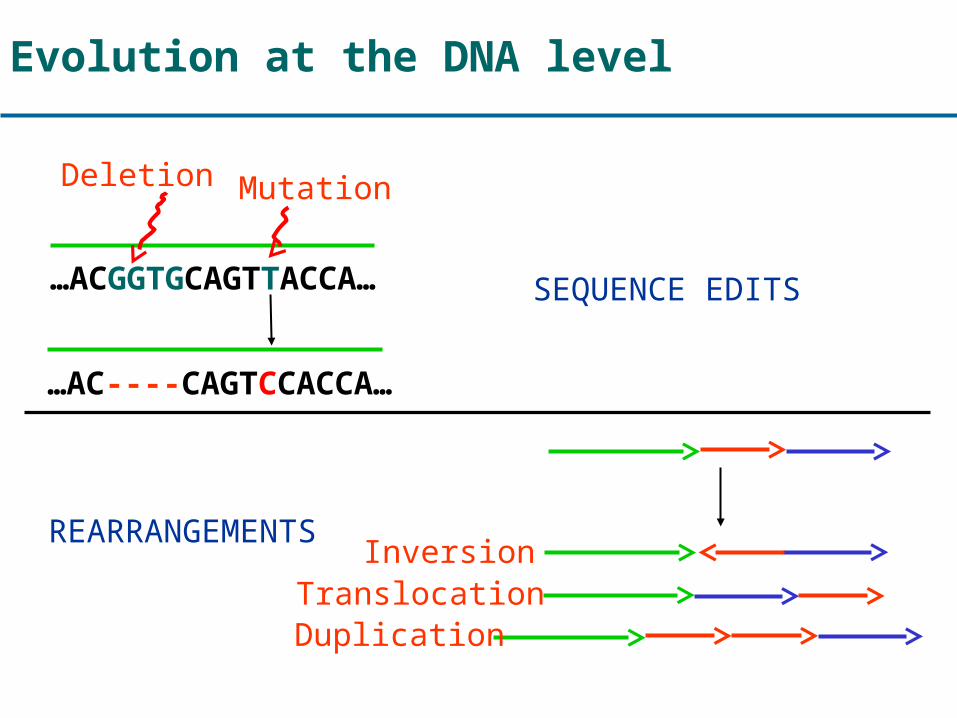
Evolution at the DNA level
…ACGGTGCAGTTACCA…
…AC----CAGTCCACCA…
Mutation
SEQUENCE EDITS
REARRANGEMENTS
Deletion
InversionTranslocationDuplication
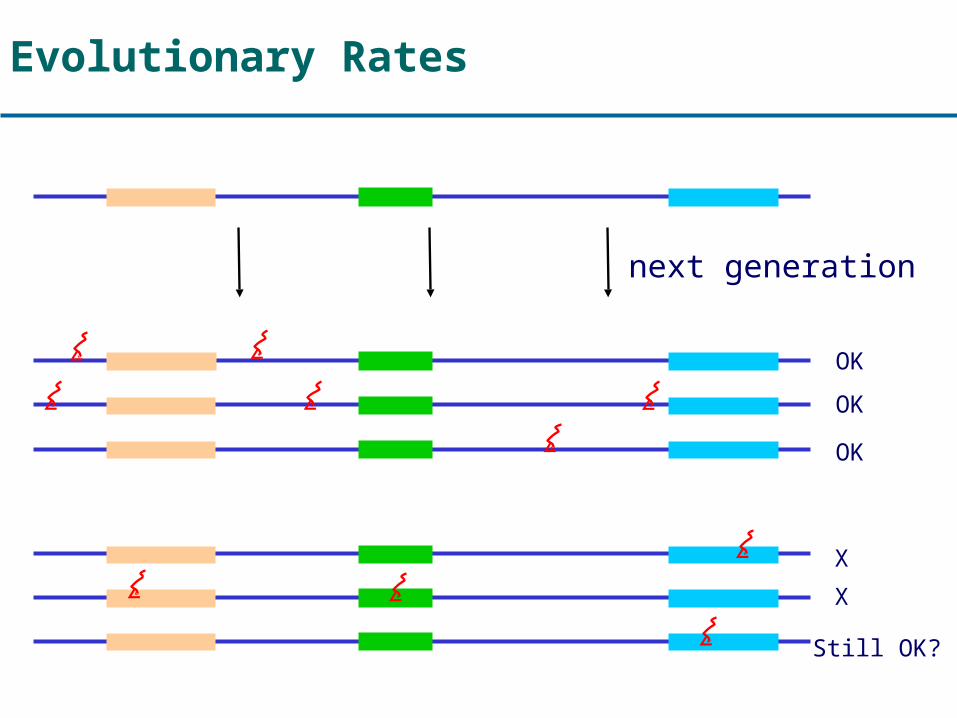
Evolutionary Rates
OK
OK
OK
X
X
Still OK?
next generation

Sequence conservation implies function
Alignment is the key to• Finding important regions• Determining function• Uncovering the evolutionary forces

Sequence Alignment
-AGGCTATCACCTGACCTCCAGGCCGA--TGCCC---TAG-CTATCAC--GACCGC--GGTCGATTTGCCCGAC
DefinitionGiven two strings x = x1x2...xM, y = y1y2…yN,
an alignment is an assignment of gaps to positions0,…, N in x, and 0,…, N in y, so as to line up each
letter in one sequence with either a letter, or a gapin the other sequence
AGGCTATCACCTGACCTCCAGGCCGATGCCCTAGCTATCACGACCGCGGTCGATTTGCCCGAC

What is a good alignment?
Alignment: The “best” way to match the letters of one sequence with those of the other
How do we define “best”?
Alignment:A hypothesis that the two sequences come from a common ancestor through sequence edits
Parsimonious explanation:Find the minimum number of edits that transform one sequence into the other

Scoring Function
• Sequence edits: AGGCCTC
Mutations AGGACTC
InsertionsAGGGCCTC
DeletionsAGG.CTC
Scoring Function:Match: +mMismatch: -sGap: -d
Score F = (# matches) m - (# mismatches) s – (#gaps) d
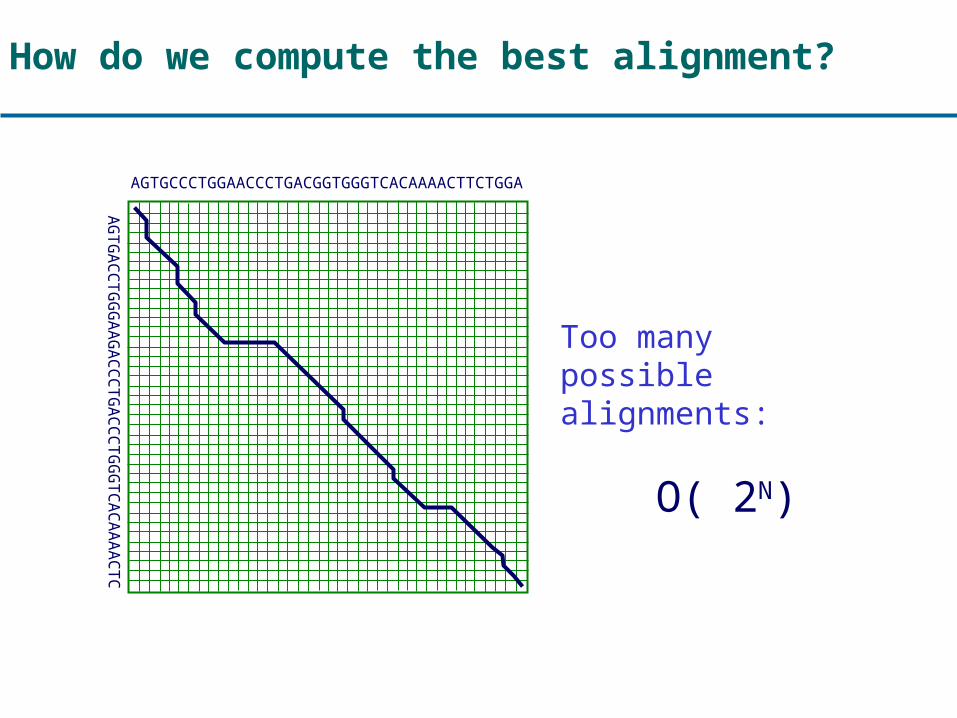
How do we compute the best alignment?
AGTGCCCTGGAACCCTGACGGTGGGTCACAAAACTTCTGGA
AGTGACCTGGGAAGACCCTGACCCTGGGTCACAAAACTC
Too many possible alignments:
O( 2N)

Dynamic Programming
• Given two sequences x = x1……xM and y = y1……yN
• Let F(i, j) = Score of best alignment of x1……xi to y1……yj
• Then, F(M, N) == Score of best alignment
Idea: Compute F(i, j) for all i and j Do this by using F(i–1 , j), F(i, j–1), F(i–1, j–1)

Dynamic Programming (cont’d)
Notice three possible cases:
1. xi aligns to yj
x1……xi-1 xi
y1……yj-1 yj
2. xi aligns to a gap
x1……xi-1 xi
y1……yj -
3. yj aligns to a gap
x1……xi -
y1……yj-1 yj
m, if xi = yj
F(i,j) = F(i-1, j-1) + -s, if not
F(i,j) = F(i-1, j) - d
F(i,j) = F(i, j-1) - d

Dynamic Programming (cont’d)
• How do we know which case is correct?
Inductive assumption:F(i, j-1), F(i-1, j), F(i-1, j-1) are optimal
Then,F(i-1, j-1) + s(xi, yj)
F(i, j) = max F(i-1, j) – dF( i, j-1) – d
Where s(xi, yj) = m, if xi = yj; -s, if not
i-1, j-1 i-1, j
i, j-1 i, j
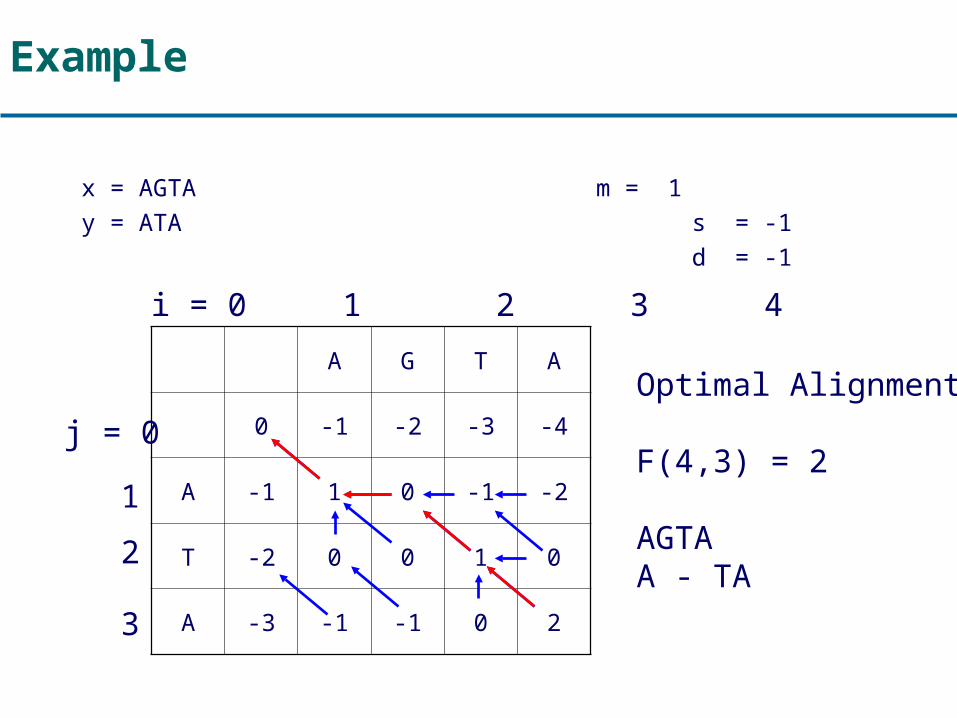
Example
x = AGTA m = 1y = ATA s = -1
d = -1
A G T A
0 -1 -2 -3 -4
A -1 1 0 -1 -2
T -2 0 0 1 0
A -3 -1 -1 0 2
F(i,j) i = 0 1 2 3 4
j = 0
1
2
3
Optimal Alignment:
F(4,3) = 2
AGTAA - TA

The Needleman-Wunsch Algorithm
1. Initialization.a. F(0, 0) = 0b. F(0, j) = - j dc. F(i, 0) = - i d
2. Main Iteration. Filling-in partial alignmentsa. For each i = 1……M
For each j = 1……N F(i-1,j) – d [case 1]
F(i, j) = max F(i, j-1) – d [case 2]
F(i-1, j-1) + s(xi, yj) [case 3]
UP if [case 1]Ptr(i,j) = LEFT if [case 2]
DIAG if [case 3]
3. Termination. F(M, N) is the optimal score, andfrom Ptr(M, N) can trace back optimal alignment

Alignment on a Large Scale
• Given a gene that we care about, how can we compare it to all existing DNA?
• Assume we use Dynamic Programming:
The entire genomic database
gene of interest
~105
~1011

Index-based Local Alignment
Main idea:
1. Construct a dictionary of all the words in the query
2. Initiate a local alignment for each word match between query and DB
Running Time:Theoretical worst case: O(MN)Fast in practice
query
DB
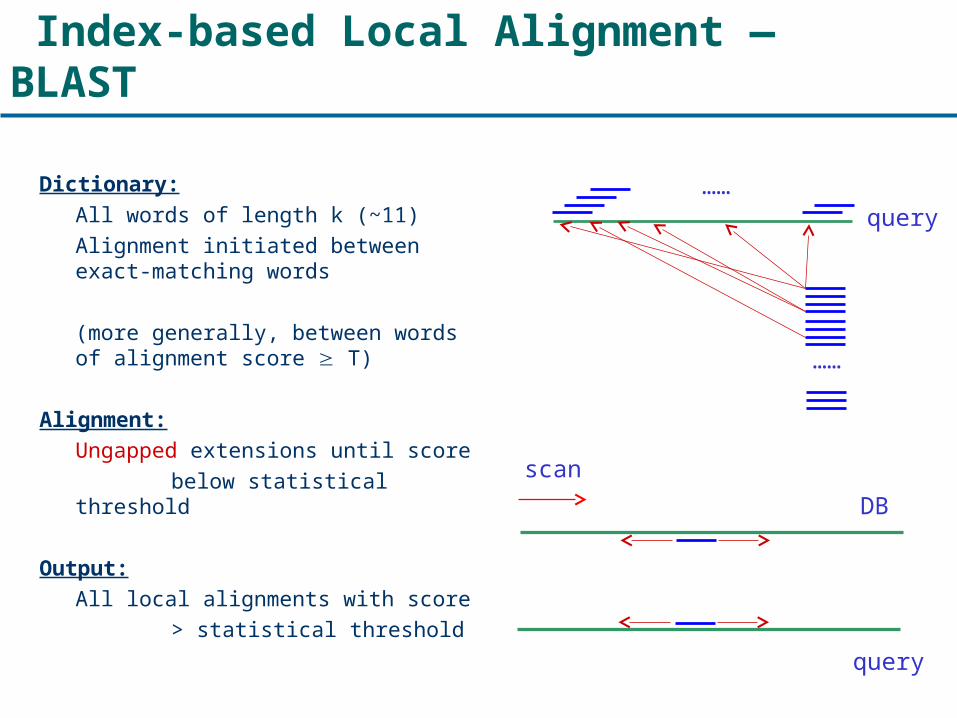
Index-based Local Alignment — BLAST
Dictionary:All words of length k (~11)Alignment initiated between exact-matching words
(more generally, between words of alignment score T)
Alignment:Ungapped extensions until score
below statistical threshold
Output:All local alignments with score
> statistical threshold
……
……
query
DB
query
scan
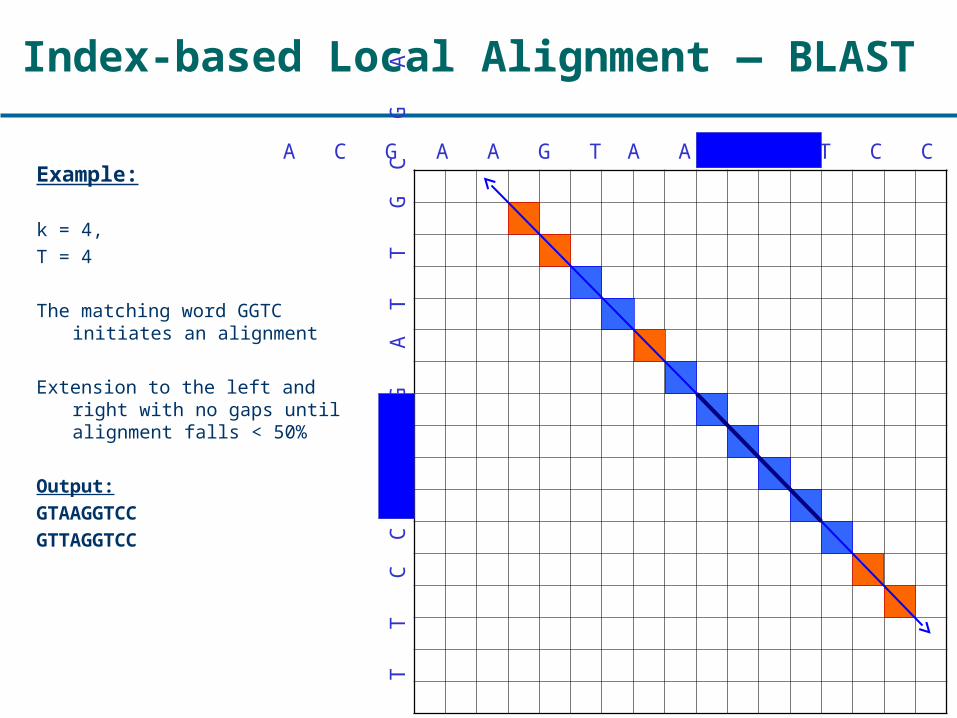
Index-based Local Alignment — BLAST
A C G A A G T A A G G T C C A G T
C
C
C
T
T
C C
T
G
G
A T
T
G
C
G
A
Example:
k = 4,T = 4
The matching word GGTC initiates an alignment
Extension to the left and right with no gaps until alignment falls < 50%
Output:GTAAGGTCC
GTTAGGTCC

Gapped BLAST
A C G A A G T A A G G T C C A G T
C
T
G
A
T
C C
T
G
G
A
T
T
G C
G
A
Added features:
• Pairs of words can initiate alignment
• Nearby alignments are merged
• Extensions with gaps until score < T below best score so far
Output:
GTAAGGTCCAGTGTTAGGTC-AGT

Example
Query: gattacaccccgattacaccccgattaca (29 letters) [2 mins]
Database: All GenBank+EMBL+DDBJ+PDB sequences (but no EST, STS, GSS, or phase 0, 1 or 2 HTGS sequences) 1,726,556 sequences; 8,074,398,388 total letters
>gi|28570323|gb|AC108906.9| Oryza sativa chromosome 3 BAC OSJNBa0087C10 genomic sequence, complete sequence Length = 144487 Score = 34.2 bits (17), Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus
Query: 4 tacaccccgattacaccccga 24 ||||||| |||||||||||||
Sbjct: 125138 tacacccagattacaccccga 125158
Score = 34.2 bits (17),
Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus
Query: 4 tacaccccgattacaccccga 24 ||||||| |||||||||||||
Sbjct: 125104 tacacccagattacaccccga 125124
>gi|28173089|gb|AC104321.7| Oryza sativa chromosome 3 BAC OSJNBa0052F07 genomic sequence, complete sequence Length = 139823 Score = 34.2 bits (17), Expect = 4.5 Identities = 20/21 (95%) Strand = Plus / Plus
Query: 4 tacaccccgattacaccccga 24 ||||||| |||||||||||||
Sbjct: 3891 tacacccagattacaccccga 3911
http://www.ncbi.nlm.nih.gov
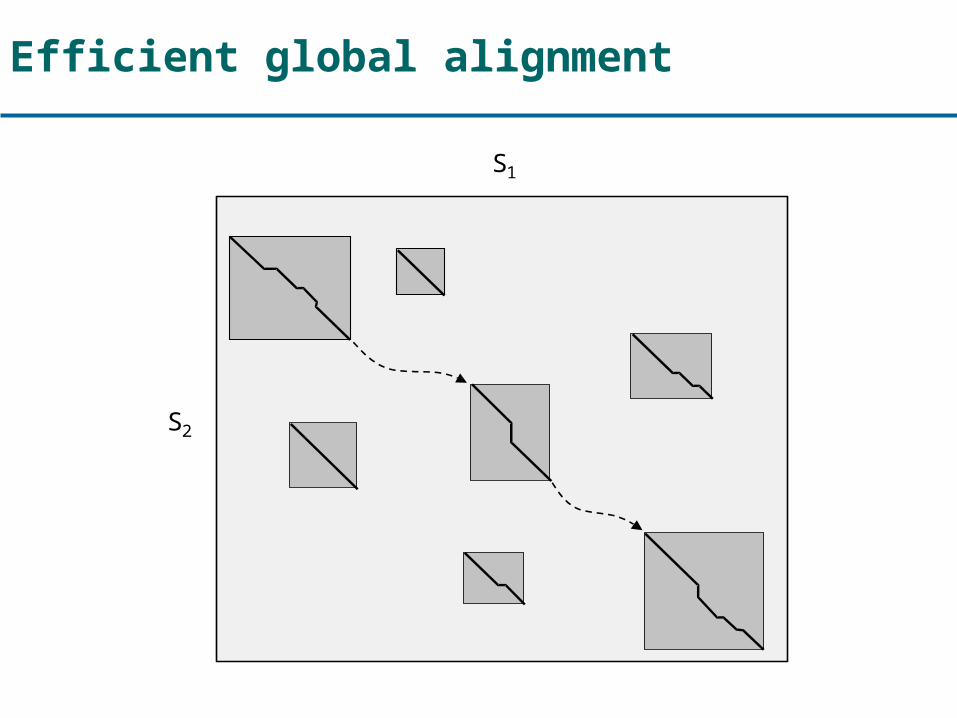
Efficient global alignment
S1
S2

Global alignment with the chaining approach
1. Find local alignments2. Chain them into a rough global map3. Align regions in-between
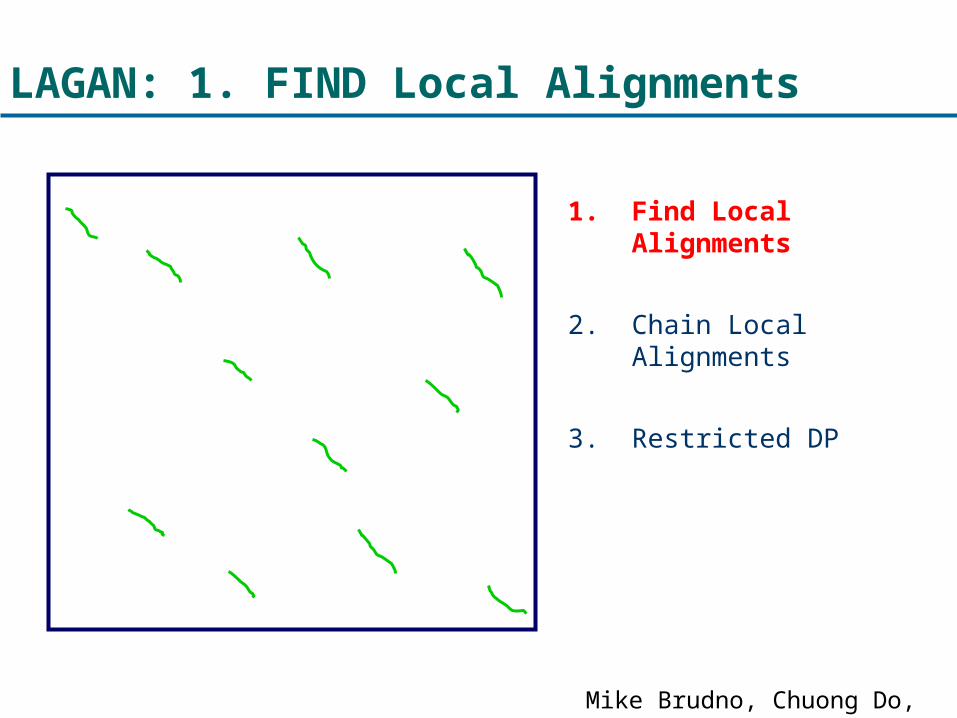
LAGAN: 1. FIND Local Alignments
1. Find Local Alignments
2. Chain Local Alignments
3. Restricted DP
Mike Brudno, Chuong Do, et al.
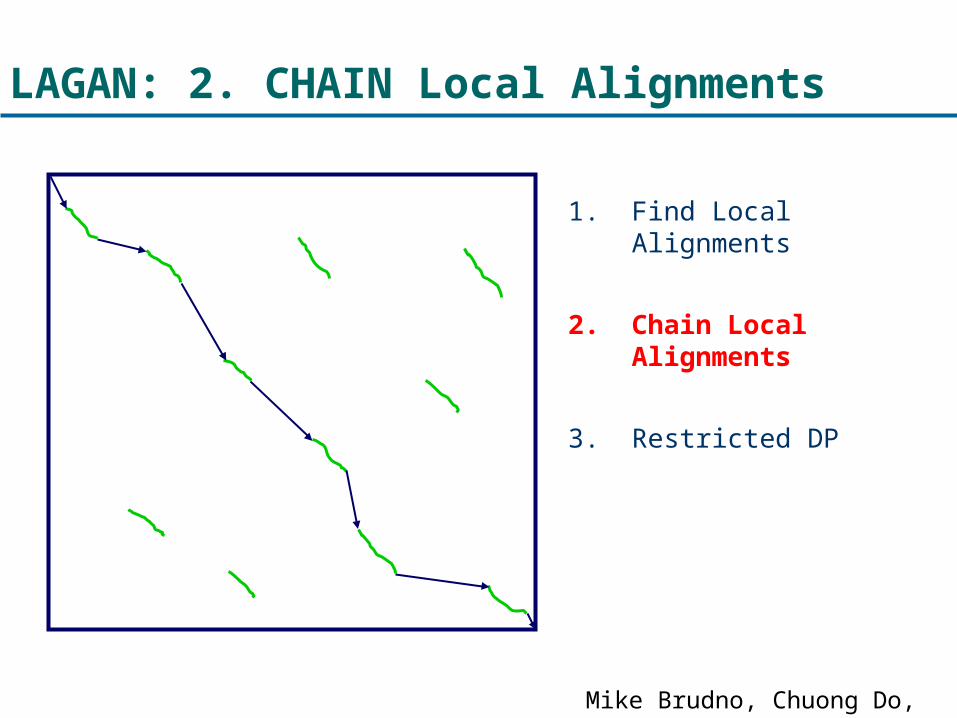
LAGAN: 2. CHAIN Local Alignments
1. Find Local Alignments
2. Chain Local Alignments
3. Restricted DP
Mike Brudno, Chuong Do, et al.
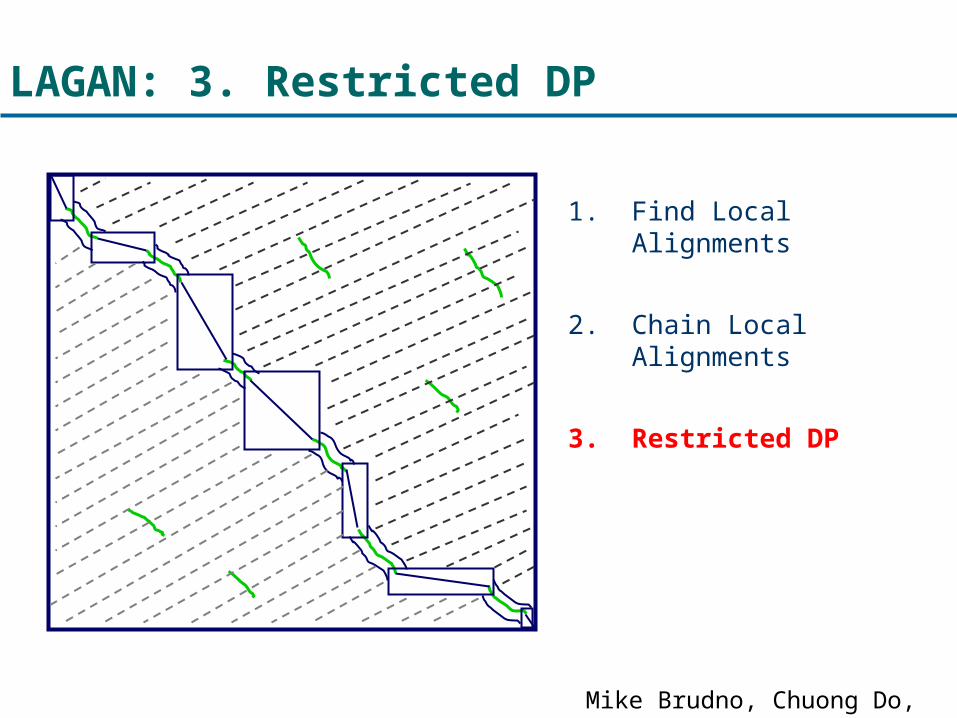
LAGAN: 3. Restricted DP
1. Find Local Alignments
2. Chain Local Alignments
3. Restricted DP
Mike Brudno, Chuong Do, et al.

Multiple Alignment
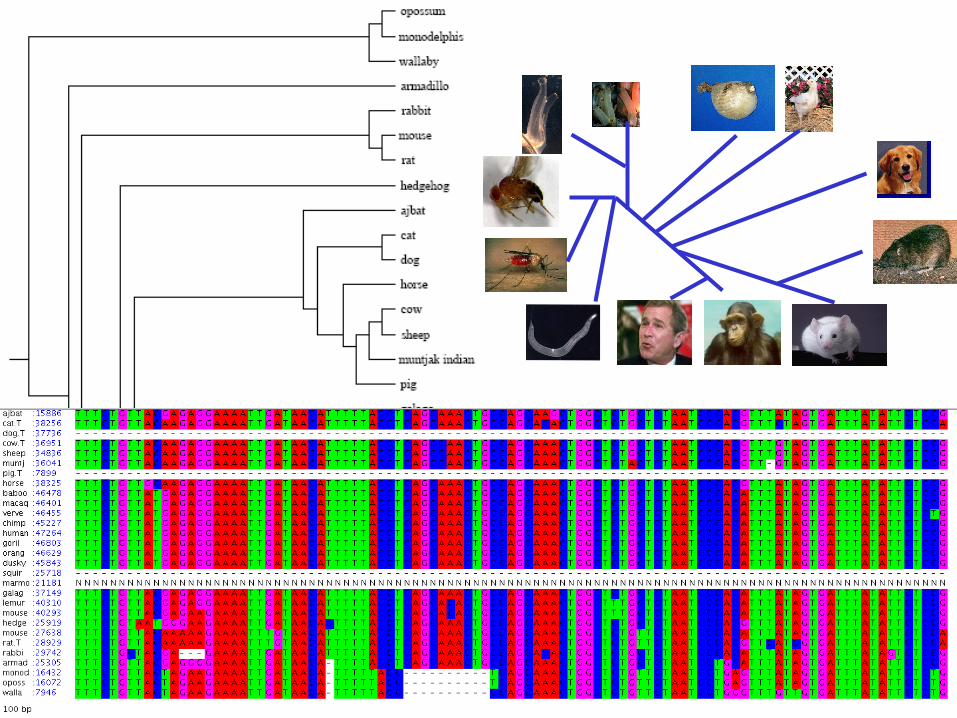

Definition
• Given N sequences x1, x2,…, xN: Insert gaps (-) in each sequence xi, such that
All sequences have the same length L Score of the global map is maximum
• A faint similarity between two sequences becomes significant if present in many
• Multiple alignments can help improve the pairwise alignments

Scoring Function: Sum Of Pairs
Definition: Induced pairwise alignmentA pairwise alignment induced by the multiple alignment
Example:
x: AC-GCGG-C y: AC-GC-GAG z: GCCGC-GAG
Induces:
x: ACGCGG-C; x: AC-GCGG-C; y: AC-GCGAGy: ACGC-GAC; z: GCCGC-GAG; z: GCCGCGAG
Given sequences x1, …, xN, aligned in a multiple alignment m,
S(m) = k<l wkl s(xk, xl)

A Profile Representation
• Given a multiple alignment M = m1…mn
Replace each column mi with profile entry pi Frequency of each letter in # gaps
Can think of this as a “likelihood” of each letter in each position
- A G G C T A T C A C C T G T A G – C T A C C A - - - G C A G – C T A C C A - - - G C A G – C T A T C A C – G G C A G – C T A T C G C – G G
A 1 1 .8 C .6 1 .4 1 .6 .2G 1 .2 .2 .4 1T .2 1 .6 .2- .2 .8 .4 .8 .4

Multiple Sequence Alignments
Algorithms

Generalization of Needleman-Wunsh:
S(m) = i S(mi)
(sum of column scores)
F(i1,i2,…,iN): Optimal alignment up to (i1, …, iN)
F(i1,i2,…,iN) = max(all neighbors of cube)(F(nbr)+S(nbr))
Multidimensional DP

• Example: in 3D (three sequences):
• 7 neighbors/cell
F(i,j,k) = max{ F(i-1,j-1,k-1)+S(xi, xj, xk),F(i-1,j-1,k )+S(xi, xj, - ),F(i-1,j ,k-1)+S(xi, -, xk),F(i-1,j ,k )+S(xi, -, - ),F(i ,j-1,k-1)+S( -, xj, xk),F(i ,j-1,k )+S( -, xj, xk),F(i ,j ,k-1)+S( -, -, xk) }
Multidimensional DP

Running Time:
1. Size of matrix: LN;
Where L = length of each sequence N = number of sequences
2. Neighbors/cell: 2N – 1
Therefore………………………… O(2N LN)
Multidimensional DP

Running Time:
1. Size of matrix: LN;
Where L = length of each sequence N = number of sequences
2. Neighbors/cell: 2N – 1
Therefore………………………… O(2N LN)
Multidimensional DP

Progressive Alignment
• When evolutionary tree is known:
Align closest first, in the order of the tree In each step, align two sequences x, y, or profiles px, py, to generate a new
alignment with associated profile presult
Weighted version: Tree edges have weights, proportional to the divergence in that edge New profile is a weighted average of two old profiles
x
w
y
z
pxy
pzw
pxyzw

Progressive Alignment
• When evolutionary tree is known:
Align closest first, in the order of the tree In each step, align two sequences x, y, or profiles px, py, to generate a new
alignment with associated profile presult
Weighted version: Tree edges have weights, proportional to the divergence in that edge New profile is a weighted average of two old profiles
x
w
y
z
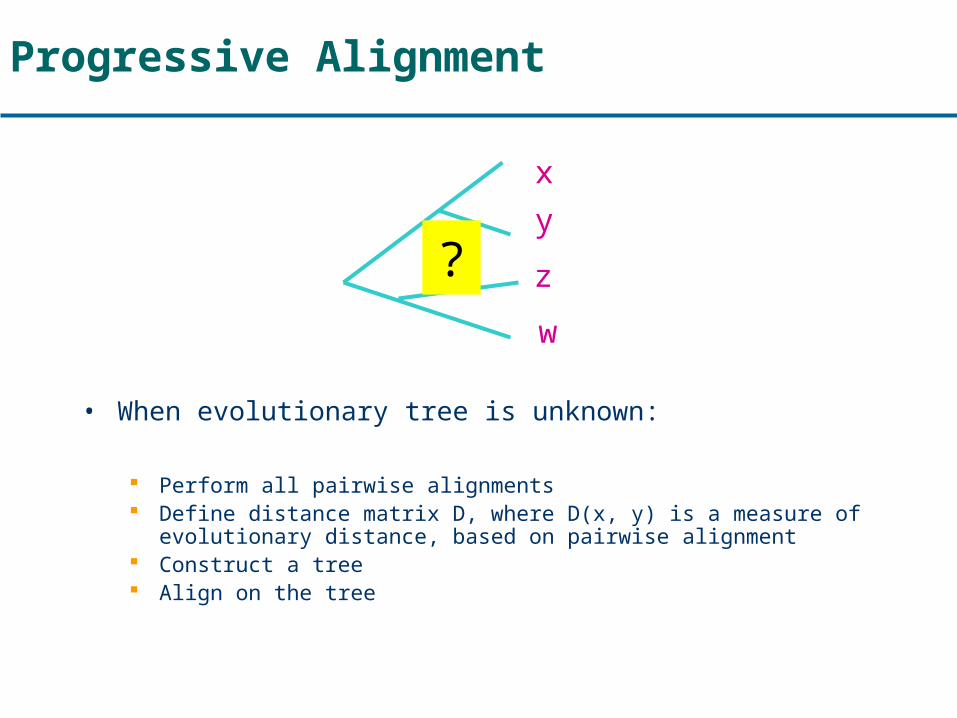
Progressive Alignment
• When evolutionary tree is unknown:
Perform all pairwise alignments Define distance matrix D, where D(x, y) is a measure of
evolutionary distance, based on pairwise alignment Construct a tree Align on the tree
x
w
y
z?

Some useful sites
• Genome browsers Ensembl: www.ensembl.org UCSC: genome.ucsc.edu/cgi-bin/hgGateway
• Genomic alignment LAGAN: lagan.stanford.edu MAVID:
baboon.math.berkeley.edu/mavid• Protein multiple alignment
MUSCLE: www.drive5.com ProbCons: probcons.stanford.edu
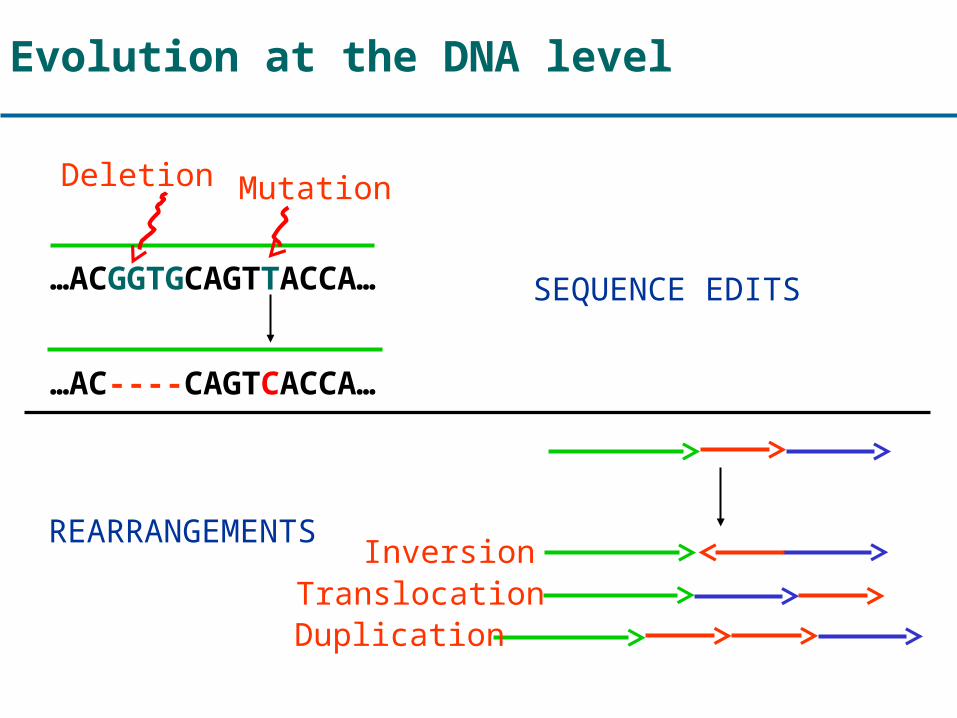
Evolution at the DNA level
…ACGGTGCAGTTACCA…
…AC----CAGTCACCA…
Mutation
SEQUENCE EDITS
REARRANGEMENTS
Deletion
InversionTranslocationDuplication
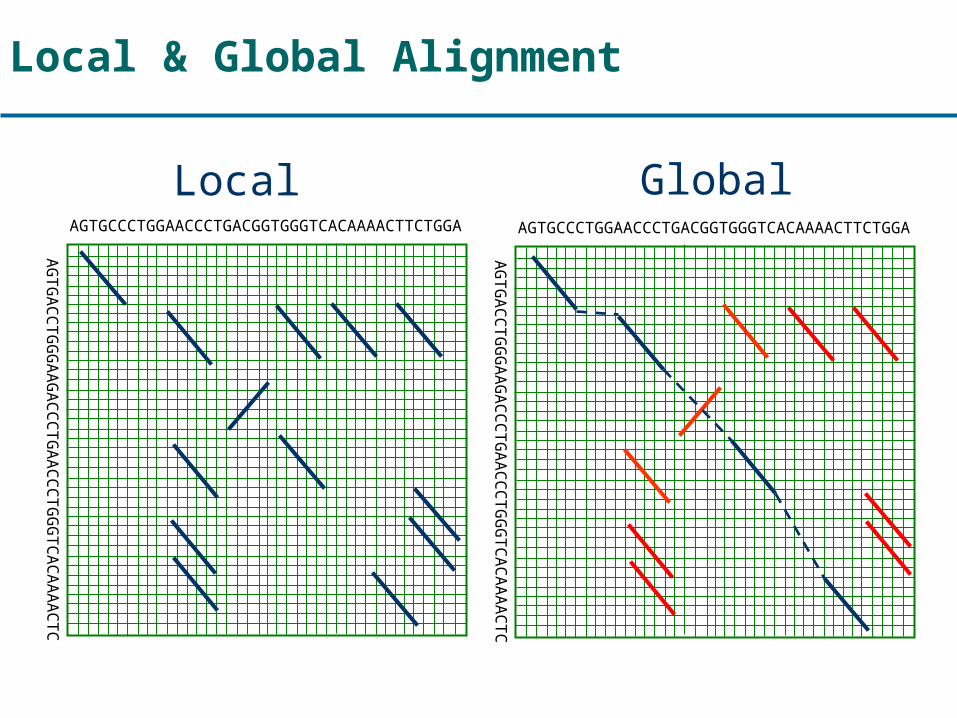
Local & Global Alignment
AGTGCCCTGGAACCCTGACGGTGGGTCACAAAACTTCTGGA
AGTGACCTGGGAAGACCCTGAACCCTGGGTCACAAAACTC
AGTGCCCTGGAACCCTGACGGTGGGTCACAAAACTTCTGGA
AGTGACCTGGGAAGACCCTGAACCCTGGGTCACAAAACTC
Local Global

Glocal Alignment Problem
Find least cost transformation of one sequence into another using shuffle operations
• Sequence edits
• Inversions
• Translocations
• Duplications
• Combinations of above
AGTGCCCTGGAACCCTGACGGTGGGTCACAAAACTTCTGGA
AGTGACCTGGGAAGACCCTGAACCCTGGGTCACAAAACTC
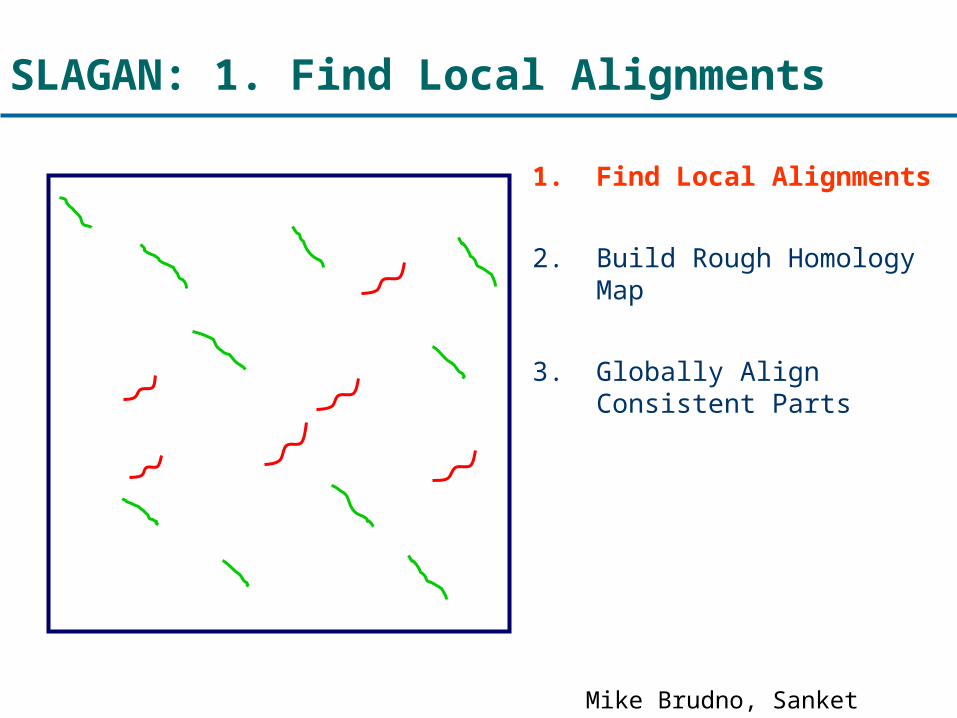
SLAGAN: 1. Find Local Alignments
1. Find Local Alignments
2. Build Rough Homology Map
3. Globally Align Consistent Parts
Mike Brudno, Sanket Malde, et al.
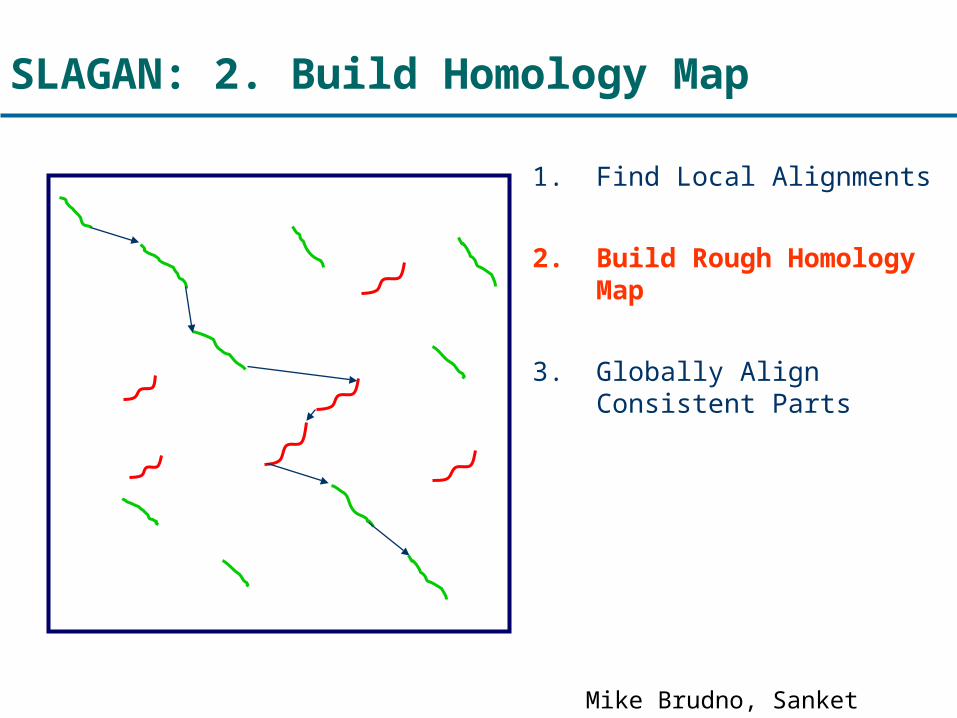
SLAGAN: 2. Build Homology Map
1. Find Local Alignments
2. Build Rough Homology Map
3. Globally Align Consistent Parts
Mike Brudno, Sanket Malde, et al.
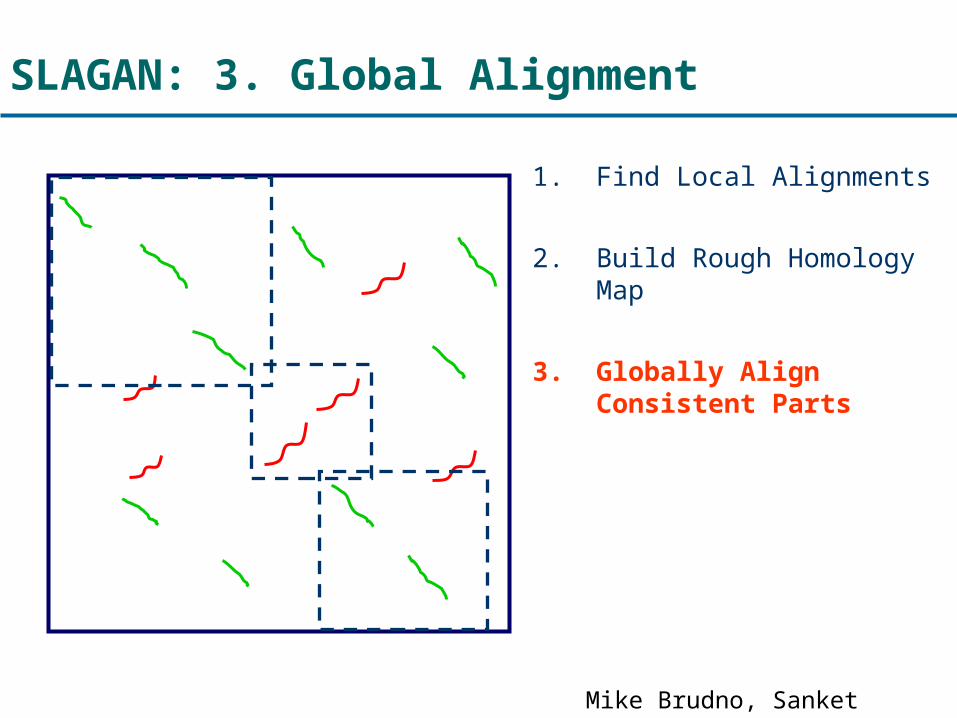
SLAGAN: 3. Global Alignment
1. Find Local Alignments
2. Build Rough Homology Map
3. Globally Align Consistent Parts
Mike Brudno, Sanket Malde, et al.
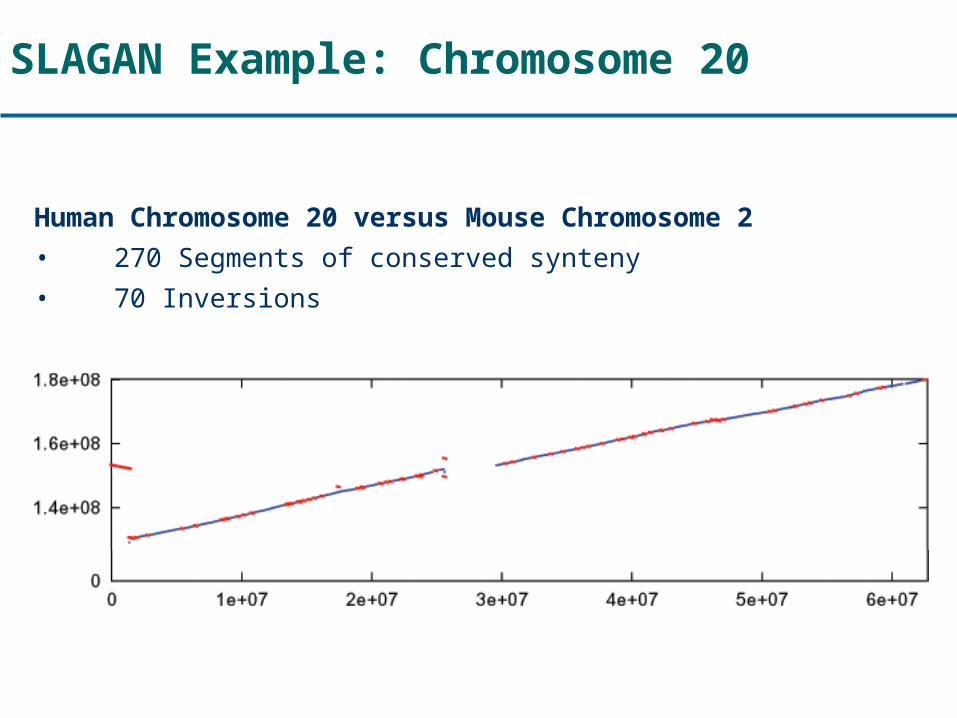
SLAGAN Example: Chromosome 20
Human Chromosome 20 versus Mouse Chromosome 2
• 270 Segments of conserved synteny
• 70 Inversions
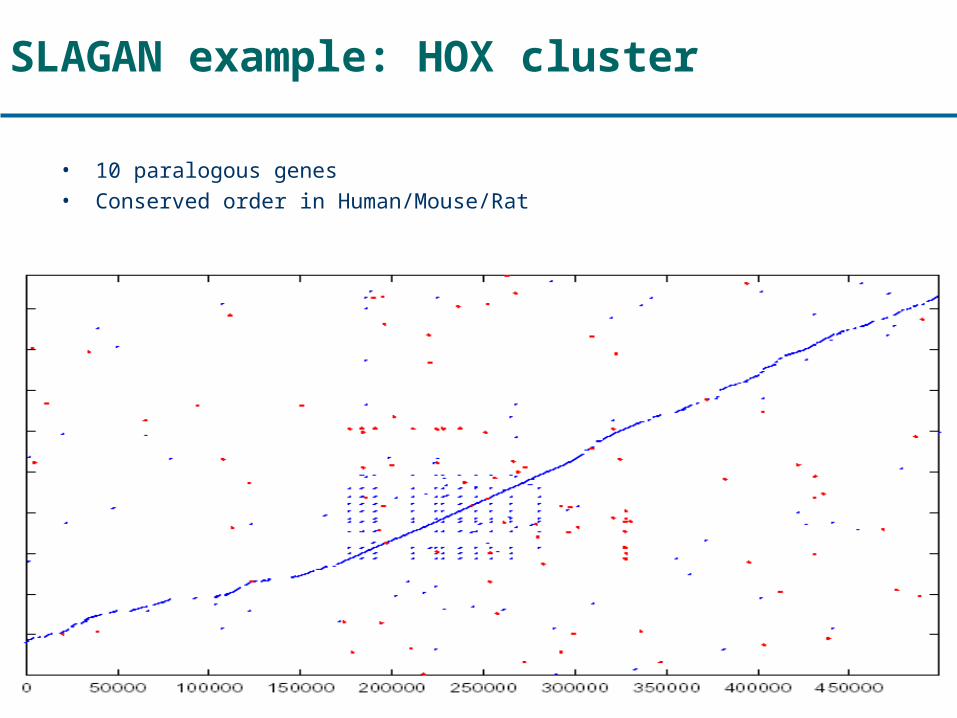
SLAGAN example: HOX cluster
• 10 paralogous genes• Conserved order in Human/Mouse/Rat
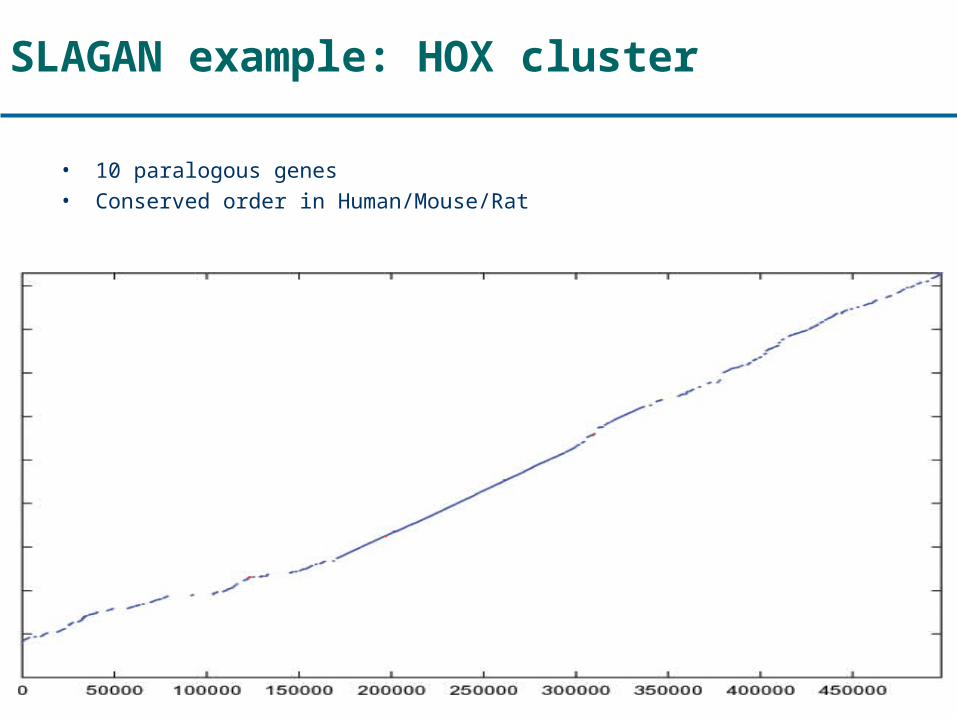
SLAGAN example: HOX cluster
• 10 paralogous genes• Conserved order in Human/Mouse/Rat
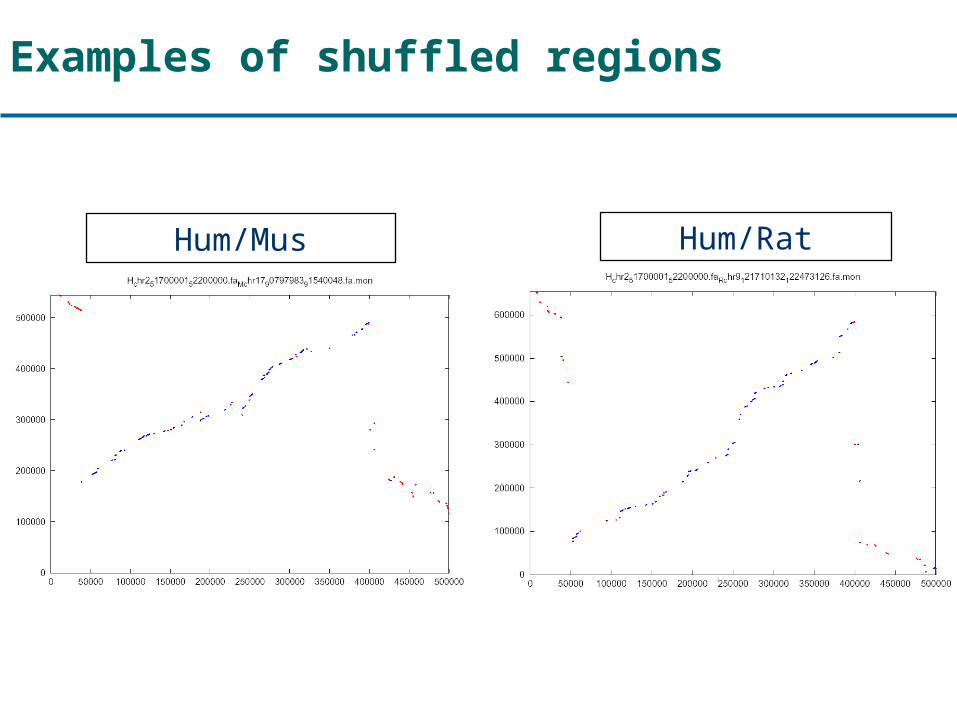
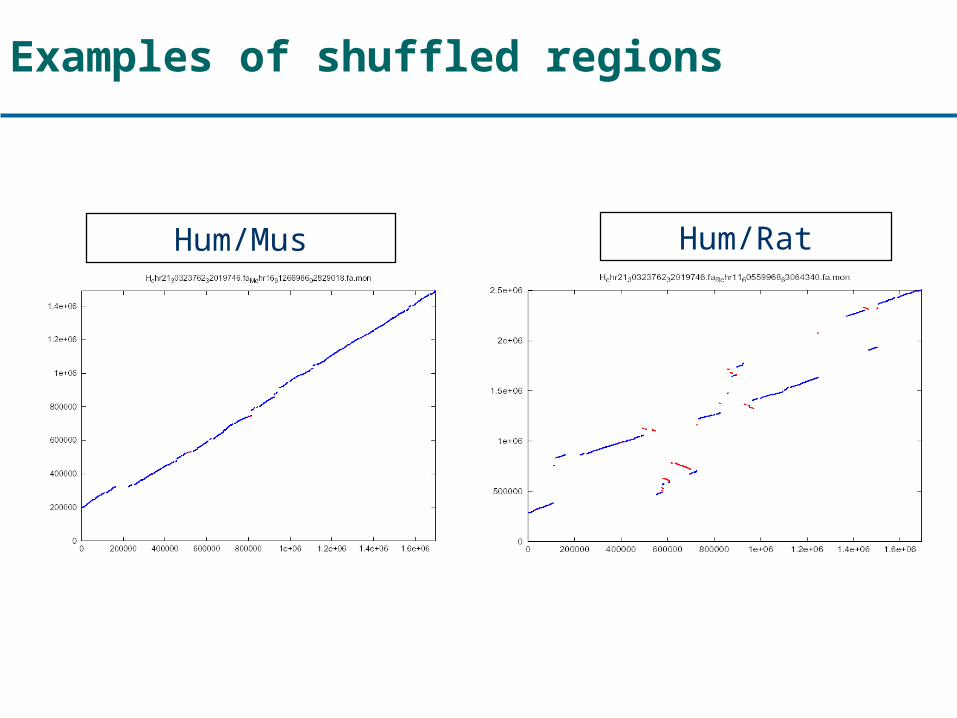
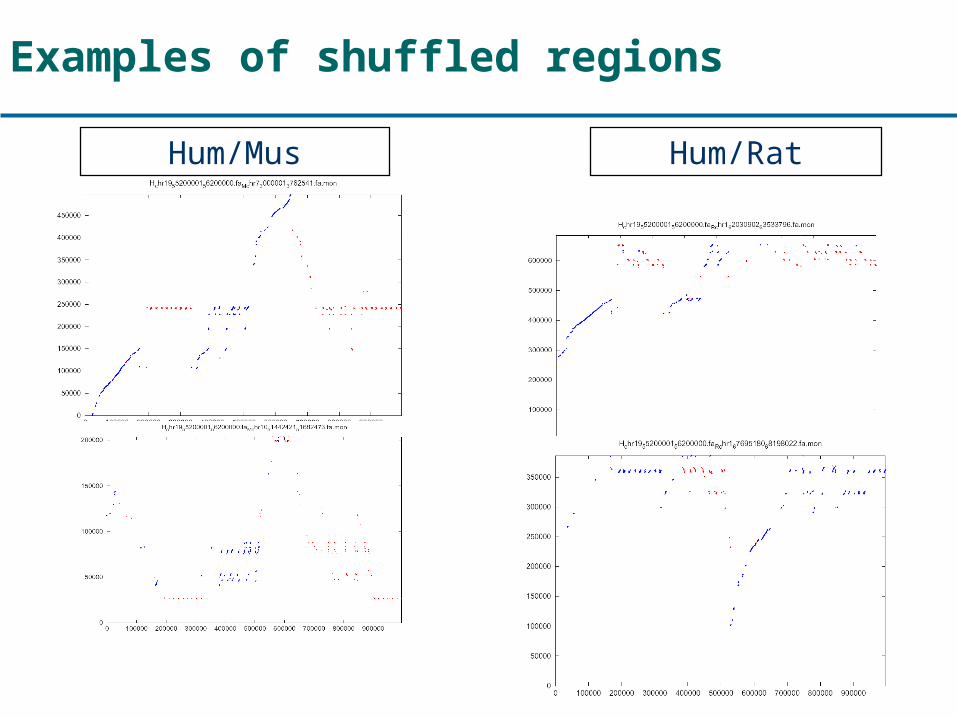
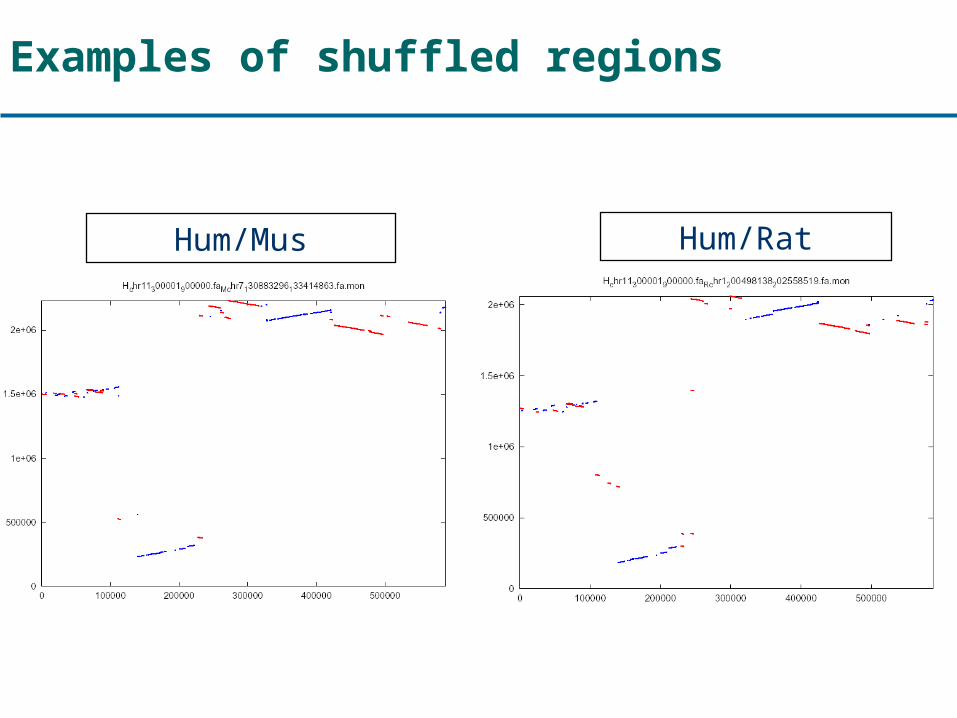
Examples of shuffled regions
Hum/Mus Hum/Rat
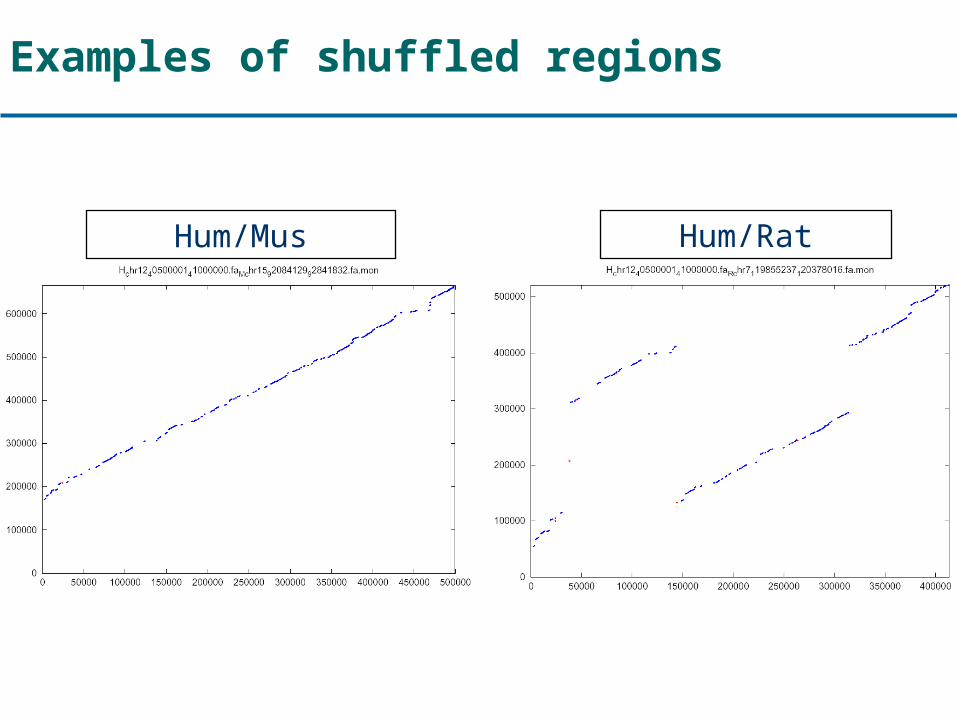
Examples of shuffled regions
Hum/Mus Hum/Rat
Examples of shuffled regions
Hum/Mus Hum/Rat
Examples of shuffled regions
Hum/Mus Hum/Rat
Examples of shuffled regions
Hum/Mus Hum/Rat

More DNA is coming…